WO2012001941A1 - 遺伝性疾患の予防・改善剤 - Google Patents
遺伝性疾患の予防・改善剤 Download PDFInfo
- Publication number
- WO2012001941A1 WO2012001941A1 PCT/JP2011/003655 JP2011003655W WO2012001941A1 WO 2012001941 A1 WO2012001941 A1 WO 2012001941A1 JP 2011003655 W JP2011003655 W JP 2011003655W WO 2012001941 A1 WO2012001941 A1 WO 2012001941A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- exon
- mutation
- dystrophin
- preventive
- gene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4707—Muscular dystrophy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4707—Muscular dystrophy
- C07K14/4708—Duchenne dystrophy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/12—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
Definitions
- the present invention relates to a preventive / ameliorating agent for a hereditary disease caused by a mutation in an exon of a gene and capable of generating a functional truncated protein by skipping an exon containing the mutation, and having a molecular weight of 1500 or less.
- the present invention relates to a preventive / ameliorating agent characterized by containing a compound.
- Muscular dystrophy is a general term for inherited muscular diseases in which muscle atrophy and muscle weakness gradually progress while repeatedly destroying / degenerating muscle fibers (muscle necrosis) and regeneration, and progressive muscular dystrophy is well known.
- Duchenne muscular dystrophy (DMD) a type of progressive muscular dystrophy, is the most common muscular dystrophy and develops due to mutations in the dystrophin gene on the X chromosome (see Non-Patent Document 1). Due to progressive muscle atrophy that begins in childhood, DMD patients are usually in their twenties and die from heart failure or respiratory failure.
- Dystrophin protein (hereinafter, also simply referred to as “dystrophin”) exists inside the cell membrane of muscle cells, and mechanical energy generated by muscle contraction by actin-myosin is transferred to the cell membrane, surrounding connective tissue, and tendons. It plays a role in maintaining the structure of muscle cells by adjusting the balance so that excessive shock is not applied.
- DMD patients due to mutations in the dystrophin gene, there is little or no dystrophin in the muscle fiber, so that muscle cell membranes are broken by muscle contraction, and more calcium ions than normal are It will flow into the fiber. Excessive calcium activates enzymes such as carpain and proteases that break muscles and induce apoptosis. As a result, fibroblasts are activated and fibrosis occurs, and tissue is scarred and muscle cells regenerate. It becomes difficult to be done and muscle atrophy progresses.
- Becker muscular dystrophy which is a type of progressive muscular dystrophy, also develops due to mutations in the dystrophin gene, but the onset is usually adult, and the progression of symptoms is slow compared to DMD. It is. Although both DMD and BMD develop due to mutations in the dystrophin gene, the degree of symptoms and the progression rate are different. This difference between DMD and BMD is explained by reading frame rules.
- dystrophin encoded by this dystrophin mRNA lacking some exons due to skipping is shorter than normal dystrophin, but has some function to maintain the structure of muscle cells, so the symptoms of muscular dystrophy are It is relatively light and the progression rate of muscle atrophy is slow.
- This treatment method uses an antisense oligonucleotide (AON) against dystrophin mRNA to induce exon skipping, thereby converting the DMD phenotype to the BMD phenotype and reducing the symptoms (Non-patent Document 3). ).
- AON antisense oligonucleotide
- Several different AONs have been designed for either splice sites or splicing facilitating elements to induce exon skipping in cells of DMD patients.
- AONs were able to repair the reading frame of dystrophin mRNA.
- exon 19's AON to exon splicing enhancer (ESE) caused exon 19 skipping in the above-mentioned DMD patient cells, and generation of truncated dystrophin was observed (see Non-Patent Documents 3 to 5).
- AON for exon 51 is also frequently used for patient cells, and these AONs are currently in the clinical research stage (see Non-Patent Documents 6 to 8).
- AON has to be regularly injected intramuscularly or intravenously, which is troublesome for the patient, and a large amount of preparation is expensive. Furthermore, when AON treatment was performed on mdx mice, which are muscular dystrophy model mice, dystrophin expression recovered to some extent in skeletal muscle, but recovery of dystrophin expression in the heart was difficult (Non-patent Document 9). Therefore, small molecules that regulate exon skipping are highly desirable clinically.
- PTC124 (registered trademark) (3- [5- (2-fluorophenyl) -1,2,4-oxadiazol-3-yl] benzoic acid), a small molecule non-aminoglycoside nonsense mutation suppressor compound, has a nonsense mutation It has been reported that some DMD patients can be treated (Non-patent Documents 10 and 11), and a phase 2b clinical trial is currently conducted in the United States and the like. PTC124 is a therapeutic agent that restores the expression of full-length functional dystrophin to some extent by inducing ribosomes to read-through a premature stop codon (PTC) during translation. The effect on the nonsense mutation-dependent mRNA degradation mechanism is still unclear.
- TG003 is a compound that affects splicing both in vitro and in vivo (see Non-Patent Documents 12 and 13).
- TG003 is a protein of SR protein. It describes that it has an action of regulating alternative splicing through an oxidation reaction, and that such action can be used to prevent or treat diseases such as cancer.
- TG003 can promote exon skipping of the dystrophin gene.
- the present invention relates to a preventive / ameliorating agent for a hereditary disease caused by a mutation in an exon of a gene and capable of generating a functional truncated protein by skipping an exon containing the mutation, and having a molecular weight of 1500 or less. It is an object of the present invention to provide a preventive / ameliorating agent characterized by containing a compound.
- the present inventors analyzed mutations of the dystrophin gene in 400 or more muscular dystrophy patients.
- a nonsense mutation that is expected to have a severe DMD phenotype is exon 31.
- the patient was found to have BMD type symptoms.
- the mRNA of the dystrophin gene of such patients was analyzed, it was found that the nonsense mutation in exon 31 induced skipping of exon 31, thereby partially producing mature mRNA encoding functional truncated dystrophin. found.
- the present inventors searched for a low molecular weight compound that promotes exon skipping, and TG003, which is a Clk-specific inhibitor, promotes exon 31 and exon 27 skipping of endogenous dystrophin gene in a dose-dependent manner. It was found that the production of functional truncated dystrophin can be enhanced in these cells, and the present invention has been completed.
- the present invention is (1) an agent for preventing / ameliorating a hereditary disease caused by a mutation in an exon of a gene and capable of generating a functional truncated protein by skipping an exon containing the mutation.
- a preventive / improving agent characterized by containing a compound having a molecular weight of 1500 or less, or (2) the preventive / improving method according to (1) above, wherein the compound is a compound having a splicing-modulating action.
- the preventive / ameliorating agent according to (1) or (2) above wherein the compound is a compound having an effect of inducing and promoting exon skipping containing a mutation
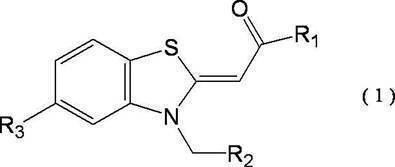
- the Cd -like kinase inhibiting compound is represented by the general formula (1) [Wherein, R 1 and R 2 each independently represent a linear or branched C 1 -C 10 hydrocarbon group; R 3 represents a methoxy group, an ethoxy group, an acetoxy group or a halogen atom.
- the preventive / ameliorating agent according to (4) above which is a compound represented by (6) above, or (6) the above mutation (1) to (5), wherein the mutation is a nonsense mutation
- the preventive / ameliorating agent according to any one of the above, and (7) the nonsense mutation is a nonsense mutation that suppresses exon splicing enhancer activity in the gene and / or increases exon splicing silencer activity in the gene.
- the gene is a dystrophin gene
- the functional truncated protein is a functional truncated dystrophin protein
- the genetic disease is Duchenne
- the 31 mutation is a nonsense mutation of guanine to thymine at nucleotide number 4303 of the polynucleotide sequence of SEQ ID NO: 1, and the mutation of exon 27 lacks the guanine at nucleotide number 3613 of the polynucleotide sequence of SEQ ID NO: 1
- the expression of a functional truncated protein is induced / increased by inducing / promoting exon skipping of a gene containing a mutation causing a genetic disease, and as a result, the genetic disease Can be prevented and improved.
- the compound having a molecular weight of 1500 or less used in the present invention is a low molecule and excellent in tissue transferability, etc., even in organs such as the heart that could not obtain a sufficient effect by conventional AON that is a polymer, Sufficient prevention and improvement effects can be expected.
- the mutations targeted by the present invention are not limited to nonsense mutations, application to various mutations causing genetic diseases can be expected.
- FIG. 1 It is a figure which shows a mode that an exon skipping arises by the point mutation in the exon 31 of a patient's dystrophin gene, and the open reading frame (ORF) of a truncated dystrophin gene is restored.
- A Point mutation found in the dystrophin gene of patient number KUCG797. Exon 31 c. The position of the 4303G> T (p.Glu1435X) mutation is indicated by a bar. The DNA sequence of the adjacent intron region is shown, as well as the codons changed by mutation.
- Panels e to g show the results of examining dystrophin expression in the patient by immunohistochemistry
- panels b to d show the results of examining dystrophin expression in healthy subjects by immunohistochemistry.
- Panels e and b show the results of staining with DYS2
- panels f and c show the results of staining with DYS3
- panels g and d show the results of staining with MANDYS1.
- Scale bar 60 ⁇ M.
- C shows the results of agarose gel analysis of RT-PCR products obtained from control and patient muscles.
- the left lane (M) represents the result using a DNA size marker digested with ⁇ X174-HaeIII
- the center lane (Control) represents the result of templating a total RNA derived from healthy subjects
- the right lane (Patient) is The result which used the total RNA derived from KUCG797 as a template is represented.
- (F) shows the results of electrophoresis on agarose gel of mutant mRNA recovered from H492-dys Ex31m / Hela and RT-PCR product of wild-type mRNA recovered from H492-dys Ex31w / Hela.
- the leftmost lane (M) shows the results using the DNA size marker digested with ⁇ X174-HaeIII, and the second lane (w) from the left is the total RNA derived from H492-dys Ex31w / Hela as a template.
- the third lane (m) from the left represents the result using the total RNA derived from H492-dys Ex31m / Hela as a template.
- the structure of these PCR products is shown schematically on the right side of the panel.
- the nucleotide lengths of the DNA size marker and the PCR product are shown on the left and right sides of the panel, respectively.
- the second and third lanes from the right represent the results of electrophoresis on an agarose gel after RT-PCR treatment except that reverse transcriptase (RT) was not added.
- the second lane from the right Lane (w) represents the result using the total RNA derived from H492-dys Ex31w / Hela as the template
- the rightmost lane (m) represents the result using the total RNA derived from H492-dys Ex31m / Hela as the template.
- FIG. 3 is a diagram predicting a binding sequence in exon 31 for an RNA binding protein that regulates splicing.
- SRp30c / SRSF9 and hnRNPA1 Two types of proteins (SRp30c / SRSF9 and hnRNPA1) whose scores greatly change depending on the presence or absence of such mutations are indicated by open boxes. Peripheral sequences showing high homology with the SELEX consensus sequence of SRp30c / SRSF9 (left panel) or hnRNPA1 (right panel) are underlined.
- B Alignment of the SELEX consensus sequence of SRp30c / SRSF9 or hnRNPA1 and the exon 31 sequence is shown. The left panel shows the homology between wild type exon 31 RNA and the SRp30c / SRSF9SELEX consensus sequence. R represents a purine residue. The right panel shows c.
- W represents A or U.
- the vertical bars in both panels connect the same residues.
- c Nucleotides mutated with 4303G> T are highlighted in open boxes. It is a figure which shows a mode that the binding property to hnRNPA1 increases in both in vitro and in vivo by the point mutation of dystrophin exon 31 found in a patient gene, and exon skipping increases.
- A It is a figure which shows the result of the gel mobility shift assay with respect to GST-hnRNPA1 and dystrophin exon 31RNA.
- Lanes 3 to 5 show the results using GST-hnRNPA1 of 100, 200 and 400 ng and dystrophin wild type exon 31 RNA, respectively, and lanes 8 to 10 use GST-hnRNPA1 of 100, 200 and 400 ng, respectively.
- the results using the dystrophin mutant exon 31 RNA are shown, and lanes 2 and 7 show the results using only GST.
- Lanes 1 and 6 show where the RNA itself has moved on the gel (shown as “Free RNA” on the right side of the panel).
- the complex of hnRNPA1 and RNA is indicated as “Bound RNA”.
- (B) shows the results of an in vitro splicing assay of an mRNA precursor containing dystrophin exon 31.
- Lanes 1 to 5 show results obtained by using linearized pCDC-dys Ex31w as a template for in vitro transcription and incubation times of 0, 15, 30, 60, and 90 minutes, respectively.
- PCDC-dys Ex31m linearized was used as a template for in vitro transcription, and the incubation times were 0, 15, 30, 60, and 90 minutes, respectively.
- the mRNA precursor and the structure of two different mRNAs are shown on the right side of the panel. Numbered boxes represent exons, and lines between boxes represent introns. Exons 14 and 15 are chicken ⁇ -crystallin exons derived from the CDC mRNA precursor. MRNA containing dystrophin exon 31 (black circle) is CDC-dys Ex31 c.
- the leftmost lane (M) represents a DNA size marker digested with ⁇ X174-HaeIII, and the second lane (mock) from the left represents the result of using total RNA derived from H492-dys Ex31m / Hela as a template.
- the third lane from the left represents the result using total RNA derived from H492-dys Ex31m • Flag-SRp30c / Hela as a template
- the second lane from the right is H492.
- the rightmost lane shows the total RNA derived from H492-dys Ex31m • Flag-hnRNPA1 plasmid / Hela. Representing the results of the plate.
- the nucleotide lengths of the DNA size marker and PCR product are shown on the left and right sides of the panel, respectively.
- the leftmost lane (M) represents a DNA size marker digested with ⁇ X174-HaeIII
- the second lane from the left (DMSO) represents the result when H492-dys Ex31m / Hela was treated with DMSO.
- the third lane from TG003 shows the result when H492-dys Ex31m / Hela is processed with TG003
- the rightmost lane (SRPIN340) shows the result when H492-dys Ex31m / Hela is processed with SRPIN340.
- the nucleotide lengths of the DNA size marker and PCR product are shown on the left and right sides of the panel, respectively.
- the graph shows the average and standard deviation of the ratios of experiments performed three times individually. * P ⁇ 0.0001 (c)
- H492-dys Ex31m / Hela in order to investigate whether TG003 that promotes skipping of mutant exon 31 promotes skipping of wild-type exon 31, H492-dys Ex31w
- the result of the splicing analysis in the cell performed using / Hela is shown.
- the first to sixth lanes from the left represent the results when H492-dys Ex31w / Hela was treated with TG003 at the concentration ( ⁇ M) at the top of the panel, and the first to sixth lanes from the right represent H492-dys.
- the first to sixth lanes from the left represent the results when H492-dys Ex27w / Hela was treated with TG003 at the concentration ( ⁇ M) at the top of the panel, and the first to sixth lanes from the right represent H492-dys.
- the result when Ex27m / Hela was treated with TG003 at the concentration ( ⁇ M) at the top of the panel is shown, and the middle lane (M) shows the DNA size marker digested with ⁇ X174-HaeIII.
- B Exon skip / inclusion ratio calculated from the results of FIG. The graph shows the average and standard deviation of the ratios of experiments performed three times individually.
- TG003 not only promotes the skipping of mutant exon 31, but also promotes the expression of truncated dystrophin (exon 31 deleted dystrophin) in patient cells.
- A The result of the splicing analysis in the cell performed using the primary culture cell of the patient muscle treated with various amounts of TG003 is shown.
- the first lane (M) from the left represents a DNA size marker digested with ⁇ X174-HaeIII, and the second to seventh lanes from the left represent the primary cultured cells with the concentration ( ⁇ M) indicated in the upper part of the panel.
- the results when processing with TG003 are shown.
- the DNA size marker is shown on the left side of the panel, and the nucleotide length and structure of the PCR product are outlined on the right side of the panel.
- the number box on the right side of the panel shows the exon number of dystrophin.
- (B) Exon skip / inclusion ratio calculated from the results of FIG. The graph shows the average and standard deviation of the ratios of experiments performed twice individually.
- (C) shows the results of Western blotting analysis of dystrophin expression in patient cells treated with TG003.
- the upper panel shows the results using an antibody against the C-terminus of dystrophin
- the middle panel (Dystrophin (Exon 31/32)) uses an antibody against the exon 31 part of dystrophin.
- the lower panel represents the results using an anti-desmin antibody.
- the left lane represents the result of positive control, the middle lane (0) represents the result using patient cells not treated with TG003, and the right lane (7) represents TG003. Results are shown using treated patient cells.
- the results in the lower panel indicated that almost the same number of cells were used for this Western blot analysis.
- FIG. 5 shows the 79 exon configuration of human dystrophin. It can also be read from this figure how many characters of the codon correspond to the boundary between exons.
- the preventive / ameliorating agent of the present invention causes an exon resulting from a mutation in an exon of a gene and containing the mutation (hereinafter also simply referred to as “mutant exon”) to be skipped.
- a preventive / improving agent for a hereditary disease capable of producing a functional truncated protein (hereinafter also referred to as “hereditary disease targeted by the present invention”) having a molecular weight of 1500 or less (hereinafter “ The compound is also referred to as “compound in the present invention.”), But is not particularly limited, but is preferably a compound having a molecular weight of 1000 or less, more preferably a compound having a molecular weight of 700 or less, and a compound having a molecular weight of 500 or less. More preferably, it is a compound having a molecular weight of 300 or less.
- two or more kinds of the compounds in the present invention may be used in combination in the preventive / ameliorating agent of the present invention.
- the compound in the present invention is not limited as long as it is a compound that exhibits a preventive effect and / or an improved effect (hereinafter also referred to as “a preventive / improved effect in the present invention”) for a genetic disease targeted by the present invention.
- a preventive / improved effect in the present invention a compound having a splicing regulating action can be exemplified, and more preferably, an effect of inducing and / or promoting the skipping of a mutant exon (hereinafter referred to as “skip induction / induction in the present invention”).
- the compound having a “promoting effect” can also be exemplified, and more preferably, the skipping of the mutant exon is induced and / or promoted to induce and / or increase the expression of the functional truncated protein. Effect (hereinafter referred to as “the expression inducing and increasing effect of the functional truncated protein in the present invention”).
- the above general formula (1) [wherein R 1 and R 2 each independently represents a linear or branched C 1 -C 10 hydrocarbon group; R 3 represents methoxy A group, an ethoxy group, an acetoxy group or a halogen atom;
- the compound represented by formula ( II ) is particularly preferred, and among them, TG003 (a compound in which R 1 and R 2 are methyl groups and R 3 is a methoxy group in the general formula (1)) is particularly preferred.
- TG003 a compound in which R 1 and R 2 are methyl groups and R 3 is a methoxy group in the general formula (1)
- the compound represented by the general formula (1) can be synthesized by the method described in Patent Document 1 described above.
- the preventive effect against a genetic disease targeted by the present invention includes the effect of suppressing the onset of the genetic disease and the effect of delaying the onset
- the improvement effect on a genetic disease targeted by the present invention is the preventive / ameliorating agent of the present invention in addition to the effect of improving the symptoms of the genetic disease. Including the effect of delaying the rate of worsening of symptoms compared to the case of not administering.
- Whether a certain compound has the preventive / ameliorating effect of the present invention is, for example, a model mammal of a genetic disease targeted by the present invention (that is, a genetic disease targeted by the present invention).
- the test compound can be confirmed by a method comprising the step D of evaluating the test compound as a compound having the preventive / ameliorating effect of the present invention.
- a certain compound has the effect of inducing or promoting skipping in the present invention is determined, for example, by a DNA fragment (minigene) containing a mutant exon and a region adjacent to the exon (exon region or intron region)
- a suitable expression vector for mammalian cells to produce a recombinant vector; a step of transfecting such a recombinant vector into a mammalian cell; a transformed cell obtained by transfection (hereinafter referred to as “in the present invention”).
- test compound in contact with the test compound the step of culturing the transformed cell: isolating RNA from the cultured transformed cell and responding to the minigene by RT-PCR Amplifying the RNA to be processed; by analyzing the amplified product (eg electrophoresis or sequencing) A step of confirming whether or not skipping of the mutant exon is induced and / or promoted; when skipping of the mutant exon is induced and / or promoted, the test compound is induced and / or promoted by skipping in the present invention. It can be confirmed by a method for determining a compound having a skipping induction / promoting effect in the present invention, which comprises a step of evaluating a compound having an effect.
- a certain compound has an effect of inducing or increasing the expression of a truncated protein is determined by culturing the transformed cell in a state where the transformed cell in the present invention is in contact with the test compound.
- signals to peptides encoded by mutant exons are detected in Western blotting analysis using antibodies against peptides encoded by mutant exons and antibodies encoded by other exons. It can be performed by confirming that a signal for a peptide encoded by other exons is not detected.
- whether or not the truncated protein is a functional truncated protein can be determined by the expression of the truncated protein in the model mammal of the genetic disease targeted by the present invention. It can be confirmed using improvement as an index.
- the “functional truncated protein” in the present invention is a truncated protein encoded by a mature mRNA from which at least one exon of a gene causing a hereditary disease has been deleted, and the gene contains It means a protein in which the function of a corresponding full-length protein of a normal gene (particularly, a function related to a genetic disease) remains at least to some extent.
- a certain compound is a Clk inhibitory compound is determined by measuring the phosphorylation activity of Clk in the presence of the test compound; Step of comparing with the value of the phosphorylation activity of Clk in the absence of the test; When the value of the phosphorylation activity of Clk in the presence of the test compound is lower than the value of the phosphorylation activity of Clk in the absence of the test compound, it can be easily confirmed by a method comprising the step of evaluating the test compound as a Clk inhibitory compound.
- the above-mentioned Clk is obtained by isolating the Clk gene from a desired mammal based on the known sequence information of Clk, integrating the gene into an appropriate expression vector, and then expressing the Clk.
- the phosphorylation activity of Clk can be measured using an antibody or the like that specifically binds to phosphorylated SR protein in which the SR protein that is a substrate of Clk is phosphorylated.
- the mammal from which the Clk gene is derived is not particularly limited, but humans, monkeys, mice, rats, hamsters, guinea pigs, cows, pigs, horses, rabbits, sheep, goats, cats, dogs and the like are preferable. Among them, humans can be more preferably exemplified.
- the type of “mutation” in the genetic disease targeted by the present invention is not particularly limited as long as it can generate a functional truncated protein by skipping an exon containing the mutation, in addition to nonsense mutation and splicing abnormality.
- a mutation that shifts the reading frame of amino acids compared to a normal wild type gene can be exemplified, among which a nonsense mutation and an out-of-frame mutation can be preferably exemplified.
- More preferred examples include nonsense mutations and out-of-frame mutations that suppress exon splicing enhancer activity in a gene causing the hereditary disease and / or increase exon splicing silencer activity in the gene.
- nonsense mutations and outs Can be particularly preferably exemplified nonsense or out-of-frame mutation as skipping of exon contains of-frame mutation is induced and promote.
- out-of-frame mutations include mutations that are gene deletions, duplications, or inversions, and that shift the reading frame of amino acids compared to normal wild-type genes.
- exon containing the mutation” to be skipped in the present invention is not limited to one exon containing such a mutation, but a plurality of adjacent exons including the exon (preferably 2 to 8, more preferably May be 2 to 5, more preferably 2 to 3, more preferably 2) exons, and the preferred number and range of such exons is such that when the exons are skipped, the remaining exons Those skilled in the art can select as appropriate by using as an index that the truncated protein encoded by the mature mRNA composed of the remaining exons is a functional truncated protein. it can. For example, when the gene is a dystrophin gene, the number and range of exons to be skipped can be determined based on known exon information (FIG.
- exon 51 if an out-of-frame mutation is present in exon 51, if exons 51 and 52 are skipped or exons 50 and 51 are skipped, the remaining exons are in-frame, and out-of-frame mutation Exists in the exon 53, the exons 53 to 58 are skipped, or the exons 52 and 53 are skipped, and the remaining exons are in-frame.
- exon skipping or “exon skipping” means that an exon disappears from an mRNA precursor when it is processed from a mRNA transcribed from a gene (mRNA precursor) into a mature mRNA. Or to eliminate the exon.
- hereditary diseases targeted by the present invention are hereditary diseases caused by mutations in exons of genes, and exons containing the mutations are skipped to produce functional truncated proteins.
- Duchenne muscular dystrophy whose exon of dystrophin gene is exon 31 or exon 27 of dystrophin gene can be illustrated more preferably.
- mutation in exon 31 of dystrophin gene which is a nonsense mutation of guanine to thymine at nucleotide number 4303 of the polynucleotide sequence of SEQ ID NO: 1 (dystrophin cDNA sequence), or a mutation in exon 27 of the dystrophin gene is a nucleotide of the polynucleotide sequence of SEQ ID NO: 1.
- a Duchenne muscular dystrophy that is an out-of-frame mutation in which the guanine in No. 3613 is deleted can be particularly preferably exemplified.
- the mammal to be administered with the preventive / ameliorating agent of the present invention is not particularly limited, but humans, monkeys, mice, rats, hamsters, guinea pigs, cattle, pigs, horses, rabbits, sheep, goats, cats, dogs, etc. Can be preferably exemplified, and among these, humans can be more suitably exemplified.
- the origin of Clk in which the Clk inhibitory compound exhibits an inhibitory action depends on the type of mammal to be administered with the preventive / ameliorating agent of the present invention It is preferable that they coincide with each other because more prevention and improvement effects of the present invention can be enjoyed.
- the transformed cell is brought into contact with the aforementioned transformed cell of the present invention and the compound of the present invention (30 ⁇ M).
- Step of culturing Isolating RNA from cultured transformed cells, and amplifying RNA corresponding to the minigene by RT-PCR: Analyzing the amplified product (for example, electrophoresis or sequencing)
- the step of calculating the ratio of the amplified product skipped by the mutant exon to the amplified product not skipped The exon skip / inclusion ratio when the exon skip / inclusion ratio calculated by the compound of the present invention is not used More than twice the ratio is preferred Three times or more, more preferably 4 times or more, more preferably can be preferably exemplified that has risen to more than 5 times.
- the effect of inducing the exon skip / inclusion ratio to a positive value larger than 0 is the skipping induction / promotion in the present invention. It can be illustrated as a particularly preferred degree of effect.
- the preventive / ameliorating agent of the present invention may contain optional components such as other hereditary disease preventive / ameliorating agents in addition to the aforementioned compound of the present invention. .
- the compound in the present invention contained in the preventive / ameliorating agent of the present invention can be made into an appropriate preparation by a conventional method.
- the dosage form of the preparation may be a solid preparation such as a powder or a granule, but from the viewpoint of obtaining the superior preventive / improving effect of the present invention, it is a liquid such as a solution, emulsion, suspension or the like. It is preferable.
- a method of mixing the compound in the present invention with a solvent, and a method of further mixing a suspending agent and an emulsifier can be preferably exemplified.
- an appropriate pharmaceutically acceptable carrier for example, an excipient, a binder, a solvent, a solubilizing agent, Suspending agent, emulsifying agent, tonicity agent, buffering agent, stabilizing agent, soothing agent, preservative, antioxidant, coloring agent, lubricant, disintegrant, wetting agent, adsorbent, sweetener, dilution
- an optional component such as an agent can be blended.
- the amount of the compound in the present invention contained in the preventive / ameliorating agent of the present invention is not particularly limited as long as the preventive / ameliorating effect of the present invention is obtained, but for example, with respect to the total amount of the preventive / ameliorating agent of the present invention.
- 0.0001 to 99.9999% by mass preferably 0.001 to 80% by mass, more preferably 0.001 to 50% by mass, and still more preferably 0.005 to 20% by mass can be exemplified. .
- the method for administering the preventive / ameliorating agent of the present invention is not particularly limited as long as the preventive / ameliorating effect of the present invention is obtained, and is intravenous, oral, intramuscular, subcutaneous, transdermal, or nasal. Examples include transpulmonary administration.
- the dose of the preventive / ameliorating agent of the present invention can be appropriately adjusted according to the condition of the genetic disease to be administered, the body weight of the subject to be administered, etc., but one adult per person in terms of the compound in the present invention. For example, 0.1 ⁇ g to 10000 mg, more preferably 1 ⁇ g to 3000 mg, and still more preferably 10 ⁇ g to 1000 mg per day can be suitably exemplified.
- the preventive / ameliorating agent of the present invention containing the compound of the present invention is also used as an exon skipping inducer / promoter for inducing / promoting exon skipping of a gene containing a mutation causing a hereditary disease. Can do.
- the use of the compound in the present invention in the production of the preventive / ameliorating agent of the present invention the method of using the compound in the present invention for the preventive / ameliorating agent of the present invention: Use of a compound in the present invention in skipping an exon of a gene containing a mutation causing a hereditary disease to induce and increase the expression of a functional truncated protein: and heritability targeted by the present invention
- Use of the compound of the present invention in prevention / amelioration of a disease a method for preventing / ameliorating a genetic disease targeted by the present invention by administering the preventive / ameliorating agent of the present invention to a subject, or the present invention
- the contents of the words in these uses and methods and the preferred embodiments thereof are as described above.
- RT-PCR reverse transcription PCR
- the amplified product was purified and subcloned into pT7 Blue-T vector (Novagen) as it was, and then the sequence of the amplified product was determined.
- an automatic DNA sequencing apparatus (Model 310; manufactured by Applied Biosystems) was used.
- the case of one of the analyzed muscular dystrophy patients is as follows. This patient is a 5-year-old boy. The boy's parents were healthy Japanese and had no family history of muscle disease. He started walking alone at 1 year and 4 months, and his motor development was normal, but a regular blood test at the age of 2 showed a serum creatine kinase (CK) value of 2567 IU / l (normal value is less than 169 IU / l) I was hospitalized. She was examined at Kobe University Hospital and examined for mutations in the dystrophin gene. Thereafter, the CK value continued to rise gently (1331 to 4740 IU / l), but no muscle weakness or abnormal gait was observed. A muscle biopsy was performed at the age of five. The above research was conducted with the approval of the Kobe University Ethics Committee.
- KUCG797 dystrophin gene a point mutation was found in exon 31.
- This mutation is the substitution of nucleotide 4303 of dystrophin cDNA from G to T (G to U in RNA) (c. 4303G> T: FIG. 1a). Because this nucleotide change was a substitution of a glutamic acid-encoding GAG codon to a stop codon-encoding TAG (p.Glu1435X), dystrophin was produced in this case according to the reading frame rules described in the background art. It was expected to be severe DMD.
- KUCG797 biopsy of rectus femoris was performed to obtain a skeletal muscle sample.
- the skeletal muscle sample was snap frozen with isopentane cooled with liquid nitrogen.
- a frozen serial section having a thickness of 10 ⁇ m was prepared from the frozen skeletal muscle sample, and the section was analyzed by immunohistochemical staining. Specifically, the above-mentioned frozen frozen sections having a thickness of 10 ⁇ m were fixed in cold acetone for 5 minutes. This section was blocked with normal goat serum and incubated overnight at 4 ° C. in the presence of anti-dystrophin antibody (primary antibody).
- primary antibody anti-dystrophin antibody
- DYS2 (manufactured by Novocastra)
- DYS3 (manufactured by Novocastra)
- MANDYS1 donated by Prof. Dr. Glenn E. ⁇ Morris).
- DYS2 recognizes an epitope of exons 77 to 79 (C-terminal side) of the dystrophin gene
- DYS3 recognizes an epitope of exons 10 to 12 (N-terminal side) of the dystrophin gene
- MANDYS1 recognizes an exon 31 of the dystrophin gene Recognizes an epitope of / 32 (rod domain).
- FIG. 1b shows the result of staining with DYS2
- panel f in FIG. 1b shows the result of staining with DYS3
- panel g in FIG. 1b shows the result of staining with MANDYS1.
- Severe DMD was expected from the genotype of the KUCG797 dystrophin gene, but the results of immunohistochemical staining were contrary to that expectation and recognized the N-terminal or C-terminal dystrophin domain as in BMD. A sparse and discontinuous signal was observed for the antibody (Panel e and Panel f in FIG. 1b). In addition, no signal was observed for the MANDYS1 antibody that recognizes exon 31/32 of the dystrophin gene (panel g in FIG. 1b).
- the region from exon 27 to exon 32 was amplified using total RNA isolated from KUCG797 as a template and using the aforementioned forward c27f and reverse 2F as primers.
- the amplification product was electrophoresed on a 2% agarose gel in Tris-borate / EDTA buffer and shown in FIG. 1c.
- the rightmost lane shows the result of using KUCG797-derived total RNA as a template
- the center lane shows the result of templated total RNA derived from healthy subjects.
- H492 vector used for splicing analysis in cells (Mol Genet Metab (2005) 85, 213-219., J Med Genet (2006) 43, 924-930., Hum Genet (2007 ) 120, 737-742.), A plasmid (H492-dys Ex31 plasmid) in which a dystrophin gene fragment (mutant minigene) containing mutant exon 31 and adjacent introns on both sides thereof is inserted is constructed. It was decided to examine the mRNA of the gene fragment in transfected Hela cells.
- the H492 vector encodes two cassette exons (A and B) and an intron sequence containing a multicloning site.
- the H492-dys Ex31 plasmid was constructed by the following method. From a genomic DNA of KUCG797, a fragment containing mutant exon 31 of the dystrophin gene and adjacent intron regions on both sides thereof was amplified by PCR. Intron 30f-NheI (GCGGCTAGCGTGATCCACCTGCCTCGAC: SEQ ID NO: 4) and intron 31r-BamHI (GCGGGATCCTCAAATCCAATCTTGCCAAT: SEQ ID NO: 5) were used as primers. A fragment obtained by digesting the above-mentioned amplification product with NheI and BamHI (manufactured by New England Biolabs) was inserted into H492 digested with both enzymes.
- NheI and BamHI manufactured by New England Biolabs
- a plasmid (H492-dys Ex31m plasmid) containing a fragment (mutant minigene) containing the mutant exon 31 derived from KUCG797 and the adjacent intron regions on both sides thereof was constructed (FIG. 1e).
- a plasmid (H492-dys Ex31w plasmid) containing a fragment (wild type minigene) containing the wild type exon 31 of the dystrophin gene and the adjacent intron regions on both sides of the wild type exon 31 by a similar method using the genomic DNA of a healthy person Constructed (FIG. 1e).
- the entire sequence of the plasmid was determined and it was confirmed that the target fragment was contained.
- H492-dys Ex31m plasmid and H492-dys Ex31w plasmid were transfected into Hela cells, respectively, to obtain H492-dys Ex31m / Hela and H492-dys Ex31w / Hela. Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's manual. In these cells obtained by transfection, the mRNA precursor is transcribed from the CMV promoter (CMVp).
- CMVp CMV promoter
- the region containing the minigene was amplified by RT-PCR using total RNA isolated from H492-dys Ex31m / Hela or H492-dys Ex31w / Hela as a template and using the aforementioned intron 30f and intron 30r as primers.
- the amplification product was electrophoresed on a 2% agarose gel in Tris-borate / EDTA buffer. The result is shown in FIG.
- the third lane (m) from the left shows the results using the total RNA derived from H492-dys Ex31m / Hela as the template, and the second lane (w) from the left shows the total RNA derived from H492-dys Ex31w / Hela.
- Mutation of the mutant exon 31 not only destroys the binding site for SRp30c / SRSF9, but also generates a high affinity binding site for hnRNPA1 (FIG. 2a). In other words, the mutation of the mutant exon 31 generates an RNA sequence having high homology with the SELEX winner sequence of hnRNPA1 (FIG. 2b) (Embo J (1994) 13, 1197-1204.). It is well known that hnRNPA1 binds to an exon splicing silencer (ESS) and causes exon skipping.
- ESS exon splicing silencer
- ESE of exon 31 of the dystrophin gene was recognized by SRp30c / SRSF9, and it was suggested that ESE changes to ESS that binds to hnRNPA1 by mutation.
- Human hnRNPA1 cDNA was subjected to PCR amplification, and the obtained amplified fragment was inserted between the BamHI site and NotI site of GST-pCDNA3 to prepare a GST-hnRNPA1 plasmid. The entire sequence of this plasmid was determined and it was confirmed that the target fragment was contained.
- This GST-hnRNPA1 plasmid was transfected into HEK293T cells, and the resulting transformed cells were cultured. A whole cell lysate was prepared from the recovered transformed cells according to the method described in the literature (Methods Mol Biol (2008) 488, 357-365.).
- the GST-hnRNPA1 protein was purified from the whole cell lysate using an anti-GST affinity resin (manufactured by SIGMA). Subsequently, a mutant mobility exon 31 RNA labeled with 32 P or wild type exon 31 RNA was mixed with the aforementioned GST-hnRNPA1 protein and incubated at 20 ° C. for 30 minutes, and a gel mobility shift assay was performed. Specifically, the following method was used.
- the gel mobility shift assay was basically performed according to the method described in the literature (Nucleic acids research (1995) 23, 3638-3641.). That is, the assay was performed by electrophoresing each of the aforementioned complexes on an 8% natural polyacrylamide gel. Band analysis was performed by autoradiography.
- the binding buffer used in this assay was 16 mM Hepes-KOH (pH 7.9), 80 mM KCl, 0.16 mM EDTA, 0.8 mM DTT, 8% glycerol, 100 ng / ⁇ l BSA, 50 ng / ⁇ l E.
- the wild type exon 31 of the dystrophin gene or the aforementioned mutant exon 31 is added to the intron region of a chicken ⁇ crystallin (CDC) mRNA precursor (Molecular cell (2000) 6, 673-682.).
- An mRNA precursor was prepared (see the construct on the right side of the panel of FIG. 3b).
- pCDC-dys Ex31w is prepared by inserting wild-type exon 31 amplified by PCR between the SacI site and the StyI site of pCDC (Molecular cell (2000) 6, 673-682.). did.
- pCDC-dys Ex31m was prepared in the same manner using mutant exon 31 instead of wild type exon 31.
- pCDC-dys Ex31w and pCDC-dys Ex31m were each linearized with SmaI and used as templates for in vitro transcription. According to the method described in the literature (Molecular cell (2000) 6, 673-682.), Transcription is performed in vitro so that the mRNA precursor is labeled with 32 P, and then the transcribed mRNA precursor is purified. It was. In addition, in vitro splicing assay was carried out according to the method described in the literature (Molecular cell (2000) 6, 673-682.). Body) and Hela cell nuclear extract (Cilbiotech) were mixed, and then incubated at 30 ° C. for the time indicated at the top of the panel (0, 15, 30, 60, 90 minutes). The mRNA product obtained by incubation was electrophoresed on a 6% denaturing polyacrylamide gel, and then the RNA band was analyzed by autoradiography. The result is shown in FIG.
- RNA binding protein (SRp30c / SRSF9 or SRp75 / SRSF4) is further overexpressed in H492-dys Ex31m / Hela (Hela cells transfected with a plasmid containing a mutant minigene) prepared in Example 1 above. It was decided to examine what kind of effect the splicing pattern of the mutant minigene would have.
- the plasmid used for overexpression of SRp30c / SRSF9 is the BamHI site of Flag-pCDNA3 (The Journal of biological chemistry (2004) 279, 7009-7013.) Obtained by PCR amplification of human SRp30c cDNA.
- the plasmid used for overexpression of SRp75 / SRSF4 was constructed by inserting the mouse SRp75 cDNA amplified by PCR into the BamHI site and the XhoI site of Flag-pCDNA3. And inserted between.
- the Flag-hnRNPA1 plasmid was prepared by the following method.
- the human hnRNPA1 cDNA was subjected to PCR amplification, and the obtained amplified fragment was inserted between the BamHI site and NotI site of Flag-pCDNA3 (Nucleic®acids®research® (2009) 37, 6515-6527.)
- To obtain the Flag-hnRNPA1 plasmid. was made. The entire sequence of this plasmid was determined and it was confirmed that the target fragment was contained.
- This Flag-hnRNPA1 plasmid was transfected into HEK293T cells, and the resulting transformed cells were cultured.
- a whole cell lysate was prepared from the recovered transformed cells according to the method described in the literature (Methods Mol Mol Biol (2008) 488, 357-365.).
- the literature is basically the same except that the resin is washed twice with buffer E (20 mM Hepes-KOH pH 7.9, 1000 mM KCl, 0.2 mM EDTA, 10% glycerol, and 1 mM DTT) before elution from the column.
- the Flag-hnRNPA1 protein was purified from the whole cell lysate using an anti-Flag-M2 affinity resin (manufactured by SIGMA) as described in Journal of biological chemistry (2002) 277, 7540-7545. .
- H492-dys Ex31m plasmid and Flag-SRp30c plasmid were co-transfected using Lipofectamine 2000 (Invitrogen) to obtain H492-dys Ex31m • Flag-SRp30c / Hela. Further, H492-dys Ex31m plasmid and Flag-SRp75 plasmid were co-transfected using Lipofectamine 2000 (Invitrogen) to obtain H492-dys Ex31m • Flag-SRp75 / Hela.
- H492-dys Ex31m plasmid and Flag-hnRNPA1 plasmid were co-transfected using Lipofectamine 2000 (manufactured by Invitrogen) to obtain H492-dys Ex31m • Flag-hnRNPA1 plasmid / Hela.
- FIG. 3 c shows the result of splicing analysis similar to “Splicing analysis 1 in cells using minigene” in Example 1 described above for each of these transformed cells.
- the leftmost lane in FIG. 3c shows the marker
- the second lane (mock) from the left shows the result using the total RNA derived from H492-dys Ex31m / Hela as a template
- the third lane from the left (SRp30c).
- / SRSF9 shows the results using the total RNA derived from H492-dys Ex31m • Flag-SRp30c / Hela as a template
- the second lane (SRp75 / SRSF4) from the right is H492-dys Ex31m • Flag-SRp75 / Hela.
- FIG. 3d shows the result of calculating the ratio of the exon 31 skipped mRNA to the mRNA containing exon 31 (Exon skip / inclusion ratio) by quantifying the band concentration in FIG. 3c.
- dystrophin exon 31 contains SRp30c / SRSF9-dependent ESE, and that this ESE was mutated and changed to ESS binding to hnRNPA1, thereby causing exon 31 skipping.
- TG003 and SRPIN340 are both known as low-molecular compounds that affect alternative splicing.
- SRPIN340 is a specific inhibitor of SR protein kinases (SRPKs) (Proc Natl Acad Sci U S A (2006) 103, 11329-11333.), Possibly regulating alternative splicing through inhibition of SR protein phosphorylation by SRPK (Biochem J (2009) 417, 15-27., Genome Biol (2009) 10, 242.).
- SRPKs SR protein kinases
- the other is an inhibitor of Clk kinase named TG003.
- TG003 has also been shown to affect alternative splicing of adenovirus E1A, SC35, and Clk itself (Non-Patent Documents 12 and 13).
- FIG. 5 shows the results of splicing analysis (RT-PCR analysis using total RNA extracted from cells) for each of the incubated cells, similar to "Splicing analysis 1 in cells using minigene" in Example 1 above. Shown in 4a. The leftmost lane in FIG.
- FIG. 4a shows a DNA size marker digested with ⁇ X174-HaeIII
- the second lane (DMSO) from the left shows the result when H492-dys Ex31m / Hela was treated with DMSO.
- the third lane (TG003) shows the result when H492-dys Ex31m / Hela is processed with TG003
- the rightmost lane (SRPIN340) shows the result when H492-dys Ex31m / Hela is processed with SRPIN340.
- FIG. 4b shows the result of calculating the exon skip / inclusion ratio by quantifying the band concentration in FIG. 4a.
- TG003 may promote not only the skipping of the mutant exon 31 but also the skipping of the wild type exon 31. Therefore, using H492-dys Ex31m / Hela (Hela cells transfected with a plasmid containing a mutant minigene) and H492-dys Ex31w / Hela (Hela cells transfected with a plasmid containing a wild-type minigene), Splicing analysis was performed in the same manner as in “Splicing analysis in cells 3 using the minigene” described above.
- FIG. 4d shows the result of calculating the exon skip / inclusion ratio by quantifying the concentration of the band in FIG. 4c.
- TG003 promoted exon 31 skipping in a dose-dependent manner in H492-dys Ex31m / Hela (Hela cells transfected with a plasmid containing a mutant minigene).
- H492-dys Ex31w / Hela Hela cells transfected with a plasmid containing a wild type minigene
- TG003 does not promote exon skipping in wild type exon 31, but specifically promotes exon 31 skipping of mutant exon 31.
- This mutation is an out-of-frame mutation that shifts the reading frame of the dystrophin cDNA (SEQ ID NO: 1), and as a result, the C41 terminal side of the mutation (nucleotides 3641 to 3643 of the dystrophin cDNA (SEQ ID NO: 1)). A stop codon is generated at (part). According to the reading frame rules described in the background art of the present specification, it was predicted from the genotype that dystrophin was not produced in this case and severe DMD was produced, but the actual symptoms were milder than that. Met.
- a plasmid (H492-dys Ex27m) containing a fragment containing the mutant exon 27 derived from this patient and the adjacent intron regions on both sides thereof was constructed.
- a plasmid (H492-dys Ex27w) containing a fragment containing the wild-type exon 27 of the dystrophin gene and adjacent intron regions on both sides thereof was constructed by the same method using the genomic DNA of healthy individuals. For both the H492-dys Ex27m plasmid and the H492-dys Ex27w plasmid, the entire plasmid sequence was determined and it was confirmed that the target fragment was contained.
- H492-dys Ex27m and H492-dys Ex27w were transfected into Hela cells in the same manner as in the above-mentioned “Splicing analysis in cells using minigene 1”, and H492-dys Ex27m / Hela was transfected respectively. And H492-dys Ex27m / Hela was obtained.
- FIG. 5b shows the result of calculating the exon skip / inclusion ratio by quantifying the concentration of the band in FIG. 5a.
- DMEM Dulbecco's modified Eagle's medium
- Gibco horse serum
- Gibco Antibiotic-Antimyotic
- the aforementioned primary myocytes were cultured for 2 weeks in the presence or absence of TG003 at a concentration (1 ⁇ M, 2 ⁇ M, 5 ⁇ M, 7 ⁇ M, 10 ⁇ M).
- the medium and TG003 were replaced every 2 days.
- both the forward c27f and reverse 2F primers described above were used. The same method as in “Splicing analysis 3” was used. Both the forward c27f and reverse 2F primers described above are primer sets that amplify the region from exon 27 to 32 of the dystrophin gene. The result of such splicing analysis is shown in FIG. 6a. 6B shows the result of calculating the exon skip / inclusion ratio by quantifying the concentration of the band in FIG. 6A.
- TG003 promoted exon 31 skipping in a dose-dependent manner even when MUCG797-derived myocytes were used. These results suggest that TG003 can promote skipping of mutant exon 31 in patient cells, and it is also strongly expected that TG003 can increase the expression of truncated dystrophin with some dystrophin function remaining. It was.
- DMEM Dulbecco's modified Eagle's medium
- Gibco horse serum
- Gibco Antibiotic-Antimyotic
- 7 ⁇ M of the above-mentioned primary muscle cells derived from KUCG797 were added.
- the cells were cultured for 2 weeks in the presence or absence of TG003.
- the medium and TG003 were replaced every 2 days.
- These cultured myocytes were washed twice with PBS and collected using 1X Cell Lysis Buffer (manufactured by Cell Signaling Technology). Total protein extracted from the collected cells was applied to a 3-10% gradient polyacrylamide gel (PAGEL, manufactured by ATTO).
- PAGEL 3-10% gradient polyacrylamide gel
- the amount of total protein applied was 4 ⁇ g for the control, 20 ⁇ g for the mutant TG003 (0 ⁇ M), and 60 ⁇ g for the mutant TG003 (7 ⁇ M).
- the protein fraction migrated on polyacrylamide gel was transferred to a HYBOND-P membrane (GE Healthcare).
- Western blotting analysis was performed on the aforementioned membrane using an ECL-advance-Western-Blotting-Detection kit (GE-Healthcare) according to the manufacturer's manual.
- An antibody against the C-terminus of dystrophin NCL-DYS2, manufactured by Leica
- an antibody corresponding to exon 31 of dystrophin 8H11, manufactured by Santa ⁇ Cruz
- the membrane was incubated as described above.
- an anti-mouse IgG antibody GE Healthcare
- Western blotting analysis of desmin was performed using the same protocol as described above.
- the desmin antibody H-76, manufactured by Santa Cruz
- an anti-rabbit IgG antibody manufactured by GE Healthcare
- the present invention can be suitably used in the field of prevention and improvement of genetic diseases. More specifically, the present invention is preferably used in the field of prevention and improvement of genetic diseases caused by mutations in exons of a gene and capable of generating functional truncated proteins by skipping exons containing the mutations. Can do.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Enzymes And Modification Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
[式中、R1及びR2は各々独立に、直鎖状、分岐鎖状のC1-C10炭化水素基を示し;R3はメトキシ基、エトキシ基、アセトキシ基又はハロゲン原子を示す。]
で表される化合物であることを特徴とする上記(4)に記載の予防・改善剤や、(6)前記変異が、ナンセンス変異であることを特徴とする上記(1)~(5)のいずれかに記載の予防・改善剤や、(7)前記ナンセンス変異が、前記遺伝子におけるエクソンスプライシングエンハンサー活性を抑制し、及び/又は、前記遺伝子におけるエクソンスプライシングサイレンサー活性を上昇させる、ナンセンス変異であることを特徴とする上記(6)に記載の予防・改善剤や、(8)前記遺伝子がジストロフィン遺伝子であり、前記機能性トランケート型タンパク質が機能性トランケート型ジストロフィンタンパク質であり、前記遺伝性疾患がデュシェンヌ型筋ジストロフィーであることを特徴とする上記(1)~(7)のいずれかに記載の予防・改善剤や、(9)前記ジストロフィン遺伝子のエクソンが、ジストロフィン遺伝子のエクソン31又はエクソン27であることを特徴とする上記(8)に記載の予防・改善剤や、(10)前記ジストロフィン遺伝子のエクソン31の変異が、配列番号1のポリヌクレオチド配列のヌクレオチド番号4303におけるグアニンのチミンへのナンセンス変異であり、エクソン27の変異が、配列番号1のポリヌクレオチド配列のヌクレオチド番号3613におけるグアニンが欠失したアウトオブフレーム変異であることを特徴とする上記(9)に記載の予防・改善剤に関する。
本発明の予防・改善剤は、遺伝子のエクソンの変異に起因し、かつ、該変異が含まれるエクソン(以下、単に「変異型エクソン」とも表示する。)をスキッピングさせて機能性トランケート型タンパク質を生成させ得る遺伝性疾患(以下、「本発明が対象とする遺伝性疾患」とも表示する。)の予防・改善剤であって、分子量1500以下の化合物(以下、「本発明における化合物」とも表示する。)を含有している限り特に制限されないが、分子量1000以下の化合物であることが好ましく、分子量700以下の化合物であることがより好ましく、分子量500以下の化合物であることがさらに好ましく、分子量300以下の化合物であることがさらにより好ましい。また、本発明の予防・改善剤には、本発明における化合物を2種類以上併用してもよい。
筋ジストロフィー患者400名以上について、以下の方法により、そのジストロフィン遺伝子の変異を解析した。
標準的なフェノール-クロロフォルム抽出法により、患者の血液試料からDNAを単離した。フィコールパーク密度勾配法(Amersham Biosciences AB社製)を用いて全血から回収した末梢リンパ球から、或いは凍結筋試料を薄片化した筋切片から、トータルRNAを単離した。逆転写PCR(RT-PCR)及びRT-nested PCR法により、骨格筋で発現したジストロフィンmRNAを分析した。骨格筋由来のジストロフィンmRNAについて、内側プライマーセット(フォワードc27f:CCTGTAGCACAAGAGGCCTTA(配列番号2)、及び、リバース2F:TCCACACTCTTTGTTTCCAATG(配列番号3))を使用してエクソン27~32を含む領域を増幅した。増幅産物を精製し、そのまま、或いはpT7 Blue-T ベクター(Novagen社製)にサブクローニングしてから、増幅産物の配列を決定した。この配列の決定には、自動DNA配列決定装置(モデル310;Applied Biosystems社製)を用いた。
次に、以下の方法で、前記患者(5歳)に骨格筋バイオプシーを行い、ジストロフィン免疫染色を行った。
KUCG797のジストロフィン遺伝子の遺伝子型(DMD型)と、免疫染色パターンとの間のこの矛盾を説明するため、本発明者らは、ジストロフィン遺伝子のエクソン31におけるナンセンス変異がエクソンスプライシングエンハンサー(ESE)を破壊し、変異型エクソン(変異を含むエクソン)のスキッピングをもたらしたと推定した。この推定の可能性を証明するため、KUCG797の骨格筋のジストロフィンmRNAをRT-PCR増幅法で解析した。
この仮説をさらに分析するため、細胞におけるスプライシング解析に用いられているH492ベクター(Mol Genet Metab (2005) 85, 213-219., J Med Genet (2006) 43, 924-930., Hum Genet (2007) 120, 737-742.)に、変異型エクソン31とその両側の隣接イントロンとを含むジストロフィン遺伝子断片(変異型ミニ遺伝子)を挿入したプラスミド(H492-dys Ex31プラスミド)を構築し、かかるプラスミドをトランスフェクトしたHela細胞において、その遺伝子断片のmRNAを調べることにした。なお、H492ベクターは、2つのカセットエクソン(A及びB)とマルチクローニングサイトを含むイントロン配列とをコードしている。
前述の実施例1の結果から、KUCG797のジストロフィン遺伝子のエクソン31の点変異が、エクソンスキッピングを引き起こすことが示されたので、エクソン31のスキッピングやインクルージョン(inclusion)を調節する候補因子の同定を試みた。
ジストロフィン遺伝子の野生型エクソン31と、変異型エクソン31のRNA配列をSpliceAidプログラム(http://www.introni.it/splicing.html)(Bioinformatics (2009) 25, 1211-1213.)で解析したところ、この変異型エクソン31の点変異は、SRタンパク質のメンバーであるSRp30c/SRSF9(SFSR9)との結合力を低下させることが分かった(図2a)。SRタンパク質は、プリンリッチであることが多いエクソンスプライシングエンハンサー(ESE)と結合することが知られている。エクソン31の配列は、SELEXで同定されたSRp30c/SRSF9に対する高親和性結合配列と高い類似性を有する(図2b)(Rna (2007) 13, 1287-1300.)。上記変異型エクソン31の変異は、SRp30c/SRSF9に対する結合部位を破壊するだけでなく、hnRNPA1高親和性結合部位をも生じさせる(図2a)。つまり、上記変異型エクソン31の変異は、hnRNPA1のSELEX winner配列と高い相同性をもつRNA配列を生じさせる(図2b)(Embo J (1994) 13, 1197-1204.)。hnRNPA1がエクソンスプライシングサイレンサー(ESS)と結合し、エクソンスキッピングを生じさせることはよく知られている。SpliceAidプログラムでの結果から、ジストロフィン遺伝子のエクソン31のESEはSRp30c/SRSF9に認識され、変異によりESEがhnRNPA1に結合するESSに変わることが示唆された。
ジストロフィン遺伝子のエクソン31のESEはSRp30c/SRSF9に認識され、変異によりESEがhnRNPA1に結合するESSに変わるという仮説を検証するため、先ず、ゲルモビリティシフトアッセイにより、hnRNPA1に対する結合活性を、変異型エクソン31RNAと野生型エクソン31RNAとで比較した。具体的には以下の方法で行った。
変異型エクソン31は、hnRNPA1とより強い結合親和性を持つようになったことで、スプライシングの際に、エクソンとして効率的に認識されなかった可能性が考えられたので、この可能性を検証するために、インビトロでのスプライシングアッセイを行った。
次に、前述の実施例1で作製したH492-dys Ex31m/Hela(変異型ミニ遺伝子を含むプラスミドをトランスフェクションしたHela細胞)において、RNA結合タンパク質(SRp30c/SRSF9又はSRp75/SRSF4)をさらに過剰発現させることによって、変異型ミニ遺伝子のスプライシングパターンにどのような影響が生じるかを調べることにした。
本発明者らは、ジストロフィン遺伝子のエクソン31のスキッピングを促進し得る低分子化合物を見つけるために、様々な低分子化合物を探索した。その代表例として、以下には、TG003とSRPIN340についての結果を記載する。
H492-dys Ex31m/Hela(変異型ミニ遺伝子を含むプラスミドをトランスフェクションしたHela細胞)を、30μMのTG003を含むDMSO溶液又は30μMのSRPIN340を含むDMSO溶液中で24時間インキュベートした。ネガティブコントロールとしてDMSO溶液を用いて同様にインキュベートした。インキュベートした各細胞について、前述の実施例1の「ミニ遺伝子を利用した、細胞におけるスプライシング解析1」と同様のスプライシング解析(細胞抽出したトータルRNAを用いたRT-PCR解析)を行った結果を図4aに示す。図4aの一番左のレーンは、φX174-HaeIIIで消化したDNAサイズマーカーを示し、左から2番目のレーン(DMSO)は、H492-dys Ex31m/HelaをDMSO処理した場合の結果を示し、左から3番目のレーン(TG003)は、H492-dys Ex31m/HelaをTG003処理した場合の結果を示し、一番右のレーン(SRPIN340)は、H492-dys Ex31m/HelaをSRPIN340処理した場合の結果を示す。また、図4aのバンド濃度を定量して、エクソンスキップ/インクルージョンレシオを算出した結果を図4bに示す。
「ミニ遺伝子を利用した、細胞におけるスプライシング解析3」の結果からは、TG003が、変異型エクソン31のスキッピングだけでなく、野生型エクソン31のスキッピングをも促進する可能性もあった。そこで、H492-dys Ex31m/Hela(変異型ミニ遺伝子を含むプラスミドをトランスフェクションしたHela細胞)と共に、H492-dys Ex31w/Hela(野生型ミニ遺伝子を含むプラスミドをトランスフェクションしたHela細胞)を用いて、前述の「ミニ遺伝子を利用した、細胞におけるスプライシング解析3」と同様の方法でスプライシング解析を行った。なお、細胞を処理する溶液としては、種々の濃度(0μM、5μM、10μM、20μM、30μM、50μM)のTG003を含むDMSO溶液を用いた。その結果を図4cに示す。また、図4cのバンドの濃度を定量して、エクソンスキップ/インクルージョンレシオを算出した結果を図4dに示す。
TG003が、ジストロフィン遺伝子のエクソン31以外のエクソンの変異に対してもスキッピングを促進させる効果を発揮するか否かを確認するために、KUCG797の患者とは別の筋ジストロフィー患者由来のジストロフィン遺伝子の変異を解析した。この患者のジストロフィン遺伝子の変異はエクソン27に認められた。この変異は、ジストロフィンcDNA(配列番号1)の3613番目のグアニンヌクレオチドが欠失したものである(c.3613delG)。この変異は、ジストロフィンcDNA(配列番号1)の読み枠をシフトさせるアウトオブフレーム変異であり、その結果、その変異の少しC末端側(ジストロフィンcDNA(配列番号1)の3641番目から3643番目のヌクレオチド部分)にストップコドン(終止コドン)を生じさせる。本明細書の背景技術に記載の読み枠ルールにしたがえば、この症例ではジストロフィンが産生されず、重篤なDMDとなることがその遺伝子型から予想されたが、実際の症状はそれより軽症であった。
次に、ジストロフィン遺伝子のスプライシングに対してTG003が及ぼす影響を、患者(KUCG797)由来の筋細胞において調べることとした。
まず、KUCG797から採取した筋細胞について初代培養を行った。具体的には、以下のような方法で行った。6ウエルプレート(Gelatin-Coated micro plate 6 well with Lid;IWAKI社製)中に、20%のFBS(Gibco社製)、4%のUltroser(登録商標)G(PALL社製)及び1%のAntibiotic-Antimyotic(Gibco社製)をコンフルエントになるまで添加したダルベッコ変法イーグル培地(DMEM)(Sigma社製)にて、患者由来の筋細胞を培養した。筋細胞を筋管に分化させるため、2%ウマ血清(Gibco社製)及び1%Antibiotic-Antimyotic(Gibco社製)を添加したダルベッコ変法イーグル培地(DMEM)(Sigma社製)において、種々の濃度(1μM、2μM、5μM、7μM、10μM)のTG003の存在下又は不在下で、前述の初代筋細胞を2週間培養した。培地とTG003は2日毎に入れ替えた。
前述のKUCG797由来の筋細胞において、実際にトランケート型ジストロフィンが発現しているかどうかを確認するために、以下のウエスタンブロッティング解析を行った。
Claims (10)
- 遺伝子のエクソンの変異に起し、かつ、該変異が含まれるエクソンをスキッピングさせて機能性トランケート型タンパク質を生成させ得る遺伝性疾患の予防・改善剤であって、分子量1500以下の化合物を含有することを特徴とする予防・改善剤。
- 前記化合物が、スプライシング調節作用を有する化合物であることを特徴とする請求項1に記載の予防・改善剤。
- 前記化合物が、変異が含まれるエクソンのスキッピングを誘導・促進させる効果を有する化合物であることを特徴とする請求項1又は2に記載の予防・改善剤。
- 前記化合物が、Cdc-likeキナーゼ阻害化合物であることを特徴とする請求項1~3のいずれかに記載の予防・改善剤。
- 前記Cdc-likeキナーゼ阻害化合物が、一般式(1)

[式中、R1及びR2は各々独立に、直鎖状、分岐鎖状のC1-C10炭化水素基を示し;R3はメトキシ基、エトキシ基、アセトキシ基又はハロゲン原子を示す。]
で表される化合物であることを特徴とする請求項4に記載の予防・改善剤。 - 前記変異が、ナンセンス変異であることを特徴とする請求項1~5のいずれかに記載の予防・改善剤。
- 前記ナンセンス変異が、前記遺伝子におけるエクソンスプライシングエンハンサー活性を抑制し、及び/又は、前記遺伝子におけるエクソンスプライシングサイレンサー活性を上昇させる、ナンセンス変異であることを特徴とする請求項6に記載の予防・改善剤。
- 前記遺伝子がジストロフィン遺伝子であり、前記機能性トランケート型タンパク質が機能性トランケート型ジストロフィンタンパク質であり、前記遺伝性疾患がデュシェンヌ型筋ジストロフィーであることを特徴とする請求項1~7のいずれかに記載の予防・改善剤。
- 前記ジストロフィン遺伝子のエクソンが、ジストロフィン遺伝子のエクソン31又はエクソン27であることを特徴とする請求項8に記載の予防・改善剤。
- 前記ジストロフィン遺伝子のエクソン31の変異が、配列番号1のポリヌクレオチド配列のヌクレオチド番号4303におけるグアニンのチミンへのナンセンス変異であり、エクソン27の変異が、配列番号1のポリヌクレオチド配列のヌクレオチド番号3613におけるグアニンが欠失したアウトオブフレーム変異であることを特徴とする請求項9に記載の予防・改善剤。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012522455A JP5950818B2 (ja) | 2010-06-28 | 2011-06-27 | 遺伝性疾患の予防・改善剤 |
| US13/805,151 US9241929B2 (en) | 2010-06-28 | 2011-06-27 | Prophylactic or ameliorating agent for genetic diseases |
| CN2011800303278A CN103096928A (zh) | 2010-06-28 | 2011-06-27 | 遗传性疾病的预防/改善剂 |
| EP11800411.8A EP2586462A4 (en) | 2010-06-28 | 2011-06-27 | PROPHYLACTIC OR ALTERNATIVE AGENT FOR GENETIC DISEASES |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010146699 | 2010-06-28 | ||
| JP2010-146699 | 2010-06-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012001941A1 true WO2012001941A1 (ja) | 2012-01-05 |
Family
ID=45401683
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/003655 Ceased WO2012001941A1 (ja) | 2010-06-28 | 2011-06-27 | 遺伝性疾患の予防・改善剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9241929B2 (ja) |
| EP (1) | EP2586462A4 (ja) |
| JP (1) | JP5950818B2 (ja) |
| CN (2) | CN106994124A (ja) |
| WO (1) | WO2012001941A1 (ja) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8450474B2 (en) | 2004-06-28 | 2013-05-28 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8637483B2 (en) | 2009-11-12 | 2014-01-28 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| DE202014005288U1 (de) | 2013-06-27 | 2014-07-11 | Nivarox-Far S.A. | Uhrfeder aus austenitischem Edelstahl |
| US8865883B2 (en) | 2008-10-24 | 2014-10-21 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| EP2924514A1 (fr) | 2014-03-24 | 2015-09-30 | Nivarox-FAR S.A. | Ressort d'horlogerie en acier inoxydable austénitique |
| US9217148B2 (en) | 2013-03-14 | 2015-12-22 | Sarepta Therapeutics, Inc. | Exon skipping compositions for treating muscular dystrophy |
| US9506058B2 (en) | 2013-03-15 | 2016-11-29 | Sarepta Therapeutics, Inc. | Compositions for treating muscular dystrophy |
| WO2017175842A1 (ja) * | 2016-04-07 | 2017-10-12 | 国立大学法人京都大学 | スプライシングの改変に関する化合物及び医薬組成物 |
| WO2018151326A1 (ja) * | 2017-02-20 | 2018-08-23 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| EP3720466A1 (en) * | 2017-12-05 | 2020-10-14 | Royal Holloway And Bedford New College | Treatment of muscular dystrophies |
| US20210091966A1 (en) * | 2019-09-24 | 2021-03-25 | Genetec Inc. | Intermediary device for daisy chain and tree configuration in hybrid data/power connection |
| USRE48960E1 (en) | 2004-06-28 | 2022-03-08 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| WO2022255411A1 (ja) * | 2021-06-01 | 2022-12-08 | 国立大学法人京都大学 | 自然免疫応答を増強させる方法 |
| US11770155B2 (en) | 2020-05-19 | 2023-09-26 | Genetec Inc. | Power distribution and data routing in a network of devices interconnected by hybrid data/power links |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4119167A4 (en) | 2020-03-11 | 2025-08-06 | Biocomber Co Ltd | SINGLE-STRANDED NUCLEIC ACID MOLECULE FOR INDUCING FRAME SHIFT-1 AND COMPOSITION |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050171026A1 (en) | 2004-01-09 | 2005-08-04 | Tokyo Medical And Dental University | Therapeutic composition of treating abnormal splicing caused by the excessive kinase induction |
| JP2009521408A (ja) * | 2005-12-02 | 2009-06-04 | サートリス ファーマシューティカルズ, インコーポレイテッド | Cdc2様キナーゼ(CLK)のモジュレータおよびその使用方法 |
| WO2009087238A2 (en) * | 2008-01-10 | 2009-07-16 | Centre National De La Recherche Scientifique (Cnrs) | Chemical molecules that inhibit the slicing mechanism for treating diseases resulting from splicing anomalies |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010535708A (ja) * | 2007-08-03 | 2010-11-25 | ビオマリン アイジーエー リミテッド | デュシェンヌ型筋ジストロフィーの治療のための薬物併用 |
-
2011
- 2011-06-27 JP JP2012522455A patent/JP5950818B2/ja not_active Expired - Fee Related
- 2011-06-27 EP EP11800411.8A patent/EP2586462A4/en not_active Withdrawn
- 2011-06-27 CN CN201611191464.0A patent/CN106994124A/zh active Pending
- 2011-06-27 US US13/805,151 patent/US9241929B2/en active Active
- 2011-06-27 CN CN2011800303278A patent/CN103096928A/zh active Pending
- 2011-06-27 WO PCT/JP2011/003655 patent/WO2012001941A1/ja not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050171026A1 (en) | 2004-01-09 | 2005-08-04 | Tokyo Medical And Dental University | Therapeutic composition of treating abnormal splicing caused by the excessive kinase induction |
| JP2009521408A (ja) * | 2005-12-02 | 2009-06-04 | サートリス ファーマシューティカルズ, インコーポレイテッド | Cdc2様キナーゼ(CLK)のモジュレータおよびその使用方法 |
| WO2009087238A2 (en) * | 2008-01-10 | 2009-07-16 | Centre National De La Recherche Scientifique (Cnrs) | Chemical molecules that inhibit the slicing mechanism for treating diseases resulting from splicing anomalies |
Non-Patent Citations (38)
| Title |
|---|
| BIOCHEM BIOPHYS RES COMMUN, vol. 226, 1996, pages 445 - 449 |
| BIOCHEM J, vol. 417, 2009, pages 15 - 27 |
| BIOINFORMATICS, vol. 25, 2009, pages 1211 - 1213 |
| CELL, vol. 53, 1988, pages 219 - 228 |
| DISSET A ET AL.: "An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements.", HUM. MOL. GENET., vol. 15, no. 6, 2006, pages 999 - 1013 * |
| EMBO J, vol. 13, 1994, pages 1197 - 1204 |
| GENES CELLS, vol. 13, 2008, pages 233 - 244 |
| GENOME BIOL, vol. 10, 2009, pages 242 |
| GENOMICS, vol. 2, 1988, pages 90 - 95 |
| HUM GENET, vol. 120, 2007, pages 737 - 742 |
| HUM MOL GENET, vol. 15, 2006, pages 999 - 1013 |
| J CLIN INVEST, vol. 95, 1995, pages 515 - 520 |
| J CLIN PHARMACOL, vol. 47, 2007, pages 430 - 444 |
| J MED GENET, vol. 43, 2006, pages 924 - 930 |
| LANCET NEUROLOGY, vol. 8, 2009, pages 873 - 875 |
| LANCET NEUROLOGY, vol. 8, 2009, pages 918 - 928 |
| MASAFUMI MATSUO: "Becker musclar dystrophy resulted from exon skipping that removes nonsense mutation", GENE & MEDICINE, vol. 3, no. 2, 1999, pages 86 - 90, XP009173686 * |
| MASATOSHI HAGIWARA ET AL.: "Chemical Biology ni yoru Nanji Shikkan eno Chosen", THE JAPANESE PHARMACOLOGICAL SOCIETY KANTO BUKAI KOEN YOSHISHU, vol. 120TH, 2009, pages 14, XP009173390 * |
| METHODS MOL BIOL, vol. 488, 2008, pages 357 - 365 |
| MOL GENET METAB, vol. 85, 2005, pages 213 - 219 |
| MOLECULAR AND CELLULAR BIOLOGY, vol. 13, 1993, pages 4023 - 4028 |
| MOLECULAR CELL, vol. 6, 2000, pages 673 - 682 |
| MURAKI M ET AL.: "Manipulation of alternative splicing by a newly developed inhibitor of Clks.", J. BIOL. CHEM., vol. 279, no. 23, 2004, pages 24246 - 24254, XP055192700, DOI: doi:10.1074/jbc.M314298200 * |
| NATURE MEDICINE, vol. 12, 2006, pages 175 - 177 |
| NATURE, vol. 447, 2007, pages 87 - 91 |
| NISHIDA A ET AL.: "Chemical treatment enhances skipping of a mutated exon in the dystrophin gene.", NAT. COMMUN., vol. 2, 10 May 2011 (2011-05-10), XP055144728, DOI: doi:10.1038/ncomms1306 * |
| NUCLEIC ACIDS RESEARCH, vol. 23, 1995, pages 3638 - 3641 |
| NUCLEIC ACIDS RESEARCH, vol. 37, 2009, pages 6515 - 6527 |
| O'LEARY DA ET AL.: "Identification of small molecule and genetic modulators of AON-induced dystrophin exon skipping by high-throughput screening.", PLOS ONE, vol. 4, no. 12, 2009, XP055083574, DOI: doi:10.1371/journal.pone.0008348 * |
| PEDIATR RES, vol. 59, 2006, pages 690 - 694 |
| PROC NATL ACAD SCI USA, vol. 103, 2006, pages 11329 - 11333 |
| RNA, vol. 13, 2007, pages 1287 - 1300 |
| See also references of EP2586462A4 * |
| THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 277, 2002, pages 7540 - 7545 |
| THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 279, 2004, pages 24246 - 24252 |
| THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 279, 2004, pages 7009 - 7013 |
| THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 357, 2007, pages 2677 - 2686 |
| YOMODA J ET AL.: "Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A.", GENES CELLS., vol. 13, no. 3, 2008, pages 233 - 244 * |
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9175286B2 (en) | 2004-06-28 | 2015-11-03 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9447415B2 (en) | 2004-06-28 | 2016-09-20 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8455634B2 (en) | 2004-06-28 | 2013-06-04 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8455636B2 (en) | 2004-06-28 | 2013-06-04 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8476423B2 (en) | 2004-06-28 | 2013-07-02 | The University of Western Austrailia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8486907B2 (en) | 2004-06-28 | 2013-07-16 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8524880B2 (en) | 2004-06-28 | 2013-09-03 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| USRE48960E1 (en) | 2004-06-28 | 2022-03-08 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10995337B2 (en) | 2004-06-28 | 2021-05-04 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10266827B2 (en) | 2004-06-28 | 2019-04-23 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| USRE47751E1 (en) | 2004-06-28 | 2019-12-03 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10968450B2 (en) | 2004-06-28 | 2021-04-06 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9018368B2 (en) | 2004-06-28 | 2015-04-28 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9024007B2 (en) | 2004-06-28 | 2015-05-05 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9035040B2 (en) | 2004-06-28 | 2015-05-19 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10781451B2 (en) | 2004-06-28 | 2020-09-22 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8455635B2 (en) | 2004-06-28 | 2013-06-04 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| USRE47769E1 (en) | 2004-06-28 | 2019-12-17 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9994851B2 (en) | 2004-06-28 | 2018-06-12 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10421966B2 (en) | 2004-06-28 | 2019-09-24 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9249416B2 (en) | 2004-06-28 | 2016-02-02 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9422555B2 (en) | 2004-06-28 | 2016-08-23 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US8450474B2 (en) | 2004-06-28 | 2013-05-28 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9441229B2 (en) | 2004-06-28 | 2016-09-13 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9605262B2 (en) | 2004-06-28 | 2017-03-28 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US10227590B2 (en) | 2004-06-28 | 2019-03-12 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| USRE47691E1 (en) | 2004-06-28 | 2019-11-05 | The University Of Western Australia | Antisense oligonucleotides for inducing exon skipping and methods of use thereof |
| US9453225B2 (en) | 2008-10-24 | 2016-09-27 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US9447416B2 (en) | 2008-10-24 | 2016-09-20 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US9447417B2 (en) | 2008-10-24 | 2016-09-20 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US9434948B2 (en) | 2008-10-24 | 2016-09-06 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US9234198B1 (en) | 2008-10-24 | 2016-01-12 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US8871918B2 (en) | 2008-10-24 | 2014-10-28 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US8865883B2 (en) | 2008-10-24 | 2014-10-21 | Sarepta Therapeutics, Inc. | Multiple exon skipping compositions for DMD |
| US9758783B2 (en) | 2009-11-12 | 2017-09-12 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| US9228187B2 (en) | 2009-11-12 | 2016-01-05 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| US10287586B2 (en) | 2009-11-12 | 2019-05-14 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| US11447776B2 (en) | 2009-11-12 | 2022-09-20 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| US8637483B2 (en) | 2009-11-12 | 2014-01-28 | The University Of Western Australia | Antisense molecules and methods for treating pathologies |
| US10781450B2 (en) | 2009-11-12 | 2020-09-22 | Sarepta Therapeutics, Inc. | Antisense molecules and methods for treating pathologies |
| US10907154B2 (en) | 2013-03-14 | 2021-02-02 | Sarepta Therapeutics, Inc. | Exon skipping compositions for treating muscular dystrophy |
| US11932851B2 (en) | 2013-03-14 | 2024-03-19 | Sarepta Therapeutics, Inc. | Exon skipping compositions for treating muscular dystrophy |
| US9217148B2 (en) | 2013-03-14 | 2015-12-22 | Sarepta Therapeutics, Inc. | Exon skipping compositions for treating muscular dystrophy |
| US10337003B2 (en) | 2013-03-15 | 2019-07-02 | Sarepta Therapeutics, Inc. | Compositions for treating muscular dystrophy |
| US9506058B2 (en) | 2013-03-15 | 2016-11-29 | Sarepta Therapeutics, Inc. | Compositions for treating muscular dystrophy |
| US10364431B2 (en) | 2013-03-15 | 2019-07-30 | Sarepta Therapeutics, Inc. | Compositions for treating muscular dystrophy |
| WO2014206582A2 (fr) | 2013-06-27 | 2014-12-31 | Nivarox-Far S.A. | Ressort d'horlogerie en acier inoxydable austenitique |
| DE202014005288U1 (de) | 2013-06-27 | 2014-07-11 | Nivarox-Far S.A. | Uhrfeder aus austenitischem Edelstahl |
| EP2924514A1 (fr) | 2014-03-24 | 2015-09-30 | Nivarox-FAR S.A. | Ressort d'horlogerie en acier inoxydable austénitique |
| WO2017175842A1 (ja) * | 2016-04-07 | 2017-10-12 | 国立大学法人京都大学 | スプライシングの改変に関する化合物及び医薬組成物 |
| US12023338B2 (en) | 2017-02-20 | 2024-07-02 | Kyoto University | Pharmaceutical composition and treatment method for genetic disease associated with splicing abnormalities |
| WO2018151326A1 (ja) * | 2017-02-20 | 2018-08-23 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| JP2022078297A (ja) * | 2017-02-20 | 2022-05-24 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| JP7129095B2 (ja) | 2017-02-20 | 2022-09-01 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| JP7360206B2 (ja) | 2017-02-20 | 2023-10-12 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| JPWO2018151326A1 (ja) * | 2017-02-20 | 2019-12-12 | 国立大学法人京都大学 | スプライシング異常に起因する遺伝性疾病のための医薬組成物及び治療方法 |
| EP3720466A1 (en) * | 2017-12-05 | 2020-10-14 | Royal Holloway And Bedford New College | Treatment of muscular dystrophies |
| US11611446B2 (en) * | 2019-09-24 | 2023-03-21 | Genetec Inc. | Intermediary device for daisy chain and tree configuration in hybrid data/power connection |
| US20210091966A1 (en) * | 2019-09-24 | 2021-03-25 | Genetec Inc. | Intermediary device for daisy chain and tree configuration in hybrid data/power connection |
| US12316468B2 (en) | 2019-09-24 | 2025-05-27 | Genetec Inc. | Intermediary device for daisy chain and tree configuration in hybrid data/power connection |
| US11770155B2 (en) | 2020-05-19 | 2023-09-26 | Genetec Inc. | Power distribution and data routing in a network of devices interconnected by hybrid data/power links |
| WO2022255411A1 (ja) * | 2021-06-01 | 2022-12-08 | 国立大学法人京都大学 | 自然免疫応答を増強させる方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9241929B2 (en) | 2016-01-26 |
| JPWO2012001941A1 (ja) | 2013-08-22 |
| EP2586462A4 (en) | 2013-12-11 |
| CN103096928A (zh) | 2013-05-08 |
| US20130102644A1 (en) | 2013-04-25 |
| JP5950818B2 (ja) | 2016-07-13 |
| EP2586462A1 (en) | 2013-05-01 |
| CN106994124A (zh) | 2017-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5950818B2 (ja) | 遺伝性疾患の予防・改善剤 | |
| Hordeaux et al. | MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates | |
| Guy et al. | Defects in tRNA anticodon loop 2′‐O‐methylation are implicated in nonsyndromic X‐linked intellectual disability due to mutations in FTSJ1 | |
| Sheriff et al. | ABE8e adenine base editor precisely and efficiently corrects a recurrent COL7A1 nonsense mutation | |
| US11866784B2 (en) | Exonic splicing enhancers and exonic splicing silencers | |
| Xu et al. | Pathogenicity of intronic and synonymous variants of ATP7B in Wilson disease | |
| JP2021526819A (ja) | Bag3遺伝子治療の最適化 | |
| Paris et al. | A polymorphism that delays fibrosis in hepatitis C promotes alternative splicing of AZIN1, reducing fibrogenesis | |
| US20240279666A1 (en) | Methods of treating alzheimer's disease | |
| JP5686730B2 (ja) | 心臓肥大を抑制、遅延、および/または予防するための方法、および医薬組成物 | |
| Yaméogo et al. | Removal of the GAA repeat in the heart of a Friedreich’s ataxia mouse model using CjCas9 | |
| KR20220021906A (ko) | 유전자 요법 모니터링 | |
| Hernández-Imaz et al. | Functional analysis of mutations in exon 9 of NF1 reveals the presence of several elements regulating splicing | |
| EP3458587A1 (en) | Means for modulating gene expression | |
| Waugh et al. | Association between regulator of G protein signaling 9–2 and body weight | |
| Gong et al. | KLF6/Sp1 initiates transcription of the tmsg‐1 gene in human prostate carcinoma cells: An exon involved mechanism | |
| Yang et al. | Liquid-liquid phase separation of RBM33 facilitates hippocampus aging by inducing microglial senescence by activating CDKN1A | |
| US20260102465A1 (en) | Compositions and methods utilizing a novel human foxo3 isoform | |
| Chao et al. | Beneficial bystander-enhanced cryptic splice rescue of cardiac-type Fabry GLA IVS4+ 919G> A by adenine base editing in patient fibroblasts | |
| US11992515B2 (en) | Method for treating pulmonary fibrosis using S100A3 protein | |
| JP7301326B2 (ja) | 新生児期~小児期発症の脳小血管病又はその保因者の検出方法 | |
| WO2018118808A1 (en) | Methods of treating autism spectrum disorders | |
| Zhang et al. | A conserved hormonal signalling–H2A. Z axis rapidly reorganizes 3D chromatin interactions in adipocyte thermogenesis | |
| EP4486450A1 (en) | Methods and compositions to regulate mapt splicing for modeling and treatment of tauopathies | |
| WO2025080779A2 (en) | Methods and compositions for altering gene expression |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180030327.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11800411 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012522455 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13805151 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011800411 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |