WO2012017328A2 - Combination pharmaceutical composition and methods of treating and preventing the infectious diseases - Google Patents
Combination pharmaceutical composition and methods of treating and preventing the infectious diseases Download PDFInfo
- Publication number
- WO2012017328A2 WO2012017328A2 PCT/IB2011/002470 IB2011002470W WO2012017328A2 WO 2012017328 A2 WO2012017328 A2 WO 2012017328A2 IB 2011002470 W IB2011002470 W IB 2011002470W WO 2012017328 A2 WO2012017328 A2 WO 2012017328A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- leu
- ser
- lys
- gin
- glu
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2815—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD8
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/40—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum bacterial
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/42—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum viral
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K41/00—Medicinal preparations obtained by treating materials with wave energy or particle radiation ; Therapies using these preparations
- A61K41/0004—Homeopathy; Vitalisation; Resonance; Dynamisation, e.g. esoteric applications; Oxygenation of blood
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/04—Drugs for disorders of the respiratory system for throat disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/241—Tumor Necrosis Factors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/249—Interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2812—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
- A61K2039/507—Comprising a combination of two or more separate antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Definitions
- the present invention relates to a pharmaceutical composition and method of treating and preventing the infectious diseases, including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- infectious diseases including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- the invention relates to the area of medicine and may be used for the treatment and preventing the infectious diseases, including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- infectious diseases including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- U.S. Patent No. 7,582,294 discloses a medicament for treating Benign Prostatic Hyperplasia or prostatitis by administration of a homeopathically activated form of antibodies to prostate specific antigen (PSA).
- PSA prostate specific antigen
- Ultra-low doses of antibodies to gamma interferon have been shown to be useful in the treatment and prophylaxis of diseases of viral etiology. See U.S. Patent No. 7,572,441 , which is incorporated herein by reference in its entirety.
- the present invention is directed to a pharmaceutical composition and methods of its use in treatment and preventing of the infectious diseases , including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- infectious diseases including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- the solution to the existing problem is presented in form of a combination pharmaceutical composition for treatment and prophylaxis (prevetion) of infectious diseases, which comprises a) an activated-potentiated form of antibodies to cytokine and b) an activated-potentiated form of antibodies to receptor.
- the invention provides a combination pharmaceutical composition
- a combination pharmaceutical composition comprising a) an activated-potentiated form of an antibody to at least one cytokine and b) an activated-potentiated form of an antibody to at least one receptor.
- the pharmaceutical composition further comprises a solid carrier, wherein said activated-potentiated forms of antibodies are impregnated onto said solid carrier.
- the pharmaceutical composition is in the form of a tablet.
- the pharmaceutical composition including said activated- potentiated forms of antibodies is in the form of a mixture of C12, C30, and C200 homeopathic dilutions. It is specifically contemplated that said mixture of C12, C30, and C200 homeopathic dilutions is impregnated onto a solid carrier.
- the activated-potentiated forms of said antibodies may be activated- potentiated forms of a monoclonal, polyclonal or natural antibody. It is specifically contemplated that the activated-potentiated form of said antibodies is activated- potentiated form of a polyclonal antibody.
- the invention provides activated- potentiated forms of antibodies to antigen(s) having sequences described in the specification and claimed in the appended claims.
- the pharmaceutical composition includes activated-potentiated forms of antibodies prepared by successive centesimal dilutions coupled with shaking of every dilution. Vertical shaking is specifically contemplated.
- the invention provides a method of treating and preventing the infectious diseases, said method comprising administering to a patient in need thereof a) an activated-potentiated form of an antibody to at least one cytokine and b) an activated-potentiated form of an antibody to at least one receptor.
- the activated-potentiated forms of antibodies are administered in the form of pharmaceutical composition.
- the pharmaceutical composition is administered in the form of a solid oral dosage form which comprises a pharmaceutically acceptable carrier and an activated-potentiated form of an antibody to at least one cytokine and activated-potentiated form of an antibody to at least one receptor, said activated- potentiated forms impregnated onto said carrier.
- said solid oral dosage form is a tablet.
- the pharmaceutical composition may be administered in one to three unit dosage forms, each of the dosage form being administered from once daily to six times daily.
- the pharmaceutical composition is administered twice daily, each administration consisting of two oral dosage forms.
- the pharmaceutical composition is administered in one to two unit dosage forms, each of the dosage forms being administered twice daily.
- the pharmaceutical composition is administered in one to two unit dosage forms, each of the dosage forms being administered four times daily. All variants and embodiments described with respect to the composition aspect of the invention may be used with the method aspect of the invention.
- antibody as used herein shall mean an immunoglobulin that specifically binds to, and is thereby defined as complementary with, a particular spatial and polar organization of another molecule.
- Antibodies as recited in the claims may include a complete immunoglobulin or fragment thereof, may be natural, polyclonal or monoclonal, and may include various classes and isotypes, such as IgA, IgD, IgE, lgG1 , lgG2a, lgG2b and lgG3, IgM, etc. Fragments thereof may include Fab, Fv and F(ab')2, Fab', and the like.
- the singular "antibody” includes plural “antibodies.”
- activated-potentiated form or “potentiated form” respectively, with respect to antibodies recited herein is used to denote a product of homeopathic potentization of any initial solution of antibodies.
- Homeopathic potentization denotes the use of methods of homeopathy to impart homeopathic potency to an initial solution of relevant substance.
- 'homeopathic potentization may involve, for example, repeated consecutive dilutions combined with external treatment, particularly vertical (mechanical) shaking. In other words, an initial solution of antibody is subjected to consecutive repeated dilution and multiple vertical shaking of each obtained solution in accordance with homeopathic technology.
- the preferred concentration of the initial solution of antibody in the solvent ranges from about 0.5 to about 5.0 mg/ml.
- the preferred procedure for preparing each component, i.e. antibody solution is the use of the mixture of three aqueous or aqueous-alcohol dilutions of the primary matrix solution (mother tincture) of antibodies diluted 100 12 , 100 30 and 100 200 times, respectively, which is equivalent to centesimal homeopathic dilutions (C12, C30, and C200) or the use of the mixture of three aqueous or aqueous-alcohol dilutions of the primary matrix solution of antibodies diluted 100 12 , 100 30 and 100 50 times, respectively, which is equivalent to centesimal homeopathic dilutions (C12, C30 and C50).
- an antibody is in the "activated-potentiated” or “potentiated” form when three factors are present.
- the "activated-potentiated” form of the antibody is a product of a preparation process well accepted in the homeopathic art.
- the "activated-potentiated” form of antibody must have biological activity determined by methods well accepted in modern pharmacology.
- the biological activity exhibited by the "activated potentiated” form of the antibody cannot be explained by the presence of the molecular form of the antibody in the final product of the homeopathic process.
- the activated potentiated form of antibodies may be prepared by subjecting an initial, isolated antibody in a molecular form to consecutive multiple dilutions coupled with an external impact, such as mechanical shaking.
- the external treatment in the course of concentration reduction may also be accomplished, for example, by exposure to ultrasonic, electromagnetic, or other physical factors.
- V. Schwabe "Homeopathic medicines", M., 1967, U.S. Patents Nos. 7,229,648 and 4,311 ,897 which are incorporated by reference in their entirety and for the purpose stated, describe such processes that are well-accepted methods of homeopathic potentiation in the homeopathic art. This procedure gives rise to a uniform decrease in molecular concentration of the initial molecular form of the antibody.
- the required homeopathic potency can be determined by subjecting the intermediate dilutions to biological testing in the desired pharmacological model.
- 'homeopathic potentization may involve, for example, repeated consecutive dilutions combined with external treatment, particularly vertical (mechanical) shaking.
- an initial solution of antibody is subjected to consecutive repeated dilution and multiple vertical shaking of each obtained solution in accordance with homeopathic technology.
- the preferred concentration of the initial solution of antibody in the solvent preferably, water or a water-ethyl alcohol mixture, ranges from about 0.5 to about 5.0 mg/ml.
- the preferred procedure for preparing each component i.e.
- antibody solution is the use of the mixture of three aqueous or aqueous-alcohol dilutions of the primary matrix solution (mother tincture) of antibodies diluted 100 12 , 100 30 and 100 200 times, respectively, which is equivalent to centesimal homeopathic dilutions C12, C30 and C200 or the mixture of three aqueous or aqueous-alcohol dilutions of the primary matrix solution (mother tincture) of antibodies diluted 100 12 , 100 30 and 100 50 times, respectively, which is equivalent to centesimal homeopathic dilutions C12, C30 and C50.
- Examples of how to obtain the desired potency are also provided, for example, in U.S. Patent Nos.
- the claimed "activated-potentiated” form of antibody encompasses only solutions or solid preparations the biological activity of which cannot be explained by the presence of the molecular form of the antibody remaining from the initial, starting solution.
- the "activated-potentiated” form of the antibody may contain traces of the initial molecular form of the antibody, one skilled in the art could not attribute the observed biological activity in the accepted pharmacological models to the remaining molecular form of the antibody with any degree of plausibility due to the extremely low concentrations of the molecular form of the antibody remaining after the consecutive dilutions.
- the biological activity of the "activated-potentiated' form of the antibodies of the present invention is not attributable to the initial molecular form of the antibody.
- Preferred is the "activated-potentiated” form of antibody in liquid or solid form in which the concentration of the molecular form of the antibody is below the limit of detection of the accepted analytical techniques, such as capillary electrophoresis and High Performance Liquid Chromatography.
- Particularly preferred is the "activated-potentiated” form of antibody in liquid or solid form in which the concentration of the molecular form of the antibody is below the Avogadro number.
- the "activated-potentiated" form of the antibodies contains molecular antibody, if any, at a concentration below the threshold dose for the molecular form of the antibody in the given biological model.
- the present invention provides a combination pharmaceutical composition that includes activated-potentiated form of antibodies to cytokine and activated- potentiated form of antibodies to receptor, prepared according to the homeopathic technology of potentiation by repeated, consistent dilution and intermediate external action of shaking as described in more detail herein below.
- the pharmaceutical composition of the invention is particularly useful in the treatment and prophylaxis of the infectious diseases, including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, flu of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- infectious diseases including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, flu of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- the pharmaceutical composition of the invention possesses unexpected synergetic therapeutic effect, which manifest itself in particular therapeutic effectiveness in treating and preventing the infectious diseases, including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- infectious diseases including bacterial infections caused by different infectious agents such as pseudotuberculosis, whooping cough, yersiniosis, pneumonitis of different etiology, and acute and chronic viral infections such as acute respiratory tract infections, influenza of different types, acute viral hepatitis A, B, C and other types of hepatitis, the diseases and conditions caused by HIV or associated with HIV, including AIDS.
- the pharmaceutical composition of the invention expands the arsenal of preparations available for the treatment prophylaxis of the infectious diseases, including bacterial infections and acute and chronic viral infections.
- the combination pharmaceutical composition in accordance with this aspect of the invention may be in the liquid form or in solid form.
- Activated- potentiated form of the antibodies included in the pharmaceutical composition is prepared from an initial molecular form of the antibody via a process accepted in homeopathic art.
- the starting antibodies may be monoclonal, or polyclonal antibodies prepared in accordance with known processes, for example, as described in Immunotechniques, G. Frimel, M., "Meditsyna", 1987, p. 9-33; "Hum. Antibodies. Monoclonal and recombinant antibodies, 30 years after" by Laffly E., Sodoyer R. - 2005 - Vol. 14. - N 1-2. P.33-55, both incorporated herein by reference.
- Monoclonal antibodies may be obtained, e.g., by means of hybridoma technology.
- the initial stage of the process includes immunization based on the principles already developed in the course of polyclonal antisera preparation. Further stages of work involve the production of hybrid cells generating clones of antibodies with identical specificity. Their separate isolation is performed using the same methods as in the case of polyclonal antisera preparation.
- Polyclonal antibodies may be obtained via active immunization of animals.
- suitable animals e.g. rabbits
- the animals' immune system generates corresponding antibodies, which are collected from the animals in a known manner. This procedure enables preparation of a monospecific antibody-rich serum.
- the serum containing antibodies may be purified, for example by using affine chromatography, fractionation by salt precipitation, or ion-exchange chromatography.
- the resulting purified, antibody-enriched serum may be used as a starting material for the preparation of the activated-potentiated form of the antibodies.
- the preferred concentration of the resulting initial solution of antibody in the solvent preferably water or a water-ethyl alcohol mixture, ranges from about 0.5 to about 5.0 mg/ml.
- the preferred procedure for preparing each component of the combination drug according to the present invention is the use of the mixture of three aqueous- alcohol dilutions of the primary matrix solution of antibodies diluted 100 12 , 100 30 and 100 50 times, respectively, which is equivalent to centesimal homeopathic dilutions C12, C30, and C50 or diluted 100 12 , 100 30 and 100 200 times, respectively, which is equivalent to centesimal homeopathic dilutions C12, C30 and C200.
- a solid carrier is treated with the desired dilution obtained via thehomeopathic process.
- the carrier mass is impregnated with each of the dilutions.
- the starting material for the preparation of the activated potentiated form that comprise the combination pharmaceutical composition of the invention is polyclonal, animal-raised antibody to the corresponding antigen.
- the desired antigen may be injected as immunogen into a laboratory animal, preferably, rabbits.
- Polyclonal antibodies to CD4 receptor may be obtained using the whole molecule of human CD4 receptor of the following sequence:
- polypeptide fragment of CD4 receptor chosen, for example, from the following amino-acid sequences:
- Lys lie Asp lie Val Val Leu Ala Phe Gin Lys Ala Ser Ser lie
- the exemplary procedure for preparation of the starting polyclonal antibodies to CD4 receptor may be described as follows. In 7-9 days before blood sampling, 1 -3 intravenous injections of the desired antigen are made to the rabbits to increase the level of polyclonal antibodies in the rabbit blood stream. Upon immunization, blood samples are taken to test the antibody level. Typically, the maximum level of immune reaction of the soluble antigen is achieved within 40 to 60 days after the first injection of the antigen. Upon completion of the first immunization cycle, rabbits have a 30-day rehabilitation period, after which re-immunization is performed with another 1 -3 intravenous injections.
- the immunized rabbits' blood is collected from rabbits and placed in a 50ml centrifuge tube.
- Product clots formed on the tube sides are removed with a wooden spatula, and a rod is placed into the clot in the tube center.
- the blood is then placed in a refrigerator for one night at the temperature of about 40°C.
- the clot on the spatula is removed, and the remaining liquid is centrifuged for 10 min at 13,000 rotations per minute. Supernatant fluid is the target antiserum.
- the obtained antiserum is typically yellow.
- 10 ml of the antiserum of rabbits is diluted twofold with 0.15 M NaCI, after which 6.26g Na 2 S0 4 is added, mixed and incubated for 12-16 hours at 4°C.
- the sediment is removed by centrifugation, diluted in 10ml of phosphate buffer and dialyzed against the same buffer during one night at ambient temperature. After the sediment is removed, the solution is applied to a DEAE-cellulose column balanced by phosphate buffer.
- the antibody fraction is determined by measuring the optical density of the eluate at 280 nm.
- the isolated crude antibodies are purified using affine chromatography method by attaching the obtained antibodies to CD4 antigen located on the insoluble matrix of the chromatography media, with subsequent elution by concentrated aqueous salt solutions.
- the resulting buffer solution is used as the initial solution for the homeopathic dilution process used to prepare the activated potentiated form of the antibodies.
- the preferred concentration of the initial matrix solution of the antigen-purified polyclonal rabbit antibodies to CD4 receptor is 0.5 to 5.0 mg/ml, preferably, 2.0 to 3.0 mg/ml.
- polyclonal antibodies to gamma interferon may also be obtained by a similar methodology to the methodology described for CD4 receptor antibodies using an adjuvant.
- Polyclonal antibodies to gamma interferon may be obtained using the whole molecule of gamma interferon of the following sequence:
- Polyclonal antibodies to gamma interferon may be obtained using the whole molecule of gamma interferon of the following sequence:
- gamma interferon fragments as antigen is also contemplated.
- the suitable sequence for such antigen is as follow:
- Polyclonal antibodies to gamma interferon may be obtained using the molecule of recombinant gamma interferon of one of the following sequences: SEQ ID NO: 18
- polyclonal antibodies to alpha interferon may also be obtained by a similar methodology to the methodology described for CD4 receptor antibodies using an adjuvant.
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 8 of the following sequence:
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 2 of the following sequence: SEQ ID NO: 21
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 1 7 of the following sequence: SEQ ID NO: 22
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 4 of the following sequence: SEQ ID NO: 23
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 21 of the following sequence: SEQ ID NO: 24
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type1/13 of the following sequence: SEQ ID NO: 25
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 10 of the following sequence: SEQ ID NO: 26
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 5 of the following sequence: SEQ ID NO: 27
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 7 of the following sequence: SEQ ID NO: 28
- Polyclonal antibodies to alpha interferon may be obtained using the whole molecule of human alpha interferon type 14 of the following sequence: SEQ ID NO: 29
- polyclonal antibodies to CD8 receptor may also be obtained by a similar methodology to the methodology described for CD4 receptor antibodies using an adjuvant.
- Polyclonal antibodies to CD8 receptor may be obtained using the whole molecule of CD8 receptor of the following sequence:
- CD8 receptor fragments as antigen is also contemplated.
- suitable sequences for such antigen are as follow:
- Polyclonal antibodies to tumor necrosis factor alpha may be obtained by the above-mentioned method of obtaining antibodies to CD4 receptor using a whole molecule of tumor necrosis factor alpha of the following sequence:
- tumor necrosis factor alpha TNF-a
- a polypeptide fragment of the tumor necrosis factor selected, for example, from the following sequences:
- polyclonal antibodies to histamine which is a biogenic amine (4(2- aminoethyl)-imidazole or beta-imidazolylethylamine with the chemical formula C5H9N3), may be obtained by the above-mentioned method of obtaining antibodies to CD4 using adjuvant and industrially produced histamine dihydrochloride as immunogen (antigen) for immunization of rabbits.
- the activated-potentiated form of an antibody to cytokine or receptor may be prepared from an initial solution by homeopathic potentization, preferably using the method of proportional concentration decrease by serial dilution of 1 part of each preceding solution (beginning with the initial solution) in 9 parts (for decimal dilution), or in 99 parts (for centesimal dilution), or in 999 parts (for millesimal dilution) of a neutral solvent, starting with a concentration of the initial solution of antibody in the solvent, preferably, water or a water- ethyl alcohol mixture, in the range from about 0.5 to about 5.0 mg/ml, coupled with external impact.
- the external impact involves multiple vertical shaking (dynamization) of each dilution.
- a 12-centesimal dilution (denoted C12) one part of the initial matrix solution of antibodies to CD4 receptor with the concentration of 3.0 mg/ml is diluted in 99 parts of neutral aqueous or aqueous-alcohol solvent (preferably, 15%-ethyl alcohol) and then vertically shaked many times (10 and more) to create the 1st centesimal dilution (denoted as C1 ).
- the 2nd centesimal dilution (C2) is prepared from the 1st centesimal dilution C1. This procedure is repeated 1 1 times to prepare the 12th centesimal dilution C12.
- the 12th centesimal dilution C12 represents a solution obtained by 12 serial dilutions of one part of the initial matrix solution of antibodies to gamma interferon with the concentration of 3.0 mg/ml in 99 parts of a neutral solvent in different containers, which is equivalent to the centesimal homeopathic dilution C12. Similar procedures with the relevant dilution factor are performed to obtain dilutions C30, C50 and C 200. The intermediate dilutions may be tested in a desired biological model to check activity.
- the preferred activated-potentiated form for the composition of the invention are a mixture of C12, C30, and C50 dilutions or C12, C30 and C200 dilutions.
- each component of the composition e.g., C12, C30, C50, C200
- the next-to-last dilution is obtained (e.g., until C1 1 , C29, and C199 respectively)
- one part of each component is added in one container according to the mixture composition and mixed with the required quantity of the solvent (e.g. with 97 parts for centesimal dilution).
- the active substance is possible to use as mixture of various homeopathic dilutions, e.g. decimal and/or centesimal (D20, C30, C100 or C 2, C30, C50 or C12, C30, C200, etc.), the efficiency of which is determined experimentally by testing the dilution in a suitable biological model, for example, in models described in the examples herein.
- various homeopathic dilutions e.g. decimal and/or centesimal (D20, C30, C100 or C 2, C30, C50 or C12, C30, C200, etc.
- the vertical shaking may be substituted for external exposure to ultrasound, electromagnetic field or any similar external impact procedure accepted in the homeopathic art.
- the pharmaceutical composition of the invention may be in the form of a liquid or in the solid unit dosage form.
- the preferred liquid carrier is water or water-ethyl alcohol mixture.
- the solid unit dosage form of the pharmaceutical composition of the invention may be prepared by impregnating a solid, pharmaceutically acceptable carrier with the mixture of the activated potentiated form aqueous or aqueous-alcohol solutions of active component.
- the carrier may be impregnated consecutively with each requisite dilution. Both orders of impregnation are acceptable.
- the pharmaceutical composition in the solid unit dosage form is prepared from granules of the pharmaceutically acceptable carrier which was previously saturated with the aqueous or aqueous-alcoholic dilutions of the activated potentiated form of antibodies to at least one cytokine and activated potentiated form of antibodies to at least one receptor.
- the solid dosage form may be in any form known in the pharmaceutical art, including a tablet, a capsule, a lozenge, and others.
- an inactive pharmaceutical ingredients one can use glucose, sucrose, maltose, amylum, isomaltose, isomalt and other mono- olygo- and polysaccharides used in manufacturing of pharmaceuticals as well as technological mixtures of the above mentioned inactive pharmaceutical ingredients with other pharmaceutically acceptable excipients, for example isomalt, crospovidone, sodium cyclamate, sodium saccharine, anhydrous citric acid etc), including lubricants, disintegrants, binders and coloring agents.
- the preferred carriers are lactose and isomalt.
- the pharmaceutical dosage form may further include standard pharmaceutical excipients, for example, microcrystalline cellulose, magnesium stearate and citric acid.
- lactose 100-300 m granules of lactose are impregnated with aqueous or aqueous-alcoholic solutions of the activated-potentiated forms of antibodies in the ratio of 1 kg of antibody solution to 5 or 10 kg of lactose (1 :5 to 1 :10).
- the lactose granules are exposed to saturation irrigation in the fluidized boiling bed in a boiling bed plant (e.g. "Huttlin Pilotlab" by Huttlin GmbH) with subsequent drying via heated air flow at a temperature below 40"C.
- the estimated quantity of the dried granules (10 to 34 weight parts) saturated with the activated potentiated form of antibodies is placed in the mixer, and mixed with 25 to 45 weight parts of "non-saturated" pure lactose (used for the purposes of cost reduction and simplification and acceleration of the technological process without decreasing the treatment efficiency), together with 0.1 to 1 weight parts of magnesium stearate, and 3 to 10 weight parts of microcrystalline cellulose.
- the obtained tablet mass is uniformly mixed, and tableted by direct dry pressing (e.g., in a Korsch - XL 400 tablet press) to form 150 to 500 mg round pills, preferably, 300 mg.
- aqueous-alcohol solution (3.0-6.0 mg/pill) of the activated-potentiated form of antibodies in the form of a mixture of centesimal homeopathic dilutions C12, C30, and C50 or a mixture of centesimal homeopathic dilutions C12, C30 and C200. While the invention is not limited to any specific theory, it is believed that the activated potentiated form of the antibodies described herein do not contain the molecular form of the antibody in an amount sufficient to have biological activity attributed to such molecular form.
- the biological activity of the combination drug (pharmaceutical composition) of the invention is amply demonstrated in the appended examples.
- the combination of the invention is administered from once daily to six times daily, preferably twice daily or four times daily, each administration including one or three combination unit dosage forms.
- the sigma-1 (o 1 ) receptor - an intracellular one which is localized in the cells of central nervous system, the cells of the most of peripheral tissues and immune competent cells.
- This receptor via control of homeostasis of intracellular calcium regulates intracellular signaling events leading to activation of the corresponding transcription factors and transcription of a whole gene family coding in particular the factors of resistance to infectious agents and cytokines.
- the ability of drugs to influence to the efficiency of interaction of ligands with sigma-1 receptor indicates the presence of antiviral and immunomodulating components in the spectrum of its pharmacological activity that allows to consider these preparations as effective ones for the treatment and prophylaxis of various infectious diseases.
- Results are represented as percentage of specific binding inhibition in control (distilled water was used as control) (Table 1 ).
- % of specific binding inhibition in control 100% - (specific binding during the test/specific binding in control) * 100%).
- TCID50 50% Tissue Culture Infective Dose
- Mononuclear cells of peripheral blood of a human being were separated from blood of healthy seronegative donor through centrifugation in density gradient ficcol-gipaque.
- the cells were activated during 3 days with use of 1 mkg/ml phytohemagglutinin P and 5 ME/ml of recombinant interleukine-2 of a human being in medium RPMI 1640 (DIFCO) with 10% fetal calf serum (complement was removed through heating during 45 minutes within temperature of 56°C), 1 % solution of antibiotics (PSN Gibco containing 50 pg/ml penicillin, 50 pg/ml streptomycin and 100 ⁇ /ml neomycin).
- combination medications were introduced to well 15-30 minutes after contamination of cells with strain HIV-1 - LAI with dose of 100 TCID50 (50 mkl inoculums of strain HIV-1 -LAI). On the 7 th day after infection of cells supernatant used for evaluation of influence of medications to inhibition of HIV replication was selected.
- Efficiency medications was defined on inhibition of HIV replication that was evaluated on enzymatic activity of HIV-reverse transcriptase in supernatants of macrophages of peripheral blood of a human being with use of HIV RT RetroSys production set INNOVAGEN (lot 10-059C). For calculation of % of inhibition of HIV replication as control supernatant of cells was used to which tested medications were not introduced (see Table 2).
- Macrophages of donor peripheral blood received from mononuclear cells of human peripheral blood were isolated from blood of two healthy seronegative donors through centrifugation in density gradient ficcol-gipaque. Mononuclear cells of human peripheral blood were grown for 3 days in medium RPMI1640 (DIFCO) that was added with 10% fetal calf serum (complement was removed through heating during 45 minutes within temperature of 56°C), 1 % solution of antibiotics (PSN Gibco containing 50 pg /ml penicillin, 50 pg/ml streptomycin and 100 pg /ml neomycin), 15 ng/ml GM-CSF (granulocyte macrophagal colony-stimulating factor).
- DIFCO medium RPMI1640
- antibiotics PSN Gibco containing 50 pg /ml penicillin, 50 pg/ml streptomycin and 100 pg /ml neomycin
- 15 ng/ml GM-CSF gran
- GM-CSF granulocyte macrophagal colony-stimulating factor
- M-CSF macrophagal colony-stimulating factor
- combination medications were introduced to well 24 hours before contamination of cells with strain HIV-1 -LAI with dose of 1000 TCID50 (100 mkl inoculums of strain HIV-1 -Ba-L) and on the 3 rd , 7 th , 10 ,h , 14 ,h , 17 th day after contamination. On the 3 rd , 7 th , 10 th , 14 th , 17 th day after infection of cells supernatant used for evaluation of influence of medications to inhibition of HIV replication was selected.
- ULD AB to IFN gamma tested as mono-component that allows to make comparison of efficiency of complex drug with its separate components.
- Azidothymidine was diluted with medium RPMI 1640 (DIFCO) till concentration of 8 nM was achieved.
- Efficiency medications was defined by inhibition of HIV replication that was evaluated on enzymatic activity of HIV-reverse transcriptase in supernatants of supernatant macrophages of peripheral blood of a human being with use of HIV RT RetroSys production set INNOVAGEN (lot 10-059C).
- Antiretroviral activity of complex medication exceeds antiretroviral activity of its separate components (ULD AB to IFN gamma and ULD AB to CD4).
- Antiretroviral activity of complex medication is lasted during the whole experiment period in contrast to antiretroviral activity of its separate components (ULD AB to IFN gamma and ULD AB to CD4).
- Example 4 (mononuclear cells; reverse transcriptase; therapy regimen)
- TCID50 50% Tissue Culture Infective Dose.
- the assessment of antiretroviral activity of a complex product consisting of ultra low-dose rabbit polyclonal antibodies to interferon alpha, ultra low-dose rabbit polyclonal antibodies to interferon gamma, ultra low-dose rabbit polyclonal antibodies to CD4 and ultra low-dose rabbit polyclonal antibodies to CD8 as 1 : 1 : 1 : 1 ratio (a mixture of homoeopathic dilutions C 12+C30+C50) (hereinafter referred to as the Complex product), was carried out using human peripheral blood mononuclear cells infected with the strain HIV-1 LAI in vitro. Azidothymidine (Sigma - AZ169-100 mg, Lot 107 K1578) was used as a comparator product.
- Human peripheral blood mononuclear cells were isolated from blood of a seronegative healthy donor by centrifugation on a Ficoll-Hypaque density gradient. The cells were stimulated for 3 days with 1 pg/mL of phytohemagglutinin P and 5 l U/mL of recombinant human interleukin-2 in RPMI 1640 (DIFCO) medium supplemented with 10% fetal calf serum (the complement was removed by heating for 45 minutes at 56°C), 1 % antibiotic solution (PSN Gibco containing 50 pg/mL of penicillin, 50 pg/mL of streptomycin and 100 pg/mL of neomycin).
- the products were placed in a well 15-30 minutes after cells infection with the strain HIV-1 - LAI at the dose of 100 TCID50 (50 pL inoculum of the strain HIV-1 -LAI). Supernatant fluids used to assess the effect of products on the inhibition of HIV replication were also collected on day 7 after infection of cells.
- the complex product was diluted with RPMI 1640 (DIFCO) medium at a 4-fold dilution (at a 1/4 dilution) to a final volume of 50 pL.
- Azidothymidine was diluted with RPMI 1640 (DIFCO) medium to yield a 8 nM concentration.
- the products' efficiency was established by the inhibition of HIV replication which was assessed by HIV-reverse transcriptase activity in the supernatant fluid from human peripheral blood mononuclear cells using the H IV RT RetroSys kit made by INNOVAGEN (Lot 10-059C).
- this experimental model demonstrated the antiretroviral activity of the complex product comprising ultra low-dose rabbit polyclonal antibodies to interferon alpha, ultra low-dose rabbit polyclonal antibodies to interferon gamma, ultra low-dose rabbit polyclonal antibodies to CD4 and ultra low-dose rabbit polyclonal antibodies to CD8 as 1 : 1 : 1 : 1 ratio (a mixture of homoeopathic dilutions C12+C30+C50).
- Example 5 (mononuclear cells; nucleocapsid protein p24; prevention and therapy regimen)
- the assessment of antiretroviral activity of ultra low-dose of rabbit polyclonal antibodies to interferon-alpha (a mixture of homoeopathic dilutions C12+C30+C50), ultra low-dose of rabbit polyclonal antibodies to interferon- gamma (a mixture of homoeopathic dilutions C12+C30+C50) ⁇ ULD /F/V-y)
- ultra low-dose of rabbit polyclonal antibodies to CD4 receptor a mixture of homoeopathic dilutions C1 2+C30+C50
- ultra low-dose of rabbit polyclonal antibodies to CD8 receptor (ULD Ab IFN-a+ IFN-y+CD4+CD8) was carried out using human peripheral blood mononuclear cells infected with the strain HIV-1 LAI in vitro.
- Human peripheral blood mononuclear cells were isolated from blood of a seronegative healthy donor by centrifugation on a Ficoll-Hypaque density gradient. The cells were stimulated for 3 days with 1 pg/mL of phytohemagglutinin P and 5 lU/mL of recombinant human interleukin-2.
- the products were placed in a well containing 100 pL of activated mononuclears 24 hours before or 15 min after cell infection with the strain HIV-1 - LAI at the dose of 100 TCID50 (50 pL inoculum of the strain HIV-1 -LAI).
- ULD Ab IFN-a+ IFN- y+CD4+CD8 ( 12.5 pL) or reference azidotimidine ( 1000 nM) were mixed with RPMI 1640 medium (DIFCO) to achive a final probe volume of 50 pL
- the supernatant fluids were collected on day 7 after infection of cells.
- the products' activity was measured by the inhibition of HIV replication which was assessed by the level of core nucleocapsid protein p24 in the supernatant fluid from human peripheral blood mononuclear cells using Retrotek Elisa kit.
- ULD Ab IFN-a+ IFN-y+CD4+CD8 inhibited HIV replication by 94 ⁇ 6% when added to a well 24 hours before the infection, a and by 46 ⁇ 13 % when added to a well 15 min after the infection of cells with the strain HIV-1 LAI.
- Azidotimidine at a dose of 1000 nM inhibited HIV replication by 99 ⁇ 0 and 99 ⁇ 1 % added to a well 24 hours before and 15 min after the infection of cells with the strain HIV-1 LAI, respectively.
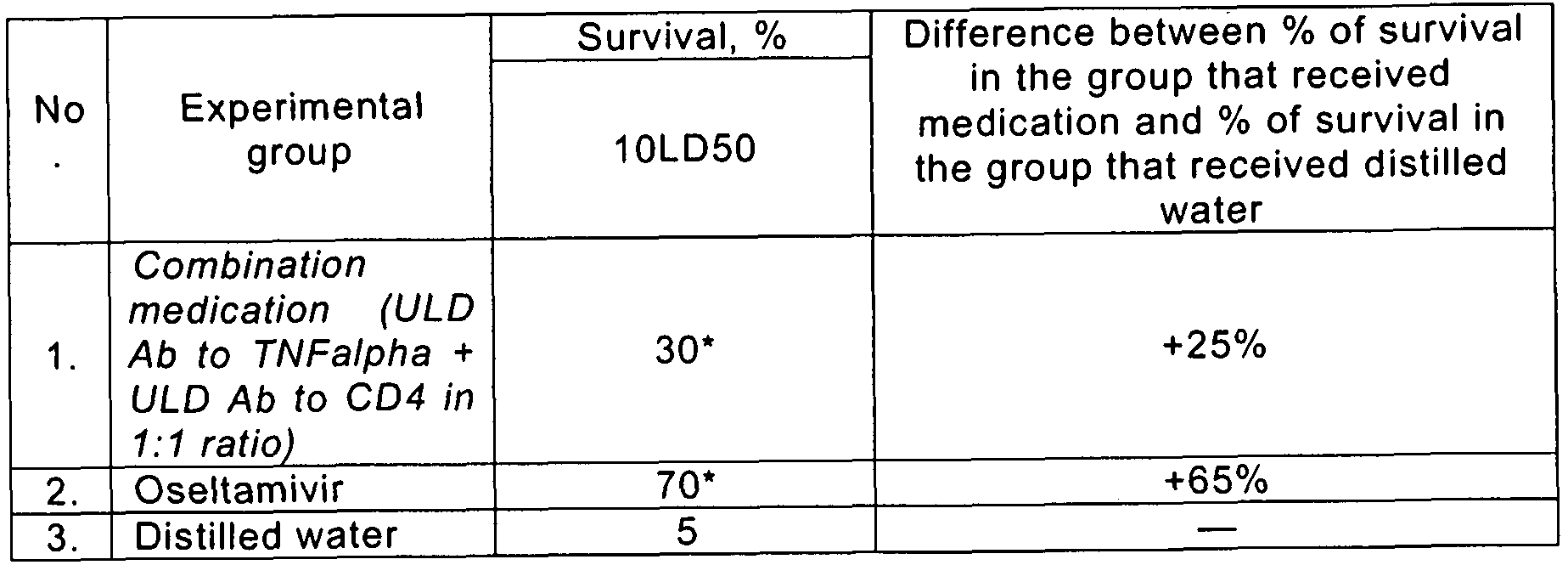
- Infectious process was simulated through intranasal introduction of influenza virus A/California/07/2009swl (H 1 N 1 ) with a dose 10LD50.
- Antiviral activity combination medication containing ULD AB to IFNalpha and ULD AB to CD4 in the model of influenza infection at female Balb/c mice infected through intranasal introduction of influenza virus A/California/07/2009swl (H 1 N 1 ) with a dose of 10LD50 (10 th day after infection).
- combination medication increased in 25% survival of experimental animals infected with influenza virus A/California/07/2009swl (H 1 N 1 ) with a dose of 10LD50 as compared with control.
- survival in the group that received ULD AB to IFNalpha was only 5% higher than the survival in control group
- survival rate in the group received ULD AB to CD4 was only 10% higher than the survival in control group.
- ULD AB to IFNalpha and ULD AB to CD4 provides more pronounced antiviral effect than separate components, in spite of the fact that the dose of ULD AB to IFNalpha and ULD AB to CD4 as part of combination medication is twice lower than the dose of ULD AB to IFNalpha and ULD AB to CD4 tested as separate medications.
- Infectious process was simulated through intranasal introduction of influenza virus A/California/07/2009swl (H 1 N 1 ) with a dose 10LD50.
- ULD AB to TNFalpha, ULD AB to CD4 and combination medication were added to drinking bowls of animals of corresponding experimental groups (free access was allowed).
- Antiviral activity combination medication containing ULD Ab to TNFalpha and ULD Ab to CD4 in the model of influenza infection at female Balb/c mice infected through intranasal introduction of influenza virus A/California/07/2009swl (H1 N 1 ) with a dose of 10LD50 (10 th day after infection).
- combination medication increased in 25% survival of experimental animals infected with influenza virus A/California/07/2009swl (H 1 N 1 ) with a dose of 10LD50 as compared with control.
- survival in the group that received ULD Ab to TNFalpha was only 5% higher than the survival in control group
- survival rate in the group received ULD Ab to CD4 was only 1 0% higher than the survival in control group.
- compositions containing activated potentiated form of ultra-low doses (ULD) antibodies to interferon gamma (Ab IFNgamma), antibodies to CD4 (Ab CD4), antibodies to histamine (Ab His), impregnated onto lactose in the form of aqueous alcoholic solution of mixture of homeopathic dilutions C12, C30, C200 of each (Ab IFNgamma + Ab CD4 +Ab to His) was used in the study.
- ULD ultra-low doses
- Ab IFNgamma antibodies to interferon gamma
- Ab CD4 antibodies to CD4
- Ab His antibodies to histamine
- catarrh signs are enrolled. Patients with body temperature 37.8 °C and higher (provided that the temperature is registered at the onset of the disease), with the duration of the disease not exceeding 48 hours by the time of the therapy onset, not having severe complications were included in the study. Express test to detect influenza virus antigens was conducted. Patients with positive test results were not included in the study. Prior to the beginning of all the procedures the patients sign Informed consent to participate in the study. The patients were given diaries, in which body temperature twice daily, concomitant therapy, etc were registered.
- the patients receive Ab IFNgamma + Ab CD4 +Ab His or placebo at a dose of 8 tablets daily on Day 1 and at a dose of 3 tablets daily on Days 2-5. If required the patients were allowed to take antipyretics. The intake of antiviral, immunomodulating, antihistamines and antibiotics is not allowed. Prior to start of therapy and at the last visit blood and urine samples are collected for assessment of laboratory parameters aimed at monitoring the safety of the conducted therapy. The overall therapy duration is 5 days, the duration of follow-up observation period is 2 days. Thus the duration of each patient's participation in the study is 7 days.
- Time to reducing body temperature down to 37.0 °C and lower was considered as the therapy efficacy criterion; besides the number of antipyretics intakes was compared.
- Table 1 Mean values of body temperature in patients depending on
- Day 4 Day 5 , Day 5 Day 6 , Day 6 Day 7 , evening morning evening morning evening morning
- compositions containing activated potentiated forms of ultra-low doses (ULD) antibodies to interferon - gamma (Ab IFNgamma), antibodies to CD4 (Ab to CD4), antibodies to histamine (Ab to His), impregnated onto lactose in the form of aqueous alcoholic mixture of homeopathic dilutions C12, C30, C200 of each (Ab IFNgamma + Ab CD4 +Ab His) was used in the study.
- ULD ultra-low doses
- CD4 antibodies to CD4
- Ab to His antibodies to histamine
- the patients were given diaries, in which body temperature twice daily, concomitant therapy, etc were registered.
- the patients receive Ab IFNgamma + Ab CD4 +Ab His at a dose of 8 tablets daily on Day 1 and at a dose of 3 tablets daily on Days 2-5 or or Tamiflu at a dose of 75 mg 2 TID according to patient's information leaflet. If required the patients were allowed to take antipyretics. The intake of antiviral, immunomodulating, antihistamines and antibiotics is not allowed.
- blood and urine samples Prior to start of therapy and at the last visit blood and urine samples are collected for assessment of laboratory parameters aimed at monitoring the safety of the conducted therapy.
- the overall therapy duration is 5 days, the duration of follow-up observation period is 2 days. Thus the duration of each patient's participation in the study is 7 days.
- Time to reducing body temperature down to 37.0 °C and lower was considered as the therapy efficacy criterion; besides the number of antipyretics intakes was compared.
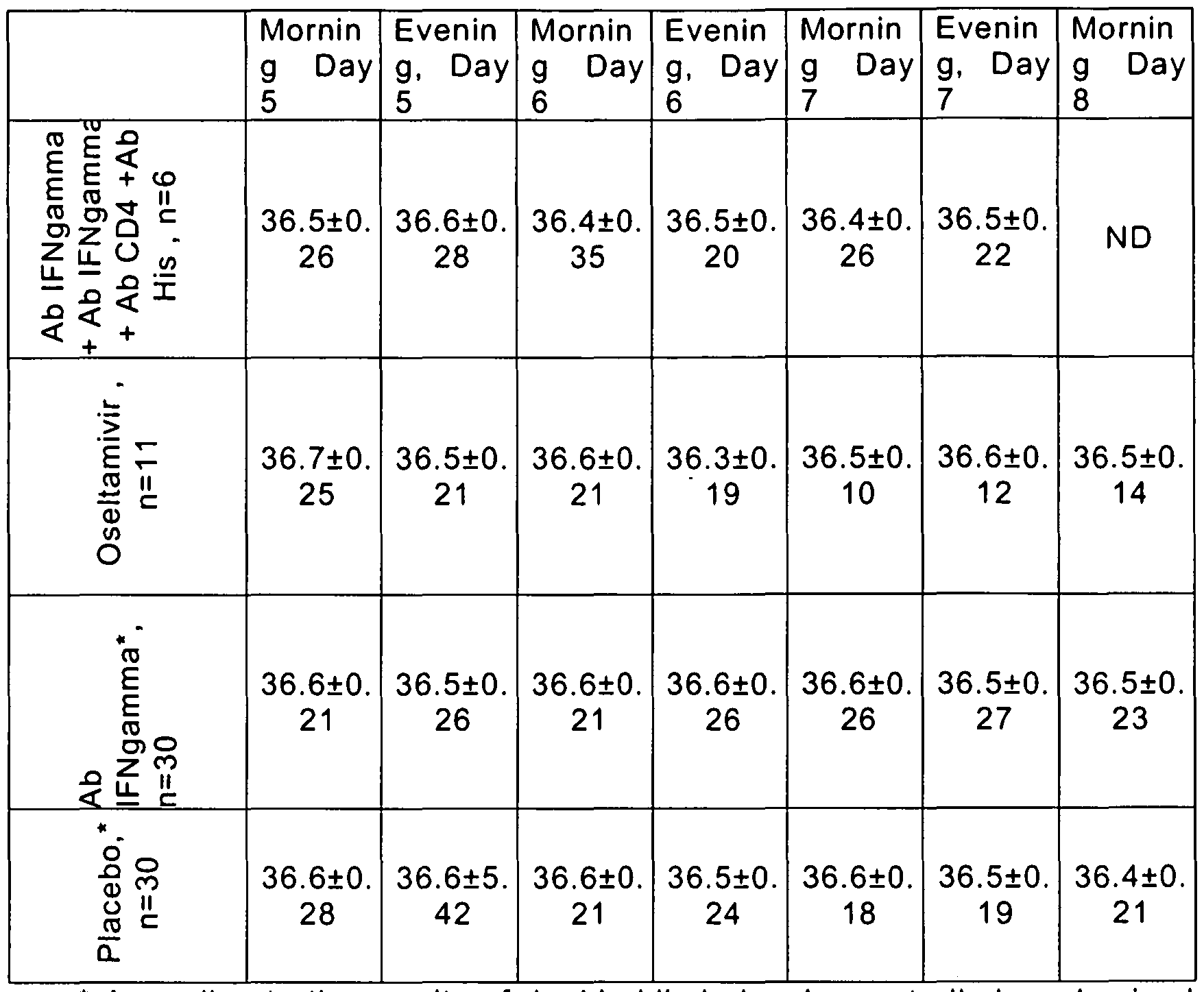
- Proportions of patients with the body temperature reduced down to 37.0°C and lower in the groups did not significantly differ in the course of therapy. As early as by Day 4 of the treatment patients of both groups practically recovered (see Figure 2). As early as by Day 2 of the treatment in 1/3 of patients of both groups normalization of body temperature was registered. The difference in mean number of antipyretic intakes also was not significant and by the morning of Day 4 of the therapy was 7.6 ⁇ 0.8 in the group receiving Ab IFNgamma + Ab CD4 +Ab His and 7.4 ⁇ 0.90 in Oseltamivir group respectively.
- Table 1 Mean values of body temperatu re in patients depending on treatment groups, °C, M ⁇ SD
- ⁇ i 38.1 ⁇ 37.3 ⁇ 37.3 ⁇ 36.9 ⁇ 36.9 ⁇ 36.7 ⁇ 36.8 ⁇ 36.5 ⁇ jS ii 0.82 0.71 0.72 0.53 0.47 0.46 0.37 0.38 % c
- compositions containing activated potentiated forms of ultra-low doses (ULD) antibodies to interferon - gamma (Ab IFNgamma), antibodies to CD4 (Ab to CD4), antibodies to histamine (Ab to His), impregnated onto lactose in the form of aqueous alcoholic mixture of homeopathic dilutions C12, C30, C200 of each (Ab IFNgamma + Ab CD4 +Ab His) was used in the study.
- ULD ultra-low doses
- CD4 antibodies to CD4
- Ab to His antibodies to histamine
- ULD anti-IFN- ⁇ + anti-CD4 and ULD anti-IFN- ⁇ + anti-CD4 + anti-His has been evaluated in the course of the open-label comparative clinical trial with participation of the human immunodeficiency virus (HIV) infected patients at Local Centre for Prevention and Fight against AIDS and Infectious Diseases.
- HIV human immunodeficiency virus
- ART antiretroviral therapy
- CD4 M CD8 lymphocytes counts, CD4/CD8 immunoregulatory index were assayed in all the patients.
- COBAS AMPLICOR HIV-1 MONITOR Kit version 1 ,5 for automatic PCR-analyzer COBAS AMPLICOR, Roche, Switzerland
- Phenotyping of peripheral blood circulating lymphocytes was carried out on flow cytofluorometer FACSCount (Becton Dickinson, USA) using FACSCount Reagent Kit, which contain FITC PE fluorochrome-labeled antibodies to CD3, CD4, CD8.
- ULD anti-IFN- ⁇ monotherapy for 6 weeks decreased the number of HIV-1 RNA copies in 36% treatment naive patients (in 4 out of 1 1 people in 3A subgroup), the average viral load decrease was 9.5%.
- the combination of ULD anti-IFN- ⁇ and ART improved the efficacy of therapy: the viral load decrease was registered in 50% of patients (in 8 out of 16 people in 3B subgroup), the average viral load decrease was 14.2%. In patients taking only ART (group No 4) the decrease in viral load were detected in 32% of patients (in 7 out of 22 patients) and an average viral load decrease 13.3%.
- the present study demonstrated antiretroviral activity of ULD anti- IFN- Y + anti-CD4 and ULD anti-IFN- ⁇ + anti-CD4 + anti-His pharmaceutical compositions, possibly mediated by the change in functional activity of CD4 receptors, which blocks HIV penetration into the cells, and also suppresses HIV replication inside the cell due to activation of transcription of mRNA of antiviral proteins.
- compositions containing ULD anti-IFN- ⁇ + anti-CD4 and ULD anti-IFN- ⁇ + anti-CD4 + anti- His makes it possible to use them for the treatment and prophylaxis of HIV infection both in treatment naive HIV-infected patients and in patients taking ART.
- Control group consisted of 5 patients with persistent viremia and stable normal levels of aminotransferases ( ⁇ 20 U/l) received no specific therapy. During the study course regular examinations, control of viral load and laboratory rates were carried out, concomitant therapy was registered as well as undesirable adverse events. Therapy efficacy was assessed on week 24 by viral load with HCV RNA and activity of alanine-aminotransferase (ALT).
- ALT alanine-aminotransferase
- Antiviral activity of the studies pharmaceutical compositions was accompanied with positive changes in ALT level registered in patients of groups 1 -3 by the end of 24-week therapy. Normalization of ALT level was found in 2 patients of ULD anti- IFN-Y+anti-CD4 group, in 1 patient of ULD anti- IFN-Y+anti-CD4+anti-His group and in 1 patient ULD anti- IFN- ⁇ group. In 1 patient of control group ALT level exceeded upper border of norm (>20 U/l) due to an increase of viral load at the end of 24-week study period. No drugs-related adverse events were registered during the study, which evidences of their good tolerance. Absence of pathological variations in blood and urine analysis including markers of renal and hepatic insufficiency confirmed safety of the treatment.
- ULD anti- IFN-y+anti-CD4 ULD anti- IFN-y+anti-CD4+anti- His and ULD anti- IFN- ⁇ was accompanied with a reduction of activity of chronic hepatitis C, which was confirmed by the reduction and even normaluzation of ALT level in some patients at the end of 24-week course of treatment.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Alternative & Traditional Medicine (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Biotechnology (AREA)
- Gastroenterology & Hepatology (AREA)
- Otolaryngology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description














Claims
Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11778688.9A EP2601219A2 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| CA2807529A CA2807529A1 (en) | 2011-07-15 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| MX2013001451A MX368313B (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases. |
| GB1303867.4A GB2503066B8 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| CN201180048456XA CN103154030A (en) | 2010-08-06 | 2011-07-15 | Compound pharmaceutical composition and method for treating and preventing infectious diseases |
| NZ606993A NZ606993A (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| JP2013522318A JP2013537532A (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and method for treating and preventing infectious diseases |
| ES201390022A ES2510940R1 (en) | 2010-08-06 | 2011-07-15 | Combined pharmaceutical composition and its use to prepare a medicine for the treatment and prevention of infectious diseases |
| PH1/2013/500232A PH12013500232A1 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| KR1020137005740A KR20140012021A (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| BR112013002296A BR112013002296A2 (en) | 2010-08-06 | 2011-07-15 | pharmaceutical composition and its use and method for treating infectious disease |
| SG2013008974A SG187732A1 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| EA201300135A EA030513B1 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and method of treating and preventing infectious diseases |
| DE112011102640T DE112011102640T5 (en) | 2010-08-06 | 2011-07-15 | Pharmaceutical combination composition and methods for the treatment and prevention of infectious diseases |
| UAA201300103A UA112748C2 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| AU2011287292A AU2011287292B2 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| IL224545A IL224545A (en) | 2010-08-06 | 2013-02-03 | Combination pharmaceutical composition for treating and preventing infectious diseases |
Applications Claiming Priority (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2010133050 | 2010-08-06 | ||
| RU2010133051/15A RU2505312C2 (en) | 2010-08-06 | 2010-08-06 | Combined therapeutic agent for treating various types of influenza |
| RU2010133052/15A RU2500422C2 (en) | 2010-08-06 | 2010-08-06 | Combined drug for treating viral infections and method of treating viral infections |
| RU2010133047/15A RU2517085C2 (en) | 2010-08-06 | 2010-08-06 | Complex medication and method of preventing hiv infection, prevention and treatment of hiv-induced and hiv-associated diseases, including aids |
| RU2010133050/15A RU2502521C2 (en) | 2010-08-06 | 2010-08-06 | Combined drug for treating bacterial infections and method of treating bacterial infections |
| RU2010133041 | 2010-08-06 | ||
| RU2010133053 | 2010-08-06 | ||
| RU2010133041/15A RU2010133041A (en) | 2010-08-06 | 2010-08-06 | INTEGRATED MEDICINE AND METHOD FOR PREVENTION OF HIV INFECTION, PREVENTION AND TREATMENT OF DISEASES CAUSED BY HIV OR ASSOCIATED WITH HIV, INCLUDING AIDS |
| RU2010133053/15A RU2521392C2 (en) | 2010-08-06 | 2010-08-06 | Combined drug for treating viral infections and method of treating viral infections |
| RU2010133052 | 2010-08-06 | ||
| RU2010133051 | 2010-08-06 | ||
| RU2010133043/15A RU2519862C2 (en) | 2010-08-06 | 2010-08-06 | Complex medication and method of prevention and treatment of infectious viral diseases |
| RU2010133047 | 2010-08-06 | ||
| RU2010133043 | 2010-08-06 | ||
| RU2011127226/15A RU2577299C2 (en) | 2011-07-04 | 2011-07-04 | Method for treating infectious diseases and complex medication for treatment of infectious diseases |
| RU2011127226 | 2011-07-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012017328A2 true WO2012017328A2 (en) | 2012-02-09 |
| WO2012017328A3 WO2012017328A3 (en) | 2012-04-05 |
Family
ID=45507292
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2011/002470 Ceased WO2012017328A2 (en) | 2010-08-06 | 2011-07-15 | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US20130045237A1 (en) |
| EP (1) | EP2601219A2 (en) |
| JP (3) | JP2013537532A (en) |
| KR (1) | KR20140012021A (en) |
| CN (1) | CN103154030A (en) |
| AU (1) | AU2011287292B2 (en) |
| BR (1) | BR112013002296A2 (en) |
| CZ (1) | CZ2013159A3 (en) |
| DE (1) | DE112011102640T5 (en) |
| EA (1) | EA030513B1 (en) |
| ES (1) | ES2510940R1 (en) |
| FR (1) | FR2963563A1 (en) |
| GB (2) | GB2548034B (en) |
| IL (1) | IL224545A (en) |
| MX (1) | MX368313B (en) |
| NZ (1) | NZ606993A (en) |
| PE (1) | PE20131185A1 (en) |
| PH (1) | PH12013500232A1 (en) |
| SG (2) | SG10201403870XA (en) |
| WO (1) | WO2012017328A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015189711A3 (en) * | 2014-06-06 | 2016-02-18 | Oleg Iliich Epshtein | Veterinary composition and method of improving livability of animals, promoting live weight gain in mammals and birds, enhancing the effectiveness of immunization, and preventing and/or treating infectious diseases (variants) |
| GB2498276B (en) * | 2010-08-06 | 2018-05-23 | Iliich Epshtein Oleg | Medicinal agent and mode of prevention of HIV contamination, prevention and treatment of diseases caused by HIV and HIV-associated diseases including AIDS. |
| WO2021040570A1 (en) * | 2019-08-29 | 2021-03-04 | Oleg Iliich Epshtein | Medicament and method for treating infectious diseases |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2181297C2 (en) | 2000-06-20 | 2002-04-20 | Эпштейн Олег Ильич | Method of treatment of pathological syndrome and medicinal agent |
| UA76638C2 (en) * | 2002-08-02 | 2006-08-15 | Oleh Illich Epshtein | Homeopathic medication based on anti-interferon antibodies and method for treating a pathological syndrome associated with interferon |
| RU2309732C1 (en) * | 2006-03-13 | 2007-11-10 | Олег Ильич Эпштейн | Pressed solid oral formulation of medicinal preparation and method for preparing solid oral formulation of medicinal preparation |
| PH12013500107A1 (en) | 2010-07-15 | 2013-03-11 | Oleg Iliich Epshtein | A method of increasing the effect of an activated-potentiated form of an antibody |
| EP2593474A2 (en) | 2010-07-15 | 2013-05-22 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating diseases or conditions associated with neurodegenerative diseases |
| EA029199B1 (en) | 2010-07-21 | 2018-02-28 | Олег Ильич Эпштейн | Combination pharmaceutical composition for treating attention deficit hyperactivity disorder and methods of treating attention deficit hyperactivity disorder |
| PH12013500142A1 (en) * | 2010-07-21 | 2013-03-11 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating diseases or conditions associated with respiratory disease or condition |
| WO2012017328A2 (en) * | 2010-08-06 | 2012-02-09 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
| RU2013111962A (en) | 2013-03-18 | 2014-09-27 | Олег Ильич Эпштейн | METHOD FOR DETERMINING THE EXPRESSION OF MODIFICATION ACTIVITY ASSOCIATED WITH A CARRIER |
| RU2013111961A (en) | 2013-03-18 | 2014-09-27 | Олег Ильич Эпштейн | METHOD FOR DETERMINING THE EXPRESSION OF MODIFICATION ACTIVITY ASSOCIATED WITH A CARRIER |
| DE102020007979A1 (en) | 2020-12-29 | 2022-06-30 | Charité Universitätsmedizin Institut für Mikrobiologie und Infektionsimmunologie | Composition for treating coronavirus infections |
| US20250319115A1 (en) * | 2022-05-25 | 2025-10-16 | Flagship Pioneering Innovations Vii, Llc | Compositions and Methods for Modulating Cytokines |
| WO2023230249A1 (en) * | 2022-05-25 | 2023-11-30 | Baruch S. Blumberg Institute | Novel hepatoselective polyadenylating polymerases inhibitors and their method of use |
| WO2025088452A1 (en) * | 2023-10-24 | 2025-05-01 | Epshtein Oleg Ilyich | Artificial substance and method of its preparation |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4311897A (en) | 1979-08-28 | 1982-01-19 | Union Carbide Corporation | Plasma arc torch and nozzle assembly |
| RU2192888C1 (en) | 2001-02-15 | 2002-11-20 | Эпштейн Олег Ильич | Medicinal agent and method of treatment of pathological syndrome |
| US7229648B2 (en) | 2003-03-14 | 2007-06-12 | Dreyer Lee R | Homeopathic formulations useful for treating pain and/or inflammation |
| US7572441B2 (en) | 2002-08-02 | 2009-08-11 | Oleg Iliich Epshtein | Media and method for treating pathological syndrome |
| US7582294B2 (en) | 2002-08-02 | 2009-09-01 | Oleg Oliich Epshtein | Medicament for treating prostate diseases |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2181297C2 (en) * | 2000-06-20 | 2002-04-20 | Эпштейн Олег Ильич | Method of treatment of pathological syndrome and medicinal agent |
| RU2001134982A (en) * | 2001-12-26 | 2004-02-20 | Олег Ильич Эпштейн | METHOD OF IMMUNE RESPONSE CORRECTION AND MEDICINE |
| UA76640C2 (en) * | 2002-08-02 | 2006-08-15 | Олєг Ільіч Епштєйн | Method for correcting pathological immune responses and homeopathic medicinal agent |
| RU2309732C1 (en) * | 2006-03-13 | 2007-11-10 | Олег Ильич Эпштейн | Pressed solid oral formulation of medicinal preparation and method for preparing solid oral formulation of medicinal preparation |
| BRPI0712540A2 (en) * | 2006-06-06 | 2012-10-16 | Oleg Iliich Epshtein | medicinal agent for treating adiposity, diabetes, and diseases associated with glucose intolerance |
| WO2008078331A1 (en) * | 2006-12-22 | 2008-07-03 | Rajesh Shah | Medicinal formulation for the treatment for hepatitis c |
| RU2332236C1 (en) * | 2007-02-02 | 2008-08-27 | Олег Ильич Эпштейн | Medicine for bird influenza treatment |
| EP2593474A2 (en) * | 2010-07-15 | 2013-05-22 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating diseases or conditions associated with neurodegenerative diseases |
| EP2593477A2 (en) * | 2010-07-15 | 2013-05-22 | Oleg Iliich Epshtein | Pharmaceutical compositions and methods of treatment |
| JP2013536174A (en) * | 2010-07-21 | 2013-09-19 | イリイチ・エプシテイン オレグ | Combination pharmaceutical composition and method of treating rotational dizziness, motion sickness and autonomic neurovascular dystonia |
| KR20180127515A (en) * | 2010-07-21 | 2018-11-28 | 올레그 일리치 엡쉬테인 | A combination pharmaceutical composition and methods of treating diabetes and metabolic disorders |
| PH12013500142A1 (en) * | 2010-07-21 | 2013-03-11 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating diseases or conditions associated with respiratory disease or condition |
| WO2012017328A2 (en) * | 2010-08-06 | 2012-02-09 | Oleg Iliich Epshtein | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases |
-
2011
- 2011-07-15 WO PCT/IB2011/002470 patent/WO2012017328A2/en not_active Ceased
- 2011-07-15 AU AU2011287292A patent/AU2011287292B2/en not_active Ceased
- 2011-07-15 CN CN201180048456XA patent/CN103154030A/en active Pending
- 2011-07-15 EA EA201300135A patent/EA030513B1/en not_active IP Right Cessation
- 2011-07-15 NZ NZ606993A patent/NZ606993A/en not_active IP Right Cessation
- 2011-07-15 JP JP2013522318A patent/JP2013537532A/en active Pending
- 2011-07-15 US US13/135,899 patent/US20130045237A1/en not_active Abandoned
- 2011-07-15 PH PH1/2013/500232A patent/PH12013500232A1/en unknown
- 2011-07-15 MX MX2013001451A patent/MX368313B/en active IP Right Grant
- 2011-07-15 EP EP11778688.9A patent/EP2601219A2/en not_active Ceased
- 2011-07-15 ES ES201390022A patent/ES2510940R1/en active Pending
- 2011-07-15 GB GB1707875.9A patent/GB2548034B/en not_active Expired - Fee Related
- 2011-07-15 PE PE2013000216A patent/PE20131185A1/en not_active Application Discontinuation
- 2011-07-15 SG SG10201403870XA patent/SG10201403870XA/en unknown
- 2011-07-15 DE DE112011102640T patent/DE112011102640T5/en not_active Withdrawn
- 2011-07-15 KR KR1020137005740A patent/KR20140012021A/en not_active Ceased
- 2011-07-15 BR BR112013002296A patent/BR112013002296A2/en not_active Application Discontinuation
- 2011-07-15 CZ CZ20130159A patent/CZ2013159A3/en unknown
- 2011-07-15 SG SG2013008974A patent/SG187732A1/en unknown
- 2011-07-15 GB GB1303867.4A patent/GB2503066B8/en not_active Expired - Fee Related
- 2011-07-15 FR FR1156483A patent/FR2963563A1/en not_active Withdrawn
-
2013
- 2013-02-03 IL IL224545A patent/IL224545A/en active IP Right Grant
-
2014
- 2014-07-07 US US14/324,575 patent/US20150023972A1/en not_active Abandoned
- 2014-07-08 US US14/325,455 patent/US20150023980A1/en not_active Abandoned
-
2016
- 2016-07-01 JP JP2016131741A patent/JP2016216478A/en active Pending
-
2018
- 2018-04-26 JP JP2018085391A patent/JP2018135370A/en not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4311897A (en) | 1979-08-28 | 1982-01-19 | Union Carbide Corporation | Plasma arc torch and nozzle assembly |
| RU2192888C1 (en) | 2001-02-15 | 2002-11-20 | Эпштейн Олег Ильич | Medicinal agent and method of treatment of pathological syndrome |
| US7572441B2 (en) | 2002-08-02 | 2009-08-11 | Oleg Iliich Epshtein | Media and method for treating pathological syndrome |
| US7582294B2 (en) | 2002-08-02 | 2009-09-01 | Oleg Oliich Epshtein | Medicament for treating prostate diseases |
| US7229648B2 (en) | 2003-03-14 | 2007-06-12 | Dreyer Lee R | Homeopathic formulations useful for treating pain and/or inflammation |
Non-Patent Citations (3)
| Title |
|---|
| G. FRIMEL, M: "Meditsyna", IMMUNOTECHNIQUES, 1987, pages 9 - 33 |
| LAFFLY E., SODOYER R., HUM. ANTIBODIES. MONOCLONAL AND RECOMBINANT ANTIBODIES, vol. 14, no. 1-2., 2005, pages 33 - 55 |
| V. SCHWABE, HOMEOPATHIC MEDICINES, 1967, pages 14 - 29 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2498276B (en) * | 2010-08-06 | 2018-05-23 | Iliich Epshtein Oleg | Medicinal agent and mode of prevention of HIV contamination, prevention and treatment of diseases caused by HIV and HIV-associated diseases including AIDS. |
| WO2015189711A3 (en) * | 2014-06-06 | 2016-02-18 | Oleg Iliich Epshtein | Veterinary composition and method of improving livability of animals, promoting live weight gain in mammals and birds, enhancing the effectiveness of immunization, and preventing and/or treating infectious diseases (variants) |
| WO2021040570A1 (en) * | 2019-08-29 | 2021-03-04 | Oleg Iliich Epshtein | Medicament and method for treating infectious diseases |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2011287292B2 (en) | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases | |
| US8637034B2 (en) | Pharmaceutical compositions comprising activated-potentiated antibodies to interferon-gamma and S100 protein | |
| RU2577299C2 (en) | Method for treating infectious diseases and complex medication for treatment of infectious diseases | |
| US20120294899A1 (en) | Pharmaceutical composition and methods of treating and preventing the diseases caused by HIV or associated with HIV | |
| AU2011286486B9 (en) | Drug and method for the prophylaxis of HIV infection and for the prophylaxis and treatment of diseases caused by or associated with HIV, including aids | |
| US20120263725A1 (en) | Pharmaceutical composition and methods of treating and preventing the diseases caused by HIV or associated with HIV | |
| US20120263726A1 (en) | Pharmaceutical composition and method of inhibiting of production or amplifying of elimination of P24 protein | |
| RU2517085C2 (en) | Complex medication and method of preventing hiv infection, prevention and treatment of hiv-induced and hiv-associated diseases, including aids | |
| CA2807529A1 (en) | Combination pharmaceutical composition and methods of treating and preventing the infectious diseases | |
| WO2012017327A2 (en) | Pharmaceutical composition and methods of treating and preventing the diseases caused by hiv or associated with hiv |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180048456.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11778688 Country of ref document: EP Kind code of ref document: A2 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2013000323 Country of ref document: CL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12013500232 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2807529 Country of ref document: CA Ref document number: 2013522318 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000216-2013 Country of ref document: PE Ref document number: 1120111026404 Country of ref document: DE Ref document number: 112011102640 Country of ref document: DE Ref document number: MX/A/2013/001451 Country of ref document: MX Ref document number: P201390022 Country of ref document: ES |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201300135 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011778688 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: GE Ref document number: GE P |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13009 Country of ref document: GE |
|
| ENP | Entry into the national phase |
Ref document number: 1303867 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20110715 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201300103 Country of ref document: UA Ref document number: 1303867.4 Country of ref document: GB Ref document number: PV2013-159 Country of ref document: CZ |
|
| ENP | Entry into the national phase |
Ref document number: 20137005740 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2011287292 Country of ref document: AU Date of ref document: 20110715 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013/03364 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13404 Country of ref document: GE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013002296 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013002296 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130130 |