WO2012024363A2 - Compounds and methods for the treatment or prevention of flaviviridae viral infections - Google Patents
Compounds and methods for the treatment or prevention of flaviviridae viral infections Download PDFInfo
- Publication number
- WO2012024363A2 WO2012024363A2 PCT/US2011/048027 US2011048027W WO2012024363A2 WO 2012024363 A2 WO2012024363 A2 WO 2012024363A2 US 2011048027 W US2011048027 W US 2011048027W WO 2012024363 A2 WO2012024363 A2 WO 2012024363A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- methyl
- thiophene
- compound
- ynyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)(C)C#Cc1cc([*+2])c(C(OC)=O)[s]1 Chemical compound CC(C)(C)C#Cc1cc([*+2])c(C(OC)=O)[s]1 0.000 description 3
- MDDXNVPRRYHPMO-UHFFFAOYSA-N CC(C)(C)C#Cc1cc(N)c(C(OC)=O)[s]1 Chemical compound CC(C)(C)C#Cc1cc(N)c(C(OC)=O)[s]1 MDDXNVPRRYHPMO-UHFFFAOYSA-N 0.000 description 1
- CZIPGOWPFOFKIK-UHFFFAOYSA-N CC(C)(C)C#Cc1cc(NCc2n[n](C)cc2)c(C(OC)=O)[s]1 Chemical compound CC(C)(C)C#Cc1cc(NCc2n[n](C)cc2)c(C(OC)=O)[s]1 CZIPGOWPFOFKIK-UHFFFAOYSA-N 0.000 description 1
- NBLSEVGWZSYUIY-UHFFFAOYSA-N CC(C)C#Cc1cc(N(C2CCOCC2)C(C2CCC(C)CC2)=O)c(C(O)=O)[s]1 Chemical compound CC(C)C#Cc1cc(N(C2CCOCC2)C(C2CCC(C)CC2)=O)c(C(O)=O)[s]1 NBLSEVGWZSYUIY-UHFFFAOYSA-N 0.000 description 1
- QMUIRCOWVKHUAH-UHFFFAOYSA-N CC(CC1)CCC1C(CN(Cc1ccccc1)c1c(C(O)=O)[s]c(C#CC(C)(C)C)c1)=O Chemical compound CC(CC1)CCC1C(CN(Cc1ccccc1)c1c(C(O)=O)[s]c(C#CC(C)(C)C)c1)=O QMUIRCOWVKHUAH-UHFFFAOYSA-N 0.000 description 1
- OQZPMWIQUJXTBE-UHFFFAOYSA-N CC(CC1)CCC1C(N(Cc1ccccc1)c1c(C(OC)=O)[s]c(C#CC(C)(C)C)c1)=O Chemical compound CC(CC1)CCC1C(N(Cc1ccccc1)c1c(C(OC)=O)[s]c(C#CC(C)(C)C)c1)=O OQZPMWIQUJXTBE-UHFFFAOYSA-N 0.000 description 1
- ABWNOEXXCMPYOH-UHFFFAOYSA-N CC(CC1)CCC1C(Nc1c(C(OC)=O)[s]c(C#CC(C)(C)C)c1)=O Chemical compound CC(CC1)CCC1C(Nc1c(C(OC)=O)[s]c(C#CC(C)(C)C)c1)=O ABWNOEXXCMPYOH-UHFFFAOYSA-N 0.000 description 1
- WFZJWUSNPZSECP-PTEKRPQWSA-N C[C@@H](CC1)CC(C(CCC2)C[C@@H]2O)[C@H]1N Chemical compound C[C@@H](CC1)CC(C(CCC2)C[C@@H]2O)[C@H]1N WFZJWUSNPZSECP-PTEKRPQWSA-N 0.000 description 1
- YYCDPZLXDFZOMG-KEAIWBDQSA-N C[C@@H](CCC1)CC1C(C[C@@H](CC1)F)C1Nc1c(CC=O)[s]c(C#CC(C)(C)C)c1 Chemical compound C[C@@H](CCC1)CC1C(C[C@@H](CC1)F)C1Nc1c(CC=O)[s]c(C#CC(C)(C)C)c1 YYCDPZLXDFZOMG-KEAIWBDQSA-N 0.000 description 1
- DYBSWDVIMGONLG-JOCQHMNTSA-N C[C@H](CC1)CC[C@@H]1NN(C(CC1)CCC1=O)c1c(C(OC)=O)[s]c(Br)c1 Chemical compound C[C@H](CC1)CC[C@@H]1NN(C(CC1)CCC1=O)c1c(C(OC)=O)[s]c(Br)c1 DYBSWDVIMGONLG-JOCQHMNTSA-N 0.000 description 1
- OYKLZQSJHWCIRB-GAZRBOARSA-N C[C@H](CC1)CC[C@@H]1NN(C1CC=CCC1)c1c(C(O)=O)[s]c(C#CC(C)(C)C)c1 Chemical compound C[C@H](CC1)CC[C@@H]1NN(C1CC=CCC1)c1c(C(O)=O)[s]c(C#CC(C)(C)C)c1 OYKLZQSJHWCIRB-GAZRBOARSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4535—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom, e.g. pizotifen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/212—IFN-alpha
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- HCV Hepatitis C virus
- HCV is believed to replicate through the production of a complementary negative-strand RNA template. Due to the lack of efficient culture replication system for the virus, HCV particles were isolated from pooled human plasma and shown, by electron microscopy, to have a diameter of about 50-60 nm.
- the HCV genome is a single-stranded, positive-sense RNA of about 9,600 bp coding for a polyprotein of 3009-3030 amino-acids, which is cleaved co and post-translationally into mature viral proteins (core, El, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B). It is believed that the structural glycoproteins, El and E2, are embedded into a viral lipid envelope and form stable heterodimers. It is also believed that the structural core protein interacts with the viral RNA genome to form the nucleocapsid.
- the nonstructural proteins designated NS2 to NS5 include proteins with enzymatic functions involved in virus replication and protein processing including a polymerase, protease and helicase.
- HCV infection The main source of contamination with HCV is blood.
- the magnitude of the HCV infection as a health problem is illustrated by the prevalence among high-risk groups. For example, 60% to 90% of hemophiliacs and more than 80% of intravenous drug abusers in western countries are chronically infected with HCV. For intravenous drug abusers, the prevalence varies from about 28% to 70% depending on the population studied. The proportion of new HCV infections associated with post-transfusion has been markedly reduced lately due to advances in diagnostic tools used to screen blood donors. [0004] Combination of pegylated interferon plus ribavirin is the treatment of choice for chronic HCV infection. This treatment does not provide sustained viral response (SVR) in a majority of patients infected with the most prevalent genotype (la and lb). Furthermore, significant side effects prevent compliance to the current regimen and may require dose reduction or discontinuation in some patients.
- SVR sustained viral response
- the present invention generally relates to compounds useful for treating or preventing Flaviviridae infections, such as HCV infections.
- the invention is directed to a compound selected from the structural formulae depicted below or in FIG. 1, or a pharmaceutically acceptable salt thereof:
- the invention is directed to a compound depicted below or or a pharmaceutically acceptable salt thereof:
- the invention is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention described herein and a pharmaceutically acceptable carrier or excipient.
- the invention provides methods of treating a HCV infection in a subject, comprising administering to the subject a therapeutically effective amount of a compound of the invention described herein.
- the invention is directed to a method of inhibiting or reducing the activity of HCV polymerase in a subject, comprising administering to the subject a therapeutically effective amount of a compound of the invention described herein.
- the invention is directed to a method of inhibiting or reducing the activity of HCV polymerase in a biological in vitro sample, comprising administering to the sample an effective amount of a compound of the invention described herein.
- the present invention also provides use of the compounds of the invention descrbed herein for the manufacture of the medicament for treating a HCV infection in a subject, or for inhibiting or reducing the activity of HCV polymerase in a subject.
- Also provided herein is use of the compounds of the invention descrbed herein for treating a HCV infection in a subject, or for inhibiting or reducing the activity of HCV polymerase in a subject.
- FIG. l shows certain compounds of the invention.
- the present invention is directed to compounds represented by the structural formulae depicted in FIG.1 and Exemplification below, or pharmaceutically acceptable salts thereof.
- the compounds are selected from the following structural formulae or pharmaceutically acceptable salts thereof:
- the compounds are selected from the following structural formulae or pharmaceutically acceptable salts thereof:
- the compound is selected from the following structural formula or a pharmaceutically acceptable salt thereof:
- compounds of the invention may optionally be substituted with one or more substituents, such as illustrated generally below, or as exemplified by particular classes, subclasses, and species of the compounds described above.
- substituents such as illustrated generally below, or as exemplified by particular classes, subclasses, and species of the compounds described above.
- the phrase “optionally substituted” is used interchangeably with the phrase “substituted or unsubstituted.”
- substituted refers to the replacement of one or more hydrogen radicals in a given structure with the radical of a specified substituent.
- an optionally substituted group may have a substituent at each substitutable position of the group.
- substituent When more than one position in a given structure can be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at each position. When the term “optionally substituted” precedes a list, said term refers to all of the subsequent substitutable groups in that list. If a substituent radical or structure is not identified or defined as “optionally substituted", the substituent radical or structure is unsubstituted. For example, if X is optionally substituted C 1 -C 3 alkyl or phenyl; X may be either optionally substituted C 1 -C 3 alkyl or optionally substituted phenyl.
- up to refers to zero or any integer number that is equal or less than the number following the phrase.
- up to 3 means any one of 0, 1, 2, and 3.
- a specified number range of atoms includes any integer therein. For example, a group having from 1 -4 atoms could have 1 , 2, 3, or 4 atoms.
- a stable compound or chemically feasible compound is one that is not substantially altered when kept at a temperature of 40°C or less, in the absence of moisture or other chemically reactive conditions, for at least a week. Only those choices and
- aliphatic or "aliphatic group”, as used herein, means a straight-chain (i.e., unbranched), or branched, hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation but is non-aromatic. Unless otherwise specified, aliphatic groups contain 1-10 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms. In other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms. Aliphatic groups may be linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups.
- Specific examples include, but are not limited to, methyl, ethyl, isopropyl, n-propyl, sec -butyl, vinyl, n-butenyl, ethynyl, and tert- butyl and acetylene.
- alkyl as used herein means a saturated straight .or branched chain hydrocarbon.
- alkenyl as used herein means a straight or branched chain hydrocarbon comprising one or more double bonds.
- alkynyl as used herein means a straight or branched chain hydrocarbon comprising one or more triple bonds.
- Each of the "alkyl”, “alkenyl” or “alkynyl” as used herein can be optionally substituted as set forth below.
- the "alkyl” is C 1 -C 6 alkyl or C 1 -C 4 alkyl.
- the "alkenyl” is C 2 -C 6 alkenyl or C 2 -C 4 alkenyl.
- the "alkynyl” is C 2 -C 6 alkynyl or C 2 -C 4 alkynyl.
- cycloaliphatic refers to a non-aromatic carbon only containing ring system which can be saturated or contains one or more units of unsaturation, having three to fourteen ring carbon atoms.
- the number of carbon atoms is 3 to 10.
- the number of carbon atoms is 4 to 7.
- the number of carbon atoms is 5 or 6.
- the term includes monocyclic, bicyclic or polycyclic, fused, spiro or bridged carbocyclic ring systems.
- the term also includes polycyclic ring systems in which the carbocyclic ring can be "fused" to one or more non-aromatic carbocyclic or heterocyclic rings or one or more aromatic rings or combination thereof, wherein the radical or point of attachment is on the carbocyclic ring.
- "Fused" bicyclic ring systems comprise two rings which share two adjoining ring atoms.
- Bridged bicyclic group comprise two rings which share three or four adjacent ring atoms.
- Spiro bicyclic ring systems share one ring atom.
- Examples of cycloaliphatic groups include, but are not limited to, cycloalkyl and
- cycloalkenyl groups include, but are not limited to, cyclohexyl, cyclopropenyl, and cyclobutyl.
- heterocycle refers to a non-aromatic ring system which can be saturated or contain one or more units of unsaturation, having three to fourteen ring atoms in which one or more ring carbons is replaced by a heteroatom such as, N, S, or O.
- non-aromatic heterocyclic rings comprise up to three heteroatoms selected from N, S and O within the ring.
- non-aromatic heterocyclic rings comprise up to two heteroatoms selected from N, S and O within the ring system.
- non-aromatic heterocyclic rings comprise up to three heteroatoms selected from N and O within the ring system. In yet other embodiments, non-aromatic heterocyclic rings comprise up to two heteroatoms selected from N and O within the ring system.
- the term includes monocyclic, bicyclic or polycyclic fused, spiro or bridged heterocyclic ring systems. The term also includes polycyclic ring systems in which the heterocyclic ring can be fused to one or more non-aromatic carbocyclic or heterocyclic rings or one or more aromatic rings or combination thereof, wherein the radical or point of attachment is on the heterocyclic ring.
- heterocycles include, but are not limited to, piperidinyl, piperizinyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, azepanyl, diazepanyl, triazepanyl, azocanyl, diazocanyl, triazocanyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, isothiazolidinyl, oxazocanyl, oxazepanyl, thiazepanyl, thiazocanyl, benzimidazolonyl, tetrahydrofuranyl, tetrahydrofuranyl, tetrahydrothiophenyl, tetrahydrothiophenyl, ⁇ , including, for example, 3-morpholino, 4-morpholino, 2-thiomorpholino, 3-thiomorpholino, 4- ⁇ , 1-pyrrolidinyl, 2-pyrazo
- aryl (or “aryl ring” or “aryl group”) used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, “aryloxyalkyl”, or “heteroaryl” refers to carbocyclic aromatic ring systems.
- aryl may be used interchangeably with the terms “aryl ring” or “aryl group”.
- Carbocyclic aromatic ring” groups have only carbon ring atoms (typically six to fourteen) and include monocyclic aromatic rings such as phenyl and fused polycyclic aromatic ring systems in which two or more carbocyclic aromatic rings are fused to one another.
- Examples include 1-naphthyl, 2-naphthyl, 1-anthracyl and 2-anthracyl.
- carbocyclic aromatic ring or “carbocyclic aromatic”, as it is used herein, is a group in which an aromatic ring is “fused” to one or more non- aromatic rings (carbocyclic or heterocyclic), such as in an indanyl, phthalimidyl, naphthimidyl, phenanthridinyl, or tetrahydronaphthyl, where the radical or point of attachment is on the aromatic ring.
- heteroaryl refers to heteroaromatic ring groups having five to fourteen members, in which one or more ring carbons is replaced by a heteroatom such as, N, S, or O.
- heteroaryl rings comprise up to three heteroatoms selected from N, S and O within the ring.
- heteroaryl rings comprise up to two heteroatoms selected from N, S and O within the ring system.
- heteroaryl rings comprise up to three heteroatoms selected from N and O within the ring system. In yet other embodiments, heteroaryl rings comprise up to two heteroatoms selected from N and O within the ring system.
- Heteroaryl rings include monocyclic heteroaromatic rings and polycyclic aromatic rings in which a monocyclic aromatic ring is fused to one or more other aromatic rings. Also included within the scope of the term "heteroaryl", as it is used herein, is a group in which an aromatic ring is "fused” to one or more non-aromatic rings (carbocyclic or heterocyclic), where the radical or point of attachment is on the aromatic ring.
- Bicyclic 6,5 heteroaromatic ring as used herein, for example, is a six membered heteroaromatic ring fused to a second five membered ring, wherein the radical or point of attachment is on the six membered ring.
- heteroaryl groups include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, imidazolyl, pyrrolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl or thiadiazolyl including, for example, 2-furanyl, 3-furanyl, N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl, 3- isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-oxadiazolyl, 5-oxadiazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-pyrazolyl, 4-pyrazolyl, 1 -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 2-pyridyl, 3-pyrazo
- cyclo As used herein, "cyclo”, “cyclic”, “cyclic group” or “cyclic moiety”, include mono-, bi-, and tri-cyclic ring systems including cycloaliphatic, heterocycloaliphatic, aryl, or heteroaryl, each of which has been previously defined.
- a "bicyclic ring system” includes 8-12 (e.g., 9, 10, or 1 1) membered structures that form two rings, wherein the two rings have at least one atom in common (e.g., 2 atoms in common).
- Bicyclic ring systems include bicycloaliphatics (e.g., bicycloalkyl or bicycloalkenyl), bicycloheteroaliphatics, bicyclic aryls, and bicyclic heteroaryls.
- bridged bicyclic ring system refers to a bicyclic
- bridged bicyclic ring systems include, but are not limited to, adamantanyl, norbornanyl, bicyclo[3.2.1]octyl, bicyclo[2.2.2]octyl, bicyclo[3.3.1]nonyl, bicyclo[3.2.3]nonyl, 2-oxa-bicyclo[2.2.2]octyl, l-aza-bicyclo[2.2.2]octyl, 3-aza- bicyclo[3.2.1]octyl, and 2,6-dioxa-tricyclo[3.3.1.03,7]nonyl.
- a bridged bicyclic ring system can be optionally substituted with one or more substituents such as alkyl (including carboxyalkyl, hydroxyalkyl, and haloalkyl such as trifluoromethyl), alkenyl, alkynyl, cycloalkyl, (cycloalkyl)alkyl, heterocycloalkyl, (heterocycloalkyl)alkyl, aryl, heteroaryl, alkoxy, cycloalkyloxy, heterocycloalkyloxy, aryloxy, heteroaryloxy, aralkyloxy, heteroaralkyloxy, aroyl, heteroaroyl, nitro, carboxy, alkoxycarbonyl, alkylcarbonyloxy, aminocarbonyl, alkylcarbonylamino, cycloalkylcarbonylamino,
- heteroarylcarbonylamino heteroaralkylcarbonylamino, cyano, halo, hydroxy, acyl, mercapto, alkylsulfanyl, sulfoxy, urea, thiourea, sulfamoyl, sulfamide, oxo, or carbamoyl.
- bridge refers to a bond or an atom or an unbranched chain of atoms connecting two different parts of a molecule.
- the two atoms that are connected through the bridge (usually but not always, two tertiary carbon atoms) are denotated as “bridgeheads”.
- spiro refers to ring systems having one atom (usually a quaternary carbon) as the only common atom between two rings.
- ring atom is an atom such as C, N, O or S that is in the ring of an aromatic group, cycloalkyl group or non-aromatic heterocyclic ring.
- a "substitutable ring atom" in an aromatic group is a ring carbon or nitrogen atom bonded to a hydrogen atom.
- the hydrogen can be optionally replaced with a suitable substituent group.
- substituted ring atom does not include ring nitrogen or carbon atoms which are shared when two rings are fused.
- substituted ring atom does not include ring carbon or nitrogen atoms when the structure depicts that they are already attached to a moiety other than hydrogen.
- heteroatom means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized form of nitrogen, sulfur, phosphorus, or silicon; the quaternized form of any basic nitrogen or; a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or NR + (as in N-substituted pyrrolidinyl)).
- an optionally substituted aralkyl can be substituted on both the alkyl and the aryl portion. Unless otherwise indicated as used herein optionally substituted aralkyl is optionally substituted on the aryl portion.
- Optional substituents on the aliphatic group of R * are selected from NH 2 , NH(C aliphatic), N(C aliphatic) 2 , halogen, C M aliphatic, OH, 0(C aliphatic), N0 2 , CN, C0 2 H, C0 2 (C 1-4 aliphatic), 0(halo C aliphatic), or halo(C 1 -4 aliphatic), wherein each of the foregoing C 1 - 4 aliphatic groups of R * is unsubstituted.
- optional substituents on the nitrogen of a heterocyclic ring include those described above.
- suitable substituents include -OH, -NH 2) -NH(C,-C 4 alkyl), -N(C,-C 4 alkyl) 2 , -CO(C,-C 4 alkyl), -C0 2 H, -C0 2 (C 1 -C 4 alkyl), -0(d-C 4 alkyl), and C1-C4 aliphatic that is optionally substituted with one or more substituents independently selected from the group consisting of halogen, oxo, -CN, -OH, -N ⁇ , -NH(C 1 - C 4 alkyl), -N(C 1 -C alkyl) 2 , -OCO(C,-C 4 alkyl), -CO(C,-C 4 alkyl), -C0 2 H, -C0 2 (C,-C 4 alkyl),
- substituents include -R + , -N(R + ) 2 , -C(0)R + , -C0 2 R + , -C(0)C(0)R + , -C(0)CH 2 C(0)R + , -S0 2 R + ,
- Optional substituents on the aliphatic group or the phenyl ring of R + are selected from NH 2> NH(C 1-4 aliphatic), N(Ci 4 aliphatic) 2 , halogen, C )-4 aliphatic, OH, 0(C 1-4 aliphatic), N0 2 , CN, C0 2 H, C0 2 (C M aliphatic), 0(halo CM aliphatic), or halo(C]. 4 aliphatic), wherein each of the foregoing C 1 - 4 aliphatic groups of R + is unsubstituted.
- an aryl (including aralkyl, aralkoxy, aryloxyalkyl and the like) or heteroaryl (including heteroaralkyl and heteroarylalkoxy and the like) group may contain one or more substituents. Suitable substituents on the unsaturated carbon atom of an aryl or heteroaryl group are selected from those described above.
- halogen -CN, -OH, -NH 2 , -NH(C 1 -C 4 alkyl), -N(C 1 -C 4 alkyl) 2 , -OCO(C 1 -C 4 alkyl), -CO(C 1 - C 4 alkyl), -C0 2 H, -C0 2 (C 1 -C 4 alkyl), -0(C C alkyl), and C r C aliphatic that is optionally substituted with one or more substituents independently selected from the group consisting of halogen, oxo, -CN, -OH, -NH 2 , -NH(C,-C 4 alkyl), -N(C 1 -C 4 alkyl) 2 , -OCO(C,-C 4 alkyl), -CO(C,-C 4 alkyl), -C0 2 H, -C0 2 (C 1 -Q alkyl), -0(C 1 -C
- substituents include: halogen; -R°; -OR 0 ; -SR°; 1 ,2- methylenedioxy; 1 ,2-ethylenedioxy; phenyl (Ph) optionally substituted with R°; -O(Ph) optionally substituted with R°; -(CH 2 )].
- each independent occurrence of R° is selected from hydrogen, optionally substituted C 1 - ⁇ aliphatic, an unsubstituted 5-6 membered heteroaryl or heterocyclic ring, phenyl, -O(Ph), or -CH 2 (Ph), or, two independent occurrences of R°, on the same substituent or different substituents, taken together with the atom(s) to which each R° group is bound, form a 5-8-membered heterocyclyl, aryl, or heteroaryl ring or a 3-8- membered cycloalkyl ring, wherein said heteroaryl or heterocycl
- Optional substituents on the aliphatic group of R° are selected from NH 2 , NH(C ]- aliphatic), N(C ]-4 aliphatic) 2 , halogen, C aliphatic, OH, 0(C )-4 aliphatic), N0 2 , CN, C0 2 H, C0 2 (C 1- aliphatic), 0(haloC, -4 aliphatic), or haloC 1-4 aliphatic, CHO, N(CO)(CM aliphatic), C(0)N(C aliphatic), wherein each of the foregoing CMaliphatic groups of R° is unsubstituted.
- Non-aromatic nitrogen containing heterocyclic rings that are substituted on a ring nitrogen and attached to the remainder of the molecule at a ring carbon atom are said to be N substituted.
- an N alkyl piperidinyl group is attached to the remainder of the molecule at the two, three or four position of the piperidinyl ring and substituted at the ring nitrogen with an alkyl group.
- Non-aromatic nitrogen containing heterocyclic rings such as pyrazinyl that are substituted on a ring nitrogen and attached to the remainder of the molecule at a second ring nitrogen atom are said to be N' substituted-N-heterocycles.
- an N' acyl N-pyrazinyl group is attached to the remainder of the molecule at one ring nitrogen atom and substituted at the second ring nitrogen atom with an acyl group.
- two independent occurrences of R° may be taken together with the atom(s) to which each variable is bound to form a 5-8-membered heterocyclyl, aryl, or heteroaryl ring or a 3-8-membered cycloalkyl ring.
- Exemplary rings that are formed when two independent occurrences of R° (or R + , or any other variable similarly defined herein) are taken together with the atom(s) to which each variable is bound include, but are not limited to the following: a) two independent occurrences of R° (or R + , or any other variable similarly defined herein) that are bound to the same atom and are taken together with that atom to form a ring, for example, N(R°)2, where both occurrences of R° are taken together with the nitrogen atom to form a piperidin-1-yl, piperazin-1-yl, or morpholin-4-yl group; and b) two independent occurrences of R° (or R + , or any other variable similarly defined herein) that are bound to different atoms and are taken together with both of those atoms to form a ring, for example
- amino refers to -NH 2 .
- hydroxyl'Or hydroxy or “alcohol moiety” refers to -OH.
- alkoxy refers to an alkyl group, as previously defined, attached to the molecule through an oxygen (“alkoxy” e.g., -O-alkyl) or sulfur (“alkylthio” e.g., -S-alkyl) atom.
- halogen means F, CI, Br, or I.
- cyano or "nitrile” refer to -CN or -C ⁇ N.
- alkoxyalkyl alkoxyalkenyl
- alkoxyaliphatic alkoxyalkoxy
- alkoxyalkoxy mean alkyl, alkenyl, aliphatic or alkoxy, as the case may be, substituted with one or more alkoxy groups.
- haloalkyl haloalkenyl
- haloaliphatic haloalkoxy
- cyclo(haloalkyl) mean alkyl, alkenyl, aliphatic, alkoxy, or cycloalkyl, as the case may be, substituted with one or more halogen atoms.
- This term includes perfluorinated alkyl groups, such as -CF 3 and -CF2CF3.
- cyanoalkyl mean alkyl, alkenyl, aliphatic or alkoxy, as the case may be, substituted with one or more cyano groups.
- the cyanoalkyl is (NC)-alkyl-.
- aminoalkyl means alkyl, alkenyl, aliphatic or alkoxy, as the case may be, substituted with one or more amino groups, wherein the amino group is as defined above.
- hydroxyalkyl means alkyl, aliphatic or alkoxy, as the case may be, substituted with one or more -OH groups.
- alkoxyalkyl means alkyl, aliphatic or alkoxy, as the case may be, substituted with one or more alkoxy groups.
- alkoxyalkyl refers to an alkyl group such as (alkyl-O)-alkyl-, wherein alkyl has been defined above.
- a protecting group and “protective group” as used herein, are interchangeable and refer to an agent used to temporarily block one or more desired functional groups in a compound with multiple reactive sites.
- a protecting group has one or more, or specifically all, of the following characteristics: a) is added selectively to a functional group in good yield to give a protected substrate that is b) stable to reactions occurring at one or more of the other reactive sites; and c) is selectively removable in good yield by reagents that do not attack the regenerated, deprotected functional group. As would be understood by one skilled in the art, in some cases, the reagents do not attack other reactive groups in the compound.
- the reagents may also react with other reactive groups in the compound.
- protecting groups are detailed in Greene, T. W., Wuts, P. G in "Protective Groups in Organic Synthesis", Third Edition, John Wiley & Sons, New York: 1999 (and other editions of the book), the entire contents of which are hereby incorporated by reference.
- the term "nitrogen protecting group”, as used herein, refers to an agent used to temporarily block one or more desired nitrogen reactive sites in a multifunctional compound.
- Preferred nitrogen protecting groups also possess the characteristics exemplified for a protecting group above, and certain exemplary nitrogen protecting groups are also detailed in Chapter 7 in Greene, T.W., Wuts, P. G in "Protective Groups in Organic Synthesis", Third Edition, John Wiley & Sons, New York: 1999, the entire contents of which are hereby incorporated by reference.
- the term "displaceable moiety” or “leaving group” refers to a group that is associated with an aliphatic or aromatic group as defined herein and is subject to being displaced by nucleophilic attack by a nucleophile.
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, cis-trans, conformational, and rotational) forms of the structure.
- isomeric e.g., enantiomeric, diastereomeric, cis-trans, conformational, and rotational
- the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers are included in this invention, unless only one of the isomers is drawn specifically.
- a substituent can freely rotate around any rotatable bonds. For example,
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13 C- or 14 C-enriched carbon are within the scope of this invention.
- Such compounds are useful, for example, as analytical tools or probes in biological assays.
- Such compounds, especially deuterium (D) analogs can also be therapeutically useful.
- the compounds of the invention are defined herein by their chemical structures and/or chemical names. Where a compound is referred to by both a chemical structure and a chemical name, and the chemical structure and chemical name conflict, the chemical structure is determinative of the compound's identity.
- the compounds described herein can exist in free form, or, where appropriate, as salts. Those salts that are pharmaceutically acceptable are of particular interest since they are useful in administering the compounds described above for medical purposes. Salts that are not pharmaceutically acceptable are useful in manufacturing processes, for isolation and purification purposes, and in some instances, for use in separating stereoisomeric forms of the compounds of the invention or intermediates thereof.
- the term "pharmaceutically acceptable salt” refers to salts of a compound, which are, within the scope of sound medical judgment, suitable for use in humans and lower animals without undue side effects, such as, toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- compositions described herein include those derived from suitable inorganic and organic acids and bases. These salts can be prepared in situ during the final isolation and purification of the compounds.
- acid addition salts can be prepared by, for example, 1) reacting the purified compound in its free-base form with a suitable organic or inorganic acid; and 2) isolating the salt thus formed.
- acid addition salts might be a more convenient form for use and use of the salt amounts to use of the free basic form.
- Examples of pharmaceutically acceptable, non-toxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid
- organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, glycolate, gluconate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, ox
- base addition salts can be prepared by, for example, 1) reacting the purified compound in its acid form with a suitable organic or inorganic base and 2) isolating the salt thus formed.
- base addition salt might be more convenient and use of the salt form inherently amounts to use of the free acid form.
- Salts derived from appropriate bases include alkali metal (e.g., sodium, lithium, and potassium), alkaline earth metal (e.g., magnesium and calcium), ammonium and N ⁇ Ci ⁇ alkyl ⁇ salts.
- alkali metal e.g., sodium, lithium, and potassium
- alkaline earth metal e.g., magnesium and calcium
- ammonium and N ⁇ Ci ⁇ alkyl ⁇ salts e.g., sodium, lithium, and potassium
- Basic addition salts include pharmaceutically acceptable metal and amine salts.
- Suitable metal salts include the sodium, potassium, calcium, barium, zinc, magnesium, and aluminium.
- the sodium and potassium salts are usually preferred.
- Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate and aryl sulfonate.
- Suitable inorganic base addition salts are prepared from metal bases which include sodium hydride, sodium hydroxide, potassium hydroxide, calcium hydroxide, aluminium hydroxide, lithium hydroxide, magnesium hydroxide, zinc hydroxide and the like.
- Suitable amine base addition salts are prepared from amines which are frequently used in medicinal chemistry because of their low toxicity and acceptability for medical use. Ammonia, ethylenediamine, N-methyl-glucamine, lysine, arginine, ornithine, choline, N, N'-dibenzylethylenediamine, chloroprocaine, dietanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine,
- the methods of the invention can be employed for preparing pharmaceutically acceptable solvates (e.g., hydrates) and clathrates of these compounds.
- solvate is a solvate formed from the association of one or more pharmaceutically acceptable solvent molecules to one of the compounds described herein.
- solvate includes hydrates (e.g., hemihydrate, monohydrate, dihydrate, trihydrate, tetrahydrate, and the like).
- hydrate means a compound described herein or a salt thereof that further includes a stoichiometric or non-stoichiometric amount of water bound by non-covalent intermolecular forces.
- clathrate means a compound described herein or a salt thereof in the form of a crystal lattice that contains spaces (e.g., channels) that have a guest molecule (e.g., a solvent or water) trapped within.
- the methods of the invention can be employed for preparing pharmaceutically acceptable derivatives or prodrugs of these compounds.
- a "pharmaceutically acceptable derivative or prodrug” includes any
- ester, salt of an ester, or other derivative or salt thereof, of a compound described herein which, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound described herein or an inhibitorily active metabolite or residue thereof.
- Particularly favoured derivatives or prodrugs are those that increase the bioavailability of the compounds when such compounds are administered to a patient (e.g., by allowing an orally administered compound to be more readily absorbed into the blood) or which enhance delivery of the parent compound to a biological compartment (e.g., the brain or lymphatic system) relative to the parent species.
- prodrug means a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide a compound described herein. Prodrugs may become active upon such reaction under biological conditions, or they may have activity in their unreacted forms.
- prodrugs contemplated in this invention include, but are not limited to, analogs or derivatives of compounds of the invention that comprise biohydrolyzable moieties such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues.
- biohydrolyzable moieties such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues.
- Other examples of prodrugs include derivatives of compounds described herein that comprise -NO, -N0 2 , -ONO, or -ON0 2 moieties.
- Prodrugs can typically be prepared using well-known methods, such as those described by BURGER'S MEDICINAL CHEMISTRY AND DRUG DISCOVERY (1995) 172-178, 949-982 (Manfred E. Wolff ed., 5th ed).
- a "pharmaceutically acceptable derivative” is an adduct or derivative which, upon administration to a patient in need, is capable of providing, directly or indirectly, a compound as otherwise described herein, or a metabolite or residue thereof.
- pharmaceutically acceptable derivatives include, but are not limited to, esters and salts of such esters.
- compositions described above include, without limitation, esters, amino acid esters, phosphate esters, metal salts and sulfonate esters.
- the compounds in accordance with the present invention can contain a chiral center.
- the compounds of formula may thus exist in the form of two different optical isomers (i.e. (+) or (-) enantiomers). All such enantiomers and mixtures thereof including racemic mixtures are included within the scope of the invention.
- the single optical isomer or enantiomer can be obtained by method well known in the art, such as chiral HPLC, enzymatic resolution and chiral auxiliary.
- the compounds of the invention are provided in the form of a single enantiomer at least 95%, at least 97% and at least 99% free of the corresponding enantiomer.
- the compounds of the invention are in the form of the (+) enantiomer at least 95 % free of the corresponding (-) enantiomer.
- the compounds of the invention are in the form of the (+) enantiomer at least 97% free of the corresponding (-) enantiomer.
- the compounds of the invention are in the form of the (+) enantiomer at least 99% free of the corresponding (-) enantiomer.
- the compounds of the invention are in the form of the (-) enantiomer at least 95% free of the corresponding (+) enantiomer.
- the compounds of the invention are in the form of the (-) enantiomer at least 97% free of the corresponding (+) enantiomer.
- the compounds of the invention are in the form of the (-) enantiomer at least 99% free of the corresponding (+) enantiomer.
- the compounds of the invention are provided as pharmaceutically acceptable salts £e.g. Handbook of Pharmaceutical Salts Properties, Selection, and Use, Wiley, 2002, (P. Heinrich Stahl, Camille G. Wermuth, ed.)) ⁇
- pharmaceutically acceptable salts can be derived from
- acids and bases examples include hydrochloric, hydrobromic, sulphuric, nitric, perchloric, fumaric, maleic, phosphoric, glycollic, lactic, salicylic, succinic, toleune-p-sulphonic, tartaric, acetic, trifluoroacetic, citric, methanesulphonic, formic, benzoic, malonic, naphthalene-2-sulphonic and benzenesulphonic acids.
- Other acids such as oxalic, while not themselves
- pharmaceutically acceptable may be useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable acid addition salts.
- Salts derived from amino acids are also included (e.g. L-arginine, L-Lysine).
- Salts derived from appropriate bases include alkali metals (e.g. sodium, lithium, potassium), alkaline earth metals (e.g. calcium, magnesium), ammonium, NR 4+ (where R is
- the pharmaceutically acceptable salt is a sodium salt.
- the pharmaceutically acceptable salt is a potassium salt.
- the pharmaceutically acceptable salt is a lithium salt.
- the pharmaceutically acceptable salt is a tromethamine salt.
- the pharmaceutically acceptable salt is an L- arginine salt.
- the pharmaceutically acceptable salt is a calcium salt.
- polymorphism is an ability of a compound to crystallize as more than one distinct crystalline or "polymorphic" species.
- a polymorph is a solid crystalline phase of a compound with at least two different arrangements or polymorphic forms of that compound molecule in the solid state.
- Polymorphic forms of any given compound are defined by the same chemical formula. or composition and are as distinct in chemical structure as crystalline structures of two different chemical compounds.
- the compounds of the invention described herein can exist in different solvate forms, for example hydrates. Solvates of the compounds of the invention may also form when solvent molecules are incorporated into the crystalline lattice structure of the compound molecule during the crystallization process.
- the terms "subject,” “host,” or “patient” includes an animal and a human (e.g., male or female, for example, a child, an adolescent, or an adult).
- a human e.g., male or female, for example, a child, an adolescent, or an adult.
- the "subject,” “host,” or “patient” is a human.
- the present invention provides a method for treating or preventing a Flaviviridae viral infection in a host comprising administering to the host a therapeutically effective amount of at least one compound according to the invention described herein.
- the viral infection is chosen from Flaviviridae infections.
- the Flaviviridae infection is Hepatitis C virus (HCV), bovine viral diarrhea virus (BVDV), hog cholera virus, dengue fever virus, Japanese encephalitis virus or yellow fever virus.
- HCV Hepatitis C virus
- BVDV bovine viral diarrhea virus
- hog cholera virus dengue fever virus
- Japanese encephalitis virus yellow fever virus.
- the Flaviviridae viral infection is hepatitis C viral infection (HCV).
- the methods of the invention are directed for treatment of HCV genotype 1 infection.
- the HCV is genotype la or genotype lb.
- the present invention provides a method for treating or preventing a Flaviviridae viral infection in a host comprising administering to the host a therapeutically effective amount of at least one compound according to the invention described herein, and further comprising administering at least one additional agent chosen from viral serine protease inhibitors, viral polymerase inhibitors, viral helicase inhibitors, immunomudulating agents, antioxidant agents, antibacterial agents, therapeutic vaccines, hepatoprotectant agents, antisense agents, inhibitors of HCV NS2/3 protease and inhibitors of internal ribosome entry site (IRES).
- at least one additional agent chosen from viral serine protease inhibitors, viral polymerase inhibitors, viral helicase inhibitors, immunomudulating agents, antioxidant agents, antibacterial agents, therapeutic vaccines, hepatoprotectant agents, antisense agents, inhibitors of HCV NS2/3 protease and inhibitors of internal ribosome entry site (IRES).
- a method for inhibiting or reducing the activity of viral polymerase in a host comprising administering a therapeutically effective amount of a compound according to the invention described herein.
- a method for inhibiting or reducing the activity of viral polymerase in a host comprising administering a therapeutically effective amount of a compound according to the invention described herein and further comprising administering one or more viral polymerase inhibitors.
- viral polymerase is a Flaviviridae viral polymerase.
- viral polymerase is a R A-dependant RNA- polymerase.
- viral polymerase is HCV polymerase.
- viral polymerase is HCV 5B polymerase.
- the compounds described above can be formulated in pharmaceutically acceptable formulations that optionally further comprise a pharmaceutically acceptable carrier, adjuvant or vehicle.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound according to the invention described herein and at least one pharmaceutically acceptable carrier, adjuvant, or vehicle, which includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- Remington's Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) discloses various carriers used in formulating pharmaceutically acceptable compositions and known techniques for the preparation thereof. Except insofar as any conventional carrier medium is incompatible with the compounds of the invention, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other
- side effects encompasses unwanted and adverse effects of a therapy (e.g., a prophylactic or therapeutic agent). Side effects are always unwanted, but unwanted effects are not necessarily adverse. An adverse effect from a therapy (e.g., prophylactic or therapeutic agent) might be harmful or uncomfortable or risky.
- a therapy e.g., prophylactic or therapeutic agent
- a pharmaceutically acceptable carrier may contain inert ingredients which do not unduly inhibit the biological activity of the compounds.
- the pharmaceutically acceptable carriers should be biocompatible, e.g., non-toxic, non-inflammatory, non-immunogenic or devoid of other undesired reactions or side-effects upon the administration to a subject. Standard pharmaceutical formulation techniques can be employed.
- Some examples of materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins (such as human serum albumin), buffer substances (such as twin 80, phosphates, glycine, sorbic acid, or potassium sorbate), partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes (such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, or zinc salts), colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene- polyoxypropylene-block polymers, methylcellulose, hydroxypropyl methylcellulose, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose
- compositions thereof can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops), bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated.
- parenteral as used herein includes, but is not limited to, subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- compositions are administered orally, intraperitoneally or intravenously.
- any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous suspensions or solutions, can be used for the oral administration.
- carriers commonly used include, but are not limited to, lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- Liquid dosage forms for oral administration include, but are not limited to, pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- inert diluents such as, for example, water or other solvents, solubilizing agents
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate, e) solution retarding agents such as paraffin, f) absorption accelerators such as quaternary ammonium compounds, g) wetting agents such as, for example, cetyl alcohol
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions that can be used include polymeric substances and waxes. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar' as well as high molecular weight polethylene glycols and the like.
- the active compounds can also be in microencapsulated form with one or more excipients as noted above.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art.
- the active compound may be admixed with at least one inert diluent such as sucrose, lactose or starch.
- Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- the dosage forms may also comprise buffering agents. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- buffering agents include polymeric substances and waxes.
- Injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- Injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- Sterile injectable forms may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or di-glycerides.
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceuticaUy-acceptable oils, such as olive oil or castor oil, especially in their polybxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- the rate of compound release can be controlled.
- biodegradable polymers include poly(orthoesters) and poly(anhydrides).
- Depot injectable formulations are also prepared by entrapping the compound in liposomes or microemulsions that are compatible with body tissues.
- compositions for rectal or vaginal administration are specifically suppositories which can be prepared by mixing the active compound with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Dosage forms for topical or transdermal administration include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches.
- the active component is admixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives or buffers as may be required.
- Ophthalmic formulation, eardrops, and r eye drops are also contemplated as being within the scope of this invention.
- transdermal patches which have the added advantage of providing controlled delivery of a compound to the body, can also be used.
- Such dosage forms can be made by dissolving or dispensing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- compositions described above and pharmaceutically acceptable compositions thereof may also be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- unit dosage form refers to physically discrete units suitable as unitary dosage for subjects undergoing treatment, with each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, optionally in association with a suitable pharmaceutical carrier.
- the unit dosage form can be for a single daily dose or one of multiple daily doses (e.g., about 1 to 4 or more times per day). When multiple daily doses are used, the unit dosage form can be the same or different for each dose.
- the amount of the active compound in a unit dosage form will vary depending upon, for example, the host treated, and the particular mode of administration, for example, from 0.01 mg/kg body weight/day to 100 mg/kg body weight/day.
- a suitable dose will be in the range of from about 0.1 to about 750 mg kg of body weight per day, for example, in the range of 0.5 to 60 mg/kg/day, or, for example, in the range of 1 to 20 mg/kg/day.
- the desired dose may conveniently be presented in a single dose or as divided dose administered at appropriate intervals, for example as two, three, four or more doses per day.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound according to the invention described herein, and further comprising one or more additional agents chosen from viral serine protease inhibitors, viral polymerase inhibitors, viral NS5A inhibitors, viral helicase inhibitors, immunomudulating agents, antioxidant agents, antibacterial agents, therapeutic vaccines, hepatoprotectant agents, antisense agent, inhibitors of HCV NS2/3 protease and inhibitors of internal ribosome entry site (IRES).
- additional agents chosen from viral serine protease inhibitors, viral polymerase inhibitors, viral NS5A inhibitors, viral helicase inhibitors, immunomudulating agents, antioxidant agents, antibacterial agents, therapeutic vaccines, hepatoprotectant agents, antisense agent, inhibitors of HCV NS2/3 protease and inhibitors of internal ribosome entry site (IRES).
- additional agents chosen from viral serine protease inhibitors, viral polymerase inhibitors, viral helicase inhibitors, immunomudulating agents, antioxidant agents, antibacterial agents, therapeutic vaccines, hepatoprotectant agents, antisense agent, inhibitors of HCV NS2/3 protease and inhibitors of internal ribosome entry site (IRES).
- compositions and combinations include, for example, ribavirin, amantadine, merimepodib, Levovirin, Viramidine, and maxamine.
- the compound and additional agent are administered sequentially.
- the compound and additional agent are administered simultaneously.
- the combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a combination as defined above together with a pharmaceutically acceptable carrier therefore comprise a further aspect of the invention.
- viral serine protease inhibitor means an agent that is effective to inhibit the function of the viral serine protease including HCV serine protease in a mammal.
- Inhibitors of HCV serine protease include, for example, those compounds described in WO 99/07733 (Boehringer Ingelheim), WO 99/07734 (Boehringer Ingelheim), WO 00/09558 (Boehringer Ingelheim), WO 00/09543 (Boehringer Ingelheim), WO 00/59929 (Boehringer Ingelheim), WO 02/060926 (BMS), WO 2006039488 (Vertex), WO
- viral polymerase inhibitors as used herein means an agent that is effective to inhibit the function of a viral polymerase including an HCV polymerase in a mammal.
- Inhibitors of HCV polymerase include non-nucleosides, for example, those compounds described in:WO 03/010140 (Boehringer Ingelheim), WO 03/026587 (Bristol Myers Squibb); WO 02/100846 Al , WO 02/100851 A2, WO 01 /85172 AI (GSK), WO 02/098424 A 1 (GSK), WO 00/06529 (Merck), WO 02/06246 Al (Merck), WO 01 /47883 (Japan Tobacco), WO 03/000254 (Japan Tobacco) and EP 1 256 628 A2 (Agouron).
- inhibitors of HCV polymerase also include nucleoside analogs, for example, those compounds described in: WO 01 /90121 A2 (Idenix), WO 02/069903 A2 (Biocryst Pharmaceuticals Inc.), and WO 02/057287 A2 (Merck/ Isis) and WO 02/057425 A2 (Merck/lsis).
- nucleoside inhibitors of an HCV polymerase include R1626, R1479 (Roche), R7128 (Roche), MK-0608 (Merck), R1656, (Roche-Pharmassef) and Valopicitabine (Idenix).
- Specific examples of inhibitors of an HCV polymerase include JTK- 002/003 and JTK- 109 (Japan Tobacco), HCV-796 (Viropharma), GS-9190(Gilead), and PF- 868,554 (Pfizer).
- viral NS5A inhibitor means an agent that is effective to inhibit the function of the viral NS5A protease in a mammal.
- Inhibitors of HCV NS5A include, for example, those compounds described in WO2010/1 17635, WO2010/117977, WO2010/117704, WO2010/1200621 , WO2010/096302, WO2010/017401 , WO2009/102633, WO2009/ 102568, WO2009/102325, WO2009/102318, WO2009020828, WO2009020825, WO2008144380, WO2008/021936, WO2008/021928, WO2008/021927, WO2006/133326, WO2004/014852, WO2004/014313, WO2010/096777, WO2010/065681 , WO2010/065668, WO2010/065674, WO2010/062821 , WO2010/099527
- HCV NS5A inhibitors include: EDP-239 (being developed by Enanta); ACH-2928 (being developed by Achillion); PPI-1301 (being developed by Presido Pharmaceuticals); PPI-461 (being developed by Presido
- AZD-7295 being developed by AstraZeneca
- GS-5885 being developed by Gilead
- BMS-824393 being developed by Bristol-Myers Squibb
- BMS-790052 being developed by Bristol-Myers Squibb
- nucleoside or nucleotide polymerase inhibitors such as PSI-661 (being developed by Pharmasset), PSI-938 (being developed by Pharmasset), PSI-7977 (being developed by Pharmasset), ⁇ -189 (being developed by Inhibitex), JTK- 853 (being developed by Japan Tobacco) , TMC-647055 (Tibotec Pharmaceuticals), RO- 5303253 (being developed by Hoffmann-La Roche), and IDX-184 (being developed by Idenix Pharmaceuticals).
- viral helicase inhibitors as used herein means an agent that is effective to inhibit the function of a viral helicase including a Flaviviridae helicase in a mammal.
- Immunomodulatory agent as used herein means those agents that are effective to enhance or potentiate the immune system response in a mammal.
- Immunomodulatory agents include, for example, class I interferons (such as alpha-, beta-, delta- and omega- interferons, x-interferons, consensus interferons and asialo-interferons), class II interferons (such as . gamma-interferons) and pegylated interferons.
- Exemplary immunomudulating agents include, but are not limited to:
- interferon including natural interferon (such as OMNIFERON, Viragen and SUMIFERON, Sumitomo, a blend of natural interferon's), natural interferon alpha (ALFERON, Hemispherx Biopharma, Inc.), interferon alpha nl from lymphblastoid cells (WELLFERON, Glaxo Wellcome), oral alpha interferon, Peg-interferon, Peg-interferon alfa 2a (PEGASYS, Roche), recombinant interferon alpha 2a (ROFERON, Roche), inhaled interferon alpha 2b (AERX, Aradigm), Peg-interferon alpha 2b (ALBUFERON, Human Genome Sciences/Novartis, PEGINTRON, Schering), recombinant interferon alfa 2b
- natural interferon such as OMNIFERON, Viragen and SUMIFERON, Sumitomo, a blend of natural interferon's
- VIRAFERONPEG Schering
- interferon beta- la REBIF, Serono, Inc. and Pfizer
- consensus interferon alpha INFERGEN, Valeant Pharmaceutical
- interferon gamma- lb ACTIMMUNE, Intermune, Inc.
- un-pegylated interferon alpha alpha interferon, and its analogs
- synthetic thymosin alpha 1 ZADAXIN, SciClone Pharmaceuticals Inc.
- class I interferon as used herein means an interferon selected from a group of interferons that all bind to receptor type 1. This includes both naturally and synthetically produced class I interferons. Examples of class I interferons include alpha-, beta-, delta- and omega- interferons, tau-interferons, consensus interferons and asialo- interferons.
- class II interferon as used herein means an interferon selected from a group of interferons that all bind to receptor type II. Examples of class II interferons include gamma-interferons.
- Antisense agents include, for example, ISIS- 14803.
- inhibitors of HCV NS3 protease include BILN-2061
- ISIS- 14803 ISIS- 14803
- the additional agent is interferon alpha, ribavirin, silybum marianum, interleukine-12, amantadine, ribozyme, thymosin, N-acetyl cysteine or cyclosporin.
- the additional agent is interferon alpha 1 A, interferon alpha 1 B, interferon alpha 2A, or interferon alpha 2B.
- Interferon is available in pegylated and non pegylated forms. Pegylated interferons include PEGASYSTM and Peg-intronTM.
- the recommended dose of PEGASYSTM monotherapy for chronic hepatitis C is 180 mg (1.0 mL vial or 0.5 mL prefilled syringe) once weekly for 48 weeks by subcutaneous administration in the abdomen or thigh.
- the recommended dose of PEGASYSTM when used in combination with ribavirin for chronic hepatitis C is 180 mg (1.0 mL vial or 0.5 mL prefilled syringe) once weekly.
- Ribavirin is typically administered orally, and tablet forms of ribavirin are currently commercially available.
- General standard, daily dose of ribavirin tablets e.g., about 200 mg tablets
- ribavirn tablets are administered at about 1000 mg for subjects weighing less than 75 kg, or at about 1200 mg for subjects weighing more than or equal to 75 kg. Nevertheless, nothing herein limits the methods or combinations of this invention to any specific dosage forms or regime.
- ribavirin can be dosed according to the dosage regimens described in its commercial product labels.
- the recommended dose of PEG-lntronTM regimen is 1.0 mg/kg/week
- the dose should be administered on the same day of the week.
- the recommended dose of PEG- lntron is 1.5 micrograms/ kg/ week.
- viral serine protease inhibitor is a flaviviridae serine protease inhibitor.
- viral polymerase inhibitor is a flaviviridae polymerase inhibitor.
- viral helicase inhibitor is a flaviviridae helicase inhibitor.
- viral serine protease inhibitor is HCV serine protease inhibitor
- viral polymerase inhibitor is HCV polymerase inhibitor
- viral helicase inhibitor is HCV helicase inhibitor.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound according to the invention described herein, one or more additional agents select from non-nucleoside HCV polymerase inhibitors (e.g., HCV-796), nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), HCV NS3 protease inhibitors (e.g., VX-950/telaprevir and ITMN-191), interferon and ribavirin, and at least one pharmaceutically acceptable carrier or excipient.
- non-nucleoside HCV polymerase inhibitors e.g., HCV-796
- nucleoside HCV polymerase inhibitors e.g., R7128, R1626, R147
- HCV NS3 protease inhibitors e.g., VX-950/telaprevir and ITMN-191
- interferon and ribavirin interferon and ribavirin
- compositions comprising a combination as defined above together with a pharmaceutically acceptable carrier therefore comprise a further aspect of the invention.
- the individual components for use in the method of the present invention or combinations of the present invention may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations.
- the present invention provides the use of a compound according to the invention described herein for treating or preventing Flaviviridae viral infection in a host.
- the present invention provides the use of a compound according to the invention described herein for the manufacture of a medicament for treating or preventing a viral Flaviviridae infection in a host.
- the present invention provides the use of a compound according to the invention described herein for inhibiting or reducing the activity of viral polymerase in a host.
- composition or combination according to the invention further comprises at least one compound according to the invention described herein; one or more additional agents select from non-nucleoside HCV polymerase inhibitors (e.g., HCV-796), nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), and HCV NS3 protease inhibitors (e.g., VX-950/telaprevir and ITMN-191); and interferon and/or ribavirin.
- non-nucleoside HCV polymerase inhibitors e.g., HCV-796
- nucleoside HCV polymerase inhibitors e.g., R7128, R1626, R1479
- HCV NS3 protease inhibitors e.g., VX-950/telaprevir and ITMN-191
- interferon and/or ribavirin interferon and/or ribavirin.
- the additional agent is interferon a 1A, interferon a IB, interferon a 2A, or interferon a 2B, and optionally ribavirin.
- the present invention provides a method for treating or preventing a HCV viral infection in a host comprising administering to the host a combined therapeutically effective amounts of at least one compound according to the invention described herein, and one or more additional agents select from non-nucleoside HCV polymerase inhibitors (e.g., HCV-796), nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), HCV NS3 protease inhibitors (e.g., VX-950/telaprevir and ITMN-191), interferon and ribavirin.
- non-nucleoside HCV polymerase inhibitors e.g., HCV-796
- nucleoside HCV polymerase inhibitors e.g., R7128, R1626
- the compound and additional agent are administered sequentially.
- the compound and additional agent are administered simultaneously.
- a method for inhibiting or reducing the activity of HCV viral polymerase in a host comprising administering to the host a combined therapeutically effective amounts of at least one compound of the invention, and one or more additional agents select from non-nucleoside HCV polymerase inhibitors (e.g., HCV-796) and nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), interferon and ribavirin.
- non-nucleoside HCV polymerase inhibitors e.g., HCV-796
- nucleoside HCV polymerase inhibitors e.g., R7128, R1626, R1479
- the present invention provides the use of at least one compound of the invention, in combination with the use of one or more additional agents select from non-nucleoside HCV polymerase inhibitors (e.g., HCV-796), nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), HCV NS3 protease inhibitors (e.g., VX- 950/telaprevir and ITMN-191), interferon and ribavirin, for the manufacture of a medicament for treating or preventing a HCV infection in a host.
- non-nucleoside HCV polymerase inhibitors e.g., HCV-796
- nucleoside HCV polymerase inhibitors e.g., R7128, R1626, R147
- HCV NS3 protease inhibitors e.g., VX- 950/telaprevir and ITMN-191
- interferon and ribavirin interferon and
- the dose of each compound may be either the same as or differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- the ratio of the amount of a compound according to the invention described herein administered relative to the amount of the additional agent will vary dependent on the selection of the compound and additional agent.
- the additional agent non-nucleoside HCV polymerase inhibitors (e.g., HCV-796), nucleoside HCV polymerase inhibitors (e.g., R7128, R1626, R1479), HCV NS3 protease inhibitors (e.g., VX-950/telaprevir and ITMN-191), interferon or ribavirin) will vary dependent on the selection of the compound and additional agent.
- the compounds according to the invention described herein can be prepared by any suitable method known in the art.
- the compounds can be prepared in accordance with procedures described in US 6,881,741, US 2005/0009804, US
- the compounds according to the invention described herein can be prepared by any suitable method known in the art, for example, US 6,881 ,741, US 2005/0009804, US 2006/0276533, WO 2002/100851, and WO 08/58393. Preparation details of some exemplary compounds are described below. Syntheses of certain exemplary compounds of the invention are described below. Generally, the compounds of the invention can be prepared as shown in those syntheses optionally with any desired appropriate modification.
- RT refers to the LCMS retention time, in minutes, associated with the compound. Unless otherwise indicated, the method employed to obtain the reported retention times is as follows:
- RT refers to the LCMS retention time, in minutes, associated with the compound.
- NMR and Mass Spectroscopy data of certain specific compounds are summarized in Table 1.
- Step III To a solution of 5-(3,3-dimethyl-but-1-ynyl)-3-(4-oxo-cyclohexylamino)-thiophene-2- carboxylic acid methyl ester (200 mg, 0.539 mmol) in toluene (5 mL) was sequentially added pyridine (85 mL, 1.08 mmol) and 2,4-dimethylbenzoyl chloride (182 mL, 1.08 mmol) under nitrogen atmosphere. The reaction mixture was heated at 1 10 °C overnight in a sealed tube, cooled to RT, and diluted with EtOAc (10 mL).
- Compound 90 3-[(2,4-Dichloro-benzoyl)-(trans-4-hydroxy-cyclohexyl)-amino]-5-(3,3- dimethyl-but-l-ynyl)-thiophene-2-carboxylic acid
- reaction mixture was extracted by EtOAc (2 x 20 mL), and the organic layer was dried over Na2S0 , filtered, and concentrated to dryness.
- the residue was purified by flash column chromatography on silica gel (0 to 50 % EtOAc in Hexanes) to give 5-(3,3-dimethyl-but-l -ynyl)-3-(ira «s-4-hydroxy- cyclohexylamino)-thiophene-2-carboxylic acid methyl ester (1.32 g, 88 %).
- the reaction mixture was deoxygenated by bubbling nitrogen gas for 10 minutes and tert-b tyl acetylene (1.37 mL, 1 1.12 mmol), 2,2'-bis(diphenylphosphino)-l,l'-binaphthyl (156 mg, 0.25 mmol) and triethylamine (1.94 mL, 13.9 mmol) were sequentially added.
- the reaction mixture was heated at 60 °C overnight, diluted with DCM, filtered over Celite and washed with DCM. The filtrate was washed with brine, dried over Na 2 S0 4 , filtered, and concentrated to dryness.
- the aqueous solution was diluted with H 2 0 (10 mL) and acidified with aqueous HCl 2M to pH 2.
- the reaction mixture was extracted by DCM (3 x 10 mL), and the organic layer was dried over Na 2 S0 4 , filtered, and concentrated to dryness.
- the residue was purified by reverse phase preparative HPLC to give 3-[(irani-4-allyloxy-cyclohexyl)-(fram-4-methyl-cyclohexanecarbonyl)-amino]-5-(3,3- dimethyl-but-1-ynyl)-thiophene-2-carboxylic acid (210 mg, 98%).
- the reaction mixture was deoxygenated by bubbling nitrogen gas for 10 minutes and iert-butyl acetylene (136 mg, 1.55 mmol), triphenylphosphine (10 mg, 10 mol %) and triethylamine (381 ⁇ , 2.75 mmol) were sequentially added.
- the reaction mixture was heated at 60 °C overnight, and concentrated to dryness.
- the reaction mixture was extracted with EtOAc, and the organic layer was washed with H 2 0, dried over Na 2 S0 4 , filtered, and concentrated to dryness.
- Step I To a solution of 5-bromo-3-[(tra «i-4-methyl-cyclohexanecarbonyl)-(4-oxo-cyclohexyl)- amino]-thiophene-2-carboxylic acid methyl ester (420 mg, 0.92 mmol) in toluene (5 mL) was added diethylaminosulphurtrifluoride (362 ⁇ , 2.76 mmol) under nitrogen atmosphere. The reaction mixture was stirred at RT overnight, diluted with DCM, washed with brine, and H 2 0. The organic phase was dried over Na 2 S0 4 , filtered, and concentrated to dryness.
- the reaction mixture was deoxygenated by bubbling nitrogen gas for 10 minutes and te -butyl acetylene (199 ⁇ , 1.61 mmol), 2,2'-bis(diphenylphosphino)-l, r-binaphthyl (20 mg, 0.03 mmol) and triethylamine (279 ⁇ , 2.0 mmol) are sequentially added.
- the reaction mixture was heated at 60 °C overnight, diluted with DCM, filtered over Celite and washed with DCM. The filtrate was washed with brine, dried over Na 2 S04, filtered, and concentrated to dryness.
- reaction mixture was extracted by ether (1 x 50 mL), and the organic layer was dried over MgSC>4, filtered, and concentrated to dryness.
- the residue was purified by flash column chromatography on silica gel (0 to 12 % EtOAc in DCM) to give 3-[Isopropyl-(ira «5-4-methyl-cyclohexanecarbonyl)-amino]-5- trifluoroprop-1-ynyl-thiophene-2-carboxylic acid methyl ester ( 1.2 mg, 11 %).
- the product is purified by silica gel chromatography (10-90% EtOAc in hexane) to give the desired 5-(2-substituted- ethyn-1-yl)-3-[(1,4-dioxaspiro[4.5]decan-8-yl)-(4-ira «i- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid methyl ester.
- the product is purified bysilica gel chromatography (10-90% ethylacetate in hexane) to give 5-(2-substituted-ethyn-1-yl)-3-[(4- oxocyclohexyl)-(4-/ra «5-methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid methyl ester
- the product is isolated by purification by silica gel chromatography and reversed phase HPLC (60-95% methanol in H 2 0 (0.1% TFA) over 30min) to give 5-(2-substituted-ethyn-1-yl)-3-[(4-rra «i-hydroxycyclohexyl)-(4-tra «5- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid.
- Step 3 5-(3-Methylbut-1-ynyl)-3-[(4-oxocyclohexyl)-(4-irans- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid methyl ester (50 mg, 0.1 1 mmol) was taken in THF (10 mL) and two drops of water, cooled the reaction mixture to - 25°C. Then added NaBH 4 (4.2 mg, 0.1 1 mmol) and stirred for 2 h. Then the reaction was quenched by addition of IN HCl, then diluted with ethyl acetate and water.
- Compound 26 was prepared in a similar manner as described above for the preparation of Compound 4 by utilizing 3-ethynyl-3-methyloxetane instead of 3-methylbut- 1 -yne.
- 3-Ethynyltetrahydrofuran was prepared from (tetrahydrofuran-3-yl)methanol by the methods described in compound 26, and used to prepare compound 27 by the general methods described above.
- Compound 28 was prepared in a similar manner as described above for the preparation of Compound 4 by utilizing 1-ethynyl-1-methylcyclopropane instead of 3-methylbut-1-yne.
- reaction mixture was diluted with ethyl acetate, washed with water, and brine and dried (Na2S04).
- the solution was concentrated, and the residue purified by silica gel chromatography (10- 90% ethyl acetate in hexane) to give 5-(trimethy lsilylethyn- 1 -y l)-3-[( 1 ,4- dioxaspiro[4.5]decan-8-yl)-(4-ira/7i-methylcyclohexanecarbonyl)amino]thiophene-2- carboxylic acid methyl ester , which was used as obtained in the subsequent step.
- reaction mixture was heated at 60°C overnight. Then the reaction mixture was diluted with ethyl acetate washed with water and brine and dried (Na 2 S04). The product was purified by silica gel chromatography (10-90% ethyl acetate in hexane) to give 5-(5,5-dimethylhexa-1,3-diyn-1-yl)-3-[(1,4-dioxaspiro[4.5]decan-8-yl)-(4-ira «j- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid methyl ester.
- Compound 32 was prepared from 5-(5,5-dimethylhexa-1,3-diyn-1-yl)-3-[(l ,4- dioxaspiro[4.5]decan-8-yl)-(4-ira «i-methylcyclohexanecarbonyl)amino]thiophene-2- carboxylic acid methyl ester using the general methods, steps 2-4 as described above.
- Step 1 Methyl 3-bromo-5-(3,3-dimethylbut-l-yn-l-yl)thiophene-2-carboxylate
- Step 2 Methyl 3-(l, 3-dimethoxypropan-2-ylamino)-5-(3, 3-dimethylbut-l-ynyl) thiophene- 2-carboxylate
- Step 3 Methyl 3-(-N-(l,3-dimethoxypropan-2-yl)-4-trans- methylcyclohexanecarboxamido)-5-(3,3-dimethylbut-l-yn-l-yl)thiophene-2-carboxylate
- trans-4-methyl cyclohexanecarboxylic acid 0.5g, 3.52mmol, leq
- 1 ,2- dichloroethane 4.5 mL
- DMF 0.005 mL, 0.07 mmol, 0.02 eq
- oxalyl chloride 0.32 mL, 3.87 mmole, l .leq
- Step 4 3-(-N-(l,3-Dimethoxypropan-2-yl)-4-trans-methylcyclohexanecarboxamido)-5-(3,3- dimethylbut-l-yn-l-yl)thiophene-2-carboxylic acid (Compound 39)
- Compound 33 was prepared from S-1-methoxypropyl-2-amine in the manner described for compound 39.
- Compound 36 was prepared from i?-1-methoxypropyl-2-amine in the manner described for compound 39.
- Compound 37 was prepared from S-1-methoxybutyl-2-amine in the manner described for compound 39.
- R cyclopropyl
- R ethyl
- Compound 35 was prepared in two steps as described for Compound 38.
- Step 1 Methyl 5-(3,3-dimethylbut-l-yn-l-yl)-3-((N-4-trans-ethoxycyclohexyl)-4-trans- methylcyclohexanecarboxamido)thiophene-2-carboxylate
- Step 2 5-(3,3-Dimethylbut-l-yn-l-yl)-3-( (N-4-trans-ethoxycyclohexyl)-4-trans- methylcyclohexanecarboxamido)thiophene-2-carboxylic acid
- the crude product was purified by silica-gel plug using 10 % ethyl acetate in hexanes as eluent to afford product ⁇ 85 % purity, which was triturated with methanol to afford methyl 3-bromo-5-iodo-thiophene-2-carboxylate (35.4 g, 45 % yield) as a light yellow solid.
- Step 4 Methyl 3-(N-(tetrahydro-2H-pyran-4-yl)-4-trans-methyl-cyclohexanecarboxamido)- 5-(3-methylbut-l-yn-l-yl)thiophene-2-carboxylate
- Methyl 5-(3-methylbut- 1-yn- 1 -yl)-3-((tetrahydro-2H-pyran-4-yl)amino)thiophene-2- carboxylate was acylated with ira «5-4-methyl cyclohexanecarbonyl chloride by the methods described above to afford the desired product, isolated after silica gel chromatography (30% EtOAc in pet. ether), 100 mg (48%).
- Step 5 3-(N-(tetrahydro-2H-pyran-4-yl)-4-trans-methyl-cyclohexanecarboxamido)-5-(3- methylbut-l-yn-l-yl)thiophene-2-carboxylic acid
- Compound 52 was prepared from trans-4-methylcyclohexylamine and methyl 3-bromo-5-(3- methylbut- 1 -yn- 1 -yl)thiophene-2 -carboxylate according to the scheme described for compound 43.
- Step 1 5-(3-Methylbut-l-yn-l-yl)-3-[(l,4-dioxaspiro[4.5]decan-8-yl)-(4-trans- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid methyl ester
- reaction mixture was warmed to RT and stirred for 2h to get dark color.
- the reaction progress was monitored by TLC.
- the reaction mixture was diluted with diethyl ether (100 mL), filtered through celite bed, washed with excess of diethyl ether (2x50 mL).
- Step 3A 5-(3-Methylbut-l-yn-l-yl)-3-((N-4-cis-(2-hydroxy)-ethoxycycloh ⁇
- Step 3B 5-(3-Methylbut-l-yn-l-yl)-3-((N-4-trans-(2-hydroxy)-ethoxycyclohexyl)-(4-t methylcyclohexane)carboxamido)thiophene-2-carboxylic acid (compound 62)
- Methyl 3-bromo-5-(3,3-dimethylbut-1-ynyl)thiophene-2-carboxylate 1000 mg, 3.320 mmol
- tert-butyl 4-aminopiperidine-1-carboxylate 797.9 mg, 3.984 mmol
- cesium carbonate 3.45 g, 9.960 mmol
- dicyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphane 136.3 mg, 0.3320 mmol
- the mixture was cooled to room temperature and 1 mL of pyridine was added to the mixture followed by the addition of 1 mL of methanol.
- the reaction was diluted with
- tert-Butyl 4-[[5-(3,3-dimethylbut- 1 -ynyl)-2-methoxycarbonyl-3-thienyl]-(4-/ "a «s- methylcyclohexanecarbonyl)amino]piperidine-1-carboxylate (860 mg, 1.579 mmol) was dissolved in 5 mL of dichloromethane and 1 mL of TFA was added. The reaction was stirred at room temperature for 1 hour. The reaction was evaporated in vacuo and the residue dissolved in dichloromethane and washed with saturated sodium bicarbonate and brine. The organic dried over anhydrous sodium sulfate, filtered, and evaporated in vacuo to afford 679 mg of the desired product.
- compound 50 Methyl 5-(3,3-dimethylbut-1-ynyl)-3-[[l-(2-methoxyacetyl)-4-piperidyl]-(4-trans- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate (360 mg, 0.70 mmol)was dissolved in 1 : 1 mixture of MeOH and water (5 ml). To the solution was added Lithium hydroxide (33.4 mg, 1.39 mmol) and the mixture was stirred overnight. Inspection of the LC/MS showed product and also slight hydrolysis of the amide.
- Compound 51 was prepared by the methods described for compound 50.
- Step 1 Methyl 5-iodo-3-(trans-4-methylcyclohexanecarboxamido)thiophene-2-carboxylate
- Methyl 3-amino-5-iodo-thiophene-2-carboxylate (50 g, 174.9 mmol) was dissolved in CH2C12 (500 mL) and pyridine (30 mL) and cooled to 0°C and then added iram-4-methyl cyclohexanecarbonyl chloride (60.2 g, 210 mmol) dropwise (neat), and rinsed with a small amount of DCM. After 5 min, removed bath, and stirred as reaction came to RT.
- 1,4-Dioxane solvent used in reaction was anhydrous and deoxygenated by purging with nitrogen gas for 30 mins.
- Methyl 3-bromo-5-(3,3-dimethylbut-1-ynyl)thiophene-2- carboxylate 500 mg, 1.660 mmol
- phenyljphosphane (68 mg, 0.17 mmol) and cesium carbonate (1.62 g, 4.98 mmol) were taken into 5 mL of toluene and argon was bubbled to the mixture for 5 minutes.
- the catalyst Pd 2 dba 3 (76 mg, 0.083 mmol) was added to the mixture and the reaction was sealed and heated at 90°C overnight for 18 hrs.
- Step 4 3-(N-((S)-l-carboxypropyl)-(4-trans-methylcyclohexane)-carboxamido)-5-(3,3- dimethylbut-l-yn-l-yl)thiophene-2-carboxylic acid
- Step 1 Methyl 5-(3,3-dimethylbut-l-yn-l-yl)-3-((l-morpholinopropan-2- yl)amino)thiophene-2-carboxylate
- Step 1 Methyl 5-(3,3-dimethylbut-l-ynyl)-3-[(l-methyl-2-pyrazol-l-yl- ethyl)amino]thiophene-2-carboxylate.
- Step 2 Methyl 5-(3,3-dimethylbut-l-ynyl)-3-[(trans-4-methylcyclohexanecarbonyl)-(l- methyl-2-pyrazol-l-yl-ethyl)amino]thiophene-2-carboxylate.
- Triethylamine (2 mL) was added to the mixture followed by the addition of 1 mL of methanol. The mixture was stirred at room temperature for 1 hour then evaporated in vacuo. The crude gum was dissolved in dichloromethane and absorbed onto Si02. This was purified by silica gel column chromatography eluting with a gradient of hexanes to 100 % ethyl acetate. The desired fractions were combined and evaporated in vacuo to afford the desired product as a clear oil that crystallized upon setting at room temperature. Yield 150 mg, 38.7 %.
- Title compound was prepared by the hydrolysis of methyl 5-(3,3-dimethylbut-1-ynyl)-3-[(4- trans-methy lcyclohexanecarbony l)-( 1 -methyl-2-pyrazol- 1 -yl-ethy l)amino]thiophene-2- carboxylate (170 mg, 0.3620 mmol) as described for the preparation of Compound 29. Yield 157 mg, 90 %.
- Step 1 Methyl S-(3,3-dimethylbut-l-ynyl)-3-fl-(3-isopropyl-l,2,4-oxadiazol-5- yl)ethylamino]thiophene-2-carboxylate.
- Methyl 3-bromo-5-(3,3-dimethylbut-1-ynyl)thiophene-2-carboxylate (373 mg, 1.24 mmol)
- l-(3-isopropyl-1,2,4-oxadiazol-5-yl)ethanamine 400 mg, 1.49 mmol
- cesium carbonate (1.61 g, 4.95 mmol) were taken into 12 mL of anhydrous dioxane and degassed with argon.
- Step 2 Methyl 5-(3,3-dimethylbut-l-ynyl)-3-[l-(3-isopropyl-l,2,4-oxadiazol-5-yl)ethyl-(4- trans-methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate.
- Step 3 5-(3,3 ⁇ imethylbut-l-ynyl)-3-[l-(3-isopropyl-l,2,4-oxadiazol-5-yl)ethyl-(4-trans- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid.
- Step 1 Methyl 5-(3,3-dimethylbut-lrynyl)-3-f(3-ethyl-l,2,4-oxadiazol-5- yl)methylamino]thiophene-2-car box late
- Methyl 3-bromo-5-(3,3-dimethylbut-l -ynyl)thiophene-2-carboxylate 400 mg, 1.33 mmol
- (3-ethyl-1,2,4-oxadiazol-5-yl)methanamine (261 mg, 1.59 mmol)
- cesium carbonate (1.73 g, 5.31 mmol) were taken into 12 ml of anhydrous dioxane and degassed with argon.
- Step 3 S-(3,3-dimethylbut-l-ynyl)-3-[(3-ethyl-l,2,4-oxadiazol-5-yl)methyl-(4-trans- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid.
- the title compound was prepared by the hydrolysis of methyl 5-(3,3-dimethylbut-1-ynyl)-3- [(3-ethyl- 1 ,2,4-oxadiazol-5-yl)methyl-(4-/ra «s- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate (90 mg, 0.1908 mmol) as described for the preparation of Compound 29. Yield 77 mg, 84 %.
- Step 1 Methyl 5-(3,3-dimethylbut-l-ynyl)-3-[l-(3-ethyl-l,2,4-oxadiazol-5- yl)ethylamino]thiophene-2-carboxylate
- the title compound was prepared by the hydrolysis of methyl 5-(3,3-dimethylbut-1-ynyl)-3- [l-(3-ethyl-1,2,4-oxadiazol-5-yl)ethyl-(4-/ra «5- methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate (120 mg, 0.2471 mmol as described for the preparation of Compound 29. Yield 1 1 1 mg, 90.5 %.
- Methyl 3-bromo-5-(3,3-dimethylbut-1-ynyl)thiophene-2-carboxylate 400 mg, 1.33 mmol
- 2-pyrazol-1-ylpropan-l -amine 200 mg, 1.59 mmol
- cesium carbonate 1.08 g, 3.32 mmol
- Step 1 Methyl 5-iodo-3-f(4-methylcyclohexanecarbonyl)aminoJthiophene-2-carboxylate 4-irans-Methylcyclohexanecarbonyl chloride (6.24 g, 38.9 mmol) was added dropwise to a cooled solution (0 °C) of methyl 3-amino-5-iodo-thiophene-2-carboxylate (10 g, 35.3 mmol) in dichloromethane (90 mL) and pyridine (6 mL). The reaction was warmed to room temperature and stirred overnight.. The reaction mixture was washed with water, IN HCl, and brine. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to provide the title compound as a pale yellow solid. Yield 14.45 g, 99%.
- Methyl 5-iodo-3-[(4-trans-methylcyclohexanecarbonyl)amino]thiophene-2-carboxylate (14.4 g, 35.4 mmol) and N,N-diisopropylamine (7.5 mL, 53 mmol) were taken into 100 mL of dioxane and degassed by bubbling nitrogen into the mixture for 10 minutes.
- step 2 To the product of step 2 (900 mg, 1.7 mmol) in dichloromethane (5 mL) was added trifluoroacetic acid (5 mL) . The reaction was stirred for 30 minutes and solvent was removed. Diluted with DCM and washed 2 x with saturated sodium bicarbonate. The organic layer was dried and concentrated to give 723 mg of desired product as an off-white solid. MS: m/z (obs.): 431.3 [M+H] + ;
- Step 1 Methyl 3-(N-((S)-l-acetylpyrrolidin-3-yl)-4-trans-methylcyclohexanecarboxamido)- 5-(3,3-dimethylbut-l-yn-l-yl)thiophene-2-carboxylate
- Step 2 3-(N-( (S)-l-A cetylpyrrolidin-3-yl)-4-trans-methylcyclohexanecarboxamido)-5-(3,3- dimethylbut-l-yn-l-yl)thiophene-2-carboxylic acid
- Step 1 Methyl 5-(3,3-dimethylbut-l-yn-l-yl)-3-(trans-4-methyl-N-((S)-l- methylsulfonyl)pyrrolidin-3-yl)cyclohexanecarboxamido)thiophene-2-carboxylate
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Zoology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description















































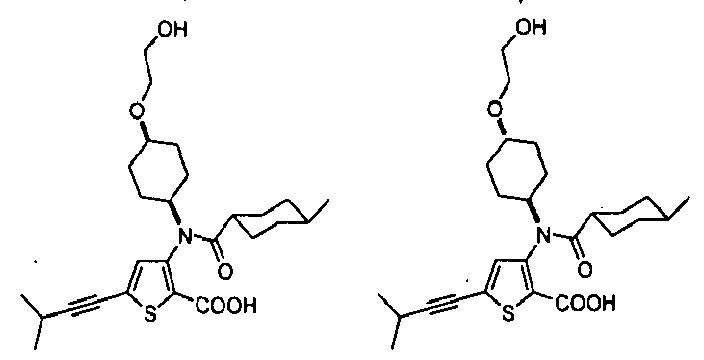


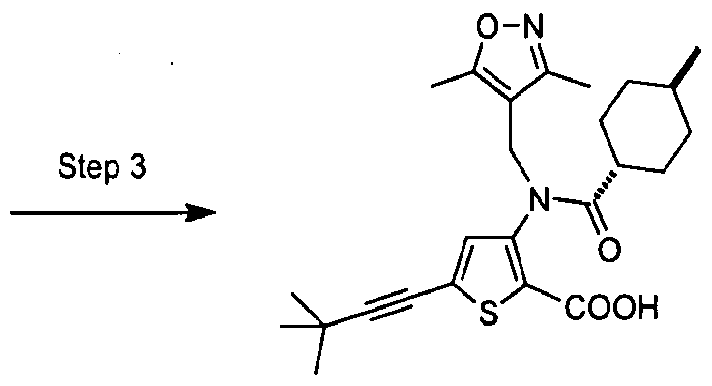
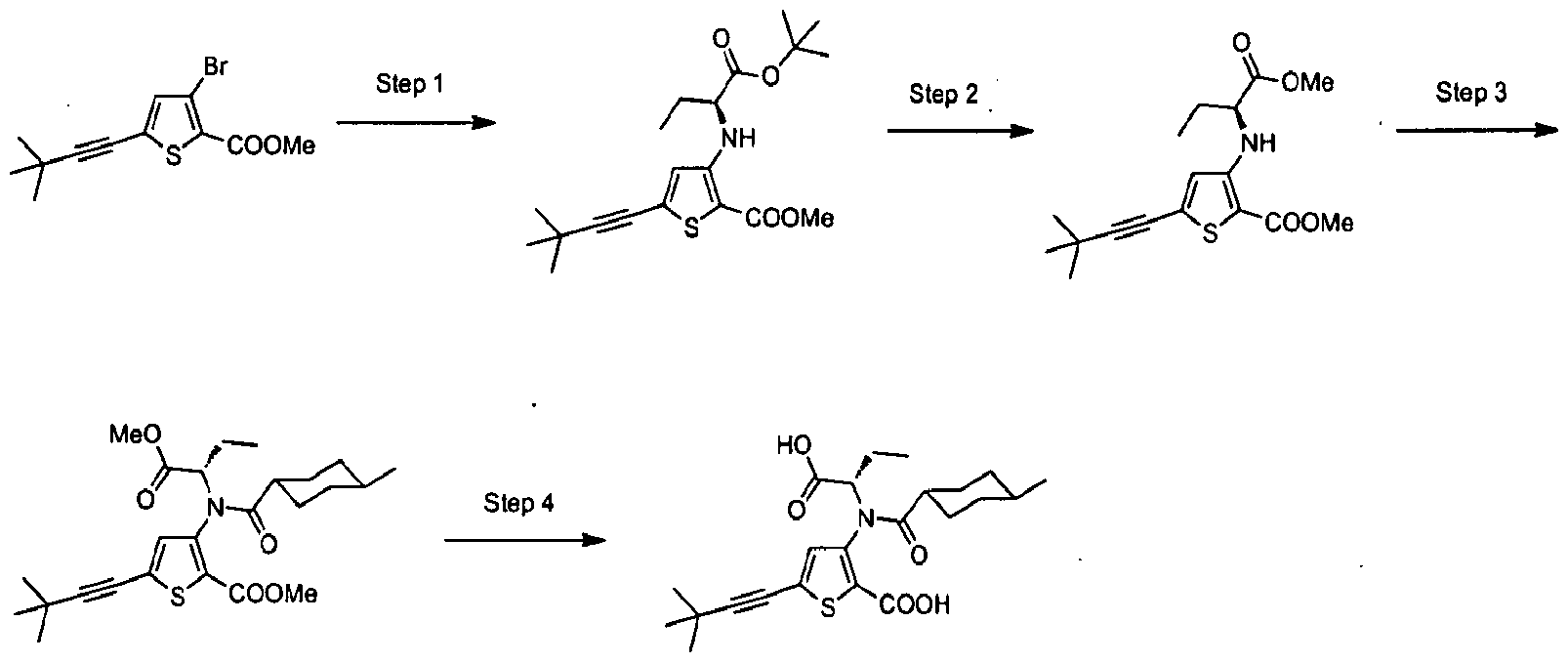












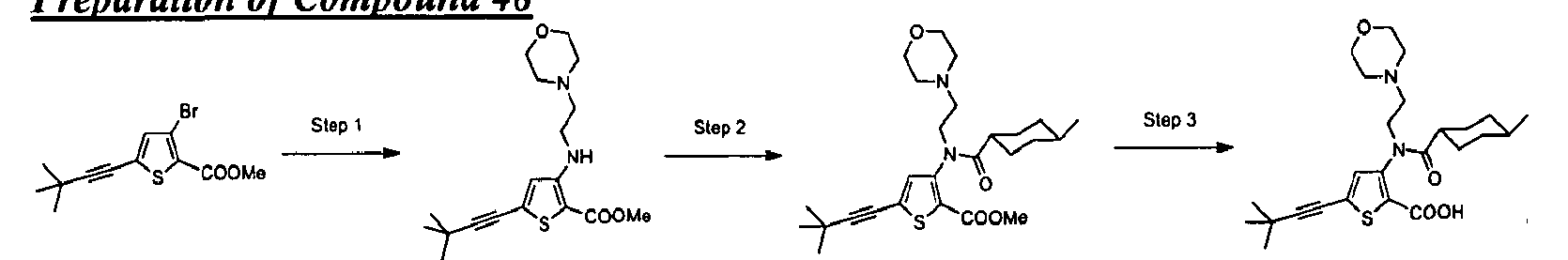
















Claims


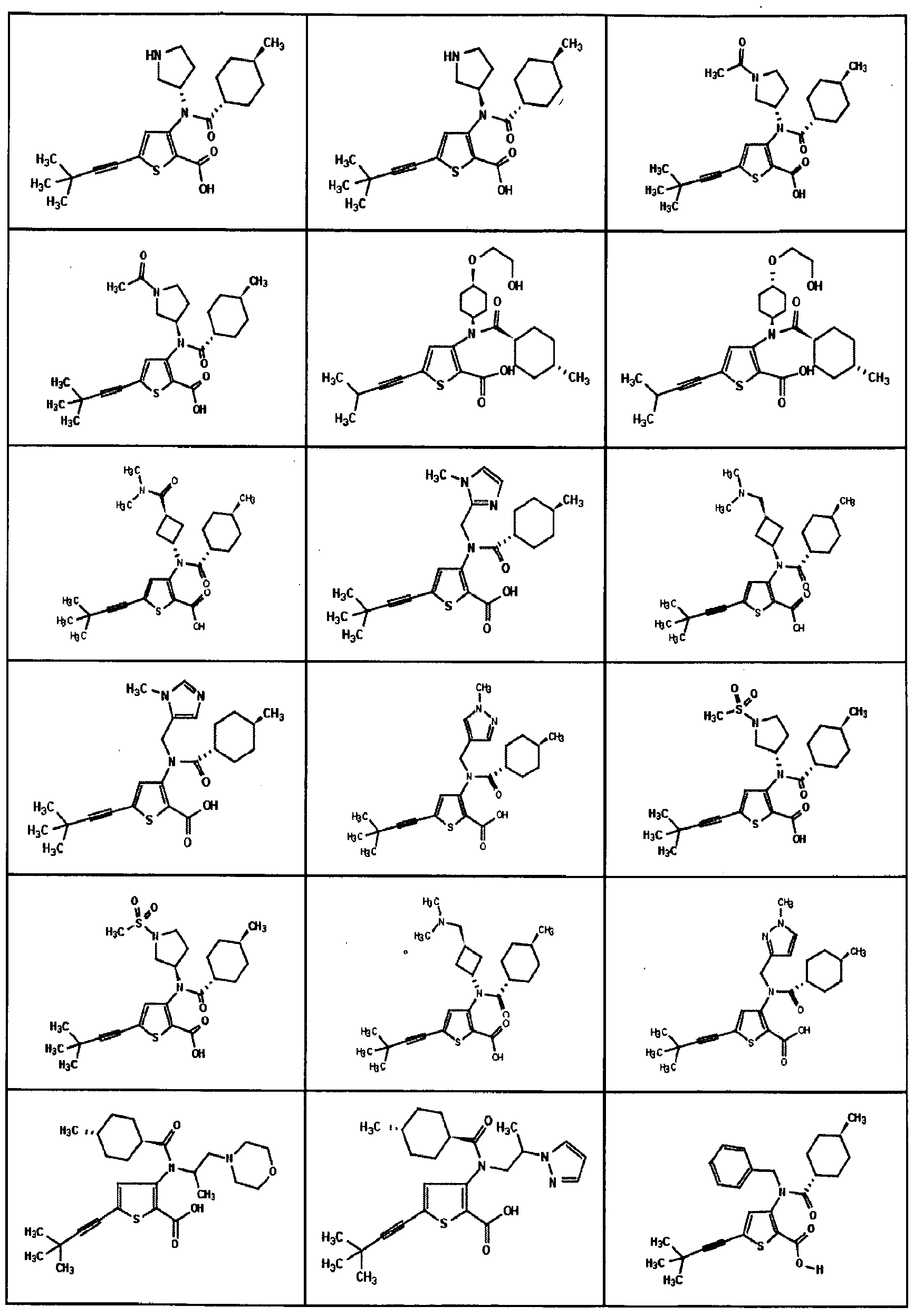





Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2808291A CA2808291A1 (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| MX2013001869A MX2013001869A (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of flaviviridae viral infections. |
| EP11749659.6A EP2606041A2 (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| AU2011292040A AU2011292040A1 (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of Flaviviridae viral infections |
| JP2013524949A JP2013534249A (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of Flaviviridae viral infections |
| CN2011800478412A CN103153978A (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| US13/767,347 US20140065103A1 (en) | 2010-08-17 | 2013-02-14 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37439610P | 2010-08-17 | 2010-08-17 | |
| US61/374,396 | 2010-08-17 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/767,347 Continuation US20140065103A1 (en) | 2010-08-17 | 2013-02-14 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012024363A2 true WO2012024363A2 (en) | 2012-02-23 |
| WO2012024363A3 WO2012024363A3 (en) | 2012-05-31 |
Family
ID=44533193
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/048027 Ceased WO2012024363A2 (en) | 2010-08-17 | 2011-08-17 | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20140065103A1 (en) |
| EP (1) | EP2606041A2 (en) |
| JP (1) | JP2013534249A (en) |
| CN (1) | CN103153978A (en) |
| AU (1) | AU2011292040A1 (en) |
| CA (1) | CA2808291A1 (en) |
| MX (1) | MX2013001869A (en) |
| WO (1) | WO2012024363A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013030750A1 (en) | 2011-09-01 | 2013-03-07 | Lupin Limited | Antiviral compounds |
| WO2013118097A1 (en) | 2012-02-10 | 2013-08-15 | Lupin Limited | Antiviral compounds with a dibenzooxaheterocycle moiety |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140206888A1 (en) * | 2011-07-26 | 2014-07-24 | Vertex Pharmaceuticals Incorporated | Methods for preparation of thiophene compounds |
| CN108096562B (en) * | 2017-12-25 | 2021-05-11 | 武汉百药联科科技有限公司 | Application of 20S proteasome inhibitor in preparation of medicine for treating Japanese encephalitis virus infection |
Citations (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998017679A1 (en) | 1996-10-18 | 1998-04-30 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| WO1999007733A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO1999007734A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| WO2000006529A1 (en) | 1998-07-27 | 2000-02-10 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti S.P.A. | Diketoacid-derivatives as inhibitors of polymerases |
| WO2000009543A2 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| WO2000009558A1 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO2000059929A1 (en) | 1999-04-06 | 2000-10-12 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| WO2001047883A1 (en) | 1999-12-27 | 2001-07-05 | Japan Tobacco Inc. | Fused-ring compounds and use thereof as drugs |
| WO2001085172A1 (en) | 2000-05-10 | 2001-11-15 | Smithkline Beecham Corporation | Novel anti-infectives |
| WO2001090121A2 (en) | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2002006246A1 (en) | 2000-07-19 | 2002-01-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Dihydroxypyrimidine carboxylic acids as viral polymerase inhibitors |
| WO2002018369A2 (en) | 2000-08-31 | 2002-03-07 | Eli Lilly And Company | Peptidomimetic protease inhibitors |
| WO2002057287A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2002060926A2 (en) | 2000-11-20 | 2002-08-08 | Bristol-Myers Squibb Company | Hepatitis c tripeptide inhibitors |
| WO2002069903A2 (en) | 2001-03-06 | 2002-09-12 | Biocryst Pharmaceuticals, Inc. | Nucleosides, preparation thereof and use as inhibitors of rna viral polymerases |
| EP1256628A2 (en) | 2001-05-10 | 2002-11-13 | Agouron Pharmaceuticals, Inc. | Hepatitis c virus (hcv) ns5b rna polymerase and mutants thereof |
| WO2002098424A1 (en) | 2001-06-07 | 2002-12-12 | Smithkline Beecham Corporation | Novel anti-infectives |
| WO2002100846A1 (en) | 2001-06-11 | 2002-12-19 | Shire Biochem Inc. | Compounds and methods for the treatment or prevention of flavivirus infections |
| WO2002100851A2 (en) | 2001-06-11 | 2002-12-19 | Shire Biochem Inc. | Thiophene derivatives as antiviral agents for flavivirus infection |
| WO2003000254A1 (en) | 2001-06-26 | 2003-01-03 | Japan Tobacco Inc. | Fused cyclic compounds and medicinal use thereof |
| WO2003010140A2 (en) | 2001-07-25 | 2003-02-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c virus polymerase inhibitors with heterobicyclic structure |
| WO2003026587A2 (en) | 2001-09-26 | 2003-04-03 | Bristol-Myers Squibb Company | Compounds useful for treating hepatitus c virus |
| WO2003035060A1 (en) | 2001-10-24 | 2003-05-01 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine protease, particularly hepatitis c virus ns3-ns4a protease, incorporating a fused ring system |
| WO2003087092A2 (en) | 2002-04-11 | 2003-10-23 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 - ns4 protease |
| WO2004014313A2 (en) | 2002-08-12 | 2004-02-19 | Bristol-Myers Squibb Company | Combination pharmaceutical agents as inhibitors of hcv replication |
| WO2004092162A1 (en) | 2003-04-11 | 2004-10-28 | Vertex Pharmaceuticals, Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2004092161A1 (en) | 2003-04-11 | 2004-10-28 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| US20050009804A1 (en) | 2002-12-10 | 2005-01-13 | Laval Chan Chun Kong | Compounds and methods for the treatment or prevention of Flavivirus infections |
| WO2005007681A2 (en) | 2003-07-18 | 2005-01-27 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005028502A1 (en) | 2003-09-18 | 2005-03-31 | Vertex Pharmaceuticals, Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005035525A2 (en) | 2003-09-05 | 2005-04-21 | Vertex Pharmaceuticals Incorporated | 2-amido-4-aryloxy-1-carbonylpyrrolidine derivatives as inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005077969A2 (en) | 2004-02-04 | 2005-08-25 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2006039488A2 (en) | 2004-10-01 | 2006-04-13 | Vertex Pharmaceuticals Incorporated | Hcv ns3-ns4a protease inhibition |
| US20060276533A1 (en) | 2005-05-13 | 2006-12-07 | Real Denis | Compounds and methods for the treatment or prevention of Flavivirus infections |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2008021927A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021936A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021928A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008058393A1 (en) | 2006-11-15 | 2008-05-22 | Virochem Pharma Inc. | Thiophene analogues for the treatment or prevention of flavivirus infections |
| WO2008144380A1 (en) | 2007-05-17 | 2008-11-27 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009020825A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Process for synthesizing compounds useful for treating hepatitis c |
| WO2009020828A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Crystalline form of methyl ((1s)-1-(((2s)-2-(5-(4'-(2-((2s)-1-((2s)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1h-imidazol-5-yl)-4-biphenylyl)-1h-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt |
| WO2009102568A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Conformationally restricted biphenyl derivatives for use as hepatitis c virus inhibitors |
| WO2009102633A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009102318A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| WO2010017401A1 (en) | 2008-08-07 | 2010-02-11 | Bristol-Myers Squibb Company | Bi-1h-benzimidazoles as hepatitis c virus inhibitors |
| WO2010062821A1 (en) | 2008-11-28 | 2010-06-03 | Glaxosmithkline Llc | Anti-viral compounds, compositions, and methods of use |
| WO2010065681A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010065674A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010091413A1 (en) | 2009-02-09 | 2010-08-12 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| WO2010096462A1 (en) | 2009-02-17 | 2010-08-26 | Enanta Pharmaceuticals, Inc | Linked diimidazole derivatives |
| WO2010096302A1 (en) | 2009-02-17 | 2010-08-26 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010094077A1 (en) | 2009-02-20 | 2010-08-26 | Bluescope Steel Limited | A high strength thin cast strip product and method for making the same |
| WO2010096777A1 (en) | 2009-02-23 | 2010-08-26 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010099527A1 (en) | 2009-02-27 | 2010-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| WO2010111483A1 (en) | 2009-03-27 | 2010-09-30 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| WO2010117977A1 (en) | 2009-04-09 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010117635A1 (en) | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010117704A1 (en) | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010120621A1 (en) | 2009-04-13 | 2010-10-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010120935A1 (en) | 2009-04-15 | 2010-10-21 | Abbott Laboratories | Anti-viral compounds |
| WO2010122162A1 (en) | 2009-04-24 | 2010-10-28 | Tibotec Pharmaceuticals | Diaryl ethers |
| WO2010126967A1 (en) | 2009-04-28 | 2010-11-04 | Boehringer Ingelheim International Gmbh | Ex-vivo treatment of immunological disorders with pkc-theta inhibitors |
| WO2010132538A1 (en) | 2009-05-12 | 2010-11-18 | Schering Corporation | Fused tricyclic aryl compounds useful for the treatment of viral diseases |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA05006196A (en) * | 2002-12-10 | 2006-01-27 | Virochem Pharma Inc | Compounds and methods for the treatment or prevention of flavivirus. |
| DE102004061746A1 (en) * | 2004-12-22 | 2006-07-06 | Bayer Healthcare Ag | Alkynyl-substituted thiophenes |
-
2011
- 2011-08-17 EP EP11749659.6A patent/EP2606041A2/en not_active Withdrawn
- 2011-08-17 WO PCT/US2011/048027 patent/WO2012024363A2/en not_active Ceased
- 2011-08-17 CN CN2011800478412A patent/CN103153978A/en active Pending
- 2011-08-17 JP JP2013524949A patent/JP2013534249A/en not_active Withdrawn
- 2011-08-17 MX MX2013001869A patent/MX2013001869A/en unknown
- 2011-08-17 CA CA2808291A patent/CA2808291A1/en not_active Abandoned
- 2011-08-17 AU AU2011292040A patent/AU2011292040A1/en not_active Abandoned
-
2013
- 2013-02-14 US US13/767,347 patent/US20140065103A1/en not_active Abandoned
Patent Citations (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998017679A1 (en) | 1996-10-18 | 1998-04-30 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| WO1999007733A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO1999007734A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| WO2000006529A1 (en) | 1998-07-27 | 2000-02-10 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti S.P.A. | Diketoacid-derivatives as inhibitors of polymerases |
| WO2000009543A2 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| WO2000009558A1 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO2000059929A1 (en) | 1999-04-06 | 2000-10-12 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| WO2001047883A1 (en) | 1999-12-27 | 2001-07-05 | Japan Tobacco Inc. | Fused-ring compounds and use thereof as drugs |
| WO2001085172A1 (en) | 2000-05-10 | 2001-11-15 | Smithkline Beecham Corporation | Novel anti-infectives |
| WO2001090121A2 (en) | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2002006246A1 (en) | 2000-07-19 | 2002-01-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Dihydroxypyrimidine carboxylic acids as viral polymerase inhibitors |
| WO2002018369A2 (en) | 2000-08-31 | 2002-03-07 | Eli Lilly And Company | Peptidomimetic protease inhibitors |
| WO2002060926A2 (en) | 2000-11-20 | 2002-08-08 | Bristol-Myers Squibb Company | Hepatitis c tripeptide inhibitors |
| WO2002057287A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2002057425A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2002069903A2 (en) | 2001-03-06 | 2002-09-12 | Biocryst Pharmaceuticals, Inc. | Nucleosides, preparation thereof and use as inhibitors of rna viral polymerases |
| EP1256628A2 (en) | 2001-05-10 | 2002-11-13 | Agouron Pharmaceuticals, Inc. | Hepatitis c virus (hcv) ns5b rna polymerase and mutants thereof |
| WO2002098424A1 (en) | 2001-06-07 | 2002-12-12 | Smithkline Beecham Corporation | Novel anti-infectives |
| WO2002100846A1 (en) | 2001-06-11 | 2002-12-19 | Shire Biochem Inc. | Compounds and methods for the treatment or prevention of flavivirus infections |
| WO2002100851A2 (en) | 2001-06-11 | 2002-12-19 | Shire Biochem Inc. | Thiophene derivatives as antiviral agents for flavivirus infection |
| US6881741B2 (en) | 2001-06-11 | 2005-04-19 | Virochem Pharma Inc. | Compounds and methods for the treatment or prevention of Flavivirus infections |
| WO2003000254A1 (en) | 2001-06-26 | 2003-01-03 | Japan Tobacco Inc. | Fused cyclic compounds and medicinal use thereof |
| WO2003010140A2 (en) | 2001-07-25 | 2003-02-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c virus polymerase inhibitors with heterobicyclic structure |
| WO2003026587A2 (en) | 2001-09-26 | 2003-04-03 | Bristol-Myers Squibb Company | Compounds useful for treating hepatitus c virus |
| WO2003035060A1 (en) | 2001-10-24 | 2003-05-01 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine protease, particularly hepatitis c virus ns3-ns4a protease, incorporating a fused ring system |
| WO2003087092A2 (en) | 2002-04-11 | 2003-10-23 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 - ns4 protease |
| WO2004014313A2 (en) | 2002-08-12 | 2004-02-19 | Bristol-Myers Squibb Company | Combination pharmaceutical agents as inhibitors of hcv replication |
| WO2004014852A2 (en) | 2002-08-12 | 2004-02-19 | Bristol-Myers Squibb Company | Iminothiazolidinones as inhibitors of hcv replication |
| US20050009804A1 (en) | 2002-12-10 | 2005-01-13 | Laval Chan Chun Kong | Compounds and methods for the treatment or prevention of Flavivirus infections |
| WO2004092162A1 (en) | 2003-04-11 | 2004-10-28 | Vertex Pharmaceuticals, Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2004092161A1 (en) | 2003-04-11 | 2004-10-28 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005007681A2 (en) | 2003-07-18 | 2005-01-27 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005035525A2 (en) | 2003-09-05 | 2005-04-21 | Vertex Pharmaceuticals Incorporated | 2-amido-4-aryloxy-1-carbonylpyrrolidine derivatives as inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005028502A1 (en) | 2003-09-18 | 2005-03-31 | Vertex Pharmaceuticals, Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2005077969A2 (en) | 2004-02-04 | 2005-08-25 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| WO2006039488A2 (en) | 2004-10-01 | 2006-04-13 | Vertex Pharmaceuticals Incorporated | Hcv ns3-ns4a protease inhibition |
| US20060276533A1 (en) | 2005-05-13 | 2006-12-07 | Real Denis | Compounds and methods for the treatment or prevention of Flavivirus infections |
| WO2006133326A1 (en) | 2005-06-06 | 2006-12-14 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2008021927A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021936A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008021928A2 (en) | 2006-08-11 | 2008-02-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008058393A1 (en) | 2006-11-15 | 2008-05-22 | Virochem Pharma Inc. | Thiophene analogues for the treatment or prevention of flavivirus infections |
| WO2008144380A1 (en) | 2007-05-17 | 2008-11-27 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009020825A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Process for synthesizing compounds useful for treating hepatitis c |
| WO2009020828A1 (en) | 2007-08-08 | 2009-02-12 | Bristol-Myers Squibb Company | Crystalline form of methyl ((1s)-1-(((2s)-2-(5-(4'-(2-((2s)-1-((2s)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1h-imidazol-5-yl)-4-biphenylyl)-1h-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt |
| WO2009102318A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009102568A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Conformationally restricted biphenyl derivatives for use as hepatitis c virus inhibitors |
| WO2009102633A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| WO2010017401A1 (en) | 2008-08-07 | 2010-02-11 | Bristol-Myers Squibb Company | Bi-1h-benzimidazoles as hepatitis c virus inhibitors |
| WO2010062821A1 (en) | 2008-11-28 | 2010-06-03 | Glaxosmithkline Llc | Anti-viral compounds, compositions, and methods of use |
| WO2010065681A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010065668A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010065674A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010091413A1 (en) | 2009-02-09 | 2010-08-12 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| WO2010096462A1 (en) | 2009-02-17 | 2010-08-26 | Enanta Pharmaceuticals, Inc | Linked diimidazole derivatives |
| WO2010096302A1 (en) | 2009-02-17 | 2010-08-26 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010094077A1 (en) | 2009-02-20 | 2010-08-26 | Bluescope Steel Limited | A high strength thin cast strip product and method for making the same |
| WO2010096777A1 (en) | 2009-02-23 | 2010-08-26 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010099527A1 (en) | 2009-02-27 | 2010-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| WO2010111483A1 (en) | 2009-03-27 | 2010-09-30 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| WO2010117635A1 (en) | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010117704A1 (en) | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010117977A1 (en) | 2009-04-09 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010120621A1 (en) | 2009-04-13 | 2010-10-21 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010120935A1 (en) | 2009-04-15 | 2010-10-21 | Abbott Laboratories | Anti-viral compounds |
| WO2010122162A1 (en) | 2009-04-24 | 2010-10-28 | Tibotec Pharmaceuticals | Diaryl ethers |
| WO2010126967A1 (en) | 2009-04-28 | 2010-11-04 | Boehringer Ingelheim International Gmbh | Ex-vivo treatment of immunological disorders with pkc-theta inhibitors |
| WO2010132538A1 (en) | 2009-05-12 | 2010-11-18 | Schering Corporation | Fused tricyclic aryl compounds useful for the treatment of viral diseases |
Non-Patent Citations (14)
| Title |
|---|
| "BURGER'S MEDICINAL CHEMISTRY AND DRUG DISCOVERY", vol. 172-178, 1995, pages: 949 - 982 |
| "Handbook of Chemistry and Physics" |
| "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS |
| "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| E. W. MARTIN: "Remington's Pharmaceutical Sciences", 1980, MACK PUBLISHING CO. |
| GAO M. ET AL., NATURE, vol. 465, 2010, pages 96 - 100 |
| GREENE, T. W. AND WUTS, P. G: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY & SONS |
| JANET S. DODD: "The ACS Style Guide: A Manual for Authors and Editors, 2nd Ed.,Washington, D.C.", 1997, AMERICAN CHEMICAL SOCIETY |
| KRIEGER, N, LOHMANN AND BARTENSCHLAGER, R: "Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations", J. VIROL., vol. 75, 2001, pages 4614 - 4624 |
| P. HEINRICH STAHL AND CAMILLE G. WERMUTH,: "Handbook ofPharmaceutical Salts Properties, Selection, and Use", 2002, WILEY |
| P. J. KOCIENSKI: "Protecting Groups", 2005, THIEME |
| S. M. BERGE ET AL.: "describe pharmaceutically acceptable salts in detail", J. PHARMACEUTICAL SCIENCES, vol. 66, 1977, pages 1 - 19 |
| T. W. GREENE AND P. G. M. WUTS: "Protective Groups in Organic Synthesis", WILEY INTERSCIENCE |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE BOOKS |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013030750A1 (en) | 2011-09-01 | 2013-03-07 | Lupin Limited | Antiviral compounds |
| WO2013118097A1 (en) | 2012-02-10 | 2013-08-15 | Lupin Limited | Antiviral compounds with a dibenzooxaheterocycle moiety |
| WO2013118102A1 (en) | 2012-02-10 | 2013-08-15 | Lupin Limited | Antiviral compounds with a heterotricycle moiety |
| US9073943B2 (en) | 2012-02-10 | 2015-07-07 | Lupin Limited | Antiviral compounds with a dibenzooxaheterocycle moiety |
| US9073942B2 (en) | 2012-02-10 | 2015-07-07 | Lupin Limited | Antiviral compounds with a heterotricycle moiety |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2011292040A1 (en) | 2013-03-07 |
| US20140065103A1 (en) | 2014-03-06 |
| EP2606041A2 (en) | 2013-06-26 |
| MX2013001869A (en) | 2013-06-28 |
| JP2013534249A (en) | 2013-09-02 |
| CA2808291A1 (en) | 2012-02-23 |
| CN103153978A (en) | 2013-06-12 |
| WO2012024363A3 (en) | 2012-05-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5841531B2 (en) | Inhibitors of Flaviviridae virus | |
| AU2011205797B2 (en) | Inhibitors of Flaviviridae viruses | |
| US20130190289A1 (en) | Compounds and methods for the treatment or prevention of flavivirus infections | |
| SG182475A1 (en) | Inhibitors of flaviviridae viruses | |
| JP2013504576A (en) | Inhibitors of Flaviviridae virus | |
| US20130136719A1 (en) | Compounds and methods for the treatment or prevention of flavivirus infections | |
| US20140065103A1 (en) | Compounds and methods for the treatment or prevention of flaviviridae viral infections | |
| US20140235704A1 (en) | Thiophene compounds | |
| US20130183266A1 (en) | Compounds and methods for the treatment or prevention of flavivirus infections | |
| WO2013016499A1 (en) | Methods for preparation of thiophene compounds | |
| US20130203706A1 (en) | Compounds and methods for the treatment or prevention of flavivirus infections | |
| HK1186184A (en) | Compounds and methods for the treatment or prevention of flaviviridae viral infections | |
| WO2012006060A1 (en) | Compounds and methods for the treatment or prevention of flavivirus infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180047841.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11749659 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2808291 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2013524949 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011749659 Country of ref document: EP Ref document number: MX/A/2013/001869 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2011292040 Country of ref document: AU Date of ref document: 20110817 Kind code of ref document: A |