WO2012060185A1 - エステルの製法 - Google Patents
エステルの製法 Download PDFInfo
- Publication number
- WO2012060185A1 WO2012060185A1 PCT/JP2011/073340 JP2011073340W WO2012060185A1 WO 2012060185 A1 WO2012060185 A1 WO 2012060185A1 JP 2011073340 W JP2011073340 W JP 2011073340W WO 2012060185 A1 WO2012060185 A1 WO 2012060185A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- nmr
- mhz
- cdcl
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D313/00—Heterocyclic compounds containing rings of more than six members having one oxygen atom as the only ring hetero atom
- C07D313/02—Seven-membered rings
- C07D313/04—Seven-membered rings not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B33/00—Oxidation in general
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/50—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by reactions decreasing the number of carbon atoms
- C07C37/56—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by reactions decreasing the number of carbon atoms by replacing a carboxyl or aldehyde group by a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/39—Preparation of carboxylic acid esters by oxidation of groups which are precursors for the acid moiety of the ester
- C07C67/42—Preparation of carboxylic acid esters by oxidation of groups which are precursors for the acid moiety of the ester by oxidation of secondary alcohols or ketones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/82—Benzo [b] furans; Hydrogenated benzo [b] furans with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D307/83—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/88—Benzo [c] furans; Hydrogenated benzo [c] furans with one oxygen atom directly attached in position 1 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/93—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems condensed with a ring other than six-membered
- C07D307/935—Not further condensed cyclopenta [b] furans or hydrogenated cyclopenta [b] furans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/16—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D309/28—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/30—Oxygen atoms, e.g. delta-lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D323/00—Heterocyclic compounds containing more than two oxygen atoms as the only ring hetero atoms
- C07D323/04—Six-membered rings
Definitions
- the present invention relates to a method for producing an ester, and more particularly to a method for producing an ester by a buyer-bilger oxidation reaction.
- the Buyer-Billiger oxidation reaction has been widely used in organic synthesis as a method for converting ketones and aldehydes to valuable esters (including lactones that are cyclic esters).
- ⁇ -caprolactone obtained from cyclohexanone is useful as a raw material for polyesters and polyamides, and development of an efficient synthesis method is important.
- the cyclohexanone Bayer-Billiger oxidation reaction is a ring expansion reaction from a stable 6-membered ring with small ring strain to an unstable 7-membered ring with large ring strain, so it is generally considered difficult because of low reactivity. Yes. Therefore, organic peracids with strong oxidizing power are often used.
- Non-Patent Documents 1 and 2 there is a method using TsOH as a catalyst in a HFIP (1,1,1,3,3,3-hexafluoro-2-propanol) solvent (for example, Non-Patent Documents 1 and 2).
- a heterogeneous oxidation method using a hydrotalcite solid catalyst such as Mg, Mg—Al, Sn or the like (for example, Non-Patent Documents 7 to 10).
- the first method since epoxidation of olefin is also promoted, there is a problem that the functional group selectivity is not good when a cyclic ketone containing olefin is used as a reaction substrate.
- the second method has a problem that the toxicity of the organic selenium reagent as a catalyst is high, and the third method has a problem that the yield of obtaining ⁇ -caprolactone from cyclohexanone is low, and the fourth method has a problem. There was a problem of lack of generality of the substrate.
- the present invention has been made to solve such problems, and has as its main object to obtain esters (including lactones) from ketones and aldehydes in a high yield using a low toxicity catalyst.
- M (BAr 4 ) n Li, Na, Ca, Sr, etc.
- the ester production method of the present invention is a method for producing an ester from a ketone or an aldehyde as a reaction substrate using hydrogen peroxide by a buyer-billiger oxidation reaction, and as a catalyst, M (BAr 4) which is a borate salt.
- M is an alkali metal, alkaline earth metal or triarylmethyl, and four Ar are aryls having an electron-withdrawing group, and all four may be the same or different, and n is M It is the same number as the valence of.
- an ester can be obtained from a ketone or aldehyde as a reaction substrate in a high yield using a low toxicity catalyst.
- hydrogen peroxide is used instead of organic peracid, the by-product derived from peroxide is only water, which can be said to be an environment-friendly buyer-bilger oxidation reaction.
- a cyclic ketone is used as the reaction substrate, the oxidation reaction proceeds efficiently under mild conditions, and a lactone can be obtained in a high yield.
- the ester production method of the present invention is a method for producing an ester from a ketone or an aldehyde as a reaction substrate using hydrogen peroxide by a buyer-billiger oxidation reaction, and M (BAr 4 ) n which is a borate salt as a catalyst.
- M is an alkali metal, alkaline earth metal or triarylmethyl, and four Ar are aryls having an electron-withdrawing group, and all four may be the same or different, and n is a valence of M. Is the same number as the number).
- M in M (BAr 4 ) n which is a borate salt is an alkali metal, an alkaline earth metal or triarylmethyl.
- the alkali metal include Li, Na, and K
- examples of the alkaline earth metal include Mg, Ca, Sr, and Ba. All three aryls in triarylmethyl may be the same or different. If they are different, they may all be separate, or the two may be the same and the rest may be different.
- Examples of triarylmethyl include triphenylmethyl (trityl) and tris (pentafluorophenyl) methyl.
- n 1 when M is an alkali metal or triarylmethyl, and n is 2 when M is an alkaline earth metal.
- Ars are aryls having an electron-withdrawing group, and all four Ars may be the same or different. If they are different, all may be separate, two may be the same and the rest may be separate, two may be the same and the remaining two may be the same, or three may be the same.
- the electron withdrawing group include a halogen atom, a trihalomethyl group, a nitro group, and a nitrile group.
- the halogen atom is preferably a fluorine atom
- the trihalomethyl group is preferably a trifluoromethyl group.
- the aryl having an electron-withdrawing group pentafluorophenyl, 3,5-bis (trifluoromethyl) phenyl, and the like are preferable.
- the amount of borate salt M (BAr 4 ) n used is not particularly limited as long as it is a catalytic amount.
- 0.01 to 20 mol% of the reaction substrate is used. preferable. If it is less than 0.01 mol%, it is not preferable because problems such as a slow reaction rate or a high by-product ratio may occur. Even if it exceeds 20 mol%, the yield is greatly improved. This is not preferable from an economic point of view.
- the reaction rate promoting effect it is preferable to adjust 0.1 mol% with respect to the reaction substrate. From an economic standpoint, the upper limit is more preferably 5 mol% with respect to the reaction substrate.
- the ketone used as the reaction substrate is not particularly limited, and examples thereof include cyclic ketones, chain ketones, and chromanones.
- examples of cyclic ketones include cyclopropanones, cyclobutanones, cyclopentanones, cyclohexanones, cycloheptanones, and condensed ring ketones.
- the reaction proceeds in high yield.
- the synthetic intermediate Monomers can also be used as reaction substrates.
- chain ketones examples include dialkyl ketones such as dipentyl ketone, arylalkyl ketones such as acetophenone, and diaryl ketones such as benzophenone.
- chromanones examples include 4-chromanone and 3-chromanone.
- aldehyde used as the reaction substrate include aromatic aldehydes such as benzaldehyde, 4-chlorobenzaldehyde, and 1-naphthylaldehyde.
- Such a reaction substrate may have a carbon-carbon double bond (that is, an olefin bond), a carbon-carbon triple bond, a halogen group, a hydroxy group, a silyl group, or a siloxy group.
- Olefins may be converted to epoxies by hydrogen peroxide, but the possibility of production of the ester of the present invention is small, and esters are selectively produced.
- the reaction substrate having an olefin bond include ketones or aldehydes having a vinyl group, an allyl group, an isopropenyl group, and the like.
- the reaction substrate having a carbon-carbon triple bond include ketones or aldehydes having an ethynyl group, a propynyl group, and the like.
- reaction substrate having a silyl group examples include ketones or aldehydes having a trimethylsilyl group, a dimethylphenylsilyl group, a dimethyl t-butylsilyl group, and the like.
- reaction substrate having a siloxy group examples include ketones or aldehydes having a trimethylsiloxy group, a dimethylphenylsiloxy group, a dimethyl t-butylsiloxy group, and the like.
- the reaction solvent may be appropriately selected depending on the reaction substrate and the catalyst.
- halogenated hydrocarbons include 1,2-dichloroethane (DCE) and 1,4-dichlorobutane (DCB).
- aromatic hydrocarbons include toluene, xylene, and benzene. Includes acetonitrile, propionitrile, butyronitrile, and examples of the ester solvent include methyl acetate and ethyl acetate.
- the reaction temperature may be appropriately set according to the reaction substrate and the catalyst. If the reaction temperature is too low, the reaction rate is slow and it may take a long time to complete the reaction, which is not preferable. If the reaction temperature is too high, the reaction substrate may decompose or side reactions may become dominant. However, the appropriate reaction temperature varies depending on the reaction substrate and the catalyst. For this reason, a suitable range for the reaction temperature cannot generally be determined, but as one guideline, it may be appropriately set between 0 ° C. and 100 ° C., preferably between 25 ° C. (room temperature) and 70 ° C.
- a Bronsted acid may be used as a co-catalyst.
- the reaction activity is further improved.
- cocatalysts include phenols having one or more OH groups on the aromatic ring, carboxylic acids, oxocarbonic acids, phosphoric acid mono- or diesters, and the like.
- Examples of the phenols include pentafluorophenol, catechol, 3-fluorocatechol, tetrafluorocatechol, tetrachlorocatechol, resorcinol, 4-fluororesorcinol, tetrafluororesorcinol, tetrachlororesorcinol and the like.
- Examples of the carboxylic acid include acetic acid, mandelic acid, oxalic acid, malonic acid, succinic acid, salicylic acid, and phthalic acid.
- Examples of the oxocarbonic acid include delta acid, squaric acid, croconic acid, rosinic acid, heptagonic acid and the like.
- Examples of phosphoric acid mono- or diesters include binaphthyl hydrogen phosphate (BP). Among these, tetrafluorocatechol and oxalic acid are preferable because of their high reaction promoting effect. Furthermore, oxalic acid is preferable in view of inexpensive points.
- BP binaphthyl hydrogen phosphate
- the cocatalyst is preferably, for example, 0.01 to 100 mol% with respect to the reaction substrate. Further, the molar amount is preferably 1 to 10 times by mole, more preferably 1 to 5 times by mole to the borate salt.
- Ca [B (3,5-CF 3 C 6 H 3 ) 4 ] 2 ⁇ 8H 2 O and Ba [B (3,5-CF 3 C 6 H 3 ) 4 ] 2 ⁇ 7H 2 O are SrCl Sr [B (3,5-CF 3 C 6 H 3 ) 4 ] 2 ⁇ 10H 2 O was synthesized in the same manner except that CaCl 2 and BaCl 2 were used instead of 2 , respectively. Furthermore, Sr [B (C 6 F 5 ) 4 ] 2 is used except that LiB (C 6 F 5 ) 4 is used instead of LiB (3,5-CF 3 C 6 H 3 ) 4 .4H 2 O. And Sr [B (3,5-CF 3 C 6 H 3 ) 4 ] 2 ⁇ 10H 2 O.
- Examples 1 to 11, Comparative Examples 1 and 2 As shown in Table 1, in Examples 1 to 11, ⁇ -caprolactone was produced from commercially available cyclohexanone by using various borate salts as catalysts and hydrogen peroxide by the Bayer-Billiger oxidation reaction. In Comparative Examples 1 and 2, ⁇ -caprolactone was produced in the same manner using TsOH and Sc (OTf) 3 as catalysts. Details of the catalysts and reaction conditions used in each Example and each Comparative Example are as shown in Table 1.
- Example 4 A detailed experimental procedure for Example 4 will be described below as a representative example. To a solution of cyclohexanone (50 mg, 0.5 mmol) and Sr [B (3,5-CF 3 C 6 H 3 ) 4 ] 2 ⁇ 10H 2 O (10 mg, 0.005 mmol) in 1,2-dichloroethane (10 mL) was added 30 % Hydrogen peroxide solution (57 ⁇ L, 0.55 mmol) was added, and the reaction vessel was placed in a 70 ° C. oil bath.
- the metal species of the borate salt used as the catalyst gave good results for both alkali metals and alkaline earth metals. Especially, the reaction activity and lactone selectivity of Ca and Sr were particularly good. Further, as shown in Example 5, the reaction proceeded efficiently even when the catalyst amount was lowered from 1 mol% to 0.1 mol%. On the other hand, as shown in Examples 7 to 9, when the counter anion of the catalyst was changed from tetrakis (3,5-bis (trifluoromethyl) phenyl) borate to tetrakis (pentafluorophenyl) borate, the reaction activity was greatly increased. Improved.
- Example 12 to 23 Comparative Examples 3 to 6
- Table 2 As shown in Table 2, in Examples 12 to 23, various borates are used as catalysts, and the corresponding ⁇ -caprolactone is produced from commercially available 4-tert-butylcyclohexanone using hydrogen peroxide by the Bayer-Billiger oxidation reaction. did.
- Comparative Example 3 no catalyst was used, and in Comparative Examples 4 to 6, NaBF 4 , NaBPh 4 , and LiNTf 2 were used as catalysts to produce the same ⁇ -caprolactone.
- the catalyst and reaction conditions shown in Table 2 were adopted, and the reaction was performed according to Example 4 described above.
- Example 12 2 molar equivalents of hydrogen peroxide in 1,2-dichloroethane was used, and the reaction was carried out using 5 mol% of NaB (3,5-CF 3 C 6 H 3 ) 4 as a catalyst with respect to the reaction substrate. As a result, 73% yield of lactone and 27% yield of spirobisperoxide as a by-product were obtained. In Example 13, when a solvent in which 1,2-dichloroethane and water were mixed at a volume ratio of 2: 1 was used as a reaction solvent, the yield of lactone was improved to 89%.
- Example 14 and Example 15 when toluene and acetonitrile were used in place of 1,2-dichloroethane of Example 13, respectively, the reaction activity decreased, but lactone was selectively obtained. In Example 14 and Example 15, the progress of the reaction is slow, but no compound other than the lactone is produced. Therefore, it is expected that the yield of the lactone is improved if the reaction time is increased. In Example 16, when a solvent in which 1,2-dichloroethane and water were mixed at a volume ratio of 10: 1 was used as the reaction solvent, the reaction activity was greatly improved compared to Example 13, and the lactone was The yield of was also improved.
- Examples 17 to 20 2 molar equivalents of hydrogen peroxide were used in the same reaction solvent as in Example 16, and 1 mol% of metal borate (the metal species were Li, Na, K, Ca, respectively) with respect to the reaction substrate. ) was used as a catalyst.
- the reaction activity was high, conversion was 90% or more, and lactone yield was 84% or more.
- the order of reaction activity was Ca, Li, Na, K from the highest.
- Example 21 1.1 mole equivalent of hydrogen peroxide was used in the same reaction solvent as in Example 16, and the reaction was carried out using 1 mol% of borate salt as a catalyst with respect to the reaction substrate. As a result, the reaction activity was high, the conversion rate was 89% or more, and the yield of lactone was 85% or more.
- the borate salt used in Example 23 the cation was a trityl cation, but good results were obtained.
- Example 24 to 30 lactones were synthesized from various cyclic ketones using hydrogen peroxide by the buyer-billiger oxidation reaction.
- Example 24 when 2-methylcyclohexanone, which is an asymmetric cyclic ketone, was used, the corresponding ⁇ -caprolactone was obtained in high yield with ordinary regioselectivity.
- Example 25 when 4-isopropenylcyclohexanone, which is a cyclic ketone having a substituent having an olefin bond, was used, the corresponding ⁇ -caprolactone was obtained in a yield of 56%, and no olefin epoxidation was observed. .
- Example 26 when 4-hydroxycyclohexanone was used, a 5-membered ring lactone having a hydroxyethyl group was obtained. This lactone is considered to have been re-wound into a 5-membered ring with less ring distortion once the corresponding ⁇ -caprolactone was formed.
- Examples 27 to 29 when a 5-membered ring ketone or 4-membered ring ketone was used, the corresponding 6-membered ring lactone or 5-membered ring lactone was obtained in high yield.
- Example 30 when a condensed ring ketone having an olefin bond in the ring was used, the corresponding condensed ring lactone was obtained in high yield with ordinary regioselectivity, and olefin epoxidation was not observed.
- Example 24 • Major product of Example 24: colorless oil.
- Example 31 to 56 Comparative Examples 7 and 8
- Table 3 shows the structural formulas of the promoters used in the examples.
- Example 39 A detailed experimental procedure for Example 39 will be described below as a representative example of the Buyer-Billiger oxidation reaction using a catalyst and a promoter. In addition, other examples and comparative examples were also reacted according to Example 39.
- cyclopentanone 84 mg, 1.0 mmol
- Li [B (C 6 F 5 ) 4 ] .2.5 Et 2 O (10.4 mg, 0.01 mmol)
- oxalic acid 4.6 mg, 0.05 mmol
- a 30% hydrogen peroxide solution (115 ⁇ L, 1.1 mmol) was placed in a 1,2-dichloroethane (10 mL) solution, and the reaction vessel was placed in an oil bath at 50 ° C.
- Example 32 to 45 the reaction was carried out using 1 mol% of LiB (C 6 H 5 ) 4 as a catalyst and 5 mol% of a promoter in a DCE solvent.
- phenols Examples 32 to 36
- carboxylic acids Examples 37 to 44
- phosphoric acid diesters Example 45
- Example 31 Example 31 in which no promoter was used.
- the reaction was faster and the conversion rate was higher.
- tetrafluorocatechol Example 36
- inexpensive oxalic acid Example 39 were the most effective.
- the reaction did not proceed at all with the Bronsted acid used in Examples 36 and 39 without using the borate salt catalyst (Comparative Examples 7 and 8).
- Examples 46 to 48 and 49 to 51 tetrafluorocatechol or oxalic acid was used as a cocatalyst, and reduction of the use amount of the catalyst and the cocatalyst was examined. As a result, it was possible to reduce the amount of catalyst used to 0.01 mol% and the amount of promoter used to 0.05 mol% (Examples 48 and 51).
- Examples 52 to 56 the effect of the cocatalyst was examined in the same manner using toluene or benzene as a solvent. As a result, it was found that Examples 54 to 56 using the promoter had an activation effect by the promoter compared to Examples 52 and 53 where the promoter was not used.
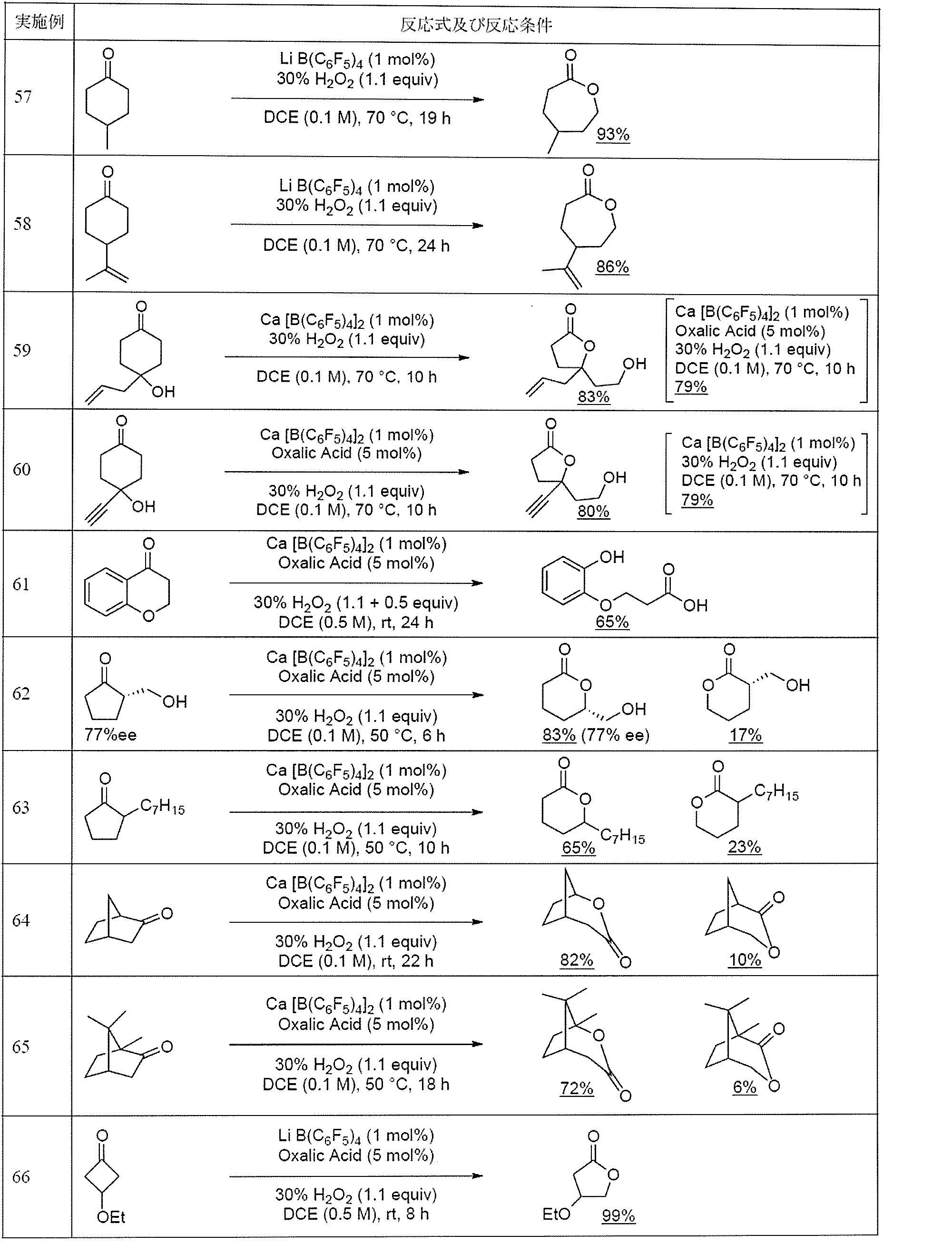
- Examples 57 to 76 As shown in Tables 5 and 6, the buyer's billiger oxidation reaction of various reaction substrates was examined.
- a borate salt of Li or Ca was used as the catalyst.
- the catalytic activity of the borate salt of Ca is higher, but the borate salt of Li is a commercial product, and since the absolute amount of use is small because of its low molecular weight, the Li borate salt was first used ( Examples 57, 58, 66-71, 73).
- the borate salt of Ca was used as a catalyst (Examples 59-65, 72, 74-76).
- Example 57 and 58 illustrated the case where oxalic acid was not used.
- the reaction substrate of Example 58 had an olefin moiety, but this olefin moiety was not oxidized.
- Examples 59 and 60 when 4-hydroxycyclohexanone was used as a reaction substrate, once the corresponding ⁇ -caprolactone was produced, a product re-rolled into a 5-membered ring with less ring distortion was obtained in high yield. Obtained. These reaction substrates have carbon-carbon double bonds or carbon-carbon triple bonds, but these sites were not oxidized.
- Table 5 an example in which no promoter was used was shown in Example 59, but an example in which oxalic acid was used as a promoter in parentheses was shown. Moreover, although the example which used oxalic acid as a promoter in Example 60 was shown, the example which did not use a promoter in parenthesis was shown.
- Example 61 when 4-chromanone was used as a reaction substrate, the produced lactone was hydrolyzed to obtain the corresponding hydroxycarboxylic acid.
- Example 62 when optically active cyclopentanone having an asymmetric point at the ⁇ -position of the ketone was used as a reaction substrate, racemization did not proceed at all, and the produced lactone maintained the asymmetric yield as it was. .
- Example 62 and Examples 63 to 65 a reaction substrate in which two kinds of rearrangement occurred from the Criegee intermediate was used, but a product was obtained with the same regioselectivity as in the prior art.
- Example 73 when a chain ketone was used as a reaction substrate, the corresponding chain ester was obtained in high yield.
- Example 74 when benzaldehyde, which is an aromatic aldehyde, was used as a reaction substrate, the corresponding formate ester was hydrolyzed in the system, and the corresponding phenol was obtained in high yield.
- Example 63 • Major product of Example 63: colorless oil.
- 1 H NMR (CDCl 3 , 400 MHz) ⁇ 0.88 (t, J 6.9 Hz, 3H), 1.21-1.40 (m, 9H), 1.43- 1.61 (m, 3H), 1.66-1.75 (m, 1H), 1.79-1.93 (m, 3H), 2.40-2.48 (m, 1H), 2.55-2.63 (m, 1H), 4.24-4.30 (m, 1H );
- Example 64 • Major product of Example 64: colorless oil.
- Example 66 Colorless oil. This is the same product as Example 29.
- Example 71 Same product as Example 30.
- Example 76 • Major product of Example 76: colorless oil.
- 1 H NMR (CDCl 3 , 400 MHz) ⁇ 1.00 (s, 6H), 1.63-1.67 (m, 2H), 2.26-2.30 (m, 1H ), 4.91-5.00 (m, 2H), 5.72 (dd, J 17.4, 11.0 Hz, 1H); 13 C NMR (CDCl 3 , 100 MHz) ⁇ 26.6, 30.0, 36.3, 36.8, 111.8, 147.0, 181.0.
- the present invention is mainly applicable to the chemical and chemical industry.
- ⁇ -caprolactone which is a lactone, is useful as a biodegradable polymer or a synthetic intermediate for nylon-6.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Furan Compounds (AREA)
- Pyrane Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description
以下の実施例及び比較例で触媒として使用したボレート塩について説明する。NaB(3,5-CF3C6H3)4,LiB(C6F5)4とPh3CB(C6F5)4は市販品をそのまま反応に用いた。LiB(3,5-CF3C6H3)4とKB(3,5-CF3C6H3)4は文献(Organomet., 1992, vol.11, p3920)に記載された方法に従って合成した。
白色粉末。1H NMR (CD3CN, 400 MHz)δ 7.65-7.70 (m, 12H); 13C NMR (CD3CN, 100 MHz) δ118.7, 125.5 (q, JC-F = 270 Hz), 129.9 (q, JC-F = 31.5 Hz), 135.7, 162.6 (q, JB-C = 49.6 Hz); 19F NMR (CD3CN) δ-63.1. Anal. Calcd. for C32H20BF24LiO4: C, 40.79; H, 2.14. Found: C, 41.00; H, 1.88.
白色粉末。 1H NMR (CD3CN, 400 MHz)δ 7.65-7.70 (m, 12H); 13C NMR (CD3CN, 100 MHz) δ118.7, 125.5 (q, JC-F = 271 Hz), 130.0 (q, JC-F = 32.4 Hz), 135.7, 162.7 (q, JB-C = 49.6 Hz); 19F NMR (CD3CN) δ-63.1. Anal. Calcd. for C32H16BF24LiO2: C, 40.96; H, 1.72. Found: C, 41.01; H, 1.70.
白色粉末。 1H NMR (CD3CN, 400 MHz)δ7.65-7.70 (m, 24H); 13C NMR (CD3CN, 100 MHz) δ118.7, 125.5 (q, JC-F = 271 Hz), 129.9 (q, JC-F = 31.5 Hz), 135.7, 162.7 (q, JB-C = 49.6 Hz); 19F NMR (CD3CN) δ-63.1. Anal. Calcd. For C64H44BF48O10Sr: C, 38.55; H, 2.22. Found: C, 38.56; H, 2.13.
白色粉末。1H NMR (CD3CN, 400 MHz)δ7.65-7.70 (m, 24H); 13C NMR (CD3CN, 100 MHz) δ118.6, 125.4 (q, JC-F = 271 Hz), 129.9 (q, JC-F = 31.5 Hz), 135.6, 162.6 (q, JB-C = 48.6 Hz); 19F NMR (CD3CN) δ-63.1. Anal. Calcd. for C64H40BCaF48O8: C,40.23; H, 2.11. Found: C, 40.23; H, 2.30.
淡茶色粉末。1H NMR (CD3CN, 400 MHz)δ77.65-7.70 (m, 24H); 19F NMR (CD3CN) δ-63.1. Anal. Calcd. For C64H38BBaF24O7: C, 38.63; H, 1.92. Found: C, 38.65; H, 2.08.
白色粉末。 19F NMR (CD3CN) δ-168.3, -163.8 (t, J = 24.6 Hz), -133.7.
表1に示すように、実施例1~11では、各種のボレート塩を触媒とし、バイヤー・ビリガー酸化反応により過酸化水素を用いて市販のシクロヘキサノンからε-カプロラクトンを製造した。比較例1,2では、それぞれTsOH及びSc(OTf)3を触媒とし、同様にしてε-カプロラクトンを製造した。各実施例、各比較例で用いた触媒及び反応条件の詳細は表1に示したとおりである。また、この反応では、ε-カプロラクトンのほか、このラクトンが加水分解したヒドロキシカルボン酸やバイヤー・ビリガー酸化反応の反応中間体であるCriegee中間体(表1の欄外の※4参照)が二量化したスピロビスペルオキシドが生成した。表1には、シクロヘキサノンから反応生成物への転換率及び各反応生成物の収率を示した。なお、表1のシクロヘキサノンの転換率や各生成物の収率は、反応溶液から少量をサンプリングし、1H NMR解析により算出した。

無色液体。 TLC, Rf = 0.11 (hexane-EtOAc = 4:1); 1H NMR (CDCl3, 400 MHz) δ1.76-1.87 (m, 6H), 2.63-2.66 (m, 2H), 4.23-4.26 (m, 2H).
無色固体。1H NMR (CDCl3, 400 MHz) δ1.41 (m, 2H), 1.63 (m, 4H), 2.36 (t, J= 7.4 Hz, 2H), 3.65 (t, J= 6.5 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ24.4, 25.1, 31.9,33.9, 62.3, 178.8.
白色固体。TLC, Rf = 0.67 (hexane-EtOAc = 4:1); 1H NMR (CDCl3, 400 MHz) δ1.47 (bs, 4H), 1.57 (bs, 12H), 2.28 (bs, 4H).
表2に示すように、実施例12~23では、各種のボレート塩を触媒とし、バイヤー・ビリガー酸化反応により過酸化水素を用いて市販の4-tert-ブチルシクロヘキサノンから対応するε-カプロラクトンを製造した。比較例3では無触媒、比較例4~6ではそれぞれNaBF4、NaBPh4、LiNTf2を触媒とし、同様のε-カプロラクトンを製造した。各実施例、各比較例では、表2に示した触媒及び反応条件を採用し、上述した実施例4に準じて反応を行った。また、この反応では、ε-カプロラクトンのほか、スピロビスペルオキシドが生成した。表2には、シクロヘキサノンから反応生成物への転換率及び各反応生成物の収率を示した。なお、表2の転換率や各生成物の収率は、反応溶液から少量をサンプリングし、1H NMR解析により算出した。

無色固体。TLC, Rf = 0.35 (hexane-EtOAc = 4:1); 1H NMR (CDCl3, 400 MHz) δ 0.89(s, 9H), 1.25-1.40 (m 2H), 1.48-1.55 (m, 1H), 2.0-2.1 (m, 2H), 2.53-2.59 (m, 1H), 2.69-2.74 (m, 1H), 4.11-4.18 (m, 1H), 4.34 (ddd, J = 1.9, 5.9, 12.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ23.6, 27.3, 30.1, 32.3, 33.3, 50.6, 69.0, 177.8.
白色固体。TLC, Rf = 0.75 (hexane-EtOAc = 4:1); 1H NMR (CDCl3, 400 MHz) δ 0.86(bs, 18H), 1.05-1.12 (m, 2H), 1.20-1.32 (m, 4H), 1.41-1.51 (m, 4H), 1.74 (bs, 6H), 3.17 (bs, 2H); 13C NMR (CDCl3, 100 MHz) δ 22.8, 23.1, 27.6, 29.7, 32.0, 32.3, 47.4, 47.5, 108.1.
実施例24~30では、化1に示すように、バイヤー・ビリガー酸化反応により過酸化水素を用いて種々の環状ケトンからラクトンを合成した。実施例24では、非対称の環状ケトンである2-メチルシクロヘキサノンを用いたところ、高収率で対応するε-カプロラクトンが通常の位置選択性でもって得られた。実施例25では、オレフィン結合を持つ置換基を有する環状ケトンである4-イソプロペニルシクロヘキサノンを用いたところ、対応するε-カプロラクトンが収率56%で得られ、オレフィンのエポキシ化は見られなかった。実施例26では、4-ヒドロキシシクロヘキサノンを用いたところ、ヒドロキシエチル基を有する5員環ラクトンが得られた。このラクトンは、一旦、対応するε-カプロラクトンが生成したあと、環の歪みの少ない5員環に巻き直したものと考えられる。実施例27~29では、5員環ケトンや4員環ケトンを用いたところ、対応する6員環ラクトンや5員環ラクトンが高収率で得られた。実施例30では、環内にオレフィン結合を有する縮合環ケトンを用いたところ、対応する縮合環ラクトンが通常の位置選択性でもって高収率で得られ、オレフィンのエポキシ化は見られなかった。
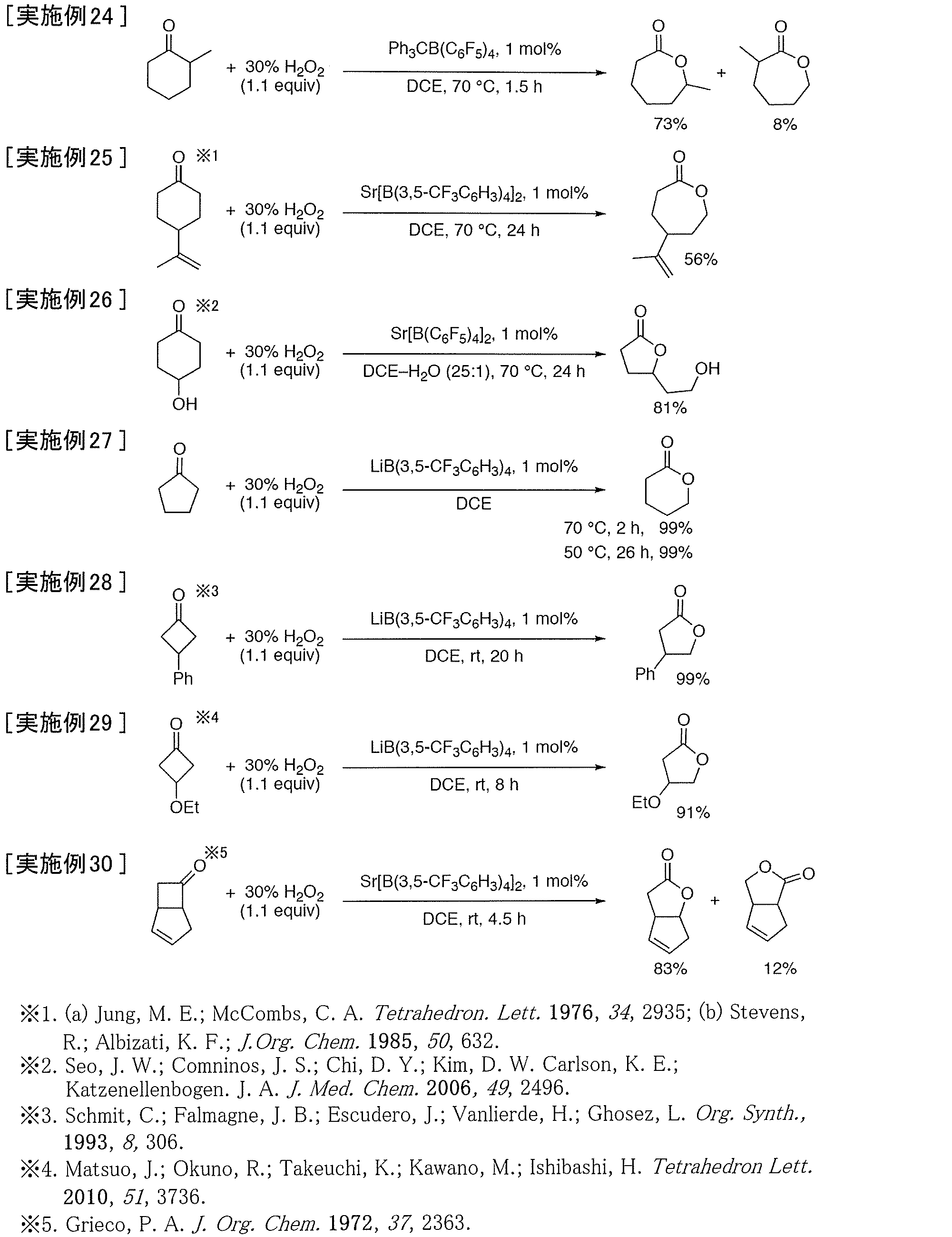
表3に示すように、実施例31では、ボレート塩触媒のみを用いてバイヤー・ビリガー酸化反応によりシクロペンタノンからδ-バレロラクトンを合成した。また、実施例32~56では、ボレート塩触媒とブレンステッド酸助触媒とを用いてバイヤー・ビリガー酸化反応によりシクロペンタノンからδ-バレロラクトンを合成した。実施例で使用した助触媒の構造式を表4に示す。


表5及び表6に示すように、様々な反応基質のバイヤービリガー酸化反応を検討した。触媒としては、Li又はCaのボレート塩を用いた。一般的に、触媒活性はCaのボレート塩の方が高いが、Liのボレート塩は市販品であり、分子量が小さいため絶対的な使用量が少ないことから、まずLiのボレート塩を使用した(実施例57,58,66-71,73)。しかし、反応基質によってはLiのボレート塩では不十分で、生成物の化学収率が低い場合には、Caのボレート塩を触媒として用いた(実施例59-65,72,74-76)。


Claims (9)
- バイヤー・ビリガー酸化反応により過酸化水素を用いて反応基質であるケトン又はアルデヒドからエステルを製造する方法であって、
触媒として、ボレート塩であるM(BAr4)n(Mはアルカリ金属、アルカリ土類金属又はトリアリールメチルであり、4つのArは電子吸引性基を有するアリールであって4つとも同じであっても異なっていてもよく、nはMの価数と同じ数である)を用いる、
エステルの製法。 - 前記ボレート塩のArは、ペンタフルオロフェニル又は3,5-ビストリフルオロメチルフェニルである、
請求項1に記載のエステルの製法。 - 前記触媒は、前記反応基質に対して0.1~5mol%使用する、
請求項1又は2に記載のエステルの製法。 - 前記反応基質は、炭素-炭素二重結合、炭素-炭素三重結合、ハロゲン基、ヒドロキシル基、シリル基又はシロキシ基を有している、
請求項1~3のいずれか1項に記載のエステルの製法。 - 前記反応基質は、環状ケトン、鎖状ケトン、クロマノン類又は芳香族アルデヒド類である、
請求項1~4のいずれか1項に記載のエステルの製法。 - 助触媒として、ブレンステッド酸を用いる、
請求項1~5のいずれか1項に記載のエステルの製法。 - 前記助触媒は、芳香環上に1以上のOH基を持つフェノール類、カルボン酸、オキソカーボン酸、リン酸モノエステル又はリン酸ジエステルである、
請求項6に記載のエステルの製法。 - 前記助触媒は、テトラフルオロカテコール又はシュウ酸である、
請求項6又は7に記載のエステルの製法。 - 前記助触媒は、前記触媒に対して1~5倍モル使用する、
請求項6~8のいずれか1項に記載のエステルの製法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11837836.3A EP2636665B1 (en) | 2010-11-02 | 2011-10-11 | Method for producing ester |
| US13/881,544 US8853426B2 (en) | 2010-11-02 | 2011-10-11 | Method for manufacturing ester |
| JP2012541793A JP5920889B2 (ja) | 2010-11-02 | 2011-10-11 | エステルの製法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010245944 | 2010-11-02 | ||
| JP2010-245944 | 2010-11-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012060185A1 true WO2012060185A1 (ja) | 2012-05-10 |
Family
ID=46024304
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/073340 Ceased WO2012060185A1 (ja) | 2010-11-02 | 2011-10-11 | エステルの製法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8853426B2 (ja) |
| EP (1) | EP2636665B1 (ja) |
| JP (1) | JP5920889B2 (ja) |
| WO (1) | WO2012060185A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102942548A (zh) * | 2012-11-20 | 2013-02-27 | 南京理工大学 | 一种δ-十二内酯的合成方法 |
| WO2015074162A1 (es) | 2013-11-22 | 2015-05-28 | Pontificia Universidad Catolica De Chile | Variantes de enzima fenilacetona monooxigenasa (pamo) capaces de catalizar conversión de ciclohexanona a caprolactona. |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11208394B2 (en) * | 2018-09-17 | 2021-12-28 | Regents Of The University Of Minnesota | Chemical process to manufacture branched-caprolactone |
| EP3983369B1 (en) | 2019-06-12 | 2023-08-02 | Nouryon Chemicals International B.V. | Process for the production of diacyl peroxides |
| ES2963382T3 (es) | 2019-06-12 | 2024-03-26 | Nouryon Chemicals Int Bv | Proceso para la producción de peróxidos de diacilo |
| WO2020249689A1 (en) | 2019-06-12 | 2020-12-17 | Nouryon Chemicals International B.V. | Process for the production of peroxyesters |
| WO2020249692A1 (en) * | 2019-06-12 | 2020-12-17 | Nouryon Chemicals International B.V. | Method for isolating carboxylic acid from an aqueous side stream |
| US11976035B2 (en) | 2019-06-12 | 2024-05-07 | Nouryon Chemicals International B.V. | Process for the production of diacyl peroxides |
| CN110452212B (zh) * | 2019-07-30 | 2020-08-14 | 浙江大学 | 一种11-十一内酯类化合物和己内酯类化合物的制备方法 |
| CN115057998B (zh) * | 2022-07-07 | 2023-07-25 | 武汉理工大学 | 一种联合生产ε-己内酯与聚丁二酸丁二醇酯的方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09104681A (ja) * | 1995-10-06 | 1997-04-22 | Nippon Peroxide Co Ltd | δ−ラクトン化合物の製造方法 |
| JP2003190804A (ja) | 2001-12-25 | 2003-07-08 | Asahi Kasei Corp | ケトン化合物の酸化触媒 |
| JP2004352636A (ja) * | 2003-05-28 | 2004-12-16 | Takasago Internatl Corp | ラクトン類又はエステル類の製造方法 |
| JP2010245944A (ja) | 2009-04-08 | 2010-10-28 | Mitsubishi Electric Corp | 高周波増幅器 |
-
2011
- 2011-10-11 EP EP11837836.3A patent/EP2636665B1/en not_active Not-in-force
- 2011-10-11 WO PCT/JP2011/073340 patent/WO2012060185A1/ja not_active Ceased
- 2011-10-11 US US13/881,544 patent/US8853426B2/en not_active Expired - Fee Related
- 2011-10-11 JP JP2012541793A patent/JP5920889B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09104681A (ja) * | 1995-10-06 | 1997-04-22 | Nippon Peroxide Co Ltd | δ−ラクトン化合物の製造方法 |
| JP2003190804A (ja) | 2001-12-25 | 2003-07-08 | Asahi Kasei Corp | ケトン化合物の酸化触媒 |
| JP2004352636A (ja) * | 2003-05-28 | 2004-12-16 | Takasago Internatl Corp | ラクトン類又はエステル類の製造方法 |
| JP2010245944A (ja) | 2009-04-08 | 2010-10-28 | Mitsubishi Electric Corp | 高周波増幅器 |
Non-Patent Citations (19)
| Title |
|---|
| ANGEW. CHEM. INT. ED., vol. 41, 2002, pages 4481 |
| APP. CATAL.B: ENVIRON., vol. 72, 2007, pages 18 |
| CATAL. LETT., vol. 131, 2009, pages 618 |
| DAISUKE NAKAJIMA ET AL.: "Alcali-Alcali Dorui Kinzoku no Kasadakai Borate-en Shokubai to Kasanka Suiso ni yoru Baeyer-Villiger Sanka Hanno", 91ST ANNUAL MEETING OF THE CHEMICAL SOCIETY OF JAPAN, 11 March 2011 (2011-03-11), KOEN YOKOSHU, pages 1221, XP008168374 * |
| DAISUKE NAKAJIMA ET AL.: "Kasanka Suisosui o Sankazai ni Mochiiru Kankyo Chowagata Shokubaiteki Baeyer-Villiger Sanka Hanno", DAI 41 KAI ANNUAL MEETING OF UNION OF CHEMISTRY- RELATED SOCIETIES IN CHUBU AREA, 6 November 2010 (2010-11-06), JAPAN KOEN, pages 199, XP008169387 * |
| FUJIKI K. ET AL.: "Evaluation of Lewis Acidity of "Naked" Lithium Ion through Diels-Alder Reaction Catalyzed by Lithium TFPB in Nonpolar Organic Solvents", CHEMISTRY LETTERS, vol. 29, 2000, pages 62 - 63, XP009097713 * |
| GREEN CHEM., vol. 5, 2003, pages 524 |
| HUDRLIK ET AL., J. AM. CHEM. SOC., vol. 102, 1980, pages 6894 |
| J. MOL. CATAL. A:CHEM., vol. 191, 2003, pages 93 |
| J. ORG. CHEM., vol. 66, 2001, pages 2429 |
| KOTSUKI ET AL., ORG. LETT., vol. 12, 2010, pages 1616 |
| LIAO B.-S. ET AL.: "An Efficient Preparation of Bis(indole)methanes Catalyzed by Tetrakis [3,5-bis(trifluoromethyl)phenyl]borate Salts in Aqueous Medium", SYNTHESIS, no. 20, 2007, pages 3125 - 3128, XP055085511 * |
| NEIMANN K. ET AL.: "Electrophilic Activation of Hydrogen Peroxide: Selective Oxidation Reactions in Perfluorinated Alcohol Solvents", ORGANIC LETTERS, vol. 2, no. 18, 2000, pages 2861 - 2863, XP008116670 * |
| ORGANIC LETTERS, vol. 2, 2000, pages 2861 |
| ORGANOMET., vol. 11, 1992, pages 3920 |
| See also references of EP2636665A4 |
| TETRAHEDRON LETTERS, vol. 42, 2001, pages 2293 |
| TETRAHEDRON LETTERS, vol. 46, 2005, pages 8665 |
| TETRAHEDRON, vol. 63, 2007, pages 1435 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102942548A (zh) * | 2012-11-20 | 2013-02-27 | 南京理工大学 | 一种δ-十二内酯的合成方法 |
| CN102942548B (zh) * | 2012-11-20 | 2015-04-22 | 南京理工大学 | 一种δ-十二内酯的合成方法 |
| WO2015074162A1 (es) | 2013-11-22 | 2015-05-28 | Pontificia Universidad Catolica De Chile | Variantes de enzima fenilacetona monooxigenasa (pamo) capaces de catalizar conversión de ciclohexanona a caprolactona. |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012060185A1 (ja) | 2014-05-12 |
| JP5920889B2 (ja) | 2016-05-18 |
| EP2636665A4 (en) | 2014-04-16 |
| EP2636665A1 (en) | 2013-09-11 |
| US8853426B2 (en) | 2014-10-07 |
| US20130217898A1 (en) | 2013-08-22 |
| EP2636665B1 (en) | 2016-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5920889B2 (ja) | エステルの製法 | |
| Thummel et al. | Base-induced rearrangement of epoxides. V. Phenyl-substituted epoxides | |
| Caron et al. | An optimized sequential kinetic resolution of trans-1, 2-cyclohexanediol | |
| Chenal et al. | Carbon monoxide as a building block in organic synthesis: Part II. One-step synthesis of esters by alkoxycarbonylation of naturally occurring allylbenzenes, propenylbenzenes and monoterpenes | |
| JP3251525B2 (ja) | 3−オキソカルボン酸エステルの製造方法 | |
| CN109956850B (zh) | 3,7-二甲基-7-辛烯醇和3,7-二甲基-7-辛烯基羧酸酯化合物的生产方法 | |
| Simpura et al. | Tandem aldol-transfer–Tischtschenko reaction of aldehydes and β-hydroxyketones catalyzed by trimethylaluminum | |
| EP2948245B1 (en) | Process for the preparation of 4-methylpent-3-en-1-ol derivatives | |
| JPS5820948B2 (ja) | デルタ 4 ,デルタ 8− トランスフアルネシルサクサン マタハ ソノエステルルイ ノ セイゾウホウ | |
| CN1218918C (zh) | 香叶基香叶醇的合成方法 | |
| Yadav et al. | InBr3-catalyzed stereoselective synthesis of trans-2, 6-disubstituted 3, 6-dihydro-2H-pyrans | |
| Yadav et al. | Chemoselective allylation of aldehydes using cerium (III) chloride: simple synthesis of homoallylic alcohols | |
| Lattanzi et al. | Synthesis of a renewable hydroperoxide from (+)-norcamphor: influence of steric modifications of the bicyclic framework on asymmetric sulfoxidation | |
| EP0676404A2 (en) | Oxotitanium complexes useful as asymmetric reaction catalysts particularly for producing beta-hydroxy ketones or alpha-hydroxy carboxylic acid esters | |
| CN106946705B (zh) | 一种合成(1r,2s)-二氢茉莉酮酸甲酯的方法 | |
| JP4361359B2 (ja) | グリオキシル酸エステルの製造方法 | |
| US3202704A (en) | Ech=che | |
| Yadav et al. | Indium (III) chloride catalyzed allylation of gem-diacetates: a facile synthesis of homoallyl acetates | |
| JP3626520B2 (ja) | 3−置換−3−メチルブタナールの製造方法 | |
| JP2838656B2 (ja) | ヒノキチオール及びその中間生成物の製造方法 | |
| RU2565059C1 (ru) | Способ получения амидов карбоновых кислот | |
| SU615056A1 (ru) | Способ получени 1,5-диметилциклооктадиена-1,5 | |
| Arnold | Phosphoramidites as ligands for copper in catalytic asymmetric CC bond formation reactions with organozinc reagents | |
| US3652603A (en) | Method for production of 2 3-di(lower alkoxy)-5-methyl-1 4-benzoquinone | |
| JPH11335319A (ja) | α−ヒドロキシカルボン酸の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11837836 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012541793 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13881544 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011837836 Country of ref document: EP |