WO2012099785A2 - Modulators of tlr3/dsrna complex and uses thereof - Google Patents
Modulators of tlr3/dsrna complex and uses thereof Download PDFInfo
- Publication number
- WO2012099785A2 WO2012099785A2 PCT/US2012/021207 US2012021207W WO2012099785A2 WO 2012099785 A2 WO2012099785 A2 WO 2012099785A2 US 2012021207 W US2012021207 W US 2012021207W WO 2012099785 A2 WO2012099785 A2 WO 2012099785A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- hydrogen
- mhz
- nmr
- tlr3
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
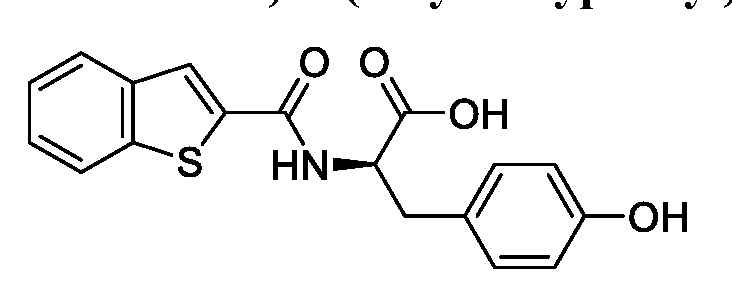
- RCYYJEHKVQJXDD-CQSZACIVSA-N OC([C@@H](Cc(cc1)ccc1O)NC(c1cc(cccc2)c2[s]1)=O)=O Chemical compound OC([C@@H](Cc(cc1)ccc1O)NC(c1cc(cccc2)c2[s]1)=O)=O RCYYJEHKVQJXDD-CQSZACIVSA-N 0.000 description 1
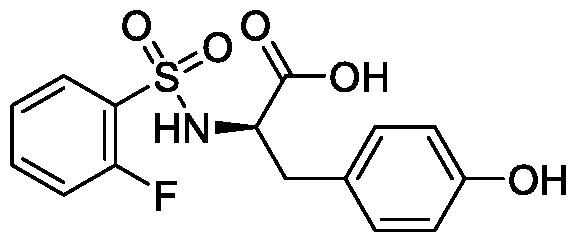
- UYBVYAVIONKCEA-CYBMUJFWSA-N OC([C@@H](Cc(cc1)ccc1O)NS(c1ccccc1F)(=O)=O)=O Chemical compound OC([C@@H](Cc(cc1)ccc1O)NS(c1ccccc1F)(=O)=O)=O UYBVYAVIONKCEA-CYBMUJFWSA-N 0.000 description 1
- ZYHDFIJDHZZJNS-CYBMUJFWSA-N OC([C@@H](Cc1ccccc1)NC(c([s]c1cc(C(F)(F)F)ccc11)c1Cl)=O)=O Chemical compound OC([C@@H](Cc1ccccc1)NC(c([s]c1cc(C(F)(F)F)ccc11)c1Cl)=O)=O ZYHDFIJDHZZJNS-CYBMUJFWSA-N 0.000 description 1
- QEQFBONYJYZBRG-AWEZNQCLSA-N OC([C@H](Cc(cc1)ccc1O)NS(c1cccc(F)c1)(=O)=O)=O Chemical compound OC([C@H](Cc(cc1)ccc1O)NS(c1cccc(F)c1)(=O)=O)=O QEQFBONYJYZBRG-AWEZNQCLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D333/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D333/70—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to compounds and compositions that can modulate formation of Toll-like receptor 3 (TLR3) and double-stranded RNA (dsRNA) complex, and methods for using the same.
- TLR3 Toll-like receptor 3
- dsRNA double-stranded RNA
- RNA-binding proteins play key roles in post-transcriptional modifications, which, along with transcriptional regulation, is believed to be a main method of controlling patterns of gene expression during development.
- TLRs Toll-like receptors
- LPS lipopolysaccharides
- TLR2 lipopeptides
- TLR5 single- stranded RNA
- TLR7 and TLR8 double stranded RNA
- TLR3 CpG motif-containing DNA
- profilin present on uropathogenic bacteria TLRl 1).
- TLR3 signaling is activated by dsRNA released from necrotic cells during inflammation or viral infection.
- TLR3 activation induces secretion of type I interferons and pro-inflammatory cytokines, such as the NF-KB-dependent genes, TNF-a, IL-1, and IL-6 and triggers immune cell activation and recruitment that are protective during certain microbial infections.
- a dominant-negative TLR3 allele has been associated with increased susceptibility to herpes simplex encephalitis, a serious illness with significant risks of morbidity and death, upon primary infection with HSV-1 in childhood. In mice, TLR3 deficiency is associated with decreased survival upon coxsackie virus challenge.
- TLR3 uncontrolled or sustained innate immune response via TLR3 has been shown to contribute to morbidity and mortality in certain viral infection models including West Nile, phlebovirus, vaccinia, and influenza A. TLR3 has also been implicated to be involved with diseases such as atherosclerosis, systemic lupus erythematosus, and rheumatoid arthritis. Thus, modulation of TLR3 pathways offers an attractive method to fight a variety of diseases. [0004] Despite this potential, the discovery of treatment agents has been slow due to the complexity associated with disrupting the protein-R A contact: immense effort is required to design individual compounds that target specific RNA-binding domains with high binding affinity, selectivity, and also functional activity in cell-based assays. Nevertheless, the information gained with regards to the residues involved in protein-RNA interactions could enable the development of specific agents to disrupt these interactions and thereby limit their signaling capacity.
- Ar 1 can be substituted with R 3 , where R 3 is alkyl, OR c , halide, NR a R b , or SR d , and where R a , R b , R c , and R d are those defined herein.
- Ar 2 can be substituted with R 1 , R 2 , and R 4 , where each of R 1 , R 2 , and R 4 is independently alkyl, OR c , halide, NR a R b , or SR d , and where R a , R b , R c , and R d are those defined herein.
- R 1 is halide, alkyl, haloalkyl, -OR c .
- R 2 is typically halide.
- R 3 is typically halide, or -OR c .
- R 4 is typically halide.
- X 1 is S.
- Z 1 and Z 2 are O.
- X 2 is NR a .
- R a is hydrogen.
- X 3 is OR c .
- R c is hydrogen
- R 1 is hydrogen, methyl, trifluoromethyl, CI, F, or methoxy.
- R 2 is hydrogen, F, or CI.
- R 3 is hydrogen, CI, or OH.
- R 4 is hydrogen or F.
- each of X 1 and X 2 is independently -NR a -, -O- or -S-; each of Z 1 , and Z 2 is
- each of R h R 2 , R 3 , and R 4 is independently hydrogen, alkyl, -OR c , halide, -NR a R b , or -SR d , provided at least one of Ri, R 2 , R 3 , and R 4 is not hydrogen; each R a is independently hydrogen, alkyl, or a nitrogen protecting group; R b is hydrogen or alkyl; each R c is independently hydrogen, alkyl, or a hydroxyl protecting group; R d is hydrogen, alkyl, or a thiol protecting group.
- Ri is hydrogen, halide, alkyl, haloalkyl, -OR c ; each of R 2 and R 4 is independently hydrogen or halide; and R 3 is hydrogen, halide, or -OR c , and wherein R c is hydrogen, alkyl, or a hydroxyl protecting group, provided at least one of Ri, R 2 , R 3 and R 4 is not hydrogen.
- Ri is hydrogen, methyl, trifluoromethyl, CI, F, or methoxy.
- R 2 is hydrogen, F, or CI.
- R 3 is hydrogen, CI, or OH.
- R 4 is hydrogen or F.
- R 5 and R 6 are independently hydrogen, halide, or -OR c , and wherein R c is hydrogen, alkyl, or a hydroxyl protecting group.
- R 5 is halide, typically fluoride.
- R c is hydrogen.
- Such methods typically comprise contacting a cell with an effective amount of a compound of Formula I.
- Still other aspects of the invention provide methods for treating a subject for a clinical condition associated with Toll-like receptor 3 (TLR3)/double-stranded RNA (dsRNA) activation.
- Such treatment methods typically include administering to the subject a compound of Formula I.
- the clinical condition comprises an infectious disease, an inflammatory disease, or a combination thereof.
- the clinical condition comprises viral infection, a clinical condition associated with viral infection, atherosclerosis, systemic lupus erythematosus, rheumatoid arthritis, or a combination thereof.
- viruses infection can be treated by methods of the invention, in some particular cases, methods of the invention are used to treat infection of virus, where virus comprises herpes simplex- 1, West Nile virus, phlebovirus, vaccinia, influenza A, or a combination thereof.
- Yet other aspects of the invention provide methods for treating a clinical condition associated with a Toll-like receptor 3 (TLR3)/double-stranded RNA (dsRNA) interaction in a subject, said method comprising administering to the subject in need of such a treatment a TLR3/dsRNA interaction inhibitor.
- TLR3 Toll-like receptor 3
- dsRNA double-stranded RNA
- the clinical condition comprises an infectious disease, an inflammatory disease, or a combination thereof.
- the clinical condition comprises viral infection, a clinical condition associated with viral infection, atherosclerosis, systemic lupus erythematosus, rheumatoid arthritis, or a combination thereof.
- methods of the invention are used to treat infection of virus, where virus comprises herpes simplex- 1, West Nile virus, phlebovirus, vaccinia, influenza A, or a combination thereof.
- compositions comprising a compound disclosed herein.
- Figure 1 shows Glide computer simulation model of binding mode of compounds
- Figure 2(a) shows dose-dependent inhibitory response graph of Poly IC-induced
- FIG. 1 shows bar graph of specificity test results for compound 4a at 27 ⁇ .
- LPS, Poly IC, FSL-1, Pam3CSK4 and R848 were used to selectively activate TLR4, TLR3, TLR2/6, TLR2/1 and TLR7 respectively.
- Figure 2(c) is a graph showing competitive binding between compound 4a and dsRNA to TLR3.
- Figure 2(d) is a graph showing the TLR3 binding competition results of compound 4a and la with dsRNA. The results show compound 4a competes with dsRNA in binding to TLR3 but the negative control compound la does not show any significant binding to TLR3.
- Figure 3 is a bar graph of ELISA assay results based on the RAW 264.7 cell showing that compound 4a inhibits TNF-a and IL- ⁇ production.
- the Poly IC dose was 15 g/mL.
- Figure 4 is a graph of the toxicity test results of compound 4a to different
- Cytochrome P450 enzymes at 25 ⁇ and 50 ⁇ . Ketoconazole, quinidine, ticlopidine and aNF selectively inhibit Cyp3A4, Cyp2D6, Cyp2C19 and CyplA2, respectively.
- Figure 5 is a graph showing viability of RAW 264.7 cells in the presence of compound 4a at different concentrations.
- Figure 6 is a graph of kinase profiling results showing that compound 4a (a TLR3 inhibitor) at 10 ⁇ did not significantly affect activities of representative kinases.
- Figure 7 is a graph showing the result of animal model in vivo test. Wild-type mice were treated with a single 100 ⁇ g poly IC i.v. injection and investigated the serum concentration of MCP-1 4 hours after treatment. As expected poly IC led to a significant increase in MCP-1, only in wild-type but not TLR3-/- mice. Pretreatment with compound 4a 30 minutes prior to poly IC application decreased MCP-1 production by about 50%.
- Figure 8 is a graph showing response of cytokine RANTES to TLR3 stimulation.
- Figure 9 is a graph showing decreasing RANTES production to TLR3 stimulation with increased pre-treatment doses of compound 4a.
- Figure 10 is a graph showing pretreatment with 27 ⁇ g of compound 4a results in decreased RANTES production with increasing poly IC doses.
- Figure 11 is an electrophoresis slide showing inhibition of XMRV-induced
- IP- 10 interferon gamma-induced protein 10 production.
- LNCaP human prostatic carcinoma cell line
- Figure 12 is an electrophoresis slide showing inhibition of XMRV-induced IP-
- RNA isolated from LNCaP cells infected with XMRV in the presence or absence of compound 4a (10 and 25 ⁇ ) was subjected to RT-PCR designed to detect IP-10/CXCLlO mRNA. GAPDH was used as the internal control.
- Figure 13 is a graph showing that compound 4a significantly improves poly IC- induced impaired re-endothelialization.
- Some aspects of the invention is based on in silico screening and discovery by the present inventors of compounds that can interact with the dsRNA binding region of TLR3.
- 1.2 million compounds from Enamine drug database
- Initial nine hits were selected for cell assay screening.
- Figure A The initial hits ( Figure A) were first evaluated using the present inventors' previously established high-throughput cell assay of TLR3 activation. See, for example, ACS Med. Chem. Lett. 2010, 1, 194 and Bioorg. Med. Chem. Lett., 2010, 20, 5411. Poly IC
- compounds T5626448 and T5260630 had IC 50 values of 153.7 ⁇ 5.9 ⁇ and 144.9 ⁇ 3.7 ⁇ , respectively. Both of these compounds are structural derivatives of D- phenylalanine.
- the homologous motif indicated that the D-phenylalanine scaffold can be used as a core structure to develop small molecule inhibitors of TLR3.
- the in silico predicted binding modes ( Figure 1) also indicated that the binding affinities of T5626448 and T5260630 can be further modified by varying the substituents on the benzene or thiophene rings.
- Scheme 1 Representative Synthetic Scheme [0047] As exemplified in Scheme 1 , synthetic routes were developed to allow structure- activity relationship (SAR) analysis. According to isosteric replacement theory, the -F group on the phenyl ring was modified to -CI or -CF 3 . To research the electronic effect, the -F group was replaced by -CH 3 or -OCH 3 . Different substitutions on the phenyl group were also examined. To inspect the impact on the activities imposed by the amino acid chiral center, both R- and S- isomers were also incorporated.
- SAR structure- activity relationship
- T5626448 An improvement of two orders of magnitude in inhibitory potency of T5626448 was achieved, with compound 4a (see Table 1) that showed a low ⁇ (3.44 ⁇ 0.41 ⁇ ) IC 5 o value.
- Other analogs of T5626448 and T5260630 compounds were synthesized and evaluated for inhibitory activity.
- IC 5 o average values and corresponding SD values (in ⁇ ) were determined from the results of at least three independent tests.
- Results showed that in general the greater the electronic withdrawing capability of the group at the Ri position, the more activity was observed. This observation was further supported by compound 6a, where a decrease in the potency by 10-fold (6a vs 4a) with a methoxyl substituent at Ri position was detected.
- the -CF 3 replacement of the -F at Ri position decreased the activity slightly (5a vs 4a). When the fluoride was relocated from the Ri position to R 4 , the activity decreased significantly (12a vs 4a).
- a secondary cell assay was used to confirm that compound 4a inhibits the downstream signaling transduction mediated by the formation of the TLR3/dsRNA complex.
- TLR3 signaling suppression the release of the proinflammatory cytokines, TNF-a and IL- ⁇ , were also studied. These results further confirmed that compound 4a suppresses TLR3-mediated inflammation response (Figure 3).
- Compound 4a was also found to have low toxicity to Cytochrome P450 (CYP450) enzymes, Cyp3A4, 2D6, 2C19 and 1A2, compared to known inhibitors to these specific enzymes ( Figure 4). Furthermore, this compound ( Figure 5) and others did not show any significant toxicity in RAW 264.7 cells as determined by WST-1 methodology.
- CYP450 Cytochrome P450
- Compounds of the invention target the dsRNA binding region of TLR3 with good specificity, high binding affinity. Accordingly, compounds of the invention can be used to selectively inhibit TLR3/dsRNA binding.
- the starting materials and reagents used in preparing these compounds generally are either available from commercial suppliers, such as Aldrich Chemical Co., or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1991, Volumes 1-15; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, Volumes 1-5 and Supplemental; and Organic Reactions, Wiley & Sons: New York, 1991, Volumes 1-40.
- the starting materials and the intermediates of the synthetic reaction schemes can be isolated and purified if desired using conventional techniques, including but not limited to, filtration, distillation, crystallization, chromatography, and the like. Such materials can be characterized using conventional means, including physical constants and spectral data.
- reaction temperature range of from about -78 °C to about 150 °C, often from about 0 °C to about 125 °C, and more often and conveniently at about room (or ambient) temperature, e.g., about 20 °C.
- substituents on the compounds of the invention can be present in the starting compounds, added to any one of the intermediates or added after formation of the final products by known methods of substitution or conversion reactions. If the substituents themselves are reactive, then the substituents can themselves be protected according to the techniques known in the art. A variety of protecting groups are known in the art, and can be employed. Examples of many of the possible groups can be found in "Protective Groups in Organic Synthesis" by Green et al, John Wiley and Sons, 1999. For example, nitro groups can be added by nitration and the nitro group can be converted to other groups, such as amino by reduction, and halogen by diazotization of the amino group and replacement of the diazo group with halogen.
- Acyl groups can be added by Friedel-Crafts acylation. The acyl groups can then be transformed to the corresponding alkyl groups by various methods, including the Wolff- Kishner reduction and Clemmenson reduction.
- Amino groups can be alkylated to form mono- and di-alkylamino groups; and mercapto and hydroxy groups can be alkylated to form corresponding ethers.
- Primary alcohols can be oxidized by oxidizing agents known in the art to form carboxylic acids or aldehydes, and secondary alcohols can be oxidized to form ketones. Thus, substitution or alteration reactions can be employed to provide a variety of substituents throughout the molecule of the starting material, intermediates, or the final product, including isolated products.
- the present invention includes pharmaceutical compositions comprising at least one compound of the invention, or an individual isomer, racemic or non-racemic mixture of isomers or a pharmaceutically acceptable salt or solvate thereof, together with at least one pharmaceutically acceptable carrier, and optionally other therapeutic and/or prophylactic ingredients.
- the compounds of the invention are administered in a therapeutically effective amount by any of the accepted modes of administration for agents that serve similar utilities.
- Suitable dosage ranges are typically 1-500 mg daily, typically 1-100 mg daily, and often 1-30 mg daily, depending on numerous factors such as the severity of the disease to be treated, the age and relative health of the subject, the potency of the compound used, the route and form of administration, the indication towards which the administration is directed, and the preferences and experience of the medical practitioner involved.
- One of ordinary skill in the art of treating such diseases is typically able, without undue experimentation and in reliance upon personal knowledge and the disclosure of this application, to ascertain a therapeutically effective amount of the compounds of the invention.
- compounds of the invention are administered as pharmaceutical formulations including those suitable for oral (including buccal and sub-lingual), rectal, nasal, topical, pulmonary, vaginal, or parenteral (including intramuscular, intraarterial, intrathecal, subcutaneous and intravenous) administration or in a form suitable for administration by inhalation or insufflation.
- Typical manner of administration is generally oral using a convenient daily dosage regimen which can be adjusted according to the degree of affliction.
- a compound or compounds of the invention, together with one or more conventional adjuvants, carriers, or diluents, can be placed into the form of pharmaceutical compositions and unit dosages.
- the pharmaceutical compositions and unit dosage forms can be comprised of conventional ingredients in conventional proportions, with or without additional active compounds or principles, and the unit dosage forms can contain any suitable effective amount of the active ingredient commensurate with the intended daily dosage range to be employed.
- compositions can be employed as solids, such as tablets or filled capsules, semisolids, powders, sustained release formulations, or liquids such as solutions, suspensions, emulsions, elixirs, or filled capsules for oral use; or in the form of suppositories for rectal or vaginal administration; or in the form of sterile injectable solutions for parenteral use.
- Formulations containing about one (1) milligram of active ingredient or, more broadly, about 0.01 to about one hundred (100) milligrams, per tablet, are accordingly suitable representative unit dosage forms.
- the compounds of the invention can be formulated in a wide variety of oral administration dosage forms.
- the pharmaceutical compositions and dosage forms can comprise a compound or compounds of the invention or pharmaceutically acceptable salts thereof as the active component.
- the pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispersible granules.
- a solid carrier can be one or more substances which can also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
- the carrier In powders, the carrier generally is a finely divided solid which is a mixture with the finely divided active component.
- the active component In tablets, the active component generally is mixed with the carrier having the necessary binding capacity in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from about one (1) to about seventy (70) percent of the active compound.
- Suitable carriers include but are not limited to magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatine, tragacanth, methylcellulose, sodium
- preparation is intended to include the formulation of the active compound with encapsulating material as carrier, providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is in association with it.
- carrier which is in association with it.
- cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges can be as solid forms suitable for oral administration.
- liquid form preparations including emulsions, syrups, elixirs, aqueous solutions, aqueous suspensions, or solid form preparations which are intended to be converted shortly before use to liquid form preparations.
- Emulsions can be prepared in solutions, for example, in aqueous propylene glycol solutions or may contain emulsifying agents, for example, such as lecithin, sorbitan monooleate, or acacia.
- Aqueous solutions can be prepared by dissolving the active component in water and adding suitable colorants, flavors, stabilizers, and thickening agents.
- Aqueous suspensions can be prepared by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, and other well known suspending agents.
- Solid form preparations include solutions, suspensions, and emulsions, and can contain, in addition to the active component, colorants, flavors, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilizing agents, and the like.
- the compounds of the invention can also be formulated for parenteral
- compositions can take such forms as
- oily or nonaqueous carriers, diluents, solvents or vehicles include propylene glycol, polyethylene glycol, vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl oleate), and can contain formulatory agents such as preserving, wetting, emulsifying or suspending, stabilizing and/or dispersing agents.
- the active ingredient can be in powder form, obtained by aseptic isolation of sterile solid or by lyophilization from solution for constitution before use with a suitable vehicle, e.g., sterile, pyrogen-free water.
- a suitable vehicle e.g., sterile, pyrogen-free water.
- the compounds of the invention can be formulated for topical administration to the epidermis as ointments, creams or lotions, or as a transdermal patch.
- Ointments and creams can, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Lotions can be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilizing agents, dispersing agents, suspending agents, thickening agents, or coloring agents.
- Formulations suitable for topical administration in the mouth include lozenges comprising active agents in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatine and glycerine or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- the compounds of the invention can be formulated for administration as suppositories.
- a low melting wax such as a mixture of fatty acid glycerides or cocoa butter is first melted and the active component is dispersed homogeneously, for example, by stirring. The molten homogeneous mixture is then poured into convenient sized molds, allowed to cool, and to solidify.
- the compounds of the invention can also be formulated for vaginal
- the compounds of the invention can be formulated for nasal administration.
- the solutions or suspensions are applied directly to the nasal cavity by conventional means, for example, with a dropper, pipette or spray.
- the formulations can be provided in a single or multidose form. In the latter case of a dropper or pipette, this can be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension. In the case of a spray, this can be achieved for example by means of a metering atomizing spray pump.
- the compounds of the invention can be formulated for aerosol administration, particularly to the respiratory tract and including intranasal administration.
- the compound will generally have a small particle size for example of the order of five (5) microns or less. Such a particle size can be obtained by means known in the art, for example by micronization.
- the active ingredient is provided in a pressurized pack with a suitable propellant such as a
- chlorofluorocarbon for example, dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, or carbon dioxide or other suitable gas.
- the aerosol can conveniently also contain a surfactant such as lecithin.
- the dose of drug can be controlled by a metered valve.
- the active ingredients can be provided in a form of a dry powder, for example, a powder mix of the compound in a suitable powder base such as lactose, starch, starch
- the powder carrier typically forms a gel in the nasal cavity.
- the powder composition can be presented in unit dose form, for example, in capsules or cartridges of e.g., gelatine or blister packs from which the powder can be administered by means of an inhaler.
- formulations can be prepared with enteric coatings adapted for sustained or controlled release administration of the active ingredient.
- the compounds of the invention can be formulated in transdermal or subcutaneous drug delivery devices. These delivery systems are advantageous when sustained release of the compound is necessary or desired and when patient compliance with a treatment regimen is crucial.
- Compounds in transdermal delivery systems are frequently attached to a skin-adhesive solid support.
- the compound of interest can also be combined with a penetration enhancer, e.g., Azone (l-dodecylazacycloheptan-2-one).
- Azone l-dodecylazacycloheptan-2-one
- Sustained release delivery systems can be inserted subcutaneously into the subdermal layer by surgery or injection.
- the subdermal implants encapsulate the compound in a lipid soluble membrane, e.g., silicone rubber, or a biodegradable polymer, e.g., polylactic acid.
- the pharmaceutical preparations are typically in unit dosage forms.
- the preparation is often subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
- compositions which include therapeutically effective mounts of compounds of Formula (I) or pharmaceutically acceptable salts thereof or a prodrug thereof, and one or more
- compositions of this disclosure comprise a combination of a compound of the present disclosure and one or more additional therapeutic or prophylactic agent
- both the compound and the additional agent are usually present at dosage levels of between about 10 to 150%, and more typically between about 10 and 80% of the dosage normally administered in a monotherapy regimen.
- compositions of the invention can be used to treat a wide variety of clinical conditions.
- the term "treat” or “treating" of a clinical condition includes: (1) preventing a clinical condition or a disease, i.e., causing the clinical symptoms of the clinical condition or disease not to develop in a mammal that may be exposed to or predisposed to the clinical condition or disease but does not yet experience or display symptoms of the clnical condition or disease; (2) inhibiting the clinical condition or disease, i.e., arresting or reducing the development of the clinical condition or disease or its clinical symptoms; or (3) relieving the clinical condition or disease, i.e., causing regression of the clinical condition or disease or its clinical symptoms.
- Typical clinical conditions related to TLR3 that can be treated by administering a therapeutically effective amount of a compound of the invention include, but not limited to, clinical conditions manifested by (i) an activation of TLR3; (ii) TLR3/dsRNA complex formation; as well as (iii) TLR3/dsRNA activation.
- therapeutically effective amount means the amount of a compound that, when administered to a mammal for treating a clinical condition or a disease, is sufficient to effect such treatment for the clinical condition or disease.
- the “therapeutically effective amount” will vary depending on the compound, the clinical condition or the disease and its severity and the age, weight, etc., of the mammal to be treated.
- Exemplary clinical conditions that can be treated include, but are not limited to, cancer such as prostate cancer, an infectious disease, an inflammatory disease, viral infection, atherosclerosis, systemic lupus erythematosus, rheumatoid arthritis, or a combination thereof. While a wide variety virus infection can be treated by methods of the invention, in some particular cases, methods of the invention are used to treat infection of virus, where virus comprises herpes simplex- 1, West Nile virus, phlebovirus, vaccinia, influenza A, XMRV, or a combination thereof. In addition, compounds of the invention can be used to treat clinical conditions associated with viral infection such as cancer. Exemplary cancers resulting from a viral infection include, but are not limited to, prostate cancer, cervical cancer, etc.
- NMR spectra were acquired on Bruker 300 spectrometer, running at 300 MHz for
- the Enamine drug database (1.2 million small molecules) was docked into the dsRNA and TLR3 binding domain (PDB: 3CIY) using Glide 5.6. The molecules are created, as appropriate, with multiple protonation and tautomeric states.
- the TLR3 conformations were prepared using standard Glide protocols. This includes addition of hydrogens, restrained energy- minimizations of the protein structure with the Optimized Potentials for Liquid Simulations-All Atom (OPLS-AA) force field, and finally setting up the Glide grids using the Protein and Ligand Preparation Module. All 1.2 million compounds were first docked and ranked using High Throughput Virtual Screening (HTVS) Glide, continued with standard precision (SP) Glide for the top 10000 compounds. The resultant top 5000 compounds were then docked using the more accurate and computationally intensive extra-precision (XP) mode. Initial top-ranked 100 compounds were selected out.
- HTVS High Throughput Virtual Screening
- SP standard precision
- Complementarity exists between the ligand and the active site of TLR3.
- Reasonable chemical structure and pose are in the active site of TLR3. Some unusually highly scored molecules were found to have many rotatable bonds (such as long aliphatic structures), which were excluded for further evaluation.
- Protonation state and the tautomeric form of the ligand have to be acceptable.
- 100 candidate compounds can meet the above criteria. In order to achieve good chemical diversity, the resulting 100 candidates were subsequently filtered by chemical diversity and binding energy. Consequently, 9 potential TLR3 inhibitors were designated and purchased for vitro assaying.
- RAW 264.7 Mae leukaemic monocyte macrophage cell line
- RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL).
- RAW cells were then planted in 96-well plates at 100,000 cells per well and grown for 24 h in the media descried previously at 37°C in a 5% C0 2 humidified incubator. After 24 h, non-adherent cells and media were removed and replaced with fresh RPMI 1640 medium (only RPMI).
- the adherent macrophages were treated with high molecular weight polyinosine-polycytidylic acid (Poly IC) (10 ⁇ g/mL) (Invivogen), an agonist of TLR3, and then added different concentrates of potential inhibitor. Two rows were only treated with Poly IC as an control. Plates were then incubated for an additional 24 h. Following incubation 100 of media was removed and added to flat black 96-well microfluor plates (Thermo Scientific, MA, USA). To each well, 10 ⁇ ⁇ of 2, 3-diaminonaphthalene (0.05 mg/mL in 0.62 M aqueous HC1 solution) was added and incubated for 15 min in the dark.
- Poly IC polyinosine-polycytidylic acid
- the reaction was quenched by addition of 5 of a 3 M aqueous NaOH solution and the plate was read on Beckman Coulter DTX880 reader (Beckman Coulter, CA, USA) with excitation at 365 nm and emission at 450 nm.
- the nitrite (a stable metabolite of nitric oxide) concentration was determined from a nitrite standard curve.
- the IC50 values for both inhibition and cytotoxicity were determined graphically using software Origin v7.5.
- RAW cells were planted in 6-well plates at 1 ,000,000 cells per well with 3 mL of medium (RPMI 1640 medium, supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL)) and grown for 24 h at 37°C in a 5% C0 2 humidified incubator. After 24 h, non-adherent cells and media were removed and replaced with fresh RPMI 1640 medium (3 mL/well). Two wells of adherent macrophages were treated with high molecular weight Poly IC (Invivogen, 10 ⁇ g/mL), only one well was treated with 27 ⁇ compound 4a.
- RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL)
- one well was treated with with only 4a (27 ⁇ ) and the other was treated with nothing. Plates were then incubated for an additional 24 h. The medium was removed, the cells were washed with PBS (3 x 1 mL), the 6 well plate was put on ice, then 500 ⁇ , of lysis buffer was added in each well (Lysis Buffer: 120 ⁇ 0.5M EDTA; 12 mL Mammalian Protein Extraction Reagent, 100 ⁇ ⁇ cocktail, 0.36 mL NaCl (5 M, aqueous) ). After 5 min, the mixture was transferred into corresponding 1.5 mL tube, spun for 20 min at 13.2 K rpm in a cold room.
- Lysis Buffer 120 ⁇ 0.5M EDTA; 12 mL Mammalian Protein Extraction Reagent, 100 ⁇ ⁇ cocktail, 0.36 mL NaCl (5 M, aqueous
- cytokine interleukin- ⁇ (IL- ⁇ ) and TNF-a were quantified with enzyme-linked immunosorbent assays (ELISA) using cytokine-specific capture antibodies, biotinylated monoclonal detection antibodies, and recombinant human cytokine standards according to commercially available ELISA kits from R&D Systems. The cytokine level in each sample was determined in duplicate.
- ELISA enzyme-linked immunosorbent assays
- LPS lipopolysaccharide
- FSL-1 ((S,R)-(2,3-bispalmitoyloxypropyl)-Cys-Gly-Asp-Pro-Lys- His-Pro-Lys-Ser-Phe)
- R848 (4-amino-2-(ethoxymethyl)-a, a-dimethyl-lH-imidazo[4,5- c]quinoline-l-ethanol)
- Pam 3 CSK 4 N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]- [R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine.3HCl
- Test agent was incubated (two wells per condition) with microsomes at 37 °C.
- Control incubations containing vehicle or reference inhibitors were run along side the test agents.
- the final assay contained test agent, probe substrates at the indicated concentration, 2 mM NADPH, 3 mM MgCl 2 in 50 mM potassium phosphate buffer, pH 7.4.
- the final microsomal concentration was 0.5 mg/mL.
- the maximum solvent concentration in the final assay was ⁇ 0.5% to minimize the inhibition of Cyps by solvent.
- NADPH was added last to start the assay.
- the assay was stopped by the addition of acetonitrile containing internal standard, the samples were centrifuged, and the amount of probe metabolite in the supernatant was determined by LC/MS/MS. (Testing was done by Apredica, Watertown, MA using subcellular fractions)
- This assay was conducted in a 96-well plate with 5,000 cells in 100 ⁇ , media
- Endothelial dysfunction and atherosclerosis are chronic inflammatory diseases characterized by activation of the innate and acquired immune system. Zimmer, S. et al., Circ. Res., 2011, 108, 1358-1366. It is believed that specialized protein receptors of the innate immune system recognize products of microorganisms and endogenous ligands such as nucleic acids. TLR3 detects long double-stranded RNA and is abundantly expressed in endothelial cells. It has been shown that TLR3 stimulation not only induces endothelial dysfunction but also enhances the formation of atherosclerotic plaques in the mice. Id.
- Monocyte chemoattractant protein-1 (MCP-1) and RANTES (Regulated upon Activation, Normal T cell Expressed and presumably Secreted) are two of the cytokines that have been shown to respond to TLR3 stimulation. MCP-1 is believed to be produced predominantly by macrophages and endothelial cells and has shown to be a potent chemotactic factor for monocytes. Expression of this proinflammatory chemokine has been shown to increase in atherosclerotic lesions. Kanda, H. et al, J. Clin. Invest., 2006, 116, 1494-1505.
- RANTES also known as CCL5 (Chemokine (C-C motif) ligand 5), is a member of the "CC" subfamily of chemokines. It has been shown to play a primary role in the inflammatory immune response via its ability to chemoattract leukocytes and modulate their function. Kim, M. O. et al., J. Neurochem. 2004, 90, 297-308. It has also been shown that antagonism of MCP-1 and RANTES receptors can reduce atherosclerotic. Kanda, H. et al; J. Clin. Invest., 2006, 116, 1494-1505 and Veillard, N. R. et al, Circ. Res., 2004, 94, 253- 261.
- Wild-type mice were treated with a single 100 ⁇ g of poly IC i.v. injection and the serum concentration of MCP-1 was investigated 4 hours after the treatment. As shown in Figure7, administration of poly IC resulted in a significant increase in MCP-1, only in wild-type but not TLR3-/- mice. Pretreatment with compound 4a at various concentrations 30 minutes prior to poly I:C application decreased MCP-1 production by about 50%.
- RANTES is another cytokine that has been shown to respond to TLR3
- RANTES peaked approximately 4h after i.v. injection of poly IC.
- pretreatment with compound 4a resulted in decrease of RANTES in a dose-dependent manner.
- Figure 9 As shown in Figure 10, RANTES production increased relatively proportionally with increasing poly IC doses ("vehicle"). However, pretreatment with 27 ⁇ g of compound 4a resulted in a significant decrease in RANTES production.
- Xenotropic murine leukemia-related retrovirus is a recently discovered retrovirus that has been linked to human prostate cancer.
- Schlaberga, R. et al. Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 16351-16356. It is estimated that XMRV infection affects a large fraction of the world population, with prostate cancer affecting one in six men.
- TLR3 inhibitors of the invention efficiently suppressed the activity of XMRV to induce interferon ⁇ -induced protein 10 (i.e., IP-10, also known as C-X-C motif chemokine 10 or CXCL10) production. Such results indicate TLR3 inhibitors of the invention can be used to treat and/or prevent prostate cancer.
- IP-10 interferon ⁇ -induced protein 10
- CXCL10 C-X-C motif chemokine 10
- RNA isolated from LNCaP human prostatic carcinoma cell line
- XMRV human prostatic carcinoma cell line
- TLR3 inhibitor compound 4a 50 ⁇
- Glyceraldehyde 3 -phosphate dehydrogenase Glyceraldehyde 3 -phosphate dehydrogenase
- compound 4a inhibits XMRV-induced IP- 10 production.
- Figure 12 shows the results of inhibition of XMRV-induced IP-10/CXCLlO production by compound 4a at 10 and 25 ⁇ .
- mice were subjected to a 4mm long electric denudation of the left common carotid artery. These C57B1/6J littermates were then treated with 100 ⁇ g of poly IC or vehicle (NaCl 0,9%) via i.v. injection every 48 hours for five days. Half of the mice in each group were also treated with 27 ⁇ g compound 4a or vehicle (DMSO) via i.p. injection every 48 hours. The mice were then sacrificed and aortic arch including both common carotid arteries excised. The remaining endothelial lesion was visualized using Evan's blue staining.
- Thianaphthene-2-carboxylic acid (356.42 mg, 2 mmol) was suspended in dry toluene (6 mL), thionyl chloride (4.4 mL, 60 mmol) and DMF (0.05 mL) were added at room temperature, and then the mixture was refluxed 8 h. 4 The volatiles were removed at reduced pressure gave benzo[b]thiophene-2-carbonyl chloride as a yellow power. Purified by flash chromatography on silica gel, using ethyl acetate/hexane (1 :9) as eluent, give 3 as a white power (393.64 mg, 94.9%).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description






















































Claims


Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/980,874 US9409880B2 (en) | 2011-01-20 | 2012-01-13 | Modulators of TLR3/dsRNA complex and uses thereof |
| EP12736857.9A EP2665708A4 (en) | 2011-01-20 | 2012-01-13 | MODULATORS OF THE TLR3 / ARNDS COMPLEX AND THEIR USES |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161434492P | 2011-01-20 | 2011-01-20 | |
| US61/434,492 | 2011-01-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012099785A2 true WO2012099785A2 (en) | 2012-07-26 |
| WO2012099785A3 WO2012099785A3 (en) | 2012-09-27 |
Family
ID=46516313
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/021207 Ceased WO2012099785A2 (en) | 2011-01-20 | 2012-01-13 | Modulators of tlr3/dsrna complex and uses thereof |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9409880B2 (en) |
| EP (1) | EP2665708A4 (en) |
| WO (1) | WO2012099785A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015016282A1 (en) * | 2013-07-30 | 2015-02-05 | 国立大学法人東京大学 | Pharmaceutical composition for preventing or treating radiation-induced gastrointestinal syndrome |
| JP2022536664A (en) * | 2019-06-12 | 2022-08-18 | フィジーン、エルエルシー | Enhancement of fibroblast therapeutic activity by RNA |
| WO2023066949A3 (en) * | 2021-10-19 | 2023-06-22 | Holfeld Johannes | Tlr3 inhibitors for use in the treatment of diseases associated with cardiovascular calcification |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69523182T2 (en) * | 1995-06-06 | 2002-02-07 | Pfizer | SUBSTITUTED N- (INDOL-2-CARBONYL) GLYCINAMIDES AND DERIVATIVES AS GLYCOGEN PHOSPHORYLASE INHIBITORS |
| ES2288871T3 (en) * | 1999-09-24 | 2008-02-01 | Genentech, Inc. | DERIVATIVES OF TYROSINE. |
| JP2004522738A (en) * | 2001-01-17 | 2004-07-29 | アミュラ テラピューティクス リミテッド | Cyclic 2-carbonylaminoketones as inhibitors of cruzipaine and other cysteine proteases |
| IL156776A0 (en) * | 2001-01-17 | 2004-02-08 | Amura Therapeutics Ltd | Inhibitors of cruzipain and other cysteine proteases |
| WO2002057270A1 (en) * | 2001-01-17 | 2002-07-25 | Amura Therapeutics Limited | Inhibitors of cruzipain and other cysteine proteases |
| CZ20032829A3 (en) * | 2001-04-16 | 2005-03-16 | Tanabe Seiyaku Co., Ltd. | Heterocyclic compounds |
| KR100575944B1 (en) * | 2001-06-28 | 2006-05-02 | 화이자 프로덕츠 인코포레이티드 | Triamide-Substituted Indole, Benzofuran and Benzothiophene as Inhibitors of Microsomal Triglyceride Delivery Protein (MTP) and / or Apofatty Protein (AP) Secretion |
| WO2004007501A1 (en) | 2002-07-16 | 2004-01-22 | Amura Therapeutics Limited | Biologically active compounds |
| WO2007030761A2 (en) * | 2005-09-08 | 2007-03-15 | Smithkline Beecham Corporation | Acyclic 1,4-diamines and uses thereof |
| WO2007138112A2 (en) * | 2006-06-01 | 2007-12-06 | Devgen N.V. | Compounds that interact with ion channels, in particular with ion channels from the kv family |
| GB0918922D0 (en) * | 2009-10-28 | 2009-12-16 | Vantia Ltd | Aminopyridine derivatives |
-
2012
- 2012-01-13 US US13/980,874 patent/US9409880B2/en active Active - Reinstated
- 2012-01-13 WO PCT/US2012/021207 patent/WO2012099785A2/en not_active Ceased
- 2012-01-13 EP EP12736857.9A patent/EP2665708A4/en not_active Withdrawn
Non-Patent Citations (15)
| Title |
|---|
| "Organic Reactions", vol. 1-40, 1991, WILEY & SONS |
| "Remington: The Science and Practice ofpharmacy", 1995, MACK PUBLISHING COMPANY |
| ACS MED. CHEM. LETT., vol. 7, 2010, pages 194 |
| BIOORG. MED. CHEM. LETT., vol. 20, 2010, pages 5411 |
| FIESER; FIESER'S: "Reagents for Organic Synthesis", vol. 1-15, 1991, WILEY & SONS |
| GREEN ET AL.: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY AND SONS |
| HAYAT, M. J. ET AL., ONCOLOGIST, vol. 12, 2007, pages 20 - 37 |
| KANDA, H. ET AL., J. CLIN. INVEST., vol. 116, 2006, pages 1494 - 1505 |
| KIM, M. O. ET AL., J. NEUROCHEM., vol. 90, 2004, pages 297 - 308 |
| RODD'S: "Chemistry of Carbon Compounds", vol. 1-5, 1989, ELSEVIER SCIENCE PUBLISHERS |
| SCHLABERGA, R. ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 106, 2009, pages 16351 - 16356 |
| See also references of EP2665708A4 |
| VEILLARD, N. R. ET AL., CIRC. RES., vol. 94, 2004, pages 253 - 261 |
| XU, H. Q.; ZHANG, A. H.; AUCLAIR, C.; XI, X. G., NUCLEIC ACIDS RES., vol. 31, 2003, pages E70 |
| ZIMMER, S. ET AL., CIRC. RES., vol. 108, 2011, pages 1358 - 1366 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015016282A1 (en) * | 2013-07-30 | 2015-02-05 | 国立大学法人東京大学 | Pharmaceutical composition for preventing or treating radiation-induced gastrointestinal syndrome |
| JP2022536664A (en) * | 2019-06-12 | 2022-08-18 | フィジーン、エルエルシー | Enhancement of fibroblast therapeutic activity by RNA |
| EP3982984A4 (en) * | 2019-06-12 | 2023-07-12 | Figene, LLC | Enhancement of fibroblast therapeutic activity by rna |
| WO2023066949A3 (en) * | 2021-10-19 | 2023-06-22 | Holfeld Johannes | Tlr3 inhibitors for use in the treatment of diseases associated with cardiovascular calcification |
Also Published As
| Publication number | Publication date |
|---|---|
| US9409880B2 (en) | 2016-08-09 |
| EP2665708A4 (en) | 2014-07-09 |
| US20140094507A1 (en) | 2014-04-03 |
| WO2012099785A3 (en) | 2012-09-27 |
| EP2665708A2 (en) | 2013-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2427166T3 (en) | Novel tricyclic heterocyclic compound | |
| Zhang et al. | Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors | |
| CN104030987B (en) | Dihydroorotate dehydrogenase inhibitors | |
| JP5828201B2 (en) | Naphthalene derivatives | |
| KR101325237B1 (en) | Quinoline derivatives as axl kinase inhibitors | |
| EP1448539A1 (en) | Hiv protease inhibitors, compositions containing the same, their pharmaceutical uses and materials for their synthesis | |
| JP2006518341A (en) | Hydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors | |
| JP2005534618A (en) | 7-azaindole as a C-JUNN-terminal kinase inhibitor for neurodegenerative diseases | |
| CN114213310B (en) | Indoline compounds and derivatives thereof, preparation methods, pharmaceutical compositions and applications | |
| EP0922035B1 (en) | Tetrahydroisoquinoline derivatives and their pharmaceutical use | |
| JP2007522142A (en) | Benzimidazole-substituted thiophene derivatives having activity against IKK3 | |
| CN107879975B (en) | Histone deacetylase inhibitor and application thereof | |
| JP5068913B2 (en) | Pharmaceutically active sulfonyl amino acid derivatives | |
| US20200131144A1 (en) | Amine or (thio)amide containing lxr modulators | |
| Zhou et al. | N-Arylsulfonylsubstituted-1H indole derivatives as small molecule dual inhibitors of signal transducer and activator of transcription 3 (STAT3) and tubulin | |
| EP1042317B1 (en) | Indole derivatives as PKC-inhiboitors | |
| US9409880B2 (en) | Modulators of TLR3/dsRNA complex and uses thereof | |
| Baumgartner et al. | Structure‐based design and synthesis of the first weak non‐phosphate inhibitors for IspF, an enzyme in the non‐mevalonate pathway of isoprenoid biosynthesis | |
| JP6987125B2 (en) | New 2,4,6-trisubstituted s-triazine compound and its production method and use | |
| Xu et al. | Design, synthesis, and evaluation of potent Wnt signaling inhibitors featuring a fused 3-ring system | |
| Boddiboyena et al. | Synthesis and biological evaluation of novel amide derivatives of 1, 2, 4-oxadiazole-imidazopyridines as anticancer agents | |
| KR20080095912A (en) | Phtharazinone pyrazole derivatives, their preparation and use as pharmaceutical preparations | |
| CN101717397B (en) | Substituted pyrido [2',1':2,3] imidazo [4,5-c ] isoquinolinone compounds, synthetic method and application thereof, and pharmaceutical composition containing the compounds | |
| WO2023061051A1 (en) | Sulfamide usp8 inhibitor and use thereof | |
| WO2025247412A1 (en) | PHENYLTHIAZOLAMINE PI4KIIIβ/HDAC DUAL-TARGET INHIBITOR, PREPARATION METHOD THEREFOR, PHARMACEUTICAL COMPOSITION THEREOF, AND USE THEREOF |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12736857 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2012207543 Country of ref document: AU Date of ref document: 20120113 Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012736857 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012736857 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13980874 Country of ref document: US |