WO2012131881A1 - ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 - Google Patents
ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 Download PDFInfo
- Publication number
- WO2012131881A1 WO2012131881A1 PCT/JP2011/057694 JP2011057694W WO2012131881A1 WO 2012131881 A1 WO2012131881 A1 WO 2012131881A1 JP 2011057694 W JP2011057694 W JP 2011057694W WO 2012131881 A1 WO2012131881 A1 WO 2012131881A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- particles
- nickel
- active material
- electrode active
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B7/00—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions
- C30B7/14—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions the crystallising materials being formed by chemical reactions in the solution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G45/00—Compounds of manganese
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/40—Complex oxides containing nickel and at least one other metal element
- C01G53/42—Complex oxides containing nickel and at least one other metal element containing alkali metals, e.g. LiNiO2
- C01G53/44—Complex oxides containing nickel and at least one other metal element containing alkali metals, e.g. LiNiO2 containing manganese
- C01G53/50—Complex oxides containing nickel and at least one other metal element containing alkali metals, e.g. LiNiO2 containing manganese of the type (MnO2)n-, e.g. Li(NixMn1-x)O2 or Li(MyNixMn1-x-y)O2
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/80—Compounds containing nickel, with or without oxygen or hydrogen, and containing one or more other elements
- C01G53/82—Compounds containing nickel, with or without oxygen or hydrogen, and containing two or more other elements
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/04—Processes of manufacture in general
- H01M4/0471—Processes of manufacture in general involving thermal treatment, e.g. firing, sintering, backing particulate active material, thermal decomposition, pyrolysis
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/04—Processes of manufacture in general
- H01M4/049—Manufacturing of an active layer by chemical means
- H01M4/0497—Chemical precipitation
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/03—Particle morphology depicted by an image obtained by SEM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/30—Particle morphology extending in three dimensions
- C01P2004/32—Spheres
- C01P2004/34—Spheres hollow
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/51—Particles with a specific particle size distribution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/40—Electric properties
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/362—Composites
- H01M4/366—Composites as layered products
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
Definitions
- the present invention relates to nickel-manganese composite hydroxide particles, a method for producing the same, a positive electrode active material for a non-aqueous electrolyte secondary battery using the nickel-manganese composite hydroxide particles as a raw material, a method for producing the same, and the non-aqueous electrolyte 2
- the present invention relates to a non-aqueous electrolyte secondary battery using a positive electrode active material for a secondary battery as a positive electrode material.
- the lithium ion secondary battery is composed of a negative electrode, a positive electrode, an electrolytic solution and the like, and a material capable of desorbing and inserting lithium is used as an active material of the negative electrode and the positive electrode.
- lithium ion secondary batteries With regard to lithium ion secondary batteries, research and development are being actively carried out at present. Among them, lithium ion secondary batteries using layered or spinel type lithium metal composite oxide as positive electrode material are 4V class The high voltage density is obtained, and the practical use is advanced as a battery having a high energy density.
- a lithium cobalt composite oxide (LiCoO 2 ) which is relatively easy to synthesize, a lithium nickel composite oxide (LiNiO 2 ) using nickel which is cheaper than cobalt, a lithium, and lithium Nickel-cobalt-manganese composite oxide (LiNi 1/3 Co 1/3 Mn 1/3 O 2 ), lithium-manganese composite oxide using manganese (LiMn 2 O 4 ), lithium-nickel-manganese composite oxide (LiNi 0.5 Mn 0.5) Lithium composite oxides such as O 2 ) have been proposed.
- LiNi 0.5 Mn 0.5 O 2 lithium nickel manganese composite oxide
- Lithium nickel manganese complex oxide LiNi 0.5 Mn 0.5 O 2
- Lithium nickel manganese complex oxide LiNi 0.5 Mn 0.5 O 2
- the composition ratio of nickel to manganese is basically 1: at transition metal site: It contains by the ratio of 1 (refer nonpatent literature 1).
- the positive electrode material is required to be constituted by particles having a uniform and appropriate particle diameter.
- a lithium nickel manganese composite oxide as a positive electrode active material so as to be particles having a small particle diameter and a uniform particle diameter.
- lithium-nickel-manganese composite oxides are usually produced from composite hydroxides, composite hydroxides that become raw materials in making lithium-nickel-manganese composite oxides into particles with small particle diameter and uniform particle diameter It is necessary to use small particle size and uniform particle size.
- a composite hydroxide to be a raw material of lithium nickel manganese composite oxide forming the positive electrode material, It is necessary to use a composite hydroxide consisting of particles having a small particle size and a narrow particle size distribution.
- Patent Document 1 shows a composite hydroxide particle substantially having a ratio of manganese: nickel of 1: 1, which is an average particle size.
- the atomic ratio of manganese to nickel is substantially reduced in the presence of a complexing agent in an aqueous solution having a pH value of 9 to 13 while controlling the degree of oxidation of manganese ions within a certain range. It is disclosed that the mixed aqueous solution of manganese salt and nickel salt, which is 1: 1, is reacted with an alkaline solution under appropriate stirring conditions to co-precipitate particles formed.
- the average particle diameter D50 is 3 to 15 ⁇ m and the minimum particle diameter is 0.
- lithium complex oxides having D10 / D90 of 0.30 to 0.70 are disclosed.
- this lithium composite oxide has high filling property, is excellent in charge and discharge capacity characteristics and high output characteristics, and hardly deteriorates even under a large condition of charge and discharge load, this lithium composite oxide is used. For example, it is described that it is possible to obtain a lithium ion non-aqueous electrolyte secondary battery having excellent output characteristics and less deterioration of cycle characteristics.
- the lithium composite oxide disclosed in Patent Document 2 has a minimum particle size of 0.5 ⁇ m or more and a maximum particle size of 50 ⁇ m or less with respect to an average particle size of 3 to 15 ⁇ m, fine particles and Coarse particles are included. And in the particle size distribution defined by the above D10 / D50 and D10 / D90, it can not be said that the range of the particle size distribution is narrow. That is, the lithium composite oxide of Patent Document 2 can not be said to be particles having sufficiently high particle size uniformity, and even if such a lithium composite oxide is adopted, the performance improvement of the positive electrode material can not be expected and sufficient. It is difficult to obtain a lithium ion non-aqueous electrolyte secondary battery having performance.
- Patent Document 3 in a method for producing a positive electrode active material for a non-aqueous electrolyte battery, an aqueous solution containing two or more transition metal salts, or an aqueous solution of two or more different transition metal salts and an alkaline solution are simultaneously treated in a reaction vessel.
- a method has been proposed in which a precursor hydroxide or oxide is obtained by charging and coprecipitating in the presence of a reducing agent or aerating an inert gas.
- Patent Document 3 since the technology of Patent Document 3 recovers the formed crystals while classifying them, it is considered that it is necessary to strictly control the production conditions in order to obtain a product of uniform particle diameter, Industrial scale production is difficult. Moreover, although crystal particles having a large particle diameter can be obtained, it is difficult to obtain particles having a small diameter.
- the reaction area without changing the particle size. That is, by making the particles porous or hollow the particle structure, the surface area contributing to the battery reaction can be increased, and the reaction resistance can be reduced.
- Patent Document 4 describes a positive electrode active material for a non-aqueous electrolyte secondary battery having at least a lithium transition metal complex oxide having a layered structure, wherein the lithium transition metal complex oxide has an outer shell and an outer shell.
- a positive electrode active material for a non-aqueous electrolyte secondary battery is disclosed, which is a lithium transition metal composite oxide comprising hollow particles having a space portion inside the outer shell portion.
- this positive electrode active material for non-aqueous-electrolyte secondary batteries is excellent in battery characteristics, such as cycling characteristics, output characteristics, and thermal stability, and it is also described that it is suitably used for a lithium ion secondary battery.
- the positive electrode active material disclosed in Patent Document 4 is a hollow particle, there is no description regarding the particle diameter although an increase in specific surface area is expected compared to a solid particle. Therefore, although the improvement of the reactivity with the electrolytic solution by the increase of the specific surface area can be expected, the effect on the migration distance of the lithium ion by the micronization is unknown, and the improvement of the output characteristics can not be expected sufficiently. Furthermore, the particle size distribution is considered to be equivalent to that of a conventional positive electrode active material, and therefore, there is a high possibility that the battery capacity is reduced due to the selective deterioration of the fine particles due to the nonuniformity of the applied voltage in the electrode.
- a lithium composite oxide that can sufficiently improve the performance of a lithium ion secondary battery and a composite hydroxide that is a raw material of such a composite oxide have not been developed.
- various methods for producing composite hydroxides have been studied, currently, on the industrial scale, composite water as a raw material of composite oxides that can sufficiently improve the performance of lithium ion secondary batteries
- No method has been developed that can produce oxides. That is, a positive electrode active material having a small particle size, high particle size uniformity, and a large reaction area, for example, having a hollow structure has not been developed, and such a positive electrode active material and its industrial manufacturing method are developed Is required.
- the present invention provides a nickel-manganese composite hydroxide which, when used as a raw material, provides a lithium-nickel-manganese composite oxide having a small particle size and high particle size uniformity and having a high specific surface area by a hollow structure. It aims at providing an object particle.
- Another object of the present invention is to provide an industrial production method of the above-mentioned nickel-manganese composite hydroxide particles and the positive electrode active material.
- the present inventors determined the particle size of nickel-manganese composite hydroxide as a raw material The distribution is controlled, and by forming a structure having a central part consisting of fine primary particles and an outer shell part consisting of primary particles larger than the primary particles outside the central part, the above-mentioned small particle diameter is high in particle diameter uniformity. It was found that a lithium-nickel-manganese composite oxide having a hollow structure was obtained.
- the nickel-manganese composite hydroxide can be obtained by controlling the atmosphere in each step while separating into a nucleation step and a particle growth step by pH control at the time of crystallization.
- the present invention has been completed based on these findings.
- Ni x Mn y Co z M t (OH) 2 + a (x + y + z + t 1, 0.3 ⁇ x) according to crystallization reaction ⁇ 0.7, 0.1 ⁇ y ⁇ 0.55, 0 ⁇ z ⁇ 0.4, 0 ⁇ t ⁇ 0.1, 0 ⁇ a ⁇ 0.5, M is Mg, Ca, Al, Ti, It is a manufacturing method which manufactures nickel manganese compound hydroxide particles denoted by one or more sorts of additional elements chosen from V, Cr, Zr, Nb, Mo, and W), Control of the aqueous solution for nucleation including at least a metal compound containing nickel and a metal compound containing manganese and an ammonium ion supplier so that the pH value becomes 12.0 to 14.0 at a liquid temperature of 25 ° C.
- the aqueous solution for particle growth containing nuclei formed in the nucleation step is controlled so that the pH value at a liquid temperature of 25 ° C. becomes 10.5 to 12.0, and particles are started from the start of the particle growth step
- the oxygen concentration in the oxidizing atmosphere is 10% by volume or more, while the oxygen concentration in the mixed atmosphere is 0.5% by volume or less.
- the particle growth step it is preferable to switch from the air atmosphere to a mixed atmosphere of an inert gas in the range of 0 to 30% of the entire particle growth step time from the start of the particle growth step.
- aqueous solution for particle growth one formed by adjusting the pH of the aqueous solution for nucleation after completion of the nucleation step can be used.
- aqueous solution for particle growth an aqueous solution containing the nucleus formed in the nucleation step added to an aqueous solution different from the aqueous solution for nucleation which formed the nucleus.
- the particle growth step it is preferable to discharge part of the liquid portion of the aqueous solution for particle growth.
- the temperature of the aqueous solution for nucleation and particle growth is preferably maintained at 20 ° C. or higher, and the ammonia concentration of each aqueous solution is in the range of 3 to 25 g / L. It is preferable to keep it inside.
- a mixed aqueous solution containing a nickel-containing metal compound and a manganese-containing metal compound in the nucleation step and the particle growth step is added by adding an aqueous solution in which a salt containing one or more additional elements is dissolved, or simultaneously mixing the aqueous solution in which a salt containing one or more additional elements is dissolved and the mixed aqueous solution. It is preferable to supply the solution into a crystallization tank in which a pre-reaction aqueous solution containing at least a body is stored to obtain the aqueous solution for nucleation.
- the nickel-manganese composite hydroxide obtained in the particle growth step can be coated with the one or more additional elements.
- an aqueous solution containing the one or more additive elements is added to the liquid in which the nickel-manganese composite hydroxide is suspended while being controlled to have a predetermined pH, and the nickel-manganese composite water is added.
- the method of depositing on the oxide surface, the method of spray-drying a slurry in which the nickel-manganese composite hydroxide and the salt containing the one or more additional elements are suspended, or the nickel-manganese composite hydroxide and the one or more kinds There is a method of mixing a salt containing an additive element by a solid phase method.
- an index indicating the spread of the particle size distribution [(d90 ⁇ d10) / average particle size] is not more than 0.55, has a central part consisting of fine primary particles, and the fine primary outside the central part It is characterized in that it has an outer shell made of plate-like primary particles larger than the particles.
- the fine primary particles preferably have an average particle size of 0.01 to 0.3 ⁇ m, and plate-like primary particles larger than the fine primary particles preferably have an average particle size of 0.3 to 3 ⁇ m, and the outer shell portion It is preferable that the thickness of (5) is 5 to 45% in proportion to the particle size of the secondary particles.
- the one or more additive elements be uniformly distributed in the inside of the secondary particles and / or uniformly coat the surface of the secondary particles.
- the nickel-manganese composite hydroxide particles of the present invention are preferably produced by the method for producing composite hydroxide particles according to the present invention described above.
- the method for producing a positive electrode active material according to the present invention has a general formula: Li 1 + u Ni x Mn y Co z M t O 2 ( ⁇ 0.05 ⁇ u ⁇ 0.50, 0.3 ⁇ x ⁇ 0.7, 0.1 ⁇ y ⁇ 0.55, 0 ⁇ z ⁇ 0.4, 0 ⁇ t ⁇ 0.1, M is selected from Mg, Ca, Al, Ti, V, Cr, Zr, Nb, Mo, W
- a positive electrode active material comprising a lithium-nickel-manganese composite oxide having a hexagonal crystal structure having a layered structure, which is represented by Heat-treating the nickel-manganese composite hydroxide particles at a temperature of 105 to 750 ° C.
- the method further comprises a mixing step of forming a lithium mixture, and a baking step of baking the mixture formed in the mixing step at a temperature of 800 to 980 ° C. in an oxidizing atmosphere.
- the lithium mixture is preferably adjusted so that the ratio of the sum of the number of atoms of metals other than lithium contained in the lithium mixture and the number of atoms of lithium is 1: 0.95 to 1.5.
- the firing step it is preferable to perform calcination at a temperature of 350 to 800 ° C. in advance before firing.
- the oxidizing atmosphere in the firing step is preferably an atmosphere containing 18 to 100% by volume of oxygen.
- the positive electrode active material of the present invention have the general formula: Li 1 + u Ni x Mn y Co z M t O 2 (-0.05 ⁇ u ⁇ 0.50,0.3 ⁇ x ⁇ 0.7,0.1 Y ⁇ 0.55, 0 ⁇ z ⁇ 0.4, 0 ⁇ t ⁇ 0.1, M is selected from Mg, Ca, Al, Ti, V, Cr, Zr, Nb, Mo, W
- a positive electrode active material comprising a lithium-nickel-manganese composite oxide represented by a hexagonal lithium-containing composite oxide having a layered structure, which is represented by An index showing the spread of particle size distribution [(d90 ⁇ d10) / average particle size] is not more than 0.60, and has a hollow structure composed of the hollow part inside the particle and the outer shell part outside it It is characterized by
- the thickness of the outer shell portion is preferably 5 to 45% as a ratio to the particle diameter of the lithium-nickel-manganese composite oxide particles.
- the positive electrode active material for a non-aqueous electrolyte secondary battery of the present invention is preferably produced by the above method for producing a positive electrode active material according to the present invention.
- the non-aqueous electrolyte secondary battery of the present invention is characterized in that the positive electrode is formed of the above-mentioned positive electrode active material for non-aqueous electrolyte secondary battery according to the present invention.
- nickel-manganese composite hydroxide particles are obtained in which the lithium-nickel-manganese composite oxide obtained when used as a raw material has a small particle size and high particle size uniformity and a high specific surface area due to the hollow structure. Further, a positive electrode active material comprising the lithium-nickel-manganese composite oxide has high capacity, good cycle characteristics when used in a non-aqueous secondary battery, and enables high output.
- the non-aqueous secondary battery configured with the positive electrode including the battery has excellent battery characteristics.
- the methods for producing the nickel-manganese composite hydroxide particles and the positive electrode active material provided by the present invention are both easy and suitable for large-scale production, and their industrial value is extremely large.
- FIG. 1 is a schematic flow chart of the process for producing the nickel-manganese composite hydroxide of the present invention.
- FIG. 2 is a schematic flow chart of another process for producing the nickel-manganese composite hydroxide of the present invention.
- FIG. 3 is a schematic flow chart of a process for producing a lithium-nickel-manganese composite oxide as a positive electrode active material from the nickel-manganese composite hydroxide of the present invention.
- FIG. 4 is a schematic flow chart from the production of the nickel manganese composite hydroxide of the present invention to the production of a non-aqueous electrolyte secondary battery.
- FIG. 1 is a schematic flow chart of the process for producing the nickel-manganese composite hydroxide of the present invention.
- FIG. 2 is a schematic flow chart of another process for producing the nickel-manganese composite hydroxide of the present invention.
- FIG. 3 is a schematic flow chart of a process for producing a lithium-nic
- FIG. 5 is a SEM photograph (1,000 ⁇ magnification) of the nickel-manganese composite hydroxide of the present invention.
- FIG. 6 is a cross-sectional SEM photograph (observation magnification of 10,000 times) of the nickel-manganese composite hydroxide of the present invention.
- FIG. 7 is a SEM photograph (1,000 ⁇ magnification) of a lithium nickel manganese composite oxide which is a positive electrode active material of the present invention.
- FIG. 8 is a cross-sectional SEM photograph (observation magnification: 10,000 times) of the lithium nickel manganese composite oxide which is a positive electrode active material of the present invention.
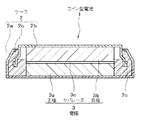
- FIG. 9 is a schematic cross-sectional view of a coin-type battery used for battery evaluation.
- FIG. 10 is a schematic explanatory view of a measurement example of impedance evaluation and an equivalent circuit used for analysis.
- the present invention provides (1) nickel-manganese composite hydroxide particles as a raw material of a positive electrode active material for a non-aqueous electrolyte secondary battery and a method for producing the same, (2) non-aqueous electrolyte using the nickel-manganese composite hydroxide particles
- the present invention relates to a positive electrode active material for a secondary battery and a method for producing the same, and (3) a non-aqueous electrolyte secondary battery using the positive electrode active material for a non-aqueous electrolyte secondary battery as a positive electrode.
- the influence of the positive electrode active material for non-aqueous electrolyte secondary battery adopted for the positive electrode is large.
- the particle size, the particle size distribution, and the specific surface area are important factors, and have a desired particle structure,
- the positive electrode active material adjusted to the desired particle size and particle size distribution is preferable.
- the said composite hydroxide particle is especially suitable as a raw material of the positive electrode active material which has a hollow structure of this invention, it demonstrates on the premise of using for the raw material of the positive electrode active material of this invention hereafter .
- the composite hydroxide particles of the present invention are substantially spherical particles as illustrated in FIG. Specifically, as illustrated in FIG. 6, a plurality of plate-like primary particles are aggregated to form substantially spherical secondary particles, and more specifically, the inside of the particles is made of fine primary particles. And a shell having a plate-like primary particle larger than the fine primary particle outside the central portion.
- the central portion has a structure having a large number of gaps in which fine primary particles are continuous, the shrinkage due to sintering is more likely to occur in the firing step as compared with the outer shell portion made of plate-like primary particles having a larger thickness. It originates from low temperature. For this reason, sintering proceeds from a low temperature at the time of firing, and shrinks from the center of the particle to the outer shell side where the progress of sintering is slow, and a space is generated in the center. In addition, since the central portion is considered to have a low density and the contraction rate is also large, the central portion is a space having a sufficient size. Thus, the positive electrode active material obtained after firing has a hollow structure.
- the plate-like primary particles are aggregated in random directions to form secondary particles.
- the aggregation of the plate-like primary particles in a random direction causes voids to be substantially uniform among the primary particles, and when mixed and fired with the lithium compound, the molten lithium compound spreads into the secondary particles, lithium Diffusion is sufficiently performed.
- the average particle diameter of the fine primary particles is preferably 0.01 to 0.3 ⁇ m, and more preferably 0.1 to 0.3 ⁇ m, in order to form a space during the baking.
- the plate-like primary particles larger than the fine primary particles preferably have an average particle diameter of 0.3 to 3 ⁇ m, more preferably 0.4 to 1.5 ⁇ m, and 0.4 to 1 It is particularly preferred that the thickness is .0 ⁇ m. If the average particle size of the fine primary particles is less than 0.01 ⁇ m, a core of a sufficient size may not be formed in the composite hydroxide particles, and if it exceeds 0.3 ⁇ m, the temperature is lowered for starting the sintering. And shrinkage may not be sufficient, and a sufficiently large space may not be obtained after firing.
- the average particle diameter of the plate-like primary particles of the outer shell portion is less than 0.3 ⁇ m, sintering at the time of firing may be cooled to a low temperature, and a sufficiently large space may not be obtained after firing. If it exceeds 3 ⁇ m, in order to obtain sufficient crystallinity of the positive electrode active material to be obtained, it is necessary to raise the firing temperature, and sintering occurs between the secondary particles to obtain the positive electrode active material obtained.
- the particle size may exceed the above range.
- the fine primary particles are preferably plate-like and / or needle-like.
- the density of the central portion becomes sufficiently low, and a large shrinkage occurs by firing to produce a sufficient amount of space.
- the thickness of the outer shell is preferably 5 to 45%, more preferably 7 to 35%, as a ratio to the particle diameter of the secondary particles.
- the positive electrode active material particles obtained using the composite hydroxide as a raw material have a hollow structure, and the ratio of the thickness of the outer shell portion to the particle diameter is substantially maintained by the ratio of the composite hydroxide secondary particles Be done. Therefore, by setting the ratio of the thickness of the outer shell portion to the secondary particle diameter in the above range, a sufficient hollow portion can be formed in the lithium-nickel-manganese composite oxide particles.
- the thickness of the outer shell portion is too thin at less than 5% as a ratio to the particle diameter of the secondary particles, the shrinkage of the composite hydroxide particles becomes large in the firing step during the production of the positive electrode active material, and Sintering may occur between secondary particles of the lithium-nickel-manganese composite oxide to deteriorate the particle size distribution of the positive electrode active material. On the other hand, if it exceeds 45%, problems such as the formation of a sufficiently large center portion may occur.
- the particle diameter of the said fine primary particle and plate-like primary particle and the ratio of the thickness of the outer shell part with respect to the said secondary particle diameter observe the cross section of nickel manganese compound hydroxide particle using a scanning electron microscope It can measure by doing.
- a plurality of nickel-manganese composite hydroxide particles are embedded in a resin or the like so that cross-sectional observation of the particles can be performed by cross-section polishing or the like.
- the particle diameter of the fine primary particles and plate-like primary particles is determined by measuring the maximum diameter of preferably 10 or more of the primary particle cross sections in the secondary particles as the particle diameter and calculating the average value. Can.
- the ratio of the thickness of the outer shell portion to the secondary particle diameter can be determined as follows. From secondary particles in the above-mentioned resin, select particles in which the cross section of the particle center can be observed, and the distance between the outer periphery of the outer shell and the inner periphery of the central part The distance between the two shortest points is measured to determine the average thickness of the outer shell of each particle. The above-mentioned ratio of the thickness of the outer shell of each particle is determined by dividing the average thickness by setting the secondary particle diameter to the distance between any two points at which the distance is maximum on the outer periphery of the secondary particle. Furthermore, the ratio of the thickness of the outer shell portion to the secondary particle diameter in the nickel-manganese composite hydroxide particles can be determined by averaging the ratio of each particle determined for 10 or more particles.
- the composite hydroxide particles of the present invention are adjusted to have an average particle size of 3 to 7 ⁇ m, preferably 3.5 to 6.5 ⁇ m.
- the positive electrode active material obtained using the composite hydroxide particles of the present invention as a raw material can be adjusted to a predetermined average particle size (2 to 8 ⁇ m).
- the particle diameter of the composite hydroxide particles correlates with the particle diameter of the obtained positive electrode active material, and thus affects the characteristics of a battery using this positive electrode active material as a positive electrode material.
- the average particle size of the composite hydroxide particles is less than 3 ⁇ m, the average particle size of the obtained positive electrode active material is also reduced, the packing density of the positive electrode is reduced, and the battery capacity per volume is reduced. descend. Conversely, when the average particle diameter of the composite hydroxide particles exceeds 7 ⁇ m, the specific surface area of the positive electrode active material decreases, and the interface with the electrolytic solution decreases, so that the resistance of the positive electrode increases. Output characteristics are degraded.
- the composite hydroxide particles of the present invention are adjusted so that [(d90 ⁇ d10) / average particle size], which is an index indicating the spread of the particle size distribution, is 0.55 or less, preferably 0.52 or less. ing.
- the particle size distribution of the positive electrode active material is strongly influenced by the composite hydroxide particles that are the raw materials, when fine particles or coarse particles are mixed in the composite hydroxide particles, similar particles also exist in the positive electrode active material. You will come to That is, when [(d90 ⁇ d10) / average particle size] exceeds 0.55 and the particle size distribution is wide, fine particles or coarse particles are also present in the positive electrode active material.
- the composite hydroxide particle of the present invention if [(d90 ⁇ d10) / average particle size] is adjusted to 0.55 or less, a positive electrode active material obtained using this as a raw material is also obtained.
- the range of particle size distribution is narrow, and the particle size can be made uniform. That is, with respect to the particle size distribution of the positive electrode active material, [(d90 ⁇ d10) / average particle diameter] can be made to be 0.60 or less.
- d10 accumulates the number of particles in each particle diameter from the side of smaller particle diameter, and the accumulated volume is the total volume of all particles Means a particle size of 10% of the Also, d90 means a particle size in which the number of particles is similarly accumulated, and the accumulated volume is 90% of the total volume of all particles.
- the method for determining the average particle diameter and d90 and d10 is not particularly limited.
- the average particle diameter and d90 and d10 can be determined from a volume integration value measured by a laser light diffraction scattering particle size analyzer.
- d50 is used as the average particle diameter
- a particle diameter at which the cumulative volume is 50% of the total particle volume may be used as in d90.
- the composite hydroxide particles of the present invention are adjusted such that the composition is represented by the following general formula. If a lithium-nickel-manganese composite oxide is produced using a nickel-manganese composite hydroxide having such a composition as a raw material, measurement is performed when an electrode having the lithium-nickel-manganese composite oxide as a positive electrode active material is used in a battery. While being able to make the value of the positive electrode resistance carried out low, battery performance can be made favorable.
- M is one or more additive elements selected from Mg, Ca, Al, Ti, V, Cr, Zr, Nb, Mo, W)
- the composition ratio (Ni: Mn: Co: M) of the composite hydroxide particles is maintained also in the obtained positive electrode active material. Therefore, the composition ratio of the composite hydroxide particles of the present invention is adjusted to be the same as the composition ratio required for the positive electrode active material to be obtained.
- the method of producing composite hydroxide particles of the present invention is a method of producing nickel-manganese composite hydroxide particles by a crystallization reaction, which comprises: The process comprises a nucleation step for producing, and b) a particle growth step for growing the nuclei produced in the nucleation step.
- the particle size distribution of the obtained composite hydroxide particles becomes wide.
- the time in which the nucleation reaction mainly occurs (nucleation step) and the time in which the particle growth reaction mainly occurs (particle growth step) are clearly separated. This is characterized in that a narrow particle size distribution is achieved in the composite hydroxide particles obtained.
- the particle structure of the composite hydroxide particles obtained is composed of a central part consisting of primary fine particles and an outer shell part consisting of primary particles larger than the central part There is a feature in the point to be.
- FIGS. 1 and 2 corresponds to a nucleation step, and (B) corresponds to a particle growth step.
- the proportion of the metal compound to be dissolved in water is adjusted so that the composition ratio of each metal in the mixed aqueous solution is the same as the composition ratio of each metal in the composite hydroxide particles of the present invention, Make a mixed aqueous solution.
- an alkaline aqueous solution such as an aqueous sodium hydroxide solution, an aqueous ammonia solution containing an ammonium ion supplier, and water are supplied to the reaction vessel and mixed to form an aqueous solution.
- the pH value of this aqueous solution (hereinafter referred to as “pre-reaction aqueous solution”) is adjusted to 12.2 to 14.0, preferably 12.3 to 14.0, based on a liquid temperature of 25 ° C., by adjusting the supply amount of the alkaline aqueous solution It is adjusted to be in the range of 13.5, more preferably 12.5 to 13.0.
- the concentration of ammonium ion in the aqueous solution before reaction is adjusted to 3 to 25 g / L, preferably 5 to 20 g / L, by adjusting the supply amount of the aqueous ammonia solution.
- the temperature of the aqueous solution before reaction is also adjusted to preferably 20 ° C. or more, more preferably 20 to 60 ° C.
- the pH value of the aqueous solution in the reaction tank and the concentration of ammonium ions can be measured by a general pH meter and an ion meter, respectively.
- the mixed aqueous solution is supplied into the reaction vessel while stirring the pre-reaction aqueous solution.
- an aqueous solution for nucleation which is a reaction aqueous solution in the nucleation step, is formed in the reaction vessel in which the aqueous solution before reaction and the mixed aqueous solution are mixed, and fine nuclei of the complex hydroxide are formed in the aqueous solution for nucleation. Will be generated.
- the pH value of the aqueous solution for nucleation is in the above-mentioned range, formation of nuclei occurs preferentially with little growth of generated nuclei.
- the aqueous solution for nucleation is supplied with the aqueous alkaline solution and the aqueous ammonia solution together with the mixed aqueous solution.
- the pH value of the aqueous solution for nucleation is controlled so as to maintain the range of 12.0-14.0 and the concentration of ammonium ion in the range of 3-25 g / L on the basis of a liquid temperature of 25 ° C.
- nucleation step is ended. Whether or not a predetermined amount of nuclei has been generated is judged by the amount of metal salt added to the aqueous solution for nucleation.
- the pH value of the aqueous solution for nucleation is adjusted to 10.5 to 12.0, preferably 11.0 to 12.0, based on the liquid temperature 25 ° C.
- An aqueous solution for particle growth which is a reaction aqueous solution in the growth step, is obtained.
- the control of the pH at the time of adjustment is performed by adjusting the supply amount of the aqueous alkali solution.
- the growth reaction of nuclei takes precedence over the reaction of generating nuclei. Therefore, in the particle growth step, new nuclei are added to the aqueous solution for particle growth. The nuclei grow (particle growth) with almost no formation, and composite hydroxide particles having a predetermined particle size are formed.
- the aqueous alkaline solution and the aqueous ammonia solution are also supplied to the aqueous solution for particle growth together with the mixed aqueous solution.
- the pH value of the aqueous solution for particle growth is controlled so as to maintain the range of 10.5 to 12.0 and the concentration of ammonium ion within the range of 3 to 25 g / L on the basis of a liquid temperature of 25 ° C.
- the particle diameter of the composite hydroxide particles can be determined in each process by obtaining the relationship between the amount of metal salt added to the reaction aqueous solution in each of the nucleation step and the particle growth step and the obtained particles by a preliminary test. It can be easily determined from the amount of metal salt added.
- nucleation occurs preferentially in the nucleation step, and growth of nuclei hardly occurs, and conversely, only nucleus growth occurs in the particle growth step, Almost no new nuclei are generated. Therefore, in the nucleation step, it is possible to form homogeneous nuclei with a narrow range of particle size distribution, and in the particle growth step, it is possible to grow nuclei homogeneously. Therefore, in the method for producing composite hydroxide particles, it is possible to obtain homogeneous nickel-manganese composite hydroxide particles with a narrow range of particle size distribution.
- metal ions are crystallized as nuclei or composite hydroxide particles in both steps, so the ratio of the liquid component to the metal component in each reaction aqueous solution increases.
- the concentration of the mixed aqueous solution to be supplied apparently decreases, and the composite hydroxide particles may not grow sufficiently, particularly in the particle growth step.
- the aqueous solution for particle growth out of the reaction tank in the middle of the particle growth step after completion of the nucleation step. Specifically, the supply and stirring of the mixed aqueous solution, the aqueous alkaline solution and the aqueous ammonia solution to the aqueous solution for particle growth are stopped to precipitate nuclei and composite hydroxide particles, and the supernatant of the aqueous solution for particle growth is discharged. Thereby, the relative concentration of the mixed aqueous solution in the aqueous solution for particle growth can be increased.
- the composite hydroxide particles can be grown in a state where the relative concentration of the mixed aqueous solution is high, the particle size distribution of the composite hydroxide particles can be further narrowed, and the secondary of the composite hydroxide particles can be obtained.
- the overall density of the particles can also be increased.
- the pH of the aqueous solution for nucleation is adjusted to form an aqueous solution for particle growth after the nucleation step is completed, and the particle growth step is performed subsequently to the nucleation step,
- the transition to the particle growth process can be performed quickly.
- the transition from the nucleation step to the particle growth step can be transferred only by adjusting the pH of the reaction aqueous solution, and the pH adjustment can be easily performed by temporarily stopping the supply of the aqueous alkali solution.
- the pH of the reaction aqueous solution can also be adjusted by adding sulfuric acid to the reaction aqueous solution in the case of an inorganic acid of the same type as the acid constituting the metal compound, for example, a sulfate.
- a component adjustment aqueous solution adjusted to a pH suitable for the particle growth step and an ammonium ion concentration is formed separately from the aqueous solution for nucleation, and An aqueous solution containing a nucleus generated by performing a nucleation step in another reaction tank (an aqueous solution for nucleation, preferably one obtained by removing a part of the liquid component from the aqueous solution for nucleation) to obtain a reaction aqueous solution,
- the particle growth step may be performed by using this reaction aqueous solution as an aqueous solution for particle growth.
- the state of the reaction aqueous solution in each step can be set as the optimum condition for each step.
- the pH of the aqueous solution for particle growth can be made the optimum condition from the start of the particle growth step.
- the nickel-manganese composite hydroxide particles formed in the particle growth step can be made more homogeneous with a narrower range of particle size distribution.
- reaction atmosphere Next, the control of the reaction atmosphere in each step, the substances and solutions used in each step, and the reaction conditions will be described in detail.
- the particle structure of the nickel-manganese composite hydroxide particles of the present invention is formed by controlling the atmosphere in the reaction vessel in the nucleation step and the particle growth step. Therefore, the atmosphere control in each process of the manufacturing method has an important meaning.
- the atmosphere in the reaction tank during the crystallization reaction controls the growth of primary particles forming nickel-manganese composite hydroxide particles, and in the oxidizing atmosphere, low density particles formed by fine primary particles and having many voids In the case of a weakly oxidizing atmosphere to a non-oxidizing atmosphere, primary particles are large and dense and dense particles are formed.
- the primary particles in the central portion are in the form of fine plates and / or needles, and the primary particles in the outer shell portion are in the form of plates.
- the primary particles of the nickel-manganese composite hydroxide may have a shape such as a rectangular parallelepiped, an ellipse, or a spheroid.
- the oxidizing atmosphere for forming the central portion in the present invention is defined as an atmosphere in which the oxygen concentration in the reaction vessel inner space exceeds 1% by volume.
- An oxidizing atmosphere having an oxygen concentration of more than 2% by volume is preferable, an oxidizing atmosphere having an oxygen concentration of more than 10% by volume is more preferable, and an atmospheric atmosphere (oxygen concentration: 21% by volume) is easy to control.
- the atmosphere By setting the atmosphere to have an oxygen concentration of more than 1% by volume, the average particle size of the primary particles can be set to 0.01 to 0.3 ⁇ m.
- the oxygen concentration is 1% by volume or less, the average particle size of the primary particles in the central part may exceed 0.3 ⁇ m.
- the upper limit of the oxygen concentration is not particularly limited, but when it exceeds 30% by volume, the average particle diameter of the primary particles may be less than 0.01 ⁇ m, which is not preferable.
- the atmosphere in the weakly oxidizing to non-oxidizing range for forming the outer shell portion in the present invention is defined as an atmosphere in which the oxygen concentration in the space in the reaction tank is 1% by volume or less.
- the mixed atmosphere of oxygen and inert gas is controlled so that the oxygen concentration is 0.5 volume% or less, more preferably 0.2 volume% or less.
- the switching of the atmosphere in the particle growth step is carried out in the center of the nickel-manganese composite hydroxide particles so that in the positive electrode active material finally obtained, fine particles are generated to obtain a hollow portion that does not deteriorate cycle characteristics.
- the timing is determined in consideration of the size of. For example, with respect to the whole particle growth process time, it is performed within a time range of 0 to 40%, preferably 0 to 30%, more preferably 0 to 25% from the start of the particle growth process. When the switching is performed at a time exceeding 40% with respect to the whole particle growth process time, the formed center becomes large, and the thickness of the outer shell to the particle diameter of the secondary particle becomes too thin. On the other hand, if the above switching is performed before the start of the particle growth step, that is, during the nucleation step, the central portion becomes too small or secondary particles having the above structure are not formed.
- the pH value of the reaction aqueous solution in the nucleation step, it is necessary to control the pH value of the reaction aqueous solution to be in the range of 12.0 to 14.0, preferably 12.3 to 13.5, based on the liquid temperature 25 ° C. There is. If the pH value is more than 14.0, the generated nuclei become too fine, and there is a problem that the reaction aqueous solution gels. In addition, if the pH value is less than 12.0, since the growth reaction of the nucleus occurs together with the nucleation, the range of the particle size distribution of the formed nucleus becomes wide and becomes inhomogeneous.
- the nucleation step by controlling the pH value of the reaction aqueous solution to the above-mentioned range, it is possible to suppress the growth of nuclei and cause almost only nucleation, and the nuclei formed are also homogeneous and the range of particle size distribution Can be narrow.
- the pH value of the reaction aqueous solution it is necessary to control the pH value of the reaction aqueous solution to be in the range of 10.5 to 12.0, preferably 11.0 to 12.0 on the basis of a liquid temperature of 25 ° C. If the pH value is more than 12.0, the number of newly generated nuclei is large and fine secondary particles are formed, so that hydroxide particles having a good particle size distribution can not be obtained. In addition, when the pH value is less than 10.5, the solubility by ammonia ion is high, and metal ions remaining in the solution without precipitation are increased, so that the production efficiency is deteriorated.
- the nickel-manganese composite hydroxide particles can be made homogeneous and have a narrow range of particle size distribution.
- the fluctuation range of pH is preferably within 0.2 above and below the set value.
- nucleation and particle growth may not be constant, and uniform manganese composite hydroxide particles having a narrow range of particle size distribution may not be obtained.
- the pH value is 12, since it is the boundary condition of nucleation and nuclear growth, depending on the presence or absence of the nucleus present in the reaction aqueous solution, it can be set as either the nucleation step or the particle growth step. .
- the growth of the nucleus is preferred because a large amount of nuclei exist in the reaction aqueous solution.
- the hydroxide particles having a narrow particle size distribution and a relatively large particle size can be obtained.
- nucleation occurs preferentially because the growing nucleus does not exist, and the pH value of the particle growth step is 12 By making the size smaller, the generated nuclei grow to obtain good hydroxide particles.
- the pH value of the particle growth step may be controlled to a value lower than the pH value of the nucleation step, and in order to clearly separate nucleation and particle growth, the pH value of the particle growth step is
- the pH value is preferably 0.5 or more lower than the pH value of the production step, and more preferably 1.0 or more lower.
- the amount of nuclei generated in the nucleation step is not particularly limited, but in order to obtain composite hydroxide particles having a good particle size distribution, the total amount, ie, to obtain composite hydroxide particles It is preferable to be 0.1% to 2% of the total metal salt to be supplied, and it is more preferable to be 1.5% or less.
- the particle diameter of the composite hydroxide particles can be controlled by the time of the particle growth step, so if the particle growth step is continued until the desired particle diameter is grown, composite hydroxide particles having the desired particle diameter are obtained. be able to.
- the particle diameter of the composite hydroxide particles can be controlled not only by the particle growth step but also by the pH value of the nucleation step and the amount of raw material added for nucleation.
- the amount of raw material to be introduced is increased and the number of nuclei to be generated is increased. Thereby, even when the particle growth step is performed under the same conditions, the particle diameter of the composite hydroxide particles can be reduced.
- the particle diameter of the obtained composite hydroxide particles can be increased.
- the conditions for the metal compound, ammonia concentration in the reaction solution, reaction temperature, etc. will be described below, but the difference between the nucleation step and the particle growth step is only by controlling the pH of the reaction solution and the atmosphere in the reaction tank.
- the conditions such as the metal compound, the ammonia concentration in the reaction solution, the reaction temperature and the like are substantially the same in both steps.
- Metal compounds As the metal compound, a compound containing a target metal is used.
- the compound to be used is preferably a water-soluble compound, and examples thereof include nitrates, sulfates and hydrochlorides.
- nickel sulfate, manganese sulfate and cobalt sulfate are preferably used.
- the additive element (one or more elements selected from Mg, Ca, Al, Ti, V, Cr, Zr, Nb, Mo, W) is preferably a water-soluble compound, for example, titanium sulfate, peroxo Ammonium titanate, potassium potassium oxalate, vanadium sulfate, ammonium vanadate, chromium sulfate, potassium chromate, zirconium sulfate, zirconium nitrate, niobium nitrate, niobium molybdate, ammonium tungstate, sodium tungstate, ammonium tungstate, etc. can be used. .
- an additive containing the additive element may be added to the mixed aqueous solution, and the additive element is uniformly dispersed in the composite hydroxide particle. It can be co-precipitated in a dispersed state.
- the composite hydroxide particles are slurried with an aqueous solution containing the additive element, and controlled to a predetermined pH
- an aqueous solution containing the one or more additive elements By adding an aqueous solution containing the one or more additive elements and depositing the additive element on the surface of the composite hydroxide particles by a crystallization reaction, the surface can be uniformly coated with the additive element.
- an alkoxide solution of the additive element may be used instead of the aqueous solution containing the additive element.
- the surface of the composite hydroxide particles can also be coated with the additional element by spraying an aqueous solution or a slurry containing the additional element onto the composite hydroxide particles to dry them. Further, the slurry in which the composite hydroxide particles and the salt containing the one or more additional elements are suspended is spray-dried, or the composite hydroxide and the salt containing the one or more additional elements are mixed by a solid phase method And the like.
- the atomic ratio of the metal ions of the composite hydroxide particles obtained is reduced by reducing the atomic ratio of the additive element ions present in the mixed aqueous solution by the coating amount. Can be matched.
- the step of coating the surface of the particles with the additive element may be performed on the particles after the composite hydroxide particles are heated.
- the concentration of the mixed aqueous solution is preferably 1 to 2.6 mol / L, preferably 1.5 to 2.2 mol / L in total of the metal compounds. If the concentration of the mixed aqueous solution is less than 1 mol / L, the amount of the crystallized material per reaction vessel decreases, which is not preferable because the productivity decreases.
- the metal compound may not necessarily be supplied to the reaction tank as a mixed aqueous solution.
- the total concentration of all metal compound aqueous solutions is in the above range and
- an aqueous metal compound solution may be separately prepared and simultaneously supplied as an aqueous solution of individual metal compounds at a predetermined ratio into the reaction vessel.
- the amount of the mixed aqueous solution or the aqueous solution of individual metal compounds supplied to the reaction vessel is such that the concentration of the crystallized material at the end of the crystallization reaction is about 30 to 200 g / L, preferably 80 to 150 g / L. It is desirable to When the concentration of the crystallized material is less than 30 g / L, the aggregation of the primary particles may be insufficient, and when it exceeds 200 g / L, the diffusion of the mixed aqueous solution to be added in the reaction tank is sufficient. However, there is a possibility that the grain growth may be biased.
- ammonia concentration The ammonia concentration in the reaction aqueous solution is maintained at a constant value preferably in the range of 3 to 25 g / L, preferably 5 to 20 g / L, in order not to cause the following problems.
- Ammonia acts as a complexing agent, so if the ammonia concentration is less than 3 g / L, the solubility of metal ions can not be kept constant, and plate-like hydroxide primary particles with a uniform shape and particle size Since the gel-like nuclei are easily formed, the particle size distribution is also easily spread.
- the concentration of ammonia fluctuates, the solubility of metal ions fluctuates, and uniform hydroxide particles are not formed, so it is preferable to keep the value constant.
- ammonium ion supplier is not particularly limited, and, for example, ammonia, ammonium sulfate, ammonium chloride, ammonium carbonate, ammonium fluoride and the like can be used.
- the temperature of the reaction solution is preferably set at 20 ° C. or higher, particularly preferably 20 to 60 ° C. If the temperature of the reaction solution is less than 20 ° C., since the solubility is low, nucleation is likely to occur and control becomes difficult. On the other hand, if the temperature exceeds 60 ° C., volatilization of ammonia is promoted, and therefore an excess ammonium ion donor must be added to maintain a predetermined ammonia concentration, resulting in high cost.
- Alkaline solution It does not specifically limit about the alkaline aqueous solution which adjusts pH in reaction aqueous solution,
- alkali metal hydroxide aqueous solution such as sodium hydroxide and potassium hydroxide, can be used.
- alkali metal hydroxide although it may be directly supplied into the reaction aqueous solution, it is preferable to add it as an aqueous solution to the reaction aqueous solution in the reaction tank because of easy pH control of the reaction aqueous solution in the reaction tank .
- the method of adding the alkaline aqueous solution to the reaction tank is not particularly limited, and the pH value of the reaction aqueous solution is predetermined by a pump such as a metering pump capable of controlling the flow rate while sufficiently agitating the reaction aqueous solution. So as to be kept in the range of
- production equipment In the method for producing composite hydroxide particles of the present invention, an apparatus of a type in which the product is not recovered until the reaction is completed is used.
- a commonly used batch reaction vessel equipped with a stirrer When such an apparatus is employed, there is no problem that growing particles are recovered simultaneously with the overflow liquid, as in a continuous crystallizer which recovers a product by a general overflow, so the particle size distribution is narrow and the particle size is narrow. It is possible to obtain uniform particles.
- an atmosphere-controllable device such as a closed type device is used.
- the composite hydroxide particles obtained can be made to have the above-described structure, and nucleation reaction and particle growth reaction can be promoted almost uniformly, so that the particle size distribution is excellent. Particles can be obtained, that is, particles with a narrow range of particle size distribution.
- Ti vanadium manganese composite oxide particles represented by at least one additive element selected from Ti, V, Cr, Zr, Nb, Mo, and W
- the positive electrode active material of the present invention is a lithium-nickel-manganese composite oxide particle, and the composition thereof is adjusted to be represented by the following general formula.
- u indicating an excess amount of lithium is in the range of -0.05 to 0.50.
- the excess amount u of lithium is less than ⁇ 0.05, the reaction resistance of the positive electrode in the non-aqueous electrolyte secondary battery using the obtained positive electrode active material is increased, and the output of the battery is lowered.
- the excess amount u of lithium exceeds 0.50, the initial discharge capacity in the case of using the above-mentioned positive electrode active material for the positive electrode of the battery decreases, and the reaction resistance of the positive electrode also increases.
- the excess amount u of lithium is preferably 0.10 or more, and preferably 0.35 or less.
- the positive electrode active material of the present invention is adjusted so that the lithium nickel manganese composite oxide particles contain an additional element.
- the above-described additive element it is possible to improve the durability and output characteristics of a battery using this as a positive electrode active material.
- the above effect can be obtained in the whole particle, and the above effect can be obtained and the reduction of the capacity can be suppressed by adding a small amount.
- the additive element M is adjusted to be in the above range at the above atomic ratio y.
- the positive electrode active material of the present invention has an average particle diameter of 2 to 8 ⁇ m.
- the average particle size is less than 2 ⁇ m, the packing density of the particles decreases when the positive electrode is formed, and the battery capacity per volume of the positive electrode decreases.
- the average particle size exceeds 8 ⁇ m, the specific surface area of the positive electrode active material is reduced, and the interface with the battery electrolyte is decreased, whereby the resistance of the positive electrode is increased and the output characteristics of the battery are reduced.
- the positive electrode active material of the present invention is adjusted to have an average particle diameter of 2 to 8 ⁇ m, preferably 3 to 8 ⁇ m, more preferably 3 to 6.5 ⁇ m, a battery using this positive electrode active material for the positive electrode
- the battery capacity per volume can be increased, and excellent battery characteristics such as high safety and high output can be obtained.
- the positive electrode active material of the present invention is an index indicating the spread of particle size distribution [(d90 ⁇ d10) / average particle size] is 0.60 or less, preferably 0.55 or less It is composed of secondary particles of lithium-nickel-manganese composite oxide having a very high uniformity.
- the particle size distribution of the positive electrode active material to 0.60 or less in the index [(d90 ⁇ d10) / average particle diameter]
- the ratio of fine particles and coarse particles can be reduced.
- the battery used for the positive electrode is excellent in safety, and has good cycle characteristics and battery output.
- the said average particle diameter and d90, d10 are the same as that of what is used for the composite hydroxide particle mentioned above, and it can measure similarly.
- the positive electrode active material of the present invention is characterized in that it has a hollow structure constituted of a hollow portion inside the secondary particle and an outer shell portion outside thereof as illustrated in FIG.
- a hollow structure With such a hollow structure, the reaction surface area can be increased, and the electrolyte penetrates from the grain boundaries or gaps between primary particles in the outer shell portion, and the primary particle surface on the hollow side of the particles inside Since lithium insertion and extraction are also performed at the reaction interface in the above, the movement of Li ions and electrons is not hindered, and the output characteristics can be enhanced.
- the thickness of the outer shell portion is preferably 5 to 45%, and more preferably 8 to 38%, as a ratio to the particle diameter of the lithium-nickel-manganese composite oxide particles.
- the absolute value is more preferably in the range of 0.5 to 2.5 ⁇ m, and particularly preferably in the range of 0.4 to 2.0 ⁇ m.
- the ratio of the thickness of the outer shell portion exceeds 45%, the amount of the electrolytic solution decreases from the above grain boundaries or voids which allow the electrolytic solution to penetrate into the hollow portion inside the particles, and the surface area contributing to the battery reaction decreases. Therefore, the positive electrode resistance is increased, and the output characteristics are degraded.
- the ratio of the thickness of the outer shell portion to the particle diameter of the lithium-nickel-manganese composite oxide can be determined in the same manner as the above-mentioned composite hydroxide particles.
- the positive electrode active material when used for the positive electrode of a 2032 type coin battery, it is 200 mAh / g or more when cobalt is not added, and cobalt is added by about 30% of all metal elements other than lithium in atomic ratio.
- a high initial discharge capacity of 150 mAh / g or more, a low positive electrode resistance and a high cycle capacity retention rate can be obtained, and the excellent characteristics as a positive electrode active material for a non-aqueous electrolyte secondary battery are exhibited.
- the method for producing a positive electrode active material of the present invention produces a positive electrode active material so as to have the above average particle diameter, particle size distribution, particle structure and composition. If possible, the method is not particularly limited, but the following method is preferable because the positive electrode active material can be produced more reliably.
- the heat treatment step is a step of heating nickel manganese composite hydroxide particles (hereinafter simply referred to as “composite hydroxide particles”) to a temperature of 105 to 750 ° C. to heat treat the composite hydroxide particles.
- composite hydroxide particles nickel manganese composite hydroxide particles
- the water contained in is removed.
- all the composite hydroxide particles need not be nickel manganese complex oxide particles (hereinafter referred to as It is not necessary to convert it simply into "complex oxide particles".
- the heating temperature when the heating temperature is less than 105 ° C., excess water in the composite hydroxide particles can not be removed, and the above variation can not be suppressed.
- the heating temperature exceeds 750 ° C., the particles are sintered by heat treatment, and composite oxide particles having a uniform particle diameter can not be obtained.
- the atmosphere in which the heat treatment is performed is not particularly limited, as long as it is a non-reducing atmosphere, but is preferably performed in an air stream that can be easily performed.
- the heat treatment time is not particularly limited, but excess water removal of the composite hydroxide particles may not be sufficiently performed in less than 1 hour, so at least 1 hour or more is preferable, and 5 to 15 hours is more preferable. .
- the equipment used for the heat treatment is not particularly limited, as long as the composite hydroxide particles can be heated in a non-reducing atmosphere, preferably in an air stream, and there is no gas generation in the electric furnace Are preferably used.
- a lithium mixture is obtained by mixing the composite hydroxide particles (hereinafter referred to as "heat-treated particles") or the like heat-treated in the above heat treatment step with a lithium-containing substance such as a lithium compound. Is a process of obtaining
- the heat-treated particles include not only the composite hydroxide particles from which residual water has been removed in the heat treatment step, but also the composite oxide particles converted into oxides in the heat treatment step or mixed particles thereof.
- the heat treatment particles and the lithium compound are the number of atoms of metals other than lithium in the lithium mixture, that is, the ratio of the sum of the number of atoms of nickel, manganese, cobalt and the additive element (Me) to the number of atoms of lithium (Li)
- the (Li / Me) is mixed so as to be 0.95 to 1.5, preferably 1 to 1.5, more preferably 1.1 to 1.35. That is, since Li / Me does not change before and after the firing step, Li / Me mixed in this mixing step becomes Li / Me in the positive electrode active material, so that Li / Me in the lithium mixture is intended to be obtained Mixed to be the same as Li / Me in
- the lithium compound used to form the lithium mixture is not particularly limited.
- lithium hydroxide, lithium nitrate, lithium carbonate, or a mixture thereof is preferable in that it is easily available.
- lithium hydroxide or lithium carbonate is more preferably used in consideration of ease of handling and stability of quality.
- the lithium mixture is preferably sufficiently mixed before firing. If mixing is not sufficient, Li / Me may vary among individual particles, and problems may occur while sufficient battery characteristics can not be obtained.
- general mixers can be used for mixing, for example, shaker mixers, Loedige mixers, Julia mixers, V blenders, etc. can be used, and complex oxidation is performed to such an extent that the shape of heat-treated particles is not destroyed.
- the substance particles and the substance containing lithium may be sufficiently mixed.
- the firing step is a step of firing the lithium mixture obtained in the mixing step to form a lithium-nickel-manganese composite oxide.
- the firing step is a step of firing the lithium mixture obtained in the mixing step to form a lithium-nickel-manganese composite oxide.
- lithium in the lithium-containing substance diffuses into the heat-treated particles, so that a lithium-nickel-manganese composite oxide is formed.
- the calcination of the lithium mixture is carried out at 800-980 ° C., more preferably at 820-960 ° C.
- the firing temperature is less than 800 ° C.
- the diffusion of lithium into the heat-treated particles is not sufficiently carried out, and surplus lithium and unreacted particles remain, or the crystal structure is not sufficiently prepared, which is used for batteries In such a case, sufficient battery characteristics can not be obtained.
- the temperature is preferably raised to the above temperature by setting the temperature rising rate to 3 to 10 ° C./min. Furthermore, the reaction can be carried out more uniformly by maintaining the temperature around the melting point of the lithium compound for about 1 to 5 hours.
- the holding time at a predetermined temperature is preferably at least 2 hours or more, and more preferably 4 to 24 hours. If it is less than 2 hours, the formation of the lithium-nickel-manganese composite oxide may not be sufficiently performed.
- the descent speed is set to 2 to 10 ° C./min to prevent deterioration of the mortar, and the temperature is 200 ° C. It is preferable to cool the atmosphere to the following.
- lithium hydroxide or lithium carbonate when used as the lithium compound, it is lower than the calcination temperature and preferably at a temperature of 350 to 800 ° C., preferably 450 to 780 ° C. for about 1 to 10 hours before calcination. It is preferable to hold and calcine preferably for 3 to 6 hours. That is, it is preferable to perform calcination at a reaction temperature of lithium hydroxide or lithium carbonate and heat-treated particles. In this case, if lithium hydroxide or lithium carbonate is maintained at around the above-mentioned reaction temperature, the diffusion of lithium into the heat-treated particles is sufficiently performed, and a uniform lithium-nickel-manganese composite oxide can be obtained.
- the heat treatment particles as the raw material may be those having the particle surface uniformly covered with the additional element M.
- the concentration of the additional element M on the surface of the lithium-nickel-manganese composite oxide particles can be increased by firing the lithium mixture containing such heat-treated particles under appropriate conditions. More specifically, if a lithium mixture containing heat-treated particles coated with additive element M is fired at a low firing temperature and a short firing time, lithium having an increased concentration of additive element M on the particle surface is obtained.
- Nickel-manganese composite oxide particles can be obtained.
- lithium-nickel-manganese composite oxide in which the additive element is uniformly distributed in the particles Object particles can be obtained. That is, lithium nickel manganese composite oxide particles having a target concentration distribution can be obtained by adjusting heat treatment particles as raw materials and firing conditions.
- the atmosphere at the time of firing is preferably an oxidizing atmosphere, more preferably an oxygen concentration of 18 to 100% by volume, and particularly preferably a mixed atmosphere of oxygen and an inert gas having the oxygen concentration. That is, the firing is preferably performed in the air or an oxygen stream. If the oxygen concentration is less than 18% by volume, the crystallinity of the lithium-nickel-manganese composite oxide may not be sufficient. In particular, in consideration of the battery characteristics, it is preferable to carry out in an oxygen stream.
- the furnace used for the firing is not particularly limited as long as it can heat the lithium mixture in the atmosphere or an oxygen stream, but there is no gas generation from the viewpoint of keeping the atmosphere in the furnace uniform. Electric furnaces are preferred, and either batch or continuous furnaces can be used.
- the lithium-nickel-manganese composite oxide particles obtained by firing may have aggregation or slight sintering. In this case, it may be crushed, whereby a lithium-nickel-manganese composite oxide, that is, the positive electrode active material of the present invention can be obtained.
- mechanical energy is injected into an aggregate consisting of a plurality of secondary particles generated by sintering necking between secondary particles at the time of firing, and the secondary particles themselves are hardly destroyed. It is an operation to separate secondary particles and loosen aggregates.
- the nonaqueous electrolyte secondary battery of the present invention employs a positive electrode in which the above-mentioned positive electrode active material for nonaqueous electrolyte secondary battery is used as a positive electrode material.
- the structure of the non-aqueous electrolyte secondary battery of the present invention will be described.
- the non-aqueous electrolyte secondary battery of the present invention has substantially the same structure as a general non-aqueous electrolyte secondary battery except that the positive electrode active material of the present invention is used as a positive electrode material.
- the secondary battery of the present invention has a structure including a case, and a positive electrode, a negative electrode, a non-aqueous electrolytic solution, and a separator housed in the case. More specifically, the positive electrode and the negative electrode are stacked via the separator to form an electrode assembly, and the obtained electrode assembly is impregnated with the non-aqueous electrolytic solution, and the positive electrode current collector of the positive electrode and the positive electrode terminal passing outside
- the secondary battery of the present invention is formed by connecting between the negative electrode current collector of the negative electrode and the negative electrode terminal leading to the outside using a current collection lead and the like and sealing in a case. Ru.
- the structure of the secondary battery of the present invention is not limited to the above-mentioned example, and the outer shape thereof can adopt various shapes such as a cylindrical shape and a laminated shape.
- the positive electrode which is a feature of the secondary battery of the present invention, will be described.
- the positive electrode is a sheet-like member, and is formed by applying and drying a positive electrode mixture paste containing the positive electrode active material of the present invention, for example, on the surface of a current collector made of aluminum foil.
- a positive electrode is suitably processed according to the battery to be used.
- a cutting process is performed to form an appropriate size according to a target battery, and a pressure compression process using a roll press or the like is performed to increase the electrode density.
- the positive electrode mixture paste is formed by adding a solvent to the positive electrode mixture and kneading.
- the positive electrode mixture is formed by mixing the positive electrode active material of the present invention in powder form, a conductive material and a binder.
- the conductive material is added to provide the electrode with appropriate conductivity.
- the conductive material is not particularly limited, and for example, carbon black-based materials such as graphite (natural graphite, artificial graphite, expanded graphite and the like) and acetylene black and ketjen black can be used.
- the binder plays a role of holding the positive electrode active material particles.
- the binder used for the positive electrode mixture is not particularly limited, and examples thereof include polyvinylidene fluoride (PVDF), polytetrafluoroethylene (PTFE), fluororubber, ethylene propylene diene rubber, styrene butadiene, cellulose resin, Polyacrylic acid or the like can be used.
- Activated carbon or the like may be added to the positive electrode mixture, and the electric double layer capacity of the positive electrode can be increased by adding activated carbon or the like.
- the solvent dissolves the binder and disperses the positive electrode active material, the conductive material, the activated carbon, and the like in the binder.
- this solvent is not particularly limited, for example, an organic solvent such as N-methyl-2-pyrrolidone can be used.
- the mixing ratio of each substance in the positive electrode mixture paste is not particularly limited.
- the content of the positive electrode active material is 60 to 95 parts by mass
- the conductive material is contained as in the positive electrode of a general non-aqueous electrolyte secondary battery.
- the amount can be 1 to 20 parts by mass
- the content of the binder can be 1 to 20 parts by mass.
- the negative electrode is a sheet-like member formed by applying a negative electrode mixture paste onto the surface of a metal foil current collector such as copper and drying.
- This negative electrode is formed by substantially the same method as that of the positive electrode although the components constituting the negative electrode mixture paste and the composition thereof, the material of the current collector, etc. are different. Is done.
- the negative electrode mixture paste is obtained by adding a suitable solvent to a negative electrode mixture obtained by mixing a negative electrode active material and a binder to form a paste.
- the negative electrode active material for example, a lithium-containing substance such as metal lithium or lithium alloy, or an occluding substance capable of occluding and desorbing lithium ions can be adopted.
- the storage material is not particularly limited, and, for example, natural graphite, artificial graphite, an organic compound fired body such as a phenol resin, and a powdery body of a carbon material such as coke can be used.
- a fluorine-containing resin such as PVDF can be used as a binder as in the positive electrode, and as a solvent for dispersing the negative electrode active material in the binder, Organic solvents such as N-methyl-2-pyrrolidone can be used.
- the separator is disposed between the positive electrode and the negative electrode, and has a function of separating the positive electrode and the negative electrode and holding an electrolyte.
- a separator is, for example, a thin film such as polyethylene or polypropylene, and a film having a large number of fine pores can be used, but it is not particularly limited as long as it has the above-mentioned function.
- Non-aqueous electrolyte solution is one in which a lithium salt as a support salt is dissolved in an organic solvent.
- organic solvent examples include cyclic carbonates such as ethylene carbonate, propylene carbonate, butylene carbonate and trifluoropropylene carbonate; linear carbonates such as diethyl carbonate, dimethyl carbonate, ethyl methyl carbonate and dipropyl carbonate; and further tetrahydrofuran, 2- Ether compounds such as methyltetrahydrofuran and dimethoxyethane; sulfur compounds such as ethylmethylsulfone and butanesultone; phosphorus compounds such as triethyl phosphate and trioctyl phosphate alone or in combination of two or more , Can be used.
- cyclic carbonates such as ethylene carbonate, propylene carbonate, butylene carbonate and trifluoropropylene carbonate
- linear carbonates such as diethyl carbonate, dimethyl carbonate, ethyl methyl carbonate and dipropyl carbonate
- 2- Ether compounds such as methyltetrahydrofuran and dim
- LiPF 6 LiBF 4 , LiClO 4 , LiAsF 6 , LiN (CF 3 SO 2 ) 2 , and complex salts thereof can be used.
- the non-aqueous electrolytic solution may contain a radical scavenger, a surfactant, a flame retardant, and the like to improve battery characteristics.
- the non-aqueous electrolyte secondary battery of the present invention has the above configuration, and has the positive electrode using the positive electrode active material of the present invention, so high initial discharge capacity and low positive electrode resistance can be obtained, and high capacity and high output. It becomes. Moreover, even in comparison with the conventional lithium nickel oxide positive electrode active material, it can be said that the thermal stability is high and the safety is also excellent.
- the secondary battery of the present invention Since the secondary battery of the present invention has the above-mentioned properties, it is suitable as a power source for small portable electronic devices (such as notebook personal computers and mobile phone terminals) which always require high capacity.
- the secondary battery of the present invention is also suitable for a battery as a motor drive power source which requires high output. If the size of the battery becomes large, it becomes difficult to ensure safety, and expensive protective circuits are indispensable. However, the secondary battery of the present invention has excellent safety, so safety is ensured. Not only is it easy, but expensive protection circuits can be simplified and less expensive. And since size reduction and high output-ization are possible, it is suitable as a power supply for transportation apparatus which receives restrictions in mounting space.
- Example 1 [Production of composite hydroxide particles] Composite hydroxide particles were produced as follows. In all of the examples, Wako Pure Chemical Industries, Ltd. reagent special grade samples were used for preparation of the composite hydroxide particles, the positive electrode active material and the secondary battery.
- Ni: Mn 50: 50.
- This mixed aqueous solution was added to the pre-reaction aqueous solution in the reaction tank at a rate of 88 ml / min to obtain a reaction aqueous solution.
- 25% by mass ammonia water and 25% by mass sodium hydroxide aqueous solution are also added to this reaction aqueous solution at a constant rate to keep the ammonia concentration in the reaction aqueous solution (aqueous solution for nucleation) at the above value
- pH Nucleation was performed by crystallization for 2 minutes and 30 seconds while controlling the value to 13.0 (nucleation pH value).
- the supply of the 25 mass% sodium hydroxide aqueous solution is restarted to the reaction aqueous solution (the aqueous solution for particle growth) again, and the ammonia concentration is maintained at the above value. Crystallization is continued for 30 minutes and particle growth is performed while controlling the value to 11.6 at a liquid temperature of 25 ° C., and then the supply liquid is temporarily stopped, and the oxygen concentration in the reaction tank space is 0.2 volume Nitrogen gas was circulated at 5 L / min until it became% or less. Thereafter, the liquid feed was resumed, and crystallization was performed for 2 hours from the start of the growth.
- the product was washed with water, filtered and dried to obtain composite hydroxide particles.
- the switching from the air atmosphere to the nitrogen atmosphere is performed at a point of 12.5% of the entire particle growth process time from the start of the particle growth process.
- the pH was controlled by adjusting the supply flow rate of the aqueous sodium hydroxide solution with a pH controller, and the fluctuation range was within the range of 0.2 above and below the set value.
- a value of [(d90-d10) / average particle size] showing an average particle size and a particle size distribution of this composite hydroxide was measured using a laser diffraction scattering type particle size distribution measuring apparatus (Microtrac HRA manufactured by Nikkiso Co., Ltd.) It calculated and calculated
- the composite hydroxide particles were heat-treated at 700 ° C. for 6 hours in a stream of air (oxygen: 21% by volume) to convert into composite oxide particles and recovered.
- the mixing was carried out using a shaker mixer apparatus (TURBULA Type T2C, manufactured by Willie et Bachkofen (WAB).
- the obtained lithium mixture was calcined at 500 ° C. for 4 hours in the atmosphere (oxygen: 21% by volume), calcined at 900 ° C. for 4 hours, cooled, and then crushed to obtain a positive electrode active material. .
- the obtained positive electrode active material is substantially spherical and the particle diameter is almost uniform. That was confirmed.
- the SEM observation result of this positive electrode active material is shown in FIG.
- the cross-sectional SEM observation result of this positive electrode active material is shown in FIG. The ratio of the thickness of the outer shell portion to the particle diameter of the positive electrode active material determined from this observation was 12%.
- the obtained positive electrode active material is analyzed by powder X-ray diffraction using Cu-K ⁇ radiation using an X-ray diffraction apparatus (manufactured by PANalytical, X'Pert PRO), the crystal structure of the positive electrode active material It was confirmed that the layer was composed of hexagonal layered crystal lithium nickel manganese composite oxide single phase.
- composition analysis of the positive electrode active material is performed by ICP emission spectroscopy in the same manner, the composition is 9.55% by mass of Li, 29.7% by mass of Ni, and 27.8% by mass of Mn. It was confirmed to be 1.36 Ni 0.50 Mn 0.50 O 2 .
- the 2032 type coin battery was used for evaluation of the obtained positive electrode active material.
- the coin battery 1 is composed of a case 2 and an electrode 3 accommodated in the case 2.
- the case 2 has a hollow cathode can 2a open at one end, and a cathode can 2b disposed in the opening of the cathode can 2a.
- a cathode can 2b disposed in the opening of the cathode can 2a.
- a space for housing the electrode 3 is formed between the negative electrode can 2b and the positive electrode can 2a.
- the electrode 3 is composed of a positive electrode 3a, a separator 3c and a negative electrode 3b, which are stacked in this order, and the positive electrode 3a is in contact with the inner surface of the positive electrode can 2a and the negative electrode 3b is in contact with the inner surface of the negative electrode can 2b. Is housed in case 2.
- the case 2 is provided with a gasket 2c, and the gasket 2c fixes the positive electrode can 2a and the negative electrode can 2b so as to maintain an electrically insulated state.
- the gasket 2 c also has a function of sealing the gap between the positive electrode can 2 a and the negative electrode can 2 b to shut off the inside of the case 2 from the outside in an airtight liquid tight manner.
- This coin battery 1 was produced as follows. First, 52.5 mg of the obtained positive electrode active material, 15 mg of acetylene black, and 7.5 mg of polytetrafluoroethylene resin (PTFE) are mixed, and pressed at a pressure of 100 MPa to a diameter of 11 mm and a thickness of 100 ⁇ m. Made. The produced positive electrode 3a was dried at 120 ° C. for 12 hours in a vacuum dryer. Using this positive electrode 3a, the negative electrode 3b, the separator 3c, and the electrolytic solution, a coin-type battery 1 was produced in a glove box under an Ar atmosphere with a dew point controlled to -80.degree.
- PTFE polytetrafluoroethylene resin
- the negative electrode 3b a negative electrode sheet in which a copper powder having a mean particle diameter of about 20 ⁇ m punched into a disk shape having a diameter of 14 mm and a polyvinylidene fluoride applied to a copper foil was used. Moreover, the polyethylene porous film with a film thickness of 25 micrometers was used for the separator 3c.
- an electrolyte solution an equivalent mixed solution of ethylene carbonate (EC) and diethyl carbonate (DEC) (made by Toyama Pharmaceutical Co., Ltd.) using 1 M LiClO 4 as a supporting electrolyte was used.
- the initial discharge capacity is left for about 24 hours after producing the coin-type battery 1, and after the open circuit voltage OCV (open circuit voltage) is stabilized, the cut-off voltage 4 with the current density to the positive electrode of 0.1 mA / cm 2.
- the initial discharge capacity was defined as the capacity when the battery was charged to 8 V and discharged to a cutoff voltage of 2.5 V after 1 hour of rest.
- the cycle capacity retention rate is a cycle of charging up to 4.5 V with a current density to the positive electrode of 2 mA / cm 2 and discharging to 3.0 V repeated 200 times, and the discharge capacity and initial discharge capacity after repeated charge and discharge The ratio of was calculated to be the capacity retention rate.
- a multichannel voltage / current generator manufactured by Advantest, R6741A was used.
- positive electrode resistance was evaluated as follows.
- the coin-type battery 1 is charged at a charge potential of 4.1 V and measured by an AC impedance method using a frequency response analyzer and a potentiogalvanostat (manufactured by Solartron, 1255B) to obtain the Nyquist plot shown in FIG. Since this Nyquist plot is expressed as the sum of solution resistance, negative electrode resistance and its capacity, and positive electrode resistance and characteristic curve showing its capacity, fitting calculation is performed using an equivalent circuit based on this Nyquist plot, and positive electrode resistance is calculated. The value of was calculated.
- initial stage discharge capacity was 208.7 mAh / g
- positive electrode resistance was 8.2 (ohm).
- capacity retention rate after 200 cycles was 85%.
- Table 1 shows the characteristics of the composite hydroxide obtained in the present example
- Table 2 shows the characteristics of the positive electrode active material and each evaluation of the coin-type battery manufactured using this positive electrode active material. The same contents are shown in Tables 1 and 2 for the following Examples 2 to 8 and Comparative Examples 1 to 5.
- the composition of the obtained positive electrode active material is a composition of 8.84% by mass of Li, 29.9% by mass of Ni, 28.0% by mass of Mn, and Li 1.25 Ni 0.50 M 0.50 O 2 confirmed.
- Example 3 The particle growth step in the composite hydroxide particle production step is the same as Example 1 except that switching from air atmosphere to nitrogen atmosphere is performed at 6.25% of the entire particle growth step time. Then, a positive electrode active material for a non-aqueous electrolyte secondary battery was obtained and evaluated.
- the composition of the obtained composite hydroxide particles and the positive electrode active material is the same as in Example 1, and the composite hydroxide particles are similar to Example 1 in needle-like fine primary particles (having a particle diameter of about 0. (3 ⁇ m) and an outer shell made of plate-like primary particles (particle diameter 0.7 ⁇ m) larger than the fine primary particles outside the central portion.
- the composition of the obtained positive electrode active material is a composition of 9.54 mass% of Li, 29.2 mass% of Ni, 27.3 mass% of Mn, and 0.93 mass% of W, and Li 1.36 It was confirmed that it was Ni 0.4925 Mn 0.4925 W 0.005 O 2 .
- the composition of the obtained positive electrode active material is a composition of 9.58 mass% of Li, 29.3 mass% of Ni, 27.5 mass% of Mn, and 0.46 mass% of Zr, and Li 1.36 It was confirmed that it is Ni 0.4925 Mn 0.4925 Zr 0.005 O 2 .
- the composition of the obtained positive electrode active material is a composition of 7.86 mass% of Li, 30.2 mass% of Ni, and 28.3 mass% of Mn, and is Li 1.10 Ni 0.50 Mn 0.50 O 2 That was confirmed.
- Example 7 In the composite hydroxide particle production step, a positive electrode active material for a non-aqueous electrolyte secondary battery is obtained in the same manner as in Example 1 except that the temperature in the tank is 50 ° C. and the ammonia concentration is 20 g / L. did.
- the composition of the obtained composite hydroxide and the positive electrode active material is the same as in Example 1, and the composite hydroxide particles are similar to Example 1 in needle-like fine primary particles (particle diameter about 0.3 ⁇ m And an outer shell composed of plate-like primary particles (having a particle diameter of about 0.7 ⁇ m) larger than the fine primary particles.
- Example 8 In the composite hydroxide particle production step, the procedure is the same as in Example 1 except that in the particle growth step, switching from the air atmosphere to the nitrogen atmosphere is performed at 25% of the entire particle growth step time.
- a positive electrode active material for a non-aqueous electrolyte secondary battery was obtained and evaluated.
- the composition of the obtained composite hydroxide and the positive electrode active material is the same as in Example 1, and the composite hydroxide particles are needle-like fine primary particles (particle diameter 0.3 ⁇ m) as in Example 1. And an outer shell made of plate-like primary particles (particle diameter 0.5 ⁇ m) larger than the fine primary particles outside the central portion.
- Example 1 Using a reaction tank for continuous crystallization equipped with an overflow pipe at the upper part, while maintaining the pH value of the reaction aqueous solution at a constant value of 11.0 at a liquid temperature of 25 ° C. in an air atmosphere, Example 1 and The same mixed aqueous solution and aqueous ammonia solution and sodium hydroxide solution were continuously added at a constant flow rate, and crystallization was performed by a general method in which the overflowing slurry was continuously collected. After the interior of the continuous tank is in equilibrium with the average residence time in the reaction tank being 10 hours, the slurry is recovered and solid-liquid separated to obtain a crystallized product as in Example 1.
- a positive electrode active material for a non-aqueous electrolyte secondary battery was obtained and evaluated.
- the compositions of the obtained composite hydroxide and positive electrode active material were the same as in Example 1. Since the entire composite hydroxide particles were composed of primary particles similar to the outer shell part of Example 1, the positive electrode active material became particles of a dense solid structure.
- Example 3 A nickel-manganese composite hydroxide was obtained in the same manner as in Example 1, except that the pH values at nucleation and particle growth were both kept constant at 12.6 based on a liquid temperature of 25 ° C. .
- nitrogen gas was allowed to flow at 5 L / min in the space in the reaction tank to keep the oxygen concentration at 0.2 volume% or less.
- the particles had a broad particle size distribution and became amorphous particles including gel-like precipitates, so that solid-liquid separation was difficult and the treatment was stopped.
- Example 4 The positive electrode active material for a non-aqueous electrolyte secondary battery was obtained and evaluated in the same manner as in Example 1 except that the firing temperature was set to 1000 ° C.
- the compositions of the obtained composite hydroxide and positive electrode active material were the same as in Example 1. However, from the result of the X-ray diffraction measurement, the crystal structure of the hexagonal crystal is broken and the performance as the positive electrode active material can not be expected, so the battery evaluation was not performed.
- Example 5 A non-aqueous electrolyte was prepared in the same manner as in Example 1, except that switching from air atmosphere to nitrogen atmosphere in the grain growth step was performed at a point of 50% of the particle growth step time from the start of the particle growth step.
- a positive electrode active material for secondary battery was obtained and evaluated. Since the thickness of the outer shell portion of the composite hydroxide was insufficient, sintering of secondary particles proceeded in the firing step to be a positive electrode active material, and an active material containing coarse particles was obtained.
- the compositions of the obtained composite hydroxide and positive electrode active material were the same as in Example 1.
- each positive electrode active material also has a structure including an outer shell portion in which aggregated primary particles are sintered, and a hollow portion inside the outer shell portion.
- a coin-type battery using these positive electrode active materials has a high initial discharge capacity, excellent cycle characteristics, and a low positive electrode resistance, and is a battery having excellent characteristics.
- Comparative Example 1 since the continuous crystallization method was used, the nucleation and the grain growth can not be separated, and the grain growth time is not constant, so that the particle size distribution is broad. For this reason, the coin battery 1 has a high initial discharge capacity, but has poor cycle characteristics.
- the non-aqueous electrolyte secondary battery using this positive electrode active material has a high initial discharge capacity, It can be confirmed that the battery has excellent cycle characteristics, low positive electrode resistance, and excellent characteristics.
- the pH value of the reaction aqueous solution at this time is controlled to 12.8, and nitrogen flow is started at the start of the particle growth process, and the atmosphere is switched to the nitrogen atmosphere (from the start of the particle growth process to the entire particle growth process time While maintaining the ammonia concentration at the above value, and controlling the pH value of the reaction aqueous solution at particle growth to 11.6, for 2 hours, and after extracting a half amount of the supernatant
- the mixture was calcined at 760 ° C. for 4 hours in the air and calcined at 950 ° C. for 10 hours in the same manner as in Example 1 except that 7.93% by mass of Li and 19.19% by mass of Ni were used.
- Example 1 the obtained composite hydroxide and the positive electrode active material for a non-aqueous electrolyte secondary battery were evaluated in the same manner as Example 1.
- Table 3 shows the characteristics of the composite hydroxide obtained in this example
- Table 4 shows the characteristics of the positive electrode active material and the evaluations of the coin-type battery produced using this positive electrode active material. The same contents are also shown in Tables 3 and 4 for the following Examples 9 to 12 and Comparative Examples 6 to 8.
- Example 10 Ni 0.33 Co 0.33 Mn 0.33 Zr 0.005 W 0.005 (OH) 2 + a (0 ⁇ a ⁇ 0) in the same manner as in Example 9 except that sodium tungstate aqueous solution was simultaneously and continuously added at the time of crystallization reaction.
- the nickel manganese composite hydroxide represented by .5) was obtained, and heat processing were performed.
- Example 9 In the same manner as in Example 9 except that the firing temperature was set to 930 ° C., 8.07% by mass of Li, 19.6% by mass of Ni, 19.7% by mass of Co, and 18.3% by mass of Mn A positive electrode active material represented by Li 1.15 Ni 0.33 Co 0.33 Mn 0.33 Zr 0.005 W 0.005 O 2 was obtained with a composition of 0.46% by mass of Zr and 0.93% by mass of W.
- Example 11 The pH value in the nucleation step was changed to 12.6, the addition time of the mixed aqueous solution was changed from 2 minutes and 30 seconds to 30 seconds, and the particle growth step was started 60 minutes after the nitrogen flow was started, nitrogen was released from the atmosphere.
- Nickel cobalt manganese composite hydroxide was obtained. Further, using the obtained composite hydroxide, heat treatment and firing were performed in the same manner as in Example 10 to obtain a positive electrode active material.
- the composition of the obtained composite hydroxide and positive electrode active material was the same as in Example 10.
- a nickel-manganese composite hydroxide was obtained. 7.19% by mass of Li, 28.7% by mass of Ni, and 14 of Co in the same manner as in Example 1 except that the Li / M ratio is 1.05 and the sintering conditions are 850 ° C. and 10 hours.
- Example 13 The positive electrode active material obtained in the same manner as in Example 9 is dried while sending hot air at 180 ° C. with a fluid drying apparatus (MP-01, manufactured by Powrex Co., Ltd.), and a lithium tungstate aqueous solution is sprayed to obtain a positive electrode active.
- the positive electrode active material in which lithium tungstate was coated on the material surface was obtained.
- the composition of the obtained active material was the same as in Example 10. From the cross-sectional SEM observation EDX analysis, it was confirmed that a large amount of tungsten was present in the vicinity of the surface of the active material particles.
- Example 14 The composite hydroxide obtained in the same manner as in Example 9 is dispersed in an ammonium tungstate solution to a concentration of 150 g / L and slurried, and then the slurry is micro-mist drier (MDL-made by Fujisaki Electric Co., Ltd.) The resultant was spray-dried using 050 M to obtain a positive electrode active material in the same manner as in Example 10 except that a composite hydroxide coated with an ammonium tungstate was obtained.
- the composition of the obtained active material was the same as in Example 10. From the cross-sectional SEM observation EDX analysis, it was confirmed that a large amount of tungsten was present in the vicinity of the surface of the active material particles.
- Example 6 A composite oxide positive electrode active material was obtained in the same manner as in Example 9 except that the firing conditions were 1050 ° C. and 10 hours. Since the sintering temperature was high and the sintering progressed, the specific surface area was low and the positive electrode resistance value was high.
- Example 7 A positive electrode active material was prepared in the same manner as in Example 10, except that nitrogen was allowed to flow through the crystallization reaction in the nucleation step and the particle growth step to form a non-oxidizing atmosphere (oxygen: 0.2% by volume or less). I got
- the entire composite hydroxide particle was composed of primary particles similar to the outer shell of Example 10, and the positive electrode active material became particles with a dense structure, so the specific surface area was low and the positive electrode resistance was high. .
- Lithium nickel cobalt was prepared in the same manner as in Example 12, except that nitrogen was allowed to flow during the crystallization reaction in the nucleation step and the particle growth step to form a non-oxidizing atmosphere (oxygen: 0.2 vol% or less). A manganese composite oxide was obtained.
- the entire composite hydroxide particle was composed of primary particles similar to the outer shell of Example 10, and the positive electrode active material became particles with a dense structure, so the specific surface area was low and the positive electrode resistance was high. .
- the non-aqueous electrolyte secondary battery of the present invention is suitable as a power source for small portable electronic devices (such as notebook personal computers and mobile phone terminals) which always require high capacity.
- non-aqueous electrolyte secondary battery of the present invention has excellent safety, and can be downsized and output at high power, so it is suitable as a power source for transportation equipment that is restricted by the mounting space.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Metallurgy (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Composite Materials (AREA)
- Secondary Cells (AREA)
Abstract
Description
少なくともニッケルを含有する金属化合物およびマンガンを含有する金属化合物とアンモニウムイオン供給体とを含む核生成用水溶液を、液温25℃基準で、pH値が12.0~14.0となるように制御して、酸素濃度が1容量%を超える酸化性雰囲気中で核生成を行う核生成工程と、
該核生成工程において形成された核を含有する粒子成長用水溶液を、液温25℃基準におけるpH値が10.5~12.0となるように制御するとともに、粒子成長工程の開始時から粒子成長工程時間の全体に対して0~40%の範囲で、前記酸化性雰囲気から酸素濃度1容量%以下の酸素と不活性ガスの混合雰囲気に切り替えて、前記核を成長させる粒子成長工程とを備えることを特徴とする。
原料として、本発明のニッケルマンガン複合水酸化物粒子を用い、該ニッケルマンガン複合水酸化物粒子を、105~750℃の温度で熱処理する工程と、前記熱処理後の粒子に対してリチウム化合物を混合して、リチウム混合物を形成する混合工程と、該混合工程で形成された前記混合物を、酸化性雰囲気中800~980℃の温度で焼成する焼成工程とを備えることを特徴とする。
本発明のニッケルマンガン複合水酸化物粒子は、一般式NixMnyCozMt(OH)2+a(x+y+z+t=1、0.3≦x≦0.8、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、0≦a≦0.5、Mは、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の添加元素)で表され、複数の板状一次粒子が凝集して形成された略球状の二次粒子であり、該二次粒子は、平均粒径が3~7μmであり、粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕が0.55以下であり、微細一次粒子からなる中心部を有し、中心部の外側に該微細一次粒子よりも大きな板状一次粒子からなる外殻部を有するものである。
本発明の複合水酸化物粒子は、図5に例示されるように、略球状の粒子である。具体的には、図6に例示されるように、複数の板状一次粒子が凝集して形成された略球状の二次粒子となっており、さらに詳細には、粒子内部は微細一次粒子からなる中心部を有し、中心部の外側に該微細一次粒子よりも大きな板状一次粒子からなる外殻部を有する構造を備えている。かかる構造により、本発明の正極活物質であるリチウムニッケルマンガン複合酸化物を形成する焼結工程において、粒子内へのリチウムの拡散が十分に行われるため、リチウムの分布が均一で良好な正極活物質が得られる。
本発明の複合水酸化物粒子は、その平均粒径が、3~7μm、好ましくは3.5~6.5μmに調整されている。平均粒径を3~7μmとすることで、本発明の複合水酸化物粒子を原料として得られる正極活物質を所定の平均粒径(2~8μm)に調整することができる。このように、複合水酸化物粒子の粒径は、得られる正極活物質の粒径と相関するため、この正極活物質を正極材料に用いた電池の特性に影響するものである。
本発明の複合水酸化物粒子は、その粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕が、0.55以下、好ましくは0.52以下となるように調整されている。
本発明の複合水酸化物粒子は、その組成が、以下の一般式で表されるように調整される。このような組成を有するニッケルマンガン複合水酸化物を原料として、リチウムニッケルマンガン複合酸化物を製造すれば、このリチウムニッケルマンガン複合酸化物を正極活物質とする電極を電池に用いた場合に、測定される正極抵抗の値を低くできるとともに、電池性能を良好なものとすることができる。
(x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、0≦a≦0.5、Mは、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の添加元素)
本発明の複合水酸化物粒子の製造方法は、晶析反応によってニッケルマンガン複合水酸化物粒子を製造する方法であって、a)核生成を行う核生成工程と、b)核生成工程において生成された核を成長させる粒子成長工程とから構成されている。
図1に示すように、まず、ニッケルおよびマンガンを含有する複数の金属化合物を所定の割合で水に溶解させ、混合水溶液を作製する。本発明の複合水酸化物粒子の製造方法では、得られる複合水酸化物粒子における上記各金属の組成比は、混合水溶液における各金属の組成比と同様となる。
核生成工程の終了後、前記核生成用水溶液のpH値を、液温25℃基準で、10.5~12.0、好ましくは11.0~12.0となるように調整して、粒子成長工程における反応水溶液である粒子成長用水溶液を得る。具体的には、この調整時のpHの制御は、アルカリ水溶液の供給量を調節することにより行う。
次に、各工程における反応雰囲気の制御、各工程において使用する物質や溶液、反応条件について、詳細に説明する。
上述のように、核生成工程においては、反応水溶液のpH値が、液温25℃基準で12.0~14.0、好ましくは12.3~13.5の範囲となるように制御する必要がある。pH値が14.0を超える場合、生成する核が微細になり過ぎ、反応水溶液がゲル化する問題がある。また、pH値が12.0未満では、核形成とともに核の成長反応が生じるので、形成される核の粒度分布の範囲が広くなり不均質なものとなってしまう。すなわち、核生成工程において、上述の範囲に反応水溶液のpH値を制御することで、核の成長を抑制してほぼ核生成のみを起こすことができ、形成される核も均質かつ粒度分布の範囲が狭いものとすることができる。
核生成工程において生成する核の量は、特に限定されるものではないが、粒度分布の良好な複合水酸化物粒子を得るためには、全体量、つまり、複合水酸化物粒子を得るために供給する全金属塩の0.1%から2%とすることが好ましく、1.5%以下とすることがより好ましい。
上記複合水酸化物粒子の粒径は、粒子成長工程の時間により制御できるので、所望の粒径に成長するまで粒子成長工程を継続すれば、所望の粒径を有する複合水酸化物粒子を得ることができる。
金属化合物としては、目的とする金属を含有する化合物を用いる。使用する化合物は、水溶性の化合物を用いることが好ましく、硝酸塩、硫酸塩、塩酸塩などがあげられる。たとえば、硫酸ニッケル、硫酸マンガン、硫酸コバルトが好ましく用いられる。
添加元素(Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の元素)は、水溶性の化合物を用いることが好ましく、たとえば、硫酸チタン、ペルオキソチタン酸アンモニウム、シュウ酸チタンカリウム、硫酸バナジウム、バナジン酸アンモニウム、硫酸クロム、クロム酸カリウム、硫酸ジルコニウム、硝酸ジルコニウム、シュウ酸ニオブ、モリブデン酸アンモニウム、タングステン酸ナトリウム、タングステン酸アンモニウムなどを用いることができる。
混合水溶液の濃度は、金属化合物の合計で1~2.6mol/L、好ましくは1.5~2.2mol/Lとすることが好ましい。混合水溶液の濃度が1mol/L未満では、反応槽当たりの晶析物量が少なくなるために生産性が低下して好ましくない。
反応水溶液中のアンモニア濃度は、以下の問題を生じさせないために、好ましくは3~25g/L、好ましくは5~20g/Lの範囲内で一定値に保持する。
反応槽内において、反応液の温度は、好ましくは20℃以上、特に好ましくは20~60℃に設定する。反応液の温度が20℃未満の場合、溶解度が低いため核発生が起こりやすく制御が難しくなる。一方、60℃を超えると、アンモニアの揮発が促進されるため、所定のアンモニア濃度を保つために、過剰のアンモニウムイオン供給体を添加しなければならならず、コスト高となる。
反応水溶液中のpHを調整するアルカリ水溶液については、特に限定されるものではなく、たとえば、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物水溶液を用いることができる。かかるアルカリ金属水酸化物の場合、直接、反応水溶液中に供給してもよいが、反応槽内における反応水溶液のpH制御の容易さから、水溶液として反応槽内の反応水溶液に添加することが好ましい。
本発明の複合水酸化物粒子の製造方法では、反応が完了するまで生成物を回収しない方式の装置を用いる。たとえば、撹拌機が設置された通常に用いられるバッチ反応槽などである。かかる装置を採用すると、一般的なオーバーフローによって生成物を回収する連続晶析装置のように、成長中の粒子がオーバーフロー液と同時に回収されるという問題が生じないため、粒度分布が狭く粒径の揃った粒子を得ることができる。
本発明の正極活物質は、一般式:Li1+uNixMnyCozMtO2(-0.05≦u≦0.50、x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、Mは、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の添加元素)で表されるリチウムニッケルマンガン複合酸化物粒子であって、層状構造を有する六方晶系の結晶構造を有するものである。
本発明の正極活物質は、リチウムニッケルマンガン複合酸化物粒子であるが、その組成が、以下の一般式で表されるように調整されるものである。
(-0.05≦u≦0.50、x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、Mは、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の添加元素)
図7に示されるように、本発明の正極活物質は、平均粒径が2~8μmである。平均粒径が2μm未満の場合には、正極を形成したときに粒子の充填密度が低下して、正極の容積あたりの電池容量が低下する。一方、平均粒径が8μmを超えると、正極活物質の比表面積が低下して、電池の電解液との界面が減少することにより、正極の抵抗が上昇して電池の出力特性が低下する。
図7に例示されるように、本発明の正極活物質は、粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕が0.60以下、好ましくは0.55以下である、きわめて均質性が高いリチウムニッケルマンガン複合酸化物の二次粒子により構成される。
本発明の正極活物質は、図8に例示するように、二次粒子内部の中空部とその外側の外殻部で構成される中空構造を有する点に特徴がある。このような中空構造とすることにより、反応表面積を大きくすることができ、かつ、外殻部の一次粒子間の粒界あるいは空隙から電解液が浸入して、粒子内部の中空側の一次粒子表面における反応界面でもリチウムの挿脱入が行われるため、Liイオン、電子の移動が妨げられず、出力特性を高くすることができる。
上記正極活物質は、たとえば、2032型コイン電池の正極に用いた場合、コバルトを添加しない場合において200mAh/g以上、コバルトを原子比でリチウム以外の全金属元素の30%程度添加した場合においても150mAh/g以上の高い初期放電容量と、低い正極抵抗および高いサイクル容量維持率が得られるものとなり、非水系電解質二次電池用正極活物質として優れた特性を示すものである。
本発明の正極活物質の製造方法は、上記平均粒径、粒度分布、粒子構造および組成となるように正極活物質を製造できるのであれば、特に限定されないが、以下の方法を採用すれば、該正極活物質をより確実に製造できるので、好ましい。
熱処理工程は、ニッケルマンガン複合水酸化物粒子(以下、単に「複合水酸化物粒子」という)を105~750℃の温度に加熱して熱処理する工程であり、複合水酸化物粒子に含有されている水分を除去している。この熱処理工程を行うことによって、粒子中に焼成工程まで残留している水分を一定量まで減少させることができる。これにより、得られる製造される正極活物質中の金属の原子数やリチウムの原子数の割合がばらつくことを防ぐことができる。
混合工程は、上記熱処理工程において熱処理された複合水酸化物粒子(以下、「熱処理粒子」という)などと、リチウムを含有する物質、たとえば、リチウム化合物とを混合して、リチウム混合物を得る工程である。
焼成工程は、上記混合工程で得られたリチウム混合物を焼成して、リチウムニッケルマンガン複合酸化物を形成する工程である。焼成工程においてリチウム混合物を焼成すると、熱処理粒子に、リチウムを含有する物質中のリチウムが拡散するので、リチウムニッケルマンガン複合酸化物が形成される。
リチウム混合物の焼成は、800~980℃で、より好ましくは820~960℃で行われる。
焼成時間のうち、所定温度での保持時間は、少なくとも2時間以上とすることが好ましく、より好ましくは、4~24時間である。2時間未満では、リチウムニッケルマンガン複合酸化物の生成が十分に行われないことがある。保持時間終了後、特に限定されるものではないが、リチウム混合物を匣鉢に積載して焼成する場合には匣鉢の劣化を抑止するため、降下速度を2~10℃/minとして、200℃以下になるまで雰囲気を冷却することが好ましい。
特に、リチウム化合物として、水酸化リチウムや炭酸リチウムを使用した場合には、焼成する前に、焼成温度より低く、かつ、350~800℃、好ましくは450~780℃の温度で1~10時間程度、好ましくは3~6時間、保持して仮焼することが好ましい。すなわち、水酸化リチウムや炭酸リチウムと熱処理粒子の反応温度において仮焼することが好ましい。この場合、水酸化リチウムや炭酸リチウムの上記反応温度付近で保持すれば、熱処理粒子へのリチウムの拡散が十分に行われ、均一なリチウムニッケルマンガン複合酸化物を得ることができる。
焼成時の雰囲気は、酸化性雰囲気とすることが好ましく、酸素濃度を18~100容量%とすることがより好ましく、上記酸素濃度の酸素と不活性ガスの混合雰囲気とすることが特に好ましい。すなわち、焼成は、大気ないしは酸素気流中で行うことが好ましい。酸素濃度が18容量%未満であると、リチウムニッケルマンガン複合酸化物の結晶性が十分でない状態になる可能性がある。特に電池特性を考慮すると、酸素気流中で行うことが好ましい。
焼成によって得られたリチウムニッケルマンガン複合酸化物粒子は、凝集もしくは軽度の焼結が生じている場合がある。この場合には、解砕してもよく、これにより、リチウムニッケルマンガン複合酸化物、つまり、本発明の正極活物質を得ることができる。なお、解砕とは、焼成時に二次粒子間の焼結ネッキングなどにより生じた複数の二次粒子からなる凝集体に、機械的エネルギを投入して、二次粒子自体をほとんど破壊することなく二次粒子を分離させて、凝集体をほぐす操作のことである。
本発明の非水系電解質二次電池は、上記非水系電解質二次電池用正極活物質を正極材料に用いた正極を採用したものである。まず、本発明の非水系電解質二次電池の構造を説明する。
まず、本発明の二次電池の特徴である正極について説明する。正極は、シート状の部材であり、本発明の正極活物質を含有する正極合材ペーストを、たとえば、アルミニウム箔製の集電体の表面に塗布乾燥して形成されている。
負極は、銅などの金属箔集電体の表面に、負極合材ペーストを塗布し、乾燥して形成されたシート状の部材である。この負極は、負極合材ペーストを構成する成分やその配合、集電体の素材などは異なるものの、実質的に前記正極と同様の方法によって形成され、正極と同様に、必要に応じて各種処理が行われる。
セパレータは、正極と負極との間に挟み込んで配置されるものであり、正極と負極とを分離し、電解質を保持する機能を有している。かかるセパレータは、たとえば、ポリエチレンやポリプロピレンなどの薄い膜で、微細な孔を多数有する膜を用いることができるが、上記機能を有するものであれば、特に限定されない。
非水系電解液は、支持塩としてのリチウム塩を有機溶媒に溶解したものである。
本発明の非水系電解質二次電池は、上記構成であり、本発明の正極活物質を用いた正極を有しているので、高い初期放電容量、低い正極抵抗が得られ、高容量で高出力となる。しかも、従来のリチウムニッケル系酸化物の正極活物質との比較においても、熱安定性が高く、安全性においても優れているといえる。
本発明の二次電池は、上記性質を有するので、常に高容量を要求される小型携帯電子機器(ノート型パーソナルコンピュータや携帯電話端末など)の電源に好適である。
[複合水酸化物粒子の製造]
複合水酸化物粒子を、以下のようにして作製した。なお、すべての実施例を通じて、複合水酸化物粒子、正極活物質および二次電池の作製には、和光純薬工業株式会社製試薬特級の各試料を使用した。
まず、反応槽(34L)内に、水を半分の量まで入れて撹拌しながら、槽内温度を40℃に設定した。このときの反応槽内は、大気雰囲気(酸素濃度:21容量%)とした。この反応槽内の水に、25質量%水酸化ナトリウム水溶液と25質量%アンモニア水を適量加えて、液温25℃基準で、槽内の反応液のpH値が13.0となるように調整した。さらに、該反応液中のアンモニア濃度を15g/Lに調節して反応前水溶液とした。
核生成終了後、反応水溶液のpH値が液温25℃基準で11.6になるまで、25質量%水酸化ナトリウム水溶液の供給のみを一時停止した。
得られた複合水酸化物について、その試料を無機酸により溶解した後、ICP発光分光法により化学分析を行ったところ、その組成は、Ni0.5Mn0.5(OH)2+a(0≦a≦0.5)であった。
上記複合水酸化物粒子を、空気(酸素:21容量%)気流中にて、700℃で6時間の熱処理を行って、複合酸化物粒子に転換して回収した。
複合水酸化物粒子と同様の方法で、得られた正極活物質の粒度分布を測定したところ、平均粒径は4.8μmであり、〔(d90-d10)/平均粒径〕値は、0.49であった。
得られた正極活物質の評価には、2032型コイン電池を使用した。図9に示すように、このコイン型電池1は、ケース2と、このケース2内に収容された電極3とから構成されている。
得られたコイン型電池1の性能を評価する、初期放電容量、サイクル容量維持率、正極抵抗は、以下のように定義した。
Li/Me=1.25となるように水酸化リチウムと複合酸化物粒子を混合したこと、焼成温度を850℃としたこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価を行った。得られた正極活物質の組成は、Liが8.84質量%、Niが29.9質量%、Mnが28.0質量%の組成であり、Li1.25Ni0.50M0.50O2であることが確認された。
複合水酸化物粒子製造工程における粒子成長工程において、大気雰囲気から窒素雰囲気への切り替えを、粒子成長工程時間全体に対して6.25%の時点で行ったこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた複合水酸化物粒子および正極活物質の組成は、実施例1と同様であり、複合水酸化物粒子は実施例1と同様に針状の微細一次粒子(粒径およそ0.3μm)からなる中心部と、該中心部の外側にこの微細一次粒子よりも大きい板状の一次粒子(粒径0.7μm)からなる外殻部とにより構成されていた。
複合水酸化物粒子製造工程において、硫酸ニッケルと硫酸マンガンに加えて、タングステン酸ナトリウムを水に溶かして1.8mol/Lの混合水溶液を形成したこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、この混合水溶液では、各金属の元素モル比が、Ni:Mn:W=49.25:49.25:0.5となるように調整した。得られた複合水酸化物の組成は、Ni0.4925Mn0.4925W0.005(OH)2+a(0≦a≦0.5)であった。また、得られた正極活物質の組成は、Liが9.54質量%、Niが29.2質量%、Mnが27.3質量%、Wが0.93質量%の組成であり、Li1.36Ni0.4925Mn0.4925W0.005O2であることが確認された。
複合水酸化物粒子製造工程において、硫酸ニッケルと硫酸マンガンに加えて、硫酸ジルコニウムを水に溶かして1.8mol/Lの混合水溶液を形成したこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、この混合水溶液では、各金属の元素モル比が、Ni:Mn:Zr=49.25:49.25:0.5となるように調整した。得られた複合水酸化物の組成は、Ni0.4925Mn0.4925Zr0.005(OH)2+a(0≦a≦0.5)であった。また、得られた正極活物質の組成は、Liが9.58質量%、Niが29.3質量%、Mnが27.5質量%、Zrが0.46質量%の組成であり、Li1.36 Ni0.4925Mn0.4925Zr0.005O2であることが確認された。
Li/Me=1.10となるように、水酸化リチウムと複合酸化物粒子を混合したこと以外は実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた正極活物質の組成は、Liが7.86質量%、Niが30.2質量%、Mnが28.3質量%の組成であり、Li1.10Ni0.50Mn0.50O2であることが確認された。
複合水酸化物粒子製造工程において、槽内温度を50℃、アンモニア濃度を20g/Lとしたこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であり、複合水酸化物粒子は実施例1と同様に針状の微細一次粒子(粒径およそ0.3μm)からなる中心部と、該中心部の外側にこの微細一次粒子よりも大きい板状の一次粒子(粒径およそ0.7μm)からなる外殻部とにより構成されていた。
複合水酸化物粒子製造工程において、粒子成長工程における、大気雰囲気から窒素雰囲気への切り替えを、粒子成長工程時間全体に対して25%の時点で行ったこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であり、複合水酸化物粒子は実施例1と同様に針状の微細一次粒子(粒径0.3μm)からなる中心部と、該中心部の外側にこの微細一次粒子よりも大きい板状の一次粒子(粒径0.5μm)からなる外殻部とにより構成されていた。
上部にオーバーフロー用配管を備えた連続晶析用の反応槽を用いて、大気雰囲気中で、反応水溶液のpH値を液温25℃基準で11.0の一定値に保ちながら、実施例1と同様の混合水溶液とアンモニア水溶液および水酸化ナトリウム溶液を一定流量で連続的に加えて、オーバーフローするスラリーを連続的に回収する、一般的な方法により晶析を行った。反応槽内の平均滞留時間を10時間として、連続槽内が平衡状態になってから、スラリーを回収して、固液分離して、晶析物を得たこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であった。複合水酸化物粒子全体が実施例1の外殻部と同様な一次粒子で構成されたため、正極活物質は緻密な中実構造の粒子となった。
核生成時と粒子成長時のpH値を、いずれも液温25℃基準で11.6の一定値に保ったこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。晶析中は、反応槽内空間に窒素ガスを5L/minで流通させて、酸素濃度が0.2容量%以下となるように保持した。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であった。複合水酸化物粒子全体が実施例1の外殻部と同様な一次粒子で構成されたため、正極活物質は緻密な中実構造の粒子となった。
核生成時と粒子成長時のpH値を、いずれも液温25℃基準で12.6の一定値に保ったこと以外は、実施例1と同様にして、ニッケルマンガン複合水酸化物を得た。晶析中は、反応槽内空間に窒素ガスを5L/minで流通させて、酸素濃度が0.2容量%以下となるように保持した。しかしながら、晶析反応全期間において新たな核が生成したために、粒度分布が広くゲル状の析出物を含む不定形の粒子となり、固液分離が困難であり処理を中止した。
焼成温度を1000℃としたこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であった。ただし、X線回折測定の結果から、六方晶の結晶構造が崩れており、正極活物質としての性能が期待できないため電池評価は行わなかった。
粒成長工程における大気雰囲気から窒素雰囲気への切り替えを、粒子成長工程開始時から粒子成長工程時間全体に対して50%の時点で行ったこと以外は、実施例1と同様にして、非水系電解質二次電池用正極活物質を得るとともに評価した。複合水酸化物の外殻部の厚さが不足していたため、正極活物質とする焼成段階において二次粒子同士の焼結が進み、粗大粒を含む活物質となった。なお、得られた複合水酸化物および正極活物質の組成は、実施例1と同様であった。


実施例1~7の複合水酸化物粒子および正極活物質は、本発明に従って製造されたため、平均粒径および粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕値のいずれもが、好ましい範囲にあり、粒径分布が良好で粒径がほぼ揃った粒子となっている。また、いずれの正極活物質も、凝集した一次粒子が焼結している外殻部と、その内側の中空部とからなる構造を備えている。これらの正極活物質を用いたコイン型電池は、初期放電容量が高く、サイクル特性に優れ、正極抵抗も低いものとなっており、優れた特性を有した電池となっている。
反応前水溶液のpH値を、液温25℃基準で12.8に、液中アンモニア濃度を10g/Lに調節するとともに、硫酸ニッケル、硫酸コバルト、硫酸マンガン、硫酸ジルコニウムを水に溶かして、各金属の元素モル比が、Ni:Co:Mn:Zr=33.2:33.1:33.3:0.5となるようにして得た1.8mol/Lの混合水溶液を用い、核生成時の反応水溶液のpH値を12.8に制御して、かつ、粒子成長工程開始時に窒素流通を開始し、大気雰囲気から窒素雰囲気に切り替える(粒子成長工程開始時から粒子成長工程時間全体に対して0%)とともに、アンモニア濃度を上記値に保持して、粒子成長時の反応水溶液のpH値を11.6に制御しつつ、2時間晶析し、上澄み液の半量抜き出し後、さらに2時間晶析したことを除き、実施例1と同様にして、Ni0.332Co0.331Mn0.332Zr0.005(OH)2+a(0≦a≦0.5)で表されるニッケルマンガン複合水酸化物を得た。
晶析反応時に、タングステン酸ナトリウム水溶液を同時に連続的に添加したこと以外は、実施例9と同様にして、Ni0.33Co0.33Mn0.33Zr0.005W0.005(OH)2+a(0≦a≦0.5)で表されるニッケルマンガン複合水酸化物を得て、かつ、加熱処理を行った。焼成温度を930℃としたこと以外は、実施例9と同様にして、Liが8.07質量%、Niが19.6質量%、Coが19.7質量%、Mnが18.3質量%、Zrが0.46質量%、Wが0.93質量%の組成で、Li1.15Ni0.33Co0.33Mn0.33Zr0.005W0.005O2で表される正極活物質を得た。
核生成工程でのpH値を12.6、混合水溶液の添加時間を2分30秒から30秒に変更し、粒子成長工程を開始して60分後から窒素流通を開始し、大気雰囲気から窒素雰囲気に切り替え(粒子成長工程開始時から粒子成長工程時間全体に対して25%)、核生成工程および粒子成長工程におけるアンモニア濃度を5g/Lに保持したこと以外は、実施例10と同様にして、ニッケルコバルトマンガン複合水酸化物を得た。また、得られた複合水酸化物を用いて、実施例10と同様にして、加熱処理と焼成を行い、正極活物質を得た。なお、得られた複合水酸化物および正極活物質の組成は、実施例10と同様であった。
Ni、Co、Mnの組成を、それぞれx=0.50、y=0.25、z=0.25となるように複合水酸化物の調製を行ったこと以外は、実施例11と同様にして、ニッケルマンガン複合水酸化物を得た。Li/M比を1.05、焼成条件を850℃、10時間としたこと以外は、実施例1と同様にして、Liが7.19質量%、Niが28.7質量%、Coが14.4質量%、Mnが13.4質量%、Zrが0.45質量%、Wが0.91質量%の組成で、Li1.05Ni0.495Co0.248Mn0.247Zr0.005W0.005O2で表される正極活物質を得た。
実施例9と同様にして得られた正極活物質を流動乾燥装置(株式会社パウレック製、MP-01)にて180℃の熱風を送りながら乾燥させると共に、タングステン酸リチウム水溶液を噴霧せしめ、正極活物質表面にタングステン酸リチウムを被覆させた正極活物質を得た。なお、得られた活物質の組成は実施例10と同様であり、断面SEM観察のEDX分析によりタングステンが活物質粒子の表面付近に多く存在することが確認された。
実施例9と同様にして得られた複合水酸化物をタングステン酸アンモニウム溶液に150g/Lとなるように分散し、スラリー化したのち、該スラリーをマイクロミストドライヤ(藤崎電機株式会社製、MDL-050M)を用いて噴霧乾燥し、タングステン酸アンモニウム塩を被覆させた複合水酸化物を得た以外は、実施例10と同様にして正極活物質を得た。なお、得られた活物質の組成は実施例10と同様であり、断面SEM観察のEDX分析によりタングステンが活物質粒子の表面付近に多く存在することが確認された。
焼成条件を1050℃、10時間としたこと以外は、実施例9と同様にして、複合酸化物正極活物質を得た。焼成温度が高く焼結が進んだために、比表面積が低く、正極抵抗値が高くなっていた。
核生成工程および粒子成長工程における晶析反応中に窒素を流通させて、非酸化性雰囲気(酸素:0.2容量%以下)としたこと以外は、実施例10と同様にして、正極活物質を得た。
核生成工程および粒子成長工程における晶析反応中に窒素を流通させて、非酸化性雰囲気(酸素:0.2容量%以下)としたこと以外は、実施例12と同様にして、リチウムニッケルコバルトマンガン複合酸化物を得た。


2 ケース
2a 正極缶
2b 負極缶
2c ガスケット
3 電極
3a 正極
3b 負極
3c セパレータ
Claims (21)
- 晶析反応によって一般式:NixMnyCozMt(OH)2+a(x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、0≦a≦0.5、Mは添加元素であり、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の元素)で表されるニッケルマンガン複合水酸化物粒子を製造する製造方法であって、
少なくともニッケルを含有する金属化合物およびマンガンを含有する金属化合物とアンモニウムイオン供給体とを含む核生成用水溶液を、液温25℃基準で、pH値が12.0~14.0となるように制御して、酸素濃度が1容量%を超える酸化性雰囲気中で核生成を行う核生成工程と、
該核生成工程において形成された核を含有する粒子成長用水溶液を、液温25℃基準におけるpH値が10.5~12.0となるように制御するとともに、粒子成長工程の開始時から粒子成長工程時間の全体に対して0~40%の範囲で前記酸化性雰囲気から酸素濃度1容量%以下の酸素と不活性ガスの混合雰囲気に切り替えて、前記核を成長させる粒子成長工程と、
を備えることを特徴とするニッケルマンガン複合水酸化物粒子の製造方法。 - 前記酸化性雰囲気の酸素濃度が10容量%以上である、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記混合雰囲気の酸素濃度が、0.5容量%以下である、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記粒子成長工程において、大気雰囲気から不活性ガスの混合雰囲気への切り替えを、粒子成長工程の開始時から粒子成長工程時間の全体に対して0~30%の範囲で行うことを特徴とする、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記粒子成長用水溶液として、前記核生成工程が終了した前記核生成用水溶液のpHを調整して形成されたものを用いることを特徴とする、請求項1記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記粒子成長用水溶液は、前記核生成工程において形成された核を含有する水溶液を、該核を形成した核生成用水溶液とは異なる水溶液に対して添加したものであることを特徴とする、請求項1記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記粒子成長工程において、前記粒子成長用水溶液の液体部の一部を排出することを特徴とする、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記核生成工程および前記粒子成長工程において、前記核生成用および粒子成長用水溶液の温度を、20℃以上に維持することを特徴とする、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 前記核生成工程および前記粒子成長工程において、各水溶液のアンモニア濃度を3~25g/Lの範囲内に維持することを特徴とする、請求項1に記載のニッケル複合水酸化物粒子の製造方法。
- 前記粒子成長工程で得られたニッケルマンガン複合水酸化物に、前記1種以上の添加元素を含む化合物を被覆することを特徴とする、請求項1に記載のニッケルマンガン複合水酸化物粒子の製造方法。
- 一般式:NixMnyCozMt(OH)2+a(x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、0≦a≦0.5、Mは添加元素であり、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の元素)で表され、複数の一次粒子が凝集して形成された略球状の二次粒子であり、該二次粒子は、平均粒径が3~7μmであり、粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕が0.55以下であり、微細一次粒子からなる中心部を有し、中心部の外側に該微細一次粒子よりも大きな板状一次粒子からなる外殻部を有することを特徴とする、ニッケルマンガン複合水酸化物粒子。
- 前記微細一次粒子は、平均粒径0.01~0.3μmであり、前記微細一次粒子よりも大きな板状一次粒子は、平均粒径0.3~3μmであることを特徴とする、請求項11に記載のニッケルマンガン複合水酸化物粒子。
- 前記外殻部の厚さは、前記二次粒子の粒径に対する比率で5~45%であることを特徴とする、請求項11に記載のニッケルマンガン複合水酸化物粒子。
- 前記1種以上の添加元素が、前記二次粒子の内部に均一に分布および/または該二次粒子の表面を均一に被覆していることを特徴とする、請求項11に記載のニッケルマンガン複合水酸化物粒子。
- 一般式:Li1+uNixMnyCozMtO2(-0.05≦u≦0.50、x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、Mは添加元素であり、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の元素)で表され、層状構造を有する六方晶系の結晶構造を有するリチウムニッケルマンガン複合酸化物からなる正極活物質の製造方法であって、
請求項11~14のいずれかに記載の前記ニッケルマンガン複合水酸化物粒子を、105~750℃の温度で熱処理する工程と、前記熱処理後の粒子に対してリチウム化合物を混合してリチウム混合物を形成する混合工程と、該混合工程で形成された前記混合物を、酸化性雰囲気中、800℃~980℃の温度で焼成する焼成工程とを備えることを特徴とする、非水系電解質二次電池用正極活物質の製造方法。 - 前記リチウム混合物は、該リチウム混合物に含まれるリチウム以外の金属の原子数の和とリチウムの原子数との比が、1:0.95~1.5となるように調整されることを特徴とする、請求項15に記載の非水系電解質二次電池用正極活物質の製造方法。
- 前記焼成工程において、焼成前に予め350℃~800℃の温度で仮焼を行うことを特徴とする、請求項15に記載の非水系電解質二次電池用正極活物質の製造方法。
- 前記焼成工程における酸化性雰囲気を、18容量%~100容量%の酸素を含有する雰囲気とすることを特徴とする、請求項15に記載の非水系電解質二次電池用正極活物質の製造方法。
- 一般式:Li1+uNixMnyCozMtO2(-0.05≦u≦0.50、x+y+z+t=1、0.3≦x≦0.7、0.1≦y≦0.55、0≦z≦0.4、0≦t≦0.1、Mは添加元素であり、Mg、Ca、Al、Ti、V、Cr、Zr、Nb、Mo、Wから選択される1種以上の元素)で表され、層状構造を有する六方晶系リチウム含有複合酸化物により構成されるリチウムニッケルマンガン複合酸化物からなる正極活物質であって、平均粒径が2~8μmであり、粒度分布の広がりを示す指標である〔(d90-d10)/平均粒径〕が0.60以下であり、凝集した一次粒子が焼結している外殻部と、その内側に存在する中空部とからなる中空構造を備えることを特徴とする、非水系電解質二次電池用正極活物質。
- 前記外殻部の厚さは、前記リチウムニッケルマンガン複合酸化物粒子の粒径に対する比率で5~45%であることを特徴とする、請求項19に記載の非水系電解質二次電池用正極活物質。
- 正極が、請求項19または20に記載の非水系電解質二次電池用正極活物質によって形成されていることを特徴とする、非水系電解質二次電池。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011530314A JP4915488B1 (ja) | 2011-03-28 | 2011-03-28 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| US13/820,110 US10017875B2 (en) | 2011-03-28 | 2011-03-28 | Nickel manganese composite hydroxide particles and manufacturing method thereof, cathode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and a non-aqueous electrolyte secondary battery |
| CN201180058151.7A CN103249678B (zh) | 2011-03-28 | 2011-03-28 | 镍锰复合氢氧化物粒子及其制造方法、非水系电解质二次电池用正极活性物质及其制造方法、以及非水系电解质二次电池 |
| EP11862712.4A EP2653447B1 (en) | 2011-03-28 | 2011-03-28 | Nickel-manganese composite hydroxide particles, method for producing same, positive electrode active material for nonaqueous electrolyte secondary batteries, method for producing positive electrode active material for nonaqueous electrolyte secondary batteries, and nonaqueous electrolyte secondary battery |
| PCT/JP2011/057694 WO2012131881A1 (ja) | 2011-03-28 | 2011-03-28 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| KR1020137008724A KR101272411B1 (ko) | 2011-03-28 | 2011-03-28 | 니켈 망간 복합 수산화물 입자와 그의 제조 방법, 비수계 전해질 이차 전지용 정극 활성 물질과 그의 제조 방법, 및 비수계 전해질 이차 전지 |
| US15/989,147 US10669646B2 (en) | 2011-03-28 | 2018-05-24 | Nickel manganese composite hydroxide particles and manufacturing method thereof, cathode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and a non-aqueous electrolyte secondary battery |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2011/057694 WO2012131881A1 (ja) | 2011-03-28 | 2011-03-28 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/820,110 A-371-Of-International US10017875B2 (en) | 2011-03-28 | 2011-03-28 | Nickel manganese composite hydroxide particles and manufacturing method thereof, cathode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and a non-aqueous electrolyte secondary battery |
| US15/989,147 Division US10669646B2 (en) | 2011-03-28 | 2018-05-24 | Nickel manganese composite hydroxide particles and manufacturing method thereof, cathode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and a non-aqueous electrolyte secondary battery |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012131881A1 true WO2012131881A1 (ja) | 2012-10-04 |
Family
ID=46170994
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/057694 Ceased WO2012131881A1 (ja) | 2011-03-28 | 2011-03-28 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US10017875B2 (ja) |
| EP (1) | EP2653447B1 (ja) |
| JP (1) | JP4915488B1 (ja) |
| KR (1) | KR101272411B1 (ja) |
| CN (1) | CN103249678B (ja) |
| WO (1) | WO2012131881A1 (ja) |
Cited By (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012252964A (ja) * | 2011-06-06 | 2012-12-20 | Sumitomo Metal Mining Co Ltd | 非水系電解質二次電池用正極活物質とその製造方法、ならびに、ニッケルコバルトマンガン複合水酸化物とその製造方法 |
| JP2014026990A (ja) * | 2013-11-01 | 2014-02-06 | Toyota Motor Corp | リチウムイオン二次電池 |
| WO2014181891A1 (ja) | 2013-05-10 | 2014-11-13 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、非水電解質二次電池用正極活物質とその製造方法および非水電解質二次電池 |
| JP2015140292A (ja) * | 2014-01-29 | 2015-08-03 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質、および、非水系電解質二次電池 |
| WO2015182665A1 (ja) * | 2014-05-29 | 2015-12-03 | 住友化学株式会社 | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 |
| WO2016013674A1 (ja) * | 2014-07-25 | 2016-01-28 | 住友金属鉱山株式会社 | ニッケルマンガン複合水酸化物粒子およびその製造方法 |
| JP2016031854A (ja) * | 2014-07-29 | 2016-03-07 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、およびそれを用いた非水系電解質二次電池用正極活物質の製造方法 |
| JP2016091626A (ja) * | 2014-10-30 | 2016-05-23 | 旭硝子株式会社 | 正極活物質、その製造方法、リチウムイオン二次電池用正極およびリチウムイオン二次電池 |
| JP2016094307A (ja) * | 2014-11-12 | 2016-05-26 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子の製造方法および非水電解質二次電池用正極活物質の製造方法 |
| EP3026740A4 (en) * | 2013-07-24 | 2016-09-21 | Sumitomo Metal Mining Co | ACTIVE MATERIAL FOR POSITIVE ELECTRODE OF A SECONDARY BATTERY WITH A WATER-FREE ELECTROLYTE AND METHOD FOR THE PRODUCTION THEREOF, AND A SECONDARY BATTERY WITH A WATER-FREE ELECTROLYTE THEREWITH |
| KR20170012248A (ko) | 2014-05-27 | 2017-02-02 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 해당 정극 활물질을 사용한 비수계 전해질 이차 전지 |
| JP2017134996A (ja) * | 2016-01-27 | 2017-08-03 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質とその製造方法、及び該正極活物質を用いた非水系電解質二次電池 |
| JP2017154915A (ja) * | 2016-02-29 | 2017-09-07 | 住友金属鉱山株式会社 | ニッケル複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに非水系電解質二次電池 |
| JP2017154916A (ja) * | 2016-02-29 | 2017-09-07 | 住友金属鉱山株式会社 | ニッケル複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに非水系電解質二次電池 |
| KR20180021681A (ko) | 2015-06-26 | 2018-03-05 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 전이 금속 함유 복합 수산화물과 그의 제조 방법, 비수전해질 이차 전지용 정극 활물질과 그의 제조 방법 및 비수전해질 이차 전지 |
| JP2018085198A (ja) * | 2016-11-22 | 2018-05-31 | 住友金属鉱山株式会社 | 遷移金属含有複合水酸化物の製造方法および非水電解質二次電池用正極活物質の製造方法 |
| JP2018104276A (ja) * | 2016-12-27 | 2018-07-05 | 住友金属鉱山株式会社 | 遷移金属含有複合水酸化物粒子およびその製造方法、並びに、非水電解質二次電池用正極活物質およびその製造方法 |
| WO2018123951A1 (ja) | 2016-12-26 | 2018-07-05 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| WO2019013053A1 (ja) | 2017-07-12 | 2019-01-17 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、及び、それを用いた非水電解質二次電池 |
| JP2019019047A (ja) * | 2017-07-12 | 2019-02-07 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、及び、それを用いた非水電解質二次電池 |
| KR20190040293A (ko) | 2016-08-31 | 2019-04-17 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 비수계 전해질 이차 전지 |
| CN109983604A (zh) * | 2016-11-22 | 2019-07-05 | 住友金属矿山株式会社 | 非水电解质二次电池用正极活性物质以及非水电解质二次电池 |
| JP2019175721A (ja) * | 2018-03-29 | 2019-10-10 | 三洋電機株式会社 | 非水電解質二次電池用正極の製造方法及び非水電解質二次電池の製造方法 |
| JP2020501329A (ja) * | 2016-12-12 | 2020-01-16 | ポスコPosco | リチウム二次電池用正極活物質、その製造方法、およびそれを含むリチウム二次電池 |
| WO2020027158A1 (ja) | 2018-07-31 | 2020-02-06 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 |
| JP2020074264A (ja) * | 2016-07-20 | 2020-05-14 | 三星エスディアイ株式会社Samsung SDI Co., Ltd. | リチウム二次電池用ニッケル系活物質、その製造方法、及びそれを含む正極を含んだリチウム二次電池 |
| WO2020195432A1 (ja) | 2019-03-27 | 2020-10-01 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質とその製造方法、及び、リチウムイオン二次電池 |
| WO2020195431A1 (ja) | 2019-03-27 | 2020-10-01 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質とその製造方法、及び、リチウムイオン二次電池 |
| US10840510B2 (en) | 2014-07-31 | 2020-11-17 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for non-aqueous electrolyte secondary battery and method for producing same |
| JP2020202193A (ja) * | 2020-09-18 | 2020-12-17 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質及び該正極活物質を用いた非水系電解質二次電池 |
| WO2021054468A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| WO2021054469A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| WO2021054466A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| US11024839B2 (en) | 2016-11-22 | 2021-06-01 | Sumitomo Metal Mining Co., Ltd. | Transition metal-containing composite hydroxide and production method thereof, and production method of positive electrode active material for nonaqueous electrolyte secondary battery |
| US11121368B2 (en) | 2015-11-27 | 2021-09-14 | Sumitomo Metal Mining Co., Ltd. | Positive electrode material for nonaqueous electrolyte secondary battery and method for producing the same, and positive electrode composite material paste, and nonaqueous electrolyte secondary battery |
| US11196048B2 (en) | 2016-09-13 | 2021-12-07 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary battery, method for producing the same, and nonaqueous electrolyte secondary battery containing the positive electrode active material |
| WO2022065443A1 (ja) | 2020-09-25 | 2022-03-31 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびその製造方法、リチウムイオン二次電池 |
| US11309542B2 (en) | 2016-12-08 | 2022-04-19 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, preparing method thereof, and lithium secondary battery including positive electrode including the same |
| US11411214B2 (en) | 2015-10-28 | 2022-08-09 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary batteries, production method thereof, positive electrode mixture material paste for nonaqueous electrolyte secondary batteries, and nonaqueous electrolyte secondary battery |
| WO2022191202A1 (ja) * | 2021-03-10 | 2022-09-15 | 茂 佐野 | 正極及び蓄電池 |
| US11456458B2 (en) | 2016-12-08 | 2022-09-27 | Samsung Sdi Co., Ltd. | Nickel-based active material precursor for lithium secondary battery, preparing method thereof, nickel-based active material for lithium secondary battery formed thereof, and lithium secondary battery comprising positive electrode including the nickel-based active material |
| US11569503B2 (en) | 2016-07-20 | 2023-01-31 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, method of preparing the same, and lithium secondary battery including positive electrode including the nickel-based active material |
| JP2024022236A (ja) * | 2022-08-05 | 2024-02-16 | トヨタ自動車株式会社 | リチウムイオン二次電池の正極活物質 |
| US12113213B2 (en) | 2013-07-24 | 2024-10-08 | Sumitomo Metal Mining Co., Ltd. | Cathode active material for non-aqueous electrolyte secondary battery and manufacturing method for same, and non-aqueous electrolyte secondary battery |
| JP2025058687A (ja) * | 2023-09-28 | 2025-04-09 | 本田技研工業株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池及びリチウムイオン二次電池用正極活物質の製造方法 |
| US12418041B2 (en) | 2016-03-24 | 2025-09-16 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for non-aqueous electrolyte secondary battery and method for manufacturing the same, positive electrode mixed material paste for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5392035B2 (ja) * | 2009-12-02 | 2014-01-22 | 住友金属鉱山株式会社 | 非水系電解質二次電池正極活物質用ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| US8945768B2 (en) | 2011-05-06 | 2015-02-03 | Toyota Jidosha Kabushiki Kaisha | Lithium-ion secondary battery |
| US20170050864A1 (en) * | 2011-05-30 | 2017-02-23 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous secondary batteries, method for producing same, and nonaqueous electrolyte secondary battery using positive electrode active material |
| WO2012164752A1 (ja) | 2011-05-30 | 2012-12-06 | 住友金属鉱山株式会社 | 非水系二次電池用正極活物質とその製造方法、ならびに該正極活物質を用いた非水系電解質二次電池 |
| JP5776996B2 (ja) * | 2011-05-30 | 2015-09-09 | 住友金属鉱山株式会社 | 非水系二次電池用正極活物質及びその正極活物質を用いた非水系電解質二次電池 |
| JP5741932B2 (ja) * | 2011-06-01 | 2015-07-01 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質の前駆体となる遷移金属複合水酸化物とその製造方法、及び非水系電解質二次電池用正極活物質の製造方法 |
| US10128501B2 (en) * | 2011-06-07 | 2018-11-13 | Sumitomo Metal Mining Co., Ltd. | Nickel composite hydroxide and manufacturing method thereof, cathode active material for nonaqueous-electrolyte secondary battery and manufacturing method thereof, and nonaqueous-electrolyte secondary battery |
| JP5370515B2 (ja) | 2012-02-22 | 2013-12-18 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極材料とその製造方法、および該正極材料を用いた非水系電解質二次電池 |
| KR101525000B1 (ko) * | 2012-09-24 | 2015-06-03 | 한국화학연구원 | 리튬이차전지의 양극 활물질용 니켈-망간 복합 수산화물의 제조방법, 이에 따라 제조된 니켈-망간 복합 수산화물 및 이를 포함하는 리튬이차전지용 양극 활물질 |
| KR101409973B1 (ko) * | 2012-11-15 | 2014-06-19 | 주식회사 포스코이에스엠 | 1차 입자의 꼭지각의 크기가 조절된 리튬 망간 복합 산화물, 및 이의 제조 방법 |
| JP6017978B2 (ja) | 2013-01-24 | 2016-11-02 | トヨタ自動車株式会社 | 正極活物質及び該活物質を用いたリチウム二次電池 |
| KR101538617B1 (ko) * | 2013-07-31 | 2015-07-22 | 전자부품연구원 | 나노크기의 이산화티탄이 포함된 구형의 니켈코발트망간수산화물을 이용한 고강도용 비수계 리튬이차전지용 양극재료 및 그의 제조 방법 |
| JP6142929B2 (ja) * | 2014-01-31 | 2017-06-07 | 住友金属鉱山株式会社 | ニッケルマンガン複合水酸化物粒子とその製造方法、非水電解質二次電池用正極活物質とその製造方法、および非水電解質二次電池 |
| JP6252384B2 (ja) * | 2014-06-27 | 2017-12-27 | 住友金属鉱山株式会社 | ニッケル複合水酸化物及びその製造方法、正極活物質及びその製造方法、並びに非水系電解質二次電池 |
| JP6658534B2 (ja) * | 2014-10-30 | 2020-03-04 | 住友金属鉱山株式会社 | ニッケル含有複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| JP6287970B2 (ja) * | 2014-10-30 | 2018-03-07 | 住友金属鉱山株式会社 | ニッケル複合水酸化物とその製造方法 |
| JP6265117B2 (ja) * | 2014-12-22 | 2018-01-24 | 住友金属鉱山株式会社 | ニッケルコバルトマンガン複合水酸化物とその製造方法 |
| KR101913906B1 (ko) * | 2015-06-17 | 2018-10-31 | 주식회사 엘지화학 | 이차전지용 양극활물질, 이의 제조방법 및 이를 포함하는 이차전지 |
| DE102015115691B4 (de) | 2015-09-17 | 2020-10-01 | Zentrum für Sonnenenergie- und Wasserstoff-Forschung Baden-Württemberg Gemeinnützige Stiftung | Lithium-Nickel-Mangan-basierte Übergangsmetalloxidpartikel, deren Herstellung sowie deren Verwendung als Elektrodenmaterial |
| JP6924657B2 (ja) * | 2017-09-11 | 2021-08-25 | 株式会社田中化学研究所 | 電池用正極活物質に用いられる遷移金属複合水酸化物粒子の製造方法 |
| WO2019087493A1 (ja) | 2017-10-31 | 2019-05-09 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質とその製造方法、及び正極活物質を用いた非水系電解質二次電池 |
| JP6495997B1 (ja) | 2017-11-20 | 2019-04-03 | 住友化学株式会社 | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 |
| US11251430B2 (en) | 2018-03-05 | 2022-02-15 | The Research Foundation For The State University Of New York | ϵ-VOPO4 cathode for lithium ion batteries |
| CN108711621B (zh) * | 2018-05-25 | 2021-05-11 | 上海应用技术大学 | 一种碳掺杂双金属氧化物材料及其制备方法 |
| JP7310117B2 (ja) * | 2018-10-26 | 2023-07-19 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、リチウムイオン二次電池用正極活物質とその製造方法、及び、それを用いたリチウムイオン二次電池 |
| WO2020152768A1 (ja) * | 2019-01-22 | 2020-07-30 | 住友金属鉱山株式会社 | ニッケルマンガンコバルト複合水酸化物、ニッケルマンガンコバルト複合水酸化物の製造方法及び、リチウムニッケルマンガンコバルト複合酸化物 |
| US12454465B2 (en) | 2019-01-22 | 2025-10-28 | Sumitomo Metal Mining Co., Ltd. | Nickel manganese cobalt composite hydroxide, method for producing nickel manganese cobalt composite hydroxide, lithium nickel manganese cobalt composite oxide, and lithium ion secondary battery |
| WO2020153093A1 (ja) * | 2019-01-22 | 2020-07-30 | 住友金属鉱山株式会社 | ニッケルマンガンコバルト複合水酸化物、ニッケルマンガンコバルト複合水酸化物の製造方法、リチウムニッケルマンガンコバルト複合酸化物及び、リチウムイオン二次電池 |
| WO2020153094A1 (ja) | 2019-01-22 | 2020-07-30 | 住友金属鉱山株式会社 | ニッケルマンガンコバルト複合水酸化物、ニッケルマンガンコバルト複合水酸化物の製造方法、リチウムニッケルマンガンコバルト複合酸化物及び、リチウムイオン二次電池 |
| WO2020152771A1 (ja) * | 2019-01-22 | 2020-07-30 | 住友金属鉱山株式会社 | ニッケルマンガンコバルト複合水酸化物、ニッケルマンガンコバルト複合水酸化物の製造方法及び、リチウムニッケルマンガンコバルト複合酸化物 |
| WO2020209239A1 (ja) * | 2019-04-11 | 2020-10-15 | Jfeミネラル株式会社 | 前駆体、前駆体の製造方法、正極材、正極材の製造方法、および、リチウムイオン二次電池 |
| CN111180689B (zh) * | 2019-12-30 | 2022-01-25 | 中南大学 | 微米空心多孔复合球状钠离子电池正极材料及其制备方法 |
| JP7392151B2 (ja) * | 2020-03-27 | 2023-12-05 | 寧徳時代新能源科技股▲分▼有限公司 | 二次電池、当該二次電池を含む電池モジュール、電池パック及び装置 |
| EP4186867A4 (en) * | 2020-07-21 | 2024-09-04 | Sumitomo Metal Mining Co., Ltd. | METHOD FOR PRODUCING NICKEL-CONTAINING HYDROXIDE, METHOD FOR PRODUCING POSITIVE ELECTRODE ACTIVE MATERIAL FOR LITHIUM ION SECONDARY BATTERIES, POSITIVE ELECTRODE ACTIVE MATERIAL FOR LITHIUM ION SECONDARY BATTERIES, AND LITHIUM ION SECONDARY BATTERY |
| CN113666434A (zh) * | 2021-08-13 | 2021-11-19 | 格林爱科(荆门)新能源材料有限公司 | 一种镍钴锰三元前驱体晶种的制备方法 |
| CN113979489B (zh) * | 2021-12-27 | 2022-03-22 | 金驰能源材料有限公司 | 一种晶面可控的中空正极材料的前驱体及其制备方法 |
| CN116014103B (zh) * | 2023-01-03 | 2026-01-02 | 宁波容百新能源科技股份有限公司 | 一种高镍三元正极材料及其制备方法和应用 |
| CN116525815B (zh) * | 2023-06-30 | 2023-11-17 | 宜宾锂宝新材料有限公司 | 球形镍锰酸锂正极材料、其制备方法和复合正极材料 |
| EP4525086A4 (en) * | 2023-07-31 | 2025-09-24 | Beijing Easpring Mat Tech Co Ltd | LITHIUM-RICH MANGANESE OXIDE POSITIVE ELECTRODE MATERIAL, PREPARATION METHOD AND USE THEREOF, AND POSITIVE ELECTRODE SHEET AND USE THEREOF |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08315822A (ja) * | 1995-05-18 | 1996-11-29 | Japan Energy Corp | リチウム2次電池正極活物質原料用Ni−Mn複合水酸化物粉末及びその製造方法 |
| JP2003017049A (ja) * | 2001-06-27 | 2003-01-17 | Toyota Central Res & Dev Lab Inc | リチウム二次電池正極活物質用リチウム遷移金属複合酸化物およびその製造方法 |
| JP2003086182A (ja) | 2001-09-13 | 2003-03-20 | Matsushita Electric Ind Co Ltd | 正極活物質、その製造方法および非水電解質二次電池 |
| JP2004193115A (ja) * | 2002-11-27 | 2004-07-08 | Nichia Chem Ind Ltd | 非水電解質二次電池用正極活物質および非水電解質二次電池 |
| JP2004210560A (ja) | 2002-12-27 | 2004-07-29 | Tanaka Chemical Corp | マンガンニッケル複合水酸化物粒子 |
| JP2004253174A (ja) | 2003-02-18 | 2004-09-09 | Nichia Chem Ind Ltd | 非水電解液二次電池用正極活物質 |
| WO2008013208A1 (en) * | 2006-07-26 | 2008-01-31 | Agc Seimi Chemical Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary battery and method for producing the same |
| JP2008120679A (ja) * | 2000-11-16 | 2008-05-29 | Hitachi Maxell Ltd | リチウム含有複合酸化物およびその製造方法、並びに非水二次電池 |
| JP2008147068A (ja) | 2006-12-12 | 2008-06-26 | Ise Chemicals Corp | 非水電解液二次電池用リチウム複合酸化物 |
| JP2008235157A (ja) * | 2007-03-23 | 2008-10-02 | Matsushita Electric Ind Co Ltd | リチウム二次電池用正極活物質 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69700735T2 (de) * | 1996-08-29 | 2000-03-02 | Murata Mfg. Co., Ltd. | Lithium-Sekundärbatterie |
| US7585435B2 (en) * | 2000-11-06 | 2009-09-08 | Tanaka Chemical Corporation | High density cobalt-manganese coprecipitated nickel hydroxide and process for its production |
| KR100628797B1 (ko) * | 2000-11-16 | 2006-09-26 | 히다치 막셀 가부시키가이샤 | 리튬 함유 복합 산화물을 포함하는 양극 활물질 |
| CN100382363C (zh) * | 2002-09-26 | 2008-04-16 | 清美化学股份有限公司 | 锂二次电池用正极活性物质及其制备方法 |
| JP2004210550A (ja) | 2002-12-26 | 2004-07-29 | Canon Inc | モールド成形金型 |
| US8728666B2 (en) * | 2005-04-28 | 2014-05-20 | Nissan Motor Co., Ltd. | Positive electrode material for lithium ion battery with nonaqueous electrolyte, and battery using the same |
| JP4781004B2 (ja) * | 2005-04-28 | 2011-09-28 | パナソニック株式会社 | 非水電解液二次電池 |
| JP4884859B2 (ja) | 2006-07-05 | 2012-02-29 | 大和製衡株式会社 | 箱詰め装置 |
| JP5137414B2 (ja) * | 2007-02-20 | 2013-02-06 | 住友金属鉱山株式会社 | 非水電解液二次電池用正極活物質およびその製造方法、ならびに、該正極活物質を用いた非水電解液二次電池 |
| JP5135843B2 (ja) * | 2007-03-26 | 2013-02-06 | 三菱化学株式会社 | リチウム遷移金属複合酸化物、および、それを用いたリチウム二次電池用正極、ならびに、それを用いたリチウム二次電池 |
| JP5481786B2 (ja) * | 2007-07-03 | 2014-04-23 | 住友化学株式会社 | リチウム複合金属酸化物 |
| US8632698B2 (en) * | 2007-07-26 | 2014-01-21 | Lg Chem, Ltd. | Electrode active material having core-shell structure |
| KR101106429B1 (ko) | 2009-12-01 | 2012-01-18 | 삼성에스디아이 주식회사 | 이차 전지 |
| JP4941617B2 (ja) * | 2009-12-02 | 2012-05-30 | 住友金属鉱山株式会社 | ニッケル複合水酸化物粒子および非水系電解質二次電池 |
-
2011
- 2011-03-28 KR KR1020137008724A patent/KR101272411B1/ko active Active
- 2011-03-28 WO PCT/JP2011/057694 patent/WO2012131881A1/ja not_active Ceased
- 2011-03-28 EP EP11862712.4A patent/EP2653447B1/en active Active
- 2011-03-28 JP JP2011530314A patent/JP4915488B1/ja active Active
- 2011-03-28 CN CN201180058151.7A patent/CN103249678B/zh active Active
- 2011-03-28 US US13/820,110 patent/US10017875B2/en active Active
-
2018
- 2018-05-24 US US15/989,147 patent/US10669646B2/en active Active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08315822A (ja) * | 1995-05-18 | 1996-11-29 | Japan Energy Corp | リチウム2次電池正極活物質原料用Ni−Mn複合水酸化物粉末及びその製造方法 |
| JP2008120679A (ja) * | 2000-11-16 | 2008-05-29 | Hitachi Maxell Ltd | リチウム含有複合酸化物およびその製造方法、並びに非水二次電池 |
| JP2003017049A (ja) * | 2001-06-27 | 2003-01-17 | Toyota Central Res & Dev Lab Inc | リチウム二次電池正極活物質用リチウム遷移金属複合酸化物およびその製造方法 |
| JP2003086182A (ja) | 2001-09-13 | 2003-03-20 | Matsushita Electric Ind Co Ltd | 正極活物質、その製造方法および非水電解質二次電池 |
| JP2004193115A (ja) * | 2002-11-27 | 2004-07-08 | Nichia Chem Ind Ltd | 非水電解質二次電池用正極活物質および非水電解質二次電池 |
| JP2004210560A (ja) | 2002-12-27 | 2004-07-29 | Tanaka Chemical Corp | マンガンニッケル複合水酸化物粒子 |
| JP2004253174A (ja) | 2003-02-18 | 2004-09-09 | Nichia Chem Ind Ltd | 非水電解液二次電池用正極活物質 |
| WO2008013208A1 (en) * | 2006-07-26 | 2008-01-31 | Agc Seimi Chemical Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary battery and method for producing the same |
| JP2008147068A (ja) | 2006-12-12 | 2008-06-26 | Ise Chemicals Corp | 非水電解液二次電池用リチウム複合酸化物 |
| JP2008235157A (ja) * | 2007-03-23 | 2008-10-02 | Matsushita Electric Ind Co Ltd | リチウム二次電池用正極活物質 |
Non-Patent Citations (2)
| Title |
|---|
| CHEMISTRY LETTERS, vol. 30, no. 8, 2001, pages 744 |
| See also references of EP2653447A4 |
Cited By (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012252964A (ja) * | 2011-06-06 | 2012-12-20 | Sumitomo Metal Mining Co Ltd | 非水系電解質二次電池用正極活物質とその製造方法、ならびに、ニッケルコバルトマンガン複合水酸化物とその製造方法 |
| JP2016154143A (ja) * | 2013-05-10 | 2016-08-25 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、非水電解質二次電池用正極活物質とその製造方法および非水電解質二次電池 |
| US11283072B2 (en) | 2013-05-10 | 2022-03-22 | Sumitomo Metal Mining Co., Ltd. | Transition metal composite hydroxide particles and production method thereof, cathode active material for non-aqueous electrolyte rechargeable battery and production method thereof, and nonaqueous electrolyte rechargeable battery |
| JPWO2014181891A1 (ja) * | 2013-05-10 | 2017-02-23 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、非水電解質二次電池用正極活物質とその製造方法および非水電解質二次電池 |
| CN105122517A (zh) * | 2013-05-10 | 2015-12-02 | 住友金属矿山株式会社 | 过渡金属复合氢氧化粒子及其制造方法、非水电解质二次电池用正极活性物质及其制造方法、以及非水电解质二次电池 |
| EP3007254A4 (en) * | 2013-05-10 | 2016-12-21 | Sumitomo Metal Mining Co | TRANSITION METAL COMPOSITE HYDROXIDE PARTICLES, METHOD FOR THE MANUFACTURE THEREOF, POSITIVE ELECTRODE ACTIVE MATERIAL FOR SECONDARY BATTERIES WITH A WATER-FREE ELECTROLYTE, METHOD FOR THE PRODUCTION THEREOF, AND A SECONDARY BATTERY WITH A WATER-FREE ELECTROLYTE |
| KR20160006172A (ko) | 2013-05-10 | 2016-01-18 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 전이 금속 복합 수산화물 입자와 그의 제조 방법, 비수전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 비수전해질 이차 전지 |
| WO2014181891A1 (ja) | 2013-05-10 | 2014-11-13 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、非水電解質二次電池用正極活物質とその製造方法および非水電解質二次電池 |
| US10424787B2 (en) | 2013-05-10 | 2019-09-24 | Sumitomo Metal Mining Co., Ltd. | Transition metal composite hydroxide particles and production method thereof, cathode active material for non-aqueous electrolyte rechargeable battery and production method thereof, and nonaqueous electrolyte rechargeable battery |
| US20160093885A1 (en) * | 2013-05-10 | 2016-03-31 | Sumitomometal Mining Co., Ltd. | Transition metal composite hydroxide particles and production method thereof, cathode active material for non-aqueous electrolyte rechargeable battery and production method thereof, and nonaqueous electrolyte rechargeable battery |
| US12113213B2 (en) | 2013-07-24 | 2024-10-08 | Sumitomo Metal Mining Co., Ltd. | Cathode active material for non-aqueous electrolyte secondary battery and manufacturing method for same, and non-aqueous electrolyte secondary battery |
| EP3026740A4 (en) * | 2013-07-24 | 2016-09-21 | Sumitomo Metal Mining Co | ACTIVE MATERIAL FOR POSITIVE ELECTRODE OF A SECONDARY BATTERY WITH A WATER-FREE ELECTROLYTE AND METHOD FOR THE PRODUCTION THEREOF, AND A SECONDARY BATTERY WITH A WATER-FREE ELECTROLYTE THEREWITH |
| JP2014026990A (ja) * | 2013-11-01 | 2014-02-06 | Toyota Motor Corp | リチウムイオン二次電池 |
| JP2015140292A (ja) * | 2014-01-29 | 2015-08-03 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質、および、非水系電解質二次電池 |
| KR102373071B1 (ko) | 2014-05-27 | 2022-03-11 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 해당 정극 활물질을 사용한 비수계 전해질 이차 전지 |
| US10256505B2 (en) | 2014-05-27 | 2019-04-09 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary batteries, production method thereof, and nonaqueous electrolyte secondary battery including said material |
| KR20170012248A (ko) | 2014-05-27 | 2017-02-02 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 해당 정극 활물질을 사용한 비수계 전해질 이차 전지 |
| WO2015182665A1 (ja) * | 2014-05-29 | 2015-12-03 | 住友化学株式会社 | リチウム二次電池用正極活物質、リチウム二次電池用正極及びリチウム二次電池 |
| US10938019B2 (en) | 2014-05-29 | 2021-03-02 | Sumitomo Chemical Company, Limited | Positive electrode active material for lithium secondary batteries, positive electrode for lithium secondary batteries, and lithium secondary battery |
| JPWO2016013674A1 (ja) * | 2014-07-25 | 2017-04-27 | 住友金属鉱山株式会社 | ニッケルマンガン複合水酸化物粒子およびその製造方法 |
| WO2016013674A1 (ja) * | 2014-07-25 | 2016-01-28 | 住友金属鉱山株式会社 | ニッケルマンガン複合水酸化物粒子およびその製造方法 |
| JP2016031854A (ja) * | 2014-07-29 | 2016-03-07 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子とその製造方法、およびそれを用いた非水系電解質二次電池用正極活物質の製造方法 |
| US10840510B2 (en) | 2014-07-31 | 2020-11-17 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for non-aqueous electrolyte secondary battery and method for producing same |
| JP2016091626A (ja) * | 2014-10-30 | 2016-05-23 | 旭硝子株式会社 | 正極活物質、その製造方法、リチウムイオン二次電池用正極およびリチウムイオン二次電池 |
| JP2016094307A (ja) * | 2014-11-12 | 2016-05-26 | 住友金属鉱山株式会社 | 遷移金属複合水酸化物粒子の製造方法および非水電解質二次電池用正極活物質の製造方法 |
| KR20180021681A (ko) | 2015-06-26 | 2018-03-05 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 전이 금속 함유 복합 수산화물과 그의 제조 방법, 비수전해질 이차 전지용 정극 활물질과 그의 제조 방법 및 비수전해질 이차 전지 |
| US11404690B2 (en) | 2015-06-26 | 2022-08-02 | Sumitomo Metal Mining Co., Ltd. | Transition metal-containing composite hydroxide and manufacturing method thereof, positive electrode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and non-aqueous electrolyte secondary battery |
| US10547052B2 (en) | 2015-06-26 | 2020-01-28 | Sumitomo Metal Mining Co., Ltd. | Transition metal-containing composite hydroxide and manufacturing method thereof, positive electrode active material for a non-aqueous electrolyte secondary battery and manufacturing method thereof, and non-aqueous electrolyte secondary battery |
| US11411214B2 (en) | 2015-10-28 | 2022-08-09 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary batteries, production method thereof, positive electrode mixture material paste for nonaqueous electrolyte secondary batteries, and nonaqueous electrolyte secondary battery |
| US11121368B2 (en) | 2015-11-27 | 2021-09-14 | Sumitomo Metal Mining Co., Ltd. | Positive electrode material for nonaqueous electrolyte secondary battery and method for producing the same, and positive electrode composite material paste, and nonaqueous electrolyte secondary battery |
| JP2017134996A (ja) * | 2016-01-27 | 2017-08-03 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質とその製造方法、及び該正極活物質を用いた非水系電解質二次電池 |
| JP2017154916A (ja) * | 2016-02-29 | 2017-09-07 | 住友金属鉱山株式会社 | ニッケル複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに非水系電解質二次電池 |
| JP2017154915A (ja) * | 2016-02-29 | 2017-09-07 | 住友金属鉱山株式会社 | ニッケル複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに非水系電解質二次電池 |
| US12418041B2 (en) | 2016-03-24 | 2025-09-16 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for non-aqueous electrolyte secondary battery and method for manufacturing the same, positive electrode mixed material paste for non-aqueous electrolyte secondary battery, and non-aqueous electrolyte secondary battery |
| US11569503B2 (en) | 2016-07-20 | 2023-01-31 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, method of preparing the same, and lithium secondary battery including positive electrode including the nickel-based active material |
| JP2022113728A (ja) * | 2016-07-20 | 2022-08-04 | 三星エスディアイ株式会社 | リチウム二次電池用ニッケル系活物質、その製造方法、及びそれを含む正極を含んだリチウム二次電池 |
| US11302919B2 (en) | 2016-07-20 | 2022-04-12 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, method of preparing the same, and lithium secondary battery including positive electrode including the nickel-based active material |
| US11742482B2 (en) | 2016-07-20 | 2023-08-29 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, method of preparing the same, and lithium secondary battery including positive electrode including the nickel-based active material |
| JP2020074264A (ja) * | 2016-07-20 | 2020-05-14 | 三星エスディアイ株式会社Samsung SDI Co., Ltd. | リチウム二次電池用ニッケル系活物質、その製造方法、及びそれを含む正極を含んだリチウム二次電池 |
| KR20190040293A (ko) | 2016-08-31 | 2019-04-17 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 비수계 전해질 이차 전지 |
| US11165062B2 (en) | 2016-08-31 | 2021-11-02 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary batteries, method for producing same, and nonaqueous electrolyte secondary battery |
| US11196048B2 (en) | 2016-09-13 | 2021-12-07 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary battery, method for producing the same, and nonaqueous electrolyte secondary battery containing the positive electrode active material |
| CN109983604A (zh) * | 2016-11-22 | 2019-07-05 | 住友金属矿山株式会社 | 非水电解质二次电池用正极活性物质以及非水电解质二次电池 |
| JP2018085198A (ja) * | 2016-11-22 | 2018-05-31 | 住友金属鉱山株式会社 | 遷移金属含有複合水酸化物の製造方法および非水電解質二次電池用正極活物質の製造方法 |
| US11024839B2 (en) | 2016-11-22 | 2021-06-01 | Sumitomo Metal Mining Co., Ltd. | Transition metal-containing composite hydroxide and production method thereof, and production method of positive electrode active material for nonaqueous electrolyte secondary battery |
| US11456458B2 (en) | 2016-12-08 | 2022-09-27 | Samsung Sdi Co., Ltd. | Nickel-based active material precursor for lithium secondary battery, preparing method thereof, nickel-based active material for lithium secondary battery formed thereof, and lithium secondary battery comprising positive electrode including the nickel-based active material |
| US11309542B2 (en) | 2016-12-08 | 2022-04-19 | Samsung Sdi Co., Ltd. | Nickel-based active material for lithium secondary battery, preparing method thereof, and lithium secondary battery including positive electrode including the same |
| US11670766B2 (en) | 2016-12-12 | 2023-06-06 | Posco Holdings Inc. | Positive electrode active material for lithium secondary battery, method for preparing same, and lithium secondary battery comprising same |
| JP2020501329A (ja) * | 2016-12-12 | 2020-01-16 | ポスコPosco | リチウム二次電池用正極活物質、その製造方法、およびそれを含むリチウム二次電池 |
| US11735726B2 (en) | 2016-12-26 | 2023-08-22 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for nonaqueous electrolyte secondary battery, method for producing the same, and nonaqueous electrolyte secondary battery |
| KR20190095302A (ko) | 2016-12-26 | 2019-08-14 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법 및 비수계 전해질 이차 전지 |
| WO2018123951A1 (ja) | 2016-12-26 | 2018-07-05 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 |
| JP2018104276A (ja) * | 2016-12-27 | 2018-07-05 | 住友金属鉱山株式会社 | 遷移金属含有複合水酸化物粒子およびその製造方法、並びに、非水電解質二次電池用正極活物質およびその製造方法 |
| JP7087381B2 (ja) | 2016-12-27 | 2022-06-21 | 住友金属鉱山株式会社 | 遷移金属含有複合水酸化物粒子およびその製造方法、並びに、非水電解質二次電池用正極活物質およびその製造方法 |
| WO2019013053A1 (ja) | 2017-07-12 | 2019-01-17 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、及び、それを用いた非水電解質二次電池 |
| JP2019019047A (ja) * | 2017-07-12 | 2019-02-07 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、及び、それを用いた非水電解質二次電池 |
| JP7293576B2 (ja) | 2017-07-12 | 2023-06-20 | 住友金属鉱山株式会社 | 金属複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、及び、それを用いた非水電解質二次電池 |
| KR102532481B1 (ko) * | 2017-07-12 | 2023-05-16 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 금속 복합 수산화물과 그의 제조 방법, 비수전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 그것을 사용한 비수전해질 이차 전지 |
| KR20200040760A (ko) | 2017-07-12 | 2020-04-20 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 금속 복합 수산화물과 그의 제조 방법, 비수전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 그것을 사용한 비수전해질 이차 전지 |
| JP2019175721A (ja) * | 2018-03-29 | 2019-10-10 | 三洋電機株式会社 | 非水電解質二次電池用正極の製造方法及び非水電解質二次電池の製造方法 |
| US12087944B2 (en) | 2018-07-31 | 2024-09-10 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for lithium ion secondary battery, method of manufacturing positive electrode active material for lithium ion secondary battery, and lithium ion secondary battery |
| WO2020027158A1 (ja) | 2018-07-31 | 2020-02-06 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極活物質の製造方法、リチウムイオン二次電池 |
| WO2020195431A1 (ja) | 2019-03-27 | 2020-10-01 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質とその製造方法、及び、リチウムイオン二次電池 |
| WO2020195432A1 (ja) | 2019-03-27 | 2020-10-01 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質とその製造方法、及び、リチウムイオン二次電池 |
| US12315916B2 (en) | 2019-09-19 | 2025-05-27 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for lithium ion secondary battery and lithium ion secondary battery |
| WO2021054468A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| US12562383B2 (en) | 2019-09-19 | 2026-02-24 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for lithium ion secondary battery and lithium ion secondary battery |
| US12315924B2 (en) | 2019-09-19 | 2025-05-27 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for lithium ion secondary battery and lithium ion secondary battery |
| WO2021054466A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| WO2021054469A1 (ja) | 2019-09-19 | 2021-03-25 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびリチウムイオン二次電池 |
| JP2020202193A (ja) * | 2020-09-18 | 2020-12-17 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質及び該正極活物質を用いた非水系電解質二次電池 |
| JP7069534B2 (ja) | 2020-09-18 | 2022-05-18 | 住友金属鉱山株式会社 | 非水系電解質二次電池用正極活物質及び該正極活物質を用いた非水系電解質二次電池 |
| US12555786B2 (en) | 2020-09-25 | 2026-02-17 | Sumitomo Metal Mining Co., Ltd. | Positive electrode active material for lithium ion secondary battery, method for producing the same, and lithium ion secondary battery |
| KR20230074480A (ko) | 2020-09-25 | 2023-05-30 | 스미토모 긴조쿠 고잔 가부시키가이샤 | 리튬 이온 이차 전지용 정극 활물질 및 그 제조 방법, 리튬 이온 이차 전지 |
| WO2022065443A1 (ja) | 2020-09-25 | 2022-03-31 | 住友金属鉱山株式会社 | リチウムイオン二次電池用正極活物質およびその製造方法、リチウムイオン二次電池 |
| JP2022140180A (ja) * | 2021-03-10 | 2022-09-26 | 茂 佐野 | 正極及び蓄電池 |
| WO2022191202A1 (ja) * | 2021-03-10 | 2022-09-15 | 茂 佐野 | 正極及び蓄電池 |
| JP2024022236A (ja) * | 2022-08-05 | 2024-02-16 | トヨタ自動車株式会社 | リチウムイオン二次電池の正極活物質 |
| JP7666445B2 (ja) | 2022-08-05 | 2025-04-22 | トヨタ自動車株式会社 | リチウムイオン二次電池の正極活物質 |
| JP2025058687A (ja) * | 2023-09-28 | 2025-04-09 | 本田技研工業株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池及びリチウムイオン二次電池用正極活物質の製造方法 |
| JP7692969B2 (ja) | 2023-09-28 | 2025-06-16 | 本田技研工業株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池及びリチウムイオン二次電池用正極活物質の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2653447B1 (en) | 2015-05-20 |
| CN103249678A (zh) | 2013-08-14 |
| JPWO2012131881A1 (ja) | 2014-07-24 |
| US10669646B2 (en) | 2020-06-02 |
| KR101272411B1 (ko) | 2013-06-07 |
| CN103249678B (zh) | 2016-06-15 |
| US20140011090A1 (en) | 2014-01-09 |
| JP4915488B1 (ja) | 2012-04-11 |
| US10017875B2 (en) | 2018-07-10 |
| KR20130044369A (ko) | 2013-05-02 |
| US20180347069A1 (en) | 2018-12-06 |
| EP2653447A4 (en) | 2014-06-11 |
| EP2653447A1 (en) | 2013-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4915488B1 (ja) | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および非水系電解質二次電池 | |
| KR101644252B1 (ko) | 니켈 복합 수산화물과 그의 제조 방법, 비수계 전해질 이차 전지용 정극 활물질과 그의 제조 방법, 및 비수계 전해질 이차 전지 | |
| JP4894969B1 (ja) | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに、非水系電解質二次電池 | |
| JP4840545B1 (ja) | ニッケル複合水酸化物粒子および非水系電解質二次電池 | |
| KR102553596B1 (ko) | 니켈 망간 함유 복합 수산화물 및 그 제조 방법 | |
| JP5708277B2 (ja) | ニッケルマンガン複合水酸化物粒子とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、ならびに非水系電解質二次電池 | |
| JP5316726B2 (ja) | ニッケル複合水酸化物とその製造方法、非水系電解質二次電池用正極活物質とその製造方法、および、非水系電解質二次電池 | |
| JP5880426B2 (ja) | ニッケル複合水酸化物及びその製造方法、並びに正極活物質の製造方法 | |
| JP5590337B2 (ja) | マンガン複合水酸化物粒子、非水系電解質二次電池用正極活物質、および非水系電解質二次電池と、それらの製造方法 | |
| JP6252383B2 (ja) | マンガンコバルト複合水酸化物及びその製造方法、正極活物質及びその製造方法、並びに非水系電解質二次電池 | |
| WO2016208413A1 (ja) | 遷移金属含有複合水酸化物とその製造方法、非水電解質二次電池用正極活物質とその製造方法、および非水電解質二次電池 | |
| JP6168004B2 (ja) | マンガン複合水酸化物及びその製造方法、正極活物質及びその製造方法、並びに非水系電解質二次電池 | |
| JP2016011227A (ja) | ニッケル複合水酸化物及びその製造方法、正極活物質及びその製造方法、並びに非水系電解質二次電池 | |
| JPWO2017119451A1 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JPWO2017119459A1 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP7224754B2 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP2016031854A (ja) | 遷移金属複合水酸化物粒子とその製造方法、およびそれを用いた非水系電解質二次電池用正極活物質の製造方法 | |
| JP7135354B2 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP7338133B2 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP2019021426A (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP2019003818A (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP7119302B2 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP7319026B2 (ja) | 非水系電解質二次電池用正極活物質前駆体、非水系電解質二次電池用正極活物質、非水系電解質二次電池用正極活物質前駆体の製造方法、非水系電解質二次電池用正極活物質の製造方法 | |
| JP2016033926A (ja) | 正極活物質及びその製造方法、並びに非水系電解質二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2011530314 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11862712 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011862712 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20137008724 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13820110 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |