WO2012155199A1 - Amine derivatives as potassium channel blockers - Google Patents
Amine derivatives as potassium channel blockers Download PDFInfo
- Publication number
- WO2012155199A1 WO2012155199A1 PCT/AU2012/000538 AU2012000538W WO2012155199A1 WO 2012155199 A1 WO2012155199 A1 WO 2012155199A1 AU 2012000538 W AU2012000538 W AU 2012000538W WO 2012155199 A1 WO2012155199 A1 WO 2012155199A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- halo
- hal
- compound according
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C1CC*CC1 Chemical compound C1CC*CC1 0.000 description 11
- JBJBXTLMDUZPDN-UHFFFAOYSA-N CC(c1nnc[n]1C1=C)NC1C1=CCC(C(F)(F)F)N=C1 Chemical compound CC(c1nnc[n]1C1=C)NC1C1=CCC(C(F)(F)F)N=C1 JBJBXTLMDUZPDN-UHFFFAOYSA-N 0.000 description 1
- MKYZDLFUMKCSMM-KTKRTIGZSA-N CCCC(C(C1CC1)N)/C=C\S(CC)(=C)=O Chemical compound CCCC(C(C1CC1)N)/C=C\S(CC)(=C)=O MKYZDLFUMKCSMM-KTKRTIGZSA-N 0.000 description 1
- SKSOZQWNDAZDHP-SDVJBAMBSA-N CCS(c1cc(C(C2CC2)N(Cc2ccc(C(F)(F)F)nc2)C([C@@H](C2)[C@H]2c(cc2)ccc2F)=O)ccc1)(=O)=O Chemical compound CCS(c1cc(C(C2CC2)N(Cc2ccc(C(F)(F)F)nc2)C([C@@H](C2)[C@H]2c(cc2)ccc2F)=O)ccc1)(=O)=O SKSOZQWNDAZDHP-SDVJBAMBSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/08—Antibacterial agents for leprosy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/78—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton
- C07C217/80—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings
- C07C217/82—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings of the same non-condensed six-membered aromatic ring
- C07C217/90—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton having amino groups and etherified hydroxy groups bound to carbon atoms of non-condensed six-membered aromatic rings of the same non-condensed six-membered aromatic ring the oxygen atom of at least one of the etherified hydroxy groups being further bound to a carbon atom of a six-membered aromatic ring, e.g. amino-diphenylethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/32—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/16—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with acylated ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/38—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/16—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom containing only one pyridine ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/70—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/08—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/26—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/12—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D411/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms
- C07D411/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D411/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen and sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Definitions
- the present invention relates to compounds useful in the modulation of potassium channel activity in cells, in particular the activity of Kv1.3 channels found in T cells.
- the invention also relates to the use of these compounds in the treatment or prevention of autoimmune and inflammatory diseases, including multiple sclerosis, pharmaceutical compositions containing these compounds and methods for their preparation.
- Potassium channels represent a complex class of voltage-gated ion channels from both functional and structural standpoints. Their functions include regulating neurotransmitter release, heart rate, insulin secretion, neuronal excitability, epithelial electrolyte transport, smooth muscle contraction, and cell volume.
- four sequence-related potassium channel genes - shaker, shaw. shab. and shal - have been identified in Drosophila, and each has been shown to have human homolog(s).
- KCNA3 encodes the voltage-gated Kv1.3 potassium channel, which is shaker-related and is expressed in lymphocytes (T and B lymphocytes), the central nervous system, fat and other tissues.
- the functional channel is composed of four identical ⁇ 1 ⁇ 3 ⁇ -sub units.
- the K v 1.3 potassium channel regulates membrane potential and thereby indirectly influences calcium signaling in human effector- memory T cells (Grissmer S. et al, Proc. Natl. Acad. Sci. U.S.A. 87(23): 9411 -5; DeCoursey T.E. et al, Nature 307 (5950): 465-8; Chandy K.G. et al. Trends Pharmacol. Sci. 25(5); 280-9; Wulff H. et al, J. Clin. Invest. 111 (1 ): 1703-13). Effector memory T cells are important mediators of multiple sclerosis, Type I diabetes mellltus, psoriasis, and rheumatoid arthritis.
- the K v 1.3 channel is expressed in T and B lymphocytes in a distinct pattern thai depends on the state of lymphocyte activation and differentiation.
- naive and central memory T cells increase expression of the Ca3.1 channel per cell
- effector- memory T cells Increase expression of the K v 1.3 channel.
- human B cells naive and early memory B cells express small numbers of K v 1.3 and KCa3.1 channels when they are quiescent, and augment KCa3.1 expression after activation.
- class- switched memory B cells express high numbers of 1.3 channels per cell (about 1500/cell) and this number increases after activation (Chandy K. G. et al, Trends Pharmacol. Sci. 25(5): 280-9; Wulff H.
- Kv1.3 The K v 1.3 channel promotes the calcium homeostasis required for T-cell receptor-mediated cell activation, gene transcription, and proliferation (Panyi, G et al (2004) Trends Immunol 25:565-569). Kv1.3 is physically coupled through a series of adaptor proteins to the T-cell receptor signaling complex and it traffics to the Immunological synapse during antigen presentation. However, blockade of the channel does not prevent immune synapse formation (Panyi G. et al. Proc. Natl. Acad. Sci.
- K V 1.3 and KCa3.1 regulate membrane potential and calcium signaling of T cells. Calcium entry through the CRAC channel is promoted by potassium efflux through the Kv1.3 and KCa3.1 potassium channels. Blockade of K v 1.3 channels in effector-memory T cells suppresses activities like calcium signaling, cytokine production (interferon-gamma, interleukin 2) and cell proliferation.
- Effector-memory T cells were originally defined by their expression of cell surface markers, and can enter sites of Inflammation in non- lymphoid tissues, while not participating in the process of lymphoid recirculation carried out by most other lymphocytes.
- TEMs have been shown to uniquely express high numbers of the K v 1.3 potassium channel and depend on these channels for their function.
- K v 1.3 blockers paralyze effector-memory T cells at the sites of inflammation and prevent their reactivation in inflamed tissues. In contrast.
- K v 1.3 blockers do not affect the homing to and motility within lymph nodes of naive and central memory T cells, most likely because these cells express the KCa3.1 channel and are therefore protected from the effect of K v 1.3 blockade. Suppressing the function of these cells by selectively blocking the Kv1 -3 channel offers the potential for highly effective therapy of autoimmune diseases with minimal effects on either beneficial immune responses or other organs (Chandy K.G. et al, Trends Pharmacol. Sci. 25(5): 280-9; Wulff H. et al, J. Clin. Invest. 1 1 1 (11): 1703-13; Beeton C et al, Proc. Natl. Acad. Sci.
- Kv1.3 has been reported to be expressed in the inner mitochondrial membrane in lymphocytes.
- the apoptotic protein Bax has been suggested to insert into the outer membrane of the mitochondria and occlude the pore of K 1.3 via a lysine residue.
- Kv1.3 blockade may contribute to apoptosie (Szabo I. et al, J. Biot. Chem. 280(13): 12790-8; Szabo I. et al., Proc. Natl. Acad Sci. U.S.A. 105(39): 14861-6).
- Autoimmune Disease is a family of disorders resulting from tissue damage caused by a malfunctioning immune system, affecting tens of millions of people worldwide. Such diseases may be restricted to a single organ, as e.g. in multiple sclerosis and Type I diabetes mellitus, or may involve multiple organs as in the case of rheumatoid arthritis and systemic lupus erythematosus. Treatment is generally palliative and typically includes anti-inflammatory and immunosuppressive drugs. The severe side effects of many of these therapies have fueled a continuing search for more effective and selective immunosuppressive drugs. Among these are those which can selectivel .
- Multiple sclerosis is a disease caused by autoimmune damage to the central nervous system including the brain, which affects roughly two and a half million people worldwide. Symptoms include muscle weakness and paralysis, and the disease can progress rapidly and unpredictably and may eventually lead to death. Treatment usually includes the use of anti-inflammatory and immunosuppressive drugs which have potentially severe side effects.
- K V 1 3 has been shown to be highly expressed in autoreactive effector memory T cells from MS patients ( ulff, H et al (2003) J Clin Invest 111 :1703-1713; Rus H et al (2005) PNAS 102:11094- 1 099).
- Animal models of multiple sclerosis have been successfully treated using blockers of the K v 1.3 potassium channel.
- disease-associated myelin-specific T cells from the blood are predominantly co-stimulation independent effector-memory T cells that express high numbers of K v 1.3 channels.
- T cells in MS lesions in postmortem brain lesions are also predominantly effector-memory T cells that express high levels of the K v 1.3 channel (Wulff H. et al, J. Clin. Invest. 111(11): 1703-13; Beeton C. et al, Proc. Natl, Acad. Sci. U.S.A. 103(46): 17414-9).
- Type 1 diabetes mellitus is a disease caused by autoimmune destruction of insulin- producing cells in the pancreas, resulting in high blood sugar and other metabolic abnormalities.
- Type 1 diabetes mellitus affects close to four hundred thousand people in the US alone, and is usually diagnosed before age 20. Its long-term consequences may include blindness, nerve damage and kidney failure, and left untreated is rapidly fatal. Treatment involves life-long administration of insulin or pancreas transplantation, both of which may entail serious side effects (Beeton C. et al, Proc. Natl. Acad. Sci. U.S.A. 103(46): 17414-9).
- v 1.3 is also considered a therapeutic target for the treatment of obesity, for enhancing peripheral insulin sensitivity in patients with type-2 diabetes mellitus, for preventing bone resorption fn periodontal disease, for rheumatoid arthritis, for inflammatory skin conditions, such as psoriasis, and for asthma (Tucker K. et al. Int. J. Obes. (Lond) 32(8): 1222-32; Xu J. et al, Hum. ol Genet. 12(5): 551-9; Xu J. et al. Proc. Natl. Acad. Sci. U.S.A. 101(9): 3112-7; Valverde P. et al, J. Dent.
- K v 1.3 blockers are thus potential therapeutic agents as immunosuppressants or Immune system modulators including for the prevention of graft rejection, and the treatment of autoimmune and inflammatory disorders.
- K v 1.3 modulators may be used alone or in conjunction with other immunosuppressants, such as selective KCa3.1 blockers or cyclosporin, in order to possibly achieve synergism and/or to reduce toxicity, especially of cyclosporin.
- immunosuppressants such as selective KCa3.1 blockers or cyclosporin
- 5,494,895 discloses the use of a thirty-one amino acid peptide, scorpion peptide margatoxin, as a selective inhibitor and probe of K v 1 3 channels present in human lymphocytes, and also as an immunosuppressant. However the use of this compound is limited by its potent toxicity.
- the present invention provides compounds of Formula (I) and related Formulae, and pharmaceutical compositions thereof.
- compounds of Formula (I) have potency and selectivity in the prevention and treatment of conditions that have been associated with autoimmune disorders, immune-mediated disorders, inflammatory disorders, or other disorders, or conditions which benefit clinically from immunosuppressants, including multiple sclerosis, type-1 diabetes mellitus, type-2 diabetes mellitus, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, contact dermatitis, obesity, graft-versus host disease, transplant rejection, and delayed type hypersensitivity.
- compounds, pharmaceutical compositions and methods provided are useful to treat, prevent or ameliorate a range of conditions In mammals such as, but not limited to, immune disorders and autoimmune diseases of various genesis or etiology, for example rheumatoid arthritis, multiple sclerosis, psoriasis, type 1 diabetes, graft-versus host disease, transplant rejection.
- compounds, pharmaceutical compositions and methods provided are useful as antiinflammatory agents for the treatment of arthritis, and as agents to treat Parkinson's Disease, Alzheimer's Disease, asthma, myocardial infarction, neurodegenerative disorders, inflammatory bowel disease and autoimmune disorders, renal disorders, obesity, eating disorders, cancer, schizophrenia, epilepsy, sleeping disorders, cognitive disorders, depression, anxiety, blood pressure, and lipid disorders.
- the present invention provides compounds of Formula (I):
- G 1 denotes a single bond
- G 2 denotes a CO group
- X is selected from a single bond, an alkylene group having 1 to 6 carbon atoms optionally substituted with 1 or 2 substituents selected from fluoro or CrCe-alkyl,
- Y is selected from an alkylene group having 1 to 6 carbon atoms optionally substituted one or two times with C 3 -C e -cycloalkyl or d-C 3 -alkyl; or a 3-8- membered cycloalkylene group,
- Q . is selected from O, NH or a single bond
- W is selected from SO, S0 2 or a single bond.
- U is cyctoalkyi, cycloalkenyi, heterocyclyl or heteroaryl, each of the above groups being optionally substituted with 1 to 3 substitutent6 selected from Hal, N0 2 , CN, - S j-C,-C e -alkyl. -S-Ci-Ce-alkyl. N e 2 .
- V is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, N0 2 . CN, SO z -Ci-Ce alkyl, NMe 2 , C C e -alkyl, O-d-Ce-alkyl, -(CH z ) m -0-Ci-C 6 -alkyl, -CrCe-halo-alkyl. 0-Ci-C e -halo-alkyl or a 5-6-mBmbered heteroaromatic group,
- T denotes phenyl, triezoly), thiazolyl, oxazolyl, oxadiazolyl. or pyrazolyt.
- R 1 is Hal, -Ci-Ce-alkyl. O-C-Ce-alkyl, -(CHz O-C-Ca-alkyl, 0-C,-C e -haio-alkyl,
- O-C-Ce-halo-alkyl O-C-Ce-halo-alkyl, -SOj-Ci-Ce-alkyl, -(CH 2 ) m -S0 2 -Ci-C e -alkyl, -SOrCi-Ce-halo- alkyl, - ⁇ CH 2 ) m -SO z -C,-C B -halo-alkyl, -S0 2 -3-8-cyeioalkyl, -(CHj SOrS-S- cycloalkyl, cyano or -Ci-Ce-halo-alkyl,
- R 2 and R a are independently from one another H, Hal, -Ci-Ce-alkyl, -0-C,-C 6 -alkyl, -(CH 2 ), «-0-Ci-C B -alkyl, 0-C C 6 -halo-alkyl, -(CH z ) m -0-C r C e -halo-alkyl, -S0 2 -C,-C 6 - alkyl, -(CH a ) m -S ⁇ VCrC 6 -alkyl, .SO r C r C e -halo-alkyl, alkyl. -S0 2 -3-8-cycloalkyl, -(CH 2 ) m -SOj-3-8-cycloalkyl, -Ci-Ce-haio-alkyl, or
- R 1 and R 2 are linked to form with the ring T to which they are attached a 7-12-membered fused heterocyclyl or 7-12-membered fused cycloalkyl. each of which may be optionally substituted with 1 to 3 Hal, - ( -VCe-halo-alkyl, N0 2 , CN, C C e -alkyl. - (CH 2 ) m -0-C,-Ce-alkyl. or -0-Ci-C 6 -alkyl,
- R 3 is CrCe-alkyl, Ci-C e -hal08lkyl,
- aloalkyl a 3-8-membered cycloalkyl group, optionally substituted with 1 to 3 substitutents independently selected from Hal, or CrCe-alkyl; or a 3-8-membered heterocyclic group, optionally substituted with 1 to 3 substitutents independently selected from Hal, -Ci-Ce-halo-alkyl, N0 2 , CN.
- Ci-Ce-alkyl -(CH 2 ) m - O-d-Ce-alkyl, -O-Ci-Ce-alkyl, -(CH 2 ) m - O-C Ci-halo-alkyl, -(CH 2 ) m -S02-Ci-C 6 -halo-alkyl, -S0 2 -Ci-C e 'halo-alkyl, -0-C C e - halo-alkyl. -C(0)-C,-C 6 -alkyl. or -C(0)0-C C e -alkyl,
- R 4 denotes H, C ⁇ Ce-alkyl, or forms together with R 3 a 3-8-membered cycloalkyl ring, optionally substituted with Hal, -d-Ce-halo-alkyl, N0 2 , CN.
- Ci-Ce-alkyl, -(CHa -O- Ci-C e -alkyl, -O-C.-Ce-alkyl. -C(0).Ci-Ce-alkyl, or -C(0)0-C,-C e -alkyl, m is selected from 1. 2, 3 or 4, preferably 1 or 2,
- Hal is F, CI, Br, or I, wherein -G 2 -Y-W together is at least 3 atoms in length, as well as pharmaceutically acceptable salts thereof, or is an enantiomeric mixture of 2 enantiomers in all ratios, and/or as a mixture of diastereoisomers in all ratios.
- the present invention provides a kit or a set comprising at least one compound of Formula (I) or related Formulae, preferably in combination with immunomodulating agents.
- the kit consists of separate packs of; (a) an effective amount of a compound of the Formula (I) and/or pharmaceutically usable derivatives, solvates, salts, hydrates and stereoisomers thereof, including mixtures thereof in all ratios, and
- the present invention relates to pharmaceutical compositions comprising a compound provided herein, and a pharmaceutical carrier, excipient or diluent.
- the pharmaceutical composition can comprise one or more of the compounds described herein. It will be understood that compounds provided herein useful in the pharmaceutical compositions and treatment methods disclosed herein, can be pharmaceutically acceptable as prepared and used.
- the present invention relates to methods for preventing, treating or ameliorating a condition from among those listed herein, particularly conditions that are associated with immune-mediated reactions, autoimmune conditions, or other conditions which are modulated by immunosuppression.
- these conditions are multiple sclerosis, type-1 diabetes mellitus. rheumatoid arthritis, p$oriasls, contact dermatitis, obesity, systemic lupus erythematosus, graft-versus host disease, and transplant rejection, which method comprises administering to a mammal in need thereof an amount of one or more of the compounds provided herein, or pharmaceutical composition thereof, effective to prevent, treat or ameliorate the condition.
- the present invention extends to the use of any of the compounds of the invention for the preparation of medicaments that may be administered for such treatments, as well as to such compounds for the treatments disclosed and specified.
- the present invention is directed to methods for synthesizing the compounds described herein, with representative synthetic protocols and pathways described below.
- the Invention provides new compounds which can modulate the activity of the voltage gated potassium channel K v 1.3, and thus avert or treat any maladies that may be causally related to aberrations in such activity.
- the Invention also provides a series of compounds that can treat or alleviate maladies or symptoms of same, such as immune-mediated disorders and autoimmune diseases, that may be causally related to the activation of the Kv1.3 channel.
- the invention also provides a series of compounds that can treat a disease or condition, wherein the disease or condition is selected from: Acute disseminated encephalomyelitis (ADE ), Addison's disease, Allopecia areata, Alzhelmers disease, Ankylosing spondylitis, Antiphospholipid antibody syndrome, Autoimmune hemolytic anemia, Autoimmune hepatitis. Autoimmune inner ear disease, Autoimmune Lymphoproliferative Syndrome (ALPS), Autoimmune polyendocrine/polyglandular syndrome, Autoimmune thrombocytoipenia purpura, Bale disease, Behcet disease. Bullous pemphigoid, Cardiomyopathy.
- ADE Acute disseminated encephalomyelitis
- Addison's disease Addison's disease
- Allopecia areata Allopecia areata
- Alzhelmers disease Ankylosing spondylitis
- Antiphospholipid antibody syndrome Autoimmune hemolytic
- Celiac sprue-dermatitis herpetiformis Chronic fatigue immune dysfunction syndrome (CFIDS), Chronic inflammatory demyelinating neuropathy, Cicatrical pemphigoid, Coeliac disease, Cold agglutinin disease.
- CIDS Chronic fatigue immune dysfunction syndrome
- Chronic inflammatory demyelinating neuropathy Cicatrical pemphigoid
- Coeliac disease Cold agglutinin disease.
- CREST syndrome Crohn's disease
- Cystic fibrosis Degos disease
- Dermatomyositis Diabetes (Type I or Juvenile onset)
- Early onset dementia Eczema.
- Endotoxin shock Essential mixed cryoglobulinemia, Familial Mediterranean fever, Fibromyalgia, Fibromyositis, Goodpasture's syndrome, Graves' disease, Guillain-Barre syndrome (GBS), Hashimoto's t yroldosis. Hidradenitis suppurativa, Idiopathic pulmonary fibrosis, Idiopathic thrombocytopenic purpura, IgA nephropathy, Lambert-Eaton Myasthenic Syndrome, Leukemia, Lichen planus, Meniere disease, Mixed connective tissue disease, Multiple sclerosis.
- Multiphasic disseminated encephalomyelitis Myasthenia gravis, Neuromyelitis Optica, Paraneoplastic Syndromes, Pemphigus, Pemphigus vulgaris, Pernicious anaemia, Polyarteritis nodosum, Polychondritis, Polymyalgia rhematica, Polymyositis, Primary agammaglobulinemia, Primary biliary cirrhosis.
- Transplant or Allograft rejection Ulcerative colitis, Uveitis, Vasculitis, Vitiligo, Graft vs Host disease, pustular psoriasis, and Wegener's granulomatosis.
- the invention further provides a series of compounds that can treat a disease or condition, wherein the disease or condition is selected from: resistance by transplantation of organs or tissue, graft- versus-host diseases brought about by medulla ossium transplantation, rheumatoid arthritis, systemic lupus, erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, type I diabetes uveitis, juvenile- onset or recent-onset diabetes mellitus.
- the disease or condition is selected from: resistance by transplantation of organs or tissue, graft- versus-host diseases brought about by medulla ossium transplantation, rheumatoid arthritis, systemic lupus, erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, type I diabetes uveitis, juvenile- onset or recent-onset diabetes mellitus.
- posterior uveitis allergic encephalomyelitis, glomerulonephritis, infectious diseases caused by pathogenic microorganisms, inflammatory and hyperprollferative skin diseases, psoriasis, atopical dermatitis, contact dermatitis, eczematous dermatitises, seborrhoeis dermatitis, Lichen planus, Pemphigus, bullous pemphigoid. Epidermolysis bullosa, urticaria angioedemas.
- vasculitides erythemas, cutaneous eosinophilias, Lupus erythematosus, acne, Alopecia areata, keratoconjunctivitis, vernal conjunctivitis, uveitis associated with Behcet's disease, keratitis, herpetic keratitis, conical cornea, dystrophia epitheliatis corneae, corneal leukoma, ocular pemphigus, Mooren's ulcer, Scleritis, Graves' opthalmopathy, Vogt- Koyanagi-Harada syndrome, sarcoidosis, pollen allergies, reversible obstructive airway disease, bronchial asthma, allergic asthma, intrinsic asthma, extrinsic asthma, dust asthma, chronic or inveterate asthma, late asthma and airway hyper-responsiveness, bronchitis, gastric ulcers, vascular damage caused by ischemic diseases and thrombosis, ischemic bowel diseases
- the invention further provides pharmaceutical compositions that are effective in the treatment or prevention of a variety of disease states, including the diseases associated with the central nervous system, cardiovascular conditions, chronic pulmonary obstructive disease (COPD), inflammatory bowel disease, rheumatoid arthritis, osteoarthritis, and other diseases where an immunological inflammatory component or autoimmune component is present.
- COPD chronic pulmonary obstructive disease
- inflammatory bowel disease rheumatoid arthritis
- osteoarthritis osteoarthritis
- compounds of the present invention are used in the treatment and prophylaxis of a condition selected from Multiple sclerosis, Rheumatoid arthritis, Psoriasis, Type 1 Diabetes, Type II Diabetes, Systemic lupus nephritis, Oncology. Glomerulonephritis. Sjogrens's syndrome, Transplant rejection, Graft versus host disease, Allergic contact dermatitis, Neointimal hyperplasia/restenosis, Periodontal disease, Leprosy, and Obesity.
- a condition selected from Multiple sclerosis, Rheumatoid arthritis, Psoriasis, Type 1 Diabetes, Type II Diabetes, Systemic lupus nephritis, Oncology. Glomerulonephritis. Sjogrens's syndrome, Transplant rejection, Graft versus host disease, Allergic contact dermatitis, Neointimal hyperplasia/restenosis, Periodontal disease, Leprosy, and Obe
- W is S0 2 .
- Q is a single bond. ln an embodiment Q is a single bond, and W is S0 2 .
- Q and W are single bonds.
- G 2 -Y-W together is from 3-6 atoms in length.
- G 2 -Y-W together is 3 atoms in length.
- G 2 -Y-W together is 4 atoms in length.
- G 2 -Y-W together is 5 atoms in length.
- G 2 -Y-W together is 6 atoms in length.
- G 2 -Y-W together is 3 atoms in length and W is a single bond.
- G a -Y-W together is 3 atoms in length and W is S0 2 .
- G 2 -Y-W together is 3 atoms in length and Q is a single bond.
- G 2 -Y-W together is 4 atoms in length and W is a single bond.
- G 2 -Y-W together is 4 atoms in length and W is S0 2 .
- G 2 -Y-W together is 4 atoms in length and Q is a single bond.
- G -Y-W together is 5 atoms in length and W is a single bond.
- G s -Y-W together is 5 atoms in length and W is S0 2 .
- G 2 -Y-W together is 5 atoms in length and Q is a single bond.
- G 2 -Y-W together is 6 atoms in length and W is a single bond. ln an embodiment G 2 -Y-W together is 6 atoms in length and W is S0 2 .
- G 2 -Y-W together Is 6 atoms in length and Q is a single bond.
- -G 2 -Y-W- is selected from one of the following:
- V is an optionally substituted phenyl group.
- V is an optionally substituted phenyl group
- Q is a single bond
- V is an optionally substituted phenyl group
- W is S0 2 .
- V is an optionally substituted phenyl group
- Q is a single bond
- W is S0 2 .
- V Is an optionally substituted phenyl group.
- Q and W are single bonds. Accordingly, in a further embodiment the invention relates to compounds of formula (la):
- X is selected from a single bond, an alkylene group having 1 to 6 carbon atoms optionally substituted with 1 or 2 substituents selected from fluoro or C-rCe-alkyl,
- Y is selected from an alkylene group having 1 to 6 carbon atoms optionally substituted one or two times with CyCa-cycloalkyl or Ci-Cs-alkyl; or a 3-8- membered cycloalkyiene group,
- Q is selected from O, NH or a single bond
- ⁇ V Is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, N0 2 , CN, S0 2 -Ci-C e alkyl, NMej. CVCs-alkyl, 0-C,-C e -alkyl.
- U is cycloalkyl. cycloalkenyl, heterocyclyl or heteroaryl, each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal, N0 2 , CN. - SOj-CrCe-a!kyl. -S-C-Ce-alkyl, NMe 4 . C,-C 6 -alkyl, -C(0)0-C r C e -alkyl. ⁇ - ⁇ ,- ⁇ - alkyl, -(CHiJm-O-C Ce-alk l, -C Ce-halo-alkyl, or a 5-6-membered heteroaromatic group being optionally substituted by Hal,
- T denotes phenyl, triazolyl, thiazolyl, oxazolyl, oxadiazoly!, or pyrazolyl
- R 1 is Hal, -C-Ce-alkyl, 0-C,-Ce-alkyl, -(CH ⁇ nrO-d-Ce-alkyl, 0-Ci-C e -halo-alkyl. -(CH z ) m - O-CrCe-halo-alkyl, -SOj-C Ce-alkyl, -(CH ⁇ -SCVCrCe-alkyl.
- R 2 and R 3 ⁇ 4 are independently from one another H, Hal, -C r C e -alkyl, -0-Ci-C e -alkyl, -(CH 2 ) rn -0-C 1 -C e -alkyl, 0-C r Ce-halo-alkyl, -(CH 2 ) m -0-Ci-C e -halo-alkyl, -S0 2 -C r C e - alkyl, -(CH 2 ) m -S0 2 -C 1 -C e -alkyl, -S0 2 -C n -C 6 -halo-alkyl,
- R 1 and R 2 are linked to form with the ring T to which they are attached a 7 ⁇ 12-membered fused heterocyclyl or 7-12-membered fused cycloalkyi, each of which may be optionally substituted with 1 to 3 Hal, d-Ce-halo-alkyl, N0 2 , CN, CrC 6 -alkyl. - (CHzJm-O-d-Ce-alkyl, or -O-C C -alkyl,
- R 3 is CVCe-alkyl, C,-C 6 -haloalkyl. -(CH ⁇ -Od-Ce-alkyl. or -(ChfeJnrO-C-Ce- haloalkyl; a 3-8-membered cycloalkyi group, optionally substituted with 1 to 3 substitutents independently selected from Hal, -Cr-Ce-halo-alkyl, or Ci-Cs-alkyl; or a 3-8-membered heterocyclic group, optionally substituted with 1 to 3 substitutents independently selected from Hal, -C C e -halo-alkyl.
- R 4 denotes H, Ci-Ce-alkyl, or forms together with R s a 3-8-membered cycloalkyi ring, optionally substituted with Hal. -Ci-Ce-halo-alkyl, N0 2 , CN, CrCe-alkyl, - ⁇ CH 2 ) m - - Ci-Ce-alkyl, -0-Ci-C e -alkyl, -C(0)-C t -Ce-alkyl.
- each m is independently selected from 1 , 2, 3, or 4 preferably 1 or 2; as well as pharmaceutically acceptable salte thereof, or is an enantiomeric mixture of 2 enantiomers In all ratios, and/or as a mixture of diastereoisomers in all ratios.
- X is an aikylene group having 1 to 4 carbon atoms, optionally substituted with 1 or 2 substituents selected from fluoro or Ci-C 8 -el yl.
- X is selected from methylene or ethylene.
- X is methylene
- X is a single bond.
- Y is an aikylene group having 1 to 4 carbon atoms, optionally substituted one or two times with C3-C 8 -cycloalkyl or Ci-C 3 -alkyl.
- Y is selected from methylene, ethylene, propylene, isopropylene, or tertbutylene.
- Y is a 3-8-membered cycloalkylene group, or 3-8-membered cycloalkenylene.
- Y is a 3-membered cycloalkylene.
- Q is a single bond.
- the compound of the invention is a compound of formula (la 1 ). In an embodiment the compound of the invention is a compound of formula (la”). In an embodiment the compound of the invention is a compound of formula (la 1 "). In an embodiment the compound of the invention is a compound of formula (la lv ). ln an embodiment the compound of the invention is a compound of formula (la v ).
- the compound of the invention Is a compound of formula (la vl ).
- the compound of the invention is a compound of formula (la 1 ), (la 111 ), (la ⁇ ), wherein Q is a single bond.
- the compound of the invention is a compound of formula (la la ).
- the compound of the invention is a compound of formula (la" la ).
- the compound of the invention is a compound of formula (la vla ).
- the invention contemplates the following further general formulae: wherein 1 , R 2 , R z , R 3 and R 4 , T, U and V are as defined above for compounds of formula (la).
- X selected from a single bond, an alkylene group having 1 to 6 carbon atoms optionally substituted with 1 or 2 substituents selected from fluoro or Ci-Ce-alkyl,
- Y is a 3-membered cycloalkylene group
- Q is selected from O. NH or a single bond
- V is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, N0 2 , CN, S0 2 -d-Ce alkyl, N e*. d-Ce-alkyl, O-d-d-alkyl, -(CH 2 ) m -0-Ci-C6-alkyl, -CrCe-halo-alkyl, 0-d-C $ -halo-alkyl or a 5-6-membered heteroaromatic group,
- U is cycloalkyl, cycloalkenyl, heterocyclyl or heteroaryl, each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal, N0 2 , CN, ⁇ SOa-CvCe-alkyl, -S-Ci-C e -alkyl.
- T denotes phenyl, triazolyl, thiazolyl, oxazolyl, oxadiazol l, or pyrazolyl
- R 1 is Hal, -C-Ce-alkyl, 0-C,-C e -alkyl, -(CHa O-C d-alkyl. 0-Ci-C 6 -halo-alkyl, - ⁇ CH 2 ) m - 0-d-C 6 -halo-alkyl. -S0 2 -Ci-C e -alkyl, - ⁇ CH 2 ) m -SCvd-C e -alkyl.
- R 2 and R are Independently from one another H, Hal, -Ci-C 6 -alkyl, -0-Ci-C e -alkyl, (CH 2 ) m -0-C 1 -C 6 -alkyl, O-Ci-C 6 -hal0-alkyl, -(CH 2 ) m -O-C C e -hal0-alkyl, -S0 2 -Ci-C e - alkyl, -(CH 2 ) m -S0 2 -Ci-C6-alkyl ( -S0 2 -C,-C 6 -halo-alkyl.
- R and R 2 are linked to form with the ring T to which they are attached a 7-12-membered fused heterocyclyl or 7-12-membered fused cycloalkyl. each of which may be optionally substituted with 1 to 3 Hal, -d-C e -halo-alkyl, C ⁇ , CN, d-Ce-alkyl, - (CH 2 ) m -0-Ci-C e -alkyl, or -O-CrCe-alkyl,
- R 3 is C-Ce-alkyl, C,-C 6 -haloalkyl, -(CH ⁇ .O-d-d-alkyl. or -(CH 2 ), negligence-0-Ci-C6- haloalkyl; a 3-8-membered cycloalkyl group, optionally substituted with 1 to 3 substitutents Independently selected from Hal. -C,-C e -halo-alkyl, or d-Cs-alkyl; or a 3-8-membered heterocyclic group, optionally substituted with 1 to 3 substitutents independently selected from Hal, -d-drhalo-alkyl.
- R* denotes H, d-Cj-alkyl, or forms together with R 3 a 3-8-membered cycloalkyi ring, optionally substituted with Hal, -C t -Ce-halo-alkyl, NO z , CiM, d-Ce-alkyl, -(CH 2 ) m -0- C-Ce-alkyl.
- each m is independently selected from 1, 2, 3, or 4, as well as pharmaceutically acceptable salts thereof, or Is an enantiomeric mixture of 2 enantiomers in all ratios, and/or as a mixture of diastereoisomers in all ratios.
- Q is a single bond.
- Q and X are single bonds.
- X is Ci-C 4 alkylene. in ah embodiment X is methylene, ethylene, propylene or isopropylene, In an embodiment X is methylene or ethylene. In an embodiment X is methylene.
- the compounds of the invention may be represented by the following general formula (lb la ):
- X is selected from a single bond, an alkylene group having 1 to 6 carbon atoms optionally substituted with 1 or 2 substituents selected from fluoro or d-d-alkyl.
- Y is an alkylene group having 1 to 6 carbon atoms
- Q is selected from O. NH or a single bond.
- V is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, N0 2l CN, S0 2 -C,-C e alkyl, NMe 2 , d-Ce-alkyl, O-d-Ce-alkyl, -(CH 2 ) m -0-Ci-C e -alkyl. -Ci-Ce-halo-alkyl, O-d-Ce-halo-alkyl or a 5-6-membered heteroaromatic group,
- U is cycloalkyl, cydoalkenyl, heterocydyl or heteraary!, each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal, NO ? , CN. - SOz-C-Ce-alkyl, -S-d-Ce-alkyl, NMe 2 , d-C e -alkyl, -C(0)0-C C e -alkyl. O-d-Ce- alkyl, -(CHi) m -0-Ci-C 6 -alkyl. -C
- T is a phenyl, a triazolyl, a thiazolyl, an oxazolyl, an oxadiazolyl, or pyrazolyl group.
- R is Hal, -d-Ce-alkyl, O-d-Ce-alkyl, -(CH 2 ) m -0-d-Ce-alkyl, 0-C r C e -halo-alkyl, -(CH 2 ) m - O-d-Ce-halo-alkyl, -SOj-d-Ce-alkyl. -(CH 2 ) ffl -S0 2 -d-Ce-a'kyl.
- R 2 and R 2 ' are independently from one another H, Hal, -d-Ce-alkyl, -0-C,-C 6 -alkyl, -(CHz O-d-Ce-alkyl, O-d-Ce-halo-alkyl, -(CHz O-d-Ce-halo-alkyl, -S0 2 -d-C e - alkyl, -(CH 2 > m -S0 2 -C 1 -C e -alkyl > -SOj-d-Ce-halo-alkyl.
- R 1 and R 2 are linked to form with the ring T to which they are attached a 7-12-membered fused heterocydyl or 7-12-membered fused cycloalkyl. each of which may be optionally substituted with 1 to 3 Hal, -d-Cg-halo-alkyl, N0 2 , CN, Ci-C 3 -alkyl. - (CH 2 ) n ,-0'Ci-CB-alkyl. or -0-d-C 6 -alkyl.
- R 3 is d-Ce-alkyl, d-Ce-haloalkyl,
- haloalkyl a 3-8-membered cydoalkyl group, optionally substituted with 1 to 3 substitutents independently selected from Hal. -Ci-Ce-halo-alkyl, or Ci-Ce-alkyl; or a 3-8-membered heterocyclic group, optionally substituted with 1 to 3 substitutents independently selected from Hal, -d-Ce-halo-atkyl, N0 2 , CN. d-Ce-alkyl.
- R 4 denotes H, d-C 6 -elkyl. or forms together with R 3 a 3-8-membered cycloalkyi ring, optionally substituted with Hal. -d-Ce-halo-alkyl, N0 2 , CN, Ci-Ce-alkyl, -(CH 2 ) m -0- d-Ce-alkyl, -0-C,-C e -alkyl, -C(0)-Ci-C e -alkyl, or -C(0)0-C,-C B -alky
- Q is a single bond.
- Q and X are single bonds.
- R 4 is H or CrC+ alkyl.
- R 4 is H.
- R 4 is C 1 -C4 alkyl.
- R 4 is methyl
- R 3 is optionally substituted C1-C4 alkyl, or optionally substituted C 3 -C e cycloalkyl.
- R 3 and R 4 are independently CrCj-alkyl.
- R 3 and R 4 are both methyl.
- R 4 is hydrogen and R 3 is tetrahydrofuranyl, azetidlnyl, piperadlnyl, or tetrahydropyranyl.
- the present invention provides compounds of Formulae (I), (la), (lb) or (Ic) and each sub formula wherein R 4 denotes M or Me and R 3 is selected from the following groups:
- substitutents independently selected from Hal, -Ci-C e -halo-alkyl, N0 2 , CN, Ci-Ce-alkyl, -(CH s ) m -0-C r Ce-a!kyl, '(CH 2 ) rn -S0 2 -C 1 -C 6 -alkyl 1 -S0 2 -C,-C e -alkyl, -O-C Cralk l.-iCH ⁇ -O-C Ce-halo- alkyl, -(CH ⁇ m-SC ⁇ -C Ce-halo-alkyl. -S0 2 -Ci-C e -halo-alkyl, or -O-CVCe-halo-alkyl, or R* forms together with R* a 3 membered cyctoalkyl ring.
- R is H and R 3 is C,-C e alkyl, cyclopropyl. or a 3-8-membered heterocyclic group.
- R 4 is H and R 1 is ( Ce alkyl or cyclopropyl. ln an embodiment with specific reference to compounds of formula (la), (lb), (Ic) and sub formula thereof, R 4 is H and R 3 is cyclopropyl.
- R* is H and R 3 is ethyl.
- R 4 is H and R 3 is isopropyl.
- R 4 is H and R J is methyl.
- U is a 5-6-membered cycloalkyl group, a .5-12-membered heterbcyclyl or a 5-6 membered heteroaryl, each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal. N0 2 . CN, SO 3 ⁇ 4 , NMe 2 .
- U is selected from pyridinyl, pyrazmyl, pyridazinyl, pyrimidinyl, imidazolyl, pyrazolyl. tetrahydropyranyl. cyclohexyl, 8-azabicyclo[3.2.1]octan-3-yl, triazolyl and piperidinyl. each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal. N0 2 . CN, S0 2> Nlvte*, Ci-Ce-alkyl, O-CrCe-alkyl, -(CH 2 ) m - O-C Ce-alkyl.
- U is selected from pyridinyl, pyridazinyl and pyrazolyl, each of the above groups being optionally substituted with 1 to 3 substitutents selected from CF 3 . - S0 2 -Ci-C6-alkyl, C C e -a'kyl or Hal.
- U is selected from pyridinyl. pyridazinyl and pyrazolyl, each of the above groups being optionally substituted with a substituted selected from CF 3 , -S0 2 Me, methyl or F.
- U is selected from pyridinyl. pyridazinyl and pyrazolyl, each of the above groups being optionally substituted with a substituted selected from CF 3 , -S0 2 Me, methyl or F.
- V is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, -C C B -halo-alkyl, 0-CrC B -halo-alkyl or S0 2 -Ci-C e alkyl.
- V is a phenyl group optionally substituted with 1 to 3 substitutents selected from Hal. N0 2l CN. SOa-CrCe alkyl, NMe3 ⁇ 4. C,-C 6 -alkyl, 0-C r C 6 -alkyl, -(CH 2 )m-0- C,-Ce-alkyl, -C t -Ce-halo-alkyl, O-CrCe-halo-alkyl or a 5-6-membered heteroaromatic group.
- V is a phenyl group optionally substituted with 1 to 3 substitutents selected from Hal. -C C 6 -halo-alkyl, O-C,-Ce-hal0-alkyl or S0 2 -C,-C e alkyl.
- V is a phenyl group optionally substituted with 1 or 2 substituents selected from F, CI. -CF 3l -OCFj, -OCHF 2 or -S0 2 Me.
- V is a phenyl group optionally substituted by F. ln a further embodiment, with specific reference to compounds of Formula (I), (la), (lb), (lc) and sub formula thereof the present invention provides wherein V is selected from:
- R denotes Hal, N(1 ⁇ 4. CN, S0 2 -Ci.C e alkyl, N e 2 , C-Ce-alkyl, 0-C r C e -alkyi, - (CHjJm-O-CrCe-alkyl, CF 3l or a 5-6-membered heteroaromatic group.
- T is phenyl, triazolyl, oxadiazolyl or diazolyl.
- T is phenyl
- R 1 is O-C t -Ce-alkyl. Hal, -(CH 2 ) m -0-Ci-C e -alkyl, -C,-C 6 -alkyl, 0-C r C e - halo-alkyl, -SCvC ⁇ Ce-alkyl, -Ci-Ce-halo-alkyl, -S0 2 -3-8-cycloalkyl, or cyano, in which m is 1.
- R 2 and R 2' are H or Hal. In an embodiment R 2 is H or Hal and R ? is H.
- R 1 is 0-Ci-C e -alkyl, Hal, -(CHi)m-0-C C a -alkyl, -C C e -alkyl, O-C Ce- halo-alkyl, -SOi-Ci-C «-alkyl, -d-Ce-halo-alkyl, -S0 2 -3-8-cydoall ⁇ yl, or cyano, in which m is I. R ⁇ s H or Hal and R ⁇ is H.
- R 1 and R 2 are linked to form with the ring T to which they are attached
- R 1 and R* are linked to form with the ring T to which they are attached
- T is phenyl, triazolyl. oxadiazolyl or diazolyl;
- R 1 is 0-Ci-C 6 -alkyl, Hal. - (CHzJm-O-CrCe-alkyl, -Ci -C h alky I. O-d-Ce-halo-alkyl, -SO_-C,-C e -alkyl ( -d-Ce-halo-alkyl, -SOi-3-8-cycloalkyl, or cyano.
- m is 1 ;
- R 2 is H or Hal and R is H; or
- R and R 2 are linked to form with the ring T to which they are attached a dihydrobenzofuranyl, an
- indanyl each of these groups being optionally substituted by 1 to 3 -Ci-C B -alkyl.
- T is phenyl
- R 1 is O-d-CVhalo-alkyl, -SOi-CrCe-alk l or Hal. and R* and R 2 are H; or R 1 and R' are linked to form with the ring T to which they are attached which is optionally substituted with 1 or 2 -0,-0 $
- T is a phenyl ring wherein at least one of R 1 , R 2 or R s ' is in para position with regard to the rest of the molecule.
- T is a phenyl ring wherein at least one of R ⁇ R a or R 2 ' is in meta position with regard to the rest of the molecule.
- T is a phenyl ring wherein at least one of R R 2 or R 2 ' is in ortho position with regard to the rest of the molecule.
- the present invention provides compounds of Formula (I) and related Formulae wherein Hal preferably denotes F, CI or Br, most preferably F. and/or
- a 3-8-membered cycloalkyl group preferably is a cyclopropyl, a cyclobutyl, or a cyclopentyl, and/or
- a 3-8-membered cycloalkylene group preferably is cyclopropylene, a cydobutylene, or a cyclopentylene, and/or
- a 3-8-membered heterocyclic group preferably has 1 to 3 carbon atoms which is replaced by a group selected from O, S, N, SO, S0 3 , CO.
- a 3-8-membered heterocyclic group preferably denotes one of the following groups:
- a 7-12 membered heterocyclic ring preferably denotes a bicyclic ring having 7 to 12 carbon atoms wherein the 2 rings are fused or bridged, and wherein 1 to 3 carbon atoms may be replaced by a group selected from O, S, N, SO, S0 2 , CO.
- a 7- 2 membered heterocyclic ring preferably denotes one of the following groups:
- a 5-6-membered heteroaromatic group denotes an aromatic ring having 5 or 6 members and containing 1 to 3 heteroatoms selected from N, O or S.
- a 5-6-membered heteroaromatic group preferably denotes one of the following groups:
- a CvC B -halo-alkyl denotes a linear or branched aikyl having 1 to 6 carbon atom wherein 1 to 6 H atom is replaced by a halogen, preferably a F atom.
- the present invention provides compounds of Formula (II):
- G 1 denotes a single bond
- G 2 denotes a CO group
- X is selected from a single bond, an alkylene group having 1 to 6 carbon atome optionally substituted with 1 or 2 substituents selected from fluoro or C r Ce-alkyl,
- Y is selected from an alkylene group having 1 to 6 carbon atoms optionally substituted one or two times with C 3 -C 3 -cycloalkyl or Ci-C 3 -alkyl; or a 3-8- membered cycloalkylene group,
- Q 16 selected from O, NH or a single bond
- W is selected from SO, SO» or a single bond
- U is cycioalkyi, cycloalkenyl. heterocyciyi or heteroaryl, each of the above groups being optionally substituted with 1 to 3 substitutents selected from Hal, N0 2l CN, - SOz-d-Ce-alkyl, -S-C C 6 -alkyl, NMe 2 , C,-C e -alkyl, -C(0)0-C,-C 6 -alkyl. 0-d-C e - alkyl, -(CHiJ m -O-C Ce-alkyl, -CVCe-halo-alkyl, or a 5- membered heteroaromatic group being optionally substituted by Hal,
- V is an aryl group optionally substituted with 1 to 3 substitutents selected from Hal, N ⁇ 3 ⁇ 4, CN, S02-C r C e alkyl, NMe 2 , C t -C e -alkyl, O-CrCe-alkyl, -(CHi) m -0'Ci-C e -a]kyl, -C,-Ce-halo-aikyi, o-C,-Ce-halo-alkyl or a 5-6-membered heteroaromatic group,
- T denotes phenyl, triazolyl. thiazolyl, oxazolyl, oxadiazolyl, or pyrazolyl,
- R 1 is H, Hal, -C-Ce-alkyl, O-CrC-alkyl. ⁇ CH ⁇ -O-C.-Ce-alkyl, 0-C,-Ce-halo-alkyl. - (CHz O-CrCe-halo-alkyl, -S0 2 -C,-C 6 -alkyl, -S0 2 -Ci-C 6 - halo-alkyl, -(CH 2 ) rt ,-S0 2 -C 1 -C e -haio-alkyl. -SO r 3-8-cycloalkyl, -(CH 2 ) m -SO s -3-S- cycloalkyl, cyano or -CVCe-hafo-alkyl,
- R 2 and R 2 ' are independently from one another H. Hal, -Ci-Ce-alkyl, -0-Ci-C e -alkyl, -(CH 2 ) m -0-C 1 -C B -alkyi, O-C,-C e -hal0-alkyl, -(CH 2 ) m -0-C C 6 -halo-alkyl, -S0 2 -C C e - alkyl, -(CH 2 ) m -S0 2 -C,-C,-alkyl, -S0 2 -CrCe-halo-alkyl, -(CH 2 ) m -S0 2 -Ci-C e' halo- alkyl, 'S0 2 -3-8-cycloalkyl, -(CH 2 ) m -S0 2 -3-8-cycloalkyl.
- R 1 and R* are linked to form with the ring T to which they are attached a 7-12-membered fused heterocyciyi or 7-12-membered fused cycioalkyi, and optionally substituted with 1 to 3 Hal, -CrCe-halo-alkyl, N0 2 , CIM, a linear or branched alkyl group having 1 to 6 carbon atoms, -(CH 2 ) m -0-C,-C e -alkyl, or -0-Ci-C 6 -alkyl,
- R 3 is Ci-Ce-alkyl
- R* is Ci-Ce-alkyl
- m is selected from 1 , 2, 3 or 4, preferably 1 or 2
- Hal is F, CI, Br. or I, wherein -G 2 -Y-W together is at least 3 atoms in length, as well as pharmaceutically acceptable salts thereof, or is an enantiomeric mixture of 2 enantiomers in all ratios, and/or as a mixture of diastereoisomers in all ratios.
- R 3 is Ci-C 3 -alkyl.
- R 4 is Ci-C 3 -alkyl.
- R 3 and R 4 are Ci-C 3 -alkyl.
- R 3 is methyl
- R 4 is methyl
- R* and R 4 are methyl.
- R 1 , R 2 , R 2 ', m and Hal are as defined above for a compound of Formula (I), (la), (lb) or (lc).
- R 3 and R 4 are methyl and T is phenyl and R , R 2 . and R z are all H.
- pharmaceutical composition, method or use of the present invention described above a compound of Formula (II) may be present or used instead of a compound of Formula (I).
- Alkyi refers to monovalent alkyl groups which may be straight chained or branched.
- an alkyl group has 1 to 6 carbon atoms (i.e., Ci-Ce-alkyl).
- an alkyl group ha 1 to 4 carbon atoms (i.e., (VCValkyl).
- an alkyl group has 1 to 3 carbon atoms (i.e., Ci-C 3 -alkyl). Examples of such alkyl groups include methyl, ethyl, n-propyl, so-propyl. n-butyl, /so-butyl. n-hexyl, and the like.
- Alkylene refers to divalent alkyl groups which may be straight chained or branch chained.
- the alkenylene group has 1 to 6 carbon atoms. In certain embodiments 1 to 4 carbon atoms. In certain other embodiments 1 to 3 carbon atoms. In still further embodiments 1 or 2 carbon atoms.
- alkylene groups include methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), and the propylene isomers (e.g., - CH 2 CH 2 CH 2 - and -CH(CHj)CH 2 -). and the like.
- Aryl refers to an aromatic carbocyclic group having a single ring (eg. phenyl) or multiple condensed rings (eg naphthyl or anthryl), preferably having from 6 to 14 carbon atoms.
- aryl groups include phenyl, naphthyl and the like.
- Cycloalkyl refers to cyclic alkyl groups having a single cyclic ring or multiple condensed or fused rings, preferably incorporating 3 to 12 carbon atoms.
- Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl. cyclooctyl. and the like, or multiple ring structures such as adamantanyl, indanyl, 1,2,3,4-tetrahydronapthalenyl and the like.
- Cycloalkenyl refers to cyclic alkenyl groups having a single cyclic ring or multiple condensed or fused rings, and at least one point of internal unsaturation, preferably incorporating 3 to 8 carbon atoms.
- suitable cycloalkenyl groups include, for instance, cyclobut-2-enyl, cyclopent-3-enyl, cyclohex-4-enyl, cyclooct-3-enyl, indenyl and the like.
- Hal or “halogen” refers to fluoro, chloro, bromo and iodo.
- Heteroaryl or “Heteroaromatic” refers to a monovalent aromatic heterocyclic group which fulfils the Hiickel criteria for aromaticity (ie. contains 4n + 2 ⁇ electrons) and preferably has from 2 to 10 carbon atoms and 1 to 4 heteroatoms selected from oxygen, nitrogen, selenium, and sulfur within the ring (and includes oxides of sulfur, selenium and nitrogen).
- Such heteroaryl groups can have a single ring (eg. pyridyl, pyrrolyl or N-oxides thereof or furyl) or multiple condensed rings (eg. indolizinyl, benzoimldazolyl, coumarinyl, quinolinyl. isoquinolinyl or benzothienyl).
- Heterocyclyl or “Heterocyclic” refers to a monovalent saturated or unsaturated group having a single ring or multiple condensed or fused rings, preferably from 1 to 8 carbon atoms and from 1 to 4 hetero atoms selected from nitrogen, sulfur, SO, SO t , oxygen, selenium or phosphorous within the ring, in one embodiment, the heteroatoms are selected from nitrogen, sulfur, SO, SO z and oxygen.
- heterocyclyl and heteroaryl groups include, but are not limited to, oxazole, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, qulnolizine, isoquinoline.
- phenanthroline isothiazole, phenazine, isoxazole, isothiazole, phenoxazine, phenothiazine, imldazolidine, imidazoline, piperidine, piperazine, indoline.
- phthalimide 1 ,2,3,4-tetrahydroisoquinoline, 4,5,6,7-tetrahydrobenzo[b]thiophene, thlazole, thiadiazoles.
- oxadiazole oxatriazole, tetrazole, thiazolidine, thiophene, benzotbjthiophene, morphollno, piperidinyl, pyrrolidine, tetrahydrofuranyl, triazole, and the like.
- “Pharmaceutically acceptable” means approved or approvable by a regulatory agency of the Federal or a state government or the corresponding agency in countries other than the United States, or that is listed in the U.S. Pharmacopoeia or other generally recognized pharmacopoeia for use in animals, and more particularly, in humans.
- “Pharmaceutically acceptable salt” refers to a salt of a compound of the invention that is pharmaceutically acceptable and that possesses the desired pharmacological activity of the parent compound.
- such salts are non-toxic may be inorganic or organic acid addition salts and base addition salts.
- such salts include: (1) acid addition salts, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like; or formed with organic acids such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, ma Ionic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl) benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1 ,2-ethane-disulforwc acid, 2- hydroxyethanesulfonic acid, benzenesulfonic acid, 4-chlorobenzenesulfonic acid, 2- naphthalenesulfonic acid, 4-toluen
- Salts further include, by way of example only, sodium, potassium, calcium, magnesium, ammonium, tetraalkylammonium, and the like; and when the compound contains a basic functionality, salts of non toxic organic or inorganic acids, such as hydrochloride, hydrobromide, tartrate, mesylate, acetate, maleate, oxalate and the like.
- non toxic organic or inorganic acids such as hydrochloride, hydrobromide, tartrate, mesylate, acetate, maleate, oxalate and the like.
- pharmaceutically acceptable cation refers to an acceptable cationic counter- ion of an acidic functional group. Such cations are exemplified by sodium, potassium, calcium, magnesium, ammonium, tetraalkylammonium cations, and the like.
- “Pharmaceutically acceptable vehicle” refers to a diluent, adjuvant, excipient or carrier with which a compound of the invention is administered.
- Prodrugs refers to compounds, including derivatives of the compounds of the invention, which have cleavable groups and become by solvolysis or under physiological conditions the compounds of the invention which are pharmaceutically active in vivo. Such examples include, but are not limited to, choline ester derivatives and the like, N-alkylmorpholine esters and the like.
- Solvate refers to forms of the compound that are associated with a solvent, usually by a solvolysis reaction. This physical association includes hydroge bonding. Conventional solvents include water, ethanol, acetic acid and the like.
- the compounds of the invention may be prepared e.g. in crystalline form and may be solvated or hydrated.
- Suitable solvates include pharmaceutically acceptable solvates, such as hydrates, and further include both stoichiometric solvates and non-stoichiometric solvates. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. 'Solvate' encompasses both solution-phase and isolable solvates. Representative solvates include hydrates, ethanolates and methanolates.
- compounds of the invention may possess asymmetric centres and are therefore capable of existing in more than one stereoisomeric form.
- the invention thus also relates to compounds in substantially pure isomeric form at one or more asymmetric centre eg., greater than about 90% ee, such as about 95% or 97% ee or greater than 99% ee, as well as mixtures in all ratios, including racemic mixtures, thereof.
- the compounds may also therefore appear as an enantiomerically enriched mixture of two enantiomers in all ratios, and/or as a mixture of diastereoisomers in all ratios.
- Such isomers may be prepared by asymmetric synthesis, for example using chiral intermediates, or mixtures may be resolved by conventional methods, eg., chromatography, or use of a resolving agent.
- the compounds of the present invention may be capable of undergoing tautomerism. Accordingly, all possible tautomers of a compound of the present invention fall within the scope and spirit of the invention.
- a hydrogen atom may be replaced with an isotope of hydrogen.
- deuterium may be used to replace a metabolically labile hydrogen to improve the pharmacokinetics.
- tritium may be incorporated into a compound for diagnostic or analytical purposes, including but not limited to biodistribution studies.
- “Therapeutically effective amount” means the amount of a compound that, when administered to a subject for treating a disease, is sufficient to effect such treatment for the disease.
- the "therapeutically effective amount” includes that amount of a compound or composition that will elicit the biologicaf or medical response of a subject that is being sought by a medical doctor or other clinician.
- the “therapeutically effective amount” can vary depending on the compound, the disease and its severity, and the age, weight, etc., of the subject to be treated.
- Preventing refers to a reduction in risk of acquiring or developing a disease or disorder (i.e., causing at least one of the clinical symptoms of the disease not to develop in a subject that may be exposed to a disease-causing agent, or predisposed to the disease in advance of disease onset.
- prophylaxis is related to "prevention”, and refers to a measure or procedure the purpose of which is to prevent, rather than to treat or cure a disease.
- 'Treating" or “treatment” of any disease or disorder refers, in one embodiment, to ameliorating the disease or disorder (i.e., arresting the disease or reducing the manifestation, extent or severity of at least one of the clinical symptoms thereof).
- 'treating' or 'treatment' refers to ameliorating at least one physical parameter, which may not be discernible by the subject.
- "treating" or “treatment” refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- “treating" or “treatment” relates to slowing the progression of the disease.
- Compounds of the present invention are meant to embrace compounds of the Formula(e) as hereinbefore described, which expression includes the prodrugs, the pharmaceutically acceptable salts, and the solvates, e.g., hydrates, where the context so permits. Similarly, reference to intermediates, whether or not they themselves are claimed, is meant to embrace their salts, and solvates, where the context so permits.
- the inhibition data shown for the compounds of the list below was calculated using the steady state current amplitude at the end of the depolarising pulse in patch clamp evaluations (as described in the Biology Protocols: 1. Electrophysiology).
- Compounds of Formula (I) can be made by the reaction of the compounds of Formula 5, wherein R 1 , R 2 , R 7 ', R 3 , R 4 , G 1 , X, Q, T and U are as above defined, with a compound of Formula 8, wherein G ⁇ Y, W and V are as above defined, and wherein LG denotes a suitable leaving group, as depicted in scheme 1.
- LG preferably denotes an halogen, preferably chlorine or bromine, a sulfonate, or denotes an activated acid derivative obtained by the reaction of a carboxylic acid in the presence of an amide coupling agent.
- the amide coupling agents include EDCl.
- BOP, PyBOP, HOBt, HATU, T3P, DCC can be used in a suitable solvent, as for example dichloromethane or dimethyiformamide at room temperature.
- the secondary amines 5 is converted to compounds of Formula (I) by reaction with an activated acid such as an acid chloride, in dichloromethane at room temperature, or a mixed anhydride or a N-succinimide ester in ethanol at room temperature or by reaction with an acid in the presence of an amide coupling reagent selected from EDCl, BOP, PyBOP, HOBt, HATU, T3P, DCC in dichloromethane or dimethyiformamide at room temperature.
- an activated acid such as an acid chloride, in dichloromethane at room temperature, or a mixed anhydride or a N-succinimide ester in ethanol at room temperature
- an amide coupling reagent selected from EDCl, BOP, PyBOP, HOBt,
- compounds of Formula (I) can be synthesised by reacting a compound of Formula 7, wherein R 1 , R 2 , R 2 ', R 5 , R 4 , G 4 , Y, W, T and V are as above defined, with a compound of Formula 9, wherein G ⁇ X. Q, U are as above define and wherein LG denotes a suitable leaving group. Examples of such alkanes having LG groups are alkyl halides or alkyl sulfonates.
- the reaction of compounds of Formula 7 with compounds of Formula 9 is preferably performed in the presence of a base such as sodium hydride, potassium tert-butoxide, potassium carbonate preferably in dimethyiformamide or acetonitrile.
- the temperature of the reaction Is between room temperature and 100 "C, preferably between 20 and 60"C.
- a phase transfer catalyst such as tetra-rt- butylammonium bromide can be used. Preferred conditions are the use of acetone or acetonitrile with heating at 45-100 °C.
- the compounds of Formula 5 can be synthesised b reacting ketones 1 , wherein R 1 . R 2 , R 2 ', R s and T are as above defined, with amines 2, wherein G 1 , X, Q and U are as above defined, according to scheme 2.
- This reaction is preferably performed by reductive amination via an imine intermediate.
- R 4 and T are as above defined, with the aldehyde of Formula 4, wherein Q and U are as above defined, and wherein X' denotes a linear or branched alkyi having 1 to 6 carbon atom or a cyclic alkyi having 3 to 8 carbon atoms wherein 1 -CH 2 - group, in this linear, branched or cyclic alkyi, is replaced by a -CO- group.
- the reductive aminatlon reaction described in scheme 2 can occur in a single pot reaction using borohydride reagants, including but not limited to sodium cyanoborohydride, sodium acetoxyboro-hydride and sodium borohydride in halogenated solvents such as dichloromethane or 1 ,2-dichloroethane, or alcohols such as methanol, typically at room temperature for 0.5-12 hours.
- borohydride reagants including but not limited to sodium cyanoborohydride, sodium acetoxyboro-hydride and sodium borohydride in halogenated solvents such as dichloromethane or 1 ,2-dichloroethane, or alcohols such as methanol, typically at room temperature for 0.5-12 hours.
- the reaction can also occur in two steps. Firstly by the formation of imine in the presence of an acid including but not limited to p-toiuenesulfonic acid, or Amberlyst resin and also a dehydrating reagent, such as but not limited to magnesium sulphate, sodium sulphate, molecular slaves or TiCU using for example dichloromethane, ethanol, ethyl acetate or dimethyl sulfoxide as solvent. This step may be performed in a Dean Stark apparatus using toluene at 90-110 °C. In a second step, the imine can be converted to the secondary amine using borohydride reagents as described above.
- an acid including but not limited to p-toiuenesulfonic acid, or Amberlyst resin and also a dehydrating reagent, such as but not limited to magnesium sulphate, sodium sulphate, molecular slaves or TiCU using for example dichloromethane, ethanol, ethy
- Secondary amines 5 may be prepared by the alkylation of primary amines 2 with a compound of Formula 6 or the amine 3 with a compound of Formula 10, wherein LG in Formulae 6 and 10 denotes a suitable leaving group.
- suitable leaving groups may be selected from halogen, preferably chlorine or bromine, or a sulfonate group, preferably selected from mesylate, tosylate, benzyl sulphonyl, a perfluoroalkyl sulfonate such as mono, di or trifluoromethyl sulfonate or triflate.
- the reaction is preferably performed In the presence of a base, such as potassium carbonate or triethylamine, in solvents preferably selected from dichloromethane, acetonitrile. dimethyl sulfoxide, at temperatures ranging from room temperature to 100 "C, As disclosed in scheme 3.
- a base such as potassium carbonate or triethylamine
- Compounds of Formula 7 may be prepared by reacting a compound of Formula 3, wherein R ⁇ R 2 , R 2 ', R 5 , R 4 and T are as above defined, with a compound of Formula 8, wherein G 2 , Y. , and V are as above defined, and wherein LG is as defined above, as mentioned In scheme 4.
- Amines of Formula 3 can be acylated with compounds of Formula 8, using techniques well known in the art.
- LG in the compounds of Formula 8 is preferably an activated acid obtained by reaction of a COOH group in the presence of an amide coupling reagent.
- the amide coupling reagents include EDCI. BOP, PyBOP, HOBt. HATU, T3P, DCC. They can be used in a suitable solvent, as for example dichloromethane or dlmethylformamide at room temperature.
- Amines 3 wherein R 4 is H can be prepared by functional group transformations well known in the art- Non-limiting examples are provided in Scheme 5.
- Ketones 11 can be condensed with hydroxylamine. for instance in ethanol at 80-100 "C for 1-2 days to give 12, which can in turn be reduced to amine 3 using for example Raney nickel in methanol at 80-100 °C for 2-6 hours, or by hydrogenation using palladium on charcoal for instance in ethanol at room temperature for 1-12 hours.
- Aldehydes 13 can be treated with metallated alkyl species such as Grignard reagents or alkyl lithiums to give secondary alcohols 14 for instance In diethylether or tetrahydrofuran at -78 "C for 1-4 hours, which can be converted to azides 15 using techniques known in the art, for instance by reaction with sodium azide in chloroform and sulfuric acid at 0 °C to room temperature.
- the azides can be reduced to the amines 3 for example by catalytic hydrogenation with palladium on charcoal in methanol at room temperature or by using PPh 3 / H 2 0 (Staudinger conditions).
- Reaction of benzonitriles 16 with Grignard reagents or alkyl lithiums for example diethyl ether or tetrahydrofuran at room temperature to 50 "C for 1-4 hours can give imines 17. which may be reduced for instance with sodium borohydride in methanol at room temperature or lithium aluminium hydride in dimethyl formamide at room temperature or borane-THF complex in tetrahydrofuran at -20 *C to room temperature to give the amines 3.
- EDC.HCI (1-ethyl-3- (3-dimethylaminopropyl)carbodiirnide hydrochloride), eq. (equivalents), EtOAc (ethyl acetate), EtOH (ethanol), g (gram). Gen. (General Procedure). cHex (cyclohexane), HPLC (high performance liquid chromatography), hr (hour), IPA (isopropyl alcohol), int. (intermediate), LCMS (liquid chromatography - mass spectrometry), MHz (Megahertz), MeOH (methanol), min (minute), mL (milliliter), mmol (millimole).
- PBS phosphate buffered saline
- PDA photodiode array
- PMB para-methoxybenzyl
- cPr cyclo-propyl
- iPr Iso-propyl
- PTLC preparative thin layer chromatography
- Rt retention time
- T room temperature
- TBAF tetra-butylammonium fluoride
- TBTU W,/V,W',W-tetramethyl-0-(benzotriazoH- yl)uronium tetrafluoroborate
- T3P propane phosphonic acid anhydride
- TEA triethyl amine
- TFA trlfluoroacetic acid
- THF tetrahydrofuran
- t triplet
- PetEther petroleum ether
- TBME terf-butyl methyl ether
- TLC thin layer chromatography
- TMS trimethylsilyl
- TMSI trimethylsilyl iodide
- UV ultraviolet
- # of iso. number of stereoisomers).
- HPLC Method 1 - Waters Alliance 2695, column Waters XBridge C8 3.5 ⁇ ⁇ 4.6x50 mm. conditions: solvent A (H 2 0 with 0.1% TFA), solvent B (ACN with 0.05% TFA), gradient 5% B to 100% B over 8 min, UV detection with PDA Water 996 (230-400 nm).
- LCMS Method 2 - Agilent 1100 Series LC/MSD, column Phenomenex Gemini-NX C18 5 ⁇ , Zorbax Eclipse XBD-C8 or Luna 5 ⁇ C8, 150 x 4.6 mm, with mobile phase 80% ACN, 15% H z O, 5% buffer (3:1 MeOH/H 2 0, 315 mg HC0 2 NH 4 , 1 mL AcOH) and MS detection (ESI method).
- UPLC Method 3 - Waters Acquity, column Waters Acquity UPLC BEH C18 1.7 ⁇ 2.1x50 mm, conditions; solvent A (10 mM ammonium acetate in water + 5% ACN), solvent B (ACN), UV detection (PDA, 230-400 nm) and MS detection (SQ detector, positive and negative ESI modes, cone voltage 30 V). Gradient 5% B to 100% B over 3 min or gradient 40 B to 100% B over 3 min.
- Triethylamine or diisopropylethylamine (2 eq.) was added to a solution of sec-amine (1 eq.) in anhydrous DCM or Et 2 0 (0.1-0.3M) and the resulting solution was stirred at RT under a nitrogen atmosphere for 5 min.
- Acid chloride 1.5-2.5 eq.
- the reaction mixture was stirred until completion (LCMS).
- the reaction mixture was diluted with EtOAc or diethyl ether and washed thoroughly with NaHC0 3 (aq.) solution followed by distilled water.
- the organic layer was separated, dried (MgSO*) and concentrated under reduced pressure.
- the crude material was purified using either PTLC or silica-gel flash chromatography using increasing gradient of EtOAc or/and MeOH in hexane or DCM as elu nt.
- Oxalyl chloride (1.5 eq.) was added slowly to a solution of anhydrous dimethylsulfoxide (3.0 eq.) in anhydrous DCM (0.17-2.0 ) at -78 "C under a nitrogen atmosphere and stirred for 30 min.
- anhydrous dimethylsulfoxide (3.0 eq.) in anhydrous DCM (0.17-2.0 ) at -78 "C under a nitrogen atmosphere and stirred for 30 min.
- To the resulting mixture was added the alcohol (1 eq.) dissolved in anhydrous DCM (0.2-Q.4M) and the reaction mixture was stirred at -78 "C for 45 min.
- Anhydrous triethylamine (6 eq.) was added drop-wise and reaction was stirred at -78 °C for 30 min and then at RT for 30 min.
- microwave irradiation of the above solution at 120°C for 5 minutes produced the desired product.
- N-tert-butoxycarbonyl derivative was dissolved in a solution of HCI in Et 2 0. After several minutes a precipitate formed and LCMS was used to monitor the reaction progress. Upon completion the precipitate was collected by vacuum filtration and the product re-crystallised from DMC/Et 2 0,
- Step i To a suspension of NaH 60% In mineral oil (1.2eq) in dry DMF (0.1M) under N 2 at RT was added the appropriate thiol (1.2eq.) drop-wise. After stirring at this temperature for 10 min. a solution of the 6-bromopyridine analogue in dry DMF (0.1 M) was added drop- wise and the reaction heated to 60°C. The reaction was monitored by LCMS/TLC and following completion was quenched with sat.NH 4 CI. The product was extracted with EtOAc (3 x) and the combined extracts dried (MgS0 4 ) and concentrated under reduced pressure. The crude material was purified by either PTLC or silica-gel flash column chromatography using the appropriate solvent to obtain the thlopyridine intermediate.
- Step ii) To a solution of the thiopyridine intermediate in dry DCM (0.1 M) under 2 at 0°C was added m-CPBA (2 eq) portion-wise. The reaction was monitored by LCMS TLC and following completion was quenched with sat NaHCOj. The product was extracted with EtOAc (3 x) and the combined extracts dried (MgS0 4 ) and concentrated under reduced pressure. The crude material was purified by either PTLC or silica-gel flash column chromatography using the appropriate solvent
- Enantiomer A of 4-chlorophenyl(cyclopropyl)met anamine purified from the racemate of 4-chlorophenyl(cyclopropyl)methanamine by preparative HPLC using a Chiralpak AYH 250x20 mm column (Daicel) (eluent BOH DEA 0.1 % v/v, flow 10 ml min, 427 mg, 2.35 mmol) and pyridine-2-carboxaldehyde (201 mg, 1.87 mmol), were reacted according to General Procedure A to afford the titled compound (433 mg, 85%) as a yellow oil.
- Enantiomer B of 4-chlorophenyl(cyclopropyl)methanamine (purified from the racemate of 4-chlorophenyl(cy opropyl)methanamine by preparative HPLC using a Chiralpak AYH 250x20 mm column (Daicel) (eluent EtOH DEA 0.1% v/v, flow 10 ml min, 419 mg, 2.31 mmol) and pyridine-2-carboxaldehyde (201 mg, 1.87 mmol), were reacted according to General Procedure A to give the titled compound (499 mg, 98%) as a yellow oil.
- 1 HN R (CDCI.) S 8.56 (d. J 4.5 H3 ⁇ 4.
- Step 1 2-chloro-/V-f(1 S)-1-(4-fluorophenyl)-2-methylproPyltecetamtde
- Step 2 ⁇ (1 S)-1-(4-fluorophenyl)-2-methylpropyn-2-(4-fluoropiperidin ⁇ 1 -v[3 ⁇ 4acetamide
- Step 3 Enantiomer A of 1 -(4-fluorophenyl)-A -f2-(4-fluoropiperldln-1-yl)ethvn-2- methvtpropan-1 -amine
- Tetraethyl orthotitanate (2.84 mL, 13.4 mmol) was added into a solution of cyclopropyl[3-(ethylsulfonyl)phenyl]methanone (2.0 9; 8.4 mmol) and 2,2-difluoro- 2-pyridin-2-yl-ethylamine benzenesulfonate (3.18 g, 10.1 mmol) in anhydrous THF (16 mL) at RT. The resulting mixture was heated at reflux for 2 hours, and then cooled to RT. Sodium borohydride (952 mg. 25.2 mmol) was added and the resulting mixture was stirred at RT until completion.
- Aluminium chloride (8.78 g, 65.8 mmol) was added into a solution of propionyl chloride (4.86 mL, 55.7 mmol) in anhydrous DCM (35 mL) cooled at 5*C. The resulting solution was stirred at 5 D C for 15 minutes, and then added dropwise over 5 minutes into a solution of (ethylthio)benzene in anhydrous DCM (35 mL) cooled at -10°C. After 1.5 hours at -10°C, the reaction mixture was poured into a mixture of a 5N aqueous solution of HCI (100 mL) and crushed ice, and then was extracted with DCM (2x100 mL).
- Step 3 allyl-[1-(4-ethanesulfonyl-Phenyl)-propyll-amine
- a mixture of bis(dibenzylideneacetone)palladium (387 mg, 0.67 mmol) and 1 ,4- bis(diphenylphosphino)butane (287 mg, 0.67 mmol) was prepared in THF (20 mL) under nitrogen and stirred at T for 15 minutes.
- the preformed catalyst and thiosalicylic acid (2.28 g, 14.8 mmol) were added into a solution of allyl-[1-(4- ethanesulfonyl-phenyl)-propyG-amine (3.60 g, 13.5 mmol) in THF (20 mL). The resulting mixture was stirred at 60 4 C for 3 hours until completion.
- Step 2 terf-Butyl 3-[(£)-f4-fluorophenyl)(hvdroxyimino)methvnazetidine-1- carboxylate
- Step 3 terf-Butyl 3-famino(4-fluorophenyl)methyllazetidine-1-carboxylate
- Step 1 3-(4-Benzyloxy-benzoyl)-azetidine-1-carboxylic aoid tert-butyl ester
- Step 3 7erf-butyl 3-rf4-(difluoromethoxy>phenvnfhvdroxy methyllazetidine-1- carboxylate
- Step 5 tert-butyl 3-f
- Step 6 tert-butyl 3-faminof4-(dlfluoromethoxy)phenyllmethyl)azetidine-1» carboxylate
- Step-1 1-r4-(dlfluoromethoxy> phenyl1-2-methylpropan-1-ol
- Step-2 1-(1-azido-2-methylpropyl)-4-fdifluoromethoxy) benzene
- Step-3 (1-f4-(difluoromethoxy3 ⁇ 4 phenyl]-2-methylpropyl ⁇ amine
- Step 2 cvclopropyhP-isopropylsulfanyl-phenvD-melhanone
- the reaction mixture was diluted with a 5N aqueous solution of HO (32.3 mL) over 20 minutes keeping temperature below 1Q e C, and then stirred at 40"C for 2 hours and at RT for 72 hours.
- the layers were separated and the organic one was washed with water, dried (Na 2 S0 4 ) and concentrated under reduced pressure to give the title compound as a yellow oil (10.38 g, 88%), used without further purification.
- UPLC/MS (max plot) 89.9%; Rt 1.97 min; ( S+) 221.3 (( ⁇ + ⁇ ).
- Step 3 C-cvclopropyl-C-f3-(propane-2-sulfonyl -phenyll-methylamine (Intermediate al) and cvctopropy 3-(propane-2-sulfonyl)-ohenylll-methanone (Intermediate az)
- Step 1 Preparation of cvclopropyl f3-(ethylthio) phenyl! methanone
- Step 2 Preparation of cyclopropyl [3-(ethylsulfonvD phenyll methanone
- Step 1 C-Cvclopropyl-C-( 3-6thylsulfanyl-phenyl)-methylarnine hydrochloride
- Step 3 -(azido(3-fethylsulfonyl)phenyl)methvnietrahvdro-2H-pyran
- Step 4 4-fazido(3-(ethylsulfonyl>phenyl')methylMetrahvdro-2H-pyran
- Step 2 Svpthesis of cvcloproDyl(2-f2.2-difluoroethoxy)phenyl)benzonltrile
- Step 1 4-i3-ethylsulfanyl-bezoyl)-piperidine-1-carboxylic acid tert-butyl ester
- Step 2 4-(3-ethanesulfonyl-bezoyl -piperidine-1-carboxyHc acid tert-butyl ester
- Step 1 1-(2.4-difluorophenyl)-N-hvdroxy-1-n-methylpiperid ⁇ l-(2,4-difluorophenyl)- /-hydroxy-1-(piperidin-4-yl)metrianimine hydrochloride was reacted according to General Procedure Q to give the title compound.
- Step 2 1-(2.4-difluorophenyl)-1 -(1-methylpiperidin-4-yl)methanamine
- Step 2 benzyl K1-f4-(difluoromethoxytohenyll-2-hydroxyethyl> carbamate
- Step 3 benzyl ⁇ 1-r4-(difluoromethoxy)phenyll-2-methoxyethyl) carbamate
- chiral acids or acid chlorides are racemic mixtures of enantiomers, otherwise chiral acids or chiral acid chlorides are enantbpure and the absolute stereochemistry is not known.
- Example 1 A/-rcvclopropyl 4-methoxyphfenyl)methvn-3-f4-fluorophenyl)-W-(pyridin- 2-ylmethyl)butanamide
- Example 5 Diastereomer A of (3ffl-A/-ri-cvclopropyl(4-chlorophenvi)methyl1-3 : (4 : f(uorophenyl)-A -(pyridin-2-ylmethyl)butanamide. and
- Example 6 Diastereomer B of (3W)-A/-ri-cvclopropyl(4-chlorophenvnmethvn-3-(4-fluorophenyl)-A/-(pyrldln-2- ylmethvQbutanamlde
- Example 3 was a diastereomeric mixture and was resolved by on Prep LC 4000 with 2777C Sample Manager PAL (loop: 5ml) and Waters Fraction collector III, equipped with Waters 2487 Dual Detector using Chiralpak ADH 250x 20 mm (Daicel) (eluent hexane EtOH DEA 85/15 01 v/v/v, flow 10 ml min) to afford compound 5 (first eluting) ant- compound 6 (second eluting).
- Example 7 Diaatereomer A of (3S)-A 1 vclopropyl(4-chIorophenyl)methyll-3-(4- fluorophenyl)-rV-(pyridin-2-ylmethyl)butanamide, and Example 8: diastereomer B of (3S)-A-ri-cyclopropyl(4-chiorophenyl)methvn-3 ' f4-fluorophenyl)- V- ⁇ pyridin-2- ylmethvDbutanamide
- Example 4 was a diastereomeric mixture and was resolved by on Prep LC 4000 with 2777C Sample Manager PAL (loop: 5ml) and Waters Fraction collector III, equipped with Waters 2487 Dual Detector using Chiralpak IA 250x20 mm (eluent hexane ISOH 50/50 v/v, flow 10 ml min) to afford compound 7 (first eluting) and compound 8 (second eluting).
- Example 10 A -f ( 4-chlorophenvn ( cvclopropynmethyll-2-f4-fluorophenyl)-/V-(pyridin- 2-ylmethyl)cvclopropanecarboxamlde fii
- Example 13 Dlastereomer C of A ⁇ -r(4-chloropher>yt)(cvclopropy»methvn-2- 4- fluorophenvn-A -(pyridin-2-ylmethv cvclopropanecarboxamide
- Example 14 Diastereomer D of A/-r(4-c lorophenyl)(cvciopropyl)methyll-2-(4- fluorophenvn-Af-ipyrldin-2»ylmetriv»cvclopropanecarboxamide
- Example 41 A/-ri-(2.3-dihvdro-1 -inder>-5-v»ethvn-3-(4-fluorophenyl)-W-rZ-(3.6- dimethylpyrazin-2-yloXv)ethyllbutanamlde
- Example 44 ⁇ -T1-f2.3-dihvdro 1/ -lnden-S-yl)ethyl1-3- ⁇ 4-fluoroDhenvn-/tf-r2-(6- chloropyridin-2-yloxy)ethvnbutanamide
- Example 48 /tf-ri-f2.3-dihvdro-iH-lnden-S-yl)etrivn-3-(4-fluoroprienyl)-W-r2-(S- chloropyridin-2-yloxytethyriproP8namide
- Example 46 A/-r -(2.3-d ' ihvdrp-1W-inden-5-vnethyll-3-(4-Huorophenyl)-W-i2-(pyridin- 2-yloxy)ethvHbutanamide
- Example 53 AW -( 2.3-dihvdro-1 ff-iriden-S-yllethyll-3-(4-fluorophenvn-A/-r2-(6- methylpyridin-2-yloxy)ethyllpropanamide
- Example 54 N-fl -f 2.2-dimethvM .3-benzoxathlol-5-yl)ethvn-3-( 4-fluoroph&nv»-A (pyridm-2-ylmethvDpropanamide
- Example 65 3-(4 ⁇ fluorophenyl) " JV-f(1S) ' 1-i2-methoxyphenvHethviyA/-(pyridin-2- vtmethvUpropanamide
- Example 67 (50 mg) was separated by chiral HPLC (AD-H, 250x20mm, 5um) using EtOH+0.1% DEA dOmL/min) to give the title compound (1st eluting pic) as a yellow oil (19 mg).
- HPLC Methodhod 1
- Rt 3.13 min Purity: 99.4%
- UPLC/MS Methodhod 3 407.2 (M+H) * .
- Example 62 ⁇ - ⁇ -f 2.2-dimethyl-1.3-bon20xathiot-5-yl)ethvH-W-iPyrldin-2-ylmethv ⁇ 3-
- Example 52 To a solution of Example 52 (70 mg, 0.14 mmol) in glacial AcOH (5 mL) at 0 *C was added H 2 0 2 (0.03 mL. 0.28 mmol). After stirring for 2 hr, H 2 0 2 (0.12 mL, 1.12 mmol) was added and the stirring continued at RT overnight. Water was added and the aqueous phase was extracted with DCM. The combined organics were then extracted with 1 M NaOH. dired (MgSCv), filtered and concentrated under reduced pressure. The crude mixlure was purified by MD Autoprep to give the title compound as a vitreous solid. 'H NMR ⁇ CDCI a ) ⁇ 8.41-8.29 (m. 1H).
- Example 54 To a solution of Example 54 (70 mg, 0.16 mmol) in glacial AcOH (5 rnL) at 0 *C was added H 2 0 2 (35 ⁇ . 0.31 mmol). After stirring for 2 nr. Hi0 2 (0.14 mL, 1.24 mmol) was added and the stirring continued at RT overnight. Water was added and the aqueous phase was extracted with DCM. The combined organics were then extracted with 1 M NaOH, dired (MgS ⁇ 3 ⁇ 4 ⁇ , filtered and concentrated under reduced pressure. The crude was purified by MD Autoprep to give the title compound as a vitreous solid. 'H NMR (CDCI 3 ) ⁇ 8.50-8.37 (m.
- Example 65 2-f(4-fluorophenyHsulfonyn-A - ⁇ 1 -f2-f methoxymethyl)pheny»ethyl)-/v- (pyridin-2-ylmethvOacetamide and
- Example 66 1-»4-fliiorophenyl)sulfonvH-W- 1-r2- (methoxymethyl)phenyllethyl - tf-(pyrldin-2-ylmethvHcvclopropanecarboxamide
- Example 65 HPLC (Method 1) Rt 2.78 min (Purity: 93.7%). UPLC/MS (Method 3) 457.1 ( +H)V
- Example 76 Diastereomer A of A -r(4-chlorophenylUcvclopropyl)methyll-3-f(4- fluorophenvnsulfonvn-/V-(pyridin-2-ylmethyl)butanamide
- Example 78 Diastereomer A of 3-H-fluorophenv0-fV-ri-(4-methyl-4H-1.2.4-triazol-3- yliethvH-fl-(pyridin-2-ylmethyl)butanamlde
- Example 80 tf-r(4-chlorophenyl>(cvclopropyl)rr>ethyll-2-f 4-fiuorophenvnsulfonyl -A/- (8-methyl-8-a3 ⁇ 4abicvclof3.2.1loctan-3-yl>acetamlde
- Example 88 W-fcvclopropyir4 ⁇ difluorometh0xy)phenyl1methyl)-3-r(4- fluorophenv»)sulfonvn-yV-f(6-methoxypyridin-3-vnmethyllbutanamide
- Example 91 Enantiomer A of JV-(cvclopropylf4- ⁇ difluoromethoxy)phenylTmethyl ' 1 - r(4-fluorophenvnsulfonyn-fV-(pyridin-2-ylmethyl)cvclopropanecarboxamlde and
- Example 92 Enantiomer B of iV-(cvclopropyl[4-(drfluoromethoxy)phenyllmethylM- r(4-fluorophenyl3 ⁇ 4sulfonvn-W-(pyridin-2-ylmethvncvclopropanecarboxamide
- Example 93 A/-f(4-c
- Example 94 Enantlomer A of V-f(1-(4-fluorophenyl)-2-methylpropylM-ri4- fluoroph-jnyl)sulfonyl -A/-f2-(4-fluoropipendin-1-yl)ethvncvclopropanecarboxamide
- Example 96 Enantiomer A of (/V-n-(3-bromophenyHcvclopropyn-3-(4-fluorophenyl)- W-(pyridln-2-ylmethyl)butanamide
- Steps 1-3 Preparation of Intermediate ak.
- HPLC 92 1 % (AUC), Rt 2.1 miri.
- Method A- 0.1% TFA in H 2 0, B-0.1% TFA in ACN, Flow 2.0 mL/min; Column: XBridge C8 (50 x 4.6 mm. 3.5 m).
- Step 8 Preparation of I-8 Trimethylsulfoxonium iodide (1B5.7 g, 0.72 mol) was suspended in dry DMSO (700 mL). To this suspension under ice-cooling, sodium hydride (60 % suspension in mineral oil) (32.4 g, 1.35 mol) was carefully added in portions over a time period of 1 h. (Note: exotherm! Then reaction mixture ' was stirred at RT for 1 h. To this suspension was added a solution of tert-Butyl (4-fluorophenyl)acrylate (100.0 g, 0.42 mol) in dry DMSO (250 mL) and the mixture was stirred at RT for 12 h.
- tert-Butyl (4-fluorophenyl)acrylate 100.0 g, 0.42 mol
- tert-butyl 2-(4-fluorophenyl)cyclopropanecarboxylate (12.0 g; 50.79 mmol; 1.00 eq.) was dissolved in DCM (60 mL). 4M HCI in dioxane (38.1 mL) was added. After stirring for 6hr.at RT an HPLC indicated 65 % conversion. More HCI in dioxane (20 mL) was added and the reaction was stirred O/N at RT after what an HPLC indicated full conversion and 100 % a/a.
- reaction mixture was stirred at 0 a C for 15 min and then 2 1 4 1 6-Tripropyl-[1 ,3 1 5,2,4,6]trioxatriphosphinane 2.4.6-trioxide (21.43 mL; 36.03 mmol; 2.00 eq.) was added dropwise (addition took 15 min).
- the cooling bath was removed and the mixture was heated to 60 "C (external temperature) for 5hr.until completion.
- the reaction mixture was cooled to RT, quenched with a saturated solution of NaHCOa (100 mL) and phases were separated.
- HPLC 99.1 % (AUC). Rt 4.5 min. Method; A- 0.1 % TFA in H 2 0, B-0.1 % TFA in ACN. Flow 2.0 mL/min; Column XBridge C8 (50 X 4. ⁇ mm, 3.5 Mm).
- Acid chloride 1-10 (300 mg, 1.51 mmol) was added to a solution (1.25 M) of amine I-4 ( 300 mg, 0.98 mmol) in pyridine at RT under ⁇ atmosphere and stirred overnight ( ⁇ 20 h).
- the reaction was diluted with diethyl ether (50 mL) and washed with H a O (30 mL x2), aqueous NH ⁇ CI (30 mL), aqueous NaHC0 3 (30 mL) and brine (15 mL).
- the organic layer was dried (MgSO ⁇ ), filtered and concentrated under reduced pressure.
- Step 2 Preparation of cvclopropyl[3-(ethylsurfanyl)phenyllmethanQl fll-2)
- Step 3 Preparation of cvclopropylf3-(ethylsulfonyl)phenyllmetrianol (II-3)
- Step 4 Preparation of -fazjdotcyclopropy methyll-S-fmethylsulfonvnbenzene
- Step 5 Preparation of 1-cvclopropyH -f3-(ethylsulfonv»Dhenyllmethanamlne fll-5)
- Step 6 Preparation of 1-cyciopropyl-H3-(ethyls )lfonyl) phenvH-N-ff6-(trifluoromethyl) pyridin-3-yll methyl) methanamine (H-6)
- Step 7 Preparation of (1R. 2R)-2-(4-Fluoro-phenyl)-cvclopropanecarboxyllc acid f(R/S)- cvclopropyl-(3-ethanesutfonyl-phenvO-methylK6-trifluoromethyl-pyridin-3-ylmethVl)-amide i!bZl
- Step 1-2 Preparation of cyclopropyl f3-(ethylthio) phenyl! methanone (int. am) Method as previously described.
- Step 3 Preparation of 1-cyclopropyl-1-f3-iethylsulfonyl) phenyll-N-(f6-(trifluoromethyl) pyridin-3- ⁇ methyl) methanamine fll-6)
- Reaction mixture was stirred at reflux for 6h until completion (a sample was treated with an excess of NaBH* at 5 e C before injection in UPLC/MS). Reaction mixture was cooled down to 0*C and N3BH (1 .59 g; 41.96 mrnol; 2.00 eq.) was added portion wise over 5 minutes. Reaction mixture was stirred at 0°C for 1h until reduction completion.
- Reaction mixture was quenched with an excess of methanol added dropwise (important foaming) then resulting suspension was filtered and filtrate was concentrated until 30mL was left.
- Sodium hydroxide 1 N (1D0mL) was added and resulting thick suspension was suspended In MTBE. Filtration was done and salts were washed with MTBE. Biphasic filtrate was separated and aqueous phase was extracted with MTBE.
- Step 4 Preparation C1 R. 2R)-2-(4-Fluoro-Phenyl)-cvclooropanecarboxylic acid UR)- cvclopropyl-(3-ethanesulfonyl-phenyl)-methylH6- ⁇
- racemic benzylamlne intermediate II-5 (synthesis described below) was synthesized and both enantiomers were separated by chiral HPLC. Then enantiomer ll-5a was coupled with 4-bromobenzoic acid and structure of resulting amide H-9 was determined by X-ray to be the (R)-enantiomer, as described below. To provide further confirmation, the (S)-enantiomer ll-5b was used to synthesize an analogue of the final compound, which was compared with pure Example 99 by chiral HPLC. The stereochemistry of Example 99 was proved to be RRR.
- Step 1 Preparation of C-CvclOpropyl-C-O-ethylaulfanyl-phenvD-methylamine hydrochloride ( ⁇ -6)
- Reaction mixture was stirred at 40"C for 4h to get lithium-bromine exchange completion. Temperature was brought down to -30 e C and cyclopropanecerbonitrile (7.64mL; 101.32 mmol; 1.1 Oeq) was added drop wise over 10 minutes. Resulting nice orange light suspension was stirred at -30*C for 2h and was then allowed to warm up to 0°C until completion (Monitoring of reaction was done by quenching sample with HCI (1 N) and following ketimine and ketone formation by UPLC/MS).
- Step 2 Preparation of C-Cvclopropyl-C-(3-ethanesulfonyl-phenyl)-methylamine (II-5)
- H-8 (12.00 g; 49.22 mmol; 1.00 eq
- perchloric acid (4.20 mL; 49.22 mmol; 1.00 eq; 70%) in one portion.
- reaction mixture was cooled down to 15°C and hydrogen peroxide (50.27 mL; 492.21 mmol; 10.00 eq; 30%) was added drop wise over 10 min (exothermic at the beginning of addition) keeping temperature at 20°C.
- solution was stirred at RT for 15 min after what exotherm brought temperature at 30°C, ice bath was used to maintain temperature at 25 * C for 5h until nearly completion.
- Step 4 Preparation of 4-Bromo-N-rtR)-cyclopropyl-f3-ethanesulfonyl-phenyl)-methyll- benzamide ⁇ -9)
- first eluting enantiomer ll-5a was R and second eluting enantiomer ll-5b was S.
- Il-5b was then used for the synthesis depicted below;
- Example 99 ethaneBulfonyl-phenyl)-methyl (6-trifluoromethyl-pyridin-3-ylmethyl)-amide (11-10) was injected by chiral SFC in the same conditions than the one used for the isolation of Example 99 (Chiralcel OD-H, 250x20mm, 5um; Co-solvent: 20% MeOH). Under these conditions Example 99 was the first eluting and product 11-10, showed to be the second eluting Example 100 (SRR).
- Example 99 (1R. 2R ⁇ 2-(4 ⁇ Fluoro-phenyl)-cyclopropanecarboxylic acid ffS)- cyclopropyM3-ethanesulfonyl-phenyl)-methyll-(6 rifl ⁇
- LCMS conditions in Tables 3-12 are denoted as: 8 LCMS (Method 2) and 0 UPLC (Method 3).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Transplantation (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Pyridine Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description


















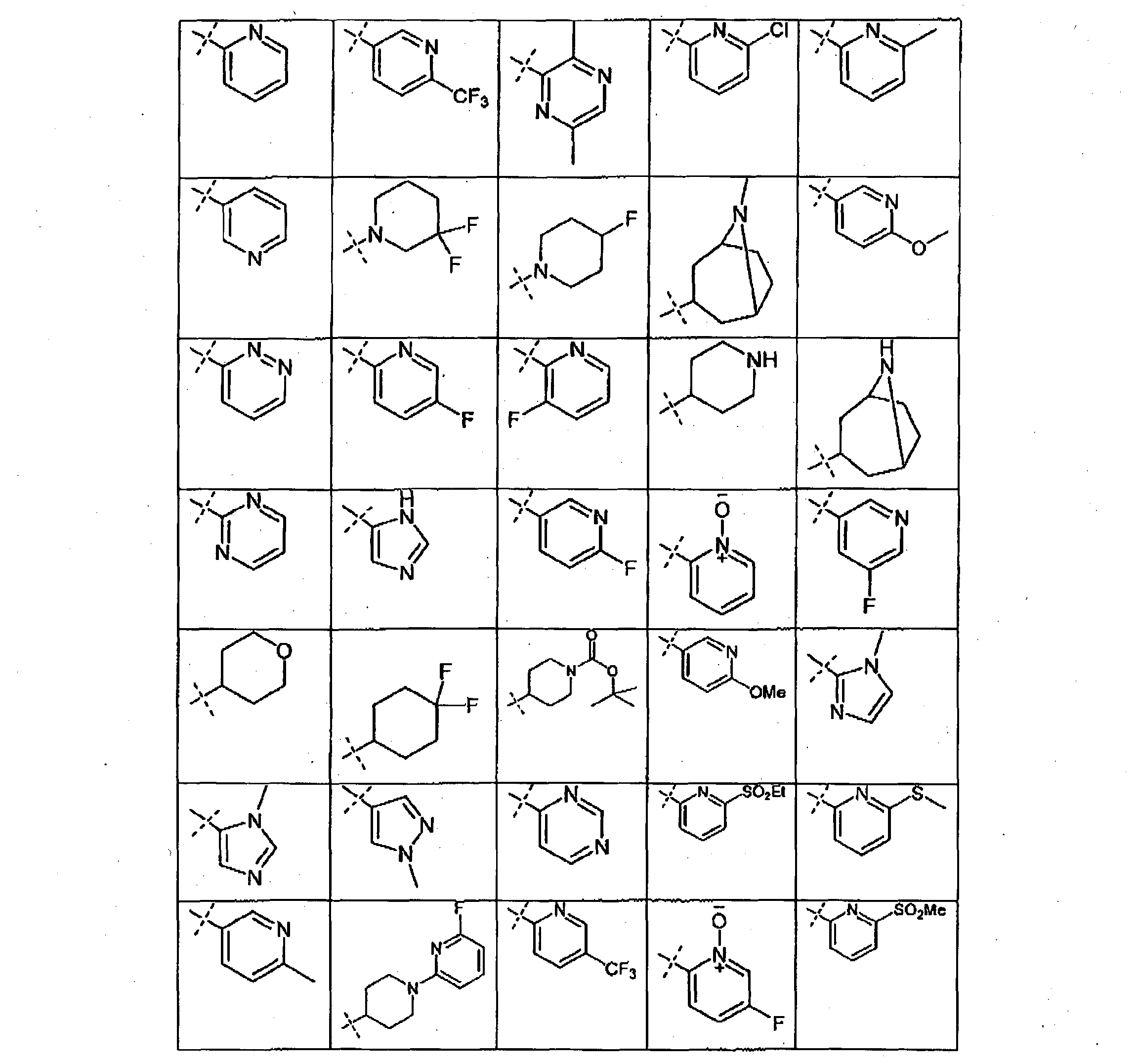






















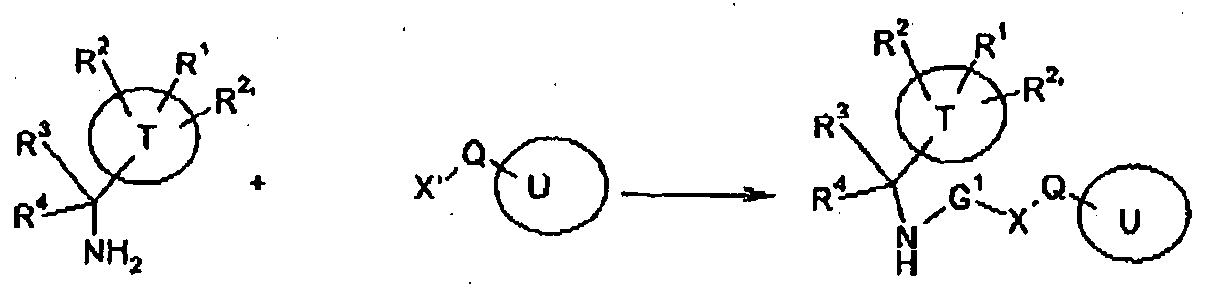




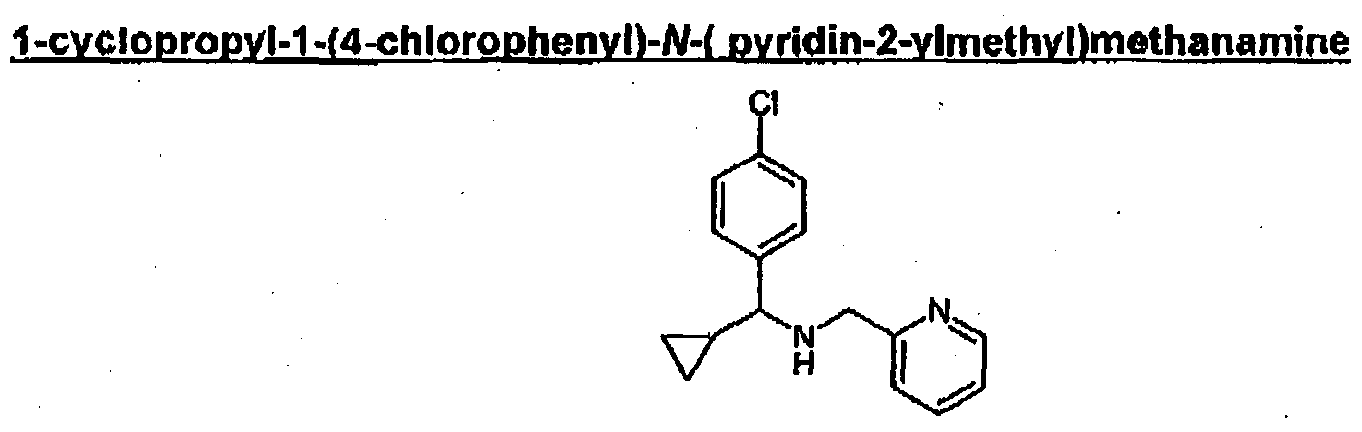




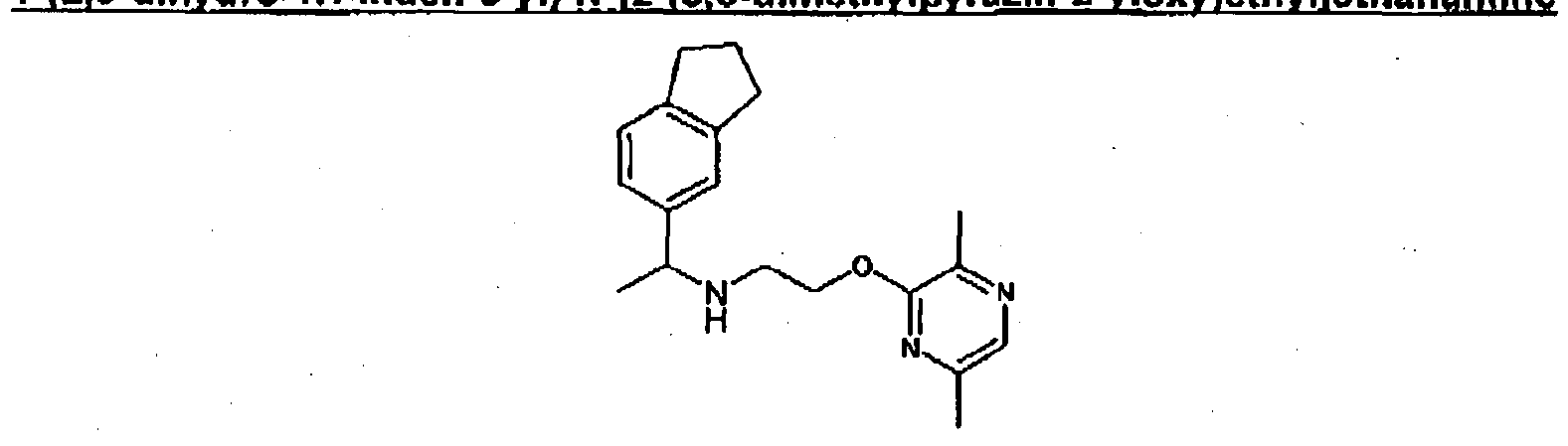


















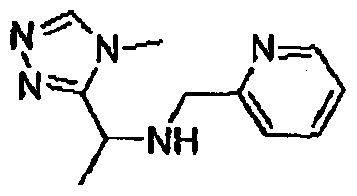
















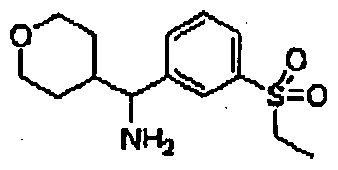












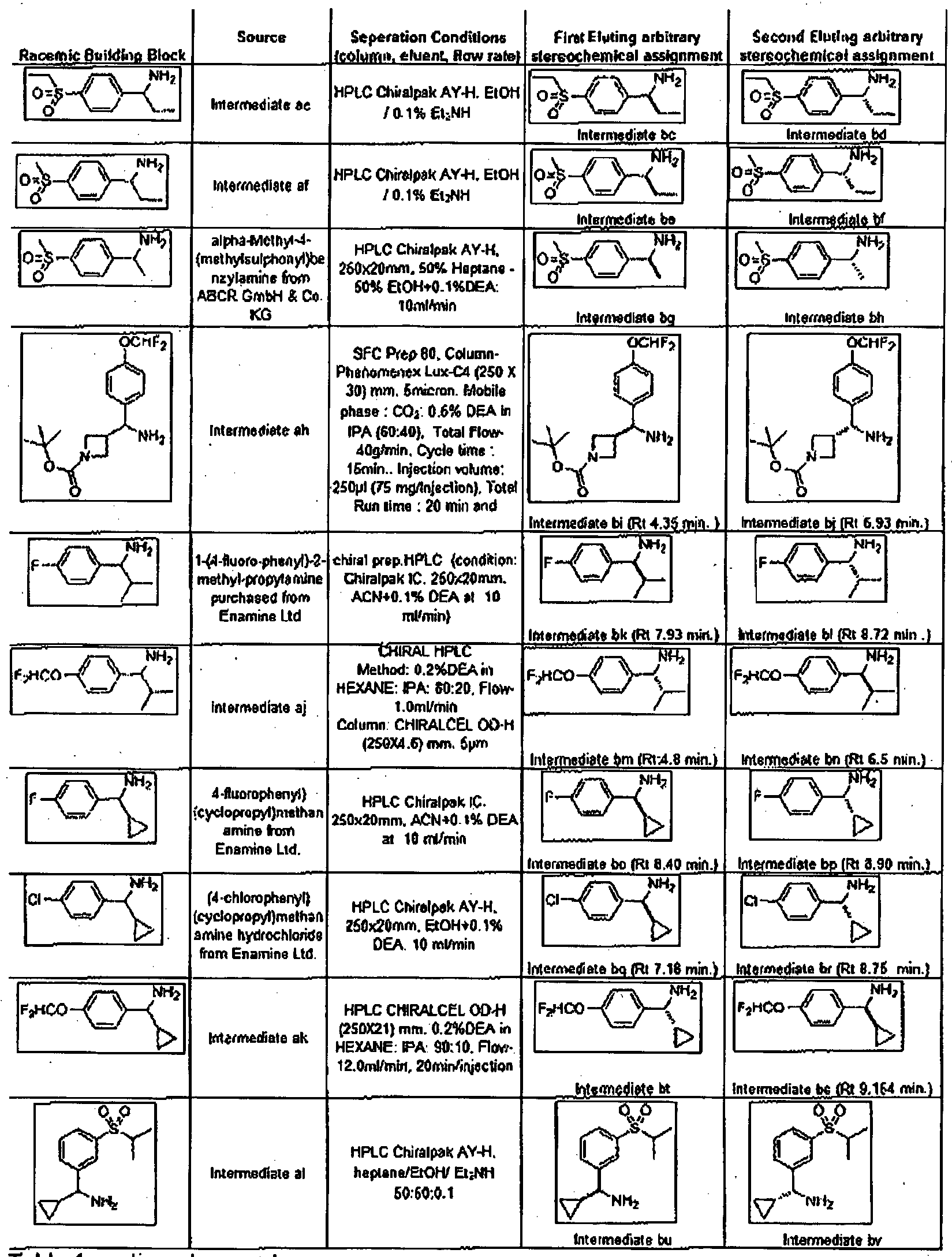






















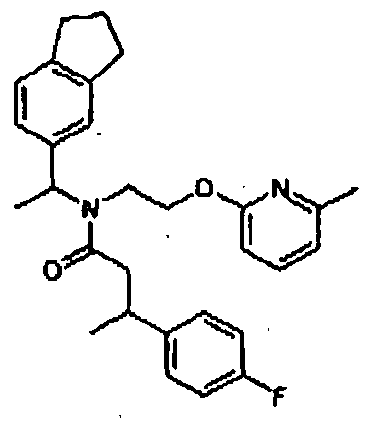






















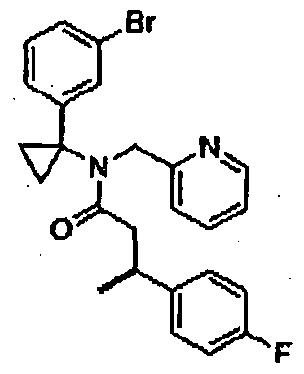



















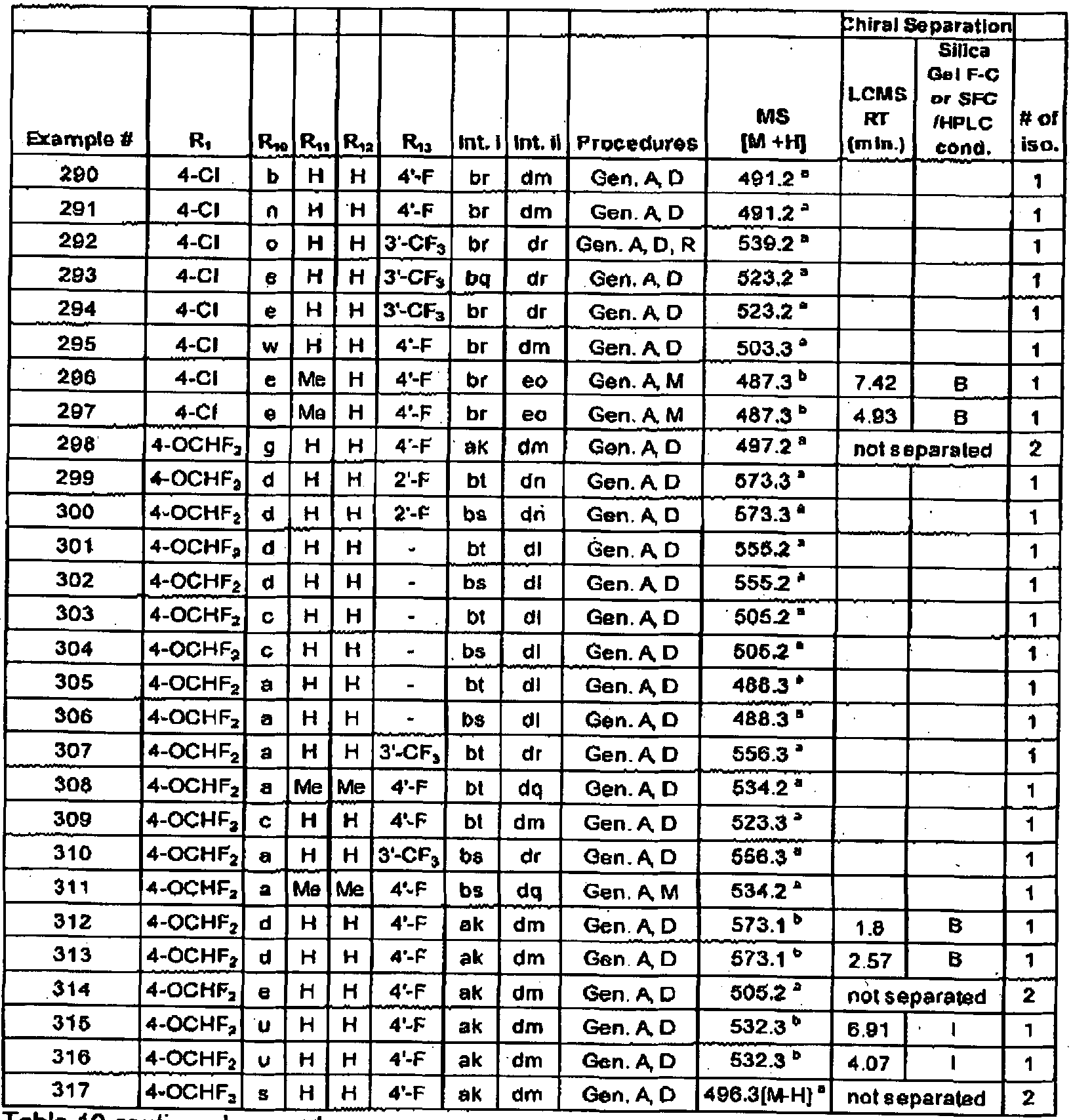





Claims












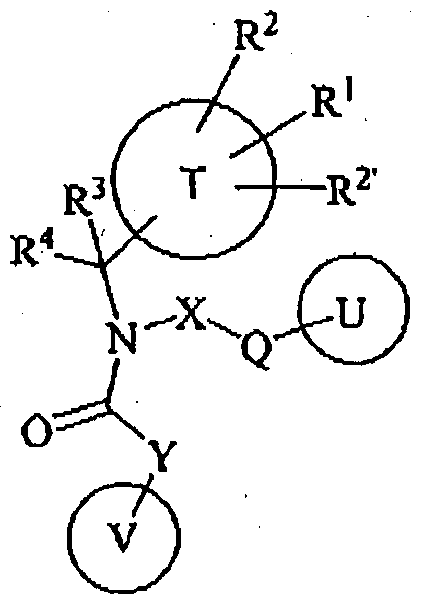








Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201280033131.9A CN103781763B (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| NZ617638A NZ617638B2 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| US14/118,170 US9493451B2 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| AU2012255690A AU2012255690B2 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| EP12785759.7A EP2709985B1 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| JP2014510611A JP6254075B2 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| CA2836311A CA2836311A1 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| IL229447A IL229447B (en) | 2011-05-16 | 2013-11-14 | Amine derivatives as potassium channel blockers |
| US14/668,272 US20150299184A1 (en) | 2011-05-16 | 2015-03-25 | Amine Derivatives as Potassium Channel Blockers |
| US15/366,513 US9914702B2 (en) | 2011-05-16 | 2016-12-01 | Amine derivatives as potassium channel blockers |
| US15/876,960 US20180273476A1 (en) | 2011-05-16 | 2018-01-22 | Amine derivatives as potassium channel blockers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161486536P | 2011-05-16 | 2011-05-16 | |
| US61/486,536 | 2011-05-16 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/118,170 A-371-Of-International US9493451B2 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
| US14/668,272 Division US20150299184A1 (en) | 2011-05-16 | 2015-03-25 | Amine Derivatives as Potassium Channel Blockers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012155199A1 true WO2012155199A1 (en) | 2012-11-22 |
Family
ID=47176042
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2012/000538 Ceased WO2012155199A1 (en) | 2011-05-16 | 2012-05-16 | Amine derivatives as potassium channel blockers |
Country Status (8)
| Country | Link |
|---|---|
| US (4) | US9493451B2 (en) |
| EP (1) | EP2709985B1 (en) |
| JP (1) | JP6254075B2 (en) |
| CN (1) | CN103781763B (en) |
| AU (1) | AU2012255690B2 (en) |
| CA (1) | CA2836311A1 (en) |
| IL (1) | IL229447B (en) |
| WO (1) | WO2012155199A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015042243A1 (en) | 2013-09-19 | 2015-03-26 | Bristol-Myers Squibb Company | Triazolopyridine ether derivatives and their use in neurological and pyschiatric disorders |
| US9493451B2 (en) | 2011-05-16 | 2016-11-15 | Bionomics Limited | Amine derivatives as potassium channel blockers |
| WO2019180185A1 (en) | 2018-03-22 | 2019-09-26 | F. Hoffmann-La Roche Ag | Oxazine monoacylglycerol lipase (magl) inhibitors |
| WO2020193419A1 (en) * | 2019-03-22 | 2020-10-01 | Saniona A/S | Novel potassium channel inhibitors |
| US10934244B2 (en) | 2015-06-15 | 2021-03-02 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| WO2021058416A1 (en) | 2019-09-23 | 2021-04-01 | F. Hoffmann-La Roche Ag | Heterocyclic compounds |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| WO2022043284A1 (en) | 2020-08-26 | 2022-03-03 | F. Hoffmann-La Roche Ag | Heterocyclic compounds useful as magl inhibitors |
| WO2022049134A1 (en) | 2020-09-03 | 2022-03-10 | F. Hoffmann-La Roche Ag | Heterocyclic compounds |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11608347B2 (en) | 2018-01-08 | 2023-03-21 | Hoffmann-La Roche Inc. | Octahydropyrido[1,2-alpha]pyrazines as MAGL inhibitors |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11802133B2 (en) | 2018-08-13 | 2023-10-31 | Hoffmann-La Roche Inc. | Heterocyclic compounds as monoacylglycerol lipase inhibitors |
| US11814375B2 (en) | 2019-09-12 | 2023-11-14 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| WO2023247670A1 (en) | 2022-06-24 | 2023-12-28 | F. Hoffmann-La Roche Ag | New heterocyclic-carbonyl-cyclic compounds as magl inhibitors |
| EP4294523A4 (en) * | 2021-02-18 | 2025-01-08 | X-Biotix Therapeutics, Inc. | ARYLTHIOETHER ACETAMIDE AND RELATED COMPOUNDS AND THEIR USE IN THE TREATMENT OF MEDICAL CONDITIONS |
| WO2025133134A1 (en) * | 2023-12-21 | 2025-06-26 | SelectION Therapeutics GmbH | Peptide for treating atopic dermatitis |
| US12440477B2 (en) | 2017-12-14 | 2025-10-14 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| EP4522583A4 (en) * | 2022-05-10 | 2026-03-11 | Relay Therapeutics Inc | PI3K-ALPHA Inhibitor and Methods for Using It |
| US12577257B2 (en) | 2018-11-22 | 2026-03-17 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112022006507A2 (en) * | 2019-10-07 | 2022-06-28 | De Shaw Res Llc | ARYLMETHYLENE HETEROCYCLIC COMPOUNDS AS KV1.3 POTASSIUM AGITATOR CHANNEL BLOCKERS |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007015775A2 (en) * | 2005-07-22 | 2007-02-08 | Merck & Co., Inc. | Potassium channel inhibitors |
| WO2007089735A2 (en) * | 2006-02-01 | 2007-08-09 | Merck & Co., Inc. | Potassium channel inhibitors |
| US20080221194A1 (en) * | 2005-03-15 | 2008-09-11 | Harvey Andrew J | Novel Potassium channel Blockers and Uses Thereof |
| WO2009043117A1 (en) * | 2007-10-04 | 2009-04-09 | Bionomics Limited | Novel aryl potassium channel blockers and uses thereof |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1469772A (en) | 1973-06-21 | 1977-04-06 | Boots Co Ltd | Fungicidal imidazole derivatives |
| JPS58103348A (en) * | 1981-12-11 | 1983-06-20 | Mitsui Toatsu Chem Inc | Preparation of n-substituted amide compound |
| JPS58131977A (en) | 1981-12-29 | 1983-08-06 | Mitsubishi Chem Ind Ltd | N-(2,3-epoxypropylene)-n-aralkylsulfonamide and selective herbicide containing said compound as active component |
| JPS5916831A (en) * | 1982-07-20 | 1984-01-28 | Mitsui Toatsu Chem Inc | Preparation of n-substituted amide compound |
| JPS59155366A (en) | 1983-02-22 | 1984-09-04 | Ube Ind Ltd | Amide derivative, its preparation, and herbicide |
| JPS59155304A (en) | 1983-02-23 | 1984-09-04 | Ube Ind Ltd | Fungicide for agricultural and horticultural purposes |
| JPS6041603A (en) | 1983-08-15 | 1985-03-05 | Hokko Chem Ind Co Ltd | Fungicide for agricultural and horticultural use |
| JPS6084204A (en) | 1983-09-20 | 1985-05-13 | Hokko Chem Ind Co Ltd | Agricultural and horticultural germicide |
| JP2612645B2 (en) * | 1991-04-05 | 1997-05-21 | 東洋インキ製造株式会社 | Method for producing carboxamide compound |
| JPH0641603A (en) | 1992-07-24 | 1994-02-15 | Hitachi Metals Ltd | Press-forming machine |
| JPH0684204A (en) | 1992-09-01 | 1994-03-25 | Nippon Columbia Co Ltd | Optical information recording medium |
| US5494895A (en) | 1993-07-22 | 1996-02-27 | Merck & Co., Inc. | Scorpion peptide margatoxin with immunosuppressant activity |
| IL117440A0 (en) | 1995-03-31 | 1996-07-23 | Pfizer | Pyrrolidinyl hydroxamic acid compounds and their production process |
| EP0891347A4 (en) | 1995-10-31 | 1999-04-07 | Merck & Co Inc | TRITERPENES DERIVATIVES HAVING IMMUNOSUPPRESSIVE ACTIVITY |
| AU708667B2 (en) | 1995-10-31 | 1999-08-12 | Merck & Co., Inc. | Triterpene derivatives with immunosuppressant activity |
| HUP0103622A3 (en) | 1998-08-20 | 2003-01-28 | Agouron Pharmaceuticals Inc La | Non-peptide gnrh agents, methods and intermediates for their preparation and medicaments containing them |
| NZ526622A (en) | 2000-12-11 | 2006-07-28 | Amgen Sf Llc | CXCR3 antagonists |
| US20050228017A1 (en) * | 2001-10-31 | 2005-10-13 | Morphochem Aktiengesellschaft Fur Kombinatorische Chemie | Novel anticancer compounds |
| US7507753B2 (en) | 2001-12-28 | 2009-03-24 | Takeda Chemical Industries Ltd. | Biaryl compound and use thereof |
| AU2005224091B2 (en) | 2004-03-15 | 2012-02-02 | Janssen Pharmaceutica, N.V. | Novel compounds as opioid receptor modulators |
| AU2005268893B2 (en) | 2004-08-05 | 2011-10-13 | F. Hoffmann-La Roche Ag | Indole, indazole or indoline derivatives |
| US20100022637A1 (en) | 2005-09-29 | 2010-01-28 | The Trustees Of Columbia University In The City Of New York | Identification of anti-cancer compounds and compounds for treating huntington's disease and methods of treatment thereof |
| US20110178326A1 (en) | 2008-08-06 | 2011-07-21 | Actavis Group Ptc Ehf | Unsaturated cinacalcet salts and processes for preparing cinacalcet hydrochloride |
| US8188079B2 (en) | 2009-08-19 | 2012-05-29 | Hoffman-La Roche Inc. | 3-amino-5-phenyl-5,6-dihydro-2H-[1,4]oxazines |
| CN103781763B (en) | 2011-05-16 | 2017-03-22 | 生态学有限公司 | Amine derivatives as potassium channel blockers |
| EP2524912A1 (en) | 2011-05-16 | 2012-11-21 | Bionomics Limited | Amine derivatives |
-
2012
- 2012-05-16 CN CN201280033131.9A patent/CN103781763B/en not_active Expired - Fee Related
- 2012-05-16 WO PCT/AU2012/000538 patent/WO2012155199A1/en not_active Ceased
- 2012-05-16 EP EP12785759.7A patent/EP2709985B1/en not_active Not-in-force
- 2012-05-16 JP JP2014510611A patent/JP6254075B2/en not_active Expired - Fee Related
- 2012-05-16 CA CA2836311A patent/CA2836311A1/en not_active Abandoned
- 2012-05-16 US US14/118,170 patent/US9493451B2/en not_active Expired - Fee Related
- 2012-05-16 AU AU2012255690A patent/AU2012255690B2/en not_active Ceased
-
2013
- 2013-11-14 IL IL229447A patent/IL229447B/en active IP Right Grant
-
2015
- 2015-03-25 US US14/668,272 patent/US20150299184A1/en not_active Abandoned
-
2016
- 2016-12-01 US US15/366,513 patent/US9914702B2/en not_active Expired - Fee Related
-
2018
- 2018-01-22 US US15/876,960 patent/US20180273476A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080221194A1 (en) * | 2005-03-15 | 2008-09-11 | Harvey Andrew J | Novel Potassium channel Blockers and Uses Thereof |
| WO2007015775A2 (en) * | 2005-07-22 | 2007-02-08 | Merck & Co., Inc. | Potassium channel inhibitors |
| WO2007089735A2 (en) * | 2006-02-01 | 2007-08-09 | Merck & Co., Inc. | Potassium channel inhibitors |
| WO2009043117A1 (en) * | 2007-10-04 | 2009-04-09 | Bionomics Limited | Novel aryl potassium channel blockers and uses thereof |
Non-Patent Citations (6)
| Title |
|---|
| DATABASE CAS [online] 5 May 2011 (2011-05-05), XP055135273, accession no. 290492-99-7 * |
| DATABASE CAS 10 April 2011 (2011-04-10), XP055135315, retrieved from N N N N accession no. 277997-05-3 * |
| DATABASE CAS 13 April 2011 (2011-04-13), XP055135311, retrieved from N N N N accession no. 279264-78-6 * |
| DATABASE CAS 4 May 2011 (2011-05-04), XP055135310, retrieved from N N N N accession no. 289770-12-2 * |
| DATABASE CAS 5 May 2011 (2011-05-05), XP055135274, retrieved from N N N N accession no. 290480-74-8 * |
| See also references of EP2709985A4 * |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9493451B2 (en) | 2011-05-16 | 2016-11-15 | Bionomics Limited | Amine derivatives as potassium channel blockers |
| US9914702B2 (en) | 2011-05-16 | 2018-03-13 | Bionomics Limited | Amine derivatives as potassium channel blockers |
| WO2015042243A1 (en) | 2013-09-19 | 2015-03-26 | Bristol-Myers Squibb Company | Triazolopyridine ether derivatives and their use in neurological and pyschiatric disorders |
| US10934244B2 (en) | 2015-06-15 | 2021-03-02 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US12440477B2 (en) | 2017-12-14 | 2025-10-14 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11608347B2 (en) | 2018-01-08 | 2023-03-21 | Hoffmann-La Roche Inc. | Octahydropyrido[1,2-alpha]pyrazines as MAGL inhibitors |
| WO2019180185A1 (en) | 2018-03-22 | 2019-09-26 | F. Hoffmann-La Roche Ag | Oxazine monoacylglycerol lipase (magl) inhibitors |
| US12281124B2 (en) | 2018-08-13 | 2025-04-22 | Hoffmann-La Roche Inc. | Heterocyclic compounds as monoacylglycerol lipase inhibitors |
| US11802133B2 (en) | 2018-08-13 | 2023-10-31 | Hoffmann-La Roche Inc. | Heterocyclic compounds as monoacylglycerol lipase inhibitors |
| US12577257B2 (en) | 2018-11-22 | 2026-03-17 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| US12006289B2 (en) | 2019-03-22 | 2024-06-11 | Saniona A/S | Potassium channel inhibitors |
| CN113631538A (en) * | 2019-03-22 | 2021-11-09 | 萨尼奥纳有限责任公司 | Novel potassium channel inhibitors |
| KR102865950B1 (en) | 2019-03-22 | 2025-09-30 | 사니오나 에이/에스 | Novel potassium channel inhibitors |
| US12391644B2 (en) | 2019-03-22 | 2025-08-19 | Saniona A/S | Potassium channel inhibitors |
| WO2020193419A1 (en) * | 2019-03-22 | 2020-10-01 | Saniona A/S | Novel potassium channel inhibitors |
| KR20210143756A (en) * | 2019-03-22 | 2021-11-29 | 사니오나 에이/에스 | Novel Potassium Channel Inhibitors |
| CN113631538B (en) * | 2019-03-22 | 2024-05-31 | 萨尼奥纳有限责任公司 | New potassium channel inhibitors |
| US11814375B2 (en) | 2019-09-12 | 2023-11-14 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| WO2021058416A1 (en) | 2019-09-23 | 2021-04-01 | F. Hoffmann-La Roche Ag | Heterocyclic compounds |
| WO2022043284A1 (en) | 2020-08-26 | 2022-03-03 | F. Hoffmann-La Roche Ag | Heterocyclic compounds useful as magl inhibitors |
| US11981661B2 (en) | 2020-09-03 | 2024-05-14 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| WO2022049134A1 (en) | 2020-09-03 | 2022-03-10 | F. Hoffmann-La Roche Ag | Heterocyclic compounds |
| EP4294523A4 (en) * | 2021-02-18 | 2025-01-08 | X-Biotix Therapeutics, Inc. | ARYLTHIOETHER ACETAMIDE AND RELATED COMPOUNDS AND THEIR USE IN THE TREATMENT OF MEDICAL CONDITIONS |
| EP4522583A4 (en) * | 2022-05-10 | 2026-03-11 | Relay Therapeutics Inc | PI3K-ALPHA Inhibitor and Methods for Using It |
| WO2023247670A1 (en) | 2022-06-24 | 2023-12-28 | F. Hoffmann-La Roche Ag | New heterocyclic-carbonyl-cyclic compounds as magl inhibitors |
| WO2025133134A1 (en) * | 2023-12-21 | 2025-06-26 | SelectION Therapeutics GmbH | Peptide for treating atopic dermatitis |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170088519A1 (en) | 2017-03-30 |
| US20140336198A1 (en) | 2014-11-13 |
| US20180273476A1 (en) | 2018-09-27 |
| IL229447B (en) | 2018-02-28 |
| JP2014524886A (en) | 2014-09-25 |
| NZ617638A (en) | 2015-09-25 |
| EP2709985A4 (en) | 2014-12-10 |
| US20150299184A1 (en) | 2015-10-22 |
| EP2709985A1 (en) | 2014-03-26 |
| CN103781763A (en) | 2014-05-07 |
| US9914702B2 (en) | 2018-03-13 |
| US9493451B2 (en) | 2016-11-15 |
| EP2709985B1 (en) | 2017-10-04 |
| CN103781763B (en) | 2017-03-22 |
| AU2012255690A1 (en) | 2013-03-14 |
| IL229447A0 (en) | 2014-01-30 |
| CA2836311A1 (en) | 2012-11-22 |
| AU2012255690B2 (en) | 2015-06-11 |
| JP6254075B2 (en) | 2017-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2709985B1 (en) | Amine derivatives as potassium channel blockers | |
| TWI761961B (en) | Fused pyridone compound and preparation method and application thereof | |
| EP3580220B1 (en) | Aminotriazolopyridines as kinase inhibitors | |
| KR102479789B1 (en) | Cyclopropyl-amide compounds as dual LSD1/HDAC inhibitors | |
| TWI911340B (en) | heteroarylmethylamine compounds | |
| JPWO2015079692A1 (en) | Urea derivative or pharmacologically acceptable salt thereof | |
| JP2009197010A (en) | Derivative having ppar agonistic activity | |
| US20180186739A1 (en) | Spiro[cyclobutane-1,3'-indolin]-2'-one derivatives as bromodomain inhibitors | |
| US12286422B2 (en) | Methods and compositions of 4-substituted benzoylpiperazine-1-substituted carbonyls as β-catenin/B-cell lymphoma 9 inhibitors | |
| IL301007A (en) | Gpr52 modulator compounds | |
| TW202229275A (en) | Autotaxin inhibitor compounds | |
| JP7168456B2 (en) | Indoline derivatives and methods of using and producing them | |
| EP2524912A1 (en) | Amine derivatives | |
| JP2008239616A (en) | HDL raising agent | |
| NZ617638B2 (en) | Amine derivatives as potassium channel blockers | |
| HK1195556B (en) | Amine derivatives as potassium channel blockers | |
| HK1195556A (en) | Amine derivatives as potassium channel blockers | |
| KR102651320B1 (en) | Novel heteroaryl substituted derivative, and pharmaceutical composition for the prevention or treatment of neurodegenerative disease, cancer, and inflammatory disease comprising thereof | |
| HK1263406B (en) | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | |
| HK1263406A1 (en) | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12785759 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012255690 Country of ref document: AU Date of ref document: 20120516 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2836311 Country of ref document: CA Ref document number: 2014510611 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012785759 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012785759 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14118170 Country of ref document: US |