WO2012169475A1 - クライゼン転位反応による二環性化合物の製造方法 - Google Patents
クライゼン転位反応による二環性化合物の製造方法 Download PDFInfo
- Publication number
- WO2012169475A1 WO2012169475A1 PCT/JP2012/064416 JP2012064416W WO2012169475A1 WO 2012169475 A1 WO2012169475 A1 WO 2012169475A1 JP 2012064416 W JP2012064416 W JP 2012064416W WO 2012169475 A1 WO2012169475 A1 WO 2012169475A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- general formula
- added
- acid
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(CC1(C2)C#N)=C[C@]1C2=O Chemical compound *C(CC1(C2)C#N)=C[C@]1C2=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/48—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation involving decarboxylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/22—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from lactams, cyclic ketones or cyclic oximes, e.g. by reactions involving Beckmann rearrangement
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/54—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition of compounds containing doubly bound oxygen atoms, e.g. esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/14—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof
- C07C227/18—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions involving amino or carboxyl groups, e.g. hydrolysis of esters or amides, by formation of halides, salts or esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/32—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/41—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by hydrogenolysis or reduction of carboxylic groups or functional derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
- C07C45/515—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups the singly bound functional group being an acetalised, ketalised hemi-acetalised, or hemi-ketalised hydroxyl group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/723—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups polycyclic
- C07C49/727—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups polycyclic a keto group being part of a condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/377—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
- C07C51/38—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups by decarboxylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/14—All rings being cycloaliphatic
- C07C2602/20—All rings being cycloaliphatic the ring system containing seven carbon atoms
Definitions
- the present invention is bicyclic ⁇ - amino acid derivative or a pharmacologically acceptable salt thereof, especially a compound having an activity as an alpha 2 [delta] ligands and methods for making intermediates.
- Non-Patent Document 1 Compounds that exhibit high affinity binding to the ⁇ 2 ⁇ subunit of the voltage-gated calcium channel have been shown to be effective, for example, in the treatment of neuropathic pain (eg, Non-Patent Document 1 and Non-Patent Document 1). Patent Document 2).
- ⁇ 2 ⁇ ligands are known as therapeutic agents for neuropathic pain, and examples of ⁇ 2 ⁇ ligands include gabapentin and pregabalin.
- ⁇ 2 ⁇ ligands such as these compounds are useful for the treatment of epilepsy, neuropathic pain and the like (for example, Patent Document 1).
- Other compounds are disclosed in, for example, Patent Document 2, Patent Document 3, and Patent Document 4.
- Patent Document 5 and Patent Document 6 have reported Patent Document 5 and Patent Document 6 as ⁇ 2 ⁇ ligands and production methods thereof.
- An object of the present invention is to provide a method for producing a bicyclic ⁇ -amino acid derivative or a pharmacologically acceptable salt thereof, particularly a compound having activity as an ⁇ 2 ⁇ ligand and an intermediate thereof.
- Patent Document 5 or Patent Document 6 reports a production method as described in Scheme 1, and the inventors of the present application (1) improved the yield of Step1-Step4, and (2) cheaper than (2). To make it possible to manufacture using various raw materials, and (3) to improve the reproducibility by facilitating the stirring at Step 4, to continue the earnest research, as a result to solve the problem, The present invention has been completed.
- R 1 a hydrogen atom or a C1-C6 alkyl group
- R 1 a hydrogen atom or a C1-C6 alkyl group
- a compound having the general formula (IV) is produced by heating a compound having the general formula (III) in the presence of an acid anhydride or an acid anhydride and an acid,
- R 1 a hydrogen atom or a C1-C6 alkyl group
- a compound having the general formula (V) is produced by heating a compound having the general formula (IV) with malonic acid in the presence of a base or a base and a catalyst,
- R 1 a hydrogen atom or a C1-C6 alkyl group
- R 1 is a methyl group, an ethyl group, or a butyl group.
- R 1 is an ethyl group.
- R 1 a hydrogen atom or a C1-C6 alkyl group
- a bicyclic ⁇ -amino acid derivative having excellent activity as an ⁇ 2 ⁇ ligand, a production intermediate thereof, and a salt thereof can be provided.
- a target compound can be produced using only inexpensive raw materials, and it is also necessary to use a reagent with high ignition risk such as sodium hydride, n-butyllithium, sodium borohydride and the like. Absent.
- the production process can be continuously performed without isolating a low-boiling compound, and the reaction in a homogeneous system is possible, the target compound can be produced efficiently.
- C1-C6 alkyl group refers to a linear or branched alkyl group having 1 to 6 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec -Butyl, tert-butyl, pentyl, isopentyl, 2-methylbutyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 4-methylpentyl, 3-methylpentyl, 2-methyl Pentyl group, 1-methylpentyl group, 3,3-dimethylbutyl group, 2,2-dimethylbutyl group, 1,1-dimethylbutyl group, 1,2-dimethylbutyl group, 1,3-dimethylbutyl group, 2 , 3-dimethylbutyl group, 2-ethylbutyl group and the like.
- R 1 a hydrogen atom or a C1-C6 alkyl group
- the acid anhydride used in this reaction is preferably acetic anhydride, propionic anhydride, butanoic anhydride, succinic anhydride, or the like, and more preferably acetic anhydride.
- a catalyst may not be used, but the reaction can be promoted by adding a catalytic amount of acid.
- the acid used as the catalyst is preferably a carboxylic acid, and more preferably maleic acid.
- it is not necessary to use a solvent but the use of an aprotic polar solvent such as dimethylacetamide can promote the progress of the reaction.
- This reaction can be preferably carried out in about 10-30 hours by heating to about 100-130 ° C.
- Knovenagel type condensation reaction Doebner type reaction, Scheme 3
- the compound having the general formula (V) is produced by reacting the compound having the general formula (IV) under the conditions of the Knovenagel type condensation reaction.
- R 1 a hydrogen atom or a C1-C6 alkyl group
- Preferable as the base used in this reaction is pyridine.
- the reaction can be smoothly renewed by adding piperidine or morpholine as a catalyst. Although this reaction proceeds by heating, it is preferably heating at 70 ° C. or higher.
- the solvent used in this reaction is preferably pyridine, acetonitrile or toluene.
- Scheme 4 A compound having the general formula (I) and a compound having the general formula (II) are produced by reacting the compound having the general formula (V) under the conditions of [2 + 2] cycloaddition reaction.
- R 1 a hydrogen atom or a C1-C6 alkyl group
- the acid anhydride used in this reaction is preferably acetic anhydride, propionic anhydride, or butyric anhydride, and more preferably acetic anhydride.
- the tertiary amine used in this reaction is preferably triethylamine, tripropylamine, tributylamine, N-methylmorpholine, and more preferably triethylamine.
- the solvent used in this reaction is preferably an aprotic solvent such as N, N-dimethylformamide, N, N-dimethylacetamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone, 1,3-dimethyl. -2-Imidazolidinone, and more preferably N, N-dimethylacetamide.
- This reaction proceeds by heating, but a suitable reaction temperature is 100 to 120 ° C., and the reaction time in this case is 5 to 10 hours.
- salt in the present invention means that a compound represented by the general formula (VI) forms a salt by reacting with an acid or a base when the structure has an amino group and / or a carboxyl group. So that salt.
- a compound having the general formula (VI) may be left in the air or recrystallized to absorb moisture and have adsorbed water to form a hydrate. Such hydrates are also included in the salts of the present invention.
- the compound having the general formula (VI), or a pharmacologically acceptable salt thereof shows activity as an ⁇ 2 ⁇ ligand and has an affinity for the ⁇ 2 ⁇ subunit of the voltage-gated calcium channel. It is useful as an active ingredient in pharmaceutical compositions used for the treatment and / or prevention of pain, central nervous system disorders, and other disorders.
- the reaction mixture was again cooled to below 10 ° C., and potassium carbonate (2.38 g) and water (400 mL) were added sequentially. The mixture was stirred until the insoluble material was dissolved, and the organic layer was separated and washed with water (100 mL). The obtained organic layer was concentrated under reduced pressure, and the residue was distilled (about 20 mmHg, 80-85 ° C.) to obtain the title compound (132.83 g, yield 91%, colorless oil).
- the aqueous layer was extracted with toluene (210 mL), combined with the organic layer described above, and then washed successively with water (102 mL) and 20% brine (102 mL).
- the organic layer was filtered to remove insolubles, and then used in the next step without concentration or purification.
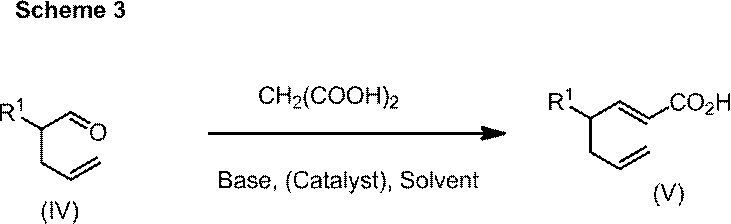
- 2-ethylpent-4-enal 58.44 g, yield 87%.
- (2E) -4-ethylhepta-2,6-dienoic acid (100.57 g) obtained by the above method was dissolved in N, N-dimethylacetamide (255 mL), and acetic anhydride (108 mL, 1 .14 mol), triethylamine (79 mL, 0.57 mol) was added.
- the reaction mixture was warmed and stirred at 115-117 ° C. for 5 hours.
- the reaction mixture was cooled to room temperature, n-hexane (510 mL) and water (714 mL) were added, and the organic layer was separated.
- the aqueous layer was extracted twice with hexane (255 mL each), all the organic layers were combined, and then washed successively with 5% aqueous sodium bicarbonate (102 mL) and water (102 mL). The obtained organic layer was concentrated under reduced pressure, and the residue was distilled (90-100 ° C., about 25 mmHg) to obtain the title compound (50.92 g, colorless oil).
- the toluene solution of 2-methylpent-4-enal obtained by the above method was added malonic acid (99.90 g, 0.96 mol), acetonitrile (285 mL), morpholine (26 mL, 0.30 mol), pyridine ( 97 mL, 1.20 mol) was sequentially added, and the mixture was heated to 70 to 80 ° C. and stirred for about 21 hours.
- the reaction mixture was cooled to room temperature, water (380 mL) was added, concentrated hydrochloric acid (130 mL) was added, and the organic layer was separated.
- the aqueous layer was extracted with toluene (140 mL), and the organic layers described above were combined, and then extracted sequentially into the aqueous layer with 2M NaOH aqueous solution (358 mL) and 1.3M NaOH aqueous solution (134 mL).
- the aqueous layers were combined, acidified with concentrated hydrochloric acid (80 mL), and extracted twice with toluene (190 mL, 95 mL).
- the organic layers were combined, washed with water (95 mL), and the organic layer was concentrated under reduced pressure to obtain the title compound (69.05 g, brown oil).
- (2E) -4-methylhepta-2,6-dienoic acid (67.74 g) obtained by the above method was dissolved in N, N-dimethylacetamide (210 mL), and acetic anhydride (91 mL, 0 mL) was dissolved. 96 mol), triethylamine (67 mL, 0.48 mol) was added. The reaction mixture was warmed and stirred at 115-120 ° C. for 6 hours. The reaction mixture was cooled to room temperature, n-hexane (360 mL) and water (540 mL) were added, and the organic layer was separated.
- the aqueous layer was extracted twice with hexane (180 mL each), and all the organic layers were combined, and then washed successively with 5% aqueous sodium bicarbonate (90 mL) and water (90 mL).
- the obtained organic layer was concentrated under reduced pressure, and the residue was distilled (74-76 ° C., about 25 mmHg) to obtain the title compound (39.09 g, colorless oil).
- the aqueous layer was extracted with toluene (180 mL), and the above-mentioned organic layers were combined, and then extracted sequentially into the aqueous layer with 2M NaOH aqueous solution (358 mL) and 1.3M NaOH aqueous solution (134 mL). After combining the aqueous layers, the mixture was acidified with concentrated hydrochloric acid (80 mL), and extracted twice with toluene (240 mL, 180 mL). The organic layers were combined, washed with water (120 mL), and the organic layer was concentrated under reduced pressure to obtain the title compound (99.11 g, brown oil).
- (2E) -4-allyloct-2-enoic acid (99.10 g) obtained by the above method was dissolved in N, N-dimethylacetamide (297 mL), and acetic anhydride (103 mL, 1.09 mol) was dissolved. ), Triethylamine (76 mL, 0.55 mol) was added. The reaction mixture was warmed and stirred at 115-120 ° C. for 6 hours. The reaction mixture was cooled to room temperature, n-hexane (480 mL) and water (720 mL) were added, and the organic layer was separated.
- the aqueous layer was extracted twice with hexane (240 mL each), and all the organic layers were combined, and then washed successively with 5% aqueous sodium bicarbonate (120 mL) and water (120 mL). The obtained organic layer was concentrated under reduced pressure, and the residue was distilled (120-128 ° C., about 25 mmHg) to obtain the title compound (61.95 g).
- N-Butyllithium (1.58 M hexane solution, 34.8 mL, 55 mmol) was added dropwise to the reaction solution, and the mixture was further stirred for 10 minutes under ice cooling, then allyl bromide (4.7 mL, 55 mmol) was added, and After stirring for an hour, the mixture was further stirred at room temperature for 4 hours.
- 1N Hydrochloric acid and saturated aqueous ammonium chloride solution were added to the reaction mixture, and the mixture was extracted with n-pentane. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the aqueous layer was extracted with ethyl acetate, and the organic layers were combined, washed with saturated brine, and dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the desired product as a pale yellow oily substance (1.32 g, 31%, E / Z mixture).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Materials Engineering (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description

[1] 一般式(I)を有する化合物及び一般式(II)を有する化合物を製造する方法であり、

(1)一般式(III)を有する化合物を、酸無水物又は、酸無水物及び酸の存在下、加熱することにより、一般式(IV)を有する化合物を製造し、

(2)一般式(IV)を有する化合物を、塩基又は、塩基及び触媒の存在下、マロン酸と加熱することにより、一般式(V)を有する化合物を製造し、

(3)一般式(V)を有する化合物を酸無水物及び3級アミンの存在下、加熱することにより、一般式(I)を有する化合物及び一般式(II)を有する化合物を製造する方法。
[2] R1が、メチル基、エチル基又はブチル基である、[1]に記載の方法。
[3] R1が、エチル基である、[1]記載の方法。
[4] (1)で用いる酸無水物が無水酢酸である、[1]-[3]のいずれか1項に記載の方法。
[5] (1)で用いる酸が、マレイン酸である、[1]-[4]のいずれか1項に記載の方法。
[6] (2)で用いる塩基がピリジンである、[1]-[5]のいずれか1項に記載の方法。
[7] (2)で用いる触媒がピペリジン又はモルホリンである、[1]-[6]のいずれか1項に記載の方法。
[8] (3)で用いる酸無水物が無水酢酸で、3級アミンがトリエチルアミンである、[1]-[7]のいずれか1項に記載の方法。
[9] 一般式(VI)を有する化合物またはその塩を製造する方法であり、

[1]に記載の製造する方法により一般式(I)を有する化合物及び一般式(II)を有する化合物を製造した後、
その一般式(I)を有する化合物及び一般式(II)を有する化合物を用いて、一般式(VI)を有する化合物またはその塩を製造する方法。
本発明によれば、安価な原料のみを使用して目的とする化合物を製造することができ、水素化ナトリウムやn-ブチルリチウムや水素化ホウ素ナトリウム等の発火危険性が高い試薬を用いる必要もない。また、低沸点の化合物を単離することなく、連続的に製造工程を行うことが可能であり、均一系での反応が可能であることから効率良く目的とする化合物を製造することができる。
(1)クライゼン転位反応(Scheme 2)
一般式(III)を有する化合物に対して、クライゼン転位反応の条件で反応を行うことにより、一般式(IV)を有する化合物を製造する。

本反応に用いる酸無水物としては、好適には、無水酢酸、無水プロピオン酸、無水ブタン酸、無水コハク酸等であり、より好適には、無水酢酸である。
本反応においては、触媒を用いなくてもよいが、触媒量の酸を添加することにより反応を促進することができる。触媒として用いる酸としては、好適にはカルボン酸であり、より好適にはマレイン酸である。
本反応においては、溶媒を使用しなくてもよいが、ジメチルアセトアミドのような非プロトン性の極性溶媒を用いると反応の進行を促進することができる。
本反応は、好適には、100-130℃程度に加熱することによって、10-30時間程度で行うことができる。
(2)Knovenagel型縮合反応(Doebner型反応、Scheme 3)
一般式(IV)を有する化合物に対して、Knovenagel型縮合反応の条件で反応を行うことにより、一般式(V)を有する化合物を製造する。
本反応に用いられる塩基として、好適には、ピリジンであり。触媒としてピペリジン又はモルホリン等を加えることによって反応を円滑に新させることができる。
本反応は、加熱することによって反応が進行するが、好適には、70℃以上の加熱である。
本反応に用いられる溶媒として、好適には、ピリジン、アセトニトリル又はトルエンである。
(3)[2+2]付加環化反応(Scheme 4)
一般式(V)を有する化合物に対して、[2+2]付加環化反応の条件で反応を行うことにより、一般式(I)を有する化合物及び一般式(II)を有する化合物を製造する。

本反応に用いられる酸無水物としては、好適には、無水酢酸、無水プロピオン酸、無水酪酸であり、より好適には、無水酢酸である。
本反応に用いられる3級アミンとしては、好適には、トリエチルアミン、トリプロピルアミン、トリブチルアミン、N-メチルモルホリンであり、より好適には、トリエチルアミンである。
本反応は、加熱することによって進行するが、好適な反応温度は、100-120℃であり、その場合の反応時間は5-10時間である。
一般式(I)を有する化合物及び一般式(II)を有する化合物を用いて、先に記載の特許文献6(WO 2010/110361)に記載の方法により製造を行うことにより、一般式(VI)を有する化合物を製造することができる。

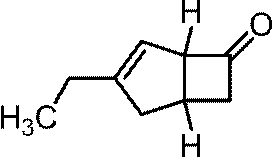
(1-a)1,1-ビス(アリルオキシ)ブタン

1H NMR (400 MHz, CDCl3) δ 0.94 (t, 3H, J=7.4 Hz), 1.35-1.45 (m, 2H), 1.61-1.66 (m, 2H), 4.01 (dd, 2H, J=5.6, 12.0 Hz), 4.09 (dd, 2H, J=5.6, 12.0 Hz), 4.61 (t, 1H, J=5.8 Hz), 5.16 (dd, 2H, J=1.6, 10.8 Hz), 5.29 (dd, 2H, J=1.6, 17.2 Hz), 5.92 (ddt, 2H, J=10.8, 17.2, 5.6 Hz).
(1-b)2-エチルペンタ-4-エナール
1H NMR (400 MHz, CDCl3) δ0.93 (t, 3H, J=7.4 Hz), 1.53-1.61 (m, 1H), 1.64-1.73 (m, 1H), 2.21-2.34 (m, 2H), 2.37-2.44 (m, 1H), 5.04-5.11 (m, 2H), 5.75 (ddt, 1H, J=10.0, 17.2, 7.0 Hz), 9.62 (d, 1H, J=2.0 Hz).
(1-c)(2E)-4-エチルヘプタ-2,6-ジエン酸

1H NMR (400 MHz, CDCl3) δ0.88 (t, 3H, J=7.4 Hz), 1.32-1.42 (m, 1H), 1.50-1.60 (m, 1H), 2.12-2.25 (m, 3H), 5.00-5.06 (m, 2H), 5.65-5.76 (m, 1H), 5.80 (d, 1H, J=15.8 Hz), 6.90 (dd, 1H, J=8.4, 15.8 Hz).
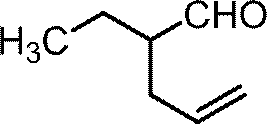
(1-d)3-エチルビシクロ[3.2.0]ヘプタ-3-エン-6オン
1H NMR (400 MHz, CDCl3) δ1.07 (t, 3H, J=7.4 Hz), 2.14 (q, 2H, J=7.4 Hz), 2.28-2.34 (m, 1H), 2.75-2.86 (m, 3H), 3.16-3.25 (m, 1H), 4.16-4.22 (m, 1H), 5.20-5.24 (m, 1H).
(実施例2)3-メチルビシクロ[3.2.0]ヘプタ-3-エン-6オン
(2-a)1,1-ビス(アリルオキシ)プロパン
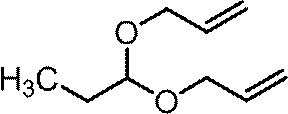
1H NMR (400 MHz, CDCl3) δ0.94 (t, 3H, J=7.4 Hz), 1.681 (dq, 2H, J=5.8, 7.4 Hz), 3.99-4.04 (m, 2H), 4.08-4.13 (m, 2H), 4.53 (t, 1H, J=5.8 Hz), 5.15-5.18 (m, 2H), 5.27-5.32 (m, 2H), 5.88-5.98 (m, 2H).
(2-b)2-メチルペンタ-4-エナール

(2-c)(2E)-4-メチルヘプタ-2,6-ジエン酸

1H NMR (400 MHz, CDCl3) δ1.08 (d, 3H, J=7.2 Hz), 2.08-2.23 (m, 2H), 2.41-2.48 (m, 1H), 5.02-5.08 (m, 2H), 5.68-5.77 (m, 1H), 5.80 (dd, 1H, J=1.2, 16.0 Hz), 7.02 (dd, 1H, J=7.4, 16.0 Hz).
(2-d)3-メチルビシクロ[3.2.0]ヘプタ-3-エン-6オン

1H NMR (400 MHz, CDCl3) δ1.80 (s, 3H), 2.26-2.32 (m, 1H), 2.72-2.86 (m, 3H), 3.16-3.26 (m, 1H), 4.15-4.23 (m, 1H), 5.20-5.24 (m, 1H).
(実施例3)
(3-a)1,1-ビス(アリルオキシ)ヘキサン

1H NMR (400 MHz, CDCl3) δ 0.90 (t, 3H, J=6.8 Hz), 1.26-1.42 (m, 6H), 1.62-1.68 (m, 2H), 4.02 (dd, 2H, J=5.6, 12.8 Hz), 4.11 (dd, 2H, J=5.6, 12.8 Hz), 4.60 (t, 1H, J=5.8 Hz), 5.17 (dd, 2H, J=1.8, 10.6 Hz), 5.29 (dd, 2H, J=1.8, 17.4 Hz), 5.92 (ddt, 2H, J=10.6, 17.4, 5.6 Hz).
(3-b)2-アリルヘキサナール

(3-c)(2E)-4-アリルオクタ-2-エン酸

1H NMR (400 MHz, CDCl3) δ0.88 (t, 3H, J=7.2 Hz), 1.16-1.39 (m, 5H), 1.45-1.54 (m, 1H), 2.09-2.36 (m, 3H), 5.00-5.05 (m, 2H), 5.65-5.76 (m, 1H), 5.79 (d, 1H, J=15.6 Hz), 6.90 (dd, 1H, J=8.6, 15.6 Hz).
(3-d)3-ブチルビシクロ[3.2.0]ヘプタ-3-エン-6オン

1H NMR (400 MHz, CDCl3) δ0.91 (t, 3H, J=7.4 Hz), 1.31 (tq, 2H, J=7.4, 7.4 Hz), 1.41-1.49 (m, 2H), 2.13 (t, 2H, J=7.6 Hz), 2.28-2.34 (m, 1H), 2.73-2.86 (m, 3H), 3.16-3.25 (m, 1H), 4.15-4.23 (m, 1H), 5.21-5.25 (m, 1H).
(参考例1)[6-アミノメチル-3-エチルビシクロ[3.2.0]へプタ-3-エン-6-イル]酢酸

3-オキソヘキサン酸エチル(7.91g、50mmol)のテトラヒドロフラン溶液(50mL)に水素化ナトリウム(>63%油性、2.09g、55mmol)を氷冷下加え、そのまま10分攪拌した。反応液にn-ブチルリチウム(1.58Mヘキサン溶液、34.8mL、55mmol)を滴下し、さらに氷冷下で10分攪拌した後、臭化アリル(4.7mL、55mmol)を加え、そのまま1時間攪拌した後、室温でさらに4時間攪拌した。反応液に1N塩酸、飽和塩化アンモニウム水溶液を加えた後、n-ペンタンで抽出した。飽和食塩水で有機層を洗浄、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。得られた残渣をエタノール(80mL)に溶解させ、氷冷下で水素化ホウ素ナトリウム(1.51g、40mmol)を加え、そのまま2時間攪拌した。1N塩酸(50mL)を加えて30分攪拌した後、飽和食塩水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィーにより精製し、目的物を淡黄色油状物質として得た(3.64g、37%、ジアステレオマーの混合物)。
1H-NMR(400MHz、CDCl3):δ ppm: 0.91 (3H, t, J=7.5Hz), 1.28 (3H, t, J=7.2Hz), 1.43-1.55 (2H, m), 1.98-2.28 (2H, m), 2.45-2.48 (2H, m), 2.88-2.93 (1H, m), 4.07-4.10 (1H, m), 4.10-4.20 (2H, m), 5.01-5.09 (2H, m), 5.75-5.86 (1H, m).
(1-b)4-エチル-3-ヒドロキシヘプタ-6-エン酸
4-エチル-3-ヒドロキシヘプタ-6-エン酸エチル(3.64g、18.2mmol)を2N水酸化カリウム メタノール溶液(120mL)に溶解させ、室温で一晩攪拌した。反応液の溶媒を減圧留去した後、1N水酸化ナトリウム水溶液(200mL)を加え、ジエチルエーテルで抽出した。水層を氷冷下にて濃塩酸を加えて酸性にした後、再びジエチルエーテルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、溶媒を減圧留去し、目的物を淡黄色油状物質として得た(3.14g、<100%、ジアステレオマーの混合物)。
1H-NMR(400MHz、CDCl3):δ ppm: 0.91-0.96 (3H, m), 1.39-1.52 (3H, m), 2.01-2.28 (2H, m), 2.52-2.55 (2H, m), 4.05-4.15 (2H, m), 5.03-5.10 (2H, m), 5.74-5.86 (1H, m).
(1-c)3-エチルビシクロ[3.2.0]へプタ-3-エン-6-イリデン酢酸tert-ブチル
4-エチル-3-ヒドロキシへプタ-6-エン酸(3.13g、18.2mmol)を無水酢酸(15mL)に溶解し、酢酸カリウム(4.27g、43.6mmol)を加え室温で1時間40分攪拌した。反応液を加熱還流し3時間半攪拌して、反応液中に「3-エチルビシクロ[3.2.0]ヘプタ-6-エン-6-オン」を生成させた後、反応液に氷水、トルエンを加えそのまま室温で一晩攪拌した。飽和食塩水(50mL)、トルエン(20mL)を加え分液後、有機層を1N水酸化ナトリウム水溶液、飽和食塩水で順次洗浄し無水硫酸マグネシウムで乾燥して、ろ過した。このろ液を、ジメトキシホスホリル酢酸tert-ブチル(4.48g、20mmol)のテトラヒドロフラン溶液(50mL)に氷冷下で水素化ナトリウム(>65%油性、761.9mg、20mmol)を加えて調製した反応液に加え、さらに1時間攪拌した。反応液に飽和塩化アンモニウム水溶液、飽和食塩水を加え分液した。水層を酢酸エチルで抽出し、有機層を併せて飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィーにより精製し、目的物を淡黄色油状物質として得た(1.32g、31%、E/Z体混合物)。
1H-NMR(400MHz、CDCl3):δ ppm:
Major isomer: 1.06 (3H, t, J=7.4Hz), 1.45 (9H, s), 2.07-2.22 (3H, m), 2.59-2.70 (2H, m), 2.87-2.96 (1H, m), 3.30 (1H, ddt, J=8.6, 18.4, 2.7Hz), 3.86-3.88 (1H, m), 5.22-5.23 (1H, m), 5.45-5.47 (1H, m).
Minor isomer: 1.08 (3H, t, J=7.3Hz), 1.49 (9H, s), 2.07-2.21 (3H, m), 2.43-2.47 (1H, m), 2.59-2.70 (1H, m), 2.75-2.85 (1H, m), 2.87-2.96 (1H, m), 4.28-4.31 (1H, m), 5.35-5.38 (1H, m), 5.45-5.47 (1H, m).
(1-d)[3-エチル-6-(ニトロメチル)ビシクロ[3.2.0]へプタ-3-エン-6-イル]酢酸tert-ブチル
[3-エチルビシクロ[3.2.0]へプタ-3-エン-6-イリデン酢酸tert-ブチル(1.32g、5.63mmol)をニトロメタン(7mL)に溶解し、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(1.2mL、7.3mmol)を加え、50-60℃で7時間加熱攪拌した。放冷後、飽和リン酸二水素カリウム水溶液を加え、酢酸エチルで抽出後、無水硫酸マグネシウムで乾燥、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィーで精製し、目的物を無色油状物質として得た(1.39g、84%)。
1H-NMR(400MHz、CDCl3):δ ppm: 1.09 (3H, t, J=7.4Hz), 1.46 (9H, s), 1.52 (1H, dd, J=7.6, 13.2Hz), 2.06(1H,d, 16.6Hz), 2.14 (2H, q, J=7.4Hz), 2.30 (1H, ddd, J=2.4, 7.6, 13.2Hz), 2.47 (2H, s), 2.49 (1H, dd, J=7.6,16.6Hz), 2.86 (1H, quint, J=7.6Hz), 3.21-3.22 (1H, m), 4.75 (1H, d, J=11.7Hz), 4.84 (1H, d, J=11.7Hz), 5.27 (1H, s).
(1-e)[6-アミノメチル-3-エチルビシクロ[3.2.0]へプタ-3-エン-6-イル]酢酸
[3-エチル-6-(ニトロメチル)ビシクロ[3.2.0]へプタ-3-エン-6-イル]酢酸tert-ブチル(1.09g、4.71mmol)をエタノール(10mL)および水(5mL)に溶解させ、鉄粉末(1.32g、23.5mmol)、塩化アンモニウム(249.6mg、4.71mmol)を加え、加熱還流下2時間攪拌した。放冷後、飽和食塩水、飽和炭酸水素ナトリウム水溶液、および酢酸エチルで希釈し、セライトろ過により不溶物を除去した。ろ液を有機層と水層に分離し、有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。残渣に4N塩酸 酢酸エチル溶液(20mL)を加え室温で1時間攪拌した後、溶媒を減圧留去した。ジクロロメタンに懸濁させトリエチルアミンを滴下し、生じた粉末をろ取、ジクロロメタンで洗浄後、乾燥し、目的物を白色粉末として得た(425.1mg、43%)。
1H-NMR(400MHz、CD3OD):δ ppm: 1.10 (3H, t, J=7.4Hz), 1.48 (1H, dd, J=7.5, 12.5Hz), 2.03-2.08 (2H, m), 2.14 (2H, q, J=7.4Hz), 2.46 (1H, d, J=16.2Hz), 2.46-2.53 (1H, m), 2.51 (1H, d, J=16.2Hz), 2.85 (1H, quint, J=7.5Hz), 3.09-3.10 (1H, m), 3.14 (1H, d, J=13.0Hz), 3.18 (1H, d, J=13.0Hz), 5.38 (1H, dd, J=1.7, 3.7Hz).
(ジアステレオマー混合物から光学分割を行う工程)
(参考例2)
[(1R,5S,6S)-6-アミノメチル-3-エチルビシクロ[3.2.0]へプタ-3-エン-6-イル]酢酸tert-ブチル D-マンデル酸塩

1H-NMR(400MHz,DMSO-d6) δ ppm: 1.04 (3H, t, J=7.6 Hz), 1.28-1.35 (1H, m), 1.39 (9H, s), 1.96-2.11 (4H, m), 2.28 (1H, d , J=15.6 Hz), 2.33 (1H, d, J=15.6 Hz), 2.36-2.40 (1H, m), 2.72 (1H, quint, J=7.6 Hz), 3.00 (1H, d, J=13.2 Hz), 3.03 (1H, d, J=13.2 Hz), 3.31(1H, br s), 4.54 (1H, s), 5.21 -5.23 (1H, m), 7.13 -7.25 (3H, m), 7.35 -7.37 (2H, m).
[α]20 D-104.4°(C=0.108, MeOH).
Anal. calcd for C24H35NO5: C, 69.04; H, 8.45; N, 3.35; Found C, 69.15; H, 8.46; N, 3.46.
Claims (9)
- 一般式(I)を有する化合物及び一般式(II)を有する化合物を製造する方法であり、
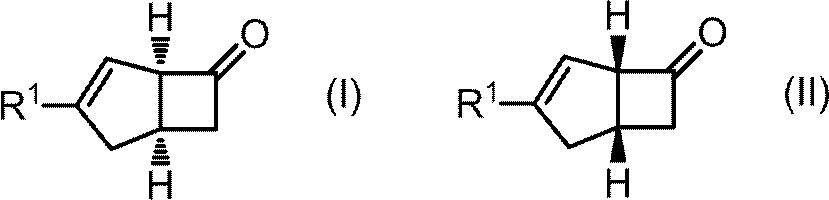
[式中、置換基は以下のように定義される。R1:水素原子またはC1-C6アルキル基]
(1)一般式(III)を有する化合物を、酸無水物又は、酸無水物及び酸の存在下、加熱することにより、一般式(IV)を有する化合物を製造し、

[式中、置換基は以下のように定義される。R1:水素原子またはC1-C6アルキル基]
(2)一般式(IV)を有する化合物を、塩基又は、塩基及び触媒の存在下、マロン酸と加熱すすることにより、一般式(V)を有する化合物を製造し、

[式中、置換基は以下のように定義される。R1:水素原子またはC1-C6アルキル基]
(3)一般式(V)を有する化合物を酸無水物及び3級アミンの存在下、加熱することにより、一般式(I)を有する化合物及び一般式(II)を有する化合物を製造する方法。 - R1が、メチル基、エチル基又はブチル基である、請求項1に記載の方法。
- R1が、エチル基である、請求項1記載の方法。
- (1)で用いる酸無水物が無水酢酸である、請求項1-3のいずれか1項に記載の方法。
- (1)で用いる酸が、マレイン酸である、請求項1-4のいずれか1項に記載の方法。
- (2)で用いる塩基がピリジンである、請求項1-5のいずれか1項に記載の方法。
- (2)で用いる触媒がピペリジン又はモルホリンである、請求項1-6のいずれか1項に記載の方法。
- (3)で用いる酸無水物が無水酢酸で、3級アミンがトリエチルアミンである、請求項1-7のいずれか1項に記載の方法。
- 一般式(VI)を有する化合物またはその塩を製造する方法であり、

[式中、置換基は以下のように定義される。
R1:水素原子またはC1-C6アルキル基]
請求項1に記載の製造する方法により一般式(I)を有する化合物及び一般式(II)を有する化合物を製造した後、
その一般式(I)を有する化合物及び一般式(II)を有する化合物を用いて、一般式(VI)を有する化合物またはその塩を製造する方法。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137032257A KR101816337B1 (ko) | 2011-06-08 | 2012-06-05 | 클라이젠 전위 반응에 의한 2 고리성 화합물의 제조 방법 |
| JP2013519490A JP5980780B2 (ja) | 2011-06-08 | 2012-06-05 | クライゼン転位反応による二環性化合物の製造方法 |
| CA2838651A CA2838651C (en) | 2011-06-08 | 2012-06-05 | Method for producing bicyclic compound via claisen rearrangement |
| CN201280027551.6A CN103562170B (zh) | 2011-06-08 | 2012-06-05 | 经由克莱森重排制备双环化合物的方法 |
| EP12796945.9A EP2719677B1 (en) | 2011-06-08 | 2012-06-05 | Method for producing bicyclic compound via claisen rearrangement |
| BR112013031365-0A BR112013031365B1 (pt) | 2011-06-08 | 2012-06-05 | Método para produzir um composto |
| ES12796945.9T ES2649968T3 (es) | 2011-06-08 | 2012-06-05 | Procedimiento de producción de un compuesto bicíclico vía transposición de Claisen |
| IL229801A IL229801A (en) | 2011-06-08 | 2013-12-05 | Manufacture method for bicyclic compound by claizan replacement |
| US14/100,402 US9162971B2 (en) | 2011-06-08 | 2013-12-09 | Methods for producing bicyclic compounds via claisen rearrangements |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011127958 | 2011-06-08 | ||
| JP2011-127958 | 2011-06-08 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/100,402 Continuation US9162971B2 (en) | 2011-06-08 | 2013-12-09 | Methods for producing bicyclic compounds via claisen rearrangements |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012169475A1 true WO2012169475A1 (ja) | 2012-12-13 |
Family
ID=47296038
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/064416 Ceased WO2012169475A1 (ja) | 2011-06-08 | 2012-06-05 | クライゼン転位反応による二環性化合物の製造方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9162971B2 (ja) |
| EP (1) | EP2719677B1 (ja) |
| JP (1) | JP5980780B2 (ja) |
| KR (1) | KR101816337B1 (ja) |
| CN (1) | CN103562170B (ja) |
| BR (1) | BR112013031365B1 (ja) |
| CA (1) | CA2838651C (ja) |
| ES (1) | ES2649968T3 (ja) |
| HU (1) | HUE035366T2 (ja) |
| IL (1) | IL229801A (ja) |
| TW (1) | TWI543964B (ja) |
| WO (1) | WO2012169475A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016146299A1 (en) | 2015-05-27 | 2016-09-22 | Novassay Sa | Separation of enantiomers of 3-ethylbicyclo[3.2.0]hept-3-en-6-one |
| WO2017183539A1 (ja) * | 2016-04-18 | 2017-10-26 | 第一三共株式会社 | メルドラム酸を用いる二環性化合物の製造方法 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2837689B1 (en) * | 2012-04-10 | 2017-02-15 | Daiichi Sankyo Company, Limited | Optical resolution method for bicyclic compound using enzyme |
| TWI635071B (zh) | 2013-07-08 | 2018-09-11 | 第一三共股份有限公司 | 光學活性二環γ-胺基酸衍生物及其製造方法 |
| CN110818582B (zh) * | 2019-11-20 | 2023-06-09 | 合肥拓锐生物科技有限公司 | 一种gaba类似物及其盐,及其合成方法、应用、药物 |
| CN114195661B (zh) * | 2021-12-21 | 2023-12-22 | 苏州楚凯药业有限公司 | 一种苯磺酸米洛巴林的制备方法 |
| WO2024134671A1 (en) * | 2022-12-21 | 2024-06-27 | Bhisaj Pharmaceuticals Pvt Ltd | Preparation of 3-ethylbicyclo[3.2.0]hept-3-en-6-one |
| CN116462576B (zh) * | 2023-04-21 | 2023-12-19 | 山东新时代药业有限公司 | 一种米洛巴林关键中间体的制备方法 |
| CN116903468B (zh) * | 2023-07-14 | 2024-01-26 | 山东新时代药业有限公司 | 一种米洛巴林中间体的制备方法 |
| CN118420477B (zh) * | 2024-07-01 | 2024-10-29 | 中节能万润股份有限公司 | 一种苯磺酸米洛巴林的制备方法 |
| DE102024130129A1 (de) | 2024-10-17 | 2026-04-23 | Technische Hochschule Nürnberg Georg Simon Ohm, in Vertretung des Freistaates Bayern | Vernetzer auf Basis von Acetalen für chemisch abbaubare und recyclingfähige Silicone, recyclingfähiges Siliconmaterial und Verfahren zu dessen Aufspaltung |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0287956A2 (de) * | 1987-04-23 | 1988-10-26 | BASF Aktiengesellschaft | Verfahren zur Herstellung von 5-Formylvaleriansäureestern oder den entsprechenden Acetalen |
| EP0521571A2 (en) * | 1991-07-04 | 1993-01-07 | ENICHEM S.p.A. | Procedure for the preparation of bicyclo [3.2.0] hept-2-en-7-ones |
| US20030078300A1 (en) | 2001-04-19 | 2003-04-24 | Pfizer Inc. | Fused bicyclic or tricyclic amino acids |
| US20030220397A1 (en) | 1997-10-27 | 2003-11-27 | Bryans Justin Stephen | Cyclic amino acids and derivatives thereof useful as pharmaceutical agents |
| US20040152779A1 (en) | 1999-10-20 | 2004-08-05 | Bryans Justin Stephen | Bicyclic amino acids as pharmaceutical agents |
| US20060154929A1 (en) | 2002-07-11 | 2006-07-13 | Anker Naomi B | Treatment of neuropathic pain with 6h-pyrrolo[3,4-d]pyridazine compounds |
| WO2006075596A1 (ja) * | 2005-01-13 | 2006-07-20 | Kuraray Co., Ltd. | 2-アリルカルボン酸化合物の製造方法 |
| WO2009041453A1 (ja) * | 2007-09-28 | 2009-04-02 | Daiichi Sankyo Company, Limited | 二環性γ-アミノ酸誘導体 |
| US20100110361A1 (en) | 2007-12-12 | 2010-05-06 | Nitto Denko Corporation | Liquid crystalline coating solution and polarizing film |
| WO2010084798A1 (ja) * | 2009-01-21 | 2010-07-29 | 第一三共株式会社 | 3環性化合物 |
| WO2010110361A1 (ja) | 2009-03-26 | 2010-09-30 | 第一三共株式会社 | 二環性γ-アミノ酸誘導体の製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2719676B1 (en) * | 2011-06-08 | 2017-12-13 | Daiichi Sankyo Company, Limited | Method for producing bicyclic compound via iminium salt |
-
2012
- 2012-06-05 WO PCT/JP2012/064416 patent/WO2012169475A1/ja not_active Ceased
- 2012-06-05 EP EP12796945.9A patent/EP2719677B1/en active Active
- 2012-06-05 JP JP2013519490A patent/JP5980780B2/ja active Active
- 2012-06-05 CA CA2838651A patent/CA2838651C/en active Active
- 2012-06-05 BR BR112013031365-0A patent/BR112013031365B1/pt active IP Right Grant
- 2012-06-05 CN CN201280027551.6A patent/CN103562170B/zh active Active
- 2012-06-05 KR KR1020137032257A patent/KR101816337B1/ko active Active
- 2012-06-05 ES ES12796945.9T patent/ES2649968T3/es active Active
- 2012-06-05 HU HUE12796945A patent/HUE035366T2/en unknown
- 2012-06-07 TW TW101120397A patent/TWI543964B/zh active
-
2013
- 2013-12-05 IL IL229801A patent/IL229801A/en active IP Right Grant
- 2013-12-09 US US14/100,402 patent/US9162971B2/en active Active
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0287956A2 (de) * | 1987-04-23 | 1988-10-26 | BASF Aktiengesellschaft | Verfahren zur Herstellung von 5-Formylvaleriansäureestern oder den entsprechenden Acetalen |
| EP0521571A2 (en) * | 1991-07-04 | 1993-01-07 | ENICHEM S.p.A. | Procedure for the preparation of bicyclo [3.2.0] hept-2-en-7-ones |
| US20030220397A1 (en) | 1997-10-27 | 2003-11-27 | Bryans Justin Stephen | Cyclic amino acids and derivatives thereof useful as pharmaceutical agents |
| US20040152779A1 (en) | 1999-10-20 | 2004-08-05 | Bryans Justin Stephen | Bicyclic amino acids as pharmaceutical agents |
| US20030078300A1 (en) | 2001-04-19 | 2003-04-24 | Pfizer Inc. | Fused bicyclic or tricyclic amino acids |
| US20060154929A1 (en) | 2002-07-11 | 2006-07-13 | Anker Naomi B | Treatment of neuropathic pain with 6h-pyrrolo[3,4-d]pyridazine compounds |
| WO2006075596A1 (ja) * | 2005-01-13 | 2006-07-20 | Kuraray Co., Ltd. | 2-アリルカルボン酸化合物の製造方法 |
| WO2009041453A1 (ja) * | 2007-09-28 | 2009-04-02 | Daiichi Sankyo Company, Limited | 二環性γ-アミノ酸誘導体 |
| US20100249229A1 (en) | 2007-09-28 | 2010-09-30 | Kousei Shimada | BICYCLIC gamma-AMINO ACID DERIVATIVE |
| US20100110361A1 (en) | 2007-12-12 | 2010-05-06 | Nitto Denko Corporation | Liquid crystalline coating solution and polarizing film |
| WO2010084798A1 (ja) * | 2009-01-21 | 2010-07-29 | 第一三共株式会社 | 3環性化合物 |
| WO2010110361A1 (ja) | 2009-03-26 | 2010-09-30 | 第一三共株式会社 | 二環性γ-アミノ酸誘導体の製造方法 |
Non-Patent Citations (4)
| Title |
|---|
| J BIOL. CHEM., vol. 271, no. 10, 1996, pages 5768 - 5776 |
| J J BEEREBOOM: "The Cyclization of Geranic Acids. Preparation of a Cyclobutanone", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 30, no. 12, 1965, pages 4230 - 4234, XP055137633 * |
| J MED. CHEM., vol. 41, 1998, pages 1838 - 1845 |
| See also references of EP2719677A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016146299A1 (en) | 2015-05-27 | 2016-09-22 | Novassay Sa | Separation of enantiomers of 3-ethylbicyclo[3.2.0]hept-3-en-6-one |
| WO2017183539A1 (ja) * | 2016-04-18 | 2017-10-26 | 第一三共株式会社 | メルドラム酸を用いる二環性化合物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2719677A1 (en) | 2014-04-16 |
| ES2649968T3 (es) | 2018-01-16 |
| TW201302694A (zh) | 2013-01-16 |
| KR20140031295A (ko) | 2014-03-12 |
| JPWO2012169475A1 (ja) | 2015-02-23 |
| EP2719677A4 (en) | 2014-12-03 |
| CN103562170A (zh) | 2014-02-05 |
| BR112013031365B1 (pt) | 2021-09-28 |
| JP5980780B2 (ja) | 2016-08-31 |
| HUE035366T2 (en) | 2018-05-02 |
| CA2838651C (en) | 2016-05-17 |
| CN103562170B (zh) | 2015-09-02 |
| TWI543964B (zh) | 2016-08-01 |
| CA2838651A1 (en) | 2012-12-13 |
| US20140094623A1 (en) | 2014-04-03 |
| IL229801A (en) | 2017-11-30 |
| US9162971B2 (en) | 2015-10-20 |
| EP2719677B1 (en) | 2017-09-06 |
| BR112013031365A2 (pt) | 2016-11-29 |
| KR101816337B1 (ko) | 2018-01-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5980780B2 (ja) | クライゼン転位反応による二環性化合物の製造方法 | |
| JP5922118B2 (ja) | イミニウム塩を経由する二環性化合物の製造方法 | |
| PL199794B1 (pl) | Związki pośrednie do wytwarzania pochodnych cyklicznych aminokwasów | |
| CN102356061A (zh) | 用于制备二环γ-氨基酸衍生物的方法 | |
| JP5683273B2 (ja) | 光学活性カルボン酸の製造方法 | |
| KR100846419B1 (ko) | 프레가발린의 신규한 제조 방법 | |
| HK1196599A (en) | Method for producing bicyclic compound via claisen rearrangement | |
| HK1196598A (en) | Method for producing bicyclic compound via iminium salt | |
| HK1196598B (en) | Method for producing bicyclic compound via iminium salt | |
| HK1196599B (en) | Method for producing bicyclic compound via claisen rearrangement | |
| WO2010071122A1 (ja) | シクロヘキセン誘導体の製造方法 | |
| HK1160105A (en) | Process for producing optically active carboxylic acid | |
| HK1199017B (en) | Optical resolution method for bicyclic compound using asymmetric catalyst |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12796945 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013519490 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012796945 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012796945 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20137032257 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2838651 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013031365 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013031365 Country of ref document: BR Kind code of ref document: A2 Effective date: 20131205 |