WO2013015245A1 - ポリウレタンフォーム - Google Patents
ポリウレタンフォーム Download PDFInfo
- Publication number
- WO2013015245A1 WO2013015245A1 PCT/JP2012/068593 JP2012068593W WO2013015245A1 WO 2013015245 A1 WO2013015245 A1 WO 2013015245A1 JP 2012068593 W JP2012068593 W JP 2012068593W WO 2013015245 A1 WO2013015245 A1 WO 2013015245A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyol
- weight
- parts
- polyurethane foam
- less
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/65—Low-molecular-weight compounds having active hydrogen with high-molecular-weight compounds having active hydrogen
- C08G18/66—Compounds of groups C08G18/42, C08G18/48, or C08G18/52
- C08G18/6629—Compounds of groups C08G18/42, C08G18/48, or C08G18/52 with compounds of group C08G18/36 or hydroxylated esters of higher fatty acids of C08G18/38
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/4244—Polycondensates having carboxylic or carbonic ester groups in the main chain containing oxygen in the form of ether groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/32—Polyhydroxy compounds; Polyamines; Hydroxyamines
- C08G18/3203—Polyhydroxy compounds
- C08G18/3206—Polyhydroxy compounds aliphatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/36—Hydroxylated esters of higher fatty acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/4009—Two or more macromolecular compounds not provided for in one single group of groups C08G18/42 - C08G18/64
- C08G18/4018—Mixtures of compounds of group C08G18/42 with compounds of group C08G18/48
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/44—Polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/65—Low-molecular-weight compounds having active hydrogen with high-molecular-weight compounds having active hydrogen
- C08G18/66—Compounds of groups C08G18/42, C08G18/48, or C08G18/52
- C08G18/6666—Compounds of group C08G18/48 or C08G18/52
- C08G18/6696—Compounds of group C08G18/48 or C08G18/52 with compounds of group C08G18/36 or hydroxylated esters of higher fatty acids of C08G18/38
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/02—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by the reacting monomers or modifying agents during the preparation or modification of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/122—Hydrogen, oxygen, CO2, nitrogen or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2101/00—Manufacture of cellular products
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0041—Foam properties having specified density
- C08G2110/0066—≥ 150kg/m3
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/06—CO2, N2 or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2205/00—Foams characterised by their properties
- C08J2205/04—Foams characterised by their properties characterised by the foam pores
- C08J2205/044—Micropores, i.e. average diameter being between 0,1 micrometer and 0,1 millimeter
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2205/00—Foams characterised by their properties
- C08J2205/06—Flexible foams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2375/00—Characterised by the use of polyureas or polyurethanes; Derivatives of such polymers
- C08J2375/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2375/00—Characterised by the use of polyureas or polyurethanes; Derivatives of such polymers
- C08J2375/04—Polyurethanes
- C08J2375/08—Polyurethanes from polyethers
Definitions
- the present invention relates to a polyurethane foam.
- silicone foam or rubber sponge is used as a sealing material around a heat source part, for example, around a battery of an electric vehicle, around an electronic control part, or a sealing part of a solar cell.
- rubber sponge has a long compressive residual strain and thus has a long-term sealability. Furthermore, the gas containing sulfur generated from the rubber sponge may corrode the electronic substrate.
- Patent Documents 1 and 2 it has been proposed to use polyurethane foam obtained by a mechanical floss method as a sealing material.
- a foaming gas is compressed and mixed into a raw material containing a polyol, an isocyanate, a foam stabilizer, and a catalyst as a polyurethane raw material.
- a polyurethane foam is formed by discharging this polyurethane raw material from an Oaks mixer or a nozzle with a narrowed tip.
- the foaming gas that has been compressed until the polyurethane raw material is discharged expands to form bubbles, and in this state, the polyol and isocyanate react and cure to form a polyurethane foam.
- polyurethane foam produced by the mechanical floss method is formed into a thin sheet suitable as a sealing material.
- polyurethane foam itself is less expensive than silicone foam, and further has better compressive residual strain than rubber sponge.
- the environmental temperature that can be used as a sealing material for polyurethane foam manufactured by the conventional mechanical floss method is about 70 ° C to 80 ° C, and since the compression residual strain is large at temperatures higher than that, it can be used as a sealing material around the heat source. It was difficult to use for a long time.
- a method for improving the heat resistance of a polyurethane foam (1) a method for increasing the intermolecular crosslinking density, and (2) a method using a compound having a highly cohesive functional group such as an ester group or a phenyl group are known. .
- the method (1) when the intermolecular crosslink density is increased, the hardness of the polyurethane foam is increased and the tear strength is lowered, which makes it unsuitable for use as a sealing material.
- a compound having a highly cohesive functional group such as an ester group or a phenyl group is often solid or waxy at room temperature and may be liquid at room temperature. Many have high raw material viscosity. For this reason, when a large amount of this compound is added to the polyurethane raw material, the mechanical floss method may cause defects such as voids (also referred to as pinholes) in the polyurethane foam.
- voids also referred to as pinholes
- an object of the present invention is to provide a polyurethane foam suitable as a sealing material around a heat source such as a battery periphery of an electric vehicle.
- the polyurethane foam of the present invention capable of solving the above problems is In a polyurethane foam obtained by a mechanical floss method from a polyurethane raw material containing polyol, isocyanate, foam stabilizer, catalyst and foaming gas,
- the polyol includes a castor oil-based polyol having a viscosity at 25 ° C.
- the castor oil-based polyol is contained in 20 parts by weight or more and 80 parts by weight or less in 100 parts by weight of the polyol
- the polyether-based polyol is contained in 20 parts by weight or more and 80 parts by weight or less in 100 parts by weight of the polyol
- the apparent density of the urethane foam is 100 kg / m 3 or more and 700 kg / m 3 or less
- the compressive residual strain at 100 ° C. is 20% or less.
- the polyurethane foam according to the present invention is The polyol contains 20 parts by weight or more and 80 parts by weight or less of a polyether polyol having an ethylene oxide ratio of 50 mol% or more, which is a ratio of oxyethylene units in polyoxyalkylene, to 100 parts by weight of the polyol.
- the total part by weight of the polyether polyol and the castor oil polyol having an ethylene oxide ratio of 50 mol% or more is preferably 50 parts by weight or more and 100 parts by weight or less.
- the polyurethane foam according to the present invention is The castor oil-based polyol is contained in 40 parts by weight or more and 70 parts by weight or less in 100 parts by weight of the polyol,
- the polyether polyol having an ethylene oxide ratio of 50 mol% or more is preferably contained in an amount of 30 parts by weight or more and 60 parts by weight or less in 100 parts by weight of the polyol.
- the thickness of the said urethane foam is 0.1 mm or more and 15 mm or less.
- the 25% compressive load of the urethane foam is preferably 0.02 MPa or more and 0.40 MPa or less.
- the urethane foam preferably has an average cell diameter of 50 ⁇ m or more and 300 ⁇ m. It is preferable that the compression residual strain of the urethane foam at 100 ° C. is 10% or less.
- the compressive residual strain of the urethane foam at 110 ° C. is preferably 10% or less.
- the compressive residual strain at 100 ° C. is 20% or less, the compressive residual strain at high temperature is small. Therefore, it is possible to provide a polyurethane foam that is also suitable as a sealing material in the vicinity of the heat source.
- FIG. 1 is a perspective view of a polyurethane foam according to an embodiment of the present invention. It is a figure which shows an example of the manufacturing apparatus of the polyurethane foam of this invention. It is a figure which shows the manufacturing apparatus of another example.
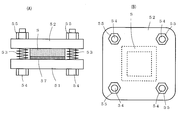
- (A) is a side view which shows the time of use of the plate-shaped jig
- (B) is the top view. It is a perspective view of the cylindrical container used for a dustproof test.
- the polyurethane foam 1 of the present invention shown in FIG. 1 is obtained from a polyurethane raw material by a mechanical floss method.
- the polyurethane raw material contains a polyol, an isocyanate, a foam stabilizer, a catalyst, and a foaming gas.
- the polyol includes a castor oil-based polyol having a viscosity at 25 ° C. (conforming to JIS Z 8803: 2011) of 2000 mPa ⁇ s or less, and a polyether-based polyol.
- the castor oil-based polyol is contained in an amount of 20 to 80 parts by weight in 100 parts by weight of the polyol.
- the polyether polyol is contained in an amount of 20 to 80 parts by weight in 100 parts by weight of the polyol.
- the compression residual strain at 100 ° C. of the polyurethane foam cannot be sufficiently reduced.
- the polyurethane foam becomes too hard even if the density of the polyurethane foam is low, so that the compression adhesion when used as a sealing material is inferior. Become.
- the polyol contains a polyether polyol from the viewpoint of improving hydrolysis resistance.
- the polyether-based polyol is less than 20 parts by weight in 100 parts by weight of the polyol, the hydrolyzability is lowered, and the compressive residual strain under high humidity cannot be sufficiently reduced.
- the amount of castor oil-based polyol added is insufficient, and the compression residual strain of polyurethane foam at 100 ° C. cannot be sufficiently reduced.
- castor oil-based polyols examples include castor oil, a reaction product of castor oil and polyol, and an esterification reaction product of castor oil fatty acid and polyol.
- examples of the polyol to be reacted with castor oil or castor oil fatty acid include divalent polyols such as ethylene glycol, diethylene glycol and propylene glycol, and trivalent or higher polyols such as glycerin, trimethylolpropane, hexanetriol and sorbitol. .
- the castor oil-based polyol one having a viscosity at 25 ° C. (based on JIS Z 8803: 2011) of 2000 mPa ⁇ s or less is used. Further, the castor oil-based polyol preferably has 2 to 3 functional groups and a number average molecular weight of 500 to 1,000 (or a hydroxyl value of 115 mgKOH / g to 225 mgKOH / g).
- Polyether polyols include ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, butylene glycol, neopentyl glycol, glycerin, pentaerythritol, trimethylolpropane, sorbitol, sucrose, and other polyhydric alcohols, ethylene oxide, propylene Polyether polyols for polyurethane such as polyether polyols to which alkylene oxides such as oxides are added can be used.
- the polyether polyol preferably has 2 to 4 functional groups and a number average molecular weight of 2,000 to 4,000. Furthermore, the polyether polyol preferably has an ethylene oxide ratio (hereinafter sometimes abbreviated as EO ratio), which is a ratio of oxyethylene units in polyoxyalkylene, of 50 mol% or more, more preferably.
- the ethylene oxide ratio is 60 mol% or more, particularly 70 mol% or more.
- the upper limit of the ethylene oxide ratio is 100 mol% or less, more preferably 90 mol% or less.
- the ethylene oxide ratio here is the ratio of the ethylene oxide adduct in the alkylene oxide adduct.
- the heat resistance means a degree to which the compressive residual strain is not easily increased in a high temperature environment.
- the polyol contains 20 parts by weight or more and 80 parts by weight or less of a polyether-based polyol having an ethylene oxide ratio of 50 mol% or more in 100 parts by weight of the polyol, and the ethylene oxide ratio is 50 mol% or more in 100 parts by weight of the polyol.
- the total weight part of the ether polyol and castor oil polyol is preferably 50 parts by weight or more and 100 parts by weight or less.
- the castor oil-based polyol is contained in 40 parts by weight or more and 70 parts by weight or less in 100 parts by weight of the polyol, and the polyether-based polyol having an ethylene oxide ratio of 50 mol% or more is 30 parts by weight in 100 parts by weight of the polyol. More than 60 parts by weight is preferable. Thereby, the heat resistance of a polyurethane foam can be improved.
- a polycarbonate-based polyol may be included as the polyol.
- the polycarbonate-based polyol include those obtained by dealcoholization reaction of polyhydric alcohols such as ethylene glycol, diethylene glycol, propylene glycol, butanediol, pentanediol, and hexanediol with dialkyl carbonate, dialkylene carbonate, diphenyl carbonate, and the like. Can be mentioned.
- polycarbonate-based polyols those having a viscosity at 60 ° C. (JIS Z 8803: 2011 compliant) are preferably 1500 mPa ⁇ s or less.
- the polycarbonate-based polyol preferably has a functional group number of 2 to 3 and a number average molecular weight of 500 to 1,000 (or a hydroxyl value of 112 mgKOH / g to 224 mgKOH / g).
- polyester-based polyol may be included as the polyol.
- the polyester polyol include low molecular polyols such as ethylene glycol, diethylene glycol, propylene glycol, butanediol, hexanediol, glycerin, trimethylolpropane, trimethylolethane, pentaerythritol, diglycerin, sorbitol, sucrose, and succinic acid.
- polyester-based polyol examples include polyols that are ring-opening condensates of caprolactone and methylvalerolactone classified as lactone esters.
- the isocyanate may be aromatic, alicyclic, or aliphatic, and may be a bifunctional isocyanate having two isocyanate groups in one molecule, or three or more in one molecule. It may be a trifunctional or higher functional isocyanate having an isocyanate group. These may be used alone or in combination.
- bifunctional isocyanate 2,4-tolylene diisocyanate (TDI), 2,6-tolylene diisocyanate (TDI), m-phenylene diisocyanate, p-phenylene diisocyanate, 4,4′-diphenylmethane diisocyanate (MDI), 2,4′-diphenylmethane diate (MDI), 2,2′-diphenylmethane diisocyanate (MDI), xylylene diisocyanate, 3,3′-dimethyl-4,4′-biphenylene diisocyanate, 3, Aromatic compounds such as 3′-dimethoxy-4,4′-biphenylene diisocyanate, cyclohexane-1,4-diisocyanate, isophorone diisocyanate, dicyclohexylmethane-4,4′-diisocyanate, methylcyclohexane diisocyanate Those alicyclic
- tri- or higher functional isocyanate examples include 1-methylbenzole-2,4,6-triisocyanate, 1,3,5-trimethylbenzole-2,4,6-triisocyanate, biphenyl-2,4,4 ′.
- Examples include phenylmethane-4,4 ′, 4 ′′ -triisocyanate, polymethylene polyphenyl isocyanate (polymeric MDI), and the like.
- the isocyanate is not limited to one type, and may be one or more types.
- one type of aliphatic isocyanate and two types of aromatic isocyanate may be used in combination.
- the isocyanate index is preferably 100 or more and 110 or less. When the isocyanate index is outside this range, the compression residual strain of the polyurethane foam increases.
- the isocyanate index is a value obtained by multiplying the number of moles of isocyanate groups by 100 with respect to 1 mole of active hydrogen groups contained in the polyurethane raw material.
- a nonionic surfactant such as polydimethylsiloxane or polyoxyalkylene is suitable.
- the foam-forming gas can form a foam structure suitable for polyurethane foam.
- the amount of the foam stabilizer is preferably 3 to 8 parts by weight per 100 parts by weight of polyol.
- an amine-based catalyst for polyurethane foam and a metal catalyst are used alone or in combination.
- the amine-based catalyst include a monoamine compound, a diamine compound, a triamine compound, a polyamine compound, a cyclic amine compound, an alcohol amine compound, an ether amine compound, and the like. One kind of these may be used, or two or more kinds may be used in combination.
- the metal catalyst include an organic tin compound, an organic bismuth compound, an organic lead compound, an organic zinc compound, and the like. One kind of these may be used, or two or more kinds may be used.
- the amount of the catalyst is preferably 0.05 parts by weight or more and 0.5 parts by weight or less per 100 parts by weight of polyol.
- the foaming gas a gas that does not adversely affect the reaction between the polyol and the isocyanate, for example, an inert gas such as dry air or nitrogen is preferable.
- the foam-forming gas is preferably included in the polyurethane raw material so that the mixing ratio in the polyurethane raw material is 31% by volume or more and 91% by volume or less.
- the mixing ratio of the foaming gas refers to the volume% of the foaming gas relative to 100 parts by volume of the polyurethane raw material excluding the foaming gas.
- crosslinking agents foaming aids, fillers, etc. are added to the polyurethane raw material as necessary.
- the crosslinking agent include low molecular compounds having 2 to 4 active hydrogen-containing groups capable of reacting with isocyanate groups and having a number average molecular weight of 50 to 800.
- Examples of the low molecular weight compound used as the crosslinking agent include ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, 1,4-butanediol, 1,6-hexanediol, neopentyl glycol, glycerin, trimethylolpropane, Examples include triethanolamine, pentaerythritol, and the like, and one or more of these can be used in combination.
- 1,4-butanediol or ethylene glycol is suitable as a crosslinking agent.
- the amount of the crosslinking agent added is preferably 5 parts by weight or more and 15 parts by weight or less per 100 parts by weight of the polyol.
- the foaming aid is added when lowering the apparent density of the polyurethane foam, and water can be mentioned as a suitable foaming aid.
- the amount of water added as a foaming aid is preferably 0.4 to 2.5 parts by weight per 100 parts by weight of polyol.
- the filler include a metal hydroxide as a viscosity modifier of a polyurethane raw material, a colorant, and an antistatic agent.
- the polyurethane foam of the present invention has a compressive residual strain at 100 ° C. of 20% or less.
- the compressive residual strain is one obtained by changing the JIS K 6400-4: 2004 A method (70 ° C.) to 100 ° C. and performing 50% compression.
- a more preferable value is 10% or less at 100 ° C., and further 110 ° C. 20% or less, particularly 10% or less at 110 ° C.
- the polyurethane foam of the present invention has a small compressive residual strain at a relatively high temperature such as 100 ° C. or 110 ° C., it can maintain a good sealing property even at a high temperature, and is suitable as a sealing material in the vicinity of the heat source part. Can be used for
- the polyurethane foam of the present invention preferably has an apparent density of 100 kg / m 3 or more and 700 kg / m 3 or less.
- the apparent density is a value measured based on JIS K 7222: 2005.
- the polyurethane foam can have a hardness suitable for the sealing material, and the adhesiveness when used as the sealing material becomes good and the sealing performance is enhanced.
- the polyurethane foam of the present invention is preferably formed to have a thickness of 0.1 mm to 15 mm. By setting it as such thickness, since a polyurethane foam can be used as a sealing material used for a narrow space, it is suitable.
- the polyurethane foam of the present invention preferably has a 25% compression load of 0.02 MPa or more and 0.40 MPa or less.
- the 25% compressive load is a compressive stress generated when a ⁇ 50 mm sample is compressed by 25% at a speed of 1 mm / min under the test conditions of JIS K 6254: 1993. Thereby, the elastic deformation of the polyurethane foam becomes good, and when the polyurethane foam is used as a sealing material, the adhesion at the time of sealing becomes high.
- the polyurethane foam of the present invention preferably has an average cell diameter of 50 ⁇ m or more and 300 ⁇ m or less.
- the average cell diameter can be calculated by dividing the cumulative cell diameter by the number of cells for cells in contact with a 25 mm straight line when the cross section of the foam layer is observed with a scanning electron microscope at a magnification of 200 times. . Thereby, the sealing property of polyurethane foam becomes better.
- the polyurethane foam of the present invention is produced by a mechanical floss method.
- a manufacturing apparatus 10 shown in FIG. 2 is an example of a polyurethane foam manufacturing apparatus of the present invention.
- the manufacturing apparatus 10 includes a mixing mechanism 31 that mixes raw materials to obtain a polyurethane raw material M, a roll mechanism that includes a supply roll 33 around which the base material 14 is wound and supplies the base material 14 by a driving source (not shown) and a product recovery roll 34. 32, a discharge nozzle 35 for supplying the polyurethane raw material M onto the base material 14, a thickness control means 36 including a doctor knife for controlling the thickness of the polyurethane raw material M on the base material 14, Heating means 38 such as a heater for heating the polyurethane raw material M.
- the polyurethane raw material M mixed in the mixing unit 31 is formed on the base material 14 made of PET film or release paper supplied from the supply roll 33. Supply from the discharge nozzle 35.
- the thickness of the polyurethane raw material M discharged onto the base material 14 is set to a predetermined thickness using the thickness control means 36. Subsequently, the polyurethane raw material M is heated by the heating means 38 to be reacted and cured.
- the laminate 20 in which the polyurethane foam 1 is formed on the base material 14 is wound around the product collection roll 34. Thereafter, the laminate 20 is cut into a predetermined size and shape, and the base material 14 is peeled off to obtain the target polyurethane foam 1.
- the product collection roll 34 that collects the stacked body 20 is described, but the present invention is not limited thereto.
- the polyurethane foam 1 and the base material 14 may be peeled off from the laminate 20, the polyurethane foam 1 may be recovered with a polyurethane foam recovery roll, and the base material 14 may be recovered with a base material recovery roll.
- FIG. 3 shows a manufacturing apparatus 10A according to a modification.
- a surface protection mechanism 41 is provided between the discharge nozzle 35 and the product collection roll 34.
- the surface protection mechanism 41 includes a surface protection film supply roll 42 and a surface protection film collection roll 43.
- the surface protection film supply roll 42 supplies the surface protection film 16 such as a PET film from a driving source (not shown).
- Reference numeral 40 denotes a thickness control means composed of a roller, which controls the thickness of the polyurethane raw material M between the base material 14 and the surface protection film 16.
- the surface protective film 16 is laminated on the upper surface of the polyurethane raw material M supplied onto the base material 14. Thereby, a polyurethane foam can be manufactured in a state where the polyurethane raw material M is sandwiched between the base material 14 and the surface protective film 16. Moreover, the surface protection film 16 can be peeled from the laminate and recovered.
- a base material collection roll is provided below the product collection roll 34, and the base material 14 is peeled off from the polyurethane foam 1 and collected. You may comprise so that only the foam 1 may be wound up and collect
- Polyurethane foams 1 of Examples 1 to 15 were produced from the polyurethane raw material M having the composition shown in Tables 1 and 2 using the production apparatus 10 shown in FIG. At this time, a PET film having a thickness of 25 ⁇ m or more and 125 ⁇ m was used as the base material 14, and the polyurethane raw material was heated to 120 to 200 ° C. by the heating means 38. Nitrogen as the foaming gas was supplied and mixed at a flow rate of 0.1 NL / min to the polyurethane raw material of the mixing unit 31 so that the mixing ratio (volume%) shown in Tables 1 and 2 was obtained. The supply speed of the base material 14 in the roll mechanism 32 was 5 m / min.
- polyurethane foams of Comparative Examples 1 to 10 were obtained from polyurethane raw materials having the formulations shown in Table 3. Comparative Examples 1 to 7 were produced by the same production method as Examples 1 to 14 described above except that the blending of the polyurethane raw material was different.
- Comparative Examples 8 to 10 a polyurethane raw material containing no foaming gas and containing water as a foaming agent is discharged onto a base material made of PET film and heated, and foamed by a chemical foaming method to produce a polyurethane foam. It has gained.
- polyurethane foams of Examples 1 to 15 and Comparative Examples 1 to 10 were obtained.
- the thickness, apparent density, 25% compressive load, compressive residual strain, dust resistance, and average cell diameter were measured for these polyurethane foams.
- the measurement methods for 25% compression load, compression residual strain, and dust resistance will be described in detail below.
- the measurement results are shown in Tables 1 to 3.
- comprehensive evaluation was set as (circle) (pass), when a compression residual strain (100 degreeC) is 20% or less and a dustproof test result is (circle). It can be said that the smaller the compressive residual strain and the higher the dustproof property, the better the sealing performance.
- the 25% compressive load is compressive stress when a ⁇ 50 mm sample is compressed 25% at a speed of 1 mm / min under the test conditions of JIS K6254: 1993.
- FIG. 4A is a side view showing a plate-shaped jig used for the dustproof test
- FIG. 4B is a plan view thereof
- FIG. 5 is a perspective view of a cylindrical container used for a dustproof test.
- the outer frame size is 50 mm square
- the inner frame size is 45 mm square
- the frame width is 2.5 mm as shown in FIG.
- the sample S formed in a frame shape is sandwiched between and set so as to be compressed by 25% in the thickness direction of the sample S.
- One surface of the sample S is joined to one plate-like jig 51 by a nonwoven fabric base acrylic double-sided adhesive tape 57 having a thickness of 160 ⁇ m.
- a spring 53 is interposed between the plate-shaped metal parts 51 and 52, and the plate-shaped metal parts 51 and 52 are fastened with bolts 54 and nuts 55.
- the plate-like jigs 51 and 52 on which the sample S is set are put together with 1 kg of powder into a cylindrical container 61 having an inner diameter of 250 mm and a length in the direction of the shaft 63 of 340 mm as shown in FIG.
- the powder is a talc powder having a particle size of 4 ⁇ m or more and 10 ⁇ m or less.
- the cylindrical container 61 is rotated around the shaft 63 at 60 rpm for 10 minutes. It is visually confirmed whether or not the powder has entered the inside of the sample S taken out from the cylindrical container 61. When the powder could not be confirmed inside the sample S, it was evaluated as “ ⁇ ”, and when the powder could be confirmed inside the sample S, it was evaluated as “ ⁇ ”.
- Examples 1 to 15 all show good heat resistance with a compressive residual strain of 20% or less at 80 ° C., 100 ° C., and 110 ° C.
- polyether polyol polyether polyol B (EO ratio 70 mol%) having a ratio of oxyethylene units in polyoxyalkylene (EO ratio) of 50 mol% or more (EO ratio 70 mol%) is used.
- Examples 3, 4, 6 and 13 in which the total weight part of the polyether polyol having a ratio of 50 mol% or more and the castor oil-based polyol is 50 parts by weight or more and 100 parts by weight or less are 80 ° C., 100 ° C. and 110 ° C.
- each compressive residual strain is smaller than the other examples, and the heat resistance is very high.
- castor oil-based polyol is contained in 40 parts by weight or more and 70 parts by weight or less in 100 parts by weight of polyol, and the ethylene oxide ratio is 50 mol% in 100 parts by weight of polyol. Since the polyether polyol is 30 parts by weight or more and 60 parts by weight or less, the heat resistance is very high.
- Comparative Examples 1 and 2 since the blending amount of castor oil-based polyol is 0 or small, the compression residual strain is 20% or less at 80 ° C., but both exceed 100% at 100 ° C. and 110 ° C. Is low.
- Comparative Example 3 since the viscosity of the castor oil-based polyol is high, voids are generated during the production of the polyurethane foam, and the dust resistance is poor.
- Comparative Example 4 does not contain castor oil-based polyol, and the polycarbonate-based polyol, which is another polyol, has a very high viscosity, resulting in insufficient stirring and mixing, resulting in low heat resistance and poor dust resistance.
- Comparative Example 5 since the amount of castor oil-based polyol is large, the viscosity of the polyurethane raw material becomes high, voids are generated during the production of the polyurethane foam, and the dust resistance is poor.
- Comparative Example 6 since the apparent density of the polyurethane foam is too low, the hardness is lowered, the cell becomes rough, and the dust resistance is inferior.
- Comparative Example 7 since the apparent density of the polyurethane foam is too high, the hardness is increased, the adhesion during compression is impaired, and the dust resistance is inferior. Since Comparative Examples 8 to 10 are based on the chemical foaming method rather than the mechanical froth method, the cells become rough and the dustproof property is inferior.
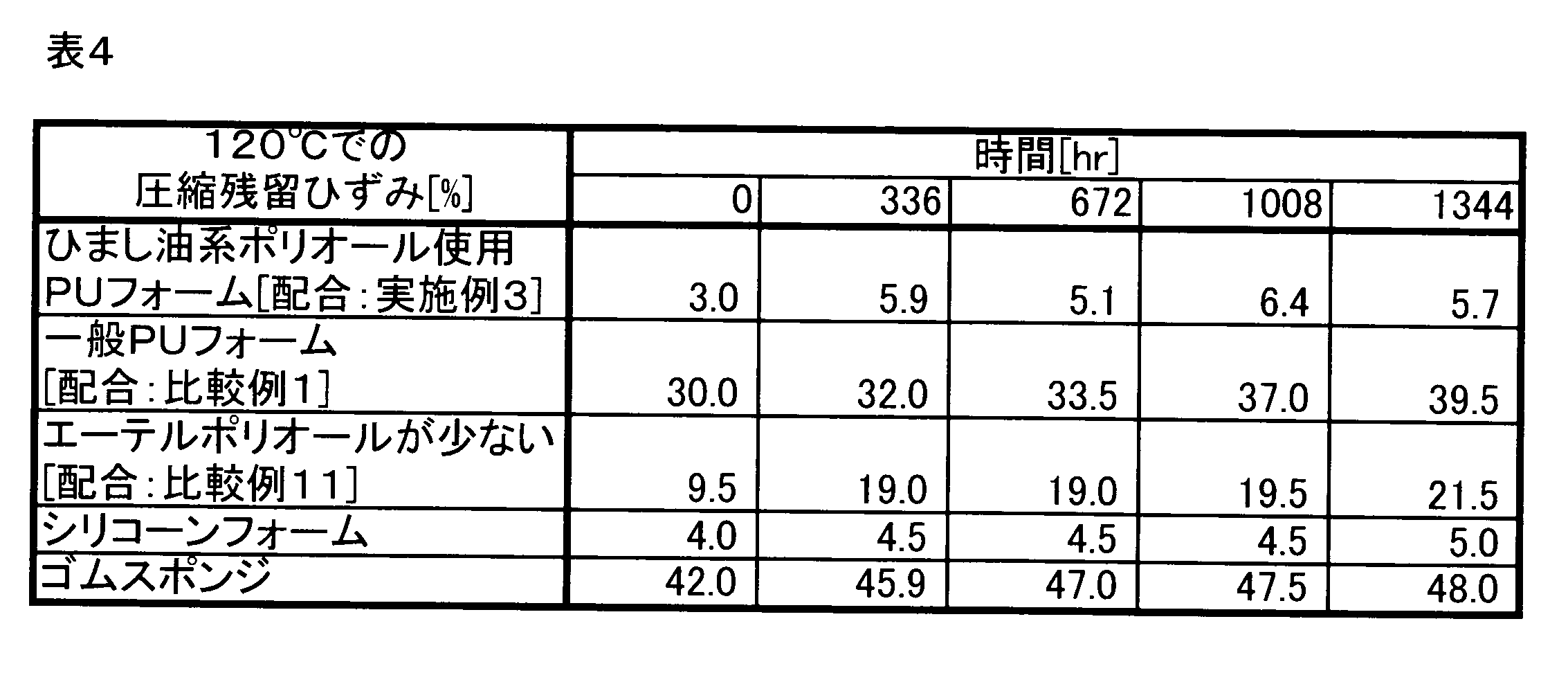
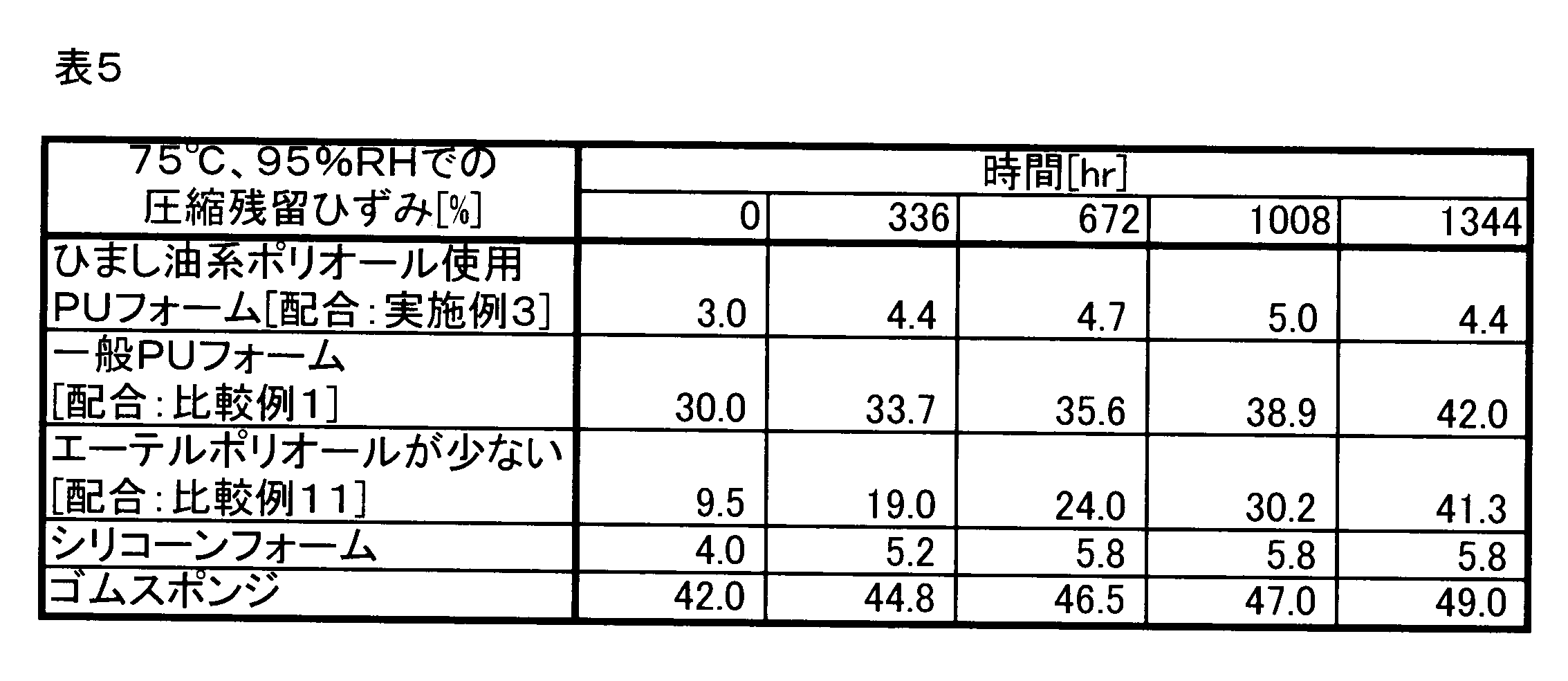
- Example 3 using castor oil-based polyol, Comparative Example 1 without using castor oil-based polyol, Comparative Example 11 in which the blending amount of polyether polyol is less than 20 parts by weight, silicone foam, rubber sponge, 120 The sample was left for a predetermined time in an environment of 70 ° C. or in an environment of humidity 95% RH at 70 ° C., and then the compression residual strain (50% compression, 100 ° C. ⁇ 22 hours) was measured.
- the amount of polyether polyol B was changed from 22 parts by weight to 18 parts by weight, and the amount of polyester polyol was changed from 56 parts by weight to 60 parts by weight. Other than that, it produced similarly to Example 4.
- Table 4 shows the measurement results of the compressive residual strain after standing in a 120 ° C. environment
- Table 5 shows the measurement results of the compressive residual strain after standing in a humidity of 95% RH at 70 ° C.
- “Time” in Tables 4 and 5 is a standing time in each environment.
- the polyurethane foam of Example 3 using castor oil-based polyol has a smaller compressive residual strain at high temperature than the polyurethane foam and rubber sponge of Comparative Example 1 not using castor oil-based polyol. Moreover, it was confirmed that the polyurethane foam using the castor oil-based polyol exhibits a compression residual strain at a high temperature equivalent to that of the silicone foam.
- Comparative Example 10 in which the blending amount of the polyether polyol is less than 20 parts by weight has a smaller compressive residual strain than Comparative Example 1 in which the castor oil-based polyol is not used and only the polyether-based polyol is used.
- the compressive residual strain after standing for 672 hours at 75 ° C. and 95% RH is larger than 20%, and the compressive residual strain under high humidity cannot be sufficiently reduced.
- the polyurethane foam of the present invention since the polyurethane foam of the present invention has a small compressive residual strain even at a high temperature, it exhibits excellent sealing properties even in a high temperature environment. Therefore, it is also suitable as a sealing material in the vicinity of the heat source. Furthermore, the polyurethane foam of the present invention is less expensive than the silicone foam and has a small compressive residual strain equivalent to that of the silicone foam under a high temperature environment. For this reason, the polyurethane foam of the present invention can be used in place of the silicone foam as a sealing material for high-temperature environments, which conventionally had to use the silicone foam from the viewpoint of heat resistance, and the cost can be reduced. is there.
- the polyurethane foam of the present invention is suitable for the following uses.
- the material having these performances is generally silicone foam, but the price is very high compared to other foamed materials. Since the buffer material is used in a large amount for one battery, it is desired to switch to an inexpensive material in order to reduce the cost of the battery. Therefore, the polyurethane foam of the present invention is less expensive than the silicone foam and has a very small compressive residual strain at 80 to 110 ° C. For this reason, the polyurethane foam of this invention can be used conveniently as a buffer material between electric vehicle battery cells.
- the buffer material of the battery pack mounted on the electric vehicle is required to have a small compressive residual strain.
- Silicone foam has a small compression residual strain, but contains a large amount of low-molecular siloxane. Silica may precipitate from the silicone foam due to the low molecular siloxane.
- silicone foam is used as a buffer material for the battery pack, the contacts on the electronic board mounted on the battery pack are contaminated by the deposition of silica, and the electrical characteristics may be deteriorated. For this reason, silicone foam is not suitable for the battery pack cushioning material. Therefore, urethane foam is generally used as the battery pack cushioning material. However, urethane foam has a large compressive residual strain.
- the polyurethane foam of the present invention has a very low amount of low molecular siloxane compared to the silicone foam. Further, the compressive residual strain is sufficiently smaller than that of conventional urethane foam. For this reason, when the polyurethane foam of the present invention is used as a buffer material for a battery pack of an electric vehicle, it has a sufficient buffer property due to a small compressive residual strain, and contact failure is unlikely to occur because silica does not precipitate. Therefore, the polyurethane foam according to the present invention can be suitably used as an electric vehicle battery pack cushioning material.
- the cushioning material used for the solar panel is required to have waterproofness, a small compressive residual strain, and a small compressive residual strain even in a high temperature environment.
- the material having these performances is generally only silicone foam, but the price is very high compared to other foamed materials.
- the polyurethane foam of the present invention is less expensive than the silicone foam, has a very small compressive residual strain at 80 to 110 ° C., and does not increase the compressive residual strain even in a high temperature environment. It is also highly waterproof. For this reason, the polyurethane foam of this invention can be used conveniently as a solar panel cushioning material.
- Medical monitors are required to have higher brightness than consumer monitors, and the internal temperature of the monitor is up to about 60 ° C.
- urethane foam or polyolefin foam is used as a cushioning material between the liquid crystal glass and the housing frame of a consumer monitor.
- conventionally used urethane foam and polyolefin foam are not sufficiently heat resistant, and a material having higher heat resistance and flexibility has been desired.
- the polyurethane foam of the present invention has a very small compressive residual strain at 80 to 110 ° C., and has a flexibility of 25% compressive load of 0.02 MPa to 0.40 MPa. For this reason, the polyurethane foam of the present invention can be suitably used as a medical monitor cushioning material.
- the polyurethane foam of the present invention includes the electric vehicle battery cell cushioning material, the electric vehicle battery pack cushioning material, the solar panel cushioning material, the medical monitor cushioning material, the electric vehicle battery cell sealing material, It can also be suitably used as an automotive battery pack sealing material, a solar panel sealing material, or a medical monitor sealing material.
- the compressive residual strain at 100 ° C. is 20% or less, the compressive residual strain at high temperature is small. Therefore, it is possible to provide a polyurethane foam that is also suitable as a sealing material in the vicinity of the heat source.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
Description
メカニカルフロス法は、まずポリウレタン原料として、ポリオール、イソシアネート、整泡剤、触媒を含む原料に造泡用気体を圧縮して混入させる。このポリウレタン原料を、オークスミキサーまたは先端を絞ったノズルから吐出することにより、ポリウレタンフォームを形成する。このポリウレタン原料の吐出時にそれまで圧縮されていた造泡用気体が膨張して気泡を形成し、その状態でポリオールとイソシアネートが反応して硬化することによりポリウレタンフォームを形成する。
ポリオール、イソシアネート、整泡剤、触媒及び造泡用気体を含むポリウレタン原料からメカニカルフロス法により得られるポリウレタンフォームにおいて、
前記ポリオールは、25℃における粘度が2000mPa・s以下のひまし油系ポリオールと、ポリエーテル系ポリオールとを含み、
前記ひまし油系ポリオールは、前記ポリオール100重量部中に、20重量部以上80重量部以下含まれ、
前記ポリエーテル系ポリオールは、前記ポリオール100重量部中に、20重量部以上80重量部以下含まれ、
前記ウレタンフォームの見掛け密度が100kg/m3以上700kg/m3以下であり、100℃での圧縮残留ひずみが20%以下であることを特徴とする。
前記ポリオールは、前記ポリオール100重量部に、ポリオキシアルキレン中のオキシエチレン単位の比率であるエチレンオキサイド比率が50モル%以上のポリエーテル系ポリオールを20重量部以上80重量部以下含み、
前記ポリオール100重量部中、前記エチレンオキサイド比率が50モル%以上であるポリエーテル系ポリオールと前記ひまし油系ポリオールとの合計重量部が、50重量部以上100重量部以下であることが好ましい。
前記ひまし油系ポリオールは、前記ポリオール100重量部中に、40重量部以上70重量部以下含まれ、
前記エチレンオキサイド比率が50モル%以上であるポリエーテル系ポリオールは、前記ポリオール100重量部中に、30重量部以上60重量部以下含まれることが好ましい。
また、前記ウレタンフォームの厚さが0.1mm以上15mm以下であることが好ましい。
前記ウレタンフォームの25%圧縮荷重が0.02MPa以上0.40MPa以下であることが好ましい。
前記ウレタンフォームの平均セル径が50μm以上300μmであることが好ましい。
100℃での前記ウレタンフォームの圧縮残留ひずみが10%以下であることが好ましい。
110℃での前記ウレタンフォームの圧縮残留ひずみが10%以下であることが好ましい。
ポリオールは、25℃における粘度(JIS Z 8803:2011準拠)が2000mPa・s以下のひまし油系ポリオールと、ポリエーテル系ポリオールとを含む。また、ひまし油系ポリオールは、ポリオール100重量部中に、20重量部以上80重量部以下含まれている。また、前記ポリエーテル系ポリオールは、前記ポリオール100重量部中に、20重量部以上80重量部以下含まれている。
このポリカーボネート系ポリオールとしては、エチレングリコール、ジエチレングリコール、プロピレングリコール、ブタンジオール、ペンタンジオール、ヘキサンジオールなどの多価アルコールと、ジアルキルカーボネート、ジアルキレンカーボネート、ジフェニルカーボネートなどとの脱アルコール反応によって得られるものが挙げられる。
このポリエステル系ポリオールとしては、エチレングリコール、ジエチレングリコール、プロピレングリコール、ブタンジオール、ヘキサンジオール、グリセリン、トリメチロールプロパン、トリメチロールエタン、ペンタエリスリトール、ジグリセリン、ソルビトール、ショ糖等の低分子ポリオールと、コハク酸、アジピン酸、マレイン酸、フマル酸、フタル酸、イソフタル酸、無水コハク酸、無水マレイン酸、無水フタル酸等との縮合により得られるものが挙げられる。更に、ポリエステル系ポリオールとしては、ラクトンエステルとして分類されるカプロラクトン、メチルバレロラクトンの開環縮合物であるポリオールが挙げられる。
図2に示す製造装置10は、本発明のポリウレタンフォームの製造装置の一例である。製造装置10は、原料を混合してポリウレタン原料Mを得る混合部31と、基材14が巻かれ図示しない駆動源により基材14を供給する供給ロール33と製品回収ロール34とよりなるロール機構32と、基材14上にポリウレタン原料Mを供給する吐出ノズル35と、基材14上のポリウレタン原料Mの厚さを制御するドクターナイフ等からなる厚さ制御手段36と、基材14上のポリウレタン原料Mを加熱するヒーター等の加熱手段38とを備えている。
・ひまし油系ポリオール1;伊藤製油社製、商品名「H-30」、粘度(25℃)690mPa・s(粘度(60℃)85mPa・s)、官能基数2.7、数平均分子量950(水酸基価160mgKOH/g)
・ひまし油系ポリオール2;伊藤製油社製、商品名「AC-005」、粘度(25℃)1150mPa・s(粘度(60℃)125mPa・s)、官能基数2、数平均分子量550(水酸基価204mgKOH/g)
・ひまし油系ポリオール3;伊藤製油社製、商品名「AC-006」、粘度(25℃)3000mPa・s(粘度(60℃)200mPa・s)、官能基数2、数平均分子量630(水酸基価178mgKOH/g)
・ポリエーテル系ポリオールA;三洋化成工業社製、商品名「GP-3000」、粘度(25℃)500mPa・s、官能基数3、数平均分子量3000、EO比率0モル%
・ポリエーテル系ポリオールB;三洋化成工業社製、商品名「FA-103」、粘度(25℃)730mPa・s、官能基数3、数平均分子量3300、EO比率70モル%
・ポリエーテル系ポリオールC;三洋化成工業社製、商品名「GL-3000」、粘度(25℃)510mPa・s、官能基数3、数平均分子量3000、EO比率20モル%
・ポリエステル系ポリオール;クラレ社製、商品名「P-510」、粘度(25℃)540mPa・s、官能基数2、数平均分子量500(水酸基価224mgKOH/g)
・ポリカーボネート系ポリオール1;クラレ社製、商品名「C-590」、粘度(60℃)170mPa・s、官能基数2、数平均分子量500(水酸基価224mgKOH/g)
・ポリカーボネート系ポリオール2;クラレ社製、商品名「C-1090」、粘度(60℃)1800mPa・s、官能基数2、数平均分子量1000(水酸基価112mgKOH/g)
・架橋剤;1,4-ブタンジオール
・金属触媒;城北化学工業社製、商品名「MRH110」、オクチル酸スズ
・整泡剤;モメンティブ社製、商品名「L-5614」、ノニオン系界面活性剤
・金属水酸化物;昭和電工社製、商品名「ハイジライトH-10」、水酸化アルミニウム
・造泡用気体;窒素
・イソシアネート:日本ポリウレタン工業社製、商品名「コロネート1130」、NCO%:31%、クルードMDI



圧縮残留ひずみ(%)=[(圧縮前の厚さ-解放後の厚さ)/圧縮前の厚さ]×100
比較例3は、ひまし油系ポリオールの粘度が高いため、ポリウレタンフォームの製造時にボイドが発生し、防塵性が劣る。
比較例4は、ひまし油系ポリオールを含まず、その他のポリオールであるポリカーボネート系ポリオールの粘度が非常に高く、攪拌混合が不十分になるため、耐熱性が低くなり、かつ防塵性が劣る。
比較例5は、ひまし油系ポリオールの配合量が多いため、ポリウレタン原料の粘度が高くなってポリウレタンフォームの製造時にボイドが発生し、防塵性が劣る。
比較例7は、ポリウレタンフォームの見掛け密度が高過ぎるため、硬度が上昇し、圧縮時の密着性が損なわれ、防塵性が劣る。
比較例8~10は、メカニカルフロス法ではなく、化学発泡法によるものであるため、セルが粗くなって防塵性が劣る。
<電気自動車バッテリーセル間緩衝材>
電気自動車に搭載されるバッテリーセルは、発電・充電時に膨張・収縮をする。このため、バッテリーセル間には一定のスペースが設けられている。このスペースには、事故などバッテリーに衝撃が加わった際にセルが破損しないように、緩衝材が設けられている。ところで、バッテリー内部のセル温度は最大80℃程度まで上がるため、緩衝材にはそれ以上の耐熱性を有する素材が求められる。また、緩衝材は衝撃吸収材の役割も果たすため、柔軟性および低圧縮残留歪性を有する素材が求められる。
また、電気自動車に搭載されるバッテリーパックの緩衝材には、圧縮残留ひずみが小さいことが求められる。シリコーンフォームの圧縮残留ひずみは小さいが、低分子シロキサンを多く含む。シリコーンフォームからは、この低分子シロキサンに起因して、シリカが析出することがある。バッテリーパックの緩衝材としてシリコーンフォームを用いると、バッテリーパックに搭載された電子基板上の接点が、このシリカの析出によって汚染されてしまい、電気特性が悪化してしまう虞がある。このため、バッテリーパック緩衝材にはシリコーンフォームは適していない。そのため、バッテリーパック緩衝材として一般的にはウレタンフォームが使用されている。しかし、ウレタンフォームは、圧縮残留ひずみが大きい。
太陽光パネルは屋外で使用されるため、太陽光パネルに用いられる緩衝材には防水性、小さい圧縮残留ひずみ、および高温環境下でも圧縮残留ひずみが小さいことが求められる。これらの性能を有する素材は一般的にはシリコーンフォームしかないが、他の発泡材料に比べ価格が非常に高い。
しかし本発明のポリウレタンフォームはシリコーンフォームと比較して、安価であり、80~110℃の圧縮残留ひずみが非常に小さく、また、高温環境下でも圧縮残留ひずみが増大しない。また、防水性も高い。このため、本発明のポリウレタンフォームは太陽光パネル緩衝材として好適に使用することができる。
医療用モニターは、民生用モニターより高輝度が求められ、モニター内部温度は最大60℃程度までになる。民生用モニターの液晶ガラスと筐体フレームの間の緩衝材には、一般的にはウレタンフォームやポリオレフィンフォームが用いられている。しかし従来一般的に用いられているウレタンフォームやポリオレフィンフォームは耐熱性が十分ではなく、より高耐熱性、柔軟性を有する素材が望まれていた。
本出願は、2011年7月25日出願の日本特許出願(特願2011-161650)に基づくものであり、その内容はここに参照として取り込まれる。
Claims (10)
- ポリオール、イソシアネート、整泡剤、触媒及び造泡用気体を含むポリウレタン原料からメカニカルフロス法により得られるポリウレタンフォームにおいて、
前記ポリオールは、25℃における粘度が2000mPa・s以下のひまし油系ポリオールと、ポリエーテル系ポリオールとを含み、
前記ひまし油系ポリオールは、前記ポリオール100重量部中に、20重量部以上80重量部以下含まれ、
前記ポリエーテル系ポリオールは、前記ポリオール100重量部中に、20重量部以上80重量部以下含まれ、
前記ウレタンフォームの見掛け密度が100kg/m3以上700kg/m3以下であり、100℃での圧縮残留ひずみが20%以下であることを特徴とするポリウレタンフォーム。 - 前記ポリオールは、前記ポリオール100重量部に、ポリオキシアルキレン中のオキシエチレン単位の比率であるエチレンオキサイド比率が50モル%以上のポリエーテル系ポリオールを20重量部以上80重量部以下含み、
前記ポリオール100重量部中、前記エチレンオキサイド比率が50モル%以上であるポリエーテル系ポリオールと前記ひまし油系ポリオールとの合計重量部が、50重量部以上100重量部以下であることを特徴とする請求項1に記載のポリウレタンフォーム。 - 前記ひまし油系ポリオールは、前記ポリオール100重量部中に、40重量部以上70重量部以下含まれ、
前記エチレンオキサイド比率が50モル%以上であるポリエーテル系ポリオールは、前記ポリオール100重量部中に、30重量部以上60重量部以下含まれることを特徴とする請求項2に記載のポリウレタンフォーム。 - 前記ウレタンフォームの厚さが0.1mm以上15mm以下であることを特徴とする請求項1~3のいずれか一項に記載のポリウレタンフォーム。
- 前記ウレタンフォームの25%圧縮荷重が0.02MPa以上0.40MPa以下であることを特徴とする請求項1~4のいずれか一項に記載のポリウレタンフォーム。
- 前記ウレタンフォームの平均セル径が50μm以上300μmであることを特徴とする請求項1~5のいずれか一項に記載のポリウレタンフォーム。
- 100℃での前記ウレタンフォームの圧縮残留ひずみが10%以下であることを特徴とする請求項1~6のいずれか一項に記載のポリウレタンフォーム。
- 110℃での前記ウレタンフォームの圧縮残留ひずみが10%以下であることを特徴とする請求項1~7のいずれか一項に記載のポリウレタンフォーム。
- 請求項1~8のいずれか一項に記載のポリウレタンフォームからなるシール材。
- 請求項1~8のいずれか一項に記載のポリウレタンフォームからなる緩衝材。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12816858.0A EP2738193B1 (en) | 2011-07-25 | 2012-07-23 | Polyurethane foam |
| JP2013525716A JP5905464B2 (ja) | 2011-07-25 | 2012-07-23 | ポリウレタンフォーム |
| CN201280036825.8A CN103703045B (zh) | 2011-07-25 | 2012-07-23 | 聚氨酯泡沫体 |
| US14/234,424 US9624336B2 (en) | 2011-07-25 | 2012-07-23 | Polyurethane foam |
| KR1020147001896A KR101605378B1 (ko) | 2011-07-25 | 2012-07-23 | 폴리우레탄 폼 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011161650 | 2011-07-25 | ||
| JP2011-161650 | 2011-07-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013015245A1 true WO2013015245A1 (ja) | 2013-01-31 |
Family
ID=47601092
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/068593 Ceased WO2013015245A1 (ja) | 2011-07-25 | 2012-07-23 | ポリウレタンフォーム |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9624336B2 (ja) |
| EP (1) | EP2738193B1 (ja) |
| JP (1) | JP5905464B2 (ja) |
| KR (1) | KR101605378B1 (ja) |
| CN (1) | CN103703045B (ja) |
| TW (1) | TWI525120B (ja) |
| WO (1) | WO2013015245A1 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103450441A (zh) * | 2013-07-29 | 2013-12-18 | 洛阳嘉冠化工产品有限公司 | 一种煤矿瓦斯抽采孔封孔用双袋发泡组分 |
| US10316156B2 (en) * | 2014-09-24 | 2019-06-11 | Nitto Denko Corporation | Foamed sheet |
| CN113861921A (zh) * | 2021-09-03 | 2021-12-31 | 惠州锂威新能源科技有限公司 | 一种聚氨酯发泡胶及其制备方法及应用、软包锂离子电池 |
| WO2022118974A1 (ja) * | 2020-12-03 | 2022-06-09 | 日本発條株式会社 | 電池セル用クッション材及び電池 |
| JPWO2022224509A1 (ja) * | 2021-04-20 | 2022-10-27 | ||
| JPWO2022244480A1 (ja) * | 2021-05-21 | 2022-11-24 | ||
| WO2023067654A1 (ja) * | 2021-10-18 | 2023-04-27 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2024059718A (ja) * | 2019-10-28 | 2024-05-01 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| WO2024171817A1 (ja) * | 2023-02-13 | 2024-08-22 | 株式会社イノアックコーポレーション | ポリウレタンフォーム及びクッション材 |
| WO2025141888A1 (ja) * | 2023-12-28 | 2025-07-03 | 株式会社イノアックコーポレーション | 植物由来原料を含むポリウレタンフォーム、衝撃吸収材 |
| WO2025141889A1 (ja) * | 2023-12-28 | 2025-07-03 | 株式会社イノアックコーポレーション | ポリウレタンフォーム、衝撃吸収材 |
| WO2025173667A1 (ja) * | 2024-02-13 | 2025-08-21 | 株式会社イノアックコーポレーション | ポリウレタンフォーム、及び内装材 |
| WO2025197147A1 (ja) * | 2024-03-22 | 2025-09-25 | 株式会社イノアックコーポレーション | ポリウレタンフォーム及び衝撃吸収材 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104449020B (zh) * | 2013-09-20 | 2019-01-11 | 陶氏环球技术有限公司 | 用于涂料组合物的反应性多官能添加剂 |
| SI3186293T1 (sl) * | 2014-08-28 | 2018-12-31 | Basf Se | Proti staranju odporno poliuretansko tesnilo |
| WO2016073396A1 (en) * | 2014-11-05 | 2016-05-12 | Dow Global Technologies Llc | Impact protection foam |
| US9853263B2 (en) | 2014-11-10 | 2017-12-26 | Ford Global Technologies, Llc | Battery assembly including structural foamed materials |
| KR101543974B1 (ko) * | 2014-12-08 | 2015-08-11 | 이상호 | 박막 폴리우레탄 폼 적층체 및 그 제조방법 |
| KR101631688B1 (ko) * | 2015-08-05 | 2016-06-17 | 주식회사 에스제이폼웍스 | 박막 폴리우레탄 폼 테이프 적층체 및 그 제조방법 |
| CN105384910A (zh) * | 2015-12-24 | 2016-03-09 | 山东一诺威新材料有限公司 | 可降解聚氨酯仿木材料及其制备方法 |
| NL2021944B1 (en) * | 2018-11-06 | 2020-05-15 | Escateq B V | A cleaning pad for an escalator or moving walkway |
| CN111269759B (zh) * | 2018-12-04 | 2022-04-19 | 北京化工大学 | 一种制备不同羟值蓖麻油基多元醇的方法 |
| CN110172174A (zh) * | 2019-06-13 | 2019-08-27 | 徐州工业职业技术学院 | 一种环保型聚氨酯泡沫保温材料及其制备方法 |
| CN110423650A (zh) * | 2019-08-12 | 2019-11-08 | 南京林业大学 | 一种烷氧基蓖麻油基多元醇及其制备方法、蓖麻油基聚氨酯硬泡 |
| US20220363858A1 (en) * | 2019-11-12 | 2022-11-17 | Huntsman International Llc | In-situ formation of low density thermoplastic polyurethane flexible foams |
| BR112023017821A2 (pt) * | 2021-03-10 | 2023-09-26 | Dow Global Technologies Llc | Composição de espuma de poliuretano, espuma de poliuretano, e, método para envasar uma bateria |
| CN115368527B (zh) * | 2022-08-15 | 2024-05-03 | 惠州亿纬锂能股份有限公司 | 一种用于圆柱电池模组的聚氨酯发泡材料及其制备方法和应用 |
| CN115505089A (zh) * | 2022-10-11 | 2022-12-23 | 力神(青岛)新能源有限公司 | 一种隔热片及其应用 |
| CN115926101B (zh) * | 2023-02-14 | 2023-06-09 | 旭川化学(苏州)有限公司 | 一种耐低温聚氨酯鞋底用树脂及其制备方法和应用 |
| CN119241801B (zh) * | 2024-09-29 | 2025-10-10 | 江南大学 | 一种一体化梯度聚氨酯泡沫材料及其制备方法 |
| CN120271445B (zh) * | 2025-06-10 | 2025-09-16 | 深圳市优和新材料有限公司 | 一种低粘度高羟值蓖麻油基多元醇及其合成方法与应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09183826A (ja) * | 1995-12-28 | 1997-07-15 | Tokyo Seat Kk | 一体スキン層付ウレタンフォーム成形品の製造方法 |
| JP2002214895A (ja) | 2001-01-17 | 2002-07-31 | Inoac Corp | シール部材 |
| JP2005227392A (ja) | 2004-02-10 | 2005-08-25 | Inoac Corp | シール部材およびその製造方法 |
| JP2008280447A (ja) * | 2007-05-11 | 2008-11-20 | Bridgestone Corp | ポリウレタン発泡体及びそれを用いた導電性ローラ |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2652731B2 (ja) | 1991-06-26 | 1997-09-10 | 東海ゴム工業株式会社 | 鋼板構造材充填用硬質ウレタンフォーム形成材料 |
| US7338983B2 (en) * | 2004-02-10 | 2008-03-04 | World Properties, Inc. | Low density polyurethane foam, method of producing, and articles comprising the same |
| WO2008073808A1 (en) | 2006-12-11 | 2008-06-19 | Dow Global Technologies Inc. | Bio-based carpet materials |
| KR100792278B1 (ko) | 2007-02-27 | 2008-01-07 | 고려상사주식회사 | 인산염 피막 냉간 압조용 스테인리스 강선 및 이를 이용한직결 나사 |
| JP2010053157A (ja) | 2008-08-26 | 2010-03-11 | Nippon Polyurethane Ind Co Ltd | 軟質ポリウレタンフォームの製造方法 |
-
2012
- 2012-07-23 US US14/234,424 patent/US9624336B2/en active Active
- 2012-07-23 EP EP12816858.0A patent/EP2738193B1/en active Active
- 2012-07-23 WO PCT/JP2012/068593 patent/WO2013015245A1/ja not_active Ceased
- 2012-07-23 CN CN201280036825.8A patent/CN103703045B/zh active Active
- 2012-07-23 KR KR1020147001896A patent/KR101605378B1/ko active Active
- 2012-07-23 JP JP2013525716A patent/JP5905464B2/ja active Active
- 2012-07-25 TW TW101126849A patent/TWI525120B/zh active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09183826A (ja) * | 1995-12-28 | 1997-07-15 | Tokyo Seat Kk | 一体スキン層付ウレタンフォーム成形品の製造方法 |
| JP2002214895A (ja) | 2001-01-17 | 2002-07-31 | Inoac Corp | シール部材 |
| JP2005227392A (ja) | 2004-02-10 | 2005-08-25 | Inoac Corp | シール部材およびその製造方法 |
| JP2008280447A (ja) * | 2007-05-11 | 2008-11-20 | Bridgestone Corp | ポリウレタン発泡体及びそれを用いた導電性ローラ |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2738193A4 |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103450441A (zh) * | 2013-07-29 | 2013-12-18 | 洛阳嘉冠化工产品有限公司 | 一种煤矿瓦斯抽采孔封孔用双袋发泡组分 |
| US10316156B2 (en) * | 2014-09-24 | 2019-06-11 | Nitto Denko Corporation | Foamed sheet |
| JP7769022B2 (ja) | 2019-10-28 | 2025-11-12 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2024059718A (ja) * | 2019-10-28 | 2024-05-01 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| WO2022118974A1 (ja) * | 2020-12-03 | 2022-06-09 | 日本発條株式会社 | 電池セル用クッション材及び電池 |
| JPWO2022118974A1 (ja) * | 2020-12-03 | 2022-06-09 | ||
| JP7748969B2 (ja) | 2020-12-03 | 2025-10-03 | 株式会社東洋クオリティワン | 電池セル用クッション材及び電池 |
| JP7373694B2 (ja) | 2021-04-20 | 2023-11-02 | 株式会社イノアックコーポレーション | ポリウレタンフォーム及びトナーシール部材 |
| JPWO2022224509A1 (ja) * | 2021-04-20 | 2022-10-27 | ||
| JPWO2022244480A1 (ja) * | 2021-05-21 | 2022-11-24 | ||
| JP7581504B2 (ja) | 2021-05-21 | 2024-11-12 | 株式会社イノアックコーポレーション | バッテリー用クッション材、防塵用クッション材、及び止水用クッション材 |
| CN113861921A (zh) * | 2021-09-03 | 2021-12-31 | 惠州锂威新能源科技有限公司 | 一种聚氨酯发泡胶及其制备方法及应用、软包锂离子电池 |
| WO2023067654A1 (ja) * | 2021-10-18 | 2023-04-27 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2023080376A (ja) * | 2021-10-18 | 2023-06-08 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP7439320B2 (ja) | 2021-10-18 | 2024-02-27 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP7439319B2 (ja) | 2021-10-18 | 2024-02-27 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP7439318B2 (ja) | 2021-10-18 | 2024-02-27 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2023080378A (ja) * | 2021-10-18 | 2023-06-08 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP7520180B2 (ja) | 2021-10-18 | 2024-07-22 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP7320158B1 (ja) * | 2021-10-18 | 2023-08-02 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2023080377A (ja) * | 2021-10-18 | 2023-06-08 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| JP2023080375A (ja) * | 2021-10-18 | 2023-06-08 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| WO2024171817A1 (ja) * | 2023-02-13 | 2024-08-22 | 株式会社イノアックコーポレーション | ポリウレタンフォーム及びクッション材 |
| JPWO2024171817A1 (ja) * | 2023-02-13 | 2024-08-22 | ||
| WO2025141889A1 (ja) * | 2023-12-28 | 2025-07-03 | 株式会社イノアックコーポレーション | ポリウレタンフォーム、衝撃吸収材 |
| WO2025141888A1 (ja) * | 2023-12-28 | 2025-07-03 | 株式会社イノアックコーポレーション | 植物由来原料を含むポリウレタンフォーム、衝撃吸収材 |
| WO2025173667A1 (ja) * | 2024-02-13 | 2025-08-21 | 株式会社イノアックコーポレーション | ポリウレタンフォーム、及び内装材 |
| WO2025197147A1 (ja) * | 2024-03-22 | 2025-09-25 | 株式会社イノアックコーポレーション | ポリウレタンフォーム及び衝撃吸収材 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101605378B1 (ko) | 2016-03-22 |
| EP2738193B1 (en) | 2019-01-09 |
| EP2738193A1 (en) | 2014-06-04 |
| KR20140041798A (ko) | 2014-04-04 |
| TWI525120B (zh) | 2016-03-11 |
| EP2738193A4 (en) | 2015-06-03 |
| JP5905464B2 (ja) | 2016-04-20 |
| CN103703045A (zh) | 2014-04-02 |
| CN103703045B (zh) | 2015-11-25 |
| TW201313767A (zh) | 2013-04-01 |
| US9624336B2 (en) | 2017-04-18 |
| US20140193631A1 (en) | 2014-07-10 |
| JPWO2013015245A1 (ja) | 2015-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5905464B2 (ja) | ポリウレタンフォーム | |
| US9982111B2 (en) | One-part moisture-curable polyurethane composition | |
| JP7052913B1 (ja) | 蓄電デバイス用包装材、蓄電デバイス用容器及び蓄電デバイス | |
| CN116891715A (zh) | 蓄电元件包装材用粘接剂、蓄电元件包装材、蓄电元件用容器及蓄电元件 | |
| JP6915728B1 (ja) | ガスバリア性ラミネート接着剤、積層体及び包装体 | |
| JP6996546B2 (ja) | 蓄電デバイス用包装材、蓄電デバイス用容器及び蓄電デバイス | |
| JP5367356B2 (ja) | 止水性ポリウレタンフォーム | |
| JP5561657B2 (ja) | 繊維性基材用無溶剤型ポリウレタン樹脂形成性接着剤組成物、該接着剤組成物を用いた積層体及び積層体の製造方法 | |
| CN101687406A (zh) | 叠层体、使用该叠层体的扁形电缆及电布线用部件 | |
| JP2023174368A (ja) | ホットメルト接着シート | |
| CN115537170A (zh) | 蓄电元件包装材用聚氨基甲酸酯粘接剂、蓄电元件用包装材、蓄电元件用容器及蓄电元件 | |
| TWI460242B (zh) | 導電構件用感壓式接著劑組成物,及使用該組成物而成之積層體 | |
| WO2024162417A1 (ja) | 硬化性熱伝導性接着剤、熱伝導性部材及びバッテリアセンブリ | |
| CN116496747A (zh) | 粘合带 | |
| KR20220166065A (ko) | 축전 디바이스용 포장재, 축전 디바이스용 용기 및 축전 디바이스 | |
| JP7336481B2 (ja) | 2液型ウレタン系接着剤組成物 | |
| JP2019044005A (ja) | 湿気硬化型ホットメルト接着剤及び積層体 | |
| KR20230107548A (ko) | 무용제형 반응성 접착제, 그 경화물 및 적층체 | |
| WO2024247905A1 (ja) | 蓄電デバイス包装材用接着剤、蓄電デバイス包装材、蓄電デバイス用容器及び蓄電デバイス | |
| JP2024106735A (ja) | 蓄電デバイス包装材用接着剤、蓄電デバイス用包装材、蓄電デバイス用容器及び蓄電デバイス | |
| CN115498342A (zh) | 蓄电元件用包装材、蓄电元件用容器及蓄电元件 | |
| CN116368200A (zh) | 聚氨酯泡沫以及调色剂密封构件 | |
| JP2022126353A (ja) | 2液型ウレタン系接着剤組成物 | |
| CN116890488A (zh) | 泡罩包装用层叠体及泡罩包装 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12816858 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013525716 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147001896 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14234424 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012816858 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |