WO2013053690A1 - Pde9i with imidazo pyrazinone backbone - Google Patents
Pde9i with imidazo pyrazinone backbone Download PDFInfo
- Publication number
- WO2013053690A1 WO2013053690A1 PCT/EP2012/069936 EP2012069936W WO2013053690A1 WO 2013053690 A1 WO2013053690 A1 WO 2013053690A1 EP 2012069936 W EP2012069936 W EP 2012069936W WO 2013053690 A1 WO2013053690 A1 WO 2013053690A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyrazin
- imidazo
- methyl
- azetidin
- fluorophenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- KYRKGOCWXBDKBD-UHFFFAOYSA-N CC(C(N1)=C[n]2c(-c3ccccc3)ncc2C1=O)N(C1)CC1Oc(cccc1)c1F Chemical compound CC(C(N1)=C[n]2c(-c3ccccc3)ncc2C1=O)N(C1)CC1Oc(cccc1)c1F KYRKGOCWXBDKBD-UHFFFAOYSA-N 0.000 description 1
- DWJFOZLYWSESSK-UHFFFAOYSA-N CC(C(N1)=C[n]2c(-c3ccccc3)ncc2C1=O)N(C1)CC1Oc1ncccn1 Chemical compound CC(C(N1)=C[n]2c(-c3ccccc3)ncc2C1=O)N(C1)CC1Oc1ncccn1 DWJFOZLYWSESSK-UHFFFAOYSA-N 0.000 description 1
- OAMBWTBJNZOALP-UHFFFAOYSA-N CCc1ccc(C2CN(CC(N3)=C[n]4c(-c5ccccc5)ncc4C3=O)C2)cc1 Chemical compound CCc1ccc(C2CN(CC(N3)=C[n]4c(-c5ccccc5)ncc4C3=O)C2)cc1 OAMBWTBJNZOALP-UHFFFAOYSA-N 0.000 description 1
- CPWSTVGPJSYNMA-UHFFFAOYSA-N O=C1NC(CN(C2)CC2Oc(cc2)ccc2F)=C[n]2c(-c3ccccc3)ncc12 Chemical compound O=C1NC(CN(C2)CC2Oc(cc2)ccc2F)=C[n]2c(-c3ccccc3)ncc12 CPWSTVGPJSYNMA-UHFFFAOYSA-N 0.000 description 1
- DXOVFJHHONEJOL-UHFFFAOYSA-N O=C1NC(CN(C2)CC2Oc(cccc2)c2F)=C[n]2c(-c3ccccc3)ncc12 Chemical compound O=C1NC(CN(C2)CC2Oc(cccc2)c2F)=C[n]2c(-c3ccccc3)ncc12 DXOVFJHHONEJOL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to cyclic guanylate monophosphate (cGMP)-specific phos- phodiesterase type 9 inhibitors (hereinafter referred to as PDE9 inhibitors) of the form 7H- imidazo[1 ,5-a]pyrazin-8-ones for the use as a medicament. Moreover the invention relates to a pharmaceutical composition comprising 7H-imidazo[1 ,5-a]pyrazin-8-ones, as well as a process for preparation of the compounds.
- cGMP cyclic guanylate monophosphate
- PDE9 inhibitors cyclic guanylate monophosphate
- PDE9 inhibitors cyclic guanylate monophosphate
- PDE9 inhibitors cyclic guanylate monophosphate (cGMP)-specific phos- phodiesterase type 9 inhibitors
- PDE9 inhibitors cyclic guanylate monophosphate (cGMP)-specific phos- phodiesterase type 9 inhibitors
- PDEs phosphodiesterases
- the phosphodiesterases are a superfamily of enzymes that metabolically inactivate the ubiquitous intracellular messengers cAMP and cGMP. This function involves the PDEs in a broad range of important cellular functions, such as immune response, memory, and vision.
- the human genome encodes for 21 PDEs that are categorized into 1 1 families (Mehats C, Andersen CB, Filopanti M, Jin SL, Conti M. "Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling.” Trends Endocrinol Metab. 2002;13:29-35).
- cGMP Cyclic guanylate monophosphate
- PDE9 Cyclic guanylate monophosphate
- PDE9 is a member of the PDE enzyme family that selectively hydrolyses cGMP over cAMP (D A Fisher et al., J. Biol. Chemistry, vol. 273, No. 25, 15559-15564 (1998)).
- PDE9 inhibitors have been reported as useful to treat cardiovascular disorders (WO 03/037899), and insulin resistance syndrome, hypertension, and/or type 2 diabetes (WO 03/037432) as well as for treatment of obesity related conditions (WO/2005/041972).
- Wunder F. et al (Mol. Pharmacol. 2005 Dec; 68(6):1775-81 , 2005) report the in vitro characterization of 1 -(2-chlorophenyl)-6-[(2R)-3,3,3-trifluoro-2-methylpropyl]-1 ,5-dihydro-4H- pyrazolo[3,4-d]pyrimidine-4-one, a selective inhibitor of phosphodiesterase 9 (PDE9), which is under development for the treatment of Alzheimer's disease.
- AD cannot be cured and is degenerative, palliative treatment of patients is essential.
- the present invention discloses novel PDE9 inhibitors for the use as a medicament, such as in the treatment of patients suffering from cognitive impairments, in particular cognitive impairments that relate to neurodegenerative diseases such as cortical dementia (e.g. Alzheimer's disease) or subcortical dementia, e.g. AIDS related dementia.
- cognitive impairments that relate to neurodegenerative diseases such as cortical dementia (e.g. Alzheimer's disease) or subcortical dementia, e.g. AIDS related dementia.
- the PDE9 inhibitors of the present invention have the structure I (i.e. a 7H-imidazo[1 ,5- a]pyrazin-8-one backbone):
- R2 is cyclized with either R1 or R3.
- the invention relates to methods of improving conditions involving PDE9, such as cognition, in particular the invention relates to a method of treating diseases involving cognitive difficul- ties, difficulties with executive functioning (such as planning, organization, mental flexibility and task coordination) as well as with pereception (agnosia).
- the methods of improving conditions involving PDE9 and/or treating diseases involving PDE9 comprises the administration of a compound of the present invention or a pharmaceutically acceptable salt, solvate or prodrug thereof to a patient in need thereof.
- the compound of the present invention or a pharmaceutically acceptable salt, solvate or prodrug thereof may be in the form of a pharmaceutical composition.
- the invention relates to an improved pharmaceutical composition
- a compound of the present invention particularly useful for the treatment of cognitive diffi- culties, difficulties with executive functioning (such as planning, organization, mental flexibility and task coordination) as well as with pereception (agnosia), in particular when associated neurodegenerative diseases, such as cortical or subcortical dementias, e.g. Alzheimer's disease (AD).
- AD Alzheimer's disease
- Cognitive impairment includes a decline in cognitive functions or cognitive domains, such as, e.g., difficulties with attention, learning, memory and executive function (relevant reactions to external stimuli). Cognitive impairment also may include: deficits in attention, disorganized thinking, slow thinking, difficulty in understanding, poor concentration, impairment of problem solving, poor memory, difficulty in expressing thoughts and/or integrating thoughts, feelings and behaviour, and/or extinction of irrelevant thoughts, and difficulty in attention and vigilance, verbal learning and memory, visual learning and memory, speed of processing, social cognition, reasoning and problem solving, e.g., executive functioning.
- guanylyl cyclase (alt. guanylate cyclase) converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which in turn activates cGMP-dependent protein kinase G (PKG).
- GTP guanosine triphosphate
- cGMP cyclic guanosine monophosphate
- PKG cGMP-dependent protein kinase G
- cGMP As a result high levels of cGMP will eventually lead to improvement of cognition via the activation of PKG.
- the level of cGMP can be increased by inhibition of PDE9, which - as mentioned above - has the highest affinity for cGMP of any of the PDEs. Accordingly, PDE9 inhibitors will improve synaptic transmission and thereby enhance cognitive performance as evidenced by the results presented in the experimental section.
- Embodiments according to the invention in a first embodiment (E1 ) the present invention relates to compounds having the structure (I) (also referred to as compounds of formula (I))
- R1 when cyclized with R2, is
- R7 is selected from the group consisting of H, -CH 3 , -C 2 H 5 , and -C 3 H wherein * denotes the cyclization point
- R1 when not cyclized, is selected from the group consisting of H
- R7 is selected from the group consisting of H, -CH 3 , -C 2 H 5 , and -C 3 H 7
- R2 is a compound selected from the group consisting of
- R8 and R12 independently are selected from the group consisting of H, -CH 3 , -C 2 H 5 , and -C 3 H 7 wherein * denotes the cyclization point, and
- R9 is selected from the group consisting of H, Ci-C 6 alkyl, branched C 3 -C 6 alkyl, C 3 -C 6 cycloalkyl, C 6 -Ci 0 aryl, substituted C 6 -Ci 0 aryl, C 3 -C 9 het- eroaryl, substituted C 3 -C 9 heteroaryl, CrC 6 alkoxy, branched C 3 -C 6 alkoxy, C 3 -C 6 cycloalkoxy, C 6 -Ci 0 aryloxy, substituted C 6 -Ci 0 aryloxy, C 3 -C 9 heteroary- loxy, substituted C 3 -C 9 heteroaryloxy; and
- R10 is selected from the group consisting of H, -CH 3 , and -C 2 H 5 ; and R1 1 is selected from the group consisting of C 6 -Cio aryl, substituted C 6 - Ci 0 aryl, C 3 -C 9 heteroaryl, substituted C 3 -C 9 heteroaryl
- R4 is selected from the group consisting of hydrogen, -CH 3 , -C 2 H 5 , -C 3 H 7 , -CF 3 , -CN, F and CI;
- R5 is selected from the group consisting of C 6 -Ci 0 aryl, substituted C 6 -Ci 0 aryl, C 3 -C 9 heteroaryl, substituted C 3 -C 9 heteroaryl, C 3 -C 6 heterocyclyl, substituted C 3 -C 6 hetero- cyclyl, C 3 -C 6 cycloalkyl, and substituted C 3 -C 6 cycloalkyl;
- R6 is selected from the group consisting of hydrogen, F, CI, CN, -CH 3 , -C 2 H 5 , -C 3 H 7 , and -CF 3 ;
- A is absent or -CH 2 - and tautomers and pharmaceutically acceptable acid addition salts thereof, and polymorphic forms thereof.
- (E4) of (E1 ) R9 is branched C C 3 alkyl.
- R9 is phenyl or napthyl.
- R9 is substituted phenyl or substituted napthyl.
- the substituent is se- lected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl.
- R9 is a C 4 -C 9 heteroaryl.
- R9 is selected from the group consisting of pyridyl, pyridazine, pyrimidinyl, pyrazinyl, quinolinyl, quinazolinyl, and quinoxalinyl.
- R9 is a substituted C 4 -C 9 heteroaryl.
- R9 is selected from the group consisting of substituted pyridyl, substituted pyridazine, substituted pyrimidinyl, substituted pyrazinyl, substituted quinolinyl, and substituted quinazolinyl, and substituted quinoxalinyl.
- R9 is selected from the group consisting of 2-pyridyl, 3-pyridyl, 2-pyridazine, 2-pyrimidinyl, 4-pyrimidinyl, 2-pyrazinyl, 2-quinolinyl, 2-quinoxalinyl, 6-quinoxalinyl, and 2-quinazolinyl.
- R9 is selected from the group consisting of substituted 2-pyridyl, substituted 3-pyridyl, substituted 2-pyridazine, substituted 2-pyrimidinyl, substituted 4-pyrimidinyl, substituted 2-pyrazinyl, substituted 2-quinolinyl, substituted 2-quinoxalinyl, substituted 6-quinoxalinyl, and substituted 2-quinazolinyl.
- the substituent of R9 is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R9 is C C 4 alkoxy.
- R9 is methoxy or ethoxy. In an embodiment (E18) of embodiment (E1 ) R9 is branched C 3 -C 4 alkoxy.
- R9 is isopropoxy or isobutoxy.
- R9 is C 6 -Ci 0 aryloxy
- R9 is selected from the group consisting of phenyloxy and naphtyloxy.
- R9 when R9 is substituted C 6 -Ci 0 aryloxy, R9 is selected from the group consisting of substituted phenyloxy and substituted naphtyloxy.
- R9 when R9 is a substituted C 6 -Ci 0 aryloxy, is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R9 is a C 4 -C 9 heteroaryloxy.
- R9 is selected from the group consisting of pyridineoxy, pyridazineoxy, pyrimidineoxy and quinoxalineoxy
- R9 is a substituted C 4 -C 9 het- eroaryloxy.
- R9 is selected from the group consisting of substituted pyridineoxy, pyridazineoxy, substituted pyrimidineoxy and quinoxalineoxy
- the substituent is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R1 1 is a C 6 -Ci 0 aryl selected from the group consisting of phenyl and naphthyl. In a preferred embodiment (E29) of embodiment (E28) R1 1 is phenyl.
- R1 1 is a substituted C 6 -Ci 0 aryl selected from the group consisting of substituted phenyl and substituted naphthyl.
- R1 1 is substituted phenyl.
- the substituent is se- lected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R1 1 is a C 4 -C 9 heteroaryl.
- R1 1 is selected from the group consisting of pyridyl, pyridazine, pyrimidinyl, pyrazinyl, quinolinyl, quinazolinyl, and quinoxalinyl.
- R1 1 is selected from the group consisting of 2-pyridyl, 3-pyridyl, 2-pyridazine, 2-pyrimidinyl, 4-pyrimidinyl, 2-pyrazinyl, 2- quinolinyl, 2-quinoxalinyl, 6-quinoxalinyl, and 2-quinazolinyl.
- R1 1 is a substituted C 4 - C 9 heteroaryl.
- R1 1 is selected from the group consisting of substituted pyridyl, substituted pyridazine, substituted pyrimidinyl, substituted pyraz- inyl, substituted quinolinyl, and substituted quinazolinyl, and substituted quinoxalinyl.
- R1 1 is selected from the group consisting of substituted 2-pyridyl, substituted 3-pyridyl, substituted 2-pyridazine, substituted 2-pyrimidinyl, substituted 4-pyrimidinyl, substituted 2-pyrazinyl, substituted 2-quinolinyl, substituted 2-quinoxalinyl, substituted 6-quinoxalinyl, and substituted 2-quinazolinyl.
- R1 1 is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R5 is selected from the group consisting of phenyl and naphthyl.
- R5 is substituted phenyl.
- the substituent is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethyl- amino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R5 is pyridyl
- R5 is substituted pyridyl.
- the substituent is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
- R5 is selected from the group consisting of tet- rahydropyranyl, tetrahydrofuranyl and piperidyl.
- R5 is selected from the group consisting of substituted tetrahydropyranyl, substituted tetrahydrofuranyl and substituted piperidyl.
- the substituent is selected from group consisting of F, CI, methyl, cyano and methoxy.
- R5 is selected from the group consisting of cyclobutyl, cyclopentyl and cyclohexyl.
- R5 is cyclopentyl or cyclohexyl.
- R5 is selected from the group consisting of substituted cyclobutyl, substituted cyclopentyl and substituted cyclohexyl.
- R5 is substituted cyclopentyl or substituted cyclohexyl.
- substituent is selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, dimethylamino, cyclopropyl, and isopropyl; in particular the substituents are selected from the group consisting of F, CI, methyl, trifluoromethyl, methoxy, trifluoromethoxy, cyano, ethyl, and dimethylamino.
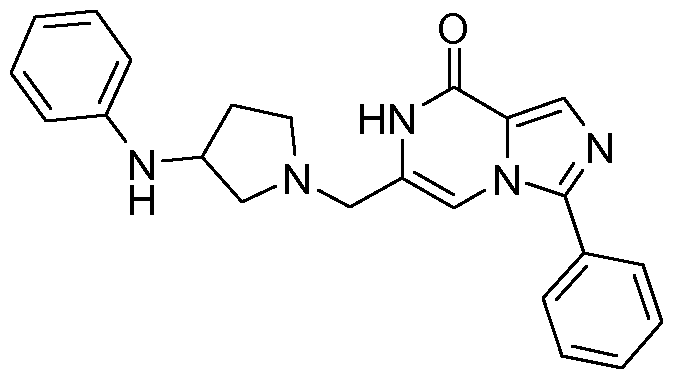
- the compound of formula (I) is selected among the compounds listed in Table 1 , in the form of the free base, one or more tautomers thereof or a pharmaceutically acceptable acid addition salt thereof.
- the compound is for use as a medicament.
- the compound is for use in the treatment of a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, ampheta- mine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, ampheta- mine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- the compound is for preparation of a medicament for use in the treatment of a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, amphetamine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, amphetamine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- Embodiment (E59) of the present invention covers a method of treating a subject suffering from a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, sub- stance-induced psychotic disorder, for example psychosis induced by alcohol, amphetamine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine, which method comprises administering to said subject a compound of any of embodiments (E1 )-(E55).
- a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, sub- stance-induced psychotic disorder, for example psychosis induced by alcohol, amphetamine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine
- the present invention covers a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of any of embodiments (E1 ) to (E55), and one or more pharmaceutically acceptable carriers, diluents and excipients.
- the pharmaceutical composition is for the treatment of a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, ampheta- mine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- a disease selected from the group consisting of Alzheimer's disease, mental retardation; CIAS, attention-deficit/hyperactivity disorder; and age-related cognitive decline, substance-induced psychotic disorder, for example psychosis induced by alcohol, ampheta- mine, cannabis, cocaine, hallucinogens, inhalants, opioids, or phencyclidine.
- Table 1 lists compounds of the invention and the corresponding IC50 values (nM) determined as described in the section "PDE9 inhibition assay". Each of the compounds constitutes an individual embodiment of the present invention:
- halo and halogen are used interchangeably and refer to fluorine, chlorine, bromine or iodine.
- Ci-C 6 alkoxy refers to a straight-chain or branched saturated alkoxy group having from one to six carbon atoms, inclusive, with the open valency on the oxygen. Examples of such groups include, but are not limited to, methoxy, ethoxy, n-butoxy, 2-methyl- pentoxy and n-hexyloxy.
- C 3 -C 8 cycloalkyi typically refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl.
- hydroxyheterocycloalkyl refers to a heterocycloalkyi as defined above which is substituted with one hydroxy group.
- CrC 6 alkyl-heterocycloalkyl refers to a heterocycloalkyi as defined above which is substituted with a Ci-C 6 alkyl group.
- examples of such groups include, but are not limited to, tetrahydropyran-4-yl-methyl and 2-morpholin-4-yl-ethyl.
- aryl refers to a phenyl ring, optionally substituted with halogen, Ci-C 6 alkyl, CrC 6 alkoxy or halo(CrC 6 )alkyl as defined above. Examples of such groups include, but are not limited to, phenyl and 4-chlorophenyl.
- heteroaryloxy refers to an aryloxy where one or more carbon atoms have been sub- stituted with one more hetero atoms, such as N, O, S.
- present invention further provides certain embodiments of the invention, which are described below. Additionally, the present invention further provides certain embodiments of the invention that are described below.
- the present invention also comprises salts of the compounds, typically, pharmaceutically acceptable salts.
- Such salts include pharmaceutically acceptable acid addition salts.
- Acid addition salts include salts of inorganic acids as well as organic acids.
- inorganic or organic acid addition salts include the pharmaceutically acceptable salts listed in Berge, S.M. et al., J. Pharm. Sci. 1977, 66, 2, the contents of which are hereby incorporated by reference.
- the compounds of this invention may exist in unsolvated as well as in solvated forms with pharmaceutically acceptable solvents such as water, ethanol and the like. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of this invention.
- compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
- compositions may be specifically formulated for administration by any suitable route such as oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) routes. It will be appreciated that the route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient.
- Pharmaceutical compositions for oral administration include solid dosage forms such as capsules, tablets, dragees, pills, lozenges, powders and granules.
- compositions may be prepared with coatings such as enteric coatings or they may be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art.
- Liquid dosage forms for oral administration include solutions, emulsions, suspensions, syrups and elixirs.
- Typical oral dosages range from about 0.001 to about 100 mg/kg body weight per day. Typical oral dosages also range from about 0.01 to about 50 mg/kg body weight per day. Typical oral dosages further range from about 0.05 to about 10 mg/kg body weight per day. Oral dosages are usually administered in one or more dosages, typically, one to three dosages per day. The exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art. The formulations may also be presented in a unit dosage form by methods known to those skilled in the art. For illustrative purposes, a typical unit dosage form for oral administration may contain from about 0.01 to about 1000 mg, from about 0.05 to about 500 mg, or from about 0.5 mg to about 200 mg.
- parenteral routes such as intravenous, intrathecal, intramuscular and similar administra- tion
- typical doses are in the order of half the dose employed for oral administration.
- the present invention also provides a process for making a pharmaceutical composition
- a process for making a pharmaceutical composition comprising admixing a therapeutically effective amount of a compound of formula (I) and at least one pharmaceutically acceptable carrier or diluent.
- the compound utilized in the aforementioned process is one of the specific com- pounds disclosed in the Experimental Section herein.
- the compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof.
- One example is an acid addition salt of a compound having the utility of a free base.
- a compound of formula (I) contains a free base such salts are prepared in a conventional manner by treating a solution or suspension of a free base of formula (I) with a molar equivalent of a pharmaceutically acceptable acid.
- suitable organic and inorganic acids are described above.
- solutions of the compounds of formula (I) in sterile aqueous so- lution, aqueous propylene glycol, aqueous vitamin E or sesame or peanut oil may be employed.
- aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose.
- the aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration.
- the compounds of formula (I) may be readily incorporated into known sterile aqueous media us- ing standard techniques known to those skilled in the art.
- Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solutions and various organic solvents.
- solid carriers include lactose, terra alba, sucrose, cyclodextrin, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and lower alkyl ethers of cellulose.
- liquid carriers include, but are not limited to, syrup, pea- nut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water.
- the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- sustained release material such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical compositions formed by combining the compounds of formula (I) and a pharmaceutically acceptable carrier are then readily administered in a variety of dosage forms suit- able for the disclosed routes of administration.
- the formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and optionally a suitable excipient.
- the orally available formu- lations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or water-in-oil liquid emulsion.
- the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it may be in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but will range from about 25 mg to about 1 g per dosage unit.
- the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
- the pharmaceutical compositions of the invention may be prepared by conventional methods in the art. For example, tablets may be prepared by mixing the active ingredient with ordinary adjuvants and/or diluents and subsequently compressing the mixture in a conventional tabletting machine prepare tablets.
- adjuvants or diluents comprise: corn starch, potato starch, talcum, magnesium stearate, gelatin, lactose, gums, and the like. Any other adjuvants or additives usually used for such purposes such as colorings, flavorings, preservatives etc. may be used provided that they are compatible with the active ingredients.
- the PDE9 inhibitors of the present invention may be used in the treatment of cognition deficiencies related to neurodegenerative disorders, such dementia, such as cortical dementia or subcortical dementia.
- Cortical dementias arise from a disorder affecting the cerebral cortex, the outer layers of the brain that play a critical role in cognitive processes such as memory and language.
- Particularly considered cortical dementias are Alzheimer's disease; vascular dementia (also known as multi-infarct dementia), including Binswanger's disease; Dementia with Lewy bodies (DLB); Alcohol-Induced Persisting Dementia, including Korsakoff's syndrome and Wernicke's encephalopathy; frontotemporal lobar degeneration (FTLD), including: Pick's disease, fronto- temporal dementia (or frontal variant FTLD), semantic dementia (or temporal variant FTLD), and progressive non-fluent aphasia; Creutzfeldt-Jakob disease; dementia pugilistica; Moya- moya disease; and posterior cortical atrophy (an Alzheimer's disease variant).
- vascular dementia also known as multi-infarct dementia
- DLB Dementia with Lewy bodies
- Subcortical dementias result from dysfunction in the parts of the brain that are beneath the cortex. Usually, the memory loss and language difficulties that are characteristic of cortical dementias are not present. Rather, people with subcortical dementias, such as Huntington's disease, Parkinson's Disease, and AIDS dementia complex, tend to show changes in their personality and attention span, and their thinking slows down.
- Particularly considered subcortical dementias are dementia due to Huntington's disease, dementia due to hypothyroidism, dementia due to Parkinson's disease, dementia due to Vitamin B1 deficiency, dementia due to Vitamin B12 deficiency, dementia due to folate deficiency, dementia due to syphilis, dementia due to subdural hematoma, dementia due to hypercalcaemia, dementia due to hypo- glycaemia, AIDS dementia complex, pseudodementia (a major depressive episode with prominent cognitive symptoms), substance-induced persisting dementia, dementia due to multiple etiologies, dementia due to other general medical conditions (i.e. end stage renal failure, cardiovascular disease etc.), dementia not otherwise specified (used in cases where no specific criteria is met).
- Compounds of formula V, where X is a halogen can be prepared by halogenation of formula IV with SOCI 2 , PBr 3 .
- Compounds of formula IV can be prepared by reduction with NaBH 4 , LAH, DIBAL, BH 3 , etc, of compounds of formula III, which can be made by carbonylation of compounds of formula II in the presence of CO, and a palladium catalyst, such as Pd(dppf)CI 2 , Pd(PPh 3 ) 2 CI 2 , Pd(PPh 3 ) 4 , and a base, such as Cs 2 C0 3 , K 2 C0 3 , or Et 3 N (Scheme 2):
- a palladium catalyst such as Pd(dppf)CI 2 , Pd(PPh 3 ) 2 CI 2 , Pd(PPh 3 ) 4
- a base such as Cs 2 C0 3 , K 2 C0 3 , or Et 3 N (Scheme 2):
- Compounds of formula XII can be prepared by acid catalyzed demethylation of compounds of formula XI, which can be made from reaction of Wein- reb amides of formula X with a Grignard reagent.
- Compounds of formula X can be made by carbonylation of compounds of formula II with CO in the presence of a palladium catalyst, such as Pd(dppf)CI 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 CI 2 , and a base, such as Cs 2 C0 3 , K 2 C0 3 , Et 3 N, KOAc, etc., in solvents, such as toluene, DMF, DMSO (Scheme 3):
- a palladium catalyst such as Pd(dppf)CI 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 CI 2
- a base such as Cs 2 C0 3 , K 2 C0 3 , Et 3 N, KOAc, etc.
- solvents such as toluene, DMF, DMSO (Scheme 3):
- Compounds of formula XX can be prepared by hydrogena- tion of compounds of formula XIX, with H 2 and in the presence of Pd/C or Pd(OH) 2 /C in solvents such as alcohol or EtOAc, etc.
- Compounds of formula XIX can be synthesized by me- sylation of compounds of formula XVI, followed by reductive deoxygenation with NaBH 4 or LiAIH 4 , and acid catalyzed demethylation.
- Compounds of formula XVI can be made by reduction with LAH, NaBH 4 , DIBAL, etc, of compounds of formula XV, which can be made by [3+2] cycloaddtion of compounds of formula XIV with benzyl-methoxymethyl- trimethylsilanylmethylamine with the aid of TFA in solvents such as toluene or CH 2 CI 2 .
- Compounds of formula XIV can be made by Suzuki reaction of compounds of formula II with bo- ronic ester XIII in the presence of a palladium catalyst, such as Pd(dppf)CI 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 CI 2 , etc., and a base, such as Cs 2 C0 3 , K 2 C0 3 , KOAc, etc., in solvent, such as toluene, DMF, DMSO, etc; or by Heck reaction of compounds of formula II with ethyl acrylate in the presence of a palladium catalyst, such as Pd(dppf)CI 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 CI 2 and a base, such as Cs 2 C0 3 , K 2 C0 3 , KOAc, etc.
- a palladium catalyst such as Pd(dppf)CI 2 , Pd(PPh 3 ) 4
- Boronic ester XIII can be generated by coupling of ethyl propiolate with bis(pinacolato)diboron in the presence of CuCI, and Xantphos as catalysts, i-BuOK as a base in the solvent of THF (Scheme 4):
- Compounds of formula II can be prepared by cyclization of compounds of formula XXVI in the presence of POCI 3 or SOCI 2 .
- Compounds of formula XXVI can be made by reduction of com- pounds of formula XXII in H 2 with Pd/C or Raney Ni to generate XXIII, which can then be converted to amides of formula XXIV by reaction with a variety of acid chlorides, or by coupling with various carboxylic acids in the presence of coupling reagents, such as HOBt and EDC, or HATU. Subsequent deprotection under acidic conditions yields compounds of formula XXV, which can undergo diazotization and iodination.
- Compounds of formula XXII can be made from Boc protection of compounds of formula XXI (Scheme 5):
- a PDE9 assay may for example, be performed as follows: The assay is performed in 60 uL samples containing a fixed amount of the relevant PDE enzyme (sufficient to convert 20-25% of the cyclic nucleotide substrate), a buffer (50 mM HEPES7.6; 10mM MgCI 2 ; 0.02% Tween20), 0.1 mg/ml BSA, 225 pCi of 3 H-labelled cyclic nucleotide substrate, tritium labeled cAMP to a final concentration of 5 nM and varying amounts of inhibitors.
- the assay was performed in 60 uL assay buffer (50 mM HEPES pH 7.6; 10mM MgCI 2 ; 0.02% Tween20) containing enough PDE9 to convert 20-25% of 10 nM 3 H-cAMP and varying amounts of inhibitors. Following a 1 hour incubation the reactions were terminated by addition of 15 uL 8 mg/mL yttrium silicate SPA beads (Amersham). The beads were allowed to settle for one hr in the dark before the plates were counted in a Wallac 1450 Microbeta counter. IC 50 values were calculated by non linear regression using XLfit (IDBS).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurosurgery (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description

















































Claims


Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11201502728WA SG11201502728WA (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone |
| EP16185105.0A EP3121178B1 (en) | 2011-10-10 | 2012-10-09 | 6-[4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7h-imidazo[1,5-a]pyrazin-8-one as pde9 inhibitor |
| MX2019014982A MX381326B (en) | 2011-10-10 | 2012-10-09 | 7H-IMIDAZO[1-5-A]PYRAZIN-8-ONE DERIVATIVES AS PHOSPHODIESTERASE 9 (PDE9) INHIBITORS AND THEIR USE IN THE TREATMENT OF NEURODEGENERATIVE DISORDERS |
| HK15111740.3A HK1211023A1 (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone |
| AU2012323085A AU2012323085B2 (en) | 2011-10-10 | 2012-10-09 | PDE9i with imidazo pyrazinone backbone |
| IN2829DEN2015 IN2015DN02829A (en) | 2011-10-10 | 2012-10-09 | |
| CA2886885A CA2886885C (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone |
| BR112015007731-5A BR112015007731B1 (en) | 2011-10-10 | 2012-10-09 | Compound, pharmaceutical composition comprising it and use thereof |
| CN201280076285.6A CN104703987B (en) | 2011-10-10 | 2012-10-09 | PDE9 inhibitors with imidazopyrazinone skeleton |
| MX2015004422A MX370433B (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone. |
| JP2015535994A JP2015531401A (en) | 2011-10-10 | 2012-10-09 | PDE9I having imidazopyrazinone skeleton |
| MA37958A MA37958B1 (en) | 2011-10-10 | 2012-10-09 | Pde9i having an imidazo pyrazinone skeleton |
| US14/434,308 US9643970B2 (en) | 2011-10-10 | 2012-10-09 | Substituted imidazo [1,5-a]pyrazines as PDE9 inhibitors |
| EP12772769.1A EP2906562B1 (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone |
| TNP2015000100A TN2015000100A1 (en) | 2011-10-10 | 2015-03-16 | Pde9i with imidazo pyrazinone backbone |
| IL237836A IL237836A (en) | 2011-10-10 | 2015-03-19 | 91pde Mother of Imidzo-Firzinone Broadcast |
| US15/452,827 US9993477B2 (en) | 2011-10-10 | 2017-03-08 | Substituted imidazo[1,5-a]pyrazines as PDE9 inhibitors |
| AU2017201873A AU2017201873C1 (en) | 2011-10-10 | 2017-03-20 | PDE9i with imidazo pyrazinone backbone |
| IL253448A IL253448A0 (en) | 2011-10-10 | 2017-07-12 | Pde9i with imidazo pyrazinone backbone |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011001692 | 2011-10-10 | ||
| CNPCT/CN2011/001692 | 2011-10-10 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/434,308 A-371-Of-International US9643970B2 (en) | 2011-10-10 | 2012-10-09 | Substituted imidazo [1,5-a]pyrazines as PDE9 inhibitors |
| US15/452,827 Continuation US9993477B2 (en) | 2011-10-10 | 2017-03-08 | Substituted imidazo[1,5-a]pyrazines as PDE9 inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013053690A1 true WO2013053690A1 (en) | 2013-04-18 |
Family
ID=47019007
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2012/069936 Ceased WO2013053690A1 (en) | 2011-10-10 | 2012-10-09 | Pde9i with imidazo pyrazinone backbone |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US9643970B2 (en) |
| EP (2) | EP2906562B1 (en) |
| JP (1) | JP2015531401A (en) |
| CN (2) | CN106905324B (en) |
| AU (2) | AU2012323085B2 (en) |
| BR (1) | BR112015007731B1 (en) |
| CA (1) | CA2886885C (en) |
| HK (1) | HK1211023A1 (en) |
| IL (2) | IL237836A (en) |
| IN (1) | IN2015DN02829A (en) |
| MA (1) | MA37958B1 (en) |
| MX (2) | MX381326B (en) |
| SG (1) | SG11201502728WA (en) |
| TN (1) | TN2015000100A1 (en) |
| WO (1) | WO2013053690A1 (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150274736A1 (en) * | 2011-10-10 | 2015-10-01 | H. Lundbeck A/S | PDE9i with imidazo pyrazinone backbone |
| WO2016174188A1 (en) * | 2015-04-30 | 2016-11-03 | H. Lundbeck A/S | Imidazopyrazinones as pde1 inhibitors |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| WO2017005786A1 (en) | 2015-07-07 | 2017-01-12 | H. Lundbeck A/S | Pde9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| WO2018009424A1 (en) | 2016-07-06 | 2018-01-11 | Imara, Inc. | Pde9 inhibitors for treatment of peripheral diseases |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| WO2018073251A1 (en) | 2016-10-18 | 2018-04-26 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as pde1 inhibitors |
| WO2018078038A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders |
| WO2018078042A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising administration of imidazopyrazinones |
| US10150771B2 (en) | 2014-10-10 | 2018-12-11 | H. Lundbeck A/S | Triazolopyrazinones as PDE1 inhibitors |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| WO2019101511A1 (en) | 2017-11-23 | 2019-05-31 | Basf Se | Substituted trifluoromethyloxadiazoles for combating phytopathogenic fungi |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| WO2019226944A1 (en) | 2018-05-25 | 2019-11-28 | Imara Inc. | Monohydrate and crystalline forms of 6-[(3s,4s)-4-methyl-1- (pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl- 7h-imid azo [1,5- a] pyrazin-8-one |
| WO2019243528A1 (en) * | 2018-06-20 | 2019-12-26 | Janssen Pharmaceutica Nv | Oga inhibitor compounds |
| US10538525B2 (en) | 2016-04-12 | 2020-01-21 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| US11370795B2 (en) | 2017-05-26 | 2022-06-28 | Imara Inc. | Process for the synthesis of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US12213975B2 (en) | 2018-08-31 | 2025-02-04 | Cardurion Pharmaceuticals, Inc. | PDE9 inhibitors for treating sickle cell disease |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201717948A (en) * | 2015-08-10 | 2017-06-01 | H 朗德貝克公司 | Combination treatment comprising administration of 2-amino-3,5,5-trifluoro-3,4,5,6-tetrahydropyridines |
| LT3374359T (en) * | 2015-11-09 | 2020-03-25 | Astrazeneca Ab | Dihydroimidazopyrazinone derivatives useful in the treatment of cancer |
| WO2018154133A1 (en) * | 2017-02-27 | 2018-08-30 | Janssen Pharmaceutica Nv | [1,2,4]-triazolo [1,5-a]-pyrimidinyl derivatives substituted with piperidine, morpholine or piperazine as oga inhibitors |
| AU2018281131B2 (en) | 2017-06-08 | 2022-01-20 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine PDE9 inhibitors |
| EP3946348A4 (en) * | 2019-04-05 | 2023-08-02 | Imara Inc. | PDE9 INHIBITORS FOR THE TREATMENT OF SICKLE CELL DISEASE |
| EP3965768A1 (en) * | 2019-05-07 | 2022-03-16 | Imara Inc. | Pde9 inhibitors for treating thalassemia |
| WO2022036111A1 (en) * | 2020-08-13 | 2022-02-17 | Imara Inc. | Methods and compositions for treating sickle cell disease |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003037899A1 (en) | 2001-11-02 | 2003-05-08 | Pfizer Limited | Pde9 inhibitors for treating cardiovascular disorders |
| WO2003037432A1 (en) | 2001-11-02 | 2003-05-08 | Pfizer Products Inc. | Treatment of insulin resistance syndrome and type 2 diabetes with pde9 inhibitors |
| WO2005041972A1 (en) | 2003-10-31 | 2005-05-12 | Pfizer Products Inc. | Phosphodiesterase 9 inhibition as treatment for obesity-related conditions |
| WO2012040230A1 (en) * | 2010-09-20 | 2012-03-29 | Envivo Pharmaceuticals, Inc. | Imidazotriazinone compounds |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2395558C (en) | 1997-11-12 | 2007-07-17 | Bayer Aktiengesellschaft | Intermediates for 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| IL144156A0 (en) | 1999-01-20 | 2002-05-23 | Dresden Arzneimittel | Use of imidazo [1,5-a]-pyrido [3,2-e] -pyrazinones as medicaments |
| GB2388111A (en) | 2002-05-01 | 2003-11-05 | Bayer Ag | Novel imidazotriazinone compounds |
| US20040220186A1 (en) | 2003-04-30 | 2004-11-04 | Pfizer Inc. | PDE9 inhibitors for treating type 2 diabetes,metabolic syndrome, and cardiovascular disease |
| JP2009507032A (en) | 2005-09-02 | 2009-02-19 | アボット・ラボラトリーズ | New imidazo heterocycle |
| TW200815436A (en) | 2006-05-30 | 2008-04-01 | Elbion Ag | 4-amino-pyrido[3,2-e]pyrazines, their use as inhibitors of phosphodiesterase 10, and processes for preparing them |
| EP2123801A4 (en) | 2006-09-08 | 2011-01-19 | Tokuyama Corp | METHOD AND EQUIPMENT FOR PRODUCING A NITRIDE OF A GROUP III ELEMENT |
| US8299080B2 (en) | 2006-12-13 | 2012-10-30 | Aska Pharmaceutical Co., Ltd. | Substituted imidazo[1,5-A] quinoxalines as a PDE9 inhibitor |
| WO2008072778A1 (en) * | 2006-12-13 | 2008-06-19 | Aska Pharmaceutical Co., Ltd. | Therapeutic agent for urinary tract disease |
| RS52166B (en) | 2007-05-11 | 2012-08-31 | Pfizer Inc. | AMINO-HETEROCYCLIC COMPOUNDS |
| DE102007032349A1 (en) | 2007-07-11 | 2009-01-15 | Bayer Healthcare Ag | Imidazo, pyrazolopyrazines and imidazotriazines and their use |
| TWI404721B (en) | 2009-01-26 | 2013-08-11 | Pfizer | Amino-heterocyclic compounds |
| WO2011028820A1 (en) | 2009-09-02 | 2011-03-10 | Concert Pharmaceuticals, Inc. | Substituted derivatives of bicyclic [4.3.0] heteroaryl compounds |
| JO3116B1 (en) | 2011-10-07 | 2017-09-20 | Eisai R&D Man Co Ltd | Pyrazolokinoline is derived as inhibitors for PDE9 |
| BR112015007731B1 (en) * | 2011-10-10 | 2022-05-31 | H. Lundbeck A/S | Compound, pharmaceutical composition comprising it and use thereof |
| BR112014018199A8 (en) | 2012-01-26 | 2021-03-02 | H Lundbeck As | pde9 inhibitors with imidazo triazinone main chain, pharmaceutical composition comprising them, use of said inhibitors and process for their preparation |
| HK1206726A1 (en) | 2012-03-19 | 2016-01-15 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
-
2012
- 2012-10-09 BR BR112015007731-5A patent/BR112015007731B1/en active IP Right Grant
- 2012-10-09 CN CN201710247417.1A patent/CN106905324B/en active Active
- 2012-10-09 CA CA2886885A patent/CA2886885C/en active Active
- 2012-10-09 AU AU2012323085A patent/AU2012323085B2/en active Active
- 2012-10-09 EP EP12772769.1A patent/EP2906562B1/en active Active
- 2012-10-09 JP JP2015535994A patent/JP2015531401A/en active Pending
- 2012-10-09 IN IN2829DEN2015 patent/IN2015DN02829A/en unknown
- 2012-10-09 MX MX2019014982A patent/MX381326B/en unknown
- 2012-10-09 HK HK15111740.3A patent/HK1211023A1/en unknown
- 2012-10-09 US US14/434,308 patent/US9643970B2/en active Active
- 2012-10-09 SG SG11201502728WA patent/SG11201502728WA/en unknown
- 2012-10-09 CN CN201280076285.6A patent/CN104703987B/en active Active
- 2012-10-09 WO PCT/EP2012/069936 patent/WO2013053690A1/en not_active Ceased
- 2012-10-09 MX MX2015004422A patent/MX370433B/en active IP Right Grant
- 2012-10-09 EP EP16185105.0A patent/EP3121178B1/en active Active
- 2012-10-09 MA MA37958A patent/MA37958B1/en unknown
-
2015
- 2015-03-16 TN TNP2015000100A patent/TN2015000100A1/en unknown
- 2015-03-19 IL IL237836A patent/IL237836A/en active IP Right Grant
-
2017
- 2017-03-08 US US15/452,827 patent/US9993477B2/en active Active
- 2017-03-20 AU AU2017201873A patent/AU2017201873C1/en active Active
- 2017-07-12 IL IL253448A patent/IL253448A0/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003037899A1 (en) | 2001-11-02 | 2003-05-08 | Pfizer Limited | Pde9 inhibitors for treating cardiovascular disorders |
| WO2003037432A1 (en) | 2001-11-02 | 2003-05-08 | Pfizer Products Inc. | Treatment of insulin resistance syndrome and type 2 diabetes with pde9 inhibitors |
| WO2005041972A1 (en) | 2003-10-31 | 2005-05-12 | Pfizer Products Inc. | Phosphodiesterase 9 inhibition as treatment for obesity-related conditions |
| WO2012040230A1 (en) * | 2010-09-20 | 2012-03-29 | Envivo Pharmaceuticals, Inc. | Imidazotriazinone compounds |
Non-Patent Citations (14)
| Title |
|---|
| "Remington: The Science and Practice of Pharmacy", 1995, MACK PUBLISHING CO. |
| BERGE, S.M. ET AL., J. PHARM. SCI., vol. 66, 1977, pages 2 |
| BLOKLAND ET AL.: "Improving memory; a role for phosphordiesterases", CURRENT PHARMACOLOGICAL DESIGN, vol. 12, 2006, pages 2511 - 2523 |
| BORON, W. F.: "Medical Physiology: A Cellular and Molecular Approach", 2005, ELSEVIER/SAUNDERS |
| BREER H; BOEKHOFF , TAREILUS E.: "Rapid kinetics of second messenger formation in olfactory transduction.", NATURE, vol. 345, 1990, pages 65 - 68 |
| COOKE ET AL.: "Plasticity in the human central nervous system", BRAIN, vol. 129, 2006, pages 1659 - 1673 |
| D A FISHER ET AL., J. BIOL. CHEMISTRY, vol. 273, no. 25, 1998, pages 15559 - 15564 |
| F. JOSEF VAN DER STAAY, NEUROPHARMACOLOGY, vol. 55, no. 5, October 2008 (2008-10-01), pages 908 - 918 |
| MATTHEW A. J., ORGANCI LETTERS., vol. 10, no. 15, 2008, pages 3259 - 3262 |
| MEHATS C; ANDERSEN CB; FILOPANTI M; JIN SL; CONTI M.: "Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling.", TRENDS ENDOCRINOL METAB., vol. 13, 2002, pages 29 - 35 |
| ORGANIC LETTERS, vol. 12, no. 5, 2010, pages 1024 - 1027 |
| PATRICK R. VERHOEST ET AL: "Identification of a Brain Penetrant PDE9A Inhibitor Utilizing Prospective Design and Chemical Enablement as a Rapid Lead Optimization Strategy", JOURNAL OF MEDICINAL CHEMISTRY, vol. 52, no. 24, 24 December 2009 (2009-12-24), pages 7946 - 7949, XP055011767, ISSN: 0022-2623, DOI: 10.1021/jm9015334 * |
| WUNDER F. ET AL., MOL. PHARMACOL., vol. 68, no. 6, December 2005 (2005-12-01), pages 1775 - 81 |
| ZHOU ET AL.: "Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation", NATURE, vol. 368, 1994, pages 635 - 639 |
Cited By (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9643970B2 (en) * | 2011-10-10 | 2017-05-09 | H. Lundbeck A/S | Substituted imidazo [1,5-a]pyrazines as PDE9 inhibitors |
| US20150274736A1 (en) * | 2011-10-10 | 2015-10-01 | H. Lundbeck A/S | PDE9i with imidazo pyrazinone backbone |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11247992B2 (en) | 2014-02-13 | 2022-02-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10300051B2 (en) | 2014-02-13 | 2019-05-28 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10717737B2 (en) | 2014-02-13 | 2020-07-21 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9994546B2 (en) | 2014-02-13 | 2018-06-12 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10676457B2 (en) | 2014-02-13 | 2020-06-09 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10513493B2 (en) | 2014-02-13 | 2019-12-24 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11155532B2 (en) | 2014-02-13 | 2021-10-26 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10174030B2 (en) | 2014-02-13 | 2019-01-08 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US10968221B2 (en) | 2014-07-10 | 2021-04-06 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10640503B2 (en) | 2014-07-10 | 2020-05-05 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US10556908B2 (en) | 2014-07-10 | 2020-02-11 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10138249B2 (en) | 2014-07-10 | 2018-11-27 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US10047086B2 (en) | 2014-07-10 | 2018-08-14 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US10112950B2 (en) | 2014-07-10 | 2018-10-30 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10125133B2 (en) | 2014-07-10 | 2018-11-13 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyridines and substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10150771B2 (en) | 2014-10-10 | 2018-12-11 | H. Lundbeck A/S | Triazolopyrazinones as PDE1 inhibitors |
| US11401272B2 (en) | 2015-04-03 | 2022-08-02 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US10800779B2 (en) | 2015-04-03 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US10011606B2 (en) | 2015-04-30 | 2018-07-03 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| RU2712219C2 (en) * | 2015-04-30 | 2020-01-27 | Х. Лундбекк А/С | Imidazopirasothines as pde1 inhibitors |
| CN107531714A (en) * | 2015-04-30 | 2018-01-02 | H.隆德贝克有限公司 | Imidazopyrazines ketone as PDE1 inhibitor |
| EA031946B1 (en) * | 2015-04-30 | 2019-03-29 | Х. Лундбекк А/С | Imidazopyrazinones as pde1 inhibitors |
| KR102605759B1 (en) | 2015-04-30 | 2023-11-23 | 하. 룬드벡 아크티에셀스카브 | Imidazopyrazinone as a PDE1 inhibitor |
| KR20170140216A (en) * | 2015-04-30 | 2017-12-20 | 하. 룬드벡 아크티에셀스카브 | Imidazopyrazinone as a PDE1 inhibitor |
| CN107531714B (en) * | 2015-04-30 | 2019-07-19 | H.隆德贝克有限公司 | Imidazopyrazines ketone as PDE1 inhibitor |
| US10858362B2 (en) | 2015-04-30 | 2020-12-08 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| WO2016174188A1 (en) * | 2015-04-30 | 2016-11-03 | H. Lundbeck A/S | Imidazopyrazinones as pde1 inhibitors |
| US11472810B2 (en) | 2015-04-30 | 2022-10-18 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| US11608342B2 (en) | 2015-07-07 | 2023-03-21 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| WO2017005786A1 (en) | 2015-07-07 | 2017-01-12 | H. Lundbeck A/S | Pde9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| IL280619B1 (en) * | 2015-07-07 | 2023-06-01 | H Lundbeck As | Pde9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| EP3865484A1 (en) | 2015-07-07 | 2021-08-18 | H. Lundbeck A/S | Pde9 inhibitor with imidazo pyrazinone backbone for treatment of peripheral diseases |
| AU2021201227B2 (en) * | 2015-07-07 | 2023-02-16 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| EP4335497A3 (en) * | 2015-07-07 | 2024-05-01 | H. Lundbeck A/S | Pde9 inhibitor with imidazo pyrazinone backbone for treatment of peripheral diseases |
| EP4335497A2 (en) | 2015-07-07 | 2024-03-13 | H. Lundbeck A/S | Pde9 inhibitor with imidazo pyrazinone backbone for treatment of peripheral diseases |
| CN107810187A (en) * | 2015-07-07 | 2018-03-16 | H.隆德贝克有限公司 | PDE9 inhibitor with imidazotriazinone skeleton and imidazopyrazinone skeleton for treating peripheral diseases |
| IL280619B2 (en) * | 2015-07-07 | 2023-10-01 | H Lundbeck As | Pde9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| CN107810187B (en) * | 2015-07-07 | 2020-09-15 | H.隆德贝克有限公司 | PDE9 inhibitors with imidazotriazinone backbone and imidazopyrazinone backbone for the treatment of peripheral diseases |
| US10513524B2 (en) | 2015-07-07 | 2019-12-24 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| AU2016289856B2 (en) * | 2015-07-07 | 2020-11-26 | H. Lundbeck A/S | PDE9 inhibitors with imidazo triazinone backbone and imidazo pyrazinone backbone for treatment of peripheral diseases |
| US10723700B2 (en) | 2015-08-12 | 2020-07-28 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US11498900B2 (en) | 2015-08-12 | 2022-11-15 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10538525B2 (en) | 2016-04-12 | 2020-01-21 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US11104680B2 (en) | 2016-04-12 | 2021-08-31 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| US20210085684A1 (en) * | 2016-07-06 | 2021-03-25 | Imara Inc. | Pde9 inhibitors for treatment of peripheral diseases |
| CN109475556A (en) * | 2016-07-06 | 2019-03-15 | 伊马拉公司 | PDE9 inhibitors for the treatment of peripheral diseases |
| CN114903900A (en) * | 2016-07-06 | 2022-08-16 | 伊马拉公司 | PDE9 inhibitors for the treatment of peripheral diseases |
| WO2018009424A1 (en) | 2016-07-06 | 2018-01-11 | Imara, Inc. | Pde9 inhibitors for treatment of peripheral diseases |
| US10633382B2 (en) | 2016-10-18 | 2020-04-28 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as PDE1 inhibitors |
| WO2018073251A1 (en) | 2016-10-18 | 2018-04-26 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as pde1 inhibitors |
| CN109803653A (en) * | 2016-10-28 | 2019-05-24 | H.隆德贝克有限公司 | Combined therapy including giving Imidazopyrazines ketone |
| US10905688B2 (en) | 2016-10-28 | 2021-02-02 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-α]pyrazinones as PDE1 inhibitors |
| US10912773B2 (en) | 2016-10-28 | 2021-02-09 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-a]pyrazinones as PDE1 inhibitors |
| WO2018078038A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders |
| WO2018078042A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising administration of imidazopyrazinones |
| US11999741B2 (en) | 2017-05-26 | 2024-06-04 | Cardurion Pharmaceuticals, Inc. | Process for the synthesis of 6-((3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-3-(tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US11370795B2 (en) | 2017-05-26 | 2022-06-28 | Imara Inc. | Process for the synthesis of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| WO2019101511A1 (en) | 2017-11-23 | 2019-05-31 | Basf Se | Substituted trifluoromethyloxadiazoles for combating phytopathogenic fungi |
| WO2019226944A1 (en) | 2018-05-25 | 2019-11-28 | Imara Inc. | Monohydrate and crystalline forms of 6-[(3s,4s)-4-methyl-1- (pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl- 7h-imid azo [1,5- a] pyrazin-8-one |
| EP4349403A2 (en) | 2018-05-25 | 2024-04-10 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3s,4s)-4-methyl-1- (pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl- 7h-imid azo [1,5- a] pyrazin-8-one |
| EP3801526A4 (en) * | 2018-05-25 | 2022-04-06 | Imara Inc. | CRYSTALLINE AND MONOHYDRATE FORMS OF 6-[(3S,4S)-4-METHYL-1-(PYRIMIDIN-2-YLMETHYL)PYRROLIDIN-3-YL]-3-TETRAHYDROPYRAN-4-YL-7H-IMIDAZO[1,5- A] PYRAZIN-8-ONE |
| EP4349403A3 (en) * | 2018-05-25 | 2024-06-05 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3s,4s)-4-methyl-1- (pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl- 7h-imid azo [1,5- a] pyrazin-8-one |
| US12006319B2 (en) | 2018-05-25 | 2024-06-11 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| US12466832B2 (en) | 2018-05-25 | 2025-11-11 | Cardurion Pharmaceuticals, Inc. | Monohydrate and crystalline forms of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-3-tetrahydropyran-4-yl-7H-imidazo[1,5-a]pyrazin-8-one |
| WO2019243528A1 (en) * | 2018-06-20 | 2019-12-26 | Janssen Pharmaceutica Nv | Oga inhibitor compounds |
| US11512064B2 (en) | 2018-08-31 | 2022-11-29 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| US12213975B2 (en) | 2018-08-31 | 2025-02-04 | Cardurion Pharmaceuticals, Inc. | PDE9 inhibitors for treating sickle cell disease |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2017201873C1 (en) | PDE9i with imidazo pyrazinone backbone | |
| EP3178820B1 (en) | Pde9- inhibitors with imidazo triazinone backbone | |
| TWI743401B (en) | Indole carboxamide compounds | |
| JP7208142B2 (en) | Tyrosinamide derivatives as RHO kinase inhibitors | |
| CN108295073B (en) | Polyfluoro compounds as bruton's tyrosine kinase inhibitors | |
| CN114466849A (en) | N-substituted-3,4-(fused 5-ring)-5-phenyl-pyrrolidin-2-one compounds as inhibitors of ISOQC and/or QC enzymes | |
| CN111051309A (en) | Triazolotriazine derivatives useful as A2A receptor antagonists | |
| CN120676947A (en) | Compounds and compositions useful as MK2 kinase degrading agents | |
| HK1256857B (en) | Polyfluorinated compounds acting as bruton's tyrosine kinase inhibitors | |
| HK40019786A (en) | Triazolotriazine derivatives as a2a receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12772769 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WPC | Withdrawal of priority claims after completion of the technical preparations for international publication |
Ref document number: PCT/CN2011/001692 Country of ref document: CN Date of ref document: 20140404 Free format text: WITHDRAWN AFTER TECHNICAL PREPARATION FINISHED |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 237836 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2015000164 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 37958 Country of ref document: MA |
|
| ENP | Entry into the national phase |
Ref document number: 2886885 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2012323085 Country of ref document: AU Date of ref document: 20121009 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P442/2015 Country of ref document: AE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015000874 Country of ref document: CL Ref document number: 15076140 Country of ref document: CO |
|
| ENP | Entry into the national phase |
Ref document number: 2015535994 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14434308 Country of ref document: US Ref document number: MX/A/2015/004422 Country of ref document: MX |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015007731 Country of ref document: BR |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012772769 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012772769 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 112015007731 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150407 |