WO2013089063A1 - バイサルファイト反応を利用したメチル化シトシンの検出方法 - Google Patents
バイサルファイト反応を利用したメチル化シトシンの検出方法 Download PDFInfo
- Publication number
- WO2013089063A1 WO2013089063A1 PCT/JP2012/081936 JP2012081936W WO2013089063A1 WO 2013089063 A1 WO2013089063 A1 WO 2013089063A1 JP 2012081936 W JP2012081936 W JP 2012081936W WO 2013089063 A1 WO2013089063 A1 WO 2013089063A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbon atoms
- general formula
- dna
- alkyl group
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C1*(*)C(*)C(*)N(*)C1* Chemical compound *C1*(*)C(*)C(*)N(*)C1* 0.000 description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N C(C1)N2CCN1CC2 Chemical compound C(C1)N2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- QXKMWFFBWDHDCB-UHFFFAOYSA-N CC1N(CC2)CCN2C1 Chemical compound CC1N(CC2)CCN2C1 QXKMWFFBWDHDCB-UHFFFAOYSA-N 0.000 description 1
- QLTAFAWFCURPFJ-UHFFFAOYSA-N CC1N(CC2)CCN2C1C Chemical compound CC1N(CC2)CCN2C1C QLTAFAWFCURPFJ-UHFFFAOYSA-N 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
Definitions
- the present invention relates to a method for converting unmethylated cytosine in single-stranded DNA to uracil by a novel bisulfite reaction, a method for amplifying single-stranded DNA in which unmethylated cytosine is converted to uracil, and single-stranded DNA.
- the present invention relates to a method for detecting methylated cytosine.
- methylation of genomic DNA in vivo occurs to suppress mRNA expression. Furthermore, it has been reported that differences in the methylation pattern on the genome are related to diseases such as development, differentiation, and cancer. It plays an important role in research and development of regenerative medicine.
- a method for measuring methylated cytosine in a DNA base sequence a method of comparing fragments by a restriction enzyme sensitive to methylation, a bisulfite method, a methylation-specific PCR method, or high performance liquid chromatography (HPLC) is used. Methods are known. Among them, the bisulfite method is widely used as a general method because it can be applied to high throughput at low cost and is effective for sequencing and screening.
- the present invention is a method for converting unmethylated cytosine in single-stranded DNA into uracil by a new bisulfite reaction that has a higher conversion rate from unmethylated cytosine to uracil as compared to the conventional bisulfite method,
- An object is to provide a method for amplifying single-stranded DNA in which unmethylated cytosine is converted to uracil, and a method for detecting methylated cytosine in single-stranded DNA.
- a method for converting methylated cytosine into uracil (hereinafter, sometimes abbreviated as uracil conversion method of the present invention), (2) A method for amplifying single-stranded DNA in which unmethylated cytosine is converted to uracil after the bisulfite reaction in (1) (hereinafter abbreviated as single-stranded DNA amplification method of the present invention). ), (3) A method for detecting methylated cytosine in the single-stranded DNA, wherein the single-stranded DNA amplified in (2) is subjected to base sequence analysis (hereinafter sometimes abbreviated as the method for detecting methylated cytosine of the present invention). (It may be abbreviated as the method of the present invention in combination with the methods (1) to (3) above).
- unmethylated cytosine in single-stranded DNA can be converted to uracil with high efficiency, and the single-stranded DNA is a relatively long single-stranded DNA of 500 to 1000 bp. Can be converted with high efficiency. Furthermore, since the methylated cytosine is hardly deaminated even in a bisulfite reaction at a high temperature, the reaction time can be shortened without degrading the detection accuracy of the methylated cytosine. .
- the methylated cytosine in the single-stranded DNA was converted into a relatively long single-stranded DNA.
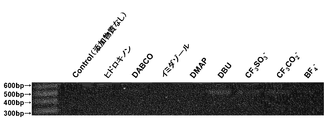
- FIG. 1 shows the results of electrophoresis of the PCR amplification products obtained in Comparative Examples 1-2 and Examples 1-2 using an agarose gel.
- FIG. 2 shows the results of electrophoresis of the PCR amplification products obtained in Comparative Examples 1-2, Example 1, and Examples 3-8 on an agarose gel.
- FIG. 3 shows the results of electrophoresis of the PCR amplification products obtained in Comparative Examples 1-2 and Examples 9-16 using an Agilent 2100 bioanalyzer.
- FIG. 4 shows the results of decoding the base sequences of the PCR amplification products of Example 1 and 2 (method with DBU added).
- FIG. 5 is a diagram showing whether the cytosine of the CpG dinucleotide for the PCR amplification product of Example 1 (method with DBU added) is unmethylated cytosine or methylated cytosine.
- FIG. 6 is a diagram showing whether the cytosine of the CpG dinucleotide for the PCR amplification product of Example 2 (method with DBN added) is unmethylated cytosine or methylated cytosine.
- the alkyl group having 1 to 6 carbon atoms in R 1 to R 6 may be linear, branched or cyclic, but preferably has 1 to 3 carbon atoms.
- R 1 to R 6 are preferably hydrogen atoms.
- N represents an integer of 1 to 3, preferably 1 or 3, and particularly preferably 3.
- Y represents a carbon atom, an oxygen atom or a nitrogen atom, preferably a carbon atom or an oxygen atom, and particularly preferably a carbon atom.
- k is 0, when Y is a nitrogen atom, k is 1, and when Y is a carbon atom, k is 2.
- Examples of the alkyl group having 1 to 6 carbon atoms in R 7 to R 12 include the same as the alkyl group having 1 to 6 carbon atoms in the above R 1 to R 6 , and preferable examples thereof are also the same.
- R 7 to R 11 are preferably hydrogen atoms, but R 12 is preferably an alkyl group having 1 to 6 carbon atoms, and more preferably an alkyl group having 1 to 3 carbon atoms.
- Examples of the compound represented by the general formula [2] include the following formulas [2-1] to [2-19], preferably the formulas [2-1] to [2-4], and the formula [2-1 ] And [2-2] are more preferable.
- the alkyl group having 1 to 6 carbon atoms in R 13 to R 15 may be linear, branched or cyclic, but is preferably linear or branched. Preferred are those having 2 to 6 carbon atoms, more preferred are those having 2 to 4 carbon atoms.
- Compounds represented by the general formula [3] include primary amines such as ethylamine, propylamine, isopropylamine, butylamine, pentylamine, hexylamine, N-ethylmethylamine, N-methylpropylamine, N-butylmethylamine Secondary amines such as N-hexylmethylamine, N-ethylpropylamine, N-butylethylamine, trimethylamine, triethylamine, tripropylamine, triisopropylamine, tributylamine, N, N-dimethylethylamine, N, N-diethyl Examples include tertiary amines such as methylamine and N-ethyldiisopropylamine.
- Tertiary amines are preferable, among which triethylamine, N-ethyldiisopropylamine and triisopropylamine are preferable, triethylamine and N-ethyldiisopropyl. Min is more preferable.
- dialkylamino group having 2 to 6 carbon atoms in R 16 , R 18 and R 20 include alkyl having 1 to 3 carbon atoms such as dimethylamino group, diethylamino group, dipropylamino group and ethylmethylamino group.
- An amino group substituted with a group is exemplified, but an amino group substituted with an alkyl group having 1 to 2 carbon atoms is preferred, and a dimethylamino group is particularly preferred.
- Examples of the compound represented by the general formula [4] include the following formulas [4-1] to [4-14], preferably the formulas [4-1] to [4-4], and the formula [4-1] [4-2] and [4-4] are more preferable.
- R 21 is preferably an alkyl group having 1 to 3 carbon atoms
- R 22 is preferably an alkyl group having 2 to 6 carbon atoms, more preferably an alkyl group having 2 to 3 carbon atoms.
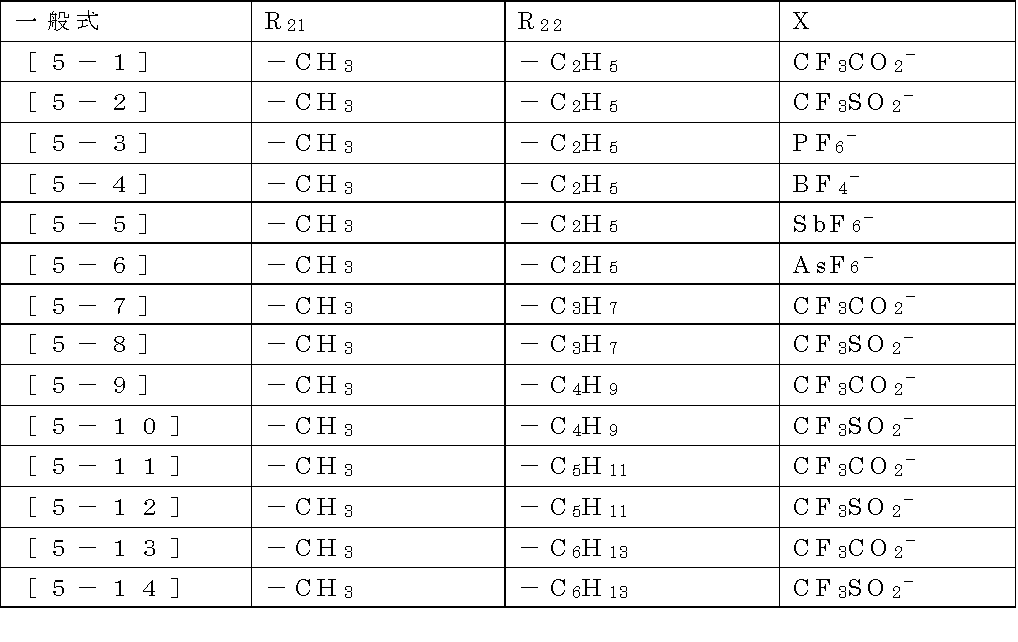
- Examples of the counter anion for X 1 in the general formula [5] include halide ions such as fluorine ion, chlorine ion, bromine ion and iodine ion. More specifically, SbF 6 ⁇ , AsF 6 ⁇ , PF 6 ⁇ , BF 4 ⁇ , CF 3 SO 2 ⁇ , CF 3 CO 2 — and the like can be mentioned. BF 4 ⁇ , CF 3 SO 2 ⁇ , CF 3 CO 2 — and the like are preferable, and CF 3 CO 2 — is particularly preferable. Particularly preferred.
- Specific examples of the compound represented by the general formula [5] include those described in the formulas [5-1] to [5-14] in the following Table 1, and the formulas [5-1] to [5-14]
- the compound described in 5-4] is preferable, and the compound described in Formula [5-1] or [5-2] is more preferable.
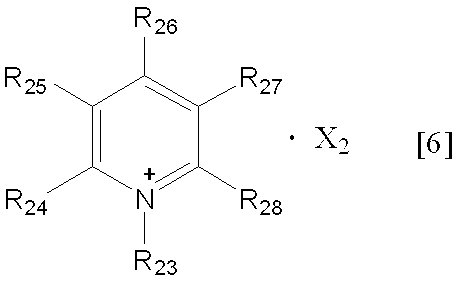
- the alkyl group having 1 to 6 carbon atoms in R 23 to R 28 in the general formula [6] may be linear, branched or cyclic. Or a branched one is preferred, and those having 1 to 4 carbon atoms are preferred.
- Examples include pentyl group, 1,2-dimethylbutyl group, 2,2-dimethylbutyl group, 1-ethylbutyl group, 2-ethylbutyl group, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, is
- R 24 to R 28 in the general formula [6] are preferably hydrogen atoms.
- Examples of X 2 in the general formula [6] include the same as X 1 in the general formula [5], and preferable examples include the same.
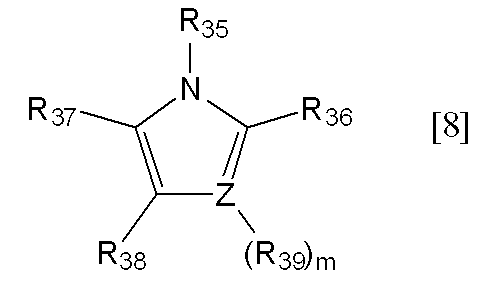
- R 36 to R 39 are preferably hydrogen atoms.
- Z represents a nitrogen atom or a carbon atom, preferably a nitrogen atom.
- m represents 0, and when Z is a carbon atom, m represents 1.
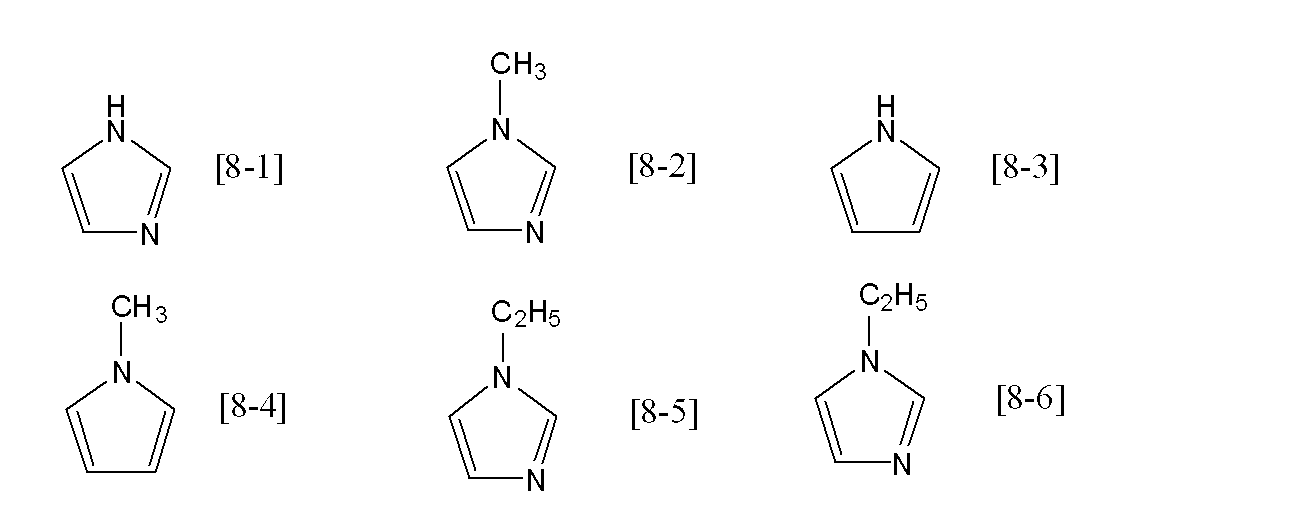
- Specific examples of the compound represented by the general formula [8] include, for example, the following formulas [8-1] to [8-8], preferably the formulas [8-1] to [8-4] [8-1] to [8-2] are more preferable, and the formula [8-1] is still more preferable.
- the single-stranded DNA according to the present invention is preferably a single-stranded DNA containing methylated cytosine, and preferably has a promoter region with a high content of methylated cytosine.
- the above single-stranded DNA may be either a known sequence or an unknown sequence, but in the case of an unknown sequence, it is necessary to perform a base sequence analysis on the single-stranded DNA before being subjected to the bisulfite reaction. For this reason, the known sequence is preferred.
- the number of bases of the single-stranded DNA is usually 80 to 1000 bases, preferably 100 to 600 bases.
- an alkali or an aqueous solution thereof is added to the DNA according to the present invention or the solution containing the DNA according to the present invention to make the solution alkaline with a pH of usually 10 to 14, preferably 12 to 14.
- the alkali include alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, alkaline earth metal hydroxides such as barium hydroxide, magnesium hydroxide and calcium hydroxide, and alkali metal carbonates such as sodium carbonate. Examples thereof include salts, ammonia, and amines.
- alkali metal hydroxides such as sodium hydroxide and potassium hydroxide are preferable, and sodium hydroxide is particularly preferable among these.
- the concentration in the reaction solution is usually An amount of 1 to 10%, preferably 3 to 10%, more preferably 3 to 8% may be used.
- the concentration in the reaction solution is usually 1 to 1000 mmol / L, preferably 1 to 500 mmol, more preferably 10 to 300 mmol.
- Hydrolysis of the sulfonated cytosine during the bisulfite reaction is not particularly limited as long as it is a method usually used in this field, but is usually 30 to 100 ° C., preferably 80 to 95 ° C., usually 60 minutes to This is done by heating for 20 hours, preferably 60 minutes to 5 hours, more preferably 60 to 120 minutes.
- the hydrolysis treatment may proceed simultaneously with the reaction between the DNA and sulfite. In that case, the reaction temperature and reaction time in the reaction between DNA and sulfite may be set in accordance with the hydrolysis conditions of the sulfonated cytosine.
- the hydrolyzed DNA is preferably subjected to purification treatment before desulfonation treatment.
- the purification treatment is performed for the purpose of removing a high concentration of sulfite used in the bisulfite reaction, and may be performed according to a DNA purification method usually used in this field.
- a chaotropic agent such as guanidine hydrochloride or sodium iodide is added to DNA or a solution containing DNA, and then separated and purified by an HPLC method or the like, for example, a phenol / chloroform / isoamyl alcohol mixture.
- HPLC method for example, a phenol / chloroform / isoamyl alcohol mixture.
- Examples include extraction purification using a solution, alcohol precipitation, purification using a column packed with silica gel, filter filtration, and the like, and alcohol precipitation is preferred.
- the alcohol precipitation method is specifically performed as follows.
- DNA can be separated and purified by removing the supernatant and washing with alcohol.
- 0.1 to 1 ⁇ L of ethatin mate or glycogen may be added to 10 ⁇ L of the solution containing DNA.
- the alcohol include ethanol, isopropanol, butanol and the like, and isopropanol is particularly preferable.
- DNA is extracted from cells using a DNA extraction kit or the like, and 1 ⁇ g of DNA is dissolved in, for example, 5 to 15 ⁇ L of sterile water.
- 3 to 5 ⁇ L of the solution is, for example, 50 to 100 ⁇ L of 2 to 5 mol / L sodium bisulfite (pH 5.0 to 7.0) and the compounds represented by the general formulas [1] to [8] are liquid
- the compounds represented by the general formulas [1] to [8] are solid so that the concentration in the reaction solution is 3 to 10%, the concentration in the reaction solution is 1 to 1000 mmol / L.
- 1 to 12 ⁇ L of a compound represented by the general formulas [1] to [8] or an aqueous solution containing them is added and heated at 80 to 100 ° C. for 60 to 120 minutes.
- the double-stranded DNA becomes single-stranded DNA, which simultaneously sulphonates cytosine in the single-stranded DNA and hydrates the sulphonated cytosine. Can be disassembled. Then, 1 mol / L Tris buffer (pH 7.0 to 8.0) and isopropanol are used at a ratio of 40:60 to 60:40, preferably 40:60 to 50:50, 5 to 10 times the hydrolyzed solution, respectively. Add the amount to precipitate the hydrolyzed DNA. At this time, if 1 to 3 ⁇ L of ethatin mate or glycogen is added, DNA precipitation can be easily confirmed.
- the single-stranded DNA amplification method of the present invention is performed by subjecting a uracilized DNA (bisulfite reaction product) obtained by the above-described uracil conversion method of the present invention to a PCR reaction, whereby unmethylated cytosine is converted into uracil. Single-stranded DNA converted to is amplified.
- the PCR reaction in the single-stranded DNA amplification method of the present invention is carried out according to a method known per se, for example, the method described in Nucleic® Acids® Research, 1991, Vol. 19, 3749, BioTechniques, 1994, Vol. 16, 1134-1137. Specifically, it is performed as follows.
- the DNA obtained by a purification method usually used in this field for example, extraction with a mixed solution of phenol / chloroform / isoamyl alcohol, alcohol precipitation, column purification, filter filtration, etc. is purified. It is preferable to do this. It is more preferable to extract DNA having the desired base pair (bp) after the purification.
- the extraction method include methods known per se, such as a method using agarose gel electrophoresis, a method using liquid chromatography, and electrophoresis such as a polyacrylamide gel described in the supplemented lab manual Genetic Engineering, 1990, 27-28. And the like.
- the double-stranded DNA according to the present invention obtained by the PCR reaction as described above may be further subjected to a PCR reaction in order to obtain more target DNA.
- the dNTPs are not particularly limited as long as they are a mixture of four types of deoxyribonucleotide triphosphates (dATP, dCTP, dGTP, dTTP) usually used in this field.
- DNA amplification method of the present invention Preferred examples of the DNA amplification method of the present invention will be described below. That is, first, as described in the section of the uracil conversion method of the present invention, single-stranded DNA is subjected to a bisulfite reaction to obtain uracilated DNA. Thereafter, the obtained uracilized DNA is subjected to a PCR reaction.
- uracilized DNA is amplified.
- the double-stranded DNA is subjected to electrophoresis using, for example, an agarose gel or a non-denaturing polyacrylamide gel, and DNA having a target chain length is extracted. If necessary, the obtained DNA may be purified by extraction with a mixed solution of phenol / chloroform / isoamyl alcohol, for example.
- the methylated cytosine detection method of the present invention is performed by subjecting the amplified uracilized DNA (PCR reaction product) obtained by the single-stranded DNA amplification method of the present invention to base sequence analysis.
- the base sequence analysis is not particularly limited as long as it is a base sequence analysis method usually used in this field.
- uracilized DNA obtained by the amplification method of the present invention is incorporated into a vector, the obtained recombinant vector is transformed into a competent cell, the competent cell is cultured, and uracil is obtained therefrom. This is done by extracting a plasmid containing the modified DNA and decoding it with, for example, a sequencer.
- Methylated cytosine can be detected by comparing the base sequence obtained in this way with the base sequence of normal DNA not subjected to bisulfite reaction. That is, in the bisulfite reaction according to the present invention, all cytosines other than methylated cytosine (unmethylated cytosine) are uracilated, and thus methylated cytosine is detected by finding cytosine that is not uracilized in the obtained base sequence. Can be detected.
- uracilized DNA For uracilized DNA to which adenine has been added, it is preferable to purify the DNA obtained by a method such as extraction with a phenol / chloroform / isoamyl alcohol mixed solution, alcohol precipitation, column purification, filter filtration and the like after the synthesis reaction. .
- E. coli transformation vector to which thymine base is added and T4 DNA ligase 300 to 3000sUnits are added to 10 to 100 ng of uracilized DNA to which adenine has been added and 30 to 90 minutes at 10 to 40 ° C. By reacting, a recombinant vector incorporating uracilized DNA can be obtained.
- Examples of methods for transforming the above recombinant vector into competent cells include, for example, a heat shock method of heating at 35 to 45 ° C. for 20 to 90 seconds, an electroporation method of applying an electric pulse of 1.5 to 2.5 kV, and the like.
- a heat shock method of heating at 35 to 45 ° C. for 20 to 90 seconds an electroporation method of applying an electric pulse of 1.5 to 2.5 kV, and the like.
- the competent cell used here any commonly used Escherichia coli, Bacillus subtilis, and the like can be used, and the amount used can be appropriately set within the range usually used.
- the competent cells are cultured, for example, by culturing at 30 to 40 ° C. for 12 to 20 hours on a medium such as LB agar medium containing 30 to 150 ⁇ g / ml ampicillin or M9 agar medium containing 30 to 150 ⁇ g / ml ampicillin.
- a medium such as LB agar medium containing 30 to 150 ⁇ g / ml ampicillin or M9 agar medium containing 30 to 150 ⁇ g / ml ampicillin.
- the above medium contains a carbon source, which is a nutrient source for microorganisms, a nitrogen source, an inorganic salt, a yeast extract as a growth factor, and the like, so long as the transformant can be cultured efficiently. Any of medium, synthetic medium, and the like may be used.
- inorganic salts include monopotassium phosphate, dipotassium phosphate, magnesium phosphate, magnesium sulfate, sodium chloride, ferrous sulfate, manganese sulfate, copper sulfate, and calcium carbonate.
- a method for extracting a plasmid containing uracilized DNA from cultured competent cells for example, first, DNA derived from a plasmid containing uracilized DNA in a colony is amplified by colony PCR. Thereafter, whether the target plasmid is amplified in the colony may be confirmed by, for example, electrophoresis, and the target plasmid may be extracted from the colony where the insertion of the target plasmid is confirmed.
- those designed to amplify the target DNA that is, those containing all or part of the uracilized DNA, or vectors located at both ends of the inserted uracilized DNA
- examples include sequences derived from vectors, and vectors derived from vectors located at both ends of uracilized DNA are preferred. That is, in the DNA amplification method of the present invention and the methylated cytosine detection method of the present invention, unmethylated cytosine is uracilized, and the uracil is read as thymine during the PCR reaction, so that all cytosine is unmethylated cytosine. If present, the uracilized DNA will be composed of 3 bases.
- a sequence derived from a vector that can be composed of 4 bases and is located at both ends of uracilized DNA is preferable.
- the number of nucleotides in the primer is usually 12-30, preferably 15-25, more preferably 18-22.
- the DNA polymerase may be any DNA polymerase that is usually used in this field, and specific examples include Taq DNA polymerase, Tth DNA polymerase, KOD DNA polymerase, etc. Among them, Taq DNA polymerase, KOD, among others. A DNA polymerase is preferred.
- single-stranded DNA is subjected to bisulfite reaction and PCR reaction in this order to obtain amplified uracilated DNA.
- 1 to 5 ⁇ L of sterilized water containing 10 to 100 ng of the uracilized DNA and 1 to 3 ⁇ L of 10 to 100 ng of E. coli transformation vector to which thymine base has been added and 1 to 3 ⁇ L of T4 DNA ligase of 300 to 3000 Units Apply and react at 10-20 ° C. for 30-240 minutes to obtain a recombinant vector incorporating the uracilated DNA.
- adenine is added to uracilized DNA before incorporation into a vector.
- adenine addition method for example, 0.5 to 1 ⁇ L of 1 to 5 Units Taq DNA polymerase is added to 5 to 10 ⁇ L of a PCR reaction solution containing 100 ng to 1 ⁇ g of uracilized DNA, and reacted at 55 to 75 ° C. for 10 to 30 minutes. What is necessary is just to add adenine to the 3 'end of uracilized DNA. Incidentally, after the adenine reaction, it is preferable to carry out a purification operation.
- 10 to 100 ng of the obtained recombinant vector is added to competent cells 10 8 to 10 9 cells, and transformation is performed by heating at 35 to 45 ° C. for 20 to 90 seconds. Further, for example, 30-40 ° C. on an agar medium containing 30-150 ⁇ g / ml ampicillin, 1% (w / v) tryptone, 0.5% (w / v) yeast extract, and 1% (w / v) sodium chloride. Incubate for 12-20 hours. The resulting culture is then subjected to colony PCR.
- dNTPs mixed deoxyribonucleotide triphosphates
- the reaction is performed by performing 30 to 40 cycles with one cycle of ⁇ 5 minutes. Thereafter, the presence of the target DNA in the colonies is confirmed by agarose gel electrophoresis, and the confirmed colonies are collected. The collected colonies are cultured with shaking in LB medium, and then the target DNA is removed from the culture solution using, for example, a commercially available plasmid extraction kit, and the base sequence of the DNA is decoded using a sequencer or the like. It is possible to detect methylated cytosine by comparing the obtained base sequence with a normal base sequence not subjected to bisulfite reaction and searching for unuracilized cytosine in the decoded base sequence of DNA. it can.
- Examples 1 to 16 and Comparative Examples 1 and 2 PCR amplification of DNA obtained by the bisulfite reaction of the uracil conversion method of the present invention and the conventional method (1) Extraction of mouse genomic DNA 1 ⁇ 10 6 mouse embryonic stem cells ( Genomic DNA was extracted from embryonic stem cells (ES cells) using QuickGene SP kit DNA tissue (Fuji Film Co., Ltd.) according to the instruction manual.
- the obtained PCR amplification product was fractionated by electrophoresis using a 1.5% agarose gel (Examples 1 to 8) or Agilent 2100 bioanalyzer (manufactured by Agilent) (Examples 9 to 16). Was confirmed to be amplified.
- the results are shown in Figs.
- the confirmation results are shown in Table 4.
- the obtained amplified product was divided into five stages based on the density of the band (amplified product amount), and the results are shown in Table 4.
- ⁇ visible band
- ⁇ weakly weak band
- ⁇ weak band
- ⁇ very weak band
- x indicates no band (amplification not seen) ).
- Example 17 Cloning and Base Sequence Analysis of PCR Amplified Product Cloning and base sequence analysis were performed on the PCR amplified product obtained in Example 1 (DBU added) and the PCR amplified product obtained in Example 2 (DBN added).
- each obtained colony PCR amplification product was fractionated by electrophoresis using a 1.5% agarose gel to confirm whether the colony had the vector inserted therein.
- the cells were cultured with shaking in LB medium at 37 ° C. overnight. Thereafter, plasmid extraction was performed using QuickGene® Plasmid® kit® SII (manufactured by FUJIFILM Corporation) using the obtained culture solution.
- a portion surrounded by a square represents a CpG dinucleotide
- an underline represents a primer portion.
- “ ⁇ ” indicates that the cytosine of the CpG dinucleotide is an unmethylated cytosine
- “ ⁇ ” indicates that the cytosine of the CpG dinucleotide is methylated cytosine.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Physics & Mathematics (AREA)
- Genetics & Genomics (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
一方、DNA塩基配列中のメチル化シトシンを測定する方法としては、メチル化感受性の制限酵素による切断片を比較する方法、バイサルファイト法、methylation-specific PCR法、高速液体クロマトグラフィー(HPLC)を用いる方法等が知られている。その中でもバイサルファイト法は、低コストで且つハイスループットへの応用もでき、シークエンシングやスクリーニングに有効であるため、一般的な方法として普及してきている。
本発明は、従来のバイサルファイト法と比較して、非メチル化シトシンからウラシルへの変換率が高い新たなバイサルファイト反応による、一本鎖DNA中の非メチル化シトシンをウラシルに変換する方法、非メチル化シトシンがウラシルに変換された一本鎖DNAの増幅方法、一本鎖DNAのメチル化シトシンの検出方法の提供を課題とする。
下記一般式[1]で示される化合物

下記一般式[2]で示される化合物

下記一般式[3]で示される化合物

下記一般式[4]で示される化合物

下記一般式[5]で示される化合物

下記一般式[6]で示される化合物
下記化合物一般式[7]で示される化合物

下記一般式[8]で示される化合物
即ち、本発明は、
(1)一本鎖DNAを、上記一般式[1]~[8]で示される化合物の少なくとも1種の存在下でバイサルファイト反応に付すことを特徴とする、前記一本鎖DNA中の非メチル化シトシンをウラシルに変換する方法(以下、本発明のウラシル変換方法と略記する場合がある)、
(2)(1)のバイサルファイト反応の後、更にPCR反応に付す、非メチル化シトシンがウラシルに変換された一本鎖DNAの増幅方法(以下、本発明の一本鎖DNA増幅方法と略記する場合がある)、
(3)(2)で増幅された一本鎖DNAを塩基配列解析に付す、前記一本鎖DNA中のメチル化シトシンの検出方法(以下、本発明のメチル化シトシン検出方法と略記する場合がある)(上記(1)~(3)の方法を合わせて本発明の方法と略記する場合がある)に関する。
R1~R6における炭素数1~6のアルキル基としては、直鎖状、分枝状或いは環状の何れでもよいが、炭素数1~3のものが好ましく、具体的には、例えばメチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、sec-ペンチル基、tert-ペンチル基、ネオペンチル基、2-メチルブチル基、1-エチルプロピル基、n-ヘキシル基、イソヘキシル基、sec-ヘキシル基、tert-ヘキシル基、ネオヘキシル基、2-メチルペンチル基、3-メチルペンチル基、1,2-ジメチルブチル基、2,2-ジメチルブチル基、1-エチルブチル基、2-エチルブチル基等が挙げられ、メチル基、エチル基、n-プロピル基、イソプロピル基が好ましく、メチル基、エチル基がより好ましく、メチル基が特に好ましい。

Yは、炭素原子、酸素原子又は窒素原子を表すが、炭素原子、酸素原子が好ましく、炭素原子が特に好ましい。尚、Yが酸素原子の場合kは0を、Yが窒素原子の場合kは1を、Yが炭素原子の場合kは2を表す



R13~R15における炭素数1~6のアルキル基としては、直鎖状、分枝状或いは環状の何れでもよいが、直鎖状又は分枝状が好ましい。炭素数2~6のものが好ましく、炭素数2~4のものがより好ましく、具体的には、例えばエチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、sec-ペンチル基、tert-ペンチル基、ネオペンチル基、2-メチルブチル基、1-エチルプロピル基、n-ヘキシル基、イソヘキシル基、sec-ヘキシル基、tert-ヘキシル基、ネオヘキシル基、2-メチルペンチル基、3-メチルペンチル基、1,2-ジメチルブチル基、2,2-ジメチルブチル基、1-エチルブチル基、2-エチルブチル基等が挙げられ、エチル基、n-プロピル基、イソプロピル基、n-ブチル基が好ましく、エチル基、n-プロピル基、イソプロピル基がより好ましく、エチル基、イソプロピル基が特に好ましい。
R16~R20における炭素数1~6のアルキル基としては、上記R1~R6における炭素数1~6のアルキル基と同じものが挙げられ、好ましいものも同じものが挙げられる。


一般式[5]中のR21又はR22における炭素数1~6のアルキル基としては、上記R1~R6における炭素数1~6のアルキル基と同じものが挙げられ、好ましいものも同じものが挙げられる。
一般式[6]中のR23~R28における炭素数1~6のアルキル基としては、直鎖状、分枝状或いは環状の何れでもよいが、直鎖状又は分枝状が好ましく、炭素数1~4のものが好ましい。具体的には、例えばメチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、sec-ペンチル基、tert-ペンチル基、ネオペンチル基、2-メチルブチル基、1-エチルプロピル基、n-ヘキシル基、イソヘキシル基、sec-ヘキシル基、tert-ヘキシル基、ネオヘキシル基、2-メチルペンチル基、3-メチルペンチル基、1,2-ジメチルブチル基、2,2-ジメチルブチル基、1-エチルブチル基、2-エチルブチル基等が挙げられ、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基が好ましく、n-プロピル基、n-ブチル基がより好ましい。

R29~R34における炭素数1~6のアルキル基としては、R1~R6における炭素数1~6のアルキル基と同じものが挙げられ、好ましいものも同じものが挙げられる。

R35~R39における炭素数1~6のアルキル基としては、R1~R6における炭素数1~6のアルキル基と同じものが挙げられ、好ましいものも同じものが挙げられる。

本発明のウラシル変換方法としては、一本鎖DNAを、上記一般式[1]~[8]で示される化合物の少なくとも1種の存在下でバイサルファイト反応に付すことによりなされればよい。
即ち、加水分解処理後の溶液又は加水分解処理後精製処理に付した溶液10μLに対して、0.5~3mol/Lのアルカリ水溶液を通常1~10μL、好ましくは1~5μL添加し、通常5~60分間、好ましくは5~30分間、通常25~70℃、好ましくは30~50℃で加温することにより脱スルホン酸化反応を行う。
即ち、例えば、DNA抽出キット等を用いて細胞等からDNAを抽出し、DNA 1μgを例えば滅菌水5~15μLに溶解する。該溶液3~5μLに、例えば2~5mol/Lの亜硫酸水素ナトリウム(pH5.0~7.0)50~100μL、及び、一般式[1]~[8]で示される化合物が液体の場合には、反応溶液中の濃度が3~10%となるように、一般式[1]~[8]で示される化合物が固体の場合には、反応溶液中の濃度が1~1000mmol/Lとなるように、一般式[1]~[8]で示される化合物又はそれらを含有する水溶液1~12μLを添加して、80~100℃で60~120分間加熱する。DNAが二本鎖の場合には、該条件で反応させることにより、二本鎖DNAは一本鎖DNAになり、該一本鎖DNA中のシトシンをスルホン酸化すると同時にスルホン酸化されたシトシンを加水分解することができる。次いで、1mol/L トリス緩衝液(pH7.0~8.0)及びイソプロパノールを、40:60~60:40、好ましくは40:60~50:50の割合でそれぞれ加水分解後の溶液の5~10倍量添加し、加水分解後のDNAを沈殿させる。この際、エタチンメイトやグリコーゲンを1~3μL添加するとDNA沈殿の確認が容易となる。その後、12,000~20,000gで10~20分間遠心分離し、上清を取り除き、得られたDNAをエタノールで洗浄する。これにより、加水分解後のDNAを抽出精製することができる。更に、得られたDNA 1μgを例えば滅菌水30~40μLに溶解し、該溶液に、1~3mol/Lの水酸化ナトリウム5~20μLを添加し、30~40℃で20~60分間反応させ、脱スルホン酸化する。その後、要すれば、例えば市販のキット等を用いて低分子量のDNAを取り除き精製する。これにより、本発明に係るバイサルファイト反応が完了し、一本鎖DNA中の非メチル化シトシンが効率よくウラシルに変換されたDNA(以下、ウラシル化DNAと略記する場合がある)が得られる。
本発明の一本鎖DNA増幅方法は、上記本発明のウラシル変換方法で得られたウラシル化DNA(バイサルファイト反応産物)をPCR反応に付すことによりなされ、該方法により、非メチル化シトシンがウラシルに変換された一本鎖DNAが増幅される。
即ち、まず、本発明のウラシル変換方法の項で記載したように、一本鎖DNAをバイサルファイト反応に付して、ウラシル化DNAを得る。その後、得られたウラシル化DNAをPCR反応に付す。即ち、バイサルファイト反応により得られた一本鎖DNA 1~100ngを含む溶液1~3μLに、1~10μmol/Lの増幅対象となるDNAの上流用のプライマー5~10μLと1~10μmol/Lの増幅対象となるDNAの下流用プライマー5~10μL、1~5mmol/Lの4種類のデオキシリボヌクレオチド三リン酸(dNTPs)の混合溶液5~10μL、並びに、1~5UnitsのKAPA DNAポリメラーゼ 10~20μLを添加して、例えば93~98℃、1~10分→93~98℃、10~30秒→50~60℃、10~30秒→68~72℃、68~72℃、30秒~5分間を1サイクルとして20~40サイクル行う。これにより、ウラシル化DNAが増幅される。なお、後述のウラシル化DNAをベクターに組み込む方法としてTAクローニング法を用いるために、DNAポリメラーゼの3’末端のアデニン付加活性を利用する場合、上記サイクル反応後、更に68~72℃、30秒~5分間反応させてもよい。次いで、該二本鎖DNAを例えばアガロースゲル又は未変性ポリアクリルアミドゲルを用いて電気泳動を行い、目的の鎖長のDNAを抽出する。尚、必要に応じて、得られたDNAを、例えばフェノール/クロロホルム/イソアミルアルコール混合溶液による抽出等で精製してもよい。以上の操作により、ウラシル化DNAを効率よく増幅することができる。
本発明のメチル化シトシン検出方法は、本発明の一本鎖DNA増幅方法で得られた、増幅されたウラシル化DNA(PCR反応産物)を塩基配列解析に付すことによりなされる。
(1)マウスゲノムDNAの抽出
1×106のマウス胚性幹細胞 (embryonic stem cell : ES細胞)よりQuickGene SP kit DNA tissue(富士フィルム(株)製)を使用して、現品説明書に従ってゲノムDNAを抽出した。
上記(1)で得られたゲノムDNA(360ng)を4μLの滅菌水に溶解した。該溶液を17個準備し、それぞれに、3.5mol/L亜硫酸水素ナトリウム水溶液(pH5.2)(和光純薬工業(株)製)90μL、及び表3中の下記化合物を6μL又は表3記載の濃度含有する水溶液6μLを加えて混合した(全量100μL)。次いで、95℃で、2時間インキュベートした。その後、1mol/Lトリス-塩酸緩衝液(pH7.0)((株)ニッポンジーン製)を400μL、グリコーゲン(和光純薬工業(株)製)を1μL、及びイソプロパノール(和光純薬工業(株)製)500μLを加えて混合し、室温、18800×g、5分間遠心分離した。上清を取り除いた後、75%エタノール(和光純薬工業(株)製)で洗浄し、それぞれ40μLの滅菌水に溶解した。

(2)で得られたバイサルファイト反応産物(ウラシル化DNA)を含む溶液1μLをそれぞれPCRチューブに添加し、更に、5×KAPA2GバッファーA(KAPA BIOSYSTEMS社製) 10μL、10mmol/L dNTPs 混合溶液(東洋紡(株)製) 1μL、滅菌水 31.5μL、並びに、5μmol/LのFgf4遺伝子用のPCRプライマーForward溶液及びReverse溶液 各3μL[Forward:5’GGTTGGGGTTTTTTTAGGTGATAGTAG3’(GenBank Accession No.AC149593 : 230224-230250に由来、塩基配列1)、Reverse: 5’ CCTTTTAAAACCCAACAAATAATCCCCTAC3’ 、GenBank Accession No.AC149593 : 230715-230744に由来、塩基配列2)]をそれぞれに添加し、氷上で緩やかに混合した。その後、94℃で30秒加熱した後、94℃で20秒→58℃で20秒→72℃で30秒を1サイクルとして35サイクル行い、最後に72℃で1分間加熱してPCR反応を行った。その後、得られたPCR増幅産物を1.5%のアガロースゲルを用いた電気泳動(実施例1~8)又はAgilent 2100 bioanalyzer(アジレント社製) (実施例9~16)で分画し、目的のDNAが増幅されているかを確認した。

一方、上記実施例中の各種化合物を添加してバイサルファイト反応を行った場合には、何れも目的とする鎖長に増幅産物を確認することができた。特に、DBU及びDBNを添加した場合には、多量の増幅産物が確認でき、ピペリジン及び1-メチルピペリジンも明確に増幅産物の確認ができた。その他の実施例中の化合物を添加した場合については、薄くはあるが増幅産物の確認はできた。このことから上記実施例中の化合物を添加したバイサルファイト反応においては、バイサルファイト反応の反応促進効果及びゲノムDNAの分解抑制効果があると予想された。
実施例1で得たPCR増幅産物(DBU添加)と実施例2で得たPCR増幅産物(DBN添加)について、クローニング及び塩基配列解析を行った。
ES細胞では、幹細胞未分化マーカーであるFgf4のプロモーター領域のDNAはメチル化されていないため、該DNAを本発明のウラシル化反応に付し、これらのCpGジヌクレオチドが全て非メチル化であれば、非メチル化シトシンがウラシルへ高い効率で変換されていることが証明できる。図5及び6の結果より、ほぼすべてのCpGジヌクレオチドのシトシン及びそれ以外のシトシンがウラシルに変換されていることが判った。即ち、本発明の方法によれば、非メチル化シトシンを高い効率でウラシルに変換できることが判った。
図5及び図6中の○は、CpGジヌクレオチドのシトシンが非メチル化シトシンのものであることを、●は、CpGジヌクレオチドのシトシンがメチル化シトシンされているものであることを表す。
Claims (13)
- 一本鎖DNAを、下記一般式[1]~[8]で示される化合物の少なくとも1種の存在下でバイサルファイト反応に付すことを特徴とする、前記一本鎖DNA中の非メチル化シトシンをウラシルに変換する方法;
下記一般式[1]で示される化合物
(式中、R1~R6は、それぞれ独立して、水素又は炭素数1~6のアルキル基を表し、nは1~3の整数を表す)、
下記一般式[2]で示される化合物
(式中、Yは、炭素原子、酸素原子又は窒素原子を表し、R7~R12は、それぞれ独立して水素原子、又は炭素数1~6のアルキル基を表し、kは0~2の整数を表すが、Yが酸素原子の場合は0を、Yが窒素原子の場合は1を、Yが炭素原子の場合は2を表す。)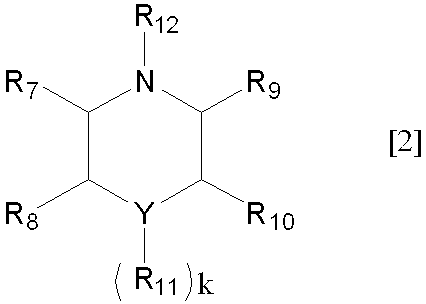
下記一般式[3]で示される化合物
(式中、R13~R15は、それぞれ独立して水素原子、又は炭素数1~6のアルキル基を表す)、
下記一般式[4]で示される化合物
(式中、R17及びR19は、それぞれ独立して水素原子、アミノ基又は炭素数1~6のアルキル基を表し、R16、R18及びR20は、それぞれ独立して水素原子、アミノ基、炭素数1~6のアルキル基又は炭素数2~6のジアルキルアミノ基を表し、R16とR17は、R16の隣りの炭素原子、R17の隣りの炭素原子と共にベンゼン環を形成してもよい。)、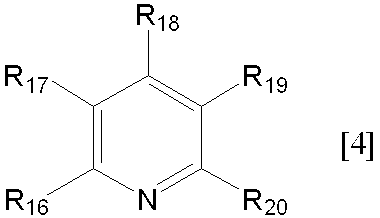
下記一般式[5]で示される化合物
(式中、R21及びR22はそれぞれ独立して炭素数1~6のアルキル基を表し、X1はカウンターアニオンを表す)、
下記一般式[6]で示される化合物
(式中、R23は炭素数1~6のアルキル基を表し、R24~R28は、それぞれ独立して水素原子、又は炭素数1~6のアルキル基を表し、X2はカウンターアニオンを表す)、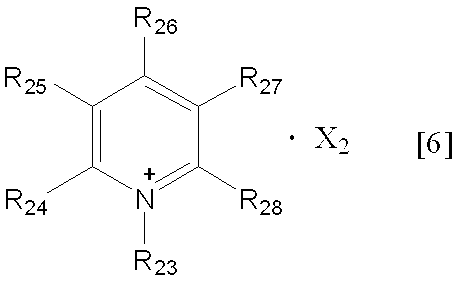
下記化合物一般式[7]で示される化合物
(式中、R29~R34は、それぞれ独立して水素原子、又は炭素数1~6のアルキル基を表す)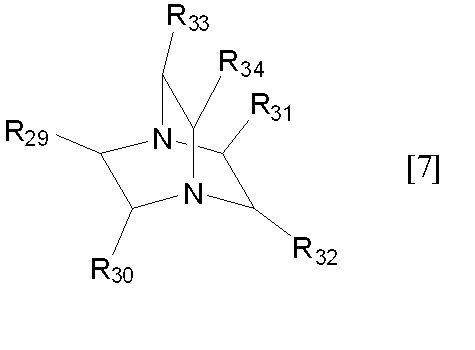
下記一般式[8]で示される化合物
[式中、Zは窒素原子又は炭素原子を表し、mは0又は1の整数を表し、Zが窒素原子の場合は0を表し、Zが炭素原子の場合は1を表す。R35~R39はそれぞれ独立して水素又は炭素数1~6のアルキル基を表す]。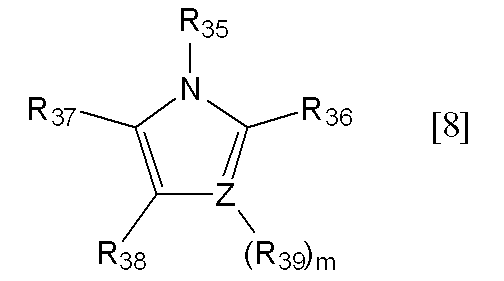
- 一般式[1]におけるR1~R6が全て水素原子である、請求項1記載の方法。
- 一般式[2]におけるYが炭素原子又は酸素原子であり、R7~R11が全て水素原子であり、R12は、炭素数1~3のアルキル基であり、Kは0又は1の整数である、請求項1記載の方法。
- 一般式[3]におけるR13~R15が炭素数1~3のアルキル基である、請求項1記載の方法。
- 一般式[4]におけるR17及びR19が水素原子であって、R16、R18及びR20の少なくとも1つが炭素数1~3のアルキル基又は炭素数1~3のジメチルアミノ基である、請求項1記載の方法。
- 一般式[5]におけるR21及びR22が炭素数1~3のアルキル基であり、Xがトリフルオロメタンスルホン酸イオン又はトリフルオロメタンカルボン酸イオンである、請求項1記載の方法。
- 一般式[6]におけるR24~R28が全て水素原子であり、R23が炭素数1~4のアルキル基であり、Xがトリフルオロメタンスルホン酸イオン又はトリフルオロメタンカルボン酸イオンである、請求項1記載の方法。
- 一般式[7]におけるR29~R32が全て水素原子である、請求項1記載の方法。
- 一般式[8]におけるR36~R39が全て水素原子であり、R35が水素原子又は炭素数1~3のアルキル基であり、Zが炭素原子又は窒素原子である、請求項1記載の方法。
- 一本鎖DNAを、前記一般式[1]~[3]で示される化合物の少なくとも1種の存在下でバイサルファイト反応に付す、請求項1記載の方法。
- 一本鎖DNAを、前記一般式[1]で示される化合物の少なくとも1種の存在下でバイサルファイト反応に付す、請求項1記載の方法。
- 請求項1のバイサルファイト反応の後、バイサルファイト反応産物をPCR反応に付す、非メチル化シトシンがウラシルに変換された一本鎖DNAの増幅方法。
- 更に、請求項12で得られた、増幅された一本鎖DNAを塩基配列解析に付す、前記一本鎖DNA中のメチル化シトシンの検出方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013549253A JP6131857B2 (ja) | 2011-12-14 | 2012-12-10 | バイサルファイト反応を利用したメチル化シトシンの検出方法 |
| US14/364,545 US9624535B2 (en) | 2011-12-14 | 2012-12-10 | Method for detecting methylated cytosine by using bisulfite reaction |
| EP12858295.4A EP2799541B1 (en) | 2011-12-14 | 2012-12-10 | Method for detecting methylated cytosine by using bisulfite reaction |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-272868 | 2011-12-14 | ||
| JP2011272868 | 2011-12-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013089063A1 true WO2013089063A1 (ja) | 2013-06-20 |
Family
ID=48612510
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/081936 Ceased WO2013089063A1 (ja) | 2011-12-14 | 2012-12-10 | バイサルファイト反応を利用したメチル化シトシンの検出方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9624535B2 (ja) |
| EP (1) | EP2799541B1 (ja) |
| JP (1) | JP6131857B2 (ja) |
| WO (1) | WO2013089063A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014103980A1 (ja) | 2012-12-28 | 2014-07-03 | 和光純薬工業株式会社 | Dna中のヒドロキシメチル化シトシンの検出方法及び検出用試薬キット |
| WO2017142070A1 (ja) * | 2016-02-17 | 2017-08-24 | 和光純薬工業株式会社 | Foxb2遺伝子を用いた癌細胞の判定方法 |
| KR20190044651A (ko) | 2016-09-02 | 2019-04-30 | 후지필름 와코 준야쿠 가부시키가이샤 | 메틸화된 dna의 증폭 방법, dna의 메틸화 판정 방법 및 암의 판정 방법 |
| WO2019207921A1 (ja) * | 2018-04-26 | 2019-10-31 | MATSUHISA Munehide | 組織特異的な細胞傷害の検査方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7371526B2 (en) | 2003-08-29 | 2008-05-13 | Applera Corporation | Method and materials for bisulfite conversion of cytosine to uracil |
| WO2006034264A2 (en) * | 2004-09-21 | 2006-03-30 | Applera Corporation | Methods of using sulfur nucleophiles as improved alternatives to sodium bisulfite for methylated dna analysis |
-
2012
- 2012-12-10 US US14/364,545 patent/US9624535B2/en active Active
- 2012-12-10 JP JP2013549253A patent/JP6131857B2/ja active Active
- 2012-12-10 WO PCT/JP2012/081936 patent/WO2013089063A1/ja not_active Ceased
- 2012-12-10 EP EP12858295.4A patent/EP2799541B1/en active Active
Non-Patent Citations (5)
| Title |
|---|
| "EpiSight Bisulfite Conversion Kit/ EpiSight BisulTaq DNA Polymerase.", WAKO PURE CHEMICAL INDUSTRIES, LTD., January 2012 (2012-01-01), pages 2 - 3, XP055155958 * |
| "EpiTect(R) Bisulfite Handbook.", SAMPLE & ASSAY TECHNOLOGIES, QIAGEN, September 2009 (2009-09-01), pages 1 - 48, XP055155957 * |
| FUMIHITO MIURA ET AL.: "Epigenetics Kaisekiho no Shin Shuho 1. Genome Morateki Methyl-ka Kaiseki", EXPERIMENTAL MEDICINE, vol. 28, no. 15, 10 September 2010 (2010-09-10), pages 2407 - 2414, XP008174394 * |
| RAIZIS, A.M. ET AL.: "A bisulfite method of 5-methylcytosine mapping that minimizes template degradation.", ANAL. BIOCHEM., vol. 226, no. 1, 20 May 1995 (1995-05-20), pages 161 - 166, XP002241348 * |
| See also references of EP2799541A4 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014103980A1 (ja) | 2012-12-28 | 2014-07-03 | 和光純薬工業株式会社 | Dna中のヒドロキシメチル化シトシンの検出方法及び検出用試薬キット |
| WO2017142070A1 (ja) * | 2016-02-17 | 2017-08-24 | 和光純薬工業株式会社 | Foxb2遺伝子を用いた癌細胞の判定方法 |
| CN108699606A (zh) * | 2016-02-17 | 2018-10-23 | 富士胶片和光纯药株式会社 | 使用foxb2基因的癌细胞的判定方法 |
| KR20190044651A (ko) | 2016-09-02 | 2019-04-30 | 후지필름 와코 준야쿠 가부시키가이샤 | 메틸화된 dna의 증폭 방법, dna의 메틸화 판정 방법 및 암의 판정 방법 |
| WO2019207921A1 (ja) * | 2018-04-26 | 2019-10-31 | MATSUHISA Munehide | 組織特異的な細胞傷害の検査方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2013089063A1 (ja) | 2015-04-27 |
| US20150176069A1 (en) | 2015-06-25 |
| EP2799541A4 (en) | 2015-07-29 |
| US9624535B2 (en) | 2017-04-18 |
| EP2799541B1 (en) | 2017-05-10 |
| EP2799541A1 (en) | 2014-11-05 |
| JP6131857B2 (ja) | 2017-05-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2024010993A1 (en) | Primer design for cell-free dna production | |
| EA006066B1 (ru) | Способы амплификации нуклеиновых кислот | |
| EP1644519A4 (en) | PROCESS FOR THE DETECTION OF ALKYLATED CYTOSIN IN DNA | |
| US7368239B2 (en) | Method and materials for polyamine catalyzed bisulfite conversion of cytosine to uracil | |
| JP6131857B2 (ja) | バイサルファイト反応を利用したメチル化シトシンの検出方法 | |
| EP1876246A1 (en) | Self-complementary primers used in LAMP gene amplification method | |
| US7371526B2 (en) | Method and materials for bisulfite conversion of cytosine to uracil | |
| JP4891764B2 (ja) | Dna中におけるアルキル化シトシンの検出方法 | |
| EP2698437B1 (en) | Method for detecting 5-hydroxymethylcytosine in nucleic acids | |
| CN114134205A (zh) | 脱氨酶介导的dna中n4-甲基胞嘧啶的单碱基分辨率定位分析方法 | |
| JP2023514422A (ja) | 一本鎖dnaポリヌクレオチドを生成するための方法および生成物 | |
| US9624530B2 (en) | Methods for preservation of genomic DNA sequence complexity | |
| CN114302965A (zh) | 引物和使用了该引物的双链dna的制造装置以及双链dna的制造方法 | |
| CN115961001A (zh) | Dna甲基转移酶结合胞嘧啶脱氨酶介导的dna中5-甲基胞嘧啶的单碱基定位分析方法 | |
| WO2016027794A1 (ja) | 癌マーカー及び癌の判定方法 | |
| EP0598832A1 (en) | Dna sequencing with a t7-type gene 6 exonuclease | |
| CN117187221A (zh) | 人工改造脱氨酶辅助的dna中5-羟甲基胞嘧啶修饰单碱基分辨率定位分析方法 | |
| EP2660321A1 (en) | Method for detecting methylated cytosine | |
| JP6176694B2 (ja) | Dna中のグアニン・脱塩基部位の検出方法 | |
| KR20240024924A (ko) | 폴리머라제 돌연변이체 및 3'-oh 비차단 가역적 종결자와의 사용 | |
| CN115786478A (zh) | 一种基因修饰研究方案及应用 | |
| Pallavi et al. | Estimation of DNA methylation in plant related to nutrient use efficiency | |
| CN113481263A (zh) | 硒原子修饰在降低dNTP亲和力和DNA解链温度中的应用 | |
| CN117778544A (zh) | 一种检测aav载体中itr结构的一代测序方法 | |
| CN114231604A (zh) | Dkk-3基因甲基化诊断试剂体系、试剂盒及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12858295 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013549253 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14364545 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012858295 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012858295 Country of ref document: EP |