WO2013093756A1 - Polymeric precursors for producing graphene nanoribbons and methods for preparing them - Google Patents
Polymeric precursors for producing graphene nanoribbons and methods for preparing them Download PDFInfo
- Publication number
- WO2013093756A1 WO2013093756A1 PCT/IB2012/057377 IB2012057377W WO2013093756A1 WO 2013093756 A1 WO2013093756 A1 WO 2013093756A1 IB 2012057377 W IB2012057377 W IB 2012057377W WO 2013093756 A1 WO2013093756 A1 WO 2013093756A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- graphene nanoribbons
- independently
- crc
- polymeric precursor
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 Cc1cccc(-c(c(-c2ccccc2)c(-c2ccc(*)cc2)c(-c2cc(-c(c(-c3ccc(*)cc3)c3)c(-c4ccccc4)c(-c4ccc(*)cc4)c3-c3cccc(-c(c(-c4ccccc4)c(-c4ccc(*)cc4)c(-c4cc(-c(c(-c5ccc(*)cc5)c5)c(-c6ccccc6)c(-c6ccc(*)cc6)c5-c5cccc(-c(c(-c6ccccc6)c(cc6C)-c7ccc(*)cc7)c6-c6ccc(*)cc6)c5)ccc4)c4)c4-c4ccc(*)cc4)c3)ccc2)c2)c2-c2ccc(*)cc2)c1 Chemical compound Cc1cccc(-c(c(-c2ccccc2)c(-c2ccc(*)cc2)c(-c2cc(-c(c(-c3ccc(*)cc3)c3)c(-c4ccccc4)c(-c4ccc(*)cc4)c3-c3cccc(-c(c(-c4ccccc4)c(-c4ccc(*)cc4)c(-c4cc(-c(c(-c5ccc(*)cc5)c5)c(-c6ccccc6)c(-c6ccc(*)cc6)c5-c5cccc(-c(c(-c6ccccc6)c(cc6C)-c7ccc(*)cc7)c6-c6ccc(*)cc6)c5)ccc4)c4)c4-c4ccc(*)cc4)c3)ccc2)c2)c2-c2ccc(*)cc2)c1 0.000 description 2
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B1/00—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors
- H01B1/04—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of carbon-silicon compounds, carbon or silicon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y30/00—Nanotechnology for materials or surface science, e.g. nanocomposites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y40/00—Manufacture or treatment of nanostructures
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/15—Nano-sized carbon materials
- C01B32/182—Graphene
- C01B32/184—Preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/647—Unsaturated compounds containing a keto groups being part of a ring having unsaturation outside the ring
- C07C49/653—Unsaturated compounds containing a keto groups being part of a ring having unsaturation outside the ring polycyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/683—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F138/00—Homopolymers of compounds having one or more carbon-to-carbon triple bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
- C08G61/10—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aromatic carbon atoms, e.g. polyphenylenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/042—Graphene or derivatives, e.g. graphene oxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L65/00—Compositions of macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/141—Side-chains having aliphatic units
- C08G2261/1412—Saturated aliphatic units
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/31—Monomer units or repeat units incorporating structural elements in the main chain incorporating aromatic structural elements in the main chain
- C08G2261/312—Non-condensed aromatic systems, e.g. benzene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/36—Oligomers, i.e. comprising up to 10 repeat units
- C08G2261/364—Oligomers, i.e. comprising up to 10 repeat units containing hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/40—Polymerisation processes
- C08G2261/46—Diels-Alder reactions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/90—Applications
- C08G2261/91—Photovoltaic applications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/90—Applications
- C08G2261/92—TFT applications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/90—Applications
- C08G2261/95—Use in organic luminescent diodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/734—Fullerenes, i.e. graphene-based structures, such as nanohorns, nanococoons, nanoscrolls or fullerene-like structures, e.g. WS2 or MoS2 chalcogenide nanotubes, planar C3N4, etc.
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/84—Manufacture, treatment, or detection of nanostructure
- Y10S977/842—Manufacture, treatment, or detection of nanostructure for carbon nanotubes or fullerenes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/932—Specified use of nanostructure for electronic or optoelectronic application
Definitions
- the present invention concerns polymeric precursors for producing graphene nanoribbons, methods for preparing them, and oligophenylene monomers for the synthesis of polymeric precursors, as well as methods for preparing the graphene nanoribbons from the polymeric precursors and the oligophenylene monomers.
- Graphene an atomically thin layer from graphite, has received considerable interest in physics, material science and chemistry since the recent discovery of its appealing electronic properties. These involve superior charge carrier mobility and the quantum Hall effect. Moreover, its chemical robustness and material strength make graphene an ideal candidate for applications ranging from transparent conductive electrodes to devices for charge and energy storage.
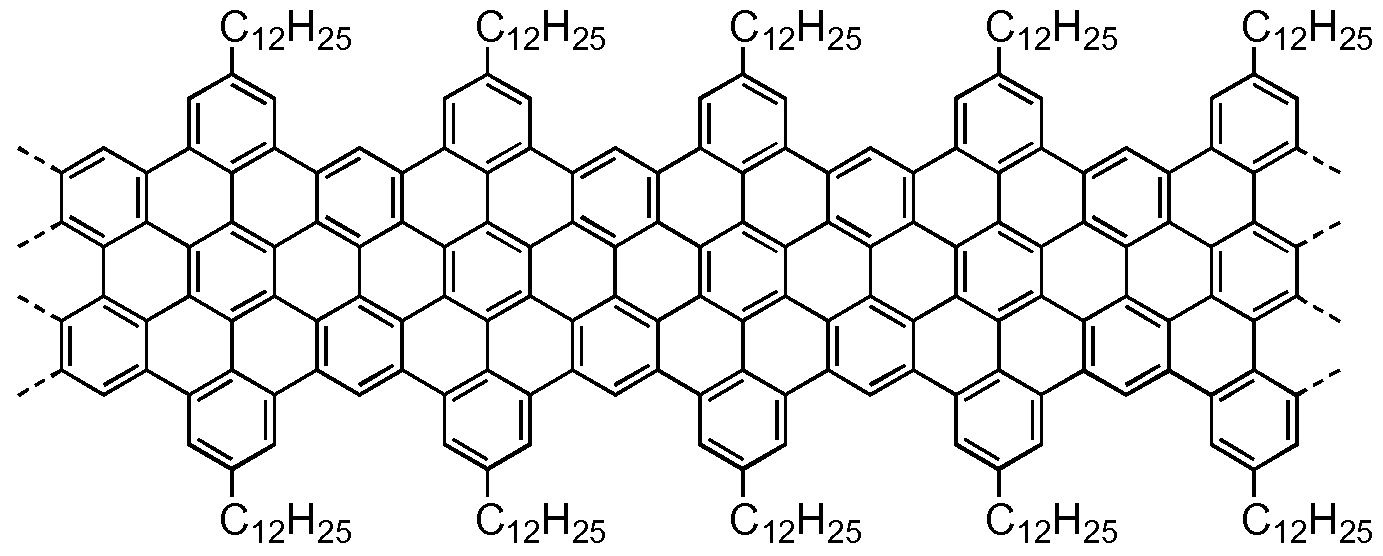
- Graphene nanoribbons are linear structures that are derived from the parent graphene lattice. Their characteristic feature is high shape-anisotropy due to the increased ratio of length over width. Currently, their usage in yet smaller, flatter and faster carbon-based devices and integrated circuits is being widely discussed in material science.
- armchair-type GNRs exhibit a band gap that can be adjusted by their width. Their length becomes relevant when GNRs are to be used in devices such as field-effect transistors (FETs) for which a minimum channel width has to be bridged. The same holds for the potential replacement of copper or gold in nanoscale conducting pathways. At the same time the edge structure of the GNRs will have a strong impact. Computational simulations and experimental results on smaller nanographenes suggest that GNRs exhibiting nonbonding 7T-electron states at zigzag edges could be used as active component in spintronic devices.
- FETs field-effect transistors
- Suzuki-Miyaura polymerization of the b/s-boronic ester with diiodobenzene furnished polyphenylenes in a strongly sterically hindered reaction.
- Intramolecular Scholl reaction of the polyphenylene with FeCI 3 as oxidative reagent provides graphene nanoribbons.
- the resulting graphene nanoribbons are ill-defined due to the statistically arranged "kinks" in their backbone. Furthermore the molecular weight is limited due to the sensitivity of the A2B2-type polymerization approach to aberrations fromstoichiometry. No lateral solubilizing alkyl chains have been introduced into the graphene nanoribbons.
- the second case suffers also from the stoichiometry issue due to the underlying A2B2- stoichiometry of the A2B2-type Suzuki protocol and the sterical hindrance of 1 ,4-diiodo- 2,3,5,6-tetraphenylbenzene.
- the third case makes use of a step-wise synthesis which provides very defined cutouts from graphene nanoribbons but is impracticable for the fabrication of high- molecular weight species.
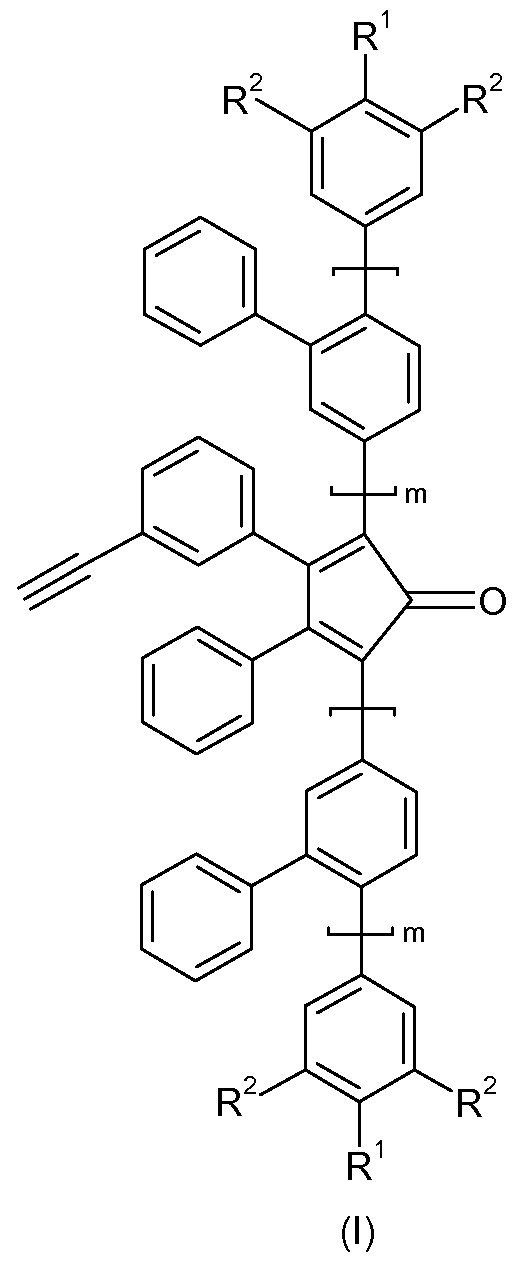
- R 1 and R 2 are independently of each other H, halogene, -OH, -NH 2 , -CN, -N0 2 or a linear or branched, saturated or unsaturated Ci-C 40 hydrocarbon residue, which can be substituted 1 - to 5-fold with halogene (F, CI, Br, I), -OR 3 , -NR 3 2 , -CN and/or -N0 2 , and wherein one or more CH 2 -groups can be replaced by -0-, -S-, -NR 4 -, -OC(O)- or - C(O)-, or an optionally substituted aryl, alkylaryl or alkoxyaryl residue;
- each R 3 is independently of each other H , C1-C30 akyl, C 2 -C 3 o alkenyl, C 2 -C 3 o alkynyl, C1-C30 haloalkyl, C 2 -C 30 haloalkenyl, C 2 -C 30 haloalkynyl or C 2 -C 30 acyl; each R 4 is independently of each other H , Ci-C 30 alkyl, C 2 -C 30 alkenyl, C 2 -C 30 alkynyl, CrC 30 haloalkyl, C 2 -C 30 haloalkenyl, C 2 -C 30 haloalkynyl or C 2 -C 30 acyl; and m represents 0, 1 or 2.
- R 1 and R 2 are independently of each other H, Ci-C 30 alkyl, CrC 30 alkoxy, Ci-C 30 alkylthio, C 2 -C 30 alkenyl, C 2 -C 30 alkynyl, CrC 30 haloalkyl, C 2 -C 30 haloalkenyl or haloalkynyl, e.g. CrC 30 perfluoroalkyl. More preferably R 1 and R 2 are independently of each other H, CrC 30 alkyl or CrC 30 alkoxy. Most preferably R 1 and R 2 and are independently of each other H or CrC 30 alkyl.
- Ci-C 30 alkyl can be linear or branched, where possible. Examples are methyl, ethyl, n-propyl, isopropyl, n-butyl, sec. -butyl, isobutyl, tert.
- Ci-C 30 alkoxy groups are straight-chain or branched alkoxy groups, e.g. methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, amyloxy, isoamyloxy, tert-amyloxy, heptyloxy, octyloxy, isooctyloxy, nonyloxy, decyloxy, undecyloxy, dode- cyloxy, tetradecyloxy, pentadecyloxy, hexadecyloxy, heptadecyloxy or octadecyloxy.
- alkylthio group means the same groups as the alkoxy groups, except that the oxygen atom of the ether linkage is replaced by a sulfur atom.
- C 2 -C 30 alkenyl groups are straight-chain or branched alkenyl groups, such as e.g.
- C 2 -30 alkynyl is straight-chain or branched and may be unsubstituted or substituted, such as, for example, ethynyl, 1-propyn-3-yl, 1 -butyn-4-yl, 1 -pentyn-5-yl, 2-methyl-3- butyn-2-yl, 1 ,4-pentadiyn-3-yl, 1 ,3-pentadiyn-5-yl, 1 -hexyn-6-yl, cis-3-methyl-2-penten- 4-yn-1 l-yl, trans-3-methyl-2-penten-4-yn-1 -yl, 1 ,3-hexadiyn-5-yl, 1 -octyn-8-yl, 1 -nonyn- 9-yl, 1 -decyn-10-yl, or 1 -tetracosyn-24-yl.
- Ci-C 3 o-perfluoroalkyl is a branched or unbranched radical such as for example -CF 3 , -CF 2 CF 3 , -CF 2 CF 2 CF 3 , -CF(CF 3 ) 2 , -(CF 2 ) 3 CF 3 or -C(CF 3 ) 3 .
- haloalkyi, haloalkenyl and haloalkynyl mean groups given by partially or wholly substituting the abovementioned alkyl group, alkenyl group and alkynyl group with halogen.
- C 2 -C 30 acyl is straight-chain or branched and may be saturated or unsaturated, such as, for example, ethanoyl, propanoyl, isobutanoyl, n-butanoyl, pentanoyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl or dodecanoyl.
- Aryl is usually C 6 -C 30 aryl, which optionally can be substituted, such as, for example, phenyl, 4-methylphenyl, 4-methoxyphenyl, naphthyl, biphenylyl, terphenylyl, pyrenyl, fluorenyl, phenanthryl, anthryl, tetracyl, pentacyl or hexacyl.
- R 1 is a linear or branched Ci-C 30 alkyl and R 2 is H.
- the problem is further solved by a polymeric precursor for producing graphene nanoribbons having repeating units of general formula (II),
- R 1 and R 2 are independently of each other H, halogene, -OH, -NH 2 , -CN, -N0 2 or a linear or branched, saturated or unsaturated Ci-C 40 hydrocarbon residue, which can be substituted 1 - to 5-fold with halogene (F, CI, Br, I), -OR 3 , -NR 3 2 , -CN and/or -N0 2 , and wherein one or more CH 2 -groups can be replaced by -0-, -S-, -NR 4 -, -OC(O)- or - C(O)-, or an optionally substituted aryl, alkylaryl or alkoxyaryl residue; each R 3 is independently of each other H, C1-C30 akyl, C 2 -C 3 o alkenyl, C 2 -C 3 o alkynyl, C1-C30 haloalkyl, C 2 -C 30 haloalkenyl, C
- R 1 and R 2 are independently of each other H, Ci-C 30 alkyl, CrC 30 alkoxy, Ci-C 30 alkylthio, C 2 -C 30 alkenyl, C 2 -C 30 alkynyl, CrC 30 haloalkyl, C 2 -C 30 haloalkenyl or haloalkynyl, e.g. CrC 30 perfluoroalkyl. More preferably R 1 and R 2 are independently of each other H, Ci-C 30 alkyl or CrC 30 alkoxy. Most preferably R 1 and R 2 are independently of each other H or CrC 30 alkyl.
- R 2 in formulae (I) and (II) is H.
- m in formulae (I) and (I I) represents 0 or 1 . More preferably, m in formulae (I) and (II) represents 0.
- R 1 is a linear or branched CrC 30 alkyl and R 2 is H.
- the oligophenylene monomer of general formula (I) is used for the preparation of the polymeric precursor for producing graphene nanoribbons having repeating units of general formula (II) by reacting it via Diels-Alder-Reaction according to Scheme 1 .
- the Diels-Alder reaction represents a well-established protocol which has been used for the build-up of functional molecules and polymers.
- the Diels-Alder-Reaction of oligophenylene monomer of general formula (I) to the polymeric precursor having repeating units of general formula (II) can be achieved under different conditions.
- the Diels-Alder-Reaction can be performed in high boiling solvents at elevated temperatures. Suitable high boiling solvents are diphenyl ether, 1 ,1 ,2,2- tetrachloroethane, 1 ,2-dichlorobenzene, 1 ,2,4-trichlorobenzene, nitrobenzene and benzophenone.
- the Diels-Alder-Reaction can be performed without using solvents at temperatures between 200 °C and 300 °C.
- the polymeric precursor having repeating units of general formula (II) contains from 2 to 900 repeating units and has a molecular weight of from 1 000 to 600 000 g/mol.
- the invention also concerns a process for the preparation of the polymeric precursor having repeating units of general formula (II) by Diels-Alder polymerization of the oligophenylene monomer of general formula (I).
- the invention also concerns a process for the preparation of graphene nanoribbons by cyclodehydrogenation of the polymeric precursor having repeating units of general formula (II) according to Scheme 2.
- the preparation of graphene nanoribbons of general formula (III) from the polymeric precursor having repeating units of general formula (II) can be performed e.g.
- reaction time is from 2 h to 10 d. If the reaction is carried out at around 20 °C, the reaction time is usually between two and four days. Preferably, a stream of an inert gas is passed through the reaction mixture during the reaction to avoid side reactions.
- the preparation of graphene nanoribbons of general formula (III) can be carried out using phenyliodine(lll)bis(trifluoroacetate) (PIFA) and boron trifluoride etherate in anhydrous DCM or molybdenum(V) pentachloride in anhydrous DCM.
- PIFA phenyliodine(lll)bis(trifluoroacetate)
- boron trifluoride etherate in anhydrous DCM or molybdenum(V) pentachloride in anhydrous DCM.
- the molecular weight of the graphene nanoribbons of general formula (III) varies from 1 000 to 600 000 g/mol.
- the oligophenylene monomer of general formula (I) can be synthesized according to Schemes 3 to 7 below, involving the two intermediates 3-bromobenzil 4 and substituted 1 ,3-bis(oligophenylenyl)propan-2-one 8, as summarized below in Scheme 7.
- the reaction conditions and solvents used are purely illustrative, of course other conditions and solvents can also be used and will be determined by the skilled in the art.
- the intermediate 3-bromobenzil 4 can be synthesized via a two-step route from commercially available 1 -bromo-3-iodobenzene 1 and ethynylbenzene 2 (Scheme 3). Sonogashira-type coupling of 1 and 2 can be used for the build-up of 3-bromodiphenylacetylene 3.
- the reaction can be achieved in a mixture of THF and triethylamine at room temperature in the presence of copper(l) iodide and a palladium(ll) catalyst.
- the second step consists in the oxidation of the acetylene group of 3- bromodiphenylacetylene 3 to yield the 3-bromobenzil 4.
- This step can be realized by stirring bromodiphenylacetylene 3 in the presence of iodine in dimethyl sulfoxide at elevated temperatures.
- Scheme 4 illustrates the synthetic route to substituted 1 ,3-bis(oligophenylenyl)propan- 2-one 8-0 starting from literature known halogenated toluene 5, wherein X and Y are independently selected from CI, Br, I or H.
- the first step consists in the bromination of the benzylic position of halogenated toluene 5 to yield the halogenated benzyl bromide 6.
- This step can be realized by heating halogenated toluene 5 under reflux in carbon tetrachloride in the presence of N-bromosuccinimide (NBS) and benzoyl peroxide.
- NBS N-bromosuccinimide
- the halogenated benzyl bromide 6 can be further reacted to the halogenated 1 ,3- bis(phenyl)propan-2-one 7. This is achieved by heating the halogenated benzyl bromide 6 under reflux in a reaction mixture of dichloromethane and water using iron(0) pentacarbonyl, potassium hydroxide and benzyltriethylammonium chloride.
- the last reaction step of this reaction sequence can be carried out with any desired substituted or non-substituted halide R 1 X' or R 2 X', wherein X' is selected from CI, Br or I and R 1 and R 2 are as defined above.
- substituted 1 ,3-bis(oligophenylenyl)propan- 2-one 8-0 is accessible using zinc(ll) iodide and a palladium(O) catalyst at room temperature. If both X and Y are H, this reaction step is omitted.
- the reaction can be achieved at an elevated temperature in a reaction mixture of toluene, ethanol and water in the presence of potassium carbonate and catalytic amounts of tetrakis(triphenylphosphine)palladium(0). Due to the higher reactivity of the iodine carbon bond, the coupling mainly proceeds at the iodine atom in the desired 1 -position.
- the next step consists in the bromination of the benzylic position of 2-bromo-5-methyl- biphenyl 11 to yield the functionalized biphenyl 12. This step can be realized by heating bromo-5-methyl-biphenyl 11 under reflux in carbon tetrachloride using N- bromosuccinimide (NBS) and benzoyl peroxide.
- the functionalized biphenyl 12 can be further reacted to 1 ,3-bis(bromobiphenyl)propan- 2-one 13. This is achieved by boiling the functionalized biphenyl 12 in a reaction mixture of dichloromethane and water in the presence of iron(0) pentacarbonyl, potassium hydroxide and benzyltriethylammonium chloride.
- the synthesis of the substituted 1 ,3-bis(oligophenylenyl)propan-2-one 8- 2 starts from commercially available 2-bromo-4-chloro-1 -iodobenzene 16 and the afore mentioned substituted phenylboronic acid pinacol ester 14. Suzuki cross coupling can be used for the build-up of the substituted 2-bromo-4-chlorobiphenyl 17.
- the reaction can be performed e.g. at an elevated temperature in a reaction mixture of toluene, ethanol and water in the presence of potassium carbonate and of tetrakis(triphenylphosphine)palladium(0).
- the substituted trisphenylenylboronic acid pinacol ester 19 can be prepared from chlorotrisphenylene 18 and bis(pinacol) ester of 1 ,4-phenyldiboronic acid under reflux in 1 ,4-dioxane in the presence of potassium acetate as a base and catalytic amounts of both tris(dibenzylideneacetone)dipalladium (0) (Pd 2 (dba) 3 ) and 2-dicyclohexyl- phosphino-2',4',6'-triisopropylbiphenyl (XPhos).
- Various articles of manufacture including electronic devices, optical devices, and optoelectronic devices, such as field effect transistors (e.g., thin film transistors), photovoltaics, organic light emitting diodes (OLEDs), complementary metal oxide semiconductors (CMOSs), complementary inverters, D flip-flops, rectifiers, and ring oscillators, that make use of the graphene nanoribbons disclosed herein also are within the scope of the present invention as are methods of making the same.
- field effect transistors e.g., thin film transistors
- OLEDs organic light emitting diodes
- CMOSs complementary metal oxide semiconductors
- CMOSs complementary inverters
- D flip-flops D flip-flops
- rectifiers and ring oscillators
- the present invention therefore, further provides methods of preparing a semiconductor material.
- the methods can include preparing a composition that includes one or more of the graphene nanoribbons of the invention disclosed herein dissolved or dispersed in a liquid medium such as a solvent or a mixture of solvents, depositing the composition on a substrate to provide a semiconductor material precursor, and processing (e.g., heating) the semiconductor precursor to provide a semiconductor material (e.g., a thin film semiconductor) that includes one or more of the graphene nanoribbons disclosed herein.
- the liquid medium can be an organic solvent, an inorganic solvent such as water, or combinations thereof.
- the composition can further include one or more additives independently selected from detergents, dispersants, binding agents, compatibilizing agents, curing agents, initiators, humectants, antifoaming agents, wetting agents, pH modifiers, biocides, and bacteriostats.
- additives independently selected from detergents, dispersants, binding agents, compatibilizing agents, curing agents, initiators, humectants, antifoaming agents, wetting agents, pH modifiers, biocides, and bacteriostats.
- surfactants and/or polymers e.g., polystyrene, polyethylene, poly-alpha-methylstyrene, polyisobutene, polypropylene, polymethylmethacrylate, and the like
- a dispersant e.g., polystyrene, polyethylene, poly-alpha-methylstyrene, polyisobutene, polypropylene, polymethylmethacrylate, and the like
- an antifoaming agent
- the depositing step can be carried out by printing, including inkjet printing and various contact printing techniques (e.g., screen-printing, gravure printing, offset printing, pad printing, lithographic printing, flexographic printing, and microcontact printing).
- the depositing step can be carried out by spin coating, drop-casting, zone casting, dip coating, blade coating, spraying or vacuum filtration.
- the present invention further provides articles of manufacture such as the various devices described herein that include a composite having a semiconductor material of the present invention and a substrate component and/or a dielectric component.
- the substrate component can be selected from doped silicon, an indium tin oxide (ITO), ITO-coated glass, ITO-coated polyimide or other plastics, aluminum or other metals alone or coated on a polymer or other substrate, a doped polythiophene, and the like.
- the dielectric component can be prepared from inorganic dielectric materials such as various oxides (e.g., Si0 2 , Al 2 0 3 , Hf0 2 ), organic dielectric materials such as various polymeric materials (e.g., polycarbonate, polyester, polystyrene, polyhaloethylene, polyacrylate), and self-assembled superlattice/self-assembled nanodielectric (SAS/SAND) materials (e.g., described in Yoon, M-H. et al., PNAS, 102 (13): 4678- 4682 (2005)), as well as hybrid organic/inorganic dielectric materials (e.g., described in US 2007/0181961 A1 ).
- the composite also can include one or more electrical contacts.
- Suitable materials for the source, drain, and gate electrodes include metals (e.g., Au, Al, Ni, Cu), transparent conducting oxides (e.g., ITO, IZO, ZITO, GZO, GIO, GITO), and conducting polymers (e.g., poly(3,4-ethylenedioxythiophene) poly(styrene- sulfonate) (PEDOT:PSS), polyaniline (PANI), polypyrrole (PPy).
- metals e.g., Au, Al, Ni, Cu
- transparent conducting oxides e.g., ITO, IZO, ZITO, GZO, GIO, GITO
- conducting polymers e.g., poly(3,4-ethylenedioxythiophene) poly(styrene- sulfonate) (PEDOT:PSS), polyaniline (PANI), polypyrrole (PPy).
- One or more of the composites described herein can be embodied within various organic electronic, optical, and optoelectronic devices such as organic thin film transistors (OTFTs), specifically, organic field effect transistors (OFETs), as well as sensors, capacitors, unipolar circuits, complementary circuits (e.g., inverter circuits), and the like.
- OFTs organic thin film transistors
- OFETs organic field effect transistors
- sensors capacitors
- unipolar circuits e.g., unipolar circuits
- complementary circuits e.g., inverter circuits
- graphene nanoribbons of the present invention are photovoltaics or solar cells.
- Compounds of the present invention can exhibit broad optical absorption and/or a very positively shifted reduction potential, making them desirable for such applications.
- the graphene nanoribbons described herein can be used as a n-type semiconductor in a photovoltaic design, which includes an adjacent p-type semiconductor material that forms a p-n junction.
- the compounds can be in the form of a thin film semiconductor, which can be deposited on a substrate to form a composite. Exploitation of compounds of the present invention in such devices is within the knowledge of a skilled artisan.
- another aspect of the present invention relates to methods of fabricating an organic field effect transistor that incorporates a semiconductor material of the present invention.
- the semiconductor materials of the present invention can be used to fabricate various types of organic field effect transistors including top-gate top-contact capacitor structures, top-gate bottom-contact capacitor structures, bottom-gate top- contact capacitor structures, and bottom-gate bottom-contact capacitor structures.
- OTFT devices can be fabricated with the present graphene nanoribbons on doped silicon substrates, using Si0 2 as the dielectric, in top-contact geometries.
- the active semiconductor layer which incorporates at least a graphene nanoribbon of the present invention can be deposited at room temperature or at an elevated temperature.
- the active semiconductor layer which incorporates at least a graphene nanoribbon of the present invention can be applied by spin-coating or printing as described herein.
- metallic contacts can be patterned on top of the films using shadow masks. The invention is illustrated in more detail by the following examples. Examples
- Figure 3 Chemical formulae, exact masses and molecular weights of tetrameric, pentameric, hexameric and heptameric polymeric precursor Ha.
- Figure 4 Molecular weight distribution of polymeric precursor Ha after fractionation by GPC (Table 1 , entry 2, THF, PSS).
- Figure 5 Raman spectrum of graphene nanoribbons Ilia (powder).
- THF 14 ⁇ g mL
- graphene nanoribbons Ilia as a film on a glass substrate drop-casted from dispersion in THF.
- Figure 7 UV-vis absorption spectrum of graphene nanoribbons Ilia in an exfoliated solution in NMP.
- Figure 8 Output characteristics of graphene nanoribbons Ilia at various gate biases
- a purple powder of 2,5-bis(4-dodecylphenyl)-3-(3-ethynylphenyl)-4-phenyl-2,4- cyclopentadienone la in a 25-mL Schlenk tube was heated to 260 °C using a heating mantle. The powder at first melted and then lost its purple color to be pale yellow. After cooling down to room temperature, the resulting polymer was sonicated in THF for 30 min, and the insoluble polymer was filtered off. The filtrate was concentrated in vacuo and fractionated by gel permeation chromatography (eluent: dichloromethane).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Nanotechnology (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- General Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Crystallography & Structural Chemistry (AREA)
- Physics & Mathematics (AREA)
- Materials Engineering (AREA)
- Composite Materials (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
- Thin Film Transistor (AREA)
- Carbon And Carbon Compounds (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description




















Claims

Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN4378CHN2014 IN2014CN04378A (en) | 2011-12-20 | 2012-12-17 | |
| CN201280062080.2A CN104114489B (en) | 2011-12-20 | 2012-12-17 | For the preparation of the polymer precursor and preparation method thereof of graphene nanobelt |
| JP2014548286A JP2015504046A (en) | 2011-12-20 | 2012-12-17 | Polymer precursor for producing graphene nanoribbon and method for producing the same |
| EP12859354.8A EP2794476A4 (en) | 2011-12-20 | 2012-12-17 | POLYMER PRECURSORS FOR THE PRODUCTION OF GRAPHENE NANORUBANS AND METHODS FOR THE PREPARATION THEREOF |
| KR1020147019888A KR20140116112A (en) | 2011-12-20 | 2012-12-17 | Polymeric precursors for producing graphene nanoribbons and methods for preparing them |
| US14/367,041 US9550678B2 (en) | 2011-12-20 | 2012-12-17 | Polymeric precursors for producing graphene nanoribbons and methods for preparing them |
| SG11201403360VA SG11201403360VA (en) | 2011-12-20 | 2012-12-17 | Polymeric precursors for producing graphene nanoribbons and methods for preparing them |
| RU2014129615A RU2014129615A (en) | 2011-12-20 | 2012-12-17 | POLYMERIC PREDATORS INTENDED FOR OBTAINING GRAPHENE NANO-TAPES, AND METHODS FOR PRODUCING THEM |
| IL233003A IL233003A0 (en) | 2011-12-20 | 2014-06-08 | Polymeric precursors for producing graphene nanoribbons and methods for preparing them |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161577689P | 2011-12-20 | 2011-12-20 | |
| US61/577,689 | 2011-12-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013093756A1 true WO2013093756A1 (en) | 2013-06-27 |
Family
ID=48667866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2012/057377 Ceased WO2013093756A1 (en) | 2011-12-20 | 2012-12-17 | Polymeric precursors for producing graphene nanoribbons and methods for preparing them |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9550678B2 (en) |
| EP (1) | EP2794476A4 (en) |
| JP (1) | JP2015504046A (en) |
| KR (1) | KR20140116112A (en) |
| CN (1) | CN104114489B (en) |
| IL (1) | IL233003A0 (en) |
| IN (1) | IN2014CN04378A (en) |
| RU (1) | RU2014129615A (en) |
| SG (1) | SG11201403360VA (en) |
| TW (1) | TWI579316B (en) |
| WO (1) | WO2013093756A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2845838A1 (en) | 2013-09-04 | 2015-03-11 | Basf Se | Purification process for graphene nanoribbons |
| CN105502351A (en) * | 2015-12-04 | 2016-04-20 | 华南理工大学 | Soluble graphene nanoribbon as well as synthetic method and application thereof |
| KR20160123335A (en) * | 2014-02-13 | 2016-10-25 | 바스프 에스이 | Graphene nanoribbons with controlled zig-zag edge and cove edge configuration |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5945501B2 (en) * | 2012-03-05 | 2016-07-05 | 本田技研工業株式会社 | Method for producing photoelectric conversion material |
| CN105399074B (en) * | 2015-12-04 | 2017-06-06 | 华南理工大学 | A kind of graphene nanobelt and its synthetic method and application |
| US10998405B2 (en) | 2015-12-17 | 2021-05-04 | Intel Corporation | Low-defect graphene-based devices and interconnects |
| GB201613012D0 (en) * | 2016-07-27 | 2016-09-07 | Price Richard J | Improvements relating to graphene nanomaterials |
| WO2018094234A1 (en) * | 2016-11-18 | 2018-05-24 | The Regents Of The University Of California | Synthesis of graphene nanoribbons from monomeric molecular precursors bearing reactive alkyne moieties |
| JP7166635B2 (en) * | 2017-05-08 | 2022-11-08 | 慶應義塾 | Polymer having a layered structure like graphite or graphene |
| CN107597196B (en) * | 2017-07-27 | 2020-01-17 | 东华大学 | A kind of preparation method of surface-modified titanium dioxide organic graphene nanotubes |
| CN108285139B (en) * | 2017-12-11 | 2021-06-18 | 昆明理工大学 | A kind of preparation method and application of nitrogen-doped graphene carbon material |
| CN114181220B (en) * | 2021-12-17 | 2023-03-10 | 中国科学技术大学 | Solenoid-shaped magnetic carbon nano material and preparation method thereof |
| CN117509629B (en) * | 2023-11-27 | 2024-10-22 | 厦门大学 | Edge fluorinated nano graphene material and preparation method and application thereof |
| CN117993513B (en) * | 2024-02-07 | 2025-03-04 | 张江国家实验室 | Ising machine core circuit, its unit and method for annealing the same |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004089862A2 (en) * | 2003-04-02 | 2004-10-21 | Dow Global Technologies Inc. | Multifunctional unsymmetrically substituted monomers and polyarylene compositions therefrom |
| WO2010147860A1 (en) * | 2009-06-15 | 2010-12-23 | William Marsh Rice University | Graphene nanoribbons prepared from carbon nanotubes via alkali metal exposure |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5965679A (en) * | 1996-09-10 | 1999-10-12 | The Dow Chemical Company | Polyphenylene oligomers and polymers |
| JP2000191752A (en) * | 1998-12-25 | 2000-07-11 | Dow Chem Co:The | Polyphenylene oligomer and polymer |
| KR100569187B1 (en) * | 2003-09-27 | 2006-04-10 | 한국과학기술연구원 | Compounds derived from cyclopentadienone, preparation methods thereof, and EL elements using the same |
| JP4005571B2 (en) * | 2004-02-04 | 2007-11-07 | 独立行政法人科学技術振興機構 | Amphiphilic hexaperihexabenzocoronene derivatives |
| JP2007019086A (en) * | 2005-07-05 | 2007-01-25 | Toyota Central Res & Dev Lab Inc | Organic semiconductor material, semiconductor device using the same, and field effect transistor |
| WO2007075748A2 (en) | 2005-12-20 | 2007-07-05 | Northwestern University | Intercalated superlattice compositions and related methods for modulating dielectric property |
| GB0622150D0 (en) * | 2006-11-06 | 2006-12-20 | Kontrakt Technology Ltd | Anisotropic semiconductor film and method of production thereof |
| GB0802912D0 (en) * | 2008-02-15 | 2008-03-26 | Carben Semicon Ltd | Thin-film transistor, carbon-based layer and method of production thereof |
| TW201012749A (en) | 2008-08-19 | 2010-04-01 | Univ Rice William M | Methods for preparation of graphene nanoribbons from carbon nanotubes and compositions, thin films and devices derived therefrom |
| WO2011112589A1 (en) * | 2010-03-08 | 2011-09-15 | William Marsh Rice University | Transparent electrodes based on graphene and grid hybrid structures |
-
2012
- 2012-12-17 KR KR1020147019888A patent/KR20140116112A/en not_active Withdrawn
- 2012-12-17 SG SG11201403360VA patent/SG11201403360VA/en unknown
- 2012-12-17 CN CN201280062080.2A patent/CN104114489B/en not_active Expired - Fee Related
- 2012-12-17 IN IN4378CHN2014 patent/IN2014CN04378A/en unknown
- 2012-12-17 JP JP2014548286A patent/JP2015504046A/en active Pending
- 2012-12-17 WO PCT/IB2012/057377 patent/WO2013093756A1/en not_active Ceased
- 2012-12-17 RU RU2014129615A patent/RU2014129615A/en not_active Application Discontinuation
- 2012-12-17 EP EP12859354.8A patent/EP2794476A4/en not_active Withdrawn
- 2012-12-17 US US14/367,041 patent/US9550678B2/en not_active Expired - Fee Related
- 2012-12-19 TW TW101148524A patent/TWI579316B/en not_active IP Right Cessation
-
2014
- 2014-06-08 IL IL233003A patent/IL233003A0/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004089862A2 (en) * | 2003-04-02 | 2004-10-21 | Dow Global Technologies Inc. | Multifunctional unsymmetrically substituted monomers and polyarylene compositions therefrom |
| WO2010147860A1 (en) * | 2009-06-15 | 2010-12-23 | William Marsh Rice University | Graphene nanoribbons prepared from carbon nanotubes via alkali metal exposure |
Non-Patent Citations (3)
| Title |
|---|
| MATTHIAS, TREIER ET AL.: "Surface-assisted cyclodehydrogenation provides a synthetic route owards easily processable and chemically tailored nanographenes.", NATURE CHEMISTRY., vol. 3, 7 November 2010 (2010-11-07), pages 61 - 67, XP055156668 * |
| MIHOV, G. ET AL.: "Toward Nanoamphiphiles: Efficient Synthesis of Desymmetrized Polyphenylene Dendrimers.", J. ORG. CHEM., vol. 69, 20 October 2004 (2004-10-20), pages 8029 - 8037, XP055072130 * |
| See also references of EP2794476A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2845838A1 (en) | 2013-09-04 | 2015-03-11 | Basf Se | Purification process for graphene nanoribbons |
| KR20160123335A (en) * | 2014-02-13 | 2016-10-25 | 바스프 에스이 | Graphene nanoribbons with controlled zig-zag edge and cove edge configuration |
| JP2017507890A (en) * | 2014-02-13 | 2017-03-23 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | Graphene nanoribbons with controlled zigzag edge and cove edge arrangement |
| US10329378B2 (en) | 2014-02-13 | 2019-06-25 | Basf Se | Graphene nanoribbons with controlled zig-zag edge and cove edge configuration |
| KR102333861B1 (en) | 2014-02-13 | 2021-12-01 | 바스프 에스이 | Graphene nanoribbons with controlled zig-zag edge and cove edge configuration |
| CN105502351A (en) * | 2015-12-04 | 2016-04-20 | 华南理工大学 | Soluble graphene nanoribbon as well as synthetic method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2014129615A (en) | 2016-02-10 |
| IL233003A0 (en) | 2014-07-31 |
| TW201331255A (en) | 2013-08-01 |
| US9550678B2 (en) | 2017-01-24 |
| EP2794476A1 (en) | 2014-10-29 |
| US20140353554A1 (en) | 2014-12-04 |
| EP2794476A4 (en) | 2015-06-03 |
| SG11201403360VA (en) | 2014-07-30 |
| JP2015504046A (en) | 2015-02-05 |
| CN104114489B (en) | 2016-04-27 |
| CN104114489A (en) | 2014-10-22 |
| TWI579316B (en) | 2017-04-21 |
| IN2014CN04378A (en) | 2015-09-04 |
| KR20140116112A (en) | 2014-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9550678B2 (en) | Polymeric precursors for producing graphene nanoribbons and methods for preparing them | |
| US20170081192A1 (en) | Ortho-terphenyls for the preparation of graphene nanoribbons | |
| Zhang et al. | Synthesis, self-assembly, and charge transporting property of contorted tetrabenzocoronenes | |
| US20160207774A1 (en) | Oligophenylene monomers and polymeric precursors for producing graphene nanoribbons | |
| JP2014501695A (en) | Diketopyrrolopyrrole semiconductor | |
| KR20150000860A (en) | Graphene nanoribbon precursors and monomers suitable for preparation thereof | |
| Lin et al. | A star-shaped electron acceptor based on 5, 5′-bibenzothiadiazole for solution processed solar cells | |
| WO2021143222A1 (en) | High-performance hole transport material, and preparation method therefor and application thereof | |
| US20150299381A1 (en) | Polymeric precursors for producing graphene nanoribbons and suitable oligophenylene monomers for preparing them | |
| Zhou et al. | Improved photovoltaic performance of star-shaped molecules with a triphenylamine core by tuning the substituted position of the carbazolyl unit at the terminal | |
| WO2020058203A1 (en) | Novel fluorescent pyrene derivatives, methods for preparing the same, and uses thereof | |
| Huang et al. | Ladder-type conjugated oligomers prepared by the Scholl oxidative cyclodehydrogenation reaction: synthesis, characterization and application in field effect transistors | |
| JP6664710B2 (en) | Polymer and method for producing the same | |
| JP2011222974A (en) | Organic semiconductor material and organic semiconductor element | |
| KR20120109597A (en) | Dibenzofluoranthene compound and organic thin-film solar cell using same | |
| JP6275117B2 (en) | Organic semiconductor material for organic transistor and organic transistor element | |
| JP6143257B2 (en) | Organic semiconductor material and organic semiconductor device using the same | |
| Fan et al. | Blue-light-emitting and hole-transporting molecular materials based on amorphous triphenylamine-functionalized twisted binaphthyl | |
| JP5650051B2 (en) | Organic semiconductor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201280062080.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12859354 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 233003 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2014548286 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14367041 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012859354 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012859354 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147019888 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014129615 Country of ref document: RU Kind code of ref document: A |