WO2013103760A1 - Compositions, methods of use, and methods of treatment - Google Patents
Compositions, methods of use, and methods of treatment Download PDFInfo
- Publication number
- WO2013103760A1 WO2013103760A1 PCT/US2013/020212 US2013020212W WO2013103760A1 WO 2013103760 A1 WO2013103760 A1 WO 2013103760A1 US 2013020212 W US2013020212 W US 2013020212W WO 2013103760 A1 WO2013103760 A1 WO 2013103760A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- beta
- lactamase inhibitor
- structures described
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)=*C(N)=C Chemical compound CC(*)=*C(N)=C 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N C1CCCCC1 Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- PQTHWHNFQHOKSK-UHFFFAOYSA-N O=C(c1c2nc[nH]c2cc(C(F)(F)F)c1)Nc1cccc(C2NNNN2)c1 Chemical compound O=C(c1c2nc[nH]c2cc(C(F)(F)F)c1)Nc1cccc(C2NNNN2)c1 PQTHWHNFQHOKSK-UHFFFAOYSA-N 0.000 description 1
- PZYXGNXJCSHONZ-UHFFFAOYSA-N O=C(c1ccc2[nH]ncc2c1)Nc1cc(C2NNNN2)ccc1 Chemical compound O=C(c1ccc2[nH]ncc2c1)Nc1cc(C2NNNN2)ccc1 PZYXGNXJCSHONZ-UHFFFAOYSA-N 0.000 description 1
- KRHCUVORUCETIF-UHFFFAOYSA-N Oc1cc(C(F)(F)F)cc(C(Nc2cc(C3NNNN3)ccc2)=O)c1 Chemical compound Oc1cc(C(F)(F)F)cc(C(Nc2cc(C3NNNN3)ccc2)=O)c1 KRHCUVORUCETIF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- Beta-lactam compounds such as penicillins are the most widely used antibiotics due to their effective inhibition of the transpeptidases required for bacterial cell wall synthesis. Beta-lactamases catalyze ⁇ -lactam hydrolysis and are primary mediators of bacterial resistance to these compounds. There are four ⁇ -lactamase families, Classes A to D, among which Classes A and C are the most commonly observed in the clinic.
- CTX-M is a new group of Class A ⁇ -lactamases that is particularly effective against the extended spectrum ⁇ - lactam antibiotics such as cefotaxime, which itself was developed to counter bacterial resistance to first-generation penicillins and cephalosporins.
- ESBL extended spectrum beta-lactamase
- CTX-M has become the most frequently observed ESBL in many regions of the world.
- ⁇ -lactamase inhibitors e.g., clavulanic acid
- ⁇ -lactam ring a ⁇ -lactam ring, making them susceptible to resistance stemming from up-regulation of ⁇ -lactamase production, selection for new ⁇ -lactamases, and other mechanisms evolved over millions of years' chemical warfare between bacteria and ⁇ -lactam producing microorganisms.
- embodiments of the present disclosure in one aspect, relate to a beta- lactamase inhibitor, pharmaceutical compositions including a beta-lactamase inhibitor, methods of treatment of a condition (e.g., infection) or disease, methods of treatment using compositions or pharmaceutical compositions, and the like.
- a composition includes: a beta-lactamase inhibitor.
- the composition also includes an antibiotic such as a beta-lactam antibiotic.
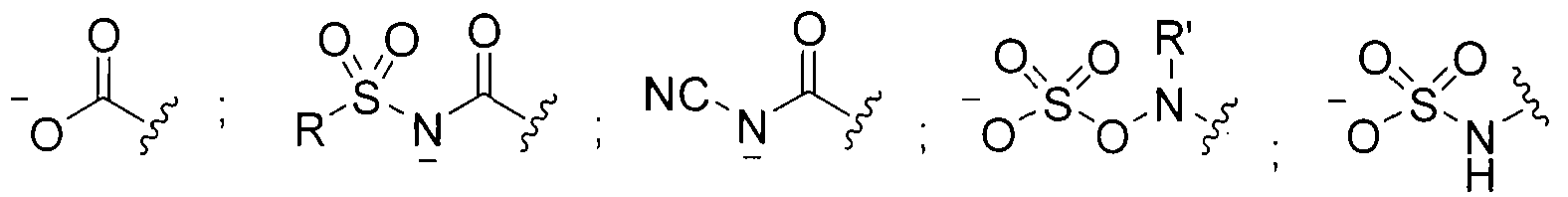
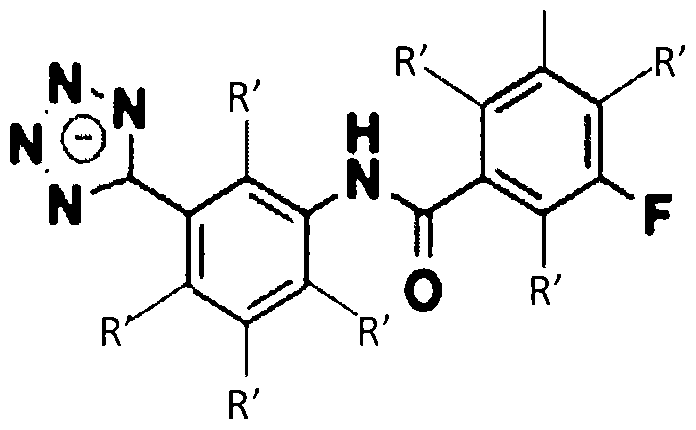
- X 1 , X 2 , and X 3 are each independently selected from C-R' or N; wherein R' is H, a halogen, a substituted or unsubstituted alky I, a substituted or unsubstituted aryl, a substituted or unsubstituted cycloalkyl, a substituted or unsubstituted cycloalkenyl, a substituted or unsubstituted heteroaryl, a substituted or unsubstituted biaryl, a substituted or unsubstituted fused aryl, a substituted or unsubstituted alkenyl, or a substituted or unsubstituted alkynyl; wherein R is an alkyl group, an aryl group, a heteroaryl group, or a cyclic or heterocyclic group; and Q is O or S.
- a pharmaceutical composition includes: a therapeutically effective amount of a beta-lactamase inhibitor, or a pharmaceutically acceptable salt of the beta-lactamase inhibitor, and a pharmaceutically acceptable carrier, to treat a condition.
- the pharmaceutical composition also includes an antibiotic such as a beta-lactam antibiotic.
- the beta-lactamase inhibitor can be represented by any one of the structures described by structures A or B as described herein.
- a method of treating a condition includes:
- a pharmaceutical composition wherein the pharmaceutical composition includes a therapeutically effective amount of a beta-lactamase inhibitor, or a pharmaceutically acceptable salt of the beta-lactamase inhibitor, and a pharmaceutically acceptable carrier, to treat the condition.
- the pharmaceutical composition also includes an antibiotic such as a beta-lactam antibiotic.
- the beta-lactamase inhibitor can be represented by any one of the structures described by structures A or B as described herein.
- Figure 1 illustrates the crystal structure of compound 1 in complex with CTX-M-9.
- the dashed lines represent hydrogen bonds with a sphere representing a water molecule.
- Figure 2 illustrates crystal complex structures with compounds targeting Prol 67: (a) Compound 4, (b) Compound 10, and (c) Compound 11.
- the gray dashed lines represent hydrogen bonds between the ligand and CTX-M-9.
- the carbon atoms of the protein are colored medium gray along with oxygens and nitrogens. Resolution for the structures ranges from 1 .2- 1 .4 A. Unbiased 2F 0 -F C densities are shown in blue at 1 .5 ⁇ .
- Figure 3 illustrates crystal complex structures with compounds targeting Asp240: (a) Compound 18, (b) Compound 16, and (c) Compound 12, in comparison to the designed pose in cyan.
- the gray dashed lines represent hydrogen bonds between the ligand and CTX-M-9. Resolution for the structures ranges from 1 .2- 1 .4 A. Unbiased 2F 0 -F C densities are shown in blue at 1 .5 ⁇ .
- Figure 6 illustrates analogs designed to target both Pro 167 and Asp240.
- Figure 7 illustrates a table describing the binding of many new inhibitors to the CTX- M active site.
- Embodiments of the present disclosure will employ, unless otherwise indicated, techniques of organic chemistry, biochemistry, molecular biology, pharmacology, medicine, and the like, which are within the skill of the art. Such techniques are explained fully in the literature.
- substituted refers to any one or more hydrogens on the designated atom that can be replaced with a selection from the indicated group, provided that the designated atom's normal valence is not exceeded, and that the substitution results in a stable compound.
- 2 hydrogens on the atom can be replaced.
- Keto substituents are not present on aromatic moieties.
- a ring system e.g., carbocyclic or heterocyclic
- aliphatic group refers to a saturated or unsaturated linear or branched hydrocarbon group and encompasses alkyl, alkenyl, and alkynyl groups, for example.
- alkyl or “alkyl group” refers to a saturated aliphatic hydrocarbon radical which can be straight or branched, having 1 to 20 carbon atoms, wherein the stated range of carbon atoms includes each intervening integer individually, as wel l as sub-ranges.
- alkyl include, but are not limited to methyl, ethyl, n-propyl, i-propyl, n-butyl, s- butyl, t-butyl, n-pentyl, and s-pentyl.
- the term "lower alkyl” means an alkyl group having less than 10 carbon atoms.
- alkenyl or “alkenyl group” refers to an aliphatic hydrocarbon radical which can be straight or branched, containing at least one carbon-carbon double bond, having 2 to 20 carbon atoms, wherein the stated range of carbon atoms includes each intervening integer individually, as well as sub-ranges.
- alkenyl groups include, but are not limited to, ethenyl, propenyl, n-butenyl, i-butenyl, 3-methylbut-2-enyl, n- pentenyl, heptenyl, octenyl, decenyl, and the like.
- arylalkyl refers to an arylalkyl group wherein the aryl and alkyl are as herein described.
- arylalkyl include, but are not limited to, -phenylmethyl, - phenylethyl, -phenylpropyl, -phenylbutyl, and -phenylpentyl.
- substituted cycloalkenyl substituted aryl
- substituted biaryl substituted fused aryl
- substituted fused aryl and the like means that the substituted group may contain in place of one or more hydrogens a group such as hydroxy, amino, halo, trifluoromethyl, cyano,— NH(lower alkyl), ⁇ N(lower alky I) 2 , lower alkoxy, lower alkylthio, or carboxy, and thus embraces the terms haloalkyi, alkoxy, fluorobenzyl, and the sulfur and phosphorous containing substitutions referred to below.
- halo refers to a fluorine, chlorine, bromine, and iodine, and radicals thereof.
- haloalkyi refers to an alkyl or alkenyl radical in which one or more hydrogens are substituted by halogen radicals. Examples of haloalkyi include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
- alkoxy represents an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge. Examples of alkoxy include, but are not limited to, methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy, n- pentoxy, and s-pentoxy.
- lower alkoxy means an alkoxy group having less than 10 carbon atoms.
- cycloalkyl refers to a non-aromatic mono-or multicyclic ring system of about 3 to about 10 carbon atoms, preferably of about 5 to about 10 carbon atoms. Preferred ring sizes of rings of the ring system include about 5 to about 6 ring atoms.
- Exemplary monocyclic cycloalkyl include cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- Exemplary multicyclic cycloalkyl include 1 -decalin, norbornyl, adamant-( l -or 2-)yl, and the like.
- cycloalkenyl refers to a non-aromatic mono-or multicyclic ring system of about 3 to about 10 carbon atoms, preferably of about 5 to about 10 carbon atoms, and which contains at least one carbon-carbon double bond. Preferred ring sizes of rings of the ring system include about 5 to about 6 ring atoms.
- Exemplary monocyclic cycloalkenyl include cyclopentenyl, cyclohexenyl, cycloheptenyl, and the like.
- An exemplary multicyclic cycloalkenyl is norbornylenyl.
- aryl refers to an aromatic monocyclic or multicyclic ring system of about 6 to about 14 carbon atoms, preferably of about 6 to about 10 carbon atoms.
- exemplary aryl groups include phenyl or naphthyl, or phenyl substituted or naphthyl substituted.
- heteroaryl is used herein to denote an aromatic ring or fused ring structure of carbon atoms with one or more non-carbon atoms, such as oxygen, nitrogen, and sulfur, in the ring or in one or more of the rings in fused ring structures.
- examples are furanyl, pyranyl, thienyl, imidazyl, pyrrolyl, pyridyl, pyrazolyl, pyrazinyl, pyrimidinyl, indolyl, indazolyl, quinolyl, isoquinolyl, quinoxalyl, and quinazolinyl.
- biasing refers to an aryl, as defined above, where two aryl groups are joined by a direct bond or through an intervening alkyl group, preferably a lower alkyl group.
- fused aryl refers to a multicyclic ring system as included in the term “aryl,” and includes aryl groups and heteroaryl groups that are condensed. Examples are naphthyl, anthryl and phenanthryl. The bonds can be attached to any of the rings.
- Alkyl and heteroarylkyl refer to aryl and heteroaryl moieties, respectively, that are linked to a main structure by an intervening alkyl group, e.g., containing one or more methylene groups.
- fluorobenzyl refers to a benzyl group wherein the phenyl moiety is substituted with one or more fluorine atoms, including 2, 3, 4 and 5 fluorine atom
- halobenzyl refers to benzyl substituted with one or more different halogens, including fluorine, chlorine, bromine, and iodine (not astatine).
- sulfuride and thioether as used herein, alone or in combination, refer to a sulfur atom covalently linked to two atoms; the formal oxidation state of said sulfur is (II). These terms may be used interchangeably.
- sulfanyl refers to the— S--R group, wherein R may be a group such as: alkyl, alkenyl, alkynyl, aryl, alicyclic, heterocyclic, aryl, heteroaryl, arylalkyl and heteroarylalkyl, wherein the alkyl, alkenyl, alkynyl, aryl, alicyclic, heterocyclic, aryl, heteroaryl, arylalkyl and heteroarylalkyl groups may be optionally substituted.
- sulfanyl groups include methylsulfanyl (-SCH 3 ) and iso-propylsulfanyl (-SCH(CH 3 ) 2 ) and the like.
- sulfoxide refers to a sulfur atom covalently linked to three atoms, at least one of which is an oxygen atom; the formal oxidation state of said sulfur atom is (IV).
- sulfinyl refers to the groups - S(0) ⁇ R, wherein R may be, but is not limited to alkyl, alkenyl, alkynyl, aryl, alicyclic, heterocyclic, aryl, heteroaryl, arylalkyl and heteroarylalkyl, wherein the alky], alkenyl, alkynyl, aryl, alicycl ic, heterocycl ic, aryl, heteroaryl, arylalkyl and heteroarylalkyl groups may be optionally substituted.
- a non-limiting example of a sulfinyl group includes methylsulfinyl (-S(0)CH 3 ) and the like.
- sulfurone refers to a sulfur atom covalently linked to four atoms, at least two of which are oxygen atoms; the formal oxidation state of said sulfur atom is (VI).
- sulfonyl refers to the groups— S(02) ⁇ R, wherein R may be, but is not limited to, alkyl, alkenyl, alkynyl, aryl, alicyclic, heterocyclic, aryl, heteroaryl, arylalkyl and heteroarylalkyl, wherein the alkyl, alkenyl, alkynyl, aryl, alicyclic, heterocyclic, aryl, heteroaryl, arylalkyl and heteroarylalkyl groups may be optionally substituted.
- a non-limiting example of a sulfonyl group includes methylsulfonyl (-S(0 2 )CH 3 ) and the like.
- phosphite refers to a phosphorus atom covalently linked to three carbon atoms, wherein the formal oxidation state of said phosphorus is (III).
- phosphinyl as used herein, alone or in combination, refers to the monoradical derived from a phosphite group, as defined above.
- phosphonate refers to a phosphorus atom covalently linked to four atoms, three of which are oxygen and one of which is carbon wherein the formal oxidation state of said phosphorus is (V).
- phosphonyl as used herein, alone or in combination, refers to the monoradical derived from a phosphonate group, as defined above.
- phosphate refers to a phosphorus atom covalently linked to four oxygen atoms, wherein the formal oxidation state of said phosphorus is (V).
- phosphatidyl as used herein, alone or in combination, refers to the monoradical derived from a phosphate group, as defined above.
- ketone ester, ether, and acyl have their art recognized meanings.
- unit dosage form refers to physically d iscrete units su itable as unitary dosages for human and/or animal subjects, each unit containing a predetermined quantity of a compound (e.g., compositions or pharmaceutical compositions, as described herein) calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier or vehicle.
- a compound e.g., compositions or pharmaceutical compositions, as described herein
- the specifications for unit dosage forms depend on the particular compound employed, the route and frequency of administration, and the effect to be ach ieved, and the pharmacodynamics associated with each compound in the host.
- a “pharmaceutically acceptable excipient,” “pharmaceutically acceptable diluent,” “pharmaceutically acceptable carrier,” or “pharmaceutically acceptable adjuvant” means an excipient, diluent, carrier, and/or adjuvant that are useful in preparing a pharmaceutical composition that are generally safe, non-toxic and neither biologically nor otherwise undesirable, and include an excipient, diluent, carrier, and adjuvant that are acceptable for veterinary use and/or human pharmaceutical use.
- “A pharmaceutically acceptable excipient, diluent, carrier and/or adjuvant” as used in the specification and claims includes one and more such excipients, diluents, carriers, and adjuvants.
- a "pharmaceutical composition” is meant to encompass a composition or pharmaceutical composition suitable for administration to a subject, such as a mammal, especially a human.
- a “pharmaceutical composition” is sterile, and preferably free of contam inants that are capable of eliciting an undesirable response within the subject (e.g., the compound(s) in the pharmaceutical composition is pharmaceutical grade).
- Pharmaceutical compositions can be designed for administration to subjects or patients in need thereof via a number of different routes of administration including oral, intravenous, buccal, rectal, parenteral, intraperitoneal, intradermal, intracheal, intramuscular, subcutaneous, inhalational and the like.
- terapéuticaally effective amount refers to that amount of an embodiment of the composition or pharmaceutical composition being administered that will relieve to some extent one or more of the symptoms of the disease, i.e., infection, being treated, and/or that amount that will prevent, to some extent, one or more of the symptoms of the d isease, i.e., infection, that the host being treated has or is at risk of developing.
- “Pharmaceutically acceptable salt” refers to those salts that retain the biological effectiveness and optionally other properties of the free bases and that are obtained by reaction with inorganic or organic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p- toluenesulfonic ac id, sal icyl ic ac id, mal ic ac id, maleic acid, succin ic acid, tartaric acid, citric acid, and the l ike.
- inorganic or organic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p- toluenesulfonic ac id, sal icyl ic ac id, mal ic ac id,
- salts are within the scope of the present disclosure.
- Reference to a compound used in the composition or pharmaceutical composition of any of the formulas herein is understood to include reference to salts thereof, unless otherwise indicated.
- the term "salt(s)”, as employed herein, denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases.
- zwitterions inner salts
- Salts of the compounds of a compound may be formed, for example, by reacting the compound with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Embodiments of the compounds of the composition or pharmaceutical composition of the present disclosure that contain a basic moiety may form salts with a variety of organic and inorganic acids.
- Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides (formed with hydrochloric acid), hydrobromides (formed with hydrogen bromide), hydroiodides
- Embodiments of the compounds of the composition or pharmaceutical composition of the present disclosure that contain an acidic moiety may form salts with a variety of organic and inorganic bases.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as benzathines, dicyclohexylamines, hydrabamines (formed with N,N-bis(dehydroabietyl)ethylenediamine), N-methyl-D-glucamines, N-methyl-D-glucamides, t-butyl amines, and salts with amino acids such as arginine, lysine, and the like.
- Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g., benzyl and phenethyl bromides), and others.
- lower alkyl halides e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates
- Solvates of the compounds of the composition or pharmaceutical composition of the present disclosure are also contemplated herein.
- All stereoisomers of the compounds of the composition or pharmaceutical composition of the present disclosure such as those that may exist due to asymmetric carbons on the various substituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons) and diastereomeric forms are contemplated within the scope of this disclosure.
- Individual stereoisomers of the compounds of the disclosure may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers.
- the stereogenic centers of the compounds of the present disclosure can have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- prodrug refers to an inactive precursor of the compounds of the composition or pharmaceutical composition of the present disclosure that is converted into a biologically active form in vivo.
- Prodrugs are often useful because, in some situations, they may be easier to administer than the parent compound. They may, for instance, be bioavailable by oral administration whereas the parent compound is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug.
- a prodrug may be converted into the parent drug by various mechanisms, including enzymatic processes and metabolic hydrolysis. Harper, N.J. (1962). Drug Latentiation in Jucker, ed. Progress in Drug Research, 4:221 -294; orozowich et al. (1977). Application of Physical Organic Principles to Prodrug Design in E. B. Roche ed. Design of Biopharmaceutical Properties through Prodrugs and Analogs, APhA; Acad. Pharm. Sci.; E. B. Roche, ed.
- administration refers to introducing a composition of the present disclosure into a host.
- One preferred route of administration of the composition is oral administration.
- Another preferred route is intravenous administration.
- any route of administration such as topical, subcutaneous, peritoneal, intraarterial, inhalation, vaginal, rectal, nasal, introduction into the cerebrospinal fluid, or instillation into body compartments can be used.
- treat refers to acting upon a condition, a disease or a disorder with a composition to affect the condition, disease or disorder by improving or altering it.
- the improvement or alteration may include an improvement in symptoms or an alteration in the physiologic pathways associated with the condition, disease, or disorder.
- Treatment covers one or more treatments of a tumor or a disease in a host (e.g., a mammal, typically a human or non-human animal of veterinary interest), and includes: (a) reducing the risk of occurrence of the disease in a subject determined to be predisposed to the condition or disease but not yet diagnosed with it (b) impeding the development of the condition or disease, and/or (c) relieving the condition disease, e.g., causing regression of the condition or disease and/or relieving one or more disease symptoms.
- a host e.g., a mammal, typically a human or non-human animal of veterinary interest
- prophylactically treat or “prophylactically treating” refers completely or partially preventing (e.g., about 50% or more, about 60% or more, about 70% or more, about 80% or more, about 90% or more, about 95% or more, or about 99% or more) a condition, a disease, or a symptom thereof and/or may be therapeutic in terms of a partial or complete cure for a condition, a disease, and/or adverse effect attributable to the disease.
- the term "host,” “subject,” or “patient,” includes humans and mammals (e.g., mice, rats, pigs, cats, dogs, and horses). Typical hosts to which compounds of the present disclosure may be administered will be mammals, particularly primates, especially humans. For veterinary applications, a wide variety of subjects will be suitable, e.g., l ivestock such as cattle, sheep, goats, cows, swine, and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and domesticated animals particularly pets such as dogs and cats.
- l ivestock such as cattle, sheep, goats, cows, swine, and the like
- poultry such as chickens, ducks, geese, turkeys, and the like
- domesticated animals particularly pets such as dogs and cats.
- living host refers to a host noted above or another organism that is alive.
- living host refers to the entire host or organism and not just a part excised (e.g., a liver or other organ) from the living host.
- compositions including a beta-lactamase inhibitor provides compositions including a beta-lactamase inhibitor, pharmaceutical compositions including a beta-lactamase inhibitor, methods of treatment of a condition (e.g., infection) or disease, methods of treatment using compositions or pharmaceutical compositions, and the like.
- An embodiment of the present disclosure can be used in combination (e.g., in the same composition or separately) to treat resistant strains of bacteria (e.g., MRSA). Additional details are described in the Examples.
- CTX-M beta-lactamases are the main resistance mechanisms against extended spectrum beta-lactam antibiotics in many regions of the world. Inhibitors against these proteins can restore the efficacy of beta-lactam antibiotics against resistant bacteria such as MRSA (superbug). Embodiments of the present disclosure describe novel inhibitors against CTX-M beta-lactamases.
- embodiments of the present disclosure include beta-lactamase inhibitors that can be used in combination with a beta-lactam antibiotic to treat resistant strands of bacteria.
- the beta-lactam antibiotic can include penicillin and penicillin derivatives, cephalosporin and cephalosporin derivatives, monobactam and monobactam derivatives, carbapenem and carbapenem derivatives, and a combination thereof.
- the derivatives described regarding a beta-lactam antibiotic derivatives are those known in the art.
- An embodiment of the present disclosure includes a composition and pharmaceutical composition including a beta-lactamase inhibitor.
- the pharmaceutical composition and the method of treatment e.g., of an infection such as one directly or indirectly caused by a bacterial infection
- the pharmaceutical composition and the method of treatment includes a therapeutically effective amount of a beta-lactamase inhibitor, or a pharmaceutically acceptable salt of the beta-lactamase inhibitor, and a pharmaceutically acceptable carrier, to treat a condition (e.g., bacterial infection).
- the bacterial infections can be caused by one or more types of bacteria, in particular, drug or multidrug resistant bacteria.
- the bacteria can include, but is not limited to, Staphylococcus aureus, Streptococcus pyogenes,
- Streptococcus pneumonia Enterococcus faecalis, Enterococcus faecium, Pseudomonas aeruginosa, Clostridium difficile, Escherichia coli, Salmonella, Acinetobacter baumannii, Mycobacterium tuberculosis, or a combination thereof.
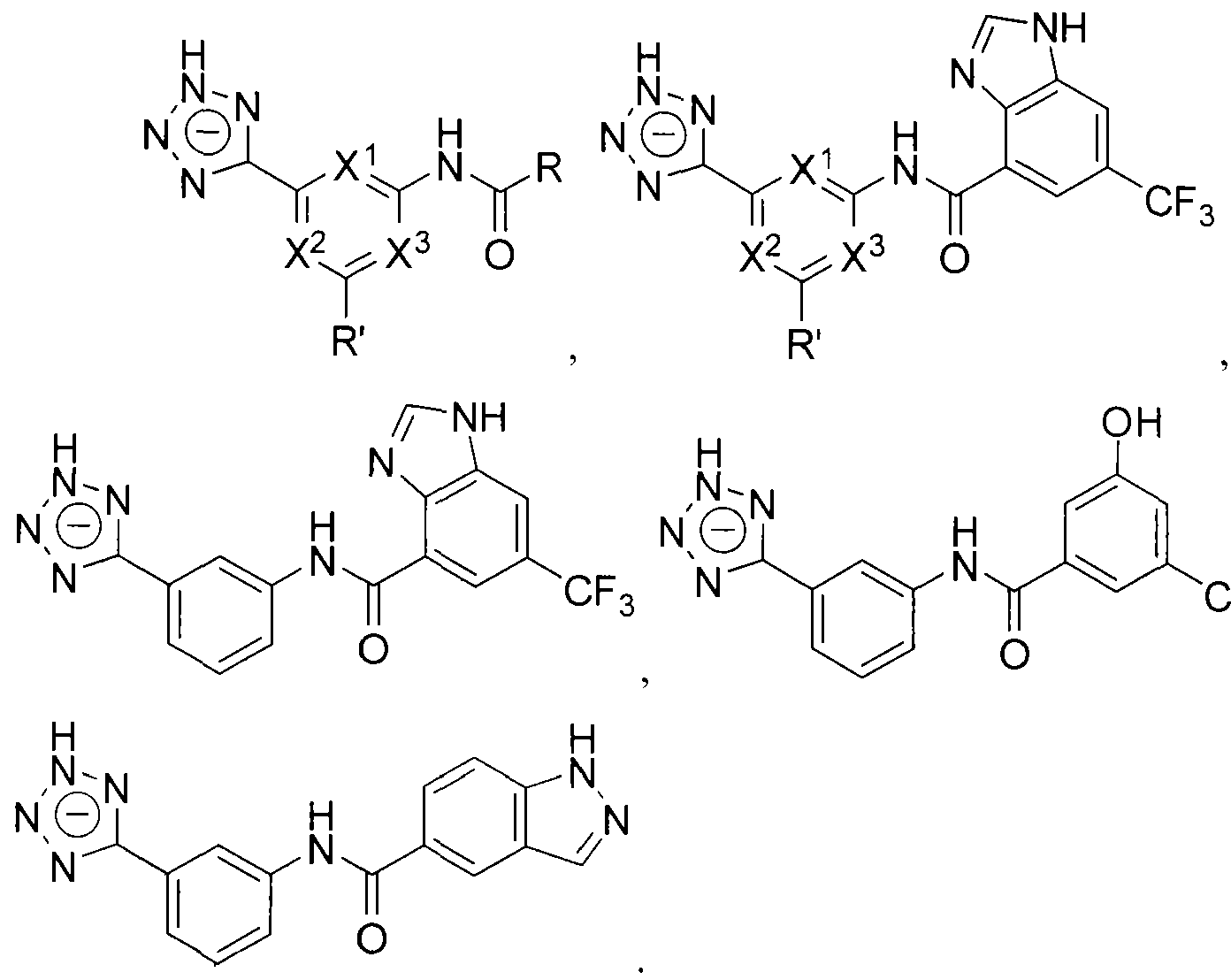
- the beta-lactamase inhibitor can include structures A, A', A", and A'" as shown below.
- Z can be selected from one of the following structures:
- R' can be H, a halogen, a substituted or unsubstituted alkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted cycloalkyi, a substituted or unsubstituted cycloalkenyl, a substituted or unsubstituted heteroaryl, a substituted or unsubstituted biaryl, a substituted or unsubstituted fused aryl, a substituted or unsubstituted alkenyl, or a substituted or unsubstituted alkynyl.
- R can be an alkyl group (e.g., C I to C5 hydrocarbons such as methyl, ethyl, and the like), an aryl group, a heteroaryl group, or a cyclic or hetero (e.g., O, N) cycl ic group (e.g., C I to C7 cyclic hydrocarbons).
- the R groups described herein can be substituted or unsubstituted.
- Q can be O or S.
- the beta- lactamase inhibitor can include structures:
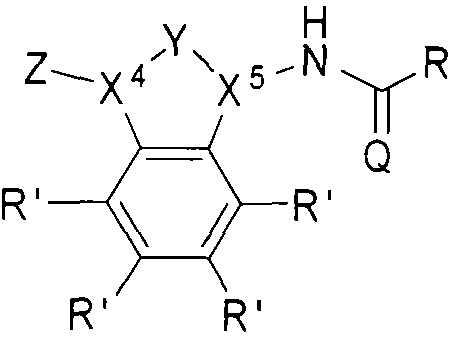
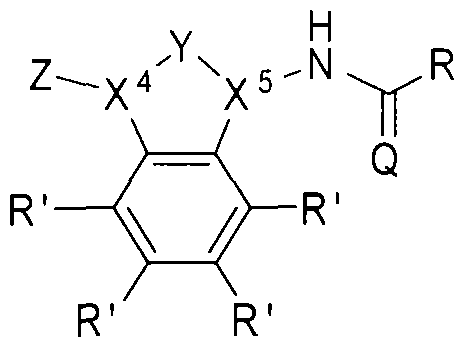
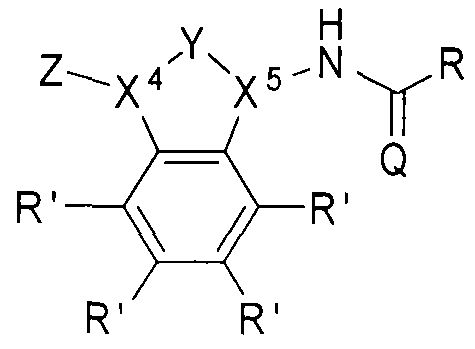
- the beta-lactamase inhibitor can include structure B, as shown below.
- Z can be selected from one of the following:
- X and X can each be independently selected from CH or .
- group Y can be absent, in which case X 4 and X 5 can each be independently selected from CH 2 or NH.
- R' can be H, a halogen, a substituted or unsubstituted alkyl, a substituted or unsubstituted aryl, a substituted or unsubstituted cycloalkyi, a substituted or unsubstituted cycloalkenyl, a substituted or unsubstituted heteroaryl, a substituted or unsubstituted biaryl, a substituted or unsubstituted fused aryl, a substituted or unsubstituted alkenyl, or a substituted or unsubstituted alkynyl.
- R can be an alkyl group (e.g., CI to C5 hydrocarbons such as methyl, ethyl, and the l ike), an aryl group, a heteroaryl group, or a cyclic or hetero (e.g., O, N) cyclic group (e.g., C I to C7 cyclic hydrocarbons).
- the R groups described herein can be substituted or unsubstituted.
- Q can be O or S.
- -lactamase inhibitor can include structures:
- R' group in any of the structures listed above can include the structures listed in Tables 1 -3 ( Figures 4-6, respectively) in Example 1 .
- the compounds covered by compound A exclude compound A'" for the composition and pharmaceutical compositions.
- methods include the compounds covered by compound A including compound A'" .
- methods include the compounds covered by compound A excluding compound A'".
- the therapeutically effective amount to result in uptake of the beta-lactamase inhibitor and/or antibiotic (e.g., each either alone or in combination with one another) into the host will depend upon a variety of factors, including for example, the age, body weight, general health, sex, and diet of the host; the time of administration; the route of administration; the rate of excretion of the specific compound employed; the duration of the treatment; the existence of other drugs used in combination or coincidental with the specific composition employed; and like factors well known in the medical arts.
- Embodiments of the present disclosure include a beta-lactamase inhibitor as identified herein and can be formulated with one or more pharmaceutically acceptable excipients, diluents, carriers and/or adjuvants.
- embodiments of the present disclosure include a beta-lactamase inhibitor formulated with one or more pharmaceutically acceptable auxiliary substances.
- beta-lactamase inhibitor can be formulated with one or more pharmaceutical ly acceptable excipients, diluents, carriers, and/or adjuvants to provide an embodiment of a composition of the present disclosure.
- the pharmaceutically acceptable excipients such as vehicles, adjuvants, carriers or diluents, are readily available to the public.
- pharmaceutically acceptable auxiliary substances such as pH adjusting and buffering agents, tonicity adjusting agents, stabilizers, wetting agents and the like, are readily available to the public.
- the beta-lactamase inhibitor can be adm inistered to the host using any means capable of resulting in the desired effect.
- the beta-lactamase inhibitor can be incorporated into a variety of formulations for therapeutic administration.
- the beta-lactamase inhibitor can be formulated into
- compositions by combination with appropriate, pharmaceutically acceptable carriers or diluents, and may be formulated into preparations in solid, semi-solid, liquid or gaseous forms, such as tablets, capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants and aerosols.
- the beta-lactamase inhibitor may be administered in the form of its pharmaceutically acceptable salts, or a subject active composition may be used alone or in appropriate association, as well as in combination, with other pharmaceutically active compounds.
- a subject active composition may be used alone or in appropriate association, as well as in combination, with other pharmaceutically active compounds.
- the following methods and excipients are merely exemplary and are in no way limiting.
- the beta-lactamase inhibitor can be used alone or in combination with appropriate additives to make tablets, powders, granules or capsules, for example, with conventional additives, such as lactose, mannitol, corn starch or potato starch; with binders, such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins; with disintegrators, such as corn starch, potato starch or sodium
- Embodiments of the beta-lactamase inhibitor can be formulated into preparations for injection by dissolving, suspending or emulsifying them in an aqueous or nonaqueous solvent, such as vegetable or other similar oils, synthetic aliphatic acid glycerides, esters of higher aliphatic acids or propylene glycol; and if desired, with conventional additives such as solubil izers, isotonic agents, suspending agents, emu lsifying agents, stabilizers and preservatives.
- Embodiments of the beta-lactamase inhibitor can be utilized in aerosol formulation to be adm inistered via inhalation.
- Embodiments of the beta-lactamase inhibitor can be formulated into pressurized acceptable propellants such as dichlorodifluoromethane, propane, nitrogen and the like.
- embodiments of the beta-lactamase inhibitor can be made into suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases.
- Embodiments of the beta-lactamase inhibitor can be administered rectally via a suppository.
- the suppository can include vehicles such as cocoa butter, carbowaxes and polyethylene glycols, wh ich melt at body temperature, yet are solidified at room temperature.
- Unit dosage forms for oral or rectal admin istration such as syrups, elixirs, and suspensions, may be provided wherein each dosage unit, for example, teaspoonful, tablespoonful, tablet or suppository, contains a predetermined amount of the composition containing one or more compositions.
- unit dosage forms for injection or intravenous administration may comprise the beta-lactamase inhibitor in a composition as a solution in sterile water, normal saline or another pharmaceutically acceptable carrier.
- Embodiments of the beta-lactamase inhibitor can be formulated in an injectable composition in accordance with the disclosure.
- injectable compositions are prepared as l iquid solutions or suspensions; sol id forms suitable for solution in, or suspension in, liquid vehicles prior to injection may also be prepared.
- the preparation may also be emulsified or the active ingredient (triamino-pyridine derivative and/or the labeled triamino- pyridine derivative) encapsulated in liposome vehicles in accordance with the present disclosure.
- the beta-lactamase inhibitor can be formulated for del ivery by a continuous delivery system.
- continuous delivery system is used interchangeably herein with “controlled delivery system” and encompasses continuous (e.g., controlled) delivery devices (e.g., pumps) in combination with catheters, injection devices, and the l ike, a wide variety of which are known in the art.
- Continuous delivery devices e.g., pumps
- Mechanical or electromechanical infusion pumps can also be suitable for use with the present disclosure. Examples of such devices include those described in, for example, U.S. Pat. Nos.
- beta-lactamase inhibitor can be in a liquid formulation in a drug-impermeable reservoir, and is delivered in a continuous fashion to the individual.
- the drug delivery system is an at least partially implantable device.
- the implantable device can be implanted at any suitable implantation site using methods and devices well known in the art.
- An implantation site is a site within the body of a subject at which a drug delivery device is introduced and positioned.
- Implantation sites include, but are not necessarily limited to, a subdermal, subcutaneous, intramuscular, or other suitable site within a subject's body. Subcutaneous implantation sites are used in some embodiments because of convenience in implantation and removal of the drug delivery device.
- Drug release devices suitable for use in the disclosure may be based on any of a variety of modes of operation.
- the drug release device can be based upon a diffusive system, a convective system, or an erodible system (e.g., an erosion-based system).
- the drug release device can be an electrochemical pump, osmotic pump, an electroosmotic pump, a vapor pressure pump, or osmotic bursting matrix, e.g., where the drug is incorporated into a polymer and the polymer provides for release of drug formulation concomitant with degradation of a drug-impregnated polymeric material ⁇ e.g. , a
- the drug release device is based upon an electrodiffusion system, an electrolytic pump, an effervescent pump, a piezoelectric pump, a hydrolytic system, etc.
- Drug release devices based upon a mechanical or electromechanical infusion pump can also be suitable for use with the present disclosure. Examples of such devices include those described in, for example, U.S. Pat. Nos. 4,692, 147; 4,360,019; 4,487,603; 4,360,019; 4,725,852, and the like.
- a subject treatment method can be accomplished using any of a variety of refillable, non-exchangeable pump systems. Pumps and other convective systems are generally preferred due to their generally more consistent, controlled release over time. Osmotic pumps are used in some embodiments due to their combined advantages of more consistent controlled release and relatively small size (see, e.g., PCT published application no. WO 97/27840 and U.S. Pat. Nos.
- Exemplary osmotically-driven devices suitable for use in the disclosure include, but are not necessarily limited to, those described in U.S. Pat. Nos. 3,760,984; 3,845,770; 3,916,899; 3,923,426; 3,987,790; 3,995,631 ; 3,916,899; 4,016,880; 4,036,228; 4, 1 1 1 ,202; 4, 1 1 1 1 ,203; 4,203,440; 4,203,442; 4,210, 139; 4,327,725; 4,627,850; 4,865,845; 5,057,318; 5,059,423; 5,1 12,614; 5, 137,727; 5,234,692; 5,234,693 ; 5,728,396; and the like.
- the drug delivery device is an implantable device.
- the drug delivery device can be implanted at any suitable implantation site using methods and devices well known in the art.
- an implantation site is a site within the body of a subject at which a drug delivery device is introduced and positioned. Implantation sites include, but are not necessarily limited to a subdermal, subcutaneous, intramuscular, or other suitable site within a subject's body.
- an active agent e.g., the beta- lactamase inhibitor
- an implantable drug delivery system e.g., a system that is programmable to provide for administration of the agent.
- implantable drug delivery system e.g., a system that is programmable to provide for administration of the agent.
- implantable drug delivery system e.g., a system that is programmable to provide for administration of the agent.
- implantable drug delivery system e.g., a system that is programmable to provide for administration of the agent.
- Exemplary programmable, implantable systems include implantable infusion pumps.
- Exemplary implantable infusion pumps, or devices useful in connection with such pumps, are described in, for example, U.S. Pat. Nos.
- a further exemplary device that can be adapted for the present disclosure is the Synchromed infusion pump (Medtronic).
- Suitable excipient vehicles for the beta-lactamase inhibitor are, for example, water, saline, dextrose, glycerol, ethanol, or the like, and combinations thereof.
- the vehicle may contain minor amounts of auxiliary substances such as wetting or emulsifying agents or pH buffering agents.
- Methods of preparing such dosage forms are known, or will be apparent upon consideration of this disclosure, to those skilled in the art. See, e.g., Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania, 17th edition, 1985.
- the composition or formulation to be administered will, in any event, contain a quantity of the beta-lactamase inhibitor adequate to achieve the desired state in the subject being treated.
- compositions of the present disclosure can include those that comprise a sustained- release or controlled release matrix.
- embodiments of the present disclosure can be used in conjunction with other treatments that use sustained-release formulations.
- a sustained-release matrix is a matrix made of materials, usually polymers, which are degradable by enzymatic or acid-based hydrolysis or by dissolution. Once inserted into the body, the matrix is acted upon by enzymes and body fluids.
- a sustained-release matrix desirably is chosen from biocompatible materials such as liposomes, polylactides (polylactic acid), polyglycolide (polymer of glycolic acid), polylactide co-glycolide (copolymers of lactic acid and glycolic acid), polyanhydrides, poly(ortho)esters, polypeptides, hyaluronic acid, collagen, chondroitin sulfate, carboxcylic acids, fatty acids, phospholipids,
- biocompatible materials such as liposomes, polylactides (polylactic acid), polyglycolide (polymer of glycolic acid), polylactide co-glycolide (copolymers of lactic acid and glycolic acid), polyanhydrides, poly(ortho)esters, polypeptides, hyaluronic acid, collagen, chondroitin sulfate, carboxcylic acids, fatty acids, phospholipids,
- polysaccharides polysaccharides, nucleic acids, polyamino acids, amino acids such as phenylalanine, tyrosine, isoleucine, polynucleotides, polyvinyl propylene, polyvinylpyrrolidone and silicone.
- Illustrative biodegradable matrices include a polylactide matrix, a polyglycolide matrix, and a polylactide co-glycolide (co-polymers of lactic acid and glycolic acid) matrix.
- the pharmaceutical composition of the present disclosure (as well as combination compositions) can be delivered in a controlled release system.
- the beta-lactamase inhibitor may be administered using intravenous infusion, an implantable osmotic pump, a transdermal patch, liposomes, or other modes of administration.
- a pump may be used (Sefton ( 1987). CRC Crit. Ref. Biomed. Eng.
- a controlled release system is placed in proximity of the therapeutic target thus requiring only a fraction of the systemic dose.
- a controlled release system is placed in proximity of the therapeutic target, thus requiring only a fraction of the systemic.
- Other controlled release systems are discussed in the review by Langer ( 1 990). Science 249: 1527- 1 533.
- compositions of the present disclosure include those formed by impregnation of the beta-lactamase inhibitor described herein into absorptive materials, such as sutures, bandages, and gauze, or coated onto the surface of solid phase materials, such as surgical staples, zippers and catheters to deliver the compositions.
- absorptive materials such as sutures, bandages, and gauze
- solid phase materials such as surgical staples, zippers and catheters.
- Embodiments of the beta-lactamase inhibitor can be administered to a host in one or more doses.
- dose levels can vary as a function of the specific the beta-lactamase inhibitor administered, the severity of the symptoms and the susceptibility of the subject to side effects.
- Preferred dosages for a given compound are readily determinable by those of skill in the art by a variety of means.
- multiple doses of the beta-lactamase inhibitor are administered.
- the frequency of administration of the beta-lactamase inhibitor can vary depending on any of a variety of factors, e.g., severity of the symptoms, and the like.
- the beta-lactamase inhibitor can be administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw), three times per week (tiw), four times per week, five times per week, six times per week, every other day (qod), daily (qd), twice a day (qid), or three times a day (tid).
- the beta-lactamase inhibitor is administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw), three times per week (tiw), four times per week, five times per week, six times per week, every other day (qod), daily (qd), twice a day (qid), or three times a day (tid).
- the beta-lactamase inhibitor is administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw),
- the duration of administration of the beta-lactamase inhibitor analogue can vary, depending on any of a variety of factors, e.g., patient response, etc.
- the beta-lactamase inhibitor in combination or separately can be administered over a period of time of about one day to one week, about two weeks to four weeks, about one month to two months, about two months to four months, about four months to six months, about six months to eight months, about eight months to 1 year, about 1 year to 2 years, or about 2 years to 4 years, or more.
- Embodiments of the present disclosure provide methods and compositions for the administration of the active agent (e.g., the beta-lactamase inhibitor) to a host (e.g., a human) using any available method and route suitable for drug delivery, including in vivo and ex vivo methods, as well as systemic and localized routes of administration.
- the active agent e.g., the beta-lactamase inhibitor
- a host e.g., a human
- Routes of administration include intranasal, intramuscular, intratracheal,
- An active agent e.g., the beta-lactamase inhibitor
- routes of administration may be combined, if desired, or adjusted depending upon the agent and/or the desired effect.
- An active agent e.g., the beta-lactamase inhibitor
- Embodiments of the beta-lactamase inhibitor can be administered to a host using available conventional methods and routes suitable for delivery of conventional drugs, including systemic or localized routes.
- routes of administration contemplated by the disclosure include, but are not limited to, enteral, parenteral, or inhalational routes.
- Parenteral routes of administration other than inhalation administration include, but are not limited to, topical, transdermal, subcutaneous, intramuscular, intraorbital,
- intracapsular, intraspinal, intrasternal, and intravenous routes i.e., any route of administration other than through the alimentary canal.
- Parenteral administration can be conducted to effect systemic or local delivery of the beta-lactamase inhibitor. Where systemic delivery is desired, administration typically involves invasive or systemically absorbed topical or mucosal administration of pharmaceutical preparations.
- the beta-lactamase inhibitor can also be delivered to the subject by enteral administration.
- Enteral routes of administration include, but are not limited to, oral and rectal (e.g., using a suppository) delivery.
- Methods of administration of the beta-lactamase inhibitor through the skin or mucosa include, but are not limited to, topical application of a suitable pharmaceutical preparation, transdermal transmission, injection and epidermal administration.
- a suitable pharmaceutical preparation for transdermal transmission, absorption promoters or iontophoresis are suitable methods.
- Iontophoretic transmission may be accomplished using commercially available "patches" that deliver their product continuously via electric pulses through unbroken skin for periods of several days or more.
- Beta-lactam compounds such as penicillins are the most widely used antibiotics due to their effective inhibition of the transpeptidases required for bacterial cell wall synthesis' "3 . Beta-lactamases catalyze ⁇ -lactam hydrolysis and are primary mediators of bacterial resistance to these compounds 4 ' 5 . There are four ⁇ -lactamase families, Classes A to D, among which Classes A and C are the most commonly observed in the clinic 6 ' 7 .
- CTX-M is a new group of Class A ⁇ -lactamases that is particularly effective against the extended spectrum ⁇ -lactam antibiotics such as cefotaxime 8" 12 , which itself was developed to counter bacterial resistance to first-generation penicillins and cephalosporins.
- ESBL extended spectrum beta-lactamase
- CTX-M has become the most frequently observed ESBL in many regions of the world.
- ⁇ -lactamase inhibitors in combination with a ⁇ -lactam antibiotic are well- established strategies to counter resistance' 3 .
- Existing ⁇ -lactamase inhibitors e.g., clavulanic acid
- ⁇ -lactam ring making them susceptible to resistance stemming from up-regulation of ⁇ -lactamase production, selection for new ⁇ -lactamases, and other mechanisms evolved over millions of years' chemical warfare between bacteria and ⁇ -lactam producing microorganisms 14"1 7 .
- these problems can be overcome by developing structurally novel ( ⁇ - ⁇ -lactam) inhibitors of ⁇ -lactamases.
- the tetrazole-based chemotype appealed to us for several reasons.
- the tetrazole group has excellent shape and electrostatic complementarity with the active site subpocket of CTX- M that usually binds the C(3)4' carboxylate of traditional ⁇ -lactam antibiotics (Fig. 1).
- the tetrazole ring is a well-known binding bioisostere for carboxylate groups that often possesses more favorable pharmacokinetics properties.
- the highest affinity compound from the series targeting Prol 67 was the 3-trifluoromethyl compound 10, with a Ki of 2.4 ⁇ (Table 1 , Figure 4).
- Analogs 6-8 bearing less hydrophobic and non-spheroid substituents had similar ligand efficiency as 1 but superior LipE values (1 .37 for 1 vs. 2.51 for 7).
- the parameter LipE (defined as logKi - clogP) 24 provides a measure of binding affinity improvement achieved while retaining favorable physiochemical properties. A surprising result was that even large substituents (2-pyrimidyl, compound 11) could be tolerated at the 3-position, although ligand efficiency suffers.
- a complex crystal structure of 11 confirmed a similar binding pose as 1 (see below) so the reduced ligand efficiency of this analog perhaps reflects a steric clash and/or unfavorable desolvation energy associated with the burial of a pyrimidine ring nitrogen atom.
- Figure 2 shows the X-ray crystal structures of compounds 4, 10, and 11 in the active site of CTX-M-9; these compounds were designed to make significant non-polar interactions with Pro 167.
- the size increase in the bulkier side substituents such as trifluoromethyl is demonstrated in their larger electron density volumes, compared with that of the fluorine atom in 1.
- the atoms of the new ligands make similar contacts with the surrounding active site atoms as does 1.
- compounds 4, 10, and 11 (Fig. 2 a-c) all form hydrogen bonds between the tetrazole ring and Thr235, Ser237, and Serl 30 from the protein, which is similar to compound 1 ( Figure 1 , Table 1 ).
- the three branched fluorine atoms of compound 10 are in close vdw contacts with these protein carbon atoms, which are approximately 3.4-3.8 A away.
- the pyrimidine ring forms a water-mediated interaction with Asp240 and induces Asp240 to adopt a new conformation (Fig. 2).
- This water-mediated contact exists in the complex structure only with partial occupancy, as suggested by the relatively weak electron density of the water (2 ⁇ ) and the presence of two Asp240 conformations including the one previously observed in apo and other complex structures.
- vdw contact observed between the carbon atoms of the pyrimidine ring in 11 and Pro l 67Cy, Thrl 68Cy, Thrl 71 Cy, which are 3.3-3.6 A distant.
- the affinity of compound 11 is less than for 4 or 10; this may be due to the burial of a polar pyrimidine ring nitrogen atom and electrostatic repulsion between this nitrogen and Pro l 670.
- the vdw contacts described above may be slightly too close for the optimal carbon-carbon distance in vdw interaction ( ⁇ 4 A) and thus suggest possible minor steric clash.
- Crystal structures were also obtained for compounds designed to establish polar interactions with Asp240, including compounds 12, 16, and 18. Again the core structure of these compounds, including the tetrazole ring and the amide bond, establishes contacts with Serl 30, Thr235, Ser237, Asn l 04 and Asn l 32 similar to compound 1 (Fig.3 a-c). Both compounds 16 and 18 form a direct hydrogen bond with Asp240 as designed. Compound 16 has in addition a favorable contact between N-1 /C-2 N-3 of the benzimidazole ring and the main chain atoms around Gly238, while compound 18 establ ishes more vdw interactions with Pro 167 and Thrl 68.
- Benzimidazole 16 and indaole 15 both contact Asp240 through a hydrogen bond from N- l .
- the additional ring nitrogen (N-3) in 16 appears to form a water- mediated hydrogen bonding contact with Ser237 (Fig.3b).
- the electron density for the water molecule contacting N-3 in 16 is weaker (2.4 ⁇ ) than other structural waters in the active site, perhaps explaining why the presence of this additional interaction in 16 does not significantly improve potency in comparison to 15.
- the modest additional affinity of compound 16 may originate from an intramolecular hydrogen bond between " N-3 and the proximal amide N-H, which stabilizes the conformation conducive to a hydrogen bond between the compound and Asp240.
- Figure 3c shows the discrepancy between the designed interaction of compound 12 with Asp240 (in cyan) and its actual interactions observed in the crystal structure (in light gray).
- compound 12 we designed compound 12 to form a salt bridge with Asp240.
- the X-ray crystal structure reveals the actual binding pose in which a water-mediated hydrogen bond is formed between the positively charged side chain and Asp240.
- the new side chain is cradled in the small pocket surrounding Pro 167, underscoring once again the potential of this binding surface in establishing new interactions with future inhibitors.
- methoxyimino group nestles comfortably in the subpocket around Pro 167.
- such interactions cause small shifts in atom positions for residues in this area (e.g., -0.5 ⁇ for Asp240Ca), a conformational change not observed in complex structures with smaller substrates such as benzylpenicillin.
- Similar hydrogen bonds with Asp240 have also been found in previous complex structures with boronic acid inhibitors 9 ' l2 .
- Pro 167 and Asp240 are conserved in other CTX-M type enzymes such as Toho- 1 25"27 .
- both Glu240 and Asp240 present similar features in the protein binding pocket, including the net negative charge and the nearly identical positioning of one oxygen atom from the carboxylate group. Comparing the complex structures between a ceftazidime- like boronic acid inhibitor and CTX-M-9 (pdb ID, 1 YLY) to that of the same compound with TEM- 1 (pdb ID 1 M40) shows that the aminothiazole ring of the inhibitor is placed in similar positions and forms a hydrogen bond with residue 240 in both structures 9 ' 28 .
- Thr235 and Gly 236 are conserved in AmpC (Thr3 16 and Gly317).
- Tyrl 50 a key catalytic residue in AmpC, places its hydroxyl group in a position similar to that of Serl 30 in CTX-M.
- Other common features shared by the active sites of Class A and C enzymes may further allow the design of inhibitors with broader spectrum. For instance, existing covalent inhibitors against both classes of enzymes almost invariantly place an oxygen atom in the oxyanion hole formed by two backbone amide groups. This binding hot spot is occupied instead by a water molecule in the complex structures of our current tetrazole-based inhibitors.
- tetrazoles-type inhibitors that suitably occupy the oxyanion hole may expand the utility of this chemotype to target a wider range of ⁇ -lactamases. Such expanded spectrum compounds may also have greater potential for cellular activity against resistant bacteria. So far the inhibitors from the current study were unable to reverse ⁇ -lactam resistance in E. coli strains expressing CTX-M ⁇ -lactamase (data not shown). Whether this reflects a lack of sufficient potency or other factors such as poor permeability or active efflux from the bacterium are questions we are actively pursuing.
- Coupl ing constants J are reported in hertz (Hz).
- the known compounds 1 , 2, 3, 4, 6, 8, 10 and 13 30 were prepared according the the general procedures and/or were obtained from commercial sources (Ryan Scientific, TimTec). All other reagents and solvents were purchased from Aldrich Chemical, Acros Organics, Enamine, Alfa Aesar, Apollo Scientific and used as received . Air and/or moisture sensitive reactions were carried out under an argon atmosphere in oven-dried glassware using anhydrous solvents from commercial suppliers. Air and/or moisture sensitive reagents were transferred via syringe or cannula and were introduced into reaction vessels through rubber septa. Solvent removal was accomplished with a rotary evaporator at ca. 10-50 Torr.
- Microwave heating was accomplished using a CEM reaction microwave. Hydrogenation reactions were carried out with a ThalesNano H-Cube hydrogenator.
- the reaction mixture is diluted with water (2 m L) and after adj usting the pH to ⁇ 2 with I N HCI, the mixture is extracted with ethyl acetate. The organic extracts are washed with brine, dried over magnesium sulfate and concentrated under reduced pressure. The crude material thus obtained is purified by reverse phase HPLC to afford the desired product.
- the reaction mixture is diluted with water (2 mL) and after approximately adjusting the pH to 4 with IN HC1, the mixture was extracted with ethyl acetate. The organic extracts were washed with brine, dried over magnesium sulfate and concentrated under reduced pressure. The crude material was purified by reverse phase HPLC to afford the product as a trifluoroacetic acid salt in 5% yield.
- 2-amino 3-nitro-5-trifluoromethylbenzoic acid 25.
- Commercially available 2- chloro-3-nitro-5-trifluoromethylbenzoic acid (0.10 g, 0.37 mmol) and aqueous ammonium hydroxide (2 mL) were heated in a sealed tube in a CEM microwave at 120 ° C for an hour. After cooling, the pH was adjusted to 2 with IN HCI. The precipitate was filtered and dried to obtain 2-amino 3-nitro-5-trifluoromethylbenzoic acid as a yellow solid (80 mg). This material was used in the next step without further purification.
- ⁇ NMR (DMSO-dis) ⁇ 8.49 (s, 1 H), 8.32 (s, 1 H).
- An oven-dried vial was charged with N-[(l R,3S)-3-cyano- 2,3-dihydro-l H-inden-l -yl]-3-(trifluoromethyl)benzamide (36 mg, 0.1 1 mmol), sodium azide (14 mg, 2.0 mmol), zinc bromide ( 12 mg, 0.055 mmol) in a 1 :2 mixture of 2-propanol/water (3 mL) was heated to reflux for 18 h.
- 3-(trifluoromethyl) benzoic acid 0.1 g, 0.52 mmol
- dichloromethane 5.0 ml
- l-Chloro-N,N,2-trimethyl-l -propenylamine 0.077 mL, 0.58 mmol
- the reaction mixture was diluted with water (2 mL) and extracted with ethyl acetate. The organic extract was washed with water, brine, dried over magnesium sulfate and concentrated under reduced pressure. The crude methyl 3-[3-(trifIuoromethyl)benzamido]benzoate was used without further purification.
- N-[3-(Hydroxycarbamoyl)phenyl]-3-(trifluoromethyl)benzamide (40) A solution of N- ⁇ 3-[(Benzyloxy)carbamoyl]phenyl ⁇ -3-(trifluoromethyl)benzamide (20 mg, 0.048 mmol) in methanol was passed through a Pd/C cartridge ( 10 wt%) at a flow rate of 1 mL/min using the H-Cube hydrogenation system. The solution was concentrated under reduced pressure and purified by reverse phase HPLC to obtain the title compound in 41 % yield. ⁇ NMR
- N-(3-Nitrophenyl)-3-(trifluoromethyl)benzamide (41) An oven-dried vial charged with 3-nitroaniline (100 mg, 0.72 mmol), 3-(trifluoromethyl) benzoic acid (138 mg, 0.72 mmol), 0-(7-azabenzotriazol- l -yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (300 mg, 0.79 mmol) and ⁇ , ⁇ '-diisopropylethylamine (0.25 mL, 1 .44 mmol) in ⁇ , ⁇ - dimethylformamide (2 mL) was stirred at room temperature for 18 h.
- CTX-M-9 a class A ⁇ -lactamase that we have previously studied, was used to represent the CTX-M family.
- the protein was purified as previously described 31 and crystallized in 1 .2-1 .6M potassium phosphate buffer (pH 8.3) from hanging drops at 20°C.
- the final concentration of the protein in the drop ranged from 6.5 mg ml "1 to 9mg ml "1 .
- the complex crystals were obtained through soaking methods. Based on the variability in terms of solubility and affinity, compound soaking times varied considerably, from 1 hour to 24 hours.
- CTX-M activity was measured using the ⁇ -lactam substrate nitrocefin in l OOmM Tris-HCl (pH 7.0, with 0.01 % v/v Triton X- 100) and monitored using an Hewlett-Packard spectrophotometer at 480 nM wavelength. Nitrocefin was 50 ⁇ in the inhibition assays. The K m of nitrocefin for CTX-M was determined to be 24 ⁇ . The compounds were synthesized as previously described or purchased from the company Chembridge, and assayed without further purification. The highest concentrations at which the compounds were tested were up to 1 -3 mM (depending on their solubility) in IC50 experiment. The reaction was initiated by adding protein to the reaction buffer last. References, each of which is incorporated herein by reference:
- ratios, concentrations, amounts, and other numerical data may be expressed herein in a range format. It is to be understood that such a range format is used for convenience and brevity, and thus, should be interpreted in a flexible manner to include not only the numerical values explicitly recited as the limits of the range, but also to include all the individual numerical values or sub-ranges encompassed within that range as if each numerical value and sub-range is explicitly recited.
- a concentration range of "about 0.1 % to about 5%” should be interpreted to include not only the explicitly recited concentration of about 0.1 wt% to about 5 wt%, but also include individual concentrations (e.g., 1 %, 2%, 3%, and 4%) and the sub-ranges (e.g., 0.5%, 1 .1 %, 2.2%, 3.3%, and 4.4%) within the indicated range.
- the term “about” can include traditional rounding according to significant figures of the numerical value.
- the phrase “about 'x' to 'y '” includes “about 'x' to about 'y” ⁇
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
Description

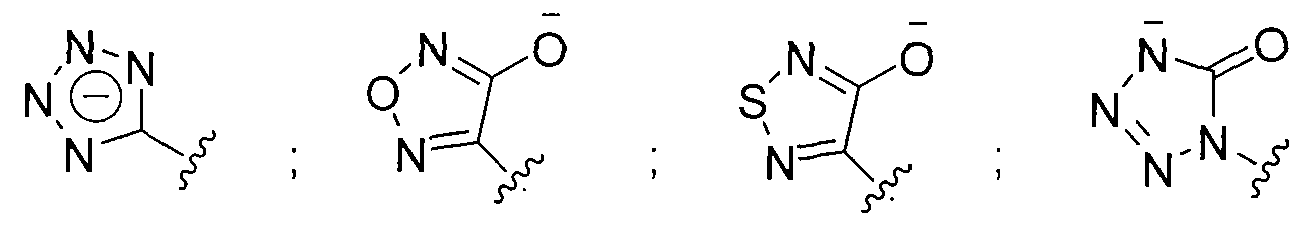







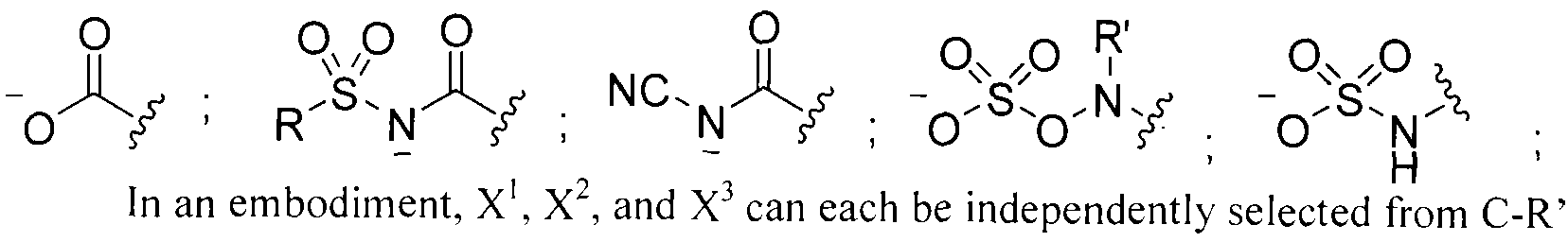








Claims






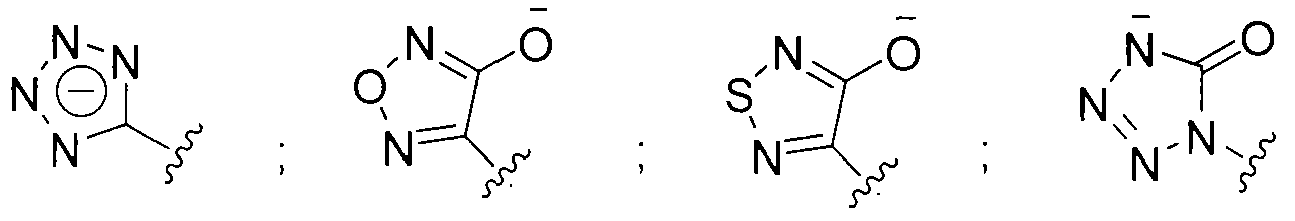






















Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380013340.1A CN104159587A (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
| US14/370,715 US9556131B2 (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
| AU2013207510A AU2013207510A1 (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
| BR112014016804A BR112014016804A8 (en) | 2012-01-06 | 2013-01-04 | compositions, methods of use and methods of treatment |
| MX2014008278A MX2014008278A (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment. |
| EP13733664.0A EP2800568A4 (en) | 2012-01-06 | 2013-01-04 | COMPOSITIONS, METHODS OF USE, AND METHODS OF TREATMENT |
| JP2014551327A JP2015506360A (en) | 2012-01-06 | 2013-01-04 | Composition, method of use and method of treatment |
| CA2860672A CA2860672A1 (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
| US15/345,619 US10604493B2 (en) | 2012-01-06 | 2016-11-08 | Compositions, methods of use, and methods of treatment |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261583679P | 2012-01-06 | 2012-01-06 | |
| US61/583,679 | 2012-01-06 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/370,715 A-371-Of-International US9556131B2 (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
| US15/345,619 Continuation US10604493B2 (en) | 2012-01-06 | 2016-11-08 | Compositions, methods of use, and methods of treatment |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013103760A1 true WO2013103760A1 (en) | 2013-07-11 |
Family
ID=48745416
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/020212 Ceased WO2013103760A1 (en) | 2012-01-06 | 2013-01-04 | Compositions, methods of use, and methods of treatment |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US9556131B2 (en) |
| EP (1) | EP2800568A4 (en) |
| JP (1) | JP2015506360A (en) |
| CN (1) | CN104159587A (en) |
| AU (1) | AU2013207510A1 (en) |
| BR (1) | BR112014016804A8 (en) |
| CA (1) | CA2860672A1 (en) |
| MX (1) | MX2014008278A (en) |
| WO (1) | WO2013103760A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015112441A1 (en) * | 2014-01-22 | 2015-07-30 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| WO2016206101A1 (en) * | 2015-06-26 | 2016-12-29 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| WO2017152033A1 (en) * | 2016-03-03 | 2017-09-08 | University Of South Florida | Compositions, methods of use, and methods of treatment |
| EP3740482A1 (en) * | 2018-01-17 | 2020-11-25 | Migal Galilee Research Institute Ltd. | New methionine metabolic pathway inhibitors |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20010033621A (en) * | 1997-12-24 | 2001-04-25 | 메틸진, 인크. | NOVEL β-LACTAMASE AND DD-PEPTIDASE INHIBITORS |
| EP1534717B1 (en) * | 2002-08-23 | 2006-12-27 | Pfizer Products Inc. | Beta-lactamase inhibitor prodrug |
| KR20100130176A (en) * | 2008-01-18 | 2010-12-10 | 머크 샤프 앤드 돔 코포레이션 | Betazalactamase inhibitor |
| US20100317621A1 (en) * | 2007-11-13 | 2010-12-16 | Burns Christopher J | Beta-lactamase inhibitors |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS591706B2 (en) * | 1980-09-05 | 1984-01-13 | わかもと製薬株式会社 | Aminophenyl tetrazole derivatives and their salts |
| US5049572A (en) * | 1987-04-06 | 1991-09-17 | Riker Laboratories, Inc. | Substituted di-t-butylphenols and anti-allergic use thereof |
| IL85896A (en) * | 1987-04-06 | 1993-01-14 | Riker Laboratories Inc | Substituted di-tert.- butylphenols and pharmaceutical compositions containing them |
| IT1232252B (en) * | 1989-02-22 | 1992-01-28 | Rotta Research Lab | DERIVATIVES OF N N PHENYL BENZAMIDE WITH ANTI-ULTER AND ANTIALLERIC ACTIVITY AND PROCEDURE FOR THEIR PREPARATION |
| IT1256870B (en) * | 1992-02-13 | 1995-12-27 | Rotta Research Lab | BIS-TETRAZOLE DERIVATIVES WITH ANTIALLERGIC AND CYTOPROTECTIVE ACTIVITY |
| ES2211965T3 (en) | 1995-08-02 | 2004-07-16 | Darwin Discovery Limited | QUINOLONAS AND ITS THERAPEUTIC USE. |
| DE19624155A1 (en) * | 1996-06-18 | 1998-01-08 | Hoechst Ag | Substituted benzoic acid derivatives, process for their preparation and the use of the compounds for the treatment of diseases |
| GB0015095D0 (en) | 2000-06-20 | 2000-08-09 | Celltech Chiroscience Ltd | Chemical compounds |
| GB0218625D0 (en) * | 2002-08-10 | 2002-09-18 | Astex Technology Ltd | Pharmaceutical compounds |
| JPWO2005108370A1 (en) * | 2004-04-16 | 2008-03-21 | 味の素株式会社 | Benzene compounds |
| AR056871A1 (en) * | 2005-10-04 | 2007-10-31 | Aventis Pharma Inc | AMIDA PYRIMIDINE COMPOUNDS AS PGDS INHIBITORS |
| CA2644910C (en) * | 2006-03-31 | 2014-01-28 | Abbott Laboratories | Indazole compounds |
| PE20080888A1 (en) * | 2006-10-18 | 2008-08-26 | Novartis Ag | HETEROCYCLIC COMPOUNDS AS ACIL-TRANSFERASE INHIBITORS OF ACIL-CoA-DIACIL-GLYCEROL 1 (DGAT1) |
| US8003692B2 (en) * | 2007-06-15 | 2011-08-23 | Board Of Regents, The University Of Texas System | Methods and compositions to inhibit edema factor and adenylyl cyclase |
| US8859620B2 (en) | 2009-08-31 | 2014-10-14 | University Of Notre Dame Du Lac | Phthalanilate compounds and methods of use |
-
2013
- 2013-01-04 CA CA2860672A patent/CA2860672A1/en not_active Abandoned
- 2013-01-04 BR BR112014016804A patent/BR112014016804A8/en not_active IP Right Cessation
- 2013-01-04 AU AU2013207510A patent/AU2013207510A1/en not_active Abandoned
- 2013-01-04 EP EP13733664.0A patent/EP2800568A4/en not_active Withdrawn
- 2013-01-04 US US14/370,715 patent/US9556131B2/en active Active
- 2013-01-04 CN CN201380013340.1A patent/CN104159587A/en active Pending
- 2013-01-04 JP JP2014551327A patent/JP2015506360A/en active Pending
- 2013-01-04 MX MX2014008278A patent/MX2014008278A/en unknown
- 2013-01-04 WO PCT/US2013/020212 patent/WO2013103760A1/en not_active Ceased
-
2016
- 2016-11-08 US US15/345,619 patent/US10604493B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20010033621A (en) * | 1997-12-24 | 2001-04-25 | 메틸진, 인크. | NOVEL β-LACTAMASE AND DD-PEPTIDASE INHIBITORS |
| EP1534717B1 (en) * | 2002-08-23 | 2006-12-27 | Pfizer Products Inc. | Beta-lactamase inhibitor prodrug |
| US20100317621A1 (en) * | 2007-11-13 | 2010-12-16 | Burns Christopher J | Beta-lactamase inhibitors |
| KR20100130176A (en) * | 2008-01-18 | 2010-12-10 | 머크 샤프 앤드 돔 코포레이션 | Betazalactamase inhibitor |
Non-Patent Citations (3)
| Title |
|---|
| CHEN, Y. ET AL.: "Molecular docking and ligand specificity in fragment-based inhibitor discovery", NATURE CHEMICAL BIOLOGY, vol. 5, no. 5, 2009, pages 358 - 364, XP055075510 * |
| See also references of EP2800568A4 * |
| YU CHEN ET AL., NATURE CHEMICAL BIOLOGY, vol. 5, no. 5, 22 March 2009 (2009-03-22), pages 358 - 364 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015112441A1 (en) * | 2014-01-22 | 2015-07-30 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| US9708336B2 (en) | 2014-01-22 | 2017-07-18 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| WO2016206101A1 (en) * | 2015-06-26 | 2016-12-29 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| US10221163B2 (en) | 2015-06-26 | 2019-03-05 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| US10227331B2 (en) | 2015-06-26 | 2019-03-12 | Merck Sharp & Dohme Corp. | Metallo-β-lactamase inhibitors |
| US10544130B2 (en) | 2015-06-26 | 2020-01-28 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
| TWI715595B (en) * | 2015-06-26 | 2021-01-11 | 美商默沙東藥廠 | Metallo-beta-lactamase inhibitors |
| WO2017152033A1 (en) * | 2016-03-03 | 2017-09-08 | University Of South Florida | Compositions, methods of use, and methods of treatment |
| US10688080B2 (en) | 2016-03-03 | 2020-06-23 | University Of South Florida | Compositions, methods of use, and methods of treatment |
| EP3740482A1 (en) * | 2018-01-17 | 2020-11-25 | Migal Galilee Research Institute Ltd. | New methionine metabolic pathway inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2800568A4 (en) | 2015-06-10 |
| US10604493B2 (en) | 2020-03-31 |
| CN104159587A (en) | 2014-11-19 |
| BR112014016804A2 (en) | 2017-06-13 |
| AU2013207510A1 (en) | 2014-07-31 |
| US20170088524A1 (en) | 2017-03-30 |
| CA2860672A1 (en) | 2013-07-11 |
| JP2015506360A (en) | 2015-03-02 |
| BR112014016804A8 (en) | 2017-07-04 |
| MX2014008278A (en) | 2014-11-10 |
| US20140378516A1 (en) | 2014-12-25 |
| EP2800568A1 (en) | 2014-11-12 |
| US9556131B2 (en) | 2017-01-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11241428B2 (en) | Heteroaryl amide derivatives as selective inhibitors of histone deacetylases 1 and/or 2(HDAC1-2) | |
| JP7546299B2 (en) | IRE1 small molecule inhibitors | |
| US9718782B2 (en) | TrkA kinase inhibitors, compositions and methods thereof | |
| TWI785770B (en) | Novel compounds as histone deacetylase 6 inhibitor, and pharmaceutical composition comprising the same | |
| US10604493B2 (en) | Compositions, methods of use, and methods of treatment | |
| AU2018304907B2 (en) | Chemical compounds | |
| JP7451569B2 (en) | 1,3,4-oxadiazole homophthalimide derivative compounds as histone deacetylase 6 inhibitors, and pharmaceutical compositions containing the same | |
| EA014080B1 (en) | Immunomodulating heterocyclic compounds | |
| JP2022502497A (en) | Inhibitors of HDAC1 and 2 | |
| KR20240130096A (en) | DHODH inhibitor containing carboxylic acid bioisostere | |
| WO2022155593A1 (en) | Inhibitors and degraders of janus kinase 2 | |
| US10688080B2 (en) | Compositions, methods of use, and methods of treatment | |
| CN109422751A (en) | One kind has the degradation active compound of tyrosine protein kinase JAK3 | |
| US12544384B2 (en) | Antibiotic conjugates | |
| KR20220033689A (en) | Novel compound having histone deacetylases inhibitory activity and use thereof | |
| ES2894657T3 (en) | Ethynyl compounds, their preparation and their therapeutic use for the treatment of malaria | |
| KR20250115893A (en) | Prodrug compound activated by fibroblast activation protein | |
| EA038487B1 (en) | Thiazole derivatives, pharmaceutical composition comprising same and uses thereof | |
| US9242938B2 (en) | Glycine reuptake inhibitor and use thereof | |
| HK40097518A (en) | Deuterated dhodh inhibitors | |
| HK40020428B (en) | Chemical compounds | |
| HK40020428A (en) | Chemical compounds | |
| EP3431474A1 (en) | Chemical compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13733664 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14370715 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2860672 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/008278 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2014551327 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2013207510 Country of ref document: AU Date of ref document: 20130104 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013733664 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014016804 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014016804 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140707 |