WO2013108836A1 - 熱可塑性エラストマー組成物 - Google Patents
熱可塑性エラストマー組成物 Download PDFInfo
- Publication number
- WO2013108836A1 WO2013108836A1 PCT/JP2013/050799 JP2013050799W WO2013108836A1 WO 2013108836 A1 WO2013108836 A1 WO 2013108836A1 JP 2013050799 W JP2013050799 W JP 2013050799W WO 2013108836 A1 WO2013108836 A1 WO 2013108836A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- weight
- temperature
- thermoplastic elastomer
- elastomer composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L53/00—Compositions of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C43/00—Compression moulding, i.e. applying external pressure to flow the moulding material; Apparatus therefor
- B29C43/003—Compression moulding, i.e. applying external pressure to flow the moulding material; Apparatus therefor characterised by the choice of material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
- C08K5/098—Metal salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2033/00—Use of polymers of unsaturated acids or derivatives thereof as moulding material
- B29K2033/04—Polymers of esters
- B29K2033/12—Polymers of methacrylic acid esters, e.g. PMMA, i.e. polymethylmethacrylate
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2067/00—Use of polyesters or derivatives thereof, as moulding material
- B29K2067/006—PBT, i.e. polybutylene terephthalate
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/0088—Blends of polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/25—Solid
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2353/00—Characterised by the use of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/05—Polymer mixtures characterised by other features containing polymer components which can react with one another
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/062—Copolymers with monomers not covered by C08L33/06
- C08L33/068—Copolymers with monomers not covered by C08L33/06 containing glycidyl groups
Definitions
- the present invention relates to a thermoplastic elastomer composition used for electrical and electronic parts, automotive parts, sealing materials, packing, vibration damping members, tubes, and the like, and a molded body obtained by thermoforming the thermoplastic elastomer composition. .
- Patent Document 1 discloses a (meth) acrylic polymer block (a) and a (meth) acrylic heavy polymer as a thermoplastic elastomer composition for a keypad having excellent flexibility, heat resistance, adhesiveness, click feeling, and recyclability.
- a thermoplastic elastomer composition for keypads is disclosed, which comprises a block copolymer (A) containing a combined block (b) (different from (a) and (b)) as an essential component.
- thermoplastic elastomer (block copolymer) that is excellent in heat decomposition and excellent in heat resistance that exhibits good rubber elasticity even at high temperatures.
- a composition comprising a block copolymer (A) comprising an acrylic polymer block and (b) an acrylic polymer block and dynamically treating them.
- Patent Document 3 provides a molded article made of a resin composition excellent in molding processability, recyclability, flexibility, rubber properties, heat resistance, oil resistance, weather resistance, transparency, adhesion to a substrate, and the like.
- the acrylic block copolymer (A) comprising the methacrylic polymer block (a) and the acrylic polymer block (b), the thermoplastic resin (B), the thermoplastic elastomer (C), and the rubber.
- a resin composition containing (D) and at least one selected from the group consisting of thermosetting resins (E) is disclosed.
- the acrylic resin (B) 1 to 100 parts by weight of the thermoplastic polyester resin (A) is provided for the purpose of providing a resin composition having excellent surface hardness and excellent rigidity in a preferred embodiment.
- a resin composition comprising 200 parts by weight, 0.1-100 parts by weight of a compatibilizing agent (C) which is a compound having at least one functional group selected from glycidyl group, acid anhydride group, and carboxyl group. It is disclosed.
- thermoplastic elastomer compositions described in Patent Documents 1 to 3 are highly dependent on molding conditions, particularly temperature, and are restricted in use.
- the resin composition described in Patent Document 4 is used for various containers such as tableware, toiletries, and flower pots, flooring, wall materials, housing equipment such as a wash bowl, and decorative items such as table tops, frames, and dolls. Is a resin composition having a high surface hardness and excellent rigidity, not an elastomer composition.
- An object of the present invention is to provide a molded article having excellent flexibility and heat resistance and good tensile strength, and the molding temperature dependence is small for the molded article to exhibit excellent flexibility and heat resistance.
- An object of the present invention is to provide a thermoplastic elastomer composition and a molded article obtained by thermoforming the thermoplastic elastomer.
- the present invention [1] (Meth) acrylic elastomer (component A), Melting point is 180-350 ° C, thermoplastic resin having functional group capable of reacting with epoxy group (component B), and solubility in tetrahydrofuran, having an average of 2 or more epoxy groups in one molecule Containing a vinyl copolymer (component C),
- the proportion of each component is (Meth) acrylic elastomer (component A) 40 to 95 parts by weight, 5 to 60 parts by weight of a thermoplastic resin (component B) and 0.1 to 30 parts by weight of a vinyl copolymer (component C),
- the total of (meth) acrylic elastomer (component A) and thermoplastic resin (component B) is 100 parts by weight
- Component C is composed of monomer units containing 50% by weight or more of monomer units (monomer units c1) having an SP value of 17.5 to 25.0,
- a thermoplastic elastomer composition having a flow initiation temperature
- thermoplastic elastomer composition according to [1] or [2] obtained by heat molding the thermoplastic elastomer composition according to [1] or [2] at 180 to 350 ° C.
- a molded article having a tensile elongation at break of 100% or more obtained by heat-molding the thermoplastic elastomer composition according to [1] or [2] at 180 to 350 ° C.
- the present invention relates to a molded article having an A hardness of 20 to 90, which is obtained by heat molding the thermoplastic elastomer composition according to [1] or [2] at 180 to 350 ° C.
- thermoplastic elastomer composition of the present invention is excellent in flexibility and heat resistance, can give a molded article having good tensile strength, and a molding temperature at which the molded article exhibits excellent flexibility and heat resistance.
- the dependency is small, and in the processing to the molded body, the limitation of the molding condition is small. That is, the temperature at which the component A that forms the continuous phase flows but the component B that forms the dispersed phase does not melt even when heat-molded at a temperature at which sufficient fluidity can be obtained for molding (temperature H described later).
- flexibility and heat resistance are not significantly changed (particularly, heat resistance is hardly lowered) as compared with the case of heat molding at (temperature L described later).
- FIG. 1 is a scanning electron microscope (SEM) photograph (5,000 times) of a cross section of a 200 ° C. press sheet obtained from the composition of Example 1.
- FIG. FIG. 2 is a scanning electron micrograph (5000 times) of a cross section of a 230 ° C. press sheet obtained from the composition of Example 1.
- 3 is a scanning electron micrograph (5000 magnifications) of a cross section of a 200 ° C. press sheet obtained from the composition of Example 4.
- FIG. FIG. 4 is a scanning electron micrograph (x5000) of a cross section of a 230 ° C. press sheet obtained from the composition of Example 4.
- FIG. 5 is a scanning electron micrograph (1000 times) of a cross section of a 230 ° C. press sheet obtained from the composition of Comparative Example 1.
- FIG. 6 is a scanning electron micrograph (1000 times) of a cross section of a 230 ° C. press sheet obtained from the composition of Comparative Example 3.
- thermoplastic elastomer composition of the present invention comprises: (Meth) acrylic elastomer (component A), A melting point of 180 to 350 ° C., a thermoplastic resin having a functional group capable of reacting with an epoxy group (component B), and solubility in tetrahydrofuran, and an average of two or more epoxy groups in one molecule A vinyl copolymer comprising a specific monomer unit (component C) In a specific ratio.
- component A forms a continuous phase and component B forms a dispersed phase.
- Component C functions as a compatibilizing agent that stabilizes the size (particle diameter) of component B (dispersed phase) in component A forming the continuous phase
- the thermoplastic elastomer composition of the present invention comprises: It is possible to give a molded article having excellent flexibility and heat resistance, and the molding article has low molding temperature dependency for exhibiting excellent flexibility and heat resistance. Is small.
- the (meth) acrylic elastomer (component A) is preferably composed of two or more (meth) acrylic monomers and, if necessary, other copolymerizable vinyl monomers, and is made to have a high molecular weight by a polymerization reaction. Is obtained.
- Examples of the (meth) acrylic monomer include, for example, methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, (meth) Cyclohexyl acrylate, (meth) acrylic acid, hydroxyethyl (meth) acrylate, hydroxypropyl (meth) acrylate, glycidyl (meth) acrylate, methoxyethyl (meth) acrylate, ethoxyethyl (meth) acrylate, ( Examples include butoxyethyl (meth) acrylate and phenoxyethyl (meth) acrylate. In this specification, when it is indicated as (meth) acryl, it means both methacryl and acryl.
- the amount of (meth) acrylic monomer is preferably 20 mol% or more, more preferably 50 mol% or more, and even more preferably 80 mol% or more in the constituent components.
- copolymerizable vinyl monomers include styrene, ⁇ -methylstyrene, vinyl acetate, ethylene, propylene, butadiene, isoprene, maleic anhydride, etc. Among these, styrene, ⁇ -methylstyrene and ethylene are included. preferable.
- Examples of the monomer polymerization method for obtaining the (meth) acryl elastomer include a radical polymerization method, a living anion polymerization method, a living radical polymerization method, and the like.
- Examples of the polymerization form include a solution polymerization method, an emulsion polymerization method, a suspension polymerization method, and a bulk polymerization method.
- Component A is preferably a block copolymer comprising two or more blocks constituting a hard segment and one or more blocks constituting a soft segment.
- the component A forming the continuous phase In order for the component A forming the continuous phase to exhibit properties as a thermoplastic elastomer, it is preferable to have a hard segment having a glass transition temperature of room temperature or higher.
- methyl methacrylate is preferable, and does not impair the purpose of the present invention (preferably 30% by weight or less, more preferably 20% by weight or less, more preferably 10% by weight or less. ), Acrylonitrile, styrene, ⁇ -methylstyrene and the like can be used.
- the glass transition temperature of the block constituting the hard segment is preferably 20 to 200 ° C, more preferably 30 to 180 ° C, and further preferably 50 to 150 ° C. If the glass transition temperature of the block constituting the hard segment is too low, the resulting composition may have insufficient heat resistance, and it is difficult to obtain raw materials for blocks having a glass transition temperature exceeding 200 ° C.
- Examples of the vinyl monomer constituting the soft segment include ethyl (meth) acrylate, butyl (meth) acrylate, methoxyethyl (meth) acrylate, ethoxyethyl (meth) acrylate, and the like.
- the glass transition temperature of the block constituting the soft segment is preferably -100 to 19 ° C, more preferably -80 to 10 ° C, and further preferably -70 to 0 ° C. If the glass transition temperature of the block constituting the soft segment is too high, the resulting composition may be insufficient in flexibility, and it is difficult to obtain raw materials for blocks having a glass transition temperature lower than ⁇ 100 ° C.
- the weight ratio of the hard segment to the soft segment in component A is preferably 10/90 to 70/30 from the viewpoint of imparting appropriate flexibility to component A, and 15/85 to 50/50. Is more preferable.
- Component A examples include Kuraray's clarity, Arkema's Nano Strength, Kaneka's Nabster, and the like.
- the weight average molecular weight of Component A is preferably 50,000 or more, more preferably 70,000 or more, and even more preferably 100,000 or more. Further, from the viewpoint of easy handling and maintaining a melt viscosity suitable for production of a molded article such as injection molding, it is preferably 1 million or less, more preferably 800,000 or less, and further preferably 700,000 or less. From these viewpoints, the weight average molecular weight of Component A is preferably 50,000 to 1,000,000, more preferably 70,000 to 800,000, and even more preferably 100,000 to 700,000.
- the A hardness of Component A is preferably 5 to 90, more preferably 10 to 80, from the viewpoint of the flexibility of the composition.
- the flow start temperature of component A is preferably 80 ° C. or higher from the viewpoint of heat resistance of the composition, and preferably 220 ° C. or lower from the viewpoint of thermoplasticity (fluidity) of the composition. From these viewpoints, the flow start temperature of component A is preferably 80 to 220 ° C, more preferably 100 to 200 ° C.
- Component B is a thermoplastic resin having a functional group capable of reacting with an epoxy group. From the viewpoint of imparting heat resistance to the composition of the present invention, it is preferably a crystalline thermoplastic resin having a high melting point. From these viewpoints, the melting point of the thermoplastic resin is 180 ° C. or higher, preferably 190 ° C. or higher, more preferably 200 ° C. or higher. Moreover, it is 350 degrees C or less, Preferably it is 330 degrees C or less, More preferably, it is 300 degrees C or less. The melting point of the thermoplastic resin is 180 to 350 ° C, preferably 190 to 330 ° C, more preferably 200 to 300 ° C.
- a crystalline thermoplastic resin refers to a thermoplastic resin whose melting point is observed by a differential scanning calorimeter (DSC).
- thermoplastic resin as the component B is one or two of these functional groups. The above is contained in the main chain or side chain of the thermoplastic resin.
- thermoplastic resin that does not have a functional group capable of reacting with an epoxy group does not react with component C, which is a compatibilizing agent, and therefore, a thermoplastic elastomer that is poorly dispersed in component A and has good flexibility and heat resistance. Can't get.
- the thermoplastic resin having a melting point of 180 ° C. or more and having a functional group capable of reacting with an epoxy group includes aromatic polyester and / or polyamide from the viewpoint of heat resistance (high melting point) and incompatibility with component A. preferable.
- aromatic polyester include polyethylene terephthalate, polybutylene terephthalate, and polyethylene naphthalate.
- polyamide include nylon 6, nylon 66, and the like.
- component B which is a thermoplastic resin having relatively high heat resistance
- component A which is an elastomer having relatively low heat resistance
- the melting point of Component B is preferably 20 ° C. or more higher than the flow start temperature of Component A, more preferably 30 ° C. or more, and further preferably 40 ° C. or more.
- Component B is poorly compatible with Component A. If the compatibility of both components is good, the composition becomes less flexible and the characteristics of the elastomer may be impaired.
- the melt viscosity can be used as an index of molecular weight as long as each resin has the same monomer composition.
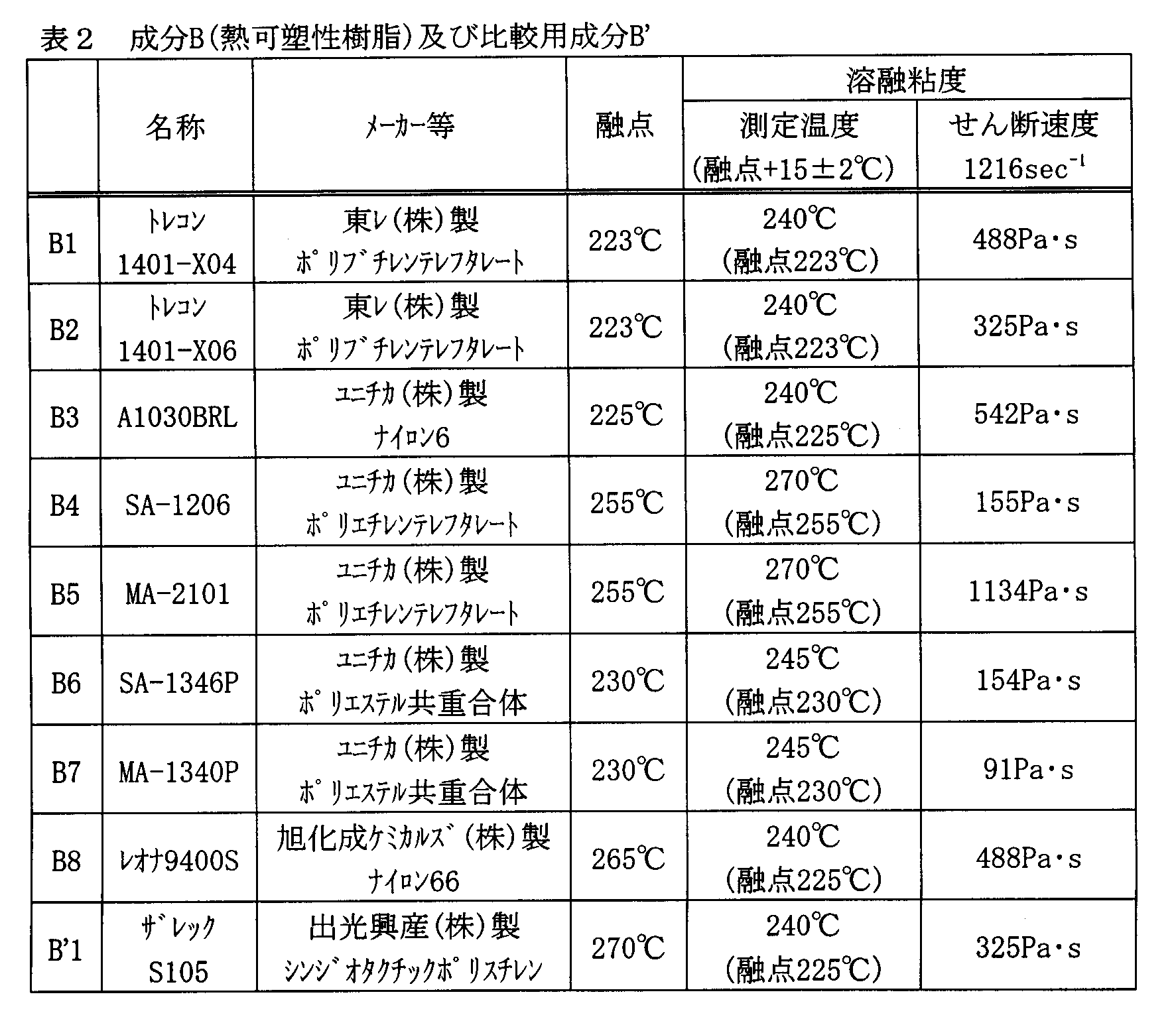
- the melt viscosity of Component B is preferably 50 Pa ⁇ s to 2000 Pa ⁇ s from the viewpoint of heat resistance at the melting point of each resin + 15 ° C. and the shear rate 1216 sec ⁇ 1 .
- the functional group at the molecular end of the polyester has a hydroxyl group and a carboxyl group.
- the abundances of hydroxyl groups and carboxyl groups at the molecular terminals of the polyester used as component B in the present invention are not particularly limited, and general ones can be used.
- component B is polyester.
- the acid value that is an index of the amount of carboxyl groups is preferably 0.1 to 100 milliequivalent / kg. More specifically, the acid value is preferably 0.1 mm equivalent / kg or more, more preferably 1 mm equivalent / kg or more, more preferably 3 mm equivalent / kg or more, and 5 mm equivalent / kg or more. Further preferred.
- the acid value of the polyester resin was determined by dissolving 200 mg of a sufficiently dried sample in 10 ml of hot benzyl alcohol, cooling the solution, adding 10 ml of chloroform and phenol red, and adding 1/25 normal alcoholic potash solution (KOH Titration with methanol solution) and measurement.
- the hydroxyl value of the polyester resin is calculated by measuring the end group concentration, which is the sum of the hydroxyl end group and the carboxyl end group of the polyester, and subtracting the acid value (carboxyl end group concentration) therefrom.
- the molecular end functional groups of polyamide are generally carboxyl groups and amino groups.
- the amount of each carboxyl group and amino group at the molecular end of the polyamide used as component B in the present invention is not particularly limited, and general ones can be used, but when component B is polyamide,
- the terminal amino group concentration is preferably 10 to 200 ⁇ mol / g. More specifically, the terminal amino group concentration is preferably 10 ⁇ mol / g or more, more preferably 15 ⁇ mol / g or more, further preferably 20 ⁇ mol / g or more, and further preferably 30 ⁇ mol / g or more.
- terminal amino group concentration and terminal carboxyl group concentration are determined from the integrated value of the characteristic signal corresponding to each terminal group by 1 H-NMR.
- the monomer unit constituting Component C is a monomer comprising 50% by weight or more of two embodiments, that is, the first embodiment: a monomer unit having an SP value of 17.5 to 25.0 (monomer unit c1).
- Unit and second aspect monomer unit containing at least 50% by weight of at least one monomer unit (monomer unit c2) selected from the group consisting of (meth) acrylic monomers, styrene and styrene derivatives
- the monomer units c1 and c2 are common in that the affinity with the component A is good.
- the first embodiment and the second embodiment are separated for convenience, and some monomers are common to both. That is, (meth) acrylic monomers, styrene, and styrene derivatives having an SP value of 17.5 to 25.0 correspond to any embodiment.
- a copolymer (block shape, graft shape, star shape, or a mixture thereof) of Component C and Component B is obtained by the reaction of the epoxy group in Component C and the functional group having reactivity with the epoxy group in Component B.
- the component B can form a fine and uniform dispersed phase due to the affinity between the monomer unit c1 or the monomer unit c2 that forms the component C and the component A that forms the continuous phase.
- the SP value for specifying the monomer unit c1 is widely known as a Solubility parameter.
- the SP value ( ⁇ ) in the present invention is determined by the following estimation formula.
- Monomer units having SP values of 17.5 to 25.0 include styrene (18.82), ⁇ -methylstyrene (19.55), methyl methacrylate (18.54), n-butyl acrylate (18.12 ), Ethyl acrylate (18.83), methyl acrylate (19.13), methoxyethyl acrylate (19.43), vinyl acetate (19.13) and the like.
- olefins having an SP value of less than 17.5, such as ethylene and propylene, are not suitable as the main monomer unit of component C in the present invention.
- examples of olefins having an SP value of less than 17.5 include ethylene (16.00) and propylene (17.04).
- the SP value of the monomer unit c1 is 17.5 or more, preferably 17.8 or more, more preferably 18.0 or more. Moreover, it is 25.0 or less, Preferably it is 23.0 or less, More preferably, it is 22.0 or less, More preferably, it is 21.0 or less, More preferably, it is 20.0 or less. From these viewpoints, the SP value of the monomer unit c1 is 17.5 to 25.0, preferably 17.8 to 23.0, more preferably 18.0 to 22.0, still more preferably 18.0 to 21.0, and still more preferably. 18.0-20.0.
- the proportion of the monomer unit c1 is more preferably 60% by weight or more, further preferably 70% by weight or more, and more preferably 80% by weight or more.
- a monomer unit whose SP value is difficult to specify can be used as the main monomer unit of Component C if it is a (meth) acrylic monomer, styrene, or a styrene derivative.
- the second embodiment of the monomer unit of component C includes at least one monomer unit (monomer unit c2) selected from the group consisting of (meth) acrylic monomers, styrene and styrene derivatives. It is a waste.
- Suitable (meth) acrylic monomers as the monomer unit c2 include methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, butyl (meth) acrylate, and cyclohexyl (meth) acrylate.
- Hydroxyethyl (meth) acrylate, hydroxypropyl (meth) acrylate, glycidyl (meth) acrylate, methoxyethyl (meth) acrylate, ethoxyethyl (meth) acrylate, butoxyethyl (meth) acrylate, (meth ) Phenoxyethyl acrylate and the like are exemplified, and among these, from the viewpoint of affinity with Component A, an alkyl (carbon number 1 to 6) ester of (meth) acrylic acid and glycidyl (meth) acrylate are preferable. Since glycidyl (meth) acrylate has a strong hydrogen bond, it is difficult to specify the SP value, but it has a high affinity with Component A and is preferable as the monomer unit c2.
- Styrene derivatives are substituted with substituents such as lower alkyl groups having 1 to 4 carbon atoms, lower alkoxy groups having 1 to 4 carbon atoms, carboxyl groups, and halogen atoms at the alpha, ortho, meta, or para positions of styrene. Means the compound formed.
- the molecular weight (atomic weight) of the substituent is preferably 60 or less, more preferably 50 or less, and still more preferably 40 or less.
- Specific examples of the styrene derivative include ⁇ -methylstyrene and p-chlorostyrene.
- the SP value deviates from the SP value required for the monomer unit c1 of the first embodiment.
- those having an SP value of less than 17.5 and more than 25.0 have low affinity with component A, and are excluded from the monomer unit c2 in the second embodiment.
- those having an SP value of the monomer unit of less than 17.5 include alkyl esters of (meth) acrylic monomers having an alkyl group having 8 or more carbon atoms.
- component C includes both component C of the first embodiment containing monomer unit c1 and component C of the second embodiment containing monomer unit c2. Applicable.
- the monomer unit constituting the vinyl copolymer (component C) preferably contains 10 to 90% by weight of (meth) acrylic monomer and 10 to 90% by weight of styrene or a styrene derivative. More preferably, it contains 15 to 80% by weight (more preferably 20 to 70% by weight) of (meth) acrylic monomer, and 20 to 85% by weight (more preferably 30 to 80% by weight) of styrene or a styrene derivative.
- component C has (meth) acrylic monomer having a high affinity with component A and styrene or a styrene derivative having a high affinity with component B as constituent units. It is preferable because it can work effectively.
- Component C must have solubility in tetrahydrofuran (THF).
- THF tetrahydrofuran
- a vinyl copolymer having no solubility in THF for example, acrylic gel
- Component B cannot be well dispersed in Component A.
- Having solubility in THF means that the solubility in THF at 25 ° C. is 2% by weight or more.
- the solubility is more preferably 5% by weight or more, and further preferably 10% by weight or more.
- Component C has an average of 2 or more, preferably 2.5 to 20, more preferably 3 to 10 epoxy groups per molecule.
- the average number of epoxy groups is less than 2
- the reactivity with the component B becomes low and the function as a compatibilizing agent is not achieved.
- the dispersion stability of component B decreases, and the problem cannot be solved. That is, a molded product having excellent flexibility and heat resistance can be provided, and a composition having a small molding temperature dependency for exhibiting excellent flexibility and heat resistance cannot be obtained.
- the epoxy value of component C is preferably 0.5 to 5 meq / g, more preferably 0.7 to 3 meq / g, from the viewpoint of the effect as a compatibilizer.
- component C examples include Alfon UG series manufactured by Toagosei Co., Ltd., Marproof G series manufactured by NOF Corporation, and Jonkrill ADR series manufactured by BASF.
- Component C needs to maintain an appropriate affinity for Component A and Component B, but when the weight average molecular weight of the vinyl copolymer (Component C) exceeds 100,000, it is dispersible or compatible with Component A. Decreases and the dispersion stabilization of component B is impaired. Moreover, since the dispersibility or compatibility to the component A becomes too high as it is less than 1000, the reaction efficiency with the component B falls, As a result, the dispersion stabilization of the component B is impaired. From these viewpoints, the weight average molecular weight of Component C is preferably 1000 or more, more preferably 3000 or more, and further preferably 5000 or more. Moreover, 100,000 or less is preferable, 80,000 or less is more preferable, and 60,000 or less is more preferable. The weight average molecular weight of Component C is preferably from 1,000 to 100,000, more preferably from 3,000 to 80,000, and even more preferably from 5,000 to 60,000.
- the number average molecular weight of Component C is preferably 500 to 50,000, more preferably 1,000 to 40,000, and still more preferably 2000 to 30,000, from the viewpoint of the dispersion stability of Component B in Component A as described above.
- the ratio of component A, component B and component C in the composition of the present invention (the total of component A and component B is 100 parts by weight) is as follows.
- the proportion of component A is 40 parts by weight or more, preferably 45 parts by weight or more, more preferably 50 parts by weight or more. Further, it is 95 parts by weight or less, preferably 90 parts by weight or less, more preferably 85 parts by weight or less.
- the proportion of component A is 40 to 95 parts by weight, preferably 45 to 90 parts by weight, more preferably 50 to 85 parts by weight.
- the proportion of component B is 5 parts by weight or more, preferably 10 parts by weight or more, more preferably 15 parts by weight or more. Further, it is 60 parts by weight or less, preferably 55 parts by weight or less, more preferably 50 parts by weight or less.
- the proportion of component B is 5 to 60 parts by weight, preferably 10 to 55 parts by weight, more preferably 15 to 50 parts by weight.
- the total amount of component A and component B in the thermoplastic elastomer composition is preferably 10.0% by weight or more, more preferably 30.0% by weight or more, further preferably 50.0% by weight or more, and further preferably 70.0% by weight or more. Moreover, 99.9 weight% or less is preferable, 99.0 weight% or less is more preferable, and 98.0 weight% or less is further more preferable. From these viewpoints, the total amount of Component A and Component B in the thermoplastic elastomer composition is preferably 10.0 to 99.9% by weight, more preferably 30.0 to 99.0% by weight, further preferably 50.0 to 98.0% by weight, and 70.0 to 98.0% by weight is more preferred.
- the proportion of component C is 0.1 parts by weight or more, preferably 0.2 parts by weight or more, more preferably 0.3 parts by weight or more.
- component C is 0.1 to 30 parts by weight, preferably 0.2 to 20 parts by weight, more preferably 0.2 to 15 parts by weight, and still more preferably 0.3 to 10 parts by weight.
- 0.1 to 15 parts by weight is preferable.
- the composition of the present invention when the reaction rate of the functional group of component B and the epoxy group of component C is slow, for example, when the composition of the present invention is produced with an extruder or the like, the degree of reaction is insufficient, The stability of the dispersion diameter of component B may be impaired. Therefore, the composition of the present invention preferably contains an aliphatic carboxylic acid metal salt (component D) as a catalyst for promoting the reaction between the functional group of component B and the epoxy group of component C.
- component D aliphatic carboxylic acid metal salt
- Examples of the aliphatic carboxylic acid metal salt include metal stearate such as sodium stearate, calcium stearate, aluminum stearate, zinc stearate, metal 12-hydroxystearate such as sodium 12-hydroxystearate and calcium 12-hydroxystearate. Examples include salts.
- the content of the aliphatic carboxylic acid metal salt (component D) is preferably 0.001 part by weight or more and more preferably 0.005 part by weight or more with respect to 100 parts by weight of the total amount of Component A and Component B. Further, it is preferably 10 parts by weight or less, and more preferably 5 parts by weight or less.
- the content of the aliphatic carboxylic acid metal salt (component D) is preferably 0.001 to 10 parts by weight, more preferably 0.005 to 5 parts by weight with respect to 100 parts by weight of the total amount of component A and component B.
- the thermoplastic elastomer composition of the present invention further contains a transesterification catalyst (component E).
- component E improves the tensile strength at break of the resulting composition. Specifically, in the tensile test, the tensile product (product of strength and elongation) is large, and the balance between strength and elongation becomes better. The reason for such an effect is that component E can increase the molecular weight of component A, and in some cases, partial crosslinking is formed. It is presumed that the component E acts as a crosslinking agent, and that the composition becomes more tough by increasing the molecular weight of the component A, which is the main component of the composition.
- a general polyester polymerization catalyst can be used as an esterification catalyst as a reaction catalyst for increasing the molecular weight of component A and partially crosslinking.
- catalysts include antimony catalysts such as antimony trioxide, tin catalysts such as butyltin, octyltin and stannoxane, titanium catalysts such as titanium aminates and titanium alkoxides, zirconium acetylacetonates and zirconium alkoxides. And zirconium-based catalysts.
- a titanium-based catalyst and a zirconium-based catalyst are preferable, and a titanium-based catalyst is more preferable.
- the content of the transesterification catalyst (component E) is preferably 0.01 parts by weight or more and more preferably 0.1 parts by weight or more with respect to 100 parts by weight of the total amount of component A and component B. Further, it is preferably 10 parts by weight or less, and more preferably 5 parts by weight or less.
- the content of the transesterification catalyst (component E) is preferably 0.01 to 10 parts by weight, more preferably 0.1 to 5 parts by weight with respect to 100 parts by weight of the total amount of component A and component B.
- the content of the transesterification catalyst (component E) is preferably 10 parts by weight or less from the viewpoint of suppressing a decrease in thermoplasticity of the composition obtained by crosslinking of the component A.
- composition of the present invention may contain a radical polymerization initiator (component F) from the viewpoint of increasing the molecular weight of component A.
- radical polymerization initiators examples include organic peroxides such as dialkyl peroxides, and azo compounds such as azobisbutyronitrile.
- the content of the radical polymerization initiator (component F) is preferably 0.01 to 15 parts by weight and more preferably 0.1 to 5 parts by weight with respect to 100 parts by weight of the total amount of component A and component B.
- Component E and Component F are both components for the purpose of increasing the molecular weight of Component A, and either component may be used or both components may be used in combination.
- component E and component F are effective to use both component E and component F when the molecular weight of component A is relatively small.
- the molecular weight of component A having a high molecular weight does not become excessive. Be careful. It is preferable that the high molecular weight component A does not exceed 1 million in weight average molecular weight, and loss of thermoplasticity such as the crosslinking of the component A must be avoided.
- the composition of the present invention preferably contains a heat stabilizer.
- heat stabilizer examples include phosphorus-containing compounds, hydrazide compounds, organic sulfur-based antioxidants, phenol-based antioxidants, amine-based antioxidants, etc., and chelate formation with other ester polymerization catalysts (component E).
- component E ester polymerization catalysts
- compounds that reduce the activity of the catalyst can also be used.
- a phosphorus containing compound and a hydrazide compound are preferable from a viewpoint which the freedom degree of use conditions becomes larger. These may be used in combination.
- Thermal aging mainly includes two phenomena, the first is a decrease in strength due to an increase in the proportion of low molecular weight components produced by thermal decomposition, and the second is free radicals produced by thermal decomposition. This is a decrease in elongation caused by the formation of active site crosslinks.
- Examples of phosphorus-containing compounds include phosphite compounds, polyphosphite compounds, phosphate ester compounds, and polyphosphate ester compounds.
- phosphite compounds include bis (2,6-di-tert-butyl-4-methylphenyl) pentaerythritol diphosphite, triphenyl phosphite, triisodecyl phosphite, and tri-2-ethylhexyl phosphite.
- Diphenylnonylphenyl phosphite Diphenylnonylphenyl phosphite, trinonylphenyl phosphite, 9,10-dihydro-9-oxa-10-phosphaphenant-10-oxide, 10-decyloxy-9,10-dihydro-9-oxa-10- Examples thereof include phosphananthrene, O-cyclohexyl phosphite, trilauryl trithiophosphite, and trioctadecyl phosphite.
- Polyphosphite compounds include diisodecyl pentaerythritol diphosphite, distearyl pentaerythritol diphosphite, dinonylphenyl pentaerythritol diphosphite, tetraphenyldipropylene glycol diphosphite, 4,4'-isobutylidenebis (3-methyl-6-tert-butylphenyl-ditridecyl phosphite), cyclic neopentanetetraylbis (2,6-di-tert-butyl-4-methylphenyl) phosphite (having pentaerythritol skeleton structure) Phosphite) and the like.
- phosphate ester compounds include trimethyl phosphate ester, triethyl phosphate ester, dibutyl phosphate ester, tributyl phosphate ester, dioctyl phosphate ester and trioctyl phosphate ester, as well as formula (I):
- R 2 represents an alkyl group having 1 to 20 carbon atoms, and n represents 1 or 2
- phosphate ester compound etc. which are represented by these are mentioned.
- Examples of the phosphoric acid ester compound represented by the formula (I) include tri (hydroxyethoxy) phosphate, tri (hydroxyethoxyethoxy) phosphate, and the like.
- Examples of the phosphoric acid ester compound represented by the formula (II) include mono-stearyl acid phosphate, di-stearyl acid phosphate and the like.
- the hydrazide compound is an acid hydrazide in which an acid and hydrazine are condensed, and examples thereof include a chain hydrazide and a cyclic hydrazide.
- R 3 , R 4 , R 5 and R 7 are each independently an aromatic monovalent carboxylic acid residue
- R 6 is an aliphatic divalent carboxylic acid residue or an aromatic divalent carboxylic acid residue.
- aromatic monovalent carboxylic acid residues represented by R 3 , R 4 , R 5 and R 7 include benzoic acid, 4-butylbenzoic acid, salicylic acid, naphthylic acid, 3- (3,5-di-t -Butyl-4-hydroxyphenyl) propionic acid, aromatic carboxylic acid residues such as phenoxypropionic acid and the like, and aliphatic divalent carboxylic acid residues represented by R 6 include succinic acid, adipic acid, Examples thereof include aliphatic dicarboxylic acid residues having 2 to 20 carbon atoms such as azelaic acid, sebacic acid, and dodecanedioic acid. Examples of the aromatic divalent carboxylic acid residue include residues of aromatic dicarboxylic acids such as phthalic acid, isophthalic acid, and terephthalic acid.
- Cyclic hydrazide has the formula (IV):
- R 8 and R 9 represent a hydrogen atom, a halogen atom, a hydroxy group, an alkyl group having 1 to 18 carbon atoms, an alkenyl group, an alkynyl group, an alkoxy group, a cycloalkyl group, or an aryl group.
- the compound etc. which are represented by these are mentioned.
- Specific examples of the compound represented by the formula (IV) include phthalic hydrazide.
- a radical polymerization initiator component F
- a radical scavenger and a radical polymerization inhibitor are also effective as a heat stabilizer.
- the content of the heat stabilizer is preferably 0.01 to 15 parts by weight, more preferably 0.05 to 10 parts by weight with respect to 100 parts by weight of the total amount of Component A and Component B.
- additives include lubricants such as heavy metal deactivators and fatty acid esters; light stabilizers such as benzotriazole compounds, benzophenone compounds, benzoate compounds and hindered phenol compounds; hydrolysis of carbodiimide compounds and oxazoline compounds Inhibitors; Plasticizers such as phthalate compounds, polyester compounds, (meth) acryl oligomers, process oils; Inorganic foaming agents such as sodium bicarbonate and ammonium bicarbonate; Organics such as nitro compounds, azo compounds, and sulfonyl hydrazides Fillers such as carbon black, calcium carbonate, talc, glass fiber; flame retardants such as tetrabromophenol, ammonium polyphosphate, melamine cyanurate, magnesium hydroxide and aluminum hydroxide; silane coupling agents, Titanate-based coupling agents, compatibilizers, such as aluminate-based coupling agent or an acid-modified polyolefin resin; other pigments or dyes.
- thermoplastic elastomer composition of the present invention comprises at least a (meth) acryl elastomer (component A), a thermoplastic resin (component B) and a vinyl copolymer (component C), and if necessary, an aliphatic carboxylic acid metal salt. It is preferable to obtain a raw material component containing (Component D), an esterification catalyst (Component E), a radical polymerization initiator (Component F) and the like by heating and kneading with an extruder or a kneader.
- the thermal stabilizer is used in combination to improve the properties of the composition.
- the heat stabilizer is added after the high molecular weight of Component A (for example, at a kneading temperature of 180 to 350 ° C., the raw materials other than the heat stabilizer are 0.5 to It is preferable that after kneading for about 30 minutes. If the heat stabilizer is added too early, component A may not be sufficiently high in molecular weight.
- the temperature of the heat-kneading is less than the melting point of component B, component B cannot be finely dispersed, and as a result, the heat resistance of the resulting composition is insufficient. Moreover, when it exceeds 350 degreeC, the (meth) acryl component of the component A will thermally decompose and the mechanical physical property of the composition obtained will fall. From these viewpoints, the preferred temperature for the heat-kneading is not lower than the melting point of Component B and not higher than 350 ° C. A more preferred lower limit temperature for the heat-kneading is (melting point of component B + 10 ° C.), and a more preferred lower limit temperature is (melting point of component B + 20 ° C.). A more preferable upper limit temperature of the heat kneading is 320 ° C., and a more preferable upper limit temperature is 300 ° C.
- extruder examples include a single screw extruder, a parallel screw twin screw extruder, a conical screw twin screw extruder, and the like.
- a twin screw extruder is preferred, and a co-rotating twin screw extruder is more preferred.
- the die attached to the discharge part of the extruder can be selected arbitrarily.
- a strand die suitable for pellet production a T die suitable for sheet or film production, a pipe die, a profile extrusion die, etc.can be mentioned.
- the extruder may be provided with a vent for venting air connected to an air release portion or a decompression device, or may be provided with a plurality of raw material inlets.
- Kneader means a batch-type mixer capable of temperature control, and includes a Banbury mixer, Brabender, and the like.
- Each component may be charged into the extruder or kneader all at once, separately, or dividedly. However, the introduction of the heat stabilizer is as described above.
- the heating and kneading time depends on the heating temperature, the type and concentration of each component, and cannot be determined unconditionally. However, the heating and kneading time is appropriately determined in consideration of the quality variation control and productivity of the obtained thermoplastic elastomer composition. It is preferable.
- a typical heat mixing time when using an extruder is, for example, 0.5 to 20 minutes, preferably 0.7 to 15 minutes, and more preferably 1 to 10 minutes.
- thermoplastic elastomer composition of the present invention thus obtained has a phase separation structure composed of a continuous phase and a dispersed phase.
- the continuous phase includes a component derived from a (meth) acrylic elastomer (component A), and the dispersed phase includes a component derived from a thermoplastic resin (component B).
- the thermoplastic elastomer composition of the present invention has a dispersed phase finely dispersed in a continuous phase, and can give a molded article excellent in flexibility and heat resistance.
- the molded article has excellent flexibility and heat resistance.
- the molding temperature dependency for exhibiting the properties is small (the allowable range of the molding temperature is wide), and the molding conditions are small in the processing of the molded body.
- the temperature at which the component A that forms the continuous phase flows but the component B that forms the dispersed phase does not melt even when heat-molded at a temperature at which sufficient fluidity can be obtained for molding (temperature H described later).
- flexibility and heat resistance are not significantly changed (particularly, heat resistance is hardly lowered) as compared with the case of heat molding at (temperature L described later).
- the lower limit of the flow starting temperature of the thermoplastic elastomer composition of the present invention is 180 ° C. from the viewpoint of heat resistance, and the upper limit is 350 ° C. from the viewpoint of workability. That is, it is 180 to 350 ° C, preferably 190 to 320 ° C, more preferably 200 to 300 ° C.
- a temperature 20 ° C. lower than the melting point of the thermoplastic resin (component B) A sheet-like molded body is produced by hot pressing in the vicinity ( ⁇ 5 ° C.) for a short time (5 minutes) to obtain a sample for measuring physical properties of the composition.
- the physical property value of the composition of the present invention does not change greatly, and the physical property value of the molded product can be regarded as the physical property value of the composition. . Not only the flow start temperature but also the A hardness, particle diameter, etc. are handled in the same manner. When obtaining a sample for measuring the physical property value of the composition by hot pressing, it is easy to obtain a sample without bubbles or the like if the composition is prepared as a lump.
- thermoplastic elastomer composition of the present invention can be prepared at two temperatures, temperature L [((the melting point of component B) ⁇ 20 ° C.) ⁇ 5 ° C.] or temperature H (((the melting point of component B) + 10 ° C.) ⁇ 5
- the molded body obtained by heating for 5 minutes with a hot press set to within [° C.] preferably has a phase separation structure composed of a continuous phase and a dispersed phase for the molded body at any temperature. .
- the maximum particle size of the dispersed phase observed with an electron microscope is preferably 5 ⁇ m or less, more preferably 3 ⁇ m or less, and even more preferably 2 ⁇ m or less, from the viewpoint of the heat resistance of the shaped body.
- the diameter of the dispersed phase is a diameter in the case of a perfect circle, and a long diameter in the case of an ellipse.
- molded product H obtained by heating for 5 minutes with a hot press set at a temperature L [((melting point of component B) ⁇ 20 ° C.) ⁇ 5 ° C.] is formed into a molded product L, temperature H [((component B The molded body obtained by heating for 5 minutes with a hot press set to the melting point of (+ 10 ° C.) ⁇ 5 ° C.) is designated as molded body H.
- the ratio of the average particle diameter of the dispersed phase observed with an electron microscope (average particle diameter of the molded body L / average particle diameter of the molded body H) is obtained by molding the composition. From the standpoint that the molding temperature dependency for exhibiting excellent flexibility and heat resistance of the molded body can be reduced (allowing a wider allowable range of molding temperature), 0.6 or more is preferable, 0.6 to 1.3 is more preferable, and 0.7 to 1.2 Is more preferable.
- thermoplastic elastomer composition of the present invention A composition in which the A hardness of each of the molded body L and the molded body H is 20 to 90 (more preferably 30 to 85) is preferable as the thermoplastic elastomer composition of the present invention.
- the ratio of the A hardness is 0.6 to 1.3 (more preferably 0.7 to 1.25, more preferably 0.8 to 1.2. ) Is preferable as the thermoplastic elastomer composition of the present invention.
- thermoplastic elastomer composition of the present invention A composition in which the flow starting temperature of the molded body H is 180 to 350 ° C. (more preferably 190 to 320 ° C., more preferably 200 to 300 ° C.) is preferable as the thermoplastic elastomer composition of the present invention. .
- thermoplastic elastomer composition of the present invention A composition having a tensile elongation at break of the molded product H of 100% or more (preferably 150 to 1000%, more preferably 200 to 800%) is preferable as the thermoplastic elastomer composition of the present invention. .
- thermoplastic elastomer composition of the present invention is particularly excellent in heat resistance and chemical resistance, and in the thermoplastic elastomer material field, it can be used in the same fields of application as thermoplastic polyesters and thermoplastic polyamide elastomers.
- thermoplastic elastomer composition of the present invention examples include the following.
- a molded body can be obtained by appropriately heat-molding the thermoplastic elastomer composition of the present invention according to a conventional method.
- the use of the molded product obtained by thermoforming the thermoplastic elastomer of the present invention is not particularly limited, and general styrene elastomers, polyolefin elastomers, polyurethane elastomers, polyamide elastomers, acrylic elastomers and polyesters It can be used in the field where the system elastomer is used.
- thermoplastic elastomer composition of the present invention Since the molded product obtained by molding the thermoplastic elastomer composition of the present invention has excellent heat resistance, for example, for applications that require heat resistance of 100 ° C. or higher (120 ° C. or higher, 150 ° C. or higher, depending on the design). Can also be suitably used.
- the temperature at the time of heat molding is preferably 180 ° C. or higher from the viewpoint of fluidity of the composition and molding processability resulting from it, from the viewpoint of preventing thermal decomposition of the (meth) acrylic component of component A in the composition, 350 ° C. or lower is preferable. From these viewpoints, the temperature at the time of thermoforming is preferably 180 to 350 ° C, more preferably 200 to 320 ° C.
- Any molding machine capable of melt-molding the composition can be used as an apparatus used for producing a molded body using the thermoplastic elastomer composition of the present invention.
- Examples thereof include a kneader, an extrusion molding machine, an injection molding machine, a press molding machine, a blow molding machine, and a mixing roll.
- the molded article of the present invention can be obtained by heat-molding the above thermoplastic elastomer composition at 180 to 350 ° C., preferably a molded article having a flow start temperature of 180 to 350 ° C., which is an indicator of heat resistance.
- a molded body at 320 ° C. is more preferable, and a molded body at 200 to 300 ° C. is a more preferable molded body.
- a molded body having a flow start temperature of less than 180 ° C may have insufficient heat resistance. There is no particular upper limit on the flow start temperature of the molded product, but it is difficult to obtain a molded product exceeding 350 ° C. from the composition of the present invention.
- the molded article of the present invention can be obtained by heat-molding the above thermoplastic elastomer composition at 180 to 350 ° C.
- the tensile elongation at break of the molded article is 100% or more as a basic characteristic required for the elastomer. Is preferable, 150 to 1000% is more preferable, and 200 to 800% is more preferable. Similarly, the tensile strength at break is preferably 2 to 20 MPa, more preferably 4 to 15 MPa.
- the molded body of the present invention can be obtained by heat-molding the above thermoplastic elastomer composition at 180 to 350 ° C., and the A hardness of the molded body that is an index of flexibility is preferably 20 to 90, preferably 30 to 85. More preferred. If the A hardness of the molded body exceeds 90, flexibility may be insufficient. There is no particular lower limit on the A hardness of the molded body, but a molded body obtained by molding the thermoplastic elastomer composition of the present invention is less likely to have an A hardness of less than 20.
- Copolymer C1 was prepared in the same manner as copolymer C3, except that a monomer mixture composed of a monomer having the composition shown in Table 3-1, xylene 15 parts by weight, and DTBP 0.3 parts by weight was used.
- C2 and comparative copolymer C′5 were prepared.
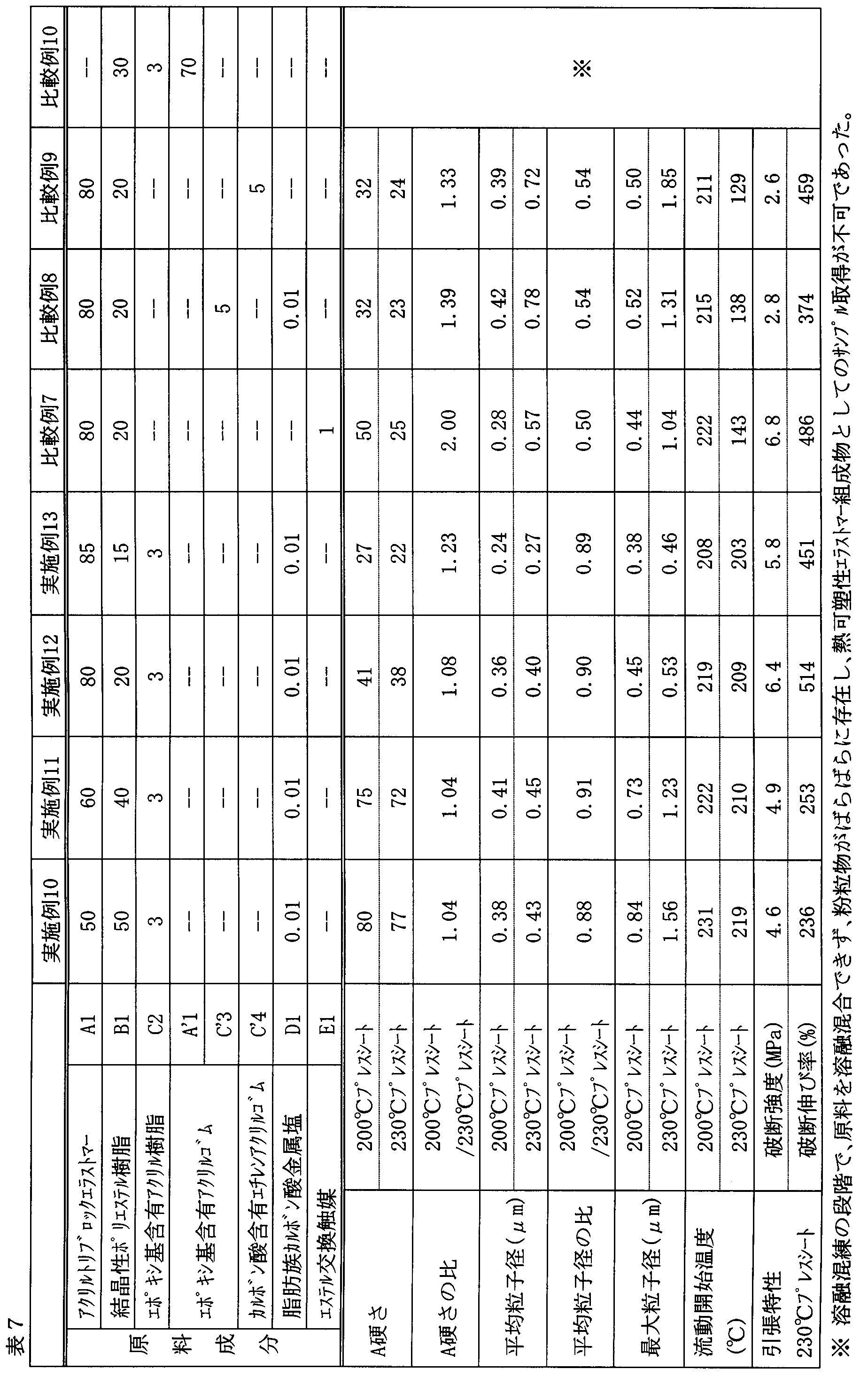
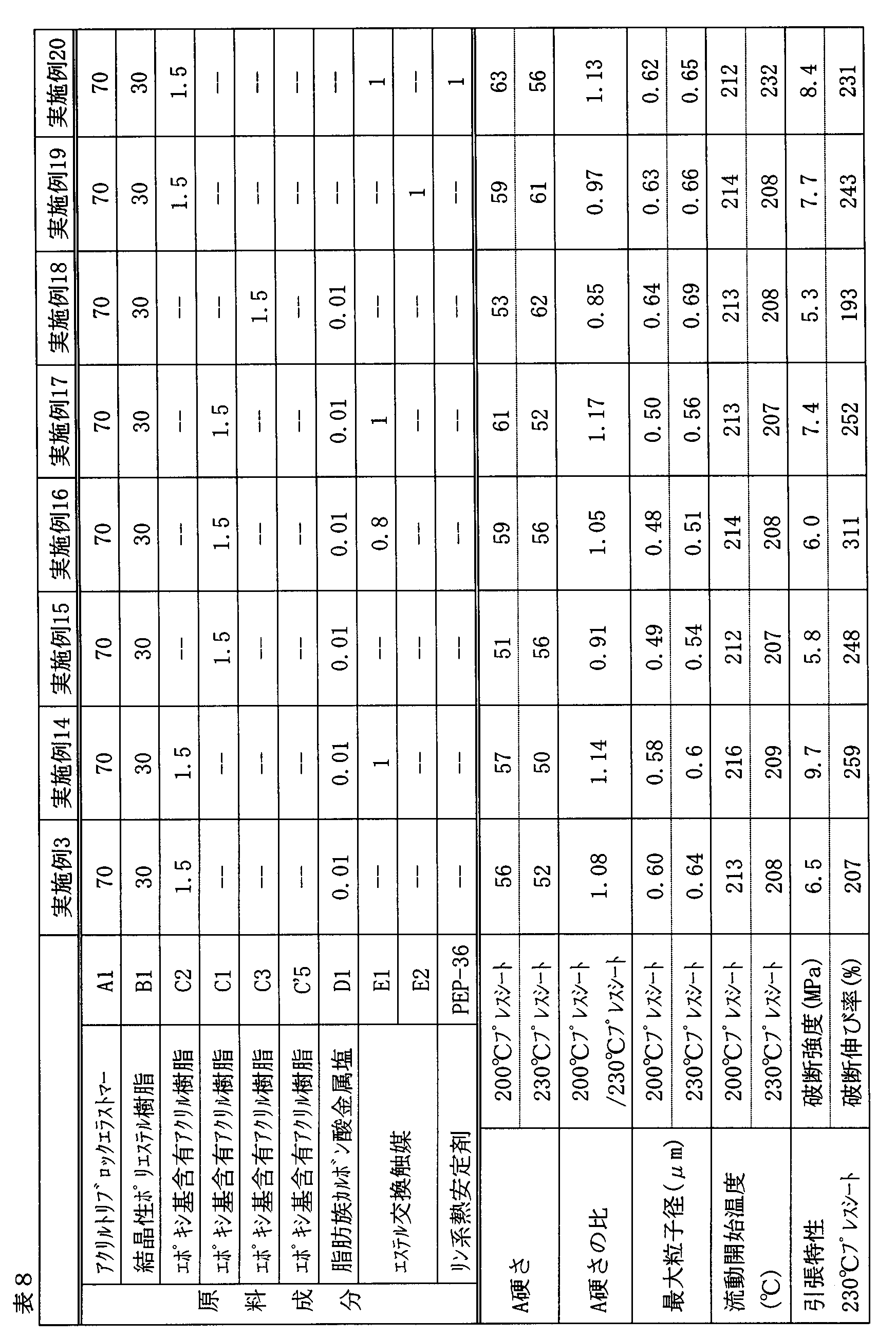
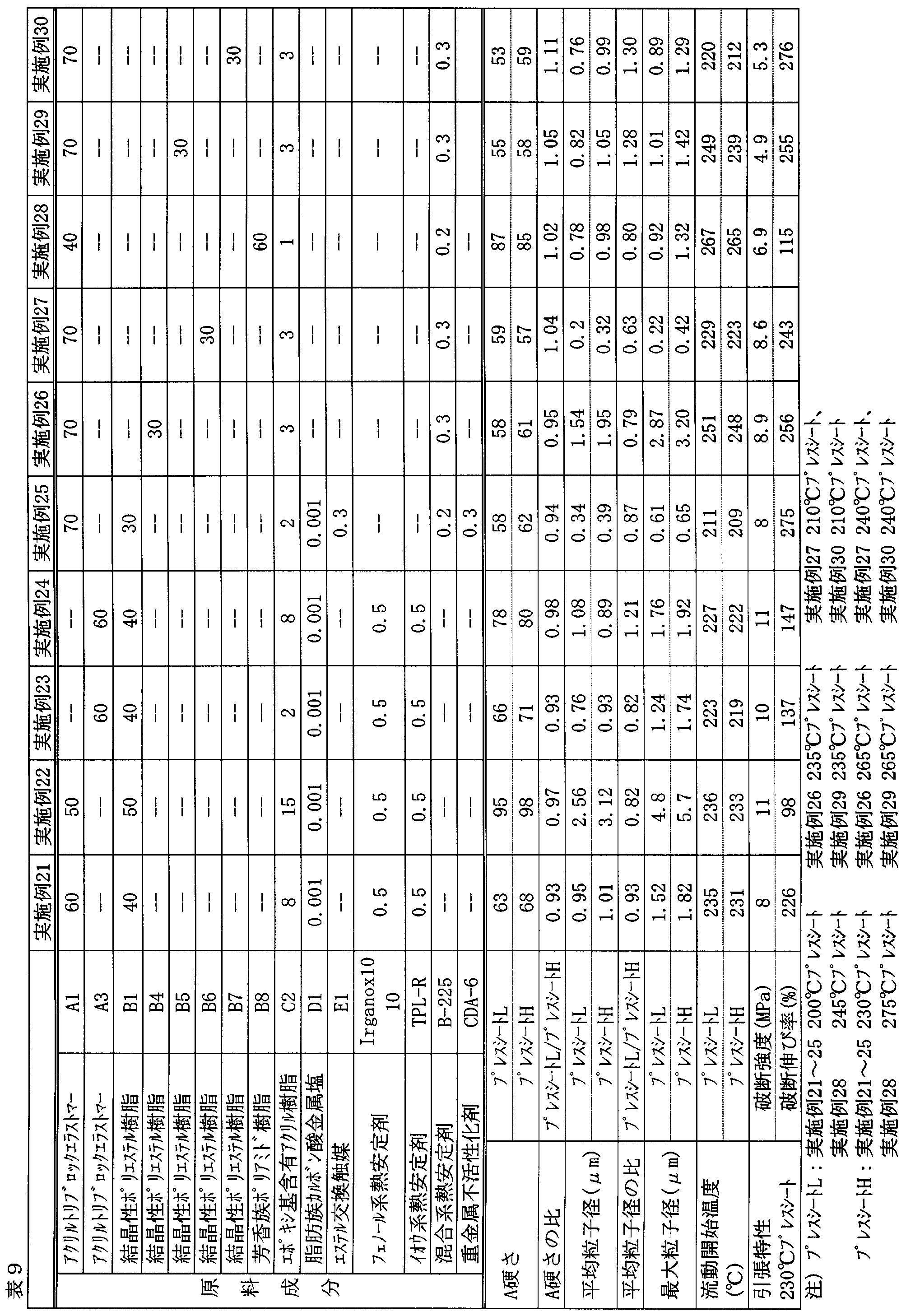
- Examples 1 to 30 and Comparative Examples 1 to 10 A batch-type kneader (Plastograph EC50 type manufactured by Brabender) heated to 260 ° C was charged with a total of 54 g of raw material components at the composition ratios (parts by weight) shown in Tables 6 to 9, and the raw material was rotated at 60 r / m. Was melt kneaded. In a kneading time of 10 minutes, the entire kneaded material in a molten state was taken out and cooled at room temperature to obtain a composition.
- Plastograph EC50 type manufactured by Brabender heated to 260 ° C was charged with a total of 54 g of raw material components at the composition ratios (parts by weight) shown in Tables 6 to 9, and the raw material was rotated at 60 r / m. Was melt kneaded. In a kneading time of 10 minutes, the entire kneaded material in a molten state was taken out and
- the physical properties of the raw material components shown in Tables 1 to 4 were measured by the following methods. Production of the raw material sheet was carried out in the same manner as the production of a press sheet of the composition described later. However, the press temperature was adjusted according to the raw material (a temperature at which a small amount of raw material was raised in advance and the temperature at which flow was visually confirmed was grasped).
- thermoplastic resin listed in Table 2 is dried in a dryer at 120 ° C. for 4 hours.
- a capillary rheometer Capillograph 1D, manufactured by Toyo Seiki Co., Ltd.
- the dried sample piece is filled into a cylinder at a temperature setting of 240 ° C. and allowed to stay for 5 minutes after completion of filling.
- the melt viscosity of the sample piece is measured under the condition of a shear rate of 1216 sec ⁇ 1 .
- THF is used as a solvent and is determined from polystyrene conversion.
- Average number of epoxy groups (Fn) a x b / 100c
- a, b and c are as follows. a: Ratio (% by weight) of vinyl monomer units having an epoxy group contained in the copolymer b: number average molecular weight of copolymer c: molecular weight of vinyl monomer having epoxy group
- Epoxy value It is the number of milliequivalents of epoxy groups contained in 1 g of sample (equivalent number of epoxy groups contained in 1 kg of sample), and corresponds to the epoxy index of JIS K7236.
- compositions obtained in Examples and Comparative Examples were heated to 200 ° C. or 230 ° C. using a mold of 2 mm thickness ⁇ 10 cm ⁇ 10 cm with a hot press machine (50 mm hot press manufactured by Toho Co., Ltd.). Two minutes of press sheets, 200 ° C. press sheet and 230 ° C. press sheet with different heating conditions were produced by applying a heat press for 5 minutes and then a cooling press for 5 minutes.
- the flow start temperature of the compositions of Examples shown below is 208 to 231 ° C.
- the press sheet forming temperature (200 ° C.) for measuring the physical properties of the composition is lower than the flow start temperature. It flows to such an extent that a sheet-like molded body free from obvious defects such as bubbles can be obtained by hot pressing.
- 200 ° C. is a temperature corresponding to a temperature L (((melting point of component B) ⁇ 20 ° C.) ⁇ 5 ° C.), and 230 ° C. is a temperature H (((component B melting point) + 10 ° C.) within ⁇ 5 ° C.].
- 250 ° C. is a temperature corresponding to the temperature L [((the melting point of component B) ⁇ 20 ° C.) ⁇ 5 ° C.]
- 280 ° C. is the temperature H [((the melting point of component B) + 10 ° C. ) Within ⁇ 5 ° C].
- the press sheet obtained by hot pressing at any of the temperature L and the temperature H also corresponds to the molded article of the present invention, but the physical properties of the press sheet obtained by hot pressing at the temperature L are as described above. It is treated as corresponding to the physical property values of the thermoplastic elastomer composition.
- the press sheet is cut with a cutter knife, and the surface of the cross section is taken at a magnification of 1000 to 5000 using a scanning electron microscope (Electron microscope VB-9800 manufactured by Keyence).
- a scanning electron microscope Electron microscope VB-9800 manufactured by Keyence.
- ImageJ image analysis software
- the size of the particles that can be observed is measured for a total of 10 particle sizes, and the average particle size is calculated.
- the particle size of the largest particle among the particles that can be observed is measured.
- the surface of the sample is treated with a solvent A that easily dissolves component A and solvent B, or solvent B that hardly dissolves component A and easily dissolves component B.
- a clear image can be obtained, which is a preferable method.
- the treatment conditions with the solvent A are adjusted, the component A that forms the continuous phase is eroded, and the presence of the component B that is the dispersed phase (dispersed particles) becomes clear. It is also possible to measure the particle size of particles composed of the component B which dissolves almost all of the component A forming the continuous phase and is released in a dispersed state in the solution.
- the component B which is the dispersed phase (dispersed particles)
- the component B which is the dispersed phase (dispersed particles)
- the solvent A or a solvent B can be selected by checking the solubility of a component A and a component B, which are raw materials, in a typical solvent in advance.
- the solvent A include acetone and methanol.
- FIGS. 1 to 6 show electron micrographs of cross sections of 200 ° C. press sheets and / or 230 ° C. press sheets of the compositions obtained in Examples 1 and 4 and Comparative Examples 1 and 3, respectively.
- an electron microscope for confirming the state of the dispersed phase either a scanning electron microscope (SEM) or a transmission electron microscope (TEM) may be used, and if a method that can be clearly observed according to the sample is selected. Good.
- Examples 10 were compared with Comparative Example 10 in which Component A was not blended, Comparative Example 6 in which Component B was not blended, and Compositions 1 to 5 and 7 to 9 in which Component C was not blended.
- Compositions 1 to 20 have excellent A tensile strength, dispersion state of dispersed phase, and flow start temperature with no significant difference between a 200 ° C. (temperature L) press sheet and a 230 ° C. (temperature H) press sheet. It can be seen that it has properties (tensile strength and elongation).
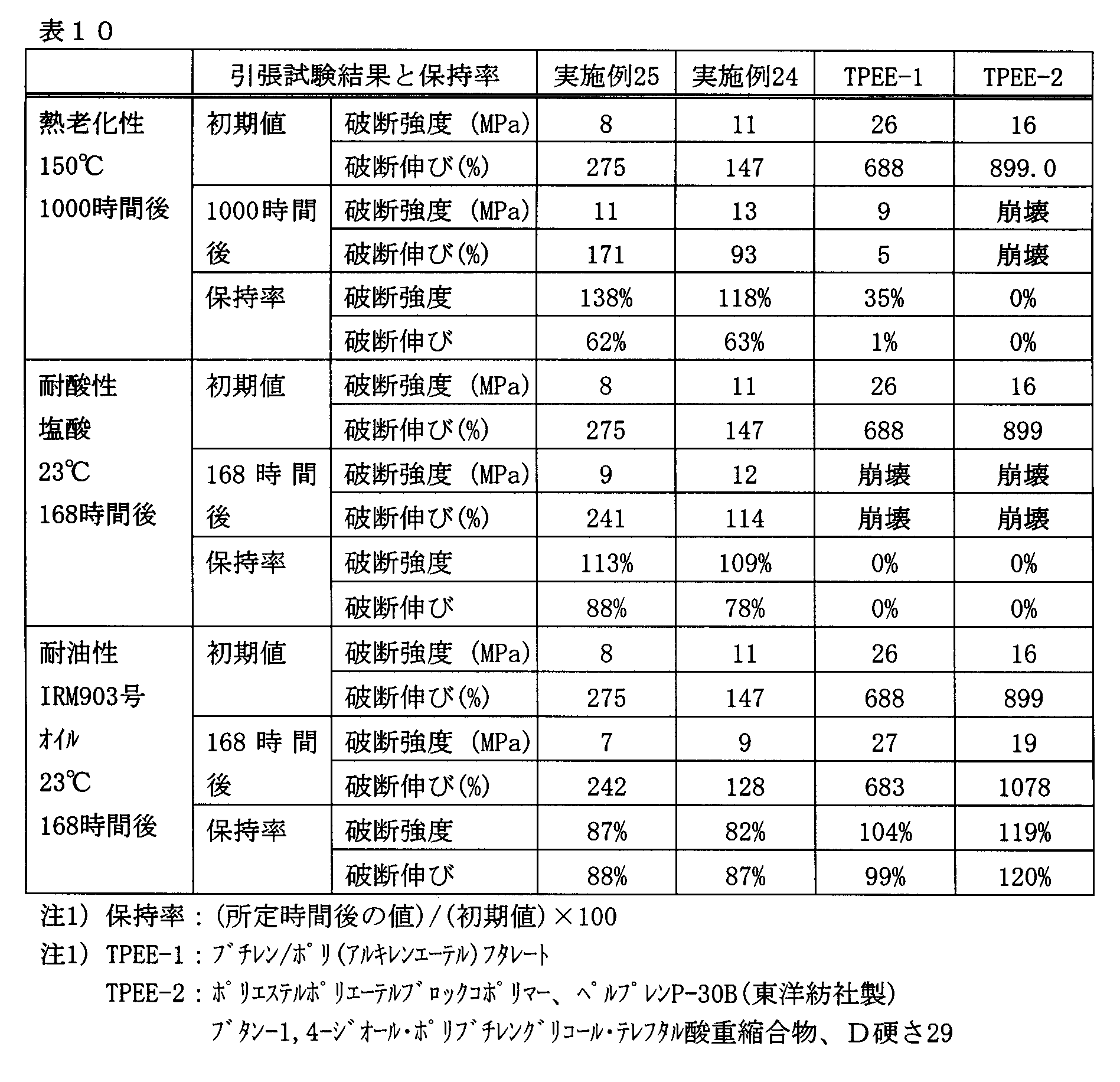
- compositions of Examples 24 and 25 and commercially available polyester elastomers were tested and evaluated for heat aging resistance, acid resistance and oil resistance by the following methods. The results are shown in Table 10.
- thermoplastic elastomer composition of the present invention has excellent heat aging properties and good acid resistance and oil resistance as compared with conventionally available polyester elastomers. It can be seen that it is excellent.
- the molded body obtained from the thermoplastic elastomer composition of the present invention can be used in various fields such as electrical and electronic parts, automotive parts, sealing materials, packings, vibration damping members, and tubes.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
〔1〕 (メタ)アクリルエラストマー(成分A)、
融点が180~350℃であり、エポキシ基と反応可能な官能基を有する熱可塑性樹脂(成分B)、及び
テトラヒドロフランへの溶解性を有し、1分子中に平均2個以上のエポキシ基を有するビニル共重合体(成分C)を含有し、
前記各成分の割合が、
(メタ)アクリルエラストマー(成分A)40~95重量部、
熱可塑性樹脂(成分B)5~60重量部、及び
ビニル共重合体(成分C)0.1~30重量部であり、
(メタ)アクリルエラストマー(成分A)と熱可塑性樹脂(成分B)の合計が100重量部であり、
成分Cが、SP値が17.5~25.0である単量体単位(単量体単位c1)を50重量%以上含む単量体単位から構成され、
流動開始温度が180~350℃である熱可塑性エラストマー組成物、
〔2〕 (メタ)アクリルエラストマー(成分A)、
融点が180~350℃であり、エポキシ基と反応可能な官能基を有する熱可塑性樹脂(成分B)、及び
テトラヒドロフランへの溶解性を有し、1分子中に平均2個以上のエポキシ基を有するビニル共重合体(成分C)を含有し、
前記各成分の割合が、
(メタ)アクリルエラストマー(成分A)40~95重量部、
熱可塑性樹脂(成分B)5~60重量部、及び
ビニル共重合体(成分C)0.1~30重量部であり、
(メタ)アクリルエラストマー(成分A)と熱可塑性樹脂(成分B)の合計が100重量部であり、
成分Cが(メタ)アクリルモノマー、スチレン及びスチレン誘導体からなる群より選ばれた少なくとも1種の単量体単位(単量体単位c2)(ただし、SP値が特定される単量体のうちSP値が17.5未満のもの及び25.0を超えるものは除く)を50重量%以上含む単量体単位から構成され、
流動開始温度が180~350℃である熱可塑性エラストマー組成物、
〔3〕 前記〔1〕又は〔2〕記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、流動開始温度が180~350℃である成形体、
〔4〕 前記〔1〕又は〔2〕記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、引張破断伸び率が100%以上である成形体、並びに
〔5〕 前記〔1〕又は〔2〕記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、A硬さが20~90である成形体
に関する。
すなわち、成形するために十分な流動性を得られる温度(後述する温度H)で加熱成形したときにも、連続相を形成する成分Aは流動するが分散相を形成する成分Bは溶融しない温度(後述する温度L)で加熱成形したときと比べて柔軟性及び耐熱性が大きく変化しない(特に耐熱性がほとんど低下しない)という効果を奏する。
(メタ)アクリルエラストマー(成分A)、
融点が180~350℃であり、エポキシ基と反応可能な官能基を有する熱可塑性樹脂(成分B)、及び
テトラヒドロフランに対して溶解性を有し、1分子中に平均2個以上のエポキシ基を有するビニル共重合体であって、特定の単量体単位から構成されるビニル共重合体(成分C)
を、特定の割合で含有するものである。本発明の熱可塑性エラストマー組成物において、成分Aが連続相を形成し、成分Bが分散相を形成する。成分Cは、連続相を形成する成分A中での成分B(分散相)のサイズ(粒子径)を安定化させる相溶化剤として機能するものであり、本発明の熱可塑性エラストマー組成物は、柔軟性及び耐熱性が優れる成形体を与えることができ、該成形体が優れた柔軟性及び耐熱性を発揮するための成形温度依存性が小さく、成形体への加工に際しても、成形条件の制限が小さいという特徴を有する。
第一の態様:SP値が17.5~25.0である単量体単位(単量体単位c1)を50重量%以上含む単量体単位、及び
第二の態様:(メタ)アクリルモノマー、スチレン及びスチレン誘導体からなる群より選ばれた少なくとも1種の単量体単位(単量体単位c2)を50重量%以上含む単量体単位
があり、単量体単位c1とc2は成分Aとの親和性が良好である点で共通している。なお、第一の態様と第二の態様は、便宜上区分したものであり、両者に共通した単量体もある。即ち、SP値が17.5~25.0である、(メタ)アクリルモノマー、スチレン及びスチレン誘導体はいずれの態様にも該当する。
[Van Krevelen] Solubility parameter : δ
δ = (Ecoh / V)0.5
Ecoh =Σ Ecoh, i
単位 : (J/cm3)0.5
推算に必要な物性値
V : 構成繰り返し単位(CRU)のモル体積 [cm3/mol]
原子団寄与法で計算される物理量
Ecoh : Hoftyzer & Van Krevelenのモル凝集エネルギー [J/mol]
原子団パラメータ
Ecoh, i :i番目のモル凝集エネルギーに対する原子団パラメータ [J/mol]

で表されるリン酸エステル化合物、式(II):
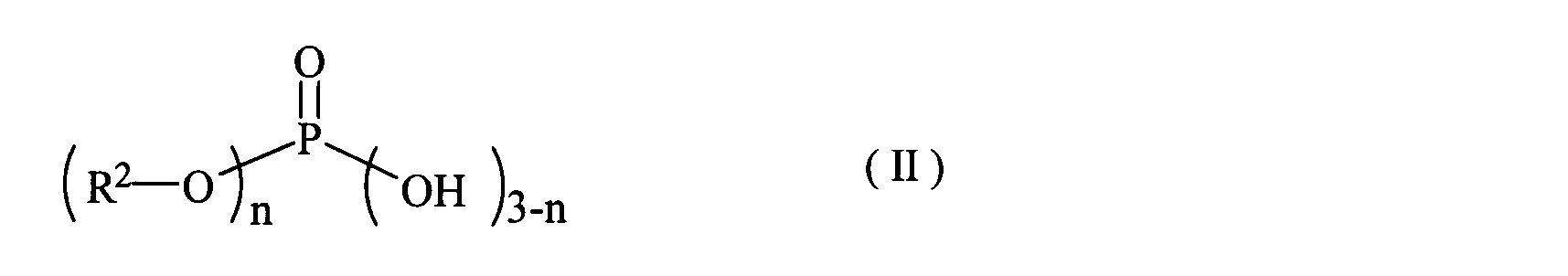
で表されるリン酸エステル化合物等が挙げられる。

で表される化合物が挙げられる。
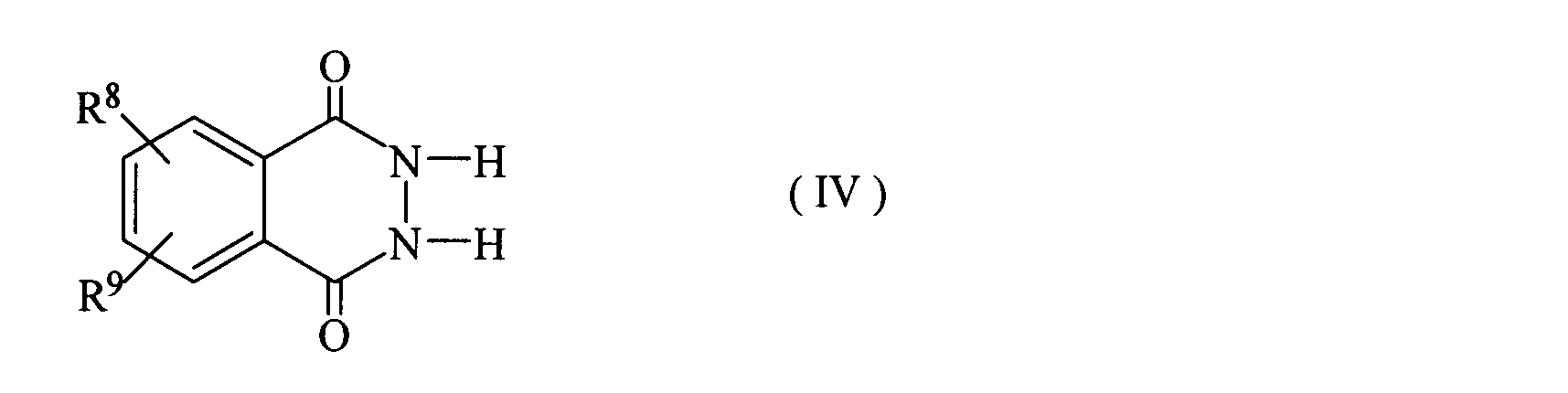
で表される化合物等が挙げられる。式(IV)で表される化合物の具体例としては、フタル酸ヒドラジド等が挙げられる。
すなわち、成形するために十分な流動性を得られる温度(後述する温度H)で加熱成形したときにも、連続相を形成する成分Aは流動するが分散相を形成する成分Bは溶融しない温度(後述する温度L)で加熱成形したときと比べて柔軟性及び耐熱性が大きく変化しない(特に耐熱性がほとんど低下しない)という効果を奏する。
調製された組成物が、ペレット、粉末、不定形塊状物等のように直接流動開始温度を測定する試料として使用できない形状である場合は、熱可塑性樹脂(成分B)の融点より20℃低い温度付近(±5℃)で短時間(5分間)の熱プレスによりシート状の成形体を作製し、組成物の物性値測定用の試料を得る。このような低い温度で短時間の熱履歴がかかっても、本発明の組成物の物性値に大きな変化が生じることはなく、上記成形体の物性値を組成物の物性値とみなすことができる。流動開始温度だけでなく、A硬さ、粒子径等も同様に扱われる。熱プレスで組成物の物性値測定用の試料を得る際には、組成物を塊状物として調製すると気泡等のない試料を得やすい。
特に、成形体L及び成形体Hの流動開始温度がいずれも、180~350℃(より好ましくは190~320℃、さらに好ましくは200~300℃)となるような組成物は、本発明の熱可塑性エラストマー組成物として好ましいものである。
・車体回り
ドアーラッチ、コントロールケーブルカバー、ブーツ、エンブレム、シャーシ・ステアリング周り、フューエルホース、
等速ジョイントブーツ、ピニオン&ラックブーツ、ストラットサスペンションブーツ、ボールジョイント用ブッシュ、ダストシール、ブレーキホース、
エアーダクトホース、エアーダクト、エアーインテークホース、エンジンコントロール系バキュームホース、インタークーラーホース、フューエルラインカバー、各種防振・制振材、ラジエータホース、ヒーターホース、オイルクーラーホース、パワーステアリングホース、各種ガスケット類、エンジンアンダーフード、エンジンルームカバー等の各種カバー・ケース類
・電子・民生機器用電線
コンピュータ・OA機器・テレビ・VTRなどの電線被覆
・通信ケーブル
通信用電線・光ファイバー用被覆材料
・絶縁電線・通信ケーブル・機器用電線・自動車用ワイヤハーネス
空・油圧ホース(チューブ)、高圧ホース(チューブ)、燃料ホース(チューブ)、コンベアベルト、V・丸ベルト、タイミングベルト、クッショングリップ、フレックスハンマー、消音ギア、フレキシブルカップリング、ガソリンタンクシート、ガスケット、パッキン、シール材、Oリング、ダイヤフラム、カールコード、アキュムレータ内装、搬送ローラ、圧縮スプリング、マンドレル、牽引ロープジャケット
ゴルフボール表皮、スキー靴・登山靴カフ、スポーツシューズ本底
オイルジャケットを備えた容量1リットルの加圧式攪拌槽型反応器のオイルジャケット温度を200℃に保った。一方、スチレン74重量部、グリシジルメタクリレート20重量部、アクリル酸n-ブチル6重量部、キシレン15重量部及び重合開始剤としてジターシャリーブチルパーオキサイド(DTBP)0.5重量部からなる単量体混合液を原料タンクに仕込んだ。一定の供給速度(48g/分、滞留時間:12分)で原料タンクから反応器に連続供給し、反応器の内容液重量が約580gで一定になるように反応液を反応器の出口から連続的に抜き出した。その時の反応器内温は、約210℃に保たれた。
反応器内部の温度が安定してから36分経過した後から、抜き出した反応液を減圧度30kPa、温度250℃に保った薄膜蒸発機により連続的に揮発成分除去処理して、揮発成分をほとんど含まない共重合体C3を回収した。180分かけて約7kgの共重合体C3を回収した。
表3-1に示す組成の単量体、キシレン15重量部、及びDTBP 0.3重量部からなる単量体混合液を用いた以外は、共重合体C3と同じ方法にて、共重合体C1、C2及び比較用共重合体C’5を製造した。
260℃に加熱されたバッチ式ニーダー(ブラベンダー社製プラストグラフEC50型)に表6~9に示す組成比(重量部)で原料成分を合計で54g投入し、60r/mの回転数で原料を溶融混練した。混練時間10分で、溶融状態の混練物を全量取り出し、室温で冷却して、組成物を得た。



プレスシートを恒温恒湿室(温度23℃、相対湿度50%)に24時間以上静置し、プレスシートの状態を安定させる。
2mm厚さのプレスシートを3枚重ね、JIS K7215「プラスチックのデュロメータ硬さ試験法」に準じて測定する。
プレスシートより幅12mm×長さ30mmの短冊状のテストピースを裁断し、動的粘弾性測定装置(TAインスツルメント社 RSA-II型)のトーションモード(10gf負荷)、周波数 10Hz、昇温速度 5℃/min、温度 0~280℃の設定で各サンプルの粘弾性特性を測定する。
得られた貯蔵弾性率の変曲点、もしくは測定不能になる温度を流動開始温度とする。なお、ガラス転移温度付近においても貯蔵弾性率の変曲点は現れるが、流動はしないため、流動開始温度には相当しない。
試料約10mgをアルミパンに入れてアルミ蓋を圧着する。アルミパンを示差走査熱量分析計(パーキンエルマー社 DSC8000)の装置測定部に設置し、空気中・昇温速度20℃/分の条件で測定する。
表2に記載の熱可塑性樹脂を120℃の乾燥機内で4時間乾燥させる。
内径1mm×長さ10mmのキャピラリーダイを有するキャピラリーレオメータ(東洋精機(株)製、キャピログラフ1D)を用い、温度設定240℃のシリンダーに乾燥させた試料片を充填し、充填完了から5分間滞留させた後、せん断速度1216sec-1の条件で試料片の溶融粘度を測定する。
ゲルパーミエーションクロマトグラフ(以下、GPCともいう)より、溶剤としてTHFを使用し、ポリスチレン換算から求める。
下記の式から算出する。
平均エポキシ基の個数(Fn)=a×b/100c
上記の式においてa、b及びcはそれぞれ以下のとおりである。
a:共重合体に含まれるエポキシ基を有するビニル単量体単位の割合(重量%)
b:共重合体の数平均分子量
c:エポキシ基を有するビニル単量体の分子量
試料1g中に含まれるエポキシ基のミリ当量数(試料1kg中に含まれるエポキシ基の当量数)であり、JIS K7236のエポキシ指数に相当するものである。
比較例6について、250℃は温度L〔((成分Bの融点)-20℃)±5℃以内〕に相当する温度であり、280℃は温度H〔((成分Bの融点)+10℃)±5℃以内〕に相当する温度である。
温度L及び温度Hのいずれの温度で熱プレスして得られるプレスシートも本発明の成形体に該当するが、温度Lで熱プレスして得られるプレスシートの物性値は前記の通り、本発明の熱可塑性エラストマー組成物の物性値にも該当するとして扱う。
プレスシートを恒温恒湿室(温度23℃、相対湿度50%)に24時間以上静置し、シートの状態を安定させる。
2mm厚さのプレスシートを3枚重ね、JIS K7215「プラスチックのデュロメータ硬さ試験法」に準じて測定する。
プレスシートをカッターナイフで切断し、その断面を走査型電子顕微鏡(キーエンス社製 電子顕微鏡VB-9800)を用いて1000倍~5000倍の倍率で表面写真を撮影する。
得られた写真について、画像解析ソフト「ImageJ」を用いて、観察できる粒子の大小合計10点の粒子サイズを測定し、平均粒子径を算出する。また、観察できる粒子のなかで最大の粒子の粒径を測定する。
このような溶剤A又は溶剤Bは、予め原料である成分A及び成分Bについて代表的溶剤への溶解性をチェックして選択することができる。溶剤Aの例としてはアセトン、メタノール等を挙げることができる。
プレスシートより幅12mm×長さ30mmの短冊状のテストピースを裁断し、動的粘弾性測定装置(TAインスツルメント社 RSA-II型)のトーションモード(10gf負荷)、周波数 10Hz、昇温速度 5℃/min、温度 0~280℃の設定で各サンプルの粘弾性特性を測定する。
得られた貯蔵弾性率の変曲点、もしくは測定不能になる温度を流動開始温度とする。なお、ガラス転移温度付近においても貯蔵弾性率の変曲点は現れるが、流動はしないため、流動開始温度には相当しない。
プレスシートから、型抜機を用いてJIS K7113に記載の3号試験片を作製し、(株)島津製作所製引張試験機(オートグラフ AG-50kND型)を用いて、23℃の温度環境下、200mm/minの速度で試験片を引っ張る。試験片破断時の応力と伸び率をそれぞれ破断強度、破断伸び率として記録する。

引張特性の試験と同じ試験片を作製し、150℃に温度設定されたギヤーオーブン(東洋精機(株)製、S45型)に試験片を吊す。試験開始から1000時間後に試験片を取り出し、23℃の恒温室内にて1日放置し、試験片の温度調整を行った後、引張試験を実施する。
引張特性の試験と同じ試験片を作製し、35%塩酸(和光純薬(株)製、特級試薬)に試験片を浸漬し、23℃の恒温室で静置する。浸漬開始から168時間後に試験片を取り出し、引張試験を実施する。
引張特性の試験と同じ試験片を作製し、ゴム耐液性試験油(日本サン石油(株)製、IRM903)に試験片を浸漬し、23℃の恒温室で静置する。浸漬開始から168時間後に試験片を取り出し、引張試験を実施する。
Claims (20)
- (メタ)アクリルエラストマー(成分A)、
融点が180~350℃であり、エポキシ基と反応可能な官能基を有する熱可塑性樹脂(成分B)、及び
テトラヒドロフランへの溶解性を有し、1分子中に平均2個以上のエポキシ基を有するビニル共重合体(成分C)を含有し、
前記各成分の割合が、
(メタ)アクリルエラストマー(成分A)40~95重量部、
熱可塑性樹脂(成分B)5~60重量部、及び
ビニル共重合体(成分C)0.1~30重量部であり、
(メタ)アクリルエラストマー(成分A)と熱可塑性樹脂(成分B)の合計が100重量部であり、
成分Cが、SP値が17.5~25.0である単量体単位(単量体単位c1)を50重量%以上含む単量体単位から構成され、
流動開始温度が180~350℃である熱可塑性エラストマー組成物。 - (メタ)アクリルエラストマー(成分A)、
融点が180~350℃であり、エポキシ基と反応可能な官能基を有する熱可塑性樹脂(成分B)、及び
テトラヒドロフランへの溶解性を有し、1分子中に平均2個以上のエポキシ基を有するビニル共重合体(成分C)を含有し、
前記各成分の割合が、
(メタ)アクリルエラストマー(成分A)40~95重量部、
熱可塑性樹脂(成分B)5~60重量部、及び
ビニル共重合体(成分C)0.1~30重量部であり、
(メタ)アクリルエラストマー(成分A)と熱可塑性樹脂(成分B)の合計が100重量部であり、
成分Cが(メタ)アクリルモノマー、スチレン及びスチレン誘導体からなる群より選ばれた少なくとも1種の単量体単位(単量体単位c2)(ただし、SP値が特定される単量体のうちSP値が17.5未満のもの及び25.0を超えるものは除く)を50重量%以上含む単量体単位から構成され、
流動開始温度が180~350℃である熱可塑性エラストマー組成物。 - ビニル共重合体(成分C)を構成する単量体単位が、(メタ)アクリルモノマーを10~90重量%含み、スチレン又はスチレン誘導体を10~90重量%含むものである請求項1又は2記載の熱可塑性エラストマー組成物。
- ビニル共重合体(成分C)の重量平均分子量が、1000~10万である、請求項1又は2記載の熱可塑性エラストマー組成物。
- (メタ)アクリルエラストマー(成分A)が、ハードセグメントを構成するブロック2個以上、及びソフトセグメントを構成するブロック1個以上を備えるブロック共重合体である、請求項1又は2記載の熱可塑性エラストマー組成物。
- ハードセグメントを構成するブロック及びソフトセグメントを構成するブロックのガラス転移温度が、それぞれ20~200℃及び-100~19℃の重合体ブロックである、請求項5記載の熱可塑性エラストマー組成物。
- (メタ)アクリルエラストマー(成分A)の重量平均分子量が、5万~100万である、請求項1又は2記載の熱可塑性エラストマー組成物。
- 熱可塑性樹脂(成分B)が、芳香族ポリエステル及び/又はポリアミドである、請求項1又は2記載の熱可塑性エラストマー組成物。
- さらに、脂肪族カルボン酸金属塩(成分D)0.001~10重量部を含有してなる、請求項1又は2記載の熱可塑性エラストマー組成物。
- さらに、エステル交換触媒(成分E)0.01~10重量部を含有してなる、請求項1又は2記載の熱可塑性エラストマー組成物。
- 少なくとも(メタ)アクリルエラストマー(成分A)、熱可塑性樹脂(成分B)及びビニル共重合体(成分C)を含有する原料成分を成分Bの融点以上350℃以下の温度で、押出機又はニーダーにより加熱混練して得られる、請求項1又は2記載の熱可塑性エラストマー組成物。
- 熱可塑性エラストマー組成物を2種類の温度、温度L〔((成分Bの融点)-20℃)±5℃以内〕又は温度H〔((成分Bの融点)+10℃)±5℃以内〕に設定された熱プレス機により5分間加熱して得られる成形体が、いずれの温度における成形体についても、
連続相と分散相とからなる相分離構造を有し、電子顕微鏡により観察される分散相の最大粒子径が5μm以下である、請求項1又は2記載の熱可塑性エラストマー組成物。 - 熱可塑性エラストマー組成物を2種類の温度、温度L〔((成分Bの融点)-20℃)±5℃以内〕又は温度H〔((成分Bの融点)+10℃)±5℃以内〕に設定された熱プレス機により5分間加熱して得られる成形体が、いずれの温度における成形体についても、
A硬さが20~90である、請求項1又は2記載の熱可塑性エラストマー組成物。 - 熱可塑性エラストマー組成物を2種類の温度、温度L〔((成分Bの融点)-20℃)±5℃以内〕又は温度H〔((成分Bの融点)+10℃)±5℃以内〕に設定された熱プレス機により5分間加熱して得られる成形体について、
A硬さの比(温度Lで加熱して得られた成形体のA硬さ/温度Hで加熱して得られた成形体のA硬さ)が、0.6~1.3である、請求項1又は2記載の熱可塑性エラストマー組成物。 - 熱可塑性エラストマー組成物を温度H〔((成分Bの融点)+10℃)±5℃以内〕に設定された熱プレス機により5分間加熱して得られる成形体の流動開始温度が180~350℃である、請求項1又は2記載の熱可塑性エラストマー組成物。
- 熱可塑性エラストマー組成物を温度H〔((成分Bの融点)+10℃)±5℃以内〕に設定された熱プレス機により5分間加熱して得られる成形体の引張破断伸び率が100%以上である、請求項1又は2記載の熱可塑性エラストマー組成物。
- ビニル共重合体(成分C)の割合が、0.1~15重量部である、請求項1又は2記載の熱可塑性エラストマー組成物。
- 請求項1~17いずれか記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、流動開始温度が180~350℃である成形体。
- 請求項1~17いずれか記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、引張破断伸び率が100%以上である成形体。
- 請求項1~17いずれか記載の熱可塑性エラストマー組成物を180~350℃で加熱成形して得られる、A硬さが20~90である成形体。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013554339A JP6087297B2 (ja) | 2012-01-19 | 2013-01-17 | 熱可塑性エラストマー組成物 |
| EP13739108.2A EP2805996B1 (en) | 2012-01-19 | 2013-01-17 | Thermoplastic elastomer composition |
| US14/371,546 US9175159B2 (en) | 2012-01-19 | 2013-01-17 | Thermoplastic elastomer composition |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012008975 | 2012-01-19 | ||
| JP2012-008975 | 2012-01-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013108836A1 true WO2013108836A1 (ja) | 2013-07-25 |
Family
ID=48799259
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/050799 Ceased WO2013108836A1 (ja) | 2012-01-19 | 2013-01-17 | 熱可塑性エラストマー組成物 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9175159B2 (ja) |
| EP (1) | EP2805996B1 (ja) |
| JP (1) | JP6087297B2 (ja) |
| WO (1) | WO2013108836A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015189844A (ja) * | 2014-03-28 | 2015-11-02 | クラレプラスチックス株式会社 | 摺動性に優れた熱可塑性樹脂組成物 |
| WO2016117586A1 (ja) * | 2015-01-20 | 2016-07-28 | 東洋紡株式会社 | 赤外光透過性ポリエステル樹脂組成物 |
| JP2016176025A (ja) * | 2015-03-20 | 2016-10-06 | アロン化成株式会社 | 熱可塑性エラストマー組成物 |
| JP2019044129A (ja) * | 2017-09-06 | 2019-03-22 | 出光興産株式会社 | ポリカーボネート系樹脂組成物及びその成形品 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6207909B2 (ja) * | 2013-07-18 | 2017-10-04 | アロン化成株式会社 | 熱可塑性エラストマー組成物 |
| JP6207915B2 (ja) * | 2013-07-19 | 2017-10-04 | アロン化成株式会社 | 熱可塑性エラストマー組成物 |
| JP6589219B2 (ja) * | 2014-02-06 | 2019-10-16 | 国立研究開発法人科学技術振興機構 | 温度センサー用樹脂組成物、温度センサー用素子、温度センサーおよび温度センサー用素子の製造方法 |
| CN107667145B (zh) * | 2015-05-26 | 2020-06-30 | Sabic环球技术有限责任公司 | 聚(对苯二甲酸丁二酯)组合物及相关制品 |
| WO2017073754A1 (ja) * | 2015-10-30 | 2017-05-04 | 矢崎総業株式会社 | 耐熱柔軟電線及びこれを用いたワイヤーハーネス |
| CN111748167A (zh) * | 2020-06-28 | 2020-10-09 | 安徽中鼎橡塑制品有限公司 | 一种热塑性硫化弹性体材料及其制备方法 |
| CN112226049B (zh) * | 2020-10-19 | 2023-07-21 | 禾创高分子技术(广东)有限公司 | 一种阻燃耐油防霉弹性体热缩管料及其母粒的制备方法 |
| US11664133B2 (en) * | 2020-10-28 | 2023-05-30 | Dell Products L.P. | Power cord assembly containing recycled and renewable polymeric compositions |
| US20250109808A1 (en) * | 2022-03-30 | 2025-04-03 | The Yokohama Rubber Co., Ltd. | Hose for refrigerant transportation and production method therefor |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03221578A (ja) * | 1990-01-26 | 1991-09-30 | Sumitomo Electric Ind Ltd | フレキシブル印刷配線板用接着剤組成物 |
| JP2003119249A (ja) * | 2001-10-17 | 2003-04-23 | Mitsui Chemicals Inc | 液晶封止用樹脂組成物 |
| JP2004035637A (ja) | 2002-07-01 | 2004-02-05 | Kanegafuchi Chem Ind Co Ltd | キーパッド用熱可塑性エラストマー組成物及びこれを用いたキーパッド |
| JP2006124724A (ja) | 2002-02-13 | 2006-05-18 | Kaneka Corp | ブロック共重合体 |
| JP2007092038A (ja) | 2005-08-30 | 2007-04-12 | Toray Ind Inc | 樹脂組成物およびそれからなる成形品 |
| JP2007525542A (ja) * | 2003-01-23 | 2007-09-06 | フロイデンベルグ−エヌオーケー ジェネラル パートナーシップ | 磁化性熱可塑性エラストマー |
| JP2009079119A (ja) | 2007-09-26 | 2009-04-16 | Kaneka Corp | 樹脂組成物からなる成形体および改質剤 |
| WO2012173241A1 (ja) * | 2011-06-17 | 2012-12-20 | 太陽インキ製造株式会社 | 難燃性硬化性樹脂組成物、それを用いたドライフィルム及びプリント配線板 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI314934B (en) | 2002-02-13 | 2009-09-21 | Kaneka Corporatio | Polymer composition |
| US20040034142A1 (en) * | 2002-05-13 | 2004-02-19 | Techno Polymer Co., Ltd., | Laser-marking thermoplastic resin composition |
| US20050275565A1 (en) | 2003-01-23 | 2005-12-15 | Daniel Nachtigal | Magnetizable polymeric compositions |
| JP5023449B2 (ja) * | 2005-08-08 | 2012-09-12 | 日油株式会社 | 熱可塑性エラストマー組成物 |
| TWI400293B (zh) * | 2008-12-24 | 2013-07-01 | Ind Tech Res Inst | 熱塑性硫化體 |
| JP2011245633A (ja) * | 2010-05-24 | 2011-12-08 | Nof Corp | 多層成形体 |
-
2013
- 2013-01-17 WO PCT/JP2013/050799 patent/WO2013108836A1/ja not_active Ceased
- 2013-01-17 US US14/371,546 patent/US9175159B2/en active Active
- 2013-01-17 EP EP13739108.2A patent/EP2805996B1/en not_active Not-in-force
- 2013-01-17 JP JP2013554339A patent/JP6087297B2/ja active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03221578A (ja) * | 1990-01-26 | 1991-09-30 | Sumitomo Electric Ind Ltd | フレキシブル印刷配線板用接着剤組成物 |
| JP2003119249A (ja) * | 2001-10-17 | 2003-04-23 | Mitsui Chemicals Inc | 液晶封止用樹脂組成物 |
| JP2006124724A (ja) | 2002-02-13 | 2006-05-18 | Kaneka Corp | ブロック共重合体 |
| JP2004035637A (ja) | 2002-07-01 | 2004-02-05 | Kanegafuchi Chem Ind Co Ltd | キーパッド用熱可塑性エラストマー組成物及びこれを用いたキーパッド |
| JP2007525542A (ja) * | 2003-01-23 | 2007-09-06 | フロイデンベルグ−エヌオーケー ジェネラル パートナーシップ | 磁化性熱可塑性エラストマー |
| JP2007092038A (ja) | 2005-08-30 | 2007-04-12 | Toray Ind Inc | 樹脂組成物およびそれからなる成形品 |
| JP2009079119A (ja) | 2007-09-26 | 2009-04-16 | Kaneka Corp | 樹脂組成物からなる成形体および改質剤 |
| WO2012173241A1 (ja) * | 2011-06-17 | 2012-12-20 | 太陽インキ製造株式会社 | 難燃性硬化性樹脂組成物、それを用いたドライフィルム及びプリント配線板 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2805996A4 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015189844A (ja) * | 2014-03-28 | 2015-11-02 | クラレプラスチックス株式会社 | 摺動性に優れた熱可塑性樹脂組成物 |
| WO2016117586A1 (ja) * | 2015-01-20 | 2016-07-28 | 東洋紡株式会社 | 赤外光透過性ポリエステル樹脂組成物 |
| JPWO2016117586A1 (ja) * | 2015-01-20 | 2017-10-26 | 東洋紡株式会社 | 赤外光透過性ポリエステル樹脂組成物 |
| JP2016176025A (ja) * | 2015-03-20 | 2016-10-06 | アロン化成株式会社 | 熱可塑性エラストマー組成物 |
| JP2019044129A (ja) * | 2017-09-06 | 2019-03-22 | 出光興産株式会社 | ポリカーボネート系樹脂組成物及びその成形品 |
| US11203664B2 (en) | 2017-09-06 | 2021-12-21 | Idemitsu Kosan Co., Ltd. | Polycarbonate resin composition and molded article thereof |
| TWI791604B (zh) * | 2017-09-06 | 2023-02-11 | 日本商出光興產股份有限公司 | 聚碳酸酯系樹脂組合物及其成形品 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2805996A1 (en) | 2014-11-26 |
| JP6087297B2 (ja) | 2017-03-01 |
| US9175159B2 (en) | 2015-11-03 |
| EP2805996B1 (en) | 2017-11-22 |
| EP2805996A4 (en) | 2015-11-11 |
| JPWO2013108836A1 (ja) | 2015-05-11 |
| US20140357775A1 (en) | 2014-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6087297B2 (ja) | 熱可塑性エラストマー組成物 | |
| WO2004013192A1 (ja) | アクリル系ブロック共重合体および熱可塑性樹脂組成物 | |
| US11891498B2 (en) | Molded article provided with a resin part | |
| JP2018529011A (ja) | 2つのセグメントを含むポリマーの、ポリマー添加剤としての使用 | |
| JP6282143B2 (ja) | セルロースエステル組成物 | |
| CN103589125A (zh) | 一种聚乳酸/聚丙烯共混物及其制备方法 | |
| JP6104744B2 (ja) | 熱可塑性エラストマー組成物 | |
| JP6207909B2 (ja) | 熱可塑性エラストマー組成物 | |
| JP5756025B2 (ja) | 熱可塑性エラストマー組成物及びその製造方法 | |
| JP2015021078A (ja) | 熱可塑性エラストマー組成物 | |
| TWI778152B (zh) | 氯乙烯樹脂用之塑化劑、氯乙烯樹脂組成物、電線及車輛內裝材料 | |
| CN104011135B (zh) | 生物降解性树脂组合物和利用其的生物降解性薄片的制备方法 | |
| JP2015021050A (ja) | 熱可塑性エラストマー組成物 | |
| JP6862709B2 (ja) | 樹脂組成物、積層体及び建装材 | |
| WO2005073269A1 (ja) | 熱可塑性エラストマー組成物および成形品 | |
| JP2015021049A (ja) | 熱可塑性エラストマー組成物 | |
| JP6207915B2 (ja) | 熱可塑性エラストマー組成物 | |
| JP2012503061A (ja) | ポリマーの相溶性を高める方法 | |
| JP2015021079A (ja) | 熱可塑性エラストマー組成物 | |
| CN1214073C (zh) | 聚丙烯树脂组合物、由其制备的模塑制品和薄膜 | |
| JP2022098911A (ja) | オレフィン系重合体組成物およびその用途 | |
| TW202003667A (zh) | 用於改良聚合物之抗環境應力龜裂性質的聚合性添加劑 | |
| BR112020024317A2 (pt) | método, composição de polímero, artigo fabricado, e, uso de uma composição de polímero reticulado reciclado. | |
| CN104945753B (zh) | 一种包装缠绕膜 | |
| JP3834582B2 (ja) | 目やにの発生が抑制された樹脂組成物及び目やに発生抑制方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13739108 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013554339 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14371546 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013739108 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013739108 Country of ref document: EP |