WO2013140705A1 - 基材担持触媒および基材担持触媒の製造方法 - Google Patents
基材担持触媒および基材担持触媒の製造方法 Download PDFInfo
- Publication number
- WO2013140705A1 WO2013140705A1 PCT/JP2013/000247 JP2013000247W WO2013140705A1 WO 2013140705 A1 WO2013140705 A1 WO 2013140705A1 JP 2013000247 W JP2013000247 W JP 2013000247W WO 2013140705 A1 WO2013140705 A1 WO 2013140705A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substrate
- supported catalyst
- thermosetting resin
- catalyst
- supported
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/06—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/40—Catalysts, in general, characterised by their form or physical properties characterised by dimensions, e.g. grain size
- B01J35/45—Nanoparticles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/54—Bars or plates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/58—Fabrics or filaments
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/58—Fabrics or filaments
- B01J35/59—Membranes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
- B01J37/0203—Impregnation the impregnation liquid containing organic compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
- B01J37/0207—Pretreatment of the support
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
- B01J37/0217—Pretreatment of the substrate before coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/40—Substitution reactions at carbon centres, e.g. C-C or C-X, i.e. carbon-hetero atom, cross-coupling, C-H activation or ring-opening reactions
- B01J2231/42—Catalytic cross-coupling, i.e. connection of previously not connected C-atoms or C- and X-atoms without rearrangement
- B01J2231/4205—C-C cross-coupling, e.g. metal catalyzed or Friedel-Crafts type
- B01J2231/4261—Heck-type, i.e. RY + C=C, in which R is aryl
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/06—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing polymers
- B01J31/063—Polymers comprising a characteristic microstructure
Definitions
- the present invention relates to a substrate-supported catalyst and a method for producing a substrate-supported catalyst.
- a catalyst supported on a carrier (hereinafter referred to as a supported catalyst) can be easily recovered and reused, and is an effective means particularly when an expensive noble metal is used as the catalyst (for example, Patent Documents 1 and 2). reference).
- the binding force between the catalyst and the carrier is not necessarily strong.
- the catalyst may be detached from the carrier during use, and the detached catalysts may aggregate with each other. In that case, the catalytic activity was gradually decreased by repeatedly using the supported catalyst.
- Patent Document 1 As a method for suppressing the deterioration of the activity of the supported catalyst due to repeated use, for example, there is a method described in Patent Document 1.
- a mixture of a thermosetting resin and a catalyst composed of a metal or a metal compound or a precursor thereof is heated to 500 ° C. or more in a non-oxidizing atmosphere to carbonize the thermosetting resin.
- a metal compound is supported on the carbide.
- the cured body of the thermosetting resin is carbonized, the cured body is carbonized while maintaining its shape although it shrinks.
- many catalysts made of metals or metal compounds are stable under the condition that the thermosetting resin is carbonized. For this reason, after mixing a catalyst etc. with a thermosetting resin, the thermosetting resin is hardened and carbonized, and the carbon material which fixed the catalyst substance firmly is obtained.
- Patent Document 2 is to support fine particles having catalytic activity on the surface of spherical resin particles and form a coating layer having a specific thickness made of a thermosetting resin so as to cover the spherical resin particles.
- a coating layer made of a thermosetting resin By providing a coating layer made of a thermosetting resin in this way, the detachment of fine particles having catalytic activity during use is suppressed, and separation and recovery from reaction products after use is facilitated. .
- the carriers may aggregate.
- the reaction product is less likely to come into contact with the catalyst, resulting in a reduction in reaction efficiency. That is, the activity per carrier is reduced.
- thermosetting resin When a thermosetting resin is carbonized as in Patent Document 1, a porous cured body is obtained.
- the catalyst When the catalyst is supported on the cured body, the catalyst may be taken into the pores of the carbide.
- the catalytic reaction proceeds on the surface of the support where the reactants can reach.
- the catalyst fine particles taken into the inside of the carbide, particularly where the reactant is difficult to reach, have a low contact efficiency with the reactant, and thus are difficult to use for the catalytic reaction.
- the present inventors have found that improving the reaction efficiency of the catalyst leads to an improvement in catalyst activity.
- the present invention has been made in view of the above circumstances, and provides a substrate-supported catalyst having excellent catalytic activity.
- the present inventors have formed an excellent catalyst by forming a cured body of a thermosetting resin having a phenolic hydroxyl group used as a catalyst carrier on the substrate surface. It has been found that a substrate-supported catalyst having activity can be provided.
- a substrate A cured body of a thermosetting resin formed on the surface of the substrate; Fine particles having catalytic activity carried on the surface of the cured body; Including A substrate-supported catalyst in which the thermosetting resin has a phenolic hydroxyl group is provided.
- catalysts have been provided that carry fine particles having catalytic activity using a cured body of a thermosetting resin as a catalyst carrier.
- the cured catalyst of a thermosetting resin having a phenolic hydroxyl group is used as a catalyst carrier, and the catalyst carrier can be dispersed on the substrate by forming the catalyst carrier on the substrate. it can.
- aggregation of catalyst fine particles can be prevented by supporting the catalyst fine particles on the cured body of the thermosetting resin as the catalyst carrier on the base material. By doing so, it is possible to prevent a decrease in the catalyst activity associated with the aggregation of the catalyst fine particles. That is, since the contact efficiency between the reactant and the catalyst can be improved, the reaction efficiency is improved, and a substrate-supported catalyst having excellent catalytic activity can be provided.
- the substrate-supported catalyst is preferably plate-shaped or sheet-shaped, and more preferably a porous body. By carrying out like this, it leads to increasing the reaction field in a base material carrying catalyst, and can improve reaction activity.
- a step of preparing a substrate Forming a cured body of a thermosetting resin on the surface of the substrate, and supporting fine particles having catalytic activity on the surface of the cured body; Including A method for producing a substrate-supported catalyst in which the thermosetting resin has a phenolic hydroxyl group is provided.
- a substrate-supported catalyst having excellent catalytic activity can be provided.
- the material which comprises the base material in this embodiment is not specifically limited, It is preferable that an interaction with a phenolic hydroxyl group is large. That is, it is preferable to use a material having good wettability with the resin. Examples thereof include cellulose, polyurethane, polyamide, and polyester. From the viewpoint of uniformly dispersing the catalyst fine particles, polyurethane is preferred. By using such a material as a substrate, it is possible to prevent the thermosetting resin covering the substrate surface from being peeled off from the substrate surface.
- the shape of the substrate is not particularly limited, but for example, a particle shape, a sheet shape or a plate shape is used. Among these, a sheet shape or a plate shape is preferable.
- the base material is preferably a porous body or a mesh structure, and more preferably a mesh structure.
- the base material is preferably a porous body or a mesh structure, and more preferably a mesh structure.
- the porous body may have a plurality of irregularities or may be provided with a plurality of holes communicating from the front surface to the back surface (hereinafter referred to as “communication holes”).
- the base material according to the present embodiment is more preferably a plate having a mesh structure. By doing so, it is possible to significantly improve the contact efficiency between the catalyst fine particles and the reactant. That is, the reaction activity can be improved, and a substrate-supported catalyst having a further excellent catalytic activity can be provided.
- the communication holes may form a honeycomb structure.
- the mechanical strength of the substrate itself can be improved. It is also possible to disperse the catalyst fine particles uniformly and at a high density.
- thermosetting resin (Hardened body of thermosetting resin)
- the cured body of the thermosetting resin is used as a catalyst carrier in the substrate-supported catalyst according to the present embodiment.
- the cured body of the thermosetting resin is formed on the surface of the substrate.
- the formation state of the cured body of the thermosetting resin is not particularly limited with respect to the base material, but may be formed in, for example, a particle shape, a film shape, or a uniform layer shape.
- the thermosetting resin before the curing treatment is not particularly limited as long as it is a thermosetting resin having a phenolic hydroxyl group, but it is particularly preferable to include a phenol resin or a derivative thereof.
- the catalyst is provided with excellent repetitive characteristics (lifetime) by forming a cured body of a thermosetting resin as a catalyst carrier so as to be dispersed on the surface of the substrate. can do.
- the reason is not necessarily clear, but is considered as follows.
- a cured body of a thermosetting resin having a phenolic hydroxyl group is formed so as to be exposed to the substrate surface. For this reason, a phenolic hydroxyl group is present at the site where the catalyst fine particles are supported, and the supported catalyst fine particles are stabilized by the phenolic hydroxyl group. Therefore, it is considered that the catalyst repetitive characteristics are improved by suppressing the detachment of the catalyst during use.
- the phenolic hydroxyl group equivalent of the thermosetting resin is 500 g / eq or less, preferably 400 g / eq or less, more preferably 350 g / eq or less. is there.
- the phenolic hydroxyl group equivalent of the thermosetting resin is within this range, it is possible to provide a substrate-supported catalyst having even more excellent catalytic activity.
- the phenolic hydroxyl group equivalent of a thermosetting resin is below the said upper limit, the phenolic hydroxyl group on the surface of a hardening body increases, and the retention of a catalyst can be maintained without weakening.
- the phenolic hydroxyl group equivalent can be quantified by a known method such as an acetylation method.
- the cured body of the thermosetting resin is formed so as to cover the surface of the substrate.
- the content of the cured body of the thermosetting resin is preferably 0.5% by weight or more based on the total amount of the base material and the cured body of the thermosetting resin. By being in this range, it is possible to provide a substrate-supported catalyst having even better catalytic activity.
- the phenol resin in this embodiment is obtained by reacting phenols and aldehydes in the presence of an alkaline or acidic catalyst, and has at least one phenolic hydroxyl group in the aromatic ring. ing.
- examples thereof include a phenol resin, a cresol resin, a resorcin resin, a xylenol resin, a naphthol resin, a bisphenol A resin, an aralkyl phenol resin, a biphenyl aralkyl phenol resin, and a modified phenol resin using cashew nut oil having a phenolic hydroxyl group.
- xylene-modified phenol resins containing substances having phenolic hydroxyl groups and various modified phenol resins such as phenol-modified rosin, oil-modified phenol resins modified with terpene oil, rubber-modified phenol resins modified with rubber, etc. can do.
- phenols used for obtaining the phenol resin those having a phenolic hydroxyl group in the molecule are preferable, and further, a substituent other than the phenolic hydroxyl group may be contained.
- cresols such as phenol, o-cresol, m-cresol, p-cresol, mixed cresol mixed with these, 2,3-xylenol, 2,4-xylenol, 2,5-xylenol, 2,6-xylenol, Xylenol such as 3,4-xylenol and 3,5-xylenol, ethylphenol such as o-ethylphenol, m-ethylphenol and p-ethylphenol, butylphenol such as isopropylphenol, butylphenol and p-tert-butylphenol, p- Halogenated phenols such as alkylphenols such as tert-amylphenol, p-oct
- aldehydes used for obtaining the phenol resin include formaldehyde, paraformaldehyde, trioxane, acetaldehyde, propionaldehyde, polyoxymethylene, chloral, hexamethylenetetramine, furfural, glyoxal, n-butyraldehyde, capro
- the method for reacting the phenols and aldehydes is not particularly limited, and a known method can be adopted.
- the catalyst for obtaining the phenol resin is not particularly limited, and examples thereof include an acid catalyst, a base catalyst, and a transition metal salt catalyst.
- the acid catalyst that can be used include inorganic acids such as hydrochloric acid, sulfuric acid, and phosphoric acids, and organic acids such as oxalic acid, p-toluenesulfonic acid, and organic phosphonic acid.
- the base catalyst include alkali metal hydroxides such as sodium hydroxide, potassium hydroxide and lithium hydroxide, alkaline earth metal hydroxides such as calcium hydroxide and barium hydroxide, ammonia, alkylamine and the like. These amines can be used.
- the transition metal salt catalyst include zinc oxalate and zinc acetate.
- the shape of the phenol resin in this embodiment is not particularly limited. Examples thereof include solid, powder, solution, and liquid, and any form can be used.
- thermosetting resin a method for curing the thermosetting resin. Although it does not specifically limit as a hardening processing method of the thermosetting resin in this embodiment, A well-known method is employable.
- thermosetting resin When a resol type phenol resin is used as the thermosetting resin, it can be cured by heating. Alternatively, a method of mixing acids such as p-toluenesulfonic acid and phenolsulfonic acid and curing at normal temperature or by heating can be used.
- a curing agent such as hexamethylenetetramine is mixed with an additive compound and cured by heating, a thermosetting resin such as an epoxy resin, a polyisocyanate, or a melamine resin. And a method of mixing with an additive compound and curing by heating.
- the curing temperature of the thermosetting resin in this embodiment is not particularly limited, it is preferably 250 ° C. or lower. When the curing temperature is not more than the above upper limit, an economical curing rate can be obtained, and decomposition of the main chain of the phenol resin can be suppressed.
- the catalyst fine particles supported on the surface of the cured body of the thermosetting resin in the present embodiment may be any metal, metal oxide, and metal compound as long as they have catalytic activity, and are particularly limited. It is not a thing.
- metals such as titanium, chromium, cobalt, nickel, copper, ruthenium, rhodium, palladium, rhenium, osmium, platinum, iron, zinc, manganese, magnesium, calcium, silver, vanadium, tin, oxides thereof, and other organic titanium And at least one selected from the group consisting of metal compounds and complexes.
- complex containing at least 2 or more types of these can also be used. Among these, palladium or platinum is particularly preferably used.
- the average particle diameter of the catalyst fine particles is preferably 1 ⁇ m or less. Further, nano-sized metal fine particles having an average particle diameter of 1 nm to 100 nm can also be used.
- the substrate-supported catalyst according to the present embodiment is not particularly limited, but is preferably a sheet shape or a plate shape. By carrying out like this, it is possible to deform
- the shape of the catalyst sheet can be changed into various shapes according to the shape of the reaction tube in the reaction apparatus, such as a folded state or a rolled state.
- the communication hole is provided in the sheet-like substrate-supported catalyst, it can be used as a catalyst filter.
- the method for forming the cured body of the thermosetting resin on the surface of the substrate can be appropriately selected depending on the shape of the substrate. For example, when the substrate is in a mesh form, a method of impregnating and curing a solid or powdered resin solution or liquid resin, a method of heating and melting a solid or powdered resin and impregnating and curing the substrate, etc. Used. On the other hand, when the substrate is particulate, a method of coating a thermosetting resin, or the like is used. By carrying out like this, it is possible to form the hardening body of a thermosetting resin uniformly with respect to a base material.
- the method for supporting the catalyst fine particles is not particularly limited.
- the catalyst fine particles are chemically deposited and supported in the liquid phase, the powder catalyst fine particles are electrostatically coated on the surface of the carrier, or the catalyst fine particles are supported by the carrier.
- the substrate-supported catalyst in this embodiment is a mode in which a nonpolar substrate is used as the substrate in the first embodiment.
- the material constituting the nonpolar substrate is formed from a compound or polymer that does not contain a polar functional group such as a carbonyl group, an imide group, an amino group, an amide group, and a hydroxy group in the chemical structure. Is preferred.
- the nonpolar substrate according to the present embodiment is not particularly limited, but preferably has a poor interaction (wetability) with the thermosetting resin.
- the nonpolar substrate include polyethylene, polypropylene, polymethylpentene, polybutene, polybutadiene, polystyrene, polyisobutylene, polytetrafluoroethylene, and other fluorine resins, natural rubber, styrene butadiene rubber, and butyl rubber.
- the reason for this is not necessarily clear, but because the wettability between the nonpolar substrate and the phenolic hydroxyl group of the thermosetting resin is poor, the phenolic hydroxyl group is other than the surface where the cured body of the thermosetting resin and the substrate contact. It is easy to orient at the location.
- catalyst fine particles having catalytic activity (hereinafter also referred to as “catalyst fine particles”) can be efficiently supported on the phenolic hydroxyl group. That is, it is considered that more catalyst fine particles can be supported on the catalyst carrier.
- polyolefin such as polyethylene and polypropylene from the viewpoint of uniformly dispersing the catalyst fine particles on the nonpolar substrate.
- polyolefin such as polyethylene and polypropylene from the viewpoint of uniformly dispersing the catalyst fine particles on the nonpolar substrate.
- the shape of the nonpolar base material is not particularly limited, but a particulate shape, a sheet shape, or a plate shape is used. Among these, a sheet shape or a plate shape is preferable.
- the nonpolar substrate is preferably made of a porous material or has a mesh structure, and more preferably has a mesh structure.
- the porous body may have a plurality of irregularities or may be provided with a plurality of holes communicating from the front surface to the back surface (hereinafter referred to as “communication holes”).
- the nonpolar base material according to the present embodiment is more preferably a plate having a mesh structure. By doing so, it is possible to significantly improve the contact efficiency between the catalyst fine particles and the reactant. That is, the reaction activity can be improved, and a substrate-supported catalyst having a further excellent catalytic activity can be provided.
- the communication holes may form a honeycomb structure.
- the mechanical strength of the substrate itself can be improved. It is also possible to disperse the catalyst fine particles uniformly and at a high density.
- the first embodiment is used as a method for forming a cured body of a thermosetting resin, catalyst fine particles, a cured body of a thermosetting resin on the surface of a substrate, and a method for supporting catalyst fine particles.
- the first embodiment is used as a method for forming a cured body of a thermosetting resin, catalyst fine particles, a cured body of a thermosetting resin on the surface of a substrate, and a method for supporting catalyst fine particles.
- the first embodiment is used as a method for forming a cured body of a thermosetting resin, catalyst fine particles, a cured body of a thermosetting resin on the surface of a substrate, and a method for supporting catalyst fine particles.
- the substrate-supported catalyst according to the present embodiment can be used for the same applications as those described in the first embodiment such as a catalyst filter.
- Example A1 (Production of cured phenol resin) Soak a polyurethane filter (Bridgestone, Everlite SF HR13) for 1 minute at room temperature in a phenolic resin solution in which liquid phenolic resin (Sumitomo Bakelite, Sumitrite Resin PR-50087) and methanol are mixed at a weight ratio of 1: 1.
- the film was naturally dried at room temperature for 30 minutes. After drying, it was heated at 90 ° C. for 30 minutes, and further heated at 150 ° C. for 30 minutes to obtain a porous sheet-like phenol resin carrier containing 30% by weight of phenol resin.
- Example A2 1 mg of palladium acetate (manufactured by Wako Pure Chemical Industries, Ltd.), 1 g of a phenol resin carrier prepared in the same manner as in Example A1, 0.3 mL of triethylamine (manufactured by Wako Pure Chemical Industries, Ltd.), and acetonitrile (manufactured by Wako Pure Chemical Industries, Ltd.) 10 mL was blended and heated in a sealed tube at 100 ° C. for 12 hours. Subsequently, the phenol resin carrier was taken out with tweezers, then washed and dried to obtain a phenol resin-supported catalyst on which palladium particles were supported. The supported amount of palladium catalyst in the phenol resin supported catalyst was 0.1% by weight.
- Comparative Example A1 In Comparative Example A1, a commercially available palladium-activated carbon (made by Wako Pure Chemical Industries, Ltd., palladium-activated carbon) was used as a catalyst carrier. The amount of palladium catalyst supported on this palladium-activated carbon was 5% by weight. The following evaluation was performed using the catalysts of Examples A1 and A2 and Comparative Example A1.
- the catalytic activity of the supported catalyst was evaluated by the reaction yield of trans-methyl cinnamate obtained by Heck reaction between iodobenzene and methyl acrylate.
- 230 ⁇ L (2.0 mmol) of iodobenzene, 230 ⁇ L (2.5 mmol) of methyl acrylate, and 350 ⁇ L (2.5 mmol) of triethylamine were dissolved in acetonitrile (20 mL).
- 500 mg of the supported catalyst was added to the obtained solution.
- the mixture was heated using an oil bath and stirred at 120 ° C. for 12 hours. After completion of the reaction, the supported catalyst was removed by tweezers or collected by filtration.
- the reaction yield was calculated from the area ratio of the chromatogram before and after the reaction.
- reaction yield is defined by (C 0 -C f ) / C 0 ⁇ 100 (%), where C 0 is the number of moles of iodobenzene before the reaction, and C f is the number of moles of iodobenzene after the reaction. It is.
- the resin-supported catalyst of the comparative example is a conventionally used resin-supported catalyst.
- the phenol resin-supported catalyst of Example A1-A2 had a higher reaction yield than Comparative Example A1.
- This result shows that, when the substrate-supported catalyst described in the examples is used, a catalytic activity superior to that of the resin-supported catalyst of the comparative example can be realized.
- the substrate-supported catalysts of the examples retain a higher activity for a long time than the catalysts of the comparative examples. That is, if the substrate-supported catalyst described in the examples is used, a catalytic activity superior to that of the resin-supported catalyst of the comparative example can be maintained for a long time without exchanging the catalyst.
- Example B1 (Production of cured phenol resin)
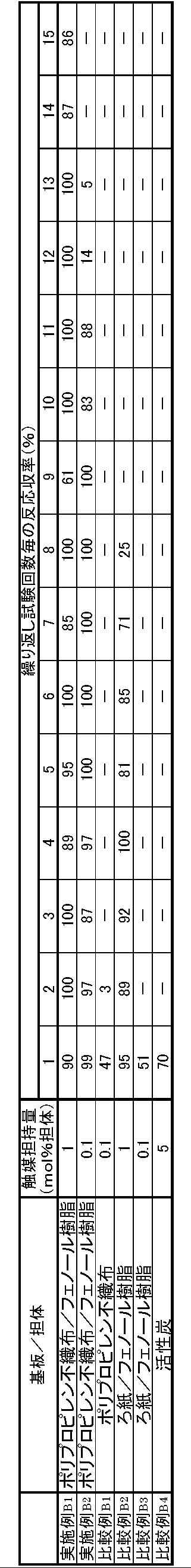
- a sheet-like polypropylene nonwoven fabric (manufactured by Japan Vilene Co., Ltd.) is immersed for 1 minute at room temperature in a phenol resin solution in which liquid phenolic resin (Sumitomo Bakelite Co., Ltd., Sumilite Resin PR-50087) and methanol are mixed at a weight ratio of 1: 1. Air-dried at room temperature for 30 minutes. After drying, it was heated at 90 ° C. for 30 minutes, and further heated at 150 ° C. for 30 minutes to obtain a plate-like phenol resin carrier having a mesh structure containing 30% by weight of phenol resin.
- liquid phenolic resin Suditomo Bakelite Co., Ltd., Sumilite Resin PR-50087
- Example B2 1 mg of palladium acetate (manufactured by Wako Pure Chemical Industries), 1 g of a phenol resin carrier prepared in the same manner as in Example 1, 0.3 mL of triethylamine (manufactured by Wako Pure Chemical Industries), and acetonitrile (manufactured by Wako Pure Chemical Industries) 10 mL was blended and heated in a sealed tube at 100 ° C. for 12 hours. Subsequently, the phenol resin carrier was taken out with tweezers, then washed and dried to obtain a phenol resin-supported catalyst on which palladium particles were supported. The amount of palladium catalyst supported in the phenol resin supported catalyst was 0.1 mmol%.
- the polypropylene non-woven fabric was taken out with tweezers, washed and dried to obtain a polypropylene-supported catalyst on which palladium particles were supported.
- the amount of palladium catalyst supported in the polypropylene supported catalyst was 0.1 mmol%.
- Comparative Example B4 A commercially available palladium-activated carbon (produced by Wako Pure Chemical Industries, Ltd., palladium-activated carbon) was used as Comparative Example 4. The amount of palladium catalyst supported on this palladium-activated carbon was 5% by weight.
- the catalytic activity of the supported catalyst was evaluated by the reaction yield of trans-methyl cinnamate obtained by Heck reaction between iodobenzene and methyl acrylate.
- 230 ⁇ L (2.0 mmol) of iodobenzene, 230 ⁇ L (2.5 mmol) of methyl acrylate, and 350 ⁇ L (2.5 mmol) of triethylamine were dissolved in acetonitrile (20 mL).
- 500 mg of the supported catalyst was added to the obtained solution.
- the mixture was heated using an oil bath and stirred at 120 ° C. for 12 hours. After completion of the reaction, the supported catalyst was removed by tweezers or collected by filtration.
- the reaction yield was calculated from the area ratio of the chromatogram before and after the reaction.
- reaction yield is defined by (C 0 -C f ) / C 0 ⁇ 100 (%), where C 0 is the number of moles of iodobenzene before the reaction, and C f is the number of moles of iodobenzene after the reaction. It is.
- the phenol resin-supported catalyst of the example had a higher reaction yield than the comparative example.
- This result shows that, when the substrate-supported catalyst described in the examples is used, a catalytic activity superior to that of the conventional supported catalyst or the substrate-supported catalyst using a substrate having a polar group can be realized.
- the substrate-supported catalyst of the examples retains a higher activity for a long period of time than a conventional catalyst or a substrate-supported catalyst using a substrate having a polar group. That is, when the substrate-supported catalyst described in the examples is used, excellent catalytic activity can be maintained for a long time without exchanging the catalyst.
- this invention includes the following aspects.
- a substrate A cured body of a thermosetting resin formed on the surface of the substrate; Fine particles having catalytic activity carried on the surface of the thermosetting resin; Including A substrate-supported catalyst in which the cured product of a thermosetting resin has a phenolic hydroxyl group.
- thermosetting resin The substrate-supported catalyst according to [1-4], wherein the substrate has a mesh shape.
- the content of the cured body of the thermosetting resin is 0.5% by weight or more based on the total amount of the cured body of the base material and the thermosetting resin [1-1] to [1-5] The substrate-supported catalyst according to any one of [1-5].
- thermosetting resin formed on the surface of the nonpolar substrate; Fine particles having catalytic activity carried on the surface of the cured body; Including A substrate-supported catalyst in which the thermosetting resin has a phenolic hydroxyl group.
- the nonpolar base material is selected from the group consisting of polyethylene, polypropylene, polymethylpentene, polybutene, polybutadiene, polystyrene, polyisobutylene, polytetrafluoroethylene and other fluororesins, natural rubber, styrene butadiene rubber and butyl rubber.
- the substrate-supported catalyst according to [2-1] comprising one or more selected.
- thermosetting resin is a phenol resin.
- thermosetting resin is a phenol resin.
- nonpolar substrate is a plate or a sheet.
- nonpolar substrate is made of a porous material.
- nonpolar substrate is made of a porous material.
- nonpolar substrate is a mesh.
- the content of the cured body of the thermosetting resin is 0.5% by weight or more based on the total amount of the cured body of the nonpolar base material and the thermosetting resin [2-1. ] To [2-6].
- thermosetting resin is cured on the surface of the nonpolar substrate by immersing the nonpolar substrate in the thermosetting resin in a solution and drying it, and then curing the thermosetting resin.
- [2-14] In the step of supporting the fine particles, The substrate support according to any one of [2-11] to [2-13], wherein the fine particles are supported by electrostatic coating or dipping on the surface of the cured body of the thermosetting resin.
- a method for producing a catalyst In the step of supporting the fine particles, The substrate support according to any one of [2-11] to [2-13], wherein the fine particles are supported by electrostatic coating or dipping on the surface of the cured body of the thermosetting resin.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Crystallography & Structural Chemistry (AREA)
- Catalysts (AREA)
- Application Of Or Painting With Fluid Materials (AREA)
- Laminated Bodies (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
前記基材の表面に形成された熱硬化性樹脂の硬化体と、
前記硬化体の表面に担持された触媒活性を有する微粒子と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する基材担持触媒が提供される。
前記基材の表面に熱硬化性樹脂の硬化体を形成するとともに、前記硬化体の表面に触媒活性を有する微粒子を担持する工程と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する基材担持触媒の製造方法が提供される。
(基材)
本実施形態における基材を構成する材料は、特に限定されないが、フェノール性水酸基との相互作用が大きいものであることが好ましい。すなわち、樹脂との濡れ性が良好である材料を用いることが好ましい。例えば、セルロース、ポリウレタン、ポリアミド、およびポリエステルなどが挙げられる。触媒微粒子を均一分散させるという観点では、ポリウレタンが好ましい。このような材料を基材として用いることによって、基材表面を覆っている熱硬化性樹脂が基材表面から剥がれることを防ぐことができる。
熱硬化性樹脂の硬化体は、本実施形態に係る基材担持触媒において触媒担体として用いられる。この熱硬化性樹脂の硬化体は、基材の表面上に形成される。熱硬化性樹脂の硬化体の形成状態は、基材に対して特に限定されるものではないが、例えば、粒子状、膜状、均一な層状に形成していても良い。硬化処理前の熱硬化性樹脂としては、フェノール性水酸基を有する熱硬化性樹脂であればとくに限定されないが、とくにフェノール樹脂またはその誘導体を含むことが好ましい。
例えば、フェノール樹脂、クレゾール樹脂、レゾルシン樹脂、キシレノール樹脂、ナフトール樹脂、ビスフェノールA樹脂、アラルキルフェノール樹脂、ビフェニルアラルキルフェノール樹脂、およびフェノール性水酸基を有するカシューナッツ油などによる変性フェノール樹脂などが挙げられる。また、フェノール性水酸基を有する物質を含む、キシレン変性フェノール樹脂、およびフェノール類とロジン、テルペン油などで変性した油変性フェノール樹脂、ゴムで変性したゴム変性フェノール樹脂などの各種変性フェノール樹脂なども使用することができる。
本実施形態における熱硬化性樹脂の硬化処理方法としてはとくに限定されないが、公知の方法を採用することができる。
本実施形態における熱硬化性樹脂の硬化体の表面に担持される触媒微粒子としては、触媒活性を有するものであれば金属、金属酸化物および金属化合物のいずれであってもよく、とくに限定されるものではない。例えばチタン、クロム、コバルト、ニッケル、銅、ルテニウム、ロジウム、パラジウム、レニウム、オスミニウム、白金、鉄、亜鉛、マンガン、マグネシウム、カルシウム、銀、バナジウム、スズなどの金属ならびにその酸化物、その他の有機チタンなどの金属化合物および錯体などの中から選ばれる少なくとも1種からなるものが挙げられる。また、これらのうちの少なくとも二種類以上を含む複合体も使用することもできる。これらの中でも、とくにパラジウムまたは白金が好適に用いられる。
本実施形態に係る基材担持触媒は、特に限定されないが、シート状あるいは板状であることが好ましい。こうすることで、反応装置の形状に合わせて、種々の形状に変形させることが可能である。触媒シートの形状は、例えば、折りたたまれた状態、丸めた状態など反応装置における反応管の形状に合わせて種々の形状に変形させることができる。シート状の基材担持触媒において連通孔が設けられている場合、触媒フィルターとして用いることも可能である。
本実施形態において熱硬化性樹脂の硬化体を基材の表面に形成する方法は、基材の形状によって、適宜方法を選択することができる。例えば、基材がメッシュ状である場合、固形や粉末の樹脂の溶液や液状樹脂を基材に含浸硬化する方法、固形や粉末の樹脂を加熱溶融して基材に含浸硬化する方法、等が用いられる。これに対し、基材が粒子状である場合、熱硬化性樹脂をコーティングする方法、等が用いられる。こうすることで、基材に対して均一に熱硬化性樹脂の硬化体を形成することが可能である。
次に、本実施形態における触媒担体である熱硬化性樹脂の硬化体を表面に分散形成させた基材への触媒微粒子の担持方法について詳細に説明する。本実施形態においては、熱硬化性樹脂の硬化体を基材の表面に形成した後、担体である熱硬化性樹脂の硬化体の表面に触媒微粒子を担持させることが好ましい。こうすることで、触媒微粒子が、触媒担体である熱硬化性樹脂の硬化体の内部に埋め込まれることを防ぐことができる。
本実施形態における基材担持触媒は、第1の実施形態において、基材として非極性基材を用いる態様となっています。ここで、非極性基材を構成する材料は、化学構造中に、カルボニル基、イミド基、アミノ基、アミド基およびヒドロキシ基等の極性官能基を含まない化合物や高分子から形成されていることが好ましい。
(実施例A1)
(フェノール樹脂の硬化体の作製)
液状フェノール樹脂(住友ベークライト社製、スミライトレジンPR-50087)とメタノールを重量比1対1で混合したフェノール樹脂溶液に、ポリウレタンフィルター(ブリヂストン社製、エバーライトSF HR13)を室温で1分間浸し、常温で30分間自然乾燥した。乾燥後、90℃で30分間加熱し、更に150℃で30分間加熱してフェノール樹脂を30重量%含む多孔質シート状のフェノール樹脂担体を得た。
酢酸パラジウム(和光純薬工業社製)10mgと、担体である上記フェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1重量%であった。パラジウム触媒の担持量は、市販の原子吸光分光光度計を用いて測定した。
酢酸パラジウム(和光純薬工業社製)1mgと、実施例A1と同様に作製したフェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は0.1重量%であった。
比較例A1では、市販のパラジウム-活性炭(和光純薬工業社製、パラジウム-活性炭素)を触媒担体として用いた。このパラジウム-活性炭のパラジウム触媒の担持量は5重量%であった。
上記実施例A1およびA2、比較例A1の触媒をそれぞれ用い、以下の評価を行った。
担持触媒の触媒活性は、ヨードベンゼンと、アクリル酸メチルとのヘック反応で得られるトランス-桂皮酸メチルの反応収率により評価した。
ヨードベンゼン230μL(2.0mmol)、アクリル酸メチル230μL(2.5mmol)、トリエチルアミン350μL(2.5mmol)をアセトニトリル(20mL)に溶解した。得られた溶液に担持触媒を500mg加えた。混合物をオイルバスを用いて加熱し、120℃で12時間攪拌した。反応終了後、担持触媒をピンセットによって取り出すか、あるいは濾別することによって回収した。
反応終了後、反応液から担持触媒を回収し、洗浄した。その後、回収した担持触媒を用いて上記のヘック反応をおこなった。これらのヘック反応の一連の操作を、トランス-桂皮酸メチルの反応収率が80%を下回るまで繰り返した。その結果を下記表1に示す。なお、比較例の樹脂担持触媒は、従来使用されている樹脂担持触媒である。

(実施例B1)
(フェノール樹脂の硬化体の作製)
液状フェノール樹脂(住友ベークライト社製、スミライトレジンPR-50087)とメタノールを重量比1対1で混合したフェノール樹脂溶液に、シート状のポリプロピレン不織布(日本バイリーン社製)を室温で1分間浸し、常温で30分間自然乾燥した。乾燥後、90℃で30分間加熱し、更に150℃で30分間加熱してフェノール樹脂を30重量%含むメッシュ構造を有した板状のフェノール樹脂担体を得た。
酢酸パラジウム(和光純薬工業社製)10mgと、担体である上記フェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1mmol%であった。パラジウム触媒の担持量は、市販の原子吸光分光光度計を用いて測定した。
酢酸パラジウム(和光純薬工業社製)1mgと、実施例1と同様に作製したフェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は0.1mmol%であった。
酢酸パラジウム(和光純薬工業社製)1mgと、シート状のポリプロピレン不織布(日本バイリーン社製、)1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、ポリプロピレン不織布をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたポリプロピレン担持触媒を得た。ポリプロピレン担持触媒中におけるパラジウム触媒の担持量は0.1mmol%であった。
(フェノール樹脂の硬化体の作製)
液状フェノール樹脂(住友ベークライト社製、スミライトレジンPR-50087)とメタノールを重量比1対1で混合したフェノール樹脂溶液に、ろ紙(アドバンテック東洋社製、No.424)を室温で1分間浸し、常温で30分間自然乾燥した。乾燥後、90℃で30分間加熱し、更に150℃で30分間加熱してフェノール樹脂を30重量%含む多孔質シート状のフェノール樹脂担体を得た。
酢酸パラジウム(和光純薬工業社製)10mgと、担体である上記フェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1mmol%であった。パラジウム触媒の担持量は、市販の原子吸光分光光度計を用いて測定した。
酢酸パラジウム(和光純薬工業社製)1mgと、比較例2と同様に作製したフェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は0.1mmol%であった。
市販のパラジウム‐活性炭(和光純薬工業社製、パラジウム‐活性炭素)を比較例4とした。このパラジウム‐活性炭のパラジウム触媒の担持量は5重量%であった。
担持触媒の触媒活性は、ヨードベンゼンと、アクリル酸メチルとのヘック反応で得られるトランス-桂皮酸メチルの反応収率により評価した。
ヨードベンゼン230μL(2.0mmol)、アクリル酸メチル230μL(2.5mmol)、トリエチルアミン350μL(2.5mmol)をアセトニトリル(20mL)に溶解した。得られた溶液に担持触媒を500mg加えた。混合物をオイルバスを用いて加熱し、120℃で12時間攪拌した。反応終了後、担持触媒をピンセットによって取り出すか、あるいは濾別することによって回収した。
反応終了後、反応液から担持触媒を回収し、洗浄した。その後、回収した担持触媒を用いて上記のヘック反応をおこなった。これらのヘック反応の一連の操作を、トランス-桂皮酸メチルの反応収率が80%を下回るまで繰り返した。その結果を下記表2に示す。なお、比較例B4の担持触媒は、従来使用されている担持触媒を用いている。
[1-1]基材と、
前記基材の表面に形成された熱硬化性樹脂の硬化体と、
前記熱硬化性樹脂の表面に担持された触媒活性を有する微粒子と、
を含み、
熱硬化性樹脂の前記硬化体がフェノール性水酸基を有する基材担持触媒。
[1-2]前記熱硬化性樹脂がフェノール樹脂である[1-1]に記載の基材担持触媒。
[1-3]前記基材が、板状あるいはシート状である[1-1]または[1-2]に記載の基材担持触媒。
[1-4]前記基材が多孔質材料である[1-1]乃至[1-3]のいずれか一つに記載の基材担持触媒。
[1-5]前記基材が、メッシュ状である[1-4]に記載の基材担持触媒。
[1-6]前記熱硬化性樹脂の硬化体の含有量が、前記基材と前記熱硬化性樹脂の硬化体の総量に対して、0.5重量%以上である[1-1]乃至[1-5]のいずれか一つに記載の基材担持触媒。
[1-7]前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である[1-1]乃至[1-6]のいずれか一つに記載の基材担持触媒。
[1-8]前記微粒子は、前記熱硬化性樹脂を硬化させた後に、前記硬化体の表面に担持したものである[1-1]乃至[1-7]のいずれか一つに記載の基材担持触媒。
[1-9]前記基材が、セルロース、ポリウレタン、ポリアミド、ポリエステルから成る群から選択される1種である[1-1]乃至[1-8]のいずれか一つに記載の基材担持触媒。
[1-10]前記微粒子が、金属、金属酸化物、および金属化合物のいずれか1種以上を含む[1-1]乃至[1-9]のいずれか一項に記載の基材担持触媒。
[1-11]基材を準備する工程と、
前記基材の表面に熱硬化性樹脂の硬化体を形成するとともに、前記熱硬化性樹脂の表面に触媒活性を有する微粒子を担持する工程と、
を含み、
前記熱硬化性樹脂の硬化体がフェノール性水酸基を有する硬化体である基材担持触媒の製造方法。
[1-12]前記基材の表面に前記熱硬化性樹脂の硬化体を形成した後に、前記微粒子を担持する[1-11]に記載の基材担持触媒の製造方法。
[1-13]前記基材を溶液状の前記熱硬化性樹脂に浸漬して乾燥させた後、前記熱硬化性樹脂を硬化させることによって、前記基材の表面に前記熱硬化性樹脂の硬化体を形成する[1-11]または[1-12]に記載の基材担持触媒の製造方法。
[1-14]前記微粒子を担持する工程において、
前記熱硬化性樹脂の硬化物の表面に対して、前記微粒子を静電塗装または浸漬して担持させている[1-11]乃至[1-13]のいずれか一つに記載の基材担持触媒の製造方法。
前記非極性基材の表面に形成された熱硬化性樹脂の硬化体と、
前記硬化体の表面に担持された触媒活性を有する微粒子と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する基材担持触媒。
[2-2]前記非極性基材が、ポリエチレン、ポリプロピレン、ポリメチルペンテン、ポリブテン、ポリブタジエン、ポリスチレン、ポリイソブチレン、ポリテトラフルオロエチレンなどのフッ素樹脂、天然ゴム、スチレンブタジエンゴムおよびブチルゴムから成る群から選択される1種以上含む[2-1]に記載の基材担持触媒。
[2-3]前記熱硬化性樹脂がフェノール樹脂である[2-1]または[2-2]に記載の基材担持触媒。
[2-4]前記非極性基材が、板状あるいはシート状である[2-1]乃至[2-3]のいずれか一つに記載の基材担持触媒。
[2-5]前記非極性基材が多孔質体からなる[2-1]乃至[2-4]のいずれか一つに記載の基材担持触媒。
[2-6]前記非極性基材が、メッシュ状である[2-1]乃至[2-4]のいずれか一つに記載の基材担持触媒。
[2-7]熱硬化性樹脂の前記硬化体の含有量が、前記非極性基材と熱硬化性樹脂の前記硬化体の総量に対して、0.5重量%以上である[2-1]乃至[2-6]のいずれか一つに記載の基材担持触媒。
[2-8]前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である[2-1]乃至[2-7]のいずれか一つに記載の基材担持触媒。
[2-9]前記微粒子は、前記熱硬化性樹脂を硬化させた後に、前記硬化体の表面に担持したものである[2-1]乃至[2-8]のいずれか一つに記載の基材担持触媒。
[2-10]前記微粒子が、金属、金属酸化物、および金属化合物のいずれか1種以上を含む材料から成る[2-1]乃至[2-9]のいずれか一つに記載の基材担持触媒。
[2-11]非極性基材を準備する工程と、
前記非極性基材の表面に熱硬化性樹脂の硬化体を形成するとともに、前記硬化体の表面に触媒活性を有する微粒子を担持する工程と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する硬化体である基材担持触媒の製造方法。
[2-12]前記非極性基材の表面に前記熱硬化性樹脂の硬化体を形成した後に、前記微粒子を担持する[2-11]に記載の基材担持触媒の製造方法。
[2-13]前記非極性基材を溶液状の前記熱硬化性樹脂に浸漬して乾燥させた後、前記熱硬化性樹脂を硬化させることによって、前記非極性基材の表面に前記熱硬化性樹脂の硬化体を形成する[2-11]または[2-12]に記載の基材担持触媒の製造方法。
[2-14]前記微粒子を担持する工程において、
前記熱硬化性樹脂の硬化体の表面に対して、前記微粒子を静電塗装または浸漬して担持させている[2-11]乃至[2-13]のいずれか一つに記載の基材担持触媒の製造方法。
Claims (17)
- 基材と、
前記基材の表面に形成された熱硬化性樹脂の硬化体と、
前記硬化体の表面に担持された触媒活性を有する微粒子と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する基材担持触媒。 - 前記基材が、セルロース、ポリウレタン、ポリアミド、ポリエステルから成る群から選択される1種以上を含む請求項1に記載の基材担持触媒。
- 前記基材が、非極性基材である請求項1に記載の基材担持触媒。
- 前記基材が、ポリエチレン、ポリプロピレン、ポリメチルペンテン、ポリブテン、ポリブタジエン、ポリスチレン、ポリイソブチレン、ポリテトラフルオロエチレンなどのフッ素樹脂、天然ゴム、スチレンブタジエンゴムおよびブチルゴムから成る群から選択される1種以上を含む請求項3に記載の基材担持触媒。
- 前記熱硬化性樹脂がフェノール樹脂である請求項1乃至4のいずれか一項に記載の基材担持触媒。
- 前記基材が、板状あるいはシート状である請求項1乃至5のいずれか一項に記載の基材担持触媒。
- 前記基材が多孔質体からなる請求項1乃至6のいずれか一項に記載の基材担持触媒。
- 前記基材が、メッシュ状である請求項1乃至6のいずれか一項に記載の基材担持触媒。
- 熱硬化性樹脂の前記硬化体の含有量が、前記基材と熱硬化性樹脂の前記硬化体の総量に対して、0.5重量%以上である請求項1乃至8のいずれか一項に記載の基材担持触媒。
- 前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である請求項1乃至9のいずれか一項に記載の基材担持触媒。
- 前記微粒子は、前記熱硬化性樹脂を硬化させた後に、前記硬化体の表面に担持されたものである請求項1乃至10のいずれか一項に記載の基材担持触媒。
- 前記微粒子が、金属、金属酸化物、および金属化合物のいずれか1種以上を含む材料からなる請求項1乃至11のいずれか一項に記載の基材担持触媒。
- 基材を準備する工程と、
前記基材の表面に熱硬化性樹脂の硬化体を形成するとともに、前記硬化体の表面に触媒活性を有する微粒子を担持する工程と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する基材担持触媒の製造方法。 - 前記基材が、非極性基材である請求項13に記載の基材担持触媒の製造方法。
- 前記基材の表面に熱硬化性樹脂の前記硬化体を形成した後に、前記微粒子を担持する請求項13または14に記載の基材担持触媒の製造方法。
- 前記基材を溶液状の前記熱硬化性樹脂に浸漬して乾燥させた後、前記熱硬化性樹脂を硬化させることによって、前記基材の表面に熱硬化性樹脂の前記硬化体を形成する請求項13乃至15のいずれか一項に記載の基材担持触媒の製造方法。
- 前記微粒子を担持する工程において、
熱硬化性樹脂の前記硬化物の表面に対して、前記微粒子を静電塗装または浸漬して担持させている請求項13乃至16のいずれか一項に記載の基材担持触媒の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2014142277A RU2014142277A (ru) | 2012-03-21 | 2013-01-21 | Катализатор, нанесенный на материал основы, и способ изготовления катализатора, нанесенного на материал основы |
| CA2866789A CA2866789A1 (en) | 2012-03-21 | 2013-01-21 | Base material-carried catalyst and method of manufacturing base material-carried catalyst |
| US14/381,812 US20150087501A1 (en) | 2012-03-21 | 2013-01-21 | Base material-carried catalyst and method of manufacturing base material-carried catalyst |
| KR1020147028875A KR20140139561A (ko) | 2012-03-21 | 2013-01-21 | 기재 담지 촉매 및 기재 담지 촉매의 제조 방법 |
| CN201380015088.8A CN104203405A (zh) | 2012-03-21 | 2013-01-21 | 基材担载催化剂以及基材担载催化剂的制造方法 |
| IN7208DEN2014 IN2014DN07208A (ja) | 2012-03-21 | 2013-01-21 | |
| EP13763933.2A EP2829323A4 (en) | 2012-03-21 | 2013-01-21 | SUBSTRATE SUPPORTED CATALYST AND METHOD FOR PRODUCING A SUBSTRATE CARRIERED CATALYST |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012063156A JP5905753B2 (ja) | 2012-03-21 | 2012-03-21 | 基材担持触媒および基材担持触媒の製造方法 |
| JP2012-063156 | 2012-03-21 | ||
| JP2012135381A JP5886145B2 (ja) | 2012-06-15 | 2012-06-15 | 基材担持触媒および基材担持触媒の製造方法 |
| JP2012-135381 | 2012-06-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013140705A1 true WO2013140705A1 (ja) | 2013-09-26 |
Family
ID=49222191
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/000247 Ceased WO2013140705A1 (ja) | 2012-03-21 | 2013-01-21 | 基材担持触媒および基材担持触媒の製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP2829323A4 (ja) |
| KR (1) | KR20140139561A (ja) |
| CN (1) | CN104203405A (ja) |
| CA (1) | CA2866789A1 (ja) |
| IN (1) | IN2014DN07208A (ja) |
| RU (1) | RU2014142277A (ja) |
| TW (1) | TW201347843A (ja) |
| WO (1) | WO2013140705A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103764283A (zh) * | 2011-09-06 | 2014-04-30 | 住友电木株式会社 | 树脂担载催化剂和树脂担载催化剂的制造方法 |
| CN110614124B (zh) * | 2018-12-20 | 2022-09-30 | 南京工程学院 | 一种具有多级结构的纳米金催化剂及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000140643A (ja) | 1998-11-13 | 2000-05-23 | Dainippon Ink & Chem Inc | 金属系触媒の固定方法 |
| JP2008110340A (ja) * | 2006-10-06 | 2008-05-15 | Kao Corp | フィルム状触媒 |
| JP2010022980A (ja) | 2008-07-23 | 2010-02-04 | Kyocera Chemical Corp | 球状触媒粒子とその製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100586763B1 (ko) * | 1998-07-02 | 2006-06-08 | 신에쓰 가가꾸 고교 가부시끼가이샤 | 포스포늄보레이트 화합물, 그의 제조방법, 에폭시 수지 조성물용경화 촉매 및 에폭시 수지 조성물 |
| JP4509286B2 (ja) * | 2000-03-17 | 2010-07-21 | 日本曹達株式会社 | 光触媒担持構造体 |
| DE10322182A1 (de) * | 2003-05-16 | 2004-12-02 | Blue Membranes Gmbh | Verfahren zur Herstellung von porösem, kohlenstoffbasiertem Material |
-
2013
- 2013-01-21 IN IN7208DEN2014 patent/IN2014DN07208A/en unknown
- 2013-01-21 WO PCT/JP2013/000247 patent/WO2013140705A1/ja not_active Ceased
- 2013-01-21 EP EP13763933.2A patent/EP2829323A4/en not_active Withdrawn
- 2013-01-21 CN CN201380015088.8A patent/CN104203405A/zh active Pending
- 2013-01-21 RU RU2014142277A patent/RU2014142277A/ru unknown
- 2013-01-21 CA CA2866789A patent/CA2866789A1/en not_active Abandoned
- 2013-01-21 KR KR1020147028875A patent/KR20140139561A/ko not_active Withdrawn
- 2013-01-28 TW TW102103079A patent/TW201347843A/zh unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000140643A (ja) | 1998-11-13 | 2000-05-23 | Dainippon Ink & Chem Inc | 金属系触媒の固定方法 |
| JP2008110340A (ja) * | 2006-10-06 | 2008-05-15 | Kao Corp | フィルム状触媒 |
| JP2010022980A (ja) | 2008-07-23 | 2010-02-04 | Kyocera Chemical Corp | 球状触媒粒子とその製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| Q.-L. YE ET AL.: "Studies on a Phenolic Resin as a Carrier for the Immobilization of Penicillin Acylase", POLYMERS FOR ADVANCED TECHNOLOGIES, vol. 8, 1997, pages 727 - 730, XP055167503 * |
| See also references of EP2829323A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104203405A (zh) | 2014-12-10 |
| KR20140139561A (ko) | 2014-12-05 |
| CA2866789A1 (en) | 2013-09-26 |
| EP2829323A4 (en) | 2015-11-18 |
| TW201347843A (zh) | 2013-12-01 |
| RU2014142277A (ru) | 2016-05-10 |
| IN2014DN07208A (ja) | 2015-04-24 |
| EP2829323A1 (en) | 2015-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9314783B2 (en) | Resin-supported catalyst and method for preparing resin-supported catalyst | |
| Lu et al. | Decoration of Pd nanoparticles with N and S doped carbon quantum dots as a robust catalyst for the chemoselective hydrogenation reaction | |
| RU2652674C2 (ru) | Тело из металлической пены с контролируемой зернистостью поверхности, способ его производства и его применение | |
| Yamazoe et al. | Controlled synthesis of carbon‐supported gold clusters for rational catalyst design | |
| JP2016032802A (ja) | 炭素触媒及びその製造方法 | |
| Cheong et al. | Au-Pd core-shell nanoparticles as alcohol oxidation catalysts: Effect of shape and composition | |
| CN107876047B (zh) | 一种α,β-不饱和醛/酮加氢用Pd/C催化剂的制备方法 | |
| WO2013140705A1 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| Liu et al. | Pd Clusters on Schiff Base–Imidazole-Functionalized MOFs for Highly Efficient Catalytic Suzuki Coupling Reactions | |
| CN108380182A (zh) | 酚醛树脂基活性炭及其制备方法和应用 | |
| JP5886145B2 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| Villa et al. | Material science for the support design: a powerful challenge for catalysis | |
| CN110152654A (zh) | 有序介孔碳-TiO2复合材料负载钯催化剂及其制备方法、应用 | |
| EP2952252B1 (en) | Selective hydrogenation catalyst, production method for selective hydrogenation catalyst, and selective hydrogenation method | |
| JP5905753B2 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| JP5931641B2 (ja) | 樹脂担持触媒の製造方法 | |
| CN110327975B (zh) | 氢甲酰化催化剂及其制备方法和应用 | |
| JP5885969B2 (ja) | 樹脂担持触媒および樹脂担持触媒の製造方法 | |
| JP2014169236A (ja) | 飽和炭化水素又は不飽和炭化水素の合成方法、合成物及び触媒複合体 | |
| JP2011177614A (ja) | 触媒担持体とその製造方法 | |
| CN110128610A (zh) | 一种壳核结构酚醛树脂微球及其制备方法 | |
| WO2023100626A1 (ja) | 水素製造用触媒及び水素製造方法 | |
| JP2014148485A (ja) | 求電子部位を有する化合物または不飽和炭化水素の水素化方法、水素化物および触媒複合体 | |
| JPH0147433B2 (ja) | ||
| JP2000140643A (ja) | 金属系触媒の固定方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13763933 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14381812 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2866789 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013763933 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147028875 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014142277 Country of ref document: RU Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |