WO2013156936A1 - Process for the preparation of rivaroxaban and intermediates thereof - Google Patents
Process for the preparation of rivaroxaban and intermediates thereof Download PDFInfo
- Publication number
- WO2013156936A1 WO2013156936A1 PCT/IB2013/053025 IB2013053025W WO2013156936A1 WO 2013156936 A1 WO2013156936 A1 WO 2013156936A1 IB 2013053025 W IB2013053025 W IB 2013053025W WO 2013156936 A1 WO2013156936 A1 WO 2013156936A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- rivaroxaban
- preparation
- iii
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
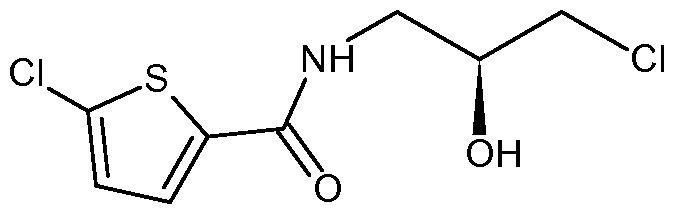
- XSGMBZPBLJKZLG-RXMQYKEDSA-N O[C@@H](CNC(c([s]1)ccc1Cl)=O)CCl Chemical compound O[C@@H](CNC(c([s]1)ccc1Cl)=O)CCl XSGMBZPBLJKZLG-RXMQYKEDSA-N 0.000 description 5
- QZLSBOVWPHXCLT-UHFFFAOYSA-N OC(c([s]1)ccc1Cl)=O Chemical compound OC(c([s]1)ccc1Cl)=O QZLSBOVWPHXCLT-UHFFFAOYSA-N 0.000 description 3
- MHCRLDZZHOVFEE-UHFFFAOYSA-N Nc(cc1)ccc1N(CCOC1)C1=O Chemical compound Nc(cc1)ccc1N(CCOC1)C1=O MHCRLDZZHOVFEE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention provides processes for the preparation of rivaroxaban and its intermediates.
- Rivaroxaban chemically is 5-chloro-N-( ⁇ (5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl) phenyl] - 1 ,3-oxazolidin-5- l ⁇ methyl)thiophene-2-carboxamide of Formula I.
- Rivaroxaban is used as an anti-thrombotic agent.
- U.S. Patent No. 7,157,456 provides rivaroxaban and processes for its preparation.
- U.S. Patent No. 8,106,192 provides a process for the preparation of N-((S)-3- bromo-2-hydroxypropyl)-5-chlorothiophene-2-carboxamide, wherein (2S)-3- aminopropane- 1 ,2-diol hydrochloride is reacted with 5-chlorothiophene-2-carbonyl chloride to provide N-((S)-2,3-dihydroxypropyl)-5-chlorothiophene-2-carboxamide.
- U.S. Publication No. 2010/0273789 provides a process for the preparation of 5- chloro-N-[(2S)-oxiran-2-ylmethyl]thiophene-2-carboxamide, wherein ((S)-3-bromo-2- hydroxypropyl)-5-chlorothiophene-2-carboxamide (50 g, 0.167 moles) is stirred with potassium carbonate (155 g, 1.12 moles) in the presence of anhydrous tetrahydrofuran (500 mL) for three days at room temperature to give 5-chloro-N-[(2S)-oxiran-2- ylmethyl]thiophene-2-carboxamide.
- 2007/0066615 provides a process for the preparation of 5- chloro-N-((2R)-2-hydroxy-3- ⁇ [4-(3-oxo-4-morpholinyl)-phenyl]amino ⁇ propyl)-2- thiophenecarboxamide, wherein a solution of 4-(4-aminophenyl)morpholin-3-one (2.6 mmol) and 5-chloro-N-[(2S)-oxiranylmethyl]-2-thiophenecarboxamide (3.1 mmol) in tetrahydrofuran is stirred overnight at 60°C in the presence of ytterbium(III)
- U.S. Publication No. 2010/0120718 provides a general method for preparing substituted N-(3-amino-2-hydroxypropyl)-5-chloro-2-thiophenecarboxamide derivatives, wherein 5-chloro-N-(2-oxiranylmethyl)-2-thiophenecarboxamide (1.0 equivalent) is stirred for 2 hours to 6 hours with a primary amine or aniline derivative (1.5 equivalents to 2.5 equivalents) in the presence of a solvent at room temperature or at temperatures up to 80°C.
- the product can be isolated from the reaction mixture by chromatography.
- PCT Publication No. WO 2012/092873 provides a process for the preparation of rivaroxaban, wherein 5-chloro-N-[(2S)-3-chloro-2-hydroxypropyl]thiophene-2- carboxamide or 5-chloro-N-[(2S)-oxiran-2-ylmethyl]thiophene-2-carboxamide is treated with substituted or unsubstituted phenyl methyl[4(3-oxo-morpholin-4-yl)phenyl] carbamate.
- the present inventors have developed simple, safe, efficient, economical, industrially feasible processes that provide rivaroxaban and its intermediates in good yield.
- the present invention provides processes for the preparation of rivaroxaban and its intermediates. Detailed Description of the Invention
- the present invention provides processes for the preparation of rivaroxaban and its intermediates.
- a first aspect of the present invention provides a process for the preparation of 5- chloro-N-[(2S)-3-chloro-2-h droxypropyl]thiophene-2-carboxamide of Formula II
- a second aspect of the present invention provides a process for the preparation of 5-chloro-N-[(2S)-3-chloro-2-h droxypropyl]thiophene-2-carboxamide of Formula II,
- R is CI, Br, or I, to obtain the compound of Formula II.
- a third aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- a fourth aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- R is CI, Br, or I, to obtain a com ound of Formula II;
- a fifth aspect of the present invention provides a process for the preparation of a compound of Formula V,
- a sixth aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- a seventh aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- R is CI, Br, or I, to obtain a compound of Formula II;
- An eighth aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- a ninth aspect of the present invention provides a process for the preparation of rivaroxaban of Formula I,
- R is CI, Br, or I, to obtain a com ound of Formula II;
- a tenth aspect of the resent invention provides a compound of Formula II.
- An eleventh aspect of the present invention provides use of the compound of Formula II
- the compound of Formula III or salts thereof, the reactive derivative of the compound of Formula IV, or the compound of Formula IVa may be prepared by any method provided in the art, for example, the methods described in U.S. Patent No.
- the salt of the compound of Formula III for example the hydrochloride salt of the compound of Formula III, may also be prepared as described herein.
- the compound of Formula III is treated with the reactive derivative of the compound of Formula IV, for example, 5- chlorothiophene-2-carbonyl chloride, to obtain the compound of Formula II in a solvent.
- the reactive derivative of the compound of Formula IV may be reacted with the compound of Formula III after isolation from the reaction mixture in which it is formed, or the reaction mixture containing the reactive derivative of the compound of Formula IV can also be used for the reaction with the compound of Formula III.
- the reactive derivative of the compound of Formula IV is reacted with the compound of Formula III in the presence of a base.
- the base may be, for example, sodium bicarbonate.
- a salt of the compound of Formula III such as the hydrochloride salt
- it may be treated with a base such as sodium bicarbonate prior to the reaction with the compound of Formula IV.
- the molar ratio of the base and the salt of a compound of Formula III may range from about 1 : 1 to about 4: 1.
- the solvent should not interfere with the reaction, and can be selected from the group comprising tetrahydrofuran, toluene, dichloromethane, ethyl acetate, or mixtures thereof.
- the compound of Formula III is treated with the compound of Formula IV in the solvent at about 0°C to about 35°C.
- the resulting mixture is stirred for about 1 hour to about 8 hours at about 0°C to about 35°C.
- the compound of Formula II may be isolated from the mixture by methods including concentration, distillation, decantation, filtration, evaporation, centrifugation, or a combination thereof, and may further be dried.
- the compound of Formula II can be converted into rivaroxaban of Formula I by following the processes mentioned herein, or processes provided in prior art, for example, U.S. Patent No. 8,106,192.
- the compound of Formula II is treated with base in solvent to obtain the compound of Formula V.
- the solvent may be 1 ,4-dioxane, methanol, ethanol, or their mixtures with water.
- the base may be sodium carbonate, potassium carbonate, calcium carbonate, sodium hydroxide, potassium hydroxide, or mixtures thereof.
- the base may be used as a solid or in solution.
- the compound of Formula II is treated with the base at about 0°C to about 30°C.
- the mixture is stirred for about 1 hour to about 8 hours at about 0°C to about 30°C.
- the product may be isolated from the mixture by methods including concentration, distillation, decantation, filtration, evaporation, centrifugation, or a combination thereof.
- the compound of Formula V is treated with the compound of Formula VI in a solvent to obtain the compound of Formula VII.
- the solvent may be ethanol, methanol, tetrahydrofuran, or their mixtures with water.
- the mixture containing the compound of Formula V and the compound of Formula VI is heated to reflux for about 0.5 hours to about 6 hours.
- the reaction mass is cooled to a temperature of about 0°C to about 35°C and stirred for about 0.5 hours to about 4 hours at about 0°C to about 35°C.
- the compound of Formula VII may be isolated from the mixture by methods including concentration, distillation, decantation, filtration, evaporation, centrifugation, or a combination thereof, and may further be dried.
- the compound of Formula VII is treated with 1 , 1 -carbonyldiimidazole in a solvent.
- the solvent may be dichloromethane.
- the mixture is stirred for about 2 hours to about 6 hours at about 25°C to about 30°C.
- the compound of Formula I may be isolated from the mixture by methods including concentration, distillation, decantation, filtration, evaporation, centrifugation, or a combination thereof, and may further be dried.
- the compound of Formula V can also be converted into rivaroxaban of
- the salt of a compound of Formula III in the present invention includes, for example, hydrochloride salts, hydrobromide salts, sulfate salts, nitrate salts, phosphate salts, formate salts, acetate salts, trifluoroacetate salts, methanesulfonate salts, and p- toluenesulfonate salts.
- the reactive derivative of a compound of Formula IV in the present invention includes acid halides, acid azides, acid anhydrides, mixed acid anhydrides, active amides, active esters, and active thio esters.
- Examples of reactive derivatives include acid chloride, acid amide of a free acid, di-ethoxyphosphoric acid ester, p-nitrophenyl ester, cyanomethyl ester, pentachlorophenyl ester, N-hydroxysuccinimide ester, imidazolyl ester, N-hydroxy phthalimide ester, 1 -hydroxybenzotriazole ester, 6-chloro- 1 - hydroxybenzotriazole ester, 1 -hydroxy- lH-2-pyridone ester, 2-pyridylthiol ester, and 2- benzothiazolylthiol ester.
- ambient temperature refers to a temperature in the range of O°C to 35°C.
- Example 1 Preparation of (2S)-l-amino-3-chloropropan-2-ol hydrochloride (Formula III) A solution of benzaldehyde (50 g, 0.540 moles) in ethanol (100 mL) was cooled to 15°C, and aqueous ammonia (25%, 57.4 mL) was added drop wise over 15 minutes to 20 minutes. Ethanol (25 mL) was added to the mixture. The mixture was stirred at 15°C to 20°C for 15 minutes to 20 minutes. (S)-Epichlorohydrin (50 g, 0.540 moles) and ethanol (50 mL) were added.
- the reaction mixture was allowed to warm to 40°C and stirred for 1 hour at 15°C to 40°C.
- the reaction mixture was again stirred at 35°C to 40°C for 6 hours, cooled to 25°C to 30°C, and further stirred for 12 hours.
- the solution was concentrated to dryness under vacuum at 50°C to 55°C.
- Ethanol 50 mL was added to the oil obtained, and the mixture was concentrated under vacuum at 50°C to 55°C.
- Toluene 125 mL was added to the oil obtained, and the mixture was heated to 35°C to 40°C.
- Aqueous hydrochloric acid (6.8 N, 129.5 mL) was added to the solution at 35°C to 40°C and stirred for 2 hours.
- the reaction mass was cooled to 25°C to 30°C, and the aqueous layer was separated.
- the organic layer was extracted with water (50 mL).
- the combined aqueous layers were concentrated under vacuum at 70°C to 75°C to get a semisolid material.
- the semisolid material was charged with ethanol (25 mL) and heated to 60°C to 65°C to get a clear solution.
- the solution was first cooled to 25°C to 30°C and then to -20°C.
- the slurry obtained was stirred for 1 hour at -20°C.
- the slurry was filtered and suck dried.
- the wet solid was dried at 45°C to 50°C under vacuum.
- the suspension was heated to 45°C to 50°C and stirred at 45°C to 50°C for 15 minutes.
- the mixture was cooled to 25°C to 30°C, and stirred at 25°C to 30°C for 2 hours.
- the slurry obtained was filtered, washed with toluene (10 mL), and the wet solid was dried at 50°C to 55°C under vacuum.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description











































Claims























Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11201406623PA SG11201406623PA (en) | 2012-04-16 | 2013-04-16 | Process for the preparation of rivaroxaban and intermediates thereof |
| AU2013250801A AU2013250801A1 (en) | 2012-04-16 | 2013-04-16 | Process for the preparation of rivaroxaban and intermediates thereof |
| US14/394,547 US20150299160A1 (en) | 2012-04-16 | 2013-04-16 | Process for the preparation of rivaroxaban and intermediates thereof |
| EP13726292.9A EP2838897A1 (en) | 2012-04-16 | 2013-04-16 | Process for the preparation of rivaroxaban and intermediates thereof |
| IN9450DEN2014 IN2014DN09450A (en) | 2012-04-16 | 2013-04-16 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1174/DEL/2012 | 2012-04-16 | ||
| IN1174DE2012 | 2012-04-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013156936A1 true WO2013156936A1 (en) | 2013-10-24 |
Family
ID=48539327
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2013/053025 Ceased WO2013156936A1 (en) | 2012-04-16 | 2013-04-16 | Process for the preparation of rivaroxaban and intermediates thereof |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20150299160A1 (en) |
| EP (1) | EP2838897A1 (en) |
| AU (1) | AU2013250801A1 (en) |
| IN (1) | IN2014DN09450A (en) |
| SG (1) | SG11201406623PA (en) |
| WO (1) | WO2013156936A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103819468A (en) * | 2013-12-05 | 2014-05-28 | 浙江天宇药业股份有限公司 | Synthesis method of Rivaroxaban and intermediate thereof |
| CN104478820A (en) * | 2014-12-22 | 2015-04-01 | 杭州瀚康生物医药科技有限公司 | Preparation method of rivaroxabanintermediate |
| WO2015104605A1 (en) | 2014-01-08 | 2015-07-16 | Wockhardt Limited | A process for preparing rivaroxaban or a pharmaceutically acceptable salt thereof |
| CN104788444A (en) * | 2015-05-12 | 2015-07-22 | 浙江天顺生物科技有限公司 | Preparation method of rivaroxaban |
| CN104817550A (en) * | 2015-05-26 | 2015-08-05 | 山东铂源药业有限公司 | Preparation method of rivaroxaban |
| CN104910141A (en) * | 2015-05-12 | 2015-09-16 | 浙江天顺生物科技有限公司 | Preparation method for Rivaroxaban intermediate 5-chloro-N-(2-oxiranylmethyl)-2-thiophene methanamide |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108707080B (en) * | 2018-06-20 | 2022-01-25 | 上海圣赢生物科技有限公司 | Environment-friendly synthesis method of linezolid and intermediate thereof |
| CN109400577B (en) * | 2019-01-07 | 2021-01-19 | 石药集团中奇制药技术(石家庄)有限公司 | Rivaroxaban related compound and preparation method and application thereof |
| CN112110910B (en) * | 2019-06-19 | 2024-03-19 | 上海特化医药科技有限公司 | Method for preparing rivaroxaban intermediate and method for preparing rivaroxaban from rivaroxaban intermediate |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6107519A (en) | 1997-11-07 | 2000-08-22 | Pharmacia & Upjohn Company | Process to produce oxazolidinones |
| WO2004060887A1 (en) * | 2003-01-07 | 2004-07-22 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| US7157456B2 (en) | 1999-12-24 | 2007-01-02 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| US20070066615A1 (en) | 2003-05-19 | 2007-03-22 | Bayer Heathcare Ag | Heterocyclic compounds |
| DE102007028320A1 (en) * | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| WO2009007027A1 (en) * | 2007-07-11 | 2009-01-15 | Bayer Schering Pharma Aktiengesellschaft | Aminoacyl prodrugs as an active pharmaceutical ingredient for thromboembolic disorders |
| US20100120718A1 (en) | 2006-11-02 | 2010-05-13 | Bayer Schering Pharma Aktiengesellschaft | Combination therapy of substituted oxazolidinones |
| US20100273789A1 (en) | 2007-07-11 | 2010-10-28 | Bayer Schering Pharma Aktiengesellschaft | Aminoacyl prodrugs |
| WO2011098501A1 (en) | 2010-02-10 | 2011-08-18 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2012092873A1 (en) | 2011-01-07 | 2012-07-12 | 浙江九洲药业股份有限公司 | Synthetic rivaroxaban intermediate and preparation method thereof |
| WO2013046211A1 (en) * | 2011-09-27 | 2013-04-04 | Symed Labs Limited | Processes for the preparation of 5-chloro-n-({(5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl) phenyl]-1,3-oxazolidin-5-yl}methyl)-2-thiophene-carboxamide and intermediates thereof |
-
2013
- 2013-04-16 EP EP13726292.9A patent/EP2838897A1/en not_active Withdrawn
- 2013-04-16 IN IN9450DEN2014 patent/IN2014DN09450A/en unknown
- 2013-04-16 WO PCT/IB2013/053025 patent/WO2013156936A1/en not_active Ceased
- 2013-04-16 US US14/394,547 patent/US20150299160A1/en not_active Abandoned
- 2013-04-16 SG SG11201406623PA patent/SG11201406623PA/en unknown
- 2013-04-16 AU AU2013250801A patent/AU2013250801A1/en not_active Abandoned
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6107519A (en) | 1997-11-07 | 2000-08-22 | Pharmacia & Upjohn Company | Process to produce oxazolidinones |
| US7157456B2 (en) | 1999-12-24 | 2007-01-02 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2004060887A1 (en) * | 2003-01-07 | 2004-07-22 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| US8106192B2 (en) | 2003-01-07 | 2012-01-31 | Bayer Pharma Aktiengesellschaft | Method for producing 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-1,3-oxazolidin-5-yl}methyl)-2-thiophenecarboxamide |
| US20070066615A1 (en) | 2003-05-19 | 2007-03-22 | Bayer Heathcare Ag | Heterocyclic compounds |
| US20100120718A1 (en) | 2006-11-02 | 2010-05-13 | Bayer Schering Pharma Aktiengesellschaft | Combination therapy of substituted oxazolidinones |
| DE102007028320A1 (en) * | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| WO2009007027A1 (en) * | 2007-07-11 | 2009-01-15 | Bayer Schering Pharma Aktiengesellschaft | Aminoacyl prodrugs as an active pharmaceutical ingredient for thromboembolic disorders |
| US20100273789A1 (en) | 2007-07-11 | 2010-10-28 | Bayer Schering Pharma Aktiengesellschaft | Aminoacyl prodrugs |
| WO2011098501A1 (en) | 2010-02-10 | 2011-08-18 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2012092873A1 (en) | 2011-01-07 | 2012-07-12 | 浙江九洲药业股份有限公司 | Synthetic rivaroxaban intermediate and preparation method thereof |
| WO2013046211A1 (en) * | 2011-09-27 | 2013-04-04 | Symed Labs Limited | Processes for the preparation of 5-chloro-n-({(5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl) phenyl]-1,3-oxazolidin-5-yl}methyl)-2-thiophene-carboxamide and intermediates thereof |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103819468A (en) * | 2013-12-05 | 2014-05-28 | 浙江天宇药业股份有限公司 | Synthesis method of Rivaroxaban and intermediate thereof |
| WO2015104605A1 (en) | 2014-01-08 | 2015-07-16 | Wockhardt Limited | A process for preparing rivaroxaban or a pharmaceutically acceptable salt thereof |
| CN104478820A (en) * | 2014-12-22 | 2015-04-01 | 杭州瀚康生物医药科技有限公司 | Preparation method of rivaroxabanintermediate |
| CN104788444A (en) * | 2015-05-12 | 2015-07-22 | 浙江天顺生物科技有限公司 | Preparation method of rivaroxaban |
| CN104910141A (en) * | 2015-05-12 | 2015-09-16 | 浙江天顺生物科技有限公司 | Preparation method for Rivaroxaban intermediate 5-chloro-N-(2-oxiranylmethyl)-2-thiophene methanamide |
| CN104788444B (en) * | 2015-05-12 | 2018-11-06 | 浙江天顺生物科技有限公司 | The preparation method of razaxaban |
| CN104817550A (en) * | 2015-05-26 | 2015-08-05 | 山东铂源药业有限公司 | Preparation method of rivaroxaban |
| CN104817550B (en) * | 2015-05-26 | 2017-06-16 | 山东铂源药业有限公司 | A kind of preparation method of razaxaban |
Also Published As
| Publication number | Publication date |
|---|---|
| IN2014DN09450A (en) | 2015-07-17 |
| US20150299160A1 (en) | 2015-10-22 |
| EP2838897A1 (en) | 2015-02-25 |
| AU2013250801A1 (en) | 2014-11-06 |
| SG11201406623PA (en) | 2014-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013156936A1 (en) | Process for the preparation of rivaroxaban and intermediates thereof | |
| CA2512504C (en) | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}methyl)-2-thiophenecarboxamide | |
| EP2521717B1 (en) | Method for the preparation of rivaroxaban | |
| CA2553237C (en) | Production method | |
| US9126990B2 (en) | Method for synthesizing rivaroxaban intermediate, 4-(4-[(5S)-(aminomethyl)-2-oxo-1,3-oxazoligdin-3-YL]phenyl)morpholin-3-one | |
| JP2024530111A (en) | Process for preparing aficamten | |
| CN114478690B (en) | Preparation method of 6, 6-dimethyl-3-azabicyclo [3.1.0] hexane derivative | |
| WO2013175431A1 (en) | Process for the preparation of rivaroxaban | |
| US9663505B2 (en) | Process for the preparation of rivaroxaban | |
| WO2015011617A1 (en) | Process for the preparation of rivaroxaban | |
| CN104817550B (en) | A kind of preparation method of razaxaban | |
| CN104788361B (en) | The synthetic method of the formic acid derivates of 5 azaspiros [2.4] heptane 6 | |
| JP2023502123A (en) | S1P1 modulator compounds and methods of preparing compounds | |
| CN103265482A (en) | Preparation method of bosutinib | |
| CN102939285B (en) | Preparation method of 5-[[2(r)-[1(r)-[3,5-bis(trifluoromethyl)phenyl]ethoxy]-3(s)-4-fluorophenyl-4-morpholinyl]methyl]-1,2-dihydro-3h-1,2,4-triazole-3-one | |
| CN105175331A (en) | Preparation method for EGFR molecule targeting antitumor drug | |
| CN104926807B (en) | A kind of razaxaban related substances " diamines " and its synthetic method | |
| CN110498771A (en) | A method of preparing the intermediate for disliking La Geli | |
| WO2012041263A2 (en) | A method of manufacturing 2-({(5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]- l,3-oxazolidin-5-yl}methyl)-lh-isoindol-l,3(2h)-dione with a high optical purity | |
| EP2895176B1 (en) | Rivaroxaban intermediate and preparation thereof | |
| CN113527117A (en) | Preparation method of AEEA | |
| CN104961736A (en) | Related substance of rivaroxaban--triamine and synthetic methods thereof | |
| CN107382897A (en) | A kind of intermediate of betrixaban and its preparation method and application | |
| WO2014020458A1 (en) | Improved process for preparation of rivaroxaban | |
| CN113943281B (en) | Synthetic method and application of isoxazole pyrimidine derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13726292 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14394547 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013726292 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013726292 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2013250801 Country of ref document: AU Date of ref document: 20130416 Kind code of ref document: A |