WO2013178788A2 - Manufacture of degarelix - Google Patents
Manufacture of degarelix Download PDFInfo
- Publication number
- WO2013178788A2 WO2013178788A2 PCT/EP2013/061264 EP2013061264W WO2013178788A2 WO 2013178788 A2 WO2013178788 A2 WO 2013178788A2 EP 2013061264 W EP2013061264 W EP 2013061264W WO 2013178788 A2 WO2013178788 A2 WO 2013178788A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- degarelix
- drug substance
- viscosity
- mannitol
- mpas
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/09—Luteinising hormone-releasing hormone [LHRH], i.e. Gonadotropin-releasing hormone [GnRH]; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/25—Growth hormone-releasing factor [GH-RF], i.e. somatoliberin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/23—Luteinising hormone-releasing hormone [LHRH]; Related peptides
Definitions
- the present invention relates to a manufacturing process for preparing Degarelix.
- Degarelix also known as FE200486, is a third generation gonadotropin releasing hormone (GnRH) receptor antagonist (a GnRH blocker) that has been developed and approved for prostate cancer patients in need of androgen ablation therapy (Doehn et al., Drugs 2006, vol. 9, No. 8, pp. 565-571; WO 09846634).
- GnRH gonadotropin releasing hormone
- Degarelix acts by immediate and competitive blockade of GnRH receptors in the pituitary and, like other GnRH antagonists, does not cause an initial stimulation of luteinizing hormone production via the
- Degarelix is a synthetic linear decapeptide containing seven unnatural amino acids, five of which are D-amino acids. It has ten chiral centers in the back bone of the decapeptide. The amino acid residue at position 5 in the sequence has an additional chiral center in the side-chain substitution giving eleven chiral centers in total. Its CAS registry number is 214766-78-6 (of free base) and it is commercially available under the Trademark
- the drug substance is chemically designated as D-Alaninamide, N-acetyl-3-(2- naphthalenyl)-D-alanyl-4-chloro-D-phenylalanyl-3-(3-pyridinyl)-D-alanyl-L-seryl-4-[[[(4S)- hexahydro-2,6-dioxo-4-pyrimidinyl]carbonyl]amino]-L-phenylalanyl-4- [(aminocarbonyl)amino]-D-phenylalanyl-L-leucyl-N6-(l-methylethyl)-L-lysyl-L-prolyl- and is represented by the chemical structure below (in the following also referred to as Formula I):
- Degarelix can also be represented as:
- AAi is D-2Nal
- AA 2 is D-4Cpa
- AA 3 is D-3Pal
- AA 4 is Ser
- AA 5 is 4Aph(L-Hor)
- AA 6 is D- Aph(Cbm)
- AA 7 is Leu
- AA 8 is Lys(iPr)
- AA 9 is Pro
- AA 10 is D-Ala.
- Degarelix can be represented as Ac-AAi-AAi 0 -NH 2 .
- Degarelix has previously been prepared using Boc-solid phase peptide synthesis (SPPS) methodology as reported in WO 98/46634 and Jiang et al., J. Med. Chem. 2001, 44, 453- 467.
- SPPS Boc-solid phase peptide synthesis
- WO2010/12835 and WO2011/066386 describe the preparation of degarelix using an Fmoc strategy.
- WO2012/055905 and WO2012/055903 describe liquid phase syntheses of degarelix.
- degarelix The physicochemical characterization of degarelix has shown that this decapeptide has the ability to self-associate, and eventually form gels in aqueous solution. Self- aggregation makes this compound build-up a depot in situ when injected subcutaneously or intramuscularly.
- the degarelix depot was shown to provide a sustained release of the active over months depending on the dosage. At present, the drug is administered in dosages of 120 mg (40 mg/ml) for first injection, and of 80 mg (20 mg/ml) for sustained release over one month.
- the present inventors have surprisingly found that viscosity, and hence sustained release properties and bioavailability, of the reconstituted drug product can be controlled through processing of the crude peptide (e.g. obtained by Fmoc strategy, liquid phase synthesis or another route) into the drug substance.
- the viscosity associated with the drug substance surprisingly correlates with the viscosity associated with the drug product, even after a further reconstitution and lyophilisation.
- the viscosity of the drug product has to be controlled within a range of up to 15 mPas, preferably within a range of 2 to 12 mPas, to obtain the desired depot formation and thus sustained release.
- the present invention provides processes that allow the manufacture of drug products that show this viscosity, as determined upon reconstitution with the reconstitution fluid at a
- the present invention thus provides a method for controlling the viscosity of a degarelix product to be no greater than 15 mPas, preferably within a range of 2 to 12 mPas, as determined upon reconstitution with water for injection in an amount of 20 mg degarelix free base/ml, comprising the steps of: 1. Providing a lyophilized degarelix drug substance which shows a viscosity of up to 3.2 mPas, as determined upon dissolution in water containing mannitol (2.5 % w/V) in an amount of 20 mg degarelix free base/ml
- the present invention provides a method for producing a lyophilized degarelix product which shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, as determined upon reconstitution with water for injection in an amount of 20 mg degarelix free base/ml, comprising the steps of:
- the present invention provides a method for producing degarelix drug product comprising a lyophilized degarelix drug product and a liquid for reconstitution (reconstitution fluid) which, upon reconstitution with said liquid in an amount of 20 mg degarelix free base/ml, shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, comprising the steps of:
- the present invention also provides a lyophilized degarelix drug substance which shows, upon dissolution in water containing 2.5 wt.% mannitol in an amount of 20 mg degarelix free base/ml, a viscosity of up to 3.2 mPas, and processes for providing this lyophilized degarelix drug substance.
- the present invention provides a degarelix drug product comprising a lyophilized degarelix drug product and a reconstitution fluid which, upon reconstitution with said reconstitution fluid in an amount of 20 mg degarelix free base/ml, shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, and contains a viscosity-reducing agent in an amount of 0.001 to 5 mg/ml.
- Figure 1 shows the relationship between drug substance viscosity and drug product viscosity.
- Figures 2 and 3 show the relationship between dynamic viscosity of degarelix dosing suspension (20 mg/ml) and degarelix plasma concentrations (rat) at day 3 ( Figure 2) and day 28 ( Figure 3).
- the methods for producing a lyophilized degarelix product both according to the first and the second aspect start with the degarelix drug substance which will be described in more detail.
- the decapeptide degarelix can be prepared by solid phase peptide synthesis, as disclosed in W098/46634, WO2010/12835 and WO2011/066386, or by liquid phase peptide synthesis, as disclosed in WO2012/055905 or WO2012/055903.
- This peptide synthesis provides crude degarelix which is further purified and then lyophilized to provide a lyophilized product which consists of degarelix, acetic acid, a residual amount of water, and minor amounts of impurities due to the production process, if any.
- This product is referred to as degarelix drug substance or merely drug substance in the present invention.
- the degarelix drug substance preferably consists of degarelix, 4.5 to 10 wt.% acetic acid (w/w), and up to 10 wt.% water (w/w).
- the manufacture of the drug substance can be divided into a first step (A) providing purified degarelix in solution, and a second step (B) providing the degarelix drug substance.
- Step (A) comprises the purification of crude degarelix in one or more steps, preferably two steps, optionally followed by a column concentration and/or salt exchange step.
- Crude degarelix as obtained by LPPS or SPPS, is first subjected to purification.
- the purification is preferably carried out by applying the peptide solution obtained by SPPS or LPPS to a column with reversed phase material, which is preferably pre-equilibrated with buffer.
- This first purification step preferably provides a purity of at least 95 %, as determined by HPLC.
- the reverse phase column chromatography is repeated to obtain a product with purity of at least 97.5 %, as determined by HPLC.
- the purified degarelix solution with a purity of at least 95 %, preferably at least 97.5% is subjected to further column chromatography step to pre-concentrate the degarelix solution and/or for salt exchange (particularly if acetic acid is used for pH adjustment of the eluents in the last purification step).
- This pre-concentration and/or salt exchange step is also preferably carried out on a reverse phase column:
- the purified degarelix solution is diluted with water (preferably 1.5 to 2.5 times) and applied to the column, pre-equilibrated with buffer.
- the column is preferably first washed with ethanol (low concentration, generally below 20 %) and aqueous ammonium acetate and subsequently with ethanol (low concentration, generally below 20 %)/acetic acid/water.
- the column is then eluted, e.g. with ethanol (high concentration, generally 20 to 60 %)/acetic acid/water to obtain a more concentrated solution of degarelix compared to the solutions after the purification step(s).
- the process is not limited to ethanol as an organic modifier in the eluent. Other solvents such as acetonitrile can also be used.
- Step (A) provides purified degarelix in solution.
- the subsequent treatment of the purified degarelix in solution to obtain the degarelix drug substance can be carried out in different ways. In the following, four preferred ways are illustrated (Steps (Bl), (B2), (B3), and (B4)).
- the purified degarelix in solution is first subjected to a concentration step in which ethanol or another organic modifier such as acetonitrile is removed by evaporation. This step is preferably carried out with a rotavap evaporator. The preferred maximum temperature during evaporation is 40 °C.
- the resulting highly concentrated, viscous degarelix product aggregated product that is usually in gel form
- acetic acid de-aggregation step
- the de-aggregation step is important to control the viscosity of the drug substance.
- the de-aggregation step is preferably carried out with one or more, most preferably all of the following conditions: • Final acetic acid concentration: 6 - 40 %, preferably 15 - 35 % (v/v)
- the lyophilization step is preferably carried out at an ice thickness of 1.2 to 2.4 cm and a secondary drying time of 1 to 17 hours.
- the secondary drying temperature is usually around 20 °C (15 to 25 °C).
- step (Bl) thus provides a lyophilized drug substance that shows a viscosity of less than 3.2 mPas, preferably between 1.15 and 2 mPas (as determined upon dissolution in an amount of 20 mg degarelix free base in 1 ml of water, containing 2.5 % (w/V) mannitol).
- the method for measuring the viscosity is described in the experimental section.
- a drug substance fulfilling this viscosity requirement is further referred to as drug substance (1), whereas a drug substance not fulfilling this viscosity requirement is further referred to as drug substance (2).
- Drug substance (2) has a preferred viscosity of 3.2 to 15 mPas, upon dissolution in an amount of 20 mg in 1 ml water. Drug substance (2) may even be gel-like, in a condition of significantly more than 3.2 mPas, even though its viscosity cannot be measured precisely.
- the drug substance, in particular drug substance (1) is preferably further characterized by an acetic acid content of 4.5 to 10.0 % (w/w) and/or a water content of 10 % or less (w/w). Additionally, the drug substance, in particular drug substance (1), preferably shows an optical density of 0.10 AU or less (at a concentration of 20 mg degarelix free base/ml in 2.5 % mannitol (aq)). The method for measuring the optical density is described in the experimental section.
- the invention provides a method for producing the degarelix drug substance (1), comprising the steps of:
- the invention further provides a method for modulating the viscosity of degarelix, such that following lyophilization and reconstitution with water, the viscosity of a 20 mg/ml degarelix solution in 2.5% w/V mannitol is no greater than 15 mPas, comprising:
- the conditions for acetic acid addition and lyophilization are as described above.
- lyophilization is 4.5 to 10.0 % (w/w).
- the degarelix solution obtained in step A (preferably after a one- or two-step purification method, e.g. without pre-concentration/salt exchange) is loaded onto a chromatographic column, ion exchange is performed and the column is rinsed (washed) with diluted acetic acid (about 1%).
- Degarelix is eluted from the column using aqueous acetic acid at a AcOH concentration of 20 to 50 wt.%, preferably 23 to 37 wt.%, preferably 23 to 27 wt.%, preferably 27 to 37 wt.%, preferably 33 to 37 wt.%, preferably 35 wt.%, and subsequently optionally diluted to appropriate AcOH concentration, filtered.
- the stationary phase in the column can be of different types.
- the stationary phase can contain functional groups like hydrocarbons (aliphatic and aromatic), alcohols, nitriles, groups with appropriate acid/base properties and ion- exchange groups but is not limited to this type of groups.
- This process provides the drug substance, preferably drug substance (1).
- the invention also provides a method for producing the degarelix drug substance, comprising the steps of:
- Step (B3) Isolation via lyophilization-reconstitution in AcOH/water-lyophilization
- Purified degarelix in solution obtained after Step A is isolated via lyophilization; the resulting lyophilized product is dissolved at a concentration between 10 and 20 g/L in 2 % acetic acid, and lyophilized again to give degarelix drug substance (1).
- Purified degarelix in solution (EtOH/water containing AcOH or ACN/water containing AcOH obtained after Step A, or as described in Step Bl except for lyophilization, or B2 except for lyophilization) is isolated via spray drying to give degarelix drug substance (1).
- a purified degarelix solution as obtained in step A is directly subjected to the spray-drying process.
- the AcOH concentration of said aqueous degarelix solution subjected to spray-drying is adjusted to 6 to 40 % (v/v), preferably 15 to 35 % (v/v).
- the lyophilized degarelix drug product comprises the degarelix drug substance and mannitol, i.e. it comprises (and preferably consists of) degarelix, acetic acid, mannitol, a residual amount of water, and minor amounts of impurities due to the production process, if any.
- Production process A is the first aspect of the invention mentioned above, i.e. a method for producing degarelix drug product which, upon reconstitution with water for injection in an amount of 20 mg degarelix free base/ml, shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, comprising the steps of:
- "upon reconstitution with water for injection in an amount of 20 mg/ml” refers to the conditions for measuring the viscosity and does not mean that the drug product is present in a solution of 20 mg/ml. Most preferably, the drug product is present in lyophilized form, optionally in combination with reconstitution liquid.
- Preferred amounts per vial are in the range of 60 to 300 mg (such as 120 mg, 80 mg, and 240 mg). Alternatively, it can be provided as reconstituted drug product, with preferred concentrations in the range of 2 to 100 mg/ml, preferably 10 to 70 mg/ml (such as 40 mg/ml, 20 mg/ml, and 60 mg/ml).
- Step b may also be referred to as compounding step.
- filtration and vial filling are carried out after compounding and before freeze-drying so that the entire preferred production process A comprises the steps of:
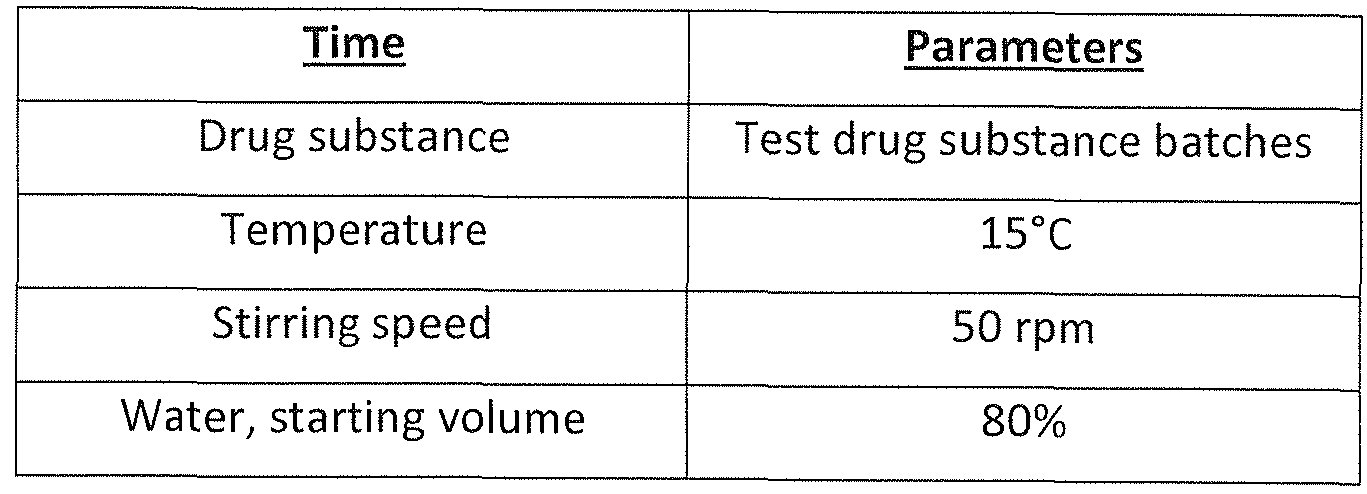
- the drug substance is subjected to a compounding step, which is generally carried out as follows:
- mannitol For the production of the unfiltered bulk drug product, drug substance and mannitol are dissolved in water (pure water; generally Milli-Q water) each in amounts of 10 - 60 g, per 1000 g batch size. Typical amounts are 20 to 50 g drug substance (as degarelix free base content as determined by HPLC and 10 to 50 g mannitol per 1000 g. The actual amount depends on the final concentration of degarelix in the drug product and the volume of the reconstitution liquid (mannitol is preferably added such that an isotonic solution with an osmolality of 300 mOsm +/-30 mOsm is obtained after reconstitution).
- water (usually approx. 80 % of the total amount of water) is added to a compounding vessel.
- the mannitol is added and dissolved by stirring.
- the drug substance is added to the stirred mannitol solution and the formulated bulk (batch) is brought to its final weight by adding the remaining water.
- This compounding is carried out in a manner so that a significant viscosity increase is avoided.
- the viscosity of the bulk product thus preferably remains below 5 mPas, preferably below 3.2 mPas during the compounding step (viscosity determined after filtration upon dissolution in an amount of 20 mg in 1 ml of 2.5 % (w/V) aqueous mannitol solution).
- the temperature is usually kept within a range of 6 - 15 °C.
- the stirrer is preferably one that provides turbulent mixing without vortex.
- the bulk drug product is then preferably sterile filtered, e.g. through two sterilizing grade filters placed in series, by pressurizing the formulated bulk with nitrogen.
- Sterilized vials are filled with the filtered bulk drug product and semi-stoppered (freeze- drying position) under aseptic conditions.
- the freeze-dryer is preferably steam sterilized before use.
- the vials are the placed on the freeze-dryer shelves.
- the subsequent freeze-drying process preferably comprises the steps of freezing, main drying (sublimation), and secondary drying. Preferred conditions are as follows:
- the freeze-drying process preferably comprises, or even consists of, three main steps, i.e. freezing, main drying (sublimation) and secondary drying.
- the vials are loaded onto refrigerated shelves maintained at 2 to 10 °C, such as 5 °C.
- the shelves are cooled from e.g. 5 °C to -30 to -40 °C, such as -35 °C.
- the shelf is cooled from e.g. 5 °C to -30 to -40 °C, such as -35 °C.
- Main drying is performed by lowering the chamber pressure (preferably to 0.100 mBar or less) and increasing the shelf temperature (preferably to 10 to 20 °C, such as +17 °C).
- the main drying time proceeds for at least 15 hours.
- chamber pressure is reduced (preferably to 0.01 mBar or less) and the shelf temperature is increased (preferably to 20 to 30 °C, such as 25°C). Secondary drying is typically completed within 7 hours.
- the lyophilized drug product is then labeled and packaged and combined with the appropriate amount of reconstitution liquid.
- the reconstitution liquid is selected depending on the viscosity of the drug substance.
- drug substance (1) is used as starting material for production process A, i.e. a lyophilized drug substance that shows a viscosity of less than 3.2 mPas, preferably between 1.15 and 2 mPas (upon dissolution in an amount of 20 mg in 1 ml of 2.5 (w/v)% aqueous mannitol solution)
- the resulting lyophilized drug product is preferably combined with water for injection (WFI) as reconstitution liquid.
- WFI water for injection
- the viscosity is generally in the range of 2 to 15 mPas (as measured upon dissolution of 20 mg degarelix (free base) in 1 ml WFI). A drug product with a viscosity within this range was found to provide a sufficient depot release of degarelix in vivo.
- Production process B is a method for producing degarelix drug product comprising a lyophilized degarelix drug product and a liquid for reconstitution (reconstitution fluid) which, upon reconstitution with said liquid in an amount of 20 mg degarelix free base/ml, shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, comprising the steps of:
- Step a may also be referred to as compounding step.
- filtration and vial filling are carried out after compounding and before freeze-drying so that the entire preferred production process B comprises the steps of:
- Production process B is identical to production process A, with the exception that in the compounding step, a viscosity-reducing agent, preferably a non-ionic surfactant is added prior to lyophilization.
- the non-ionic surfactant is preferably added in an amount of 0.0003 to 1.5 mg/ml to the bulk solution, corresponding to an amount of 0.001 to 5 mg/ml, more preferably 0.1 to 1 mg/ml, in the reconstituted drug product (e.g. when reconstituted to a degarelix concentration of 60 mg degarelix free base/ml).
- Preferred non-ionic surfactants are those with a linear alkyl chain having at least 8 carbon atoms (preferably without double bonds) and a carbohydrate moiety.
- drug substance (2) is preferably used, i.e. a lyophilized drug substance that shows a viscosity of at least 3.2 mPas (upon dissolution in an amount of 20 mg degarelix free base in 1 ml of 2.5 wt.% aqueous mannitol solution).
- the resulting lyophilized drug product is typically combined with WFI as reconstitution liquid.
- the viscosity is generally in the range of 2 to 15 mPas (as measured upon dissolution of 20 mg degarelix free base in 1 ml WFI). A drug product with a viscosity within this range was found to provide a sufficient depot formation for delayed release of degarelix in vivo.
- Production process B is particularly preferred for degarelix products that have a relatively high degarelix concentration upon reconstitution, such as 50 mg degarelix free base/ml or more, e.g. 60 mg degarelix free base/ml (240 mg drug product).
- Production process B provides a novel degarelix drug product that differs from known drug products in that the reconstituted drug product contains a viscosity-reducing agent.
- the viscosity-reducing agent is the one used in step B, preferably a non-ionic surfactant.
- the present invention thus provides a degarelix drug product which comprising a lyophilized degarelix drug product and a liquid for reconstitution which, upon
- reconstitution with said liquid in an amount of 20 mg degarelix free base /ml shows a viscosity of up to 15 mPas, preferably within a range of 2 to 12 mPas, and contains a viscosity-reducing agent in an amount of 0.001 to 5 mg/ml, more preferably 0.1 to 1 mg/ml .
- Example 1 Purification, deaggregation and lyophilisation Crude Degarelix was synthesized as described in WO2012/055905 Al, up to Step 12 in Example 5. Step 13 as disclosed in WO2012/055905 Al was replaced by the following steps, In summary, the purification process of crude degarelix drug substance, obtained after the last deprotection step, consists of three preparative reversed phase
- RPC chromatography
- Step 13 Purification step:
- Step 14 Purification step:
- the main pool obtained in the step 13 was diluted twice with water and applied to the column packed with reversed phase material, pre-equilibrated with buffer (90% of 1% AcOH and 10%of EtOH). Load: ⁇ 25 g/L column volume. The column was first washed with a first buffer (10% of EtOH and 90% of 0.5 mol/L AcONH4) and then with a second buffer (90% of 1% AcOH and 10% of EtOH).
- the column was then eluted with a mixture of buffer and ethanol (76% of 1% AcOH and 24% of EtOH). The fractions obtained were analyzed, and combined in such a way that the purity in the main pool fulfilled the acceptance criteria for the process control.
- the main pool obtained in the step 14 was diluted twice with water and applied to the column packed with reversed phase material, pre-equilibrated with buffer (90% of 1% AcOH and 10% of EtOH). Load: ⁇ 18 g/L column volume.
- the column was first washed with a first buffer (10% of EtOH and 90% of 0.5 mol/L AcONH4) and then with a second buffer (90% of 1% AcOH and 10% of EtOH).
- the column was then eluted with a mixture of buffer and ethanol (50% of 1% AcOH and 50% of EtOH).
- degarelix can be eluted with a solution of AcOH/MeCN/water, such as 12% AcOH and 22% MeCN in water.
- Step 16 (Concentration-de-aggregation-lyophilization):
- the pool of pure degarelix solutions from step 15 was concentrated below 40°C. Aqueous acetic acid and water were added to the concentrated solution, to give a concentration below 15 g/L, and an acetic acid concentration of 30%. This solution was then filtered and lyophilised to yield degarelix drug substance.
- the viscosity of the drug substance obtained by the sequence of steps was below 2.5 mPas, as determined in a concentration of 20mg/ml in 2.5 (w/V) % mannitol solution.
- Example 2 Method for measuring viscosity for drug substance and drug product
- the drug substance vial is reconstituted 2.5 % aqueous mannitol solution in water (w/V).
- the drug product vial is reconstituted with water for injection. Dispense the solvent into the vial and swirl the vial until reconstitution is complete or use a vortex for a few seconds. The liquid should look clear and no undissolved powder or particles are visible. Keep the vial upright and do not shake. The sample is measured at 350 nm, 120 minutes after addition of the solvent.
- the sample has to be homogenized by gently turning the cuvette five times back and forth through approximately 180 degrees. The four minute delay allows any air bubbles to disperse before the reading.
- the absorption caused by the cuvette and by the water for injection has to be deducted from the read-off of the sample.
- the reconstituted peptide was filtered into a measuring flask through a 0.20 ⁇ Sartoriuos Ministar filter prior to spray-drying.
- the inlet temperature and the feed rate were adjusted prior to spray-drying.
- the tubing of the pump was placed in the feed solution, and the drying was initiated.
- the inlet temperature was allowed to drop to 700°C before the cyclone and the collection vessel were dismantled for powder collection.
- the powder was collected with brushes into Petri dishes, which were weighed before and after collection to determine the yield.
- Example 2 The pool from step 14 of Example 1 was diluted with water and applied to a column packed with reversed phase material. After rinsing the column with 1% AcOH in water degarelix was eluted with 35% AcOH in water. Fractions were adjusted to contain 35 g degarelix/l, 27% AcOH (Sample 1) and 15 g degarelix/l, 30% AcOH (Sample 2). The adjusted fractions were freeze-dried and analysed. Results: see Table below:
- the temperature was set to 5, 20, 25, or 35°C
- concentration of degarelix drug substance in stock solutions during the experiments The concentration of degarelix drug substance was 5, 15, 25, or 35 mg/ml.
- the concentration of acetic acid is 13, 15, 18, 20, 22, 26, 30, 35 or 40%.
- Optical Density A sample ( ⁇ 1 ml) should be taken at the end of each experiment for measurement of the final optical density.
- the vials were stored in the freeze dryer at 5°C until de-loading.
- Viscosity was measured on samples of 20 mg/ml free base
- Degarelix bulk solutions containing 20 mg/g degarelix and 25 mg/g mannitol are compounded using different batches of drug substance and different settings of temperature.
- the compounding is performed with the stirrer positioned at the centre. Before commencing the experiments the centre position should be fixed.
- the bulks will not be filtered because it is expected that at least one of the batches will be highly aggregated and therefore very difficult to filter. Also, the batches will not be used for purposes where sterility is required.
- Viscosity DS Viscosity
- Viscosity DP (20 mg/ml)
- Figure 1 shows a correlation between drug substance viscosity and drug product viscosity.
- Degarelix batch reconstituted using WFI (as control) and using WFI containing th surfactants.
- the vials are reconstituted to 60 mg/ml degarelix.
- the drug product batch used for these experiments had a bulk viscosity of approx. 3.8 mPas.
- TPGS and Tween 20 were prepared in solutions containing 1 mg/ml (0.1% solutions)
- Example 9 Drug product viscosity and bioavailability
- Degarelix drug product is manufactured as a freeze-dried product containing mannitol.
- the products are used as investigational medicinal products in clinical studies.
- Several formulations were produced, containing various amounts of degarelix per vial, namely 10 mg, 20 mg, 40 mg, 88 mg, 128 mg, 120 mg (40mg/ml), 180 mg (60mg/ml) and 240 mg (60mg/ml) and different ratios of degarelix/mannitol. o Materials
- degarelix concentration in the bulk is 20 g/l unless otherwise stated.
- Plasma concentrations were initially measured at 2 hrs, lday, 7day and 28 day and partial AUC calculated as AUC 0-7 days and AUC 7-28 days. The design was then changed with measurement of plasma concentration (Cp)-2hrs being skipped and being replaced by Cp- 3days. Likewise, AUC 0-7days was replaced by AUC l-7days. This explains the
- a first model was calculated based on 2 components.
- the viscosity of constituted degarelix product appears to be the prominent parameter with some ability to predict in vitro release and in vivo performance of the depot.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Endocrinology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Chemistry (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Dermatology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Reproductive Health (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
Description






Claims
Priority Applications (31)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MYPI2014703537A MY182320A (en) | 2012-06-01 | 2013-05-31 | Downstream process |
| RU2014146272A RU2657444C2 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| CN201380028448.8A CN104334182B (en) | 2012-06-01 | 2013-05-31 | Made in Garek |
| MX2014014662A MX364010B (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix. |
| DK13726219.2T DK2854831T3 (en) | 2012-06-01 | 2013-05-31 | MANUFACTURE OF DEGARELIX |
| PL13726219.2T PL2854831T3 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| HK15109772.8A HK1209046B (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| SI201332090T SI2854831T1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| ES13726219T ES2988481T3 (en) | 2012-06-01 | 2013-05-31 | Manufacturing of degarelix |
| FIEP13726219.2T FI2854831T3 (en) | 2012-06-01 | 2013-05-31 | PREPARATION OF DEGARELIX |
| EP24172290.9A EP4385517A3 (en) | 2012-06-01 | 2013-05-31 | Degarelix drug product |
| KR1020147035636A KR102140982B1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| LTEPPCT/EP2013/061264T LT2854831T (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| JP2015514527A JP6226966B2 (en) | 2012-06-01 | 2013-05-31 | Production of degarelix |
| EP25151068.1A EP4512390B1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| US14/403,775 US9592266B2 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| AU2013269523A AU2013269523B2 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| BR112014029495-0A BR112014029495B1 (en) | 2012-06-01 | 2013-05-31 | ACTIVE PRINCIPLE OF FREEZE DRIED DEGARELIX AND ITS PRODUCTION METHODS, METHOD FOR PRODUCING A DEGARELIX MEDICINE, AND METHOD FOR CONTROLLING THE VISCOSITY OF A FREEZE DRIED DEGARELIX PRODUCT |
| SG11201407679PA SG11201407679PA (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| RS20241121A RS66065B1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| EP25151066.5A EP4512389B1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| NZ701978A NZ701978A (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| HRP20241284TT HRP20241284T1 (en) | 2012-06-01 | 2013-05-31 | PRODUCTION OF DEGARELIX |
| CA2874927A CA2874927A1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| EP13726219.2A EP2854831B1 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| IL235856A IL235856B (en) | 2012-06-01 | 2014-11-23 | Manufacture of degarelix |
| PH12014502682A PH12014502682B1 (en) | 2012-06-01 | 2014-12-01 | Manufacture of degarelix |
| US15/420,156 US10172906B2 (en) | 2012-06-01 | 2017-01-31 | Manufacture of Degarelix |
| US16/224,843 US10765721B2 (en) | 2012-06-01 | 2018-12-19 | Manufacture of Degarelix |
| US16/947,382 US11260102B2 (en) | 2012-06-01 | 2020-07-30 | Manufacture of Degarelix |
| US17/154,690 US11510962B2 (en) | 2012-06-01 | 2021-01-21 | Manufacture of degarelix |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12170454.8 | 2012-06-01 | ||
| EP12170454 | 2012-06-01 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/403,775 A-371-Of-International US9592266B2 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
| US15/420,156 Continuation US10172906B2 (en) | 2012-06-01 | 2017-01-31 | Manufacture of Degarelix |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013178788A2 true WO2013178788A2 (en) | 2013-12-05 |
| WO2013178788A3 WO2013178788A3 (en) | 2014-02-27 |
Family
ID=48539181
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/061264 Ceased WO2013178788A2 (en) | 2012-06-01 | 2013-05-31 | Manufacture of degarelix |
Country Status (28)
| Country | Link |
|---|---|
| US (5) | US9592266B2 (en) |
| EP (4) | EP2854831B1 (en) |
| JP (1) | JP6226966B2 (en) |
| KR (1) | KR102140982B1 (en) |
| CN (2) | CN107569456A (en) |
| AR (2) | AR092840A1 (en) |
| AU (1) | AU2013269523B2 (en) |
| CA (2) | CA3228586A1 (en) |
| DE (1) | DE25151068T1 (en) |
| DK (1) | DK2854831T3 (en) |
| ES (3) | ES3037194T1 (en) |
| FI (3) | FI2854831T3 (en) |
| HR (2) | HRP20241284T1 (en) |
| HU (1) | HUE068579T2 (en) |
| IL (1) | IL235856B (en) |
| JO (1) | JO3586B1 (en) |
| LT (1) | LT2854831T (en) |
| MX (1) | MX364010B (en) |
| MY (1) | MY182320A (en) |
| NZ (2) | NZ701978A (en) |
| PH (1) | PH12014502682B1 (en) |
| PL (3) | PL4512390T1 (en) |
| RS (1) | RS66065B1 (en) |
| RU (1) | RU2657444C2 (en) |
| SG (1) | SG11201407679PA (en) |
| SI (1) | SI2854831T1 (en) |
| TW (1) | TWI580443B (en) |
| WO (1) | WO2013178788A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112125956A (en) * | 2019-06-25 | 2020-12-25 | 深圳市健元医药科技有限公司 | A kind of preparation method of degarelix |
| EP2854831B1 (en) | 2012-06-01 | 2024-07-10 | Ferring B.V. | Manufacture of degarelix |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI539959B (en) | 2008-02-11 | 2016-07-01 | 菲瑞茵國際中心股份有限公司 | Method for treating prostate cancer during metastasis |
| CN107693496A (en) * | 2017-10-23 | 2018-02-16 | 天津双硕医药科技有限公司 | A kind of injection Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2 freeze-dried powder and preparation technology |
| EP3590526A1 (en) * | 2018-07-05 | 2020-01-08 | Antev Limited | A lyophilization process and a teverelix-tfa lyophilizate obtained thereby |
| CN111036078B (en) * | 2018-10-14 | 2022-04-26 | 深圳市健元医药科技有限公司 | Post-treatment method of GnRH antagonist |
| CN114938646B (en) * | 2019-12-05 | 2026-03-27 | 费森尤斯卡比有限责任公司 | Methods for analyzing degarelk and its related products |
| CN114460179B (en) * | 2020-11-09 | 2024-10-18 | 深圳市健翔生物制药有限公司 | Determination method of degarelix acetate in-vitro release degree for injection |
| CN114671927A (en) * | 2020-12-24 | 2022-06-28 | 深圳翰宇药业股份有限公司 | Preparation method of degarelix crude peptide |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998046634A1 (en) | 1997-04-11 | 1998-10-22 | Ferring B.V. | GnRH ANTAGONISTS BEING MODIFIED IN POSITIONS 5 AND 6 |
| WO2010012835A1 (en) | 2008-07-31 | 2010-02-04 | Andrea Peruffo | Rollerski or skate with braking system and method for braking said sports item |
| WO2011066386A1 (en) | 2009-11-25 | 2011-06-03 | Novetide, Ltd. | Process for production of degarelix |
| WO2012055903A1 (en) | 2010-10-27 | 2012-05-03 | Ferring B.V. | Process for the manufacture of degarelix and its intermediates |
| WO2012055905A1 (en) | 2010-10-27 | 2012-05-03 | Ferring B.V. | Process for the manufacture of degarelix and its intermediates |
Family Cites Families (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| DE2862341D1 (en) | 1977-12-26 | 1983-11-24 | Ihara Chemical Ind Co | Process for producing aromatic monocarboxylic acid |
| DE69214818T2 (en) | 1991-04-25 | 1997-02-27 | Romano S.-Cergue Deghenghi | LHRH antagonists |
| EP0556034B2 (en) | 1992-02-12 | 2004-07-21 | Daikyo Gomu Seiko Ltd. | A medical instrument |
| SI9300468A (en) | 1992-10-14 | 1994-06-30 | Hoffmann La Roche | Injectable composition for the sustained release of biologically active compounds |
| US6828415B2 (en) | 1993-02-19 | 2004-12-07 | Zentaris Gmbh | Oligopeptide lyophilisate, their preparation and use |
| US5506207A (en) | 1994-03-18 | 1996-04-09 | The Salk Institute For Biological Studies | GNRH antagonists XIII |
| US5595760A (en) | 1994-09-02 | 1997-01-21 | Delab | Sustained release of peptides from pharmaceutical compositions |
| US5710246A (en) | 1996-03-19 | 1998-01-20 | Abbott Laboratories | Process for intermediates for the synthesis of LHRH antagonists |
| US5860957A (en) | 1997-02-07 | 1999-01-19 | Sarcos, Inc. | Multipathway electronically-controlled drug delivery system |
| US5821230A (en) | 1997-04-11 | 1998-10-13 | Ferring Bv | GnRH antagonist decapeptides |
| US5977302A (en) | 1997-11-20 | 1999-11-02 | Ortho-Mcneil Pharmaceutical, Inc. | Liquid phase process for the preparation of GnRH peptides |
| FR2776520B1 (en) | 1998-03-25 | 2000-05-05 | Sod Conseils Rech Applic | NOVEL PHARMACEUTICAL COMPOSITIONS FOR THE SUSTAINED RELEASE OF PEPTIDES AND THEIR PREPARATION PROCESS |
| CN1243543C (en) * | 1998-07-20 | 2006-03-01 | 派普泰克有限公司 | Bioimplant Formulation |
| AU2002213441B2 (en) | 2000-10-12 | 2006-10-26 | Genentech, Inc. | Reduced-viscosity concentrated protein formulations |
| US20020103131A1 (en) | 2001-01-26 | 2002-08-01 | Jacobson Jill D. | Prevention of diabetes by administration of GnRH antagonists |
| GB0117057D0 (en) | 2001-07-12 | 2001-09-05 | Ferring Bv | Pharmaceutical composition |
| US7214662B2 (en) | 2001-11-27 | 2007-05-08 | Zentaris Gmbh | Injectable solution of an LHRH antagonist |
| SE0104463D0 (en) | 2001-12-29 | 2001-12-29 | Carlbiotech Ltd As | Intermediates for Synthesis of LHRH Antagonists, Methods of Preparation and Methods for Preparation of LHRH Antagonists |
| CN1411803A (en) | 2002-08-29 | 2003-04-23 | 四川大学 | Method for preparing precursor liposome and its device |
| BRPI0314546B8 (en) * | 2002-09-27 | 2021-05-25 | Zentaris Gmbh | pharmaceutical gel composition comprising pharmaceutically active peptides with sustained release, method for producing the same kit. |
| AR042815A1 (en) | 2002-12-26 | 2005-07-06 | Alza Corp | ACTIVE AGENT SUPPLY DEVICE THAT HAS COMPOUND MEMBERS |
| US8236292B2 (en) * | 2004-06-04 | 2012-08-07 | Camurus Ab | Liquid depot formulations |
| EP1674082A1 (en) * | 2004-12-22 | 2006-06-28 | Zentaris GmbH | Process for the manufacture of sterile suspensions or lyophilisates of low-soluble basic peptide complexes, pharmaceutical formulations comprising these complexes and their use as medicament |
| WO2006069779A1 (en) | 2004-12-30 | 2006-07-06 | F. Hoffmann-La Roche Ag | Preparing of peptides with excellent solubility |
| WO2007130809A2 (en) | 2006-05-06 | 2007-11-15 | Volodymyr Brodskyy | An automatic injectable drug mixing device |
| EP1891964A1 (en) | 2006-08-08 | 2008-02-27 | AEterna Zentaris GmbH | Application of initial doses of LHRH analogues and maintenance doses of LHRH antagonists for the treatment of hormone-dependent cancers and corresponding pharmaceutical kits |
| EP1967202A1 (en) | 2007-03-05 | 2008-09-10 | AEterna Zentaris GmbH | Use of LHRH Antagonists for the Treatment of Lower Urinary Tract Symptoms, in particular Overactive Bladder and/or Detrusor Overactivity |
| IL182922A0 (en) | 2007-05-02 | 2007-09-20 | Medimop Medical Projects Ltd | Automatic liquid drug reconstitution apparatus |
| CA2711984A1 (en) | 2008-01-15 | 2009-07-23 | Abbott Gmbh & Co. Kg | Powdered protein compositions and methods of making same |
| ES2589322T3 (en) | 2008-02-11 | 2016-11-11 | Safety Syringes, Inc. | Syringe with needle and clip safety guard to prevent the release of the protector during a reconstitution process |
| TWI539959B (en) | 2008-02-11 | 2016-07-01 | 菲瑞茵國際中心股份有限公司 | Method for treating prostate cancer during metastasis |
| RU2517135C2 (en) * | 2008-05-07 | 2014-05-27 | Меррион Рисерч Iii Лимитед | Peptide compositions and methods for production thereof |
| WO2010121835A1 (en) | 2009-04-24 | 2010-10-28 | Polypeptide Laboratories A/S | Method for the manufacture of degarelix |
| MX2011011431A (en) | 2009-05-01 | 2012-02-23 | Ferring Bv | Composition for the treatment of prostate cancer. |
| TW201043221A (en) | 2009-05-06 | 2010-12-16 | Ferring Int Ct Sa | Kit and method for preparation of a Degarelix solution |
| US20110039787A1 (en) * | 2009-07-06 | 2011-02-17 | Ferring International Center S.A. | Compositions, kits and methods for treating benign prostate hyperplasia |
| US20110066386A1 (en) * | 2009-09-16 | 2011-03-17 | Chien-Chong Hong | Anesthetic sensing optical microfluidic chip system |
| ES2385240B1 (en) * | 2010-07-26 | 2013-09-23 | Gp-Pharm, S.A. | CAPSULES OF ACTIVE PHARMACEUTICAL PRINCIPLES AND POLYINSATURATED FATTY ACIDS FOR THE TREATMENT OF PROSTATE DISEASES. |
| JO3755B1 (en) | 2011-01-26 | 2021-01-31 | Ferring Bv | Testosterone formulations |
| CN102204889B (en) * | 2011-05-23 | 2013-09-18 | 蚌埠丰原涂山制药有限公司 | Degarelix acetate lyophilized powder injection and preparation method thereof |
| KR20140063602A (en) | 2011-07-15 | 2014-05-27 | 훼링 비.브이. | Method for timing a colonoscopy wherein a picosulate composition is administered |
| CN102329373B (en) | 2011-09-29 | 2014-10-22 | 深圳翰宇药业股份有限公司 | Solid-phase synthetic process for degarelix |
| PL4512390T1 (en) | 2012-06-01 | 2025-09-01 | Ferring B.V. | Manufacture of degarelix |
-
2013
- 2013-05-31 PL PL25151068.1T patent/PL4512390T1/en unknown
- 2013-05-31 KR KR1020147035636A patent/KR102140982B1/en active Active
- 2013-05-31 FI FIEP13726219.2T patent/FI2854831T3/en active
- 2013-05-31 EP EP13726219.2A patent/EP2854831B1/en active Active
- 2013-05-31 CN CN201710709810.8A patent/CN107569456A/en active Pending
- 2013-05-31 ES ES25151066T patent/ES3037194T1/en active Pending
- 2013-05-31 US US14/403,775 patent/US9592266B2/en active Active
- 2013-05-31 FI FIEP25151068.1T patent/FI4512390T1/en unknown
- 2013-05-31 EP EP25151066.5A patent/EP4512389B1/en active Active
- 2013-05-31 CA CA3228586A patent/CA3228586A1/en active Pending
- 2013-05-31 SI SI201332090T patent/SI2854831T1/en unknown
- 2013-05-31 MY MYPI2014703537A patent/MY182320A/en unknown
- 2013-05-31 EP EP25151068.1A patent/EP4512390B1/en active Active
- 2013-05-31 RU RU2014146272A patent/RU2657444C2/en active
- 2013-05-31 AU AU2013269523A patent/AU2013269523B2/en active Active
- 2013-05-31 WO PCT/EP2013/061264 patent/WO2013178788A2/en not_active Ceased
- 2013-05-31 TW TW102119333A patent/TWI580443B/en active
- 2013-05-31 JP JP2015514527A patent/JP6226966B2/en active Active
- 2013-05-31 RS RS20241121A patent/RS66065B1/en unknown
- 2013-05-31 DE DE25151068.1T patent/DE25151068T1/en active Pending
- 2013-05-31 LT LTEPPCT/EP2013/061264T patent/LT2854831T/en unknown
- 2013-05-31 PL PL13726219.2T patent/PL2854831T3/en unknown
- 2013-05-31 CA CA2874927A patent/CA2874927A1/en active Pending
- 2013-05-31 DK DK13726219.2T patent/DK2854831T3/en active
- 2013-05-31 AR ARP130101926A patent/AR092840A1/en not_active Application Discontinuation
- 2013-05-31 HU HUE13726219A patent/HUE068579T2/en unknown
- 2013-05-31 PL PL25151066.5T patent/PL4512389T1/en unknown
- 2013-05-31 NZ NZ701978A patent/NZ701978A/en unknown
- 2013-05-31 EP EP24172290.9A patent/EP4385517A3/en active Pending
- 2013-05-31 FI FIEP25151066.5T patent/FI4512389T1/en unknown
- 2013-05-31 CN CN201380028448.8A patent/CN104334182B/en active Active
- 2013-05-31 HR HRP20241284TT patent/HRP20241284T1/en unknown
- 2013-05-31 SG SG11201407679PA patent/SG11201407679PA/en unknown
- 2013-05-31 ES ES13726219T patent/ES2988481T3/en active Active
- 2013-05-31 NZ NZ727679A patent/NZ727679A/en unknown
- 2013-05-31 ES ES25151068T patent/ES3037257T1/en active Pending
- 2013-05-31 HR HRP20260331TT patent/HRP20260331T1/en unknown
- 2013-05-31 MX MX2014014662A patent/MX364010B/en active IP Right Grant
- 2013-06-02 JO JOP/2013/0167A patent/JO3586B1/en active
-
2014
- 2014-11-23 IL IL235856A patent/IL235856B/en active IP Right Grant
- 2014-12-01 PH PH12014502682A patent/PH12014502682B1/en unknown
-
2017
- 2017-01-31 US US15/420,156 patent/US10172906B2/en active Active
-
2018
- 2018-12-19 US US16/224,843 patent/US10765721B2/en active Active
-
2020
- 2020-07-30 US US16/947,382 patent/US11260102B2/en active Active
-
2021
- 2021-01-21 US US17/154,690 patent/US11510962B2/en active Active
- 2021-09-27 AR ARP210102681A patent/AR123619A2/en not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998046634A1 (en) | 1997-04-11 | 1998-10-22 | Ferring B.V. | GnRH ANTAGONISTS BEING MODIFIED IN POSITIONS 5 AND 6 |
| WO2010012835A1 (en) | 2008-07-31 | 2010-02-04 | Andrea Peruffo | Rollerski or skate with braking system and method for braking said sports item |
| WO2011066386A1 (en) | 2009-11-25 | 2011-06-03 | Novetide, Ltd. | Process for production of degarelix |
| WO2012055903A1 (en) | 2010-10-27 | 2012-05-03 | Ferring B.V. | Process for the manufacture of degarelix and its intermediates |
| WO2012055905A1 (en) | 2010-10-27 | 2012-05-03 | Ferring B.V. | Process for the manufacture of degarelix and its intermediates |
Non-Patent Citations (5)
| Title |
|---|
| DOEHN ET AL., DRUGS, vol. 9, no. 8, 2006, pages 565 - 571 |
| JAMES, E.F. ET AL., DRUGS, vol. 69, no. 14, 2009 |
| JIANG ET AL., J. MED. CHEM., vol. 44, 2001, pages 453 - 467 |
| VAN POPPEL ET AL., UROLOGY, vol. 71, no. 6, 2008, pages 1001 - 1006 |
| VAN POPPEL, CANCER MANAGEMENT AND RESEARCH, vol. 2, 2010, pages 39 - 52 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2854831B1 (en) | 2012-06-01 | 2024-07-10 | Ferring B.V. | Manufacture of degarelix |
| CN112125956A (en) * | 2019-06-25 | 2020-12-25 | 深圳市健元医药科技有限公司 | A kind of preparation method of degarelix |
| WO2020259714A1 (en) * | 2019-06-25 | 2020-12-30 | 深圳市健元医药科技有限公司 | Method for preparing degarelix |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11510962B2 (en) | Manufacture of degarelix | |
| JP7177859B2 (en) | Freeze-drying method and resulting teverellix-TFA lyophilisate | |
| CN102688202B (en) | Temozolomide freeze-dried preparation | |
| HK40111407A (en) | Degarelix drug product | |
| HK40118404A (en) | Manufacture of degarelix | |
| HK1209046B (en) | Manufacture of degarelix | |
| EP2106788A1 (en) | Liquid and freeze dried formulations | |
| BR112014029495B1 (en) | ACTIVE PRINCIPLE OF FREEZE DRIED DEGARELIX AND ITS PRODUCTION METHODS, METHOD FOR PRODUCING A DEGARELIX MEDICINE, AND METHOD FOR CONTROLLING THE VISCOSITY OF A FREEZE DRIED DEGARELIX PRODUCT |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13726219 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 235856 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14403775 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2014000695 Country of ref document: DZ |
|
| ENP | Entry into the national phase |
Ref document number: 2874927 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P1313/2014 Country of ref document: AE |
|
| ENP | Entry into the national phase |
Ref document number: 2015514527 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/014662 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 20147035636 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013726219 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014146272 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2013269523 Country of ref document: AU Date of ref document: 20130531 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014029495 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014029495 Country of ref document: BR Kind code of ref document: A2 Effective date: 20141126 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2024/1121 Country of ref document: RS |
|
| WWG | Wipo information: grant in national office |
Ref document number: P-2024/1121 Country of ref document: RS |