WO2014021329A1 - カワハギ由来の細胞株 - Google Patents
カワハギ由来の細胞株 Download PDFInfo
- Publication number
- WO2014021329A1 WO2014021329A1 PCT/JP2013/070627 JP2013070627W WO2014021329A1 WO 2014021329 A1 WO2014021329 A1 WO 2014021329A1 JP 2013070627 W JP2013070627 W JP 2013070627W WO 2014021329 A1 WO2014021329 A1 WO 2014021329A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- cell line
- fish
- present
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
- C12N2510/04—Immortalised cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2517/00—Cells related to new breeds of animals
- C12N2517/02—Cells from transgenic animals
Definitions
- the present invention relates to a cell line derived from a living part of Monacanthidae fish, which exhibits a fibroblast-like morphology, or a substrain thereof.
- Fish cells derived from fish have many properties similar to animal cells derived from mammals.
- fish cells can generally be cultured at a relatively low temperature, and the temperature range in which they can grow is wide. Therefore, fish cells are regarded as promising as cultured cells in industrial applications and research applications such as production of biological substances as an alternative to animal cells.
- Known cultured cells of fish include cell lines derived from sturgeon eyeball epithelial cells and cell lines derived from scorpion tailfish (see, for example, Patent Documents 1 and 2 below, which are described here) Incorporated herein by reference). These established cell lines are expected not only to replace animal cells but also to elucidate the ecology and culture of sturgeons and scorpions, respectively.
- the fishes of Monacanthidae including frogfish (Stephanolepis cirrhifer), are covered with strong skin throughout the body, and the entire skin can be peeled off at the time of cooking.
- frogfish Stephanolepis cirrhifer
- the riverfish is widely preferred as a raw or cooking ingredient.
- fish cell lines may contribute to diagnosis of viral infections, production of vaccines effective against viral infections, cytotoxicity tests, and the like. Therefore, the present inventors thought that if there was an immortalized cell line of eelfish, it would be possible to find a clue to solve the problem of moribundity of cultivated cultivated fish, and that significant progress could be made in the development of cultivated eelfish.
- fish cell lines can be cultured at low temperatures, but have a low growth rate compared to mammalian cell lines. Therefore, an immortalized cellfish cell line that can be cultured at a low temperature and has a growth rate comparable to that of a mammalian cell line can be cultured easily and quickly. We can expect rapid progress in development.
- pluripotent stem cells of fish not only kingfishes, tissue cells that were previously difficult to collect can be obtained by inducing differentiation into specific cells, and the pathogenesis of fish diseases And study the onset mechanism.
- cells that have developed a specific disease after differentiation induction are used, it is possible to evaluate the effect and toxicity of the drug against the disease. Therefore, fish pluripotent stem cells can also be expected to contribute to the development of fish culture techniques.
- an immortalized cell line or a pluripotent stem cell may significantly advance the research and development of the technique for cultivating the river moth.
- established cell lines and pluripotent stem cells are not known.
- a first object of the present invention is to provide an immortalized cell line derived from a river snail.
- the second object of the present invention is to provide a pluripotent stem cell derived from a river moth.
- the third object of the present invention is to provide a method for producing and utilizing a cell line derived from brookfish.
- the inventors examined the establishment of fish cells, focusing on the fact that fish cells can be cultured at low temperatures. Therefore, we tried to grow various tissue cells using fish from various fish such as Thailand, horse mackerel and shark. However, in addition to obtaining cells with a high growth rate, most of them could not be subcultured. Therefore, the present inventors changed the type of fish and the subculture method as appropriate, and as a result of further earnest research, they have produced an immortalized cell line that can be subcultured from the cormorant's dorsal fin. Successful. Moreover, surprisingly, the obtained cell line can be cultured at a growth rate comparable to or higher than that of a mammalian cell line even when cultured at a temperature of 25 ° C.
- the cell line derived from the river moth obtained by the present inventors expresses a part of cell markers exhibiting pluripotency, and further changes in various forms by changing the culture conditions. It changed to cells.
- the present invention has been completed based on these successful examples and findings.
- the present invention preferably, it further has the following property (4).
- the maximum value is 66, the minimum value is 32, and the mode has 33 chromosomes according to the frequency distribution
- the frequency of the 33 chromosomes as a mode is about 90% of the total frequency in the frequency distribution.
- the present invention preferably, it further has the following property (5).
- the initial cell number is about 1.0 ⁇ 10 6 cells / ml, and Leibovitz's L-15 medium containing 10% FBS is used in a culture vessel having a bottom area of 75 cm 2 , and CO 2 is absent.
- the doubling time when cultured at 25 ° C. is about 14 to 28 hours
- a fish of the river coral fish that is positive for at least one cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3.
- a cell line derived from a biological site or a passage thereof is provided.
- the cell line or its passage is a cell line exhibiting a fibroblast-like morphology or its passage.
- the cell line or its subculture line is a myocyte and a myocyte-like cell, an epithelial cell and an epithelial cell-like cell, a nerve cell and a neuron-like cell, an adipocyte and an adipocyte-like cell, an immunity It has the ability to differentiate into at least one cell selected from the group consisting of cells and immune cell-like cells and liver cells and liver cell-like cells.
- the fishes of the genus Sphaerophyceae are Stephanolepis, Thamnaconus, Cantherhines, Aluterus and Rudus rus. It is a fish selected from a group.
- the fishes of the family Saccharidae are smeltfish (Stephanolepis cirrhifer).
- the biological part is a heel located on the back, chest, abdomen, buttocks or tail.
- a method for producing the cell line of the present invention or a subculture thereof comprising a step of subjecting a cell isolated from a living part of a river fish species to subculture for 70 times or more. Provided.
- a transformant obtained by introducing a foreign gene into the cell line of the present invention or its passage.
- a method for producing a transformant comprising a step of obtaining a transformant by introducing a foreign gene into the cell line of the present invention or its passage.
- an expression product of a foreign gene comprising a step of obtaining an expression product of the foreign gene from a transformant obtained by introducing the foreign gene into the cell line of the present invention or its passage.
- a method of manufacturing is provided.
- a kit for producing a transformant comprising the cell line of the present invention or its passage, a vector, and equipment for transfection.
- a kit for producing differentiated cells comprising the cell line of the present invention or its passage, a culture medium, and a culture vessel.
- serum is further included.
- the serum is at least one serum selected from the group consisting of mammalian serum, fish serum and serum replacement.
- the medium is at least one medium selected from the group consisting of a medium for mammalian cells, a medium for insect cells, and a medium for fish cells.
- the culture vessel is at least one type of culture vessel selected from the group consisting of a culture vessel whose bottom surface is coated with cell adhesion molecules and a culture vessel whose bottom surface is not coated with cell adhesion molecules. .
- the cell line of the present invention or a passage thereof is cultured in a culture vessel whose bottom surface is coated with a cell adhesion molecule or a culture vessel whose bottom surface is not coated with a cell adhesion molecule.
- a method for producing differentiated cells comprising a step of culturing using a medium for mammalian cells, a medium for insect cells, or a medium for fish cells that does not contain serum or contains mammalian serum, fish serum or serum substitutes Is provided.
- a cultured cell sheet comprising the cell line of the present invention or its passage.
- cells that are distinguished from the cell line of the present invention or its passage by staining for alkaline phosphatase activity and are positive for NANOG, OCT4, TRA-1-60 and SSEA-3 Alternatively, a method for producing a giant rod-shaped colony comprising a step of producing a giant rod-shaped colony formed by the cells is provided.
- the cell line of the present invention or its subculture includes a cell line that can be cultured at a growth rate comparable to that of a mammalian cell line. Since the cell line of the present invention or its passages can be cultured at room temperature, it can be used for research use, industrial production use, etc. in place of mammalian cell lines. Can be expected.
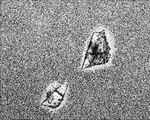
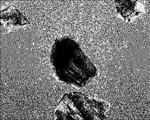
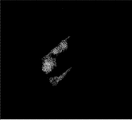
- FIG. 1A is a diagram showing cells migrating from the dorsal fin piece of a river snail.
- FIG. 1B is a diagram showing cells migrating from the tail of a hawkfish.
- FIG. 1C is a diagram showing cells migrating from the buttock piece of a river snail.
- FIG. 1D is a view showing cells migrating from the flank of a riverfish.
- FIG. 2 shows the primary cell (at the time of primary culture), the first passage cell (passage 1), the tenth passage cell (passage 10), the 20th passage cell (passage 20) and the 30th passage cell. It is the figure which showed the cell viability when each cell of (passage 30) was frozen and thawed.
- FIG. 1A is a diagram showing cells migrating from the dorsal fin piece of a river snail.
- FIG. 1B is a diagram showing cells migrating from the tail of a hawkfish.
- FIG. 1C is a diagram showing cells migrating
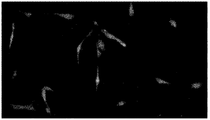
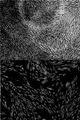
- FIG. 3A is a view of the morphology of KSC cells observed using an optical microscope at a magnification of x460.
- FIG. 3B is a view of the morphology of KSC cells forming two layers, observed using an optical microscope at a magnification of x460.
- FIG. 3C is a view obtained by observing the morphology of KSC cells in which multiple layers were formed using an optical microscope at a magnification of x460.
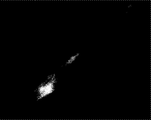
- FIG. 4 shows cells expressing human-AP.
- FIG. 5 shows the activity of human-AP expression products.
- FIG. 6 is a diagram showing expression by a fusion protein of PON and Halotag.
- FIG. 7A is a view observing cells expressing the fluorescent fusion proteins cyan fluorescent protein-yellow fluorescent protein and calmodulin.
- FIG. 7B is a view observing cells expressing a fluorescent fusion protein, red fluorescein protein and vesicular stomatitis virus G-glycoprotein.
- FIG. 7C is a diagram observing cells in which green fluorescent protein, which is a fluorescent protein, is expressed.
- FIG. 8 is a diagram in which the cytotoxicity after each cell was transfected by the lipofectamine method was verified.
- FIG. 9A is a diagram showing that KSC cells are TRA-1-60 positive cells, which are cell markers unique to pluripotent stem cells. The upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9A is a diagram showing that KSC cells are TRA-1-60 positive cells, which are cell markers unique to pluripotent stem cells. The upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9B is a diagram showing that KSC cells are OCT4-positive cells, which are cell markers unique to pluripotent stem cells.
- the upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9C is a diagram showing that KSC cells are SSEA-3 positive cells, which are cell markers unique to pluripotent stem cells.
- the upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
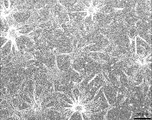
- FIG. 10A shows that KSC cells are fibroblast-like.
- FIG. 10B shows that KSC cells differentiated into myocyte-like cells.
- FIG. 10C shows that KSC cells have differentiated into epithelial cell-like cells.
- FIG. 10D shows that KSC cells have differentiated into neuron-like cells.
- FIG. 10A shows that KSC cells are fibroblast-like.
- FIG. 10B shows that KSC cells differentiated into myocyte-like cells.
- FIG. 10C shows that KSC cells have differentiated
- FIG. 10E shows that KSC cells have differentiated into liver-like cells.
- FIG. 10F shows that KSC cells differentiated into fibrocyte-like cells.
- FIG. 10G shows that KSC cells have differentiated into adipose-like cells.
- FIG. 10H is a diagram showing that KSC cells have differentiated into rose branch cells that may be immune cells.
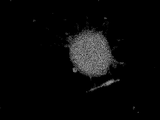
- FIG. 10I shows that KSC cells have differentiated into giant rod-shaped cells.
- FIG. 11A is a diagram showing that the giant cocoon-shaped fused cells derived from KSC cells are positive for the activity of alkaline phosphatase, a cell marker unique to undifferentiated cells.
- FIG. 11A is a diagram showing that the giant cocoon-shaped fused cells derived from KSC cells are positive for the activity of alkaline phosphatase, a cell marker unique to undifferentiated cells.
- FIG. 11B is a diagram showing that the giant rod-shaped fusion cells derived from KSC cells are NANOG positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11C shows that the giant rod-shaped fusion cells derived from KSC cells are TRA-1-60 positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11D shows that the giant rod-shaped fused cells derived from KSC cells are OCT4-positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11E shows that the giant rod-shaped fused cells derived from KSC cells are SSEA-3-positive cells, which are cell markers peculiar to undifferentiated cells.
- FIG. 12 is a view showing a cell sheet obtained by culturing KSC cells.
- the first cell line of the present invention or its passage strain is derived from a living part of a fish of the family Monacanthidae.
- the term “cell line” is understood in the broadest sense by those skilled in the art, and is also referred to as a cell line, an established cell line or the like, and is particularly a cell line that can be continuously subcultured. It is not limited.
- the “passage strain” means all cells obtained by subculturing the cell line.
- a cell line for convenience, a cell that can be evaluated as a cell line is called a cell line, and a cell line obtained after subculture is used. It is called a passage stock.
- the cell line and the passage line are collectively referred to simply as “cell line”.
- the first cell line of the present invention can be produced on the basis of cells isolated from a biological site such as a tissue or an organ excised from a fish of the family Saccharidae.
- the fish species are fishes that have the feature that the skin (outer skin) covering the entire body can be easily peeled off. For example, if a part of the skin is cut with a knife or the like, the skin can be easily peeled off by hand without using a special instrument.
- the organism from which the first cell line of the present invention is derived is not particularly limited as long as it is a riverfish having the characteristics described above. Examples include, ) Fish, Aluterus fish and Rudarius fish.
- suitable specific examples of the otterfish can include kingfish (Stephanolepis cirrhifer), horse mackerel (Thamnoconus modestus), and euph olgi, and more preferred is terfoli. is there.
- the living body part of the fish species which is used for producing the first cell line of the present invention, does not indicate only a specific part of the fish body, but includes two or more parts of tissues or organs in the fish body. Including the whole fish body.
- a suitable living body part in the smeltfish is a part corresponding to a heel, for example, a heel on the back, chest, abdomen, buttocks or tail is preferable, and a ridge on the back is more preferable .
- the method for producing the first cell line of the present invention from the living part of the fish of the family Saccharidae is not particularly limited.
- a specific example of the step of obtaining primary cultured cells comprises, for example, the following procedure. That is, the buttocks of the snapper are cut out as square pieces of an appropriate size without peeling off the skin. The excised square pieces are washed in an appropriate manner and then treated with various concentrations of antibiotics in one to several times. The treated section is aseptically minced into small pieces in the presence of a proteolytic enzyme solution such as trypsin. The operation so far is carried out at a low temperature, preferably under ice cooling. Subsequently, the micro piece immersed in the trypsin solution is left still at room temperature.
- a proteolytic enzyme solution such as trypsin
- the fine pieces after standing are collected by centrifugation or the like, and the collected fine pieces are repeatedly washed several times using a medium suitable for cell growth in a non-CO 2 environment such as Leibovitz's L-15 medium.
- a medium suitable for cell growth in a non-CO 2 environment such as Leibovitz's L-15 medium.
- a standard mammalian cell culture medium such as RPMI 1640 medium or IMDM medium (both Life Technologies) can be used.
- the cells then cover about 80% or more of the bottom surface of the culture container under appropriate culture conditions such as seeding the collagen-coated culture container containing the serum-containing medium and placing it on the bottom of the culture container. Incubate until The cells collected in the minute pieces at this time are used as primary cultured cells.
- the culture medium in the culture vessel is removed, and the cultured cells adhering to the bottom surface of the vessel are washed by an appropriate method.
- the cells seeded in a new culture container are used as the first subculture cells.
- a specific example of the step of subculturing primary cultured cells to obtain the first cell line of the present invention comprises, for example, the following procedure. That is, the subculture for obtaining the first cell line of the present invention from the primary cultured cells is achieved under appropriate culture conditions using a serum-containing medium, for example, in the same manner as the step of obtaining the primary cultured cells.
- the number of cells at the start of the culture is 4 ⁇ 10 5 cells / ml
- the standard at the end of the culture is when the confluency is 90% or more
- the medium is replaced when the culture medium becomes dirty with dead cells
- these are not particularly limited.
- the first cell line of the present invention can be stored and grown by a conventional method known by those skilled in the art, and the method is not particularly limited.
- the method for cryopreserving the first cell line of the present invention includes, for example, adding a commercially available medium to the cell line, collecting the cell by centrifugation, and then collecting the cell into a commercially available serum-free cultured cell. It can be achieved by adding a cell cryopreservation solution for use and cryopreserving.
- the frozen cell line of the present invention can be thawed by placing it in a half-thawed state at room temperature and replacing it with a fresh serum-free medium an appropriate number of times.
- the above subculture can be performed by adding a serum-containing medium to the thawed cells.
- the method for determining that the first cell line of the present invention has been produced is not particularly limited, but primary cultured cells are used 30 times or more, preferably 50 times or more, more preferably 70 times or more, and further preferably. Can be determined to be the first cell line of the present invention by subjecting to subculture for 100 times or more.
- the first cell line of the present invention is not particularly limited as long as it is a cell line that can be continuously subcultured, but is preferably one that can be subcultured substantially without limitation. That is, one aspect of the first cell line of the present invention is one that can be repeatedly cultured for many generations if cultured according to the above-described subculture method with appropriate attention and technical skills possessed by those skilled in the art. It is.
- the first cell line of the present invention may have various properties, but preferably has a fibroblast-like morphology. Fibroblasts have a distinctive morphology such as a flat and long profile, often irregular protrusions, and an elliptical nucleus. When the first cell line of the present invention exhibits a fibroblast-like morphology, the first cell line of the present invention is particularly limited as long as it has any form characteristic to fibroblasts. However, it is preferably at least a flat and long outer shape. The fact that the first cell line of the present invention exhibits a fibroblast-like morphology can be confirmed by various methods for observing the morphology of cells known by those skilled in the art. This can be confirmed by observing the first cell line of the present invention under a microscope.
- the number of divisions is 30 times or more, preferably 50 times or more, more preferably 70 times or more, More preferably, it can be 100 times or more.
- the first cell line of the present invention can be regarded as an immortalized cell. Whether or not the first cell line of the present invention can be subcultured substantially without limitation is determined by, for example, subjecting the cell line of the present invention to the subculture described above, It can be determined by confirming that the number of times is reached.
- the doubling time refers to the time for the cell number to double, as is known by those skilled in the art.
- the doubling time is in principle consistent with the cell cycle for cells in the growth phase.
- the method for measuring the doubling time of the first cell line of the present invention is not particularly limited. For example, when subjected to the above-mentioned subculture, that is, using Leibovitz's L-15 medium containing 10% FBS. When cultured at 25 ° C., it can be determined from the number of cells measured by selecting 2 to several points, preferably 3 points or more, for cells in the growth phase.
- the doubling time of the first cell line of the present invention is not particularly limited.
- the initial cell number is about 1.0 ⁇ 10 6 cells / ml, preferably 0.1 to 1.5 ⁇ 10 6 cells / ml, More preferably 0.4 to 1.0 ⁇ 10 6 cells / ml using Leibovitz's L-15 medium containing 10% FBS in a culture vessel having a bottom area of 75 cm 2 in the absence of CO 2
- When cultured at 25 ° C. at a temperature shorter than the primary cultured cells preferably 10 to 48 hours, more preferably 12 to 36 hours, more preferably 14 to 28 hours, still more preferably 16 to 24 hours, particularly preferably.
- the method for calculating the doubling time from the number of cells is not particularly limited.
- the cell is measured by, for example, attaching an index (marker) to the bottom surface of the culture container in advance, and subjecting the first cell line of the present invention to culturing, and then measuring the number of cells in the vicinity of the marker by an inverted routine microscope. It can be measured by photographing and observing.
- an average of the number of cells in a plurality of squares (for example, 1 mm ⁇ 1 mm) attached to the eyepiece may be used as the number of cells.
- a cell growth curve is obtained. From the growth curve, it is possible to know the growth phase of the delayed phase, logarithmic growth phase, stationary phase and death phase in the cell growth process.
- the time required for doubling the number of cells in the logarithmic growth phase (doubling time) can be calculated from the following formula.
- Double time (t ⁇ t 0 ) log 2 / (log N ⁇ log N 0 ) t: time [h], N: number of cells at time t 0 : initial time [h], N 0 : number of cells at time t 0
- the cultured cells When the cultured cells are cultured in a state where they are adhered to the bottom surface of the culture vessel, the cultured cells usually stop growing when they reach a confluent state covering the entire bottom surface. That is, the cultured cells can grow until a single cell layer is formed on the bottom surface of the culture container.
- another cell layer can be formed on the monolayer cell layer by continuing to grow after the monolayer cell layer is formed. Yes, and preferably a culture in which a multilayer structure is formed.
- the first cell line of the present invention includes those capable of culturing in a three-dimensional structure in which cell layers overlap.
- the fact that the first cell line of the present invention can form a multilayer structure indicates that the culture is further performed after the first cell line of the present invention becomes confluent and cultured by a method known by those skilled in the art. This can be confirmed by observing the cell structure.
- the multilayer structure is not particularly limited as long as another cell layer is formed on a single cell layer, and is a layer structure of two layers or three or more layers.
- the number of chromosomes of the first cell line of the present invention is not particularly limited because it varies depending on the species and male and female of the fish species that are origin organisms. For example, when the number is obtained from the female dorsal fin, The number of chromosomes can be 66, 33 and 32.
- the first cell line of the present invention may be regarded not only as one independent cell but also as a group of cell populations. In that case, when the first cell line of the present invention is obtained from the female dorsal fin of a river moth, it may be a population of cell lines having 66, 33 or 32 chromosomes.
- the first cell line of the present invention preferably has a frequency distribution in which the maximum value is 66 cells, the minimum value is 32 cells, and the mode (mode) is 33 cells. More preferably, it is a population of cell lines having the number of chromosomes, and in the frequency distribution, it is a population of cell lines in which the frequency of 33 chromosomes which are modes is about 90% of the total frequency.
- a method for analyzing the number of chromosomes of the first cell line of the present invention a method for analyzing the number of chromosomes of cells known by those skilled in the art can be adopted without any particular limitation. Can be analyzed by the method described in.
- a specific example of the first cell line of the present invention is a cell line derived from a living part of a cormorant fish, and has one or more of the following properties (1) to (5):
- it has the following property (1) and has at least one of the following properties (2) to (5), more preferably the following properties (1) and (2):
- the second cell line of the present invention is at least one selected from the group consisting of TRA-1-60, OCT4 and SSEA-3. It is a cell line derived from a living part of a cormorant fish that has a positive cell marker or its passage.
- the second cell line of the present invention can be produced from a living part of a fish species of the fish species, by the same method as the first cell line of the present invention.
- the second cell line of the present invention preferably exhibits a fibroblast-like morphology.
- a cell line that is positive for TRA-1-60, OCT4, or SSEA-3 refers to a cell line that expresses an epitope recognized by an antibody against human TRA-1-60, human OCT4, or human SSEA-3, respectively.
- Antibodies to TRA-1-60 react with proteins (epitope) expressed in undifferentiated human embryonic stem (ES) cells, embryonic cancer (EC) cells and embryonic germ (EG) cells, the epitopes being cells It is known to be lost by differentiation.
- OCT4 is a transcription factor highly expressed in ES cells, and is said to be involved in maintaining the undifferentiation of ES cells and maintaining pluripotency.
- SSEA-3 is known as a cell marker for pluripotent stem cells such as ES cells.
- the gene sequences encoding TRA-1-60, OCT4, and SSEA-3 are, for example, ACC_SION No.
- NM_001018111 or NM_001018111 or TRA_ 1-60 as ACCESSION numbers, respectively, by GenBank, a public database of National Center for Biotechnology Information (NCBI).
- GenBank a public database of National Center for Biotechnology Information
- NM_005397, OCT4 are registered as NM_002701
- SSEA-3 is registered as NM_033149 (ACCESSION number of the amino acid sequence is Q9JI67).
- TRA-1-60, OCT4 and SSEA-3 are cell markers exhibiting pluripotency, and are common in that they are not found on the cell surface after differentiation. From these, TRA-1-60, OCT4, SSEA-3, or cell lines that are positive for two or three of these cell markers have pluripotency, the ability to differentiate into various cells. Can be inferred. Therefore, the second cell line of the present invention is highly likely to be a pluripotent cell line.
- pluripotency is not limitedly interpreted, and in addition to the meaning of pluripotency and multipotency as an academic term, totipotency It may be interpreted as having a meaning, having pluripotency or totipotency, or having a probability, and it is interpreted in the broadest sense regardless of this.
- a primary antibody reaction is carried out by adding a primary antibody against TRA-1-60, OCT4 or SSEA-3.
- a secondary antibody against the fluorescently labeled primary antibody is added to carry out a secondary antibody reaction.
- the cells labeled with TRA-1-60 positive, OCT4 positive or SSEA-3 positive can be confirmed by observing the fluorescently labeled cells.
- the primary antibody structure globulin type, fragmented structure, etc.
- the primary antibody recognition specificity of the secondary antibody the wavelength of the fluorescent label, etc. -1-60, OCT4 and SSEA-3 can be confirmed to be positive, but the cells are divided into three populations and TRA-1-60, OCT4 or SSEA-3 are positive for each population It is preferable to confirm.
- the second cell line of the present invention may be capable of expressing a cell marker that is specifically expressed in pluripotent stem cells other than TRA-1-60, OCT4, and SSEA-3.
- the second cell line of the present invention has a low affinity for human anti-NANOG antibodies, and it can be said that NANOG is weakly positive or substantially negative depending on background conditions and image processing methods.
- one aspect of the second cell line of the present invention is, for example, myocytes and myocyte-like cells, epithelial cells and epithelial cell-like cells, nerve cells and neuron-like cells, adipocytes and adipocyte-like cells, liver A cell line that can differentiate into cells such as cells and liver cell-like cells.
- cell differentiation means that expression of at least one cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3 disappears, and a cell marker peculiar to differentiated cells is expressed.
- cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3
- myocyte-like cells, epithelial cell-like cells, neuronal cell-like cells, adipocyte-like cells, and liver cell-like cells are not completely differentiated into muscle cells, but are in the middle of differentiation or differentiation. It may include cells that exhibit a morphology similar to cells.
- a preferred embodiment of the second cell line of the present invention is a myocyte and myocyte-like cell, epithelial cell and epithelial cell-like cell, nerve cell and neuron-like cell, fat cell and adipocyte-like cell, immune cell and immunity
- a cell line having the ability to differentiate into one, preferably two or more, more preferably three or more, and even more preferably four or more cells selected from the group consisting of cell-like cells and liver cells and liver cell-like cells. is there.
- these differentiated cells are exemplary, and those having the ability to differentiate into other cells are also included in the second cell line of the present invention.
- Differentiation into each cell in the second cell line of the present invention can be achieved by a technique for differentiating pluripotent stem cells known to those skilled in the art. This can also be achieved by variously setting the type and presence, the type of medium, and the like.
- a myocyte or myocyte-like cell, epithelial cell or epithelial cell-like cell, nerve cell or neuron-like cell, liver cell or liver from the second cell line of the invention.
- Cell-like cells, fiber cells or fiber cell-like cells, adipocytes or adipocyte-like cells, immune cells or immune cell-like cells can be obtained.
- the cell shown in FIG. 10B is a myocyte-like cell having an elongated spindle-like or fibrous form and capable of proliferating.
- the cells shown in FIG. 10C are epithelial cell-like cells in which individual cells have a flat shape with an indefinite periphery, and the cells grow densely.
- the cell shown in FIG. 10D is a neuron-like cell that exhibits a cell form having an extensible axon or dendrite and has no proliferative ability.
- the cell shown in FIG. 10E is a liver-cell-like cell that has a form that looks like a lipid droplet in the cytoplasm and that can finitely divide.
- the cell shown in FIG. 10B is a myocyte-like cell having an elongated spindle-like or fibrous form and capable of proliferating.
- the cells shown in FIG. 10C are epithelial cell-like cells in which individual cells have a flat shape with an indefinite periphery, and
- FIG. 10F is a fibrous cell-like cell having a fibrous form comparable to or higher than that of muscle cells and having a proliferation ability.
- the cell shown in FIG. 10G is an adipocyte-like cell having a number of cavities, a form having extensible axons, and no proliferation ability.
- the cell shown in FIG. 10H is a dendritic immune cell-like cell that exhibits a branch-like form of a rose having an extensible tip with a sharp tip, has no proliferation ability, and is linked to a neighboring cell. is there. Further, from the second cell line of the present invention, there is a fused cell that stretches like rubber and fuses cells having proliferative ability to form a giant cocoon-like structure as shown in FIG. 10I. can get.
- This fused cell is positive for alkaline phosphatase activity as shown in FIG. 11A and positive for NANOG as shown in FIG. 11B. Furthermore, the fusion cells are also positive for TRA-1-60, OCT4 and SSEA-3 as shown in FIGS. 11C-E, respectively.
- NANOG is registered as ACCESS — NM_024865, for example, by GenBank, a public database of NCBI.
- Differentiation of the cell line can be confirmed by a change in morphology by comparing with a fibroblast-like morphology at the time of subculture, for example, a morphology as shown in FIG. 10A. It can also be confirmed by a cell determination method. For example, it may be confirmed that a cell line has differentiated using a cell marker of each differentiated cell or a substance produced by each differentiated cell as an index.
- a method for producing each differentiated cell from the second cell line of the present invention is provided as another aspect of the present invention. That is, the method for producing differentiated cells of the present invention comprises the second cell line of the present invention in a culture container whose bottom surface is coated with a cell adhesion molecule or a culture container whose bottom surface is not coated with a cell adhesion molecule. Culturing using a mammalian cell culture medium, insect cell culture medium or fish cell culture medium that does not contain serum or contains mammalian serum, fish serum or serum replacement.
- kits for producing differentiated cells comprising the second cell line of the present invention.
- a kit for producing differentiated cells preferably contains serum, a culture medium, a culture vessel, and the like, and more preferably contains a buffer (buffer), a pH adjuster, and the like. Since the second cell line of the present invention has the possibility of differentiating into cells other than the differentiated cells described above, the serum, medium, culture vessel, buffer and pH adjuster are not particularly limited. Can be used.
- mammalian serum, fish serum and serum substitute can be used, fetal bovine serum (Gibco; No. 26140-087 etc.), horse Serum (Gibco; No. 16050-130, etc.), goat serum (Gibco; No. 16210-064, etc.), rabbit serum (Gibco; No. 16120-099, etc.), mouse (KAC; No. AS3054, etc.), chicken serum (Gibco; No. 16110-082, etc.), Lamb serum (Gibco; No. 16070-096, etc.), swine serum (Gibco; No. 26250-084, etc.), dogs (KAC; No.
- the serum contained in the kit for producing the differentiated cell of the present invention may be one type or two or more types.
- the method for obtaining serum is not particularly limited, and commercially available products may be used.
- a serum substitute can be used as the serum contained in the kit for producing the differentiated cell of the present invention.
- Serum substitutes include, for example, Knockout (registered trademark) Serum Replacement (KSR), lactalbumin hydrolyzate, fish-derived cell culture medium additive Hy-Fish (Maruhachi Muramatsu), Nu-Serum (BD; No. BSE 355100) ), SERUM PLUS (Sigma; No. 14008C-500ML), L-Glutamine solution (Sigma; No. 59202C-100ML), etc., and one or more of these serum substitutes are used. be able to.
- KSR Knockout (registered trademark) Serum Replacement
- lactalbumin hydrolyzate lactalbumin hydrolyzate
- fish-derived cell culture medium additive Hy-Fish Maruhachi Muramatsu
- Nu-Serum BD; No. BSE 355100
- SERUM PLUS Sigma; No. 14008C-500ML
- L-Glutamine solution Sigma; No.
- kits for producing differentiated cells of the present invention examples include L-15 (Gibco; No. 11415-064), AIM V (Life Technologies; No. 087-0112DK), IMDM (Gibco; No. 12440-053), RPMI (Gibco; No. 11835030), EX-CELL 420 with L-glutamine (Nichirei; No. 14420C, for insects), D-MEM (Gibco), mTeSR1 (stem cell; No. ST- 05850, for stem cells), Ham's F-12 (Gibco; No. 11765-054), primate ES / iPS cell culture medium (Reprocell; No. RCHEMD001), one of these or Two or more types of media can be used.
- the kit for producing the differentiated cell of the present invention can contain a growth factor or a medium additive as a constituent component.
- growth factors and medium additives include epithelial cell growth factor, fibroblast growth factor, nerve growth factor, insulin-like growth factor, platelet-derived growth factor, vascular endothelial growth factor, transforming growth factor, cytokine, stem cell Factors, T-STIM culture supplements, IL-3 culture supplements, vascular endothelial cell growth supplements, MITO + Sealam extender, ITS culture additives, and one or more of these media can be used be able to.
- Methods for obtaining growth factors and medium additives are not particularly limited, and commercially available products may be used.
- the bottom surface of the culture container included in the kit for producing differentiated cells of the present invention is coated with cell adhesion molecules such as collagen, fibronectin, laminin, poly-D-lysine, poly-L-lysine, gelatin, and streptavidin.
- the culture vessel is a culture vessel whose bottom surface is not coated with a cell adhesion molecule, or two or more of these culture vessels.
- a method for obtaining a culture vessel whose bottom surface is coated with a cell adhesion molecule is not particularly limited, but a commercially available coated culture vessel may be used, or a non-coated culture vessel and a cell adhesion molecule may be separately prepared and used by those skilled in the art.
- the method for obtaining the cell adhesion molecule is not particularly limited.
- collagen such as collagen I (thermo: 132706), IV (cosmobio: 354534); poly-D-lysine (thermo: 132703); gelatin (CR: 354654) Fibronectin (BD: 356242); poly-L-lysine (thermo); poly-L-ornithine (biocoat); laminin (BD); matrigel biocoat (BD); three-dimensional culture scaffold AteloCell (Koken; CSM-50) or the like can be used.
- the shape and material of the culture vessel are not particularly limited, but a polystyrene flask, a petri dish and a multiwell plate are preferable.
- kits for producing differentiated cells of the present invention includes commercially available media (liquid and solid), serum (liquid and solid), serum substitutes, growth factors, media additives (various amino acids)
- a culture vessel is used, but is not particularly limited.
- a medium prepared with reference to the composition disclosed by the manufacturer may be used.
- a more preferable embodiment of the first cell line and the second cell line of the present invention is a cell line having the characteristics of the first cell line and the second cell line of the present invention.
- a cultured cell sheet comprising these cell lines obtained by culturing the first cell line and / or the second cell line of the present invention.
- the cultured cell sheet of the present invention can be expected to be applied to the treatment of high-grade fish.
- the cultured cell sheet of the present invention can contribute to the reduction of injury lethality of cultured fish.
- the cultured cell sheet of the present invention is expected to be used as a biologically-derived selective membrane to overcome this problem because artificial substances such as synthetic polymers and cellophane limit the selective permeability of substances. it can.
- the cultured cell sheet of the present invention is used in vivo from the viewpoint of use as a scaffold when forming a human cultured cell sheet because individual cells produce collagen and fibronectin, and immunity and antibacterial properties derived from the cells. It can be expected to be used for medical use of material transport capsule materials, adhesive bandages, patches, etc., for use in cancer cell trend tests, for use in immune tests, and as a beauty pack for cosmetics that secrete fish components. .
- the cultured cell sheet of the present invention is peeled off from the bottom during sheet formation and can be cultured in a floating state.
- the cultured cell sheet of the present invention has an advantage that it can be cultured in a floating state.
- the production method of the cell line of the present invention is a method of producing the first cell line of the present invention and the second cell line of the present invention.
- the method includes a step of subjecting the cells isolated from the above to subculture for 30 times or more, preferably 50 times or more, more preferably 70 times or more (hereinafter also referred to as subculture step).
- the subculture process is not particularly limited as long as the cell line of the present invention is finally obtained.
- the above-mentioned “1. First cell line of the present invention” and “2. Based on the matter described in the “second cell line of the invention”, determination of materials, methods, conditions, products to be used, and the like can be carried out.
- the cell line production method of the present invention is capable of subculture, for example, by storing and restoring cells during the subculture process as long as the object of producing the cell line of the present invention can be achieved. Appropriate steps can be provided before, during and after the steps.
- the transformant of the present invention is obtained by introducing a foreign gene into the cell line of the present invention.
- the method for producing a transformant of the present invention is a method including a step of obtaining a transformant by introducing a foreign gene into the cell line of the present invention.
- the method for producing a foreign gene product of the present invention is a method including a step of obtaining an expression product of a foreign gene from the transformant of the present invention.
- the method for introducing a foreign gene into the cell line of the present invention is not particularly limited.
- a transfer known by a person skilled in the art such as a lipofection method, an electroporation method, a microinjection method, a calcium phosphate method, a method using a baculovirus, or the like.
- the foreign gene can be introduced into the cell line by introducing the foreign gene itself into the nucleus of the cell line of the present invention, or by introducing a foreign gene, for example, using a gene introduction substance such as a vector.
- the recombinant vector may be introduced into the cell line of the present invention.
- the vector is not particularly limited as long as it is an autonomously replicable vector.
- vectors for mammalian cells more specifically pcDNA3.1, Flexi HaloTag, pcDNA3.2, pcDNA4.
- Plasmid vectors such as pcDNA5, pcDNA6, and pCMV; viral vectors such as ⁇ phage and RSV; amphibian-derived vectors having the EF1 ⁇ promoter derived from Xenopus.
- the foreign gene to be introduced is not particularly limited as long as it is a gene encoding a desired protein such as a functional protein, edible protein, enzyme, marker protein, or labeling protein. Since the cell line of the present invention is a fish-derived cell line, a gene encoding a fish-specific protein can be used as a foreign gene.
- the foreign gene can be prepared based on information on the amino acid sequence of a desired protein, and a commercially available gene can be appropriately modified and used.
- the protein encoded by the foreign gene may be one type or two or more types. For example, in the case of a foreign gene encoding a desired protein and an antibiotic resistance marker, it is possible to efficiently obtain only a transformant into which the foreign gene has been introduced due to antibiotic selectivity after gene introduction.
- a transformant has been obtained by the method for producing a transformant of the present invention can be confirmed using the presence or absence of an expression product of the introduced foreign gene as an indicator. Similarly, whether or not a foreign gene product was obtained by the method for producing a foreign gene product of the present invention can be confirmed using the presence or absence of an expression product of the foreign gene as an indicator.
- a foreign gene product may be obtained by expressing a foreign gene by culturing the transformant of the present invention, or the transformant of the present invention is cultured and proliferated. Thereafter, foreign gene expression may be induced to obtain a foreign gene product.
- the foreign gene product can be obtained as an expression product accumulated in the transformant of the present invention or an expression product accumulated in the culture solution, depending on the kind of the foreign gene.
- the confirmation of the foreign gene product is not particularly limited as long as it is a technique known by a person skilled in the art to know the presence of a specific protein. For example, Western blotting using a molecular weight of the foreign gene product or an antibody against the foreign gene product, an immunoassay Method, chromatography method and the like can be employed.
- the kit for producing the transformant of the present invention is not particularly limited as long as the cell line of the present invention is included, but in addition to the cell line of the present invention, a vector for introducing a foreign gene and a transfection vector It is preferable to include equipment such as instruments, reagents, and devices.
- the equipment for transfection is not particularly limited as long as it is used when performing each transfection technique.
- the transfection technique is a lipofection technique
- liposomes liposomes, culture media, buffers, etc.
- Instruments such as reagents and culture vessels are preferred.
- the uses of the cell line of the present invention are not particularly limited.
- fish cell physiology, fish genes and their expression systems, fish disease viruses, water environment pollution assessment In addition to appropriate research such as experimental systems, it can also be used for evaluation of physiologically active substances, screening for antiviral agents in combination with fish viruses, and the like.
- the cell line of the present invention comprises a step of introducing a test substance into the culture system of the cell line of the present invention and then evaluating the influence of the test substance on living cells;
- a virus propagation method comprising inoculating a culture system with a virus and growing the virus in a nutrient medium suitable for the growth of the virus; introducing the test substance into the virus-growing medium in the virus growth method; It can be used in a method for screening a virus therapeutic agent, including a step of evaluating the action of a test substance on a virus.
- the cell line of the present invention can be used in methods for diagnosing viral infections, methods for producing vaccines effective against viral infections, methods for evaluating cytotoxicity, and the like.
- the cytotoxicity of the test substance is evaluated by comparing the survival number of the cell line of the present invention with the survival number of the cell line after contacting the test substance.
- the step of bringing the test substance into contact with the cell line of the present invention and the step of determining the test substance as a differentiation-inducing substance by detecting muscle cells, skin cells, nerve cells or fat cells a differentiation-inducing substance Can be expected to be screened.
- the piece after standing was replaced three times with PBS containing 1% penicillin / streptomycin under ice-cooling.
- trypsin EDTA MP Biomedicals
- the tissue pieces are minced into 1 mm squares under ice cooling, and then at room temperature. Allowed to stand for 20 minutes.
- the tissue piece after standing was subjected to a centrifuge for several seconds, and then the supernatant was removed.
- the precipitated tissue pieces were treated several times with Leibovitz's L-15 medium (Life Technologies). That is, the tissue piece in the tube was spun down and the supernatant was removed, and then a fresh medium was added and stirred.
- FIGS. 1A to 1D show the appearance of cells that have migrated throughout the flask centering on each piece. Below, the subculture procedure of the cell extracted from the dorsal fin piece is described.
- the medium in the subculture culture flask was removed, and the cells adhering to the bottom of the flask were washed 3 times with PBS. Cells were detached and collected by treatment with Tryp Express (Gibco) for 10 minutes at 38 ° C. Leibovitz's L-15 medium containing 10% FBS was added to the collected cells and cultured at 25 ° C.
- the mixture was centrifuged at 1,100 rpm for 4 minutes at room temperature, then the supernatant was removed, and then the cell bunker 2 (Juji Field) was used at ⁇ 80 ° C. saved.
- the cell bunker 2 Juji Field
- the liquid portion was taken from a semi-thawed tube, transferred to a 15 ml centrifuge tube, 4 ml of L-15 medium was added, and the mixture was stirred and centrifuged.
- KSC cell As a result of performing the above 1 and 2, the present inventors have succeeded in obtaining a cell having a passage number of 70 or more and a division number of 276 or more. This cell was named KSC cell. KSC cells were almost the same as the first passage cells in terms of doubling time. Therefore, KSC cells were evaluated as established. This is in accordance with the definition of the JRCB cell bank (http://cellbank.nibio.go.jp/visitcenter/whatsculture/cellculture03.html, the description of which is incorporated herein by reference).
- CHO cell data is the second largest cell bank in the world, and the only DSMZ (http://www.dsmz.de) in Germany that seems to be highly reliable with complete cell data in the world. (The description of this document is incorporated herein by reference).
- CHO cells reached confluence (cells proliferated until they expanded to the whole flask bottom) in 72 to 96 hours when cells of 10 6 cells / ml were seeded at about 80 cm 2 , and the doubling time Is calculated as 24 hours.
- KSC cells reached confluence in 48 to 72 hours when 10 6 cells / ml cells were seeded at about 80 cm 2 , and the doubling time was calculated as 18.34 hours. Therefore, it was found that KSC cells are cells having the same or better proliferative ability as CHO cells.
- KSC cells about 0.4 ⁇ 10 6 cells / ml of cells were seeded in an area of 75 cm 2 and Leibovitz's L-15 medium containing 10% FBS was used at 25 ° C. in the absence of CO 2.
- the total number of cells when cultured until 48 hours after reaching confluence was measured using a TC10 Automated Cell Counter (BioRad).
- the number of cells after 24 hours of culture was about 1.0 ⁇ 10 6 cells / cell.
- the number of cells after 48 hours of culture was about 2.5 ⁇ 10 6 cells / ml.
- the doubling time is calculated from the number of cells immediately after seeding and the number of cells after 48 hours of culture. Was calculated to be 18.16 hours.
- the doubling time calculated from the number of cells immediately after seeding and the number of cells after 24 hours of culture is 18.16 hours, and is calculated from the number of cells after 24 hours of culture and the number of cells after 48 hours of culture. The doubling time performed was 18.16 hours.
- FIG. 2 shows that the KSC cells after thawing had a survival rate of 75% or more regardless of the number of passages.
- pdf # search % 27 FemtoJetMicroinjector% 27; http: // probes. invitrogen. com / media / pis / mp10582.
- pdf # search % 27celllight% 20pdf% 27, the description of this document is incorporated herein by reference.
- Cytotoxicity (resistance) in gene transfer by the Lipofectamine method is KSC cells, rat mesenchymal stem cells (MSC), human acute T-cell leukemia cell line (Jurkat), and mice. Comparative verification was performed using embryonic tumor cells (P19). As a result, KSC cells have a survival rate of 90% or more after gene introduction (transfected) compared to before introduction of foreign genes (control), and show superior cell stability compared to other cells. ( Figure 8).
- the cell detachment solution was put into a 15 ml tube, centrifuged at 1,100 rpm for 4 minutes, the supernatant was removed, and the cells were collected. This operation was performed quickly because the cells affected the colcemid treatment time.
- hypotonic solution was added to the 15 ml tube containing the collected cells with a pipette and gently pipetted to disperse the cells.
- a hypotonic solution was added to a 15 ml tube containing the dispersed cells until the final volume was 1.5 ml, and then the cells were dispersed again by pipetting. The cells dispersed in the hypotonic solution were allowed to stand at room temperature for 20 minutes for hypotonic treatment.
- the fixative was slowly added so that the total amount was about 10 ml, and gently agitated to immobilize the cells. Further, the supernatant was removed by centrifugation at 1,100 rpm for 4 minutes, and a few drops of the new fixative solution were added to the centrifuged cells, and the cells were dispersed by pipetting. Further, about 10 ml of fixative was added, and the whole was stirred. This operation was repeated two more times to completely fix the cells. The fixed cells were stained with Hoechst quinacrine (Wako Pure Chemical Industries) and observed with an optical microscope.
- the cell population of KSC cells includes three types of cell lines having 32, 33 and 66 chromosomes.
- a normal riverfish has 33 female and 34 male chromosomes.
- Ninety percent of the KSC cell population had 33 chromosomes, similar to the kawaha female. However, the remaining 10% of the cell population were cells with 32 and 66 chromosomes.
- Multipotency analysis using stem cell markers (1) Method According to the following procedure, KSC cells were immunochemically stained. As a blocking buffer, a PBS solution containing 10% FBS and 0.1% Triton X-100 was used. As an antibody dilution buffer, a PBS solution containing 3% goat serum and 0.1% Triton X-100 was used.
- KSC cells were cultured to confluence in a culture flask, the medium was removed, and the cells were washed twice with PBS at room temperature. 4% paraformaldehyde was added to the cells, and the cells were fixed by allowing to stand at room temperature for 20 minutes. Paraformaldehyde was removed and the immobilized cells were washed 3 times with PBS at 10 minute intervals. Blocking buffer was added and allowed to stand at room temperature for 1 hour. The blocking buffer was removed, and the cells after the blocking treatment were washed once with PBS. A primary antibody (ES / iPS Cell Charactarization Kit; Funakoshi) diluted 100 times using an antibody dilution buffer was added, and a primary antibody reaction was performed overnight at 4 ° C.
- ES / iPS Cell Charactarization Kit Funakoshi
- the cells after the reaction were washed with PBS 5 times at 10-minute intervals.
- a secondary antibody diluted with an antibody dilution buffer was added, and left at room temperature for 1 hour to carry out a secondary antibody reaction.
- the plate on which the cells were immobilized was protected from light.
- the secondary antibody was removed, and the reacted cells were washed 4 times with PBS at 10 minute intervals. Again, the plate was protected from light. The washed cells were observed using a FLoid cell imaging station (Life Technologies).
- KSC cells were presumed to be a pluripotent cell line
- differentiation into various cells was attempted by changing the culture medium and culture conditions.
- the cell morphology was observed with a light microscope at a magnification of 460 times.
- KSC cell morphology (before differentiation induction) KSC cells were cultured in a collagen-coated flask using Leibovitz's L-15 medium containing 10% (v / v) FBS. The observation result of the cells after the culture is shown in FIG. 10A.
- the fibroblast-like morphology shown in FIG. 10A is presumed to be a morphology before the KSC cells differentiate into other cells.
- FIGS. 10E-I when KSC cells were cultured using various culture containers, combinations of serum and medium, or differentiation-induced for KSC cells, adipocyte-like cells, fiber cell-like cells, liver cell-like cells, rose cells, Branched immune cell-like cells and giant rod-like fusion cells could be obtained (see FIGS. 10E-I).
- the fused cells shown in FIG. 10I were positive for alkaline phosphatase activity (see FIG. 11A) and positive for NANOG (see FIG. 11B).
- this fusion cell was also positive for TRA-1-60, OCT4 and SSEA-3 (see FIGS. 11C-E).
Landscapes
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
本出願は、2012年7月31日出願の日本特願2012-169378号の優先権を主張し、その全記載はここに開示として援用される。
本発明は、線維芽細胞様の形態を呈する、カワハギ科(Monacanthidae)魚類の生体部位に由来する細胞株またはその継代株に関する。
魚類に由来する魚類細胞は、哺乳動物に由来する動物細胞と類似する性質が多い。また、魚類細胞は、一般的に、比較的低い温度で培養が可能であり、増殖可能な温度範囲が広い。したがって、魚類細胞は、動物細胞に代替するものとして、生体由来物質の生産などの工業的用途や研究用途における培養細胞として有望視されている。
魚類の培養細胞としては、チョウザメの眼球上皮細胞由来の株化細胞やカサゴの尾鰭由来の株化細胞などが知られている(たとえば、下記特許文献1および2を参照、該文献の記載はここに開示として援用される)。これらの株化された細胞株は、動物細胞に代替可能な細胞としてだけではなく、それぞれチョウザメやカサゴの生態の解明や養殖の実現に資することが期待されている。
カワハギ(Stephanolepis cirrhifer)を含むフグ目カワハギ科(Monacanthidae)の魚類は、全身が丈夫な皮膚で覆われており、調理に際しては皮膚全体を一度に剥がすことができる。カワハギ科の魚類の中でもカワハギは生食用または調理用の食材として広く嗜まれている。
カワハギは、食材として広く愛用されているが、現在市場におかれているものは天然のカワハギであり、その取れ高は季節により変動する。また、カワハギの養殖技術はこれまでに確立されておらず、現在に至るまで種々の試みがなされている。
魚類の養殖では、細菌やウイルスなどの感染症、化学物質や重金属などの細胞毒性などに起因する養殖魚の斃死が問題となっている。そこで、養殖魚の斃死の問題を解決するために、魚類の細胞株は、ウイルス感染症の診断、ウイルス感染症に対して有効なワクチンの製造、細胞毒性試験などに寄与する可能性がある。そこで、本発明者らは、カワハギの不死化細胞株があれば、カワハギ養殖魚の斃死の問題を解決する糸口を探り出すことができ、カワハギの養殖開発において著しい進展を遂げるのではないかと考えた。特に、魚類の細胞株は、一般的に、低温での培養が可能であるものの、哺乳動物の細胞株に比べて増殖速度が小さいという性質を有する。したがって、低温培養が可能であり、かつ、増殖速度が哺乳動物の細胞株と同程度であるカワハギの不死化細胞株は、簡便かつ迅速に培養ができるものであることから、カワハギの養殖技術の開発において、急速な進歩をもたらすことが期待できる。
一方、カワハギに限らず、魚類の多能性幹細胞があれば、特定の細胞へ分化誘導することにより、従前は採取することが困難であった組織細胞を得ることができ、魚類の疾病の病因や発症メカニズムを研究することができるようになる。また、分化誘導後に特定の疾病を発症させた細胞を用いれば、その疾病に対する薬剤の効果や毒性を評価することが可能となる。したがって、魚類の多能性幹細胞もまた、魚類の養殖技術の発展に寄与することが期待できる。
上記したように、カワハギの不死化細胞株や多能性幹細胞を用いれば、カワハギの養殖技術の研究開発を格段に進められる可能性がある。しかし、これまでに、樹立されたカワハギの細胞株や多能性幹細胞は知られていない。
そこで、本発明の第1の目的は、カワハギに由来する不死化細胞株を提供することにある。また、本発明の第2の目的は、カワハギに由来する多能性幹細胞を提供することにある。さらに、本発明の第3の目的は、カワハギに由来する細胞株の製造方法および利用方法を提供することにある。
本発明者らは、まず、魚類細胞は低温下で培養が可能という点に着目して、魚類細胞の株化を検討した。そこで、タイ、アジ、サメなどの種々の魚類の鰭を用いて、様々な組織細胞の増殖を試みた。しかし、増殖速度の大きい細胞を得るどころか、継代培養さえできないものがほとんどであった。そこで、本発明者らは、魚類の種類や継代培養方法などを適宜変更して、さらに鋭意研究を積み重ねた結果、カワハギの背鰭から継代培養が可能な不死化細胞株を製造することに成功した。しかも、驚くべきことに、得られた細胞株は、25℃という室温と変わらない温度で培養した場合であっても、哺乳動物の細胞株と同程度又はそれ以上の増殖速度で培養が可能なものであった。さらに驚くべきことには、本発明者らが得たカワハギ由来の細胞株は、分化多能性を示す細胞マーカーの一部を発現するものであり、さらに培養条件を変えることにより形態の異なる種々の細胞へ変化するものであった。本発明は、これらの成功例や知見に基づいて完成された発明である。
したがって、本発明によれば、下記(1)の性質を有する、カワハギ科(Monacanthidae)魚類の生体部位に由来する細胞株またはその継代株が提供される。
(1)実質的に制限なく継代培養が可能である
(1)実質的に制限なく継代培養が可能である
本発明において、好ましくは、さらに下記(2)の性質を有する。
(2)線維芽細胞様の形態を呈する
(2)線維芽細胞様の形態を呈する
本発明において、好ましくは、さらに下記(3)の性質を有する。
(3)多層構造を形成した培養が可能である
(3)多層構造を形成した培養が可能である
本発明において、好ましくは、さらに下記(4)の性質を有する。
(4)最大値は66本であり、最小値は32本であり、およびモードは33本である度数分布に従った染色体数を有する
(4)最大値は66本であり、最小値は32本であり、およびモードは33本である度数分布に従った染色体数を有する
本発明において、好ましくは、前記(4)の性質において、モードである33本の染色体の度数が度数分布における全度数の約90%である。
本発明において、好ましくは、さらに下記(5)の性質を有する。
(5)初期細胞数を約1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した際の倍加時間は、約14~28時間である
(5)初期細胞数を約1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した際の倍加時間は、約14~28時間である
本発明の別の側面によれば、または本発明の好ましい態様として、TRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーが陽性である、カワハギ科魚類の生体部位に由来する細胞株またはその継代株が提供される。
本発明において、好ましくは、前記細胞株またはその継代株は、線維芽細胞様の形態を呈する細胞株またはその継代株である。
本発明において、好ましくは、前記細胞株またはその継代株は、筋細胞および筋細胞様細胞、上皮細胞および上皮細胞様細胞、神経細胞および神経細胞様細胞、脂肪細胞および脂肪細胞様細胞、免疫細胞および免疫細胞様細胞ならびに肝臓細胞および肝臓細胞様細胞からなる群から選ばれる少なくとも1種の細胞に分化する能力を有する。
本発明において、好ましくは、前記カワハギ科魚類が、カワハギ属(Stephanolepis)魚類、ウマヅラハギ属(Thamnaconus)魚類、メガネウマヅラハギ属(Cantherhines)魚類、ウスバハギ属(Aluterus)魚類およびアミメハギ属(Rudarius)魚類からなる群から選ばれる魚類である。
本発明において、好ましくは、前記カワハギ科魚類が、カワハギ(Stephanolepis cirrhifer)である。
本発明において、好ましくは、前記生体部位は、背部、胸部、腹部、尻部または尾部にある鰭である。
本発明の別の側面によれば、または本発明の好ましい態様として、受託番号がNITE BP-1369である、カワハギの背鰭に由来する細胞株またはその継代株が提供される。
本発明の別の側面によれば、カワハギ科魚類の生体部位から単離した細胞を70回以上の継代培養に供する工程を含む、本発明の細胞株またはその継代株を製造する方法が提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株に外来遺伝子を導入してなる、形質転換体が提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株に外来遺伝子を導入して形質転換体を得る工程を含む、形質転換体を製造する方法が提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株に外来遺伝子を導入してなる形質転換体から該外来遺伝子の発現産物を得る工程を含む、外来遺伝子の発現産物を製造する方法が提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株と、ベクターと、トランスフェクションのための器材とを含む、形質転換体を製造するためのキットが提供される。
本発明の別の側面によれば、または本発明の好ましい態様として、本発明の細胞株またはその継代株と、培地と、培養容器とを含む、分化細胞を製造するためのキットが提供される。
本発明において、好ましくは、さらに血清を含む。
本発明において、好ましくは、前記血清が、哺乳類血清、魚類血清および血清代替物からなる群から選ばれる少なくとも1種の血清である。
本発明において、好ましくは、前記培地は、哺乳動物細胞用培地、昆虫細胞用培地および魚類細胞用培地からなる群から選ばれる少なくとも1種の培地である。
本発明において、好ましくは、前記培養容器が、底面が細胞接着分子でコーティングされた培養容器および底面が細胞接着分子でコーティングされていない培養容器からなる群から選ばれる少なくとも1種の培養容器である。
本発明の別の側面によれば、本発明の細胞株またはその継代株を、底面が細胞接着分子でコーティングされた培養容器または底面が細胞接着分子でコーティングされていない培養容器の中で、血清を含有していない、または哺乳類血清、魚類血清もしくは血清代替物を含有した哺乳動物細胞用培地、昆虫細胞用培地または魚類細胞用培地を用いて培養する工程を含む、分化細胞を製造する方法が提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株からなる培養細胞シートが提供される。
本発明の別の側面によれば、本発明の細胞株またはその継代株から、アルカリフォスファターゼ活性の染色により識別され、かつ、NANOG、OCT4、TRA-1-60およびSSEA-3陽性である細胞または該細胞によって形成される巨大繭状コロニーを製造する工程を含む、巨大繭状コロニーを製造する方法が提供される。
本発明の細胞株またはその継代株によれば、カワハギの生体を用いることなく、かつ、カワハギの生体から特定の細胞を単離することなく、カワハギの生態解明のための研究や実験を繰り返し実施することが可能である。そのような研究等として期待されるものは、細菌やウイルスなどの感染性微生物に対するカワハギの感受性(感染性)の検査、化学物質や重金属などの細胞毒性物質に対するカワハギの影響を調べるための細胞毒性試験、環境変化要因に対するカワハギの応答性試験、感染性微生物に感染したカワハギの治療または予防に有効な薬剤およびワクチンの開発などが含まれる。本発明の細胞株またはその継代株によれば、カワハギの生態解明を通して、カワハギの養殖技術のさらなる発展に寄与することが期待できる。
本発明の細胞株またはその継代株は、哺乳動物の細胞株と同程度の増殖速度で培養可能な細胞株を包含する。このような本発明の細胞株またはその継代株は、室温下での培養が可能なものであることから、哺乳動物の細胞株に代替して、研究用途や工業生産用途などに使用することが期待できる。
以下、本発明の詳細について説明する。
1.本発明の第1の細胞株
本発明の第1の細胞株またはその継代株は、カワハギ科(Monacanthidae)魚類の生体部位に由来するものである。
1.本発明の第1の細胞株
本発明の第1の細胞株またはその継代株は、カワハギ科(Monacanthidae)魚類の生体部位に由来するものである。
本明細書において「細胞株」とは、当業者によって最も広義に理解されるものであり、株化細胞、樹立細胞系などともよばれ、連続的に継代培養が可能な細胞系であれば特に限定されない。また、本明細書において「継代株」とは、細胞株を継代培養して得られるすべての細胞を意味する。本明細書において、細胞株と継代株との間には厳密な区別はなく、便宜上、株化したと評価できる細胞を細胞株とよび、その後細胞株を継代培養して得られるものを継代株とよぶ。以下では、細胞株および継代株をまとめた総称として、単に「細胞株」とよぶ。
本発明の第1の細胞株は、カワハギ科魚類から摘出した組織や臓器などの生体部位から単離した細胞に基づいて製造することができる。カワハギ科魚類は、その名が示すとおり、身体全体を覆う皮(外皮)を容易に剥がすことができるという特徴を有する魚類である。たとえば、皮の一部に包丁などで切り込みを入れると、特殊な器具を用いることなく、手などで簡単に皮を剥がすことができる。本発明の第1の細胞株の由来生物は、上記した特徴を有するカワハギ科魚類であれば特に限定されないが、たとえば、カワハギ属(Stephanolepis)魚類、ウマヅラハギ属(Thamnaconus)魚類、メガネウマヅラハギ属(Cantherhines)魚類、ウスバハギ属(Aluterus)魚類およびアミメハギ属(Rudarius)魚類を挙げることができる。本発明の目的において、カワハギ科魚類の好適な具体例としては、カワハギ(Stephanolepis cirrhifer)、ウマヅラハギ(Thamnaconus modestus)およびウスバハギ(Aluterus monoceros)を挙げることができ、より好ましいのはカワハギ(Stephanolepis cirrhifer)である。
本発明の第1の細胞株を製造するために使用されるカワハギ科魚類の生体部位は、魚体中の特定の一部分のみを指すものではなく、魚体中の2以上の組織や臓器などの部位を含み、さらには魚体全部を含み得る。本発明の目的において、カワハギ科魚類における好適な生体部位は、鰭にあたる部位であり、たとえば、背部、胸部、腹部、尻部または尾部にある鰭が好ましく、背部にある鰭である背鰭がより好ましい。
本発明の第1の細胞株をカワハギ科魚類の生体部位から製造する方法は特に限定されるものではなく、当業者により知られる生体から細胞株を取得する方法、たとえば、生体から組織や臓器を摘出する手法、切除した組織等から細胞を単離する手法、単離した細胞を培養して初代培養細胞を得る手法、初代培養細胞を継代培養して細胞株およびその継代株を得る手法などを適宜組み合わせた方法を実施することにより製造することができる。
本発明の第1の細胞株を製造する方法のうち、初代培養細胞を得る工程の具体例は、たとえば、次の手順からなる。すなわち、カワハギの鰭部を、皮を剥ぐことなく、適当な大きさの角片として切除する。切除した角片を適当な方法で洗浄し、次いで種々の濃度の抗生物質を用いて1~数回に分けて処理する。処理後の切片を、無菌的にトリプシンなどのタンパク質分解酵素溶液の存在下で微小片となるように細かく刻む。ここまでの操作は低温下、好ましくは氷冷下で実施する。次いで、トリプシン溶液に浸漬させた微小片を室温で静置する。静置後の微小片を遠心分離等によって回収し、回収した微少片をLeibovitz’s L-15培地などの非CO2環境下での細胞増殖に適した培地を用いて数回繰り返し洗浄する。この際、微小片をCO2環境下で洗浄する場合は、RPMI1640倍地やIMDM培地(ともにライフテクノロジーズ社)などの標準的な哺乳動物細胞用培地が使用可能である。次いで、血清含有培地を含むコラーゲンコートされた培養容器に微小片を播き、底着させた後、25℃にするなどの適当な培養条件で、細胞が培養容器の底面の約80%以上を覆うようになるまで培養する。この際の微小片に集まった細胞を初代培養細胞とする。次いで、培養容器中の培地を除去し、容器底面に付着している培養細胞を適当な方法で洗浄した後、適当な方法で底面から培養細胞を剥離および回収し、2~3等分して新しい培養容器にて継代培養を実施する。新しい培養容器に播いた細胞を第1継代培養細胞とする。
本発明の第1の細胞株を製造する方法のうち、初代培養細胞を継代培養して本発明の第1の細胞株を得る工程の具体例は、たとえば、次の手順からなる。すなわち、初代培養細胞から本発明の第1の細胞株を得るための継代培養は、たとえば、初代培養細胞を得る工程と同じようにして、血清含有培地を用いて適当な培養条件により達成される。培養開始時の細胞数は4×105cells/mlであり、培養終了時の目安は90%以上のコンフルエントになったときであり、および培地の交換時期は死滅細胞により培養液が汚れたときなどとすることができるが、これらは特に限定されるものではない。
本発明の第1の細胞株は、当業者により知られる通常の方法で保存および増殖させることができ、その方法は特に限定されるものではない。本発明の第1の細胞株を凍結保存する方法は、たとえば、細胞株に市販の培地を加えた後、細胞を遠心分離するなどして回収し、次いで回収した細胞へ市販の無血清培養細胞用の細胞凍結保存液を加えて、凍結保存することにより達成し得る。凍結させた本発明の細胞株は、室温で半解凍状態にし、新鮮な無血清培地で適当な回数置換することにより細胞を融解させることができる。融解後の細胞に血清含有培地を加えることによって、上記の継代培養を実施できる。
本発明の第1の細胞株が製造できたことを判定する方法は特に限定されるものではないが、初代培養細胞を30回以上、好ましくは50回以上、より好ましくは70回以上、さらに好ましくは100回以上の継代培養に供することによって、そのような継代培養後の細胞を本発明の第1の細胞株であると判定できる。
本発明の第1の細胞株は、連続的に継代培養が可能な細胞系ではあれば特に限定されないが、実質的に制限なく継代培養が可能であるものであることが好ましい。すなわち、本発明の第1の細胞株の一態様は、当業者が有する相応の注意力および技術力をもって、上記した継代培養の方法に従って培養すれば、何世代にも渡って繰り返し培養できるものである。
本発明の第1の細胞株は、種々の性質を有し得るが、線維芽細胞様の形態を呈するものであることが好ましい。線維芽細胞は、扁平で長目の外形、しばしば見られる不規則な突起、楕円形の核などの特有の形態を呈する。本発明の第1の細胞株が線維芽細胞様の形態を呈するものである場合、本発明の第1の細胞株は、線維芽細胞に特有のいずれかの形態を有するものであれば特に限定されないが、少なくとも扁平で長目の外形を有するものであることが好ましい。本発明の第1の細胞株が線維芽細胞様の形態を呈することは、当業者により知られる細胞の形態を観察する種々の方法により確認することができ、たとえば、適当な倍率にセットした光学顕微鏡下で本発明の第1の細胞株を観察することにより確認することができる。
本発明の第1の細胞株が、実質的に制限なく継代培養が可能であるものである場合は、たとえば、分裂回数を30回以上、好ましくは50回以上、より好ましくは70回以上、さらに好ましくは100回以上とすることができるものである。この場合の本発明の第1の細胞株は、不死化細胞とみなし得る。本発明の第1の細胞株が、実質的に制限なく継代培養が可能であるものか否かは、たとえば、本発明の細胞株を上記した継代培養に供して、継代回数が上記の回数に達することを確認することにより判定できる。
倍加時間は、当業者により知られているとおり、細胞数が2倍に達する時間をいう。倍加時間は、増殖期にある細胞については、原則的に細胞周期と一致する。本発明の第1の細胞株の倍加時間を測定する方法は特に限定されないが、たとえば、上記した継代培養に供した場合、すなわち、10% FBSを含むLeibovitz’s L-15培地を用いて25℃で培養した場合に、増殖期にある細胞について2~数点、好ましくは3点以上を選んで計測した細胞数から求められ得る。
本発明の第1の細胞株の倍加時間は特に限定されないが、たとえば、初期細胞数を約1.0×106cells/ml、好ましくは0.1~1.5×106cells/ml、より好ましくは0.4~1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した場合、初代培養細胞より短く、好ましくは10~48時間、より好ましくは12~36時間、さらに好ましくは14~28時間、なおさらに好ましくは16~24時間、特に好ましくは約18時間である。細胞数からの倍加時間を算出する方法は特に限定されないが、たとえば、TC10 Automated Cell Counter(バイオラッド)などの細胞数計測機器を用いて計測した細胞数に基づいて、観測点(時間)と細胞数とから単位時間あたりの細胞量の増加を計測するソフトウェア(http://www.doubling-time.com/compute.php?lang=enなど)を用いて算出する方法を挙げることができる。また、細胞の計測は、たとえば、培養容器の底面にあらかじめ指標(マーカー)を付し、本発明の第1の細胞株を培養に供した後、該マーカー付近における細胞の数を倒立型ルーチン顕微鏡で撮影観察することにより計測することができる。この際、接眼レンズに付された複数の四方マス(たとえば、1mm×1mmのマス)の中の各細胞数の平均を細胞数としてもよい。培養した細胞を経時的に観察すると、細胞の増殖曲線が得られる。増殖曲線からは、細胞の増殖過程における遅延期、対数増殖期、定常期、死滅期の増殖サイクルを知ることができる。対数増殖期の細胞数が2倍になる為に必要な時間(倍加時間)を以下の計算式より算出することができる。
倍加時間=(t-t0)log2/(logN-logN0)
t:時間[h]、N:t時の細胞数
t0:初期時間[h]、N0:t0時の細胞数
倍加時間=(t-t0)log2/(logN-logN0)
t:時間[h]、N:t時の細胞数
t0:初期時間[h]、N0:t0時の細胞数
培養細胞を培養容器の底面に接着させた状態で培養すると、通常は、培養細胞は底面全体を覆うコンフルエントな状態に達すると増殖を停止する。すなわち、培養細胞は、培養容器の底面の上に単層の細胞層を形成するまで増殖し得る。しかし、本発明の第1の細胞株の一態様は、単層の細胞層を形成した後もさらに増殖を続けることにより、単層の細胞層の上に別の細胞層を形成し得るものであり、好ましくは多層構造を形成した培養が可能であるものである。したがって、本発明の第1の細胞株は、細胞層が重なり合った三次元的構造を形成した培養が可能であるものを含む。本発明の第1の細胞株が多層構造を形成し得るものであることは、本発明の第1の細胞株がコンフルエントな状態になった後さらに培養を進め、当業者により知られる方法で培養細胞の構造を観察することにより確認することができる。なお、多層構造とは、単層の細胞層の上に別の細胞層を形成した構造であれば特に限定されず、2層または3層以上の層構造である。
本発明の第1の細胞株の染色体数は、由来生物であるカワハギ科魚類の種類や雌雄によって変化することから特に限定されないが、たとえば、カワハギのメスの背鰭から取得したものである場合、その染色体数は66本、33本および32本であり得る。また、本発明の第1の細胞株は、一個の独立した細胞だけではなく、一群の細胞集団としてみなせる場合がある。その場合、本発明の第1の細胞株が、カワハギのメスの背鰭から取得したものである場合は、染色体数が66本、33本または32本である細胞株の集団であり得る。また、この場合の本発明の第1の細胞株は、好ましくは最大値が66本であり、最小値が32本であり、およびモード(最頻値)が33本である度数分布に従った染色体数を有する細胞株の集団であり、該度数分布において、モードである33本の染色体の度数が全度数の約90%である細胞株の集団であることがより好ましい。本発明の第1の細胞株の染色体数を解析する方法としては、当業者により知られる細胞の染色体数を解析する方法を特に限定することなく採用することができるが、たとえば、後述する実施例に記載されている方法により解析することができる。
本発明の第1の細胞株の具体例は、カワハギ科魚類の生体部位に由来する細胞株であって、下記(1)~(5)の性質のいずれか1つ以上の性質を有するもの、好ましくは下記(1)の性質を有し、かつ、下記(2)~(5)のいずれか1つ以上の性質を有するもの、より好ましくは下記(1)および(2)の性質を有し、かつ、下記(3)~(5)のいずれか1つ以上の性質を有するもの、さらに好ましくは下記(1)~(3)の性質を有し、かつ、下記(4)および(5)のいずれか1つ以上の性質を有するもの、なおさらに好ましくは下記(1)~(5)の性質を有するものである。
(1)実質的に制限なく継代培養が可能である
(2)線維芽細胞様の形態を呈する
(3)多層構造を形成した培養が可能である
(4)最大値は66本であり、最小値は32本であり、およびモードは33本である度数分布に従った染色体数を有する、ただし、該度数分布はモードである33本の染色体の度数が全度数の約90%であることが好ましい
(5)初期細胞数を約1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した際の倍加時間は、約14~28時間である
(1)実質的に制限なく継代培養が可能である
(2)線維芽細胞様の形態を呈する
(3)多層構造を形成した培養が可能である
(4)最大値は66本であり、最小値は32本であり、およびモードは33本である度数分布に従った染色体数を有する、ただし、該度数分布はモードである33本の染色体の度数が全度数の約90%であることが好ましい
(5)初期細胞数を約1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した際の倍加時間は、約14~28時間である
2.本発明の第2の細胞株
本発明の細胞株の別の態様として、本発明の第2の細胞株は、TRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーが陽性である、カワハギ科魚類の生体部位に由来する細胞株またはその継代株である。本発明の第2の細胞株は、本発明の第1の細胞株と同様の方法によって、カワハギ科魚類の生体部位から製造することができる。本発明の第2の細胞株は、線維芽細胞様の形態を呈することが好ましい。
本発明の細胞株の別の態様として、本発明の第2の細胞株は、TRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーが陽性である、カワハギ科魚類の生体部位に由来する細胞株またはその継代株である。本発明の第2の細胞株は、本発明の第1の細胞株と同様の方法によって、カワハギ科魚類の生体部位から製造することができる。本発明の第2の細胞株は、線維芽細胞様の形態を呈することが好ましい。
TRA-1-60、OCT4またはSSEA-3が陽性である細胞株とは、それぞれヒトTRA-1-60、ヒトOCT4またはヒトSSEA-3に対する抗体が認識するエピトープを発現している細胞株をいう。
TRA-1-60に対する抗体は、未分化ヒト胚性幹(ES)細胞、胚性癌(EC)細胞および胚性生殖(EG)細胞に発現するタンパク質(エピトープ)と反応し、該エピトープは細胞分化により失われることが知られている。OCT4はES細胞に高発現する転写因子であり、ES細胞の未分化性の維持や多分化能の維持に関与しているといわれている。SSEA-3はES細胞などの多能性幹細胞の細胞マーカーとして知られている。TRA-1-60、OCT4およびSSEA-3をコードする遺伝子配列は、たとえば、National Center for Biotechnology Information(NCBI)の公共のデータベースであるGenBankにより、それぞれACCESSION番号として、TRA-1-60はNM_001018111またはNM_005397、OCT4はNM_002701、およびSSEA-3はNM_033149(アミノ酸配列のACCESSION番号はQ9JI67)として登録されている。
TRA-1-60、OCT4およびSSEA-3は、それぞれ多能性を示す細胞マーカーであり、分化後の細胞表面には見られないものであることで共通する。これらのことから、TRA-1-60、OCT4、SSEA-3またはこれらの細胞マーカーの2種もしくは3種が陽性である細胞株は、種々の細胞に分化する能力である多能性を備えたものであると推測され得る。したがって、本発明の第2の細胞株は、多能性を有する細胞株である蓋然性が高い。ただし、本明細書においては「多能性」という用語は、限定的に解釈されるものではなく、学術用語としての多能性(pluripotency、multipotency)の意味に加えて、全能性(totipotency)の意味をも含み、多能性や全能性があること、またはその蓋然性があることなどと解釈すればよく、このことに係わらず最も広義に解釈されるものである。
細胞株がTRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーが陽性であることは、当業者により知られている特定の細胞マーカーを調べる手法を応用して確認できるが、たとえば、次の手順による免疫化学的手法により確認できる。すなわち、培養細胞を培養容器の底面に固定化し、ブロッキング処理などした後に、TRA-1-60、OCT4またはSSEA-3に対する一次抗体を加えて一次抗体反応を実施する。次いで、未反応の一次抗体を除いた後に、蛍光標識された一次抗体に対する二次抗体を加えて二次抗体反応を実施する。次いで、未反応の二次抗体を除去した後に、蛍光標識された細胞を観察することにより、TRA-1-60陽性、OCT4陽性またはSSEA-3陽性の細胞を確認することができる。なお、一次抗体の構造(グロブリンタイプや断片化構造など)、二次抗体についての一次抗体の認識特異性、蛍光標識の波長などを適宜変えることにより、一度または二度の操作で、細胞がTRA-1-60、OCT4およびSSEA-3について陽性であることを確認できるが、細胞を3つの集団に分割してそれぞれの集団についてTRA-1-60、OCT4またはSSEA-3が陽性であることを確認することが好ましい。また、本発明の第2の細胞株は、TRA-1-60、OCT4およびSSEA-3以外の多能性幹細胞において特異的に発現する細胞マーカーを発現し得るものであってもよい。
本発明の第2の細胞株は、ヒト抗NANOG抗体に対する親和性が小さく、バックグラウンドの状況や画像処理法によっては、NANOGについて弱陽性ともいえるし、実質的に陰性であるともいえる。
本発明の第2の細胞株は、上記細胞マーカーを発現するものであることから、多能性を有する細胞株である蓋然性が高い。したがって、本発明の第2の細胞株の一態様は、たとえば、筋細胞および筋細胞様細胞、上皮細胞および上皮細胞様細胞、神経細胞および神経細胞様細胞、脂肪細胞および脂肪細胞様細胞、肝臓細胞および肝臓細胞様細胞などの細胞に分化し得る細胞株である。
本発明において、細胞の分化とは、TRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーの発現が消失すること、分化細胞に特有の細胞マーカーを発現することなどに加えて、単なる形態変化も含まれる。また、筋細胞様細胞、上皮細胞様細胞、神経細胞様細胞、脂肪細胞様細胞および肝臓細胞様細胞とは、筋細胞等に完全には分化していないが、分化の途中にある細胞や分化細胞に類似する形態を呈する細胞を包含し得る。
本発明の第2の細胞株の好ましい一態様は、筋細胞および筋細胞様細胞、上皮細胞および上皮細胞様細胞、神経細胞および神経細胞様細胞、脂肪細胞および脂肪細胞様細胞、免疫細胞および免疫細胞様細胞ならびに肝臓細胞および肝臓細胞様細胞からなる群から選ばれる1種、好ましくは2種以上、より好ましくは3種以上、さらに好ましくは4種以上の細胞に分化する能力を有する細胞株である。ただし、これらの分化細胞は例示であり、他の細胞に分化する能力を有するものも本発明の第2の細胞株に含まれる。
本発明の第2の細胞株における各細胞への分化は、当業者により知られる多能性幹細胞を各細胞へ分化させる手法により達成できるが、たとえば、培養容器のコーティングの有無および種類、血清の種類および有無、培地の種類などを種々設定することによっても達成し得る。たとえば、図10B~Hに示されている通り、本発明の第2の細胞株から筋細胞または筋細胞様細胞、上皮細胞または上皮細胞様細胞、神経細胞または神経細胞様細胞、肝臓細胞または肝臓細胞様細胞、繊維細胞または繊維細胞様細胞、脂肪細胞または脂肪細胞様細胞、免疫細胞または免疫細胞様細胞を得ることができる。
これらの細胞について、図10Bに示す細胞は、細長い紡錘状または線維状の形態を呈し、かつ、増殖能がある筋細胞様の細胞である。図10Cに示す細胞は、個々の細胞は周縁が不定の扁平な形態を呈し、かつ、細胞同士が密になって増殖する上皮細胞様の細胞である。図10Dに示す細胞は、伸長性のある軸索または樹状突起を有する細胞形態を呈し、かつ、増殖能がない神経細胞様の細胞である。図10Eに示す細胞は、細胞質内に脂肪滴らしきものを有する形態を呈し、かつ、有限的に分裂可能な肝臓細胞様の細胞である。図10Fに示す細胞は、筋細胞と同程度またはそれ以上の繊維状の形態を呈し、かつ、増殖能を有する繊維細胞様の細胞である。図10Gに示す細胞は、多数の空洞を有し、中には伸展性の軸索を有する形態を呈し、かつ、増殖能がない脂肪細胞様の細胞である。図10Hに示す細胞は、伸長性がある先端が尖った突起物を有するバラの枝状の形態を呈し、増殖能がなく、かつ、近接する細胞と連結する樹状の免疫細胞様の細胞である。また、本発明の第2の細胞株からは、図10Iに示すような、ゴム様に伸縮し、増殖能を有する細胞同士が融合して、巨大な繭様の構造体を形成する融合細胞が得られる。本融合細胞は、図11Aに示すようにアルカリフォスファターゼ活性陽性であり、かつ、図11Bに示すようにNANOG陽性である。さらに、本融合細胞は、図11C~Eにそれぞれ示すように、TRA-1-60、OCT4およびSSEA-3についても陽性である。上記のTRA-1-60、OCT4およびSSEA-3をコードする遺伝子配列に加えて、NANOGは、たとえば、NCBIの公共のデータベースであるGenBankにより、それぞれACCESSION番号として、NM_024865として登録されている。
細胞株が分化したことは、継代培養時の線維芽細胞様の形態、たとえば、図10Aに示すような形態と見比べることによる形態の変化により確認することができるが、当業者により知られる分化細胞の判定手法によっても確認することができる。たとえば、各分化細胞の細胞マーカーや各分化細胞が産生する物質を指標として、細胞株が分化したことを確認してもよい。
本発明の第2の細胞株から各分化細胞を製造する方法が、本発明の別の態様として提供される。すなわち、本発明の分化細胞を製造する方法は、本発明の第2の細胞株を、底面が細胞接着分子でコーティングされた培養容器または底面が細胞接着分子でコーティングされていない培養容器の中で、血清を含有していない、または哺乳類血清、魚類血清もしくは血清代替物を含有した哺乳動物細胞用培地、昆虫細胞用培地または魚類細胞用培地を用いて培養する工程を含む。
本発明の別の態様として、本発明の第2の細胞株を含む、分化細胞を製造するためのキットが提供される。分化細胞を製造するためのキットには、血清、培地および培養容器などを含むことが好ましく、これらに加えて緩衝剤(バッファー)やpH調整剤などを含むことがより好ましい。本発明の第2の細胞株は、上記した分化細胞以外の細胞に分化する可能性があることから、血清、培地、培養容器、緩衝剤およびpH調整剤は特に限定されず、種々のものが用いられ得る。
たとえば、本発明の分化細胞を製造するためのキットに含まれる血清としては、哺乳類血清、魚類血清および血清代替物を用いることができ、ウシ胎児血清(ギブコ;No.26140-087など)、ウマ血清(ギブコ;No.16050-130など)、ヤギ血清(ギブコ;No.16210-064など)、ウサギ血清(ギブコ;No.16120-099など)、マウス(ケーエーシー;No.AS3054など)、ニワトリ血清(ギブコ;No.16110-082など)、仔ヒツジ血清(ギブコ;No.16070-096など)、ブタ血清(ギブコ;No.26250-084など)、イヌ(ケーエーシー;No.AS3070など)、サル血清(ケーエーシー;No.AS3076など)、サケ血清SeaGrow(イーストコーストバイオ;JJ80-N2751など)などに加えて、タイやブリなどの他の魚類の血清を用いることができる。また、本発明の分化細胞を製造するためのキットに含まれる血清は、1種であっても2種以上であってもよい。血清を入手する方法は特に限定されず、市販されているものを用いてもよい。
本発明の分化細胞を製造するためのキットに含まれる血清として、血清代替物を用いることができる。血清代替物としては、たとえば、Knockout(登録商標) Serum Replacement(KSR)、ラクトアルブミン水解物、魚由来細胞培養培地用添加剤Hy-Fish(マルハチ村松)、Nu-Serum(BD;No.BSE 355100)、SERUM PLUS(シグマ;No.14008C-500ML)、L-Glutamine溶液(シグマ;No.59202C-100ML)などを挙げることができ、これらの中の1種または2種以上の血清代替物を用いることができる。
本発明の分化細胞を製造するためのキットに含まれる培地としては、たとえば、L-15(ギブコ;No.11415-064)、AIM V(ライフテクノロジーズ;No.087-0112DK)、IMDM(ギブコ;No.12440-053)、RPMI(ギブコ;No.11835030)、EX-CELL 420 with L-glutamine(ニチレイ;No.14420C、昆虫用)、D-MEM(ギブコ)、mTeSR1(ステムセル;No.ST-05850、幹細胞用)、Ham’s F-12(ギブコ;No.11765-054)、霊長類ES/iPS細胞用培地(リプロセル;No.RCHEMD001)を挙げることができ、これらの中の1種または2種以上の培地を用いることができる。
本発明の分化細胞を製造するためのキットには、培地に加えて、増殖因子や培地添加剤を構成成分として含めることができる。増殖因子や培地添加剤としては、たとえば、上皮細胞増殖因子、線維芽細胞増殖因子、神経成長因子、インスリン様増殖因子、血小板由来増殖因子、血管内皮細胞増殖因子、トランスフォーミング増殖因子、サイトカイン、幹細胞因子、T-STIM培養添加物、IL-3カルチャーサプリメント、血管内皮細胞グロースサプリメント、MITO+ シーラム・エクステンダー、ITS培養添加物を挙げることができ、これらの中の1種または2種以上の培地を用いることができる。増殖因子や培地添加剤を入手する方法は特に限定されず、市販されているものを用いてもよく、たとえば、日本ベクトン・ディッキンソン株式会社(BD;http://www.bdj.co.jp/falcon/products/1f3pro00000c64gx.html、該文献の記載はここに開示として援用される)などの製造業者によって市販されているものを使用することができる。
本発明の分化細胞を製造するためのキットに含まれる培養容器は、底面がコラーゲン、フィブロネクチン、ラミニン、ポリ-D-リシン、ポリ-L-リシン、ゼラチン、ストレプトアビジンなどの細胞接着分子でコーティングされた培養容器、底面が細胞接着分子でコーティングされていない培養容器またはこれらの中の2種以上の培養容器であることが好ましい。底面が細胞接着分子でコーティングされた培養容器を入手する方法は特に限定されないが、市販のコーティング済培養容器を用いてもよいし、非コーティング培養容器および細胞接着分子を別途用意し、当業者により知られる手法で培養容器の底面を細胞接着分子でコーティングすることにより作製したものを用いてもよい。細胞接着分子の入手方法は特に限定されず、たとえば、コラーゲンI(サーモ:132706)、IV(コスモバイオ:354534)などのコラーゲン;ポリ-D-リシン(サーモ:132703);ゼラチン(CR:354654);フィブロネクチン(BD:356242);ポリ-L-リシン(サーモ);ポリ-L-オルニチン(バイオコート);ラミニン(BD);マトリゲルバイオコート(BD);3次元培養用足場AteloCell(コーケン;No.CSM-50)などを用いることができる。培養容器の形状や素材は特に限定されないが、ポリスチレン製のフラスコ、シャーレおよびマルチウェルプレートが好ましい。
本発明の分化細胞を製造するためのキットに含まれる各構成成分は、市販されている培地(液体および固形)、血清(液体および固形)、血清代替物質、増殖因子、培地添加剤(各種アミノ酸含む)、培養容器が一般的に用いられるが、特に限定されるものではない。たとえば、培地は、製造業者により開示されている組成を参考にして作製したものを用いてもよい。
本発明の第1の細胞株および第2の細胞株のより好ましい態様は、本発明の第1の細胞株および第2の細胞株の特性を併せ持った細胞株であり、たとえば、後述する実施例に記載のカワハギ(Stephanolepis cirrhifer)の背鰭に由来するKSC細胞である。KSC細胞は、微生物の識別の表示を「KSC」とし、かつ、受託番号を「NITE BP-1369」として、独立行政法人製品評価技術基盤機構の特許微生物寄託センター(〒292-0818 千葉県木更津市かずさ鎌足2-5-8)に2012年6月1日付けの受託日により寄託されている。
本発明の別の側面によれば、本発明の第1の細胞株および/または第2の細胞株を培養することにより得られる、これらの細胞株からなる培養細胞シートが提供される。本発明の培養細胞シートは、高級魚類の治療への応用が期待できるものである。特に、養殖魚は免疫が弱く傷害致死率が高いことから、本発明の培養細胞シートは、養殖魚の傷害致死率の低下に寄与し得るものである。また、本発明の培養細胞シートは、合成高分子やセロファンなどの人工的な物質は物質の選択的透過性が制限されることから、これを克服するものとして生体由来選択膜としての利用が期待できる。さらに、本発明の培養細胞シートは、個々の細胞がコラーゲンやフィブロネクチンを産生することからヒト培養細胞シートを形成する際の足場としての利用、細胞に由来する免疫性および抗菌性の観点から生体内への物質輸送カプセル素材、絆創膏、貼付剤などの医療的利用、がん細胞動向の試験への利用、免疫試験への利用、魚類成分を分泌する化粧品としての美容パックとしての利用などに期待できる。また、本発明の培養細胞シートは、シート形成時に底面から剥がれ、浮遊状態で培養を継続できる。本発明の培養細胞シートは、浮遊状態で培養ができるという利点を有する。
3.本発明の細胞株の製造方法
本発明の細胞株の製造方法は、本発明の第1の細胞株および本発明の第2の細胞株を製造する方法であり、たとえば、カワハギ科魚類の生体部位から単離した細胞を30回以上、好ましくは50回以上、より好ましくは70回以上の継代培養に供する工程(以下、継代培養工程ともよぶ。)を含む方法が挙げられる。継代培養工程は、最終的に本発明の細胞株が得られる工程であれば特に限定されるものではなく、たとえば、上記した「1.本発明の第1の細胞株」および「2.本発明の第2の細胞株」に記載の事項に基づいて、使用する材料、方法、条件、製造物の判定などを決定して実施することができる。また、本発明の細胞株の製造方法は、本発明の細胞株を製造するという目的を達成し得る限り、継代培養工程を実施している途中で細胞を保存および復帰するなど、継代培養工程の前、途中および後に適当な工程を設けることができる。
本発明の細胞株の製造方法は、本発明の第1の細胞株および本発明の第2の細胞株を製造する方法であり、たとえば、カワハギ科魚類の生体部位から単離した細胞を30回以上、好ましくは50回以上、より好ましくは70回以上の継代培養に供する工程(以下、継代培養工程ともよぶ。)を含む方法が挙げられる。継代培養工程は、最終的に本発明の細胞株が得られる工程であれば特に限定されるものではなく、たとえば、上記した「1.本発明の第1の細胞株」および「2.本発明の第2の細胞株」に記載の事項に基づいて、使用する材料、方法、条件、製造物の判定などを決定して実施することができる。また、本発明の細胞株の製造方法は、本発明の細胞株を製造するという目的を達成し得る限り、継代培養工程を実施している途中で細胞を保存および復帰するなど、継代培養工程の前、途中および後に適当な工程を設けることができる。
4.本発明の形質転換体、製造方法およびキット
本発明の形質転換体は、本発明の細胞株に外来遺伝子を導入してなるものである。また、本発明の形質転換体の製造方法は、本発明の細胞株に外来遺伝子を導入して形質転換体を得る工程を含む方法である。本発明の外来遺伝子産物の製造方法は、本発明の形質転換体から外来遺伝子の発現産物を得る工程を含む方法である。
本発明の形質転換体は、本発明の細胞株に外来遺伝子を導入してなるものである。また、本発明の形質転換体の製造方法は、本発明の細胞株に外来遺伝子を導入して形質転換体を得る工程を含む方法である。本発明の外来遺伝子産物の製造方法は、本発明の形質転換体から外来遺伝子の発現産物を得る工程を含む方法である。
本発明の細胞株に外来遺伝子を導入する手法は特に限定されず、たとえば、リポフェクション法、エレクトロポレーション法、マイクロインジェクション法、リン酸カルシウム法、バキュロウィルスを用いる手法などの当業者により知られているトランスフェクション手法を採用できる。また、細胞株への外来遺伝子の導入は、外来遺伝子そのものを本発明の細胞株の核内へ導入してもよいし、ベクターなどの遺伝子導入用物質を利用して、たとえば、外来遺伝子を導入した組換えベクターの形で本発明の細胞株内へ導入してもよい。
ベクターは、自律的に複製することが可能なベクターであれば特に限定されないが、たとえば、市販されている哺乳類細胞用のベクター、より具体的にはpcDNA3.1、Flexi HaloTag、pcDNA3.2、pcDNA4、pcDNA5、pcDNA6、pCMVなどのプラスミドベクターや;λファージ、RSVなどのウイルスベクター;アフリカツメガエル由来EF1αプロモーターを有する両生類由来のベクターなどが挙げられる。
導入すべき外来遺伝子は特に限定されず、たとえば、機能性タンパク質、食用タンパク質、酵素、マーカー用タンパク質、標識用タンパク質などの、所望のタンパク質をコードする遺伝子であればよい。本発明の細胞株は、魚類由来の細胞株であることから、魚類特有のタンパク質をコードする遺伝子を外来遺伝子とすることもできる。外来遺伝子は、所望のタンパク質のアミノ酸配列の情報に基づいて作製でき、市販のものを適宜改変して使用することができる。外来遺伝子がコードするタンパク質は1種類でも2種類以上でもよい。たとえば、所望のタンパク質と抗生物質耐性マーカーとをコードする外来遺伝子であれば、遺伝子導入後に抗生物質選択性により外来遺伝子が導入された形質転換体のみを効率よく取得することが可能である。
本発明の形質転換体の製造方法および本発明の外来遺伝子産物の製造方法の具体例は、特に限定されるものではないが、後述する実施例の「6.KSC細胞への外来遺伝子の導入および発現」に記載の方法を挙げることができる。本発明の方法を実施するために用いられる遺伝子工学的手法や分子生物学的手法は、これまでに知られている手法を制限なく用いることができ、たとえば、Molecular Cloning:A laboratory Manual,2nd Ed., Cold Spring Harbor Laboratory,Cold Spring Harbor,NY.,1989やCurrent Protocols in Molecular Biology,Supplement 1~38,John Wiley&Sons(1987-1997)(これらの文献の記載はここに開示として援用される)などに記載されている方法を参照することができる。
本発明の形質転換体の製造方法によって形質転換体が得られたか否かは、導入した外来遺伝子の発現産物の有無を指標に確認できる。同様に、本発明の外来遺伝子産物の製造方法によって外来遺伝子産物が得られたか否かは、外来遺伝子の発現産物の有無を指標に確認できる。本発明の外来遺伝子産物の製造方法では、本発明の形質転換体を培養することにより外来遺伝子を発現させて外来遺伝子産物を得てもよいし、本発明の形質転換体を培養して増殖させた後、外来遺伝子発現を誘導して外来遺伝子産物を得てもよい。また、外来遺伝子産物は、外来遺伝子の種類に応じて、本発明の形質転換体内に蓄積した発現産物または培養液中に蓄積した発現産物として得ることができる。外来遺伝子産物の確認は、当業者により知られる特定のタンパク質の存在を知る手法であれば特に限定されないが、たとえば、外来遺伝子産物の分子量や外来遺伝子産物に対する抗体などを用いたウエスタンブロット法、イムノアッセイ法、クロマトグラフィー法などを採用することができる。
本発明の形質転換体を製造するためのキットは、本発明の細胞株を含めば特に限定されないが、本発明の細胞株に加えて、外来遺伝子を導入するためのベクターとトランスフェクションのための器具、試薬、装置などの器材とを含むことが好ましい。
トランスフェクションのための器材は、各トランスフェクション手法を実施する際に使用されるものであれば特に限定されず、たとえば、トランスフェクション手法がリポフェクション法である場合は、リポソーム、培地、緩衝液などの試薬および培養容器などの器具であることが好ましい。
5.本発明の細胞株の用途
本発明の細胞株の用途は特に限定されず、たとえば、上記した用途以外にも、魚類の細胞生理、魚類遺伝子とその発現系、魚病ウイルス、水環境汚染評価の実験系などの合目的な研究に加え、生理活性物質の評価、魚類ウイルスとの組合せによる抗ウイルス剤のスクリーニングなどにも利用することができる。たとえば、本発明の細胞株は、本発明の細胞株の培養系に被験物質を導入し、次いで被験物質の生細胞に対する影響を評価する工程を含む、被験物質評価方法;本発明の細胞株の培養系にウイルスを接種し、当該ウイルスの増殖に適した栄養培地でウイルスを増殖させる工程を含む、ウイルス増殖方法;ウイルス増殖方法においてウイルスの増殖している培地中に被験物質を導入し、次いで被験物質のウイルスに対する作用を評価する工程を含む、ウイルス治療剤をスクリーニングする方法などに利用できる。さらにこれらの方法を応用して、ウイルス感染症を診断する方法、ウイルス感染症に対して有効なワクチンを製造する方法、細胞毒性を評価する方法などに本発明の細胞株を利用できる。たとえば、本発明の細胞株の生存数と、被験物質を接触させた後の細胞株の生存数とを比較することにより、被験物質の細胞毒性を評価することが期待できる。また、被験物質と本発明の細胞株とを接触させる工程および筋細胞、皮膚細胞、神経細胞または脂肪細胞を検出することにより、被験物質を分化誘導物質と判定する工程によれば、分化誘導物質をスクリーニングすることが期待できる。
本発明の細胞株の用途は特に限定されず、たとえば、上記した用途以外にも、魚類の細胞生理、魚類遺伝子とその発現系、魚病ウイルス、水環境汚染評価の実験系などの合目的な研究に加え、生理活性物質の評価、魚類ウイルスとの組合せによる抗ウイルス剤のスクリーニングなどにも利用することができる。たとえば、本発明の細胞株は、本発明の細胞株の培養系に被験物質を導入し、次いで被験物質の生細胞に対する影響を評価する工程を含む、被験物質評価方法;本発明の細胞株の培養系にウイルスを接種し、当該ウイルスの増殖に適した栄養培地でウイルスを増殖させる工程を含む、ウイルス増殖方法;ウイルス増殖方法においてウイルスの増殖している培地中に被験物質を導入し、次いで被験物質のウイルスに対する作用を評価する工程を含む、ウイルス治療剤をスクリーニングする方法などに利用できる。さらにこれらの方法を応用して、ウイルス感染症を診断する方法、ウイルス感染症に対して有効なワクチンを製造する方法、細胞毒性を評価する方法などに本発明の細胞株を利用できる。たとえば、本発明の細胞株の生存数と、被験物質を接触させた後の細胞株の生存数とを比較することにより、被験物質の細胞毒性を評価することが期待できる。また、被験物質と本発明の細胞株とを接触させる工程および筋細胞、皮膚細胞、神経細胞または脂肪細胞を検出することにより、被験物質を分化誘導物質と判定する工程によれば、分化誘導物質をスクリーニングすることが期待できる。
以下、本発明を実施例によりさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。
1.カワハギ組織からの細胞の単離
カワハギ(Stephanolepis cirrhifer)の各鰭部を、皮を剥ぐことなく5mm角に切り取ったものを1.5mlの滅菌したチューブに入れて、氷冷下で水道水、PBS(和光純薬)の順で洗浄した。洗浄した鰭片を、10% ペニシリン/ストレプトマイシン(MPバイオメディカルズ)含有PBSを用いて氷冷下で3度置換し、30分静置した。なお、「置換」とは、PBSが適量入ったチューブを用意し、そのPBS含チューブに鰭片を入れて攪拌した後、鰭片を別のPBS含チューブに入れる操作をいう。
カワハギ(Stephanolepis cirrhifer)の各鰭部を、皮を剥ぐことなく5mm角に切り取ったものを1.5mlの滅菌したチューブに入れて、氷冷下で水道水、PBS(和光純薬)の順で洗浄した。洗浄した鰭片を、10% ペニシリン/ストレプトマイシン(MPバイオメディカルズ)含有PBSを用いて氷冷下で3度置換し、30分静置した。なお、「置換」とは、PBSが適量入ったチューブを用意し、そのPBS含チューブに鰭片を入れて攪拌した後、鰭片を別のPBS含チューブに入れる操作をいう。
静置後の鰭片を、1% ペニシリン/ストレプトマイシン含有PBSを用いて氷冷下で3度置換した。処理後の鰭片を、クリーンベンチ内で、1% ペニシリン/ストレプトマイシン含有PBSの代わりに、トリプシンEDTA(MPバイオメディカルズ)を加え、氷冷下で組織片を1mm角に細かく刻み、次いで室温で20分間静置した。静置後の組織片を遠心分離機に数秒間かけた後、上清を取り除いた。沈殿した組織片にLeibovitz’s L-15培地(ライフテクノロジーズ)で数回処理した。すなわち、チューブ内の組織片を遠沈させ、上清を除いた後、新鮮培地を加えて攪拌した。その後、組織片を再度遠沈させ、上清を除いた後、再度新鮮培地を加えて攪拌した。この操作を2~3回繰り返した。Leibovitz’s L-15培地の組成は表1のとおりである(http://ja.invitrogen.com/site/jp/ja/home/support/Product-Technical-Resources/media_formulation.80.html、該文献の記載はここに開示として援用される)。

処理後の組織片を、10% FBS(ギブコ)を含むLeibovitz’s L-15培地の入った25cm2 CollagenIカルチャーフラスコ(サーモ)内に播き、底着させた後、25℃で培養した。図1A~Dは、各鰭片を中心として、フラスコ全体に遊走した細胞が展開した様相を示す。以下では、背鰭片から摘出した細胞の継代培養手順について述べる。
2.継代培養
カルチャーフラスコ中の培地を除去し、フラスコ底面に付着している細胞をPBSで3回洗浄した。TrypLE Express(ギブコ)を用いて38℃で10分間処理して、細胞を剥離および回収した。回収した細胞に、10% FBSを含むLeibovitz’s L-15培地を加え、25℃で培養した。
カルチャーフラスコ中の培地を除去し、フラスコ底面に付着している細胞をPBSで3回洗浄した。TrypLE Express(ギブコ)を用いて38℃で10分間処理して、細胞を剥離および回収した。回収した細胞に、10% FBSを含むLeibovitz’s L-15培地を加え、25℃で培養した。
細胞の保存は、回収した細胞に培地を加えた後、室温、1,100rpmで4分間遠心し、次いで上清を除去した後、セルバンカー2(十慈フィールド)を用いて、-80℃で保存した。また、凍結細胞を起こす手順として、まず、凍結細胞を室温で半解凍状態にし、Leibovitz’s L-15培地で2回培地の置換を行った。この置換は、半解凍状態のチューブから液体部分をとり、15mlの遠沈管にうつした後、L-15培地4mlを加え撹拌し遠心分離を行った。次いで、上清を除き再度L-15培地を4ml加え撹拌し遠心分離を行った。次いで、上清を除いた後、細胞に10% FBSを含むLeibovitz’s L-15培地を加えて、継代培養に供した。
3.細胞の株化の評価および25cm2フラスコにおける増殖速度
図1に示す組織周縁に集まった初代培養細胞は、通常、50~80回の分裂後に細胞の分裂限界に達する(Hayflick limit;Hayflick L,Moorhead PS,1961,Exp Cell Res.25:585-621を参照、該文献の記載はここに開示として援用される)。しかし、中には、上記分裂回数を過ぎても、倍加時間(Doubling time)を低下させず、分裂を繰り返す細胞がある。そのような細胞が、不死化細胞(株化細胞、細胞株など)と定義されている。
図1に示す組織周縁に集まった初代培養細胞は、通常、50~80回の分裂後に細胞の分裂限界に達する(Hayflick limit;Hayflick L,Moorhead PS,1961,Exp Cell Res.25:585-621を参照、該文献の記載はここに開示として援用される)。しかし、中には、上記分裂回数を過ぎても、倍加時間(Doubling time)を低下させず、分裂を繰り返す細胞がある。そのような細胞が、不死化細胞(株化細胞、細胞株など)と定義されている。
本発明者らは、上記1および2を実施した結果、継代回数が70回以上であり、かつ、分裂回数が276回以上である細胞を得ることに成功した。この細胞をKSC細胞と名づけた。KSC細胞は、倍加時間について、第1継代培養細胞とほとんど変化がなかった。したがって、KSC細胞は、株化されたと評価した。これは、JRCB細胞バンクの定義(http://cellbank.nibio.go.jp/visitercenter/whatsculture/cellculture03.html、該文献の記載はここに開示として援用される)に従うものである。
細胞の増殖率は、細胞の密度(濃度)や培養面積など条件によって大きく変動することが知られている。そこで、KSC細胞と、哺乳類細胞の株化細胞の中でも比較的増殖率の高いCHO細胞との倍加時間を比較した。CHO細胞のデータは、細胞バンク規模で世界第2位であり、かつ、世界で唯一詳細な細胞データを完備して信頼性が高いと思われるドイツのDSMZ(http://www.dsmz.de/、該文献の記載はここに開示として援用される)のデータを利用した。
DSMZの細胞データより、CHO細胞は、約80cm2に106cells/mlの細胞を播種すると72~96時間でコンフルエンス(細胞がフラスコ底面全体に展開するまでに増殖した状態)に達し、倍加時間は24時間として計算される。それに対して、KSC細胞は、約80cm2に106cells/mlの細胞を播種すると、48~72時間でコンフルエンスに達し、倍加時間は18.34時間として算出された。したがって、KSC細胞は、CHO細胞と同程度またはそれより優れた増殖能をもった細胞であることがわかった。
また、KSC細胞について、約0.4×106cells/mlの細胞を75cm2の面積に播き、10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて、コンフルエントに達する48時間後まで培養した際の全細胞数をTC10 Automated Cell Counter(バイオラッド)を用いて計測したところ、培養24時間後の細胞数は約1.0×106cells/mlであり、培養48時間後の細胞数は約2.5×106cells/mlであった。ウェブ上の倍加時間計測ソフトウェア(http://www.doubling-time.com/compute.php?lang=en)を用いて、播いた直後の細胞数と培養48時間後の細胞数とから倍加時間を算出したところ、18.16時間であった。同様に、播いた直後の細胞数と培養24時間後の細胞数とから算出された倍加時間は18.16時間であり、培養24時間後の細胞数と培養48時間後の細胞数とから算出された倍加時間は18.16時間であった。
4.凍結状態からの復帰
セルバンカー2を用いて凍結保存したKSC細胞について、解凍後の細胞生存率を評価した。図2は、継代回数に係わらず、解凍後のKSC細胞は75%以上の生存率であったことを示す。
セルバンカー2を用いて凍結保存したKSC細胞について、解凍後の細胞生存率を評価した。図2は、継代回数に係わらず、解凍後のKSC細胞は75%以上の生存率であったことを示す。
5.細胞の形態観察
株化したKSC細胞は、線維芽細胞様の形態を示した(図3Aを参照)。また、KSC細胞をフラスコ底面で培養すると、コンフルエントになった後、2層構造を形成した(図3Bを参照)。さらに、この状態で培養を続けると多層構造を形成した(図3Cを参照)。
株化したKSC細胞は、線維芽細胞様の形態を示した(図3Aを参照)。また、KSC細胞をフラスコ底面で培養すると、コンフルエントになった後、2層構造を形成した(図3Bを参照)。さらに、この状態で培養を続けると多層構造を形成した(図3Cを参照)。
6.KSC細胞への外来遺伝子の導入および発現
(1)方法
哺乳類細胞で使われている各種遺伝子導入法(トランスフェクション法)がKSC細胞で適用可能であることを、リポフェクタミン法を含むリポフェクション法、エレクトロポレーション法、マイクロインジェクション法およびバキュロウィルスを用いた手法により検証した。各手法の詳細は、下記に示す各製造業者の指示に準じた:http://tools.invitrogen.com/content/sfs/manuals/lipofectamine2000_man.pdf;http://www.invitrogen.jp/transfection/pdf/Neon_quickguide_JPN.pdf;http://www.eppendorf.de/int/img/na/lit/pdf/8301-C109F-07.pdf#search=%27FemtoJetMicroinjector%27;http://probes.invitrogen.com/media/pis/mp10582.pdf#search=%27celllight%20pdf%27、該文献の記載はここに開示として援用される。
(1)方法
哺乳類細胞で使われている各種遺伝子導入法(トランスフェクション法)がKSC細胞で適用可能であることを、リポフェクタミン法を含むリポフェクション法、エレクトロポレーション法、マイクロインジェクション法およびバキュロウィルスを用いた手法により検証した。各手法の詳細は、下記に示す各製造業者の指示に準じた:http://tools.invitrogen.com/content/sfs/manuals/lipofectamine2000_man.pdf;http://www.invitrogen.jp/transfection/pdf/Neon_quickguide_JPN.pdf;http://www.eppendorf.de/int/img/na/lit/pdf/8301-C109F-07.pdf#search=%27FemtoJetMicroinjector%27;http://probes.invitrogen.com/media/pis/mp10582.pdf#search=%27celllight%20pdf%27、該文献の記載はここに開示として援用される。
(2)材料
外来遺伝子を導入する際に使用した組換え用遺伝子として、Flexi HaloTag Vector(プロメガ)、Flexi HaloTag Clone(プロメガ)およびpcDNA3.1 Vector(ライフテクノロジーズ)を用いた。
外来遺伝子を導入する際に使用した組換え用遺伝子として、Flexi HaloTag Vector(プロメガ)、Flexi HaloTag Clone(プロメガ)およびpcDNA3.1 Vector(ライフテクノロジーズ)を用いた。
各トランスフェクション法では、次の試薬等を用いた:Lipofectamine 2000 ReagentおよびPLUS Reagent(ライフテクノロジーズ);Neon Transfection System(ライフテクノロジーズ);FemtoJet-Microinjector(エッペンドルフ);CellLight(ライフテクノロジーズ)。
(3)遺伝子導入結果
哺乳動物の細胞系で通常に使用されている上記方法を用いることにより、KSC細胞へ外来遺伝子の導入が可能であることが確認された。
哺乳動物の細胞系で通常に使用されている上記方法を用いることにより、KSC細胞へ外来遺伝子の導入が可能であることが確認された。
(4)外来遺伝子産物であるヒトアルカリホスファターゼ(ヒト-AP)の発現
KSC細胞へ導入した外来遺伝子の遺伝子産物であるFlexi HaloTag ヒト-APの発現をHaloTag Oregon Green Ligandにより蛍光検出した(図4を参照)。生産されたヒト-APの活性は、Ziva Ultra SEAP Plus Detection Kit(フナコシ)を用いて測定した(図5を参照)。図4および図5が示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認され、さらに発現産物は活性があるものであった。
KSC細胞へ導入した外来遺伝子の遺伝子産物であるFlexi HaloTag ヒト-APの発現をHaloTag Oregon Green Ligandにより蛍光検出した(図4を参照)。生産されたヒト-APの活性は、Ziva Ultra SEAP Plus Detection Kit(フナコシ)を用いて測定した(図5を参照)。図4および図5が示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認され、さらに発現産物は活性があるものであった。
(5)外来遺伝子産物であるヒトパラオキソナーゼ(PON)の発現
KSC細胞へ導入した外来遺伝子の遺伝子産物であるFlexi HaloTag PONの発現をTMR-Ligandにより蛍光検出した(図6を参照)。図6が示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認された。
KSC細胞へ導入した外来遺伝子の遺伝子産物であるFlexi HaloTag PONの発現をTMR-Ligandにより蛍光検出した(図6を参照)。図6が示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認された。
(6)外来遺伝子産物である蛍光タンパク質の発現
KSC細胞へ導入した外来遺伝子の遺伝子産物である蛍光タンパク質のシアン・フルオレセント・プロテイン-イエロー・フルオレセント・プロテイン(Cyan Fluorescent Protein-Yellow Fluorescent Protein)およびカルモジュリン;レッド・フルオレセント・プロテイン(Red Fluorescent Protein)およびベシキュラー・ストマティティス・ウイルス(Vesicular Stomatitis Virus)のG糖タンパク質;ならびにグリーン・フルオレセント・プロテイン(Green Fluorescent Protein)を発現させた細胞を蛍光顕微鏡により観察した(図7A~Cを参照)。図7A~Cが示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認された。
KSC細胞へ導入した外来遺伝子の遺伝子産物である蛍光タンパク質のシアン・フルオレセント・プロテイン-イエロー・フルオレセント・プロテイン(Cyan Fluorescent Protein-Yellow Fluorescent Protein)およびカルモジュリン;レッド・フルオレセント・プロテイン(Red Fluorescent Protein)およびベシキュラー・ストマティティス・ウイルス(Vesicular Stomatitis Virus)のG糖タンパク質;ならびにグリーン・フルオレセント・プロテイン(Green Fluorescent Protein)を発現させた細胞を蛍光顕微鏡により観察した(図7A~Cを参照)。図7A~Cが示すとおり、KSC細胞へ導入された外来遺伝子の発現が確認された。
7.トランスフェクション(遺伝子導入)における細胞安定性の評価
上記Lipofectamine法による遺伝子導入における細胞毒性(耐性)をKSC細胞、ラット間葉系幹細胞(MSC)、ヒト急性T細胞性白血病細胞株(Jurkat)およびマウス胚性腫瘍細胞(P19)を用いて比較検証した。その結果、KSC細胞は、外来遺伝子導入前(コントロール)との比較から、遺伝子導入後(トランスフェクト済)の生存率は90%以上であり、他の細胞に比べて優れた細胞安定性を示すことがわかった(図8)。
上記Lipofectamine法による遺伝子導入における細胞毒性(耐性)をKSC細胞、ラット間葉系幹細胞(MSC)、ヒト急性T細胞性白血病細胞株(Jurkat)およびマウス胚性腫瘍細胞(P19)を用いて比較検証した。その結果、KSC細胞は、外来遺伝子導入前(コントロール)との比較から、遺伝子導入後(トランスフェクト済)の生存率は90%以上であり、他の細胞に比べて優れた細胞安定性を示すことがわかった(図8)。
8.KSC細胞の高密度培養の可能性検証
KSC細胞の浮遊培養の可能性を検証した。その結果、スピナーフラスコを用いて、10% FBSを含むLeibovitz’s L-15培地で、CO2非存在下、100rpm、25℃で培養したところ、細胞の増殖が確認された。本条件の浮遊培養では、TC10 Automated Cell Counter(バイオラッド)を用いて細胞数を計測することにより、KSC細胞は約26時間に1回の割合で細胞分裂することが確認された。
KSC細胞の浮遊培養の可能性を検証した。その結果、スピナーフラスコを用いて、10% FBSを含むLeibovitz’s L-15培地で、CO2非存在下、100rpm、25℃で培養したところ、細胞の増殖が確認された。本条件の浮遊培養では、TC10 Automated Cell Counter(バイオラッド)を用いて細胞数を計測することにより、KSC細胞は約26時間に1回の割合で細胞分裂することが確認された。
9.無血清培養の可能性検証
KSC細胞の無血清培養における倍加時間を検証した。25cm2 CollagenIカルチャーフラスコを用いて、Leibovitz’s L-15培地のみの培養(無血清培養)およびLeibovitz’s L-15培地および血清代替添加物(KSR;ライフテクノロジーズ)の培養(血清代替物培養)について、KSC細胞の倍加時間を検証した。無血清培養では、FBS血清を添加する通常培養と比べて、倍加時間が大きくなるものの、KSC細胞の増殖は確認できた。また、KSC細胞は、血清代替添加物を使用することによって、通常培養と変わらない増殖速度での培養が可能であった。
KSC細胞の無血清培養における倍加時間を検証した。25cm2 CollagenIカルチャーフラスコを用いて、Leibovitz’s L-15培地のみの培養(無血清培養)およびLeibovitz’s L-15培地および血清代替添加物(KSR;ライフテクノロジーズ)の培養(血清代替物培養)について、KSC細胞の倍加時間を検証した。無血清培養では、FBS血清を添加する通常培養と比べて、倍加時間が大きくなるものの、KSC細胞の増殖は確認できた。また、KSC細胞は、血清代替添加物を使用することによって、通常培養と変わらない増殖速度での培養が可能であった。
10.染色体解析
(1)試薬
コルセミド溶液として、KaryoMAX-COLCEMID-PBS溶液(10μg/ml)(ライフテクノロジーズ)を用いた。低張液として、0.075M KClを用いた。固定液として、メタノール:酢酸=3:1溶液を用時調製して用いた。
(1)試薬
コルセミド溶液として、KaryoMAX-COLCEMID-PBS溶液(10μg/ml)(ライフテクノロジーズ)を用いた。低張液として、0.075M KClを用いた。固定液として、メタノール:酢酸=3:1溶液を用時調製して用いた。
(2)染色体標本の作成
コルセミドは細胞周期をM期で止めることから、コルセミド処理によりM期の細胞数が増える。しかし、処理時間を長くすると、染色体が短縮して、解析ができなくなる可能性がある。そこで、80~90%程度のコンフルエンスに達した細胞の中から、対数増殖期の細胞に対して0.02μg/mlになるようにコルセミド溶液を添加した後、約5時間培養を継続してコルセミド処理を実施した。
コルセミドは細胞周期をM期で止めることから、コルセミド処理によりM期の細胞数が増える。しかし、処理時間を長くすると、染色体が短縮して、解析ができなくなる可能性がある。そこで、80~90%程度のコンフルエンスに達した細胞の中から、対数増殖期の細胞に対して0.02μg/mlになるようにコルセミド溶液を添加した後、約5時間培養を継続してコルセミド処理を実施した。
次いで、処理後の細胞をトリプシン処理して剥離した後、細胞剥離液を15mlチューブに入れて、1,100rpmで4分間遠心し、上清を除去して細胞を回収した。本操作は、細胞がコルセミド処理時間に影響することから、迅速に行った。
次いで、回収した細胞を含む15mlチューブに、ピペットで少量の低張液を加え、静かにピペッティングすることにより、細胞を分散させた。分散後の細胞を含む15mlチューブに、最終容量が1.5mlになるまで低張液を加えた後、ピペッティングにより再度細胞を分散させた。低張液に分散した細胞を、室温にて20分間放置して低張処理を行った。
次いで、総量が10ml程度になるようにゆっくりと固定液を加え、静かに撹拌し細胞を固定化した。さらに1,100rpmで4分間遠心して上清を除去し、新たな固定液を遠心沈降した細胞に数滴加えて、ピペッティングにより細胞を分散させた。さらに10ml程度の固定液を加えた後、全体を撹拌した。この作業をさらに2回繰り返し、細胞を完全に固定した。固定した細胞を、ヘキスト・キナクリン(和光純薬)を用いて染色し、光学顕微鏡で観察した。
観察の結果、KSC細胞の細胞集団として、染色体が32個、33個および66個の3タイプの細胞株を含むことが分かった。通常のカワハギは、メス33個、オス34個の染色体を有する。KSC細胞の細胞集団の90%は、カワハギのメスと同じく、染色体数が33個であった。しかし、細胞集団の残り10%の細胞は、32個および66個の染色体を持つ細胞であった。
11.幹細胞マーカーによる多分化能解析
(1)方法
以下の手順に従い、KSC細胞を免疫化学染色した。なお、ブロッキング・バッファーには、10% FBSおよび0.1% Triton X-100を含むPBS溶液を用いた。また、抗体希釈バッファーには、3% ヤギ血清および0.1% Triton X-100を含むPBS溶液を用いた。
(1)方法
以下の手順に従い、KSC細胞を免疫化学染色した。なお、ブロッキング・バッファーには、10% FBSおよび0.1% Triton X-100を含むPBS溶液を用いた。また、抗体希釈バッファーには、3% ヤギ血清および0.1% Triton X-100を含むPBS溶液を用いた。
まず、KSC細胞を培養フラスコ内でコンフルエントにまで培養した後、培地を除き、細胞を室温にてPBSで2回洗浄した。細胞に4% パラホルムアルデヒドを加えて、20分間室温で静置して細胞を固定化した。パラホルムアルデヒドを除き、10分間隔で3回PBSを用いて固定化細胞を洗浄した。ブロッキング・バッファーを加えて、1時間室温で静置した。ブロッキング・バッファーを除き、ブロッキング処理後の細胞をPBSで1回洗浄した。抗体希釈バッファーを用いて100倍に希釈した一次抗体(ES/iPS Cell Characterization Kit;フナコシ)を加えて、4℃にてオーバーナイトで一次抗体反応を実施した。翌日、一次抗体を除いた後、10分間隔で5回、反応後の細胞をPBSで洗浄した。次いで、抗体希釈バッファーで希釈した二次抗体を加えて、室温で1時間静置して二次抗体反応を実施した。二次抗体について、抗TRA-1-60抗体に対してAnti-IgG+IgM(H+L),Mouse,Goat-Poly,FITC;抗SSEA-3抗体に対してAnti-RAT IgM(mu chain)(GOAT),DyLight 488;抗NANOG抗体に対してAnti-IgG(H+L),Rabbit,Goat-Poly,DyLight 488;抗OCT4抗体に対してAnti-IgG(H+L),Rabbit,Goat-Poly,DyLight 488を用いた。この際、細胞を固定化させたプレートを光から保護した。次いで、二次抗体を取り除き、反応後の細胞をPBSにより10分間隔で4回洗浄した。この際にも、プレートを光から保護した。洗浄後の細胞をFLoidセルイメージングステーション(ライフテクノロジーズ)を用いて観察した。
(2)結果
観察結果を図9A~Dに示す。KSC細胞の大部分は、TRA-1-60、OCT4およびSSEA-3を発現しており陽性であった。発現強度は、TRA-1-60、OCT4およびSSEA-3の順であった。それに対して、KSC細胞はNANOG陰性または発現していたとしても非常に微かな量であった。
観察結果を図9A~Dに示す。KSC細胞の大部分は、TRA-1-60、OCT4およびSSEA-3を発現しており陽性であった。発現強度は、TRA-1-60、OCT4およびSSEA-3の順であった。それに対して、KSC細胞はNANOG陰性または発現していたとしても非常に微かな量であった。
若尾昌平らの文献(Shohei Wakao et al.、PNAS、June 14,2011、vol.108、no.24、pp.9875-9880、該文献の記載はここに開示として援用される)には、多分化能を有するヒト皮膚線維芽細胞(Muse細胞)の存在が報告されている。以上の結果より、KSC細胞は、TRA-1-60、OCT4およびSSEA-3といった多能性幹細胞に特有の細胞マーカーを発現する線維芽細胞様細胞であることから、多能性を有する細胞株であると推測される。
12.分化誘導実験
KSC細胞が多能性を有する細胞株であると推測されたことから、培地や培養条件を変えることにより、種々の細胞への分化を試みた。細胞の形態は、光学顕微鏡により460倍の倍率で観察した。
KSC細胞が多能性を有する細胞株であると推測されたことから、培地や培養条件を変えることにより、種々の細胞への分化を試みた。細胞の形態は、光学顕微鏡により460倍の倍率で観察した。
(1)KSC細胞の形態(分化誘導前)
KSC細胞をコラーゲンコートフラスコにて10%(v/v) FBSを含むLeibovitz’s L-15培地を用いて培養した。培養後の細胞の観察結果を図10Aに示す。図10Aに示す線維芽細胞様の形態は、KSC細胞が他の細胞へ分化する前の形態であるものと推測される。
KSC細胞をコラーゲンコートフラスコにて10%(v/v) FBSを含むLeibovitz’s L-15培地を用いて培養した。培養後の細胞の観察結果を図10Aに示す。図10Aに示す線維芽細胞様の形態は、KSC細胞が他の細胞へ分化する前の形態であるものと推測される。
(2)筋細胞様細胞への分化
KSC細胞を、25cm2または75cm2 のコラーゲンコートフラスコにて、10%(v/v) FBSを含むAIM V培地(ライフテクノロジーズ;No.087-0112DK)を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、筋細胞様の形態を呈するものであった(図10Bを参照)。また、培養後の細胞は、増殖能を有するものであった。
KSC細胞を、25cm2または75cm2 のコラーゲンコートフラスコにて、10%(v/v) FBSを含むAIM V培地(ライフテクノロジーズ;No.087-0112DK)を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、筋細胞様の形態を呈するものであった(図10Bを参照)。また、培養後の細胞は、増殖能を有するものであった。
(3)上皮細胞様細胞への分化
KSC細胞を、25cm2または75cm2 の非コーティングフラスコにて、10%(v/v) FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、上皮細胞様の形態を呈するものであった(図10Cを参照)。また、培養後の細胞は、増殖可能なものであった。
KSC細胞を、25cm2または75cm2 の非コーティングフラスコにて、10%(v/v) FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、上皮細胞様の形態を呈するものであった(図10Cを参照)。また、培養後の細胞は、増殖可能なものであった。
(4)神経細胞様細胞への分化
KSC細胞を、25cm2または75cm2 の非コーティングフラスコにて、無血清のLeibovitz’s L-15培地を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、神経様軸策および融合した細胞体を形成するものであった(図10Dを参照)。また、培養後の細胞は、増殖性がなく、かつ、伸長性を有するものであった。さらに、培養後の細胞は、神経細胞に特有の細胞マーカー陽性の細胞であった。
KSC細胞を、25cm2または75cm2 の非コーティングフラスコにて、無血清のLeibovitz’s L-15培地を用いて、CO2非存在下で、初期細胞数を1~4×105cells/mlとして、25℃にて90%以上のコンフルエント状態に達するまで培養した。培養後の細胞は、神経様軸策および融合した細胞体を形成するものであった(図10Dを参照)。また、培養後の細胞は、増殖性がなく、かつ、伸長性を有するものであった。さらに、培養後の細胞は、神経細胞に特有の細胞マーカー陽性の細胞であった。
同様にして、KSC細胞を種々の培養容器、血清および培地の組み合わせを用いて培養することや、KSC細胞について分化誘導したところ、脂肪細胞様細胞、繊維細胞様細胞、肝臓細胞様細胞、バラの枝状の免疫細胞様細胞および巨大繭状の融合細胞を得ることができた(図10E~Iを参照)。特に、図10Iに示される融合細胞は、アルカリフォスファターゼ活性陽性であり(図11Aを参照)、かつ、NANOG陽性であった(図11Bを参照)。さらに、本融合細胞は、TRA-1-60、OCT4およびSSEA-3についても陽性であった(図11C~Eを参照)。
13.培養細胞シートの作製
10% FBS含有Leibovitz’s L-15培地を用いて、コラーゲンIコートフラスコにKSC細胞(1×105cells/ml)を播いた。3日に1度程度の培地交換を繰り返して、1~3カ月間培養を続けたところ、薄膜状の培養細胞シートが形成された(図12を参照)。また、培地に増殖因子・培地添加剤として分化誘導剤Noggin(ヒューマンザイム)またはG-CSF(ヒューマンザイム)を終濃度500pg/mlとなるように添加したところ、シート形成効率を高めることができ、培養細胞シートの形成速度が促進された。形成された培養細胞シートは、細胞同士が結合してシート状または薄膜状の形態であり、キメが細かく目視では細胞の形を認識できないという特徴を有するものであった。
10% FBS含有Leibovitz’s L-15培地を用いて、コラーゲンIコートフラスコにKSC細胞(1×105cells/ml)を播いた。3日に1度程度の培地交換を繰り返して、1~3カ月間培養を続けたところ、薄膜状の培養細胞シートが形成された(図12を参照)。また、培地に増殖因子・培地添加剤として分化誘導剤Noggin(ヒューマンザイム)またはG-CSF(ヒューマンザイム)を終濃度500pg/mlとなるように添加したところ、シート形成効率を高めることができ、培養細胞シートの形成速度が促進された。形成された培養細胞シートは、細胞同士が結合してシート状または薄膜状の形態であり、キメが細かく目視では細胞の形を認識できないという特徴を有するものであった。
Stephanolepis cirrhifer KSC NITE BP-1369
Claims (26)
- 下記(1)の性質を有する、カワハギ科(Monacanthidae)魚類の生体部位に由来する細胞株またはその継代株。
(1)実質的に制限なく継代培養が可能である - さらに下記(2)の性質を有する、請求項1に記載の細胞株またはその継代株。
(2)線維芽細胞様の形態を呈する - さらに下記(3)の性質を有する、請求項1または2に記載の細胞株またはその継代株。
(3)多層構造を形成した培養が可能である - さらに下記(4)の性質を有する、請求項1~3のいずれか1項に記載の細胞株またはその継代株。
(4)最大値は66本であり、最小値は32本であり、およびモードは33本である度数分布に従った染色体数を有する - 前記(4)の性質において、モードである33本の染色体の度数が度数分布における全度数の約90%である、請求項4に記載の細胞株またはその継代株。
- さらに下記(5)の性質を有する、請求項1~5のいずれか1項に記載の細胞株またはその継代株。
(5)初期細胞数を約1.0×106cells/mlとして、75cm2の底面積を有する培養容器内で10% FBSを含むLeibovitz’s L-15培地を用いて、CO2非存在下で25℃にて培養した際の倍加時間は、約14~28時間である - TRA-1-60、OCT4およびSSEA-3からなる群から選ばれる少なくとも1種の細胞マーカーが陽性である、カワハギ科魚類の生体部位に由来する細胞株またはその継代株。
- 前記細胞株またはその継代株は、線維芽細胞様の形態を呈する細胞株またはその継代株である、請求項7に記載の細胞株またはその継代株。
- 前記細胞株またはその継代株は、筋細胞および筋細胞様細胞、上皮細胞および上皮細胞様細胞、神経細胞および神経細胞様細胞、脂肪細胞および脂肪細胞様細胞、免疫細胞および免疫細胞様細胞ならびに肝臓細胞および肝臓細胞様細胞からなる群から選ばれる少なくとも1種の細胞に分化する能力を有する、請求項7または8に記載の細胞株またはその継代株。
- 前記カワハギ科魚類が、カワハギ属(Stephanolepis)魚類、ウマヅラハギ属(Thamnaconus)魚類、メガネウマヅラハギ属(Cantherhines)魚類、ウスバハギ属(Aluterus)魚類およびアミメハギ属(Rudarius)魚類からなる群から選ばれる魚類である、請求項1~9のいずれか1項に記載の細胞株またはその継代株。
- 前記カワハギ科魚類が、カワハギ(Stephanolepis cirrhifer)である、請求項1~9のいずれか1項に記載の細胞株またはその継代株。
- 前記生体部位は、背部、胸部、腹部、尻部または尾部にある鰭である、請求項1~11のいずれか1項に記載の細胞株またはその継代株。
- 受託番号がNITE BP-1369である、カワハギの背鰭に由来する細胞株またはその継代株。
- カワハギ科魚類の生体部位から単離した細胞を70回以上の継代培養に供する工程を含む、
請求項1~13のいずれか1項に記載の細胞株またはその継代株を製造する方法。 - 請求項1~13のいずれか1項に記載の細胞株またはその継代株に外来遺伝子を導入してなる、形質転換体。
- 請求項1~13のいずれか1項に記載の細胞株またはその継代株に外来遺伝子を導入して形質転換体を得る工程を含む、形質転換体を製造する方法。
- 請求項1~13のいずれか1項に記載の細胞株またはその継代株に外来遺伝子を導入してなる形質転換体から該外来遺伝子の発現産物を得る工程
を含む、外来遺伝子の発現産物を製造する方法。 - 請求項1~13のいずれか1項に記載の細胞株またはその継代株と、
ベクターと、
トランスフェクションのための器材と
を含む、形質転換体を製造するためのキット。 - 請求項7~13のいずれか1項に記載の細胞株またはその継代株と、
培地と、
培養容器と
を含む、分化細胞を製造するためのキット。 - さらに血清を含む、請求項19に記載のキット。
- 前記血清が、哺乳類血清、魚類血清および血清代替物からなる群から選ばれる少なくとも1種の血清である、請求項20に記載のキット。
- 前記培地は、哺乳動物細胞用培地、昆虫細胞用培地および魚類細胞用培地からなる群から選ばれる少なくとも1種の培地である、請求項19~21のいずれか1項に記載のキット。
- 前記培養容器が、底面が細胞接着分子でコーティングされた培養容器および底面が細胞接着分子でコーティングされていない培養容器からなる群から選ばれる少なくとも1種の培養容器である、請求項19~22のいずれか1項に記載のキット。
- 請求項7~13のいずれか1項に記載の細胞株またはその継代株を、底面が細胞接着分子でコーティングされた培養容器または底面が細胞接着分子でコーティングされていない培養容器の中で、血清を含有していない、または哺乳類血清、魚類血清もしくは血清代替物を含有した哺乳動物細胞用培地、昆虫細胞用培地または魚類細胞用培地を用いて培養する工程を含む、
分化細胞を製造する方法。 - 請求項1~13のいずれか1項に記載の細胞株またはその継代株からなる培養細胞シート。
- 請求項1~13のいずれか1項に記載の細胞株またはその継代株から、アルカリフォスファターゼ活性の染色で識別され、かつ、NANOG、OCT4、TRA-1-60およびSSEA-3陽性である細胞または該細胞によって形成される巨大繭状コロニーを製造する工程を含む、
巨大繭状コロニーを製造する方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/418,781 US9777254B2 (en) | 2012-07-31 | 2013-07-30 | Cell line derived from thread-sail filefish (Stephanolepis cirrhifer) |
| EP13825193.9A EP2881460B1 (en) | 2012-07-31 | 2013-07-30 | Cell strain derived from monacanthidae |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-169378 | 2012-07-31 | ||
| JP2012169378A JP5846384B2 (ja) | 2012-07-31 | 2012-07-31 | カワハギ由来の細胞株 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014021329A1 true WO2014021329A1 (ja) | 2014-02-06 |
Family
ID=50028002
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/070627 Ceased WO2014021329A1 (ja) | 2012-07-31 | 2013-07-30 | カワハギ由来の細胞株 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9777254B2 (ja) |
| EP (1) | EP2881460B1 (ja) |
| JP (1) | JP5846384B2 (ja) |
| WO (1) | WO2014021329A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017176005A (ja) * | 2016-03-29 | 2017-10-05 | テルモ株式会社 | 細胞培養物およびその製造方法 |
| CN108893474A (zh) * | 2018-02-07 | 2018-11-27 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113637632A (zh) * | 2021-07-20 | 2021-11-12 | 宁波大学 | 一种银鲳肌肉组织细胞系 |
| CN115595297B (zh) * | 2022-09-22 | 2023-10-13 | 中国水产科学研究院南海水产研究所 | 一种卵形鲳鲹肌肉细胞系及构建方法和应用 |
| WO2026068840A1 (en) * | 2024-09-30 | 2026-04-02 | Bluu Gmbh | Applications of cultured fish cells for the production and enhancement of food |
| WO2026068839A1 (en) * | 2024-09-30 | 2026-04-02 | Bluu Gmbh | Large scale fish cell production |
| CN119979723B (zh) * | 2025-03-13 | 2025-10-03 | 中国水产科学研究院北戴河中心实验站 | 鉴定绿鳍马面鲀性别的特异性dna片段、引物及其应用和鉴定绿鳍马面鲀性别的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004024192A (ja) | 2002-06-28 | 2004-01-29 | Fujikin Inc | チョウザメ由来の新規な株化細胞 |
| JP2008220198A (ja) | 2007-03-09 | 2008-09-25 | Japan Agengy For Marine-Earth Science & Technology | カサゴ細胞株 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7132230B2 (en) | 2002-06-28 | 2006-11-07 | Fujikin Incorporated | Cytotoxic assay and new established cell line of sturgeon origin |
-
2012
- 2012-07-31 JP JP2012169378A patent/JP5846384B2/ja not_active Expired - Fee Related
-
2013
- 2013-07-30 US US14/418,781 patent/US9777254B2/en not_active Expired - Fee Related
- 2013-07-30 WO PCT/JP2013/070627 patent/WO2014021329A1/ja not_active Ceased
- 2013-07-30 EP EP13825193.9A patent/EP2881460B1/en not_active Not-in-force
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004024192A (ja) | 2002-06-28 | 2004-01-29 | Fujikin Inc | チョウザメ由来の新規な株化細胞 |
| JP2008220198A (ja) | 2007-03-09 | 2008-09-25 | Japan Agengy For Marine-Earth Science & Technology | カサゴ細胞株 |
Non-Patent Citations (11)
| Title |
|---|
| "Current Protocols in Molecular Biology", 1987, JOHN WILEY & SONS |
| "Hayflick L. Moorhead POSITION", EXPOSURE CELL RES., vol. 25, 1961, pages 585 - 621 |
| "Molecular Cloning: A laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| EBITANI N ET AL.: "An established marine fish cell line with high plating efficiency.", ZOOL SCI, vol. 5, no. 1, 1988, pages 183 - 186, XP008176430 * |
| FAN TING-JUN ET AL.: "Establishment of a turbot fin cell line and its susceptibility to turbot reddish body iridovirus.", CYTOTECHNOLOGY, vol. 62, no. 3, 2010, pages 217 - 223, XP019816403 * |
| HASEGAWA S ET AL.: "A Cell Line (CFK) from Fin of Isogeneic Ginbuna Crusian Carp.", FISH PATHOLOGY, vol. 32, no. 2, 1997, pages 127 - 128, XP003034322 * |
| MAKIKO KIKUCHI ET AL.: "Kasago Saibokabu no Sangyo Riyo o Mezashita Baiyo Joken no Kento", KANTO GAKUIN UNIVERSITY KOGAKUBU KENKYU HAPPYO KOEN RONBUNSHU, vol. 2009, 2009, pages 170 - 171, XP008176398 * |
| MEGURO Y ET AL.: "A Cell Line Derived from the Fin of Japanese Flounder, Paralichthys olivaceus.", FISH PATHOLOGY, vol. 26, no. 2, 1991, pages 69 - 75, XP003034323 * |
| NOBUO EGAMI: "Gyorui no Bunshi Seibutsugaku Sakana no Saibo Baiyo", PROTEIN, NUCLEIC ACID AND ENZYME, vol. 34, no. 3, 1989, pages 186 - 192, XP008176429 * |
| SHOHEI WAKAO ET AL., PNAS, vol. 108, no. 24, 14 June 2011 (2011-06-14), pages 9875 - 9880 |
| YUKI NAKATANI: "Shu no Kotonaru Gyoruikan de Harabire no Tayosei o Motarasu Mechanism no Kaimei", THE ZOOLOGICAL SOCIETY OF JAPAN DAI 81 KAI TAIKAI YOKOSHU PROGRAM, 2010, pages 24, XP055191256, Retrieved from the Internet <URL:http://www.zoology.or.jp/annual-meeting/2/img/f_users/r_1005407img20100813185906.pdf> [retrieved on 20131024] * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017176005A (ja) * | 2016-03-29 | 2017-10-05 | テルモ株式会社 | 細胞培養物およびその製造方法 |
| CN108893474A (zh) * | 2018-02-07 | 2018-11-27 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
| CN108893474B (zh) * | 2018-02-07 | 2021-11-16 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014027888A (ja) | 2014-02-13 |
| EP2881460B1 (en) | 2019-01-02 |
| EP2881460A1 (en) | 2015-06-10 |
| US9777254B2 (en) | 2017-10-03 |
| JP5846384B2 (ja) | 2016-01-20 |
| EP2881460A4 (en) | 2015-12-30 |
| US20150175958A1 (en) | 2015-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5846384B2 (ja) | カワハギ由来の細胞株 | |
| KR101582483B1 (ko) | 심근 세포의 세포괴의 제조 방법 및 상기 심근 세포괴의 용도 | |
| Jin et al. | Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling | |
| KR100812856B1 (ko) | 영장류의 배아 줄기 세포로부터 배상체를 제조하는 방법 | |
| US11801267B2 (en) | Cardiac cell culture material | |
| WO2016127908A1 (zh) | 一种三维复合细胞聚集体模型及其制备方法与应用 | |
| KR20190112090A (ko) | 다능성줄기세포의 분화 제어 방법 | |
| US20190185816A1 (en) | Cardiac microtissue and uses thereof | |
| CN111263807A (zh) | 人工脂肪组织及其制造方法、人工皮肤的制造方法以及脂肪细胞的培养试剂 | |
| CN101056973B (zh) | 以细胞内线粒体为指标的心肌细胞的选择方法 | |
| Weinstein et al. | Isolation and purification of primary Schwann cells | |
| Morrison et al. | Expression patterns of Oct4, Cdx2, Tead4, and Yap1 proteins during blastocyst formation in embryos of the marsupial, Monodelphis domestica Wagner | |
| Dash et al. | Derivation and characterization of embryonic stem‐like cells of Indian major carp Catla catla | |
| KR102241349B1 (ko) | 각막상피세포 집단의 제조 방법 | |
| WO2017150294A1 (ja) | 多能性幹細胞様スフェロイドの製造方法および多能性幹細胞様スフェロイド | |
| WO2025063278A1 (ja) | コアシェル構造の三次元構造物 | |
| WO2017073761A1 (ja) | 多能性幹細胞の胚様体形成方法および多能性幹細胞の胚様体形成用組成物 | |
| EP4605520A1 (en) | Compositions and methods relating to adipocyte production | |
| Abdullah et al. | Differentiation of Mouse Bone-Marrow Mesenchymal Stem Cells into Motor Neuron Cells in vitro. | |
| JP2015198645A (ja) | 細胞構造体の作製方法 | |
| Elbaghdady et al. | Isolation and Molecular Characterization of Spermatogonial Stem Cells in Nile Tilapia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13825193 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14418781 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013825193 Country of ref document: EP |