WO2014107803A1 - Gem-difluorinated c-glycoside compounds as anti-cancer agents - Google Patents
Gem-difluorinated c-glycoside compounds as anti-cancer agents Download PDFInfo
- Publication number
- WO2014107803A1 WO2014107803A1 PCT/CA2014/000029 CA2014000029W WO2014107803A1 WO 2014107803 A1 WO2014107803 A1 WO 2014107803A1 CA 2014000029 W CA2014000029 W CA 2014000029W WO 2014107803 A1 WO2014107803 A1 WO 2014107803A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- group
- compound
- formula
- hydrogen atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *[C@@]1[C@](*)[C@@](C(C(O)O*)(F)F)O[C@](CO)[C@]1* Chemical compound *[C@@]1[C@](*)[C@@](C(C(O)O*)(F)F)O[C@](CO)[C@]1* 0.000 description 1
- IXEBJCKOMVGYKP-BMURFIBDSA-N COC([C@@H]([C@H]1OCc2ccccc2)OCc2ccccc2)O[C@H](COCc2ccccc2)[C@H]1OCc1ccccc1 Chemical compound COC([C@@H]([C@H]1OCc2ccccc2)OCc2ccccc2)O[C@H](COCc2ccccc2)[C@H]1OCc1ccccc1 IXEBJCKOMVGYKP-BMURFIBDSA-N 0.000 description 1
- WZBXZMRKNGJPGY-FKHKFOQBSA-N COc1cc([C@@H]([C@H]([C@H](CO2)[C@@H](c3c4)NCC([C@@H]([C@@H]([C@H]5O)O)O[C@H](CO)[C@H]5O)(F)F)C2=O)c3cc2c4OCO2)cc(OC)c1OC Chemical compound COc1cc([C@@H]([C@H]([C@H](CO2)[C@@H](c3c4)NCC([C@@H]([C@@H]([C@H]5O)O)O[C@H](CO)[C@H]5O)(F)F)C2=O)c3cc2c4OCO2)cc(OC)c1OC WZBXZMRKNGJPGY-FKHKFOQBSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the subject matter disclosed generally relates to novel chemical compounds and methods. More particularly, the invention provides novel derivatives of podophyllotoxin with a gem-difluoronated compound, having improved conformational and chemical stability, and having improved cytotoxicity, and methods of synthesizing and using such compounds. Preferred compounds are useful for the treatment of abnormal cell growth, such as cancers.
- Podophyllotoxin 1 is a lignan isolated from the roots of two plants Podophyllum peltatum (North America) and Podophyllum emodi (Asia). It has strong antimitotic activity by inhibiting polymerization of tubulin. Too toxic to be used in chemotherapy, it has given rise to many antitumoral compounds after structural modifications. Among them, glycosylated derivatives, compounds which are usually less toxic and more water-soluble, have emerged. For example, etoposide 2 is used in the treatment of small-cell lung cancer, bladder and testicular cancer, lymphomas, acute leukemias, and Kaposi sarcomas.
- Nitrogen-containing derivatives of podophyllotoxin such as GL-331, NPF 6 or TOP-53 7 also show very interesting activities.
- Nitrogen-containing compounds of podophyllotoxin, with an amine function substituted with a gem-difluorinated glycoside have been reported in US patent no. 8,236,935. These compounds, 8 and 9, have significant cytotoxicity and the presence of a glycoside moiety improves the solubility of these compounds in aqueous medium and the gem-difluoromethylene group not only mimics the oxygen atom, but also prevents the hydrolysis of the glycoside moiety from the core structure, thus improving the pharmacological activity of these molecules.
- this class of molecules exists as an anomeric mixture with unstable conformations which may suffer from degradation driven by ring- opening process, thus resulting in loss of biological activities, and potentially poor pharmacokinetics properties.
- the present invention describes a novel class of derivatives of podophyllotoxin with a gem-difluoronated and a fixed glycoside ring.
- the novel class of compounds of the present invention also unexpectedly exhibits significantly improved cytotoxicity against several cancer cell-lines, especially against those that are drug-resistant.
- the present invention also describes novel synthetic methods and processes to synthesize these novel compounds, which are not accessible previously via published procedures. Such compounds could be used as chemotherapy agents in the treatment of different types of cancer, either alone or associated with other treatments such as chemotherapies.
- R may be a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, acetyl, or benzoyl group
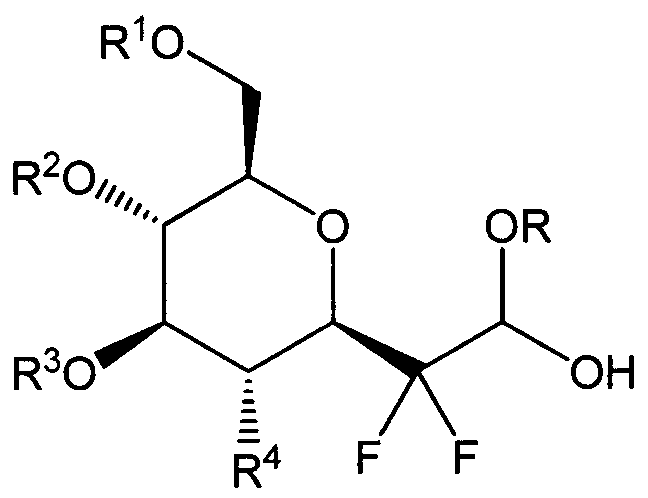
- R 1 and R 2 are identical or different, and are a hydrogen atom or a protective group for a hydroxyl group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group, or an acetal group of the CR'R" type, where R' and R" are identical or different and are a hydrogen atom or a group chosen from a linear or branched alkyl, aryl, or alkyl-aryl group,
- R 3 may be a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group,
- R 4 represents OR'", NGR'GR", N 3 , or a phthalimide, where R'" may be a hydrogen atom or a protective group for a hydroxyl group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group, and GR' and GR" are identical or different, and are a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, benzyol, acetyl, alkyloxycarbonyl, allyloxycarbonyl, or benzyloxycarbonyl group,
- R 5 may be a hydrogen atom or a group chosen from a linear or branched alkyl, acetyl, benzyl, PO 3 H, or PO 3 Na group.
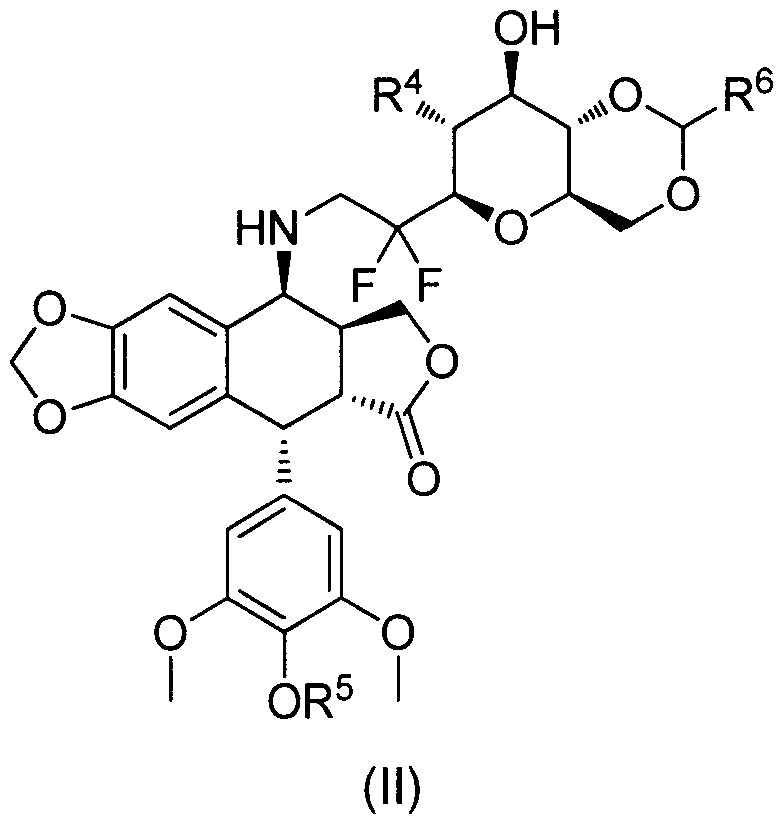
- R 4 , R 5 are as defined above,
- R 6 may be a hydrogen atom or a group chosen from an alkyi, an aryl, an alkyl- aryl, a heteroaryl, or an alkyl-heteroaryl group.
- R 5 may be a hydrogen atom, or a group chosen from an alkyi, -P0 3 H or -PO 3 Na.
- R 5 may be as defined in Formula I to III
- Ft 1 , R 2 , R 3 , R 4 are as defined in Formula I,
- R may be a C1-C12 alkyl
- said compound of formula IV may be obtained by epimerization and then by substituting the alcohol function in position 4 of podophyllotoxin or demethylated podophyllotoxin by an azido group subsequently reduced into an amine group.
- R may be a group chosen from methyl, ethyl, butyl, or isopropyl group.
- the use may be in combination with radiotherapy.
- the use may be in combination with one or more other anti-cancer agents.
- the cancer may be chosen from a bladder cancer, a brain cancer, a breast cancer, an uterus cancer, a chronic lymphoid leukemia, a colon cancer, an esophagus cancer, a liver cancer, a testicular cancer, a lymphoblastic leukemia, a follicular lymphomas, a melanomas, a malignant homeopathies, a myelomas, an ovarian cancer, a non-small-cell lung cancer, a prostate cancer, a small-cell lung cancer, an acute leukemia, a Kaposi sarcoma, and a lymphoid malignancy.
- a method of treating a patient afflicted with cancer by administering to the patient a therapeutically effective amount of a compound of the present invention.
- a method of treating a patient afflicted with cancer by administering to the patient a therapeutically effective amount of a compound of the present invention in combination with radiotherapy.
- a method of treating a patient afflicted with cancer by administering to the patient a therapeutically effective amount of a compound of the present invention in combination with one or more other anti-cancer agents.
- the cancer may be chosen from a cancer of bladder, a cancer of brain, a cancer of breast, a cancer of uterus, a chronic lymphoid leukemia, a colon cancer, an esophagus cancer, a liver cancer, a lymphoblastic leukemia, a follicular lymphomas, a melanomas, a malignant homeopathies, a myelomas, an ovarian cancer, a non-small-cell lung cancer, a prostate cancer, a small-cell lung cancer, and a lymphoid malignancy.
- alkyi by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e. unbranched) or branched chain, or cyclic hydrocarbon radical, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di-and multivalent radicals, having the number of carbon atoms designated (i.e. C Cio means one to ten carbons).
- saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec- butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- An unsaturated alkyi group is one having one or more double bonds or triple bonds. Examples of unsaturated alkyi groups include, but are not limited to, vinyl, 2-propenyl, crotyl,
- Alkyi groups which are limited to hydrocarbon groups are termed "homoalkyl”.
- Fluoroalkyl means alkyi as defined above wherein one or more hydrogen atoms are replaced by fluoro atoms.
- Alkylene by itself or as part of another substituent means a divalent radical derived from an alkyi, as exemplified, but not limited, by
- alkyi (or alkylene) group has from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention.
- a "lower alkyi” or “lower alkylene” is a shorter chain alkyi or alkylene group, generally having eight or fewer carbon atoms.
- Alkynyl means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched or combinations thereof. Examples of alkynyl include ethynyl, propargyl, 3-methyl-1 -pentynyl, 2- heptynyl and the like.
- Cycloalkyl means mono- or bicyclic saturated carbocyclic rings, each of which having from 3 to 10 carbon atoms.
- a "fused analog" of cycloalkyl means a monocyclic rings fused to an aryl or heteroaryl group in which the point of attachment is on the non-aromatic portion. Examples of cycloalkyl and fused analogs thereof include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronaphthyl, decahydronaphthyl, indanyl, and the like.
- Alkoxy means alkoxy groups of a straight or branched chain having the indicated number of carbon atoms.
- Ci -6 alkoxy for example, includes methoxy, ethoxy, propoxy, isopropoxy, and the like.
- Heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of at least one carbon atoms and at least one heteroatom selected from the group consisting of O, N, P, Si and S, and wherein the nitrogen, phosphorus, and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) O, N, P and S and Si may be placed at any interior position of the heteroalkyl group or at the position at which alkyl group is attached to the remainder of the molecule.
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH 2 -CH 2 -S -CH 2 -CH 2 - and -CH 2 -S-CH 2 -CH 2 -NH- CH 2 -.
- heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxo, alkylenedioxo, alkyleneamino, alkylenediamino, and the like). Still further, for alkylene and heteroalkylene linking groups, no orientation of the linking group is implied by the direction in which the formula of the linking group is written. For example, the formula - C(0)OR'- represents both -C(0)OR'- and -R'OC(O)-.
- heteroalkyi groups include those groups that are attached to the remainder of the molecule through a heteroatom, such as -C(0)R', -C(0)NR', - NR'R", -OR', -SR ⁇ and/or -S0 2 R'.
- heteroalkyi is recited, followed by recitations of specific heteroalkyi groups, such as -NR'R” or the like, it will be understood that the terms heteroalkyi and -NR'R" are not redundant or mutually exclusive. Rather, the specific heteroalkyi groups are recited to add clarity. Thus, the term “heteroalkyi” should not be interpreted herein as excluding specific heteroalkyi groups, such as -NR'R” or the like.
- Cycloalkoxy means cycloalkyi as defined above bonded to an oxygen atom, such as cyclopropyloxy.
- Fluoroalkoxy means alkoxy as defined above wherein one or more hydrogen atoms are replaced by fluoro atoms.
- Aryl means mono- or bicyclic aromatic rings containing only carbon atoms.
- a "fused analog" of aryl means an aryl group fused to a monocyclic cycloalkyi or monocyclic heterocyclyl group in which the point of attachment is on the aromatic portion. Examples of aryl and fused analogs thereof include phenyl, naphthyl, indanyl, indenyl, tetrahydronaphthyl, 2,3- dihydrobenzofuranyl, dihydrobenzopyranyl, 1 ,4-benzodioxanyl, and the like.
- Heteroaryl means a mono- or bicyclic aromatic ring containing at least one heteroatom selected from N, O and S, with each ring containing 5 to 6 atoms.
- a "fused analog" of heteroaryl means a heteroaryl group fused to a monocyclic cycloalkyi or monocyclic heterocyclyl group in which the point of attachment is on the aromatic portion.
- heteroaryl examples include pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, furo(2,3-b)pyridyl, quinolyl, indolyl, isoquinolyl, and the like.
- alkyl groups, cycloalkyi, alkynyl, alkenyl, aryl groups and heteroaryl groups referred to in the definitions are unsubstituted or are substituted by at least one substituent selected from the group consisting of substituents.
- the said substituents are selected from the group consisting of halogen atoms, alkyl groups having from 1 to 4 carbon atoms, alkoxy groups having from 1 to 4 carbon atoms, haloalkyl groups having from 1 to 4 carbon atoms, haloalkoxy groups having from 1 to 4 carbon atoms, cyano groups, alkynyl groups having from 2 to 6 carbon atoms, alkanoyl groups having from 1 to 5 carbon atoms, cycloalkyi groups having from 3 to 7 ring atoms, heteroaryl groups, aryl groups, aralkoxy groups having from 7 to 10 carbon atoms, arylcarbonyl groups, two adjacent-x groups are optionally joined together to form an alkylene or an alkenylene chain having 3 or 4 carbon atoms, aminocarbonyl groups, alkenyl groups having from 2 to 5 carbon atoms, alkylthio groups having from 1 to 4 carbon atoms, aminosulfinyl groups, aminos
- Heterocyclyl means mono- or bicyclic saturated rings containing at least one heteroatom selected from N, S and O, each of said ring having from 3 to 10 atoms in which the point of attachment may be carbon or nitrogen.
- a "fused analog" of heterocyclyl means a monocyclic heterocycle fused to an aryl or heteroaryl group in which the point of attachment is on the non- aromatic portion.
- heterocyclyl and fused analogs thereof include pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, 2,3-dihydrofuro(2,3-b)pyridyl, benzoxazinyl, tetrahydrohydroquinolinyl, tetrahydroisoquinolinyl, dihydroindolyl, and the like.
- the term also includes partially unsaturated monocyclic rings that are not aromatic, such as 2- or 4-pyridones attached through the nitrogen or N- substituted-(1 H,3H)-pyrimidine-2,4-diones (N-substituted uracils).
- halo or halogen
- haloalkyl by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom.
- terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl.
- halo(CrC 4 )alkyl is meant to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4- chlorobutyl, 3-bromopropyl, and the like.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, ⁇ , ⁇ '-dibenzyl ethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydramine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
- Fig. 1 illustrates the inhibition of human topoisomerase II DNA decatenation in the absence of BSA
- Fig. 2 illustrates the inhibition of human topoisomerase II DNA decatenation in the presence of BSA
- Fig. 3A illustrates the time-dependent growth of MCF-7/mdr cancer cells treated with 0.4% DMSO
- Fig. 3B illustrates the cell growth of MCF-7/mdr cancer cells upon treatment with etoposide
- Fig. 3C illustrates the cell growth of MCF-7/mdr cancer cells upon treatment with the compound of Example 1 ;
- Fig. 3D illustrates the cell growth of MCF-7/mdr cancer cells upon treatment with the compound of Example 2.
- Fig. 4A illustrates time-dependent growth of H69AR cancer cells treated with 0.4% DMSO
- Fig. 4B illustrates cell growth of H69AR cancer cells upon treatment with etoposide
- Fig. 4C illustrates cell growth of H69AR cancer cells upon treatment with the compound of Example 1 ;
- Fig. 5A illustrates time-dependent growth of MES-SA/Dx5 cancer cells treated with 0.4% DMSO
- Fig. 5B illustrates cell growth of MES-SA/Dx5 cancer cells upon treatment with etoposide
- Fig. 5C illustrates cell growth of MES-SA/Dx5 cancer cells upon treatment with the compound of Example 1 ;
- R is a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, acetyl, or benzoyl group,
- R 1 and R 2 are identical or different, and are a hydrogen atom or a protective group for a hydroxyl group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group, or an acetal group of the CR'R" type, where R' and R" are identical or different and are a hydrogen atom or a group chosen from a linear or branched alkyl, aryl, or alkyl-aryl group, R 3 is a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group,
- R 4 represents OR'", NGR'GR", N 3 , or a phthalimide, where R'" is a hydrogen atom or a protective group for a hydroxyl group chosen from a linear or branched alkyl, benzyl, benzoyl, acetyl, or pivaloyl group, and GR' and GR" are identical or different, and are a hydrogen atom or a group chosen from a linear or branched alkyl, benzyl, benzyol, acetyl, alkyloxycarbonyl, allyloxycarbonyl, or benzyloxycarbonyl group,
- R 5 is a hydrogen atom or a group chosen from a linear or branched alkyl, acetyl, benzyl, PO 3 H, or PO 3 Na group.
- the invention also includes derivatives in the state of a base, of a mineral or organic acid addition salt or a hydrate or a pharmaceutically acceptable solvate of the compound of formula (I).
- R 4 , R 5 are as defined above in Formula I,
- R 6 is a hydrogen atom or a group chosen from an alkyl, an aryl, an alkyl-aryl, a heteroaryl, or an alkyl-heteroaryl group.
- the invention also includes derivatives in the state of a base, of a mineral or organic acid addition salt or a hydrate or a pharmaceutically acceptable solvate of the compound of formula (II).
- R 5 is a hydrogen atom, or a group chosen from an alkyl, -P0 3 H or - PO 3 Na.
- the alkyl group may be linear or branched alkyl groups having 1 to 10 carbon atoms.
- the alkyl group may be substituted with one or more halogen atom(s), alkyloxy, alkylthio, -OC(0)alkyl -OC(0)Oalkyl, - OC(0)aryl and 0(0)Oaryl.
- the aryl group means mono- or bicyclic aromatic rings containing only carbon atoms.
- the heteroaryl means a mono- or bicyclic aromatic ring containing at least one heteroatom selected from N, O and S, with each ring containing 5 to 6 atoms.
- Compounds of the Formula I may be separated into diastereoisomeric pairs of enantiomers by, for example, fractional crystallization from a suitable solvent, for example MeOH or EtOAc or a mixture thereof.
- the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active amine or acid as a resolving agent or on a chiral HPLC column.
- One or more than one of the protons in compounds of Formula I to III can be replaced with deuterium atom(s), thus providing deuterated analogs that may have improved pharmacological activities.
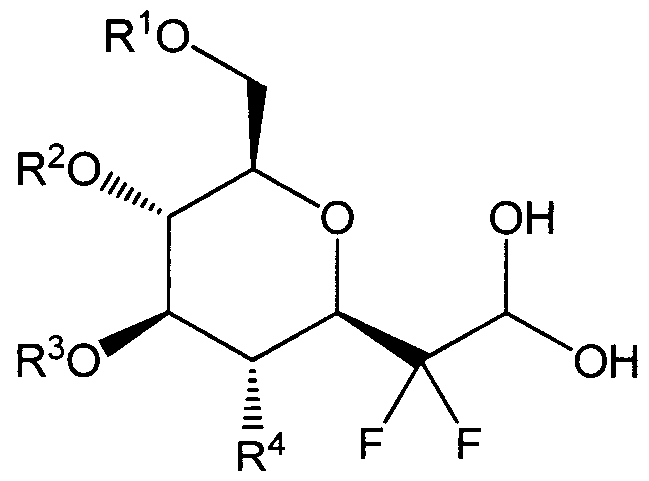
- a method for the preparation of the compounds of the present invention comprises a coupling step between a compound of Formula IV:
- R 5 is as defined in Formula I to III
- R 1 , R 2 , R 3 , R 4 are as defined in Formula I,
- R is a C1-C12 alkyl
- the compound of formula IV is obtained by epimerization and then by substituting the alcohol function in position 4 of podophyllotoxin or demethylated podophyllotoxin by an azido group subsequently reduced into an amine group.
- the present invention relates to the use of at least one compound of general formula I to III as defined above for preparing drugs/compositions for treating cancers such as for example a bladder cancer, a brain cancer, a breast cancer, a uterus cancer, a chronic lymphoid leukemia, a colon cancer, a esophagus cancer, a liver cancer, a testicular cancer, a lymphoblastic leukemia, a follicular lymphomas, a melanomas, a malignant homeopathies, a myelomas, an ovarian cancer, a non-small-cell lung cancer, a prostate cancer, a small-cell lung cancer, an acute leukemia, a Kaposi sarcoma, and a lymphoid malignancy, etc.
- cancers such as for example a bladder cancer, a brain cancer, a breast cancer, a uterus cancer, a chronic lymphoid leukemia, a colon cancer, a esophagus cancer
- Another object of the invention relates to a composition comprising at least one compound of formula I to III as defined above.
- composition according to the invention may comprise compounds of formula I to III as defined above, alone or in a mixture and in any proportions.
- compositions according to the present invention for administration via an oral, sublingual, inhalation, subcutaneous, intramuscular, intravenous, transdermal, local or rectal route, the active ingredients may be administered as unit administration forms, in a mixture with standard pharmaceutically acceptable supports/carriers.
- suitable unit administration forms include oral forms such as tablets, gelatin capsules, powders, granules and oral solutions or suspensions, topical administration forms, implants, subcutaneous, intramuscular, intravenous, intranasal or intraocular administration forms and rectal administration forms.
- non-toxic and pharmaceutically acceptable excipients such as distilled water, glucose, starch lactose, talc, vegetable oils, ethylene glycol, etc., may be added.
- compositions may also contain preservatives.
- compositions [0081] Other active ingredients may be added into the compositions.
- compositions may vary depending on the applications, the age, and the weight of the patient, if necessary.
- R is a group chosen from methyl, ethyl, butyl, or isopropyl group.
- a method of treating a patient afflicted with cancer by administering to the patient a therapeutically effective amount of a compound of the present invention.
- the method may be effected in combination with radiotherapy, chemotherapy, and/or with one or more other anti-cancer agents.
- Non-limiting examples of other anti-cancer agents include but are not limited to, angiogenesis inhibitors, antiproliferative agents, other kinase inhibitors, other receptor tyrosine kinase inhibitors, aurora kinase inhibitors, pololike kinase inhibitors, bcr-abl kinase inhibitors, growth factor inhibitors, antimitotic agents, alkylating agents, antimetabolites, platinum containing agents, growth factor inhibitors, ionizing radiation, cell cycle inhibitors, topoisomerase inhibitors, biologic response modifiers, immunomodulators, immunologicals, antibodies, hormonal therapies, retinoids/deltoids plant alkaloids, proteasome inhibitors, HSP-90 inhibitors, histone deacetylase inhibitors (HDAC) inhibitors, purine analogs, pyrimidine analogs, MEK inhibitors, CDK inhibitors, ErbB2 receptor inhibitors, mTOR inhibitors, Bel inhibitors, Mcl inhibitors and combinations thereof as well as other anti-cancer
- Angiogenesis inhibitors include, but are not limited to, EGFR inhibitors, PDGFR inhibitors, VEGFR inhibitors, TIE2 inhibitors, IGFIR inhibitors, matrix metalloproteinase 2 (MMP-2) inhibitors, matrix metalloproteinase 9 (MMP- 9) inhibitors, thrombospondin analogs such as thrombospondin- 1 and N-Ac-Sar- Gly-Val-D-allolle-Thr-Nva-He-Arg-Pro- NHCH 2 CH 3 or a salt thereof and analogues of N-Ac-Sar-Gly-Val-D-allolle-Thr-Nva-lle-Arg- PrO-NHCH 2 CH 3 such as N-Ac-GlyVal-D-alle-Ser-Gln-lle-Arg-ProNHCH 2 CH 3 or a salt thereof.
- Examples of EGFR inhibitors include, but are not limited to, Iressa (gefitinib),Tarceva (erlotinib or OSI-774), lcotinib, Erbitux (cetuximab), EMD- 7200, ABX-EGF, HR3, IgA antibodies, TP-38 (IVAX), EGFR fusion protein, EGF- vaccine, anti-EGFr immunoliposomes and Tykerb (lapatinib).
- PDGFR inhibitors include, but are not limited to, CP- 673,451 and CP- 868596.
- VEGFR inhibitors include, but are not limited to, Avastin (bevacizumab), Sutent (sunitinib, SU1 1248), Nexavar (sorafenib, BAY43- 9006), regorafenib, CP-547,632, axitinib (AG 13736), Apatinib, cabozantinib, Zactima (vandetanib, ZD-6474), AEE788, AZD-2171 , VEGF trap, Vatalanib (PTK-787, ZK-222584), Macugen, M862, Pazopanib (GW786034), BC-00016, ABT-869 and angiozyme.
- thrombospondin analogs include, but are not limited to, ABT- 510.
- BCL inhibitors include, but not limited to, obatoclax and navitoclax, ABT199.
- aurora kinase inhibitors include, but are not limited to, VX-680, AZD- 1152 and MLN-8054.
- Example of polo-like kinase inhibitors include, but are not limited to, BI-2536.
- Examples of bcr-abl kinase inhibitors include, but are not limited to, Gleevec (imatinib), ponatinib nilotinib and Dasatinib (BMS354825).
- platinum containing agents includes, but are not limited to, cisplatin, Paraplatin (carboplatin), eptaplatin, lobaplatin, nedaplatin, Eloxatin (oxaliplatin) or satraplatin.
- mTOR inhibitors includes, but are not limited to, CCI- 779, rapamycin, temsirolimus, everolimus, RAD001 , INK-128 and ridaforolimus.
- HSP-90 inhibitors includes, but are not limited to, geldanamycin, radicicol, 17-AAG, KOS-953, 17-DMAG, CNF-101 , CNF-1010, 17- AAG-nab, NCS-683664, Mycograb, CNF-2024, PU3, PU24FC1 , VER49009, IPI- 504, SNX-2112 and STA-9090.
- HDAC histone deacetylase inhibitors
- SAHA Suberoyianilide hydroxamic acid
- MS-275 valproic acid
- TSA valproic acid
- LAQ-824 Trapoxin, tubacin, tubastatin, ACY-1215 and Depsipeptide.
- Examples of MEK inhibitors include, but are not limited to, PD325901 , ARRY-142886, ARRY-438162 and PD98059.
- CDK inhibitors include, but are not limited to, flavopyridol, MCS-5A, CVT-2584, seliciclib (CYC-202, R-roscovitine), ZK- 304709, PHA-690509, BMI-1040, GPC-286199, BMS-387,032, PD0332991 and AZD-5438.
- Exambles of ErbB2 receptor inhibitors include, but are not limited to, CP-724-714, CI-1033, (canertinib), Herceptin (trastuzumab), Omitarg (2C4, petuzumab), TAK-165, GW- 572016 (lonafarnib), GW-282974, EKB-569, PI-166, dHER2 (HER2 Vaccine), APC8024 (HER2 Vaccine), anti-HER/2neu bispecific antibody, B7.her2lgG3, AS HER2 trifunctional bispecfic antibodies, mAB AR-209 and mAB 2B-1.
- alkylating agents include, but are not limited to, nitrogen mustard N- oxide, cyclophosphamide, ifosfamide, trofosfamide, Chlorambucil, melphalan, busulfan, mitobronitol, carboquone, thiotepa, ranimustine, nimustine, temozolomide, AMD-473, altretamine, AP-5280, apaziquone, brostallicin, bendamustine, carmustine, estramustine, fotemustine, glufosfamide, KW-2170, mafosfamide, and mitolactol, carmustine (BCNU), lomustine (CCNU), Busulfan, Treosulfan, Decarbazine and Temozolomide.
- antimetabolites include but are not limited to, methotrexate, 6- mercaptopurine riboside, mercaptopurine, uracil analogues such as 5-fluorouracil (5-FU) alone or in combination with leucovorin, tegafur, UFT, doxifluridine, carmofur, cytarabine, cytarabine, enocitabine, S-l, Alimta (premetrexed disodium, LY231514, MTA), Gemzar (gemcitabine), fludarabine, 5- azacitidine, capecitabine, cladribine, clofarabine, decitabine, eflornithine, ethnylcytidine, cytosine arabinoside, hydroxyurea, TS-I, melphalan, nelarabine, nolatrexed, ocfosate, disodium premetrexed,
- 5-FU 5-
- topoisomerase inhibiting agents include, but are not limited to, one or more agents selected from the group consisting of aclarubicin, amonafide, belotecan, camptothecin, 10-hydroxycamptothecin, 9- aminocamptothecin, diflomotecan, irinotecan HCL (Camptosar), edotecarin, epirubicin (Ellence), etoposide, exatecan, gimatecan, lurtotecan, orathecin (Supergen), BN-80915, mitoxantrone, pirarbucin, pixantrone, rubitecan, sobuzoxane, SN-38, tafluposide and topotecan.
- antibodies include, but are not limited to, Rituximab, Cetuximab, Bevacizumab, Trastuzimab, specific CD40 antibodies and specific IGFIR antibodies.
- hormonal therapies include, but are not limited to, exemestane (Aromasin), leuprolide acetate, anastrozole (Arimidex), fosrelin (Zoladex), goserelin, doxercalciferol, fadrozole, formestane, tamoxifen citrate (tamoxifen), Casodex, Abarelix, Trelstar, finasteride, fulvestrant, toremifene, raloxifene, lasofoxifene, letrozole, flutamide, bicalutamide, megesterol, mifepristone, nilutamide, dexamethasone, predisone and other glucocorticoids.
- retinoids/deltoids include, but are not limited to, seocalcitol (EB 1089, CB 1093), lexacalcitrol (KH 1060), fenretinide, Aliretinoin, Bexarotene and LGD-1550.
- Examples of plant alkaloids include, but are not limited to, vincristine, vinblastine, vindesine and vinorelbine.
- Examples of proteasome inhibitors include, but are not limited to, bortezomib (Velcade), MGI 32, NPI-0052 and PR-171.
- immunologicals include, but are not limited to, interferons and numerous other immune enhancing agents.
- Interferons include interferon alpha, interferon alpha-2a, interferon, alpha-2b, interferon beta, interferon gamma- 1a, interferon gamma- 1 b (Actimmune), or interferon gamma- nl and combinations thereof.
- agents include filgrastim, lentinan, sizofilan, TheraCys, ubenimex, WF-10, aldesleukin, alemtuzumab, BAM-002, decarbazine, daclizumab, denileukin, gemtuzumab ozogamicin, ibritumomab, imiquimod, lenograstim, lentinan, melanoma vaccine (Corixa), molgramostim, OncoVAC- CL, sargaramostim, tasonermin, tecleukin, thymalasin, tositumomab, Virulizin, Z-100, epratuzumab, mitumomab, oregovomab, pemtumomab (Y-muHMFGI), Provenge (Dendreon), CTLA4 (cytotoxic lymphocyte antigen 4) antibodies and agents capable of blocking CTLA4 such as MDX-010
- Examples of biological response modifiers are agents that modify defense mechanisms of living organisms or biological responses, such as survival, growth, or differentiation of tissue cells to direct them to have anti-tumor activity.
- Such agents include krestin, lentinan, sizofrran, picibanil and ubenimex.
- pyrimidine analogs include, but are not limited to, 5- Fluorouracil, Floxuridine, Doxifluridine, Ratitrexed, cytarabine (ara C), Cytosine arabinoside, Fludarabine, and Gemcitabine.
- purine analogs include, but are not limited to, Mercaptopurine and thioguanine.
- immunomodulators include but not limited to, thalidomide and lenalidomide.
- antimitotic agents include, but are not limited to, paclitaxel, docetaxel, ABRAXANE, epothilone D (KOS-862) and ZK-EPO.
- cancers that may be treated with the present invention include a bladder cancer, a brain cancer, a breast cancer, a uterus cancer, a chronic lymphoid leukemia, a colon cancer, a esophagus cancer, a liver cancer, a testicular cancer, a lymphoblastic leukemia, a follicular lymphomas, a melanomas, a malignant homeopathies, a myelomas, an ovarian cancer, a non-small-cell lung cancer, a prostate cancer, a small-cell lung cancer, an acute leukemia, a Kaposi sarcoma, and a lymphoid malignancy.
- DBU means 1 ,8-diazabicyclo[5.4.0]undec-7-ene
- DIBAL means diisobutylaluminum hydride
- DIEA means diisopropylethylamine
- DMAP means N,N-dimethylaminopyridine
- DME means 1 ,2-dimethoxyethane
- DMF means ⁇ , ⁇ -dimethylformamide
- dmpe means 1 ,2-bis(dimethylphosphino)ethane
- DMSO means dimethylsulfoxide
- dppb means 1 ,4-bis(diphenylphosphino)butane
- dppe means 1 ,2-bis(diphenylphosphino)ethane
- dppf means 1 ,1 '- bis(diphenylphosphino)ferrocene
- dppm means 1 ,1 '- bis(diphenylphosphino)methane
- DIAD
- Step 3 (3R,4S,5R,6R)-3,4,5-tris(benzyloxy)-6- ((benzyloxy)methyl)tetrahydro-2H-pyran-2-one
- Step 4 ethyl 2,2-difluoro-2-((3R,4S,5R,6R)-3,4,5- tris(benzyloxy)-6-((benzyloxy)methyl)-2-hydroxytetrahydro-2H-pyran-2-yl)acetate
- Step 5 ethyl 2,2-difluoro-2-((3R,4S,5R,6R)-3,4,5- tris(benzyloxy)-6-((benzyloxy)methyl)-2-chlorotetrahydro-2H-pyran-2-yl)acetate
- Thionyl chloride (2.25 mL, 30.85 mmol, 1.5 equiv) is then added dropwise and maintained the temperature not over 5°C. After addition, the solution is stirred for 10 min and ice-bath is removed. The solution is allowed to warm to room temperature (about 20 min). TLC indicated that starting material is consumed. A solution of HCI (1.0 N, 150 mL) is added. DCM is added (380 mL). The layers are separated.
- Step 6 ethyl 2,2-difluoro-2-((3R,4S,5R,6R)-3,4,5- tris(benzyloxy)-6- ((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)acetate
- Step 9 2,2-difluoro-2-((2R,3R,4S,5R,6R)-3,4,5- tris(benzyloxy)-6-(benzyloxymethyl)tetrahydro-2H-pyran-2-yl)ethane-1 , 1 -diol
- the suspension is stirred at 0°C and allowed to warm to room temperature gradually. The mixture is stirred at room temperature for 40 hrs. TLC indicated that starting material is consumed.
- a solution of saturated NaHC0 3 (20 mL) is added dropwise and H20 (10 mL).
- DCM (20 mL) is added. The mixture is stirred at room temperature for 30 min. The layers are separated. The aqueous phase is extracted with DCM (3x30 mL). The combined organic solution is passed through a pad of celite®, eluted with DCM (30 mL).
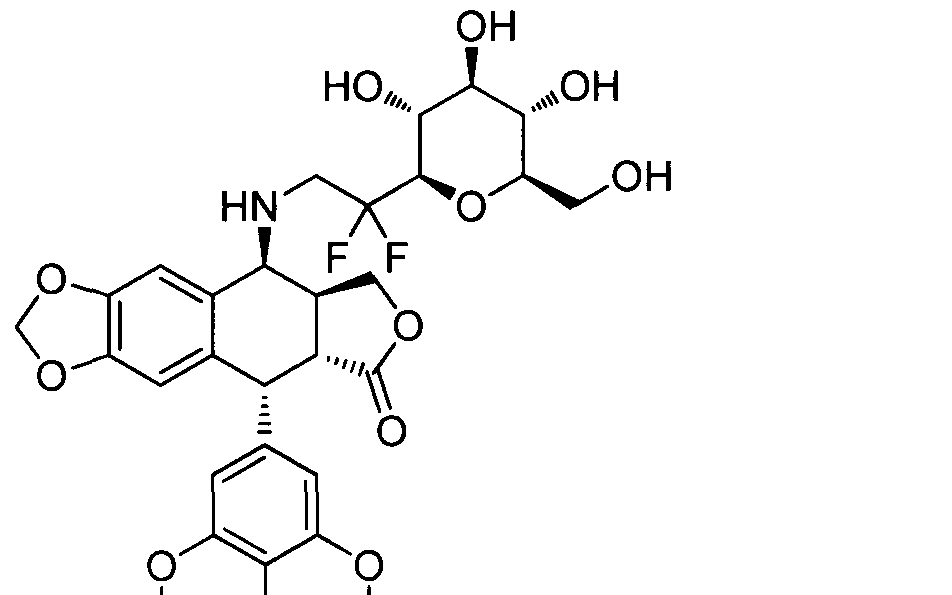
- Step 11 (5R,5aR,8aS,9S)-9-((2,2-difluoro-2- ((2R,3R,4S,5S,6R)-3,4 ) 5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2- yl)ethyl)amino)-5-(4-hydroxy-3,5- dimethoxyphenyl)-5,5a,8a,9- tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1 ,3]dioxol-6(8H)- one
- Step l 1 -ethoxy-2,2-difluoro-2-((2R,3R,4S,5R,6R)-3,4,5- tris(benzyloxy)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)ethanol
- Step 2 (5R,5aR,8aS,9S)-9-azido-5-(4-(benzyloxy)-3,5- dimethoxyphenyl)-5,5a,8a,9-tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1 ,3]dioxol-
- Step 3 (5R,5aR,8aS,9S)-9-amino-5-(4-(benzyloxy)-3,5- dimethoxypheny -S.Sa.Sa ⁇ -tetrahydrofuro ⁇ ' ⁇ 'ieyinaphtho ⁇ .S-dltl .Sldioxol-
- Step 4 (5R,5aR,8aS,9S)-5-(4-(benzyloxy)-3,5- dimethoxyphenyl)-9-((2,2-difluoro-2-((2R,3R,4S,5R,6R)-3,4 1 5-tris(benzyloxy)-6- ((benzyloxy)methyl)tetrahydro-2H-pyran-2-yl)ethyl)amino)-5,5a,8a,9- tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1 ,3]dioxol-6(8H)-one
- Step 5 (5R,5aR,8aS,9S)-9-((2,2-difluoro-2- ((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)ethyl)amino)-5-(4-hydroxy-3,5-dimethoxyphenyl)-5,5a,8a,9- tetrahydrofuroIS' ⁇ ' ⁇ Jjnaphtho ⁇ .S-dlfl . ⁇ dioxol-eiSHJ-one
- Step 1 ((5R,5aR,8aS,9S)-9-((2,2-difluoro-2- ((2R,3R,4S,5R,6R)-3,4,5-tris(benzyloxy)-6- ((benzyloxy)methyl)tetrahydro-2H- pyran-2-yl)ethyl)amino)-5-(3,4,5-trimethoxyphenyl)- 5,5a,8a,9- tetrahydrofuro[3' ) 4 l :6,7]naphtho[2,3-d][1 ,3]dioxol-6(8H)-one
- Step 2 (5R,5aR,8aS,9S)-9-((2,2-difluoro-2- ((2R,3R,4S,5S,6R)-3,4 I 5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2- yl)ethyl)amino)-5-(3,4,5-trimethoxyphenyl)- 5,5a,8a,9- tetrahydrofuro ⁇ ' ⁇ 'ie. ⁇ naphtho ⁇ .S-dltl .Sldioxol-eieHJ-one
- 100 mM stock solutions of compounds are prepared by dissolving the solid samples in 100% DMSO.
- the 10 mM stock solutions are prepared by 10-fold dilution of the 100 mM solutions with DMSO. Two-fold serial dilutions are performed in a V-shape 96-well plate.
- a negative control is the assay reaction with 20 mM EDTA.
- a positive control is the assay without any inhibitor.
- the background fluorescence of compounds is measured using the same compound concentrations with DNA and fluorescence dye.
- the human topo II DNA decatenation assay kit 96-Well Human Topo II DNA Decatenation Assay Kit Plus (from MoBiTec®, Catalog No. HDD96KE), is used for measurement of the inhibition.
- each reaction mixture is 50 ⁇ .
- 1 ⁇ of inhibitor and 24 ⁇ of premix are mixed.
- the reaction is initiated with 25 ⁇ of 20 mM MgCI 2 .
- the premix is prepared by mixing 870 ⁇ of H 2 O, 300 ⁇ of 10x assay buffer, 300 ⁇ of 20 ⁇ g/ml concatenated DNA, 30 ⁇ of 100 mM ATP and 1.5 ⁇ of 10 U/ ⁇ human topoisomerase II alpha enzyme (topo II).
- the reaction mixture is incubated at room temperature for 60 min. Then 0.2 M EDTA (5 ⁇ ) is added to stop the reaction.
- the final concentrations for the human topo II assay are 50 mM Tris-HCI, pH 8.0, 125 mM NaCI, 0.5 mM EDTA, 10 mM MgCI 2 , 2 ⁇ g/ml concatenated DNA, 1 mM ATP and 5 U/ml human topo II alpha enzyme.
- the sample (50 ⁇ ) is load onto a TDD filter plate on a vacuum manifold. Then a vacuum (80 kPa or 600 mmHg) is applied until the solution went through the filter. The filter is rinsed with 150 ⁇ of the Rinse Buffer (10 mM Tris-HCI, pH 7.5, 10 mM NaCI). Finally the 1x fluorescence dye (50 ⁇ ) is added and the fluorescence intensity at 535 nm using the excitation wavelength at 485 nm is measured.
- a negative control is the assay reaction with 20 mM EDTA.
- a positive control is the assay without any inhibitor.
- the background fluorescence of compounds is measured using the same compound concentrations with DNA and fluorescence dye.
- the human topo II DNA decatenation assay kit 96-Well Human Topo II DNA Decatenation Assay Kit Plus (from MoBiTec®, Catalog No. HDD96KE), is used for measurement of the inhibition.
- the total volume of each reaction mixture is 50 ⁇ .
- 1 ⁇ of inhibitor and 24 ⁇ of premix are mixed.
- the reaction is initiated with 25 ⁇ of 20 mM MgCI 2 .
- the premix is prepared by mixing 810 ⁇ of H 2 O, 300 ⁇ of 10x assay buffer, 300 ⁇ of 20 ⁇ g/ml concatenated DNA, 30 ⁇ of 100 mM ATP and 30 ⁇ of 500 U/ ml human topoisomerase II alpha enzyme (topo II) in a enzyme dilution buffer containing 0.5 mg/ml BSA.
- the reaction mixture is incubated at room temperature for 40 min.
- 0.2 M EDTA (5 ⁇ ) is added to stop the reaction.
- the final concentrations for the human topo II assay are 50 mM Tris-HCI, pH 8.0, 125 mM NaCI, 0.5 mM EDTA, 10 mM MgCI 2 , 2 ⁇ / ⁇ ⁇ ⁇ concatenated DNA, 1 mM ATP, 5 U/ml human topo II alpha enzyme and 5 ⁇ g/ml BSA.
- the sample (50 ⁇ ) is loaded onto a TDD filter plate on a vacuum manifold. Then a vacuum (80 kPa or 600 mmHg) is applied until the solution went through the filter. The filter is rinsed with 150 ⁇ of the Rinse Buffer (10 mM Tris-HCI, pH 7.5, 10 mM NaCI). Finally the 1 x fluorescence dye (50 ⁇ ) is added and the fluorescence intensity at 535 nm using the excitation wavelength at 485 nm is measured.
- MCF-7/mdr Human breast carcinoma cell line with multiple-drug resistance
- MCF7/mdr cells are cultured in 37 C CO2 incubator in RPMI1640 media with 10% FBS, with addition of glutamine (2 mM), penicillin (100 I.U.) and streptomycin (100 ⁇ g/ml) and HEPES (10 mM) are added to the media.
- Cells are thawed and kept in 0.8 ⁇ Doxorubicin (from Sigma- Aldrich® Canada, catalog number D1515) for a week prior to the study. During the course of study, there is no Doxorubicin in the culture media.
- MCF-7/mdr cells are plated at -18000 cells/well in 12-well tissue culture plates on Day -1 . Twenty-four (24) hours post-plating (DO), cells are treated with 0.4% DMSO, or test articles (Example 1 , Example 2 and etoposide) at 3, 6, 9, 12, and 24 ⁇ . The final concentration of DMSO is 0.4% for all wells treated with test articles.
- H69AR Human lung carcinoma cell line with multiple-drug resistance
- H69AR cells are cultured in 37 C CO 2 incubator in RPMI1640 media with 20% FBS, with addition of glutamine (2 mM), penicillin (100 I.U.) and streptomycin (100 ⁇ g/ml) and HEPES (10 mM) are added to the media.
- Cells are thawed and kept in increasing concentration of 0.1 -0.5 ⁇ Doxorubicin (from Sigma-Aldrich® Canada, catalog number D1515) for about one week prior to the study. During the course of study, there is no Doxorubicin in the culture media.
- H69AR cells are plated at -50000 cells/well in 12-well tissue culture plates on Day -1 . Twenty-four (24) hours post-plating (DO), cells are treated with 0.4% DMSO, or test articles (Example 1 and etoposide) at 3, 6, 9, 12, and 24 ⁇ . The final concentration of DMSO is 0.4% for all wells treated with test articles. [00200] At 0 (Day 0), 24 (Day 1 ), 48 (Day 2), 72 (Day 3), 96 (Day 4) and 120 (Day 5) hours post-treatment, one plate is retrieved and study terminated for time course analysis. Culture media are gently aspirated and cells washed once with 2 ml sterile PBS.
- Trypsin-0.25% EDTA (0.4 ml) is used to detach cells from the plate and 1 ml media is added to inactivate trypsin. Cells are transferred to 1.5 ml Eppendorf® tubes and spun at 2500 rpm for 2 minutes to collect cell pellets. Cell pellets are resuspended in 50 ⁇ of PBS. Cells (20 ⁇ ) are mixed with 0.4% trypan blue staining solution (20 ⁇ ) for cell counting using a hemocytometer.
- MES-SA/Dx5 Human uterine sarcoma cell line with multiple-drug resistance
- MES-SA/Dx5 Human uterine sarcoma cell line with multiple-drug resistance
- McCoy 5A media with 10% FBS, with addition of glutamine (2 mM), penicillin (100 I.U.) and streptomycin (100 ⁇ g/ml).
- MES-SA/Dx5 cells are plated at -300000 cells/well in 12-well tissue culture plates on Day -1. Twenty-four (24) hours post-plating (DO), cells are treated with 0.4% DMSO, or test articles (Example 1 and etoposide) at 3, 6, 9, 12, and 24 ⁇ . The final concentration of DMSO is 0.4% for all wells treated with test articles.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description




































Claims






Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201480015506.8A CN105246899A (en) | 2013-01-14 | 2014-01-13 | Novel organic electroluminescent compounds and organic electroluminescent device comprising the same |
| US14/760,021 US10272065B2 (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated C-glycoside compounds as anti-cancer agents |
| EP14738145.3A EP2943497A4 (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated c-glycoside compounds as anti-cancer agents |
| EP18204825.6A EP3530663B1 (en) | 2013-01-14 | 2014-01-13 | Process for preparing gem-difluorinated c-glycoside compounds |
| CA2897768A CA2897768C (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated c-glycoside compounds as anti-cancer agents |
| KR1020157022179A KR20150108391A (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated c-glycoside compounds as anti-cancer agents |
| JP2015551941A JP2016504394A (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated C-glycoside compounds as anticancer agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361752075P | 2013-01-14 | 2013-01-14 | |
| US61/752,075 | 2013-01-14 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP18204825.6A Previously-Filed-Application EP3530663B1 (en) | 2013-01-14 | 2014-01-13 | Process for preparing gem-difluorinated c-glycoside compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014107803A1 true WO2014107803A1 (en) | 2014-07-17 |
Family
ID=51166456
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2014/000029 Ceased WO2014107803A1 (en) | 2013-01-14 | 2014-01-13 | Gem-difluorinated c-glycoside compounds as anti-cancer agents |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10272065B2 (en) |
| EP (2) | EP3530663B1 (en) |
| JP (1) | JP2016504394A (en) |
| KR (1) | KR20150108391A (en) |
| CN (1) | CN105246899A (en) |
| CA (1) | CA2897768C (en) |
| WO (1) | WO2014107803A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112961135B (en) * | 2021-02-05 | 2021-11-26 | 安庆奇创药业有限公司 | Method for continuously synthesizing benzyl substituted gluconolactone by adopting microchannel reaction device |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2650384A1 (en) * | 2006-04-25 | 2007-11-08 | Institut National Des Sciences Appliquees De Rouen (Insa) | Novel gem-difluorinated c-glycoside compounds derived from podophyllotoxin, their preparation and their applications |
| CA2822097A1 (en) * | 2010-12-22 | 2012-06-28 | Tfchem | Derivatives of glyco-cf2-serine and glyco-cf2-threonine |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5036055A (en) * | 1989-06-07 | 1991-07-30 | Bristol-Myers Company | Acylated derivatives of etoposide |
| CN1235897C (en) * | 2000-06-26 | 2006-01-11 | 国家医药管理局上海医药工业研究院 | 4-beta-amino-saccharidoid side chain-4-dehydro-4'-demethylpodoph-yllotoxin derivative with antineoplastic activity and its synthesizing process |
| US8378195B2 (en) * | 2010-04-16 | 2013-02-19 | Andrew Jay Willoughby | Chord teaching apparatus |
-
2014
- 2014-01-13 WO PCT/CA2014/000029 patent/WO2014107803A1/en not_active Ceased
- 2014-01-13 JP JP2015551941A patent/JP2016504394A/en active Pending
- 2014-01-13 US US14/760,021 patent/US10272065B2/en active Active
- 2014-01-13 CA CA2897768A patent/CA2897768C/en active Active
- 2014-01-13 CN CN201480015506.8A patent/CN105246899A/en active Pending
- 2014-01-13 EP EP18204825.6A patent/EP3530663B1/en active Active
- 2014-01-13 KR KR1020157022179A patent/KR20150108391A/en not_active Withdrawn
- 2014-01-13 EP EP14738145.3A patent/EP2943497A4/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2650384A1 (en) * | 2006-04-25 | 2007-11-08 | Institut National Des Sciences Appliquees De Rouen (Insa) | Novel gem-difluorinated c-glycoside compounds derived from podophyllotoxin, their preparation and their applications |
| CA2822097A1 (en) * | 2010-12-22 | 2012-06-28 | Tfchem | Derivatives of glyco-cf2-serine and glyco-cf2-threonine |
Non-Patent Citations (2)
| Title |
|---|
| A.MERZ,OLTKI ET AL.: "« Adva-27a, a novel podophyllotoxin derivative found to be effective against multidrug- resistant human cancer cells »", ANTICANCER RES, vol. 32, no. 10, 2012, pages 4423 - 4432, XP055268394 * |
| See also references of EP2943497A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105246899A (en) | 2016-01-13 |
| JP2016504394A (en) | 2016-02-12 |
| EP2943497A1 (en) | 2015-11-18 |
| KR20150108391A (en) | 2015-09-25 |
| EP3530663A1 (en) | 2019-08-28 |
| US20150353573A1 (en) | 2015-12-10 |
| US10272065B2 (en) | 2019-04-30 |
| EP3530663B1 (en) | 2021-08-11 |
| EP2943497A4 (en) | 2017-05-03 |
| CA2897768C (en) | 2021-06-01 |
| CA2897768A1 (en) | 2014-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6272773B2 (en) | Heterocyclic amide compounds for HDAC6 inhibitors and antitumor agents | |
| JP5985658B2 (en) | Cyclic molecules as breton tyrosine kinase inhibitors | |
| WO2013185202A1 (en) | Apoptosis inducers | |
| KR20230044320A (en) | Treatment of b-cell malignancies by a combination jak and pi3k inhibitor | |
| US9226923B2 (en) | Spirocyclic molecules as protein kinase inhibitors | |
| JP2022527306A (en) | Macrocycle as a STING agonist | |
| CN113710671B (en) | Cyclic molecules as inhibitors of bruton's tyrosine kinase | |
| US8946445B2 (en) | Heterocyclic molecules as apoptosis inducers | |
| EP3530663B1 (en) | Process for preparing gem-difluorinated c-glycoside compounds | |
| HK40056349B (en) | Cyclic molecules as bruton's tyrosine kinase inhibitor | |
| NZ787908A (en) | Modulators of the integrated stress pathway | |
| HK1233180B (en) | Treatment of b-cell malignancies by a combination jak and pi3k inhibitor | |
| HK1233180A1 (en) | Treatment of b-cell malignancies by a combination jak and pi3k inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14738145 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2897768 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14760021 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015551941 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014738145 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20157022179 Country of ref document: KR Kind code of ref document: A |