WO2014121327A1 - Radical orbital switching - Google Patents
Radical orbital switching Download PDFInfo
- Publication number
- WO2014121327A1 WO2014121327A1 PCT/AU2014/000085 AU2014000085W WO2014121327A1 WO 2014121327 A1 WO2014121327 A1 WO 2014121327A1 AU 2014000085 W AU2014000085 W AU 2014000085W WO 2014121327 A1 WO2014121327 A1 WO 2014121327A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- radical
- rad
- neg
- anion
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)(CC(CC1(C)C)C(*)=O)*1O Chemical compound CC(C)(CC(CC1(C)C)C(*)=O)*1O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
- C07H19/207—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids the phosphoric or polyphosphoric acids being esterified by a further hydroxylic compound, e.g. flavine adenine dinucleotide or nicotinamide-adenine dinucleotide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/06—Free radical scavengers or antioxidants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
- C07D209/46—Iso-indoles; Hydrogenated iso-indoles with an oxygen atom in position 1
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/94—Oxygen atom, e.g. piperidine N-oxide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/96—Sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/04—Ortho- or peri-condensed ring systems
- C07D221/06—Ring systems of three rings
- C07D221/10—Aza-phenanthrenes
- C07D221/12—Phenanthridines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/091—Esters of phosphoric acids with hydroxyalkyl compounds with further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/3804—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)] not used, see subgroups
- C07F9/3808—Acyclic saturated acids which can have further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/50—Organo-phosphines
- C07F9/53—Organo-phosphine oxides; Organo-phosphine thioxides
- C07F9/5325—Aromatic phosphine oxides or thioxides (P-C aromatic linkage)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/572—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6568—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms
- C07F9/65685—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms the ring phosphorus atom being part of a phosphine oxide or thioxide
Definitions
- the present invention relates to orbital switching in radicals; in particular to the application of controlled and reversible switching of the Singly Occupied Molecular Orbital (SOMO) from a high energy level (e.g. the Highest Occupied Molecular Orbital (HOMO)) to a lower energy level by direct physical or chemical means.
- SOMO Singly Occupied Molecular Orbital
- HOMO Highest Occupied Molecular Orbital
- the molecular orbital (MO) accommodating the unpaired electron (the singly-occupied molecular orbital, or SOMO) is usually energetically the highest-occupied molecular orbital (HOMO).
- These 'converted' species include a radical nitronyl nitroxide ( ⁇ ) bonded to a tetrathiafulvalene (TTF) (Sugawara; Chem. Soc. Rev. 40, 3105; 2011).
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- DOMO Doubly-Occupied Molecular Orbital
- protons or other cations
- the addition of protons (or other cations) to the structure of Formula (I) causes the radical to be 'activated' and so to become more reactive (activating the radical for reaction, for example for use in radical-based chemistry).
- Reversing the above process, i.e. removing protons or other cations stabilises the structure of Formula (I), and if suitably stabilised will have reduced reactivity, and any bond formed between the radical of Formula (I) and another moiety can be more easily broken (e.g. by homolytic cleavage), releasing both the structure of Formula (I) and any moiety it had previously been bonded to; or at least the process and/or conditions of removing the structure of Formula (I) from a bonded moiety can be made more moderate (e.g. by using lower temperatures).
- the structure of Formula (I) can be used in chemical synthesis and/or in physical or chemical analysis or in sensors, by reversibly switching the levels of the molecular orbitals of the Structure of Formula (I), to take advantage of the switchable reactivity of the radical of RAD in the system of the invention.
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation; L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion of NEG is bonded to a proton or other cation.
- SOMO Molecular Orbital
- RAD is a group comprising a radical
- NEG comprises a negative point charge, which is capable of being neutralised
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the negative point charge of NEG; and wherein, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the negative point charge of NEG is neutralized.
- SOMO Singly-Occupied Molecular Orbital
- the ability to directly and reversibly switch/tune the energy levels of the orbitals of the radical in the structure of Formula (I) allows user control of the chemistry of structure of Formula (I).
- NEG is not a chemical group but rather is a functional physical equivalent, which provides the same function as the anion of NEG. That is, the functional physical equivalent comprises a negative point charge, wherein that negative point charge is capable of being neutralized, and wherein the energy level of the unpaired electron of RAD is lowered when the functional physical equivalent of NEG together with RAD-L forms the (corresponding) structure of Formula (I), and wherein the energy level of the unpaired electron of RAD is the highest energy level when the functional physical equivalent is electrically neutralized.
- NEG is a surface or structure capable of bearing and/or carrying a negative point charge, and where the negative point charge is capable of being neutralised.
- a structure of Formula (I), wherein the surface or structure capable of bearing and/or carrying a negative point charge is metallic or non-metallic.
- a structure of Formula (I), wherein the surface or structure capable of bearing and/or carrying a negative point charge comprises graphene.
- the negative point charge is a group comprising an anion, which is capable of being neutralised by bonding to a proton or other cation.
- a radical protecting group having the structure of Formula (I); wherein the radical of RAD is capable of forming a bond to a radical to be protected to give the protected radical; and wherein the bond to the radical to be protected is weakened when the negative point charge of NEG is not neutralised.
- a process of deprotecting a radical protected with the radical protecting group as defined previously comprising the removal the negative point charge; allowing the negative point charge to dissipate; or inverting the negative point charge to a positive point charge.
- a positive point charge could for example be used to have substantially the opposite effect as a negative point charge, e.g. to destabilise the radical of RAD, and/or to deprotect a radical protected by a structure of Formula (I).
- NEG comprises an anion
- references to the following aspects and embodiments where NEG comprises an anion also includes within its meaning a NEG that comprises a negative point charge, where this takes the form of a physical functional equivalent (as detailed hereinabove in the fourth aspect of the invention).
- references to the anion of NEG bonding to a proton or other cation encompasses within its meaning neutralisation of the negative point charge (as detailed herein above).
- the removal of a proton or other cation from an anion of NEG encompasses within its meaning the removal of the cause of neutralisation (i.e.
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation; and wherein the radical of RAD is capable of forming a bond to a radical to be protected when the anion of NEG is bonded to a proton or other cation; and wherein the radical to be protected is de-protectable when the anion of NEG is not bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- DOMO Doubly-Occupied Molecular Orbital
- protons or other cations
- the addition of protons (or other cations) to the structure of Formula (I) causes the radical to be 'activated' and so to become more reactive and so that it can form a bond to the radical to be protected.
- Reversing the above process, i.e. removing protons or other cations stabilises the structure of Formula (I), and if suitably stabilised, the bond formed between the radical to be protected and the structure of Formula (I) breaks (i.e. by homolytic cleavage), releasing both the radical to be protected and the structure of Formula (I); or at least the process and/or conditions of removing the structure of Formula (I) from the structure to be protected can be made more moderate (e.g. using lower temperatures).
- a portion of a structure can be protected with the structure of Formula (I) e.g. R-RAD-L-NEG (where the 'R-RAD' bond comprises the radical electron of RAD and the radical election of R), while various other reactions are conducted on the unprotected portions of that structure.
- the structure of Formula (I) can be selectively released (e.g. by increasing the pH) from that structure, allowing that portion which had been protected to be unprotected e.g. to give R « and RAD-L-NEG (where NEG is not bonded to a proton or other cation).
- the radical to be protected once deprotected, is free to participate in further reactions. 5
- the skilled person will be aware of those reactions, typically being radical type reactions.
- each protecting group of the invention could be used on the same structure to be protected, whereby each protecting group is deprotected under different reaction conditions, for example each responsive in a certain pH range or in the presence i o of different cations, or to progressively harsher deprotecting conditions such as increasing temperature. It is also possible that more than one protecting group can be removed (from a structure to be protected) under the same conditions.
- the two resulting radicals on the same structure can be arranged such that they can recombine.
- the two resultant radicals could be arranged such that the i s cyclization of the structure bearing these radicals occurs when they are deprotected (e.g. an intramolecular Wurtz reaction).
- radical protecting group as defined previously to protect a radical, wherein the anion of NEG is bonded to a proton or other cation.
- a process of deprotecting a radical protected with the radical protecting group as defined previously by increasing 25 the pH of the reaction medium to remove the proton bonded to the anion of NEG.
- a process of deprotecting a radical protected with the radical protecting group as defined previously by the addition of anions to the reaction medium, the added anions forming a precipitate with cations present in the reaction medium, thereby removing those cations from bonding with the anions of NEG.
- reaction medium such that protons or other cations in the reaction medium are bonded to the anion of NEG.
- the radical reaction is selected from the group comprising: radical coupling; Wurtz reaction; nitroxide mediated polymerization; nitroxide radical coupling; double bond addition; cyclization reactions; atom abstraction; and oxidation.
- a process as defined previously wherein the polymerization is conducted in the temperature range of up to 120 °C. Preferably, in the temperature range 25 to 80 °C According to an embodiment of the invention, there is provided a process as defined previously, wherein the reaction medium is buffered.
- the cation is a metal ion or metal containing ionic species or is an ammonium or phosphonium ion.
- the senor comprises a profluorescent probe.
- profluorescent probe comprises a nitroxide and a fluorophore (i.e. a fluorescent group), wherein the RAD group of the structure of Formula (I) comprises the nitroxide, and wherein the nitroxide radical quenches the fluorescence of the fluorophore.
- a fluorophore i.e. a fluorescent group
- the RAD group of the structure of Formula (I) comprises the nitroxide
- the nitroxide radical quenches the fluorescence of the fluorophore.
- the skilled person is aware of suitable fluorophore groups. When the nitroxide is engaged in a bond and thus the profluorescent probe is in a closed-shell state, the fluorescence is not quenched.
- the fluorophore of the profluorescent probe can be part of the RAD or NEG group and/or part of L of the structure of Formula (I).
- the senor is useful in determining pH; medical imaging; degree of oxidation or reduction; detecting and quantifying free radical species that may be present.
- the sensor is useful in determining pH.
- the sensor is a nitroxide profluorescent probe and when the anion of NEG is protonated (or bonded to another cation) the radical of RAD will be destabilised and will react with a suitable in situ species, such that the radical of RAD bonds to that species. In doing so the fluorescence of the corresponding fluorophore group is no longer quenched and fluorescence is therefore detectable. Calibration of the fluorescent response to change in pH (or other cation concentration) gives a sensor to measure pH (or other cation concentration).
- a process of monitoring the pH of a medium by measuring the concentration of one or more of the protected radical, the radical to be protected, or the radical protecting group as defined previously; or one or more products resulting from those species.
- the senor is useful in medical imaging; measuring degree of oxidation or reduction; detecting and quantifying free radical species that may be present; detecting anions, wherein the sensor is turned on by the addition of protons (or other cations), or turned off by the removal of protons (or other cations), and wherein this process can be reversible.
- a process as defined previously wherein the sensor is useful in detecting oxidative stress in a cell.
- a process as defined previously wherein the sensor is useful in detecting oxidative stress in non-biological systems, such as in machinery (e.g. aircraft parts), or similar object subjected to oxidative stress.
- a process as defined previously wherein one or more of the above defined profluorescent probes are used to detect oxidative stress in polymers.
- a profluorescent probe in radical form
- the radical form of the profluorescent probe can be released in response to a change in pH.
- a paint incorporating one or more of the above defined profluorescent probes, or a polymer as defined above which comprises the profluorescent probes.
- an anion sensor which results from a structure of Formula (I) forming, where the anion to be detected constitutes the anion of NEG, L is a through space interaction from NEG to RAD.
- the anion sensor may for example comprise a profluorescent probe which could be located on the RAD group.
- the anion concentration (of the anion to be detected) increases, statistically the formation of the structure of Formula (I) will also increase (that is RAD and NEG are more likely to become close enough to form a structure of Formula (I)).
- the radical of RAD is stabilised by the proximate anion.
- the radical to be protected or the RAD-L-NEG group, is an industrial antioxidant, inclusive of light stabilisers.
- the radical to be protected is a biologically active antioxidant.
- radical to be protected and/or the radical protecting group as defined previously as a radical scavenger.
- a process as defined previously wherein one or more of the radical to be protected or the radical protecting group; a resultant metabolite thereof; or a resultant product thereof is biologically active.
- a process as defined previously wherein biologically active species is released in the body.
- the biologically active species is selected from the group: nitric oxide; nitrogen dioxide; a structure comprising a nitric oxide and/or nitrogen dioxide radical; or non-steroidal anti-inflammatory drug (NSAID).
- NSAID non-steroidal anti-inflammatory drug
- a process of oxidation of a structure of Formula (1) as defined previously, to remove an electron to give a biradical species wherein the reaction is conducted in vacuum, gas phase, a low polarity solvent or in the solid state.
- a process wherein one or more of the processes as defined previously are used.
- a process as defined previously wherein the species to be oxidised in claim 53 and the species to be oxidized in claim 54 are interconvertible by the introduction or removal of a negative point charge.
- the species to be oxidised to a biradical species as defined previously and the species to be oxidised to remove an unpaired electron as defined previously are interconvertible by the addition or removal of protons or other cations.
- This aspect of the invention allows for the screening of candidate structures by various methods of calculations inclusive of single-reference Hartree-Fock (HF), Density
- DFT Functional Theory
- MP2, CC post-HF ab initio
- MCSCF multi-reference methods
- radical stability can be assessed. For example, this may be measured in terms of bond dissociation energy of RAD-R where R is a leaving group R « , such as methyl ('CF ⁇ ). Calculations can be performed on a single molecule, part of a molecule, complex or could be done on a composite structure incorporating one or more elements which when taken together make up the structure of Formula (I).
- a method of stabilising a radical bearing structure comprising the introduction of negative point charge to that structure, wherein the resultant structure formed is Formula (I):
- a seventh aspect of the invention there is provided a method of stabilising a radical bearing structure, comprising the incorporation of an anion into that structure, wherein the resultant structure formed is Formula (I):
- RAD is a group comprising the radical
- NEG is a group comprising the anion, which is capable of bonding to a proton or other cation
- an orbital switching structure of the invention is obtained.
- This might be for example used to make a superior agent for use in radical polymerization reactions, such as in nitroxide mediated polymerization.
- Such reagent could be designed to have lower activation thresholds.
- a known structure could be modified to include a radical in an arrangement such that an orbital switching structure of the invention is obtained.
- This might be for example used to make an acid group present in the structure more acidic, or to make superior agent for use in radical polymerization reactions, such as in nitroxide mediated polymerization.
- Such reagent could be designed to have lower activation thresholds.
- a ninth aspect of the invention there is provided a method of lowering the energy level of the Singly-Occupied Molecular Orbital of RAD, by the removal of the cause of neutralisation from a point charge of NEG that has been neutralised, in a structure of Formula (I): RAD-L-NEG (I) wherein RAD, NEG and L are defined in accordance with the third aspects of the invention.
- this method could be employed were it is desirable to switch the orbitals from the 'converted' electronic configuration to the 'normal' accumulated-type configuration.
- This may for example be used to activate a structure of Formula (I) to a more reactive state.
- the stabilised radical of Formula (I) is in solution but substantively inactive under the reaction conditions, treatment with proton acid could protonate the anion of NEG causing the orbitals to revert to the more normal imposed electronic configuration.
- the radical is thus destabilised, and is so more reactive, and for example could be used to react with other radicals in solution or to cap or protect a reactive group.
- the resultant structure according to the method as defined previously.
- a method of modifying a biological macromolecule comprising the incorporation of a radical into that biological macromolecule, wherein the resultant structure formed is Formula (I):
- RAD is a group comprising the radical
- NEG is a group comprising an anion, which is capable of bonding to a 10 proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion i s of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- RAD is a group comprising the radical
- NEG is a group comprising an anion, which is capable of bonding to a 25 proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- radical of RAD is released in situ (in the body in situ would be understood to be in vivo).
- an Activating Enzyme might release the radical from the radical precursor in situ.
- RAD is a group comprising a radical
- NEG is a group comprising the anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- RAD is a group comprising a radical
- NEG is a group comprising the anion, which is capable of bonding to a proton or other cation
- the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- SOMO Doubly-Occupied Molecular Orbital
- a method of modifying a biological macromolecule comprising the incorporation of a radical and an anion into that biological macromolecule, wherein the resultant structure formed is Formula (I): RAD-L-NEG
- RAD is a group comprising the radical
- NEG is a group comprising the anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- RAD is a group comprising the radical
- NEG is a group comprising the anion, which is capable of bonding to a proton or other cation
- the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton ⁇ other cation.
- SOMO Singly-Occupied Molecular Orbital
- RAD Doubly-Occupied Molecular Orbital
- SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton ⁇ other cation.
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- a new bond could be formed within the structure of the biological macromolecule such that two formerly distant parts of the biological macromolecule are brought closer together (or to tether an Activating Enzyme (or fragment thereof) to part of the enzyme to be activated) to give a structure of Formula (I).
- a part or sub-unit of the biological macromolecule could be removed or substituted such that two formerly distant parts of the biological macromolecule are brought closer together (i.e. to brought into closer proximity) to give a structure of Formula (I).
- transformations could be chemically or biologically driven.
- the skilled person will be aware of ways in which this could be done.
- a method of modifying a biological macromolecule such that the resultant biological macromolecule will form a complex with a substrate, wherein that resultant substrate-complex formed is Formula (I):
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation; 5 L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- DOMO Doubly-Occupied Molecular Orbital
- the modification of a biological macromolecule such as an enzyme
- a biological macromolecule such as an enzyme
- the resultant biological macromolecule will form a complex with a substrate
- that resultant substrate-complex formed is Formula (I).
- the biological macromolecule may contain one of the RAD/NEG groups and the substrate may contain the other RAD NEG group.
- the Linker may be the body of the biological macromolecule or simply one or more hydrogen bonds (or a combination of the biological macromolecule, substrate and hydrogen bonds). It may however, be that it is the substrate that brings the units of RAD and NEG (of the modified biological macromolecule) together in space to form the structure of Formula (I) i.e. when the substrate is bound.
- the biological macromolecule could be structurally changed by chemical reaction (addition, substitution, elimination, removal, rearrangement or some other modification) such that the group to be added is added to the biological macromolecule.
- a sub-unit of the biological unit could be swap for a new sub-unit bearing the required group. The skilled person will be aware of ways in which this could be done.
- Biologically driven transformations are also envisioned, in which the
- RAD-L-NEG (I) wherein: RAD is a group comprising a radical;
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- a method of modifying a substrate such that the resultant modified substrate will form a complex with a biological macromolecule, wherein that resultant substrate-complex formed is
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- SOMO Doubly-Occupied Molecular Orbital
- a substrate i.e. one that is involved in a biological process
- that resultant substrate- complex formed is Formula (I).
- the substrate may contain one of the RAD/NEG groups and the biological macromolecule may contain the other RAD/NEG group.
- the Linker may be the body of the biological macromolecule or simply one or more hydrogen bonds (or a combination of the biological macromolecule, substrate and hydrogen bonds). It may however, be that it is the modified substrate that brings the units of RAD and NEG together in space to form the structure of Formula (I) i.e. when the substrate is bound.
- the substrate could be structurally changed by chemical reaction (addition, substitution, elimination, removal, rearrangement or some other modification) such that the group to be added is added to the substrate.
- chemical reaction addition, substitution, elimination, removal, rearrangement or some other modification
- the modification is brought about for example by an enzyme. Again, the skilled person will be aware of ways in which this could be done.
- a method of modifying a biological macromolecule and a substrate such that the resultant modified biological macromolecule and substrate will form a complex, wherein that resultant substrate-complex formed is Formula (I):
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- L links NEG and RAD; the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after the modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than in the corresponding structure of Formula (I) when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- a method of modifying a biological macromolecule and a substrate such that the resultant modified biological macromolecule and substrate will form a complex, wherein that resultant substrate-complex formed is Formula (I):
- RAD is a group comprising a radical
- NEG is a group comprising an anion, which is capable of bonding to a proton or other cation
- the radical of RAD is not ⁇ -conjugated to the anion of NEG; and wherein after modification, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- SOMO Doubly-Occupied Molecular Orbital
- the modification of a substrate i.e. one that is involved in a biological process
- a biological macromolecule such that a biological macromolecule will form a complex with that substrate, wherein that resultant substrate-complex formed is Formula (I).
- the substrate may contain one of the RAD NEG groups and the biological macromolecule (so modified) may contain the other RAD/NEG group.
- the Linker may be the body of the biological macromolecule or simply one or more hydrogen bonds (or a combination of the biological macromolecule, substrate and hydrogen bonds). It may however, be that it is the modified substrate that brings the units of RAD and NEG together in space to form the structure of Formula (I) i.e. when the substrate is bound.
- the biological macromolecule and or substrate could be structurally changed by chemical reaction (addition, substitution, elimination, removal, rearrangement or some other modification) such that the group to be added is added to the biological macromolecule or substrate.
- a sub-unit of the biological unit could be swapped for a new sub-unit bearing the required group.
- F ogically driven transformations are also envisioned, in which the modification to the biological macromolecule or substrate is brought about for example by an enzyme. Again, the skilled person will be aware of ways in which this could be done.
- a method of modifying a biological macromolecule or substrate as defined previously wherein that modification changes the stability of the radical to alter the reactivity of the modified enzyme.
- the stability of the radical of RAD is changed such that (i) an Activating Enzyme (such as Pyruvate Formate Lyase Activating Enzyme (PFL-AE)) will act on the radical precursor to form the radical of RAD in situ, or such that the radical of RAD is formed more or less readily in situ, or that the Activating Enzyme cannot form the radical of RAD in situ; (ii) the radical of RAD will not participate in one or more further biochemical reactions (i.e.
- an Activating Enzyme such as Pyruvate Formate Lyase Activating Enzyme (PFL-AE)
- PFL-AE Pyruvate Formate Lyase Activating Enzyme
- the stability of the radical of RAD can be altered by changing the spatial arrangement (e.g. separation) of the anion of NEG to the radical of RAD in three dimensional space.
- the anion of NEG can be arranged in three dimensional space (e.g. by the placement of this on residues proximate to RAD) such that the stabilising effect on the radical is altered.
- An optimum or desired through- space distance might first be estimated through calculations. Further calculations could be used to determine which residues could be modified to give the calculated separation. Without being bound by theory, the stabilising through-space effect appears to be dependent on an inverse of the distance between the anion of NEG and the radical of RAD.
- a modification could be used to stop the activity of an enzyme which relies on the formation of a structure of Formula (I).
- this modification could be used to turn off the activity of an enzyme.
- Such a modification of the biological macromolecule or substrate could be the removal of one or more of the radical of RAD or the anion of NEG, or wherein the said modification prevents the radical of RAD and the anion of NEG from acting through-space together to provide the orbital conversion, i.e. give SOMO ⁇ HOMO configuration as defined previously.
- the G734 glycine residue could be altered to prevent hydrogen abstraction (by the wild type Pyruvate Formate Lyase Activating Enzyme (PFL-AE)) to give the radical of RAD, or for example any anions on NEG could be changed to be non-anionic.
- the carboxylic group of the aspartate on D16 on the wild type of PFL-AE could be replaced with a non-anionic group.
- PFL can be useful in the catalyses of the condensation of CoA and pyruvate to form acetyl-CoA and formate (e.g. see Fig. 13).
- a modified biological macromolecule and/or modified substrate as defined previously.
- a method of medical treatment of a medical condition comprising the administration of a therapeutically effective amount of a compound comprising a structure of Formula (I), as defined previously.
- a compound comprising a structure of Formula (I), as defined previously, for use in therapy.
- a twenty ninth aspect of the invention there is provided a method, use, process, structure, protecting group, metabolite, biological macromolecule or substrate as defined previously, substantially as hereinbefore described with reference to any one of the examples or the description.
- radical stability can be assessed. For example, this may be measured in terms of bond dissociation energy of RAD-R where R is a leaving group R*, such as methyl (*CH 3 ).
- a captodative effect i.e. simultaneous resonance with a lone-pair donor and a ⁇ -acceptor, as for example on the backbone of a protein or a peptide.
- RAD comprises a steric and/or electronically stabilised radical.
- RAD is electronically and/or sterically stabilised by groups which are proximate to that radical.
- RAD comprises an electronically stabilised radical.
- RAD comprises a radical selected from the group:
- DNA/RNA-base based radical or is an amino acid based radical.
- the DNA/RNA-base is selected from guanine (G) adenine (A), cytosine (C), thymine (T) or uracil (U).
- RAD comprises a radical group selected from the group comprising 2,2 ⁇ 6,6'-tetramethylpiperidine-N-oxy] (TEMPO),
- TIPNO 2,2,5,5-tetramethyl-4-phenyl-3-azahexane-3-oxyl
- SG 1 guanine-based or glycyl-based radical.
- NEG comprises a sterically and/or electronically destabilised anion
- a method, use, process, structure or protecting group as defined previously, wherein the anion of NEG is destabilised by an electron rich group (e.g. a carbonyl group) which is proximate to the anion of NEG, in particular where that anion is a carboxylate group, for example in a structure comprising the group -CO-C0 2 " .
- an electron rich group e.g. a carbonyl group
- NEG comprises an electronically destabilised anion
- NEG comprises an anion selected from the group comprising
- NEG comprises an anion which is a carboxylate group
- carboxylate group is a constituent part of an amino acid residue.
- amino acid residues e.g. residue based on any one of: alanine, arginine, asparagine, aspartate, cysteine, glutamine, glutamate, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine; or a modified or unnatural amino acid).
- NEG comprises an anion which is a carboxylate group, and wherein that carboxylate group is a constituent part of an amino acid residue, wherein that residue is an aspartate, or the anion of NEG comprises a pyruvate structure.
- NEG comprises an anion which is a phosphate, and wherein that phosphate is a mono, di or triphosphate.
- a method, use, process, structure or protecting group as define ⁇ previously wherein L is comprised of two or more portions, and wherein at least two of said portions are non-covalently bonded together.
- non-covalent bonding is hydrogen bonding or electrostatic bonding.
- the one or more portions comprise one or more DNA/RNA-bases, amino acids, peptides, cofactors, enzymes, enzyme fragments, activating enzymes, biological macromolecules or enzyme substrates, wherein said portions are naturally occurring, modified from those naturally occurring, or are synthetic.
- L is comprised of two or more portions, and wherein at least two of said portions bond to one or more metal centres.
- Alkali metal cations for example including lithium, sodium and potassium.
- Transitional metals for example including those which are mono-, di- or tri-cationic.
- L comprises one or more polymeric portions.
- L comprises one or more of: a bond, a hydrogen bond; a non-covalent bond; an electrostatic bond; metal bonding; alkyl, cyclic alkyl, aryl, alkene, alkyne, heterocyclic, heteroaromatic, sugar, metal complex, or is a through-space interaction.
- the separation of an atom bearing the anion of NEG and an atom bearing the radical of RAD is sufficient to allow the formation of a structure of Formula (I). That is, at this separation, the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly-Occupied Molecular Orbital (DOMO) of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- DOMO Doubly-Occupied Molecular Orbital
- L is selected from the group comprising: a bond; phenyl; biphenyl; Ci to C 20 alkyl, C 3 to C 20 cycloalkyl, RNA-based sugar; DNA-based sugar.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 15 A or less.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 10 A or less.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 5 A or less.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 5 to 15 A.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 5 to 10 A.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 10 to 15 A.
- a method, use, process, structure or protecting group as defined previously, wherein an atom bearing the negative charge of the anion of NEG and an atom bearing the unpaired electron of the radical of RAD are separated by a through-space distance of about 5 to 7 A.
- the Formula (I) comprises an ionic resin, wherein the resin may comprise negative or positively charged groups.
- the anion of NEG is a neutral functional equivalent, which provides the same function as the anion of NEG.
- the neutral functional equivalent could comprise an electron lone pair, wherein that lone pair is capable of bonding to a proton or other cation; and wherein the lowest Singly-Occupied Molecular Orbital (SOMO) of RAD is lower in energy than a Doubly- Occupied Molecular Orbital (DOMO) of the neutral functional equivalent of NEG; and wherein the SOMO of RAD is higher in energy than the DOMOs of the neutral functional equivalent of NEG when the lone pair of the neutral functional equivalent of NEG is bonded to a proton or other cation.
- SOMO Singly-Occupied Molecular Orbital
- RAD Doubly- Occupied Molecular Orbital
- the neutral functional equivalent of NEG could be selected from the group: carbenes, stable carbenes (e.g. N-heterocyclic carbenes and diaminocarbenes), phosphines or amines.
- carbenes stable carbenes (e.g. N-heterocyclic carbenes and diaminocarbenes), phosphines or amines.
- the neutral functional equivalent is reacted with a proton (or other cation) the resultant group is positively charged.
- Such deactivation of the lone pair(s) can have an effect the stability of the radical.
- strong bases such as LDA (lithium diisopropylamide) are required to deprotonate stable carbenes, and these stable carbenes are known to reversibly coordinate to alkali metals such as lithium, sodium and potassium.
- the anion of NEG is not a chemical group but rather is a functional physical equivalent, which provides the same function as the anion of NEG.
- the functional physical equivalent comprises a negative point charge, wherein that negative point charge is capable of being neutralized, and wherein the energy level of the unpaired electron of RAD is lowered when the functional physical equivalent of NEG together with RAD-L forms the (corresponding) structure of Formula (I), and wherein the energy level of the unpaired electron of RAD is the highest energy level when the functional physical equivalent is electrically
- the negative point charge could be simply electronically neutralized.
- an electrode could comprise the negative point charge, and wherein a surface could comprise a plurality of such electrodes. Where the electrode comprises a negative point charge, that point charge could be neutralized by turning off the electrode or by reversing the polarity of the electrode.
- an electrode could be used to manipulate the nature of such a negative point charge.
- the effect of the negative point charge may be tuned by the
- UV, IR, ESR and Mass Spectra will be different in the SOMO ⁇ HOMO and SOMOHOMO forms of the structure of Formula (I). Such differences could be used in the quantification of such species, detection of orbital conversion in them, and/or could be used to alter the range in which a functional group is normally detectable into a more useful detectable range.
- a method, use, process, structure or protecting group as defined previously wherein the structure of Formula (I) is selected from the group comprising the intermediates and/or products of the oxidative degradation of lipids, phospholipids, wherein such species comprise a structure selected from:
- Fig. 1 is a schematic summary of the chemical aspects and molecular orbitals considered in the present invention.
- Fig. 2 shows distonic radical anions with the orbital conversion.
- Fig. 3 shows pH-switching of radical stability.
- Fig. 4 shows the assessment of the ⁇ -assistance effect.
- Fig. 5 shows experimental evidence of the BDE-switching by pH-induced orbital conversion.
- Fig. 6 shows SOMO-HOMO conversion in various substrates.
- Fig. 7 shows a SEC traces of PMA-Br ( ⁇ ), PMA-Br with HOOC-TEMPO.
- Fig. 8 shows a SEC traces of PMA-Br ( ⁇ ), PMA-Br with HOOC.
- Fig. 9 shows a Gaussian simulation of PMA-Br with HOOC-TEMPO.
- Fig. 10 shows SEC traces of purified PMA-ON-COOH.
- Fig. 1 1 shows a Gaussian simulation of PMA-ON-COOH for 6 hours.
- Fig. 12 shows a Gaussian simulation of PMA-ON-COOH for 20 hours.
- Fig. 13 shows a proposed mechanism for PFL (activated by PFL-EA) in the catalytic condensation of CoA and pyruvate to from acetyl-CoA and formate; (b) shows a summary of the overall transformation.
- Fig. 14 shows an exemplary schematic potential energy profile of reactions with and without switching in the transition states and products.
- Fig. 15 plots the pH switch in solution (various solvents) versus the pH switch in the gas phase.
- Fig. 16 shows proof of pH switching in low polarity solution.
- Fig. 17 shows a repeat of the experiment as detailed in Fig. 16 except that a base 1 ,8- diazabicylo[5.4.0]undec-7-ene (DBU, 33nM) was included.
- DBU diazabicylo[5.4.0]undec-7-ene
- Fig. 18 shows pH switching in aqueous solution.
- Fig. 1 shows a schematic summary of the chemical aspects and molecular orbitals considered in the present invention.
- a molecule in which an acid group (COOH) is linked to a group X by a linker, and group X is bonded to a group R.
- the acid group broadly conforms to the NEG group of the invention
- the linker structure broadly conforms to the L group of the invention
- the X group (when in the unbound state, i.e. ⁇ ) corresponds to the group RAD.
- R-X COO " can dissociate to give the radicals R « and ⁇ COO " , this is because the radical formed «X COO " is stabilised by the orbital 'conversion', i.e. wherein the singly-occupied molecular orbital is not the highest-occupied molecular orbital (i.e. SOMO ⁇ HOMO).
- a representation of the molecular orbitals is shown next to the ⁇ COO " species, together with an energy level diagram of the orbitals.
- orbital diagram of ⁇ COO " it can be seen that there is an unfilled orbital (i.e. shown as ⁇ ) which is not the highest orbital, in disconformity with the established accumulated principle.
- Fig. 2 shows distonic radical anions - the new class of species with the SOMO-HOMO conversion. Shown are M06-2X/6-31+G(d) optimized geometries, spin density plots (for open-shell species only) and dipole moments ( ⁇ , in Debye), along with the
- Calculated relative energies (E r , in kJ mol "1 ) are given for the oxidation products of distonic radical anions only. See Example 1 to Example 4 for the orbital plots and configurations, and see Example 6 for the detailed description of the oxidation products.
- Fig. 3 shows pH-switching of radical stability
- a Two resonance forms of an aminoxyl radical
- b Switching of aminoxyl stability by pH-induced polar effects (the underlined numbers are the distances between the two coloured atoms in the M06-2X/6-31 +G(d) optimized geometries, A), c, Structures, M06-2X/6-31+G(d) spin density plots (for open- shell species only) and dipole moments ( ⁇ , in Debye), as well as MCSCF(9,5)/6-31 +G(d) molecular orbital configurations of the reference radicals in protonated and deprotonated forms, d-e, Energy decomposition of the calculated BDE-switches (G4(MP2)-6X for acyclic series and ONIOM approximation for the cyclic series, gas phase, kJ mol "1 ) in the pairs of homologue series of trial and reference radicals, plotted against the number of methylene units
- Fig. 4 shows assessment of the ⁇ -assistance effect, a, M06-2X/6-31+G(d) optimized geometries and BDE-switches (in electronic energy terms, kJ mol "1 ) of non-substituted TEMPO (— ), carboxy-TEMPO homologues in extended chain ⁇ ) and lowest energy (® ) conformations, and the corresponding non-bonded TEMPO... CH 3 COOH complex ( ⁇ ).
- Fig. 5 shows Experimental proof of the BDE-switching by pH-induced orbital conversion
- Optimized COO-H bond lengths in the dimer are equal to 1.33 A for A and 1.33 A for B. c and d, Calculated BDE- switches (gas-phase, 25 °C) plotted against experimentally measured GPA-switches for carboxy-TEMPO vs. its alkoxyamines (O ), carboxy-PROXYL vs. its alkoxyamines ( ⁇ ), carboxy-PROXYL vs. carboxy-TEMPO ( ⁇ ) and different combinations of alkoxyamines (O , only one example is shown), for details see Examples).
- Fig. 6 shows SOMO-HOMO conversion in various substrates.
- Calculated BDE-switches italics, in electronic energy terms, kJ mol "1 ) in TEMPO-CH 3 substituted with different anionic groups (a, using G3(MP2,CC)(+) method) and in model DNA 22 and RNA 23 sugar and base (guanine) moieties (b, using ONIOM approximation to the G3(MP2)RAD energies at the MP2/6-31 1+G(3df,2p) level of theory); see Table 14 and Table 15 for details.
- Fig. 7 shows a SEC traces of PMA-Br ( ⁇ ), PMA-Br with HOOC-TEMPO reaction in Toluene with CuBr /PMDETA for 30 min ( ⁇ ), and 20 h ( ⁇ ) at R.T.
- Fig. 8 shows a SEC traces of PMA-Br ( ⁇ ), PMA-Br with HOOC-TEMPO reaction in DMSO with CuBr /Me 6 TREN for 30 min ( ⁇ ) and 20 h ( ⁇ ) at R.T.
- Fig. 9 shows a Gaussian simulation of PMA-Br with HOOC-TEMPO reaction in DMSO with CuBr /Me 6 TREN (-) and LMD simulation (--). It showed 27.3 % of coupling product.
- Fig. 10 shows a SEC traces of purified PMA-ON-COOH kept in THF overnight ( ⁇ ), PMA-ON-COOH in DMSO with Me 6 TREN for 6 h (#)and 20 h ( ⁇ ) at R.T.
- Fig. 13 shows (a) a proposed mechanism for the condensation of CoA and pyruvate to acetyl-CoA and formate, an important reaction in the anaerobic glucose metabolism of bacteria. The process shown occurs via a radical mechanism - homolytic cleavage of the C1 -C2 bond in pyruvate.
- PFL-AE a 28 kDa monomeric enzyme, a member of the radical S-adenosylmethionine family
- PFL-AE a 28 kDa monomeric enzyme, a member of the radical S-adenosylmethionine family
- the active site of PFL-AE contains a [4Fe-4S]+ cluster which reduces G734 on PFL via an S- adenosylmethionine (AdoMet) cofactor.
- AdoMet S- adenosylmethionine
- the carboxylate group of pyruvate (e.g. corresponding to the anion of NEG) can stabilise the radical on G734 (e.g. corresponding to the radical of RAD) to give a structure of Formula (I), where the radical and anion are sufficiently close (i.e. proximate) to have a through-space interaction to give the SOMO ⁇ HOMO configuration.
- the carboxylic group e.g.
- AS is change of entropy
- ⁇ ⁇ is change of electronic energy
- ⁇ ( ⁇ ⁇ + ZPVE) is change of electronic energy corrected for zero-point vibration (enthalpy at 0 K)
- ⁇ is change of enthalpy (previous term including thermal correction, or enthalpy at a given temperature)
- AG is a change of Gibbs free energy (energy including thermal and entropic contributions at a given temperature)
- ⁇ denotes activation (kinetic) parameters
- rxn denotes reaction
- the activation barrier AG ⁇ of such reaction can be affected by the switching. In this example, it is larger in the case of non-switched species compared to the switched species forming in the reaction.
- Fig. 15 plots the pH switch in solution (various solvents) versus the pH switch in the gas phase for a big test set of species.
- the pH switches were obtained using accurate quantum chemistry. Details of the species represented in this figure are provided in Table 21. These quantum chemical calculations reveal that the presence of polar solvents reduces the underlying stabilizing effect substantially, which is consistent with its electrostatic origin. Nonetheless they show clearly that substantial pH switches are retained in low polarity solvents, and that the switches in polar solvents, while smaller, do remain synthetically useful for some combinations of leaving groups where solvent interactions help to favour dissociation in deprotonated versus protonated form.
- Fig. 16 shows proof of pH switching in low polarity solution (in this case bulk styrene monomer).
- the plot shows the molecular weight distribution of a polymeric alkoxyamine comprising a polystyryl chain bound to a 4-carboxy-TEMPO nitroxide (1 1.6mg/mL) as it is heated to 100°C in the presence of a free-radical initiator tert-butyl hydroperoxide (BHP) and styrene monomer but no base.
- BHP free-radical initiator tert-butyl hydroperoxide
- Fig. 17 shows an exact repeat of the experiment as detailed in Fig. 16 except that a base l ,8-diazabicylo[5.4.0]undec-7-ene (DBU, 33nM) was included to deprotonate the carboxylic acid of the 4-carboxy-TEMPO.
- DBU base l ,8-diazabicylo[5.4.0]undec-7-ene
- Fig. 18 shows proof of pH switching in aqueous solution.
- a standard potentiometric titration was used to measure the pKa of 4-carboxy-TEMPO nitroxide and various non-radical forms of it (i.e. alkoxyamines and hydroxyl amine) to show that formation of the radical leads to increased acidity of the carboxylic acid group.
- the hydroxylamine has a pH switch of 1.5 pKa units in a 75% ethanol / 25% water solvent.
- the pKa switch corresponds directly to a switch on radical stability of 1.5 orders of magnitude.
- a pKa switches of 0.5 unit were determined for 4-carboxy-TEMPO with a benzyl leaving group.
- the smaller switch for this leaving group is due to the differing solute-solvent interactions which are less favourable for dissociation of the deprotonated alkoxyamine versus protonated.
- the present invention provides a way in which the electronic configuration of the structure of Formula (I) can be switched between the SOMO ⁇ HOMO and
- the present invention allows the molecular orbitals in the structure of Formula (I) to be 'switched' from one electronic configuration to another physically different configuration, and this being brought about by the simple addition or removal of protons or other cations.
- This change in electronic configuration fundamentally changes the nature of the chemistry of the resultant species.
- Direct access to the 'switching' of molecular orbitals in this way by direct chemical means has hitherto been unknown. This work has been published in Nature Chemistry ( Gryn'ova G., Marshall D., Blanksby, SJ, Coote M.L. Nature Chem.(2013), 5, 474-481) and is incorporated herein by reference.
- composition of Formula (I) that can be modified comprise:
- the magnitude of that energy difference can be manipulated by the arrangement of the various groups in Formula (I). For example, this can be changed by changing the RAD, L and NEG groups, in terms of the stability of those groups, their connectivity and their spatial arrangement in three dimensional space. In that way the difference between energy states when the
- SOMO HOMO as compared to the state where SOMO ⁇ HOMO can be manipulated, and so the resultant chemical properties of the 'normal' and 'converted' species can be changed.
- the resultant chemical properties of the 'normal' and 'converted' species can be tuned to be sensitive to the reaction conditions or environment in which they are to be used.
- weakly acidic NEG groups (or strongly basic groups) will tend to bond protons (and by extension other cations) more strongly, whereas more acidic NEG groups (or weakly basic groups) will tend to bond protons (and other cations) more weakly.
- the nature of the anion of NEG can allow access to the control of the 'switch' that causes the interconversion of the 'normal' and 'converted' arrangements. Therefore, the anion can be selected to be responsive to certain reaction conditions, or could be tailored to bind more selectively to one cation in preference to another. This could be for example used to allow the switching from the 'normal' electronic configuration to the 'converted' electronic configuration to occur in a certain pH range.
- reaction conditions like the polarity of the solvent will affect how strongly the anion of NEG will bond to protons and/or other cations (such as metal ions and quaternary amines).
- reaction conditions in combination with the nature of the anion of NEG, could be used to moderate the reactivity of the structure of Formula (I). In particular these could be used to control the switching between SOMO ⁇ HOMO and SOMOHOMO states.
- the radical of RAD can be more or less stable, and this stability can be used to affect the magnitude of the energy difference between the SOMO orbitals in the 'normal' and 'converted' configurations.
- the invention also provides access to the manipulation of the acidity of the anion of NEG through the control of nature of the radical.
- radical can be used to influence the acidity of the anion of NEG (for example where the anion is a carboxylic acid).
- the stability of the resultant radical of RAD is found to affect the acidity of the (acidic) anion of NEG.
- the radical of RAD is stabilised, there is a greater propensity for the protons associated with the anion of NEG to then dissociate.
- the radical of RAD is less stabilised by the structure of Formula (I), the protons associated with the anion of NEG are less likely to dissociate.
- the anion of NEG i.e. the conjugate base of the acid
- the p a of the protonated anion of NEG i.e. the conjugate acid of NEG will correspondingly be higher in the same fashion.
- the NEG group in Formula (I) is more acidic in nature as compared to the corresponding structure where the radical is not present, and the more stable the resulting radical the more acidic the resultant anion of NEG will be.
- the binding properties of the anion of NEG can be manipulated by the nature (and/or inclusion) of the radical of RAD.
- the inclusion of a radical into an acid bearing structure could be used for the purpose of increasing that acid's acidity, where the other requirements of the structure of Formula (I) are satisfied.
- linker L allows further manipulation of the structure of Formula (I).
- L could be a group selected to provide certain physical properties to the structure of Formula (I), for example to give the resulting structure a certain solubility profile (e.g. increased solubility in non-polar solvents by the inclusion of fatty acid chains).
- L can be used to define the spatial arrangement of RAD and NEG in Formula (I), both in terms of connectivity and in terms of a three dimensional spatial arrangement.
- the radical of RAD and the anion (i.e. the negative charge) of NEG engage in a long range interaction which provides the effect which stabilises the radical of RAD, and so gives the SOMO ⁇ HOMO electronic configuration.
- L could be space i.e. having a through-space interaction between RAD and NEG causing orbital conversion and related effect on RAD stability does not require a chemical link between RAD and NEG.
- L can be used to stabilise (or destabilise) the radical of RAD and/or to destabilise (or stabilise) the anion of NEG.
- the magnitude of the difference between the energy states of SOMOHOMO and SOMO ⁇ HOMO can be further manipulated.
- That stabilisation (or destabilisation) of the radical of RAD and/or the destabilisation (or stabilisation) of the anion of NEG could be brought about by steric or electronic effects.
- Various groups could be introduced into L which could be situated proximate (i.e. close enough to have an effect) to RAD and NEG. These groups can be selected to have a steric and/or an electronic impact on the RAD and/or NEG groups.
- one or more steric groups could be used to sterically protect a group, or these could be used to 'twist' a group into an unfavourable position destabilising the RAD and/or NEG groups, or bring two mutually incompatible groups into closer proximity.
- Electronic effects that could be used include delocalisation, ⁇ -conjugation, ⁇ -assistance, hyper-conjugation, aromaticity and long range polar effects. Application of these means would be appreciated by the skilled person.
- this radical is less likely to participate in bond forming reactions, and by extension, is more likely to (homolytically) cleave from bonds which it is participating in.
- the stabilised SOMO ⁇ HOMO radical structure can be made less stable by the addition of protons or other cations to the anion of NEG. These cations switch the orbitals from SOMO ⁇ HOMO to the more 'normal' (but less stable)
- SOMO HOMO arrangement. In that way the radical becomes destabilised and so more reactive, and thereby is activated to participate in bond forming reactions, and so could be 'activated' to react with a structure to be protected.
- protons or other cations
- the addition of protons (or other cations) to the structure of Formula (I) causes the radical to be 'activated' and so to become more reactive and so that it can form a bond to a radical to be protected.
- Reversing the above process, i.e. removing protons or other cations stabilises the structure of Formula (I), and if suitably stabilised the stabilised radical can dissociate from the radical structure (i.e. by homolytic cleavage); or at least the process and/or conditions of removing the structure of Formula (I) from the structure to be protected can be made more moderate (e.g. using lower temperatures).
- a portion of a structure can be protected with the structure of Formula (I) e.g. R-RAD-L-NEG, while various other reactions are conducted on the unprotected portions of that structure.
- the structure of Formula (I) can be selectively released (e.g. by increasing the pH) from that structure, allowing that portion which had been protected to be unprotected e.g. to give R » and RAD-L-NEG (where NEG bonds to a proton or other cation).
- the radical to be protected once deprotected, is free to participate in further reactions.
- the skilled person will be aware of those reactions, typically being radical type reactions.
- a radical of Formula (I), in which the SOMO is lower in energy than one or more DOMOs is said to be SOMO-HOMO converted.
- bond energies that a radical of Formula (I) forms with other radicals e.g. Rv
- a compound of Formula (I), where NEG is protonated or bonded to a different cation can be used to "protect" R « and prevent it from undergoing other types of radical reactions through covalently bonding to RAD-L-NEG (where NEG is bonded to a proton or other cation) to give R-RAD-L-NEG (where NEG is bonded to a proton or other cation).
- R « and RAD-L-NEG one simply removes the added protons (or cations) from NEG group.
- each protecting group of the invention could be used on the same structure to be protected, whereby each protecting group is deprotected under different reaction conditions, for example each responsive in a certain pH range or other cation concentration range, or to progressively harsher deprotecting conditions such as increasing temperature. It is also possible that more than one protecting group can be removed (from a structure to be protected) under the same conditions.
- the two resulting radicals on the same structure can be arranged such that they can recombine.
- the two resultant radicals could be arranged such that the cyclization of the structure bearing these radicals occurs when they are deprotected (e.g. an intramolecular Wurtz reaction).
- the present invention provides new SOMO-HOMO converted compounds with an alternative source of high-energy HOMO(s) that enables the switching between the 'regular' and 'converted' orbital configurations.
- NEG in the structure of Formula (I) provides high energy HOMO(s) but restores up (i.e. regular) configuration upon protonation to the conjugate acid form.
- the manifestation of this unique electronic structure is not limited to the redox behaviour and that an unprecedented long-range interaction between an unpaired electron and a negative charge results in a dramatic increase in the stability of the radical and acidity of the conjugate acid.
- the acid-base motif provides an exceptional instrument for switching the orbital configuration, and thus the radical stability, by pH. It is contemplated that such switching has potential applications in biological and industrial fields.
- Radical stability Having designed free radicals with pH-controlled SOMO-HOMO energy-level conversion, there was explored their stability in the switched and non- switched forms.
- a representative practical measure of radical stability is its bond dissociation free energy (BDFE) with simple carbon-centred radicals such as methyl, •CH 3 . Therefore calculated were the methyl BDFEs of the carboxy- aminoxyl and peroxyl radicals in their neutral and deprotonated forms in the gas phase at 25 °C and found that deprotonation of the carboxylic group (and thus switching from regular to converted orbital configuration) weakens their bonds with »CH 3 .
- BDFE bond dissociation free energy
- Radical stability is influenced by polar effects, which, at long range, act primarily through-space rather than through-bond.
- the stability of the aminoxyl radical is affected by resonance between its two forms I and II (see Fig. 3a).
- pH-switchable nitroxide mediated polymerization (NMP) in agent 8 employs polar effects by introducing several basic groups (see Fig. 3b), which upon protonation destabilize II and thus the radical overall. This results in slower decomposition of the corresponding alkoxyamines, indicating an increased strength of their NO-R bonds.
- the magnitude of this pH switch ( ⁇ 15 fold) is significantly smaller than the pH effects observed in the SOMO-HOMO converted species (> 2000 fold), despite the fact that the charge in 4 is more distant from the radical centre than in 8 (see Fig. 3b).
- carboxylate anion is expected to stabilize II in the trial aminoxyl, and possibly act in a similar way in the peroxyl radical. Therefore, to determine whether this standard polar effect could account for the observed BDFE-switching, there was employed the procedure used to study the effect of amino group protonation on the COOH pK a in the homologue series + R 3 (CH 2 ) n COOH via comparison to a reference system CR 3 (CH 2 ) n COOH that is structurally similar but lacks a polar contribution.
- ⁇ -assistance represents another possible type of through-bond interaction between the remote negative charge and radical centre.
- the calculated absolute methyl BDEs of the deprotonated alkoxyamines follow the Mr dependence (see Fig. 4b).
- We also constructed a complex of TEMPO and acetic acid 16, structurally resembling the n 4 extended chain conformer 15, but clearly free of any through-bond interaction. Its calculated absolute BDE also sits on the Mr line (see Fig.
- SOMO-HOMO conversion was also investigated in the deprotonated nucleic acid radicals, say for example formed as a result of oxidative damage via hydrogen atom abstraction by, for example, hydroxyl radical.
- thermochemical parameters e.g., BDE- and GPA-switches
- their relative differences e.g., BDE- and GPA-switches
- Alkoxyamines were synthesized from aminoxyl radical precursors by a standard literature procedure. Equimolar mixtures of aminoxyl radical and alkoxyamine were prepared pairwise in methanol to a final concentration of ca. 10 ⁇ . Proton-bound dimers were generated by negative ion electrospray ionization upon infusion of these methanolic solutions at a rate of 5 iL min "1 into the ion source of a Waters QuattroMicro (Manchester, UK) triple quadrupole mass spectrometer. The mass spectrometer was operated in the negative ion mode, and controlled by Micromass MassLynx software (Version 4.1 ).
- the capillary voltage was set to -3.0 kV, cone voltage -20 V, source temperature 80 °C and desolvation temperature 1 10 °C, to optimize the production of desired proton-bound cluster ions.
- Nitrogen was used as the drying gas, at a flow rate of 300 L h "1 while collision-induced dissociation (CID) experiments used laboratory frame energies of 5-25 V as required and argon as the collision gas (3.0 ⁇
- MCSCF orbital plots were built using MacMolPlt v.7.4.2; optimized MCSCF and MRPT2 configurations for the two trial species are shown in Example 2.
- CASSCF(7,8)/STO-3G as implemented in Gaussian 09.
- the active space included four occupied orbitals (SOMO and three orbitals of the carboxylate, described above) and four corresponding virtual orbitals; orbitals were built using GaussView 5.0. Obtained results again confirmed converted configuration to be the dominant one for the trial compounds (see Example 3).
- Example 1 Single-reference molecular orbitals of the trial distonic radical anions.
- Example 2 j MCSCF and MRFT2 molecular orbttals of u- rrial distontc radfeai mio .
- Plots of MCSO (9,5V&31 +0( ⁇ optimized ' motemiar or ltals an#
- Example 6 Properties of the one-electron oxidation open- and closed-shell products.
- standard G3(MP2) methods approximate CCSD(T) calculations with a large triple- ⁇ basis from calculations with a double- ⁇ basis set via basis set corrections carried out at the R(0)MP2 (in G3(MP2)-RAD) and UMP2 (in G3(MP2,CC) levels.
- URCCSD(T) energies calculated using the Molpro code
- HHC high level correction
- PROXYL- 120.501 2 1 .61 1 16.684 13.885 49.812
- the inventions has shown that orbital conversion results in an increased radical stability, e.g. if such converted radical is a product of some chemical reaction, the Gibbs free energy (AG ⁇ ) of this reaction is lower than the Gibbs free energy of an identical reaction but involving formation of the radical species in their regular orbital configuration.
- AG ⁇ Gibbs free energy
- Fig. 14 shows that the AG ⁇ and the the AGTM are both lower for the switched species than for the non-switched species. Where AG ⁇ is smaller the reaction will be quicker. Where AG rxn is lower (i.e. more exothermic) the reaction will favour products over reagents where these two species are in equilibrium.
- Table 3 shows that ionic bonding of a cation (e.g. lithium, sodium and potassium) stabilizes the negative charge of NEG (in a structure corresponding to Formula (I)), to the extent that the SOMO of RAD is higher in energy than the DOMOs of NEG when the anion of NEG is bonded to that cation. That is, this shows that the orbital conversion in a structure corresponding to Formula (I) can be switched off by the addition of cations to the anion of NEG.
- Table 4 Calculated BDE-switches in the gas phase and in solvents of different polarity.
- the calculations in Table 4 indicate that in polar solvents such as water orbital switching is quenched, whereas in less polar solvents like toluene it is preserved.
- the polarity of the solvent can therefore be used to control orbital switching of the invention.
- the polarity of a solvent can be changed by changing the constituents of the solvent, e.g. adding or removing different solvents or adding or removing polar components e.g. ammonium salts or ionic liquids.
- B3LYP results are off by 10s of kJ mol " 1 compared to the composite methods.
- BDEs of all the protonated species are generally independent on the separation (n) between the radical and carboxylic moieties; the same is largely true for the deprotonated reference alkoxyl radicals, whereas BDEs of the SOMO-HOMO converted species in the deprotonated state increase slowly with separation.
- Example 8 thus confirming the primarily through-space nature of the orbital conversion switching effect and only a very minor contribution from the through-bond ⁇ -assistance.
- Optimized M06-2X/6-31 +G(d) geometries of the lowest energy c, and extended chain, d (top), conformers of the aminoxyl homologue with n 4, as well as the supermolecule of TEMPO and acetic acid, d (bottom); shown are the radical species and the corresponding methyl ethers in the deprotonated and protonated forms of the carboxylic group.
- Example 8 BDEs of the aminoxyl homologues. Calculated M06-2X/6-31 +G(d) bond dissociation electronic energies of the aminoxyl homologues series: extended chain conformers (o), lowest gas-phase energy conformer of n-4 ( ⁇ ) and supermolecule consisting of TEMPO and acetic acid (0).
- Example 10 Test set. Aminoxyls and alkoxyamines, studies in the mass-spec experiments. HO
- the kinetic method relationship relies on: ( ) the absence of reverse activation barriers; ( ) the absence of any isomer forms of the cluster; (Hi) a negligible entropy difference for the competitive dissociation channels (i. e. , A(A ac idG) simplifies to A(A ac jdH)).
- Assumptions ( ) and ( ) are generally considered as robust for dissociation of simple proton bound dimers and for the systems studied here only simple dimer dissociation was observed suggesting (Hi) also holds for the experiments.
- Ion abundance ratios (Ii :I 2 ), obtained from the collision-induced dissociation of proton-bound dimers at a collision energy of 10 V (in the laboratory frame) in a triple quadrupole mass spectrometer using argon as the target gas. Reported ratios are the average of at least 200 cumulative scans and include replicate infusions of samples solutions. A ratio greater than unity implies A ac idHi > A ac jdH 2 . Ratios of primary importance are those comparing aminoxyl radicals (A & E) to alkoxyamines. Selected additional ratios are reported between alkoxyamines pairs as a cross-reference for internal consistency.
- Equation 1 and kj are dissociation rate constants; ⁇ ( ⁇ 5) is the difference in entropy between the competing dissociation pathways; R is the universal gas constant, and T e ff ⁇ s the effective temperature parameter.
- the latter is an empirical scaling factor that accounts for the non-Boltzmann energy distribution within the activated dimer population and is not a true temperature.
- Example 1 1 Determination of the effective temperature. Effective temperature dependence on collision energy in the centre of mass frame for the described instrument setup, using multiple benzoic acids as reference compounds.
- ⁇ R A + R B ) 2 and RA and RB are the molecular radii of cyclohexyl radical and dioxygen taken as 1 .74 A and 2.90 A, respectively, kg is the Boltzmann constant, T is absolute temperature and ⁇ is the reduced mass f mA*mB ⁇ Table 13 Methyl BDEs of TEMPO with various anionic groups. Calculated bond dissociation electronic and free energies (in the gas phase at 25°C, kJ mol-1) for the species in Fig. 6.
- Protonaiion of the two phosphate groups is denoted as ( ⁇ - ⁇ ) tor deproionated and (1:1) ibr protonated: state:; i; two-layer ONiOM appj3 ⁇ 4 imation to the CB(MP2)-RA D energies using ti3 ⁇ 4MP2/6 ⁇ 1 1 - ⁇ -0:(3di;2p) method.
- the radical has been partially activated.
- the activated radical is in equilibrium with the (deactivated form, i.e. SOMO ⁇ HOMO) allowing for the PMA radicals to combine.
- the deactivated (i.e. deprotonated) radical coupled to PMA dissociates into the stabilised radical and the PMA radical.
- the PMA radical so deprotected is free to react, in this case with a further PMA radical to give PMA-PMA.
- the PMA radicals react with each other.
- Open-shell triplet - -669.71 1 (adiabatic) 0.37505 671 .334 669.528 670.265 670.:
- Tl diagnostics from CCSD(T)/6-31+G(d) calculations; b Dispersion correction to B3LYP, calculated using DFT-D3 procedure with BJ damping; c Calculated using 6- 31+G(d) basis set; d Calculated using GTMP2Large basis set; e Alpha and beta exchange contributions to the HF/CBS energies.
- Table 18 Cyclic homologues series. All values, except for Tl diagnostics, are in Hartrees.
- Homologues are named as follows: first letter denotes alkoxyl (O) and aminoxyl (N) species, second state denotes anionic (A) and protonated (H) carboxylic group, number corresponds to n in Example 7a, R denotes radical species and M - the corresponding methyl ethers.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Toxicology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description




















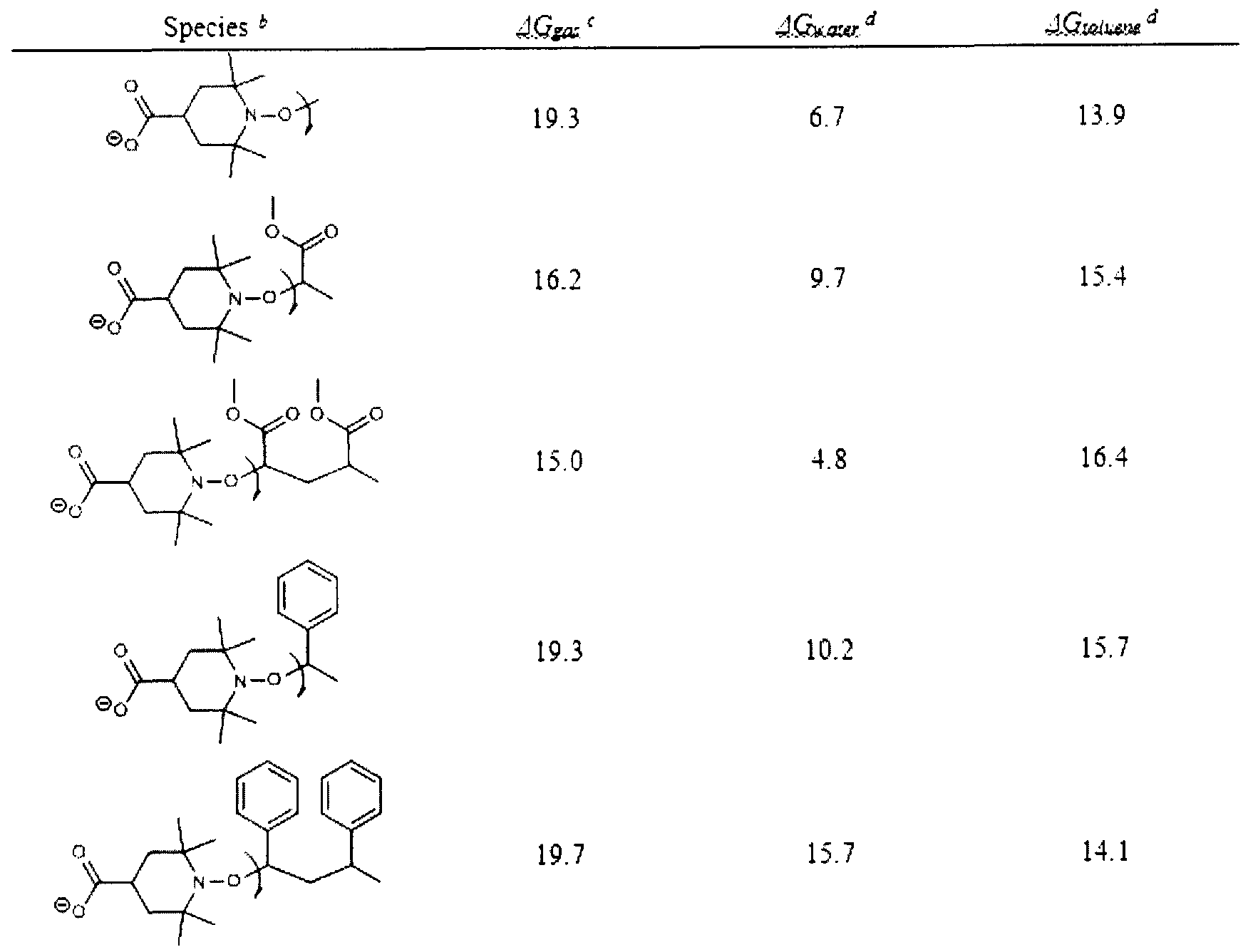
















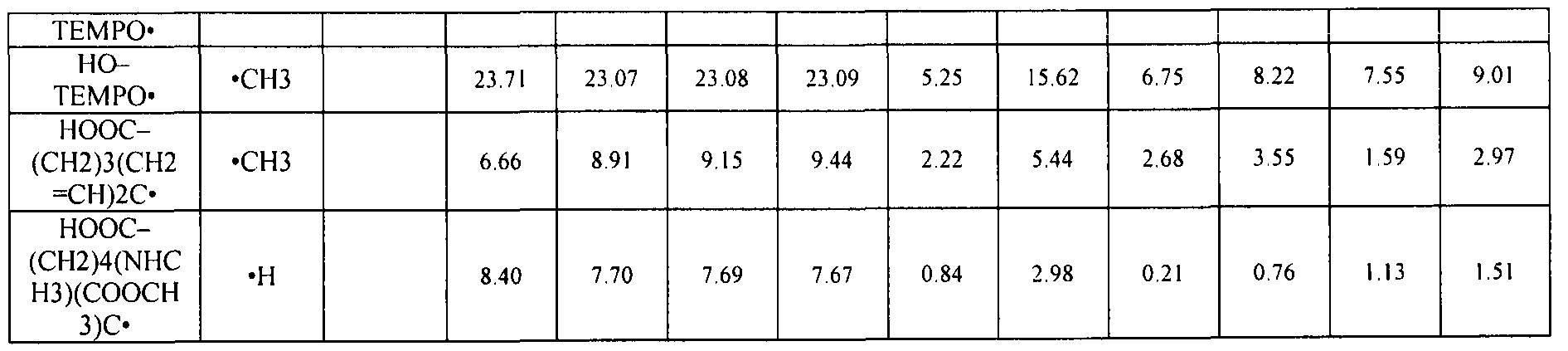
Claims




Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2014214532A AU2014214532B2 (en) | 2013-02-06 | 2014-02-06 | Radical orbital switching |
| CN201480019844.9A CN105209410A (en) | 2013-02-06 | 2014-02-06 | Radical orbital switching |
| EP14748562.7A EP2953916A4 (en) | 2013-02-06 | 2014-02-06 | ORBITAL RADICAL SWITCHING |
| US14/765,967 US20160075732A1 (en) | 2013-02-06 | 2014-02-06 | Radical orbital switching |
| CA2900272A CA2900272A1 (en) | 2013-02-06 | 2014-02-06 | Radical orbital switching |
| JP2015555501A JP2016508488A (en) | 2013-02-06 | 2014-02-06 | Radical orbit switching |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2013900371A AU2013900371A0 (en) | 2013-02-06 | Radical orbital switching | |
| AU2013900373 | 2013-02-06 | ||
| AU2013900373A AU2013900373A0 (en) | 2013-02-06 | Radical orbital switching | |
| AU2013900371 | 2013-02-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014121327A1 true WO2014121327A1 (en) | 2014-08-14 |
Family
ID=51299075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2014/000085 Ceased WO2014121327A1 (en) | 2013-02-06 | 2014-02-06 | Radical orbital switching |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20160075732A1 (en) |
| EP (1) | EP2953916A4 (en) |
| JP (1) | JP2016508488A (en) |
| CN (1) | CN105209410A (en) |
| AU (1) | AU2014214532B2 (en) |
| CA (1) | CA2900272A1 (en) |
| WO (1) | WO2014121327A1 (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12227457B2 (en) * | 2018-10-29 | 2025-02-18 | Universiteit Gent | Admixture for a cementitious material to influence the rheology properties of the cementitious material |
| CN110265646B (en) * | 2019-06-25 | 2022-09-16 | 西南大学 | Nitrogen-doped graphene-like activated carbon material and preparation method and application thereof |
| CN113707226B (en) * | 2021-08-24 | 2024-08-06 | 西安热工研究院有限公司 | Method for establishing micro-dynamics model during interconversion of xylose-xylulose |
| CN118609730A (en) * | 2024-06-13 | 2024-09-06 | 中国长城科技集团股份有限公司 | Lithium cobalt oxide performance analysis method and related equipment for lithium battery positive electrode materials |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2757865B1 (en) * | 1996-12-26 | 1999-04-02 | Atochem Elf Sa | METHOD FOR CONTROLLED RADICAL POLYMERIZATION OR COPOLYMERIZATION OF (METH) ACRYLIC, VINYLIC, VINYLIDENIC AND DIENE MONOMERS AND (CO) POLYMERS OBTAINED |
| TW593347B (en) * | 1997-03-11 | 2004-06-21 | Univ Carnegie Mellon | Improvements in atom or group transfer radical polymerization |
| SE0101153D0 (en) * | 2001-03-30 | 2001-03-30 | Zipat Ab | New material |
| JP2003096054A (en) * | 2001-09-25 | 2003-04-03 | Nof Corp | Alkoxyamine and method for producing the same |
| CN102083863B (en) * | 2008-03-07 | 2014-10-22 | 卡内基梅隆大学 | Improved Controlled Radical Polymerization Method |
| CN102746515B (en) * | 2012-07-16 | 2013-09-18 | 浙江大学 | Method for preparing block copolymers |
-
2014
- 2014-02-06 AU AU2014214532A patent/AU2014214532B2/en not_active Ceased
- 2014-02-06 US US14/765,967 patent/US20160075732A1/en not_active Abandoned
- 2014-02-06 CA CA2900272A patent/CA2900272A1/en not_active Abandoned
- 2014-02-06 JP JP2015555501A patent/JP2016508488A/en active Pending
- 2014-02-06 EP EP14748562.7A patent/EP2953916A4/en not_active Withdrawn
- 2014-02-06 CN CN201480019844.9A patent/CN105209410A/en active Pending
- 2014-02-06 WO PCT/AU2014/000085 patent/WO2014121327A1/en not_active Ceased
Non-Patent Citations (14)
Also Published As
| Publication number | Publication date |
|---|---|
| CN105209410A (en) | 2015-12-30 |
| AU2014214532A1 (en) | 2015-08-20 |
| EP2953916A4 (en) | 2016-10-26 |
| EP2953916A1 (en) | 2015-12-16 |
| US20160075732A1 (en) | 2016-03-17 |
| CA2900272A1 (en) | 2014-08-14 |
| AU2014214532B2 (en) | 2018-02-22 |
| JP2016508488A (en) | 2016-03-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Escobar et al. | Molecular recognition in water using macrocyclic synthetic receptors | |
| Kneeland et al. | Bis (alkylguanidinium) receptors for phosphodiesters: effect of counterions, solvent mixtures, and cavity flexibility on complexation | |
| Pagire et al. | Convergent deboronative and decarboxylative phosphonylation enabled by the phosphite radical trap “BecaP” | |
| WO2014121327A1 (en) | Radical orbital switching | |
| KR20120123612A (en) | Ionic liquids comprising nitrogen containing cations | |
| Glasovac et al. | Toward extension of the gas-phase basicity scale by novel pyridine containing guanidines | |
| Fener et al. | Scope and Limitations of the s‐Block Metal‐Mediated Pudovik Reaction | |
| Wang et al. | A systematic theoretical study on the acidities for cations of ionic liquids in dimethyl sulfoxide | |
| Fischer et al. | One-step methylation of aromatic phosphorus heterocycles: synthesis and crystallographic characterization of a 1-methyl-phosphininium salt | |
| Huang et al. | Uv–vis photodissociation action spectroscopy reveals cytosine–guanine hydrogen transfer in dna tetranucleotide cation radicals upon one-electron reduction | |
| LaPierre et al. | Synthesis of carbene-stabilized PNPN fragments and their carbene-dependent redox properties | |
| Tintaru et al. | Conformational sensitivity of conjugated poly (ethylene oxide)-poly (amidoamine) molecules to cations adducted upon electrospray ionization–A mass spectrometry, ion mobility and molecular modeling study | |
| Meddeb-Limem et al. | Computational study of the dimerization of glyphosate: mechanism and effect of solvent | |
| Chen et al. | Isolation of an acetylide-CuI3-tris (triazolylmethyl) amine complex active in the CuAAC reaction | |
| Darii et al. | First direct evidence of interpartner hydride/deuteride exchanges for stored sodiated arginine/fructose-6-phosphate complex anions within salt-solvated structures | |
| Jun et al. | Theoretical exploration of gas-phase conformers of proton-bound non-covalent heterodimers of guanine and cytosine rare tautomers: structures and energies | |
| Nifantiev et al. | Formation of benzooxaphosphole oxide heterocyclic system by the ring-contractive Arbuzov-Michaelis isomerization of alkoxy-substituted benzodioxaphosphorins | |
| CN116284117A (en) | A kind of thiophosphoramide derivatives and its three-component electrochemical synthesis method | |
| de la Cruz et al. | N‐Arylation of Pyrrolidino [3′, 4′: 1, 2][60] fullerene: Synthesis under Solvent‐Free Conditions and Electrochemistry of New C60–Acceptor Dyads | |
| Camanyes et al. | Theoretical study of the effect of Lewis acids on dihydrogen elimination from niobocene trihydrides | |
| Burck et al. | Phosphazene vs. diazaphospholene PN-bond cleavage in spirocyclic cyclodiphosphazenes | |
| Becherer et al. | Mass spectrometric study of oligourea macrocycles and their anion binding behavior | |
| Goswami et al. | An Imidazolium Salt That Uncharacteristically Avoids the Imminent Deprotonation of Its Acidic 2-H Proton by KN (SiMe3) 2 | |
| Bagdasaryan et al. | Synthesis and transformations of triphenylpropargylphosphonium bromide | |
| Grossert et al. | Fragmentation reactions of protonated α, ω‐diamino carboxylic acids: The importance of functional group interactions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14748562 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2015555501 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2900272 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14765967 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2014214532 Country of ref document: AU Date of ref document: 20140206 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014748562 Country of ref document: EP |