WO2014151409A1 - Organic compounds - Google Patents
Organic compounds Download PDFInfo
- Publication number
- WO2014151409A1 WO2014151409A1 PCT/US2014/025666 US2014025666W WO2014151409A1 WO 2014151409 A1 WO2014151409 A1 WO 2014151409A1 US 2014025666 W US2014025666 W US 2014025666W WO 2014151409 A1 WO2014151409 A1 WO 2014151409A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- free
- salt form
- halogen
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *N(C(C=C(N1c2ccccc2)Cl)=O)C1=O Chemical compound *N(C(C=C(N1c2ccccc2)Cl)=O)C1=O 0.000 description 2
- RKEUHNQOPBPRQD-UHFFFAOYSA-N CC(C)(CN1c2n[n](Cc(cc3)ccc3C(C)=O)c(Nc(cc3)ccc3F)c22)N=C1N(C)C2=O Chemical compound CC(C)(CN1c2n[n](Cc(cc3)ccc3C(C)=O)c(Nc(cc3)ccc3F)c22)N=C1N(C)C2=O RKEUHNQOPBPRQD-UHFFFAOYSA-N 0.000 description 1
- PSUHAEXBWCRQED-UHFFFAOYSA-N CC(c1ccc(C[n](c(Nc(cc2)ccc2F)c2C(N3C)=O)nc2N2C3=NC(C)(C)C2)cc1)O Chemical compound CC(c1ccc(C[n](c(Nc(cc2)ccc2F)c2C(N3C)=O)nc2N2C3=NC(C)(C)C2)cc1)O PSUHAEXBWCRQED-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/557—Eicosanoids, e.g. leukotrienes or prostaglandins
- A61K31/5575—Eicosanoids, e.g. leukotrienes or prostaglandins having a cyclopentane, e.g. prostaglandin E2, prostaglandin F2-alpha
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
Definitions
- the present invention relates to PDE1 inhibitory compounds of Formula I as described below, processes for their production, their use as pharmaceuticals and pharmaceutical compositions comprising them. These compounds are useful e.g., in the treatment of diseases involving disorders of the dopamine Dl receptor intracellular pathway, such as, among others, Parkinson's disease, depression, narcolepsy, psychosis, damage to cognitive function, e.g., in schizophrenia, or disorders that may be ameliorated through enhanced progesterone-signaling pathway, e.g., female sexual dysfunction.
- diseases involving disorders of the dopamine Dl receptor intracellular pathway such as, among others, Parkinson's disease, depression, narcolepsy, psychosis, damage to cognitive function, e.g., in schizophrenia, or disorders that may be ameliorated through enhanced progesterone-signaling pathway, e.g., female sexual dysfunction.
- PDEs phosphodiesterases
- CaM- PDEs Ca 2+ -calmodulin-dependent phosphodiesterases
- PDEIA is expressed in the brain with high levels in the CA1 to CA3 layers of the hippocampus and cerebellum and at a low level in the striatum.
- PDE1B is predominately expressed in the striatum, dentate gyrus, olfactory tract and in the prefrontal cortex co localized with the dopamine Dl receptor. Its expression generally correlates with brain regions having high levels of dopaminergic innervation.
- PDE1B is primarily expressed in the central nervous system, it is present in neutrophils. PDE1C is more ubiquitously expressed in the brain and is expressed in the heart and vascular smooth muscle.
- Cyclic nucleotide phosphodiesterases decrease intracellular cAMP and cGMP signaling by hydrolyzing these cyclic nucleotides to their respective inactive 5 '-monophosphates (5 'AMP and 5'GMP).
- CaM-PDEs play a critical role in mediating signal transduction in brain cells, particularly within an area of the brain known as the basal ganglia or striatum.
- NMDA-type glutamate receptor activation and/or dopamine D2 receptor activation result in increased intracellular calcium concentrations, leading to activation of effectors such as calmodulin- dependent kinase II (CaMKII) and calcineurin and to activation of CaM-PDEs, resulting in reduced cAMP and cGMP.
- CaMKII calmodulin- dependent kinase II
- CaMKII calmodulin- dependent kinase II
- CaM-PDEs calcineurin
- Dopamine Dl receptor activation leads to activation of adenylate cyclases, resulting in increased cAMP. This cyclic nucleotide in turn activates protein kinase A (PKA; cAMP-dependent protein kinase).
- PKA protein kinase A
- cGMP protein kinase G
- PKG and PKA phosphorylate downstream signal transduction pathway elements such as DARPP-32 (dopamine and cAMP-regulated phosphoprotein) and cAMP responsive element binding protein (CREB).
- DARPP-32 diopamine and cAMP-regulated phosphoprotein
- CREB cAMP responsive element binding protein
- Phosphorylated DARPP-32 in turn inhibits the activity of protein phosphates- 1 (PP-1), thereby increasing the state of phosphorylation of substrate proteins such as progesterone receptor (PR), leading to induction of physiologic responses.
- PP-1 protein phosphates- 1
- PR progesterone receptor
- Dl receptor signaling is disrupted in schizophrenia, contributing to cognitive impairment in the disease.
- the role of cAMP and cGMP in cognitive function has been well established in animal studies. Studies in rodents also have suggested that inducing cAMP and cGMP synthesis through activation of dopamine Dl or progesterone receptor enhances progesterone signaling associated with various physiological responses, including the lordosis response associated with receptivity to mating in some rodents. See Mani, et al, Science (2000) 287: 1053, the contents of which are incorporated herein by reference.
- CaM-PDEs can therefore affect dopamine -regulated and other
- intracellular signaling pathways in the basal ganglia including but not limited to nitric oxide, noradrenergic, neurotensin, CCK, VIP, serotonin, glutamate (e.g., NMDA receptor, AMPA receptor), GABA, acetylcholine, adenosine (e.g., A2A receptor), cannabinoid receptor, natriuretic peptide (e.g., ANP, BNP, CNP), DARPP- 32, and endorphin intracellular signaling pathways.
- nitric oxide e.g., noradrenergic, neurotensin, CCK, VIP, serotonin
- glutamate e.g., NMDA receptor, AMPA receptor
- GABA e.g., NMDA receptor, AMPA receptor
- acetylcholine e.g., adenosine (e.g., A2A receptor)
- cannabinoid receptor e.g.,
- PDE phosphodiesterase
- PDE1 phosphodiesterase 1
- functions in brain tissue as a regulator of locomotor activity and learning and memory functions in brain tissue as a regulator of locomotor activity and learning and memory.
- PDE1 is a therapeutic target for regulation of intracellular signaling pathways, preferably in the nervous system, including but not limited to a dopamine Dl receptor, dopamine D2 receptor, nitric oxide, noradrenergic,
- neurotensin CCK, VIP, serotonin, glutamate (e.g., NMD A receptor, AMP A receptor), GAB A, acetylcholine, adenosine (e.g., A2A receptor), cannabinoid receptor, natriuretic peptide (e.g., ANP, BNP, CNP), endorphin intracellular signaling pathway and progesterone signaling pathway.
- glutamate e.g., NMD A receptor, AMP A receptor
- GAB A acetylcholine
- adenosine e.g., A2A receptor
- cannabinoid receptor natriuretic peptide
- endorphin intracellular signaling pathway e.g., ANP, BNP, CNP
- endorphin intracellular signaling pathway e.g., endorphin intracellular signaling pathway and progesterone signaling pathway.
- inhibition of PDEIB should act to potentiate the effect of a
- PDE1 inhibitors are therefore potentially useful in diseases characterized by reduced dopamine Dl receptor signaling activity, such as Parkinson's disease, restless leg syndrome, depression, narcolepsy and cognitive impairment such as cognitive impairment associated with schizophrenia.
- PDE1 inhibitors are also useful in diseases that may be alleviated by the enhancement of progesterone-signaling such as female sexual dysfunction.
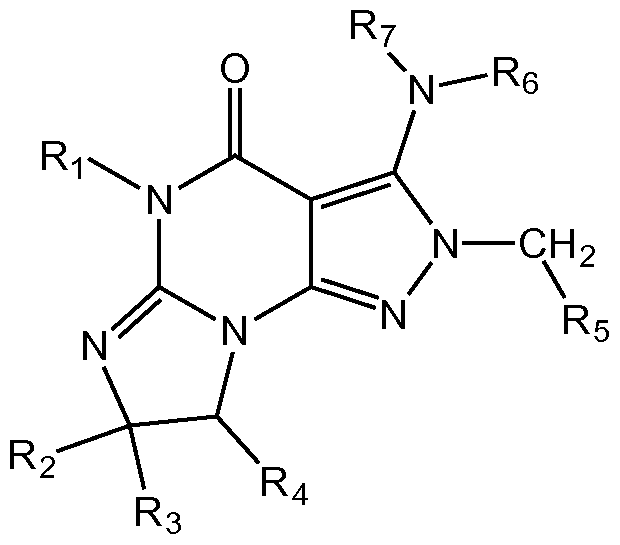
- Pvi is H or Ci_4 alkyl (e.g., methyl or ethyl);
- R 2 and R 3 are independently H or Ci_ 6 alkyl (e.g., methyl or ethyl);
- R4 is H or Ci_4 alkyl (e.g., methyl or ethyl);
- R6 and R 7 are independently H or aryl (e.g., phenyl) optionally substituted with one or more groups independently selected from Ci_6 alkyl (e.g., methyl or ethyl) and halogen (e.g., F or CI), for example unsubstituted phenyl or phenyl substituted with one or more halogen (e.g., F) or phenyl substituted with one or more Ci_ 6 alkyl and one or more halogen or phenyl substituted with one Ci_ 6 alkyl and one halogen, for example 4- fluorophenyl or 3,4-difluorophenyl or 4-fluoro-3-methylphenyl; and
- n 1, 2, 3, or 4
- the compound of Formula I as described above is a compound of Formula I(i):
- Ri is H or Ci_4 alkyl (e.g., methyl or ethyl);
- R 2 and R 3 are independently H or Ci_ 6 alkyl (e.g., methyl or ethyl);
- R4 is H or Ci_4 alkyl (e.g., methyl or ethyl);
- Re and R 7 are independently H or aryl (e.g., phenyl) optionally substituted with one or more groups independently selected from Ci_6 alkyl (e.g., methyl or ethyl) and halogen (e.g., F or CI), for example unsubstituted phenyl or phenyl substituted with one or more halogen (e.g., F) or phenyl substituted with one or more Ci_ 6 alkyl and one or more halogen or phenyl substituted with one Ci_ 6 alkyl and one halogen, for example 4- fluorophenyl or 3,4-difluorophenyl or 4-fluoro-3-methylphenyl; and
- n 1, 2, 3, or 4
- the compound of Formula I as described above is a compound of Formula I(ii):
- Ri is H or Ci_4 alkyl (e.g., methyl or ethyl);
- R 2 and R 3 are independently H or Ci_ 6 alkyl (e.g., methyl or ethyl);
- R4 is H or Ci_4 alkyl (e.g., methyl or ethyl);
- Re and R 7 are independently H or aryl (e.g., phenyl) optionally substituted with one or more groups independently selected from Ci_6 alkyl (e.g., methyl or ethyl) and halogen (e.g., F or CI), for example unsubstituted phenyl or phenyl substituted with one or more halogen (e.g., F) or phenyl substituted with one or more Ci_ 6 alkyl and one or more halogen or phenyl substituted with one Ci_ 6 alkyl and one halogen, for example 4- fluorophenyl or 3,4-difluorophenyl or 4-
- the invention further provides compounds of Formula I, I(i), and I(ii) as follows:
- Ci_ 5 alkyl e.g., methyl or ethyl
- Ci_ 4 alkyl e.g., methyl
- R 5 is aryl substituted with one or more Ci_ 6 -hydroxy alkyl (e.g., Ci_ 4 -hydroxyalkyl, e.g., 1- hydroxyethyl);
- R 5 is aryl substituted with one or more Ci_ 4 -hydroxy alkyl (e.g., 1 -hydroxy ethyl);
- R 5 is aryl substituted with one Ci_ 6 -hydroxyalkyl (e.g., Ci_ 4 -hydroxyalkyl, e.g., 1- hydroxyethyl);
- R 5 is aryl substituted with one Ci_ 4 -hydroxyalkyl (e.g., 1-hydroxyethyl);
- R 5 is phenyl substituted with one Ci_ 4 -hydroxyalkyl (e.g., 1 -hydroxy ethyl);
- R ⁇ and R 7 are independently H or aryl (e.g., phenyl) substituted with one or more groups independently selected from Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl or ethyl) and halogen (e.g., F or CI), for example phenyl substituted with one or more (e.g., two) halogen (e.g., F) or phenyl substituted with one or more Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and one or more halogen (e.g., F) or phenyl substituted with one Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and one halogen (e.g., F), for example 4-fluorophenyl or 3,4-
- any of Formulae I, I(i), I(ii), or 1.1-1.41 wherein R 7 is H and 5 is aryl (e.g., phenyl) substituted with one or more groups independently selected from Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and halogen (e.g., F or CI), for example 5 is phenyl substituted with one or more (e.g., two) halogen (e.g., F) or phenyl substituted with one Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and one halogen (e.g., F), for example wherein 5 is 4-fluorophenyl or 3,4-difluorophenyl or 4-fluoro-3- methylphenyl; Formulae I, I(i), I(ii), or 1.1-1.41, wherein R 7 is H and R 6 is aryl (e.
- R 7 is H and 5 is aryl (e.g., phenyl) substituted with one or more Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and one or more halogen (e.g., F);
- Ci_ 6 alkyl e.g., Ci_ 4 alkyl, e.g., methyl
- halogen e.g., F
- R 7 is H and 5 is aryl (e.g., phenyl) substituted with one or more Ci_ 4 alkyl (e.g., methyl) and one or more halogen (e.g., F);
- R 7 is H and 5 is aryl (e.g., phenyl) substituted with one Ci_ 6 alkyl (e.g., Ci_ 4 alkyl, e.g., methyl) and one halogen (e.g., F);
- Ci_ 6 alkyl e.g., Ci_ 4 alkyl, e.g., methyl
- halogen e.g., F
- Ci_ 6 alkyl e.g., Ci_ 4 alkyl, e.g., methyl
- halogen e.g., F
- R 2 and R 3 are independently Ci_ 4 alkyl (e.g., methyl);
- phosphodiesterase-mediated e.g., PDE 1 -mediated hydrolysis of cGMP, e.g., with an IC 50 of less than 1 ⁇ , preferably less than 500 nm, more preferably less than 50 nM, still more preferably less than 10 nM, most preferably less than or equal to 5 nM in an immobilized- metal affinity particle reagent PDE assay, for example, as described in Example 5,
- Alkyl as used herein is a saturated or unsaturated hydrocarbon
- Hydrocarbon moiety preferably having one to six carbon atoms, preferably having one to four carbon atoms, which may be linear or branched, and is mono-, di- or tri- substituted with hydroxy.
- Haloalkyl as used herein is a saturated hydrocarbon moiety
- halogens may be the same (e.g., dichloromethyl) or different (e.g., chlorofluoromethyl) .
- Aryl as used herein is a mono or bicyclic aromatic hydrocarbon, preferably phenyl, which may be optionally substituted, e.g., optionally substituted with one or more groups independently selected from Ci_6 alkyl (e.g., methyl), halogen (e.g., CI or F), Ci_ 6 -haloalkyl (e.g., trifluoromethyl), hydroxy, and carboxy.
- aryl in addition to being substituted with the groups disclosed herein, is further substituted with an aryl or a heteroaryl to form, e.g., biphenyl or pyridylphenyl.
- Heteroaryl as used herein is an aromatic moiety wherein one or more of the atoms making up the aromatic ring is sulfur or nitrogen rather than carbon, e.g., pyridyl or thiadiazolyl, which may be optionally substituted, e.g., optionally substituted with one or more groups independently selected from Ci_ 6 alkyl (e.g., methyl), halogen (e.g., CI or F), Ci_6-haloalkyl (e.g., trifluoromethyl), hydroxy, and carboxy.
- Ci_ 6 alkyl e.g., methyl
- halogen e.g., CI or F
- Ci_6-haloalkyl e.g., trifluoromethyl
- Halogen as used herein is F, CI, Br, or I.
- I(ii), e.g., any of Formulae 1.1-1.73 may exist in free or salt form, e.g., as acid addition salts.
- language such as "Compounds of the Invention” is to be understood as embracing the compounds in any form, for example free or acid addition salt form, or where the compounds contain acidic substituents, in base addition salt form.
- the Compounds of the Invention are intended for use as pharmaceuticals, therefore pharmaceutically acceptable salts are preferred. Salts which are unsuitable for pharmaceutical uses may be useful, for example, for the isolation or purification of free Compounds of the Invention or their pharmaceutically acceptable salts, are therefore also included.
- Compounds of the Invention may in some cases also exist in prodrug form.
- a prodrug form is compound which converts in the body to a Compound of the Invention.
- these substituents may form physiologically hydrolysable and acceptable esters.
- physiologically hydrolysable and acceptable ester means esters of Compounds of the Invention which are hydrolysable under physiological conditions to yield acids (in the case of Compounds of the Invention which have hydroxy substituents) or alcohols (in the case of Compounds of the Invention which have carboxy substituents) which are themselves physiologically tolerable at doses to be administered.
- the Compound of the Invention contains a hydroxy group, for example, Compound-OH
- the acyl ester prodrug of such compound i.e., Compound-0-C(0)-Ci_ 4 alkyl
- the acid ester prodrug of such compound i.e., Compound-C(0)0-Ci_ 4 alkyl
- the term thus embraces conventional pharmaceutical prodrug forms.
- the invention also provides methods of making the Compounds of the
- Parkinson's disease Tourette's Syndrome, autism, fragile X syndrome, ADHD, restless leg syndrome, depression, cognitive impairment, e.g., cognitive impairment of schizophrenia, narcolepsy and diseases that may be alleviated by the enhancement of progesterone-signaling such as female sexual dysfunction or a disease or disorder such as psychosis or glaucoma).
- progesterone-signaling such as female sexual dysfunction or a disease or disorder such as psychosis or glaucoma.
- the invention further provides a
- composition comprising a Compound of the Invention, in free, pharmaceutically acceptable salt, or prodrug form, in admixture with a
- the Compounds of the Invention and their pharmaceutically acceptable salts may be made using the methods as described and exemplified herein and by methods similar thereto and by methods known in the chemical art. Such methods include, but are not limited to, those described below. If not commercially available, starting materials for these processes may be made by procedures, which are selected from the chemical art using techniques which are similar or analogous to the synthesis of known compounds. Various starting materials, intermediates and/or Compounds of the Invention may be prepared using methods described or similarly described in WO 2006/133261, WO 2009/075784, WO 2010/065148, WO
- the Compounds of the Invention include their enantiomers, diastereoisomers and racemates, as well as their polymorphs, hydrates, solvates and complexes.
- Some individual compounds within the scope of this invention may contain double bonds. Representations of double bonds in this invention are meant to include both the E and the Z isomer of the double bond.
- some compounds within the scope of this invention may contain one or more asymmetric centers. This invention includes the use of any of the optically pure stereoisomers as well as any combination of stereoisomers.
- the Compounds of the Invention encompass their stable and unstable isotopes.
- Stable isotopes are nonradioactive isotopes which contain one additional neutron compared to the abundant nuclides of the same species (i.e., element). It is expected that the activity of compounds comprising such isotopes would be retained, and such compound would also have utility for measuring pharmacokinetics of the non-isotopic analogs.
- the hydrogen atom at a certain position on the Compounds of the Invention may be replaced with deuterium (a stable isotope which is non-radioactive). Examples of known stable isotopes include, but not limited to, deuterium, 13 C, 15 N, 18 O.
- unstable isotopes which are radioactive isotopes which contain additional neutrons compared
- isotope of the compound of the invention is the n C isotope.
- radio isotopes are useful for radio-imaging and/or pharmacokinetic studies of the compounds of the invention. Methods of making isotopes of PDE1 inhibitors disclosed in WO 2011/043816, the contents of which are incorporated by reference in their entirety, may be used for making the isotopes of the compounds of the current invention. [0020] Melting points are uncorrected and (dec) indicates decomposition.
- NMR data is in the delta values of major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. Conventional abbreviations for signal shape are used. Coupling constants (J) are given in Hz. For mass spectra (MS), the lowest mass major ion is reported for molecules where isotope splitting results in multiple mass spectral peaks. Solvent mixture compositions are given as volume percentages or volume ratios. In cases where the NMR spectra are complex, only diagnostic signals are reported.
- BOP benzotriazole-l-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- DIPEA diisopropylethylamine
- LiHMDS lithium bis(trimethylsilyl)amide
- NBS N-bromosuccinimide
- NCS N-chlorosuccinimide
- NMP N-methyl-2-pyrrolidone
- NaHC03 sodium bicarbonate
- NH 4 OH ammonium hydroxide
- POCI 3 phosphorous oxychloride
- TFA trifluoroacetic acid
- TFMSA trifluoromethanesulfonic acid
- THF tetrahydrofuran
- intermediate compounds of formula lib can be synthesized by reacting a compound of formula Ila with malonic acid and acetic anhydride in acetic acid with heating, e.g., to about 90°C for about 3 hours, and then cooled:
- Intermediate lie can be prepared by for example reacting intermediate lib with for example a chlorinating compound such as POCI 3 , sometimes with small amounts of water and heat, e.g., heating to about 80°C for about 4 hours, and then cooled:
- a chlorinating compound such as POCI 3
- Intermediate lid may be formed by reacting intermediate lie with for example P'-L in a solvent such as DMF and a base such as K 2 C0 3 , sodium bicarbonate, cesium carbonate, sodium hydroxide, triethylamine,
- P 1 is a protective group [e.g., /?-methoxybenzyl group (PMB) or
- L is a leaving group such as a halogen, mesylate, or tosylate.
- P 1 is
- Intermediate He may be prepared by reacting intermediate Hd with hydrazine or hydrazine hydrate in a solvent such as methanol and with heating, e.g. refluxed for about 4 hours, and then cooled:
- Intermediate IVa may be formed by for example reacting intermediate
- Ri is as defined previously for any of Formulae I, I(i), I(ii), or 1 . 1 -1 .73, e.g., such as a methyl group.
- Intermediate IVb may be formed by reacting intermediate IVa with for example F J -X in a solvent such as DMF with a base such as K 2 CO 3 at room temperature (Reaction 1):
- F is for example benzyl substituted with a halogen such as 4-bromobenzyl and X is a halogen (e.g., Br).
- Intermediate IVc may be synthesized from intermediate IVb by removing the protective group P 1 with an appropriate method. For example, if P 1 is a PMB group, then it can be removed with CAN or TFA/TFMSA at room temperature (Reaction 2):
- the compound may be deprotected by using acid such as hydrochloric acid or TFA.
- Intermediate IVd can be prepared by reacting intermediate IVc with for example a chlorinating compound such as POCI 3 and optionally with heating, e.g., reflux for about 2 days or more, or heated at 150 ⁇ 200°C for about 5-10 minutes in a sealed vial with a microwave instrument and then cooled (Reaction 3):
- a chlorinating compound such as POCI 3
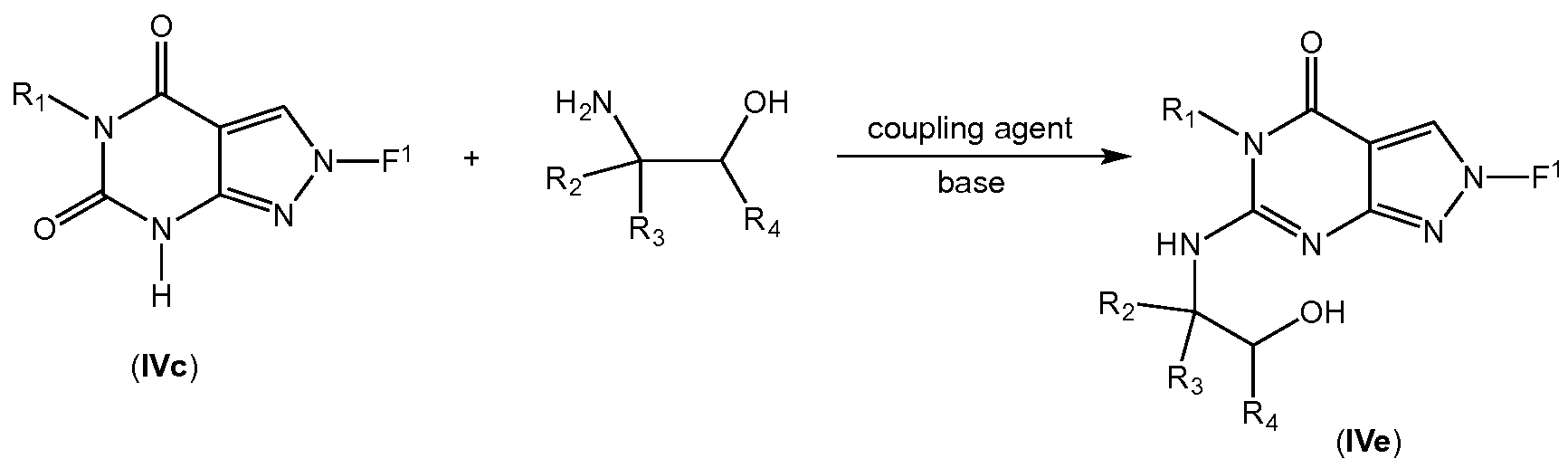
- Intermediate IVe can be formed by reacting intermediate IVd with an amino alcohol under basic condition in a solvent such as DMF or NMP and heated then cooled (Reaction 4A): — F
- R l s R 2 , R3, and R 4 are as defined previously for any of Formulae I, I(i), I(ii), or 1.1-1.73.
- intermediate IVe can be synthesized directly from intermediate IVe by reacting with an amino alcohol and a coupling reagent such as BOP in the presence of a base such as DBU (Reaction 4B):
- R l s R 2 , R3, and R 4 are as defined previously for any of Formulae I, I(i), I(ii), or 1.1-1.73.
- Intermediate IVf may be formed by reacting a compound of IVe with, for example, a dehydrating/halogenating agent such as SOCl 2 in a solvent such as CH 2 C1 2 at room temperature or heated at 35 °C for several hours, and then cooled
- Intermediate IVg may be formed by reacting intermediate IVf with, for example, catalysts such as a copper salt and 2,2,6, 6-tetramethylheptane-3,5-dione and a base such as cesium carbonate in a solvent such as NMP with heat for several hours (Reaction 6):
- catalysts such as a copper salt and 2,2,6, 6-tetramethylheptane-3,5-dione and a base such as cesium carbonate in a solvent such as NMP with heat for several hours
- F 2 is a diaryl ether.
- Intermediate IVh may be formed by reacting intermediate IVg with, for example, TFA and TFMSA in a solvent such as CH 2 CI 2 at room temperature (Reaction 7):
- Intermediate IVi may be formed by reacting intermediate IVh with
- R 5 and n are as defined previously for any of Formulae I, I(i), I(ii), or 1.1- 1.73 and L is a leaving group such as a halogen (e.g., Br).
- a halogen e.g., Br
- Intermediate IVj wherein X is halogen may be formed by reacting intermediate IVi with, for example, a halogenating agent such as
- Compounds of the Invention may be formed by reacting intermediate
- intermediate Ilf may be prepared by reacting intermediate lie with hydrazine or hydrazine hydrate in a solvent such as methoxyethanol and refluxed for about 30 minutes and then cooled:
- Intermediate Va can be synthesized by reacting a compound of formula He with for example an aryl isothiocyanate or isocyanate in a solvent such as DMF and heated at 110° C for about 2 days and then cooled:
- Intermediate Vb may be formed by removing the protective group P 1 with an appropriate method. For example, if P 1 is a PMB group, then it can be removed with A1C1 3 or TFA/TFMSA at room temperature. Intermediate Vb may also be prepared directly from a compound of Ilf using the similar methods, but the yields are relatively low.
- Intermediate Vc can be prepared by for example reacting intermediate
- reaction 11 a chlorinating compound such as POCI 3 .
- the reaction may be carried out at atmospheric pressure and refluxed for about 2 days or heated at 150 ⁇ 200°C for about 10 minutes in a sealed vial with a microwave instrument and then cooled (Reaction 11):
- Intermediate Vd can be prepared by reacting intermediate Vc with an amino alcohol under basic condition in a solvent such as DMF. The reaction may be heated overnight and then cooled (Reaction 12):
- R l s R 2 , R3, R4, and R 6 are as defined previously for any of Formulae I, I(i), I(ii), or 1.1-1.73.
- Intermediate Ve can be formed by reacting intermediate Vd with for example a dehydrating agent such as SOCl 2 in a solvent such as CH 2 C1 2 at room temperature overnight or heated at 35° C for about 4 hours, and then cooled
- Compounds of the Invention may be formed by reacting intermediate
- R 5 is as defined previously for any of Formulae I, I(i), I(ii), or 1.1-1.73 and L is a leaving group such as a halogen, mesylate, or tosylate.
- the Compounds of the Invention are useful in the treatment of diseases characterized by disruption of or damage to cAMP and cGMP mediated pathways, e.g., as a result of increased expression of PDEl or decreased expression of cAMP and cGMP due to inhibition or reduced levels of inducers of cyclic nucleotide synthesis, such as dopamine and nitric oxide (NO).
- inducers of cyclic nucleotide synthesis such as dopamine and nitric oxide (NO).
- the invention provides methods of treatment of any one or more of the following conditions:
- Neurodegenerative diseases including Parkinson's disease, restless leg, tremors, dyskinesias, Huntington's disease, Alzheimer's disease, and drug-induced movement disorders;
- Circulatory and cardiovascular disorders including cerebrovascular disease, stroke, congestive heart disease, hypertension, pulmonary hypertension, e.g., pulmonary arterial hypertension, and sexual dysfunction, including cardiovascular diseases and related disorders as described in International Application No.
- Respiratory and inflammatory disorders including asthma, chronic obstructive pulmonary disease, and allergic rhinitis, as well as autoimmune and inflammatory diseases;
- a disease or disorder such as psychosis, glaucoma, or elevated intraocular pressure
- Traumatic brain injury Any disease or condition characterized by low levels of cAMP and/or cGMP (or inhibition of cAMP and/or cGMP signaling pathways) in cells expressing PDEl; and/or
- a Compound of the Invention comprising administering an effective amount of a Compound of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, to a human or animal patient in need thereof.
- the invention provides methods of treatment or prophylaxis for narcolepsy.
- PDEl Inhibitors may be used as a sole therapeutic agent, but may also be used in combination or for coadministration with other active agents.
- the invention further comprises a method of treating narcolepsy comprising administering simultaneously, sequentially, or contemporaneously therapeutically effective amounts of
- a PDEl Inhibitor e.g., a compound according to any of Formulae I, I(i), I(ii) or
- a compound to promote wakefulness or regulate sleep e.g., selected from (a) central nervous system stimulants-amphetamines and amphetamine like compounds, e.g., methylphenidate, dextroamphetamine, methamphetamine, and pemoline; (b) modafinil, (c) antidepressants, e.g., tricyclics (including imipramine, desipramine, clomipramine, and protriptyline) and selective serotonin reuptake inhibitors (including fluoxetine and sertraline); and/or (d) gamma hydroxybutyrate (GHB),
- central nervous system stimulants-amphetamines and amphetamine like compounds e.g., methylphenidate, dextroamphetamine, methamphetamine, and pemoline
- modafinil e.g., methylphenidate, dextroamphetamine, methamphetamine, and pe
- the invention further provides methods of treatment or prophylaxis of a condition which may be alleviated by the enhancement of the progesterone signaling comprising administering an effective amount of a Compound of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, to a human or animal patient in need thereof.
- a Compound of the Invention e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form
- Diseases or conditions that may be ameliorated by enhancement of progesterone signaling include, but are not limited to, female sexual dysfunction, secondary amenorrhea (e.g., exercise amenorrhoea, anovulation, menopause, menopausal symptoms, hypothyroidism), pre-menstrual syndrome, premature labor, infertility, for example infertility due to repeated miscarriage, irregular menstrual cycles, abnormal uterine bleeding, osteoporosis, autoimmmune disease, multiple sclerosis, prostate enlargement, prostate cancer, and hypothyroidism.
- secondary amenorrhea e.g., exercise amenorrhoea, anovulation, menopause, menopausal symptoms, hypothyroidism
- pre-menstrual syndrome e.g., premature labor
- infertility for example infertility due to repeated miscarriage, irregular menstrual cycles, abnormal uterine bleeding, osteoporosis, autoimmmune disease,
- the PDE1 inhibitors may be used to encourage egg implantation through effects on the lining of uterus, and to help maintain pregnancy in women who are prone to miscarriage due to immune response to pregnancy or low progesterone function.
- the novel PDE1 inhibitors e.g., as described herein, may also be useful to enhance the effectiveness of hormone replacement therapy, e.g., administered in combination with
- estrogen/estradiol/estriol and/or progesterone/progestins in postmenopausal women and estrogen-induced endometrial hyperplasia and carcinoma.
- the methods of the invention are also useful for animal breeding, for example to induce sexual receptivity and/or estrus in a nonhuman female mammal to be bred.
- PDE1 Inhibitors may be used in the foregoing methods of treatment or prophylaxis as a sole therapeutic agent, but may also be used in combination or for co-administration with other active agents, for example in conjunction with hormone replacement therapy.
- the invention further comprises a method of treating disorders that may be ameliorated by enhancement of progesterone signaling comprising administering simultaneously, sequentially, or contemporaneously therapeutically effective amounts of
- a PDE1 Inhibitor e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, and
- a hormone e.g., selected from estrogen and estrogen analogues (e.g., estradiol, estriol, estradiol esters) and progesterone and progesterone analogues (e.g., progestins)
- estrogen and estrogen analogues e.g., estradiol, estriol, estradiol esters
- progesterone and progesterone analogues e.g., progestins
- the invention also provides a method for enhancing or potentiating dopamine Dl intracellular signaling activity in a cell or tissue comprising contacting said cell or tissue with an amount of a Compound of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, sufficient to inhibit PDE1 activity.
- a Compound of the Invention e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, sufficient to inhibit PDE1 activity.
- the invention also provides a method for treating a PDE1 -related disorder, a dopamine Dl receptor intracellular signaling pathway disorder, or disorders that may be alleviated by the enhancement of the progesterone signaling pathway in a patient in need thereof comprising administering to the patient an effective amount of a Compound of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, that inhibits PDE1, wherein PDE1 activity modulates phosphorylation of DARPP-32 and/or the GluRl AMPA receptor.
- a Compound of the Invention e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt or prodrug form, that inhibits PDE1, wherein PDE1 activity modulates phosphorylation of DARPP-32 and/or the GluRl AMPA receptor
- the invention also provides a method for the treatment for glaucoma or elevated intraocular pressure comprising topical administration of a therapeutically effective amount of a PDE1 Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt form, in an ophthalmically compatible carrier to the eye of a patient in need thereof.
- treatment may alternatively include a systemic therapy.
- Systemic therapy includes treatment that can directly reach the bloodstream, or oral methods of administration, for example.
- the invention further provides a pharmaceutical composition for topical ophthalmic use comprising a PDE1 inhibitor; for example an ophthalmic solution, suspension, cream or ointment comprising a PDE1 Inhibitor of the
- Invention e.g., a compound according to any of Formulae I, I(i), I(ii) or 1.1-1.73, in free or ophthalmologically acceptable salt form, in combination or association with an ophthalmologically acceptable diluent or carrier.
- the PDE1 inhibitor may be administered sequentially or simultaneously with a second drug useful for treatment of glaucoma or elevated intraocular pressure.
- the therapeutically effective amount of each agent may be below the amount needed for activity as monotherapy.
- a subthreshold amount i.e., an amount below the level necessary for efficacy as monotherapy
- an advantage of administering different agents with different mechanisms of action and different side effect profiles may be to reduce the dosage and side effects of either or both agents, as well as to enhance or potentiate their activity as monotherapy.
- the invention thus provides the method of treatment of a condition selected from glaucoma and elevated intraocular pressure comprising administering to a patient in need thereof an effective amount, e.g., a subthreshold amount, of an agent known to lower intraocular pressure concomitantly, simultaneously or sequentially with an effective amount, e.g., a subthreshold amount, of a PDE1 Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii) or 1.1-1.73, in free or pharmaceutically acceptable salt form, such that amount of the agent known to lower intraocular pressure and the amount of the PDE1 inhibitor in combination are effective to treat the condition.
- an effective amount e.g., a subthreshold amount
- an agent known to lower intraocular pressure concomitantly, simultaneously or sequentially with an effective amount, e.g., a subthreshold amount, of a PDE1 Inhibitor of
- one or both of the agents are administered topically to the eye.
- the invention provides a method of reducing the side effects of treatment of glaucoma or elevated intraocular pressure by administering a reduced dose of an agent known to lower intraocular pressure concomitantly, simultaneously or sequentially with an effective amount of a PDE1 inhibitor.
- methods other than topical administration, such as systemic therapeutic administration may also be utilized.
- PDE1 inhibitor may, for example, be selected from the existing drugs comprise typically of instillation of a prostaglandin, pilocarpine, epinephrine, or topical beta- blocker treatment, e.g. with timolol, as well as systemically administered inhibitors of carbonic anhydrase, e.g. acetazolamide.
- Cholinesterase inhibitors such as
- physostigmine and echothiopate may also be employed and have an effect similar to that of pilocarpine.
- Drugs currently used to treat glaucoma thus include, e.g.,
- Prostaglandin analogs such as latanoprost (Xalatan), bimatoprost (Lumigan) and travoprost (Travatan), which increase uveoscleral outflow of aqueous humor.
- Bimatoprost also increases trabecular outflow.
- Topical beta-adrenergic receptor antagonists such as timolol, levobunolol (Betagan), and betaxolol, which decrease aqueous humor production by the ciliary body.
- Alpha 2 -adrenergic agonists such as brimonidine (Alphagan), which work by a dual mechanism, decreasing aqueous production and increasing uveo-scleral outflow.
- Miotic agents parasympathomimetics
- pilocarpine work by contraction of the ciliary muscle, tightening the trabecular meshwork and allowing increased outflow of the aqueous humour.
- Physostigmine is also used to treat glaucoma and delayed gastric emptying.
- the invention provides pharmaceutical compositions comprising a PDE1 Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt form, and an agent selected from (i) the prostanoids, unoprostone, latanoprost, travoprost, or bimatoprost; (ii) an alpha adrenergic agonist such as brimonidine, apraclonidine, or dipivefrin and (iii) a muscarinic agonist, such as pilocarpine, in combination or association with a pharmaceutically acceptable diluent or carrier.
- the invention provides ophthalmic formulations comprising a PDE-1 Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, together with bimatoprost, abrimonidine, brimonidine, timolol, or combinations thereof, in free or ophthamalogically acceptable salt form, in combination or association with an ophthamologically acceptable diluent or carrier.
- a PDE-1 Inhibitor of the Invention e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, together with bimatoprost, abrimonidine, brimonidine, timolol, or combinations thereof, in free or ophthamalogically acceptable salt form, in combination or association with an ophthamologically acceptable diluent or carrier.
- a person of ordinary skill in the art can select an appropriate selective receptor sub
- alpha adrenergic agonist one can select an agonist selective for an alpha 1 adrenergic receptor, or an agonist selective for an alpha 2 adrenergic receptor such as brimonidine, for example.
- a beta-adrenergic receptor antagonist one can select an antagonist selective for either ⁇ , or ⁇ 2 , or ⁇ 3 ⁇ depending on the appropriate therapeutic application.
- a muscarinic agonist selective for a particular receptor subtype such as Mi-M 5 .
- the PDE1 inhibitor may be administered in the form of an ophthalmic composition, which includes an ophthalmic solution, cream or ointment.
- the ophthalmic composition may additionally include an intraocular-pressure lowering agent.
- the PDE1 Inhibitors disclosed may be combined with a subthreshold amount of an intraocular pressure-lowering agent which may be a bimatoprost ophthalmic solution, a brimonidine tartrate ophthalmic solution, or brimonidine tartrate/timolol maleate ophthalmic solution.
- PDE1 inhibitors are useful to treat psychosis, for example, any conditions characterized by psychotic symptoms such as hallucinations, paranoid or playful delusions, or disorganized speech and thinking, e.g.,
- PDE1 inhibitors primarily act to enhance signaling at the dopamine Dl receptor. By enhancing Dl receptor signaling, PDE1 inhibitors can increase NMD A receptor function in various brain regions, for example in nucleus accumbens neurons and in the prefrontal cortex. This
- enhancement of function may be seen for example in NMDA receptors containing the NR2B subunit, and may occur e.g., via activation of the Src and protein kinase A family of kinases.
- the invention provides a new method for the treatment of psychosis, e.g., schizophrenia, schizoaffective disorder, schizophreniform disorder, psychotic disorder, delusional disorder, and mania, such as in acute manic episodes and bipolar disorder, comprising administering a therapeutically effective amount of a phosphodiesterase- 1 (PDE1) Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt form, to a patient in need thereof.
- PDE1 phosphodiesterase- 1
- PDE 1 Inhibitors may be used in the foregoing methods of treatment prophylaxis as a sole therapeutic agent, but may also be used in combination or for co-administration with other active agents.
- the invention further comprises a method of treating psychosis, e.g., schizophrenia, schizoaffective disorder, schizophreniform disorder, psychotic disorder, delusional disorder, or mania, comprising administering simultaneously, sequentially, or contemporaneously therapeutically effective amounts of: (i) a PDEl Inhibitor of the invention, in free or pharmaceutically acceptable salt form; and
- Typical antipsychotics e.g.,
- Butyrophenones e.g. Haloperidol (Haldol, Serenace), Droperidol (Droleptan);
- Phenothiazines e.g., Chlorpromazine (Thorazine, Largactil), Fluphenazine (Prolixin), Perphenazine (Trilafon),
- Prochlorperazine Compazine
- Thioridazine Mellaril, Melleril
- Trifluoperazine Stelazine
- Mesoridazine Mesoridazine
- Thioxanthenes e.g., Chlorprothixene, Flupenthixol (Depixol, Fluanxol), Thiothixene (Navane), Zuclopenthixol (Clopixol, Acuphase);
- Atypical antipsychotics e.g.,
- Clozapine (Clozaril), Olanzapine (Zyprexa), Risperidone
- the Compounds of the Invention are particularly useful for the treatment or prophylaxis of schizophrenia.
- Compounds of the Invention are particularly useful for the treatment of Parkinson's disease, schizophrenia, narcolepsy, glaucoma and female sexual dysfunction.
- the invention provides a method of lengthening or enhancing growth of the eyelashes by administering an effective amount of a prostaglandin analogue, e.g., bimatoprost, concomitantly, simultaneously or sequentially with an effective amount of a PDEl inhibitor of the Invention, in free or pharmaceutically acceptable salt form, to the eye of a patient in need thereof.
- a prostaglandin analogue e.g., bimatoprost
- the invention provides a method for the treatment or prophylaxis of traumatic brain injury comprising administering a therapeutically effective amount of a PDE1 Inhibitor of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, in free or pharmaceutically acceptable salt form, to a patient in need thereof.
- Traumatic brain injury encompasses primary injury as well as secondary injury, including both focal and diffuse brain injuries.
- Secondary injuries are multiple, parallel, interacting and interdependent cascades of biological reactions arising from discrete subcellular processes (e.g., toxicity due to reactive oxygen species, overstimulation of glutamate receptors, excessive influx of calcium and inflammatory upregulation) which are caused or exacerbated by the inflammatory response and progress after the initial (primary) injury.
- discrete subcellular processes e.g., toxicity due to reactive oxygen species, overstimulation of glutamate receptors, excessive influx of calcium and inflammatory upregulation
- the present invention also provides
- a Compound of the Invention e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, as hereinbefore described, in free or pharmaceutically acceptable salt form for example for use in any method or in the treatment of any disease or condition as hereinbefore set forth,
- the invention provides use of a Compound of the Invention, e.g., a compound according to any of Formulae I, I(i), I(ii), or 1.1-1.73, as
- any one or more of the following diseases Parkinson's disease, restless leg, tremors, dyskinesias, Huntington's disease, Alzheimer's disease, and/or drug-induced movement disorders; depression, attention deficit disorder, attention deficit hyperactivity disorder, bipolar illness, anxiety, sleep disorder, narcolepsy, cognitive impairment, e.g., cognitive impairment of schizophrenia, dementia, Tourette's syndrome, autism, fragile X syndrome, psychostimulant withdrawal, and/or drug addiction; cerebrovascular disease, stroke, congestive heart disease, hypertension, pulmonary hypertension, e.g., pulmonary arterial hypertension, and/or sexual dysfunction; asthma, chronic obstructive pulmonary disease, and/or allergic rhinitis, as well as autoimmune and inflammatory diseases; and/or female sexual dysfunction, exercise amenorrhoea, anovulation, menopause, menopausal symptoms,
- hypothyroidism pre -menstrual syndrome, premature labor, infertility, irregular menstrual cycles, abnormal uterine bleeding, osteoporosis, multiple sclerosis, prostate enlargement, prostate cancer, hypothyroidism, and/or estrogen-induced endometrial hyperplasia and/or carcinoma; and/or any disease or condition characterized by low levels of cAMP and/or cGMP (or inhibition of cAMP and/or cGMP signaling pathways) in cells expressing PDE1, and/or by reduced dopamine Dl receptor signaling activity; and/or any disease or condition that may be ameliorated by the enhancement of progesterone signaling.
- the invention also provides use of a Compound of the Invention, in free or pharmaceutically acceptable salt form, (the manufacture of a medicament) for the treatment or prophylactic treatment of any one or more of:
- psychosis for example, any conditions characterized by psychotic symptoms such as hallucinations, paranoid or playful delusions, or disorganized speech and thinking, e.g., schizophrenia, schizoaffective disorder, schizophreniform disorder, psychotic disorder, delusional disorder, and mania, such as in acute manic episodes and bipolar disorder,
- Invention encompasses any and all of the compounds disclosed herewith, e.g., compounds according to any of Formulae I, I(i), I(ii) or 1.1-1.73, as hereinbefore described, in free or salt form.
- the words “treatment” and “treating” are to be understood accordingly as embracing prophylaxis and treatment or amelioration of symptoms of disease as well as treatment of the cause of the disease.
- the invention provides a method for the treatment of the disease or disorder disclosed herein.
- the invention provides a method for the prophylaxis of a disease or disorder as disclosed herein.
- the word "effective amount” is intended to encompass a therapeutically effective amount to treat a specific disease or disorder.
- pulmonary hypertension is intended to encompass pulmonary arterial hypertension.
- patient includes human or non-human (i.e., animal) patient.
- the invention encompasses both human and nonhuman. In another embodiment, the invention encompasses nonhuman. In other embodiment, the term encompasses human.
- Compounds of the Invention are in particular useful for the treatment of Parkinson's disease, narcolepsy and female sexual dysfunction.
- Compounds of the Invention potentiate the activity of Dl agonists, such as dopamine, they may be simultaneously, sequentially, or contemporaneously administered with conventional dopaminergic medications, such as levodopa and levodopa adjuncts (carbidopa, COMT inhibitors, MAO-B inhibitors), dopamine agonists, and
- novel PDE1 inhibitors e.g., as described herein, may also be
- progesterone/progestins to enhance the effectiveness of hormone replacement therapy or treatment of estrogen-induced endometrial hyperplasia or carcinoma.
- Dosages employed in practicing the present invention will of course vary depending, e.g. on the particular disease or condition to be treated, the particular Compound of the Invention used, the mode of administration, and the therapy desired.
- Compounds of the Invention may be administered by any suitable route, including orally, parenterally, transdermally, or by inhalation, but are preferably administered orally. In general, satisfactory results, e.g. for the treatment of diseases as
- an indicated daily dosage for oral administration will accordingly be in the range of from about 0.75 to 150 mg, conveniently administered once, or in divided doses 2 to 4 times, daily or in sustained release form.
- Unit dosage forms for oral administration thus for example may comprise from about 0.2 to 75 or 150 mg, e.g. from about 0.2 or 2.0 to 50, 75 or 100 mg of a Compound of the Invention, together with a
- compositions comprising Compounds of the Invention may be prepared using conventional diluents or excipients and techniques known in the galenic art.
- oral dosage forms may include tablets, capsules, solutions, suspensions and the like.
- TFA 500 mL is slowly added into a suspension of 2-(4- bromobenzyl)-7-(4-methoxybenzyl)-5-methyl-2H-pyrazolo[3,4-d]pyrimidine- 4,6(5H,7H)-dione (226 g, 496 mmol) in methylene chloride (320 mL), and then TFMSA (160 mL is added slowly. The reaction mixture is stirred at room
- TFA 600 mL is added into a suspension of 2-(4-phenoxybenzyl)-7,8- dihydro-5,7,7-trimethyl-[2H]-imidazo-[l,2-a]pyrazolo[4,3-e]pyrimidin-4(5H)-one (103 g, 257 mmol) in methylene chloride (210 mL) to give a tan solution, and then TFMSA (168 mL) is added. The reaction mixture is stirred at room temperature until the starting material disappears. The mixture is poured into cold water (3 L).
- NaBH 4 (18 mg, 0.48 mmol) is slowly added to a solution of 7,8- dihydro-2-(4-acetylbenzyl)-3-(4-fluorophenylamino)-5,7,7-trimethyl-[2H]-imidazo- [l,2-a]pyrazolo[4,3-e]pyrimidin-4(5H)-one (22 mg, 0.048 mmol) in methanol (1 mL) at -20 °C. The reaction mixture is stirred at -10 °C for 3 h, and then quenched with water (0.5 mL).
- Phosphodiesterase 1 is a calcium/calmodulin dependent phosphodiesterase enzyme that converts cyclic guanosine monophosphate (cGMP) to 5'-guanosine monophosphate (5'-GMP).
- PDEl can also convert a modified cGMP substrate, such as the fluorescent molecule cGMP-fluorescein, to the corresponding GMP-fluorescein.
- the generation of GMP-fluorescein from cGMP-fluorescein can be quantitated, using, for example, the IMAP (Molecular Devices, Sunnyvale, CA) immobilized-metal affinity particle reagent.
- the IMAP reagent binds with high affinity to the free 5 '- phosphate that is found in GMP-fluorescein and not in cGMP-fluorescein.
- the resulting GMP-fluorescein - IMAP complex is large relative to cGMP-fluorescein.
- Small fluorophores that are bound up in a large, slowly tumbling, complex can be distinguished from unbound fluorophores, because the photons emitted as they fluoresce retain the same polarity as the photons used to excite the fluorescence.
- the following phosphodiesterase enzymes may be used: 3',5'-cyclic- nucleotide-specific bovine brain phosphodiesterase (Sigma, St. Louis, MO) and recombinant full length human PDEl A and PDE1B (r-hPDElA and r-hPDElB, respectively) which may be produced e.g., in HEK or SF9 cells by one skilled in the art.
- the PDEl enzyme is reconstituted with 50% glycerol to 2.5 U/ml. One unit of enzyme will hydrolyze 1.0 ⁇ of 3',5'-cAMP to 5'-AMP per min at pH 7.5 at 30°C.
- reaction buffer (30 ⁇ CaCl 2 , 10 U/ml of calmodulin (Sigma P2277), lOmM Tris-HCl pH 7.2, lOmM MgCl 2 , 0.1% BSA, 0.05% NaN 3 ) to yield a final concentration of 1.25mU/ml.
- 99 ⁇ of diluted enzyme solution is added into each well in a flat bottom 96-well polystyrene plate to which 1 ⁇ of test compound dissolved in 100% DMSO is added.
- the test compounds are mixed and pre-incubated with the enzyme for 10 min at room temperature.
- the FL-GMP conversion reaction is initiated by combining 4 parts enzyme and inhibitor mix with 1 part substrate solution (0.225 ⁇ ) in a 384-well microtiter plate. The reaction is incubated in dark at room temperature for 15 min. The reaction is halted by addition of 60 ⁇ of binding reagent (1 :400 dilution of IMAP beads in binding buffer supplemented with 1 : 1800 dilution of antifoam) to each well of the 384-well plate. The plate is incubated at room temperature for 1 hour to allow IMAP binding to proceed to completion, and then placed in an Envision multimode microplate reader (PerkinElmer, Shelton, CT) to measure the fluorescence polarization (Amp).
- Envision multimode microplate reader PerkinElmer, Shelton, CT
- IC 50 values are determined by measuring enzyme activity in the presence of 8 to 16 concentrations of compound ranging from 0.0037 nM to 80,000 nM and then plotting drug concentration versus AmP, which allows IC 50 values to be estimated using nonlinear regression software (XLFit; IDBS, Cambridge, MA).
- Compounds of the Invention may be selected and tested in an assay as described or similarly described herein for PDE1 inhibitory activity.
- Exemplified Compounds of the Invention e.g., compounds of Examples 1, 2, 3, and 4 have IC 50 values of less than or equal to 5 nm. K; values for Exemplified
- the candidate compounds may be evaluated in a Novel Object Recognition (NOR) assay.
- NOR Novel Object Recognition
- This assay protocol is described in detail in Ennaceur et al, Behav. Brain Res. (1988) 31 :47-59 and Prickaerts et al, Eur. J. Pharmacol. (1997) 337: 125- 136, the contents of each of which are incorporated by reference in their entirety.
- the rats are introduced to a chamber at time Tl and allowed to interrogate two identical "familiar objects" for six minutes. Twenty-four hours later, they are re-introduced to this chamber, where one of the familiar objects has been replaced with a novel object.
- the "discrimination index” a measure of the time spent in close proximity to the novel over the familiar object, may then be measured. Since rodents will forget the original experiment at Tl within 4 hours, this test with a 24h interval is a measure of strong cognitive enhancement.
- This assay protocol can be modified in order to evaluate different phases of memory.
- the rats may be dosed with the candidate compound two hours before Tl and tested 24h later without additional dosing. This is a test of the acquisition process.
- administration at various other times after the Tl test may be done to understand the compound's effectiveness in memory consolidation and recall. Specifically, these dosing times represent acquisition (Tl - 2h), early consolidation (Tl + 0.1 h), late consolidation (Tl + 3h), and retrieval (T2 - 2h).
- Example 1 Using the protocol described above or similarly described above, the compound of Example 1 is shown to have a minimal effective dose of 0.1 mg/kg PO when administering to a rat 2 hours before Tl .
- the compounds of the invention may be tested in a reversal of catalepsy model in which motor freezing, or catalepsy, is induced by potent dopamine D2 receptor antagonists such as haloperidol or risperidone.
- the method uses the "bar grip test", in which the front paws of the mouse are placed so as to grip a 3mm-diameter, suspended wooden bar. A “step down latency” is measured by recording the time until the mouse removes its paws from the wooden bar to the floor surface.
- Catalepsy is a freezing of the musculature that prevents the mouse from moving off the bar. Reduction in the catalepsy induced in this model will indicate that the compound will have a beneficial effect both in schizophrenia where extrapyramidal side effects are frequent and in Parkinson's disease.
- mice A total of seventeen (17) eight week-old, male C57BL/6 mice (Jackson Laboratories) are used in a typical experiment testing the effect of the compound of Example 1.
- a catalepsy score is recorded for each mouse at 2, 3, 4, and 6 hours after administration of drugs.
- the chamber used for measuring catalepsy is comprised of a Plexiglas cage outfitted with a 3mm-diameter wooden bar fixed horizontally 4 cm above the floor of cage. For each test session, both forepaws of the mouse are placed on the bar while the hind paws are on the Plexiglas floor. The latency until the mouse steps both paws down from the bar to the floor surface (i.e., step down latency) is recorded up to 120 sec. If the mouse steps off immediately (less than 10 sec after placement), another attempt is made up to a maximum of 10 attempts. If none of the 10 attempts are beyond 10 sec, the longest duration recorded is used as the catalepsy score.
- the initial cataleptic duration (>10sec) is recorded during the 120 sec testing time.
- Mean step down latency is calculated for each treatment group.
- the effect of the compound of Example 1 on step down latency after haloperidol treatment is statistically evaluated by comparing group differences by analysis of variance (ANOVA, F 5i l6 ) followed by application of Newman-Keuls post-hoc multiple comparison tests at each time point across all doses tested.
- Example 1 By using the protocol described or similarly described in this example, the compound of Example 1 is shown to be active in a catalepsy model with a minimal effective dose of 0.1 mg/Kg.
- the compound of Example 1 is shown to have a Ti /2 of 171 minutes
- the compound of Example 3 is shown to have a Ti /2 of 78 minutes
- the compound of Example 4 is shown to have a Ti /2 of 67 minutes.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Epidemiology (AREA)
- Psychiatry (AREA)
- Endocrinology (AREA)
- Ophthalmology & Optometry (AREA)
- Diabetes (AREA)
- Addiction (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Reproductive Health (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Dermatology (AREA)
Abstract
Description































Claims








Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112015023592-1A BR112015023592B1 (en) | 2013-03-15 | 2014-03-13 | PDE1-INHIBITOR ORGANIC COMPOUNDS, PHARMACEUTICAL COMPOSITION INCLUDING THEM AND THERAPEUTIC USE OF SAID COMPOUNDS AND SAID COMPOSITION |
| HK16108653.3A HK1220688B (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| KR1020157029692A KR102240326B1 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| RU2015143672A RU2679142C2 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| DK14768690.1T DK2970279T3 (en) | 2013-03-15 | 2014-03-13 | ORGANIC COMPOUNDS |
| EP14768690.1A EP2970279B1 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| PL14768690T PL2970279T3 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| AU2014234990A AU2014234990B2 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| CA2906640A CA2906640C (en) | 2013-03-15 | 2014-03-13 | Substituted imidazo-[1,2-a]pyrazolo[4.3-e]pyrimidin-4[5h]-one compounds and pharmaceutical compositions and use therof as pde1 inhibitors |
| JP2016501936A JP6437519B2 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| MX2015013042A MX378194B (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| US14/777,446 US9598426B2 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| ES14768690T ES2836129T3 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
| CN201480026736.4A CN105377846B (en) | 2013-03-15 | 2014-03-13 | Organic compound |
| IL241573A IL241573B (en) | 2013-03-15 | 2015-09-10 | Derivatives of substituted [2h]imidazo-[1,2-a]pyrazolo[4,3-e]pyrimidin-4(5h)-one, pharmaceutical composition comprising them and their use as medicaments |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361788551P | 2013-03-15 | 2013-03-15 | |
| US61/788,551 | 2013-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014151409A1 true WO2014151409A1 (en) | 2014-09-25 |
Family
ID=51529966
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/025666 Ceased WO2014151409A1 (en) | 2013-03-15 | 2014-03-13 | Organic compounds |
Country Status (17)
| Country | Link |
|---|---|
| US (4) | US9598426B2 (en) |
| EP (1) | EP2970279B1 (en) |
| JP (2) | JP6437519B2 (en) |
| KR (1) | KR102240326B1 (en) |
| CN (1) | CN105377846B (en) |
| AR (1) | AR095347A1 (en) |
| AU (1) | AU2014234990B2 (en) |
| CA (1) | CA2906640C (en) |
| DK (1) | DK2970279T3 (en) |
| ES (1) | ES2836129T3 (en) |
| IL (1) | IL241573B (en) |
| MX (1) | MX378194B (en) |
| PL (1) | PL2970279T3 (en) |
| RU (1) | RU2679142C2 (en) |
| TW (1) | TWI659032B (en) |
| UY (1) | UY35467A (en) |
| WO (1) | WO2014151409A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9403836B2 (en) | 2007-12-06 | 2016-08-02 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9468637B2 (en) | 2009-05-13 | 2016-10-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2016170064A1 (en) * | 2015-04-22 | 2016-10-27 | H. Lundbeck A/S | Imidazotriazinones as pde1 inhibitors |
| US9556186B2 (en) | 2013-03-15 | 2017-01-31 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9624230B2 (en) | 2005-06-06 | 2017-04-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9763948B2 (en) | 2010-05-31 | 2017-09-19 | Intra-Cellular Therapies, Inc. | PDE1 inhibitory compounds and methods |
| US9801882B2 (en) | 2013-02-17 | 2017-10-31 | Intra-Cellular Therapies, Inc. | Phosphodiesterase-1 inhibitors and their use in treatment of cardiovascular diseases |
| US9849132B2 (en) | 2014-01-08 | 2017-12-26 | Intra-Cellular Therapies, Inc. | Products and pharmaceutical compositions |
| EP3193878A4 (en) * | 2014-09-17 | 2018-03-21 | Intra-Cellular Therapies, Inc. | Compounds and methods |
| US10011606B2 (en) | 2015-04-30 | 2018-07-03 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| US10105349B2 (en) | 2014-12-06 | 2018-10-23 | Intra-Cellular Therapies, Inc. | Organic compounds |
| EP3395806A1 (en) * | 2017-04-24 | 2018-10-31 | IGM Group B.V. | Simple oxidative functionalization of alkyl aryl ketones |
| US10131671B2 (en) | 2014-08-07 | 2018-11-20 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10150771B2 (en) | 2014-10-10 | 2018-12-11 | H. Lundbeck A/S | Triazolopyrazinones as PDE1 inhibitors |
| JP2019510039A (en) * | 2016-03-28 | 2019-04-11 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Novel compositions and methods |
| US10285992B2 (en) | 2014-08-07 | 2019-05-14 | Intra-Cellular Therapies, Inc. | Combinations of PDE1 inhibitors and NEP inhibitors and associated methods |
| US10300064B2 (en) | 2014-12-06 | 2019-05-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10538525B2 (en) | 2016-04-12 | 2020-01-21 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US10633382B2 (en) | 2016-10-18 | 2020-04-28 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as PDE1 inhibitors |
| WO2020210614A1 (en) | 2019-04-12 | 2020-10-15 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10905688B2 (en) | 2016-10-28 | 2021-02-02 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-α]pyrazinones as PDE1 inhibitors |
| US10912773B2 (en) | 2016-10-28 | 2021-02-09 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-a]pyrazinones as PDE1 inhibitors |
| US11291666B2 (en) | 2016-09-12 | 2022-04-05 | Intra-Cellular Therapies, Inc. | Uses |
| US11759465B2 (en) | 2018-01-31 | 2023-09-19 | Intra-Cellular Therapies, Inc. | Uses |
| EP4284806A4 (en) * | 2021-01-27 | 2025-02-26 | Intra-Cellular Therapies, Inc. | SALT CRYSTALS |
| EP4520399A2 (en) | 2018-05-25 | 2025-03-12 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US12364695B2 (en) | 2020-06-02 | 2025-07-22 | Intra-Cellular Therapies, Inc. | Methods of treating inflammatory disease |
| US12410175B2 (en) | 2019-09-03 | 2025-09-09 | Intra-Cellular Therapies, Inc. | Compounds |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201206937A (en) | 2010-05-31 | 2012-02-16 | Intra Cellular Therapies Inc | Organic compounds |
| EP2717877B1 (en) | 2011-06-10 | 2017-11-08 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9545406B2 (en) | 2013-03-15 | 2017-01-17 | Intra-Cellular Therapies, Inc. | Method of treating a CNS injury with a PDE1 inhibitor |
| JP2022502501A (en) | 2018-09-25 | 2022-01-11 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | New use |
| US12396992B2 (en) | 2019-01-07 | 2025-08-26 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US11628171B2 (en) | 2019-03-13 | 2023-04-18 | Children's Medical Center Corporation | Method for treating brain or nerve injury |
| AU2020343329C1 (en) | 2019-09-03 | 2026-02-12 | Intra-Cellular Therapies, Inc. | Methods of treatment |
| MX2024010993A (en) * | 2022-03-11 | 2024-09-17 | Intra Cellular Therapies Inc | Organic compounds. |
| WO2025091037A1 (en) * | 2023-10-26 | 2025-05-01 | Intra-Cellular Therapies, Inc. | Organic compounds |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006133261A2 (en) | 2005-06-06 | 2006-12-14 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2009075784A1 (en) | 2007-12-06 | 2009-06-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065148A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065149A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065151A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2011043816A1 (en) | 2009-10-08 | 2011-04-14 | Intra-Cellular Therapies, Inc. | Phosphodiesterase 1-targeting tracers and methods |
| US20120053190A1 (en) | 2009-05-13 | 2012-03-01 | Fienberg Allen A | Organic compounds |
| WO2012171016A1 (en) | 2011-06-10 | 2012-12-13 | Intra-Cellular Therapies, Inc. | Organic compounds |
Family Cites Families (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6032638B2 (en) | 1976-09-01 | 1985-07-29 | 武田薬品工業株式会社 | 3-Aminopyrazolo[3,4-d]pyrimidine derivative |
| EP0077372A1 (en) | 1981-04-22 | 1983-04-27 | Byk Gulden Lomberg Chemische Fabrik GmbH | NEW DERIVATIVES OF PYRAZOLO(3,4-d)PYRIMIDINE, PREPARATION METHOD THEREOF AND REMEDY CONTAINING THEM |
| US4469868A (en) | 1982-05-24 | 1984-09-04 | Warner-Lambert Company | Alkylimidazo[1,2-c]pyrazolo[3,4-e]pyrimidines |
| US4666908A (en) | 1985-04-05 | 1987-05-19 | Warner-Lambert Company | 5-Substituted pyrazolo[4,3-d]pyrimidine-7-ones and methods of use |
| KR920004437B1 (en) | 1989-09-12 | 1992-06-05 | 삼성전자 주식회사 | How to manage customers in cash register |
| ZA914727B (en) | 1990-06-21 | 1992-03-25 | Schering Corp | Polycyclic guanine derivatives |
| US5202328A (en) | 1991-03-06 | 1993-04-13 | Merck & Co., Inc. | Substituted fused pyrimidinones |
| US5294612A (en) | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| WO1994019351A1 (en) | 1993-02-26 | 1994-09-01 | Schering Corporation | 2-benzyl-polycyclic guanine derivatives and process for preparing them |
| GB9304919D0 (en) | 1993-03-10 | 1993-04-28 | Celltech Ltd | Chemical compounds |
| GB9315017D0 (en) | 1993-07-20 | 1993-09-01 | Glaxo Lab Sa | Chemical compounds |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| US5824683A (en) | 1995-11-28 | 1998-10-20 | Schering Corporation | 2'- 4'-halo- 1,1'-biphenyl!-4-yl!methyl!-5'-methyl-spiro cyclopentane-1,7' (8'H)- 3H! imidazo 2,1-b!purin!-4' (5'H)-ones |
| GB9526245D0 (en) | 1995-12-21 | 1996-02-21 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9622363D0 (en) | 1996-10-28 | 1997-01-08 | Celltech Therapeutics Ltd | Chemical compounds |
| US6057329A (en) | 1996-12-23 | 2000-05-02 | Celltech Therapeutics Limited | Fused polycyclic 2-aminopyrimidine derivatives |
| SE9701398D0 (en) | 1997-04-15 | 1997-04-15 | Astra Pharma Prod | Novel compounds |
| IT1291372B1 (en) | 1997-05-21 | 1999-01-07 | Schering Plough S P A | USE OF HETEROCYCLIC ANALOGS OF 1,2,4-TRIAZOLE (1,5-C) PYRIMIDINS FOR THE PREPARATION OF MEDICATIONS USEFUL FOR THE TREATMENT OF DISEASES |
| US6013621A (en) | 1997-10-17 | 2000-01-11 | The Rockfeller University | Method of treating psychosis and/or hyperactivity |
| GB9722520D0 (en) | 1997-10-24 | 1997-12-24 | Pfizer Ltd | Compounds |
| DE69943144D1 (en) | 1998-03-31 | 2011-03-03 | Kyowa Hakko Kirin Co Ltd | NITROGEN-CONTAINING HETEROCYCLIC COMPOUNDS |
| US6133273A (en) | 1998-05-08 | 2000-10-17 | American Home Products Corporation | Pyrazolopyrimidine-2,4-dione sulfonamides |
| GB9907658D0 (en) | 1999-04-06 | 1999-05-26 | Zeneca Ltd | Chemical compounds |
| WO2001000214A1 (en) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| AU5636900A (en) | 1999-06-30 | 2001-01-31 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| DE19931206A1 (en) | 1999-07-07 | 2001-01-11 | Stief Christian | Relaxing, or increasing cyclic adenosine monophosphate concentration in smooth muscular tissue, e.g. by administration of cAMP phosphodiesterase inhibitors, dipyridamole or sildenafil |
| DE60023926T2 (en) | 1999-09-10 | 2006-07-20 | Merck & Co., Inc. | TYROSINE KINASE INHIBITORS |
| JP2003510325A (en) | 1999-09-30 | 2003-03-18 | ニューロジェン・コーポレーション | Certain alkylenediamine-substituted pyrazolo [1,5-a] -1,5-pyrimidines and pyrazolo [1,5-a] -1,3,5-triazines |
| TWI265925B (en) | 1999-10-11 | 2006-11-11 | Pfizer | Pyrazolo[4,3-d]pyrimidin-7-ones useful in inhibiting type 5 cyclic guanosine 3',5'-monophosphate phosphodiesterases(cGMP PDE5), process and intermediates for their preparation, their uses and composition comprising them |
| AP2002002455A0 (en) | 1999-10-11 | 2002-06-30 | Pfizer | 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)-dIhydropyrazolo[4,3-D] pyrimidin-7-ones as phosphodiesterase inhibitors. |
| IL139073A0 (en) | 1999-10-21 | 2001-11-25 | Pfizer | Treatment of neuropathy |
| MY125533A (en) | 1999-12-06 | 2006-08-30 | Bristol Myers Squibb Co | Heterocyclic dihydropyrimidine compounds |
| GB0004890D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| JP2004500425A (en) | 2000-04-19 | 2004-01-08 | リリー アイコス リミテッド ライアビリティ カンパニー | Use of cyclic GMP-specific phosphodiesterase inhibitors for the treatment of Parkinson's disease |
| US6693099B2 (en) | 2000-10-17 | 2004-02-17 | The Procter & Gamble Company | Substituted piperazine compounds optionally containing a quinolyl moiety for treating multidrug resistance |
| EP1372656B1 (en) | 2001-03-16 | 2005-06-22 | Pfizer Limited | Pyrazolo[4,3-d]pyrimidinone compounds as cgmp pde inhibitors |
| WO2002088079A2 (en) | 2001-05-01 | 2002-11-07 | Bristol-Myers Squibb Company | Dual inhibitors of pde 7 and pde 4 |
| SE0102315D0 (en) | 2001-06-28 | 2001-06-28 | Astrazeneca Ab | Compounds |
| WO2003020724A1 (en) | 2001-08-28 | 2003-03-13 | Schering Corporation | Polycyclic guanine phosphodiesterase v inhibitors |
| EP1575916B1 (en) | 2001-08-31 | 2013-05-22 | The Rockefeller University | Phosphodiesterase activity and regulation of phosphodiesterase 1-b-mediated signaling in brain |
| CA2465893A1 (en) | 2001-11-09 | 2003-05-22 | Schering Corporation | Polycyclic guanine derivative phosphodiesterase v inhibitors |
| AU2003219770B2 (en) | 2002-02-15 | 2008-10-09 | Merckle Gmbh | Conjugates of biologically active compounds, methods for their preparation and use, formulation and pharmaceutical applications thereof |
| IL163575A0 (en) | 2002-02-21 | 2005-12-18 | Univ Rockefeller | Compositions and method for regulation of calcium-dependent signalling in brain |
| CN1668759A (en) * | 2002-05-17 | 2005-09-14 | 协和发酵工业株式会社 | Search method for substances with antidiabetic effect |
| GB0230045D0 (en) | 2002-12-23 | 2003-01-29 | Glaxo Group Ltd | Compounds |
| EP1613747A1 (en) | 2003-03-31 | 2006-01-11 | Pfizer Products Inc. | Crystal structure of 3 ,5 -cyclic nucleotide phosphodiesterase 1b (pde1b) and uses thereof |
| MXPA05010373A (en) | 2003-04-01 | 2005-12-05 | Applied Research Systems | Inhibitors of phosphodiesterases in infertility. |
| US20050113379A1 (en) | 2003-09-05 | 2005-05-26 | Ping Ge | Heteroaryl fused pyridines, pyrazines and pyrimidines as CRF1 receptor ligands |
| EP1919287A4 (en) | 2005-08-23 | 2010-04-28 | Intra Cellular Therapies Inc | ORGANIC COMPOUNDS FOR TREATING A REDUCED DOPAMINE RECEPTOR ACTIVITY SIGNAL |
| JP5453086B2 (en) * | 2006-06-06 | 2014-03-26 | イントラ−セルラー・セラピーズ・インコーポレイテッド | Organic compounds |
| US20070286890A1 (en) | 2006-06-07 | 2007-12-13 | John Garnett Walt | Eyelash applicator and method |
| JP2010509399A (en) | 2006-11-13 | 2010-03-25 | イントラ−セルラー・セラピーズ・インコーポレイテッド | Organic compounds |
| US9006258B2 (en) | 2006-12-05 | 2015-04-14 | Intra-Cellular Therapies, Inc. | Method of treating female sexual dysfunction with a PDE1 inhibitor |
| ES2588238T3 (en) | 2007-12-06 | 2016-10-31 | Intra-Cellular Therapies, Inc. | Derivatives of pyrazolopyrimidin-4,6-dione and its use as a pharmaceutical product |
| AU2009322904A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065147A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| KR20110098732A (en) | 2008-12-06 | 2011-09-01 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | Organic compounds |
| JP2012518685A (en) | 2009-02-25 | 2012-08-16 | イントラ−セルラー・セラピーズ・インコーポレイテッド | PDE1 inhibitor for eye disorders |
| TW201206937A (en) | 2010-05-31 | 2012-02-16 | Intra Cellular Therapies Inc | Organic compounds |
| WO2011153135A1 (en) | 2010-05-31 | 2011-12-08 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9434730B2 (en) | 2010-05-31 | 2016-09-06 | Intra-Cellular Therapies, Inc. | PDE1 inhibitor compounds |
| US9763948B2 (en) * | 2010-05-31 | 2017-09-19 | Intra-Cellular Therapies, Inc. | PDE1 inhibitory compounds and methods |
| AR091507A1 (en) | 2012-06-21 | 2015-02-11 | Intra Cellular Therapies Inc | SALTS OF (6aR, 9aS) -5.6a, 7,8,9,9a-HEXAHYDRO-5-METHYL-3- (PHENYLAMINE) -2 - ((4- (6-FLUOROPIRIDIN-2-IL) PHENYL) METAL ) -CICLOPENT [4,5] IMIDAZO [1,2-a] PIRAZOLO [4,3-e] PYRIMIDIN-4 (2H) -ONA |
| CA2906640C (en) | 2013-03-15 | 2021-07-20 | Intra-Cellular Therapies, Inc. | Substituted imidazo-[1,2-a]pyrazolo[4.3-e]pyrimidin-4[5h]-one compounds and pharmaceutical compositions and use therof as pde1 inhibitors |
-
2014
- 2014-03-13 CA CA2906640A patent/CA2906640C/en active Active
- 2014-03-13 JP JP2016501936A patent/JP6437519B2/en active Active
- 2014-03-13 TW TW103108972A patent/TWI659032B/en active
- 2014-03-13 AU AU2014234990A patent/AU2014234990B2/en active Active
- 2014-03-13 RU RU2015143672A patent/RU2679142C2/en active
- 2014-03-13 WO PCT/US2014/025666 patent/WO2014151409A1/en not_active Ceased
- 2014-03-13 US US14/777,446 patent/US9598426B2/en active Active
- 2014-03-13 MX MX2015013042A patent/MX378194B/en unknown
- 2014-03-13 DK DK14768690.1T patent/DK2970279T3/en active
- 2014-03-13 PL PL14768690T patent/PL2970279T3/en unknown
- 2014-03-13 CN CN201480026736.4A patent/CN105377846B/en active Active
- 2014-03-13 ES ES14768690T patent/ES2836129T3/en active Active
- 2014-03-13 KR KR1020157029692A patent/KR102240326B1/en active Active
- 2014-03-13 EP EP14768690.1A patent/EP2970279B1/en active Active
- 2014-03-13 US US14/209,258 patent/US9073936B2/en active Active
- 2014-03-14 AR ARP140101051A patent/AR095347A1/en active IP Right Grant
- 2014-03-14 UY UY0001035467A patent/UY35467A/en not_active Application Discontinuation
-
2015
- 2015-06-04 US US14/731,233 patent/US9556186B2/en active Active
- 2015-09-10 IL IL241573A patent/IL241573B/en active IP Right Grant
-
2017
- 2017-01-30 US US15/419,902 patent/US20170231994A1/en not_active Abandoned
-
2018
- 2018-08-17 JP JP2018153710A patent/JP6638036B2/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006133261A2 (en) | 2005-06-06 | 2006-12-14 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20080188492A1 (en) | 2005-06-06 | 2008-08-07 | Intra-Cellular Therapies, Inc | Organic Compounds |
| WO2009075784A1 (en) | 2007-12-06 | 2009-06-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20120238589A1 (en) * | 2007-12-06 | 2012-09-20 | Peng Li | Organic compounds |
| WO2010065148A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065149A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2010065151A1 (en) | 2008-12-06 | 2010-06-10 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20120071450A1 (en) * | 2008-12-06 | 2012-03-22 | Peng Li | Organic compounds |
| US20120053190A1 (en) | 2009-05-13 | 2012-03-01 | Fienberg Allen A | Organic compounds |
| WO2011043816A1 (en) | 2009-10-08 | 2011-04-14 | Intra-Cellular Therapies, Inc. | Phosphodiesterase 1-targeting tracers and methods |
| WO2012171016A1 (en) | 2011-06-10 | 2012-12-13 | Intra-Cellular Therapies, Inc. | Organic compounds |
Non-Patent Citations (3)
| Title |
|---|
| ENNACEUR ET AL., BEHAV. BRAIN RES., vol. 31, 1988, pages 47 - 59 |
| MANI ET AL., SCIENCE, vol. 287, 2000, pages 1053 |
| PRICKAERTS ET AL., EUR. J. PHARMACOL., vol. 337, 1997, pages 125 - 136 |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9624230B2 (en) | 2005-06-06 | 2017-04-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9403836B2 (en) | 2007-12-06 | 2016-08-02 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10010553B2 (en) | 2009-05-13 | 2018-07-03 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10238660B2 (en) | 2009-05-13 | 2019-03-26 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9468637B2 (en) | 2009-05-13 | 2016-10-18 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9763948B2 (en) | 2010-05-31 | 2017-09-19 | Intra-Cellular Therapies, Inc. | PDE1 inhibitory compounds and methods |
| US9801882B2 (en) | 2013-02-17 | 2017-10-31 | Intra-Cellular Therapies, Inc. | Phosphodiesterase-1 inhibitors and their use in treatment of cardiovascular diseases |
| US10398698B2 (en) | 2013-02-17 | 2019-09-03 | Intra-Cellular Therapies, Inc. | Uses |
| US9556186B2 (en) | 2013-03-15 | 2017-01-31 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US9849132B2 (en) | 2014-01-08 | 2017-12-26 | Intra-Cellular Therapies, Inc. | Products and pharmaceutical compositions |
| US11166956B2 (en) | 2014-08-07 | 2021-11-09 | Intra-Cellular Therapies, Inc. | Combinations of PDE1 inhibitors and NEP inhibitors |
| US10285992B2 (en) | 2014-08-07 | 2019-05-14 | Intra-Cellular Therapies, Inc. | Combinations of PDE1 inhibitors and NEP inhibitors and associated methods |
| US10131671B2 (en) | 2014-08-07 | 2018-11-20 | Intra-Cellular Therapies, Inc. | Organic compounds |
| EP3193878A4 (en) * | 2014-09-17 | 2018-03-21 | Intra-Cellular Therapies, Inc. | Compounds and methods |
| AU2015317527B2 (en) * | 2014-09-17 | 2020-08-20 | Intra-Cellular Therapies, Inc. | Compounds and methods |
| US10150774B2 (en) | 2014-09-17 | 2018-12-11 | Intra-Cellular Therapies, Inc. | Compounds and methods |
| US10150771B2 (en) | 2014-10-10 | 2018-12-11 | H. Lundbeck A/S | Triazolopyrazinones as PDE1 inhibitors |
| US10300064B2 (en) | 2014-12-06 | 2019-05-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10105349B2 (en) | 2014-12-06 | 2018-10-23 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US10543194B2 (en) | 2014-12-06 | 2020-01-28 | Intra-Cellular Therapies, Inc. | Organic compounds |
| JP2018513184A (en) * | 2015-04-22 | 2018-05-24 | ハー・ルンドベック・アクチエゼルスカベット | Imidazotriazinone as a PDE1 inhibitor |
| WO2016170064A1 (en) * | 2015-04-22 | 2016-10-27 | H. Lundbeck A/S | Imidazotriazinones as pde1 inhibitors |
| CN107531713A (en) * | 2015-04-22 | 2018-01-02 | H.隆德贝克有限公司 | Imidazolotriazinones as PDE1 inhibitors |
| CN107531713B (en) * | 2015-04-22 | 2019-07-19 | H.隆德贝克有限公司 | Imidazotriazinones as PDE1 Inhibitors |
| US11472810B2 (en) | 2015-04-30 | 2022-10-18 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| US10011606B2 (en) | 2015-04-30 | 2018-07-03 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| US10858362B2 (en) | 2015-04-30 | 2020-12-08 | H. Lundbeck A/S | Imidazopyrazinones as PDE1 inhibitors |
| JP2019510039A (en) * | 2016-03-28 | 2019-04-11 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | Novel compositions and methods |
| EP3436083A4 (en) * | 2016-03-28 | 2019-11-27 | Intra-Cellular Therapies, Inc. | NEW COMPOSITIONS AND METHODS |
| US10682354B2 (en) | 2016-03-28 | 2020-06-16 | Intra-Cellular Therapies, Inc. | Compositions and methods |
| US11104680B2 (en) | 2016-04-12 | 2021-08-31 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US10538525B2 (en) | 2016-04-12 | 2020-01-21 | H. Lundbeck A/S | 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-ones and 1,5-dihydro-4H-pyrazolo[4,3-c]pyridin-4-ones as PDE1 inhibitors |
| US11291666B2 (en) | 2016-09-12 | 2022-04-05 | Intra-Cellular Therapies, Inc. | Uses |
| US10633382B2 (en) | 2016-10-18 | 2020-04-28 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as PDE1 inhibitors |
| US10905688B2 (en) | 2016-10-28 | 2021-02-02 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-α]pyrazinones as PDE1 inhibitors |
| US10912773B2 (en) | 2016-10-28 | 2021-02-09 | H. Lundbeck A/S | Combinations comprising substituted imidazo[1,5-a]pyrazinones as PDE1 inhibitors |
| WO2018197324A1 (en) * | 2017-04-24 | 2018-11-01 | Igm Group B.V. | Simple oxidative functionalization of alkyl aryl ketones |
| US11203580B2 (en) | 2017-04-24 | 2021-12-21 | Igm Group B.V. | Simple oxidative functionalized of alkyl aryl ketones |
| EP3395806A1 (en) * | 2017-04-24 | 2018-10-31 | IGM Group B.V. | Simple oxidative functionalization of alkyl aryl ketones |
| US11759465B2 (en) | 2018-01-31 | 2023-09-19 | Intra-Cellular Therapies, Inc. | Uses |
| US11839614B2 (en) | 2018-01-31 | 2023-12-12 | Intra-Cellular Therapies, Inc. | Methods for treating or mitigating cardiotoxicity characterized by inhibition of adenosine A2 signaling and/or adenosine A2 receptor expression |
| US12605384B2 (en) | 2018-01-31 | 2026-04-21 | Intra-Cellular Therapies, Inc. | Uses |
| EP4520399A2 (en) | 2018-05-25 | 2025-03-12 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2020210614A1 (en) | 2019-04-12 | 2020-10-15 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US12410175B2 (en) | 2019-09-03 | 2025-09-09 | Intra-Cellular Therapies, Inc. | Compounds |
| US12364695B2 (en) | 2020-06-02 | 2025-07-22 | Intra-Cellular Therapies, Inc. | Methods of treating inflammatory disease |
| EP4284806A4 (en) * | 2021-01-27 | 2025-02-26 | Intra-Cellular Therapies, Inc. | SALT CRYSTALS |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2970279B1 (en) | Organic compounds | |
| EP2717877B1 (en) | Organic compounds | |
| EP2576550B1 (en) | Organic compounds | |
| EP2367429B1 (en) | Organic compounds | |
| EP2358723B1 (en) | 4,5,7,8-tetrahydro-2H-imidazo[1,2-a]pyrrolo[3,4-e]pyrimidine compounds as PDE1 inhibitors | |
| EP2358204B1 (en) | 4,5,7,8-tetrahydro-4-oxo-2H-imidazo[1,2-a]pyrrolo[3,4-e]pyrimidine compounds as PDE1 inhibitors. | |
| WO2011153138A1 (en) | Organic compounds | |
| HK1220688B (en) | Organic compounds | |
| BR112015023592B1 (en) | PDE1-INHIBITOR ORGANIC COMPOUNDS, PHARMACEUTICAL COMPOSITION INCLUDING THEM AND THERAPEUTIC USE OF SAID COMPOUNDS AND SAID COMPOSITION |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14768690 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 241573 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2906640 Country of ref document: CA Ref document number: 2016501936 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2015/013042 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14777446 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014768690 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2015143672 Country of ref document: RU Kind code of ref document: A Ref document number: 20157029692 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014234990 Country of ref document: AU Date of ref document: 20140313 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015023592 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015023592 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150915 |