WO2014152733A1 - A method of providing ocular neuroprotection - Google Patents
A method of providing ocular neuroprotection Download PDFInfo
- Publication number
- WO2014152733A1 WO2014152733A1 PCT/US2014/027672 US2014027672W WO2014152733A1 WO 2014152733 A1 WO2014152733 A1 WO 2014152733A1 US 2014027672 W US2014027672 W US 2014027672W WO 2014152733 A1 WO2014152733 A1 WO 2014152733A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- purin
- bicyclic
- monocyclic
- dihydroxytetrahydrofuran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
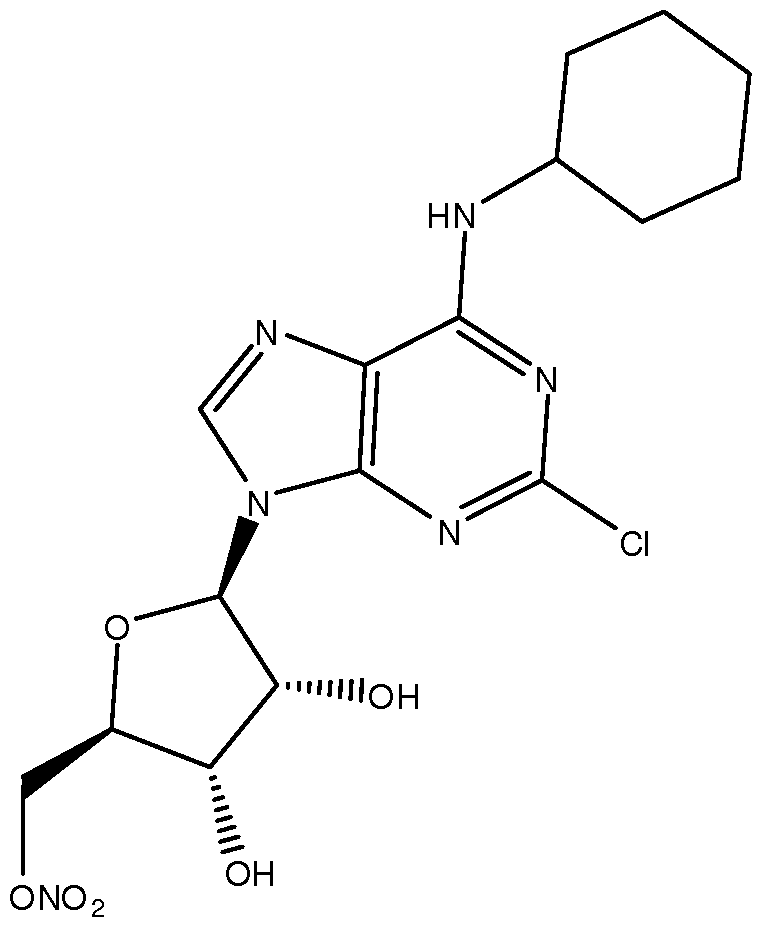
- 0 *C(C(C(O)O1)N)C1P Chemical compound *C(C(C(O)O1)N)C1P 0.000 description 9
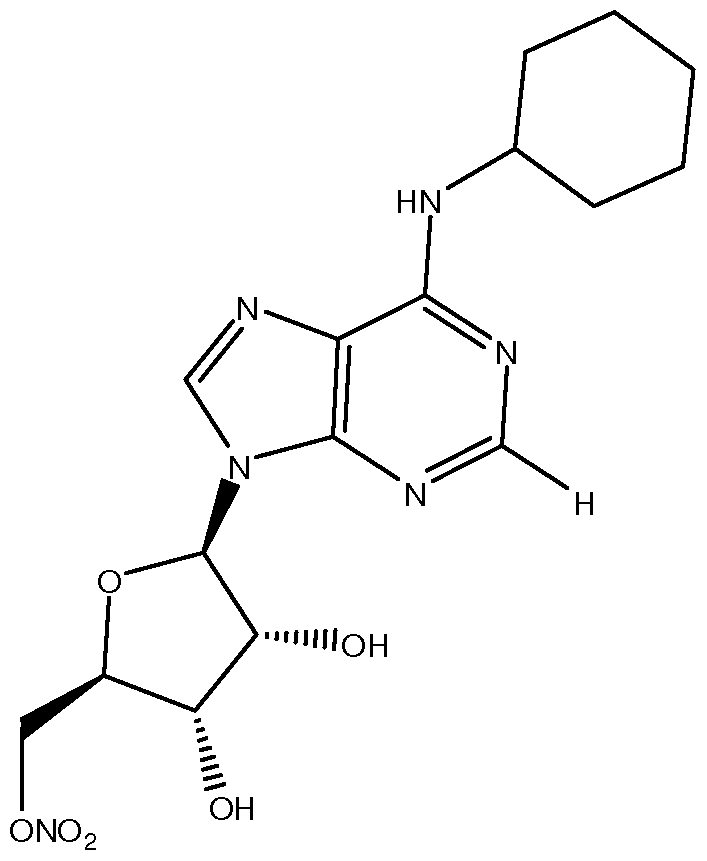
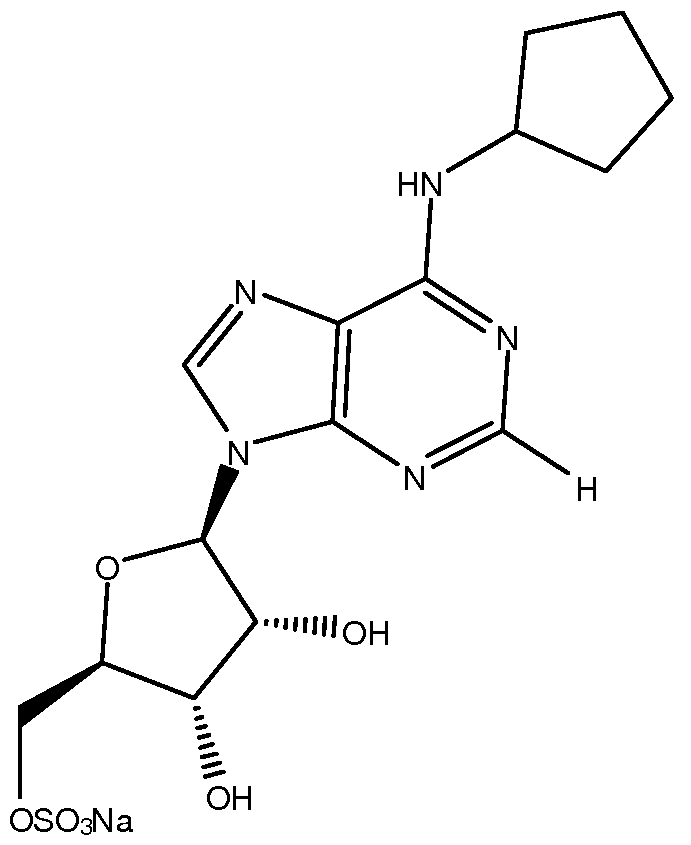
- SZBULDQSDUXAPJ-AARXTDBFSA-N OC[C@H]([C@H]([C@H]1O)O)OC1[n]1c(ncnc2NC3CCCCC3)c2nc1 Chemical compound OC[C@H]([C@H]([C@H]1O)O)OC1[n]1c(ncnc2NC3CCCCC3)c2nc1 SZBULDQSDUXAPJ-AARXTDBFSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- retinal ganglion cells The loss of retinal ganglion cells is a hallmark of certain ophthalmic diseases including ischemic optic neuropathy, (ION) and glaucoma.
- ischemic optic neuropathy ION
- glaucoma glaucoma
- the retinal ganglion cells each have an axion that extends into the brain comprising the optic nerve.
- Retinal ganglion cell damage is any kind of injury or damage to the retinal ganglion cells brought about by ocular compression, ocular ischemia, ocular trauma, ocular inflammation, ocular infection, normal tension glaucoma, elevated intraocular pressure, diabetes, interruption in the blood circulation to the retinal ganglion cells, ocular malignancy, ocular disease or general ocular deterioration.
- Optic nerve damage is also called optic nerve atrophy or optic neuropathy.
- Optic nerve and retinal ganglion cell damage can lead to vision distortion, vision loss and blindness.
- Acute and chronic animal models of optic nerve degeneration have shown the neuroprotective potential of the alpha2 adrenergic agonist brimonidine. These models include direct injury of the optic nerve (nerve crush) and models of acute and chronic ocular hypertension (Yoles et al 1999; Donello et al 2001; WoldeMussie et al 2001; Mayor-Torroglosa et al 2005; Lambert et al 2011). Brimonidine improved RGC survival in each of these models. 1. Donello JE, Padillo EU, Webster ML, et al. alpha2- Adrenoceptor agonists inhibit vitreal glutamate and aspartate accumulation and preserve retinal function after transient ischemia. J Pharmacol Exp Ther. 2001;296:216-23
- brimonidine given topically in a 0.2% solution was shown to reduce the loss in thickness of the nerve fiber layer of the retina (RNFL), where retinal ganglion cells are found, compared to treatment with timolol.
- This protective effect was attributed to neuroprotection due to the similar IOP changes seen between treatment groups (Tsai JC, Chang HW.
- Barriers to the clinical use of 0.2% brimonidine to prevent retinal ganglion cell death and consequent vision loss in patients with optic nerve neuropathies include the required dosing frequency (three times a day) and the high rate of side effects of the therapy.
- the FDA labeling for 0.2% brimonidine tartrate lists oral dryness, ocular hyperemia, burning and stinging, headache, blurring, foreign body sensation, fatigue/drowsiness, conjunctival follicles, ocular allergic reactions, and ocular pruritus as occurring in 10 to 30% of patients.
- 28.3% of study participants receiving 0.2% brimonidine discontinued the study due to adverse events, compared to 11.4% of timolol-treated subjects. This poor ocular tolerability means that compliance with chronically administered 0.2% brimonidine is a major impediment to the long-term treatment required to benefit from the neuroprotective effects of the drug.
- compositions comprising such compounds, and methods of using such compounds to treat, reduce or prevent retinal ganglion cell damage or to provide ocular neuroprotection.
- a method of preventing retinal ganglion cell damage in a subject comprising applying an effective amount of an ophthalmic pharmaceutical composition comprising a selective adenosine Ai agonist to an eye of the subject.
- the present invention provides a method of reducing retinal ganglion cell damage in a subject by administering an effective amount of an ophthalmic pharmaceutical composition comprising a selective Ai agonist to an affected eye of the subject.
- the present invention provides a method of providing ocular neuroprotection in a subject in need thereof, comprising the step of: applying a pharmaceutical composition comprising an effective amount of a selective Ai agonist to an eye of the subject.
- the methods of the ophthalmic composition comprises an effective amount of a selective adenosine Ai agonist compound according to Formula I,
- A is -CH 2 ON0 2 , -CH 2 OH, or -CH 2 OS0 3 H;
- the adenosine Ai agonist is Compound A.
- the methods of the invention are useful for preventing or reducing retinal ganglion cell damage or providing ocular neuroprotection in subjects having or at risk for developing diseases and conditions giving rise to the retinal ganglion cell damage including, but not limited to, ocular compression, ocular ischemia, ocular trauma (e.g., Purtsher' s retinopathy), ocular inflammation, ocular infection, elevated intraocular pressure, diabetes, interruption in the blood circulation to the retinal ganglion cells, ocular malignancy, ocular disease or general ocular deterioration, glaucoma (e.g., normal tension glaucoma, pseudo-exfoliative and pigment dispersion glaucoma, and closed angle glaucoma), ocular ischemic syndrome, retinal ischemia (e.g., retinal hypoxia ischemia), retinal vein occlusion, retinal artery occlusion, diabetic
- ocular compression e.g., ocular ischemia,
- retinopathy age-related macular degeneration, visual loss from retinal detachment, conditions resulting in increased permeability of the blood-retinal barrier (BRB) resulting in fluid accumulation and retinal edema, or combinations thereof.
- BRB blood-retinal barrier
- the diseases or conditions giving rise to the retinal ganglion cell damage is not caused solely by elevated intraocular pressure.
- the selective adenosine Al agonist can be administered in drops, e.g., 1 to 2 drops.
- the IOP of the affected eye is reduced by at least 10%, e.g., at least 10-20%, e.g. , by 20% or more.
- the IOP of the affected eye is reduced by at least 10% for more than 3 hours, e.g., at least 10-20% for more than 3 hours, e.g., by 20% or more for more than 3 hours.
- the IOP of the affected eye is reduced by at least 10% for at least 6 hours.
- the IOP of the affected eye is reduced by at least 20% for at least 12 hours.
- the IOP of the affected eye is reduced by at least 20% for about 12 to about 24 hours.
- the effective amount of the selective adenosine Ai agonist applied to the eye is about 20 ⁇ g to about 7.0 mg. In some embodiments, the effective amount of the selective adenosine Ai agonist is from about 30 ⁇ g to 1 mg. In some embodiments the effective amount of selective adenosine Ai agonist is at least 20 ⁇ g.
- the effective amount of the selective adenosine Ai agonist is between 60 ⁇ g and 1500 ⁇ g; is about 100 ⁇ g, about 200 ⁇ g, about 250 ⁇ g, about 300 ⁇ g, about 350 ⁇ g, about 400 ⁇ g, about 450 ⁇ g, about 500 ⁇ g, about 550 ⁇ g or about 600 ⁇ g or about 500 to 1500 ⁇ g. In certain embodiments, the effective amount of the selective adenosine Ai agonist is about 500 In one embodiment the selective adenosine Ai agonist is administered at an effective dose of about 0.1 to about 5.0% (w/v).
- the selective adenosine Ai agonist is administered at an effective dose of about 0.5 to about 1.5% (w/v). In one embodiment, the selective adenosine Ai agonist is administered at an effective dose of about 1.0% to about 3.0% (w/v). In one embodiment, selective adenosine Ai agonist is administered at an effective dose of about 3.0% (w/v).
- the effective amount of the selective adenosine AI agonist is administered as a single dose. In another embodiment, the effective amount of the selective adenosine Ai agonist is administered as a twice daily dose. In another embodiment, the selective adenosine AI agonist is administered 1 to 4 times daily.
- the method may further comprise administering a second ophthalmic agent in addition to a compound of Formula I as defined above.
- the second ophthalmic agent can be selected from the group comprising: ⁇ -blockers, prostaglandin analogs, carbonic anhydrase inhibitors, rho-kinase inhibitors, a 2 adrenergic agonists, miotics, neuroprotectants, adenosine A 3 antagonists, adenosine A 2A agonists, ion channel modulators and combinations thereof.
- the selective adenosine AI agonist administered is selected from the group consisting of:
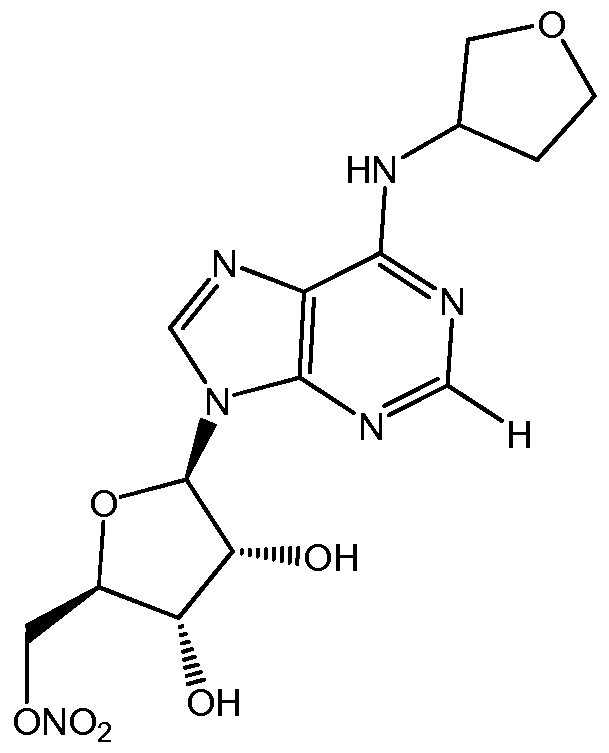
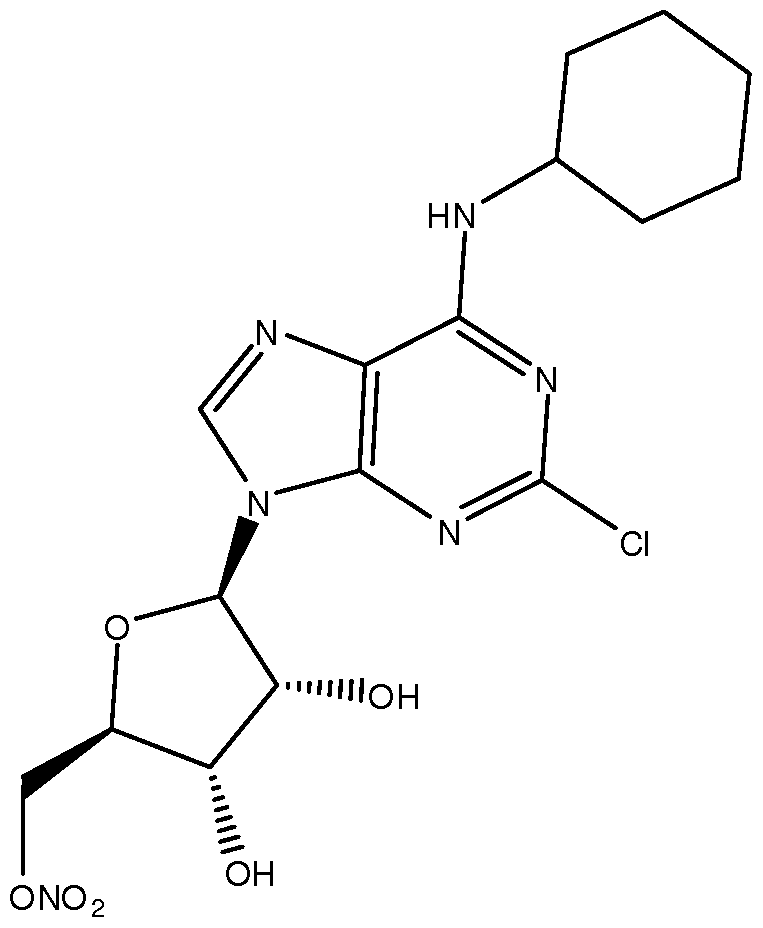
- the formulation comprises about 7mg/ml of a compound of Formula I selected from: ((2R,3S,4R,5R)-5-(2-chloro-6-(cyclopentylamino)-9H- purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl nitrate; and
- Figure 1 shows schematically a retinal cross section depicting the layers and cells that make up the retina.
- Figure 2 shows the histology data showing the degree of retinal thinning in the various ganglion cell layers along with a comparison between the effects of brimonidine and Compound A (designated as 'Drug' in the Figure).
- Figure 3 shows histology data for the percentage of degree of protection across the various ganglion cell layers, along with a comparison between the effects of brimonidine and Compound A (designated as 'Drug' in the Figure).
- Figure 4 shows the percent protection gained from the presence of Compound A or brimonidine on RGC from an ischemic insult versus an assumption of no protection in the vehicle treated group.
- Embodiments of the present invention provide compounds useful for preventing, reducing or treating retinal ganglion cell damage.
- Adenosine is a purine nucleoside that modulates many physiologic processes.
- Cellular signaling by adenosine occurs through four adenosine receptor subtypes: A 1; A 2A , A 2B , and A 3 as reported by Ralevic and Burnstock (Pharmacol Rev. 50:413-492, 1988) and Fredholm BB et al. (Pharmacol Rev. 53:527-552, 2001).
- adenosine Ai receptor agonists lower IOP in mice, rabbits and monkeys (Tian B et al. Exp Eye Res. 64:979-989, 1997; Crosson CE. J Pharmacol Exp Ther.
- compounds of Formula I e.g., Compounds A, B, C, D, E, F, G, H, I, J or K
- a subject e.g., a human
- ocular neuroprotection in a subject in need thereof.
- A is -CH 2 ON0 2 , -CH 2 OH or -CH 2 OS0 3 H;
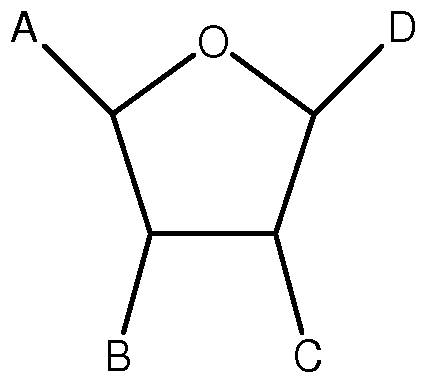
- a and B are trans with respect to each other
- R 1 is -H, -C Cio alkyl, -aryl, -3- to 7-membered monocyclic heterocycle, -8- to 12-membered bicyclic heterocycle, -C 3 -C 8 monocyclic cycloalkyl, -C 3 -C 8 monocyclic cycloalkenyl, -Cg-C 12 bicyclic cycloalkyl, -Cg-C 12 bicyclic cycloalkenyl - (CH 2 ) n -(C 3 -Cg monocyclic cycloalkyl), -(CH 2 ) n -(C 3 -Cg monocyclic cycloalkenyl), - (CH 2 ) n -(Cg-C 12 bicyclic cycloalkyl), -(CH 2 ) n -(Cg-C 12 bicyclic cycloalkyl), -(CH 2 ) n -(Cg-C 12 bicyclic cycl
- R 4 is -Q-Q5 alkyl, -aryl, -(CH 2 ) n -aryl, -(CH 2 ) n -(3- to 7-membered monocyclic heterocycle), -(CH 2 ) n -(8- to 12-membered bicyclic heterocycle), -(CH 2 ) n -(C 3 -C 8 monocyclic cycloalkyl), -(CH 2 ) n -(C 3 -C8 monocyclic cycloalkenyl), -(CH 2 ) n -(C
- R 6 is -C Cio alkyl, -aryl, -(CH 2 ) n -aryl, -(CH 2 ) n -(3- to 7-membered monocyclic heterocycle), -(CH 2 ) n -(8- to 12-membered bicyclic heterocycle), -(CH 2 ) n -(C 3 -C 8 monocyclic cycloalkyl), -(CH 2 ) n -(C 3 -C 8 monocyclic cycloalkenyl), -(CH 2 ) n -(C 8 -C 12 bicyclic cycloalkyl), -(CH 2 ) n -(C 8 -C 12 bicyclic cycloalkenyl), -(CH 2 ) n -(C 8 -C 12 bicyclic cycloalkenyl), -(CH 2 ) n -(C 3 -C 8 monocyclic cycloalkenyl),
- R 7 is -H, -C Cio alkyl, -aryl, -(CH 2 ) n -aryl, -(CH 2 ) n -(3- to 7-membered monocyclic heterocycle), -(CH 2 ) n -(8- to 12-membered bicyclic heterocycle), -(CH 2 ) n - (C 3 -C 8 monocyclic cycloalkyl), -(CH 2 ) n -(C 3 -C 8 monocyclic cycloalkenyl), -(CH 2 ) n - (C 8 -C 12 bicyclic cycloalkenyl) or -(CH 2 ) n -(C 8 -C 12 bicyclic cycloalkyl); and
- each n is independently an integer ranging from 1 to 5, and a pharmaceutically acceptable vehicle.
- the compounds for use in the invention are compounds having the formula
- A is -CH 2 ON0 2 , -CH 2 OH or -CH 2 OS0 3 H;
- a and B are trans with respect to each other
- R 1 is -C3-C8 monocyclic cycloalkyl, -3- to 7-membered monocyclic heterocycle, or -C 8 -C 12 bicyclic cycloalkyl;
- R is -H or -halo.
- the compounds for use in the invention are compounds having the formula
- A is -CH 2 ON0 2 ;
- a and B are trans with respect to each other
- B and C are cis with respect to each other;
- C and D are cis or trans with respect to each other;
- R 1 is -C3-C8 monocyclic cycloalkyl, -3- to 7-membered monocyclic heterocycle, or -C 8 -C 12 bicyclic cycloalkyl;
- R is -H or -halo.
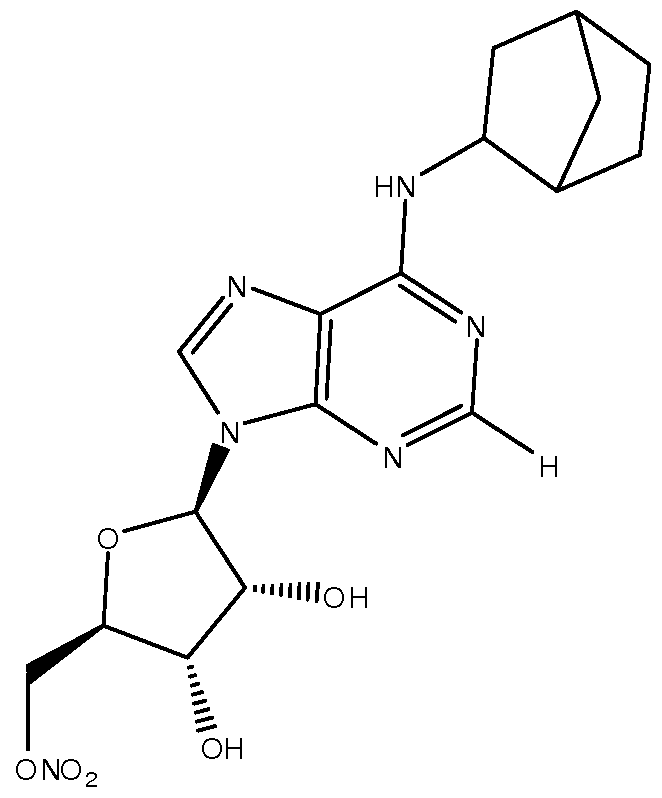
- the compound of Formula I is one of the following compounds:
- CCPA 2-chlorocyclopentyladenosine
- retinal ganglion cells damage to retinal ganglion cells can be measured using the following techniques:
- Visual field loss and its progression are hallmarks in glaucoma, including normal tension glaucoma, and high IOP glaucoma, optic neuritis and retinal ganglion cell damage.
- Visual field loss can be measured using various perimetery techniques. Visual field loss measurements can be very useful in finding early changes in vision caused by RGC damage.
- electroretinogram (ERG) or electroretinography measurements provide information on damage to RGC. Electroretinography measures electrical activity generated by the photoreceptor cells in the retina when the eye is stimulated by certain light sources. The measurement is captured by electrodes placed on the front surface of the eye (e.g. cornea) and the skin near the eye and a graphic record called an electroretinogram (ERG) is produced.
- EMG electroretinogram
- Electroretinography is useful in diagnosing several hereditary and acquired disorders of the retina, damage to retinal ganglion cells by conditions such as but not limited retinitis pigmentosa, a detached retina or functional changes caused by arteriosclerosis or diabetes.
- the photopic negative response (PhNR) of an ERG is thought to measure the presence of intact, functioning RGCs (Viswanathan S, Frishman LJ, Robson JG, et al.
- the photopic negative response of the macaque electroretinogram reduction by experimental glaucoma. Invest Ophthalmol Vis Sci.
- retinal nerve fiber layer thickness (RNFL) measurements measured by optical coherence tomography or scanning laser polarimetry as reported in Tsai JC, Chang HW. Comparison of the effects of brimonidine 0.2% and timolol 0.5% on retinal nerve fiber layer thickness in ocular hypertensive patients: a prospective, unmasked study. J OculPharmacolTher. 2005; 21:475-82.
- Subjects that are susceptible to or at risk of developing RGC damage or requiring ocular neuroprotection would be candidates for employing the preventative methods of the invention are subjects having a family history of glaucoma (e.g., normal tension glaucoma, pseudo-exfoliative and pigment dispersion glaucoma, and closed angle glaucoma),; subjects that have a family history of visual field loss;
- glaucoma e.g., normal tension glaucoma, pseudo-exfoliative and pigment dispersion glaucoma, and closed angle glaucoma
- retinal ischemia e.g., retinal hypoxia ischemia
- retinal vein occlusion e.g., retinal artery occlusion
- diabetic retinopathy e.g., age-related macular degeneration
- BRB blood-retinal barrier
- subjects that are to face ocular surgery or have experienced ocular trauma e.g., subjects that have ocular diseases or diseases associated with the development of retinal ganglion cell damage including glaucoma (e.g., normal tension glaucoma, pseudo-exfoliative and pigment dispersion glaucoma, and closed angle glaucoma), diabetes, malignancy, infection, ocular ischemia, ocular inflammation, ocular compression, elevated intraocular pressure, interruption in the blood circulation to the retinal ganglion cells, ocular ischemic syndrome, retinal ischemia (e.g., retinal hypoxia ischemia), retinal vein occlusion, retinal artery occlusion, diabetic
- provided herein is a method of preventing retinal ganglion cell damage, comprising administering an effective amount of a compound of Formula I to an eye of a subject.
- provided herein is a method of reducing or treating retinal ganglion cell damage, comprising applying an effective amount of a compound of Formula I to an affected eye of a subject.
- a method of preventing, reducing or treating retinal ganglion cell damage comprising applying an effective amount of a compound of Formula I to an eye of a subject.
- about 0.1 to 3.0 % (w/v) of a compound of Formula I is applied to an eye of a subject from 1 to 4 times daily.
- about 0.5 to about 1.5% (w/v) of a compound of Formula I is applied to an eye of a human from 1 to 4 times daily.
- about 1.5% (w/v) of a compound of Formula I is applied to an eye of a human from 1 to 4 times daily.
- the compound of Formula I is applied twice daily.
- the compound of Formula I is applied once daily.
- a compound of Formula I can be administered in drops, e.g., 1 to 2 drops.
- provided herein is a method of preventing, reducing or treating retinal ganglion cell damage, comprising administering an effective amount of Compound A to a subject.
- a method of preventing, reducing or treating retinal ganglion cell damage comprising applying an effective amount of Compound A to an eye of a subject.
- about 0.5 to about 1.5% (w/v) of Compound A is applied to an eye of a subject from 1 to 4 times daily.
- about 0.5 to about 1.5% (w/v) of Compound A is applied to an eye of a subject from 1 to 4 times daily.
- about 1.5% (w/v) of Compound A is applied to an eye of a subject from 1 to 4 times daily. In one embodiment, the compound of Formula I is applied twice daily. In one embodiment, the compound of Formula I is applied once daily.
- the Compound A can be administered in drops, e.g. , 1 to 2 drops.
- provided herein is the use of a compound of Formula I for the manufacture of a medicament for preventing or treating retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of a compound of Formula I for the manufacture of a medicament for reducing retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of a compound of Formula I for the manufacture of a medicament for treating retinal ganglion cell damage in a subject.
- provided herein is the use of a compound of Formula I for the manufacture of a medicament for providing ocular neuroprotection in a subject.
- provided herein is the use of a compound of Formula I for preventing retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of a compound of Formula I for reducing retinal ganglion cell damage associated with glaucoma in a subject. In another embodiment, provided herein is the use of a compound of Formula I for providing ocular neuroprotection in a subject.
- a compound of Formula I for treating retinal ganglion cell damage in a subject.
- provided herein is the use of Compound A for preventing retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of Compound A for reducing retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of Compound A for treating retinal ganglion cell damage in a subject. In another embodiment, provided herein is the use of Compound A for providing ocular neuroprotection in a subject. It is recognized that compounds of Formula I can contain one or more chiral centers. This invention contemplates all enantiomers, diastereomers, and mixtures of Formulas I thereof.
- certain embodiments of the present invention comprise pharmaceutically acceptable salts of compounds according to Formula I.
- Pharmaceutically acceptable salts comprise, but are not limited to, soluble or dispersible forms of compounds according to Formula I that are suitable for treatment of disease without undue undesirable effects such as allergic reactions or toxicity.
- Representative pharmaceutically acceptable salts include, but are not limited to, acid addition salts such as acetate, citrate, benzoate, lactate, or phosphate and basic addition salts such as lithium, sodium, potassium, or aluminum.
- selective adenosine Ai agonist means an Ai agonist that has a high affinity to the Ai receptor while simultaneously having a low affinity for the A 2 A , and A 3 adenosine receptors.
- Compounds of Formula I e.g. , Compounds A to K
- affinities to the Ai receptor considerably greater than their respective affinities to the A 2 A and A 3 receptors.
- the Ai selectivity data for compounds A to K is summarized in the Table below.
- alkyl refers to a fully saturated branched or unbranched hydrocarbon moiety.
- the alkyl comprises 1 to 20 carbon atoms, more preferably 1 to 16 carbon atoms, 1 to 10 carbon atoms, 1 to 7 carbon atoms, or 1 to 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, w-propyl, wo-propyl, w-butyl, sec-butyl, wo-butyl, iert-butyl, w-pentyl, isopentyl, neopentyl, w-hexyl, 3-methylhexyl, 2,2- dimethylpentyl, 2,3- dimethylpentyl, w-heptyl, w-octyl, w-nonyl, w-decyl and the like.
- C x -C y -alkyl indicates a particular alkyl group (straight- or branched-chain) of a particular range of carbons.
- Ci-C4-alkyl includes, but is not limited to, methyl, ethyl, propyl, butyl, isopropyl, tert-butyl and isobutyl.
- alkyl includes, but is not limited to, Q- Cis alkyl, Ci-Cio alkyl and Ci-C 6 alkyl.
- Q-Q5 alkyl refers to a straight or branched chain, saturated hydrocarbon having from 1 to 15 carbon atoms.
- Representative C Qs alkyl groups include, but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec- butyl, tert-buty, pentyl, isopentyl, neopentyl, hexyl, isohexyl, neohexyl, heptyl, isoheptyl, neoheptyl, octyl, isooctyl, neooctyl, nonyl, isononyl, neononyl, decyl, isodecyl, neodecyl, undecyl, dodecyl, tridecyl, tetradecyl and pentadecyl.
- the C Qs alkyl group is substituted with one or more of the following groups: -halo, -0-(d-C 6 alkyl), -OH, -CN, -COOR', -OC(0)R', -N(R') 2 , -NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -Ci-C 6 alkyl. Unless indicated, the C Qs alkyl is unsubstituted.
- C Cio alkyl refers to a straight or branched chain, saturated hydrocarbon having from 1 to 10 carbon atoms.
- Representative C Qo alkyl groups include, but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec- butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, neohexyl, heptyl, isoheptyl, neoheptyl, octyl, isooctyl, neooctyl, nonyl, isononyl, neononyl, decyl, isodecyl and neodecyl.
- the C Qo alkyl group is substituted with one or more of the following groups: -halo, -0-(C -C alkyl), -OH, -CN, -COOR', - OC(0)R', -N(R') 2 , -NHC(0)R' or -C(0)NHR' groups wherein each R' is
- C Qo alkyl independently -H or unsubstituted -Q-C 6 alkyl. Unless indicated, the C Qo alkyl is unsubstituted. C Qo alkyl includes, but is not limited to, Q-C 6 alkyl.
- C ⁇ Ce alkyl refers to a straight or branched chain; saturated hydrocarbon having from 1 to 6 carbon atoms.
- Representative Q-C 6 alkyl groups include, but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec- butyl, tert-buty, pentyl, isopentyl, neopentyl, hexyl, isohexyl, and neohexyl. Unless indicated, the C1-C6 alkyl is unsubstituted.
- aryl refers to a phenyl group or a naphthyl group.
- the aryl group is substituted with one or more of the following groups: -halo, -0-(d-C 6 alkyl), -OH, -CN, -COOR', -OC(0)R', -N(R') 2 , - NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -Ci-C 6 alkyl. Unless indicated, the aryl is unsubstituted.
- C3-C8 monocyclic cycloalkyl as used herein is a 3-, 4-, 5-, 6-, 7- or 8-membered saturated non-aromatic monocyclic cycloalkyl ring.
- Representative C 3 - Cg monocyclic cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- the C 3 -C8 monocyclic cycloalkyl group is substituted with one or more of the following groups: -halo, -0-(d-C 6 alkyl), -OH, -CN, -COOR', -OC(0)R', -N(R') 2 , - NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -C -C alkyl. Unless indicated, the C 3 -C 8 monocyclic cycloalkyl is unsubstituted.
- C 3 -C 8 monocyclic cycloalkenyl as used herein is a 3-, 4-, 5-, 6-, 7- or 8-membered non-aromatic monocyclic carbocyclic ring having at least one endocyclic double bond, but which is not aromatic. It is to be understood that when any two groups, together with the carbon atom to which they are attached form a C 3 - C 8 monocyclic cycloalkenyl group, the carbon atom to which the two groups are attached remains tetravalent.
- Representative C 3 -C 8 monocyclic cycloalkenyl groups include, but are not limited to, cyclopropenyl, cyclobutenyl, 1,3-cyclobutadienyl, cyclopentenyl, 1,3- cyclopentadienyl, cyclohexenyl, 1,3-cyclohexadienyl,
- cycloheptenyl 1,3- cycloheptadienyl, 1,4-cycloheptadienyl, -1,3,5-cycloheptatrienyl, cyclooctenyl, 1,3- cyclooctadienyl, 1,4-cyclooctadienyl, -1,3,5-cyclooctatrienyl.
- the C 3 -Cs monocyclic cycloalkenyl group is substituted with one or more of the following groups: -halo, -O-id-Ce alkyl), -OH, -CN, -COOR', -OC(0)R', -N(R') 2 , -NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -Q-C 6 alkyl. Unless indicated, the C 3 -C 8 monocyclic cycloalkenyl is unsubstituted.
- Cs-C 12 bicyclic cycloalkyl as used herein is a 8-, 9-, 10-, 11- or 12- membered saturated, non-aromatic bicyclic cycloalkyl ring system.
- Representative C 8 -C 12 bicyclic cycloalkyl groups include, but are not limited to,
- the Cs-C 12 bicyclic cycloalkyl group is substituted with one or more of the following groups: -halo, -0-(Ci-C 6 alkyl), -OH, - CN, -COOR', -OC(0)R', -N(R') 2 , -NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -Q-C 6 alkyl. Unless indicated, the Cg-C 12 bicyclic cycloalkyl is unsubstituted.
- C 8 -C 12 bicyclic cycloalkenyl as used herein is a 8-, 9-, 10-, 11- or 12-membered non-aromatic bicyclic cycloalkyl ring system, having at least one endocyclic double bond. It is to be understood that when any two groups, together with the carbon atom to which they are attached form a Cg-C 12 bicyclic cycloalkenyl group, the carbon atom to which the two groups are attached remains tetravalent.
- Representative Cs-C 12 bicyclic cycloalkenyl groups include, but are not limited to, octahydronaphthalene, hexahydronaphthalene, hexahydroindene, tetrahydroindene, octahydrobenzocycloheptene, hexahydrobenzocycloheptene,
- the C 8 -C 12 bicyclic cycloalkyl group is substituted with one or more of the following groups: - halo, -0-(Ci- C 6 alkyl), -OH, -CN, -COOR', -OC(0)R', -N(R') 2 , -NHC(0)R' or - C(0)NHR' groups wherein each R' is independently -H or unsubstituted -C C 6 alkyl. Unless indicated, the C 8 -C 12 bicyclic cycloalkenyl is unsubstituted.
- halo refers to -F, -CI, -Br or -I.
- 3- to 7-membered monocyclic heterocycle refers to: (i) a 3- or 4- membered non-aromatic monocyclic cycloalkyl in which 1 of the ring carbon atoms has been replaced with an N, O or S atom; or (ii) a 5-, 6-, or 7-membered aromatic or non-aromatic monocyclic cycloalkyl in which 1-4 of the ring carbon atoms have been independently replaced with a N, O or S atom.
- the non-aromatic 3- to 7-membered monocyclic heterocycles can be attached via a ring nitrogen, sulfur, or carbon atom.
- the aromatic 3- to 7-membered monocyclic heterocycles are attached via a ring carbon atom.
- a 3- to 7-membered monocyclic heterocycle group include, but are not limited to furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, isothiazolyl, isoxazolyl, morpholinyl, oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, piperazinyl, piperidinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, quinuclidinyl,
- the 3- to 7-membered monocyclic heterocycle group is substituted with one or more of the following groups: -halo, -O- (Ci-C 6 alkyl), -OH, -CN, -COOR', - OC(0)R', -N(R') 2 , -NHC(0)R' or -C(0)NHR' groups wherein each R' is independently -H or unsubstituted -Q-C 6 alkyl. Unless indicated, the 3- to 7-membered monocyclic heterocycle is unsubstituted.
- 8- to 12-membered bicyclic heterocycle refers to a bicyclic 8- to 12-membered aromatic or non-aromatic bicyclic cycloalkyl in which one or both of the of the rings of the bicyclic ring system have 1-4 of its ring carbon atoms independently replaced with a N, O or S atom. Included in this class are 3- to 7- membered monocyclic heterocycles that are fused to a benzene ring. A non-aromatic ring of an 8- to 12-membered monocyclic heterocycle is attached via a ring nitrogen, sulfur, or carbon atom. An aromatic 8- to 12-membered monocyclic heterocycles are attached via a ring carbon atom.
- Examples of 8- to 12-membered bicyclic heterocycles include, but are not limited to, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrzolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, cinnolinyl, decahydroquinolinyl, lH-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, isobenzofuranyl, isoindazolyl, isoindolyl, isoindolinyl, isoquinolinyl, naphthyridinyl, octahydroisoquinolinyl, phthalazinyl, pteridinyl, purinyl, quinoxal
- each ring of a the -8- to 12- membered bicyclic heterocycle group can substituted with one or more of the following groups: -halo, -0-(C -C alkyl), -OH, -CN, -COOR', - OC(0)R', -N(R') 2 , -NHC(0)R'. or -C(0)NHR' groups wherein each R' is
- phrases "pharmaceutically acceptable salt,” as used herein, is a salt of an acid and a basic nitrogen atom of a purine compound.
- Illustrative salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucaronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, and pamoate (i.e., l,l'-methylene-bis-
- the pharmaceutically acceptable salt can also be a camphorsulfonate salt.
- pharmaceutically acceptable salt also refers to a salt of a purine compound having an acidic functional group, such as a carboxylic acid functional group, and a base.
- Suitable bases include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc; ammonia, and organic amines, such as unsubstituted or hydroxy- substituted mono-, di-, or tri- alkylamines, dicyclohexylamine; tributyl amine; pyridine; N-methyl, N-ethylamine; diethylamine; triethylamine; mono-, bis-, or tris-(2-OH-lower alkylamines), such as mono-; bis-, or tris-(2- hydroxyethyl)amine, 2-hydroxy-tert-butylamine, or tris- (hydroxymethyl)methylamine, N,N-di-lower alkyl-N-(hydroxyl-lower alkyl)-amines, such as N,N-dimethyl-N-(2-
- a bold line indicates that a substituent is above the plane of the carbon atom to which it is attached and a dashed line indicates that a substituent is below the plane of the carbon atom to which it is attached.
- an effective amount refers to an amount of a selective adenosine Al agonist that is effective for: (i) preventing retinal ganglion cell damage (ii) reducing retinal ganglion cell damage or (iii) treating retinal ganglion cell damage in a subject.
- subject is intended to include organisms which are at risk of developing or are afflicted with a disease, disorder or condition associated with retinal ganglion cell damage.
- subjects include mammals, e.g., humans, dogs, cows, horses, pigs, sheep, goats, cats, mice, rabbits, rats, and transgenic non-human animals.
- the subject is a human, e.g., a human suffering from, at risk of developing or potentially capable of suffering from retinal ganglion cell damage.
- the term “treat” may mean to reduce or prevent further damage or loss of retinal ganglion cells.
- treatment can be diminishment of one or several symptoms of a disorder or complete eradication of a disorder.
- the terms "protect” or “prevent” are used interchangeably herein to delay the onset (i.e., the period prior to clinical manifestation of a disease) and/or to reduce the likelihood of a subject developing or worsening of a disease (e.g., a subject at risk of developing a disease).
- the formulations of the invention may be used to prevent elevated intraocular pressure, and/or may be used as a neuroprotective composition to prevent retinal ganglion cell damage and/or retinal ganglion cell loss.
- use includes any one or more of the following embodiments of the invention, respectively: the use in the treatment of retinal ganglion cell damage; the use for the manufacture of pharmaceutical compositions for use in the treatment of the diseases or conditions giving rise to retinal ganglion cell damage, e.g. , in the manufacture of a medicament; methods of use of compounds of the invention in the treatment of such diseases or conditions; pharmaceutical preparations having compounds of the invention for the treatment of diseases or conditions causing retinal ganglion cell damage; and compounds of the invention for use in the treatment of conditions and diseases that cause retinal ganglion cell damage; as appropriate and expedient, if not stated otherwise.
- diseases or conditions to be treated and are thus preferred for use of a compound of the present invention include but are not limited to brought about by ocular compression, ocular ischemia, ocular trauma, ocular inflammation, ocular infection, glaucoma, elevated intraocular pressure, interruption in the blood circulation to the retinal ganglion cells, ocular malignancy, ocular disease or general ocular deterioration or combinations thereof.
- the term “about” or “approximately” usually means within 20%, more preferably within 10%, and most preferably still within 5% of a given value or range. Alternatively, especially in biological systems, the term “about” means within about a log (i.e., an order of magnitude) preferably within a factor of two of a given value.
- a drop refers to a quantity of ophthalmically acceptable fluid that resembles a liquid drop.
- a drop refers to a liquid volume equivalent to about 5 ⁇ to about 200 ⁇ , e.g. , about 30 ⁇ to about 80 ⁇ .
- CCPA is 2-chloro- N6-cyclopentyladenosine
- CPA is N6- cyclopentyladenosine
- NECA is adenosine-5'-(N-ethyl)carboxamido
- NMR nuclear magnetic resonance
- R-PIA is N6- (2-phenyl-isopropyl) adenosine, R-isomer
- HPpCD is hydroxypropyl ⁇ -cyclodextrin.
- GCL is ganglion cell layer
- IPL is inner plexiform layer
- INL is inner nuclear layer
- OPL is outer plexiform layer
- ONL is outer nuclear layer
- TR is total retinal thickness.
- Scheme 1 shows methods for making nucleoside intermediates that are useful for making the compounds of the invention.
- R 2 is as defined above.
- the protected ribose compound of Formula 1 can be coupled with a purine compound of Formula 2 using lithium hexamethyldisilazide and trimethylsilyl triflate, followed by acetonide removal using trifluoroacetic acid to provide nucleoside intermediates of Formula 3 and their corresponding other anomers of Formula 4.
- the ribose diacetate of Formula 5 can be coupled with a compound of Formula 2 using lithium hexamethyldisilazide and trimethylsilyl triflate to provide acetonide-protected nucleoside intermediates of Formula 6 and their corresponding other anomers of Formula 7.
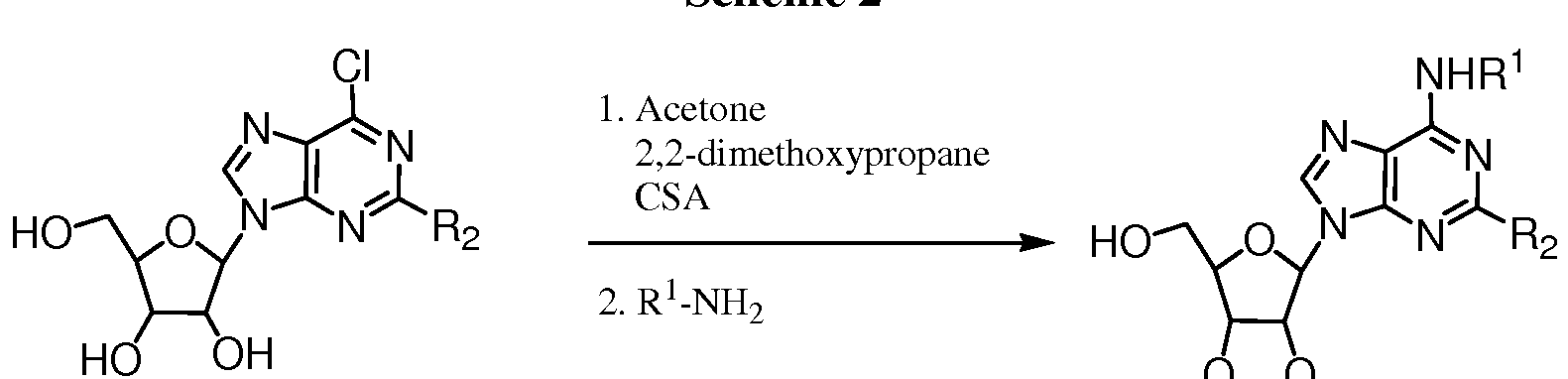
- Scheme 2 shows a method useful for making the adenosine intermediates of Formula 8 which are useful for making the compounds of the invention.
- R 1 and R 2 are defined above.
- 6-chloroadenosine derivative of formula 3a is converted to its 2' ,3'- acetonide using acetone and 2,2-dimethoxypropane in the presence of
- camphorsulfonic acid The acetonide can be futher derviatized using an amine of formula in the presence of base to provide compounds of formula 8.
- R 1 and R 2 are defined above.
- adenosine intermediates of formula 8 can be converted to their 5 '-nitrate analogs using nitric acid in the presence of acetic anhydride, or other nitrating agents, such as MSCI/ONO 3 or nitrosonium tetrafluoroborate.
- Acetonide removal using TF A/water provides compounds of the invention.
- R 1 and R 2 are defined above.
- the adenosine intermediates of formula 8 can be treated with sulfur trioxide- pyridine complex to provide the corresponding 5 '-sulfonic acid pyridine salt intermediate.
- the pyridine salt intermediate can then be neutralized using NaOH or KOH, followed by acetonide removal using TF A/water to provide the corresponding sodium or potassium salt, respectively, of the Purine Derivatives of Formula (Id) wherein A is -CH 2 OSO 3 H.
- Treatment of the sodium or potassium salt with strong aqueous acid, such as sulfuric or hydrochloric acid provides compounds of the invention wherein A is - CH 2 OSO 3 H.
- Formula I can be incorporated into various types of ophthalmic compositions or formulations for delivery.
- Formula I compounds may be delivered directly to the eye (for example: topical ocular drops or ointments; slow release devices such as pharmaceutical drug delivery sponges implanted in the cul-de- sac or implanted adjacent to the sclera or within the eye; periocular, conjunctival, sub- tenons, intracameral, intravitreal, or intracanalicular injections) or systemically (for example: orally, intravenous, subcutaneous or intramuscular injections; parenterally, dermal or nasal delivery) using techniques well known by those of ordinary skill in the art. It is further contemplated that the agents of the invention may be formulated in intraocular insert or implant devices.
- the compounds of Formula I are preferably incorporated into topical ophthalmic formulations with a pH of about 4-8 for delivery to the eye.
- Various formulations of Compound A in particular are described in PCT/US2010/033112, PCT/US2010/054040, and co-pending U.S. Provisional Application No. 61/793273 entitled "Ophthalmic Formulations,” filed on March 15, 2013, the contents of which are herein incorporated as if individually set forth.
- the compounds may be combined with ophthalmologically acceptable preservatives, surfactants, viscosity enhancers, penetration enhancers, particle stabilizers, buffers, sodium chloride, and water to form an aqueous, sterile ophthalmic suspension or solution.
- Ophthalmic solution formulations may be prepared by dissolving a compound in a physiologically acceptable isotonic aqueous buffer. Further, the ophthalmic solution may include an ophthalmologically acceptable surfactant to assist in dissolving the compound. Furthermore, the ophthalmic solution may contain an agent to increase viscosity or solubility such as hydroxypropyl ⁇ -Cyclodextrin (HPpCD), hydroxymethylcellulose, hydroxyethylcellulose,
- HPpCD hydroxypropyl ⁇ -Cyclodextrin
- hydroxypropylmethylcellulose, methylcellulose, polyvinylpyrrolidone, or the like to improve the retention of the formulation in the conjunctival sac.
- Gelling agents can also be used, including, but not limited to, gellan and xanthan gum.
- the active ingredient may be combined with a preservative in an appropriate vehicle such as mineral oil, liquid lanolin, or white petrolatum.
- Sterile ophthalmic gel formulations may be prepared by suspending the compound in a hydrophilic base prepared from the combination of, for example, carbopol-974, or the like, according to the published formulations for analogous ophthalmic preparations; preservatives and tonicity agents can be incorporated.
- Compounds in preferred embodiments are contained in a composition in amounts sufficient to prevent, reduce or treat retinal ganglion cell damage in patients experiencing elevated IOP and/or maintaining normal IOP levels in POAG or OHT patients. Such amounts are referred to herein as "an amount effective to prevent, reduce or treat retinal ganglion cell damage," or more simply “an effective amount.”
- the compounds will normally be contained in these formulations in an amount of between about 0.1% and 3.0% (w/v), or between about 0.5 to about 1.5% (w/v).
- 1 to 2 drops of these formulations would be delivered to the surface of the eye from 1 to 4 times per day, according to the discretion of a skilled clinician.
- the compounds of Formula I can also be used in combination with other oculartreatment agents, such as, but not limited to, ⁇ -blockers, prostaglandin analogs, carbonic anhydrase inhibitors, a 2 adrenergic agonists, miotics, and neuroprotectants adenosine A 3 antagonists, adenosine A 2A agonists and combinations thereof.
- oculartreatment agents such as, but not limited to, ⁇ -blockers, prostaglandin analogs, carbonic anhydrase inhibitors, a 2 adrenergic agonists, miotics, and neuroprotectants adenosine A 3 antagonists, adenosine A 2A agonists and combinations thereof.
- proparacaine hydrochloride single drop
- a single drop of treatment (0.2% brimonidine tartrate, Compound A 2.5% ophthalmic suspension, or Compound A vehicle) was applied to the cornea, with a second drop administered 5 minutes after the first.
- the anterior chamber of the right eye was cannulated with a 30G needle connected to an elevated reservoir of sterile Hanks balanced salt solution with a fluid level 59 inches above eye level (equivalent to a hydrostatic pressure of 110 mmHg) behind a closed stopcock.
- the stopcock was opened, and the rise in pressure was directly visualized by inflation of the globe. Animals that suffered an injury to the iris or cornea (beyond the single entry point) were excluded from the study. Sixty minutes after inducing the rise in pressure, the ischemic injury was terminated by closing the stopcock and removing the cannula. The needle entry wound was sealed with veterinary tissue adhesive and the animals allowed to recover from anesthesia before being returned to housing.
- ischemic injury animals were sacrificed by C0 2 inhalation and globes dissected with the proximal optic nerves intact.
- the anterior chamber was removed, and the eyes were fixed for 30 minutes in 4% paraformaldehyde/PBS, followed by overnight incubation in 30% sucrose.
- Fixed eyes were trimmed of additional connective tissue and cut along a sagittal plane to generate a flat face for embedding, and the exposed retinal edge was attached to the sclera with tissue adhesive.
- the eye was infiltrated with increasing strengths of JB-4 embedding medium (glycol methacrylate; GMA), followed by embedding in JB-4 at 4° C for sagittal sectioning.
- Optic nerves were co-embedded with their paired retina for transverse sectioning. Embedded tissues were sectioned through or near the optic nerve head at a thickness of 2 ⁇ , and collected on slides for further analysis.
- Images were captured using cellSens Dimension software on an Olympus BX61 upright microscope with a motorized stage, using a 20X objective and a resolution of 169 nm/pixel, and stitched together using the multiple image alignment procedure of the software.
- GCL ganglion cells
- IPL inner plexiform layer
- INL inner nuclear layer
- OPL outer plexiform layer
- ONL outer nuclear layer
- total retinal thickness TR distance from retinal surface above nerve fiber layer to edge of ONL
- Photoreceptor outer segments beyond the ONL were not included in measurement because of possible damage from postmortem artifactual retinal detachment.
- Distances and counts in ischemic eyes were normalized to the mean of the naive contralateral eye and compared by Student' s t-test.
- Optic nerves were stained with toluidine blue as above, and imaged with a 60x oil objective at a resolution of 56 nm/pixel and stitched together using the multiple image alignment capabilities of the software. Five regions (100 ⁇ 2 each) were selected from unoriented optic nerves - one in each of four arbitrary quadrants plus one centrally located region - and optic nerves stained pink by toluidine blue were counted.
- optic nerve sections were measured across perpendicular axes (longest and shortest), and density scaled to assume circularity along the short axis.
- Total optic nerve counts were estimated by using the scaled density and circular area predicted by the short axis, which is numerically equivalent to using unsealed density and area of an oval defined by the two axes.
- the thickness of the retinal layers from eyes subjected to a period of high IOP were assessed by histological analysis.
- the thickness of the same retinal layers were compared between eyes subjected to high IOP and the same retinal layer from the healthy contralateral eye of the same animal, which did not receive an ischemic insult with high IOP.
- the degree of retinal thinning is more pronounced in the inner retina (including the GCL where retinal ganglion cells are found, and the IPL) in eyes subjected to high IOP, and this thinning its blocked by treatment with either brimonidine or Compound A.
- Compound A is greater relative to the vehicle, and slightly greater than the effects of brimonidine.
- 100% of the RGC were protected by Compound A subjected to ischemic insult as compared to around 80% of the RGC protected by brimonidine.
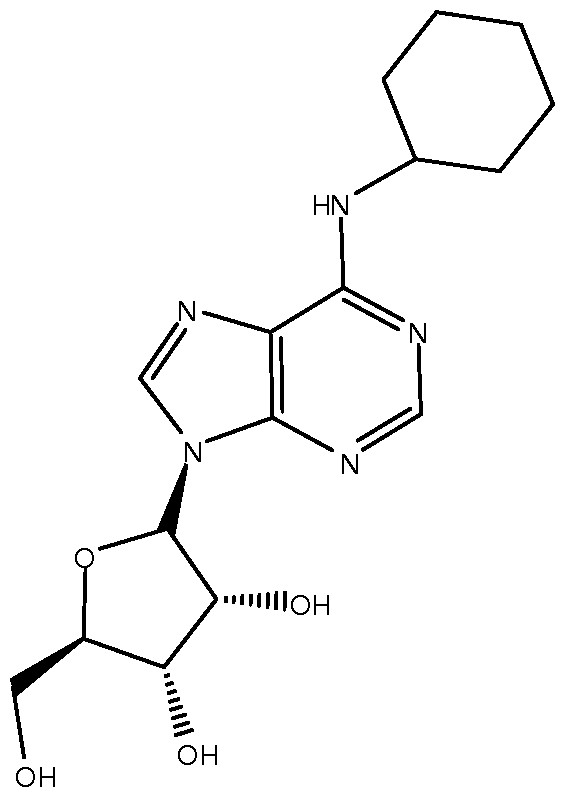
- EXAMPLE 2 Compound Synthesis 2', 3'-Isopropylidene-N 6 -cyclohexyladenosine: A solution of 6- chloroadenosine (2.58 g) and cyclohexylamine (5 g) in ethanol (20 ml) was heated at reflux for 6 hours then cooled to room temperature. The reaction mixture was concentrated in vacuo and the resultant residue was diluted with water (50 ml) and ethyl acetate (300 ml). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 x 50 ml).
- N 6 -cyclohexyladenosine as a white solid (2.600 g).
- N 6 - Cyclohexyladenosine (2.6 g) was diluted with acetone (30 ml) and to the resultant solution was added 2, 2-dimethoxypropane (12 ml), followed by D-camphorsulphonic acid (3.01 g) and the mixture was allowed to stir at room temperature for 18 hours.
- the reaction mixture was concentrated in vacuo and the resultant residue was diluted with ethyl acetate (150 ml), then neutralized to pH 8.0 using saturated aqueous NaHC0 3 .
- the organic layer was separated, dried over sodium sulfate, concentrated in vacuo.
- the residue was purified twice on the silica gel column using MeOH- CH 2 CI 2 (4:96) as an eluent to provide 2', 3'-isopropylidene-N 6 -cyclohexyladenosine (3.16 g).
- N 6 -Cyclohexyladenosine-5'-0-nitrate (Compound E): Acetic anhydride (6 ml) was slowly added to a stirred solution of nitric acid (2 g, 63%) at -25° C (CCI 4 - CO 2 bath used for cooling) and the reaction temperature maintained at -7.5 to 0° C for additional 1 hr. A solution of 2', 3'-isopropylidene-N 6 -cyclohexyladenosine (1.0 g) in acetic anhydride (3 mL) was added slowly.
- Isopropylidene-N 6 -exo-norbornyladenosine was prepared following the procedure of 2', 3'-isopropylidene-N 6 -cyclohexyladenosine and used for the subsequent reaction.
- Acetic anhydride (6 ml) was slowly added to a stirred solution of nitric acid (2 g, 63%) at -25° C (CCI 4 -CO 2 bath used for cooling) and the reaction temperature maintained at -7.5 to 0° C for additional 1 hr.
- a solution of 2', 3'-isopropylidene-N 6 - exo-norbornyladenosine (1.2 g) in acetic anhydride (3 mL) was added slowly.
- 2-Chloro-N 6 -cyclohexyladenosine A mixture of 2,6-dichloroadenosine (1.0 g) and cyclohexylamine (0.926 g) in ethanol (30 ml) was heated at reflux for 6 hours then cooled to room temperature. The mixture was concentrated under vacuo. The residue was purified on the silica gel column using MeOH - CH 2 CI 2 (1:6 to 1:5). The combined fractions were concentrated and dried under vacuum to provide 2-chloro- N 6 -cyclohexyladenosine as a white solid (2.600 g).
- 2-Chloro-2', 3'-isopropylidene-N 6 -cyclohexyladenosine 2-Chloro-N 6 - cyclohexyladenosine (0.5 g) was diluted with acetone (30 ml) and to the mixture was added 2,2-dimethoxypropane (2.04 g), followed by D-camphorsulphonic acid (CSA, 0.272 g). The resultant reaction mixture was allowed to stir at room temperature for 2 hours. Additional CSA (0.2 g) was added and stirred for 2 hours. The mixture was concentrated in vacuo and the resultant residue was diluted with ethyl acetate, then neutralized to pH 8.0 using concentrated aqueous NaHC0 3 .
- CSA 2,2-dimethoxypropane
- 2-Chloro-N 6 -cyclohexyladenosine-5'-0-nitrate (Compound H): Following the nitration and the TFA water deprotection reactions, 2-chloro-N 6 - cyclohexyladenosine-5'-0-nitrate was prepared from 2-chloro-2', 3'-isopropylidene- N 6 -cyclohexyladenosine.
- N 6 -Cyclopentyladenosine (Compound I): A solution of 6-chloroadenosine (43 g) and cyclopentylamine (5 eq.) in ethanol (50 eq.) was heated at reflux for 3 hours then cooled to room temperature. The resultant reaction mixture was concentrated in vacuo and the resultant residue was diluted with water (400 ml) and ethyl acetate (400 ml). The organic layer was separated and the aqueous layer was extracted into ethyl acetate (2 x 400 ml).
- N 6 -cyclopentyladeno sine (43 g) was diluted with acetone (75 eq.) and to the resultant solution was added 2,2- dimethoxypropane (5 eq.), followed by D-camphorsulphonic acid (1 eq) and the resultant reaction was allowed to stir at room temperature for 3 hours.
- the resultant reaction mixture was concentrated in vacuo and the resultant residue was diluted with ethyl acetate, then neutralized to pH 7.0 using concentrated aqueous NaHC0 3 .
- Acetic anhydride (22 eq) was slowly added to a stirred solution of nitric acid (5 eq., 63%) at -10° C (acetonitrile-C0 2 bath used for cooling) over a period of 4 hours with the reaction temperature maintained at -5 to 5° C during the addition.
- the resultant solution was cooled to -20° C and a solution of 2',3'-isopropylidene-N 6 -cyclopentyladenosine (18.250 gm, 0.048 mol) in acetic anhydride (37 mL, 8 eq.) was added slowly.
- the resultant reaction was allowed to stir at -15 to -5° C for 1 hour and the resultant reaction mixture was slowly poured slowly into an ice-cold solution of aqueous NaHCC"3 (168 gm in 800 mL water) and ethyl acetate (350 mL) and the resultant solution was allowed to stir for 5 minutes.
- Compound A 2',3'-isopropylidene-N 6 -cyclopentyladenosine-5'-nitrate (4.8 g) was diluted with a mixture of TFA (20 mL) and water (5 mL) and the resultant reaction was allowed to stir for 30 minutes at room temperature. The resultant reaction mixture was concentrated in vacuo and the resultant residue was diluted with water (10 mL) and concentrated in vacuo.
- reaction mixture was then cooled to -30 °C and then a solution of 2',3'-Isopropylidene-2- chloro-N 6 -cyclopentyladenosine (655 mg, 0.0016 mol, as prepared in the previous step) in acetic anhydride (8.0 mL) was added slowly. When addition was complete, the resulting reaction was allowed to warm to -5 °C and monitored using TLC
- CHO cells stably transfected with human adenosine A ⁇ receptor are grown and maintained in Dulbecco's Modified Eagles Medium with nutrient mixture F12 (DMEM/F12) without nucleosides, containing 10% fetal calf serum, penicillin (100 U/mL), streptomycin (100 ⁇ g/mL), L-glutamine (2 mM) and Geneticin (G-418, 0.2 mg/mL; A 2B , 0.5 mg/mL) at 37°C in 5% C0 2 /95% air. Cells are then split 2 or 3 times weekly at a ratio of between 1:5 and 1:20.
- DEM/F12 Dulbecco's Modified Eagles Medium with nutrient mixture F12
- Membranes for radioligand binding experiments are prepared from fresh or frozen cells as described in Klotz et al., Naunyn-Schmiedeberg's Arch. Pharmacol., 357: 1-9 (1998). The cell suspension is then homogenized in ice-cold hypotonic buffer (5 mM Tris/HCl, 2 mM EDTA, pH 7.4) and the homogenate is spun for 10 minutes (4°C) at 1,000 g.
- the membranes are then sedimented from the supernatant for 30 minutes at 100,000 g and resuspended in 50 mM Tris/HCl buffer pH 7.4 (for A 3 adenosine receptors: 50 mM Tris/HCl, 10 mM MgCl 2 , 1 mM EDTA, pH 8.25), frozen in liquid nitrogen at a protein concentration of 1-3 mg/mL and stored at -80°C.
- the affinities of selected Purine Compounds for the adenosine Ai receptor can be determined by measuring the displacement of specific [ 3 H] 2-chloro-N 6 - cyclopentyl adenosine binding in CHO cells stably transfected with human recombinant Ai adenosine receptor expressed as Ki (nM).
- Dissociation constants of unlabeled compounds are determined in competition experiments in 96- well microplates using the Ai selective agonist 2- chloro-N 6 -[ 3 H]cyclopentyladenosine ([ 3 H]CCPA, InM) for the characterization of Ai receptor binding.
- Nonspecific binding is determined in the presence of 100 ⁇ R- PIA and 1 mM theophylline, respectively.
- Binding data can be calculated by non-linear curve fitting using the program SCTFIT (De Lean et al. Mol. Pharm. 1982, 21 :5-16).
- the Ai and A 3 receptor-mediated inhibition of f or skolin- stimulated adenylyl cyclase activity was tested in membranes prepared from CHO cells stably transfected with the human Ai and A adenosine receptors.
- the A 2a and A 2 receptor- mediated stimulation of basal cyclase activity was tested in membranes prepared from CHO cells stably transfected with the human A 2a and A 3 adenosine receptors.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Ophthalmology & Optometry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Saccharide Compounds (AREA)
Abstract
Description





















Claims





















































Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020157023932A KR20150138182A (en) | 2013-03-15 | 2014-03-14 | A method of providing Ocular neuroprotection |
| EA201591434A EA201591434A1 (en) | 2013-03-15 | 2014-03-14 | METHOD OF PROVIDING THE EYE NEUROPROTECTION |
| MX2015013240A MX2015013240A (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection. |
| EP14769358.4A EP2968388A4 (en) | 2013-03-15 | 2014-03-14 | METHOD FOR PROVIDING OCULAR NEUROPROTECTURE |
| AU2014239232A AU2014239232A1 (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection |
| CN201480015327.4A CN105188713A (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection |
| CA2902888A CA2902888A1 (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection |
| JP2016502511A JP2016513707A (en) | 2013-03-15 | 2014-03-14 | How to give optic nerve protection |
| NZ630760A NZ630760A (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection |
| BR112015022044A BR112015022044A2 (en) | 2013-03-15 | 2014-03-14 | a method of providing ocular neutroprotection |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361798412P | 2013-03-15 | 2013-03-15 | |
| US61/798,412 | 2013-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014152733A1 true WO2014152733A1 (en) | 2014-09-25 |
Family
ID=51529965
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/027672 Ceased WO2014152733A1 (en) | 2013-03-15 | 2014-03-14 | A method of providing ocular neuroprotection |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20140275128A1 (en) |
| EP (1) | EP2968388A4 (en) |
| JP (1) | JP2016513707A (en) |
| KR (1) | KR20150138182A (en) |
| CN (1) | CN105188713A (en) |
| AU (1) | AU2014239232A1 (en) |
| BR (1) | BR112015022044A2 (en) |
| CA (1) | CA2902888A1 (en) |
| EA (1) | EA201591434A1 (en) |
| MX (1) | MX2015013240A (en) |
| NZ (1) | NZ630760A (en) |
| WO (1) | WO2014152733A1 (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA201290958A1 (en) | 2010-03-26 | 2013-04-30 | Инотек Фармасьютикалз Корпорейшн | A METHOD FOR REDUCING INTRAGLASE PRESSURE IN PEOPLE USING N6-CYCLOPENTILADAINOSINE (CPA), CPA DERIVATIVES OR THEIR CLEARANCE |
| BR112014018413A8 (en) | 2012-01-26 | 2017-07-11 | Inotek Pharmaceuticals Corp | METHYL NITRATE ANHYDRO POLYMORPHS [(2R,3S,4R,5R)-5-(6-(CYCLOPENTYLAMINO)-9H-PURIN-9-IL)-3,4-DIHYDROXITETRAHYDROFURAN-2-IL)] AND PREPARATION PROCESSES THE SAME |
| CA2903114A1 (en) | 2013-03-15 | 2014-09-25 | Inotek Pharmaceuticals Corporation | Ophthalmic formulations |
| JP2018501219A (en) * | 2014-12-03 | 2018-01-18 | イノテック ファーマシューティカルズ コーポレイション | How to prevent, reduce or treat macular degeneration |
| DE102017008072A1 (en) * | 2017-08-28 | 2019-02-28 | Henkel Ag & Co. Kgaa | New anionic surfactants and detergents and cleaners containing them |
| KR102007640B1 (en) | 2017-11-29 | 2019-08-07 | 퓨쳐메디신 주식회사 | The pharmaceutical compositions for the prevention and treatment of retinal diseases or optic nerve diseases containing adenosine derivatives |
| WO2025090106A1 (en) * | 2023-10-23 | 2025-05-01 | Ophthalmic Therapeutic Innovation Llc | Compositions and methods for treating neural degeneration in glaucoma and related conditions |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090220516A1 (en) * | 2005-06-22 | 2009-09-03 | Alan Laties | Neuroprotection of retinal ganglion cells |
| US20110123622A1 (en) * | 2009-10-26 | 2011-05-26 | Inotek Pharmaceuticals Corporation | Ophthalmic formulation and method of manufacture thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2001278708A1 (en) * | 2000-09-08 | 2002-03-22 | Toa Eiyo Ltd. | Adenosine derivatives and use thereof |
| WO2007111954A2 (en) * | 2006-03-23 | 2007-10-04 | Inotek Phamaceuticals Corporation | Purine compounds and methods of use thereof |
| SG182285A1 (en) * | 2010-01-11 | 2012-08-30 | Inotek Pharmaceuticals Corp | Combination, kit and method of reducing intraocular pressure |
| ES2613255T3 (en) * | 2010-03-19 | 2017-05-23 | Inotek Pharmaceuticals Corporation | Combined compositions of A1 adenosine agonists and non-selective β-adrenergic receptor blockers to reduce intraocular pressure |
-
2014
- 2014-03-14 US US14/211,567 patent/US20140275128A1/en not_active Abandoned
- 2014-03-14 BR BR112015022044A patent/BR112015022044A2/en active Search and Examination
- 2014-03-14 JP JP2016502511A patent/JP2016513707A/en not_active Withdrawn
- 2014-03-14 MX MX2015013240A patent/MX2015013240A/en unknown
- 2014-03-14 CN CN201480015327.4A patent/CN105188713A/en active Pending
- 2014-03-14 EP EP14769358.4A patent/EP2968388A4/en not_active Withdrawn
- 2014-03-14 KR KR1020157023932A patent/KR20150138182A/en not_active Withdrawn
- 2014-03-14 EA EA201591434A patent/EA201591434A1/en unknown
- 2014-03-14 WO PCT/US2014/027672 patent/WO2014152733A1/en not_active Ceased
- 2014-03-14 NZ NZ630760A patent/NZ630760A/en not_active IP Right Cessation
- 2014-03-14 CA CA2902888A patent/CA2902888A1/en not_active Abandoned
- 2014-03-14 AU AU2014239232A patent/AU2014239232A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090220516A1 (en) * | 2005-06-22 | 2009-09-03 | Alan Laties | Neuroprotection of retinal ganglion cells |
| US20110123622A1 (en) * | 2009-10-26 | 2011-05-26 | Inotek Pharmaceuticals Corporation | Ophthalmic formulation and method of manufacture thereof |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2968388A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| NZ630760A (en) | 2017-09-29 |
| CN105188713A (en) | 2015-12-23 |
| CA2902888A1 (en) | 2014-09-25 |
| AU2014239232A1 (en) | 2015-10-01 |
| EP2968388A1 (en) | 2016-01-20 |
| BR112015022044A2 (en) | 2017-07-18 |
| US20140275128A1 (en) | 2014-09-18 |
| JP2016513707A (en) | 2016-05-16 |
| EP2968388A4 (en) | 2016-10-19 |
| MX2015013240A (en) | 2016-04-07 |
| EA201591434A1 (en) | 2016-03-31 |
| KR20150138182A (en) | 2015-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5778663B2 (en) | Pharmaceutical composition for reducing human intraocular pressure | |
| US20140275128A1 (en) | Method of providing ocular neuroprotection | |
| DK2523669T3 (en) | COMBINATION, KIT AND PROCEDURE TO REDUCE INTRAOCULAR PRESSURE | |
| US20160158267A1 (en) | Methods of preventing, reducing or treating macular degeneration | |
| HK1167599B (en) | Method of reducing intraocular pressure in humans | |
| HK1171194A (en) | Combination, kit and method of reducing intraocular pressure |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480015327.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14769358 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2902888 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201591434 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20157023932 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2016502511 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2015/013240 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 241652 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2014239232 Country of ref document: AU Date of ref document: 20140314 Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014769358 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014769358 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015022044 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015022044 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150909 |