WO2014157185A1 - 改変型カルボニル還元酵素およびその遺伝子 - Google Patents
改変型カルボニル還元酵素およびその遺伝子 Download PDFInfo
- Publication number
- WO2014157185A1 WO2014157185A1 PCT/JP2014/058248 JP2014058248W WO2014157185A1 WO 2014157185 A1 WO2014157185 A1 WO 2014157185A1 JP 2014058248 W JP2014058248 W JP 2014058248W WO 2014157185 A1 WO2014157185 A1 WO 2014157185A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- seq
- methionine
- arginine
- polypeptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0006—Oxidoreductases (1.) acting on CH-OH groups as donors (1.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
- C12P7/04—Preparation of oxygen-containing organic compounds containing a hydroxy group acyclic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y101/00—Oxidoreductases acting on the CH-OH group of donors (1.1)
- C12Y101/01—Oxidoreductases acting on the CH-OH group of donors (1.1) with NAD+ or NADP+ as acceptor (1.1.1)
- C12Y101/01184—Carbonyl reductase (NADPH) (1.1.1.184)
Definitions
- the present invention relates to a modified carbonyl reductase, a gene thereof, a vector containing the gene, and a transformant transformed with the vector.
- a method of asymmetric reduction of the carbonyl group of a carbonyl compound with a microorganism or an enzyme is known.
- Asymmetric enzymes that reduce carbonyl compounds (hereinafter referred to as carbonyl reductases) are useful in the production of various optically active alcohols.
- the enzyme is deactivated by a substrate, a product, an acid or alkali used for pH adjustment, a surfactant or an organic solvent added to improve the reaction liquid property, etc. And the enzyme reaction may be inhibited. Therefore, a carbonyl reductase capable of avoiding enzyme inactivation or reaction inhibition by an organic solvent or the like leads to shortening of the reaction time and improvement of the reaction yield, and is further useful for industrial production of optically active alcohols.
- Patent Document 1 For example, attempts have been made to acquire resistance to organic solvents by random mutation introduction, and a reductase having resistance to 2-propanol and dimethyl sulfoxide has been reported (Patent Document 1, Non-Patent Document 1).
- ezetimibe and montelukast are more soluble in dimethylformamide than 2-propanol or dimethylsulfoxide. If a dimethylformamide resistant enzyme can be used for the production of such a compound, the reaction solution property can be improved, and higher productivity can be expected than when other organic solvents are used.
- an object of the present invention is to modify a wild-type enzyme whose reactivity decreases in the presence of dimethylformamide, and to improve the reactivity in the presence of an organic solvent and to improve the reactivity of the wild-type enzyme and / or It is to provide a transformant that produces the enzyme.
- the present inventors From the mutant enzyme library prepared by randomly introducing mutations into the wild-type enzyme gene, the present inventors have modified carbonyl reductase with improved reactivity in the presence of an organic solvent compared to the wild-type enzyme. As a result, the present invention has been completed.
- the present invention provides the following (a) to (c): (A) having 78% or more sequence identity with the amino acid sequence set forth in SEQ ID NO: 1 in the sequence listing; (B) reducing 2-pentanone to produce 2-pentanol, and (C) Compared with the carbonyl reductase consisting of the amino acid sequence set forth in SEQ ID NO: 1 in the Sequence Listing, the reactivity with the carbonyl compound is high in the presence of an organic solvent and / or the thermal stability is high. It relates to a polypeptide exhibiting the properties of
- the organic solvent is preferably dimethylformamide.
- amino acid sequence set forth in SEQ ID NO: 1 in the Sequence Listing; 2nd, 22nd, 25th, 39th, 42nd, 45th, 51st, 56th, 71st, 87th, 90th, 102nd, 109th, 124th, 135th, 138th, 155th 159, 175, 177, 183, 190, 195, 212, 220, 226, 228, 236, 238, 250, 254, 257, 259, 265
- an amino acid substitution is introduced into one or more amino acids selected from the 1st, 267th, 270th, 279th, 298th, 300th, 301st, and 331st amino acids.
- the amino acid substitution is the following group in the amino acid sequence set forth in SEQ ID NO: 1 in the Sequence Listing; 2nd is isoleucine, 22nd is arginine, 25th is phenylalanine, 39th is arginine, 42nd is arginine, 45th is aspartic acid, 51st is alanine, 56th is lysine, 71st is asparagine or arginine, 87th Is isoleucine, 90 is glycine, 102 is isoleucine, 109 is glycine, 124 is leucine, 135 is alanine, 138 is asparagine, 155 is leucine or arginine, 159 is phenylalanine, 175 is aspartic acid 177th phenylalanine, 183th threonine, 190th serine, 195th leucine, 212th phenylalanine, threonine or tyrosine, 220
- the amino acid substitution is the following group in the amino acid sequence set forth in SEQ ID NO: 1 in the Sequence Listing; 2nd isoleucine, 45th aspartic acid, 71st asparagine or arginine, 102th isoleucine, 124th leucine, 175th aspartic acid, 177th phenylalanine, 183th threonine, 195th leucine, 220th Valine, 226 is glycine, 236 is asparagine, 238 is isoleucine, 257 is serine, 259 is glutamic acid, 265 is lysine, 267 is proline, 270 is methionine, 300 is aspartic acid, And an amino acid substitution in which position 301 is replaced with cysteine, One or more amino acid substitutions selected from Compared with the carbonyl reductase consisting of the amino acid sequence described in SEQ ID NO: 1 in the sequence listing, it is preferable that the stability to organic solvents is improved.
- Amino acid substitutions are the following groups: 22nd arginine, 39th arginine, 51st alanine, 87th isoleucine, 90th glycine, 259th glutamic acid, and 270th amino acid substitution with methionine,
- the amino acid substitution is represented by the following (1) to (7) in the amino acid sequence set forth in SEQ ID NO: 1 in the Sequence Listing; (1) The 22nd is arginine, (2) 22nd is arginine, 87th is isoleucine, (3) 39th is arginine, (4) 39th is arginine, 51st is alanine, (5) 51st is alanine, (6) amino acid substitution in which position 87 is replaced with isoleucine, and (7) position 90 is replaced with glycine, One or more amino acid substitutions selected from are preferred.
- the present invention also relates to a polynucleotide encoding the polypeptide.
- the present invention also relates to a vector comprising the polynucleotide.
- the polypeptide having reducible coenzyme regeneration ability is preferably glucose dehydrogenase.
- the present invention also relates to a transformant obtained by transforming a host cell with the vector.
- the host cell is preferably E. coli.
- the present invention also relates to a method for producing an alcohol compound, characterized in that the polypeptide or the transformant and / or a processed product thereof is allowed to act on a carbonyl compound.
- the carbonyl compound is an asymmetric ketone and the alcohol compound is an optically active alcohol.
- the carbonyl compound is represented by the following formula (1): (Wherein R 1 and R 2 are a hydrogen atom, a halogen atom, an optionally substituted alkyl group, an optionally substituted aralkyl group, an optionally substituted aryl group, or an optionally substituted alkoxy; A group, an amino group, or a nitro group, or R 1 and R 2 may be bonded to each other to form a ring, provided that R 1 and R 2 have different structures).
- the alcohol compound is represented by the following formula (2): (Wherein R 1 and R 2 are the same as described above, * represents an asymmetric carbon), and is preferably an optically active alcohol.
- a modified carbonyl reductase whose reactivity in the presence of an organic solvent is improved as compared to the wild type and its gene, a vector containing the gene, a transformant transformed with the vector, a transformant of the transformant A method for producing a processed product can be provided.
- the polypeptide of the present invention is characterized by exhibiting the following properties (a) to (c): (A) having 78% or more sequence identity with the amino acid sequence set forth in SEQ ID NO: 1 in the sequence listing, (b) reducing 2-pentanone to produce 2-pentanol, and (c) coordination Compared with the carbonyl reductase consisting of the amino acid sequence described in SEQ ID NO: 1 in the table, the reactivity with the carbonyl compound is high and / or the thermal stability is high in the presence of an organic solvent.
- amino acids, peptides, and proteins are represented using abbreviations adopted by the IUPAC-IUB Biochemical Nomenclature Committee (CBN) shown below.
- CBN Biochemical Nomenclature Committee
- amino acid residue sequences of peptides and proteins are expressed from the N-terminus to the C-terminus from the left end to the right end.
- CBN Biochemical Nomenclature Committee
- substitution of tyrosine to aspartic acid at position 64 is expressed as “Y64D”.
- Multiple mutations are expressed by separating them with a hyphen symbol “-”. For example, “S41A-Y64D” indicates substitution of serine at position 41 with alanine and tyrosine at position 64 with aspartic acid.
- sequence identity of a polypeptide or polynucleotide is an optimal alignment of the two polypeptides or polynucleotides to be compared and the amino acid or nucleobase (eg, A, T, C, G, U, or I) Is divided by the total number of comparison bases and the result is multiplied by 100.
- amino acid or nucleobase eg, A, T, C, G, U, or I
- Sequence identity can be calculated, for example, using the following sequence analysis tools: GCG Wisconsin Package (University of Wisconsin), the ExPASy World Wide Web server for molecular biology (Swiss Bioinformatics Institute), BLAST ( US Biotechnology Information Center), GENETYX (Genetics).
- the origin of the polypeptide is not limited, but is preferably a microorganism belonging to the genus Saccharomycesaceae , more preferably a genus Vanderwaltozyma , more preferably a species belonging to Vanderwaltozyma polyspora ( Vanderwaltozyma polyspora ) Carbonyl reductase derived from microorganisms, particularly preferably Vanderwaltozyma polyspora NBRC0996.
- the microorganism can be obtained from the Biological Genetic Resource Department (NBRC: 292-0818, Kisarazu City, Chiba Prefecture).
- the wild-type enzyme gene can be prepared by amplifying the polynucleotide represented by 2.
- polypeptide of the present invention may be obtained by modifying the amino acid sequence shown in SEQ ID NO: 1.
- sequence identity between the amino acid sequence after modification and the amino acid sequence shown in SEQ ID NO: 1 is 85% or more, preferably 90% or more, more preferably 92% or more, still more preferably 95% or more, 97% or more, 98% or more, 98.5% or more, 99% or more.
- polypeptide of the present invention has the following groups in the amino acid sequence shown in SEQ ID NO: 1 in the sequence listing; the second is isoleucine, the 45th is aspartic acid, and the 71st is asparagine from the viewpoint of improving the stability to organic solvents.
- (1) to (35) In the amino acid sequence shown in SEQ ID NO: 1 in the sequence listing, the following (1) to (35); (1) 71st asparagine, 195th leucine, (2) 71st is arginine, 259th is glutamic acid, (3) 71st is arginine, 270th is methionine, (4) 71st is arginine, 300th is aspartic acid, (5) 102nd is isoleucine, 270th is methionine, (6) 177th is phenylalanine, 220th is valine, (7) Glycine at 226th, methionine at 270th, (8) 257th is serine, 259th is glutamic acid, (9) 257th is serine, 270th is methionine, (10) 259th is glutamic acid, 270th is methionine, (11) 259th is glutamic acid, 300th is aspartic acid, (12) 267th is proline,
- polypeptide of the present invention is the following group of amino acid sequences shown in SEQ ID NO: 1 in the sequence listing; from the viewpoint of improving resistance to reaction inhibition by an organic solvent; 22nd is arginine; 39th is arginine; It is preferable that one or more amino acid substitutions selected from amino acid substitutions in which the number is alanine, the number 87 is isoleucine, the number 90 is glycine, the number 259 is glutamic acid, and the number 270 is substituted with methionine are introduced.
- the enzyme of the present invention has high reactivity with carbonyl compounds even in the presence of an organic solvent.
- the presence of the organic solvent may be a state in which the enzyme-containing liquid and the organic solvent are mixed, but the enzyme-containing liquid and the organic solvent may be in a non-uniform state, which is physically stirred and mixed. But it ’s okay.
- Enzyme highly reactive in the presence of an organic solvent means that after treatment for a certain period of time in the presence of the organic solvent, compared to the wild-type enzyme described in SEQ ID NO: 1 in the sequence listing, or the presence of the organic solvent below, it shows that the reduction activity of the enzyme with respect to 2-pentanone is high.
- the enzyme has high stability to the organic solvent or high resistance to reaction inhibition by the organic solvent.
- the stability with respect to the organic solvent is improved means that the residual activity with respect to 2-pentanone or 2-hexanone after incubating the enzyme with the organic solvent is described in Example 4 or 5 below.
- the residual activity is 1% or more higher than that of the wild-type enzyme, preferably 5% or more, more preferably 10% or more, and most preferably 20% or more.
- the resistance to reaction inhibition by the organic solvent is improved
- the relative activity with respect to 2-hexanone in the presence of the organic solvent was measured by the method described in Example 31 described later.
- the relative activity is 1% or more higher than that of the wild-type enzyme, preferably 5% or more, more preferably 7% or more, still more preferably 10% or more, and most preferably 20% or more.
- the stability of the enzyme with respect to the organic solvent can be evaluated by, for example, the following method.
- a buffer solution containing an organic solvent at an arbitrary concentration (eg, 0.5% to 50%) (preferably 0.01 to 1M phosphate buffer solution having a pH of 5 to 8) is added to the cell-free extract containing the enzyme to add any Incubate at a temperature (eg, 4-40 ° C.). If the organic solvent and buffer are not homogeneous, incubate with shaking or stirring. Samples without addition of organic solvent and treatment liquid with addition of organic solvent were sampled 0.1 to 48 hours later, diluted with 0.1 M aqueous potassium phosphate (pH 7.0), and the diluted solution was used.
- the activity of the enzyme is measured by the following [Method for evaluating reducing ability for carbonyl compounds].
- the modified carbonyl reductase having improved stability to organic solvents has a residual activity in the wild type when evaluated as described above.
- the enzyme is 1% or more higher than that, preferably 5% or more, more preferably 10% or more, and most preferably 20% or more.
- the peptide of the present invention has the ability to reduce the carbonyl compound to be evaluated.
- the reduction ability with respect to the evaluation carbonyl compound is so high that the rate of decrease in absorbance is high.
- the reducing ability of the polypeptide can also be quantified, and the reducing activity 1U is defined as the amount of enzyme that catalyzes the consumption of 1 ⁇ mol NADPH per minute.
- the modified carbonyl reductase of the present invention can be searched for by the following method. Using the error-prone PCR method (Leung et al., Technique 1, 11-15 (1989)) or a kit based on the same principle, the nucleotide sequence shown in SEQ ID NO: 2 (wild type enzyme gene) A DNA fragment into which substitution, insertion, deletion or addition of one or more base sequences is introduced can be obtained.
- the wild-type enzyme gene can be used as a template, and the 240th T can be replaced with C by a conventional method to destroy the NdeI recognition site without changing the amino acid sequence of the wild-type enzyme (SEQ ID NO: 3 in the sequence listing).
- Primer 1 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the Sequence Listing) and Primer 2: 5′-ATACGCGTCACTACTACTTTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the Sequence Listing)
- a random mutation is introduced into the entire length of the gene encoding the wild type enzyme, an Nde I recognition site is added to the start codon, and a Sal I recognition site is added immediately after the stop codon.
- a kind of double-stranded DNA (mutant enzyme gene) can be obtained.
- the amplified fragment was digested with Nde I and Sal I, the 185th T of plasmid pUCN18 (manufactured pUC18 (Takara Bio Inc. by PCR) was modified to A destroy Nde I site, further 471-472 th By inserting GC into TG and inserting the Nde I site downstream of the lac promoter of the plasmid into which the Nde I site was newly introduced) and Sal I recognition site, and using this plasmid, Escherichia coli ( Escherichia coli) ) Transform HB101 strain (hereinafter, E. coli HB101). The transformed E.
- mutant enzyme gene obtained by the above method in place of the wild type gene, a mutant enzyme library into which mutations are further introduced can be prepared by the same operation.
- the modified carbonyl reductase of the present invention can be selected from the above library. Although it does not specifically limit as a selection method, Preferably it is the following method. [Selection Method 1 by Enzyme Plate Evaluation with Improved Stability to Organic Solvents] Each recombinant bacterium of the mutant enzyme library and a recombinant bacterium producing a wild-type enzyme (for example, E. coli HB101 (pNKP) shown in Reference Example 3) are mixed with an appropriate medium (for example, 2 ⁇ containing 200 ⁇ g / ml ampicillin). YT medium (tryptone 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0) is inoculated, and cultured with shaking at 37 ° C.
- an appropriate medium for example, 2 ⁇ containing 200 ⁇ g / ml ampicillin.
- YT medium tryptone 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0
- each treated cell-free extract is dispensed into a 96-well plate (manufactured by AGC Techno Glass), and NADPH (preferably 1.5 mM) and a carbonyl compound (preferably 10 mM)
- NADPH preferably 1.5 mM
- a carbonyl compound preferably 10 mM
- a phosphate buffer solution pH 5 to 7 containing 2,3-butanedione
- the NADPH fluorescence is observed over time with a UV sample photographing device FAS-III (manufactured by Toyobo Co., Ltd.).
- NADPH fluorescence remains in the enzyme solution in which the reaction does not proceed, but the cell-free extract in which the reaction has progressed disappears as NADPH decreases.
- Enzymes that disappeared in a short time compared with the wild-type enzyme as a control were selected as enzymes with improved stability against chloride.
- a plasmid is extracted from the culture medium of the selected enzyme, and the base of the modified carbonyl reductase gene using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems Japan) The sequence can be determined and the mutation site can be identified.
- recombinant bacteria are placed in an appropriate liquid medium (for example, 2 ⁇ YT medium (200% of ampicillin / 200 ⁇ g / ml) (tripton 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0). Inoculate and incubate with shaking for 20 hours at 37 ° C. For each of the obtained cultures, the cells are collected by centrifugation and suspended in a buffer (preferably 50 mM 3- (N-morpholino) propanesulfonic acid (MOPS) buffer). This is crushed using a UH-50 type ultrasonic homogenizer (manufactured by SMT), and the cell residue is removed by centrifugation to obtain a cell-free extract.
- a buffer preferably 50 mM 3- (N-morpholino) propanesulfonic acid (MOPS) buffer
- a buffer preferably 50 mM 3- (N-morpholino) propanesulphonic acid (MOPS) buffer
- dimethylformamide so that the final concentration of dimethylformamide is preferably 0.1 to 60%
- the mixture is heated (preferably at 40 to 80 ° C. for 0.1 minute to 24 hours) and then cooled on ice.
- a buffer preferably containing NADP + (preferably 1 mM), nitroblue tetrazolium (preferably 200 ⁇ M), 1-methyl-5-methylphenazinium methylsulfate (preferably 10 ⁇ M), 2-propanol (eg 0.1 to 50%) Is mixed with 50 mM of 3- (N-morpholino) propanesulfonic acid (MOPS) buffer, transferred to a 96-well plate (manufactured by AGC Techno Glass), and observed.
- the dyed product can be selected as a modified carbonyl reductase with improved stability to organic solvents.
- a plasmid is extracted from the selected recombinant bacterial culture, and the mutant RKP gene base using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems Japan) The sequence can be determined and the mutation site can be identified.
- NADPH fluorescence is observed with a Benchmark Plus microplate spectrophotometer (manufactured by BIO-RAD).
- the NADPH fluorescence remains in the enzyme solution in which the reaction does not proceed, but the cell-free extract in which the reaction has progressed disappears as NADPH decreases.
- NADPH is consumed as a result of reduction of the carbonyl compound, and the one that disappears in a short time is selected as a recombinant carbonyl reductase having improved resistance to reaction inhibition against organic solvents.
- Plasmids are extracted from the selected recombinant bacterial culture, and using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl genetic analyzer (Applied Biosystems Japan) mutant RKP gene base The sequence can be determined and the mutation site can be identified.
- Modifications that combine the properties of multiple mutations by combining multiple mutations that can increase reactivity with carbonyl compounds and / or increase thermal stability in the presence of organic solvents by site-directed mutagenesis Type carbonyl reductase can be produced.
- the polynucleotide of the present invention is not particularly limited as long as it encodes the polypeptide of the present invention.
- the polynucleotide comprising the base sequence encoding the wild-type enzyme shown in SEQ ID NO: 2 of the sequence listing, or this And polypeptides obtained by modifying the above.
- a method for modifying a wild-type enzyme gene a known method described in, for example, Current Protocols in Molecular Biology (Frederic M. Ausubel, Green Publishing Associates and Wiley-Interscience (1989)) can be used. That is, one or a plurality of bases of the wild-type enzyme gene (for example, 40, preferably 20, more preferably 10, more preferably 5, 4, 3, or 2 bases) By substituting, adding, inserting or deleting, a polynucleotide in which the amino acid sequence of the wild-type enzyme is modified can be produced.
- a mutagenesis method using PCR such as the error-prone PCR method (Leung et al., Technique 1, 11-15 (1989)), or a commercially available kit Diversity PCR Random Mutagenesis Kit (Clontech), TransformerMutageMeter.
- Kit Clontech
- EXOIII / Mung Bean Selection Kit Stratagene
- QuickChange Site Directed Mutagenesis Kit (Stratagene)
- site-directed mutagenesis methods include, for example, Olft Landt et al. (Gene, 96, 125-128 (1990)), Smith et al. (Genetic Engineering, 3, 1 , Setlow, J. Plenum Press), Vlasuk et al. (Experimental Manipulation of Gene Expression, Inouye, M. Academic Press), Hos. N. Examples include the method of Hunt et al. (Gene, 77, 51 (1989)) and the use of a commercial kit of QuikChange II Kit (manufactured by Stratagene).
- the target polynucleotide of the present invention when introducing a mutation at two positions, can be obtained by repeating the method according to the above method twice. Even when a plurality of other positions are substituted with other amino acids, the target polynucleotide of the present invention can be obtained by this method.
- the polynucleotide encoding the polypeptide of the present invention includes, for example, a carbonyl reductase having the activity of reducing 2-pentanone to produce 2-pentanol and comprising the amino acid sequence shown in SEQ ID NO: 1 in the Sequence Listing.
- a polynucleotide that encodes a polypeptide that is highly reactive to a carbonyl compound in the presence of an organic solvent and that includes a complementary nucleotide sequence to the polynucleotide consisting of the nucleotide sequence shown in SEQ ID NO: 2 in the Sequence Listing; Polynucleotides that hybridize under stringent conditions are preferred.
- a polynucleotide that hybridizes under stringent conditions with a polynucleotide having a base sequence complementary to the polynucleotide shown in SEQ ID NO: 2 in the sequence listing is shown in SEQ ID NO: 2 in the sequence listing.
- Hybridization can be performed according to the method described in Molecular Cloning 2nd Edition (Joseph Sambrook, Cold Spring Harbor Press (1989)).
- the “polynucleotide hybridizing under stringent conditions” refers to, for example, 65 in the presence of 0.7 to 1.0 M sodium chloride using a filter on which colony or plaque-derived polynucleotide is immobilized. After hybridization at 0 ° C., the filter is washed under conditions of 65 ° C. using a 3-fold concentration SSC solution (the composition of the 1-fold concentration SSC solution is 150 mM sodium chloride and 15 mM sodium citrate). Can be obtained.
- an SSC solution having a 1-fold concentration at 65 ° C. More preferably, it is washed with an SSC solution having a 0.7-fold concentration at 65 ° C., and further preferably, 0.5-fold, 0.45-fold,. It is a polynucleotide that can be obtained by washing with a 25-fold, 0.2-fold, or 0.15-fold concentrated SSC solution.
- hybridization conditions have been described as described above, the conditions are not particularly limited.
- a plurality of factors such as temperature and salt concentration can be considered as factors affecting the stringency of hybridization, and those skilled in the art can realize optimum stringency by appropriately selecting these factors.
- the sequence identity with the polynucleotide shown in SEQ ID NO: 2 is preferably 78% or more, more preferably 84% or more, still more preferably 87% or more, Preferred examples include 89%, 90%, 94%, 95%, 97% or more of the polynucleotide, and the encoded polypeptide is included in the polynucleotide as long as it has the properties of the polypeptide of the present invention.
- a polypeptide expression vector can be prepared by inserting a polynucleotide encoding the polypeptide of the present invention into an expression vector.
- the expression vector used above is not particularly limited as long as it can express the polypeptide encoded by the polynucleotide in a suitable host organism.
- examples of such vectors include plasmid vectors, phage vectors, cosmid vectors, and shuttle vectors that can exchange genes with other host strains can also be used.
- such a vector in the case of Escherichia coli, such a vector usually contains regulatory elements such as lacUV5 promoter, trp promoter, trc promoter, tac promoter, lpp promoter, tufB promoter, recA promoter, pL promoter, etc., and operates with the DNA of the present invention. It can be suitably used as an expression vector comprising expression units that are ligated together. Examples thereof include pUCN18 (see Reference Example 2), pSTV28 (manufactured by Takara Bio Inc.), pUCNT (International Publication No. 94/03613) and the like.
- regulatory element refers to a base sequence having a functional promoter and any associated transcription element (eg, enhancer, CCAAT box, TATA box, SPI site, etc.).
- operably linked means that a gene is operably linked to various regulatory elements such as promoters and enhancers that regulate the expression of the gene. It is well known to those skilled in the art that the type and kind of the control factor can vary depending on the host.
- the vector may further contain a polynucleotide encoding a polypeptide having the ability to regenerate reduced coenzyme.
- polypeptide having reducible coenzyme regeneration ability include glucose dehydrogenase.
- a transformant can be obtained by transforming a host cell with a vector.
- a transformant obtained by introducing a polynucleotide encoding the polypeptide of the present invention into a chromosome can also be mentioned as a transformant.
- Host cells for transformation with vectors are cells that can be transformed with a polypeptide expression vector containing a polynucleotide encoding each polypeptide and can express the polypeptide encoded by the introduced polynucleotide. There is no particular restriction.
- the available microbial as the host cell e.g., Escherichia (Escherichia) genus Bacillus (Bacillus) genus Pseudomonas (Pseudomonas) genus Serratia (Serratia) genus Brevibacterium (Brevibacterium) genus Corynebacterium Rii um (Corynebacterium ) genus Streptococcus (Streptococcus) genus, and Lactobacillus (Lactobacillus) bacteria have been developed host-vector systems such as the genus Rhodococcus (Rhodococcus) genus and Streptomyces (Streptomyces) genus Streptomyces which have been developed in the host-vector systems such as bacteria, Saccharomyces (Saccharomyces) genus, Kuraiberoma Isis (Kluyveromyces) sp., Schizosacc
- the vector of the present invention can be introduced into a host microorganism by a known method.
- the plasmid of the present invention in which a polynucleotide encoding a modified carbonyl reductase is introduced into the above-described expression vector pUCN18 as a polypeptide expression vector (pNKPm01--shown in Examples 2-3, 6-15, 17-27, 30).
- E. A transformant for example, E. coli HB101 (pNKPm50) shown in Example 27
- the vector is introduced into a host cell by operating according to the protocol using an E. coli HB101 competent cell (manufactured by Takara Bio Inc.) or the like. ) Is obtained.
- a transformant in which both the polypeptide of the present invention and the polypeptide having the ability to regenerate reduced coenzyme described below are expressed in the same cell can also be bred.
- the polynucleotide encoding the polypeptide of the present invention and the polynucleotide encoding the polypeptide having the ability to regenerate reduced coenzyme are incorporated into the same vector and introduced into a host cell. It can also be obtained by incorporating two types of DNA into two different vectors of different incompatibility groups and introducing them into the same host cell.
- the transformant thus obtained includes, for example, a recombinant vector in which a nucleotide encoding a modified carbonyl reductase is introduced into the expression vector pUCN18 (for example, pNKPm01 shown in Example 2) and a reduced complement.
- An alcohol compound can be produced by allowing the polypeptide or transformant of the present invention and / or a processed product thereof to act on a carbonyl compound.
- the carbonyl compound used as a substrate is not particularly limited.
- asymmetric ketones are preferable because their reduction products become useful optically active alcohols.
- Examples of the compound having a carbonyl group include the following formula (1): (Wherein R 1 and R 2 are a hydrogen atom, a halogen atom, an optionally substituted alkyl group, an optionally substituted aralkyl group, an optionally substituted aryl group, or an optionally substituted alkoxy; A group, an amino group, or a nitro group, or R 1 and R 2 may be bonded to each other to form a ring, provided that R 1 and R 2 have different structures). Examples of the product include the following formula (2): (Wherein R 1 and R 2 are the same as described above, * represents an asymmetric carbon).
- R 1 and R 2 are each an alkyl group having 1 to 14 carbon atoms, an aryl group having 6 to 14 carbon atoms, a heteroaryl group having 4 to 14 carbon atoms, an alkoxy group having 1 to 5 carbon atoms, or an alkyl group having 2 to 5 carbon atoms.
- a hydrogen atom, a halogen atom, a hydroxyl group, an amino group, or a nitro group is preferable.
- alcohol compounds produced using the polypeptide of the present invention include 2-pentanol, 2-hexanol, 2,3-butanediol, 3-hydroxy-2-butanone, 1-phenylethyl alcohol, [3- [ (5R)-(4-Fluoro-phenyl) -5-hydroxypentanoyl]-(4S) -phenyl-1,3-oxazolidine-2-one, 1-phenyl-1-propanol, 1-phenyl-1-butanol 1-phenyl-1-pentanol, 1-phenyl-1-hexanol, 1-phenyl-2-butanol, 4-phenyl-2-butanol, 2,5-hexanediol, 5-hydroxy-2-hexanone, 2 , 3-hexanediol, 2-hydroxyhexane-3-one, 3-hydroxy-2-hexanone, 3,4-hexane Ol, 4-hydroxy-3-hexanone, and 1-phenoxy-2
- An appropriate solvent for example, 100 mM phosphate buffer (pH 6.5), etc.
- a substrate that is a carbonyl compound for example, 2-pentanone or acetophenone
- NADPH or oxidized nicotinamide / adenine dinucleotide phosphate hereinafter, referred to as “NPTA”.
- a coenzyme such as NADP + ), a culture of the transformant and / or a processed product thereof, and the like are added, and the mixture is reacted with stirring under pH adjustment.
- the treated product is, for example, a crude extract, cultured cells, freeze-dried organisms, acetone-dried organisms, disrupted cells, or immobilized products thereof, and the enzyme catalytic activity of the polypeptide remains. Means what you are doing.
- the reaction temperature is preferably 5 to 80 ° C., more preferably 10 to 60 ° C., and further preferably 20 to 40 ° C.
- the pH of the reaction solution is preferably 3 to 10, more preferably 4 to 9, and still more preferably 5 to 8.
- the reaction can be carried out batchwise or continuously.
- the reaction substrate is 0.01 to 100% (w / v), preferably 0.1 to 70% (w / v), more preferably 0.5 to 50% (w / v). It can be added at a charge concentration. Further, a substrate may be newly added during the reaction.
- the treated product of the transformant means, for example, a cell-free extract, cultured cells, freeze-dried cells, acetone-dried cells, or a ground product thereof, or a mixture thereof. Furthermore, they can be used by immobilizing them with a known means in the form of polypeptides or cells. .
- NADPH or NADH is required as a coenzyme.
- the reduction reaction can be carried out even if NADPH or NADH is added to the reaction system in a necessary amount.
- a coenzyme regeneration system comprising an enzyme having the ability to convert the oxidized coenzyme (NADP + or NAD + ) into reduced NADPH or NADH (hereinafter referred to as reduced coenzyme regeneration ability) and its substrate, By performing the reaction in combination with the polypeptide of the present invention, the amount of expensive coenzyme used can be greatly reduced.
- Examples of the enzyme having the ability to regenerate reduced coenzyme include hydrogenase, formate dehydrogenase, carbonyl reductase, glucose-6-phosphate dehydrogenase, and glucose dehydrogenase. Preferably, glucose dehydrogenase is used.
- Such a reaction can also be carried out by adding a coenzyme regeneration system into the asymmetric reduction reaction system, but it encodes a polynucleotide encoding the enzyme of the present invention and a polypeptide having reduced coenzyme regeneration ability.
- the transformant transformed with both of the polynucleotides to be used is used as a catalyst, the reaction can be carried out efficiently without separately preparing an enzyme capable of regenerating reduced coenzyme and adding it to the reaction system. Can do.
- Such a transformant can be obtained by the method for producing a transformant described above.
- the method for recovering alcohol from the reaction solution after the reaction is not particularly limited, but after separating the cells or the like directly from the reaction solution or if necessary, ethyl acetate, toluene, t-butyl methyl ether, hexane, methylene chloride, etc.
- the product can be extracted with a solvent, and dehydrated, and then purified by distillation, recrystallization, silica gel column chromatography, or the like. By this method, a high-purity alcohol compound can be easily obtained.
- double-stranded DNA (RKP gene) in which an Nde I recognition site is added to the start codon portion of the gene consisting of the base sequence shown in SEQ ID NO: 2 in the sequence listing and a Sal I recognition site is added immediately after the termination codon. )was gotten.
- PCR reaction was further performed, and the 240th T was changed to C by a conventional method.
- the double-stranded DNA (Nde I site consisting of the nucleotide sequence shown in SEQ ID NO: 3 of Sequence Listing NdeI recognition site in the gene is destroyed RKP gene) was obtained.
- PCR was performed using PrimeSTAR HS DNA polymerase (manufactured by Takara Bio Inc.) as a DNA polymerase, and the reaction conditions were in accordance with the instruction manual.
- Example 2 The RKP gene was disrupted NdeI site obtained in constructing Example 1 recombinant vector pNKP was digested with Nde I and Sal I, 185 of plasmid pUCN18 (manufactured pUC18 (Takara Bio Inc. by PCR) The Nde I recognition site downstream of the lac promoter of the plasmid in which the Nde I site was newly introduced by changing the T to A to destroy the Nde I site and further changing the 471 to 472st GC to TG And a Sal I recognition site to construct a recombinant vector pNKP.
- the acetophenone reduction activity of each recombinant organism is shown below.
- E. coli HB101 pUCN18
- the acetophenone reduction activity was 0.1 U / mg or less. Meanwhile, E was expressed RKP.
- the acetophenone reduction activity of E. coli HB101 (pNKP) was 5 U / mg.
- the recombinant organism obtained in Reference Example 3 had a reducing activity against acetophenone, and RKP expression was observed.
- Reaction inhibition of wild type enzyme RKP with organic solvent A cell-free extract of wild type enzyme was obtained in the same manner as in Reference Example 4. To this cell-free extract, dimethylformamide having a final concentration of 30% was added, adjusted to pH 6.5 with sulfuric acid or sodium hydroxide, and incubated at 30 ° C. for 2 hours. In addition, a cell-free extract to which nothing was added was also incubated as a control. After 2 hours, each cell-free extract was diluted. The 2-hexanone reducing activity of these cell-free extracts was measured.
- the reaction was performed by adding 30% dimethylformamide, 10 mM 2-hexanone, 0.25 mM coenzyme NADPH, and a cell-free extract to a 100 mM phosphate buffer (pH 6.5).
- the consumption rate of NADPH was determined from the decrease rate of NADPH fluorescence, and the 2-hexanone reduction activity was calculated.
- the relative activity at the time of solvent addition was calculated from the following formula, and this value was used as an index of stability for various compounds.
- the activity (relative activity) of the wild-type enzyme under the solvent addition condition was 24% under the solvent non-addition condition.
- Relative activity (%) [Enzyme activity (with solvent)] ⁇ [Enzyme activity (without solvent)] ⁇ 100
- Example 1 Preparation of Mutant Enzyme Library 1 Using the plasmid pNKP containing the RKP gene prepared in Reference Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTATAGGG-3 ′ (SEQ ID NO: 4 in the sequence listing) and primer 2: 5′-ATACGCGTCGCACTTACTTGTTCTTGAACCTCCA-3 ′ (sequence listing) Using the error-prone PCR method (Leung et al. Technique 1, 11-15 (1989)), a DNA amplified fragment in which a random mutation was introduced into the entire RKP gene was obtained.
- Example 2 Selection 1 of modified carbonyl reductase A modified carbonyl reductase having improved stability against organic solvents was selected from the mutant enzyme library 1.
- E. coli HB101 (pNKP) (control) was cultured in the same manner as in Reference Example 4, respectively.
- a phosphate buffer solution (pH 6.5) containing dimethylformamide having a final concentration of 10 to 30% was added to 200 ⁇ l of each cell-free extract and incubated at 30 ° C. for 2 hours (dimethylformamide treatment).
- 50 ⁇ l of each cell-free extract treated with dimethylformamide was dispensed into a 96-well plate (manufactured by AGC Techno Glass), containing 50 ⁇ l of phosphate buffer (pH 6.5) containing 6 mM NADPH and 133 mM 2,3-butanedione.
- 100 ⁇ l of phosphate buffer (pH 6.5) was added and reacted at 30 ° C.
- a plasmid is extracted from the culture solution of the selected enzyme, and the base sequence of the mutant RKP gene using the Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems Japan) And the mutation site was identified.
- Table 2 shows the modified carbonyl reductase having improved stability with respect to the organic solvent.
- Example 3 Selection 2 of modified carbonyl reductase A modified carbonyl reductase having improved stability against organic solvents was selected from the mutant enzyme library 1.
- E. coli HB101 (pNKP) (control) was inoculated on LB plate medium containing 100 ⁇ g / mL ampicillin. The obtained colony was transferred to a nylon membrane (Biodyne A, 0.45 ⁇ m) that had been heated at 40 ° C., and the nylon membrane was then added with 50 mM 3- (N-morpholino) propanesulfonic acid (containing 30% dimethylformamide).
- MOPS metal-organic chemical vapor deposition
- the nylon membrane was then placed in 50 mM MOPS buffer containing 1 mM NADP + , 200 ⁇ M nitroblue tetrazolium, 10 ⁇ M 1-methyl-5-methylphenylsulfurate and 10% (v / v) 2-propanol at room temperature for 30 minutes. Soaked. Thereafter, the nylon membrane was washed with distilled water, and the stained colony was selected as a recombinant carbonyl reductase candidate having improved stability to dimethylformamide.
- the candidate strain was inoculated into 5 ml of 2 ⁇ YT medium (tripton 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0) containing 200 ⁇ g / ml ampicillin and cultured for 20 hours. About each obtained culture solution, the microbial cell was collected by centrifugation and suspended in 100 mM phosphate buffer (pH 6.5) of 1/6 amount of the culture solution. This was crushed using a UH-50 ultrasonic homogenizer (manufactured by SMT), and then the cell residue was removed by centrifugation to obtain a cell-free extract.
- 2 ⁇ YT medium tripton 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0
- Dimethylformamide having a final concentration of 20, 23, 26, and 30% and 50 mM MOPS buffer (pH 7.0) were added to 20 ⁇ l of the cell-free extract so that the total liquid volume became 40 ⁇ l, and the mixture was heated at 40 ° C. for 30 minutes. After cooling on ice for 1 minute, 200 ⁇ M of 50 mM MOPS buffer containing 1 mM NADP + , 200 ⁇ M nitroblue tetrazolium, 10 ⁇ M 1-methyl-5-methylphenylsulfurylate and 10% (v / v) 2-propanol was added. . The reaction solution was transferred to a 96-well plate (manufactured by AGC Techno Glass) and observed for 1 hour.
- the stained product was selected as a recombinant carbonyl reductase having improved stability to dimethylformamide.
- a plasmid is extracted from the selected recombinant bacterial culture, and the base of the mutant RKP gene using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems Japan) The sequence was determined and the mutation site was identified.
- Table 3 shows the modified carbonyl reductase having improved stability with respect to the organic solvent.
- Enzymes with improved stability against 29 organic solvents shown in Table 3 were obtained. Six of these were the same mutant enzymes as the enzyme obtained in Example 2.
- Relative activity (%) [enzyme activity at 3 hours (solvent added)] ⁇ [enzyme activity at 3 hours (no solvent added)] ⁇ 100
- Table 4 shows the relative activities of the wild-type enzyme and the modified carbonyl reductase evaluated in the presence of 40% dimethylformamide.
- the modified carbonyl reductase shown in Table 4 was more stable against organic solvents than the wild type enzyme.
- Example 5 Evaluation 2 of modified carbonyl reductase Recombinant bacteria of the modified carbonyl reductase obtained in Example 3 and E. coli prepared in Reference Example 3.
- E. coli HB101 (pNKP) (control) was cultured in the same manner as in Reference Example 4, respectively. About each obtained culture solution, the microbial cell was collected by centrifugation, and it suspended in the 100 mM phosphate buffer (pH 6.5) of the volume equal to 1/5 with a culture solution. This was crushed using a UH-50 ultrasonic homogenizer (manufactured by SMT), and then the cell residue was removed by centrifugation to obtain a cell-free extract.
- the modified carbonyl reductase shown in Table 5 was more stable against organic solvents than the wild type enzyme.
- Example 6 Production of Multiple Mutation Modified Carbonyl Reductase 1 Using the plasmid pNKPm02 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 3: 5′-GTTAATTTTCATTAGCGCGATTTTTAATTACATG-3 ′ (SEQ ID NO: 6 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of T257S in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of H71R and K259E in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm30. Using this pNKPm30, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase H71R-K259E E. coli HB101 (pNKPm30) was obtained.
- Example 7 Production of Multiple Mutation Modified Carbonyl Reductase 2 Using the plasmid pNKPm02 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 5: 5′-GATCACACCTTTCACCCGCTTAACTCATCATTATG-3 ′ (SEQ ID NO: 8 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of T257S in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of T257S and K259E in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm31. Using this pNKPm31, E. E. coli HB101 competent cell (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase T257S-K259E E. coli HB101 (pNKPm31) was obtained.
- Example 8 Production of Multiple Mutation Modified Carbonyl Reductase 3 Using the plasmid pNKPm02 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 7: 5′-CTCGTTATGACACAGATCTCCTTGAGGTACTC-3 ′ (SEQ ID NO: 10 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of K259E or G300D in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of K259E and G300D in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm32. Using this pNKPm32, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase K259E-G300D E. coli HB101 (pNKPm32) was obtained.
- Example 9 Production of multiple mutation-modified carbonyl reductase 4 Using the plasmid pNKPm04 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 9: 5′-GTTAATTTTCATTAGCGCGATTTTTAATTACATG-3 ′ (SEQ ID NO: 12 in the sequence listing) PCR was carried out to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of H71R or K270M in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- Example 10 Production of Multiple Mutation Modified Carbonyl Reductase 5 Using the plasmid pNKPm04 obtained in Example 2 as a template, Primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the Sequence Listing), Primer 11: 5′-GGATACCTTTAGTACCAATAACAGCTGGGATATAAG-3 ′ (SEQ ID NO: 14 in the Sequence Listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an N102I amino acid substitution in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of N102I and K270M in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm34.
- E. E. coli HB101 competent cells manufactured by Takara Bio Inc.
- pNKPm34 E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase N102I-K270M E. coli HB101 (pNKPm34) was obtained.
- Example 11 Production of Multiple Mutation Modified Carbonyl Reductase 6
- primer 1 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing)
- primer 13 5′-TTTTCAATTTTACTACTGCCTGTTGGAGCAACATAGAG-3 ′ (SEQ ID NO: 16 in the sequence listing)
- PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of E226G in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- primer 14 5′-GCTAGTTTGCTCCCAACAGCGCGTGAAATTGATAAA-3 ′
- primer 2 5′-ATACGCGTCGAACTTACACTGTTTCTTGAACCTTCA-3 ′
- double-stranded DNA encoding a C-terminal polypeptide having an NE226G amino acid substitution in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of E226G and K270M in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm35. Using this pNKPm35, E. E. coli HB101 competent cell (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase E226G-K270M E. coli HB101 (pNKPm35) was obtained.
- Example 12 Production of Multiple Mutation Modified Carbonyl Reductase 7 Using the plasmid pNKPm04 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 15: 5′-GATCACACTTTTACCGCTTAACTCATCATTATG-3 ′ (SEQ ID NO: 18 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of T257S in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of T257S and K270M in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm36. Using this pNKPm36, E. E. coli HB101 competent cell (manufactured by Takara Bio Inc.) to produce a modified carbonyl reductase T257S-K270M E. coli HB101 (pNKPm36) was obtained.
- Example 13 Production of Multiple Mutation Modified Carbonyl Reductase 8 Using the plasmid pNKPm04 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 17: 5′-GACAAGATCAACCTTTCACCAGTTTACACTCAT-3 ′ (SEQ ID NO: 20 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of K259E in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of K259E and K270M in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm37. Using this pNKPm37, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase K259E-K270M E. coli HB101 (pNKPm37) was obtained.
- Example 14 Production of Multiple Mutation Modified Carbonyl Reductase 9 Using the plasmid pNKPm04 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 19: 5′-TTTGCATAGTAGGAACGGAGCATTTGACAAG-3 ′ (SEQ ID NO: 22 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of S267P in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- PCR was performed using primer 20: 5′-CTTGTCCAAATGCTCCCGTTCACTATGCAA-3 ′ (SEQ ID NO: 23 in the sequence listing) and primer 2: 5′-ATACGCGTCCGACTTACACTTTGTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing). Then, double-stranded DNA encoding a C-terminal polypeptide having S267P and K270M amino acid substitutions in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of S267P and K270M in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm38. Using this pNKPm38, E. E. coli HB101 competent cell (manufactured by Takara Bio Inc.) to produce a modified carbonyl reductase S267P-K270M E. coli HB101 (pNKPm38) was obtained.
- Example 15 Production of Multiple Mutation Modified Carbonyl Reductase 10 Using the plasmid pNKPm04 obtained in Example 2 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 21: 5′-CTCGTTATGACACAGATCTCCTTGAGGTACTC-3 ′ (SEQ ID NO: 24 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of K270M or G300D in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- PCR was performed using primer 22: 5′-GAGTTACCTCAAGGAGATCGTGTCCATAAACGAG-3 ′ (SEQ ID NO: 25 in the sequence listing) and primer 2: 5′-ATACGCGTCGAACTTACTTATTGTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing). Then, double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of G300D in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and a polypeptide having amino acid substitutions of K270M and G300D in the amino acid sequence shown in SEQ ID NO: 1 is obtained.
- a double-stranded DNA encoding was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm39. Using this pNKPm39, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase K270M-G300D E. coli HB101 (pNKPm39) was obtained.
- Example 16 Evaluation of Multiple Mutation Modified Carbonyl Reductase 1
- Recombinant bacteria of the multiple mutation-modified carbonyl reductase obtained in Examples 6 to 15 and E. coli prepared in Reference Example 3 were used.
- E. coli HB101 (pNKP) (control) was cultured in the same manner as in Reference Example 4, respectively.
- the stability of multiple mutation-modified carbonyl reductase against dimethylformamide was evaluated.
- Table 6 shows the relative activities of the wild-type enzyme and the modified carbonyl reductase evaluated in the presence of 40% dimethylformamide.
- the modified carbonyl reductase shown in Table 6 was more stable against organic solvents than the wild type enzyme.
- Example 17 Production of Multiple Mutation Modified Carbonyl Reductase 11 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 23: 5′-CCAGTAGCCACCTGTAACTAAAACAATCAT-3 ′ (SEQ ID NO: 26 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an S2I amino acid substitution in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- PCR was performed using plasmid pNKPm37 as a template, primer 24: 5′-ATGATTGTTTTAGTTTACGGGTCTACTGG-3 ′ (SEQ ID NO: 27 in the sequence listing), primer 2: 5′-ATACGCGTCACTACTACTTTGTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing). Then, double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of S2I, K259E, and K270M in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and the amino acid sequence shown in SEQ ID NO: 1 has S2I, K259E, and K270M amino acid substitutions.
- Double-stranded DNA encoding the peptide was obtained.
- This was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm40.
- E. E. coli HB101 competent cells manufactured by Takara Bio Inc.
- coli HB101 (pNKPm40) was obtained.
- Example 18 Production of Multiple Mutation Modified Carbonyl Reductase 12 Using the plasmid pNKPm37 obtained in Example 7 as a template, Primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the Sequence Listing), Primer 25: 5′-GGCAGGCAATTGAAGAAAGTCAACAAATTTTTCTCAC-3 ′ (SEQ ID NO: 28 in the Sequence Listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of I124L in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- primer 26 5′-GTGAAGAAATTTTGTCTGAACTTCTTCAATTGCTGCC-3 ′ (SEQ ID NO: 29 in the sequence listing)
- primer 2 5′-ATACGCGTCGCACTTACACTGTTTCTTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing)
- double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of I124L, K259E, K270M in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and polyamino acids having amino acid substitutions of I124L, K259E, and K270M among the amino acid sequences shown in SEQ ID NO: 1.
- Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm41.
- E. E. coli HB101 competent cells manufactured by Takara Bio Inc.
- to produce modified carbonyl reductase I124L-K259E-K270M E. coli HB101 (pNKPm41) was obtained.
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and polymorphisms having amino acid substitutions of L177F, K259E, K270M among the amino acid sequences shown in SEQ ID NO: Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm42. Using this pNKPm42, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase L177F-K259E-K270M E. coli HB101 (pNKPm42) was obtained.
- Example 20 Production of Multiple Mutation Modified Carbonyl Reductase 14 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 29: 5′-GGACCAAAGACCCAGAACTGGGTTGATCG-3 ′ (SEQ ID NO: 32 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of F195L in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- PCR was performed using primer 30: 5′-CGATCAACCCAGTTCTGGTCTTTGGTCC-3 ′ (SEQ ID NO: 33 in the sequence listing) and primer 2: 5′-ATACGCGTCACTACTACTTTGTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing). Then, double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of F195L, K259E, and K270M in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA). N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template.
- Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm43. Using this pNKPm43, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase F195L-K259E-K270M E. coli HB101 (pNKPm43) was obtained.
- Example 21 Production of Multiple Mutation Modified Carbonyl Reductase 15 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 31: 5′-CGTTGGAGCAAAACATCATTCCTTGATGATTTC-3 ′ (SEQ ID NO: 34 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of A220V in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm44. Using this pNKPm44, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce a modified carbonyl reductase A220V-K259E-K270M. E. coli HB101 (pNKPm44) was obtained.
- Example 22 Production of Multiple Mutation Modified Carbonyl Reductase 16 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 33: 5′-CGTACCATCAACATAGTTACAAAAACAGATTTATC-3 ′ (SEQ ID NO: 36 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of S236N in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- Example 23 Production of Multiple Mutation Modified Carbonyl Reductase 17 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 35: 5′-GCTACCATCACGTACCATCAATATACACTACAAAAAAC-3 ′ (SEQ ID NO: 38 in the sequence listing) PCR was carried out to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of V238I in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- primer 36 5′-GTTTTTGGTAGTTATATTGATGTACGTGATGTAGGC-3 ′ (SEQ ID NO: 39 in the sequence listing)
- primer 2 5′-ATACGCGTCCGACTTACTATTGTTCTTGAACCTTCA-3 ′ (SEQ ID NO: 5 using the sequence listing)
- C-terminal DNA double-stranded DNA encoding a C-terminal polypeptide having amino acid substitutions of V238I, K259E, and K270M in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template.
- Example 24 Production of Multiple Mutation Modified Carbonyl Reductase 18 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 37: 5′-GATCACACTTTTACCGCTTAACTCATCATTATG-3 ′ (SEQ ID NO: 40 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of T257S in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- primer 38 5′-CATAATGATGAGTTAAGCGGTAAAAGGTTGATC-3 ′
- primer 2 5′-ATACGCGTCACTACTACTTTGTTCTTGAACCTTCA-3 ′
- C-terminal DNA double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of T257S, K259E, or K270M in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal DNA and C-terminal DNA are mixed, PCR is performed using primer 1 and primer 2 as a template.
- Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm47. Using this pNKPm47, E. E. coli HB101 competent cell (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase T257S-K259E-K270M E. coli HB101 (pNKPm47) was obtained.
- Example 25 Production of Multiple Mutation Modified Carbonyl Reductase 19 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 39: 5′-GCATAGGTGAATGAAGCTTTTTGACAAGATCAACCTTTCACC-3 ′ (SEQ ID NO: 42 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having amino acid substitutions of K259E and N265K in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and polymorphs having amino acid substitutions of K259E, N265K, and K270M among the amino acid sequences shown in SEQ ID NO: 1 Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm48. Using this pNKPm48, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase K259E-N265K-K270M E. coli HB101 (pNKPm48) was obtained.
- Example 27 Production of Multiple Mutation Modified Carbonyl Reductase 21 Using the plasmid pNKPm37 obtained in Example 7 as a template, primer 1: 5′-GGGAATTCCATATGAGTGTTTTAGTTTACAGG-3 ′ (SEQ ID NO: 4 in the sequence listing), primer 43: 5′-CTTCTCGTTATGGACGCCACTCCCTTGAGGTACTACTCG-3 ′ (SEQ ID NO: 46 in the sequence listing) PCR was performed to obtain double-stranded DNA encoding an N-terminal polypeptide having an amino acid substitution of K259E, K270M, or R301C in the amino acid sequence shown in SEQ ID NO: 1 (N-terminal DNA).
- primer 44 5′-CGAGTTACCCTCAAGGAGGTTGCCGTCCATAACGAGAAG-3 ′ (SEQ ID NO: 47 in the sequence listing)
- primer 2 5′-ATACGCGTCCGACTTACACTGTTTCTTTGAACCTTCA-3 ′ (SEQ ID NO: 5 in the sequence listing)
- double-stranded DNA encoding a C-terminal polypeptide having an amino acid substitution of K259E, K270M, R301C in the amino acid sequence shown in SEQ ID NO: 1 was obtained (C-terminal DNA).
- N-terminal side DNA and C-terminal side DNA are mixed, PCR is performed using primer 1 and primer 2 as a template, and the amino acid sequence of K259E, K270M, R301C among the amino acid sequences shown in SEQ ID NO: 1 Double-stranded DNA encoding the peptide was obtained. It was introduced into pUCN18 in the same manner as in Reference Example 2 to prepare pNKPm50. Using this pNKPm50, E. E. coli HB101 competent cells (manufactured by Takara Bio Inc.) to produce modified carbonyl reductase K259E-K270M-R301C coli HB101 (pNKPm50) was obtained.
- Example 28 Evaluation of Multiple Mutation Modified Carbonyl Reductase 2
- E. coli HB101 pNKP
- Table 7 shows the relative activities of the wild-type enzyme and the modified carbonyl reductase evaluated in the presence of 50% dimethylformamide.
- the modified carbonyl reductase shown in Table 7 was more stable against organic solvents than the wild type enzyme.
- Example 29 Preparation of Mutant Enzyme Library 2 Using the plasmid pNKPm37 containing the mutant RKP gene of K259E-K270M mutant enzyme obtained in Example 15 as a template, a mutant enzyme library was prepared in the same manner as in Example 1, and this was designated as mutant enzyme library 2.
- modified carbonyl reductase 3 A modified carbonyl reductase having improved resistance to reaction inhibition by dimethylformamide, one of organic solvents, was selected from the mutant enzyme library 2. Recombinant bacteria of the mutant enzyme library 2 prepared in Example 29 and E. coli prepared in Reference Example 3. E. coli HB101 (pNKP) (control) was inoculated on LB plate medium containing 100 ⁇ g / mL ampicillin.
- the nylon membrane was transferred to 30-60 in 50 mM 3- (N-morpholino) propansulfuric acid (MOPS) buffer containing 40% dimethylformamide. Dipped for a minute. The nylon membrane was then placed in 50 mM MOPS buffer containing 1 mM NADP + , 200 ⁇ M nitroblue tetrazolium, 10 ⁇ M 1-methyl-5-methylphenylsulfuric acid and 5% (v / v) 2-propanol at room temperature for 30 minutes. Soaked.
- MOPS N-morpholino propansulfuric acid
- the nylon membrane was washed with distilled water, and the stained colony was selected as a recombinant carbonyl reductase candidate having improved resistance to reaction inhibition by an organic solvent.
- These candidate strains were inoculated into 5 ml of 2 ⁇ YT medium (tripton 1.6%, yeast extract 1.0%, sodium chloride 0.5%, pH 7.0) containing 200 ⁇ g / ml ampicillin and cultured for 20 hours. . The obtained culture broth was crushed and centrifuged, and the precipitate was removed to obtain a cell-free extract.
- Dispense 200 ⁇ l of cell-free extract into a 96-well plate (manufactured by AGC Techno Glass), add 50 ⁇ l of 0.1 M phosphate buffer (pH 6.5) containing 0.625 mM NADPH and 10 mM 2,3-butanedione, and mix did.
- the NADPH fluorescence was observed with a Benchmark Plus microplate spectrophotometer (manufactured by BIO-RAD) over time. Those having lost NADPH due to the reduction of 2,3-butanedione and having no fluorescence were selected as recombinant carbonyl reductase recombinant bacteria having further improved resistance to reaction inhibition by organic solvents.
- a plasmid is extracted from the selected recombinant bacterial culture, and the base of the mutant RKP gene using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems Japan) and Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems Japan) The sequence was determined and the mutation site was identified.
- Table 8 shows the modified carbonyl enzymes with improved resistance to reaction inhibition by the organic solvent.
- Example 31 Evaluation 3 of modified carbonyl reductase Recombinant bacteria of the modified carbonyl reductase obtained in Example 30 and the E. coli prepared in Reference Example 3.
- E. coli HB101 (pNKP) (control) was cultured in the same manner as in Reference Example 4, respectively.
- the microbial cell was collected by centrifugation and suspended in 100 mM phosphate buffer (pH 6.5) of 1/5 amount of the culture solution. This was crushed using a UH-50 ultrasonic homogenizer (manufactured by SMT), and then the cell residue was removed by centrifugation to obtain a cell-free extract.
- Enzymes with improved resistance to reaction inhibition by the three types of organic solvents shown in Table 9 were obtained.
- the modified carbonyl reductase shown in the table has improved resistance to reaction inhibition by dimethylformamide, which is one of organic solvents, compared to the wild-type enzyme.
- Example 32 Production of 3-[(5R)-(4-fluoro-phenyl) -5-hydroxypentanoyl]-(4S) -phenyl-1,3-oxazolidine-2-one Obtained in Reference Example 4
- 12.5 U of glucose dehydrogenase (trade name: GLUCDH “Amano” II, manufactured by Amano Enzyme), 80 mg of glucose, NADP + 0.6 mg, dimethylformamide or 300 ⁇ l of 0.1 M phosphate buffer (pH 7), (S) -1- (4
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
(a)配列表の配列番号1に記載のアミノ酸配列と78%以上の配列同一性を有し、
(b)2-ペンタノンを還元して2-ペンタノールを生成し、かつ、
(c)配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤存在下でカルボニル化合物に対する反応性が高い、および/または熱安定性が高い、
の性質を示すポリペプチドに関する。
2番目、22番目、25番目、39番目、42番目、45番目、51番目、56番目、71番目、87番目、90番目、102番目、109番目、124番目、135番目、138番目、155番目、159番目、175番目、177番目、183番目、190番目、195番目、212番目、220番目、226番目、228番目、236番目、238番目、250番目、254番目、257番目、259番目、265番目、267番目、270番目、279番目、298番目、300番目、301番目、および331番目から選択される1つ以上のアミノ酸に、アミノ酸置換が導入されていることが好ましい。
2番目がイソロイシン、22番目がアルギニン、25番目がフェニルアラニン、39番目がアルギニン、42番目がアルギニン、45番目がアスパラギン酸、51番目がアラニン、56番目がリジン、71番目がアスパラギンもしくはアルギニン、87番目がイソロイシン、90番目がグリシン、102番目がイソロイシン、109番目がグリシン、124番目がロイシン、135番目がアラニン、138番目がアスパラギン、155番目がロイシンもしくはアルギニン、159番目がフェニルアラニン、175番目がアスパラギン酸、177番目がフェニルアラニン、183番目がスレオニン、190番目がセリン、195番目がロイシン、212番目がフェニルアラニン、スレオニンもしくはチロシン、220番目がバリン、226番目がグリシン、228番目がバリン、236番目がアスパラギン、238番目がイソロイシン、250番目がプロリン、254番目がアスパラギン、257番目がセリン、259番目がグルタミン酸、265番目がリジン、267番目がプロリン、270番目がメチオニン、279番目がアルギニン、298番目がプロリン、300番目がアスパラギン酸、301番目がシステイン、および、331番目がフェニルアラニンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であることが好ましい。
2番目がイソロイシン、45番目がアスパラギン酸、71番目がアスパラギンもしくはアルギニン、102番目がイソロイシン、124番目がロイシン、175番目がアスパラギン酸、177番目がフェニルアラニン、183番目がスレオニン、195番目がロイシン、220番目がバリン、226番目がグリシン、236番目がアスパラギン、238番目がイソロイシン、257番目がセリン、259番目がグルタミン酸、265番目がリジン、267番目がプロリン、270番目がメチオニン、300番目がアスパラギン酸、および、301番目がシステインに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であり、
配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤に対する安定性が向上していることが好ましい。
(1)71番目がアスパラギン、195番目がロイシン、
(2)71番目がアルギニン、259番目がグルタミン酸、
(3)71番目がアルギニン、270番目がメチオニン、
(4)71番目がアルギニン、300番目がアスパラギン酸、
(5)102番目がイソロイシン、270番目がメチオニン、
(6)177番目がフェニルアラニン、220番目がバリン、
(7)226番目がグリシン、270番目がメチオニン、
(8)257番目がセリン、259番目がグルタミン酸、
(9)257番目がセリン、270番目がメチオニン、
(10)259番目がグルタミン酸、270番目がメチオニン、
(11)259番目がグルタミン酸、300番目がアスパラギン酸、
(12)267番目がプロリン、270番目がメチオニン、
(13)270番目がメチオニン、300番目がアスパラギン酸、
(14)2番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(15)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(16)102番目がイソロイシン、226番目がグリシン、267番目がプロリン、
(17)124番目がロイシン、259番目がグルタミン酸、270番目がメチオニン、
(18)177番目がフェニルアラニン、259番目がグルタミン酸、270番目がメチオニン、
(19)220番目がバリン、259番目がグルタミン酸、270番目がメチオニン、
(20)236番目がアスパラギン、259番目がグルタミン酸、270番目がメチオニン、
(21)238番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(22)257番目がセリン、259番目がグルタミン酸、270番目がメチオニン、
(23)257番目がセリン、259番目がグルタミン酸、300番目がアスパラギン酸、
(24)259番目がグルタミン酸、265番目がリジン、270番目がメチオニン、
(25)259番目がグルタミン酸、270番目がメチオニン、300番目がアスパラギン酸、
(26)259番目がグルタミン酸、270番目がメチオニン、301番目がシステイン、
(27)2番目がイソロイシン、238番目がイソロイシン、
(28)71番目がアスパラギン、195番目がロイシン、
(29)109番目がグリシン、331番目がフェニルアラニン、
(30)124番目がロイシン、236番目がアスパラギン、
(31)159番目がフェニルアラニン、259番目がグルタミン酸、
(32)42番目がアルギニン、155番目がアルギニン、279番目がアルギニン、
(33)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(34)155番目がロイシン、250番目がプロリン、298番目がプロリン、および
(35)56番目がリジン、138番目がアスパラギン、190番目がセリン、254番目がアスパラギンに置換されるアミノ酸置換、
から選択されるアミノ酸置換が導入されていることが好ましい。
22番目がアルギニン、39番目がアルギニン、51番目がアラニン、87番目がイソロイシン、90番目がグリシン、259番目がグルタミン酸、および、270番目がメチオニンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であり、
配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤による反応阻害に対する抵抗性が向上していることが好ましい。
(1)22番目がアルギニン、
(2)22番目がアルギニン、87番目がイソロイシン、
(3)39番目がアルギニン、
(4)39番目がアルギニン、51番目がアラニン、
(5)51番目がアラニン、
(6)87番目がイソロイシン、および
(7)90番目がグリシンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であることが好ましい。

前記アルコール化合物が下記式(2):

(a)配列表の配列番号1に記載のアミノ酸配列と78%以上の配列同一性を有し、(b)2-ペンタノンを還元して2-ペンタノールを生成し、かつ、(c)配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤存在下でカルボニル化合物に対する反応性が高い、および/または熱安定性が高い。
なお、本明細書において、アミノ酸、ペプチド、タンパク質は下記に示すIUPAC-IUB生化学命名委員会(CBN)で採用された略号を用いて表される。また、特に明示しない限り、ペプチドおよびタンパク質のアミノ酸残基の配列は、左端から右端にかけてN末端からC末端となるように表される。また、参照を容易にするため、一般的に用いられている下記の命名法を適用する。1つは、“もとのアミノ酸;位置;置換したアミノ酸”と記述する方法であり、例えば、位置64におけるチロシンのアスパラギン酸への置換は「Y64D」と表される。多重変異については、ハイフン記号“-”により分けることで表記する。例えば、「S41A-Y64D」とは、位置41のセリンをアラニンへ、かつ、位置64のチロシンをアスパラギン酸へ置換することを示す。
A=Ala=アラニン、C=Cys=システイン、
D=Asp=アスパラギン酸、E=Glu=グルタミン酸、
F=Phe=フェニルアラニン、G=Gly=グリシン、
H=His=ヒスチジン、I=Ile=イソロイシン、
K=Lys=リシン、L=Leu=ロイシン、
M=Met=メチオニン、N=Asn=アスパラギン、
P=Pro=プロリン、Q=Gln=グルタミン、
R=Arg=アルギニン、S=Ser=セリン、
T=Thr=スレオニン、V=Val=バリン、
W=Trp=トリプトファン、Y=Tyr=チロシン
ポリペプチドやポリヌクレオチドの「配列同一性」とは、対比される2つのポリペプチドまたはポリヌクレオチドを最適に整列させ、アミノ酸または核酸塩基(例えば、A、T、C、G、U、またはI)が両方の配列で一致した位置の数を比較塩基総数で除し、そして、この結果に100を乗じた数値で表される。
(1)71番目がアスパラギン、195番目がロイシン、
(2)71番目がアルギニン、259番目がグルタミン酸、
(3)71番目がアルギニン、270番目がメチオニン、
(4)71番目がアルギニン、300番目がアスパラギン酸、
(5)102番目がイソロイシン、270番目がメチオニン、
(6)177番目がフェニルアラニン、220番目がバリン、
(7)226番目がグリシン、270番目がメチオニン、
(8)257番目がセリン、259番目がグルタミン酸、
(9)257番目がセリン、270番目がメチオニン、
(10)259番目がグルタミン酸、270番目がメチオニン、
(11)259番目がグルタミン酸、300番目がアスパラギン酸、
(12)267番目がプロリン、270番目がメチオニン、
(13)270番目がメチオニン、300番目がアスパラギン酸、
(14)2番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(15)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(16)102番目がイソロイシン、226番目がグリシン、267番目がプロリン、
(17)124番目がロイシン、259番目がグルタミン酸、270番目がメチオニン、
(18)177番目がフェニルアラニン、259番目がグルタミン酸、270番目がメチオニン、
(19)220番目がバリン、259番目がグルタミン酸、270番目がメチオニン、
(20)236番目がアスパラギン、259番目がグルタミン酸、270番目がメチオニン、
(21)238番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(22)257番目がセリン、259番目がグルタミン酸、270番目がメチオニン、
(23)257番目がセリン、259番目がグルタミン酸、300番目がアスパラギン酸、
(24)259番目がグルタミン酸、265番目がリジン、270番目がメチオニン、
(25)259番目がグルタミン酸、270番目がメチオニン、300番目がアスパラギン酸、
(26)259番目がグルタミン酸、270番目がメチオニン、301番目がシステイン、
(27)2番目がイソロイシン、238番目がイソロイシン、
(28)71番目がアスパラギン、195番目がロイシン、
(29)109番目がグリシン、331番目がフェニルアラニン、
(30)124番目がロイシン、236番目がアスパラギン、
(31)159番目がフェニルアラニン、259番目がグルタミン酸、
(32)42番目がアルギニン、155番目がアルギニン、279番目がアルギニン、
(33)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(34)155番目がロイシン、250番目がプロリン、298番目がプロリン、および
(35)56番目がリジン、138番目がアスパラギン、190番目がセリン、254番目がアスパラギンに置換されるアミノ酸置換
から選択されるアミノ酸置換が導入されているポリペプチドであることがより好ましい。
(1)22番目がアルギニン、
(2)22番目がアルギニン、87番目がイソロイシン、
(3)39番目がアルギニン、
(4)39番目がアルギニン、51番目がアラニン、
(5)51番目がアラニン、
(6)87番目がイソロイシン、および
(7)90番目がグリシンに置換されるアミノ酸置換
のいずれかで示されるアミノ酸置換が導入されていることがより好ましい。
[有機溶剤に対する酵素の安定性の評価方法]
酵素を含む無細胞抽出液に任意の濃度(例えば0.5%~50%)の有機溶剤を含む緩衝液(好ましくはpH5~8の0.01~1Mリン酸緩衝液)を加えて任意の温度(例えば4~40℃)でインキュベートする。有機溶剤と緩衝液が不均一な場合は振とう、または攪拌しながらインキュベートする。有機溶剤を非添加のサンプルと有機溶剤を添加した処理液を0.1~48時間後にサンプリングし、それを0.1Mのリン酸カリウム水溶液(pH7.0)で希釈し、その希釈液を用いて下記の [カルボニル化合物に対する還元能力の評価方法]で酵素の活性を測定する。相対活性は下記式で算出することができる。
相対活性(%)=[溶剤添加条件での酵素活性]÷[溶剤非添加条件での酵素活性]×100
100mMリン酸カリウム緩衝液(pH6.5)にNADPHもしくは還元型ニコチンアミド・アデニンジヌクレオチド(以下、NADH)0.25mM、還元活性を評価したいカルボニル化合物(例えば2-ペンタノン、2-ヘキサノン、2,3-ブタンジオン)1~50mMおよび本発明のポリペプチドを含む反応液を30℃で反応させ、NADPHもしくはNADH量の減少に伴う波長340nmの吸光度の減少を測定することにより、還元反応の進行を容易に評価することができる。吸光度が減少した場合、本発明のペプチドは評価対象のカルボニル化合物を還元する能力を有する、と判断することができる。なお、吸光度の減少速度が速いほど、評価対象のカルボニル化合物に対する還元能力が高いといえる。また、ポリペプチドの還元能力は数値化することも可能であり、還元活性1Uは1分間に1μmolのNADPHの消費を触媒する酵素量とした。
[有機溶剤による反応阻害に対する抵抗性の評価方法1]
100mMリン酸カリウム緩衝液(pH6.5)にNADPHもしくは還元型ニコチンアミド・アデニンジヌクレオチド(以下、NADH)3mM、還元活性を評価したいカルボニル化合物(例えば2-ペンタノン、2-ヘキサノン、2,3-ブタンジオン)1%、有機溶剤0.01~60%(v/v)または有機溶剤非添加、および本発明のポリペプチドを含む反応液を30℃で0.01~5時間反応させ、ガスクロマトグラフィーなどにより分析し、カルボニル化合物からアルコールへの変換率を求める。
相対活性(%)=[有機溶剤存在下での変換率]÷[有機溶剤非存在下での変換率]×100
エラープローンPCR法(Leung et al., Technique 1, 11-15(1989))、あるいは同様の原理に基づいたキットを用いて、配列表の配列番号2に示す塩基配列(野生型酵素遺伝子)に1つ以上の塩基配列の置換、挿入、欠失、付加が導入されたDNA断片を得ることができる。例えば、野生型酵素遺伝子をテンプレートにし、定法により、240番目のTをCに置換し、野生型酵素のアミノ酸配列を変えずにNdeI認識部位を破壊できる(配列表の配列番号3)。これをテンプレートにプライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、およびプライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)とDiversify PCR Random Mutagenesis Kit(Clontech社製)を用いて、野生型酵素をコードする遺伝子の全長にランダムに変異が導入され、かつ開始コドンにNdeI認識部位が付加され、終始コドンの直後にSalI認識部位が付加された複数種類の二本鎖DNA(変異型酵素遺伝子)を得ることができる。この増幅断片をNdeIおよびSalIで消化し、プラスミドpUCN18(PCR法によりpUC18(タカラバイオ社製)の185番目のTをAに改変してNdeIサイトを破壊し、更に471-472番目のGCをTGに改変することにより新たにNdeIサイトを導入したプラスミド)のlacプロモーターの下流のNdeI認識部位とSalI認識部位の間に挿入し、このプラスミドを用いてエシェリヒア・コリ(Escherichia coli)HB101株(以下、E.coli HB101)を形質転換する。形質転換した大腸菌を100μg/mLのアンピシリンを含むLBプレート培地に塗布し、シングルコロニーの大腸菌を得る。また、野生型遺伝子の代わりに前記方法で得られた変異型酵素遺伝子を用いて、同様の操作でさらに変異を導入した変異酵素ライブラリーを作製することもできる。
[有機溶剤に対する安定性が向上した酵素のプレート評価による選抜法1]
変異酵素ライブラリーの各組換え菌および野生型酵素を生産する組換え菌(例えば、参考例3に示すE.coli HB101(pNKP))を適当な培地(例えば200μg/mlのアンピシリンを含む2×YT培地(トリプトン1.6%、イーストエキス1.0%、塩化ナトリウム 0.5%、pH7.0))に接種し、37℃で24時間振盪培養する。得られた各培養液を菌体破砕処理し、遠心分離後、沈殿を除去し、無細胞抽出液を得る。各酵素を含む無細胞抽出液に適当な濃度の有機溶剤(好ましくは終濃度10~30%のジメチルホルムアミド)を含む緩衝液(好ましくはpH5~8の0.01~1Mリン酸緩衝液)を加えて適当な温度(例えば4~40℃)でインキュベートする。0.1~48時間程度インキュベートした後、処理した各無細胞抽出液を96穴プレート(AGCテクノグラス社製)に分注し、NADPH(好ましくは1.5mM)とカルボニル化合物(好ましくは10mMの2,3-ブタンジオン)を含むリン酸緩衝液(pH5~7)を加えて、10~40℃で反応する。経時的にUVサンプル撮影装置FAS-III(東洋紡績社製)でNADPHの蛍光を観察する。この時、反応が進行しない酵素液はNADPHの蛍光が残存するが、反応が進行した無細胞抽出液はNADPHの減少に伴い、蛍光がなくなる。対照である野生型酵素に比べ、短時間で蛍光がなくなった酵素を塩化物に対する安定性が向上した酵素として選抜した。選抜した酵素の培養液からプラスミドを抽出し、BigDye Terminator Cycle Sequencing Kit(アプライドバイオシステムズジャパン社製)およびApplied Biosystems 3130xlジェネティックアナライザ(アプライドバイオシステムズジャパン社製)を用いて改変型カルボニル還元酵素遺伝子の塩基配列を決定し、変異部位を特定することができる。
変異酵素ライブラリーの各組換え菌および野生型酵素を生産する菌(例えば、参考例3に示すE.coli HB101(pNKP))を適当な培地)を適当な寒天培地(例えば、100μg/mLのアンピシリンを含むLBプレート培地)に植菌し、30℃で24時間培養する。得られたコロニーをナイロン膜(BiodyneA、0.45μm)に転写した後、そのナイロン膜を有機溶剤(好ましくは40%のジメチルホルムアミド)を含むバッファー(好ましくは50mMの3-(N-morpholino)propanesulfonic acid(MOPS)バッファー中に0.1分~24時間浸漬する。このバッファーは40~80℃に加温されていることが好ましい。その後、このナイロン膜をNADP+(好ましくは1mM)、nitroblue tetrazolium(好ましくは200μM)、1-methoxy-5-methylphenazinium methylsulfate(好ましくは10μM)、2-プロパノール(例えば0.1~50%)を含むバッファー(好ましくは50mMの3-(N-morpholino)propanesulfonic acid(MOPS)バッファー)中に適当な温度(例えば4~40℃)で0.1分~24時間浸漬する。その後、ナイロン膜を蒸留水で洗浄し、4染色されたコロニーを有機溶剤に対する安定性が向上した改変型カルボニル還元酵素の組換え菌として選抜することができる。
変異酵素ライブラリーの各組換え菌および野生型酵素を生産する菌(例えば、参考例3に示すE.coli HB101(pNKP))を適当な寒天培地(例えば、100μg/mLのアンピシリンを含むLBプレート培地)に植菌し、30℃で24時間培養する。得られたコロニーをナイロン膜(BiodyneA、0.45μm)に転写した後、NADP+(好ましくは1mM)、nitroblue tetrazolium(好ましくは200μM)、1-methoxy-5-methylphenazinium methylsulfate(好ましくは10μM)、2-プロパノール(例えば0.1~50%)と有機溶剤(好ましくは0.1~80%のジメチルホルムアミド)含むバッファー(好ましくは50mMの3-(N-morpholino)propanesulfonic acid(MOPS)バッファー)中に適当な温度(例えば4~40℃)で0.1分~10時間浸漬する。その後、ナイロン膜を蒸留水で洗浄し、染色されたコロニーを有機溶剤による反応阻害に対する抵抗性が向上した改変型カルボニル還元酵素の組換え菌の候補として選抜することができる。


Molecular Cloning 2nd Edition(Joseph Sambrook,Cold Spring Harbor Laboratory Press(1989))、Current Protocols in Molecular Biology(Frederick M.Ausubel,Greene Publishing Associates and Wiley-Interscience(1989))。
ヴァンデルワルトザイマ・ポリスポラ(Vanderwaltozyma polyspora)NBRC0996株よりカルボニル化合物に対する還元活性を有するポリペプチド(以下、本ポリペプチドをRKPと呼ぶ)をコードするDNAをPCRにより取得した。
500ml容坂口フラスコに、バクトトリプトン16g、酵母エキス10g、塩化ナトリウム5g、アデカノールLG-109(日油社製)0.1g(いずれも1L当たり)の組成からなる液体培地(pH7)50mlを調製し、120℃で20分間蒸気殺菌をおこなった。この培地に、予め同培地にて前培養しておいたヴァンデルワルトザイマ・ポリスポラ(Vanderwaltozyma polyspora)NBRC0996株の培養液を5ml接種し、30℃で18時間振盪しながら培養を行った。この培養液から、Murray等の方法(Nucl.Acids Res.8,4321(1980))に記載の方法に従って染色体DNAを抽出した。
プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いて、ヴァンデルワルトザイマ・ポリスポラ(Vanderwaltozyma polyspora)NBRC0996株の染色体DNAを鋳型としてPCRを行った。
参考例1で得られたNdeI部位を破壊したRKP遺伝子をNdeIおよびSalIで消化し、プラスミドpUCN18(PCR法によりpUC18(タカラバイオ社製)の185番目のTをAに改変してNdeIサイトを破壊し、更に471-472番目のGCをTGに改変することにより新たにNdeIサイトを導入したプラスミド)のlacプロモーターの下流のNdeI認識部位とSalI認識部位の間に挿入し、組換えベクターpNKPを構築した。
参考例2で構築した組換えベクターpNKPを用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、組換え生物E.coli HB101(pNKP)を得た。また、pUCN18を用いてE.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、組換え生物E.coli HB101(pUCN18)を得た。
参考例3で得た2種類の組換え生物(E.coli HB101(pUCN18)、E.coli HB101(pNKP))を、200μg/mlのアンピシリンを含む2×YT培地(トリプトン1.6%、イーストエキス1.0%、塩化ナトリウム 0.5%、pH7.0)5mlに接種し、37℃で24時間振盪培養した。上記の培養で得られたそれぞれ培養液について、遠心分離により菌体を集め、5mlの100mMリン酸緩衝液(pH6.5)に懸濁した。これを、UH-50型超音波ホモゲナイザー(SMT社製)を用いて破砕した後、遠心分離により菌体残渣を除去し、無細胞抽出液を得た。これら無細胞抽出液のアセトフェノン還元活性を測定した。アセトフェノンに対する還元活性は、100mMリン酸緩衝液(pH6.5)に、アセトフェノン10mM、補酵素NADPH 0.25mM、および無細胞抽出液を添加して30℃で1分間反応を行い、波長340nmにおける吸光度の減少速度より算出した。この反応条件において、1分間に1μmolのNADPHをNADPに酸化する酵素活性を1Uと定義した。それぞれの組換え生物のアセトフェノン還元活性を以下に示す。E.coli HB101(pUCN18)については、アセトフェノン還元活性は0.1U/mg以下であった。一方、RKPを発現させたE.coli HB101(pNKP)のアセトフェノン還元活性は5U/mgであった。以上のように、参考例3で得られた組換え生物はアセトフェノンに対する還元活性を有し、RKPの発現が認められた。
参考例4と同様の方法で野生型酵素の無細胞抽出液を得た。この無細胞抽出液に終濃度30、40、50%のジメチルホルムアミドを添加し、硫酸もしくは水酸化ナトリウムでpH6.5に調整後、30℃で3時間インキュベートした。また、対照として何も添加しない無細胞抽出液も同様にインキュベートした。3時間後、各無細胞抽出液を希釈した。これら無細胞抽出液の2-ペンタノン還元活性を参考例4と同様の方法で測定した。下記式より溶剤添加時の相対活性を算出し、この値を各種化合物に対する安定性の指標とした。結果を表1に示す。
相対活性(%)=[3時間目の酵素活性(溶剤添加)]÷[3時間目の酵素活性(溶剤非添加)]×100

参考例4と同様の方法で野生型酵素の無細胞抽出液を得た。この無細胞抽出液に終濃度30%のジメチルホルムアミドを添加し、硫酸もしくは水酸化ナトリウムでpH6.5に調整後、30℃で2時間インキュベートした。また、対照として何も添加しない無細胞抽出液も同様にインキュベートした。2時間後、各無細胞抽出液を希釈した。これら無細胞抽出液の2-ヘキサノン還元活性を測定した。100mMリン酸緩衝液(pH6.5)に、2-ヘキサノン10mM、補酵素NADPH 0.25mM、および無細胞抽出液を添加して30℃で1分間反応を行い、波長340nmにおける吸光度の減少速度を測定した。この減少速度より2-ヘキサノン還元活性を算出した。下記式より溶剤添加時の相対活性を算出し、この値を各種化合物に対する安定性の指標とした。溶剤添加条件下の野生型酵素の活性(相対活性)は溶剤非添加条件下の8%であった。
相対活性(%)=[2時間目の酵素活性(溶剤添加)]÷[2時間目の酵素活性(溶剤非添加)]×100
参考例4と同様の方法で野生型酵素の無細胞抽出液を得た。この無細胞抽出液に終濃度30%のジメチルホルムアミドを添加し、硫酸もしくは水酸化ナトリウムでpH6.5に調整後、30℃で2時間インキュベートした。また、対照として何も添加しない無細胞抽出液も同様にインキュベートした。2時間後、各無細胞抽出液を希釈した。これら無細胞抽出液の2-ヘキサノン還元活性を測定した。100mMリン酸緩衝液(pH6.5)に、30%ジメチルホルムアミド、2-ヘキサノン10mM、補酵素NADPH 0.25mM、および無細胞抽出液を添加して反応を行った。NADPHの蛍光の減少速度からNADPHの消費速度を求め、2-ヘキサノン還元活性を算出した。下記式より溶剤添加時の相対活性を算出し、この値を各種化合物に対する安定性の指標とした。溶剤添加条件下の野生型酵素の活性(相対活性)は溶剤非添加条件下の24%であった。
相対活性(%)=[酵素活性(溶剤添加)]÷[酵素活性(溶剤非添加)]×100
参考例2で作製したRKP遺伝子を含むプラスミドpNKPをテンプレートに、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)およびプライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてエラープローンPCR法(Leung et al.Technique 1,11-15(1989))を用いて、RKP遺伝子全長にランダムな変異を導入したDNA増幅断片を得た。この増幅断片を制限酵素NdeIおよびSalIで消化したのち、同酵素で処理した高発現ベクターpUCN18に組み込み、複数の変異酵素発現プラスミドを作製した。このプラスミドを用いてE.coli HB101を形質転換し、100μg/mLのアンピシリンを含むLBプレート培地に塗布した。生育したコロニーは変異導入されたRKP遺伝子を有する組換え大腸菌であり、この組換え菌群を変異酵素ライブラリー1とした。
変異酵素ライブラリー1より有機溶剤に対する安定性が向上した改変型カルボニル還元酵素を選抜した。実施例1で作製した変異酵素ライブラリー1の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。得られた各培養液60μlに10mM EDTA・2Naおよび1% Triton X-100を含むリン酸緩衝液(pH7.0)240μlを加え、37℃で1時間インキュベートした。各処理液を遠心し、上清を無細胞抽出液とした。各無細胞抽出液200μlに終濃度10~30%のジメチルホルムアミドを含むリン酸緩衝液(pH6.5)を加え、30℃で2時間インキュベートした(ジメチルホルムアミド処理)。ジメチルホルムアミド処理した各無細胞抽出液を96穴プレート(AGCテクノグラス社製)に50μl分注し、6mMのNADPHを含むリン酸緩衝液(pH6.5)50μlと133mM 2,3-ブタンジオンを含むリン酸緩衝液(pH6.5)100μlを加え30℃で反応した。経時的にUVサンプル撮影装置FAS-III(東洋紡績社製)でNADPHの蛍光を観察した。反応が進行しない酵素液はNADPHの蛍光が残存するが、反応が進行した無細胞抽出液はNADPHの減少に伴い、蛍光がなくなった。対照であるE.coli HB101(pNKP)の無細胞抽出液(野生型酵素)に比べ、早く蛍光がなくなった酵素をジメチルホルムアミド存在下で反応性の高い酵素、すなわち有機溶剤に対する安定性が向上した改変型カルボニル還元酵素として選抜した。選抜した酵素の培養液からプラスミドを抽出し、Big Dye Terminator Cycle Sequencing Kit(アプライドバイオシステムズジャパン社製)およびApplied Biosystems 3130xlジェネティックアナライザ(アプライドバイオシステムズジャパン社製)を用いて変異型RKP遺伝子の塩基配列を決定し、変異部位を特定した。得られた有機溶剤に対する安定性の向上した改変型カルボニル還元酵素を表2に示す。
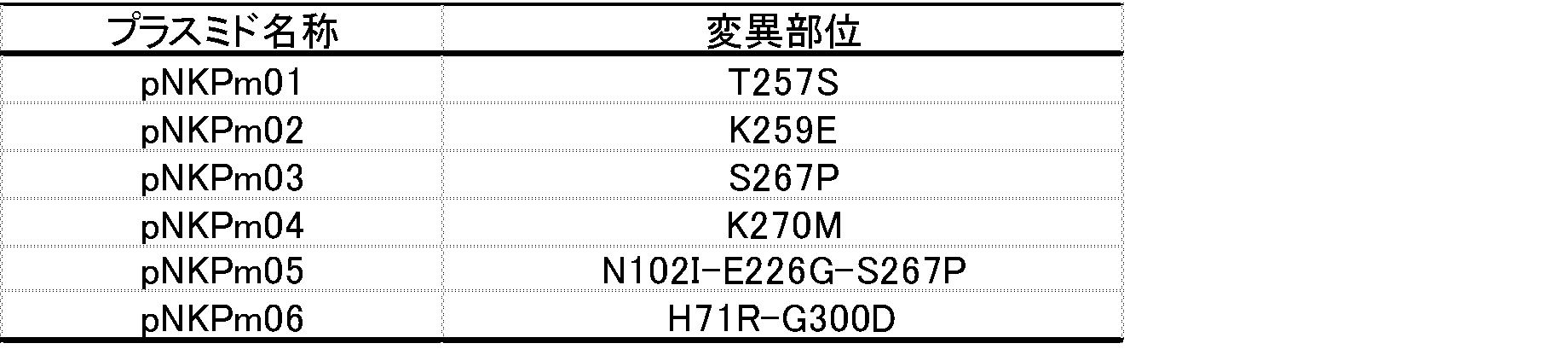
変異酵素ライブラリー1より有機溶剤に対する安定性が向上した改変型カルボニル還元酵素を選抜した。実施例1で作製した変異酵素ライブラリー1の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)を100μg/mLのアンピシリンを含むLBプレート培地上に植菌した。得られたコロニーを40℃で加熱しておいたナイロン膜(BiodyneA、0.45μm)に転写した後、そのナイロン膜を30%のジメチルホルムアミドを含む50mMの3-(N-morpholino)propanesulfonic acid(MOPS)バッファー中に40℃で30分浸漬した。その後、このナイロン膜を1mMのNADP+、200μMのnitroblue tetrazolium、10μMの1-methoxy-5-methylphenazinium methylsulfateおよび10%(v/v)の2-プロパノールを含む50mMのMOPSバッファー中に室温で30分浸漬した。その後、ナイロン膜を蒸留水で洗浄し、染色されたコロニーをジメチルホルムアミドに対する安定性が向上した改変型カルボニル還元酵素の組換え菌候補として選抜した。候補株を200μg/mlのアンピシリンを含む2×YT培地(トリプトン1.6%、イーストエキス1.0%、塩化ナトリウム 0.5%、pH7.0)5mlに接種し、20時間培養した。得られた各培養液について、遠心分離により菌体を集め、培養液の1/6量の100mMリン酸緩衝液(pH6.5)に懸濁した。これを、UH-50型超音波ホモゲナイザー(SMT社製)を用いて破砕した後、遠心分離により菌体残渣を除去し、無細胞抽出液を得た。この無細胞抽出液20μlに終濃度20、23、26、30%のジメチルホルムアミドと50mM MOPSバッファー(pH7.0)を総液量が40μlになるように添加し、40℃で30分間加熱した。氷上で1分間冷却した後、1mMのNADP+、200μMのnitroblue tetrazolium、10μMの1-methoxy-5-methylphenazinium methylsulfateおよび10%(v/v)の2-プロパノールを含む50mMのMOPSバッファーを200μ加えた。反応液を96穴プレート(AGCテクノグラス社製)に移し、1時間観察した。染色されたものをジメチルホルムアミドに対する安定性がさらに向上した改変型カルボニル還元酵素の組換え菌として選抜した。選抜した組換え菌の培養液からプラスミドを抽出し、BigDye Terminator Cycle Sequencing Kit(アプライドバイオシステムズジャパン社製)およびApplied Biosystems 3130xlジェネティックアナライザ(アプライドバイオシステムズジャパン社製)を用いて変異型RKP遺伝子の塩基配列を決定し、変異部位を特定した。得られた有機溶剤に対する安定性の向上した改変型カルボニル還元酵素を表3に示す。

実施例2で取得した改変型カルボニル還元酵素の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。得られた各培養液について、遠心分離により菌体を集め、培養液と等量から1/5量の100mMリン酸緩衝液(pH6.5)に懸濁した。これを、UH-50型超音波ホモゲナイザー(SMT社製)を用いて破砕した後、遠心分離により菌体残渣を除去し、無細胞抽出液を得た。無細胞抽出液60μlに終濃度30、40%または50%のジメチルホルムアミドを含むリン酸緩衝液(pH7.0)60μlを加え、30℃でインキュベートした(ジメチルホルムアミド処理)。3時間目のジメチルホルムアミド処理液をサンプリングし、希釈して参考例4に示した方法で2-ペンタノンに対する還元活性を測定した。下記式より残存活性は算出し、この値をジメチルホルムアミドに対する安定性の指標とした。
相対活性(%)=[3時間目の酵素活性(溶剤添加)]÷[3時間目の酵素活性(溶剤非添加)]×100
ジメチルホルムアミド40%存在下で評価した野生型酵素および改変型カルボニル還元酵素の相対活性を表4に示す。

実施例3で取得した改変型カルボニル還元酵素の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。得られた各培養液について、遠心分離により菌体を集め、培養液と等量から1/5量の100mMリン酸緩衝液(pH6.5)に懸濁した。これを、UH-50型超音波ホモゲナイザー(SMT社製)を用いて破砕した後、遠心分離により菌体残渣を除去し、無細胞抽出液を得た。25μlの無細胞抽出液に等量の80%ジメチルホルムアミド溶液を加え、30℃で30分静置した。これに100mMリン酸緩衝液(pH6.5)200μlを加え、混合した。この溶液15μlに終濃度0.625mMのNADPHと12.5mMの2,3-ブタンジオンを溶解した100mMリン酸緩衝液(pH6.5)を250μl加え、混合した。5秒振とうし、NADPHの吸光(340nm)をBenchmark Plus microplate spectrophotometer(BIO-RAD社製)で15秒間測定し、その減少速度から2,3-ブタンジオンに対する還元活性を求めた。下記式よりジメチルホルムアミド非添加時に対する添加時の酵素活性を算出し、この値をジメチルホルムアミドに対する安定性の指標とした。
相対活性(%)=[酵素活性(ジメチルホルムアミド添加)]÷[酵素活性(ジメチルホルムアミド非添加)]×100
野生型酵素および改変型カルボニル還元酵素の相対活性を表5に示す。

実施例2で取得したプラスミドpNKPm02を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー3:5’-GTTAATTTCATTAGCGCGATTTTTAATTACATG-3’(配列表の配列番号6)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257Sのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm02を鋳型として、プライマー4:5’-CATGTAATTAAAAATCGCGCTAATGAAATTAAC-3’(配列表の配列番号7)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちH71R、K259Eのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちH71R、K259Eのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm30を作製した。このpNKPm30を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素H71R-K259Eを生産する組換え生物E.coli HB101(pNKPm30)を得た。
実施例2で取得したプラスミドpNKPm02を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー5:5’-GATCAACCTTTCACCGCTTAACTCATCATTATG-3’(配列表の配列番号8)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257Sのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm02を鋳型として、プライマー6:5’-CATAATGATGAGTTAAGCGGTGAAAGGTTGATC-3’(配列表の配列番号9)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K259Eのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K259Eのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm31を作製した。このpNKPm31を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素T257S-K259Eを生産する組換え生物E.coli HB101(pNKPm31)を得た。
実施例2で取得したプラスミドpNKPm02を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー7:5’-CTCGTTATGGACACGATCTCCTTGAGGTAACTC-3’(配列表の配列番号10)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、G300Dのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm02を鋳型として、プライマー8:5’-GAGTTACCTCAAGGAGATCGTGTCCATAACGAG-3’(配列表の配列番号11)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちG300Dのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、G300Dのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm32を作製した。このpNKPm32を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K259E-G300Dを生産する組換え生物E.coli HB101(pNKPm32)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー9:5’-GTTAATTTCATTAGCGCGATTTTTAATTACATG-3’(配列表の配列番号12)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちH71R、K270Mのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm02を鋳型として、プライマー10:5’-CATGTAATTAAAAATCGCGCTAATGAAATTAAC-3’(配列表の配列番号13)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちH71R、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちH71R、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm33を作製した。このpNKPm33を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素H71R-K270Mを生産する組換え生物E.coli HB101(pNKPm33)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー11:5’-GGATACCTTTAGTACCAATAACAGCTGGGATTAAG-3’(配列表の配列番号14)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちN102Iのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー12:5’-CTTAATCCCAGCTGTTATTGGTACTAAAGGTATCC-3’(配列表の配列番号15)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちN102I、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちN102I、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm34を作製した。このpNKPm34を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素N102I-K270Mを生産する組換え生物E.coli HB101(pNKPm34)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー13:5’-TTTATCAATTTCACTGCCTGTTGGAGCAAACATAGC-3’(配列表の配列番号16)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちE226Gのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー14:5’-GCTATGTTTGCTCCAACAGGCAGTGAAATTGATAAA-3’(配列表の配列番号17)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちNE226Gのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちE226G、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm35を作製した。このpNKPm35を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素E226G-K270Mを生産する組換え生物E.coli HB101(pNKPm35)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー15:5’-GATCAACCTTTTACCGCTTAACTCATCATTATG-3’(配列表の配列番号18)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257Sのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー16:5’-CATAATGATGAGTTAAGCGGTAAAAGGTTGATC-3’(配列表の配列番号19)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm36を作製した。このpNKPm36を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素T257S-K270Mを生産する組換え生物E.coli HB101(pNKPm36)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー17:5’-GACAAGATCAACCTTTCACCAGTTAACTCATC-3’(配列表の配列番号20)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259Eのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー18:5’-GATGAGTTAACTGGTGAAAGGTTGATCTTGTC-3’(配列表の配列番号21)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm37を作製した。このpNKPm37を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm37)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー19:5’-TTTGCATAGTGAACGGAGCATTTGACAAG-3’(配列表の配列番号22)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS267Pのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー20:5’-CTTGTCAAATGCTCCGTTCACTATGCAAA-3’(配列表の配列番号23)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS267P、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS267P、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm38を作製した。このpNKPm38を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素S267P-K270Mを生産する組換え生物E.coli HB101(pNKPm38)を得た。
実施例2で取得したプラスミドpNKPm04を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー21:5’-CTCGTTATGGACACGATCTCCTTGAGGTAACTC-3’(配列表の配列番号24)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK270M、G300Dのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm04を鋳型として、プライマー22:5’-GAGTTACCTCAAGGAGATCGTGTCCATAACGAG-3’(配列表の配列番号25)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちG300Dのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK270M、G300Dのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm39を作製した。このpNKPm39を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K270M-G300Dを生産する組換え生物E.coli HB101(pNKPm39)を得た。
実施例6~15で取得した多重変異改変型カルボニル還元酵素の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。実施例4と同様に多重変異改変型カルボニル還元酵素のジメチルホルムアミドに対する安定性を評価した。ジメチルホルムアミド40%存在下で評価した野生型酵素および改変型カルボニル還元酵素の相対活性を表6に示す。

実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー23:5’-CCAGTAGCACCTGTAACTAAAACAATCAT-3’(配列表の配列番号26)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS2Iのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー24:5’-ATGATTGTTTTAGTTACAGGTGCTACTGG-3’(配列表の配列番号27)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS2I、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS2I、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm40を作製した。このpNKPm40を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素S2I-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm40)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー25:5’-GGCAGCAATTGAAGAAGTCAGAACAAATTTCTTCAC-3’(配列表の配列番号28)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちI124Lのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー26:5’-GTGAAGAAATTTGTTCTGACTTCTTCAATTGCTGCC-3’(配列表の配列番号29)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちI124L、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちI124L、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm41を作製した。このpNKPm41を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素I124L-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm41)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー27:5’-CCTTTATTCTCTTCAAAGAAGTTCCAAGCAGC-3’(配列表の配列番号30)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちL177Fのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー28:5’-GCTGCTTGGAACTTCTTTGAAGAGAATAAAGG-3’(配列表の配列番号31)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちL177F、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちL177F、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm42を作製した。このpNKPm42を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素L177F-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm42)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー29:5’-GGACCAAAGACCAGAACTGGGTTGATCG-3’(配列表の配列番号32)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちF195Lのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー30:5’-CGATCAACCCAGTTCTGGTCTTTGGTCC-3’(配列表の配列番号33)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちF195L、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちF195L、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm43を作製した。このpNKPm43を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素F195L-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm43)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー31:5’-CTGTTGGAGCAAACATCACTTCCTTGATGATTTC-3’(配列表の配列番号34)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちA220Vのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー32:5’-GAAATCATCAAGGAAGTGATGTTTGCTCCAACAG-3’(配列表の配列番号35)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちA220V、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちA220V、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm44を作製した。このpNKPm44を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素A220V-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm44)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー33:5’-CGTACATCAACATAGTTACCAAAAACAGATTTATC-3’(配列表の配列番号36)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS236Nのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー34:5’-GATAAATCTGTTTTTGGTAACTATGTTGATGTACG-3’(配列表の配列番号37)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS236N、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちS236N、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm45を作製した。このpNKPm45を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素S236N-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm45)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー35:5’-GCTACATCACGTACATCAATATAACTACCAAAAAC-3’(配列表の配列番号38)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちV238Iのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー36:5’-GTTTTTGGTAGTTATATTGATGTACGTGATGTAGC-3’(配列表の配列番号39)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちV238I、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちV238I、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm46を作製した。このpNKPm46を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素V238I-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm46)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー37:5’-GATCAACCTTTTACCGCTTAACTCATCATTATG-3’(配列表の配列番号40)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257Sのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー38:5’-CATAATGATGAGTTAAGCGGTAAAAGGTTGATC-3’(配列表の配列番号41)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K259E、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちT257S、K259E、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm47を作製した。このpNKPm47を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素T257S-K259E-K270Mを生産する組換え生物E.coli HB101(pNKPm47)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー39:5’-GCATAGTGAATGAAGCTTTTGACAAGATCAACCTTTCACC-3’(配列表の配列番号42)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、N265Kのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー40:5’-GGTGAAAGGTTGATCTTGTCAAAAGCTTCATTCACTATGC-3’(配列表の配列番号43)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、N265K、K270Mのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、N265K、K270Mのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm48を作製した。このpNKPm48を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K259E-N265K-K270Mを生産する組換え生物E.coli HB101(pNKPm48)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー41:5’-CTCGTTATGGACACGATCTCCTTGAGGTAACTC-3’(配列表の配列番号44)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、G300Dのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー42:5’-GAGTTACCTCAAGGAGATCGTGTCCATAACGAG-3’(配列表の配列番号45)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、G300Dのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、G300Dのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm49を作製した。このpNKPm49を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K259E-K270M-G300Dを生産する組換え生物E.coli HB101(pNKPm49)を得た。
実施例7で取得したプラスミドpNKPm37を鋳型として、プライマー1:5’-GGGAATTCCATATGAGTGTTTTAGTTACAGG-3’(配列表の配列番号4)、プライマー43:5’-CTTCTCGTTATGGACGCAACCTCCTTGAGGTAACTCG-3’(配列表の配列番号46)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、R301Cのアミノ酸置換を有するN末端側のポリペプチドをコードする二本鎖DNAを得た(N末端側DNA)。同様にプラスミドpNKPm37を鋳型として、プライマー44:5’-CGAGTTACCTCAAGGAGGTTGCGTCCATAACGAGAAG -3’(配列表の配列番号47)、プライマー2:5’-ATACGCGTCGACTTACTATTGTTCTTGAACCTTCA-3’(配列表の配列番号5)を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、R301Cのアミノ酸置換を有するC末端側のポリペプチドをコードする二本鎖DNAを得た(C末端側DNA)。N末端側DNAとC末端側DNAを混合し、これを鋳型として、プライマー1、プライマー2を用いてPCRを行い、配列番号1に示すアミノ酸配列のうちK259E、K270M、R301Cのアミノ酸置換を有するポリペプチドをコードする二本鎖DNAを得た。参考例2と同様の方法でpUCN18に導入し、pNKPm50を作製した。このpNKPm50を用いて、E.coli HB101コンピテントセル(タカラバイオ社製)を形質転換し、改変型カルボニル還元酵素K259E-K270M-R301Cを生産する組換え生物E.coli HB101(pNKPm50)を得た。
実施例17~27で取得した多重変異改変型カルボニル還元酵素の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。実施例4と同様に多重変異改変がたカルボニル還元酵素のジメチルホルムアミドに対する安定性を評価した。ジメチルホルムアミド50%存在下で評価した野生型酵素および改変型カルボニル還元酵素の相対活性を表7に示す。

実施例15で取得したK259E-K270M変異酵素の変異型RKP遺伝子を含むプラスミドpNKPm37をテンプレートに実施例1と同様の方法で変異酵素ライブラリーを作成し、これを変異酵素ライブラリー2とした。
変異酵素ライブラリー2より有機溶剤の1つであるジメチルホルムアミドによる反応阻害に対する抵抗性が向上した改変型カルボニル還元酵素を選抜した。実施例29で作製した変異酵素ライブラリー2の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)を100μg/mLのアンピシリンを含むLBプレート培地上に植菌した。

実施例30で取得した改変型カルボニル還元酵素の各組換え菌および参考例3で作製したE.coli HB101(pNKP)(対照)をそれぞれ参考例4と同様の方法で培養した。得られた各培養液について、遠心分離により菌体を集め、培養液の1/5量の100mMリン酸緩衝液(pH6.5)に懸濁した。これを、UH-50型超音波ホモゲナイザー(SMT社製)を用いて破砕した後、遠心分離により菌体残渣を除去し、無細胞抽出液を得た。無細胞抽出液100μlに1Mリン酸緩衝液(pH7.0)400μl、水または60%DMF溶液500μl、2-ヘキサノン1%、NADPH5%、グルコース3.4%を混合し30℃で2時間撹拌し、反応した。反応後、反応液を酢酸エチルで抽出した。抽出液を下記の[ガスクロマトグラフィーによる分析条件]に記載の条件で分析することにより、2-ヘキサノールの生成を確認した。2-ヘキサノールと2-ヘキサノンのピーク面積から変換率を算出した。
カラム:InertCapI キャピラリーカラム(30m、内径0.25mm、ジーエル サイエンス社製)
検出器:水素炎イオン化検出器
注入部温度:250℃
カラム温度:50℃
検出器温度:250℃
キャリアーガス:ヘリウム、流量150kPa
相対活性(%)=[ジメチルホルムアミド存在下での変換率]÷[ジメチルホルムアミド非存在下での変換率]×100
得られた有機溶剤による反応阻害に対する抵抗性が向上した改変型カルボニル還元酵素を表9に示す。
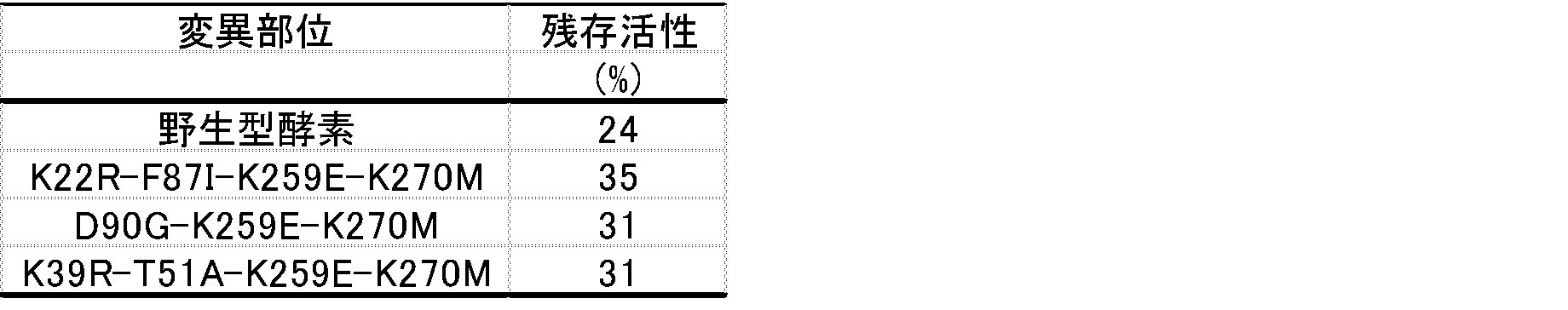
参考例4で取得したヴァンデルワルトザイマ・ポリスポラ(Vanderwaltozyma polyspora)NBRC0996株由来のカルボニル還元酵素RKP(野生型)を発現する組換え大腸菌の培養液、および実施例24で取得した改変型カルボニル還元酵素T257S-K259E-K270Mを生産する組換え大腸菌を参考例4と同様の方法を用いて培養した培養液700μlに、グルコース脱水素酵素(商品名:GLUCDH“Amano”II、天野エンザイム社製)12.5U、グルコース80mg、NADP+0.6mg、ジメチルホルムアミドまたは0.1Mリン酸緩衝液(pH7)300μl、(S)-1-(4-フルオロ-フェニル)-5-(2-オキソ-4-フェニル-オキサゾリジン-3-イル)-ペンタン-1,5-ジオン10mgを加えて、30℃で20時間撹拌した。反応液をジメチルスルホキシドで希釈し、下記の条件で高速液体クロマトグラフィー分析し、3-[(5R)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4S)-フェニル-1,3-オキサゾリジン-2-オンへの変換率、その光学純度を測定した。その結果を表10に示す。
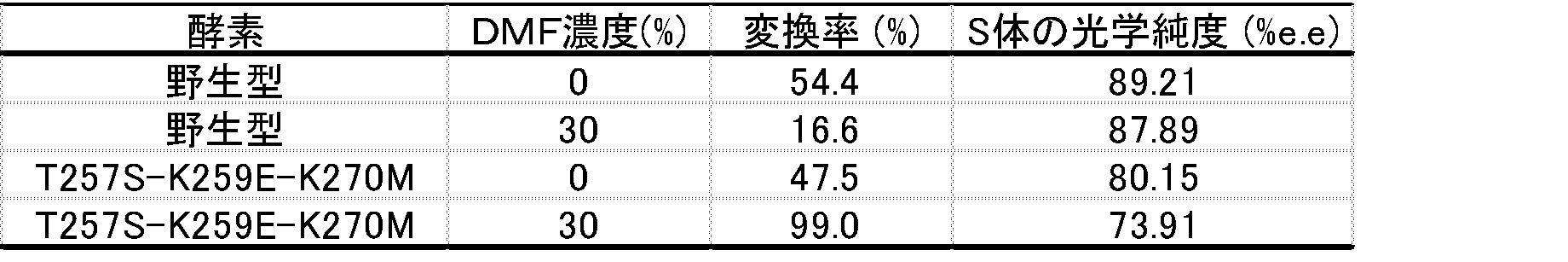
カラム:COSMOSIL 5C8-MS(250mm、内径4.6μm、ナカライテスク社製)
カラム温度:40℃
検出波長:254mm
移動相:水/アセトニトリル=1/1
保持時間:[3-[(5)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4)-フェニル-1,3-オキサゾリジン-2-オン 約8.5分、1-(4-フルオロ-フェニル)-5-(2-オキソ-4-フェニル-オキサゾリジン-3-イル)-ペンタン-1,5-ジオン 約12.9分
変換率=[3-[(5)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4)-フェニル-1,3-オキサゾリジン-2-オンの生成量/([3-[(5)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4)-フェニル-1,3-オキサゾリジン-2-オンの生成量+1-(4-フルオロ-フェニル)-5-(2-オキソ-4-フェニル-オキサゾリジン-3-イル)-ペンタン-1,5-ジオンの残存量)×100
[3-[(5R)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4S)-フェニル-1,3-オキサゾリジン-2-オンの光学純度の算出方法と分析条件]
カラム:CHIRALCEL OD-H(250mm、内径4.6μm、ダイセル化学工業社製)
カラム温度:30℃
検出波長:254mm
移動相:ヘキサン/エタノール=8/2
保持時間:3-[(5R)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4S)-フェニル-1,3-オキサゾリジン-2-オン 約18.1分、3-[(5S)-(4-フルオロ-フェニル)-5-ヒドロキシペンタノイル]-(4S)-フェニル-1,3-オキサゾリジン-2-オン 約21.7分
R体の光学純度(%e.e.)={(R体のピーク面積)-(S体のピーク面積)}÷{(R体のピーク面積)+(S体のピーク面積)}×100
Claims (17)
- 以下の(a)~(c);
(a)配列表の配列番号1に記載のアミノ酸配列と78%以上の配列同一性を有し、
(b)2-ペンタノンを還元して2-ペンタノールを生成し、かつ、
(c)配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤存在下でカルボニル化合物に対する反応性が高い、および/または熱安定性が高い、
の性質を示すポリペプチド。 - 前記有機溶剤がジメチルホルムアミドである、請求項1に記載のポリペプチド。
- 配列表の配列番号1に記載のアミノ酸配列において次の群;
2番目、22番目、25番目、39番目、42番目、45番目、51番目、56番目、71番目、87番目、90番目、102番目、109番目、124番目、135番目、138番目、155番目、159番目、175番目、177番目、183番目、190番目、195番目、212番目、220番目、226番目、228番目、236番目、238番目、250番目、254番目、257番目、259番目、265番目、267番目、270番目、279番目、298番目、300番目、301番目、および331番目から選択される1つ以上のアミノ酸に、アミノ酸置換が導入されている、請求項1または2に記載のポリペプチド。 - アミノ酸置換が、配列表の配列番号1に記載のアミノ酸配列において次の群;
2番目がイソロイシン、22番目がアルギニン、25番目がフェニルアラニン、39番目がアルギニン、42番目がアルギニン、45番目がアスパラギン酸、51番目がアラニン、56番目がリジン、71番目がアスパラギンもしくはアルギニン、87番目がイソロイシン、90番目がグリシン、102番目がイソロイシン、109番目がグリシン、124番目がロイシン、135番目がアラニン、138番目がアスパラギン、155番目がロイシンもしくはアルギニン、159番目がフェニルアラニン、175番目がアスパラギン酸、177番目がフェニルアラニン、183番目がスレオニン、190番目がセリン、195番目がロイシン、212番目がフェニルアラニン、スレオニンもしくはチロシン、220番目がバリン、226番目がグリシン、228番目がバリン、236番目がアスパラギン、238番目がイソロイシン、250番目がプロリン、254番目がアスパラギン、257番目がセリン、259番目がグルタミン酸、265番目がリジン、267番目がプロリン、270番目がメチオニン、279番目がアルギニン、298番目がプロリン、300番目がアスパラギン酸、301番目がシステイン、および、331番目がフェニルアラニンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換である、請求項3に記載のポリペプチド。 - アミノ酸置換が、配列表の配列番号1に記載のアミノ酸配列において次の群;
2番目がイソロイシン、45番目がアスパラギン酸、71番目がアスパラギンもしくはアルギニン、102番目がイソロイシン、124番目がロイシン、175番目がアスパラギン酸、177番目がフェニルアラニン、183番目がスレオニン、195番目がロイシン、220番目がバリン、226番目がグリシン、236番目がアスパラギン、238番目がイソロイシン、257番目がセリン、259番目がグルタミン酸、265番目がリジン、267番目がプロリン、270番目がメチオニン、300番目がアスパラギン酸、および、301番目がシステインに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であり、
配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤に対する安定性が向上している、請求項4に記載のポリペプチド。 - アミノ酸置換が、配列表の配列番号1に記載のアミノ酸配列において下記の(1)~(35);
(1)71番目がアスパラギン、195番目がロイシン、
(2)71番目がアルギニン、259番目がグルタミン酸、
(3)71番目がアルギニン、270番目がメチオニン、
(4)71番目がアルギニン、300番目がアスパラギン酸、
(5)102番目がイソロイシン、270番目がメチオニン、
(6)177番目がフェニルアラニン、220番目がバリン、
(7)226番目がグリシン、270番目がメチオニン、
(8)257番目がセリン、259番目がグルタミン酸、
(9)257番目がセリン、270番目がメチオニン、
(10)259番目がグルタミン酸、270番目がメチオニン、
(11)259番目がグルタミン酸、300番目がアスパラギン酸、
(12)267番目がプロリン、270番目がメチオニン、
(13)270番目がメチオニン、300番目がアスパラギン酸、
(14)2番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(15)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(16)102番目がイソロイシン、226番目がグリシン、267番目がプロリン、
(17)124番目がロイシン、259番目がグルタミン酸、270番目がメチオニン、
(18)177番目がフェニルアラニン、259番目がグルタミン酸、270番目がメチオニン、
(19)220番目がバリン、259番目がグルタミン酸、270番目がメチオニン、
(20)236番目がアスパラギン、259番目がグルタミン酸、270番目がメチオニン、
(21)238番目がイソロイシン、259番目がグルタミン酸、270番目がメチオニン、
(22)257番目がセリン、259番目がグルタミン酸、270番目がメチオニン、
(23)257番目がセリン、259番目がグルタミン酸、300番目がアスパラギン酸、
(24)259番目がグルタミン酸、265番目がリジン、270番目がメチオニン、
(25)259番目がグルタミン酸、270番目がメチオニン、300番目がアスパラギン酸、
(26)259番目がグルタミン酸、270番目がメチオニン、301番目がシステイン、
(27)2番目がイソロイシン、238番目がイソロイシン、
(28)71番目がアスパラギン、195番目がロイシン、
(29)109番目がグリシン、331番目がフェニルアラニン、
(30)124番目がロイシン、236番目がアスパラギン、
(31)159番目がフェニルアラニン、259番目がグルタミン酸、
(32)42番目がアルギニン、155番目がアルギニン、279番目がアルギニン、
(33)45番目がアスパラギン酸、175番目がアスパラギン酸、183番目がスレオニン、
(34)155番目がロイシン、250番目がプロリン、298番目がプロリン、および
(35)56番目がリジン、138番目がアスパラギン、190番目がセリン、254番目がアスパラギンに置換されるアミノ酸置換、
から選択されるアミノ酸置換が導入されている、請求項5に記載のポリペプチド。 - アミノ酸置換が、次の群;
22番目がアルギニン、39番目がアルギニン、51番目がアラニン、87番目がイソロイシン、90番目がグリシン、259番目がグルタミン酸、および、270番目がメチオニンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換であり、
配列表の配列番号1に記載のアミノ酸配列からなるカルボニル還元酵素と比較して、有機溶剤による反応阻害に対する抵抗性が向上している、請求項4に記載のポリペプチド。 - アミノ酸置換が、配列表の配列番号1に記載のアミノ酸配列において下記の(1)~(7);
(1)22番目がアルギニン、
(2)22番目がアルギニン、87番目がイソロイシン、
(3)39番目がアルギニン、
(4)39番目がアルギニン、51番目がアラニン、
(5)51番目がアラニン、
(6)87番目がイソロイシン、および
(7)90番目がグリシンに置換されるアミノ酸置換、
から選択される1つ以上のアミノ酸置換である、請求項7に記載のポリペプチド。 - 請求項1~8のいずれか1項に記載のポリペプチドをコードするポリヌクレオチド。
- 請求項9に記載のポリヌクレオチドを含むベクター。
- 還元型補酵素再生能を有するポリペプチドをコードするポリヌクレオチドをさらに含む、請求項10に記載のベクター。
- 還元型補酵素再生能を有するポリペプチドがグルコース脱水素酵素である、請求項11に記載のベクター。
- 請求項10~12のいずれか1項に記載のベクターにより宿主細胞を形質転換して得られる形質転換体。
- 前記宿主細胞が大腸菌である請求項13に記載の形質転換体。
- 請求項1~8のいずれか1項に記載のポリペプチド、または、請求項13または請求項14に記載の形質転換体および/またはその処理物を、カルボニル化合物に作用させることを特徴とする、アルコール化合物の製造方法。
- 前記カルボニル化合物が非対称ケトンであり、前記アルコール化合物が光学活性アルコールである、請求項15に記載の製造方法。
- 前記カルボニル化合物が、下記式(1):

前記アルコール化合物が下記式(2):

Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015508537A JP6442399B2 (ja) | 2013-03-28 | 2014-03-25 | 改変型カルボニル還元酵素およびその遺伝子 |
| US14/780,419 US10294461B2 (en) | 2013-03-28 | 2014-03-25 | Modified carbonyl reducing enzyme and gene |
| SG11201508053WA SG11201508053WA (en) | 2013-03-28 | 2014-03-25 | Modified carbonyl reducing enzyme and gene |
| EP14772782.0A EP2980213B1 (en) | 2013-03-28 | 2014-03-25 | Modified carbonyl reducing enzyme and gene |
| CN201480018198.4A CN105073991B (zh) | 2013-03-28 | 2014-03-25 | 改变型羰基还原酶及其基因 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-070170 | 2013-03-28 | ||
| JP2013070170 | 2013-03-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014157185A1 true WO2014157185A1 (ja) | 2014-10-02 |
Family
ID=51624155
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/058248 Ceased WO2014157185A1 (ja) | 2013-03-28 | 2014-03-25 | 改変型カルボニル還元酵素およびその遺伝子 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10294461B2 (ja) |
| EP (1) | EP2980213B1 (ja) |
| JP (1) | JP6442399B2 (ja) |
| CN (1) | CN105073991B (ja) |
| SG (1) | SG11201508053WA (ja) |
| WO (1) | WO2014157185A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107677753B (zh) * | 2017-11-24 | 2021-03-16 | 中山奕安泰医药科技有限公司 | 一种依替米贝中间体的检测方法 |
| CN109852592B (zh) * | 2019-01-14 | 2022-05-31 | 中国科学院成都生物研究所 | 耐热性提高的羰基还原酶突变体 |
| CN109837254B (zh) * | 2019-03-28 | 2022-05-31 | 中国科学院成都生物研究所 | 一种热稳定性提高的羰基还原酶突变体 |
| CN110004119B (zh) * | 2019-04-18 | 2020-10-20 | 华东理工大学 | ε-酮酯还原酶突变体及其催化合成(R)-α-硫辛酸前体的应用 |
| CN113201511B (zh) * | 2021-04-15 | 2022-08-30 | 华东理工大学 | (R)-5-羰基癸酸(酯)还原酶突变体及其在制备(R)-γ/δ-内酯中的应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994003613A1 (fr) | 1992-08-10 | 1994-02-17 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Adn codant pour la decarbamylase a thermostabilite accrue et utilisation de cet adn |
| WO2005075651A1 (ja) * | 2004-02-04 | 2005-08-18 | Api Corporation | 光学活性を有するアルコール及びカルボン酸の製造方法 |
| JP2009225773A (ja) | 2008-03-25 | 2009-10-08 | Sumitomo Chemical Co Ltd | 還元酵素及びその利用 |
| WO2011090753A2 (en) * | 2009-12-29 | 2011-07-28 | Butamax(Tm) Advanced Biofuels Llc | Alcohol dehydrogenases (adh) useful for fermentive production of lower alkyl alcohols |
| WO2011142865A2 (en) * | 2010-02-12 | 2011-11-17 | Gevo, Inc. | Yeast microorganisms with reduced by-product accumulation for improved production of fuels, chemicals, and amino acids |
| WO2012129555A2 (en) * | 2011-03-24 | 2012-09-27 | Butamax (Tm) Advanced Biofuels Llc | Host cells and methods for production of isobutanol |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60323215D1 (de) * | 2002-03-19 | 2008-10-09 | Mitsubishi Chem Corp | Neue carbonylreduktase, diese codierendes gen und verfahren zur produktion optisch-aktiver alkohole damit |
| WO2005066341A1 (en) * | 2004-01-01 | 2005-07-21 | Council Of Scientific & Industrial Research | A method for the preparation of cross linked protein crystals |
| CN102016042B (zh) * | 2008-04-30 | 2014-12-10 | 诺维信公司 | 具有磷脂酶抑制活性的分离的肽 |
| CN101407780B (zh) * | 2008-08-30 | 2010-08-25 | 江南大学 | 利用定点突变改变辅酶特异性和立体选择性制备(r)-苯基乙二醇的方法 |
| WO2011132444A1 (ja) * | 2010-04-22 | 2011-10-27 | 株式会社カネカ | 改変型カルボニル還元酵素、その遺伝子、およびそれらを利用した光学活性アルコールの製造方法 |
| CN102618513B (zh) | 2012-05-04 | 2014-06-11 | 华东理工大学 | 一种羰基还原酶、基因和突变体及在不对称还原羰基化合物中的应用 |
-
2014
- 2014-03-25 EP EP14772782.0A patent/EP2980213B1/en active Active
- 2014-03-25 CN CN201480018198.4A patent/CN105073991B/zh active Active
- 2014-03-25 SG SG11201508053WA patent/SG11201508053WA/en unknown
- 2014-03-25 WO PCT/JP2014/058248 patent/WO2014157185A1/ja not_active Ceased
- 2014-03-25 JP JP2015508537A patent/JP6442399B2/ja active Active
- 2014-03-25 US US14/780,419 patent/US10294461B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994003613A1 (fr) | 1992-08-10 | 1994-02-17 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Adn codant pour la decarbamylase a thermostabilite accrue et utilisation de cet adn |
| WO2005075651A1 (ja) * | 2004-02-04 | 2005-08-18 | Api Corporation | 光学活性を有するアルコール及びカルボン酸の製造方法 |
| JP2009225773A (ja) | 2008-03-25 | 2009-10-08 | Sumitomo Chemical Co Ltd | 還元酵素及びその利用 |
| WO2011090753A2 (en) * | 2009-12-29 | 2011-07-28 | Butamax(Tm) Advanced Biofuels Llc | Alcohol dehydrogenases (adh) useful for fermentive production of lower alkyl alcohols |
| WO2011142865A2 (en) * | 2010-02-12 | 2011-11-17 | Gevo, Inc. | Yeast microorganisms with reduced by-product accumulation for improved production of fuels, chemicals, and amino acids |
| WO2012129555A2 (en) * | 2011-03-24 | 2012-09-27 | Butamax (Tm) Advanced Biofuels Llc | Host cells and methods for production of isobutanol |
Non-Patent Citations (15)
| Title |
|---|
| CHEMSUSCHEM, vol. 1, 2008, pages 431 - 436 |
| FREDERICK M. AUSUBEL: "Current Protocols in . Molecular Biology", 1989, GREENE PUBLISHING ASSOCIATES AND WILEY-INTERSCIENCE |
| FREDERICK M. AUSUBEL: "Current Protocols in Molecular Biology", 1989, GREENE PUBLISHING ASSOCIATES AND WILEY-INTERSCIENCE |
| HOS. N. HUNT ET AL., GENE, vol. 77, 1989, pages 51 |
| JOSEPH SAMBROOK: "Molecular Cloning", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| JUAN LUCAS ARGUESO ET AL.: "Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production", GENOME RESEARCH, vol. 19, 2009, pages 2258 - 2270, XP055263693 * |
| LEUNG ET AL., TECHNIQUE, vol. 1, 1989, pages 11 - 15 |
| MARTA GORETTI ET AL.: "Production of Flavours and Fragrances via Bioreduction of (4R)-(-)- Carvone and (lR)-(-)-Myrtenal by Non- Conventional Yeast Whole-Cells", MOLECULES, vol. 18, 16 May 2013 (2013-05-16), pages 5736 - 5748, XP055286513 * |
| MURRAY ET AL., NUCL. ACIDS RES., vol. 8, 1980, pages 4321 |
| NATURE, vol. 315, 1985, pages 592 - 594 |
| OLFERT LANDT ET AL., GENE, vol. 96, 1990, pages 125 - 128 |
| See also references of EP2980213A4 |
| SMITH ET AL.: "Genetic Engineering", vol. 3, J. PLENUM PRESS, pages: 1 |
| TADAHIKO ANDO: "Biseibutsugaku Kiso Koza", 1987, KYORITSU SHUPPAN |
| VLASUK ET AL.: "Experimental Manipulation of Gene Expression", INOUYE, M. ACADEMIC PRESS |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2980213B1 (en) | 2019-03-13 |
| EP2980213A1 (en) | 2016-02-03 |
| US20160040138A1 (en) | 2016-02-11 |
| EP2980213A4 (en) | 2016-09-14 |
| US10294461B2 (en) | 2019-05-21 |
| JPWO2014157185A1 (ja) | 2017-02-16 |
| CN105073991B (zh) | 2019-04-05 |
| CN105073991A (zh) | 2015-11-18 |
| SG11201508053WA (en) | 2015-10-29 |
| JP6442399B2 (ja) | 2018-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8361765B2 (en) | Enzymic method for the enantioselective reduction of keto compounds | |
| US7575909B2 (en) | Oxidoreductase from pichia capsulata | |
| US7202069B2 (en) | (R)-2-octanol dehydrogenases, methods for producing the enzymes, DNA encoding the enzymes, and methods for producing alcohols using the enzymes | |
| US20090203096A1 (en) | Process for Production of Optically Active Alcohol | |
| CN104099305A (zh) | 一种羰基还原酶突变体及其基因和应用 | |
| JP2007523617A6 (ja) | ピキア・カプスラータ由来酸化還元酵素 | |
| JP6442399B2 (ja) | 改変型カルボニル還元酵素およびその遺伝子 | |
| JP2009502148A (ja) | ケト化合物の立体選択的還元に用いる酸化還元酵素 | |
| KR100998235B1 (ko) | 신규 카보닐 환원 효소 및 이것을 코딩하는 유전자, 및이들을 이용한 광학 활성 알콜의 제조 방법 | |
| CN111057686B (zh) | 一种醇脱氢酶突变体及应用 | |
| JP5889785B2 (ja) | 改変型カルボニル還元酵素、その遺伝子、およびそれらを利用した光学活性アルコールの製造方法 | |
| US8129163B2 (en) | Gene suitable for alcohol dehydrogenase, vector and transformant | |
| KR101780510B1 (ko) | 케토 화합물의 입체선택적 효소적 환원 방법 | |
| CN114729374A (zh) | 羰基还原酶、编码该酶的核酸、以及利用它们的光学活性化合物的制造方法 | |
| Richter et al. | Biochemical characterisation of a NADPH-dependent carbonyl reductase from Neurospora crassa reducing α-and β-keto esters | |
| JP2003033185A (ja) | エノン還元酵素 | |
| EP1236796B1 (en) | Novel enone reductases isolated from Kluyveromyces lactis, methods for producing same, and methods for selectively reducing a carbon-carbon double bond of an Alpha, Beta-unsaturated ketone using the reductases | |
| JP4270918B2 (ja) | 新規カルボニル還元酵素及びこれをコードする遺伝子、ならびにこれらを利用した光学活性アルコールの製造方法 | |
| JP4884996B2 (ja) | 新規な(s)−n−ベンジル−3−ピロリジノール脱水素酵素、その製造方法、及びこれを利用したアルコールの製造方法 | |
| CN106754800B (zh) | 一种氧化酶及其应用 | |
| JP2010279272A (ja) | 新規カルボニル還元酵素、その遺伝子、ベクター、形質転換体、およびそれらを利用した光学活性アルコールの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480018198.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14772782 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015508537 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14780419 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014772782 Country of ref document: EP |