WO2014188888A1 - 細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体 - Google Patents
細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体 Download PDFInfo
- Publication number
- WO2014188888A1 WO2014188888A1 PCT/JP2014/062495 JP2014062495W WO2014188888A1 WO 2014188888 A1 WO2014188888 A1 WO 2014188888A1 JP 2014062495 W JP2014062495 W JP 2014062495W WO 2014188888 A1 WO2014188888 A1 WO 2014188888A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- cells
- particles
- culture
- particle
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M25/00—Means for supporting, enclosing or fixing the microorganisms, e.g. immunocoatings
- C12M25/16—Particles; Beads; Granular material; Encapsulation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/067—Hepatocytes
- C12N5/0671—Three-dimensional culture, tissue culture or organ culture; Encapsulated cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/50—Proteins
- C12N2533/54—Collagen; Gelatin
Definitions
- the present invention relates to a particulate culture carrier, and more particularly to a cell culture technique using a particle-containing cell condensate in a culture in which cells are adhered to a culture vessel.
- Patent Document 1 describes a particle-containing cell aggregate composed of hydrogel particles and cells and a method for producing the same.
- Patent Document 2 describes a gelatinizable cell culture substrate using gelatin and boron as essential raw materials and a method for using the same, and an appropriate particle size is 5 to 100 ⁇ m in diameter, more preferably. It is described that the diameter is 10 to 70 ⁇ m.
- Patent Document 3 which is another background art, gelatin spheres having a predetermined size (10 to 50 ⁇ m, etc.) are arranged on a flow path surface partitioned by micro-sized pillars, and cells are three-dimensionally cultured. Are listed.
- Patent Document 4 which is another background art, a method of recovering a three-dimensional tissue by culturing cells in a culture substrate composed of fine particle-containing polymer gel particles and an aggregate of the fine particle-containing polymer gel particles Is described.
- Patent Document 5 describes an example in which cells are supported and cultured on the surface of a microcarrier made of gelatin hydrogel having a particle diameter of 0.1 to 500 ⁇ m.
- Patent Document 1 reports a method of incorporating a polymer hydrogel into cell aggregates as a method for improving substance permeability into cell aggregates in which cells are gathered three-dimensionally. Since hydrogel particles are permeable to nutrients and oxygen, it is considered that the lack of nutrients and oxygen in the internal cells and the problem of waste accumulation are improved in the three-dimensional aggregate in which the particles are dispersed. . Patent Document 1 reports a method of preparing an aggregate having a diameter of about 1 mm incorporating particles by mixing proliferative cells and hydrogel particles in a non-adhesive culture vessel and then seeding them.
- the object of the present invention is to improve the permeability of nutrients, oxygen, drugs, reagents, and the like from a culture medium while adhering a three-dimensional aggregate having a cell function close to that of a living body to a culture vessel.
- the object is to provide a cell culture method, a particulate culture carrier, and a particle-containing cell aggregate.
- cells that form a cell aggregate including particles by seeding isolated cells in a culture container and administering particles having cell adhesion and substance permeability are provided.
- a culture method is provided.
- a particulate culture carrier is provided.
- the present invention provides a particle-containing cell aggregate formed by seeding cells in a culture vessel and administering particles having cell adhesion and substance permeability on the cells. To do.
- a culture method that can maintain a cultured cell having a function similar to that of a living body for a long period of time and can be used for a cell test in a test tube.
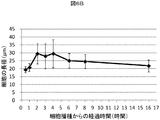
- Example 1 It is a figure which concerns on Example 1 and shows the example which measured the metabolic enzyme cytochrome P450 (molecular species CYP3A) activity of the rat hepatocyte aggregate which administered the particle
- FIG. 2 is a diagram showing each culture condition of the measurement example in FIGS. 1A to 1C according to Example 1.
- Example 1 It is a figure which concerns on Example 1 and shows the example of a time-dependent change of a cell aggregate formation when the particle
- Example 3 It is a figure which concerns on Example 3 and shows the example of a time-dependent change by microscopic observation when a rat hepatocyte is seed
- FIG. 4 is a diagram showing an example in which rat hepatocytes were cultured and mitochondrial dehydrogenase activity was measured by WST-1 assay according to Example 4.
- Example 5 It is a figure which concerns on Example 5 and shows the example of the time-dependent change of cell aggregate formation when administering a particle
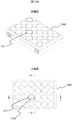
- FIG. 10 is a partial enlarged view of a multiwell plate for cell culture used in each example according to Example 6.
- Example 6 It is a figure which concerns on Example 6 and shows the example of the scanning electron micrograph of the culture surface by which the assembly area
- the present invention relates to a cell culture method for forming a cell aggregate including particles by seeding isolated cells in a culture vessel and administering particles having cell adhesion and substance permeability, a particulate culture carrier, and It is an invention of particle-containing cell aggregates.
- the particle having cell adhesion and substance permeability is a micro-scaffold particle that is a cell scaffold particle, and the administration of the particle is performed after the seeded cells settle on the culture surface. More specifically, after seeding the isolated cells in the culture vessel, the cells start to adhere to the culture surface, and then the cells move and the adjacent cells adhere to each other to start the formation of cell aggregates. By carrying out the process until it is completed, adhesion of the cells to the culture container, adhesion between the cells and the particles can be ensured.
- the cell culture method using microscaffold particles which is a preferred embodiment of the present invention is a culture method using fine particles obtained by forming chemical crosslinks between gelatin molecules by heat treatment or the like.
- a chemical crosslinking agent such as glutaraldehyde can be used.
- the concentration of the gelatin aqueous solution when preparing the gelatin particles is preferably 10% by weight, but is not limited thereto.
- One method of making gelatin particles is to prepare an emulsion by stirring an aqueous gelatin solution with an oil such as olive oil, cooling the emulsion, degreasing and dehydrating with cold acetone, etc. A procedure for obtaining fine particles is mentioned. By sifting at this stage, it is possible to divide the particle size. The dried particles are heat treated to crosslink the molecules.
- a microreactor for example, a microprocess server, manufactured by Hitachi Plant Technology
- a microprocess server manufactured by Hitachi Plant Technology
- the micro-scaffold particles may be any particles made of any material as long as they have two properties: cell adhesion on the particle surface and water-soluble substance permeability into the particles.
- it may be made of a cell adhesive substance, or the surface may be covered with a substance to which cells adhere.
- the cell adhesive substance include collagen, fibronectin, laminin, glycosaminoglycan, antibodies that bind to proteins and sugar chains on the cell membrane surface, sugar chains that bind to the cell membrane surface, and poly-L-lysine. However, it is not limited to these.
- hydrogels containing a large amount of water, porous ceramics, and porous metals, but are not limited thereto.
- Hydrogels include polyacrylic acid, polyacrylamide, polyvinylpyrrolidone, polyvinyl alcohol, alginic acid, starch, polylactic acid, agarose, pectin, cellulose, glucosaminoglycan, collagen, gelatin, fibronectin, vitronectin, laminin, proteoglycan, etc.
- quality ceramics include, but are not limited to, hydroxyapatite.
- Particle-containing cell aggregates such as microscaffold particles of the present invention are prepared by seeding cells exhibiting adhesiveness to a culture surface in a culture vessel having an adhesion region on the culture surface, and after the cells settle on the vessel. Is a cell aggregate formed by administration. When cells adhere to the raw material gelatin, cell aggregates including particles are formed in a state where the cells adhere to the culture surface.
- the particle administration time, particle size, and number of administration it is possible to obtain cell aggregates without detaching the cell aggregates from the culture equipment and without inhibiting cell-cell adhesion in the aggregates. is there.
- the cell function to be maintained may differ, so that it is desirable to adjust the appropriate particle administration time, particle diameter, and number of administration according to the purpose.
- the present invention including particles in a state in which cell aggregates are adhered to a culture container is a culture method for obtaining useful substances by floating culture by attaching cells to the surface of a conventional microcarrier. This is different from the technique of forming a cell aggregate including a culture carrier in a container.
- the present invention which includes particles in a state where cell aggregates are adhered to a culture vessel, can perform drug treatment and cell activity measurement in the same manner as in the usual test using monolayer cultured cells. .
- the particle inclusion cell aggregate which maintained activity can be easily used for cell tests such as drug screening in drug discovery research and examination of differentiation induction conditions in regenerative medicine research.
- drugs and reagents can easily penetrate into internal cells through substance-permeable microscaffold particles, the problem of drug and reagent permeability that is concerned with cell aggregates that do not contain particles is solved. Can do.
- Example 1 is an example relating to a particulate culture carrier for culturing hepatocytes, a particle-containing cell aggregate, and a cell culture method.
- the hepatocytes used in this example are mature hepatocytes that do not proliferate, and their function tends to decrease during the culture period. For this reason, generally, the period during which hepatocytes can be donated to drug tests and the like is from the start of culture to about one week.
- gelatin particles are administered to rat hepatocytes and cultured to extend the period during which the metabolic enzyme cytochrome P450 (CYP3A) activity can be measured to 10 days or more.
- CYP3A metabolic enzyme cytochrome P450
- FIG. 1A, FIG. 1B, and FIG. 1C show that in this example, four types of gelatin particles having different diameters are administered to rat hepatocytes, enzyme activity is measured over time, and the enzyme activity maintaining effect varies depending on the particle size. This is an example.
- the culture conditions 1 to 7 shown in these figures are as shown in 1 to 7 of FIG. 1D.
- FIG. 2 shows changes over time in the formation of cell aggregates when particles of different diameters were administered to rat hepatocytes under conditions 1 to 5 and cultured for 11 days.
- the enzyme activity measurement in FIGS. 1A, 1B, and 1C was performed. It is an example of the microscope observation image of the sample to be used.
- FIG. 3 shows an example of a microscopic observation image of gelatin particle suspensions having different diameters swollen in a medium.
- the rat hepatocytes of this example were prepared from Fisher 344 rats or SD rats male, 5-8 weeks old (both Charles River Japan) by the two-stage collagenase reflux method.
- Cell concentration 5 ⁇ 10 5 cells / ml in Williams E medium (8.6 nM insulin (Sigma Aldrich), 255 nM dexamethasone (Nacalai Tesque), 50 ng / ml EGF, 5 KIV / ml aprotinin) containing 10% fetal calf serum
- the isolated hepatocytes were suspended in and seeded at a density of 1 ⁇ 10 5 cells / cm 2 per culture area of the culture vessel.
- the culture plate was placed in an incubator at 37 ° C. and a carbon dioxide gas concentration of 5% to culture the cells.
- the culture vessel used in this example has a large number of hole structures with a diameter of 200 ⁇ m on the culture surface, and a fine 2 ⁇ m diameter and 1 ⁇ m height at the center of the bottom surface of each hole.
- a 96-well nanopillar culture plate with processed protrusions was used.
- the former describes a culture sheet on which nanopillars capable of controlling cell adhesion and migration are formed.
- the latter describes the conditions under which hepatocytes can easily form cell aggregates on the nanopillar sheet, and describes that the function of the three-dimensional aggregate is closer to that of a living body than the two-dimensional culture method.
- the material of the culture plate is polystyrene, and the culture surface having holes and fine protrusions is hydrophilized by surface modification and has cell adhesion.
- the nanopillar portion which is a fine protrusion, has an effect of capturing cells.
- BD biocoat
- Non-Patent Document 1 In vitro Proliferation and condrogenic differentiation of rat bone marrow stem cells cultured with gelatin hygrogel microspheres for TGF- ⁇ 1 release. Ogawa T. et al. Journal of Biomaterias Science, 21, 609-621 (2010)
- Dry gelatin powder (isoelectric point 5) was weighed, purified water was added to a concentration of 10%, and the mixture was swollen and dissolved at 37 degrees with stirring to prepare an aqueous gelatin solution.
- Olive oil was stirred while warming in a 40 ° water bath, and the gelatin solution was added thereto, and emulsified by stirring with a homogenizer (manufactured by Polytron).
- the emulsion was cooled with stirring to gelatinize the gelatin. After cooling acetone was added and further stirred, particles were collected by centrifugation. The particles were passed through a sieve while washing with cold acetone, and the particles that passed through the sieve were collected, and dried gelatin particles were obtained by vacuum drying.
- Thermal dehydration crosslinking was performed by heating at 140 ° C. for 48 hours under reduced pressure using a vacuum oven. Sterilized with ethylene oxide gas and stored in sterile dry particles.
- the average particle size and standard deviation of the particle size of the gelatin particles were obtained by swelling the particles after thermal crosslinking with water and measuring the size under a microscope.
- four types of gelatin particles having different sizes were used, as shown in FIG. 1D, with a diameter of 4.5 ⁇ 1.7 ⁇ m, 10.2 ⁇ 4.3 ⁇ m, 16.8 ⁇ 6.7 ⁇ m, 23.7. ⁇ 10.3 ⁇ m was used.
- FIG. 3 shows microscopic images of four types of gelatin particles swollen in the medium.
- the particle concentration of the particle suspension was adjusted to 3 mg / ml by dry particle weight.
- the medium in which the cells are cultured is changed from Williams E medium containing 10% fetal bovine serum to serum-free Williams E medium in which the particles are suspended, whereby the particles are administered to the cultured cells. did.
- Matrigel (BD) was replaced with serum-free Williams E medium containing 0.25 mg / ml of total protein, and thereafter culture medium was replaced with serum-free Williams E medium every 24 hours. .
- monolayer culture as a conventional method was performed. Isolated hepatocytes were suspended at a cell concentration of 5 ⁇ 10 5 cells / ml and seeded on the aforementioned culture plate at a density of 1 ⁇ 10 5 cells / cm 2 per culture area. The culture plate was placed in an incubator at 37 ° C. and a carbon dioxide gas concentration of 5% to culture the cells. Monolayer culture was replaced with serum-free Williams E medium 24 hours after cell seeding, and thereafter replaced with serum-free Williams E medium every 24 hours.
- the activity of the hepatocyte metabolic enzyme cytochrome P450 was measured using P450-Glo TM Assay (Luciferin-IPA, Promega). The measurement procedure is as follows.
- the medium in which hepatocytes were cultured was removed, replaced with 60 ⁇ l of Luciferin-IPA solution (final concentration 3 ⁇ M) diluted to 1/1000 with the medium, and reacted for 1 hour in an incubator at 37 ° C. and 5% carbon dioxide concentration. .
- 50 ⁇ l of the culture supernatant of each well was dispensed into a white 96-well plate, 50 ⁇ l of luciferin detection reagent (Promega) was added, and the mixture was allowed to react at room temperature for 20 minutes while protected from light.
- the white plate after the reaction was placed in a plate reader (SH-8000 Lab, Corona Electric), and luminescence was measured at a gate time of 1 second.
- FIG. 1A, FIG. 1B, and FIG. 1C show the average values and standard deviations of 8 wells for each culture condition.
- the activity of the cells not administered with the particles was high, and therefore the effect of the particles was not observed in the early stage of the culture.
- the activity of the cells of the aggregate administered with particles having a diameter of 4.5 ⁇ 1.7 ⁇ m and the activity of the cells of the aggregate not administered with particles were high.
- the activity of the aggregate cells administered with particles of 10.2 ⁇ 4.3 ⁇ m, 16.8 ⁇ 6.7 ⁇ m, and 23.7 ⁇ 10.3 ⁇ m was different. Compared to the above conditions, the activity was low in particles having a diameter of 4.5 ⁇ 1.7 ⁇ m, no particles, and monolayer sandwich culture, and the activity was below the detection limit in the most common monolayer culture.
- FIG. 2 shows a micrograph of the cultured hepatocytes observed with a phase-contrast inverted microscope, and photographing an arbitrary cell aggregate near the center of the well of the culture plate.
- cells administered with particles having a diameter of 4.5 ⁇ 1.7 ⁇ m promoted adhesion between cells more than cells not administered with particles on the third and fifth days of culture. There was a tendency to agglomerate. Thereafter, the shape of the aggregates on day 7 and 11 of the culture was similar to the cells to which particles were not administered, and the aggregates adhered to the area where the fine protrusions gathered at the center of the hole of the culture plate.
- the diameter of hepatocytes is about 15-30 ⁇ m
- particles with a diameter of 4.5 ⁇ 1.7 ⁇ m are about 1/10 to 1/5 the size of the cells. It did not have a big impact. Since the size of the agglomerates on day 11 is not significantly different from that of no particles, the particle dosage is 3 mg / ml dry particle weight for any particle, but the diameter is 4.5 ⁇ 1. In the case of 7 ⁇ m particles, the total amount of particles taken into the aggregates was considered to be small.
- particles having a diameter of 10.2 ⁇ 4.3 ⁇ m having a particle size of 1/5 or more of the cells formed aggregates including the particles.
- a slightly planar irregular aggregate was formed. Assuming that the diameter of the hepatocytes is about 15 to 30 ⁇ m, it was considered that the particles having a diameter of 23.7 ⁇ 10.3 ⁇ m have the same particle size as that of the cells to about twice the size of the cells.
- cytochrome P450 molecular species CYP3A
- Example 2 when gelatin particles having different diameters were administered to rat hepatocytes and cultured, depending on the particle size, the particles included in the cell aggregates were distributed only inside the aggregates, or only inside the aggregates. An example in the case where it is also present in the portion in contact with the medium will be described.
- FIG. 4 is an example of a micrograph showing a cross section of the gelatin particle-containing cell aggregate in Example 2.
- the schematic diagram at the bottom of the photo shows the outline of the cells that can be identified in the upper photo with lines, and the location of the included particles with the US marks.
- the culture vessel has a large number of hole structures with a diameter of 200 ⁇ m on the culture surface described in JP-A-2011-004612 described above, and fine protrusions with a diameter of 2 ⁇ m and a height of 1 ⁇ m are processed at the center of the bottom of each hole.
- the nanopillar culture sheet was used by adhering to the well bottom of the 2-well slide chamber.
- the culture sheet is made of polystyrene, and the culture surface having holes and fine protrusions is hydrophilized by surface modification and has cell adhesion.
- the nanopillar portion which is a fine protrusion, has an effect of capturing cells.
- Isolated hepatocytes were suspended in Williams E medium containing 10% fetal bovine serum at a cell concentration of 5 ⁇ 10 5 cells / ml and seeded at a density of 1 ⁇ 10 5 cells / cm 2 per culture area of the culture vessel.
- the slide chamber was placed in an incubator at 37 ° C. and a carbon dioxide gas concentration of 5%, and the cells were cultured.
- particles with a diameter of 4.5 ⁇ 1.7 ⁇ m were suspended in Williams E medium containing 10% fetal calf serum. Since the particles were fine and could not be counted with a cell counter, a suspension having the same concentration as the particles having a diameter of 16.8 ⁇ 6.7 ⁇ m was prepared by dry particle weight.
- the particles were administered to the cells by replacing them with Williams E medium containing 10% fetal bovine serum in which the particles were suspended. 24 hours after cell seeding, the medium was replaced with serum-free Williams E medium. After 48 hours, Matrigel (BD) was replaced with serum-free Williams E medium containing 0.25 mg / ml of total protein, and then serum-free Williams E every 24 hours. The medium was changed to E medium and the culture was continued for 5 days.
- Williams E medium containing 10% fetal bovine serum in which the particles were suspended.
- serum-free Williams E medium After 48 hours, Matrigel (BD) was replaced with serum-free Williams E medium containing 0.25 mg / ml of total protein, and then serum-free Williams E every 24 hours. The medium was changed to E medium and the culture was continued for 5 days.
- the cells cultured for 5 days on the nanopillar culture sheet were pre-fixed with 0.1 M phosphate buffer (pH 7.4) containing 2.5% glutaraldehyde and post-fixed with phosphate buffer containing 1% osmium tetroxide.
- the nanopillar sheet to which the cells were adhered was embedded in low melting point agar (Sea plaquer agarose, Lonza) with a concentration of 1.5%, dehydrated in an ethanol dilution series, and polystyrene was dissolved with propylene oxide.
- the Epon-Alardite mixed resin was infiltrated into the agar in which the cells were embedded, and polymerized by heating.
- a quasi-ultra thin section having a thickness of 400 mm was prepared from a resin-embedded sample.
- the sections were adhered to a slide glass, stained with 1% toluidine blue, and observed with an upright light microscope.
- Example 3 describes an example showing that the effect of maintaining the activity of rat liver cell metabolic enzyme cytochrome P450 (CYP3A) varies depending not only on the particle diameter but also on the dose of the particles.
- cytochrome P450 CYP3A
- FIG. 5A and FIG. 5B are diagrams showing examples in which the basal activity and drug-inducing activity of CYP3A were measured in this example.
- FIG. 5A shows the ratio of administering particles to the number of cells, and the time for administering the particles.
- 5B shows an example in which rat liver cells were cultured for 10 days and the basal activity of the metabolic enzyme cytochrome P450 (molecular species CYP3A) was measured.
- FIG. 5B shows the ratio of the number of particles administered to the number of cells and the time for administering the particles.
- rat hepatocytes were seeded using 96-well nanopillar culture equipment. Particles having a diameter of 16.8 ⁇ 6.7 ⁇ m were swollen in Williams E medium containing 10% fetal bovine serum, and after 2.5 hours or 22 hours after cell seeding, the cell number: particle number was 1: 1.
- the particle suspension was administered onto the cells in a ratio of 4: 1, 10: 1, 20: 1, 50: 1. 24 hours after cell seeding, the serum-free Williams E medium was replaced. After 48 hours, Matrigel (BD) was replaced with serum-free Williams E medium containing 0.25 mg / ml of total protein, and serum-free every 24 hours thereafter. The medium was changed to Williams E medium and cultured for 10 days.
- cytochrome P450 molecular species CYP3A
- FIG. 6A shows an example in which changes in cell morphology (time-dependent change) over time after seeding rat hepatocytes in a low-adhesion culture vessel are shown by phase contrast microscopy and F-actin distribution in this example.
- 6A is a photomicrograph of the cell morphology observed with an inverted microscope over time after cell seeding, with the top row being a phase contrast microscope image and the bottom row being a fluorescence microscope image in which F-actin is stained with rhodamine-labeled phalloidin. is there.
- the phase contrast image shows the whole image, and the outline of each cell is shown by the F-actin fluorescent staining image.
- FIG. 6B is an example in which the major axis of 60 cells arbitrarily selected in the photomicrograph after cell seeding was measured, the average value of the major axis and the standard deviation were obtained, and the change with time was shown.
- the procedure for rhodamine-labeled phalloidin staining in FIG. 6A is as follows. The cells fixed with 4% paraformaldehyde were washed with 0.1 M phosphate buffer (pH 7.4). To prevent non-specific staining, treatment with protein block (DAKO) for 30 minutes, followed by treatment with rhodamine-labeled phalloidin (Invitrogen) diluted 1:40 with PBS for 30 minutes, 0.075% Brij35j (Sigma Aldrich) ) Washed with PBS containing sputum.
- DAKO protein block
- rhodamine-labeled phalloidin Invitrogen
- PBS 0.075% Brij35j
- the cells 0.5 hours after seeding were isolated and had a round shape and an average diameter of 20 ⁇ m or less. After 1.0 hour, the two-dimensional image of the cell shown in the photograph was slightly larger, and the average diameter slightly exceeded 20 ⁇ m.
- This example describes another example showing that the effect of maintaining the activity of rat hepatocytes varies depending on the dose of particles.
- the WST-1 test is a method for measuring the activity of mitochondrial respiratory chain enzyme (succinate tetrazolium reductase) in living cells, and the higher the measured value, the higher the respiratory activity of spheroids.
- rat hepatocytes were seeded using 96-well nanopillar culture equipment. Particles having a diameter of 16.8 ⁇ 6.7 ⁇ m were swollen in Williams E medium containing 10% fetal bovine serum, and after 2.5 hours or 22 hours after cell seeding, the cell number: particle number was 1: 4, Cell suspension of the particle suspension to be 1: 2, 1: 1, 2: 1, 4: 1, 8: 1, 10: 1, 20: 1, 40: 1, 50: 1, 80: 1 Was administered on top of. 24 hours after cell seeding, the serum-free Williams E medium was replaced. After 48 hours, Matrigel (BD) was replaced with serum-free Williams E medium containing 0.25 mg / ml of total protein, and serum-free every 24 hours thereafter. The medium was changed to Williams E medium and cultured for 5 days.
- BD Matrigel
- Premix WST-1 (Takara Bio) was used. After culturing hepatocytes for 5 days, the entire medium was removed from each well of the 96-well plate containing the cells, and washed twice with Hank's solution. After removing the washing solution, 65 ⁇ l of Hank's solution containing 10% (v / v) concentration of WST-1 was added and reacted in an incubator maintained at 37 ° C. and 5% carbon dioxide concentration for 1 hour. The 96 wells after the reaction were set in a plate reader, and the absorbance at two wavelengths (440 nm, 650 nm) was measured at a half width of 5 nm. A value obtained by subtracting the value of 650 nm, which is the reference wavelength, from the value of 440 nm was obtained.
- FIG. 7 shows that rat hepatocytes were cultured for 5 days while changing the ratio of administration of particles to the number of cells and the administration time of particles, and the activity of mitochondrial dehydrogenase was measured by WST-1 assay.
- the results of calculating the average value and standard deviation of 3 wells for the group were shown.
- the number of cells: number of particles is 1:80, that is, the effect of particles is recognized even when the number of particles is more than 1/80 with respect to the number of cells.
- Administering more particles than 4: 1 significantly increased the WST-1 measurement, and further increasing the dose beyond 1: 1 further increased the value.
- the above results indicate that the intercellular particles improve the nutrient and oxygen supply from the medium to maintain the cell activity, and increase the permeability of the detection reagent WST-1 into the cells. This is considered to be the reason that the detection efficiency of respiratory activity is improved.
- Example 1 the description of the isolation and culture of rat hepatocytes already described in Example 1, the method for producing gelatin particles, and the procedure for swelling the particles in the medium are omitted.
- This example shows an example of the cell aggregate formation process and the shape of the formed cell aggregate when the particles are administered in different amounts. Furthermore, an example will be described that shows that the efficiency of forming a capillary bile duct that is a liver tissue-specific structure varies depending on the dose of particles.
- FIG. 8 is a diagram showing an example of changes over time in the formation of cell aggregates when particles are administered at a different ratio to the number of cells and cultured for 5 days.
- the phase contrast image shows the whole image of the particles and cells
- the F-actin fluorescent staining image shows the distribution of cells. That is, to a rat hepatocyte, particles having a diameter of 16.8 ⁇ 6.7 ⁇ m were administered so that the cell number: particle number was 1: 1, 4: 1, 10: 1, 20: 1, 50: 1.
- the process of cell aggregate formation when cultured for 5 days is shown.
- the upper row of each column is a phase contrast microscope image, and the lower row is a fluorescence microscope image of F-actin stained with rhodamine-labeled phalloidin.
- F-actin forms a membrane lining structure and cells have a polygonal three-dimensional structure. By observing the distribution of F-actin, it is possible to verify whether the tissue is close to living cells. So far, in the cell aggregate formed in the nanopillar culture vessel, F-actin has formed a membrane lining structure close to that of a living body (see WO2010 / 079962).
- the aggregate formed with the cell number: particle number 50: 1 or without the particles had F-actin forming a membrane backing structure close to that of a living body.
- the particles are administered at a cell number: particle number of 20: 1, 10: 1, 4: 1, or 1: 1, the cell surface that adheres to the gelatin particles is linear or dotted. An image showing strong fluorescence was observed, suggesting the possibility of adhesion points or stress fibers. These are structures formed when cells adhere to the extracellular matrix. Therefore, it was considered that the cells formed an adhesion structure on the surface of the gelatin particles, similar to the adhesion with the extracellular matrix.
- the cell number: particle number is preferably 50: 1 or less in order to form a membrane backing structure close to a living body.
- FIG. 9 is a diagram showing an example of fluorescent reagent uptake and excretion into the capillary bile duct when particles are administered at different ratios to the number of cells and cultured for 5 days. Specifically, this is an example in which the excretion ability of the capillary bile duct formed on the intercellular adhesion surface of hepatocytes is shown on the 5th day of culture using a fluorescent labeling reagent carbodichlorofluorescein-diacetate (CDFDA, Molecular Probe).
- CDFDA fluorescent labeling reagent carbodichlorofluorescein-diacetate
- CDFDA administered into the medium is taken up into cells and metabolized to become carbodichlorofluorescein (CDF), which is excreted into the capillaries through the transporter multidrug resistance-associated protein 2 (MRP2) of the cell membrane facing the capillaries.
- CDF carbodichlorofluorescein
- MRP2 transporter multidrug resistance-associated protein 2
- the procedure of the CDFDA process of this embodiment is as follows.
- the medium of hepatocytes cultured in a 24-well nanopillar culture plate was removed and washed twice with Krebs-Henseleit buffer (KH buffer).
- KH buffer Krebs-Henseleit buffer
- 400 ⁇ l of KH buffer containing 5 ⁇ M CDFDA was added per well, placed in a 37 ° incubator for 20 minutes, washed twice with KH buffer, and observed with an inverted fluorescence microscope.
- capillary bile duct Since the capillary bile duct is formed on the cell-cell adhesion surface, cell-cell interaction is required. In addition, it is known that proteins present inside the cell membrane on the intercellular adhesion surface play an important role in the expression of transporter MRP2, which excretes drugs from the cell into the capillary bile duct. For this reason, it was thought that the capillary bile duct was difficult to regenerate when there were many particles because the particles that existed between cells excessively inhibited adhesion between cells.
- the number of cells: number of particles was 50: 1 or less.
- the cell number: particle number is desirably 50: 1 to 4: 1, and more desirably 50: 1 to 10: 1. This is because, in order to improve both the uptake and excretion of drugs, not only intercellular adhesion is promoted, but a surface in contact with the medium through particles must be formed. It was.
- Example 4 the inclusion of a large number of particles showed the possibility of improving the supply of medium components to the inside of cell aggregates.
- the presence of excessive particles caused liver It became clear that the excretion function peculiar to a tissue fell. Although it is a contradictory condition in particle administration, it is desirable to obtain an appropriate particle amount according to the purpose of use of the cell aggregate.
- Example 3 the isolation and culture of rat hepatocytes already explained in Example 1, the method for producing gelatin particles, the procedure for swelling particles in the medium, and the procedure for rhodamine-labeled phalloidin staining shown in Example 3 are explained. Omitted.
- a culture container provided with a cell adhesion region for capturing cell aggregates by molding a plurality of protrusions locally on the culture surface with low cell adhesion used in each of the examples described above.
- An example will be described.
- FIG. 10A shows a perspective view and an upper surface of the cell culture multiwell plate 100 at the upper and lower stages.
- the culture surface of the bottom surface 102 of the well 101 indicated by AA and BB is subjected to fine three-dimensional processing.
- FIG. 10B shows an enlarged view of the AA and BB portions, an enlarged view of the CC and DD portions, and an end surface of the line EE in FIG. 10A at the upper, middle, and lower portions.
- the fine three-dimensional processing applied to the culture surface is enlargement of the CC and DD portions and the hole 103 shown on the end surface of the EE line, and the collective region 105 of the protrusion 104 is formed at the center of the hole 103. It is a figure which shows the example currently shape
- molded shows the example currently shape
- FIG. 11 is an example of a scanning electron micrograph of the culture surface in which the aggregation region 105 of the protrusions 104 is molded at the center of the hole 103 described above.
- FIG. 12 shows that cells 106 are seeded in the holes 103 on the culture surface shown in FIG. 10B and FIG. 11 and gelatin particles 107 are administered when the cells 106 adhere to the bottom surface, so that cell aggregates containing the particles can be obtained.
- molded in the center of the hole 103 is shown.
- a local cell adhesion region that captures cells on the culture surface is not limited to the protrusions of this example, extracellular matrix components such as collagen and laminin, antibodies that bind to the cell membrane surface, It can also be formed by optionally patterning a cell-adhesive substance such as a sugar chain that binds to the cell membrane surface or poly-L-lysine.
- the culture container of this example described above holds the particle-containing cell aggregates in the culture container by forming a cell adhesion region capable of capturing the cell aggregates on the low adhesion culture surface. Further, preferably, by providing a partition on the culture surface, the number of cells and the number of particles forming the aggregate are controlled to a desired number, and a particle-containing cell aggregate having a desired size is formed. Such particle-containing cell aggregates adhered to the culture container of this Example can be used as a material for cell tests.
- Example 1 an example of a human cell test method involving particles will be described.
- Example 3 in cell aggregates containing particles, the activity of the metabolic enzyme cytochrome P450 (molecular species CYP3A) is maintained for a longer period than aggregates not containing particles and conventional monolayer culture methods. Showed the effect.
- cytochrome P450 molecular species CYP3A
- cytochrome P450 is an enzyme that metabolizes drugs taken into hepatocytes from the blood, in drug discovery research, metabolism in pharmacokinetics, drug interaction by CYP enzyme induction, liver toxicity by CYP metabolites, etc. It has become.
- cytochrome P450 since there are species differences in enzyme substrate properties and metabolic activities, not only animal experiments but also human tests are required, and test methods using isolated human hepatocytes have been developed. Therefore, by using cell aggregates containing the above-mentioned microscaffold particles for cell tests, it is possible to carry out tests using isolated human hepatocytes that tend to lose their activity stably for a longer period of time than before. It becomes possible.
- the cryopreserved human hepatocytes are thawed, suspended in Williams E medium containing 10% fetal bovine serum at a cell concentration of 5 ⁇ 10 5 cells / ml, and 1 ⁇ 10 5 cells / cm 2 per culture area of the culture vessel. Seeded at a density of The culture plate was placed in an incubator at 37 ° C. and a carbon dioxide gas concentration of 5% to culture the cells. Cell aggregates containing the particles are obtained by swelling particles having a diameter of 16.8 ⁇ 6.7 ⁇ m in a medium and administering the particle suspension onto the cells so that the number of particles is larger than the number of cells. Formed.
- the drug metabolized by CYP was administered to the medium, and the cells and medium were collected after 2 and 7 days.
- the cell lysate and the medium were deproteinized, and the collected sample solution was analyzed by mass spectrometry to identify drug metabolites in the cell or in the medium.
- cryopreserved human hepatocytes were cultured, and the particles were administered to form cell aggregates containing the particles.
- the activity of CYP enzyme was measured using a detection reagent that specifically reacts with each molecular species of CYP.
- the induction rate was calculated by dividing the induced activity value by the basal activity value, with the CYP enzyme activity of cells not treated with the subject drug as the basal activity and the CYP enzyme activity of cells treated with the subject drug as the inductive activity.
- CYP enzyme activity for example, the P450-Glo TM Assay series (Promega) shown in Example 1 and Example 3 is used to determine the CYP induction activity by the test drug by quantifying luminescence. Can be measured.
- CYP-inducing activity can be measured by quantifying a metabolite by mass spectrometry using a known substance that is metabolized specifically for each molecular species of CYP.
- a method for estimating the degree of drug interaction by the drug to be tested can be cited.
- liver toxicity analysis using CYP metabolites will be described. Similar to the metabolite analysis example, cryopreserved human hepatocytes were cultured, and the particles were administered to form cell aggregates containing the particles. The toxicity of the drug was evaluated by changing the concentration of the test drug and administering it to the medium, and measuring the change in the number of cells over time. For example, the WST-1 method or the ATP quantification method shown in Example 4 can be used for measuring the number of cells. As mentioned above, although the some test example was given as description of Example 7, it is not restricted to this.
- the cell culture method according to the present invention described in detail above is a culture method characterized in that cells are adhered to the culture surface of a culture vessel, and has been performed by a conventional monolayer culture method so far. Cell tests can be applied in the same procedure.
- the particulate culture alone and the particle-containing cell aggregate according to the present invention are three-dimensional cell aggregates having a structure closer to that of a living body than monolayer culture, and supply nutrients and oxygen in the medium via the particles.
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Sustainable Development (AREA)
- Gastroenterology & Hepatology (AREA)
- Cell Biology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
細胞の機能が生体に近い3次元凝集体を培養容器に接着させながら、培地からの栄養、酸素、薬物、ならびに試薬の浸透性を向上させ、細胞の機能を維持できる培養法を提供する。単離細胞を培養容器に播種して、細胞が培養面へ沈降した以降に、細胞の上に、細胞接着性と物質浸透性を有するマイクロスキャフォールド粒子を投与することによって、細胞を培養容器に接着させながら、粒子を包含する細胞凝集体を形成することができる。例えば、細胞凝集体に包含させる培養担体のサイズが、細胞直径の1/5以上であるゼラチン粒子を、単位培養面積当たりの培養細胞の個数に対して細胞数:粒子数が80:1以上で、投与することによって、粒子を包含する細胞凝集体を形成する。
Description
本発明は、粒子状の培養担体に係り、特に、細胞を培養容器に接着させる培養において粒子包含細胞凝縮体を用いる細胞培養技術に関する。
本技術分野の背景技術として、特許文献1がある。特許文献1には、ハイドロゲル粒子と細胞から構成される粒子含有細胞凝集体およびその製造方法が記載されている。また、特許文献2には、ゼラチンおよびホウ素を必須の原料とするゲル化可能な細胞培養基材およびその使用方法が記載されており、適当な粒子サイズは5~100μm直径であり、より好ましくは10~70μm直径であることが記載されている。
別の背景技術である特許文献3には、マイクロサイズのピラーで区画された流路面に、所定の大きさ(10~50μm等)のゼラチン球を配置して、細胞を3次元培養することが記載されている。
別の背景技術である特許文献4には、微粒子含有高分子ゲル粒子からなる培養基材と、微粒子含有高分子ゲル粒子の集合体の中で細胞を培養し、3次元組織体を回収する方法について記載されている。また、特許文献5には、ゼラチンハイドロゲルからなる粒子径0.1から500μmのマイクロキャリアの表面に細胞を支持し、培養する例が記載されている。
創薬研究や、再生医療関連技術の産業化などにおいて、培養する細胞の機能を生体に近い状態で長期間維持する培養技術の開発が求められている。
体の基本単位は細胞である。そして生物機能の最小構成単位は、細胞-細胞間接着を基本構造として形成される細胞凝集体である。このため、体外における細胞の生物医学的研究ならびに細胞の産業利用においては、試験管内で細胞を集合させた細胞凝集体作製が、重要な要素技術となっている。たとえば細胞を用いた薬物スクリーニングにおいても、遊離した単独細胞に比べて、細胞凝集体の生物機能が高く、薬物に対する感度が高くなることが知られ、生体に近い細胞凝集体を用いた薬物作用の研究や毒物評価が可能になると考えられている。
体の基本単位は細胞である。そして生物機能の最小構成単位は、細胞-細胞間接着を基本構造として形成される細胞凝集体である。このため、体外における細胞の生物医学的研究ならびに細胞の産業利用においては、試験管内で細胞を集合させた細胞凝集体作製が、重要な要素技術となっている。たとえば細胞を用いた薬物スクリーニングにおいても、遊離した単独細胞に比べて、細胞凝集体の生物機能が高く、薬物に対する感度が高くなることが知られ、生体に近い細胞凝集体を用いた薬物作用の研究や毒物評価が可能になると考えられている。
このような中で、細胞凝集体を効率よく作製して、生物機能を高める研究がおこなわれてきた。しかしながら、この分野における問題として、細胞凝集体内部への栄養、酸素の供給と、内部細胞からの老廃物除去の効率が低下することが懸念される。供給や排泄効率が低下した結果として、細胞の機能が低下し、細胞凝集体の利用が困難となる。一方、薬物スクリーニングなどを目的として細胞の反応を解析する場合には、細胞凝集体内部への標的薬物ならびに分析試薬の浸透性低下が問題となる。
これらの問題を解決するためには、細胞凝集体内での細胞-細胞間接着を損なうことなく、凝集体内部と外部との間での物質浸透性を向上させるための技術が必要不可欠である。上述の特許文献1には、細胞が3次元的に集合した細胞凝集体内部への物質透過性を向上させる方法として、高分子ハイドロゲルを細胞の凝集体に包含させる方法が報告されている。ハイドロゲル粒子は、栄養、酸素の透過性を有することから、粒子が分散している3次元凝集体では、内部細胞における栄養と酸素の不足や、老廃物蓄積の問題が改善されると考えられる。特許文献1に、非接着性の培養容器に、増殖性の細胞とハイドロゲル粒子を混合してから播種することにより、粒子を取り込んだ直径1mm近い凝集体を作製する方法が報告されている。
一方、薬物スクリーニングなど、体外における細胞試験に細胞凝集体を用いる場合には、試験溶液の処理、洗浄などの間、細胞凝集体を容器内に保持しておくことが必要である。しかしながら、接着性の容器で形成された細胞凝集体の培養において、凝集体を培養容器内に接着させた状態で、凝集体内部に物質透過性の粒子を包含させる方法については、これまでに報告がない。
本発明の目的は、以上の観点に鑑み、細胞の機能が生体に近い3次元凝集体を培養容器に接着させながら、培地からの栄養、酸素、薬物、ならびに試薬等の浸透性を向上させる、細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体を提供することにある。
上記の目的を達成するため、本発明においては、単離細胞を培養容器に播種し、細胞接着性と物質透過性を有する粒子を投与することにより、粒子を包含する細胞凝集体を形成する細胞培養方法を提供する。
また、上記の目的を達成するため、本発明においては、培養容器に播種した細胞上に投与されることによって生成される細胞凝集体に包含される、細胞接着性と物質透過性を有する粒子からなる粒子状培養担体を提供する。
更に、上記の目的を達成するため、本発明においては、細胞を培養容器に播種し、細胞上に細胞接着性と物質透過性を有する粒子を投与することにより形成した粒子包含細胞凝集体を提供する。
本発明によれば、細胞の培養において、生体に近い機能を持つ培養細胞を長期間維持し、試験管内での細胞試験に利用できる培養法を提供できる。
本発明は、単離細胞を培養容器に播種し、細胞接着性と物質透過性を有する粒子を投与することにより、粒子を包含する細胞凝集体を形成する細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体の発明である。好適には、細胞接着性と物質透過性を有する粒子とは、細胞足場粒子であるマイクロスキャフォールド(micro scaffold)粒子であり、この粒子の投与は、播種した細胞が培養面に沈降した後に行う、より具体的には単離細胞を培養容器に播種後、細胞が培養面への接着を開始した時から、細胞が移動して近接した細胞同士が接着し、細胞凝集体の形成が開始されるまでの間に行うことにより、細胞の培養容器への接着、細胞と粒子の接着等を確実にすることができる。
本発明の好適な実施態様であるマイクロスキャフォールド粒子を用いた細胞培養法は、熱処理などによってゼラチン分子間に化学架橋を形成させて得られる微粒子を用いた培養法である。化学架橋の方法として、グルタルアルデヒドなどの化学的架橋剤を用いることも可能である。
ゼラチン粒子を調製する時のゼラチン水溶液の濃度は、10%重量濃度が望ましいが、これに限定されるものではない。ゼラチン粒子を作製する方法の一つとして、ゼラチン水溶液を、例えばオリーブオイルなどの油と一緒に撹拌することによりエマルジョンを調製し、このエマルジョンを冷却し、冷却アセトンなどで脱脂および脱水することにより、微粒子を得る手順が挙げられる。この段階でふるいにかけることにより、粒子径を分けることが可能である。乾燥させた粒子を熱処理することにより、分子を架橋する。
この他に粒子を作製する方法の例として、マイクロリアクター(例えばマイクロプロセスサーバー、日立プラントテクノロジー社製)を用いて、ゼラチン水溶液とオリーブオイルを均一に乳化させて、サイズを制御した粒子を得ることも可能である。
マイクロスキャフォールド粒子は、粒子表面の細胞接着性と、粒子内への水溶性物質透過性の2つの性質を持つものであれば、いずれの材料から作製された粒子でも用いることができる。細胞接着性を有するためには、細胞接着性物質で作製されるか、あるいは細胞が接着する物質で表面がおおわれているなどでもよい。細胞接着性物質としては、例えばコラーゲン、フィブロネクチン、ラミニン、グリコサミノグリカン、細胞膜表面のタンパク質や糖鎖に結合する抗体、細胞膜表面に結合する糖鎖、あるいはポリ-L-リジンなどが挙げられるが、これらに限定されない。
水溶性物質透過性を有するためには、大量の水分を含むハイドロゲルか、あるいは多孔質セラミックスや多孔質金属などが例に挙げられるが、これらに限定されない。ハイドロゲルとしては、ポリアクリル酸、ポリアクリルアミド、ポリビニルピロリドン、ポリビニルアルコール、アルギン酸、でんぷん、ポリ乳酸、アガロース、ペクチン、セルロース、グルコサミノグリカン、コラーゲン、ゼラチン、フィブロネクチン、ビトロネクチン、ラミニン、プロテオグリカンなど、多孔質セラミックスとしてはハイドロキシアパタイトが例に挙げられるが、これらに限定されない。
本発明のマイクロスキャフォールド粒子などの粒子包含細胞凝集体は、培養面への接着性を示す細胞を、培養面に接着領域を有する培養容器に播種し、細胞が容器に沈降した後に上述の粒子を投与することによって形成される細胞凝集体である。原料であるゼラチンに細胞が接着することにより、細胞が培養面に接着した状態で、粒子を包含する細胞凝集体が形成される。粒子投与時期、粒子径、投与数を選ぶことにより、細胞凝集体が培養器材から剥離することなく、また凝集体内の細胞-細胞間接着を阻害することなく、細胞凝集体を得ることが可能である。粒子包含細胞凝集体の利用目的によって、特に維持するべき細胞機能が異なる場合があるため、目的に応じて、適切な粒子投与時期、粒子径、投与数を調節することが望ましい。
このように、培養容器に細胞凝集体が接着した状態で粒子を包含する本発明は、従来のマイクロキャリアの表面に細胞を接着させて浮遊培養により有用物質を得る培養方法や、非接着性の容器で培養担体を包含する細胞凝集体を形成する技術とは異なる。
培養容器に細胞凝集体が接着した状態で粒子を包含する本発明は、通常行われる単層培養細胞を用いた試験と同様の操作で、薬物処理や細胞の活性計測を行うことが可能である。このため、創薬研究における薬物スクリーニングや、再生医療研究における分化誘導条件の検討などの細胞試験に、活性を維持した粒子包含細胞凝集体を、容易に用いることができる。このとき、物質透過性マイクロスキャフォールド粒子を介して薬物や試薬が内部細胞へ浸透しやすくなることから、粒子を包含しない細胞凝集体で懸念される薬物や試薬の浸透性の問題を解決することができる。
以下、図面を用いて本発明の各種の実施態様を説明する。
実施例1は、肝細胞を培養する粒子状培養担体、粒子包含細胞凝集体、及び細胞培養方法に関する実施例である。
本実施例で用いる肝細胞は、増殖しない成熟した肝実質細胞であり、培養期間中に機能が低下しやすい。このため、一般に、薬物試験などに肝細胞を供与できる期間は、培養開始から1週間程度までである。本実施例では、ゼラチン粒子をラット肝細胞に投与して培養することにより、代謝酵素チトクロームP450(CYP3A)活性の計測が可能な期間を、10日以上に延長した例を説明する。
本実施例で用いる肝細胞は、増殖しない成熟した肝実質細胞であり、培養期間中に機能が低下しやすい。このため、一般に、薬物試験などに肝細胞を供与できる期間は、培養開始から1週間程度までである。本実施例では、ゼラチン粒子をラット肝細胞に投与して培養することにより、代謝酵素チトクロームP450(CYP3A)活性の計測が可能な期間を、10日以上に延長した例を説明する。
図1A、図1B、図1Cは、本実施例において、直径の異なる4種類のゼラチン粒子をラット肝細胞に投与し、酵素活性を経時的に測定し、粒子径によって酵素活性維持効果が異なることを示した例である。なお、これらの図に示された培養条件1~7は、図1Dの1~7に示す通りである。
図2は、ラット肝細胞に条件1~5で直径の異なる粒子を投与して11日間培養した時の細胞凝集体形成の経時変化を示し、図1A、図1B、図1Cの酵素活性測定に用いる試料の顕微鏡観察像の例である。図3は、培地で膨潤させた直径の異なるゼラチン粒子懸濁液の顕微鏡観察像の例を示す。
本実施例のラット肝細胞は、Fisher344ラット、あるいはSDラットのオス、5-8週齢(ともに日本チャールスリバー)より、2段階コラゲナーゼ還流法により調製した。10%ウシ胎児血清をふくむウィリアムズE培地(8.6nMインシュリン(シグマアルドリッチ)、255nMデキサメタゾン(ナカライテスク)、50ng/ml EGF、5KIV/mlアプロチニンを添加)に、細胞濃度5×105細胞/mlで単離肝細胞を懸濁し、培養容器の培養面積当たり1×105細胞/cm2の密度で、細胞を播種した。培養プレートを、37度、炭酸ガス濃度5%のインキュベーターに入れて、細胞を培養した。
本実施例で用いる培養容器には、後で図11を用いて説明するように、培養面に直径200μmの多数のホール構造を持ち、各ホール底面の中心部分に、直径2μm高さ1μmの微細な突起を加工した、96ウェルナノピラー培養プレートを用いた。例えば、本発明者等による特開2011-004612号公報、WO2010/079602号公報参照。前者には、細胞の接着性や遊走性を制御できるナノピラーが形成された培養シートについて記載されている。後者には、ナノピラーシート上で肝細胞が細胞凝集体を形成しやすい条件について記載され、3次元凝集体の機能が2次元培養法より生体に近いことが記載されている。
この培養プレートの材質はポリスチレンであり、ホールならびに微細な突起を有する培養面は、表面改質により親水化処理され、細胞接着性を有している。特に微細な突起であるナノピラー部分は、細胞を捕捉する効果を有している。一方、単層培養には、I型コラーゲンを塗布したバイオコート(BD)96ウェル培養プレートを培養容器に用いた。
この培養プレートの材質はポリスチレンであり、ホールならびに微細な突起を有する培養面は、表面改質により親水化処理され、細胞接着性を有している。特に微細な突起であるナノピラー部分は、細胞を捕捉する効果を有している。一方、単層培養には、I型コラーゲンを塗布したバイオコート(BD)96ウェル培養プレートを培養容器に用いた。
細胞に投与する粒子として、下記の非特許文献1を一部改変した方法で作製されたゼラチン粒子を用いた。
<非特許文献1>
In vitro Proliferation and condrogenic differentiation of rat bone marrow stem cells cultured with gelatin hygrogel microspheres for TGF-β1 release.Ogawa T. et al.、Journal of Biomaterials Science、21、609-621 (2010)
<非特許文献1>
In vitro Proliferation and condrogenic differentiation of rat bone marrow stem cells cultured with gelatin hygrogel microspheres for TGF-β1 release.Ogawa T. et al.、Journal of Biomaterials Science、21、609-621 (2010)
乾燥ゼラチン粉末(等電点5)を秤量し、10%重量濃度になるように精製水を加え、撹拌しながら37度で膨潤、溶解させることにより、ゼラチン水溶液を作製した。オリーブオイルを40度のウォーターバスで加温しながら撹拌し、この中に上記のゼラチン溶液を加え、ホモジナイザー(ポリトロン社製)で撹拌することにより乳化させた。乳化液を撹拌しながら冷却して、ゼラチンをゲル化させた。冷却アセトンを加えてさらに撹拌した後、遠心分離により粒子を回収した。粒子を冷却アセトンで洗浄しながらふるいに通し、ふるいを通過した粒子を回収し、真空乾燥により乾燥ゼラチン粒子を得た。真空オーブンを用いて、減圧下140度で48時間加熱することにより、熱脱水架橋を行った。エチレンオキサイドガスで滅菌処理し、無菌の乾燥粒子の状態で保存した。
ゼラチン粒子の粒径は、熱架橋後の粒子を水で膨潤させた後、顕微鏡下で大きさを計測することにより、平均値と標準偏差を求めた。本実施例では、大きさの異なる4種類のゼラチン粒子として、図1Dに示した、直径4.5±1.7μm、10.2±4.3μm、16.8±6.7μm、23.7±10.3μmを用いた。
細胞に粒子を投与する際は、乾燥ゼラチン粒子10mgを50μlのエタノールで分散させ、血清を含まないウィリアムズE培地を加えて懸濁し、エタノールの最終濃度を1%以下とした。撹拌しながら1時間置くことにより、粒子を培地で膨潤させた。図3に培地で膨潤した4種類のゼラチン粒子の顕微鏡像を示した。
粒子懸濁液の粒子濃度を、乾燥粒子重量で3mg/mlに調製した。細胞播種から24時間後に、細胞を培養している培地を、10%ウシ胎児血清をふくむウィリアムズE培地から、粒子を懸濁した無血清ウィリアムズE培地に交換することにより、粒子を培養細胞に投与した。細胞播種から48時間後に、マトリゲル(BD)を総蛋白量で0.25mg/ml含む無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、培養を継続した。
比較実験として、従来法である単層培養を実施した。細胞濃度5×105細胞/mlで単離肝細胞を懸濁し、培養面積当たり1×105細胞/cm2の密度で、前述の培養プレートに播種した。培養プレートを、37度、炭酸ガス濃度5%のインキュベーターに入れて、細胞を培養した。単層培養は、細胞播種から24時間後に、無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、培養を継続した。単層サンドイッチ培養は、細胞播種から24時間後に、マトリゲル(BD)を総蛋白量で0.25mg/ml含む無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、培養を継続した。
播種から5日目、7日目、11日目に、肝細胞の代謝酵素チトクロームP450(分子種CYP3A)の活性を、P450-GloTMAssay(Luciferin-IPA、プロメガ)を用いて測定した。測定の手順は以下のとおりである。
肝細胞を培養している培地を除き、培地で1/1000に希釈したLuciferin-IPA溶液60μl(最終濃度3μM)に交換し、37度、炭酸ガス濃度5%のインキュベーター内で1時間反応させた。各ウェルの培養上清50μlを、白色の96ウェルプレートに分取し、luciferin検出試薬(プロメガ)を50μl加え、遮光して室温で20分間反応させた。反応後の白色プレートを、プレートリーダー(SH-8000Lab、コロナ電気)に設置し、ゲート時間1秒で発光を測定した。
図1A、図1B、図1Cに、各培養条件について、8ウェルの平均値と標準偏差を求めて示した。培養5日目には、粒子を投与していない細胞の活性が高かったことから、培養初期には粒子の効果が見られなかった。培養7日目には、直径4.5±1.7μmの粒子を投与した凝集体の細胞と、粒子を投与していない凝集体の細胞の活性が高かった。
これに対して、培養11日目には、直径10.2±4.3μm、16.8±6.7μm、ならびに23.7±10.3μmの粒子を投与した凝集体の細胞の活性が他の条件に比較して高く、直径4.5±1.7μmの粒子、粒子なし、単層サンドイッチ培養では活性が低く、最も一般的な単層培養では、活性が検出限界以下であった。
図2に、培養している肝細胞を位相差倒立顕微鏡で観察し、培養プレートのウェル中央付近の任意の細胞凝集体を撮影した顕微鏡写真を示した。本実施例の粒子投与条件では、直径4.5±1.7μmの粒子を投与した細胞は、培養3日目と5日目は粒子を投与していない細胞よりも細胞間の接着が促進され、凝集傾向が見られた。その後、培養7日目、11目の凝集体の形状は粒子を投与していない細胞と類似しており、培養プレートのホール中央部分の微細な突起が集合した領域に、凝集体が接着した。
直径10.2±4.3μmの粒子を投与した細胞は、粒子に覆われた状態で、凝集体を形成し、培養プレートのホール中央部分の微細な突起が集合した領域に、凝集体が接着した。粒子なしの培養よりも、大きい凝集体を形成したことから、細胞凝集体内に粒子が取り込まれていると考えられた。
直径16.8±6.7μm、ならびに23.7±10.3μmの粒子を投与した細胞は、粒子と混在して、培養プレートのホールに充填された状態になり、培養11日目には、細胞が粒子とともに凝集する傾向が認められた。
肝細胞の径を約15~30μm程度とみなすと、直径4.5±1.7μmの粒子は、粒径が細胞の約1/10~1/5程度であり、細胞凝集体の機能と形状に、大きい影響を与えなかった。11日目の凝集体の大きさが、粒子なしと比較して著しく違わないことから、粒子の投与量はいずれの粒子でも乾燥粒子重量で3mg/mlであるが、直径4.5±1.7μmの粒子では、凝集体内部に取り込まれている粒子の総量が、少ないと考えられた。
これに比較して、粒径が細胞の1/5以上である直径10.2±4.3μmの粒子では、粒子を包含する凝集体を形成した。また、直径16.8±6.7μm、ならびに23.7±10.3μmでは、やや平面的な不定形の凝集体が形成された。肝細胞の径を約15~30μm程度とみなすと、直径23.7±10.3μmの粒子は、粒径が細胞と同程度から細胞の2倍程度でと考えられた。
前述したように、本実施例においては、これら3種類の粒子を投与することにより、代謝酵素チトクロームP450(分子種CYP3A)の活性を10日以上計測可能に保つ効果が認められたことから、粒径が細胞の大きさの1/5より大きい粒子を包含する細胞凝集体を形成することにより、肝細胞の代謝酵素チトクロームP450活性を維持する効果があることがわかった。
実施例2では、直径の異なるゼラチン粒子をラット肝細胞に投与して培養した時、粒子径により、細胞凝集体に包含された粒子が凝集体内部にのみ分布する場合と、凝集体内部だけでなく培地に接する部分にも存在する場合の例を説明する。
図4は、実施例2におけるゼラチン粒子包含細胞凝集体の断面を示す顕微鏡写真の例である。写真の下側の模式図は、上部の写真で識別できる細胞の輪郭を線で示し、包含された粒子の位置を米印で示した。
培養容器には、上述した特開2011-004612号公報に記載の、培養面に直径200μmの多数のホール構造を持ち、各ホール底面の中心部分に直径2μm高さ1μmの微細な突起を加工したナノピラー培養シートを、2ウェルスライドチャンバーのウェル底面に接着して用いた。培養シートの材質はポリスチレンであり、ホールならびに微細な突起を有する培養面は、表面改質により親水化処理され、細胞接着性を有している。特に微細な突起であるナノピラー部分は、細胞を捕捉する効果を有している。
単離肝細胞を、細胞濃度5×105細胞/mlで10%ウシ胎児血清をふくむウィリアムズE培地懸濁し、培養容器の培養面積当たり1×105細胞/cm2の密度で、播種した。スライドチャンバーを、37度、炭酸ガス濃度5%のインキュベーターに入れて、細胞を培養した。
細胞に粒子を投与する際は、乾燥ゼラチン粒子10mgを、1mlの10%ウシ胎児血清を含むウィリアムズE培地に懸濁し、粒子を培地で膨潤させた。直径16.8±6.7μmの粒子の懸濁液の粒子濃度を、セルカウンター(TC10 system、BioRad)で計数し、投与する粒子数が、細胞数の1/16になるように調節した。実施例1で直径16.8±6.7μmの粒子を投与した時の乾燥粒子重量3mg/mlの懸濁液に比較して、1/16の希釈では、約0.2mg/mlとなった。同様に、直径4.5±1.7μmの粒子を、10%ウシ胎児血清を含むウィリアムズE培地に懸濁した。粒子が細かくセルカウンターで計数できないため、乾燥粒子重量で、直径16.8±6.7μmの粒子と同じ濃度の懸濁液を調製した。
細胞播種から3時間後に、粒子を懸濁した10%ウシ胎児血清をふくむウィリアムズE培地に交換することにより、粒子を細胞に投与した。細胞播種から24時間後に無血清ウィリアムズE培地に交換し、48時間後にマトリゲル(BD)を総蛋白量で0.25mg/ml含む無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、5日間培養を継続した。
ナノピラー培養シート上で5日間培養した細胞を、 2.5%グルタルアルデヒドを含む0.1Mリン酸バッファー(pH7.4)で前固定、1 % 四酸化オスミウムを含むリン酸バッファーで後固定した。細胞が接着したナノピラーシートを1.5 %濃度の低融点寒天(Sea plaquer agarose、 Lonza)で包埋し、エタノール希釈系列で脱水処理した後、プロピレンオキサイドでポリスチレンを溶解させた。Epon-Alardite混合樹脂を、細胞を包埋した寒天に浸透させ、加温重合した。ウルトラミクロトームを用いて、樹脂包埋した試料から、厚さ400 nmの準超薄切片を作製した。切片をスライドガラスに接着して1%トルイジンブルーで染色し、正立型光学顕微鏡で観察した。
凝集体の断面を前述の準超薄切片で観察すると、直径16.8±6.7μmの粒子を投与した細胞凝集体では、多数の粒子が凝集体内部に包含されており、その一部が凝集体外に露出していることから、粒子を介して、培地成分が凝集体内部に供給されていると考えられた。一方、直径4.5±1.7μmの粒子の場合は、細胞凝集体内部に粒子が包含され、培地に接する面が認められなかった。この結果から、粒子の大きさによって、凝集体内部細胞へ培地成分が浸透する効率が異なることが示唆された。
なお、実施例1で既に説明したラット肝細胞の単離と培養、ならびにゼラチン粒子の作製方法については、ここでは説明を省略する。
実施例3では、粒子の直径だけでなく、粒子を投与する量によっても、ラット肝細胞の代謝酵素チトクロームP450(CYP3A)活性維持の効果が異なることを示す例を説明する。
図5A、図5Bは、本実施例において、CYP3Aの基底活性ならびに薬物誘導活性を計測した例を示す図であり、図5Aは細胞数に対して粒子を投与する割合と、粒子を投与する時間を変えてラット肝細胞を10日間培養し、代謝酵素チトクロームP450(分子種CYP3A)の基底活性を測定した例を、図5Bは細胞数に対して粒子を投与する割合と、粒子を投与する時間を変えてラット肝細胞を10日間培養し、誘導薬物デキサメタゾンを処理して、代謝酵素チトクロームP450(分子種CYP3A)の誘導活性を測定した例を示す図である。
本実施例においては、実施例1と同様に、96ウェルナノピラー培養器材を用い、ラット肝細胞を播種した。直径16.8±6.7μmの粒子を、10%ウシ胎児血清をふくむウィリアムズE培地で膨潤させ、細胞播種から2.5時間後、あるいは22時間後に、細胞数:粒子数が1:1、4:1、10:1、20:1、50:1になるように、粒子懸濁液を細胞の上に投与した。細胞播種から24時間後に無血清ウィリアムズE培地に交換し、48時間後に、マトリゲル(BD)を総蛋白量で0.25mg/ml含む無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、10日間培養した。
培養8日目と9日目の培地交換の際に、薬物による酵素誘導試験区の細胞には、CYP3Aの発現を誘導するデキサメタゾン(50 μM)を含む無血清ウィリアムズE培地を用いることにより、肝細胞をデキサメタゾンに48時間暴露した。CYP3Aの基底活性試験区の細胞には、無血清ウィリアムズE培地を用いた。
デキサメタゾン処理を開始してから48時間後に、肝細胞の代謝酵素チトクロームP450(分子種CYP3A)の活性を、P450-GloTMAssay(Luciferin-IPA、プロメガ)を用いて測定した。
各試験区について、4ウェルの平均値と標準偏差を求め、図5A,図5Bに示した。図に明らかなように、単位培養面積おける細胞数に対して、細胞数:粒子数が20:1より多く粒子を投与した細胞では、基底活性試験区と誘導活性試験区の両方において、明らかに粒子なしの凝集体よりも高いCYP3A活性を検出した。さらに粒子の割合を多くすることによって、より高い活性を検出した。特に基底活性については、細胞数:粒子数が4:1以上のときに、高い活性を検出した。誘導活性については、細胞数:粒子数が50:1でも粒子の効果が認められた。この結果より、細胞数:粒子数が50:1より多ければ粒子の効果が認められるが、細胞数:粒子数が4:1以上であることが望ましい。
以上の結果は、細胞間の粒子によって、培地からの栄養、酸素の供給が向上して細胞の活性を保ったことに加えて、培地に投与した薬物デキサメタゾンへの細胞の暴露が増えて酵素誘導が強くなるとともに、検出試薬であるLuciferin-IPAの細胞内への浸透性が高まったことによって酵素活性の検出効率が向上したことが理由であると考えられる。
図6Aは、本実施例において、ラット肝細胞を低接着性培養容器に播種した後の経過時間による細胞の形態変化(経時変化)を、位相差顕微鏡観察ならびにF-アクチンの分布によって示した例であり、図6Aは、細胞播種後、細胞形態を経時的に倒立顕微鏡で観察した顕微鏡写真であり、上段は位相差顕微鏡像、下段はF-アクチンをローダミン標識ファロイジンで染色した蛍光顕微鏡像である。位相差像は全体像を示し、F-アクチン蛍光染色像により個々の細胞の輪郭を示す。一方、図6Bは、細胞播種後の顕微鏡写真において、任意に選んだ60細胞の長径を計測し、長径の平均値と標準偏差を求め、経時変化を示した例である。
図6Aのローダミン標識ファロイジン染色の手順は以下のとおりである。4%パラフォルムアルデヒドで固定した細胞を、0.1Mリン酸バッファー(pH 7.4)で洗浄した。非特異的な染色を防止するため、プロテインブロック(DAKO)で30分間処理した後、PBSで1/40に希釈したローダミン標識ファロイジン(インビトロジェン)で30分間処理し、0.075 % Brij35 (シグマアルドリッチ) を含むPBSで洗浄した。
播種0.5時間後の細胞は、細胞が単離しており、形は丸く、平均直径が20μm以下であった。1.0時間後は、写真に示される細胞の2次元像がやや大きくなり、平均直径が20μmを若干超えた。
播種2.0時間後には、細胞が培養面に接着し、写真に示される細胞の長径平均が30μmに近づき、細胞ごとの長径のばらつきが大きくなった。F-アクチンの像より、細胞が器材に接着して、3次元的には薄くなっていることが示された。3.0時間、4.0時間にも同様の形状が認められ、さらに細胞ごとの長径のばらつきが大きくなった。4.0時間には、細胞間相互作用による細胞間接着も認められるようになった。
播種6.0時間には、多くの細胞の間で細胞間接着形成され、細胞が器材に接着して3次元的に薄くなっている像が減少した。これにより、細胞長径の平均は25μm程度となり、ばらつきも減少した。8.0時間、16.0時間も同様の傾向にあった。
本実施例では、図5A、図5Bで示したように、播種2.5時間後に粒子を投与した細胞と、22時間後に投与した細胞で、CYP3A活性に、顕著な差は見られなかった。したがって、本発明において、播種した細胞が培養器材の培養面に沈降した後であれば、細胞の上に粒子を投与することによって、粒子による細胞活性維持の効果が得られることがわかった。
なお、ここでは、実施例1で既に説明したラット肝細胞の単離と培養、ゼラチン粒子の作製方法、ならびにCYP3A活性測定手順については、説明を省略する。
本実施例では、粒子を投与する量によって、ラット肝細胞の活性維持の効果が異なることを示す別の例を説明する。
5日間培養した肝細胞のミトコンドリア脱水素酵素の活性を、WST-1アッセイにより測定したとき、粒子を細胞に投与する量の増加によって、測定値が上昇する例を説明する。WST-1試験は,生細胞のミトコンドリア呼吸鎖酵素(コハク酸塩テトラゾリウム還元酵素)の活性を測定する方法であり,この測定値が高ければ,スフェロイドの呼吸活性が高いことを示す。
実施例1と同様に、96ウェルナノピラー培養器材を用い、ラット肝細胞を播種した。直径16.8±6.7μmの粒子を、10%ウシ胎児血清をふくむウィリアムズE培地で膨潤させ、細胞播種から2.5時間後、あるいは22時間後に、細胞数:粒子数が1:4、1:2、1:1、2:1、4:1、8:1、10:1、20:1、40:1、50:1、80:1になるように、粒子懸濁液を細胞の上に投与した。細胞播種から24時間後に無血清ウィリアムズE培地に交換し、48時間後に、マトリゲル(BD)を総蛋白量で0.25mg/ml含む無血清ウィリアムズE培地に交換し、以降24時間ごとに無血清ウィリアムズE培地に交換して、5日間培養した。
活性測定には,Premix WST-1 (タカラバイオ)を用いた。肝細胞を5日間培養した後に,細胞の入った96ウェルプレートの各ウェルから培地を全量除去し,ハンクス液で2回洗浄した。洗浄液を除いてから、10%(v/v)濃度のWST-1を添加したハンクス液を65μl 入れ,37度,5%炭酸ガス濃度に保ったインキュベーターで1時間反応させた。反応後の96ウェルをプレートリーダーにセットし、2波長(440 nm, 650 nm)の吸光度を、半値幅5 nmで測定した。440 nmの値から対照波長である650 nmの値を引いた値を求めた。
図7に、細胞数に対して粒子を投与する割合と、粒子を投与する時間を変えてラット肝細胞を5日間培養し、ミトコンドリア脱水素酵素の活性をWST-1アッセイにより測定し、各試験区について、3ウェルの平均値と標準偏差を求めた結果を示した。同図から明らかなように、測定の結果,細胞数:粒子数が1:80、すなわち、細胞の個数に対して、粒子の数が1/80より多い場合でも粒子の効果が認められるが、4:1よりも多くの粒子を投与することによってWST-1の測定値が著しく上がり,さらに1:1よりも投与量を増やすことによって,値がさらに上昇した。以上の結果は、細胞間の粒子によって、培地からの栄養、酸素の供給が向上して細胞の活性を保ったことに加えて、検出試薬であるWST-1の細胞内への浸透性が高まったことによって、呼吸活性の検出効率が向上したことが理由であると考えられる。
なお、ここでは実施例1で既に説明したラット肝細胞の単離と培養、ゼラチン粒子の作製方法、ならびに培地で粒子を膨潤させる手順については、説明を省略する。
本実施例では、粒子を異なる量で投与した時の、細胞凝集体形成の過程と、形成された細胞凝集体形状の例を示す。さらに、粒子の投与量によって、肝組織特異的な構造である毛細胆管を形成する効率が異なることを示す例を説明する。
創薬研究における薬物動態試験では、肝細胞に取り込まれた薬物のうち胆管に排泄される割合を計測する細胞試験系の開発が望まれている。これに応じるために、毛細胆管が効率よく形成され、排泄能を検証しやすい培養系を提供する。
図8は、細胞数に対して異なる割合で粒子を投与し、5日間培養した時の細胞凝集体形成の経時変化の例を示す図である。位相差像は粒子と細胞の全体像を示し、F-アクチン蛍光染色像は細胞の分布を示す図である。すなわち、ラット肝細胞に、直径16.8±6.7μmの粒子を、細胞数:粒子数が1:1、4:1、10:1、20:1、50:1になるように投与して、5日間培養した時の細胞凝集体形成の過程を示す。各カラムの上段は位相差顕微鏡像、下段はF-アクチンをローダミン標識ファロイジンで染色した蛍光顕微鏡像である。
粒子を投与した場合には、培養3日目に細胞凝集体が形成されていることが分かった。粒子を投与しない場合は、培養3日目までは細胞が培養容器に接着していることから、粒子を投与することにより、1日程度早く細胞凝集体を形成することが示された。
生体の肝臓では,F-アクチンは膜の裏打ち構造を形成し,細胞は多角形の立体構造をとる。F-アクチンの分布を観察することによって,生体の細胞に近い組織であるかどうかを検証することが可能である。これまでに、ナノピラー培養容器で形成した細胞凝集体では、F-アクチンが、生体に近い膜の裏打ち構造を形成した(WO2010/079602号公報参照)。
本実施例では、細胞数:粒子数が50:1、あるいは粒子なしで形成された凝集体は、F-アクチンが、生体に近い膜の裏打ち構造を形成していると考えられた。これに比較して,細胞数:粒子数が20:1、10:1、4:1あるいは1:1で粒子を投与した場合には,ゼラチン粒子に接着する細胞表面に、線状あるいは点状の強い蛍光を示す像が見られ、接着点あるいはストレスファイバーの可能性が考えられた。これらは、細胞が細胞外マトリクスに接着した時に形成される構造である。したがって,細胞は、ゼラチン粒子表面に、細胞外マトリクスとの接着と同様に、接着構造を形成していると考えられた。
この結果より、生体に近い膜の裏打ち構造を形成するには、細胞数:粒子数が50:1以下であることが望ましいと考えられた。
図9は、細胞数に対して異なる割合で粒子を投与し、5日間培養した時の蛍光試薬の取り込みと毛細胆管への排泄の例を示す図である。具体的には、肝細胞の細胞間接着面に形成される毛細胆管の排泄能を、培養5日目に、蛍光標識試薬carboxydichlorofluorescein diacetate(CDFDA、Molecular Probe)を用いて示した例である。培地中に投与したCDFDAは,細胞内に取り込まれて代謝され,carboxydichlorofluorescein(CDF)となり、毛細胆管に面する細胞膜のトランスポーターmultidrug resistance-associated protein 2(MRP2)を介して、毛細胆管に排泄される。培養している細胞の胆管排泄機能が高いほど,多くの薬物が毛細胆管内に排泄されて、細胞間に溜まるため,蛍光顕微鏡下では、点状あるいは線状の強い蛍光が観測される。
本実施例のCDFDA処理の手順は次のとおりである。24ウェルナノピラー培養プレートで培養している肝細胞の培地を除去し、Krebs-Henseleitバッファー(K-Hバッファー) で2回洗浄した。5μMのCDFDAを含むK-Hバッファーを、1ウェル当たり400μl入れて、37度のインキュベーターに20分間置いたのち、K-Hバッファーで2回洗浄し、倒立型蛍光顕微鏡で観察した。
観察の結果、細胞数:粒子数が50:1、20:1、10:1の時に、毛細胆管に排泄されたCDFと考えられる点状あるいは線状の強い蛍光が認められた。一方、4:1では、細胞凝集体が不定型で扁平であり、線状の蛍光像が認められたことから、毛細胆管が平面的に広がったと考えられた。
これに対して、細胞数:粒子数が1:1の場合は、粒子の自家蛍光を除くと、蛍光は細胞内に顆粒状に観察されたことから、CDFは毛細胆管に排泄されたのではなく、細胞内に取り込まれて、蓄積したと見られた。
毛細胆管は細胞間接着面に形成されるため、細胞間相互作用が必要である。また,細胞内から毛細胆管に薬物を排泄するトランスポーターMRP2の発現には,細胞間接着面の細胞膜内側に存在する蛋白質が,重要な役割を果たすことが知られている。このため、粒子が多い時に毛細胆管が再生されにくいのは,細胞間に過剰に存在する粒子が,細胞間の接着を阻害していることが原因なのではないかと考えられた。
図8で示したように、細胞間接着によりF-アクチンの細胞膜裏打ち構造を形成するには、細胞数:粒子数が50:1以下であることが望ましかったが、毛細胆管への排泄を検出するには、細胞数:粒子数が50:1から4:1であることが望ましく、さらに50:1から10:1であることが望ましい。これは、薬剤の取り込みと排泄の両方が向上するには、細胞間接着のみが促進されるのではなく、粒子を介して培地に接する面が形成される必要があることが理由であると考えられた。
実施例4において,多数の粒子を包含することにより、細胞凝集体内部への培地成分の供給が向上する可能性を示したが,一方で,本実施例により、過多な粒子の存在により,肝臓組織特有の排泄機能が低下することが明らかとなった。粒子投与においては相反する条件であるが,細胞凝集体の利用目的に応じて,適切な粒子量を求めることが望ましい。
なお、実施例1で既に説明したラット肝細胞の単離と培養、ゼラチン粒子の作製方法、ならびに培地で粒子を膨潤させる手順、実施例3で示したローダミン標識ファロイジン染色の手順については、説明を省略する。
本実施例では、上述した各実施例で使用される、細胞低接着性の培養表面の局所に、複数の突起を成型することにより、細胞凝集体を捕捉する細胞接着領域を設けた培養容器の一例を説明する。
図10Aは、その上段と下段に、細胞培養用マルチウェルプレート100の斜視と、上面を示す。A-A、B-Bで示したウェル101の底面102の培養面には、微細な立体加工が施されている。図10Bは、その上段、中段、下段に、図10AのA-A、B-B部分の拡大、C-C、D-D部分の拡大、E-E線端面を示す。培養面に施した微細な立体加工とは、C-C、D-D部分の拡大、ならびにE-E線端面に示したホール103であり、ホール103の中心部に突起104の集合領域105が成型されている例を示す図である。
図11は、上述のホール103の中心部に突起104の集合領域105が成型されている培養面の、走査型電子顕微鏡写真の例である。
図12は、図10B、図11に示した培養面のホール103に、細胞106を播種し、細胞106が底面に接着した時にゼラチン粒子107を投与することにより、粒子を包含する細胞凝集体がホール103の中心に成型された突起104の集合領域105に接着することを示す模式図の例を示す。
なお、培養面に細胞を捕捉する局所的な細胞接着領域の形成については、本実施例の突起に限定されるものではなく、コラーゲンやラミニンなどの細胞外マトリクス成分、細胞膜表面に結合する抗体、細胞膜表面に結合する糖鎖、あるいはポリ-L-リジンなどを例とする細胞接着性の物質を、任意にパターンニングすることによっても形成することができる。
以上説明した本実施例の培養容器は、低接着性の培養表面に、細胞凝集体を捕捉することができる細胞接着領域を形成することにより、粒子包含細胞凝集体を培養容器内に保持する。さらに、好適には培養表面に仕切りを設けることにより、凝集体を形成する細胞数と粒子数を所望の数に制御し、所望の大きさの粒子包含細胞凝集体を形成する。このような本実施例の培養容器に接着した粒子包含細胞凝集体は、細胞試験の材料として用いることができる。
本実施例では、粒子を包含するヒト細胞試験方法の実施例を説明する。
実施例1ならびに実施例3において、粒子を包含する細胞凝集体では、代謝酵素チトクロームP450(分子種CYP3A)の活性を、粒子を含まない凝集体や従来の単層培養法よりも、長期に維持する効果を示した。
実施例1ならびに実施例3において、粒子を包含する細胞凝集体では、代謝酵素チトクロームP450(分子種CYP3A)の活性を、粒子を含まない凝集体や従来の単層培養法よりも、長期に維持する効果を示した。
チトクロームP450は、血中から肝細胞に取り込まれた薬物を代謝する酵素であるため、創薬研究において、薬物動態における代謝、CYP酵素誘導による薬物相互作用、CYP代謝産物による肝毒性などが研究対象となっている。特に、酵素の基質特性や代謝活性に種差があることから、動物実験だけではなくヒトの試験が必要とされており、単離ヒト肝細胞を用いた試験方法が開発されている。そこで、上述したマイクロスキャフォールド粒子を包含する細胞凝集体を細胞試験に用いることにより、活性を失いやすい単離ヒト肝細胞を用いた試験を、これまでよりも長期に安定して実施することが可能になる。
まず、CYP代謝産物解析の例を説明する。凍結保存されたヒト肝細胞を解凍し、10%ウシ胎児血清をふくむウィリアムズE培地に、細胞濃度5×105細胞/mlで懸濁し、培養容器の培養面積当たり1×105細胞/cm2の密度で播種した。培養プレートを、37度、炭酸ガス濃度5%のインキュベーターに入れて、細胞を培養した。直径16.8±6.7μmの粒子を、培地で膨潤させ、粒子数が細胞数よりも多くなるように、粒子懸濁液を細胞の上に投与することにより、粒子を包含する細胞凝集体を形成した。CYPによって代謝される薬剤を培地に投与し、2日後と7日後に細胞ならびに培地を回収した。細胞溶解液ならびに培地の脱タンパク質処理をして、回収した試料溶液を質量分析法で解析することにより、細胞内あるいは培地中の薬物代謝産物を同定した。
次に、薬物相互作用の原因となる、薬物によるCYP酵素誘導試験の例を説明する。代謝産物解析の例と同様に、凍結保存されたヒト肝細胞を培養し、粒子を投与することにより、粒子を包含する細胞凝集体を形成した。試験対象の薬物を含む培地で48時間から72時間培養し、その後に、CYPの各分子種特異的に反応する検出試薬を用いて、CYP酵素の活性を測定した。対象薬物で処理していない細胞のCYP酵素活性を基底活性とし、対象薬物で処理した細胞のCYP酵素活性を誘導活性として、誘導活性値を基底活性値で割ることにより、誘導率を算出した。
CYP酵素活性の検出試薬としては、例えば実施例1、ならびに実施例3にも示したP450-GloTMAssayのシリーズ(プロメガ)などを用いて、発光の定量により、試験対象薬物によるCYP誘導活性を測定することができる。あるいは、CYPの各分子種特異的に代謝される既知の物質を用いて、代謝産物を質量分析法によって定量することにより、CYP誘導活性を測定することができる。
薬物相互作用の予測には、CYPを強く誘導することがわかっている薬物、例えばリファンピシンによる誘導率を試験対象薬物と並行して計測、算出し、試験対象薬物による誘導率と比較することにより、試験対象薬物による薬物相互作用の程度を推測する方法を例に挙げることができる。
さらに、CYP代謝産物による肝毒性解析の例を説明する。代謝産物解析の例と同様に、凍結保存されたヒト肝細胞を培養し、粒子を投与することにより、粒子を包含する細胞凝集体を形成した。試験対象薬剤の濃度を変えて培地に投与し、細胞数の変動を経時的に測定することにより、薬物の毒性を評価した。細胞数の計測には、実施例4にも示したWST-1法、あるいはATP定量法などを例に挙げることができる。以上、実施例7の説明として複数の試験例を挙げたが、これに限るものではない。
以上詳述した、本発明に係る細胞培養方法は、細胞が培養容器の培養面に接着していることを特徴とする培養法であり、これまでに従来の単層培養法で行われている細胞試験を、同じ手順で適用することが可能である。
更に、本発明による粒子状培養単体、粒子包含細胞凝集体は、単層培養よりも生体に近い構造を持つ3次元細胞凝集体であり、また粒子を介して培地中の栄養や酸素を供給することによって細胞の機能を維持し、細胞試験では粒子を介して培地中に投与した薬物を浸透性させるため、細胞の薬物反応を長期に安定して検出する、再現性の高い試験系を提供することができる。
100 細胞培養用マルチウェルプレート
101 ウェル
102 培養面
103 培養面に成型されたホール
104 ホール底面中央部に成型された突起
105 ホール底面中央部に成型された突起の集合領域
106 肝細胞
107 ゼラチン粒子
101 ウェル
102 培養面
103 培養面に成型されたホール
104 ホール底面中央部に成型された突起
105 ホール底面中央部に成型された突起の集合領域
106 肝細胞
107 ゼラチン粒子
Claims (15)
- 細胞を培養容器に播種し、細胞接着性と物質透過性を有する粒子を投与することにより、当該粒子を包含する細胞凝集体を形成する、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記細胞凝集体に包含させる粒子の粒径が、前記細胞の1/10から10倍の範囲である、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記細胞凝集体に包含させる粒子の粒径が、前記細胞の1/5以上である、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記粒子を細胞に投与するとき、単位培養面積当たりの前記細胞の個数に対して、粒子数が1/80より多い、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記粒子を前記細胞に投与するとき、単位培養面積当たりの前記細胞の個数に対して、粒子数を1/50より多く投与する、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
低接着性の培養容器の中に、前記細胞凝集体を捕捉することができる、少なくとも一つ以上の細胞接着領域を形成することにより、前記細胞凝集体を当該培養容器内に保持する、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記細胞が接着する培養表面に仕切りを設けることにより、当該仕切り内で前記細胞凝集体を構成するする細胞の数と、前記仕切り内に沈降した細胞上に投与する粒子の数を制御し、所望の数の粒子を包含する、所望の大きさの粒子包含細胞凝集体を形成する、
ことを特徴とする細胞培養方法。 - 請求項1に記載の細胞培養方法であって、
前記細胞が単離肝細胞、あるいは幹細胞から分化誘導した肝細胞ならびに肝前駆細胞、あるいは肝がん由来細胞である、ことを特徴とする細胞培養方法。 - 培養容器に播種した細胞上に投与されることによって生成される細胞凝集体に包含される、細胞接着性と物質透過性を有する粒子からなる、
ことを特徴とする粒子状培養担体。 - 請求項9に記載の粒子状培養担体であって、
前記細胞凝集体に包含させる前記粒子の粒径が、細胞の1/10から10倍の範囲である、
ことを特徴とする粒子状培養担体。 - 請求項9に記載の粒子状培養担体であって、
前記細胞凝集体に包含させる粒子の粒径が、細胞の1/5以上である、
ことを特徴とする粒子状培養担体。 - 請求項9に記載の粒子状培養担体であって、
前記細胞が単離肝細胞あるいは幹細胞から分化誘導した肝細胞ならびに肝前駆細胞、あるいは肝がん由来細胞である、
ことを特徴とする粒子状培養担体。 - 細胞を培養容器に播種し、当該細胞上に細胞接着性と物質透過性を有する粒子を投与することにより形成される、
ことを特徴とする粒子包含細胞凝集体。 - 請求項13に記載の粒子包含細胞凝縮体であって、
前記粒子を前記細胞に投与するとき、単位培養面積当たりの前記細胞の個数に対して、1/50より多く投与した、
ことを特徴とする粒子包含細胞凝集体。 - 請求項13に記載の粒子包含細胞凝縮体であって、
前記細胞が単離肝細胞あるいは幹細胞から分化誘導した肝細胞ならびに肝前駆細胞、あるいは肝がん由来細胞である、
ことを特徴とする粒子包含細胞凝集体。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/891,155 US20160168525A1 (en) | 2013-05-23 | 2014-05-09 | Cell Culturing Method, Particulate Culture Carrier, and Particle-Encompassing Cell Aggregate |
| EP14800833.7A EP3000869A1 (en) | 2013-05-23 | 2014-05-09 | Cell culturing method, particulate culture carrier, and particle-encompassing cell aggregate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-109154 | 2013-05-23 | ||
| JP2013109154A JP2014226097A (ja) | 2013-05-23 | 2013-05-23 | 細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014188888A1 true WO2014188888A1 (ja) | 2014-11-27 |
Family
ID=51933451
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/062495 Ceased WO2014188888A1 (ja) | 2013-05-23 | 2014-05-09 | 細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20160168525A1 (ja) |
| EP (1) | EP3000869A1 (ja) |
| JP (1) | JP2014226097A (ja) |
| WO (1) | WO2014188888A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018000081A (ja) * | 2016-06-30 | 2018-01-11 | 株式会社新菱 | ガス供給機能付き容器、細胞培養容器、運搬用容器、保存用容器、細胞培養方法、内容物運搬方法および内容物保存方法 |
| EP3395830A4 (en) * | 2015-12-25 | 2018-12-26 | Konica Minolta, Inc. | Gelatin particles, method for producing gelatin particles, gelatin particle-encapsulating cells, and method for producing gelatin particle-encapsulating cells |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019058156A (ja) * | 2017-09-28 | 2019-04-18 | オリンパス株式会社 | 画像処理装置および細胞観察システム |
| JP2021153441A (ja) * | 2020-03-26 | 2021-10-07 | 昭和電工マテリアルズ株式会社 | 細胞培養用担体、及び細胞培養方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0833486A (ja) * | 1994-07-25 | 1996-02-06 | Res Dev Corp Of Japan | 細胞培養担体と、細胞培養方法 |
| JP2005027532A (ja) | 2003-07-09 | 2005-02-03 | Fuji Xerox Co Ltd | 細胞培養基材及びその製造方法、並びに細胞培養方法 |
| US20050054101A1 (en) | 2003-07-17 | 2005-03-10 | Global Cell Solutions Llc | Automated cell culture system and process |
| WO2010079602A1 (ja) | 2009-01-08 | 2010-07-15 | 株式会社日立製作所 | 動物肝細胞の培養方法 |
| JP2010524442A (ja) * | 2007-04-20 | 2010-07-22 | フラウンホファー ゲセルシャフト ツール フェールデルンク ダー アンゲヴァンテン フォルシュンク エー.ファオ. | ナノ粒子を含む改善された三次元生体適合性骨格構造 |
| JP2011004612A (ja) | 2009-06-23 | 2011-01-13 | Hitachi Ltd | 培養基材、培養シート、及び細胞培養方法 |
| WO2011059112A1 (ja) | 2009-11-13 | 2011-05-19 | 株式会社日立ハイテクノロジーズ | 粒子含有細胞集合体 |
| JP2011130720A (ja) | 2009-12-25 | 2011-07-07 | Medgel Corp | 細胞培養基材およびその使用方法 |
| US20110256574A1 (en) | 2008-08-08 | 2011-10-20 | Agency For Science, Technology And Research | Microfluidic Continuous Flow Device |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH078273A (ja) * | 1993-06-14 | 1995-01-13 | Kurabo Ind Ltd | 接着性動物細胞の無血清培養法 |
| JP2004275122A (ja) * | 2003-03-18 | 2004-10-07 | Kitakyushu Foundation For The Advancement Of Industry Science & Technology | 培養床、培養床コーティング剤、培養床製造方法並びにこれらを用いた細胞培養方法及び培養細胞 |
| JP5263756B2 (ja) * | 2005-09-30 | 2013-08-14 | 国立大学法人 岡山大学 | 細胞の培養方法および細胞培養物 |
-
2013
- 2013-05-23 JP JP2013109154A patent/JP2014226097A/ja active Pending
-
2014
- 2014-05-09 US US14/891,155 patent/US20160168525A1/en not_active Abandoned
- 2014-05-09 WO PCT/JP2014/062495 patent/WO2014188888A1/ja not_active Ceased
- 2014-05-09 EP EP14800833.7A patent/EP3000869A1/en not_active Withdrawn
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0833486A (ja) * | 1994-07-25 | 1996-02-06 | Res Dev Corp Of Japan | 細胞培養担体と、細胞培養方法 |
| JP2005027532A (ja) | 2003-07-09 | 2005-02-03 | Fuji Xerox Co Ltd | 細胞培養基材及びその製造方法、並びに細胞培養方法 |
| US20050054101A1 (en) | 2003-07-17 | 2005-03-10 | Global Cell Solutions Llc | Automated cell culture system and process |
| JP2010524442A (ja) * | 2007-04-20 | 2010-07-22 | フラウンホファー ゲセルシャフト ツール フェールデルンク ダー アンゲヴァンテン フォルシュンク エー.ファオ. | ナノ粒子を含む改善された三次元生体適合性骨格構造 |
| US20110256574A1 (en) | 2008-08-08 | 2011-10-20 | Agency For Science, Technology And Research | Microfluidic Continuous Flow Device |
| WO2010079602A1 (ja) | 2009-01-08 | 2010-07-15 | 株式会社日立製作所 | 動物肝細胞の培養方法 |
| JP2011004612A (ja) | 2009-06-23 | 2011-01-13 | Hitachi Ltd | 培養基材、培養シート、及び細胞培養方法 |
| WO2011059112A1 (ja) | 2009-11-13 | 2011-05-19 | 株式会社日立ハイテクノロジーズ | 粒子含有細胞集合体 |
| JP2011130720A (ja) | 2009-12-25 | 2011-07-07 | Medgel Corp | 細胞培養基材およびその使用方法 |
Non-Patent Citations (1)
| Title |
|---|
| OGAWA T ET AL., JOURNAL OF BIOMATERIALS SCIENCE, vol. 21, 2010, pages 609 - 621 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3395830A4 (en) * | 2015-12-25 | 2018-12-26 | Konica Minolta, Inc. | Gelatin particles, method for producing gelatin particles, gelatin particle-encapsulating cells, and method for producing gelatin particle-encapsulating cells |
| JP2018000081A (ja) * | 2016-06-30 | 2018-01-11 | 株式会社新菱 | ガス供給機能付き容器、細胞培養容器、運搬用容器、保存用容器、細胞培養方法、内容物運搬方法および内容物保存方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014226097A (ja) | 2014-12-08 |
| EP3000869A1 (en) | 2016-03-30 |
| US20160168525A1 (en) | 2016-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2022095914A (ja) | 操作した肝臓組織、そのアレイ、およびそれを製造する方法 | |
| Yamada et al. | Cell-sized condensed collagen microparticles for preparing microengineered composite spheroids of primary hepatocytes | |
| Lea | Caco-2 cell line | |
| Pampaloni et al. | Three-dimensional tissue models for drug discovery and toxicology | |
| Pampaloni et al. | Three-dimensional cell cultures in toxicology | |
| Ryoo et al. | Advances in high throughput cell culture technologies for therapeutic screening and biological discovery applications | |
| Cheng et al. | Three-dimensional polymer scaffolds for high throughput cell-based assay systems | |
| JP5818001B2 (ja) | 肝細胞の培養方法 | |
| US20140154735A1 (en) | Tumour cell and tissue culture | |
| WO2016123474A1 (en) | Methods to generate gastrointestinal epithelial tissue constructs | |
| Wang et al. | Paper supported long-term 3D liver co-culture model for the assessment of hepatotoxic drugs | |
| US20160123960A1 (en) | Method for preparing three-dimensional, organotypic cell cultures and uses thereof | |
| WO2014188888A1 (ja) | 細胞培養方法、粒子状培養担体、及び粒子包含細胞凝集体 | |
| Agarwal et al. | Inexpensive and versatile paper-based platform for 3D culture of liver cells and related bioassays | |
| Aalders et al. | Liquid marble technology to create cost-effective 3D cardiospheres as a platform for in vitro drug testing and disease modelling | |
| KR20180049000A (ko) | 세포 배양 | |
| Cheng et al. | Transparent, biocompatible nanostructured surfaces for cancer cell capture and culture | |
| Wu et al. | In situ self-assembling liver spheroids with synthetic nanoscaffolds for preclinical drug screening applications | |
| Ma et al. | A novel perspective on bone tumors: advances in organoid research | |
| KR20190035524A (ko) | 나노섬유 기반 장기간 초대 간세포 3차원 배양시스템 및 배양방법 | |
| Phillips et al. | Developing HiPSC derived serum free embryoid bodies for the interrogation of 3-D stem cell cultures using physiologically relevant assays | |
| Al Hosni et al. | Reprogramming bone progenitor identity and potency through control of collagen density and oxygen tension | |
| Liang et al. | Chemically defined stem cell microniche engineering by microfluidics compatible with iPSCs’ growth in 3D culture | |
| JP2024539503A (ja) | 癌微小環境 | |
| Cook et al. | Investigating the Impact of various 2D and 3D cell culture platforms on the production of extracellular vesicles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14800833 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14891155 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014800833 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014800833 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |