THERAPEUTICALLY ACTIVE COMPOUNDS AND USE THEREOF
CLAIM OF PRIORITY
This application claims priority from International Application Serial No.
PCT/CN2013/080105filed July 25, 2013, which is incorporated herein by reference in its entirety.
BACKGROUND OF INVENTION
Isocitrate dehydrogenases (JDHs) catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate {i.e., a-ketoglutarate). These enzymes belong to two distinct subclasses, one of which utilizes NAD(+) as the electron acceptor and the other NADP(+). Five isocitrate dehydrogenases have been reported: three NAD(+)-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP(+)-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. Each NADP(+)-dependent isozyme is a homodimer.
IDH1 (isocitrate dehydrogenase 1 (NADP+), cytosolic) is also known as JDH; IDP;
IDCD; IDPC or PICD. The protein encoded by this gene is the NADP(+)-dependent isocitrate dehydrogenase found in the cytoplasm and peroxisomes. It contains the PTS-1 peroxisomal targeting signal sequence. The presence of this enzyme in peroxisomes suggests roles in the regeneration of NADPH for intraperoxisomal reductions, such as the conversion of 2, 4-dienoyl- CoAs to 3-enoyl-CoAs, as well as in peroxisomal reactions that consume 2-oxoglutarate, namely the alpha-hydroxylation of phytanic acid. The cytoplasmic enzyme serves a significant role in cytoplasmic NADPH production.
The human IDHl gene encodes a protein of 414 amino acids. The nucleotide and amino acid sequences for human IDHl can be found as GenBank entries NM 005896.2 and
NP 005887.2 respectively. The nucleotide and amino acid sequences for IDHl are also described in, e.g., Nekrutenko etal, Mol. Biol. Evol. 15:1674-1684(1998); Geisbrecht et a/., J. Biol. Chem. 274:30527-30533(1999); Wiemann et al, Genome Res. 11 :422-435(2001); The MGC Project Team, Genome Res. 14:2121-2127(2004); Lubec et al, Submitted (DEC-2008) to UniProtKB; Kullmann etal, Submitted (JUN-1996) to the EMBL/GenBank/DDBJ databases; and Sjoeblom eta/., Science 314:268-274(2006).
Non-mutant, e.g., wild type, IDHl catalyzes the oxidative decarboxylation of isocitrate to a-ketoglutarate thereby reducing NAD+ (NADP+) to NADH (NADPH), e.g., in the forward reaction:
Isocitrate + NAD+ (NADP+)→ a-KG + C02 + NADH (NADPH) + H+.
It has been discovered that mutations of IDHl present in certain cancer cells result in a new ability of the enzyme to catalyze the NAPH-dependent reduction of α-ketoglutarate to R(-)- 2-hydroxyglutarate (2HG). The production of 2HG is believed to contribute to the formation and progression of cancer (Dang, L et al, Nature 2009, 462:739-44).
The inhibition of mutant IDHl and its neoactivity is therefore a potential therapeutic treatment for cancer. Accordingly, there is an ongoing need for inhibitors of IDHl mutants having alpha hydroxyl neoactivity.
SUMMARY OF INVENTION
Described herein are methods of treating a cancercharacterized by the presence of a mutant allele of IDHl or IDH2. The methods comprise the step of administering to a subject in need thereof a compound of formula I, or a pharmaceutically acceptable salt, tautomer, isotopologueor hydrate thereof, wherein:
R1 is optionally substituted C4-C6 carbocyclyl;
eachR2and R3 is independently selected from optionally substituted aryl or optionally substituted heteroaryl;
R4 is alkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, or optionally substituted heteroaralkyl;
ring A is 4-6 membered non-aromatic ring having 0-1 additional heteroatoms selected from N, O or S, wherein ring A is optionally substituted with one or two R5 groups;
each R5 is independently halo; -CF3;-CN; -OR6;-N(R6)2; -C(0)Ci-C4 alkyl; Ci-
C4haloalkyl; d-C4alkyl optionally substituted with -OR6 or -N(R6)2; -0-Ci-C4alkyl optionally substituted with halo, -OR6 or -N(R6)2; -S02N(R6)2; -S02(Ci-C4alkyl); -NR6S02R6; C3-C5
carbocyclyl optionally substituted with one or two R groups; -0-(C3-C6 carbocyclyl optionally substituted with one or two R6 groups); 5-6 membered heteroaryl; -Ci-C4alkyl-C(0)0-Ci- C4alkyl; or -C(0)0-Ci-C4alkyl;or
each R6 is independently H or C1-C3 alkyl.
The compound of formula Iinhibitsmutant IDH1/2, particularly mutant IDH1 having alpha hydroxyl neoactivity. Also described herein are pharmaceutical compositionscomprising a compound of formula I.
DETAILED DESCRIPTION OF THE INVENTION
The details of construction and the arrangement of components set forth in the following description or illustrated in the drawings are not meant to be limiting. Other embodiments and different ways to practice the invention are expressly included. Also, the phraseology and terminology used herein is for the purpose of description and should not be regarded as limiting. The use of "including," "comprising," or "having," "containing", "involving", and variations thereof herein, is meant to encompass the items listed thereafter and equivalents thereof as well as additional items.
Definitions:
The term "halo" or "halogen" refers to any radical of fluorine, chlorine, bromine or iodine.
The term "alkyl" refers to a hydrocarbon chain that may be a straight chain or branched chain, containing the indicated number of carbon atoms. For example, C1-C12 alkyl indicates that the group may have from 1 to 12 (inclusive) carbon atoms in it. The term "haloalkyl" refers to an alkyl in which one or more hydrogen atoms are replaced by halo, and includes alkyl moieties in which all hydrogens have been replaced by halo (e.g., perfluoroalkyl).The terms "arylalkyl" or "aralkyl" refer to an alkyl moiety in which an alkyl hydrogen atom is replaced by an aryl group. Arylalkyl oraralkyl includes groups in which more than one hydrogen atom has been replaced by an aryl group. Examples of "arylalkyl" or "aralkyl" include benzyl, 2- phenylethyl, 3-phenylpropyl, 9-fluorenyl, benzhydryl, and trityl groups. The terms
"heteroarylalkyl" or "heteroaralkyl" refer to an alkyl moiety in which an alkyl hydrogen atom is
replaced by a heteroaryl group. Heteroarylalkyl or heteroaralkyl includes groups in which more than one hydrogen atom has been replaced by a heteroaryl group.
The term "alkylene" refers to a divalent alkyl, e.g., -CH2-, -CH2CH2-, and -CH2CH2CH2-.
The term "alkenyl" refers to a straight or branched hydrocarbon chain containing 2-12 carbon atoms and having one or more double bonds. Examples of alkenyl groups include, but are not limited to, allyl, propenyl, 2-butenyl, 3-hexenyl and 3-octenyl groups. One of the double bond carbons may optionally be the point of attachment of the alkenyl substituent. The term "alkynyl" refers to a straight or branched hydrocarbon chain containing 2-12 carbon atoms and characterized in having one or more triple bonds. Examples of alkynyl groups include, but are not limited to, ethynyl, propargyl, and 3-hexynyl. One of the triple bond carbons may optionally be the point of attachment of the alkynyl substituent.
The term "alkoxy" refers to an -O-alkyl radical. The term "haloalkoxy" refers to an alkoxy in which one or more hydrogen atoms are replaced by halo, and includes alkoxy moieties in which all hydrogens have been replaced by halo (e.g., perfluoroalkoxy).
The term "carbocyclyl" refers to a monocyclic, bicyclic or tricyclic, hydrocarbon ring systemthat is not fully aromatic, wherein any ring atom capable of substitution can be substituted by one or more substituents. A carbocyclyl can be fully or partially saturated. A bicyclic or tricylic carbocyclyl may contain one (in the case of a bicycle) or up to two (in the case of a tricycle) aromatic rings, as long as at least one ring in the carbocyclyl is non-aromatic. Unless otherwise specified, any ring atom capable of substitution in a carbocyclyl can be substituted by one or more substituents.
The term "aryl" refers to a fully aromatic monocyclic, bicyclic, or tricyclic hydrocarbon ring system. Examples of aryl moieties are phenyl, naphthyl, and anthracenyl.Unless otherwise specified, any ring atom in an aryl can be substituted by one or more substituents.
The term "cycloalkyl" as employed herein refers to a saturated cyclic, bicyclic, tricyclic,or polycyclic hydrocarbon group. Unless otherwise specified, any ring atom can be substituted by one or more substituents. The cycloalkyl groups can contain fused rings. Fused rings are rings that share a common carbon atom. Examples of cycloalkyl moieties include, but are not limited to, cyclopropyl, cyclohexyl, methylcyclohexyl, adamantyl, and norbomyl. Unless otherwise specified, any ring atom can be substituted by one or more substituents.
The term "heterocyclyl" refers to a monocyclic, bicyclic or tricyclic, ring structure that is not fully aromatic and includes one to four heteroatoms independently selected from N, O, or S in one or more of the rings. A heterocyclyl can be fully or partially saturated. A bicyclic or tricylic heterocyclyl may contain one (in the case of a bicycle) or up to two (in the case of a tricycle) aromatic rings, as long as at least one ring in the heterocyclyl is non-aromatic. Unless otherwise specified, any ring atom capable of substitution in a heterocyclyl can be substituted by one or more substituents. Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, oxolane, thiolane, oxazole, piperidine, piperazine, morpholine, lactones, lactams such as azetidinones and pyrrolidinones, sultams, sultones, and the like.
The term "heteroaryl" refers to amonocyclic, bicyclic, or tricyclic ring system having 1 -3 heteroatoms if monocyclic, 1-6 heteroatoms if bicyclic, or 1-9 heteroatoms if tricyclic, said heteroatoms independently selected from O, N, or S, wherein each ring in a heteroaryl is fully aromatic. Unless otherwise specified, any ring atom capable of substitution in a heteroaryl can be substituted by one or more substituents. The terms "hetaralkyl" and "heteroaralkyl", as used herein, refers to an alkyl group substituted with a heteroaryl group. The ring heteroatoms of the compounds provided herein include N-O, S(O), and S(0)2.
The term "substituted" refers to the replacement of a hydrogen atom with another moiety. Typical substituentsinclude alkyl (e.g., CI, C2, C3, C4, C5, C6, C7, C8, C9, CIO, CI 1, C12 straight or branched chain alkyl), cycloalkyl, haloalkyl (e.g., perfluoroalkyl such as CF3), aryl, heteroaryl, aralkyl, heteroaralkyl, heterocyclyl, alkenyl, alkynyl, cycloalkenyl,
heterocycloalkenyl, alkoxy, haloalkoxy (e.g., perfluoroalkoxy such as OCF3), halo, hydroxy, carboxy, carboxylate, cyano, nitro, amino, alkyl amino, SO3H, sulfate, phosphate,
methylenedioxy (-0-CH2-0- wherein oxygens are attached to vicinal atoms), ethylenedioxy, oxo (not a substituent on heteroaryl), thioxo (e.g., C=S) (not a substituent on heteroaryl), imino (alkyl, aryl, aralkyl), S(0)nalkyl (where n is 0-2), S(0)n aryl (where n is 0-2), S(0)n heteroaryl (where n
is 0-2), S(0)n heterocyclyl (where n is 0-2), amine (mono-, di-, alkyl, cycloalkyl, aralkyl, heteroaralkyl, aryl, heteroaryl, and combinations thereof), ester (alkyl, aralkyl, heteroaralkyl, aryl, heteroaryl), amide (mono-, di-, alkyl, aralkyl, heteroaralkyl, aryl, heteroaryl, and combinations thereof), sulfonamide (mono-, di-, alkyl, aralkyl, heteroaralkyl, and combinations thereof). In one aspect, the substituents on a group are independentlyany one single, or any subset of the aforementioned substituents. In another aspect, a substituent may itself be substituted with any one of the above substituents.
The term "tautomer" refers to each of two or more isomers of a compound (e.g., a compound described herein) that exist together in equilibrium, and are readily interchangeable by migration of a hydrogen atom or proton, accompanied by a switch of a single bond and an adjacent double bond.
As used herein, the term "elevated levels of 2HG"means 10%, 20% 30%, 50%, 75%, 100%, 200%, 500% or more 2HG thanis present in a subject that does not carry a mutant IDHl or IDH2 allele. The term "elevated levels of 2HG" may refer to the amount of 2HG within a cell, within a tumor, within an organ comprising a tumor, or within a bodily fluid.
The term"bodily fluid" includes one or more of amniotic fluid surrounding a fetus, aqueous humour, blood (e.g., blood plasma), serum, Cerebrospinal fluid, cerumen, chyme, Cowper's fluid, female ejaculate, interstitial fluid, lymph, breast milk, mucus (e.g., nasal drainage or phlegm), pleural fluid, pus, saliva, sebum, semen, serum, sweat, tears, urine, vaginal secretion, or vomit.
As used herein, the terms "inhibit" or "prevent" include both complete and partial inhibition and prevention. An inhibitor may completely or partially inhibit.
The term "treat" means decrease, suppress, attenuate, diminish, arrest, or stabilize the development or progression of a cancer (e.g., a cancer delineated herein), lessen the severity of the cancer or improve the symptoms associated with the cancer.
As used herein, an amount of a compound effective to treat a disorder, or a
"therapeutically effective amount" refers to an amount of the compound which is effective, upon single or multiple dose administration to a subject, in treating a cell, or in curing, alleviating, relieving or improving a subject with a disorder beyond that expected in the absence of such treatment.
As used herein, the term "subject" is intended to include human and non-human animals. Exemplary human subjects include a human patient having a disorder, e.g., a disorder described herein or a normal subject. The term "non-human animals" of the invention includes all vertebrates, e.g., non-mammals (such as chickens, amphibians, reptiles) and mammals, such as non-human primates, domesticated and/or agriculturally useful animals, e.g., sheep, dog, cat, cow, pig, etc.
Compounds
Provided is a compound having formula I or a pharmaceutically acceptable salt, tautomer, isotopologue or hydrate thereof, wherein:
R1 is optionally substituted C -C6 carbocyclyl;
each R2 and R3 is independently selected from optionally substituted aryl or optionally substituted heteroaryl;
R4 is alkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, or optionally substituted heteroaralkyl;
ring A is 4-6 membered non-aromatic ring having 0-1 additional heteroatoms selected from N, O or S, wherein ring A is optionally substituted with one or two R5 groups;
each R5 is independently halo; -CF3; -CN; -OR6;-N(R6)2; -C(0)Ci-C4 alkyl; Ci-C4 haloalkyl; C1 -C4 alkyl optionally substituted with -OR6 or -N(R6)2; -0-C1-C4 alkyl optionally substituted with halo, -OR6 or -N(R6)2; -S02N(R6)2; -S02(Ci-C4 alkyl); -NR6S02R6; C3-C5 carbocyclyl optionally substituted with one or two R6 groups; -0-(C3-C6 carbocyclyl optionally substituted with one or two R6 groups); 5-6 membered heteroaryl; -Ci-C alkyl-C(0)0-Ci-C alkyl; or -C(0)0-C C4 alkyl; or
each R6 is independently H or C1-C3 alkyl.
Provided is also a compound having formula I or a pharmaceutically acceptable salt or hydrate thereof, wherein:
R1 is optionally substituted C4-C6 carbocyclyl;
eachR2and R3 is independently selected from optionally substituted aryl or optionally substituted heteroaryl;
R4 is alkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, or optionally substituted heteroaralkyl;
ring A is 4-6 membered non-aromatic ring having 0-1 additional heteroatoms selected from N, O or S, wherein ring A is optionally substituted with one or two R5 groups;
eachR5 is independently halo, -CF3, -CN, -OR6, -N(R6)2, -C(0)CH3; Ci-C3haloalkyl, Ci- C3alkyl optionally substituted with -OR6 or -N(R6)2; or
each R6 is independently H or Ci-C3 alkyl.
Provided is also a compound having formula lor a pharmaceutically acceptable salt, tautomer, isotopologue or hydrate thereof, wherein:
R1 is C4-C6 carbocyclyloptionally substituted with one to three R7 groups;
each R2 and R3 is independently selected from aryl or heteroaryl, wherein said aryl or heteroaryl is independently optionally substituted with one to three R7 groups;
R4 is alkyl, aryl, heteroaryl, aralkyl, or heteroaralkyl, wherein said aryl, heteroaryl, aralkyl, and heteroaralkyl are each independently optionally substituted with one to three R7 groups;
ring A is 4-6 membered non-aromatic ring having 0-1 additional heteroatoms selected from N, O or S, wherein ring A is optionally substituted with one or two R5 groups;
each R5and R7is independently halo;-CF3;-CN;-OR6;-N(R6)2;-C(0)Ci-C4alkyl; d- C4haloalkyl;Ci-C4alkyl optionally substituted with -OR6 or -N(R6)2; -0-Ci-C4alkyl optionally substituted with halo, -OR6 or -N(R6)2; -S02N(R6)2;-S02(Ci-C4alkyl);-S(0)-Ci-4
alkyl, -NR S02R ;C3-C5 carbocyclyl optionally substituted with one or two R groups; -0-(C3-C6 carbocyclyl optionally substituted with one or two R6 groups);5-6 membered heteroaryl;-Ci- C4alkyl-C(0)0-Ci-C4alkyl; or -C(0)0-Ci-C4alkyl; or
each R6 is independently H or C1-C4 alkyl.
Provided is also a compound having formula lor a pharmaceutically acceptable salt, tautomer, isotopologue or hydrate thereof, wherein:
R1 is C4-C6 carbocyclyloptionally substituted with one to three R7 groups;
each R2 and R3 is independently selected from aryl or heteroaryl, wherein said aryl or heteroaryl is independently optionally substituted with one to three R7 groups;
R4 is alkyl, aryl, heteroaryl, aralkyl, or heteroaralkyl, wherein said aryl, heteroaryl, aralkyl, and heteroaralkyl are each independently optionally substituted with one to three R7 groups;
ring A is 4-6 membered non-aromatic ring having 0-1 additional heteroatoms selected from N, O or S, wherein ring A is optionally substituted with one or two R5 groups;
eachR5and R7is independently halo, -CF3, -CN, -OR6, -N(R6)2, -C(0)CH3; C C3haloalkyl, Ci-C3alkyl optionally substituted with -OR6 or -N(R6)2; or
each R6 is independently H or Ci-C3 alkyl.
In one embodiment, R1 is optionally substituted C4-C6cycloalkyl. In one aspect of this embodiment, R1 is C -C6cycloalkyl optionally substituted with one to three R7 groups. In another aspect of this embodiment, R1 is C4, C5, or C6cycloalkyloptionally substituted with one to two R7 groups and R7 is halo. In another aspect of this embodiment, R1 is C4 or
C6cycloalkyloptionally substituted with one to two R7 groups and R7 is halo. In yet another aspect of this embodiment, R1 is
In yet another aspect of this embodiment, R
1 is
In another embodiment, R2 is optionally substituted aryl. In one aspect of this
embodiment, R2 is aryl optionally substituted with one to three R7 groups. In another aspect of this embodiment, R2 is phenyl optionally substituted with one to two R7 groups and R7 is -CI.
In another embodiment, R3 is optionally substituted aryl or optionally substituted aryl heteroaryl. In one aspect of this embodiment, R3 is optionally substituted heteroaryl. In another aspect of this embodiment, R3 is heteroaryl optionally substituted with one to three R7 group. In yet another aspect of this embodiment, R3 is pyridinyl, indazolyl, benzoimidazolyl, indolyl, or N- methylindolyl, wherein each R3 is optionally substituted with one R7 wherein R7 is -F.In another aspect of this embodiment, R3 is optionally substituted aryl. In another aspect of this
embodiment, R3 is aryl optionally substituted with one to three R7 groups. In yet another aspect of this embodiment, R3 is phenyl optionally substituted with one R7 wherein R7 is -F. In yet another aspect of this embodiment, R3 is phenyl optionally substituted with one or two
R7swherein each R7is independently halo; -CN; -N(R6)2; C1-C4 alkyl optionally substituted with -OR6; -O-C1-C4 alkyl optionally substituted with halo, or -OR6; -S02N(R6)2; -S02(d-C4 alkyl); -S(0)-Ci-4 alkyl, -NR6S02R6; C3-C5 carbocyclyloptionally substituted with one R6; -O- (C3-C6 carbocyclyl); 5-membered heteroaryl. In yet another aspect of this embodiment, R3 is phenyl optionally substituted with one or two R7swherein each R7 is
independently -F, -S02NH2, -S02CH3, -S(0)CH3, -CN,
methoxy, -OCH2OH, -CH2OH, -S02N(CH3)2, -S02NHCH3, -NHS02CH3, -CH2CH2OH, -N(CH3) 2, t-butyl, cyclopropyl, -C(OH)(CH3)2, -OCF3, -OCHF2, -O-cyclopropyl, -1-methyl-cyclopropyl, or pyrazolyl.
In another embodiment, R4 is optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, or optionally substituted heteroaralkyl. In one aspect of this embodiment, R4 is aryl, heteroaryl, aralkyl, or heteroaralkyl, wherein said aryl, heteroaryl, aralkyl, and heteroaralkyl are each independently optionally substituted with one to three R7
groups. In another aspect of this embodiment, R4 is aryl or heteroaryl, each aryl or heteroaryl is optionally substituted with one to three R7 groups. In another aspect of this embodiment, R4 is 6- membered aryl or 5-6 membered heteroaryl, wherein said aryl or heteroaryl is optionally substituted with one to three R7 groups. In yet another aspect of this embodiment, R4 is:
each member of R4 is optionally substituted with one or two R7groups and each R7 is
independently F, CI, methyl, CF3, CN, OMe, or N(R6)2. In yet another aspect of this
embodiment, R4 is:
R
1UU is independently H, methyl, F, CI, CF
3, CN, OCH
3, or N(R
6)
2. In yet another aspect of this embodiment R
4 is:
wherein R
100 is H, methyl, CI, CF
3, CN, OCH
3, or N(R
&)
2 and R
m is H, F or methyl.
wherein "» denotes ring A's attachment to the amide moiety of formula and * denotes ring A's attachment to R
4; and each member of ring A is optionally substituted with one or two R
5
groups. In another embodiment, ring A is
wherein *> denotes ring A's attachment to the amide moiety of formula and ^ denotes ring A's attachment to R
4; and each member of ring A is optionally substituted with one or two R
5 groups. In one aspect of this embodiment, each R
5 is independently halo; -OR
6; -C(0)d-C
4alkyl; Ci-C
4alkyl optionally substituted with -OR
6; -C
3-C
5 carbocyclyl optionally substituted with one or two R
6 groups; -Ci-C
4alkyl-C(0)0-Ci-C
4alkyl; or -C(0)0-Ci-
C
4alkyl. In one aspect of this embodiment, each R is
independently -OH, -F, -CH2CH2OH, -CH2C(0)OCH2CH3, -C(0)0-t-butyl, cyclopropyl, methyl or -C(0)CH3. In one aspect of this embodiment, each R5 is independently methyl or-C(0)CH3. In another aspect of this embodiment, ring A is:
In another aspect of this embodiment, ring A is:
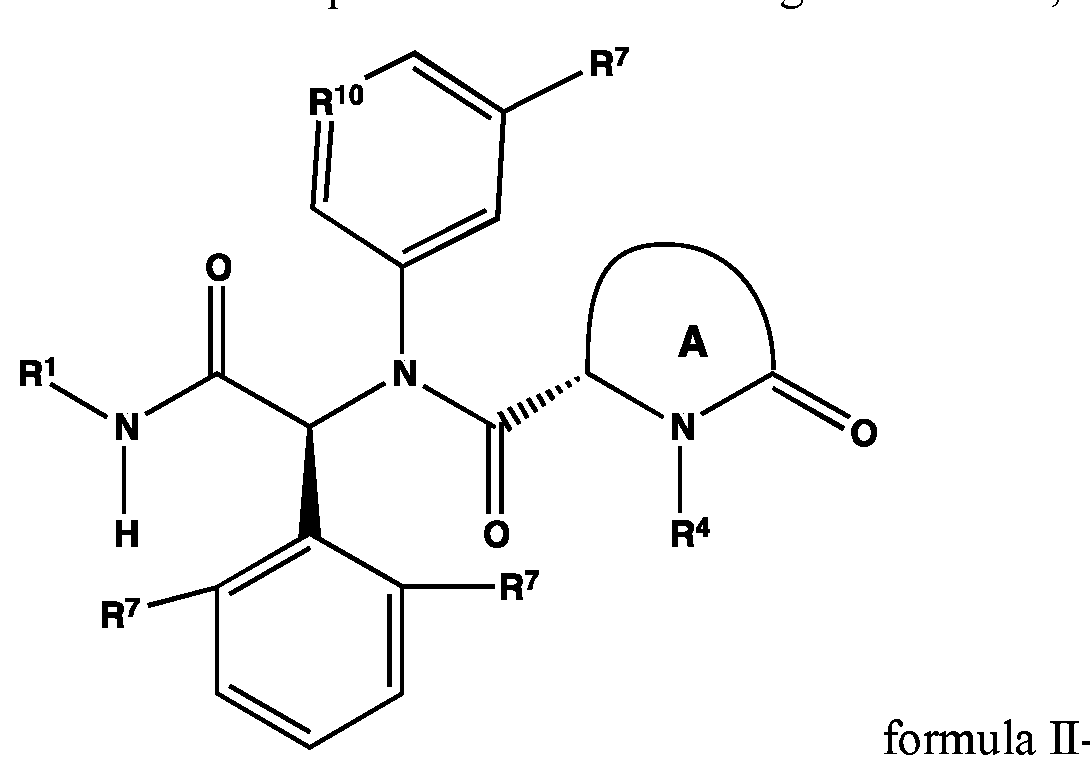
Provided is also a compound having formula Ilor a pharmaceutically acceptable salt or hydrate thereof, wherein R1, R2, R3, ring A and R4 are as defined in formula I or any one of the above embodiments.
Provided is also a compound having formula Π-a or a pharmaceutically acceptable salt or hydrate thereof, wherein R1, R4, ring A and R7 are as defined in formula I or any one of the above embodiments.
Provided is also a compound having formula Π-a-l or a pharmaceutically acceptable salt or hydrate thereof, wherein R1, R4, ring A and R7 are as defined in formula I or any one of the above embodiments and R10 is CR11 or N wherein R11 is -F, -S02NH2, -S02CH3, -CN, methoxy, -OCH2OH, -CH2OH, -S02N(CH3)2, -S02NHCH3, -NHS02CH3, -CH2CH2OH, -N(CH3) 2, t-butyl, cyclopropyl, -C(OH)(CH3)2, -OCF3, -OCHF2, -O-cyclopropyl, -1-methyl-cyclopropyl, or pyrazolyl.
Provided is also a compound having formula Π-b or a pharmaceutically acceptable salt or hydrate thereof, wherein R1, R4, and ring A are as defined in formula I or any one of the above embodiments; Rr is H or CI; and and R10 is CR11 or N wherein R11
is -F, -S02NH2, -S02CH3, -CN,
methoxy, -OCH2OH, -CH2OH, -S02N(CH3)2, -S02NHCH3, -NHS02CH3, -CH2CH2OH, -N(CH3)
2, t-butyl, cyclopropyl, -C(OH)(CH3)2, -OCF3, -OCHF2, -O-cyclopropyl, -1-methyl-cyclopropyl, or pyrazolyl.
Provided is also a compound having formula Π-b-l or a pharmaceutically acceptable salt or hydrate thereof, wherein R1, R4, and ring A are as defined in formula I or any one of the above embodiments and R7 is H or CI.
In another embodiment of formula II, Π-a, II-a-1, Il-b, or II-b-1,
R1 is:
Further embodiments provided herein include combinations of one or more of the particular embodiments set forth above.
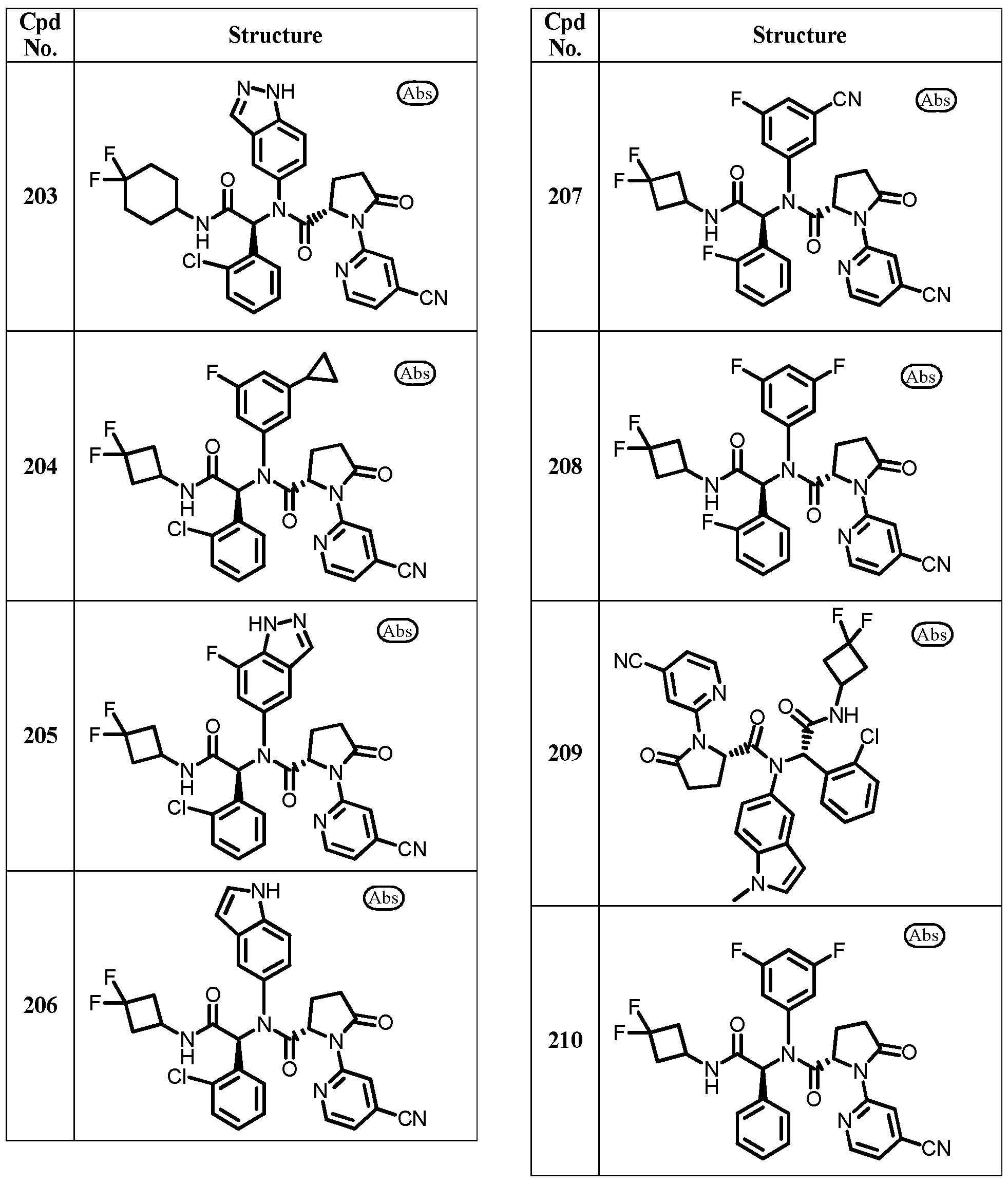
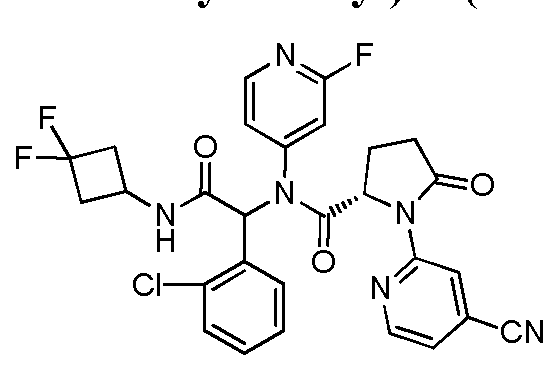
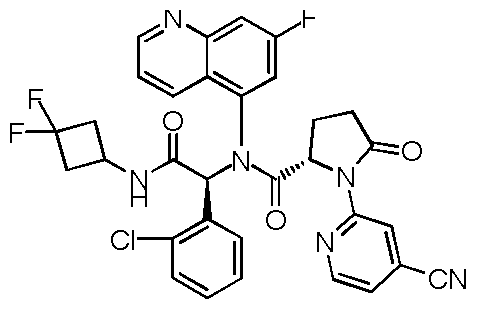
In another embodiment, exemplary compounds of formula I are depicted below in Table
30
31
32
33
40
Included herein are also methods for making compounds of Formula I or a compound of any one of the embodiments described herein comprising reacting R NC with R2CHO, R3NH2
and
, wherein R
4 is H or R
4 and R
1, R
2, R
3, R
4 and ring A as defined in Formula I or in any of the embodiments described herein. In one aspect of the preceding methods, R
4 is alkyl.
Also included herein are methods for making compounds of Formula I or a compound of any one of the embodiments described herein comprising (1) reacting R NC with R2CHO,
wherein R4 is optionally substituted aryl or optionally substituted heteroaryl; and R1, R2, R3, R4and ring A as defined in Formula I or in any of the embodiments described herein. In one aspect of the preceding methods, R4 is aryl or heteroaryl, each independently substituted with one to three R groups. In another aspect of the preceding method, R , R , R , R , R , R , R and ring A are as defined in any of the embodiments herein.
The compounds of this invention may contain one or more asymmetric centers and thus occur as racemates, racemic mixtures, scalemic mixtures,anddiastereomeric mixtures, as well as single enantiomers or individual stereoisomersthat are substantially free from another possible enantiomer or stereoisomer. The term "substantially free of other stereoisomers" as used herein means a preparation enriched in a compound having a selected stereochemistry at one or more selected stereocentersby at least about 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99%. The term "enriched" means that at least the designated percentage of a
preparation is the compound having a selected stereochemistry at one or more selected stereocenters. Methods of obtaining or synthesizing an individual enantiomer or stereoisomer for a given compound are known in the art and may be applied as practicable to final compounds or to starting material or intermediates.
In one embodiment, the compound is enriched in a specific stereoisomer by at least about 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99%.
The compounds of formula I, II, Il-a, Π-a-l, Il-b or II-b-1 may also comprise one or more isotopic substitutions. For example, H may be in any isotopic form, including 1H, 2H (D or deuterium), and 3H (T or tritium); C may be in any isotopic form, including nC, 12C, 13C, and 14C; N may be in any isotopic form, including13N,14N and 15N; O may be in any isotopic form, including 150,160 and 180; F may be in any isotopic form, including18F; and the like. For example, the compound is enriched in a specific isotopic form of H, C, N, O and/or F by at least about 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99%.
Unless otherwise indicated when a disclosed compound is named or depicted by a structure without specifying the stereochemistry and has one or more chiral centers, it is understood to represent all possible stereoisomers of the compound.
The compounds of this invention may also be represented in multiple tautomeric forms, in such instances, the invention expressly includes all tautomeric forms of the compounds described herein, even though only a single tautomeric form may be represented (e.g., alkylation of a ring system may result in alkylation at multiple sites, the invention expressly includes all
such reaction products). All such isomeric forms of such compounds are expressly included in the present invention.
Compounds described herein may be prepared following procedures detailed in the examples and other analogous methods known to one skilled in the art. Compounds produced by any of the schemes set forth below may be further modified (e.g., through the addition of substituents to rings, etc.) to produce additional compounds. The specific approaches and compounds shown herein are not intended to be limiting. The suitability of a chemical group in a compound structure for use in the synthesis of another compound is within the knowledge of one of ordinary skill in the art. Synthetic chemistry transformations and protecting group methodologies (protection and deprotection) useful in synthesizing the applicable compounds are known in the art and include, for example, those described in Larock R, Comprehensive Organic Transformations, VCH Publishers (1989); Greene, TW et al., Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); Fieser, L et al., Fieser and Fieser 's Reagents for Organic Synthesis, John Wiley and Sons (1994); and Paquette, L, ed., Encyclopedia of Reagents or Organic Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
Combinations of substituents and variables envisioned by this invention are only those that result in the formation of stable compounds.
It may be convenient or desirable to prepare, purify, and/or handle a corresponding salt of the active compound, for example, a pharmaceutically acceptable salt. Examples of
pharmaceutically acceptable salts are discussed in Berge etal, 1977, "Pharmaceutically
Acceptable Salts." J. Pharm. Sci. Vol. 66, pp. 1-19.
For example, if the compound is anionic, or has a functional group which may be anionic {e.g., -COOH may be -COO"), then a salt may be formed with a suitable cation. Examples of suitable inorganic cations include, but are not limited to, alkali metal ions such as Na+ and K+, alkaline earth cations such as Ca2+ and Mg2+, and other cations such as Al3+. Examples of suitable organic cations include, but are not limited to, ammonium ion {i.e., NH4 +) and substituted ammonium ions {e.g., NH3R+, NH2R2+, NHR3+, NR4+). Examples of some suitable substituted ammonium ions are those derived from: ethylamine, diethylamine,
dicyclohexylamine, triethylamine, butylamine, ethylenediamine, ethanolamine, diethanolamine, piperazine, benzylamine, phenylbenzylamine, choline, meglumine, and tromethamine, as well as
amino acids, such as lysine and arginine. An example of a common quaternary ammonium ion is
N(CH3)4 +.
If the compound is cationic, or has a functional group that may be cationic (e.g., -NH2 may be -NH3 +), then a salt may be formed with a suitable anion. Examples of suitable inorganic anions include, but are not limited to, those derived from the following inorganic acids:
hydrochloric, hydrobromic, hydroiodic, sulfuric, sulfurous, nitric, nitrous, phosphoric, and phosphorous.
Examples of suitable organic anions include, but are not limited to, those derived from the following organic acids: 2-acetyoxybenzoic, acetic, ascorbic, aspartic, benzoic,
camphorsulfonic, cinnamic, citric, edetic, ethanedisulfonic, ethanesulfonic, fumaric,
glucoheptonic, gluconic, glutamic, glycolic, hydroxymaleic, hydroxynaphthalene carboxylic, isethionic, lactic, lactobionic, lauric, maleic, malic, methanesulfonic, mucic, oleic, oxalic, palmitic, pamoic, pantothenic, phenylacetic, phenylsulfonic, propionic, pyruvic, salicylic, stearic, succinic, sulfanilic, tartaric, toluenesulfonic, and valeric. Examples of suitable polymeric organic anions include, but are not limited to, those derived from the following polymeric acids: tannic acid, carboxymethyl cellulose.
Unless otherwise specified, a reference to a particular compound also includes salt forms thereof.
Compositions and routes of administration
The compounds utilized in the methods described herein may be formulated together with a pharmaceutically acceptable carrier or adjuvant into pharmaceutically acceptable compositions prior to be administered to a subject. In another embodiment, such pharmaceutically acceptable compositions further comprise additional therapeutic agents in amounts effective for achieving a modulation of disease or disease symptoms, including those described herein.
The term "pharmaceutically acceptable carrier or adjuvant" refers to a carrier or adjuvant that may be administered to a subject, together with a compound of this invention, and which does not destroy the pharmacological activity thereof and is nontoxic when administered in doses sufficient to deliver a therapeutic amount of the compound.
Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers,
alumina, aluminum stearate, lecithin, self-emulsifying drug delivery systems (SEDDS) such as d- a-tocopherol polyethyleneglycol 1000 succinate, surfactants used in pharmaceutical dosage forms such as Tweens or other similar polymeric delivery matrices, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
Cyclodextrins such as α-, β-, and γ-cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2- and 3-hydroxypropyl- -cyclodextrins, or other solubilized derivatives may also be advantageously used to enhance delivery of compounds of the formulae described herein.
The pharmaceutical compositions of this invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir, preferably by oral administration or administration by injection. The pharmaceutical compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers, adjuvants or vehicles. In some cases, the pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance the stability of the formulated compound or its delivery form. The term parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intraarticular, intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and intracranial injection or infusion techniques.
The pharmaceutical compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride
solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, or carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms such as emulsions and or suspensions. Other commonly used surfactants such as Tweens or Spans and/or other similar emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
The pharmaceutical compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, emulsions and aqueous suspensions, dispersions and solutions. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions and/or emulsions are administered orally, the active ingredient may be suspended or dissolved in an oily phase is combined with emulsifying and/or suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
The pharmaceutical compositions of this invention may also be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
Topical administration of the pharmaceutical compositions of this invention is useful when the desired treatment involves areas or organs readily accessible by topical application. For application topically to the skin, the pharmaceutical composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier. Carriers
for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxyethylene
polyoxypropylene compound, emulsifying wax and water. Alternatively, the pharmaceutical composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier with suitable emulsifying agents. Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water. The pharmaceutical compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation. Topically-transdermal patches are also included in this invention.
The pharmaceutical compositions of this invention may be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
When the compositions of this invention comprise a combination of a compound of the formulae described herein and one or more additional therapeutic or prophylactic agents, both the compound and the additional agent should be present at dosage levels of between about 1 to 100%, and more preferably between about 5 to 95% of the dosage normally administered in a monotherapy regimen. The additional agents may be administered separately, as part of a multiple dose regimen, from the compounds of this invention. Alternatively, those agents may be part of a single dosage form, mixed together with the compounds of this invention in a single composition.
The compounds described herein can, for example, be administered by injection, intravenously, intraarterially, subdermally, intraperitoneally, intramuscularly, or subcutaneously; or orally, buccally, nasally, transmucosally, topically, in an ophthalmic preparation, or by inhalation, with a dosage ranging from about 0.5 to about 100 mg/kg of body weight, alternatively dosages between 1 mg and 1000 mg/dose, every 4 to 120 hours, or according to the requirements of the particular drug. The methods herein contemplate administration of an effective amount of compound or compound composition to achieve the desired or stated effect.
Typically, the pharmaceutical compositions of this invention will be administered from about 1 to about 6 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy. The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. A typical preparation will contain from about 5% to about 95% active compound (w/w). Alternatively, such preparations contain from about 20% to about 80% active compound.
Lower or higher doses than those recited above may be required. Specific dosage and treatment regimens for any particular subject will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health status, sex, diet, time of administration, rate of excretion, drug combination, the severity and course of the disease, condition or symptoms, the subject's disposition to the disease, condition or symptoms, and the judgment of the treating physician.
Upon improvement of a subject's condition, a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary. Subsequently, the dosage or frequency of administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level. Subjects may, however, require intermittent treatment on a long- term basis upon any recurrence of disease symptoms.
The pharmaceutical compositions described above comprising a compound of formula I, II, Il-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments herein, may further comprise another therapeutic agent useful for treating cancer.
Methods of Use
Provided is a method for inhibiting a mutant IDHl or IDH2 activity comprising contacting a subject in need thereof with a compound (including its tautomers and/or
isotopologues) of structural formula I, II, Π-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments herein, or a pharmaceutically acceptable salt thereof. In one embodiment, the cancer to be treated is characterized by a mutant allele of IDHl or IDH2 wherein the IDHl or IDH2 mutation results in a new ability of the enzyme to catalyze the NAPH-dependent reduction of a-ketoglutarate to i?(-)-2-hydroxyglutarate in a subject. In one
aspect of this embodiment,the mutant IDH1 has an R132X mutation. In one aspect of this embodiment, the R132X mutation is selected from R132H, R132C, R132L, R132V, R132S and Rl 32G. In another aspect, the Rl 32X mutation is Rl 32H or Rl 32C. In yet another aspect, the R132X mutation is R132H.
Also provided are methods of treating a cancer characterized by the presence of a mutant allele of IDH1 comprising the step of administering to subject in need thereof (a) a compound of formula I, II, Π-a, II-a-1 , Π-b, or II-b-1 , or a compound described in any one of the embodiments herein, or a pharmaceutically acceptable salt thereof, or (b) a pharmaceutical composition comprising (a) and a pharmaceutically acceptable carrier.
In one embodiment, the cancer to be treated is characterized by a mutant allele of IDH1 wherein the IDHl mutation results in a new ability of the enzyme to catalyze the NAPH- dependent reduction of a-ketoglutarate to i?(-)-2-hydroxyglutarate in a patient. In one aspect of this embodiment, the IDHl mutation is an R132X mutation. In another aspect of this
embodiment, the R132X mutation is selected from R132H, R132C, R132L, R132V, R132S and R132G. In another aspect, the R132X mutation is R132 H or R132C. A cancer can be analyzed by sequencing cell samples to determine the presence and specific nature of (e.g., the changed amino acid present at) a mutation at amino acid 132 of IDHl .
Without being bound by theory, applicants believe that mutant alleles of IDHl wherein the IDHl mutation results in a new ability of the enzyme to catalyze the NAPH-dependent reduction of a-ketoglutarate to i?(-)-2-hydroxyglutarate, and in particular R132H mutations of IDHl, characterize a subset of all types of cancers, without regard to their cellular nature or location in the body. Thus, the compounds and methods of this invention are useful to treat any type of cancer that is characterized by the presence of a mutant allele of IDHl imparting such acitivity and in particular an IDHl R132H or R132C mutation.
In one aspect of this embodiment, the efficacy of cancer treatment is monitored by measuring the levels of 2HG in the subject. Typically levels of 2HG are measured prior to treatment, wherein an elevated level is indicated for the use of the compound of formula I, Π, Il-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments described herein to treat the cancer. Once the elevated levels are established, the level of 2HG is determined during the course of and/or following termination of treatment to establish efficacy. In certain
embodiments, the level of 2HG is only determined during the course of and/or following termination of treatment. A reduction of 2HG levels during the course of treatment and following treatment is indicative of efficacy. Similarly, a determination that 2HG levels are not elevated during the course of or following treatment is also indicative of efficacy. Typically, the these 2HG measurements will be utilized together with other well-known determinations of efficacy of cancer treatment, such as reduction in number and size of tumors and/or other cancer- associated lesions, improvement in the general health of the subject, and alterations in other biomarkers that are associated with cancer treatment efficacy.
2HG can be detected in a sample by LC MS. The sample is mixed 80:20 with methanol, and centrifuged at 3,000 rpm for 20 minutes at 4 degrees Celsius. The resulting supernatant can be collected and stored at -80 degrees Celsius prior to LC-MS MS to assess 2-hydroxyglutarate levels. A variety of different liquid chromatography (LC) separation methods can be used. Each method can be coupled by negative electrospray ionization (ESI, -3.0 kV) to triple-quadrupole mass spectrometers operating in multiple reaction monitoring (MRM) mode, with MS parameters optimized on infused metabolite standard solutions. Metabolites can be separated by reversed phase chromatography using 10 mM tributyl-amine as an ion pairing agent in the aqueous mobile phase, according to a variant of a previously reported method (Luo etal.J Chromatogr A 1147, 153-64, 2007). One method allows resolution of TCA metabolites: t = 0, 50% B; t = 5, 95% B; t= 7, 95% B; t= 8, 0% B, where B refers to an organic mobile phase of 100% methanol. Another method is specific for 2-hydroxyglutarate, running a fast linear gradient from 50% -95% B (buffers as defined above) over 5 minutes. A Synergi Hydro-RP, 100mm x 2 mm, 2.1 μιτι particle size (Phenomonex) can be used as the column, as described above. Metabolites can be quantified by comparison of peak areas with pure metabolite standards at known concentration. Metabolite flux studies from 13C-glutamine can be performed as described, e.g., in Munger et al. Nat Biotechnol 26, 1179-86, 2008.
In one embodiment 2HG is directly evaluated.
In anotherembodiment a derivative of 2HGformed in process of performing the analytic method is evaluated. By way of example such a derivative can be a derivative formed in MS analysis. Derivatives can include a salt adduct, e.g., a Na adduct, a hydration variant, or a hydration variant which is also a salt adduct, e.g., a Na adduct, e.g., as formed in MS analysis.
In anotherembodiment a metabolic derivative of 2HG is evaluated. Examples include species that build up or are elevated, or reduced, as a result of the presence of 2HG, such as glutarate or glutamate that will be correlated to 2HG, e.g., R-2HG.
Exemplary 2HG derivatives include dehydrated derivatives such as the compounds provided below or a salt adduct thereof:
In one embodiment the cancer is a tumor wherein at least 30, 40, 50, 60, 70, 80 or 90% of the tumor cells carry an IDHl mutation, and in particular an IDHl R132H or R132C mutation, at the time of diagnosis or treatment.
IDHl R132X mutations are known to occur in certain types of cancers as indicated in Table 2, below.
Table 2. IDH mutations associated with certain cancers
Acute lymphoblastic leukemia R132C primary tumor
(ALL)
paragangliomas R132C primary tumor
IDHl R132H mutations have been identified in glioblastoma, acute myelogenous leukemia, sarcoma, melanoma, non-small cell lung cancer, cholangiocarcinomas,
chondrosarcoma, myelodysplastic syndromes (MDS), myeloproliferative neoplasm (MPN), colon cancer, and angio-immunoblastic non-Hodgkin's lymphoma (NHL). Accordingly, in one embodiment, the methods described herein are used to treat glioma (glioblastoma), acute myelogenous leukemia, sarcoma, melanoma, non-small cell lung cancer (NSCLC) or cholangiocarcinomas, chondrosarcoma, myelodysplastic syndromes (MDS), myeloproliferative neoplasm (MPN), colon cancer, or angio-immunoblastic non-Hodgkin's lymphoma (NHL) in a patient.
Accordingly in one embodiment, the cancer is a cancer selected from any one of the cancer types listed in Table 2, and the IDH R132X mutation is one or more of the IDHl R132X mutations listed in Table 2 for that particular cancer type.
In another embodiment, the methods described herein are used to treat glioma
(glioblastoma), acute myelogenous leukemia, sarcoma, melanoma, non-small cell lung cancer (NSCLC), cholangiocarcinomas (e.g., intrahepatic cholangiocarcinoma (IHCC)),
chondrosarcoma, myelodysplastic syndromes (MDS), myeloproliferative neoplasm (MPN), prostate cancer, chronic myelomonocytic leukemia (CMML), B-acute lymphoblastic leukemias (B-ALL), B-acute lymphoblastic leukemias (B-ALL), myeloid sarcoma, multiple myeloma, lymphoma colon cancer, or angio-immunoblastic non-Hodgkin's lymphoma (NHL) in a patient. In another embodiment, the cancer to be treated is an advanced hematologic malignancy selected from lymphoma (e.g., Non-Hodgkin lymphoma (NHL) such B-cell lymphoma (e.g., Burkitt lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), diffuse large B-cell lymphoma, follicular lymphoma, immunoblastic large cell lymphoma, precursor B- lymphoblastic lymphoma, and mantle cell lymphoma) and T-cell lymphoma (e.g., mycosis fungoides, anaplastic large cell lymphoma, and precursor T-lymphoblastic lymphoma).
Also provided are methods of treating a disease selected from Maffucci syndrome and Oilier disease, characterized by the presence of a mutant allele of IDH1 comprising the step of administering to subject in need thereof (a) a compound of Formula I, II, Il-a, II-a-1, Il-b, or Il-b- 1 or a compound described in any one of the embodiments herein, or a pharmaceutically acceptable salt thereof, or (b) a pharmaceutical composition comprising (a) and a
pharmaceutically acceptable carrier.
Treatment methods described herein can additionally comprise various evaluation steps prior to and/or following treatment with a compound of formula I, II, Il-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments described herein.
In one embodiment, prior to and/or after treatment with a compound of Structural formula I, II, Π-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments described herein, the method further comprisesthe step of evaluating the growth, size, weight, invasiveness, stage and/or other phenotype of the cancer.
In one embodiment, prior to and/or after treatmentwith a compound of formula I, II, Il-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments described herein, the method further comprisesthe step of evaluating the IDH1 genotype of the cancer. This may be achieved by ordinary methods in the art, such as DNA sequencing, immuno analysis, and/or evaluation of the presence, distribution or level of 2HG.
In one embodiment, prior to and/or after treatmentwith a compound of formula I, II, Il-a, II-a-1, Il-b, or II-b-1 or a compound described in any one of the embodiments described herein, the method further comprises the step of determining the 2HG level in the subject. This may be achieved by spectroscopic analysis, e.g., magnetic resonance-based analysis, e.g., MRI and/or MRSmeasurement, sample analysis of bodily fluid, such as serum or spinal cord fluid analysis, or by analysis of surgical material, e.g., by mass-spectroscopy.
Combination therapies
In some embodiments, the methods described herein comprise the additional step of coadministering to a subject in need thereof a second therapy e.g., an additional cancer therapeutic agent or an additional cancer treatment. Exemplary additional cancer therapeutic agents include for example, chemotherapy, targeted therapy, antibody therapies, immunotherapy,and hormonal
therapy. Additional cancer treatments include, for example: surgery, and radiation therapy. Examples of each of these treatments are provided below.
The term "co-administering" as used herein with respect to an additional cancer therapeutic agents means that the additional cancer therapeutic agent may be administered together with a compound of this invention as part of a single dosage form (such as a
composition of this invention comprising a compound of the invention and an second therapeutic agent as described above) or as separate, multiple dosage forms. Alternatively, the additional cancer therapeutic agent may be administered prior to, consecutively with, or following the administration of a compound of this invention. In such combination therapy treatment, both the compounds of this invention and the second therapeutic agent(s) are administered by
conventional methods. The administration of a composition of this invention, comprising both a compound of the invention and a second therapeutic agent, to a subject does not preclude the separate administration of that same therapeutic agent, any other second therapeutic agent or any compound of this invention to said subject at another time during a course of treatment.The term "co-administering" as used herein with respect to an additional cancer treatment means that the additional cancer treatment may occurprior to, consecutively with, concurrently with or following the administration of a compound of this invention.
In some embodiments, the additional cancer therapeutic agent is a chemotherapy agent. Examples of chemotherapeutic agents used in cancer therapy include, for example,
antimetabolites (e.g., folic acid, purine, and pyrimidine derivatives), alkylating agents (e.g., nitrogen mustards, nitrosoureas, platinum, alkyl sulfonates, hydrazines, triazenes, aziridines, spindle poison, cytotoxic agents, topoisomerase inhibitors and others)and hypomethylating agents (e.g., decitabine (5-aza-deoxycytidine), zebularine, isothiocyanates, azacitidine (5- azacytidine, 5-flouro-2'-deoxycytidine, 5,6-dihydro-5-azacytidine and others). Exemplary agents include Aclarubicin, Actinomycin, Alitretinoin, Altretamine, Aminopterin, Aminolevulinic acid, Amrubicin, Amsacrine, Anagrelide, Arsenic trioxide, Asparaginase, Atrasentan, Belotecan, Bexarotene, bendamustine, Bleomycin, Bortezomib, Busulfan, Camptothecin, Capecitabine, Carboplatin, Carboquone, Carmofur, Carmustine, Celecoxib, Chlorambucil, Chlormethine, Cisplatin, Cladribine, Clofarabine, Crisantaspase, Cyclophosphamide, Cytarabine, Dacarbazine, Dactinomycin, Daunorubicin, Decitabine, Demecolcine, Docetaxel, Doxorubicin, Efaproxiral,
Elesclomol, Elsamitrucin, Enocitabine, Epirubicin, Estramustine, Etoglucid, Etoposide,
Floxuridine, Fludarabine, Fluorouracil (5FU), Fotemustine, Gemcitabine, Gliadel implants, Hydroxycarbamide, Hydroxyurea, Idarubicin, Ifosfamide, Irinotecan, Irofulven, Ixabepilone, Larotaxel, Leucovorin, Liposomal doxorubicin, Liposomal daunorubicin, Lonidamine,
Lomustine, Lucanthone, Mannosulfan, Masoprocol, Melphalan, Mercaptopurine, Mesna, Methotrexate, Methyl aminolevulinate, Mitobronitol, Mitoguazone, Mitotane, Mitomycin, Mitoxantrone, Nedaplatin, Nimustine, Oblimersen, Omacetaxine, Ortataxel, Oxaliplatin,
Paclitaxel, Pegaspargase, Pemetrexed, Pentostatin, Pirarubicin, Pixantrone, Plicamycin, Porfimer sodium, Prednimustine, Procarbazine, Raltitrexed, Ranimustine, Rubitecan, Sapacitabine, Semustine, Sitimagene ceradenovec, Strataplatin, Streptozocin, Talaporfin, Tegafur-uracil, Temoporfin, Temozolomide, Teniposide, Tesetaxel, Testolactone, Tetranitrate, Thiotepa, Tiazofurine, Tioguanine, Tipifarnib, Topotecan, Trabectedin, Triaziquone, Triethylenemelamine, Triplatin, Tretinoin, Treosulfan, Trofosfamide, Uramustine, Valrubicin, Verteporfin, Vinblastine, Vincristine, Vindesine, Vinflunine, Vinorelbine, Vorinostat, Zorubicin, and other cytostatic or cytotoxic agents described herein.
Because some drugs work better together than alone, two or more drugs are often given at the same time. Often, two or more chemotherapy agents are used as combination chemotherapy.
In some embodiments, the additional cancer therapeutic agent is a differentiation agent. Such differentiation agent includes retinoids (such as all-trans-retinoic acid (ATRA), 9-cis retinoic acid, 13-cis-retinoic acid (13-cRA) and 4 -hydroxy -phenretinamide (4-HPR)); arsenic trioxide; histone deacetylase inhibitors HDACs (such as azacytidine(Vidaza) and butyrates (e.g., sodium phenylbutyrate)); hybrid polar compounds (such as hexamethylene bisacetamide
((HMBA)); vitamin D; and cytokines (such as colony-stimulating factors including G-CSF and GM-CSF, and interferons).
In some embodiments the additional cancer therapeutic agent is a targeted therapy agent. Targeted therapy constitutes the use of agents specific for the deregulated proteins of cancer cells. Small molecule targeted therapy drugs are generally inhibitors of enzymatic domains on mutated, overexpressed, or otherwise critical proteins within the cancer cell. Prominent examples are the tyrosine kinase inhibitors such as Axitinib, Bosutinib, Cediranib, dasatinib, erlotinib, imatinib, gefitinib, lapatinib, Lestaurtinib, Nilotinib, Semaxanib, Sorafenib, Sunitinib, and Vandetanib,
and also cyclin-dependent kinase inhibitors such as Alvocidib and Seliciclib. Monoclonal antibody therapy is another strategy in which the therapeutic agent is an antibody which specifically binds to a protein on the surface of the cancer cells. Examples include the anti- HER2/neu antibody trastuzumab (HERCEPTIN®) typically used in breast cancer, and the anti- CD20 antibody rituximab and Tositumomab typically used in a variety of B-cell malignancies. Other exemplary antibodies include Cetuximab, Panitumumab, Trastuzumab, Alemtuzumab, Bevacizumab, Edrecolomab, and Gemtuzumab. Exemplary fusion proteins include Aflibercept and Denileukin diftitox. In some embodiments, the targeted therapy can be used in combination with a compound described herein, e.g., a biguanide such as metformin or phenformin, preferably phenformin.
Targeted therapy can also involve small peptides as "homing devices" which can bind to cell surface receptors or affected extracellular matrix surrounding the tumor. Radionuclides which are attached to these peptides {e.g., RGDs) eventually kill the cancer cell if the nuclide decays in the vicinity of the cell. An example of such therapy includes BEXXAR®.
In some embodiments, the additional cancer therapeutic agent is an immunotherapy agent. Cancer immunotherapy refers to a diverse set of therapeutic strategies designed to induce the subject's own immune system to fight the tumor. Contemporary methods for generating an immune response against tumors include intravesicular BCG immunotherapy for superficial bladder cancer, and use of interferons and other cytokines to induce an immune response in renal cell carcinoma and melanoma subjects.
Allogeneic hematopoietic stem cell transplantation can be considered a form of immunotherapy, since the donor's immune cells will often attack the tumor in a graft-versus- tumor effect. In some embodiments, the immunotherapy agents can be used in combination with a compound or composition described herein.
In some embodiments, the additional cancer therapeutic agent is a hormonal therapy agent. The growth of some cancers can be inhibited by providing or blocking certain hormones. Common examples of hormone-sensitive tumors include certain types of breast and prostate cancers. Removing or blocking estrogen or testosterone is often an important additional treatment. In certain cancers, administration of hormone agonists, such as progestogens may be
therapeutically beneficial. In some embodiments, the hormonal therapy agents can be used in combination with a compound or a composition described herein.
Other possible additional therapeutic modalities include imatinib, gene therapy, peptide and dendritic cell vaccines, synthetic chlorotoxins, and radiolabeled drugs and antibodies.
EXAMPLES
The chemical name of each compound described below is generated by ChemBioOffice software.
DCM = dichloromethane TEA = triethylamine
DPPA = diphenylphosphoryl azide TFA :::: trifluoroacetic acid
DIPEA = A^V-Diisopropyieih iamine TFAA = trifluoroacetic anhydride
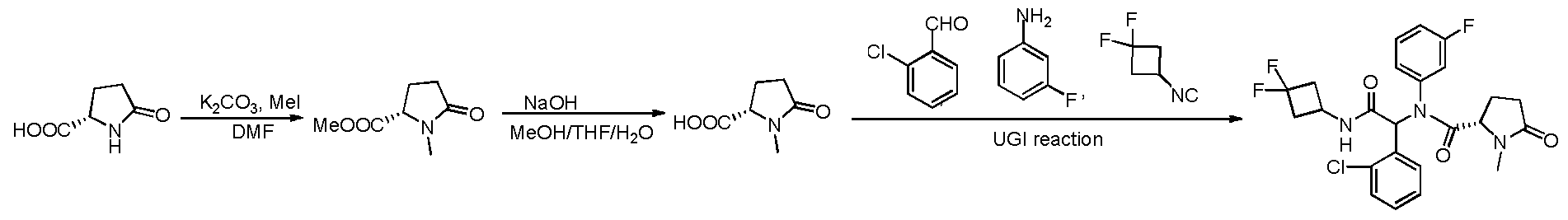
General procedures for the preparation of l,l-difluoro-3-isocyanocyclobutane
Method A:
Step A: Tert-butyl 3-oxocyclobutylcarbamate.To a solution of 3-oxocyclobutanecarboxylic acid (10 g, 88 mmol) in dry DCM (60 mL) at0°C, SOCl2 (20 mL) was added dropwise. The mixture was heated to reflux for 1.5 hand then evaporated in vacuo. The resulting mixture was co- evaporated twice with toluene (2x8 mL) and the residue was dissolved in acetone (30 mL), followed by adding dropwise to a solution of NaN3 (12g, 185.0mmol) in H20 (35 mL) at 0 °C. After addition, the mixture was stirred for another lh and then quenched with ice (110 g). The resulting mixture was extracted with Et20 (2 xl 00 mL). Combined organic layers were washed with brine, dried over anhydrous Mg2S04and concentrated to about 15mL solution. Toluene (2 x 30 mL) was added into the residue and the mixture was co-evaporated twice to remove
Et20(about 30 mL solution left each time to avoid explosion). The resulting toluene solution was heated to 90 °C until the evolution of N2 ceased. Next, 40mL of t-BuOH was added into the
reaction mixture and the resulting mixture was stirred overnight at 90 °C. The mixture was cooled and concentrated. The residue was purified by column chromatography using petroleum ether / EtO Ac (V:V, 7:1 to 5:1) as eluent to afford the desired product asa white solid. MS: 186.1 (M+l)+.
Step B:Tert-butyl 3,3-difluorocyclobutylcarbamate.To a solution of fert-butyl-3-oxocyclo - butylcarbamate (2.56g, 111.07mmol) in dry DCM (190mL), DAST (diethylaminosulfur trifluoride) (41.0 mL, 222.14mmol) was added dropwise at 0 °C under the atmosphere of N2. The mixture was then allowed to warm up to r.t and stirred overnight. The resulting mixture was slowly added into a pre-cooled saturated aq. NaHC03 solution and extracted with DCM (3 x 200 mL). Combined organic layers were washed with brine, dried over anhydrous MgS04, and concentrated in vacuo. The residue was purified by column chromatography using petroleum ether / EtO Ac (V:V, 15:1) as eluent to afford the desired product. Ή NMR (400 MHz, DMSO-d6): 54.79 (s, 1H), 4.07 (s, 1H), 2.98 (s, 2H), 2.58 - 2.29 (m, 2H), 1.46 (s, 9H). MS: 208.1 (M+l)+. Step C:N-(3,3-difluorocyclobutyl)formamide. To asolution of MeOH (170mL) and
CH3C0C1(65 mL), fert-butyl 3,3-difluoro -cyclobutylcarbamate (12. lg, 58.42mmol) was added in one portion dropwise at 0 °C. The reaction mixture was stirred at 0°Cfor 20 min, and then allowed to warm up to r.t and stirred for another 1.5 h. The reaction mixture was concentrated and dissolved in H20 (200mL). The resulting mixture was extracted by Et20 (150 mL) and the aqueous layer was adjusted to pH=l 1 with solid Na2C03and extracted by DCM (2 x 150 mL). The combined organic layers were dried over anhydrous MgS04, filtered and concentrated in vacuousing a cold-water bath (<20°C). The residue was dissolved in HCOOEt (90 mL), and transferred into a sealed pressure tube. This reaction mixture was heated to 80 °C and stirred overnight. The solvent was removed, and the residue was purified by column chromatography using petroleum ether / EtO Ac (V: V, 1 :1 to 1 :3) as eluent to afford the desired product.MS :
136.1 (M+l)+.
Step D:l,l-Difluoro-3-isocyanocyclobutane.To a solution ofN-(3,3-difluorocyclobutyl) - formamide (2.0g, 14.81mmol) and PPh3(4.27g, 16.29mmol) in DCM (35 mL) were added CC14 (1.43 mL, 14.81mmol) and TEA (2.06 mL, 14.81mmol). The reaction mixture was stirred at 45 °C overnight under a N2 atmosphere. The resulting mixture was evaporated in vacuo at 0 °C. The residue was suspended in Et20 (25 mL) at 0 °C for 30 min and then filtered. The filtrate was
evaporated to about 5mL at 0 °C under reduced pressure. The residue was purified by column chromatography using Et20 as eluent to afford the desired product which was used directly in the next step.
Method B:
Step A:Benzyl 3-oxocyclobutanecarboxylate. A mixture of 3-oxocyclobutanecarboxylic acid (5 g, 44 mmol), potassium carbonate (12 g, 88 mmol) and benzyl bromide (11.2 g, 66 mmol) in acetone (50 mL) was refluxed for 16 h. The solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. Combined organic layers were driedover anhydrous MgS04, filtered and concentrated. The residue was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane/ EtOAc to
givethedesired compound. ¾ NMR (400 MHz, CDC13): δ 7.45 - 7.27 (m, 5H), 5.19 (s, 2H), 3.55 - 3.36 (m, 2H), 3.33 - 3.11 (m, 3H).
Step B:Benzyl 3,3-difluorocyclobutanecarboxylate.To a solution of benzyl 3- oxocyclobutanecarboxylate (1.23g, 6.03 mmol) in DCM (35 mL) was added DAST (0.8 mL, 6.03 mmol) dropwise under nitrogen. The mixture was stirred at room temperature for 16 hand then diluted with DCM. After successive washes with saturated sodium bicarbonate, IN aq. hydrochloride acid, and brine, the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by silica gel chromatography with 93% hexane/ EtOAc as eluent to give the desired compound as an oil. Ή NMR (400 MHz, CDC13): δ 7.47 - 7.27 (m, 5H), 5.16 (s, 2H), 3.09 - 2.95 (m, 1H), 2.90 - 2.60 (m, 4H).
Step C:3,3-Difluorocyclobutanecarboxylic acid. Benzyl 3,3-difluorocyclobutanecarboxylate (0.84 g, 3.72 mol) was dissolved in ethanol (40 mL), and approximately 0.02 g palladium on activated carbon was added. The mixture was stirred at room temperature for 12 h under the
atmosphere of H2and then filtered through a pad of Celite. The filtrates were concentrated and dried in vacuo to give the desired compound. XH NMR (400 MHz, CDC13): 53.16 - 2.55 (m, 5H). Step D: Tert-butyl 3, 3-difluorocyclobutylcarbamate. Benzyl 3,3-difluorocyclobutanecarboxylic acid (3.7 g, 27.3 mmol), DPPA (7.87 g, 27 mmol) and TEA (2.87 g, 28.4 mmol) were dissolved in t-BuOH (25 mL). The mixture was refluxed for 5h and then diluted with ethyl acetate (about 200 mL). The organic phase was washed twice with 5% citric acid and saturated sodium hydrogen carbonate respectively, dried over anhydrous Mg2S04and evaporated under reduced pressure. The residue was purified by silica gel chromatography with 50% hexane/ EtOAc to give the desired product. MS: 208.1 (M+l)+.
Step E: 3,3-Difluorocyclobutanamine hydrochloride. To a cold solution of MeOH (170 mL) and CH3COCI (65 mL) was added tert-butyl 3, 3-difluorocyclobutylcarbamate (12.1 g, 58.4 mmol) dropwise at 0°C. After completion of the addition, the mixture was stirred at 0°C for 20 min and then allowed to warm up to room temperature. The reaction mixture was stirred for another 1.5 h and then concentrated to give the crude product which was precipitated in ether to give the desired product as a whitesolid.MS: 108.1 (M+l)+.
Step F:N-(3,3-difluorocyclobutyl)formamide. The mixture of 3,3-difluorocyclobutanamine hydrochloride^.5 g, 60.7 mmol) and TEA (3 eq) in HCOOEt (90 mL) was stirred at 80°C overnightin a sealed pressure tube. The solvent was removed invacuo and the residue was purified by column chromatography with 50% petroleum ether /EtOAc to 25% petroleum ether / EtOAc to give the desired product. 1H NMR (400 MHz, DMSO-d6): δ 8.54 (s, 1H), 8.01 - 7.89 (m, 1H), 4.16 - 3.84 (m, 1H), 3.06 - 2.73 (m, 2H), 2.72 - 2.33 (m, 2H).MS: 136.1 (M+l)+. Step G:l,l-Difluoro-3-isocyanocyclobutane. The compound was synthesized as outlined in step D of method A set forth above.
General procedures for the preparation of l-fluoro-3-isocyanocyclobutane
NC
Step A:Tert-butyl 3-hydroxycyclobutylcarbamate. To a solution oftert-butyl 3- oxocyclobutylcarbamate (2 g, 10.8 mmol, 2 eq) in EtOH (20 mL) was added NaBH4 (204 mg, 1 eq) at 0 °C. The mixture was then allowed to warm to room temperature and stirred for 30 min.
The mixture was concentrated in vacuo and the residue was purified by column chromatography using petroleum ether / EtOAc (V:V, 2:1 to pure EtOAc) as eluent to afford the desired product as a white solid. MS: 188.1 (M+l)+.
Step B:Tert-butyl 3-fluorocyclobutylcarbamate. To a solutiontert-butyl 3-hydroxycyclobutyl - carbamate (lg, 5.35 mmol) in dry DCM (20 mL) at -70°C was added DAST dropwise (lg, 0.85 mL, 1.17eq) under the atmosphere of N2. The mixture was then slowly warmed to room temperature and stirred overnight. The resulting mixture was washed with diluted aq. NaHCC . The organic layer was dried over anhydrous Mg2S04and concentrated. The residue was purified by flash chromatography using petroleum ether / EtOAc (V:V, 20:1 to 2:1) as eluent to afford a white solid as the desired product. MS: 190.1 (M+l)+.
Step C:3-Fluorocyclobutanamine. The compound was synthesized as outlined in step E of method A set forth above.
Step D:N-(3-fluorocyclobutyl)formamide. The compound was synthesized as outlined in step F of method A set forth above. 1H NMR (400 MHz, CDC13):5 8.10 (s, 1H), 5.94-5.89 (brs, 1H), 5.32-5.25 (m, 0.5H), 5.18-5.11 (m, 0.5H), 4.63-4.42 (m, 1H), 2.76-2.62 (m, 2H), 2.44-2.31 (m,
2H).
Step E:l-Fluoro-3-isocyanocyclobutane. The compound was synthesized via the general procedure as the step G in method A set forth above.
General procedures for the preparation of l,l-difluoro-4-isocyanocyclohexane
Step A:Tert-butyl 4-hydroxycyclohexylcarbamate. To a solution of 4-aminocyclohexanol (23 g, 0.2 mol) and Et3N (60 g, 0.6 mol) in THF (230 mL) was added (Boc)20 (87 g, 0.4 mol). The resulting solution was stirred at room temperature overnight. The solvent was removed under reduced pressure and the residue was extracted with EtOAc (3 x 200 mL). The combined organic layers were washed with water (2 x 200 mL) and brine (200 mL), dried over anhydrous Na2S04
and concentrated. The residue was purified by column chromatography on silica gel using DCM/ MeOH (V:V, 20:1) to afford the desired product as a white solid. MS: 216.2 (M+l)+.
Step B: Tert-butyl 4-oxocyclohexylcarbamate To a solution of tert-butyl 4-hydroxycyclohexyl- carbamate (10.0 g, 46.5 mmol) in DCM (100 mL) was added Dess-Martin periodinane (39.4 g, 92.9 mmol) portionwise. The resulting solution was stirred at room temperature overnight, quenched with aq. Na2S203solution and extracted with DCM (3 x 100 mL). The combined organic layers were washed with water (2 x 100 mL) and brine (100 mL), dried over anhydrous Na2S04, and concentrated. The residue was purified by column chromatography on silica gel using petroleum ether / EtOAc (V:V, 10:1) to afford desired product as a white solid.
Step C: Tert-butyl 4, 4-difluorocyclohexylcarbamate.To a solution of tert-butyl 4-oxocyclohexylcarbamate (2.13g, 10 mmol) in dry DCM (25 mL) was added DAST (2.58g, 16 mmol) dropwise at -5 °C under nitrogen. After addition, the reaction mixture was stirred at r.t overnight. The reaction mixture was poured into ice water slowly and extracted with DCM ( 3 x
100mL).The combined organic layerswere washed with 2N aq.NaHCC and brine, dried overanhydrous Na2S04, filtered and concentrated in vacuo. The residue was purified by column chromatography usingpetroleum ether / EtOAc (V:V, 5:l)as eluent to afford a mixture of the title compound(~70%) and the byproduct tert-butyl 4-fluorocyclohex-3-enylcarbamate (-30%) as a light-yellow solid.
To the above mixtures (2.52g, 10.7 mmol) in DCM (25 mL) was added m-CPBA (2.20g, 12.9 mmol) portionwise at 0°C while keeping the internal temperature below 5 °C. After addition, the reaction mixture was stirred at room temperature ovemight.To the reaction mixture was added saturated aq.Na2S203 (8.0 mL) at 0°C.The resulting mixture was stirred at 0°C for 40min, and then extracted by DCM (3 x 5.0mL).The combined organic layerswere washed with brine, dried overanhydrous Na2SC>4, and evaporated in vacuo. The residue was used directly in the next step without further purification.
To the aboveresidue in MeOH (15 mL) was added NaBH4(0.202g, 5.35 mmol) at 0°C. The reaction mixture was stirred at room temperature overnight. Water (0.38g) was added dropwise to quench the reactionat 0°C. The resulting mixture was stirred at 0 °C for 30min, and concentrated in vacuo. The residue was purified by column chromatography using DCM as
eluent to afford the pure compound as a white solid.1H NMR (400 MHz, CDC13): δ 4.46 (s, 1H), 3.59 (s, 1H), 2.25 - 1.69 (m, 6H), 1.61 - 1.20 (m, 11H).MS: 236.2 (M+l)+.
Step D: 4,4-Difluorocyclohexanamine hydrochloride. A mixture of fert-butyl 4,4-difluorocyclo- hexylcarbamate (6.0 g, 25.5 mmol) and 6N HCl/MeOH (60 mL) was stirred at room temperature for 2 h. The reaction mixture was concentrated to give the crude product which was directly used in next step without further purification. XH NMR (400 MHz, CD3OD): δ 4.89 (s, 2H), 3.32-3.26 (m, 1H), 2.14-2.01 (m, 4H), 2.02-1.85 (m, 2H), 1.74-1.65 (m, 2H). MS: 136.1 (M+l)+.
Step E: N-(4,4-difluorocycIohexyI)form mide. A mixture of 4,4-difluorocyclohexanamine (crude 3.4 g, 25.2 mmol), TEA (3 eq) and ethyl formate (35 mL) was stirred at 110 °C overnight in a sealed tank. The solvent was removed and the residue was purified by column
chromatography using DCM / MeOH (V:V, 10:1) as eluent to afford the desired product. H NMR (400 MHz, CDC13): δ 8.14 (s, 1H), 5.98 (s, 1H), 3.93 (m, 1H), 2.54 - 2.19 (m, 1H), 2.15 - 1.39 (m, 7H).MS: 164.1 (M+l)+.
Step F: l,l-Difluoro-4-isocyanocyclohexane. A mixture of N-(4,4-difluorocyclohexyl) - formamide (2.5 g, 15.3 mmol), PPh3 (4.4 g, 16.8 mmol), CC14 (2.3 g, 15.1 mmol), Et3N (1.5 g, 14.9 mmol) and DCM (50 mL) was heatedto 45 °C and stirred overnight. The resulting mixture was evaporated in vacuo and the residue was suspended in Et20 (125 mL) at 0 °C. The filtrate was concentrated andthe residue was purified by column chromatography on silica gel eluting with Et20 to afford the desired product as a yellow oil which was used directly in the next step.
General procedures for the preparation of 2-(3-aminophenoxy)ethanol
Step A: 2-(3-Nitrophenoxy)ethanol.A suspension of 3-nitrophenol (1 g, 7.2 mmol), 2- bromoethanol (1.2 g, 9.6 mmol) and K2C03 (2 g, 14.4 mmol) in MeCN (12 mL) was stirred at 90°C overnight. The precipitate was collected by filtration to give the first batch of product. The filtrate was concentrated and the residue was purified by column chromatography to afford another batch of the desired product as a yellow solid.
Step B: 2-(3-Aminophenoxy)ethanol To a solution of 2-(3-nitrophenoxy)ethanol(500 mg, 2.7 mmol) and NH4C1(720 mg, 13.5 mmol) in EtOH (10 mL) was added iron powder (900 mg, 16.2 mmol) at room temperature. The reaction was then stirred at 90 °C for 2hr and
subsequentlycooled. The mixture was filtered and thefiltrate was concentrated. The resulting residue was purified by column chromatography to afford the desired product as a yellow solid.MS: 154.1 (M+l)+.
General procedures for the preparation of 3-(lH-pyrazol-4-yl)aniline
Step A: Tert-butyl 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole-l- carboxylate.To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (500 mg, 2.57 mmol) and (Boc)20 (672 mg, 3.08 mmol) in DMF (1.0 mL)was addedDMAP (63 mg, 0.52 mmol)in one portion. The mixture was stirred at room temperature overnight, and then partitioned between EtOAc and saturated aq. NH4C1. The organic layer was separated, washed with brine, dried over anhydrous Na2S04, and concentrated to afford the crude product.
Step B: 4-(3-Nitrophenyl)-lH-pyrazole.To a solution of tert-butyl 4-(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)-lH-pyrazole-l -carboxylate(300 mg, 0.82 mmol), l -bromo-3 -nitrobenzene (137 mg, 0.68 mmol) and Na2C03 (216 mg, 2.04 mmol) in DME/H20 (5mL/lmL) under N2, was added Pd(PPh3)2Cl2(24 mg,0.034 mmol). The mixture was stirred at 85°C overnight, and then quenched with H20. The resulting mixture was extracted with EtOAc (3 x 25 mL). The organic layer was separated, washed with brine, dried over anhydrous Na2S04, and concentrated. The resulting residue was purified by column chromatography to afford the desired product. MS: 190.2 (M+l)+.
Step C: 3-(lH-pyrazol-4-yl)aniline.Iron powder (296 mg, 5.30 mmol) was added to a solution of 4-(3-nitrophenyl)-lH-pyrazole(200 mg, 1.06mmol) in AcOH EtOH (2mL/3mL). The reaction mixture was stirred at 90 °C for 2hr and then cooled to room temperature. The reaction mixture was filtered through Celite. The filter cake was washed with H2O.The filtrate was neutralized
with 1 N NaOHto pH=8 and extracted with EtOAc (3 x 30 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated. The resulting residue was purified by column chromatography to afford the desired product. MS: 160.2 (M+l)+.
General procedures for the preparation of 2-(3-aminophenyl)propan-2-ol
Step A: Ethyl 3-(dibenzylamino)benzoate 2. To a solution of ethyl 3-aminobenzoate (2 g, 0.012 mmol) and Et3N (5.26 mL, 0.036 mmol) in CH3CN (30 mL), was added BnBr (4.32 mL, 0.036 mmol) in one portion. The reaction mixture was heated to reflux for 18 hrand then cooled to room temperature. The mixture was concentrated to dryness in vacuo and the residue was purified by column chromatography (PE:EtOAc = 10 : 1 as eluent) to afford the desired product as a white solid. MS: 346.1 (M+l)+.
Step B: 2-(3-(dibenzyl mino)phenyl)propan-2-ol. To a solution of ethyl 3- (dibenzylamino)benzoate (1.85 g, 5.58 mmol) in anhydrous THF (15 mL) at 0 °C under nitrogen atmosphere was added MeMgBr (3M sol. in THF, 5.58 mL, 16.7 mmol) dropwise over 30 min. The reaction was stirred at room temperature overnight and quenched by addition ofsaturated NH4C1. The resulting mixture was extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with NaHCC , water and brine, dried over anhydrous Na2S04, filtered and then concentrated to dryness. The residue was purified by column chromatography (PE:EtOAc = 2 : 1 as eluent) to afford the desired product as a colorless oil. MS: 332.1 (M+l)+. Step C: 2-(3-aminophenyl)propan-2-ol.To a solution of 2-(3-(dibenzylamino)phenyl)propan-2- ol (268 mg, 0.81 mmol) in MeOH (5 mL) was added 10% Pd/C (27 mg) in one portion. The reaction mixture was hydrogenated at room temperatureovernightunder hydrogen atmosphere. The catalyst was filtered off through Celite and the filtrate was concentrated to dryness. The residue was purified by column chromatography (PE:EtOAc = 1 : 2 as eluent) to afford the desired product as a yellow solid. MS: 152.1 (M+l)+.
General procedures for the preparation of 2-(3-amino-5-fluorophenyl)propan-2-ol
Step A.Methyl 3-fluoro-5-nitrobenzoate. Thionyl chloride (488 mg, 4.1 mmol) was added dropwise to a solution of 3-fluoro-5-nitrobenzoic acid (500 mg, 2.7 mmol) in dry methanol (10 mL) at 0 °C under nitrogen atmosphere. The reaction was warmed to room temperature and stirred for 6hr. The reaction mixture was concentrated under reduced pressure to obtain the corresponding methyl ester hydrochloride as a waxy solidwhich was used directly in the next step. MS: 200 (M+l)+.
Step B. Methyl 3-amino-5-fluorobenzoate. To a solution of methyl 3-fluoro-5-nitrobenzoate (400 mg, 2 mmol) in ethanol (10 mL) was added iron powder (560 mg, 10 mmol) and ammonium chloride (540 mg, 10 mmol) in one portion. The reaction mixture was stirred at 80 °Cfor 1 hr. After cooling the reaction, the mixture was filtered through Celite. The filtrate was concentrated under reduced pressureto give the desired product. MS: 170 (M+l)+.
Step C.Methyl 3-(dibenzylamino)-5-fluorobenzoate.To a solution of methyl 3-amino-5- fluorobenzoate (440mg, 2.6 mmol) in dry DMF (10 mL) was added NaH (187 mg, 7.8 mmol) portionwise, followed by addition of benzyl bromide (1.1 g, 6.5 mmol). The reaction mixture was stirred at 40 °C for 16hr and concentrated. The resulting residue was purified by column chromatography to give the desired product. MS: 350 (M+l)+.
Step D.2-(3- ibenzylamino)-5-fluorophenyl)propan-2-ol.Methy\magnesium bromide (1 M in THF, 2.4 mL, 2.4 mmol) was dissolved in THF (5 mL)and placed in an ice-water bath. Methyl 3- (dibenzylamino)-5-fluorobenzoate (280 mg, 0.8 mmol) in THF (5 mL) was then slowly added to the reaction mixture. This mixture was stirred for 3hr while maintaining an internal temperature range between 15 to 25 °C. Then the mixture was cooled to 0 °C and treated with ammonium chloride solution, then extracted with ethyl acetate (3 x 30 mL). The combined organic layers
were dried over anhydrous Na2S04, andconcentrated in vacuo. The residue was purified by column chromatography on silica gel to give the desired product. MS: 350 (M+l)+.
Step E.2-(3-Amino-5-fluorophenyl)propan-2-ol.To a solution of 2-(3-(dibenzylamino)-5- fluorophenyl)propan-2-ol (150 mg, 0.43 mmol) in ethanol (5 mL) was added 10 % Pd/C (15 mg) under a hydrogenatmosphere. The reaction mixture was stirred at room temperaturefor 16hr. The suspension was then filtered through Celite and the filtrate was concentrated invacuo. The residue was purified by column chromatography to givethe desired product. MS :170 (M+l)+.
General procedures for the preparation of ethyl l-(3-aminophenyl)cyclopropanol
Step A.Ethyl 3-(dibenzylamino)benzoate.To a solution of ethyl 3-aminobenzoate (2 g, 0.012 mmol) and Et3N (5.26 mL, 0.036 mmol) in CH3CN (30 mL) was added BnBr (4.32 mL, 0.036 mmol) in one portion. The reaction mixture was heated to reflux for 18 hand cooled down to room temperature. The mixturewas concentrated in vacuo and the resulting residue was purified by column chromatography to afford the desired product as a white solid. MS: 346.1 (M+l)+. Step B.l-(3-(Dibenzylamino)phenyl)cyclopropanolTo a solution of ethyl 3- (dibenzylamino)benzoate (1.85 g, 5.58 mmol) in anhydrous THF (20 mL) at room temperature under N2 was added titanium tetraisopropoxide (0.25 mL, 0.84 mmol) dropwise over 10 min. After one hour of stirring, EtMgBr (THF solution, 4.1 mL, 12.3 mmol) was added dropwise over 30 min. The reaction mixture was stirred at room temperature for 3 h. The resulting mixture was quenched byaddition of saturated aq. NH4C1, and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with NaHC03, water and brine, dried over anhydrous Na2S04, and concentrated in vacuo. The residue was purified by column chromatography (PE: EtOAc = 5 : 1 as eluent) to afford the desired product as a colorless oil. 1H NMR (400 MHz,
CDC13) : δ 7.33 - 7.28 (m, 5H), 7.25 - 7.18 (m, 5H), 7.11 (t, J= 8.0 Hz, 1H), 6.80 - 6.75 (m, 1H), 6.61 - 6.56 (m, 2H), 4.65 (s, 4H), 1.17 - 1.13 (m, 2H), 0.93 - 0.90 (m, 2H). MS: 330.1 (M+l)+.
Step C.Ethyl l-(3-aminophenyl)cyclopropanol. To a solution of 1 -(3- (dibenzylamino)phenyl)cyclopropanol (1.8 g, 5.45 mmol) in MeOH (10 mL) at room
temperature was added 10% Pd/C (200 mg) in one portion. The reaction mixture was stirred at room temperatureunder a hydrogen atmosphere overnight. The suspensino was filteredthrough Celite, and the filtrate was concentrated in vacuo. The residue was purified by column
chromatography (PE: EtOAc = 2 : 1 as eluent) to afford the desired product as a yellow solid. H NMR (400 MHz, CDC13) : δ 7.10 (t, J= 7.8 Hz, 1H), 6.69 (t, J= 2.0 Hz, 1H), 6.63 - 6.60 (m, 1H), 6.56 - 6.53 (m, 1H), 1.22 - 1.19 (m, 2H), 1.01 - 0.98 (m, 2H). MS: 150.1 (M+l)+.
General procedures for the preparation of 3-fluoro-5-(methylthio)aniline
Step A.(3-Fluoro-5-nitrophenyl)(methyl)sulfane.A solution of 3-fluoro-5-nitroaniline (200 mg, 1.28 mmol), 1 ,2-dimethyldisulfane (121 mg, 1.29 mmol) and CH3CN (3 mL) was stirred at 30 °C. Neatisoamyl nitrite (150 mg, 1.28 mmol) was slowly added via syringe over 5 min. The reaction mixture was slowly heated to reflux over 10 min and maintained at a gentle reflux until N2 evolution ceased (30-60 min). The reaction mixture was cooled and the solvent was removed in vacuo to afford a dark oil. The resulting oil was purified by column chromatography to give the desired product as a pale yellow solid.
Step B:3-Fluoro-5-(methylthio)aniline. To a solution of (3-fluoro-5- nitrophenyl)(methyl)sulfane (90 mg, 0.48 mmol) in MeOH (10 mL) was added 10%Pd/C (9 mg) in one portion. The resulting mixture was purged with H2three times and stirred at room temperature for 1 h. The suspension wasfiltered through Celite, and the filter cake was washed with MeOH (5 mL).The filtrate was concentrated in vacuo to afford the desired product which was used directly in next step.MS: 158.0(M+1)+.
General procedure for the preparation of(S)-2-oxo-l,3-oxazinane-4-carboxylic acid
To a mixture of (S)-2-amino-4-hydroxybutanoic acid (10 g, 84.0 mmol) and 250 mL of aq. NaOH (2 mol/L, 20.4 g, 510 mmol) at 0°C was added a solution of triphosgene in dioxane (25.3 g in 125 mL dioxane) drop wise over Ih.Theintemal temperaturewas kept below 5 °C during the addition. The mixture was then stirred at room temperature for 2 days. The reaction mixture was then concentrated in vacuo, followed by addition of 200 mL of CH3CN. The resulting mixture was then heated to 60 °C and stirred vigorously for 0.5 h. The hot mixture was filtered immediately. The filtrate was then concentrated to 100 mL and the desired product was precipitated out. The crude product was collected by filtration and used directly in the next step without further purification. MS: 146.0(M+1)+.
General procedure for the preparation of(S)-4-(tert-butoxycarbonyl)-6-oxopiperazine-2- carboxylic acid
Br •COOBn
DIPEA
Step A: (S)-3-Amino-2-(((benzyloxy)carbonyl)amino)propanoic acid.To a mixture of (S)-4- amino-2-(((benzyloxy)carbonyl)amino)-4-oxobutanoic acid (3 g, 11.3 mmol) in MeCN (20 mL), EtOAc (20 mL ) and H20 (10 mL), was added PIAD (4.38 g, 13.5 mmol) in one portion. The
reaction mixture was stirred at room temperature overnight. The resulting mixture was filtered, and the filtrate was concentrated in vacuo to afford the desired product. MS: 239.1 (M+l)+.
Step B:(S)-Methyl 3-amino-2-(((benzyloxy)carbonyl)amino)propanoate hydrochloride. To a stirred solution of MeOH(50 mL) was added SOCl2 (5 mL)dropwise at 0°C. The resulting mixture was stirred at 0°C for 0.5 h before (S)-3-amino-2-
(((benzyloxy)carbonyl)amino)propanoic acid (2.6 g, 10 mmol) was added. Then the reaction mixture was stirred at room temperature overnight and concentrated in vacuo to afford the desired product.MS: 253.1 (M+l)+.
Step C:(S)-Methyl 3-((2-ftenzyloxy)-2-oxoethyl)amino)-2-(((benzyloxy)carbonyl)amino)pro- paneate.To a solution of (S)-methyl 3-amino-2-(((benzyloxy)carbonyl)amino)propanoate hydrochloride (2.6 g, 0.01 mol) in THF (40 mL) was added DIPEA (4.0 g, 0.03 mol) at 0°C. The mixture was stirred at 0°C for 5 min, followed by addition of benzyl 2-bromoacetate(4.7 g, 0.02 mol). Then the mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture wasquenched by addition of H2Oand then extracted with EtOAc (3 x 40 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated. The resulting residue was purified by column chromatography toafford the desired product. MS: 401.2 (M+l)+.
Step D:(S)-Methyl 3-((2-(benzyloxy)-2-oxoethyl)(tert-butoxycarbonyl)amino)-2- (((benzyloxy)carbonyl)amino)propanoate. To a solution of (S)-methyl 3-((2-(benzyloxy)-2- oxoethyl)amino)-2-(((benzyloxy)carbonyl)amino)propanoate (3.0 g, 7.5 mmol) in THF (40 mL) was added DIPEA (2.9 g, 22.5 mmol) at 0°C. The mixture was stirred at 0°C for 5 minfollowed by addition of di-tert-butyl dicarbonate(3.27 g, 15 mmol). Then the mixture was allowed to warm to room temperature and stirred overnight. After quenching with a saturated. NaHC03solution, the resulting mixture was extracted with EtOAc (3 x 60 mL)and concentrated. The resulting residue was purified by column chromatography to afford the desired product.MS: 501.2 (M+l)+. Step E:(S)-2-((2-Amino-3-methoxy-3-oxopropyl)(tert-butoxycarbonyl)amino)acetic acid. To a solution of (S)-methyl 3 -((2-(benzyloxy)-2-oxoethyl)(tert-butoxycarbonyl)amino)-2-(( (benzyl - oxy)carbonyl)amino)propanoate (2.5 g, 5 mmol) in MeOH (30 mL) was added 10% Pd/C (250 mg). The mixture was stirred under hydrogen atmosphere at room temperature overnight. The
resulting suspension was filtered through Celite, and the filtrate was concentrated in vacuo to afford the desired product. MS: 277.1 (M+l)+.
Step E:(S)-l-tert-Butyl 3-methyl 5-oxopiperazine-l,3-dicarboxylate. To a solution of (S)-2-((2- amino-3-methoxy-3-oxopropyl)(tert-butoxycarbonyl)amino)acetic acid(1.2 g, 4 mmol) in DCM (100 mL) was added DCC (1.34 g, 6 mmol) at 5°C. The mixture was stirred at 10°C for 4h followed by addition ofEt3N (0.88g, 8 mmol). The resulting mixture was stirred at room temperature for 18h and then concentrated. The residue was added to EtOAc (20 mL) and the precipitate was filtered. The filtrate was concentrated and the residue was purified by column chromatography to afford the desired product.MS: 259.1 (M+l)+.
Step F:(S)-4-(tert-Butoxycarbonyl)-6-oxopiperazine-2-carboxylic acid. To a mixture of (S)-l - tert-butyl 3-methyl 5-oxopiperazine-l ,3-dicarboxylate (500 mg, 1.9 mmol) in MeOH (20 mL) and THF (20 mL) was added a solution of LiOH*H20 (159 mg, 3.8 mmol) in H20 (10 mL) at 0°C. The mixture was stirred at room temperature for 2h and then partitioned betweenEtOAc(25 mL) and H20. The aqueous layer was acidified with2N HC1 to pH 3-4 and then extracted with EtOAc(3 x 20 mL).The combined organic layers were washed with brine, dried over anhydrous Na2S04and concentrated to afford the desired productwhich was used directly in the next reaction. MS: 245.1 (M+l)+.
General procedure for the preparation of 2-bromopyrimidine-4-carbonitrile
Step A: 2-Hydroxy-4-carboxyaldehyde oxime. 2-Hydroxy-4-methyl pyrimidine hydrochloride (25.0 g 171 mmol) and sodium nitrate (17.7 mg, 260 mmol) were slowly added to 200 mL of 50% acetic acid at 0°C. The reaction mixture was stirred at room temperature for 3 h. The resulting suspension and the solids were filtered, washed with water and dried to afford the desired product. XH NMR (400 MHz, DMSO-d6): δ 12.42 (s, 1H), 1 1.89 (s, 1H), 7.92 (d, J = 6.4 Hz, 1H), 7.75 (s, 1H), 6.43 (d, J = 6.4 Hz, 1H). MS: 140.0 (M+l)+.
Step B: 2-Bromopyrimidine-4-carbonitrile. A mixture of 2-hydroxy-4-carboxyaldehyde oxime (9 g, 28.8 mmol), tetrabutyl ammonium bromide (10 g, 71.9 mmol) and phosphorus pentoxide (2 g, 14.4 mmol) in toluene (300 mL) was stirred at 120 °C for 2 h. The resulting mixture was filtered and the filtrate was concentrated. The resulting residuewas purified by column
chromatographyto give the desired compound as a yellow solid. ¾ NMR (400 MHz, CDC13): δ 8.82 (d, J= 4.8 Hz, 1H), 7.66 (d, J= 4.8 Hz, 1H). MS: 185.0 (M+l)+.
General synthetic procedures for making compounds of formula I: aryl)
YV R
2V O I.
n=1 ,2
X=CH2,0,NH,CH(OH)(S and R),F
General procedures for the UGI reaction:
A mixture of aldehyde (3.5 mmol) and aniline (3.5 mmol) in MeOH (8 mL) was stirred at room temperature for 30 min.Then the acid (3.5 mmol) was added and the reaction mixture was stirred for another 30 min, followed by addition of the isocyanide (3.5 mmol). The resulting mixture was then stirred at room temperature overnight and quenched with H20. The resulting mixture was partitioned betweenEtOAc and H20. The organic layer was washed with brine, dried over anhydrous Na2S04, and then concentrated. The resulting residue was purified by a standard method to afford the desired product.
General procedures for the Buchwald reaction:
A mixture of amine (0.30 mmol), aryl bromide (0.30 mmol), Cs2CC>3 (129 mg, 0.39 mmol), Pd2(dba)3 (18 mg, 0.02 mmol) and Xant-Phos (9.4 mg, 0.02 mmol) in 1,4-dioxane (10 mL) was stirred under N2at 80°C overnight. After filtration, the filtrate was concentrated in vacuo and the residue was purified by a standard method to give the desired products.
Example LPreparation of (S)-methyl l-methyl-5-oxopyrrolidine-2-carboxylate.
Compound 2 was prepared according to the following scheme, using the following protocol.
Step A: (S)-Methyl l-methyl-5-oxopyrrolidine-2-carboxylate. To a mixture of (5 -5-oxopyro- lidine-2-carboxylic acid (5.0 g, 38.8 mmol) in DMF (50 mL) were added anhydrous K2C03 (16 g, 116 mmol) and iodomethane (16.4 g, 116 mmol) at room temperature The resulting mixture was warmed to 40°C,stirred for 24h and concentrated in vacuo. The residue was precipitated with EtOAc (80 mL) and filtered.The filter cake was washed with EtOAc (2 x 10 mL). The combined filtrates were concentrated and the residue was purified by column chromatography on silica gel to give the desired product. 1H-NMR (400 MHz, CDC13): δ 4.18 - 4.1 1 (m, 1H), 3.70 (s, 3H), 2.87 (s, 3H), 2.56 - 2.29 (m, 3H), 2.16 - 2.04 (m, 1H). MS: 158.1 (M+l)+.
Step B: (S)-l-Methyl-5-oxopyrrolidine-2-carboxylic acid. To a solution of (^-methyl l-methyl- 5-oxopyrrolidine-2-carboxylate (0.6 g, 3.8 mmol) in MeOH (6 mL) were added THF (2 mL), H20 (2 mL) and NaOH (0.45 g, 11.4 mmol) at room temperature The resulting mixture was stirred at room temperature for 18 h and then acidified with 2 N HC1 to pH=3-4 at 0°C. The mixture was extracted with EtOAc (3 x 30 mL),the combined organic layers were dried over anhydrous Na2S04and concentrated to give the crude product as a yellow solid (0.8 g) which was used directly in the next step. MS: 142.1 (M-l)".
Step C: Compound 2. 2-Chlorobenzaldehyde (117 mg, 0.83 mmol), 3-fluoroaniline (92.5 mg, 0.83 mmol), crude (S)-l-methyl-5-oxopyrrolidine-2-carboxylic acid (200 mg, -60% purity, 0.83 mmol) and l,l-difluoro-3-isocyanocyclobutane (119 mg, 90% purity, 1.0 mmol) were used in the UGI reaction to give the desired product (diastereomeric mixture). H NMR (400 MHz, CDC13): δ 8.52 (d, J= 4.9 Hz, 0.2H), 8.16 (m, 0.3H), 7.87 - 7.47 (m, 2H), 7.42 - 7.31 (m, 1H), 7.25 - 7.11 (m, 2H), 7.08 - 6.89 (m, 3.3H), 6.74 (d, J = 6.0 Hz, 0.7H), 6.57 (m, 2H), 4.42 - 4.26 (m, 1.3H), 4.20 - 4.08 (m, 0.5H), 4.00 (m, 1H), 3.00 (m, 2H), 2.74 (m, 3H), 2.63 - 1.82 (m, 6H). MS: 494.1 (M+l)+.
Example 2.Preparation of (S)-N-(l-(2-chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2- oxoethyl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide.
Compounds3 and 4were prepared according to the following scheme, using the following protoco
Compound 4
Step A. (S)-N-(l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)andno)-2-oxoethyl)-N-(3 - phenyl)-5-oxopyrrolidine-2-carboxamide.3- \uoroam\me (86mg, 0.78mmol), 2- chlorobenzaldehyde (109mg, 0.78mmol), (S)-5-oxopyrrolidine-2-carboxylic acid (lOOmg, 0.78mmol)and l,l-difluoro-4-isocyanocyclohexane (135mg, 0.91mmol) were used in the UGI reaction to give the desired product. MS: 508.1 (M+l)+.
Step B. (S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocychhexyl)amino)-2-oxoethyl)-N-(3 - fluorophenyl)-5-oxo-l-^yrirmdin-2-yl)pyrrolidine-2-carboxarmde and (S)-N-((R)-l-(2 - chfarophenyl)-2-((4,4-difluorocycfahexyl)amino)-2-oxoethyl)-N-(3-fluow
(pyrimidin-2-yl)pyrrolidine-2-carboxamide.A mixture of (S)-N-(l -(2-chlorophenyl) -2-((4,4- difluorocyclohexyl)amino)-2-oxoethyl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide
(100 mg, 0.20 mmol), 2-bromopyrimidine (47 mg, 0.30 mmol), Cs2C03 (129 mg, 0.39 mmol),
Pd2(dba)3 (18 mg, 0.02 mmol) and Xant-Phos (9.4 mg, 0.02 mmol) in 1,4-dioxane (10 mL) was stirred under N2 at 80 °C overnight. After filtration, the filtrate was concentrated in vacuo and the residue was purified by a standard method to give the desired products.
(S)-N-((S)-l-(2-chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-^ - phenyl)-5-oxo-l^yrimidin-2-yl)pyrrolidine-2-carboxamide. Compound4 Ή NMR (400 MHz,
CDC13): δ 8.71 (d, J = 4.8 Hz, 2H), 7.75 (m, 1H), 7.33 (m, 2H), 7.18 (m, 1H), 7.09 - 6.87 (m,
5H), 6.47 (s, 1H), 5.61 (d, J = 7.6 Hz, 1H), 4.86 (d, J= 6.6 Hz, 1H), 3.98 (m, 1H), 3.01 - 2.84 (m,
2H), 2.58 (m, 1H), 2.30 - 2.20 (m, 1H), 1.93 (m, 7H), 1.47 (m, 2H); MS: 586.2 (M+l)+.
(S)-N-((R)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-o
fluorophenyl)-5-oxo-l- yrimidin-2-yl)pyrrolidine-2-carboxamide. Compound3. I I NMR (400 MHz, CDCI3): δ 8.75 (dd, J= 4.8, 2.0 Hz, 2H), 7.40 (d, J = 7.8 Hz, 1H), 7.23 (s, 3H), 7.08 (dt, J = 11.3, 6.3 Hz, 3H), 6.99 (d, J= 3.7 Hz, 1H), 6.27 (s, lH), 6.13 - 5.92 (m, 1H), 5.02 (m, 1H), 4.76 (m, 1H), 3.92 (m, 1H), 2.88 (m, 1H), 2.67 - 2.46 (m, 1H), 2.44 - 2.19 (m, 2H), 2.00 (m, 8H). MS: 586.1 (M+l)+.
The following analogs were synthesized viatheprocedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substituted-aromatic ring or heteroaromatic ring using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
Compound6
Ή NMR (400 MHz, CDC13): δ 8.75 (d, J= 4.8 Hz, 2H), 7.35 (m, 3H), 7.25 - 6.81 (m, 5H), 6.28 (s, 1H), 5.84 (d, J = 7.5 Hz, 1H), 4.76 (m, 1H), 3.98 - 3.59 (m, 1H), 2.92 (m, 1H), 2.58 (m, 1H), 2.35 - 2.20 (m, 1H), 2.07 (m, 1H), 1.83 (m, 2H), 1.57 (m, 4H), 1.46 - 1.17 (m, 4H). MS: 550.2 (M+l)+.
Compound 7
Ή NMR (400 MHz, CDC13): δ 8.73 (m, 2H), 7.80 (s, 1H), 7.35 (s, 1H), 7.23 - 6.72 (m, 6H), 6.47 (s, 1H), 5.49 (d, J = 7.7 Hz, 1H), 4.87 (d, J = 6.6 Hz, 1H), 4.74 - 4.42 (m, 1H), 3.86 (d, J = 8.0 Hz, 1H), 3.19 - 2.77 (m, 1H), 2.56 (m, 1H), 2.44 - 2.21 (m, 1H), 2.13 - 1.73 (m, 4H), 1.60 (s, 2H), 1.26 (m, 4H). MS: 550.2 (M+l)+.
Compound 49
Ή NMR (400 MHz, CDC13): δ 8.69 (s, 2H), 7.76 (s, 1H), 7.49 - 6.68 (m, 7H), 6.44 (s, 1H), 6.19 (s, 1H), 4.93 (m, 3H), 2.23 (m, 8H). MS: 540.1 (M+l)+.
Compound 51
Ή NMR (400 MHz, CDC13): δ 8.81 (d, J= 4.9 Hz, 1H), 8.66 (d,J= 2.7 Hz, 1H), 8.04 - 7.79 (m, 1H), 7.49-7.31 (m, 1H), 7.13-6.92 (m, 6H), 6.60 (m, 1H), 6.25-5.95 (m, 1H), 5.68 (m, 1H), 4.73 (dd,J=16.0, 6.9 Hz, 1H), 4.39 (m, 1H), 2.98 (m, 3H), 2.53 (m, 4H), 2.14 - 1.93 (m, 1H).MS: 592.1 (M+l)+.
Compounds
Ή NMR (400 MHz, CDC13): δ 8.46 - 8.32 (m, 1.7H), 7.78-7.61 (m, 1.5H), 7.39 (m, 1.5H), 7.23 (m, 1.6H), 7.13-6.88 (m, 4H), 6.40 (m, 1H), 6.11 (m, 1H), 5.01-4.77 (m, 1H), 4.26 (m, 1H), 3.51 (d, J = 5.5 Hz, 0.3H), 3.13-2.75 (m, 3H), 2.61 -2.22 (m, 3H), 2.17-1.90 (m, 1H). MS: 557.1 (M+l)+.
Compound 10
1H NMR (400 MHz, CDC13): δ 8.56 (m, 2H), 8.16 (s, 1.3H), 7.74 (s, IH), 7.36 (s, 2.6H), 7.19 (s, IH), 7.12 - 6.82 (m, 3H), 6.52 (m, 2H), 6.19 (m, IH), 4.65 - 4.48 (m, IH), 4.26 (m, 1.3H), 3.90 - 3.82 (m, 0.3H), 2.87 (m, 3H), 2.64 - 1.98 (m, 6H). MS: 557.1 (M+l)+.
Compound 41
1H NMR (400 MHz, CDC13): δ 7.98 (m, IH), 7.65 (m, 2H), 7.44 - 7.30 (m, 2H), 7.03 (m, 6H), 6.51 (m, IH), 6.36 (s, IH), 5.12 (d, J = 6.3 Hz, IH), 4.33 (s, IH), 3.97 (s, 3H), 3.10 - 2.63 (m, 3H), 2.60 - 2.00 (m, 5H). MS: 587.1 (M+l)+.
Compound 26
Ή NMR (400 MHz, CDC13): δ 8.32 (m, IH), 8.05 (t, J= 8.6 Hz, IH), 7.69 (s, IH), 7.45 - 7.30 (m, IH), 7.25 - 6.78 (m, 6H), 6.38 (m, 2H), 4.88 (m, IH), 4.33 (s, IH), 3.89 (s, 3H), 3.1 1 - 2.72 (m, 3H), 2.66 - 2.29 (m, 3H), 2.23 - 1.86 (m, 2H). MS: 587.1 (M+l)+.
Compound 17
Ή NMR (400 MHz, CDC13): δ 7.93 (m, IH), 7.56 (m, 2H), 7.21 (m, 3H), 7.10 - 6.87 (m, 3H), 6.42 (m, 3H), 5.04 (m, IH), 4.25 (m, IH), 3.97 (d, J = 6.1 Hz, 3H), 3.10 - 2.69 (m, 3H), 2.60 - 2.15 (m, 4H), 2.12 - 1.87 (m, 1H).MS: 587.2 (M+l)+.
Compound 28
Ή NMR (400 MHz, CDC13): δ 8.19 (m, IH), 7.79 - 7.33 (m, 3H), 7.28 - 7.06 (m, 4H), 7.06 - 6.83 (m, 4H), 6.47 - 6.32 (m, 2H), 5.09 - 4.91 (m, IH), 4.25 (m, IH), 3.09 - 2.60 (m, 4H), 2.57 (s, 3H), 2.53 - 1.99 (m, 5H). MS: 571.0 (M+l)+.
Compound 21
1H NMR (400 MHz, CDC13): δ 8.26 (d, J= 8.5 Hz, IH), 8.15 (s, IH), 7.64 (s, IH), 7.48 (m, IH), 7.32 (d, J = 7.5 Hz, IH), 7.14 (m, 2H), 7.04 - 6.83 (m, 3H), 6.40 (s, IH), 6.04 (s, IH), 4.89 (m, IH), 4.31 (s, IH), 2.89 (m, 3H), 2.48 (m, 2H), 2.40 - 2.27 (m, 3H), 2.26 - 1.84 (m, 3H). MS: 571.2 (M+l)+.
Compound 27
Ή NMR (400 MHz, CDC13): δ 8.30 - 8.15 (m, 2H), 7.68 (s, 1H), 7.38 (m, 1H), 7.24 - 6.85 (m, 6H), 6.46 - 6.16 (m, 2H), 4.94 (d, J = 6.0 Hz, 1H), 4.32 (s, 1H), 3.10 - 2.74 (m, 3H), 2.60 - 2.43 (m, 2H), 2.36 (m, 4H), 2.23 - 1.91 (m, 2H). MS: 571.2 (M+l)+.
Compound 15
1H NMR (400 MHz, CDC13): δ 8.17 (d, J = 8.3 Hz, 1H), 7.56 (m, 2H), 7.25 - 6.96 (m, 5H), 6.89 (m, 2H), 6.42 (s, 1H), 6.21 (s, 1H), 5.12 - 4.96 (m, 1H), 4.31 (m, 1H), 3.14 - 2.74 (m, 3H), 2.55 (s, 3H), 2.51 - 2.28 (m, 3H), 2.20 (m, 1H), 2.05 - 1.87 (m, 1H). MS: 571.2 (M+l)+.
Compound 25
1H NMR (400 MHz, CDC13): δ 8.72 (m, 1H), 7.88 (m, 1H), 7.65 (s, 1H), 7.57 - 7.30 (m, 2H), 7.23 - 7.09 (m, 2H), 7.02 (s, 2H), 6.96 - 6.83 (m, 1H), 6.44 (s, 1H), 6.05 (d, J= 6.5 Hz, 1H), 5.31 - 4.93 (m, 1H), 4.33 (s, 1H), 3.02 (m, 2H), 2.86 (m, 1H), 2.63 - 2.45 (m, 2H), 2.44 - 2.23 (m, 2H), 2.01 (m, 1H). MS: 625.1(M+1)+.
1H NMR (400 MHz, CDC13): δ 8.91 - 8.34 (m, 2H), 8.03 (s, 1H), 7.79 - 7.34 (m, 3H), 7.22 6.75 (m, 5H), 6.46 (s, 1H), 6.02 (d, J = 6.5 Hz, lH), 4.95 (dd, J= 9.4, 3.1 Hz, 1H), 4.35 (s, 3.13 - 2.76 (m, 3H), 2.68 - 1.83 (m, 5H). MS: 625.1(M+1)+.
Compound39
Ή NMR (400 MHz, CDC13): δ 8.65 (d, J= 23.6 Hz, 2H), 7.87 (s, 1H), 7.59 - 7.29 (m, 3H), 7.26 - 6.71 (m, 5H), 6.59 (s, 1H), 6.28 (s, 1H), 4.83 (d, J= 8.2 Hz, 1H), 4.12 (s, 1H), 3.10 - 2.62 (m, 3H), 2.56 (m, 1H), 2.36 - 1.84 (m, 4H). MS :625.1(M+1)+.
Compound 40
1H NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.53 (s, 1H), 7.71 (s, 1H), 7.31 (d, J= 8.3 Hz, 1H), 7.25 - 6.80 (m, 6H), 6.44 (s, 1H), 6.08 (s, 1H), 4.95 (m, 1H), 4.35 (s, 1H), 3.15 - 2.76 (m, 3H), 2.66 - 2.17 (m, 4H), 2.03 (s, 1H). MS : 625.1 (M+1)+.
¾ NMR (400 MHz, CDC13): δ 8.29 (dd, J= 8.1, 2.0 Hz, 1H), 7.74 (m, 2H), 7.31 (m, 2H), 7.22 - 7.12 (m, 2H), 7.00 (s, 2H), 6.93 (m, 1H), 6.67 (dd, J = 7.9, 2.4 Hz, 1H), 6.46 (m, 1H), 6.06 (m, 1H), 4.86 (m, 1H), 4.35 (m, 1H), 2.93 (m, 3H), 2.59 - 2.39 (m, 2H), 2.23 (m, 1H), 2.02 (m, 1H). MS: 575.1 (M+l)+.
Compound 29
1H NMR (400 MHz, CDC13): δ 8.40 (m, 1H), 8.24 (m, 1H), 7.71 (d, J= 7.7 Hz, 1H), 7.49 - 7.30 (m, 2H), 7.28 - 7.21 (m, 1H), 7.12 (m, 2H), 7.04 - 6.88 (m, 3H), 6.67 (m, 1H), 6.42 (s, 2H), 4.90 (m, 1H), 4.27 (m, 1H), 3.07 - 2.76 (m, 3H), 2.58 - 2.29 (m, 3H). MS: 575.0 (M+l)+.
Compound 12
1H NMR (400 MHz, CDC13): δ 8.27 (m, 1H), 7.64 - 7.30 (m, 3H), 7.27 - 6.62 (m, 7H), 6.47 - 6.30 (m, 1H), 6.28 - 6.07 (m, 1H), 5.00 - 4.55 (m, 1H), 4.26 (m, 1H), 3.12 - 2.67 (m, 3H), 2.65 - 2.36 (m, 3H), 2.22 (m, 2H). MS: 575.1(M+1)+.
1H NMR (400 MHz, CDC13): δ 8.37 (t, J = 8.9 Hz, IH), 7.63 (m, 2H), 7.49 - 6.84 (m, 8H), 6.44 (s, IH), 5.94 (m, IH), 5.07 - 4.74 (m, IH), 4.25 (d, J= 51.6 Hz, IH), 3.10 - 2.67 (m, 3H), 2.63 - 1.85 (m, 5H), 1.25 (s, IH). MS: 591.1 (M+1)+.
Compound 35
Ή NMR (400 MHz, DMSO-d6) : δ 8.38 (d, J = 8.2 Hz, IH), 7.86 - 7.34 (m, 4H), 7.25 - 6.79 (m, 6H), 6.46 (s, IH), 5.99 (s, IH), 4.95 (d, J = 9.2 Hz, IH), 4.34 (s, IH), 3.12 - 2.70 (m, 3H), 2.63 - 1.87 (m, 6H).MS: 591.1(M+1)+.
Compound 48
Ή NMR (400 MHz, CDC13): δ 8.59 - 8.19 (m, 2H), 7.82 - 7.57 (m, 2H), 7.45 - 7.34 (m, 2H), 7.01 (m, 4H), 6.45 (s, IH), 5.94 (s, IH), 4.89 (dd, J = 9.3, 3.1 Hz, IH), 4.30 (m, IH), 3.21 - 2.69 (m, 3H), 2.61 - 1.88 (m, 5H). MS: 591.1(M+1)+.
Ή NMR (400 MHz, CDC13): δ 8.63 - 8.03 (m, 2H), 7.67 (s, IH), 7.23 - 6.65 (m, 8H), 6.45 - 5.93 (m, 2H), 4.84 (m, IH), 4.23 (m, IH), 3.04 - 2.65 (m, 4H), 2.65 - 1.83 (m, 5H). MS:
591.1(M+1)+.
Compound 36
H NMR (400 MHz, CDC13): δ 8.79 - 8.51 (m, 2H), 7.88 (s, IH), 7.51 - 7.29 (m, 2H), 7.22 (m, 2H), 7.08 (t, J= 7.3 Hz, IH), 6.99 (t, J = 7.2 Hz, IH), 6.78 (s, IH), 6.51 (d, J= 5.8 Hz, IH), 6.28 (s, IH), 4.79 (m, IH), 4.14 (s, IH), 3.02 - 2.66 (m, 3H), 2.55 (m, IH), 2.33 - 1.99 (m, 4H). MS: 582.1(M+1)+.
Compound 37
Ή NMR (400 MHz, CDC13): δ 8.74 (s, IH), 8.52 (s, IH), 7.85 - 7.30 (m, 3H), 7.24 - 6.79 (m, 5H), 6.43 (s, IH), 6.12 (s, IH), 4.92 (d, J = 6.8 Hz, IH), 4.34 (s, IH), 2.90 (m, 3H), 2.64 - 2.46 (m, IH), 2.46 - 2.11 (m, 3H), 1.97 (m, IH). MS : 582.1(M+1)+.
Ή NMR (400 MHz, CDC13): δ 8.66 - 8.38 (m, 2H), 7.90 (d, J= 7.0 Hz, 1H), 7.68 (s, 1H), 7.37 (m, 1H), 7.25 - 6.80 (m, 6H), 6.44 (s, 1H), 5.97 (d, J= 6.6 Hz, 1H), 4.91 (d, J= 6.7 Hz, 1H), 4.32 (s, 1H), 3.30 - 2.78 (m, 4H), 2.41 (m, 4H), 2.02 (s, 1H). MS : 582.1(M+1)+.
Compound 16
1H NMR (400 MHz, CDC13): δ 8.58 (d, J = 9.3 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.97 (d, J = 8.5 Hz, 1H), 7.86 - 7.59 (m, 3H), 7.48 (m, 2H), 7.18 (m, 3H), 6.97 (m, 3H), 6.38 (s, 1H), 6.11 (s, 1H), 5.20 (s, 1H), 4.30 (s, 1H), 3.09 - 2.77 (m, 3H), 2.67 - 2.44 (m, 2H), 2.36 - 2.21 (m, 2H), 2.10 - 1.92 (m, 1H). MS: 607.2 (M+l)+.
Compound 1
1H NMR (400 MHz, CDC13): δ 8.69 (d, J= 4.8 Hz, 2H), 7.71 (s, 1H), 7.31 (m, 1H), 7.18 (m, 7.13 - 6.77 (m, 6H), 6.46 (s, 1H), 6.22 (s, 1H), 5.00 - 4.62 (m, 1H), 4.35 (s, 1H), 3.19 - 2.71 3H), 2.69 - 1.83 (m, 5H).MS: 451.2 (M+l)+.
1H NMR (400 MHz, DMSO-d6): δ 8.15-8.01 (m, 1H), 7.62-7.52 (m, 1H), 7.31-6.69 (m, 9H), 6.24 (s, 1H), 5.65-4.66 (m, 1H), 2.60 (m, 1H), 2.20-2.05 (m, 3H), 1.76-0.83 (m, 4H). MS: 451.2 (M+l)+.
Compound 18
Ή NMR (400 MHz, DMSO-d6): δ 9.70 (s, 1H), 8.48 - 8.26 (m, 2H), 7.72 (s, 1H), 7.46 - 7.31 (m, 1H), 7.28 - 7.15 (m, 2H), 7.13 - 6.89 (m, 3H), 6.55 - 6.14 (m, 2H), 4.82 (m, 1H), 4.26 (m, 1H), 2.90 (m, 3H), 2.64 - 2.40 (m, 2H), 2.34 - 1.99 (m, 3H). MS: 558.1 (M+l)+.
Compound 13
Ή NMR (400 MHz, CDC13): δ 7.54 (d, J= 3.5 Hz, 1H), 7.45 - 7.29 (m, 3H), 7.28 - 6.95 (m, 6H), 6.44 (d, J = 6.0 Hz, 1H), 6.24 (s, 1H), 4.92 (m, 1H), 4.25 (s, 1H), 3.1 1 - 2.79 (m, 3H), 2.61 (m, 1H), 2.43 (m, 1H), 2.39 - 2.27 (m, 2H), 2.27 - 2.11 (m, 1H). MS: 563.1 (M+l)+.
Ή NMR (400 MHz, CDC13): δ 7.66 (s, 1H), 7.48 (s, 1H), 7.35 (s, 1H), 7.26 - 6.82 (m, 8H), 6.43 (s, 1H), 6.09 (d, J= 6.3 Hz, 1H), 4.98 (d, J= 8.7 Hz, 1H), 4.34 (s, 1H), 3.08 - 2.84 (m, 2H), 2.63 - 2.36 (m, 4H), 2.32 (m, 1H), 2.15 (m, 1H). MS: 563.1 (M+l)+.
Compound 23
Ή NMR (400 MHz, CDC13): δ 7.78 - 7.49 (m, 2H), 7.39 (m, 4H), 7.24 - 6.82 (m, 4H), 6.38 (m, 3H), 5.94 (m, 1H), 4.50 (m, 1H), 4.22 (m, lH), 3.10 - 2.59 (m, 3H), 2.59 - 1.99 (m, 6H). MS: 556.2 (M+l)+.
Example 3.Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2- oxoethyl)-N-(3-fluorophenyl)-5-oxo-l-(thiazol-4-yl)pyrrolidine-2-carboxamide
Compounds42 and 43 were prepared according to the following scheme, using the following rotocol.
A mixture(25r)-N-(l -(2-chlorophenyl)-2-(3 ,3 -difluorocyclobutylamino)-2-oxoethyl)-N-(3 - fluorophenyl)-5-oxopyrrolidine-2-carboxamide (200 mg, 0.417 mmol), 4-bromothiazole (0.045
mL, 0.626 mmol, 1.5 eq), K3P04 (124 mg, 0.585 mmol, 1.4 eq), Cul (8 mg, 0.1 eq) and trans-1,2- diaminocyclohexane (0.24 eq) in dioxane (2 mL) was stirred at 110 °C under microwave for 30 min. The resulting mixture was filtered through a Celite pad. The filtrate was concentrated and the residue was purified by a standard method to give the desired product.
(S)-N-((R)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-N-(3- fluorophenyl)-5-oxo-l-(thiazol-4-yl)pyrrolidine-2-carboxamide(Co/np<>M«</ 2
1H NMR (400 MHz, CDC13): δ 8.68 (d, J = 2.1 Hz, 1H), 7.65 (m, 5H), 7.30 - 6.90 (m, 4H), 6.47 (s, 1H), 6.23 (s, 1H), 4.88 (dd, J = 9.3, 3.0 Hz, 1H), 4.20 (s, 1H), 3.17 - 2.63 (m, 3H), 2.58 - 1.99 (m, 5H). MS: 563.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylarmno)-2-oxoethyl)-N-(3- fluorophenyl)-5-oxo-l-(thiazol-4-yl)pyrrolidine-2-carboxamide(Conipound 43)
H NMR (400 MHz, CDC13): δ 8.60 (s, 1H), 8.06 - 7.56 (m, 2H), 7.35 (s, 1H), 7.22 - 6.79 (m, 5H), 6.42 (s, 1H), 6.13 (s, 1H), 4.96 (d, J = 7.8 Hz, 1H), 4.25 (m, 1H), 3.14 - 2.70 (m, 4H), 2.63 - 2.21 (m, 4H).MS: 563.1 (M+l)+.
Example 4.Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2 -oxoethyl)-N-(3-fluorophenyl)-5-oxo-l-(pyridin-2-ylmethyl)pyrrolidine-2-carboxamide
Compound 44 was prepared according to the following scheme, using the following protocol.
Compound 44. To a solution of (2S)-N-(l -(2-chlorophenyl)-2-(3,3-difluorocyclobutyl -amino)- 2-oxoethyl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (200 mg, 0.42 mmol) in dry DMF (20 mL) was added NaH (20 mg, 0.84 mmol) at 0 °C. The mixture was stirred at this 0 °C for 0.5 h followed by addition of 2-(bromomethyl)pyridine (106 mg, 0.42 mmol). The mixture was then allowed warm to room temperature and stirred overnight. The resulting mixture was slowly added dropwise to 100 mL of water, and then extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with saturated aq. LiCl, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by a standard methodto afford the desired product. Ή NMR (400 MHz, CDC13): δ 8.51 (s, 1H), 7.88 - 7.37 (m, 3H), 7.19 - 5.95 (m, 10H), 5.14 (m, 1H), 4.34 (m, 1H), 4.10 (m, 2H), 3.00 (m, 2H), 2.81 - 1.57 (m, 6H). MS: 571.2 (M+l)+.
Example 5.Preparation of(S)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2- oxoethyl)-N-(3-fluorophenyl)-3-hydroxy-2-(pyrimidin-2-ylamino)propanamide.
Compound 9 was prepared according to the following scheme, using the following protocol.
Step A: (S)-2-Oxooxazolidine-4-carboxylic acid. To a solution of NaOH (0.8 g, 20 mmol) in water (4 mL) was added (S)-2-(benzyloxycarbonylamino)-3-hydroxypropanoic acid (1 g, 4.2 mmol) portionwise at 0°C over 3 min. The resulting solution was warmed to r.t and stirred for 2
h. After cooling to 0°C, the solution was adjusted to pH=l -2 with 2 NHCl. The mixture was extracted with EtOAc (4 x 10 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated in vacuo to give the desired product as a white solid. 1H NMR (400 MHz, DMSO-de): δ 13.93 - 12.30 (m, 1H), 8.15 (s, 1H), 4.49 (t, J= 8.6 Hz, 1H), 4.32 (m, 2H); MS: 130.0 (M-l)".
Step B: (4S)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylandno)-2-oxoethyl)-N-(3-fluoro - phenyl)-2-oxooxazolidine-4-carboxamide. 2-Chlorobenzaldehyde (160 mg, 1.14 mmol), 3- fluoroaniline (127 mg, 1.14 mmol), (S)-2-oxooxazolidine-4-carboxylic acid (150 mg, 1.14 mmol) and l,l-difluoro-3-isocyanocyclobutane (181 mg, 90% of purity, 1.37 mmol) were used in the UGI reaction to give the desired product as a white solid. ¾ NMR (400 MHz, CDC13): δ 8.15- 8.01 (m, 1H), 7.62-7.52 (m, 1H), 7.31 -6.69 (m, 9H), 6.24 (s, 1H), 5.65-4.66 (m, 4H), 2.60 (m, 1H), 2.20-2.05 (m, 3H), 1.76-1.51 (m, 5H), 1.29-0.83 (m, 5H); MS: 482.1 (M+l)+.
Step C: (S)-N-((R)-l-(2-Chlorophenyl)-2-(3,3-difluorocychbutylamino)-2-oxoethyl)-N-(3- - fluorophenyl)-2-oxo-3-(pyrirmdin-2-yl)oxazolidine-4-carboxamide and (S)-N-((S)-l-(2-chloro ^henyl)-2-(3,3-difluorocyclobutylandno)-2-oxoethyl)-N-(3-fluorophenyl)-2-oxo-3^yri 2-yl)oxazolidine-4-carboxamide. A mixture of (4<S)-N-(l -(2-chlorophenyl)-2-(3,3-difluorocyclo- butylamino)-2-oxoethyl)-N-(3-fluorophenyl)-2-oxooxazolidine-4-carboxamide (350 mg, 0.73 mmol), 2-bromopyrimidine (150 mg, 0.94 mmol), Cs2C03 (500 mg, 1.52 mmol), Pd2(dba)3 (66 mg, 0.07 mmol) and Xant-Phos (42 mg, 0.07 mmol) in 1 ,4-dioxane (15 mL) was stirred under N2 at 80°C for 18 h and then filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue was purified a standard method to give (5 -N-((i?)-l-(2-chlorophenyl)-2-(3,3- difluorocyclobutylamino)-2-oxoethyl)-N-(3-fiuorophenyl)-2-oxo-3-(pyrimidin-2-yl)oxazolidine- 4-carboxamide(S).1H NMR (400 MHz, CDC13): δ 8.73 (d, J = 4.8 Hz, 2H), 7.95 (s, 0.8H), 7.74 (s, 0.2H), 7.41 (d, J= 7.5 Hz, 1.6H), 7.24 (t, J = 7.2 Hz, 1H), 7.17 - 6.94 (m, 4.3H), 6.73 (d, J= 6.7 Hz, 1H), 6.48 (d, J = 73.8 Hz, 2H), 4.93 (s, 1H), 4.41 (dd, J = 8.6, 4.8 Hz, 1H), 4.29 (t, J = 8.6 Hz, 1H), 4.14 (m, 1H), 2.80 (m, 2H), 2.21 (s, 1H), 2.18 - 2.07 (m, 1H); MS: 560.1 (M+l)+,and (5 -N-(()S -l -(2-chloro-phenyl)-2-(3,3-difluorocyclo-butylamino)-2-oxoethyl)-N-(3-fiuoro - phenyl)-2-oxo-3-(pyrimidin-2-yl)oxazolidine-4-carboxamide {9) lYl NMR (400 MHz, CDC13): δ 8.68 (d, J = 4.8 Hz, 2H), 7.65 (s, 1H), 7.30 (s, 1H), 7.18 (s, 1H), 7.13 - 6.86 (m, 5H), 6.50 (s,
1H), 6.38 (m, 1H), 5.00 (m, 1H), 4.43 (dd, J= 8.7, 4.8 Hz, 1H), 4.32 (m, 1H), 4.20 (m, 1H), 2.99 (m, 2H), 2.50 (m, 2H). MS: 560.1 (M+l)+.
Example 6.Preparation of(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl) -amino)- 2-oxoethyl)-N-(3-fluorophenyl)-6-oxo-l-(pyrimidin-2-yl)piperidine-2-carboxamide
Compounds 19 and 20 were prepared according to the following scheme, using the following protoc
Step A.(S)-6-Oxopiperidine-2-carbox lic acid. A solution of (S)-2-aminohexanedioic acid (470 mg, 2.9 mmol) in 20% AcOH (5 mL) was stirred at 110 °C overnight. The solvent was removed in vacuo and the residue was dissolved in EtOH (lOmL). The unreacted amino acid was precipitated and filtered off. The filtrate was concentrated to give the crude desired product which was used directly in the next step. MS: 142.1 (M-l)".
Step B. (S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)andno)-2-oxoethyl)-N-(3-fluoro - phenyl)-6-oxopiperidine-2-carboxamide. 3-Fluoroaniline (217mg, 1.96mmol), 2-chlorobenzal- dehyde (274mg, 1.96mmol), (-S -6-oxopiperidine-2-carboxylic acid (280 mg, 1.96 mmol) and l,l-difluoro-3-isocyanocyclobutane (280mg, 1.96mmol) were used in the UGI reaction to give the desired product. MS: 494.1 (M+l)+ .
Step C. (S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocychbutyl)amino)-2-oxoethyl)-N-(3 - fluorophenyl )- 6-oxo- 1 -(pyrimidin -2-yl)piperidin e-2-carboxamide and (S)-N-((R)-J -(2-chloro - phenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-fluorophenyl)-6-oxo-l- (pyrimidin-2-yl)piperidine-2-carboxamideA mixture consisting of (li?)-N-(l-(2-chlorophenyl) - 2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-fluorophenyl)-3-oxo-2-(pyrimidin-2- yl)cyclohexanecarboxamide (250 mg, 0.51 mmol), 2-bromopyrimidine (121 mg, 0.76 mmol),
Cs2C03 (331 mg, 1.01 mmol), Pd2(dba)3 (46 mg, 0.05 mmol) and Xant-Phos (29 mg, 0.04 mmol) in 1,4-dioxane (15 mL) was stirred under N2 at 80 °C overnight and then filtered. The filtrate was concentrated in vacuo and the residue was purified by a standard method to give the desired products.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocychbuiyl)amino)-2-oxoethyl)-N-(3—fluoro - phenyl)-6-oxo-l-(pyrimidin-2-yl)piperidine-2-carboxamide (Compound 19). Ή NMR (400 MHz, CDCI3): δ 8.73 (m, 2H), 7.70 (s, 1H), 7.26 - 6.95 (m, 6H), 6.87 (t, J= 7.2 Hz, 1H), 6.53 (s, 1H), 6.33 (s, 1H), 4.77 (d, J= 5.3 Hz, 1H), 4.33 (s, 1H), 3.01 (d, J= 5.5 Hz, 2H), 2.85 - 2.28 (m, 4H), 2.05 (m, 2H), 1.81 (s, 2H). MS: 571.1 (M+l)+.
(S)-N-((R)-l-(2-Chloro^henyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoe - phenyl)-6-oxo-l-(pyrimidin-2-yl)piperidine-2-carboxamide (Compound 20). Ή NMR (400 MHz, CDCI3): δ 8.74 (d, J = 4.8 Hz, 2H), 7.99 (m, 1H), 7.56 - 7.32 (m, 1H), 7.27 - 6.85 (m, 6H), 6.72 (s, 1H), 6.51 (m, 1H), 4.67 - 4.48 (m, 1H), 4.34 - 4.01 (m, 1H), 2.95 - 2.60 (m, 2H), 2.59 - 2.40 (m, 1H), 2.40 - 2.19 (m, 2H), 2.15 - 2.00 (m, 2H), 1.97 - 1.59 (m, 4H). MS: 571.1 (M+l)+.
Example 7. Preparationof(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)- 2-oxoethyl)-N-(3-fluorophenyl)-5-oxo-4-(pyrimidin-2-yl)morpholine-3-carboxamide
C mpound 30 was prepared according to the following scheme, using the following protocol.
Step A: (S)-3-Hydroxy-2-(4-methoxybenzylamino)propanoic acid. (<S -2-amino-3 -hydroxy - propanoic acid (8.4 g, 80 mmol) was dissolved in a solution of NaOH (3.2g, 80 mmol) in H20 (40mL). After cooling to 10°C, 4-methoxybenzaldehyde(21.7g, 160 mmol)was added dropwise over 10 min. The mixture was stirred at room temperature for 30 min and then cooled to 0°C. NaBH4 (1.67g, 44 mmol)was added portionwise and the resulting mixture was warmed slowly to
r.t and stirred for 2 h. The mixture was washed with Et20(2 x 50mL).The aqueous phase was adjusted topH 4.5 with 2 N HC1 at 0 °C. The precipitate was filtered, washed with petroleum ether (20mL) and dried in vacuo to give the desired product as a white solid. MS: 226.1 (M+l)+. Step B: (S)-Benzyl4-(4-methoxybenzyl)-5-oxomorpholine-3-carboxylate. (<S -3-Hydroxy-2-((4- methoxybenzyl)amino)propanoic acid (5.0g,22 mmol) was dissolved in a solution of NaOH (1.15g, 29 mmol) in H20 (60mL). After cooling to 0°C, 2-chloroacetyl chloride (3.6mL, 44 mmol) was added dropwise followed by aq. NaOH(30% wt)to keep pH=13. After stirring for another 4 h, the reaction was cooled to 0°C and acidified with 2 N HC1 to adjustpH=2~3. The resulting mixture was extracted with EtOAc (2 x 30 mL). The combined organic layers were dried over anhydrous Na2S04and concentrated. The residue wasdissolved in acetone (150mL) and then treated with BnBr(9.7g,51 mmol) andDIPEA(19mL,l 11 mmol).The reaction mixture was stirred for 24 h at room temperature and concentrated in vacuo. The residue was purified by column chromatography to afford the desired product as a white solid. MS: 356.1 (M+l)+.
Step C: (S)-Benzyl 5-oxomorpholine-3-carboxylate. To a solution of (5)-benzyl4-(4- methoxybenzyl)-5-oxomorpholine-3-carboxylate (200mg, 0.56 mmol) in CH3CN(5mL) and H20 (5mL) was added CAN(ceric ammonium nitrate) (1.5 g, 2.8 mmol) at 0°C. The resulting mixture was stirred at 0°C for 1 h. DIPEAwas added at 0°C to adjust thepH to6~7 and the mixture was concentrated in vacuo. The residue was purified by columnchromatography to afford the desired product as a white solid. MS: 236.1 (M+l)+.
Step D: (S)-5-Oxomorpholine-3-carboxylic acid. To a mixture of (S^-benzyl 5-oxomorpholine- 3-carboxylate (160mg,0.7mmol) in MeOH (8mL) was added 10% Pd/C (about 5 mg). The reaction was stirred under an atmosphere of hydrogen for 30 min at room temperature. The reaction mixture was filtered through a Celite pad and concentrated in vacuo to afford the desired product as a white solid. MS: 146.1 (M+l)+.
Step E: (S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3- fluorophenyl)-5-oxomorpholine-3-carboxamide. 3-Chlorobenzaldehyde (104 mg, 0.74 mmol), 3-fluoroaniline (83 mg, 0.74 mmol), ()S -5-oxomo holine-3-carboxylic acid (108 mg, 0.74 mmol)and l ,l -difluoro-3-isocyanocyclobutane (248 mg, 1.48 mmol) were used in the UGI reaction to afford the desired product. MS: 496.1 (M+l)+.
Step F: Compound 30. Amixtureof(5)-iV-((5)-l-(2-chlorophenyl)-2-((3,3-difluoro - cyclobutyl)amino)-2-oxoethyl)-N-(3-fluorophenyl)-5-oxomo holine-3-carboxamide (100 mg, 0.2mmol), 2-bromopyrimidine (36 mg, 0.22 mmol),Pd2(dba)3 (28 mg, 0.03 mmol), XantPhos (16 mg, 0.03 mmol) and Cs2CO3(160 mg, 0.5 mmol) in 1 ,4-dioxane (4 mL) was stirred at 100°C for 3.5hunder N2. The reaction mixture was then cooled to room temperatureand filtered. The solid was washed with DCM (2 x 20 mL). The filtrate was evaporatedand the residue was purifiedby a standard methodto afford the desired product. 1H NMR (400 MHz, CDC13): δ 8.77 (m, 2H), 7.85 (m, 1H), 7.41 (s, 1H), 7.28 - 7.21 (m, 1H), 7.21 - 7.10 (m, 2H), 7.09 - 6.90 (m, 3H), 6.87 (m, 1H), 6.68 - 6.33 (m, 2H), 4.80 (m, 1H), 4.43 - 4.22 (m, 2H), 4.13 (m, 2H), 3.94 (m, 1H), 2.99 (m, 1H), 2.86 (m, 1H), 2.63 - 2.26 (m, 2H). MS: 474.1 (Μ+1)+'
Example 8.
The following analogs were synthesized viatheprocedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic (heteroaromatic) ring using the reagents and solvents set forth above, and purified via standard methods.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclopentyl)amino)-2-oxoethyl)-N-(3-fluoro phenyl)-5-oxo-l-(pyrimidin-2-yl)pyrrolidine-2-carboxamide (racemic) - Compound 73
1H NMR (400 MHz, CDC13): δ 8.71 (d, J = 4.8 Hz, 2H), 7.72 (s, 1H), 7.37 (s, 1H), 7.18 (s, 1H), 7.11 - 6.85 (m, 5H), 6.47 (s, 1H), 5.70 (d, J= 7.3 Hz, 1H), 4.86 (d, J = 7.0 Hz, 1H), 4.53 (d, J = 6.3 Hz, 1H), 3.51 (s, 1H), 2.95-2.88 (m, 1H), 2.64 - 2.47 (m, 2H), 2.40 - 1.65 (m, 8H). MS: 572.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 64
¾ NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.52 (s, 1H), 7.72 (d, J= 7.1 Hz, 1H), 7.43 - 7.33 (m, 1H), 7.25 - 7.17 (m, 1H), 7.13 - 6.81 (m, 4H), 6.43 (s, 1H), 6.12 (s, lH), 4.92 (d, J= 6.8 Hz, 1H), 4.37-4.28 (m, 1H), 3.10 - 2.82 (m, 3H), 2.59-2.49 (m, 2H), 2.42-2.36 (m, 1H), 2.31 - 2.22 (m, 1H), 2.06-1.88 (m, 2H). MS: 582.1 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl)amino)-2-oxo-l-phenylethyl)- -(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 138
1H NMR (400 MHz, CDC13): δ 8.78 (s, 1H), 8.44 (d, J = 4.9 Hz, 1H), 7.65 (s, 1H), 7.39 - 7.15 (m, 6H), 7.14 - 6.92 (m, 4H), 6.65 (m, 1H), 6.16 (s, 1H), 5.82 (s, 1H), 4.86 (d, J= 6.8 Hz, 1H), 4.31 (s, 1H), 3.15 - 2.77 (m, 3H), 2.68 - 1.91 (m, 5H). MS: 548.2 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl)amino)-l-(2-fluorophenyl)-2- oxoethyl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 149
1H NMR (400 MHz, CDC13): δ 8.74 (m, 1H), 8.50 (d, J = 4.2 Hz, 1H), 7.65 (s, 1H), 7.45 - 7.14 (m, 4H), 7.13 - 6.69 (m, 5H), 6.25 (m, 2H), 4.88 (dd, J= 9.2, 3.1 Hz, 1H), 4.33 (s, 1H), 3.21 - 2.72 (m, 3H), 2.65 - 1.88 (m, 5H).MS: 566.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri midin-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 68
¾ NMR (400 MHz, CDC13): δ 8.95 (d, J= 4.7 Hz, 1H), 7.68 (s, 1H), 7.34 (d, J= 4.6 Hz, 2H), 7.16 (s, 1H), 7.04 (d, J = 3.6 Hz, 3H), 6.92 (s, 2H), 6.51 (s, 1H), 5.92 (s, 1H), 4.81 (d, J= 9.5 Hz, 1H), 4.33 (s, 1H), 2.91 (m, 3H), 2.64 - 2.26 (m, 4H), 2.01 (s, 1H). MS: 583.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopy rimidin-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 85
Ή NMR (400 MHz, CDC13): δ 8.98 (d, J= 4.7 Hz, 1H), 7.74 (s, 1H), 7.38 (dd, J= 11.2, 5.7 Hz, 2H), 7.06 (m, 5H), 6.52 (s, 1H), 5.47 (d, J = 7.7 Hz, 1H), 4.85 (d, J = 9.2 Hz, 1H), 3.99 (s, 1H), 2.93 (dd, J= 18.6, 8.9 Hz, 1H), 2.62 (d, J = 9.5 Hz, 1H), 2.36 (s, 1H), 1.97 (m, 7H), 1.57 - 1.38 (m, 2H). MS: 611.2 (M+l)+ .
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3,5- difluoro -phenyl)-5-oxo-l-(pyrimidin-2-yl)pyrrolidine-2-carboxamide (single enantiomer) - Compound 70
¾ NMR (400 MHz, CDC1
3): δ 8.70 (d, J= 4.8 Hz, 2H), 7.60 (s, 1H), 7.37 (d, J= 8.0 Hz, 1H), 7.26 - 7.19 (m, 1H), 7.13 - 7.04 (m, 2H), 7.03 - 6.97 (m, 1H), 6.86 (s, 1H), 6.69 (dd, J = 9.8, 7.6 Hz, 1H), 6.46 (s, 1H), 6.07 (d, J= 6.7 Hz, 1H), 4.87 (dd, J = 9.1, 3.1 Hz, lH), 4.36 (s, 1H), 3.11 - 2.83 (m, 3H), 2.64 - 2.34 (m, 3H), 2.21 (m, 1H), 2.10 - 1.97 (m, 1H). MS: 576.1 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri din-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 71
H NMR (400 MHz, CDC13): δ 8.73 (d, J= 7.1 Hz, 1H), 8.60-8.46 (m, 1H), 7.56 (d, J= 7.7 Hz, 1H), 7.38-7.32 (m, 1H), 7.31 -7.27 (m, 1H), 7.26-7.18 (m, 1H), 7.14-7.00 (m, 1H), 6.96 (m, 1H), 6.85 (s, 1H), 6.69 (m, 1H), 6.40 (s, 1H), 6.02 (d, J= 6.6 Hz, 1H), 4.98-4.74 (m,lH), 4.39-4.10 (m, 1H), 3.11-2.67 (m, 3H), 2.64-1.95 (m, 5H). MS : 600.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopy rimidin-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 86
Ή NMR (400 MHz, CDC13): δ 8.98 (d, J= 4.8 Hz, 1H), 7.56 (s, 1H), 7.40 (m, 2H), 7.23 (t, J = 7.0 Hz, 1H), 7.08 (t, J= 7.6 Hz, 1H), 7.01 - 6.84 (m, 2H), 6.71 (t, J= 8.6 Hz, 1H), 6.51 (s, 1H), 6.00 (d, J = 6.7 Hz, 1H), 4.85 (dd, J = 9.3, 2.7 Hz, 1H), 4.36 (s, 1H), 3.15 - 2.80 (m, 3H), 2.67 - 2.26 (m, 4H), 2.08 (dt, J= 9.7, 8.1 Hz, 1H). MS: 601 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri - din-2-yl)-5-oxo-N-(3-sulfamoylphenyl)pyrrolidine-2-carboxamide (single enantiomer) - Compound 53
1HNMR(400 MHz, CDC13): δ 8.74 (s, 1H), 8.50 (s, 1H), 7.73 (d,J= 7.5 Hz, 1H), 7.33 (d,J = 9.3 Hz, 1H), 7.25 - 6.80 (m, 6H), 6.40 (s, 1H), 5.61 (d,J= 6.9 Hz, 1H), 4.91 (d, J = 8.0 Hz, 1H), 3.97 (s, 1H), 2.99 - 2.79 (m, 1H), 2.55 (dd, J = 13.7, 9.9 Hz, 1H), 2.25 (t, J = 11.3 Hz, 1H), 2.03 - 1.74 (m, 5H), 1.56 - 1.36 (m, 2H). MS: 610.2 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 81
1HNMR(400MHz, CDC13): δ 8.75 (s, 1H), 8.51 (d,J= 5.0 Hz, 1H), 7.62 (d,J=9.0Hz, 1H), 7.37(d,J=7.9Hz, 1H), 7.27 (d,J=5.1 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H),6.99 (d,J=6.9Hz, 1H), 6.88 (d,J=7.4Hz, 1H), 6.69 (t, J = 8.6 Hz, 1H), 6.41 (s, 1H), 5.69 (d,J=7.8Hz, 1H), 4.95 (dd, J = 9.3, 3.2 Hz, lH),3.98(m, 1H), 2.95-2.84 (m, 1H), 2.65 - 2.55 (m, 1H), 2.30 - 2.20 (m, 1H), 2.05 - 2.12 (m, 1H), 2.03 (s, 2H), 1.94 - 1.78 (m, 2H), 1.68 - 1.35 (m, 3H), 0.85 - 0.95 (m, 1H). MS: 628.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopy rimidin-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 87
H NMR (400 MHz, CDC13): δ 8.97 (d, J= 4.8 Hz, 1 H), 7.60 (d, J= 8.7 Hz, 1 H), 7.46 - 7.34 (m, 2 H), 7.22 (t, J = 7.8Hz, 1 H), 7.06 (t, J = 7.6Hz, 1 H), 7.00 - 6.87 (m, 2 H), 6.70 (t, J =
8.6Hz, 1 H), 6.48 (s, 1 H), 5.64 (d, J= 7.7 Hz, 1 H), 4.86 (dd, J= 9.3, 2.7 Hz, 1 H), 3.98 (d, J = 7.7 Hz, 1 H), 2.96 - 2.86 (m, 1 H), 2.63 - 2.55 (m, 1 H), 2.37 - 2.29 (m, 1 H), 2.15 - 1.99 (m, 5 H), 1.96 - 1.77 (m, 2 H), 1.61 - 1.34 (m, 2 H). MS: 629.2 (M+l)+ .
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-l-(2,4-dichlorophenyl)-2-((3,3-difluorocyclobutyl)amino) -2-oxoethyl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 196
1H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.49 (d, J= 5.1 Hz, 1H), 7.56 (s, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.30 (s, 1H), 7.08 (dd, J= 8.4, 2.1 Hz, 1H), 6.97 (d, J= 8.4 Hz, 1H), 6.90 (s, 1H), 6.79 - 6.72 (m, 1H), 6.35 (s, 1H), 5.99 (d, J= 6.6 Hz, 1H), 4.93 (dd, J = 9.3, 3.1 Hz, lH), 4.33 (s, 1H), 3.12 - 2.95 (m, 2H), 2.95 - 2.83 (m, 1H), 2.66 - 2.32 (m, 3H), 2.24-2.18 (m, 1H), 2.12 - 1.99 (m, 1H).MS: 634.1 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-l-(2,5-dichlorophenyl)-2-((3,3-difluorocyclobutyl)amino) -2-oxoethyl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 201
1H NMR (400 MHz, CDC13): δ 8.76 (s, 1H), 8.49 (dd, J= 5.0, 0.6 Hz, 1H), 7.58 (s, 1H), 7.30 (t, J = 5.2 Hz, 2H), 7.22 (dd, J = 8.6, 2.5 Hz, 1H), 7.02 (d, J= 2.4 Hz, 1H), 6.88 (s, 1H), 6.76 (tt, J = 8.6, 2.3 Hz, 1H), 6.34 (s, 1H), 6.14 (d, J = 6.8 Hz, 1H), 4.94 (dd, J = 9.3, 3.2 Hz, 1H), 4.43 - 4.28 (m, 1H), 3.09-3.02 (m, 2H), 2.93-2.84 (m, 1H), 2.65 - 2.32 (m, 3H), 2.27 - 2.16 (m, 1H), 2.14 - 2.00 (m, 1H).MS: 634.1 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-l-(2,6-dichlorophenyl)-2-(3,3-difluorocyclobutylamino)- 2-oxoethyl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) -
Compound 63
1H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.45 (t, J= 5.6 Hz, 1H), 7.88 (t, J= 10.0 Hz, 1H), 7.40 - 7.32 (m, 1H), 7.26 - 7.21 (m, 2H), 7.10 - 7.05 (m, 2H), 6.92 (d, J= 2.4 Hz, 1H), 6.62 (d, J = 8.6 Hz, 1H), 5.53 (d, J = 5.3 Hz, 1H), 4.84 - 4.75 (m, 1H), 4.40 (s, 1H), 3.06 - 2.92 (m, 3H), 2.65 - 2.42 (m, 4H), 2.18 - 2.02 (m, 1H). MS: 616.1 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-l-(2,6-dichlorophenyl)-2-((3,3-difluorocyclobutyl) amino) -2-oxoethyl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 199
1H NMR (400 MHz, CDC13): δ 8.78 (s, 1H), 8.44 (d, J = 5.0 Hz, 1H), 7.80 - 7.22 (m, 5H), 6.91 (s, 1H), 6.81 (tt, J= 8.7, 2.3 Hz, 1H), 6.45 (d, J= 8.5 Hz, 1H), 5.56 (d, J= 6.8 Hz, 1H), 4.83 (dd, J= 9.4, 2.7 Hz, 1H), 4.40 (d, J = 8.0 Hz, 1H), 3.23 - 2.92 (m, 3H), 2.69 - 2.39 (m, 4H), 2.23 - 2.02 (m, 1H).MS: 634.2 (M+l)+.
(2S)-l-(4-Cyanopyridin-2-yl)-N-(l-(2,3-dichlorophenyl)-2-(3,3-difluoro-cyclobutylamino)-2- oxoethyl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(racemic) - Compound
Ή NMR (400 MHz, CDC1
3): δ 8.72 (s, 1H), 8.57 (s, 1H), 7.44 (d, J= 7.9, 1H), 7.32 - 7.29 (m, 1H), 7.17 - 6.68 (m, 4H), 6.53 - 6.41 (m, 1H), 6.32 - 6.12 (m, 1H), 4.90 - 4.65 (m, 1H), 4.41 - 4.05 (m, 1H), 3.13 -2.01 (m, 8H). MS: 634.1 (M+l)
+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-(3,3-difluorocyclobutylamino)-l-(2-fluorophenyl)-2- oxoethyl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 208
1H NMR (400 MHz, CDC13): δ 8.63 (s, 1H), 8.40 (d, J = 4.9 Hz, 1H), 7.43 (s, lH), 7.20 (s, 1H), 7.16 (d, J= 5.0 Hz, 1H), 6.90 (t, J = 8.2 Hz, 3H), 6.62 (t, J= 8.7 Hz, 2H), 6.20 (s, 1H), 6.14 (d, J = 6.4 Hz, 1H), 4.81 (dd, J = 9.1, 2.9 Hz, 1H), 4.25 (s, 1H), 2.92 (s, 2H), 2.85 - 2.70 (m, 1H), 2.56 - 2.22 (m, 3H), 2.15 (m, 1H), 2.04 - 1.90 (m, 1H). MS: 584.2 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-(3,3-difluorocyclobutylamino)-2-oxo-l-phenylethyl)- N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound
Ή NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.41 (d, J= 5.1 Hz, 1H), 7.49 (s, 1H), 7.27 (dd, J = 8.2, 5.0 Hz, 2H), 7.24 (d, J= 5.4 Hz, 2H), 7.04 (d, J = 6.7 Hz, 2H), 6.71 (t, J= 8.8 Hz, 1H), 6.44 (s, 1H), 6.15 (s, 1H), 5.70 (d, J= 6.3 Hz, 1H), 4.86 (dd, J= 9.3, 2.8 Hz, 1H), 4.29 (s, 1H), 2.99 (m, 2H), 2.90 (m, 1H), 2.62 - 2.52 (m, 1H), 2.45 (m, 1H), 2.38 - 2.25 (m, 2H), 2.07 (m, 1H). MS: 566.2 (M+l)+.
(S)-N-((S)-l-(3-Chloropyridin-2-yl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 198
¾ NMR (400 MHz, CDC13): 58.75 (s, 1H), 8.49 (d, J= 5.0 Hz, 1H), 8.31 (d, J = 3.4 Hz, 1H), 7.65 - 7.56 (m, 2H), 7.27 (m, 1H), 7.19 - 7.15 (m, 1H), 6.98 (m, 1H), 6.76 - 6.56 (m, 2H), 6.11 (d, J= 6.8 Hz, 1H), 5.04 - 5.01 (m, 1H), 4.38 (m, 1H), 3.05 - 2.98 (m, 2H), 2.92 - 2.83 (m, 1H), 2.60 - 2.52 (m, 1H), 2.51 - 2.37 (m, 2H), 2.37 - 2.27 (m, 1H), 2.07 - 2.02 (m, 1H). MS: 601.1 (M+l)+. (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri -din-2-yl)-5-oxo-N-(3-sulfamoylphenyl)pyrrolidine-2-carboxamide(single enantiomer) - Compound 84
H NMR (400 MHz, CDC13): δ 8.73 (d, J = 10.0 Hz, 1H), 8.57 - 8.45 (d, J= 8.0 Ηζ,ΙΗ), 8.12 (d, J= 7.7 Hz, 1H), 7.83-7.76 (m, 2H), 7.61-7.56 (m, 1H), 7.48-7.32 (m, 1H), 7.19 (t, J= 7.1 Hz, 1H), 7.05 - 6.87 (m, 2H), 6.82 - 6.81 (m, 1H), 6.55 - 6.43 (m, 1H), 6.27 (d, J= 6.7 Hz, 1H), 5.24 (s, 1H), 4.84 (d, J= 7.2 Hz, 1H), 4.69 (s, 1H), 4.33 (s, 1H), 2.98-2.87 (m, 3H), 2.63 - 2.24 (m, 4H), 2.09 - 2.00 (m, 1H) . MS: 643.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-cyano- phenyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 128
¾ NMR (400 MHz, CDC1
3): δ 8.76 (s, 1H), 8.51 (s, 1H), 8.23 (m, 1H), 7.58-7.27 (m, 4H), 6.93 (m, 3H), 6.43 (s, 1H), 5.85 (s, 1H), 4.78 (s, 1H), 4.34 (s, 1H), 3.10 -2.82 (m, 3H), 2.37-2.52(m, 3H), 2.21-2.23 (m, 1H), 1.89-1.99 (m, 1H). MS: 589.1 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-N-(3-cyano phenyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 166
1H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.49 (d, J= 13.9 Hz, lH), 8.22-8.32 (m, 1H), 7.61 - 7.27 (m, 4H), 7.17-7.19 (m, 2H), 6.90-7.00 (m, 2H), 6.42 (s, 1H), 5.50 (s, 1H), 4.80 (d, J= 9.5 Hz, 1H), 3.97 (s, 1H), 2.99 - 2.80 (m, 1H), 2.56-2.58 (m, 1H), 2.21-2.24 (m, 1H), 1.70-2.10 (m, 6H), 1.41-1.44 (m, 2H). MS: 617.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-cyano phenyl)-l-(4-cyanopyrimidin-2-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 167
Ή NMR (400 MHz, CDC13): δ 8.91 -9.00 (m, 1H), 8.33 - 8.17 (m, 1H), 7.62 - 7.32 (m, 5H), 7.20 (t, J= 7.0 Hz, 1H), 7.02-7.06 (m, 1H), 6.95 - 6.83 (m, 1H), 6.55 (s, 1H), 6.05 - 5.88 (m, 1H), 4.72 (d, J = 9.3 Hz, 1H), 4.37 (s, 1H), 2.91-3.05 (m, 3H), 2.70 - 2.25 (m, 4H), 2.13 - 1.92 (m, 1H). MS: 590.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-N-(3- cyanophenyl)-l-(4-cyanopyrimidin-2-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 178
1H NMR (400 MHz, CDC13): δ 8.99 (s, 1H), 8.32 (s, 1H), 7.57 (m, 1H), 7.54 - 7.28 (m, 2H), 7.19 (t, J = 7.2 Hz, 3H), 7.04 (t, J = 6.8 Hz, 1H), 6.93 (d, J= 7.7 Hz, 1H), 6.53 (s, 1H), 5.64 - 5.44 (m, 1H), 4.74 (d, J= 9.3 Hz, 1H), 3.99 (s, 1H), 2.94 (dd, J= 17.8, 9.4 Hz, 1H), 2.62 (m, 1H), 2.41 - 2.24 (m, 1H), 2.10 - 1.82 (m, 7H).MS: 618.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-N-(3-cyano-5- fluorophenyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer)
- Compound 177
Ή NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.50 (s, 1H), 8.13-8.08 (m, 1H), 7.44 - 7.27 (m, 2H), 7.23 (dd, J= 12.6, 6.3 Hz, 2H), 7.07 (t, J = 7.3 Hz, 1H), 6.93 (t, J= 6.4 Hz, 1H), 6.43 (d, J= 6.1 Hz, 1H), 6.14 (dd, J= 13.9, 6.7 Hz, 1H), 4.81 (dd, J= 9.0, 2.3 Hz, 1H), 4.42 - 4.28 (m, 1H), 3.12 - 2.94 (m, 2H), 2.94 - 2.80 (m, 1H), 2.67 - 2.29 (m, 3H), 2.23 - 1.92 (m, 2H). MS: 607.1
(M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-N-(3-cyano-5- fluorophenyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 184
Ή NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.50 (s, 1H), 8.25 - 8.03 (m, 1H), 7.52 - 7.28 (m, 2H), 7.22 (t, J = 7.7 Hz, 2H), 7.01 (dt, J = 14.1, 10.1 Hz, 2H), 6.42 (d, J= 6.9 Hz, 1H), 5.58 (t, J
= 9.9 Hz, 1H), 4.83 (dd, J= 9.1, 2.3 Hz, 1H), 4.05 - 3.86 (m, 1H), 3.04 - 2.81 (m, 1H), 2.59 (m, 1H), 2.36 - 1.70 (m, 7H), 1.58 - 1.31 (m, 3H). MS: 636.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-N-(3-cyano-5- fluorophenyl)-l-(4-cyanopyrimidin-2-yl)-5-oxopyrrolidine-2-carboxamide(single nantiomer) - Compound 185
Ή NMR (400 MHz, CDC13): δ 8.97 (d, J= 4.4 Hz, 1H), 8.12 (m, 1H), 7.50 - 7.32 (m, 3H), 7.23 (d, J= 6.7 Hz, 2H), 7.06 (m, 1H), 6.95 (s, 1H), 6.50 (d, J= 8.6 Hz, 1H), 5.60 (d, J= 7.5 Hz, 1H), 4.74 (d, J= 8.8 Hz, 1H), 3.98 (s, 1H), 2.90 (m, 1H), 2.72 - 2.49 (m, 1H), 2.28 (s, 1H), 2.17 - 1.67 (m, 7H), 1.43 (m, 2H). MS: 637.2 (M+l)+.
(S)-N-(3-Cyano-5-fluorophenyl)-l-(4-cyanopyridin-2-yl)-N-((S)-2-(3,3-difluorocyclobutyl - amino)-2-oxo-l-phenylethyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 211
1H NMR (400 MHz, CDC13): δ 8.71 (d, J= 10.1 Hz, 1H), 8.38 (s, 1H), 8.02 (m, 1H), 7.23 (m, 5H), 6.97 (d, J= 7.3 Hz, 3H), 6.20 (s, 1H), 5.97 (s, 1H), 4.70 (dd, J = 9.2, 2.4 Hz, 1H), 4.27 (s, 1H), 2.93 (m, 2H), 2.85 (t, J= 8.9 Hz, 1H), 2.59 - 2.48 (m, 1H), 2.49 - 2.29 (m, 2H), 2.29 - 2.20 (m, 1H), 2.08 - 1.99 (m, 1H). MS: 573.2 (M+l)+.
(S)-N-(3-Cyano-5-fluorophenyl)-l-(4-cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl) amino)-l-(2-fluorophenyl)-2-oxoethyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer)
- Compound 207
¾ NMR (400 MHz, DMSO-d6): δ 8.78 (s, 1H), 8.62 (d, J= 5.1 Hz, 1H), 8.48 (s, 1H), 8.04 - 7.83 (m, 1H), 7.78 (s, 1H), 7.57 (s, 1H), 7.23 (m, 2H), 7.14 (d, J = 9.9 Hz, 1H), 6.95 (t, J= 7.5 Hz, 1H), 6.84 (s, 1H), 6.20 (s, 1H), 4.72 (s, 1H), 4.04 (s, 1H), 4.00 - 3.82 (m, 1H), 3.09 - 2.67 (m, 2H), 2.33 (m, 1H), 1.91 (s, 2H), 1.83 (s, 1H), 1.27 - 1.05 (m, 1H). MS: 591.2 (M+l)+ .
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyri - din-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide (racemic)- Compound
H NMR (400 MHz, CDC13): δ 9.10 - 8.03 (m, 4H), 7.47-7.39 (m, 2H), 7.27 - 6.84 (m, 3H), 6.51 -6.01 (m, 2H), 4.84-4.70 (m, 1H), 4.36-4.20 (m, 1H), 3.25 - 1.86 (m, 8H). MS: 583.1 (M+l)+ .
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 176
1H NMR (400 MHz, CDC13): δ 8.95-8.70 (m, 1H), 8.49 (d, J= 4.7 Hz, 1H), 8.36-8.11 (m, 1H), 8.12(d, J= 8.6 Hz, 1H), 7.33 (d, J= 8.0 Hz, 1H), 7.21 (t, J= 7.8 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.48-6.41(m, 1H), 6.30-6.21 (m, 1H), 4.84-6.79 (m, 1H), 4.38-4.30 (m, 1H), 3.1 1 - 2.74 (m, 3H), 2.65 - 1.91 (m, 5H). MS: 583.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4-cyanopyri - din-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 193
1H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.49 (d, J= 5.2 Hz, 1H), 8.40 - 8.27 (m, 1H), 8.21 - 8.04 (m, 1H), 7.41 - 7.36 (m, 1H), 7.26 - 7.23 (m, 1H), 7.20 (t, J= 6.9 Hz, 1H), 7.04 (t, J = 7.2 Hz, 1H), 6.93 (m, 1H), 6.52 - 6.34 (m, 1H), 5.49 (s, 1H), 4.84 (d, J = 7.4 Hz, 1H), 4.01 - 3.94 (m, 1H), 2.99-2.91 (m, 1H), 2.62-2.54 (m, 1H), 2.22-1.71 (m, 7H), 1.31 (s, 3H). MS: 611.2 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl)amino)-2-oxo-l-phenylethyl)- N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound
1H NMR (400 MHz, CDC13): δ 8.86 (m, 1H), 8.39 (m, 2H), 8.03 (m, 1H), 7.28 (d, J= 5.9 Hz, 4H), 6.98 (m, 2H), 6.29 (s, 1H), 5.85 (s, 1H), 4.85 (m, 1H), 4.33 (s, 1H), 3.26 - 2.82 (m, 3H), 2.69 - 1.88 (m, 5H). MS: 549.2 (M+l)+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl)amino)-l-(2-fluorophenyl)-2- oxoethyl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 148
Ή NMR (400 MHz, CDC1
3): δ 8.99 - 8.60 (m, 1H), 8.55 - 7.97 (m, 3H), 7.35 - 7.19 (m, 3H), 7.07 - 6.89 (m, 3H), 6.36 (m, 1H), 6.12 (s, 1H), 4.80 (s, 1H), 4.35 (s, 1H), 3.22 - 2.79 (m, 3H), 2.64 - 1.85 (m, 5H). MS: 567.2 (M+l)
+.
(S)-l-(4-Cyanopyridin-2-yl)-N-((S)-2-((3,3-difluorocyclobutyl)amino)-2-oxo-l-phenylethyl)- N-(5-isocyanopyridin-3-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 212
Ή NMR (400 MHz, CDC13): δ 9.34 (s, 1H), 8.87 - 8.56 (m, 4H), 8.41 (s, 2H), 8.27 (s, 1H), 7.54 (s, 7H), 7.01 (d, J= 6.9 Hz, 3H), 6.35 (s, 2H), 5.73 (s, 2H), 4.66 (s, 2H), 4.35 (s, 2H), 2.99 (m, 5H), 2.73 - 2.20 (m, 7H), 2.07 (s, 2H). MS: 556.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(3-cyano - phenyl)-N-(lH-indazol-7-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 186
1H NMR (400 MHz, CDC13): δ 8.72 - 8.71 (m, 1H), 8.66 (s, 1H), 8.08 (s, 1H), 7.69 (s, 1H), 7.67 (s, 1H), 7.50 - 7.49 (m, 1H), 7.36 - 7.34 (m, 1H), 7.11-7.07 (m, 1H), 7.00-6.96 (m, 1H), 6.83- 6.76 (m, 2H), 6.48 (s, 1H), 5.07 - 5.07 (m, 1H), 4.38 - 4.33 (m, 1H), 3.05 - 2.91(m, 2H),2.80 - 2.71 (m, 1H), 2.65 - 2.60(m, 1H),2.53 - 2.46(m, 2H), 2.03- 1.99(m, 1H), 1.75 - 1.67(m, 1H). MS: 603.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-3-(3-cyano phenyl)-N-(lH-indazol-7-yl)-2-oxooxazolidine-4-carboxamide(single enantiomer) - Compound 142
1H NMR (400 MHz, CDC13): δ 13.03 (s, 1H), 8.73 (s, 1H), 8.55 - 8.54 (m, 1H), 8.02 (s, 1H), 8.58-8.56 (m, 1Η),8.50-8.48 (m, 1H), 7.27-7.24 (m, 2H), 7.03 - 6.99 (m, 1H), 6.91 - 6.87 (m, 1H), 6.80-6.78 (m, 1H), 6.72-6.68 (m, 1H), 6.33 (s, 2H), 5.70 - 5.69 (m, 1H), 4.99 - 4.97 (m, 1H), 4.05 - 4.03 (m, 1H), 2.78 - 2.95(m, 1H), 2.47 - 2.40 (m, 1H), 2.08 - 4.99 (m, 6H), 1.90- 1.82 (m, 2H), 1.67 - 1.63(m, 1H), 1.58 - 1.62 (m, 1H). MS: 633.2 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopy ridin-2-yl)-N-(lH-indazol-4-yl)-5-oxopyrrolidine-2-carboxamide (racemic) - Compound 152
Ή NMR (400 MHz, DMSO-d6): δ 13.05 (m, 1H), 8.70 (m, 2H), 8.54 (d, J= 6.7 Hz, 1H), 8.21 (s, 1H), 7.80 (d, J= 6.9 Hz, 1H), 7.63 (d, J = 5.0 Hz, 1H), 7.36 (m,2H), 7.24 (d, J= 8.0 Hz, 1H), 7.18 - 6.97 (m, 1H), 6.92 - 6.79 (m,lH), 6.77 - 6.70 (m,lH), 6.35 (d, 1H), 4.66 (m, 1H), 4.20 - 4.01 (m, 1H), 3.05 - 2.78 (m, 2H), 2.68 - 2.52 (m, 2H), 2.49 - 2.26 (m,2H), 2.22 - 1.53 (m, 2H). MS : 604.2 (M+l)+.
(S)-N-(3-(lH-Pyrazol-4-yl)phenyl)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylami - no)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 200
1H NMR (400 MHz, MeOD): δ 8.73-8.54 (m, 2H), 8.14 - 7.91 (m, 1H), 7.71 (d, J= 7.6 Hz, 1H), 7.56 - 7.28 (m, 4H), 7.25 - 6.92 (m, 4H), 6.70 (d, J = 7.6 Hz, 1H), 6.54 - 6.39 (m, 1H), 5.03 (dd,
J = 9 A, 2.9 Hz, 1H), 4.31 - 4.05 (m, 1H), 3.00 - 2.73 (m, 3H), 2.64 - 2.00 (m, 5H). MS: 630.2 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyri - midin-2-yl)-5-oxo-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-2-carboxamide (racemic)- Compound 180
Ή NMR (400 MHz, CDC13): δ 8.96 (t, J= 5.5 Hz, 1H), 7.88 (s, 1H), 7.44 - 7.32 (m, 2H), 7.21 (m, 2H), 7.10 (t, J = 7.3 Hz, 1H), 7.04 - 6.95 (m, 1H), 6.91 (m, 1H), 6.52 (m, 1H), 6.18 (m, 1H), 4.89 - 4.67 (m, 1H), 4.31 (m, 1H), 3.22 - 2.75 (m, 3H), 2.70 - 1.92 (m, 5H). MS: 649.1 (M+l)+. (S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-(difluoromethoxy)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 181
1H NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.44 (m, 1H), 7.76 (d, J= 9.0 Hz, 1H), 7.33 (m, 2H), 7.21 - 6.83 (m, 6H), 6.44 (t, J = 8.8 Hz, 1H), 6.28 - 6.13 (m, 1H), 4.91 (m, 1H), 4.34 (s, 1H), 3.10 - 2.66 (m, 3H), 2.65 - 1.84 (m, 5H). MS: 630.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyrimidin-2-yl)-N-(3-(difluoromethoxy)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 194
¾ NMR (400 MHz, CDC1
3): δ 9.04 - 8.59 (m, 1H), 7.74 (s, 1H), 7.43 - 7.26 (m, 4H), 6.96 (m, 3H), 6.36 (m, 2H), 4.81 (t, J = 9.3 Hz, 1H), 4.55 (m, 1H), 4.33 (s, 1H), 4.06 - 3.89 (m, 1H), 3.15 - 2.69 (m, 2H), 2.69 - 1.86 (m, 5H). MS: 631.1 (M+l)
+.
(S)-N-((S)-l-(2C)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N- (3-methoxyphenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 129
1H NMR (400 MHz, CDC13): δ 8.75 (s, 1H), 8.51 (d, J= 5.0 Hz, 1H), 7.47 (m, 1H), 7.38 - 7.08 (m, 3H), 6.99 (d, J = 6.7 Hz, 3H), 6.89 - 6.66 (m, 2H), 6.41 (s, 1H), 6.09 (d, J= 6.6 Hz, 1H), 4.97 (dd, J = 9.3, 3.2 Hz, 1H), 4.34 (s, 1H), 3.72 (m, 3H), 3.01 (dd, J= 7.5, 4.0 Hz, 3H), 2.65 - 2.23 (m, 4H), 2.04 (d, J = 9.0 Hz, 1H). MS: 594.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutyl-amino)-2-oxoethyl)-l-(4-cyanopyri midin-2-yl)-N-(3-methoxyphenyl)-5-oxopyrrolidine-2-carboxamid(single enantiomer)- Compound 164
Ή NMR (400 MHz, CDC13): δ 8.92 (s, 1H), 7.48 - 7.39 (m, 1H), 7.33 - 7.26 (m, 2H), 7.22 - 7.08 (m, 2H), 7.04 - 6.82 (m, 3H), 6.73 (s, 2H), 6.48 (d, J= 9.5 Hz, 1H), 6.18 (m, 1H), 4.88 - 4.85 (m, 1H), 4.32 (s, 1H), 3.78 (s, 1H), 3.62 (s, 2H), 3.01 - 2.81 (m, 3H), 2.58 - 2.49 (m, 2H), 2.42 - 2.30 (m, 2H), 2.09 - 1.98 (m, 1H). MS: 595 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyrimi -din-2-yl)-N-(3-cyclopropoxyphenyl)-5-oxopyrrolidine-2-carboxamide (racemic)- Compound 192
1H NMR (400 MHz, CDC13): δ 9.06 - 8.88 (m, 1H), 7.61 - 7.30 (m, 4H), 7.27 - 7.22 (m, 1H), 7.18 (t, J = 7.4 Hz, 2H), 7.08 - 6.92 (m, 1H), 6.87 (dd, J= 8.7, 2.1 Hz, 1H), 6.78 (t, J= 9.5 Hz, 1H), 6.50 (s, 1H), 6.04 (m, 3H), 5.57 - 5.14 (m, 2H), 4.88 (m, 1H), 4.77 - 4.10 (m, 3H), 3.15 - 2.75 (m, 3H), 2.68 - 2.47 (m, 2H), 2.45 - 2.21 (m, 3H), 2.20 - 1.90 (m, 1H). MS: 621.1 (M+l)+. (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri - din-2-yl)-N-(3-(hydroxymethyl)phenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compoundl31
Ή NMR (400 MHz, CDC13): δ 8.73 (s, 1H), 8.53 (s, 1H), 7.94 - 7.70 (m, 1H), 7.31 (s, 1H), 7.26 (dd, J= 5.1, 1.3 Hz, 1H), 7.22-7.10(m, 4H), 7.02 - 6.87 (m, 2H), 6.44 (d, J= 10.5 Hz, 1H), 6.12 (d, J= 6.4 Hz, 1H), 4.91 (dd, J = 9.3, 3.2 Hz, 1H), 4.69 (s, 1H), 4.48 (s, 1H), 4.42 - 4.26 (m, 1H), 3.07-2.85 (m, 3H), 2.65 - 2.17 (m, 4H), 2.01 (s, 2H). MS: 594.2 (M+l)+.
(S)-N-((S)-l-(2-Xhlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-(l-hydroxycyclopropyl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 140
1H NMR (400 MHz, CDC13): δ 1H NMR (400 MHz, CDC13) : δ 8.73 (s, 1H), 8.52 - 8.44 (m, 1H), 7.64 - 7.30 (m, 3H), 7.22 - 6.90 (m, 5H), 6.42 - 6.38 (m, 1H), 6.03 (m, 1H), 4.87 (m, 1H), 4.30 (m, 1H), 3.05 - 2.82 (m, 3H), 2.60 - 1.88 (m, 5H), 1.21 (d, J= 3.2 Hz, 4H). MS: 620.2 (M+l)+.
(S)-N-((S)-l-(2C)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N- (3-(2-hydroxypropan-2-yl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 179
¾ NMR (400 MHz, CDC13): δ 8.69 (s, 1H), 8.54 (d, J= 5.0 Hz, 1H), 7.93 - 7.70 (m, 1H), 7. 7.19 (m, 4H), 7.1 1 (m, 2H), 7.01 - 6.72 (m, 2H), 6.45 (m, 2H), 5.05 - 4.76 (m, 1H), 4.33 (s, 3.13 - 2.58 (m, 3H), 2.42 (m, 4H), 2.09 - 1.83 (m, 1H), 1.33 (s,6H). MS: 622.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoeth-yl)-l-(4- cyanopyri-din-2-yl)-N-(3-fluoro-5-(2-hydroxypropan-2-yl)phenyl)-5-oxopyrrolidine-2- carboxamide(single enantiomer) - Compound 150
H NMR (400 MHz, CDC13): δ 8.66 (s, 1H), 8.49 (d, J= 4.8 Hz, 1H), 7.73 - 7.48 (m, 1H), 7.26 - 6.83 (m, 7H), 6.53 - 6.42 (m, 2H), 4.91 (d, J= 6.4 Hz, 1H), 4.32 (s, 1H), 3.02 - 2.72 (m, 3H), 2.58 - 1.85 (m, 6H), 1.63 (s, 2H), 1.51 (d, J= 7.0 Hz, 2H), 1.29 (d, J= 8.6 Hz, 4H).MS: 640.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri din-2-yl)-N-(3-fluoro-5-(2-hydroxypropan-2-yl)phenyl)-5-oxopyrrolidine-2- carboxamide(single enantiomer) - Compound 155
¾ NMR (400 MHz, CDC1
3): δ 8.80 (s, 1H), 8.43 (s, 1H), 7.51 (d, 1H), 7.24 (m, 4H), 7.06 (s, 3H), 6.64 (m, 1H), 6.15 (m, 1H), 5.73 (s, 1H), 4.86 (s, 1H), 4.32 (s, 1H), 3.01 (m, 3H), 2.68 - 2.27 (m, 4H), 2.12 (s, 1H), 1.44 (s, 1H), 1.29 (d, J= 9.0 Hz, 6H). MS: 639.2 (M+l)
+
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri -din-2-yl)-N-(3-(2-hydroxyethyl)phenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 160
1H NMR (400 MHz, CDC13): δ 8.76 (s, 1H), 8.52 (d, J= 5.0 Hz, 1H), 7.74 (s, 1H), 7.32-7.36 (m, 1H), 7.27 - 7.11 (m, 2H), 7.09 - 6.87 (m, 4H), 6.39-6.45 (m, 1H), 6.05 (d, J= 6.9 Hz, 1H), 4.33 (s, 1H), 3.82 (s, 1H), 3.59 (s, 1H), 3.12 - 2.79 (m, 4H), 2.74 - 2.16 (m, 5H), 1.99-2.07 (m, 1H). MS: 608.2 (M+l)+.
(S)-N-((S)-l-(2-Clorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(3-(2-hydroxyethoxy)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 130
H NMR (400 MHz, CDC13): δ 8.72 (s, 1H), 8.48 (d, J= 5.0 Hz, 1H), 7.54 - 7.28 (m, 2H), 7.18- 7.21 (m, 2H), 7.01 - 6.94 (m, 2H), 6.75-6.77 (m, 2H), 6.39 (s, 1H), 5.99 (s, 1H), 4.94 (dd, J= 9.3, 3.4 Hz, 1H), 4.31 (s, 1H), 3.79-4.06 (m, 4H), 3.07 - 2.80 (m, 3H), 2.58 - 2.21 (m, 4H), 1.87-2.00 (m, 2H). MS: 624.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-fluoro-5-((S)-methylsulfinyl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 190
1H NMR (400 MHz, CDC13): δ 8.75 (s, 1H), 8.54 (m, 1H), 8.02 - 7.78 (m, 1H), 7.33 (s, 3H), 7.21 (m 1H), 7.06 (t, J = 7.4 Hz, 1H), 6.96 (m, 1H), 6.45 (m, 1H), 6.27 (m, 1H), 4.86 (m, lH), 4.35 (m, 1H), 3.16 - 2.82 (m, 3H), 2.71 (s, 1H), 2.65 - 2.47 (m, 2H), 2.41 (m, 3H), 2.22 (m, 1H), 2.09 (m, 1H). MS: 644.1 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 96
1H NMR (400 MHz, CDC13): δ 8.84 - 8.11 (m, 3H), 7.93 - 7.35 (m, 4H), 7.25 - 6.75 (m, 2H), 6.64 - 5.94 (m, 2H), 4.89 - 4.69 (m, 1H), 4.28 (d, J= 5.7 Hz, 1H), 3.13 - 2.74 (m, 6H), 2.68 - 2.48 (m, 2H), 2.46 - 2.15 (m, 3H), 2.04 (s, 1H). MS: 642.1 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyri midin-2-yl)-N-(3-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide (racemic)- Compound 102
Ή NMR (400 MHz, CDC13): δ 8.93 (t, J= 5.3 Hz, 1H), 8.50 - 8.15 (m, 1H), 7.94 - 7.71 (m, 2H), 7.66 - 7.46 (m, 1H), 7.38 (t, J= 6.4 Hz, 1H), 7.28 (t, J= 3.6 Hz, 1H), 7.20 - 7.07 (m, 1H), 7.05 - 6.87 (m, 2H), 6.74 (m, 1H), 6.52 (m, 1H), 4.72 (dd, J = 9.2, 2.5 Hz, 1H), 4.34 (d, J = 6.4 Hz, 1H), 3.00 (s, 3H), 2.90 - 2.75 (m, 3H), 2.56 - 2.19 (m, 5H), 1.98 (m, 1H). MS: 643.1 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide (racemic)- Compound 95
1H NMR (400 MHz, CDC13): δ 8.87 - 8.13 (m, 3H), 8.02 - 7.37 (m, 4H), 7.24 - 6.87 (m, 2H), 6.51 - 6.39 (m, 1H), 5.77 - 5.28 (m, 1H), 4.89 - 4.65 (m, 1H), 3.94 (d, J= 5.2 Hz, 1H), 3.16 - 2.73 (m, 4H), 2.68 - 2.53 (m, 1H), 2.44 - 2.20 (m, 1H), 2.03 (m, 8H), 1.44 (m, 2H). MS: 670.2
(M+l)+.
(S)-N-((S)-l-(2C)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyri midin-2-yl)- N-(3-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 103
H NMR (400 MHz, CDC13): δ 8.94 (dd, J= 7.9, 4.8 Hz, 1H), 8.56 - 8.15 (m, 1H), 7.97 - 7.62 (m, 2H), 7.56 - 7.29 (m, 3H), 7.13 (t, J = 7.6 Hz, 1H), 7.06 - 6.84 (m, 2H), 6.51 (d, J= 4.2 Hz, 1H), 6.10 (dd, J= 3.2, 7.4 Hz, 1H), 4.74 (d, J = 6.6 Hz, 1H), 3.98 (s, 1H), 3.01 (s, 1H), 2.93 - 2.72 (m, 3H), 2.52 (d, J= 9.6 Hz, 1H), 2.37 - 2.20 (m, 1H), 2.13 - 1.78 (m, 7H), 1.63 - 1.40 (m, 2H). MS: 671 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopy ridin-2-yl)-N-(3-fluoro-5-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide
¾ NMR (400 MHz, CDC13): δ 8.45-8.79 (m, 2H), 8.40 - 8.13 (s, 1H), 8.09-7.67 (m, 1H), 7.63- 7.30 (m, 2H), 7.23-6.87 (m, 3H), 6.55-6.30 (m, 1H), 6.22 - 5.94 (m, 1H), 4.96 - 4.61 (m, 1H), 4.26 (m, 4H), 3.16-1.87 (m, 7H), 1.27 (d, 1H). MS: 660.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri midin-2-yl)-N-(3-fluoro-5-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carbox- amide(single enantiomer) - Compound 109
¾ NMR (400 MHz, CDC13): δ 8.96 (d, J= 4.6 Hz, 3H), 7.99 (d, J= 8.5 Hz, 2H), 7.75 (s, 2H), 7.52 (d, J = 7.0 Hz, 3H), 7.37 (d, J = 4.9 Hz, 5H), 7.19 (t, J = 7.7 Hz, 3H), 7.01 (dt, J= 7.1 Hz, 6H), 6.40-6.60(m, 3H), 6.06 (d, J= 6.5 Hz, 3H), 4.76 (d, J = 9.2 Hz, 1H), 4.35 (m, 4H), 3.14- 1.87 (m, 8H). MS: 661.1 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-fluoro-5-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide
(racemic)- Compound 105
1H NMR (400 MHz, CDC13): δ 8.96 (t, J = 4.6 Hz, 1H), 7.53-7.36 (m, 3H), 7.23 (m, J= 7.8, 1.5 Hz, 1H), 7.14-6.94 (m, 3H), 6.68 (m, J = 8.6, 2.3 Hz, 1H), 6.60 (d, J= 3.1 Hz, 1H), 6.07 (d, J =
6.7 Hz, 1H), 4.75 (q, J= 4.0, 2.1 Hz, 1H), 4.38 (d, J= 6.7 Hz, 1H), 3.78-3.67 (m, 2H), 3.39 (m, 1H), 3.26-2.92 (m, 3H), 2.67-2.36 (m, 2H). MS: 688.1 (M+l)+.
(2S)-N-(l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyri midin-2-yl)-N-(3-fluoro-5-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 108
Ή NMR (400 MHz, CDC13): δ 8.97 (s, 1H), 8.20-8.60 (m, 1H), 8.09 -7.68 (m, 1H), 7.63-7.32 (m,5H), 7.22-6.93 (m, 3H), 6.64-6.03 (m, 2H), 5.62 (s, 1H), 4.60-4.85 (m, 1H), 3.21-1.70 (m, 12H), 1.50-1.14 (m, 2H). MS: 689.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyano pyrimidin-2-yl)-N-(3-fluoro-5-(methylsulfonyl)phenyl)-5-oxopyrrolidine-2- rboxamide(single enantiomer) - Compound 168
H NMR (400 MHz, CDC13): δ 9.0 (s, 1H), 8.05-8.02 (m, 1H), 7.80 (m, 1H), 7.56-7.00 (m, 7H), 6.58 (m, 1H), 5.65 (m, 1H), 4.80 (m, 1H), 4.14 (m, 1H), 3.00-0.88 (m, 15H). MS: 689.1 (M+l)+. (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(3-(methylsulfonamido)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 159
¾ NMR (400 MHz, DMSO-d
6): δ 9.78 (s, 1H), 8.84 - 8.61 (m, 2H), 8.56 (s, 1H), 7.66 (m, 2H), 7.49 - 7.15 (m, 3H), 7.15 - 6.79 (m, 4H), 6.25 (m, 1H), 4.89 - 4.74 (m, 1H), 4.19 - 4.04 (m, 1H), 3.03 - 2.83 (m, 3H), 2.72 - 2.59 (m, 3H), 2.54 (m, 2H), 2.44 - 2.28 (m, 1H), 1.99 (m, 2H). MS : 657.1 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri- din-2-yl)-N-(3-(dimethylamino)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 161
¾ NMR (400 MHz, CDC13): δ 8.71 (d, J= 9.9 Hz, 1H), 8.50 - 8.41 (m, 1H), 7.29 (d, J= 7.8 Hz, 1H), 7.22 (dd, J= 5.0, 1.3 Hz, 1H), 7.18 - 7.05 (m, 2H), 6.99-6.86 (m, 3H), 6.56-6.47 (m, 2H), 6.37 (d, J= 6.6 Hz, 1H), 6.11 (s, 1H), 5.01 (d, J= 9.2 Hz, 1H), 4.34-4.28 (m, 1H), 3.07 - 2.70 (m, 8H), 2.61 - 2.42 (m, 2H), 2.35-2.25 (m, 2H), 2.01-1.97 (m, 1H). MS: 607.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri din-2-yl)-N-(2-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 187
H NMR (400 MHz, CDC13): δ 8.74 (m, 1H), 8.48 (m, 1H), 7.96-7.92 (m, 1H), 7.40 (m, 1H), 7.28 - 6.72 (m, 7H), 6.59-5.79 (m, 2H), 4.86-4.78 (m, 1H), 4.28 (s, 1H), 3.04-2.90 (m, 3H), 2.66 - 2.01 (m, 5H). MS: 582.1 (M+l)+.
(S)-N-((S)-l-(2-Clorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyri din-2-yl)-N-(2,3-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 188
Ή NMR (400 MHz, CDC13): δ 8.73 (m, 1H), 8.47 (d, J= 5.0 Hz, 1H), 7.84 - 7.73 (m, 1H), 7.43 (d, J= 8.1 Hz, 1H), 7.28 - 7.20 (m, 2H), 7.13 (dd, J= 8.2, 4.4 Hz, 2H), 7.01-6.83 (m, 2H), 6.62 (s, 1H), 6.42 - 5.85 (m, 1H), 4.85-4.77 (m, 1H), 4.20 (m, 1H), 3.13 - 2.78 (m, 3H), 2.68 - 2.28 (m, 4H), 2.25 - 2.04 (m, 1H). MS: 600.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri din-2-yl)-N-(2,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 197
Ή NMR (400 MHz, CDC13): δ 8.73 (m, 1H), 8.54 - 8.41 (m, 1H), 7.83-7.78 (m, 1H), 7.44 - 7.39 (m, 1H), 7.28 - 7.21 (m, 2H), 7.13 - 6.88 (m, 3H), 6.81 -6.80 (m, 1H), 6.61 - 6.31 (m, 1H), 5.91 (d, J= 6.5 Hz, 1H), 4.86-4.79 (m,lH), 4.29 (dd, J= 8.2, 6.7 Hz, 1H), 3.51 (s, 1H), 3.12 - 2.85 (m, 3H), 2.68 - 2.56 (m, 1H), 2.54 - 2.45 (m, 1H), 2.43 - 2.24 (m, 2H), 2.23 - 2.06 (m, 1H). MS: 600.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(lH-indazol-5-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 203
H NMR (400 MHz, CDC1
3): δ 8.78 (s, 1H), 8.56 (m, 1H), 8.39 (s, 1H), 8.13 - 7.88 (m, 1H), 7.44-7.32(m, 2H), 7.28 - 7.00 (m, 4H), 6.99 - 6.79 (m, 2H), 6.48 (m, 1H), 5.75 - 5.48 (m, 1H), 5.06 - 4.75 (m, 1H), 4.00 (s, 1H), 3.10 - 2.77 (m, 1H), 2.63 - 2.44 (m, 1H), 2.37 - 2.20 (m, 1H), 2.15 - 1.77 (m, 7H), 1.42 (m, 2H). MS: 632.2 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri din-2-yl)-N-(lH-indazol-6-yl)-5-oxopyrrolidine-2-carboxamide(single
enantiomer) - Compound 205
Ή NMR (400 MHz, CDC13): δ 8.78 (s, 1H), 8.57 (t, J= 5.0 Hz, 1H), 8.23 - 7.76 (m, 2H), 7.54 - 7.30 (m, 2H), 7.16 (s, 1H), 7.04 - 6.86 (m, 3H), 6.47 (d, J = 11.7 Hz, 1H), 6.02 (d, J= 6.1 Hz, 1H), 4.92 (m, 1H), 4.36 (s, 1H), 2.97 (m, 3H), 2.65 - 2.20 (m, 4H), 1.99 (m, 1H). MS: 604.2
(M+l)
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopy ridin-2-yl)-N-(lH-indazol-6-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 136
Ή NMR (400 MHz, CDC13): δ 10.41 - 9.94 (m,lH), 8.79 (s,lH), 8.57 (t, J = 5.1 Hz, 1H), 8.28 - 8.09 (m, 1H), 7.93 (m, 1H), 7.52 (s, 1H), 7.40 (d, J= 7.9 Hz, 1H), 7.33 (d, J= 7.5 Hz, 1H), 7.15 - 6.98 (m, 1H), 6.46 (d, J = 12.7 Hz, 1H), 5.50 (d, J = 7.9 Hz, 1H), 5.06 - 4.76 (m,lH), 4.02 (s, 1H), 2.92 (dd, 1H), 2.63 - 2.49 (m, 1H), 2.31 (s, 1H), 2.03 (m, 6H), 1.45 (s, 2H). MS: 632.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri -din-2-yl)-N-(lH-indazol-5-yl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 175
¾ NMR (400 MHz, CDC13): δ 8.75 (s, 1H), 8.64 - 8.46 (m, 1H), 8.34 (s, 1H), 8.09 (s, 1H), 7.94-7.92 (m, 1H), 7.42-7.32 (m, 2H), 7.24 - 7.02 (m, 2H), 6.94-6.85 (m, 2H), 6.49-6.45 (m, 6.08-6.06 (m, 1H), 5.00 - 4.76 (m, 1H), 4.35-4.31 (s, 1H), 3.00-2.85 (m, 3H), 2.64 - 2.11 (m, 4H), 2.01-1.93 (m, 1H). MS: 604.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(lH-indol-5-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 206
¾ NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.55 (m, 1H), 8.12 (d, J = 13.8 Hz, 2H), 7.52 - 7.29 (m, 2H), 7.18 - 6.80 (m, 5H), 6.46 (m, 2H), 5.83 (s, 1H), 5.83 (s, 1H), 5.08 - 4.81 (m, 1H), 4.33 (s, 1H), 2.92 (m, 3H), 2.64 - 2.16 (m, 4H), 2.01 (m, 1H). MS: 603.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(l-methyl-lH-indol-5-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 209
Ή NMR (400 MHz, CDC13): δ 8.83 - 8.39 (m, 1H), 8.01 (m, 1H), 7.68 - 7.32 (m, 1H), 7.28 - 6.72 (m, 8H), 6.55 - 6.38 (m, 1H), 5.90 (m, 1H), 5.00 - 4.73 (m, 1H), 4.33 (s, 1H), 3.80 - 3.62 (m, 3H), 2.91 (m, 3H), 2.62 - 1.78 (m, 5H). MS: 617.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri -din-2-yl)-N-(3-cyclopropylphenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 173
1H NMR (400 MHz, CDC13): δ 8.76 (s, 1H), 8.59 (d, J = 4.8 Hz, 1H), 7.50-7.60 (m, 1H), 7.41 (d, J= 7.8 Hz, 1H), 7.28 - 7.19 (m, 2H), 7.14 - 6.94 (m, 2H), 6.62-6.79 (m, 1H), 6.26 - 6.07 (m, 2H), 4.86 (dd, J= 9.3, 2.9 Hz, 1H), 4.16-4.19 (m, 1H), 3.02 - 2.76 (m, 3H), 2.57-2.59 (m, 1H), 2.40 - 2.16 (m, 3H), 2.02-2.12 (m, 1H), 1.28-1.29(m, 2H), 0.90 (t, J= 6.9 Hz, 2H). MS: 604.2 (M+l)+.
(S)-N-((S)-l-(2-cChlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri -midin-2-yl)-N-(3-cyclopropylphenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 182
Ή NMR (400 MHz, CDC13): δ 8.94 (d, J= 4.5 Hz, 1H), 7.57 - 7.49 (m, 1H), 7.43 - 7.28 (m, 2H), 7.19-7.14 (m, 2H), 7.05 - 6.79 (m, 4H), 6.51-6.46 (m, 1H), 6.00-5.97 (m, 1H), 4.82-4.80 (m,
1H), 4.32-4.33 (m, 1H), 3.09 - 2.81 (m, 3H), 2.64 - 2.24 (m, 4H), 2.05-1.72 (m, 2H), 0.99 - 0.76 (m, 4H). MS: 605.2 (M+l)+.
(S)-N-(3-(tert-Butyl)phenyl)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2- oxoethyl)-l-(4-cyanopyrimidin-2-yl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 165
Ή NMR (400 MHz, CDC13): δ 8.94 (d, J= 4.8 Hz, 1H), 8.00 - 7.54 (m, 1H), 7.41 - 7.32 (m, 2H), 7.24 - 7.15 (m, 2H), 7.14 - 7.02 (m, 2H), 6.97 - 6.81 (m, 2H), 6.53 (s, 1H), 6.20 (dd, J = 12.7, 6.8 Hz, 1H), 4.86 (m, 1H), 4.34 (s, 1H), 3.15 - 2.80 (m, 3H), 2.63 - 2.27 (m, 4H), 2.13 - 1.92 (m, 1H), 1.29 (s, 9H). MS: 621.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyri -din-2-yl)-N-(3-cyclopropyl-5-fluorophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 204
1H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.50 (s, 1H), 7.50 - 7.33 (m, 2H), 7.24 - 7.17 (m, 1H), 7.01 (m, 2H), 6.68 (m, 2H), 6.39 (m, 1H), 6.00 (s, 1H), 4.93 (s, 1H), 4.34 (s, 1H), 3.15 - 2.83 (m, 3H), 2.59-2.53 (m, 2H), 2.40-2.37 (m, 2H), 2.07 (s, 1H), 1.27 (s, 1H), 1.05 (s, 1H), 0.91 (d, J= 6.7 Hz, 1H), 0.67 (s, 1H), 0.43 (m, 1H). MS: 622.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(3-cyclopropyl-5-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 202
1H NMR (400 MHz, CDC13): δ 8.79 (s, 1H), 8.50 (s, 1H), 7.40 (m, 2H), 7.15 (m, 1H), 7.01 (m, 3H), 6.84 - 6.56 (m, 2H), 6.38 (m, 1H), 5.50 (s, 1H), 4.94 (s, 1H), 3.99 (s, 1H), 2.90 (m, 1H), 2.57 (m, 1H), 2.28 (s, 1H), 2.05 (m, 5H), 1.92 - 1.77 (m, 2H), 1.30 (m, 2H), 0.91 (t, J= 6.7 Hz, 2H), 0.67 (s, 2H).MS: 650.2 (M+l) +.
((S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4- cyanopyri-din-2-yl)-N-(3-(N-methylsulfamoyl)phenyl)-5-oxopyrrolidine-2- carboxamide(single enantiomer) - Compound 157
H NMR (400 MHz, CD3OD): δ 8.89 - 8.59 (m, 3H), 8.50 - 8.01 (m, 2H), 7.69 - 7.31 (m, 5H), 7.17 (t, J = 7.6 Hz, 2H), 7.03 (t, J = 7.6 Hz, 2H), 6.95 (t, J = 7.9 Hz, 2H), 6.51 (s, lH), 4.98 (s, 1H), 4.24 (s, 2H), 3.01 - 2.45 (m, 7H), 2.35 (s, 3H), 2.10 -2.05 (m, 1H). MS: 657.1 (M+l)+. (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri- din-2-yl)-N-(3-(N,N-dimethylsulfamoyl)phenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 156
1H NMR (400 MHz, CDC13):5 8.70 (s, 1H), 8.60 (d, J = 4.9 Hz, 1H), 8.17 (d, J = 7.7 Hz, 1H), 7.86 (s, 1H), 7.63 - 7.55 (m, 1H), 7.49 (t, J = 7.8 Hz, 1H), 7.27 (s, 1H), 7.20 - 6.92 (m, 4H), 6.50 (d, J = 6.9 Hz, 2H), 4.79 (d, J = 7.0 Hz, 1H), 4.32 (s, 1H), 3.05 - 2.75 (m, 4H), 2.60 - 1.90 (m, 10H). MS: 671.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(3- cyanopyri -din-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single
enantiomer) - Compound 69
1H NMR (400 MHz, CDC13): δ 8.14 (d, J = 8.0 Hz, 1H), 7.93 (d, J= 4.0 Hz, 1H), 7.92 (m, 1H), 7.17-7.28 (m, 4H), 6.91-7.04 (m, 4H), 6.42 (s, 1H), 6.31 (s, 1H), 4.87-4.91 (m, 1H), 4.35 (m, 1H), 2.97-3.02 (m, 2H), 2.79 - 2.86 (m, 1H), 2.45-2.57 (m, 3H), 2.23 - 2.26 (m, 1H) , 2.09 - 2.11 (m, 1H). MS: 582.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano-3- fluoropyridin-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer)
- Com ound 82
H NMR (400 MHz, DMSO-d6): δ 8.36 (d, J= 4.7 Hz, 1H), 7.70 (s, 1H), 7.39 (m, 2H), 7.25-6.63 (m, 5H), 6.39 (s, 1H), 5.96 (s, 1H), 4.85 (s, 1H), 4.34 (s, 1H), 3.12-2.69 (m, 3H), 2.64-2.01 (m, 5H). MS : 600.0 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyano-3- fluoropyridin-2-yl)-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer)
- Com ound 83
¾ NMR (400 MHz, DMSO-d
6): δ 8.37 (d, J = 4.6 Hz, 1H), 7.75 (s, 1H), 7.39 (m, 2H), 7.24-6.89 (m, 4H), 6.87-6.65 (d, 1H), 6.50-6.27 (m, 1H), 5.59-5.40 (m, 1H), 4.92-4.75 (m, 1H), 4.05-3.87 (m, 1H), 2.95-2.68 (m, 1H), 2.62-2.43 (m, 1H), 2.41 -2.25 (m, 1H), 2.25-2.09 (m, 2H), 2.05-1.74 (m, 4H), 1.59-1.24 (m, 3H). MS : 628.0 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano-3- fluoropyridin-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 88
1H NMR (400 MHz, DMSO-d6): δ 8.73 (s, 1H), 8.49 (m, 1H), 7.96 (s, 1H), 7.59-7.30 (m, 3H), 7.26-6.68 (m, 6H), 6.52-6.12 (m, 1H), 5.96 (d, J = 10.5 Hz, 1H), 4.95 (s, 1H), 4.63 (m, 1H), 4.49 (m, 1H), 4.22 (s, 1H), 4.14 -4.02 (m, 1H), 3.46-2.65 (m, 4H), 2.55-2.00 (m, 2H), 1.69-1.49 (m, 2H). MS : 618.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-N-(3- fluorophen yl)-5-oxo-l-(pyrazin-2-yl)pyrrolidine-2-carboxamide(single enantiomer) - Compound 58
Ή NMR (400 MHz, CDC13): δ 9.74 (d, J = 1.5 Hz, 1H), 8.32 (m, 2H), 7.71 (s, 1H), 7.36 (m, 1H), 7.16 (m, 1H), 6.97 (m, 4H), 6.41 (s, 1H), 5.44 (d, J = 7.0 Hz, 1H), 4.85 (d, J = 6.0 Hz, 1H), 3.96 (m, 1H), 2.98 - 2.82 (m, 1H), 2.61 - 2.48 (m, 1H), 2.35 - 2.21 (m, 1H), 2.02 (m, 5H), 1.88 (m, 2H), 1.47 - 1.19 (m, 2H). MS : 586.2 (M+l )+.
2-(((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)(3-fluorophenyl) carbamoyl)-4-hydroxypyrrolidine-l-carboxylate (single enantiomer) - Compound 74
1H NMR (400 MHz, CDC13): δ 8.60 (s, 1H), 7.89 (s, 1H), 7.71 (s, 1H), 7.45 - 7.29 (m, 2H), 7.25 - 6.86 (m, 5H), 6.41 (s, 1H), 5.54 (s, 1H), 4.98 (s, 1H), 3.98 (s, 1H), 3.16 - 2.66 (m, 2H), 2.51 (s, 1H), 2.26 (s, 1H), 1.98 (m, 7H), 1.55 (m, 3H). MS: 591.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-N-(3-fluoro - phenyl)-2-oxo-3-(pyrimidin-2-yl)oxazolidine-4-carboxamide (single enantiomer) - Compound 76
Ή NMR (400 MHz, CDC13): δ 8.70 (d, J = 4.7 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.43 - 7.31 (m, 1H), 7.19 (d, J = 7.3 Hz, 1H), 7.13 - 6.86 (m, 5H), 6.46 (s, 1H), 5.58 (d, J = 6.8 Hz, 1H), 5.02 (d, J = 4.4 Hz, 1H), 4.47 (dd, J = 8.7, 5.0 Hz, 1H), 4.24-4.13 (m, 1H), 3.98 (s, 1H), 2.14 - 1.79 (m, 6H), 1.57-1.41 (m, 2H). MS: 588.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-3-(4-cyanopyri - din-2-yl)-N-(3-fluorophenyl)-2-oxooxazolidine-4-carboxamide(single enantiomer) - Compound 77
Ή NMR (400 MHz, CDC13): δ 8.48 (s, 1H), 7.62 (d, J= 7.9 Hz, 1H), 7.33 (d, J = 8.9 Hz, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.10 - 6.85 (m, 5H), 6.44 (d, J= 5.1 Hz, 1H), 6.20 - 6.08 (m, 1H), 5.01 (m, 1H), 4.46 (dd, J = 8.7, 4.7 Hz, 1H), 4.31-4.20 (m, 2H), 3.09 - 2.91 (m, 2H), 2.58 - 2.30 (m, 2H). MS : 584.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-3-(4-cyano - pyridin-2-yl)-N-(3-fluorophenyl)-2-oxooxazolidine-4-carboxamide (single enantiomer) - Compound 78
1H NMR (400 MHz, CDC13): δ 8.55 (s, 1H), 8.50 (t, J= 5.8 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.43 - 7.29 (m, 2H), 7.20 (d, J= 7.6 Hz, 1H), 7.15 - 6.89 (m, 4H), 6.43 (d, J= 4.4 Hz, 1H), 5.54 (d, J = 7.9 Hz, 1H), 5.06 (d, J = 4.7 Hz, 1H), 4.51 (dd, J = 8.8, 5.0 Hz, 1H), 4.25 (m, 1H), 3.98 (s, 1H), 2.19 - 1.74 (m, 6H), 1.49 (m, 2H). MS: 612.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-N-(3-cyano-5- fluorophenyl)-3-(3-cyanophenyl)-2-oxooxazolidine-4-carboxamide(single enantiomer) - Compound 134
1HNMR (400 MHz, CDC13): δ 8.51 - 8.47 (m, 1H), 8.39-8.37 (d, 0.5H), 8.07-7.99 (m, 1H), 7.38 (s, 0.5H),7.33 - 7.31 (m, 1H), 7.26 - 7.22 (m, 1H), 7.08-7.07 (m, 1H), 6.90-6.87 (m, 1H), 6.53- 6.46 (m, 2H), 4.94 - 4.91 (m, 1H), 4.44 - 4.40 (m, 1H). 4.34 - 4.32(m, 1H), 4.28 - 4.23(m, 1H), 3.00 - 2.99(m, 2H), 2.50 - 2.43(m, 2H). MS: 608.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-3-(4- cyanopyri -din-2-yl)-N-(5-fluoropyridin-3-yl)-2-oxooxazolidine-4-carboxamide(single enantiomer) - Compound 135
¾ NMR (400 MHz, CDC1
3): δ 8.58 - 8.28 (m, 3H), 8.08 (d, J= 8.5 Hz, 1H), 7.32 (dd, J= 5.1 , 1.0 Hz, 2H), 7.28 - 7.20 (m, 1H), 7.07 (m, 1H), 6.91 (m, 1H), 6.66 - 6.22 (m, 2H), 5.05 - 4.85 (m, 1H), 4.57 - 4.09 (m, 3H), 3.02 (m, 2H), 2.69 - 2.30 (m, 2H). MS: 585.1 (M+l)
+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-3-(4- cyanopyri -din-2-yl)-N-(5-fluoropyridin-3-yl)-2-oxooxazolidine-4-carboxamide (single enantiomer) - Compound 132
1H NMR (400 MHz, CDC13): δ 8.91 (s, 1H), 8.41 (m, 4H), 8.11 (s, 1H), 7.23 (s, 1H), 7.05 (s, 1H), 6.91 (s, 1H), 6.52 (m, 1H), 6.05 (m, 1H), 4.95 (m, 1H), 4.37 (m, 2H), 3.95 (s, 1H), 1.71 (m, 10H). MS: 613.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-fluoro phenyl)-2-oxo-3-(thiazol-4-yl)oxazolidine-4-carboxamide(single enantiomer) - Compound
1H NMR (400 MHz, CDC13): δ 8.70-8.47 (m, 1H), 7.69-7.52 (m, 1H), 7.49 (d, J= 2.0 Hz, 1H), 7.42-7.26 (m, 1H), 7.25-6.84 (m, 5H), 6.42 (s, 1H), 6.21-6.02 (m, 1H), 5.03 (d, J= 4.6 Hz, 1H), 4.42 (m, 1H), 4.38- 4.05 (m, 2H), 2.98 (m, 2H), 2.64-2.29 (m, 2H). MS : 565.1 (M+l)+.
(4S)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-3-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(2-hydroxypropan-2-yl)phenyl)-2-oxooxazolidine-4- carboxamide (racemic)- Compound 145
1H NMR (400 MHz, CDC13): SS.63 - 8.50 (m, 1H), 8.42 (m, 1H), 7.48 - 7.40 (m, 1H), 7.29 (d, J = 7.0 Hz, 2H), 7.25 - 7.19 (m, 2H), 7.14 - 6.95 (m, 3H), 6.89 (m, 1H), 6.67 (d, J= 6.9 Hz, 1H), 6.54 - 6.42 (m, 1H), 5.11 - 4.96 (m, 1H), 4.51 - 4.40 (m, 1H), 4.32 (d, J= 9.1 Hz, 1H), 4.24 - 4.09 (m, 1H), 3.12 - 2.73 (m, 2H), 1.52 (m, 2H), 1.32 (d, J = 9.0 Hz, 4H). MS: 642.2 (M+l)+. (4S)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-3-(4- cyanopyridin-2-yl)-N-(3-fluorophenyl)-2-oxo-l,3-oxazinane-4-carboxamide (racemic)- Compound 90
Ή NMR (400 MHz, CDC13): δ 8.57 (s, 1H), 8.40(s, 1H), 7.68 (d, J= 8.0 Hz, 1H), 7.25 - 6.91 (m, 8H), 6.48 (s, 1H), 6.25 (s, 1H), 5.08 (s, 1H), 4.51 - 4.46 (m, 1H), 4.31 (m, 2H), 3.01 (m, 2H), 2.53 - 2.50 (m, 2H), 2.29 - 2.13 (m, 2H). MS: 598.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-3-(4-cyanopy - ridin-2-yl)-N-(3,5-difluorophenyl)-2-oxo-l,3-oxazinane-4-carboxamide (single enantiomer) - Compound 133
Ή NMR (400 MHz, CDC13): δ 8.55 (d, J= 5.0 Hz, 1H), 8.34 (s, 1H), 7.54 (d, J= 8.4 Hz, 1H), 7.31 (dd, J= 5.0, 1.1 Hz, 1H), 7.26 - 7.16 (m, 2H), 7.13 - 7.04 (m, 1H), 6.98 (t, J= 6.6 Hz, 2H), 6.72 - 6.63 (m, 1H), 6.49 (s, 1H), 6.44 (d, J= 6.9 Hz, 1H), 5.1 1 (dd, J = 6.4, 3.5 Hz, 1H), 4.51 - 4.22 (m, 3H), 2.98-3.04 (m, 2H), 2.67 - 2.41 (m, 2H), 2.33 - 2.09 (m, 2H).MS: 627.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-3-(4-cyano pyridin-2-yl)-N-(3,5-difluorophenyl)-2-oxo-l^-oxazinane-4-carboxamide(single enantiomer) - Compound 139
¾ NMR (400 MHz, CDC13): δ 8.64 (d, J= 5.0 Hz, 1H), 8.47 (s, 1H), 7.45 (d, J= 7.4 Hz, 1H), 7.38 - 7.30 (m, 2H), 7.24 (d, J= 7.1 Hz, 1H), 7.15-7.12 (m, 1H), 6.81-6.77 (m, 1H), 6.06 (s, 1H), 5.51 (d, J = 7.5 Hz, 1H), 5.05 - 4.88 (m, 1H), 4.62-4.56(m, 1H), 4.42 - 4.30 (m, 1H), 3.87 (s, 1H), 2.35 - 2.15 (m, 2H), 1.97-1.79 (m, 5H), 1.40 (m, 2H). MS: 643.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-3-(4-cyano pyrimidin-2-yl)-N-(3,5-difluorophenyl)-2-oxo-l,3-oxazinane-4-carboxamide(single enantiomer) - Compound 144
H NMR (400 MHz, CDC13): δ 8.96 (d, J= 4.7 Hz, 1H), 7.56 (d, J= 10.0 Hz, 1H), 7.41 (dd, J = 9.7, 6.4 Hz, 2H), 7.24-7.22 (m, 1H), 7.14 - 6.95 (m, 3H), 6.70 (t, J= 8.6 Hz, 1H), 6.52 (s, 1H), 5.53 (d, J= 7.6 Hz, 1H), 4.96 (dd, J = 7.8, 4.0 Hz, 1H), 4.46 (d, J= 8.8 Hz, 1H), 4.31 (dd, J = 10.7, 5.1 Hz, 1H), 3.99 (s, 1H), 2.49 - 2.31 (m, 1H), 2.29 - 2.01 (m, 5H), 1.98 - 1.78 (m, 2H), 1.49 (dd, J= 17.9, 8.5 Hz, 1H). MS: 645.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-3-(4-cyanopy - rimidin-2-yl)-N-(3,5-difluorophenyl)-2-oxo-l,3-oxazinane-4-carboxamide(single enantiomer) - Compound 154
1H NMR (400 MHz, CDC13): δ 8.89 (d, J = 4.8 Hz, 1H), 7.52 (d, J= 8.9 Hz, 1H), 7.40 (d, J = 4.8 Hz, 1H), 7.22 (dd, J= 8.0, 1.2 Hz, 1H), 7.16-7.15 (m, 1H), 7.08 - 6.97 (m, 2H), 6.94 (dd, J= 7.7, 1.5 Hz, 1H), 6.66 (dd, J= 9.7, 7.4 Hz, 1H), 6.56 (s, 1H), 6.43 (d, J= 6.8 Hz, 1H), 4.91 (dd, J = 8.3, 4.5 Hz, 1H), 4.41-4.33 (m, 2H), 4.24-4.20 (m, 1H), 3.06 - 2.86 (m, 2H), 2.66 - 2.42 (m, 2H), 2.39 - 2.25 (m, 1H), 2.24 - 2.12 (m, 1H). MS: 617.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-3-(4-cyano pyridin-2-yl)-N-(5-fluoropyridin-3-yl)-2-oxo-l,3-oxazinane-4-carboxamide(single
enantiomer) - Compound 143
Ή NMR (400 MHz, CDC13): δ 9.08 - 7.79 (m, 3H), 7.62 - 6.70 (m, 5H), 6.50 (m, 2H), 4.95 (m, 1H), 4.62 - 4.03 (m, 3H), 2.99 (s, 2H), 2.51 (s, 2H), 2.18 (m, 2H). MS: 599.1 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-3-(4-cyano - pyridin-2-yl)-N-(5-fluoropyridin-3-yl)-2-oxo-l,3-oxazinane-4-carboxamide (single enantiomer) - Compound 137
1H NMR (400 MHz, CDC13): δ 8.43-8.90 (m, 3H), 8.30 (s, 1H), 7.49-8.13 (m, 1H), 7.29-7.31 (m, 2H), 7.17-7.21 (m, 1H), 6.94-7.08 (m, 2H), 6.45-6.53 (m, 1H), 5.80-593 (m, 1H), 4.96-5.00 (m,
1H), 4.47-4.51 (m, 1H), 4.30 - 4.33 (m, 1H), 3.96-3.98 (m, 1H), 2.09 - 2.28 (m, 6H) , 1.83 - 1.95 (m, 2H), 1.49 - 1.63 (m, 2H). MS: 627.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-3-(4-cyanopyri din-2-yl)-N-(3-fluoro-5-(2-hydroxypropan-2-yl)phenyl)-2-oxo-l,3-oxazinane-4-carboxamide (single enantiomer) - Compound 146
Ή NMR (400 MHz, CDC13): δ 8.56 (t, J= 6.0 Hz, 1H), 8.36 (s, 1H), 7.72 - 7.45 (m, 1H), 7.23 -
7.16 (m, 1H), 7.12 (t, J= 7.1 Hz, 1H), 7.06 - 6.86 (m, 3H), 6.38 (s, 1H), 6.28 (d, J = 6.9 Hz, 1H),
5.17 - 5.01 (m, 1H), 4.50 - 4.44 (m, 1H), 4.30 (m, 2H), 2.99 (d, J= 7.8 Hz, 2H), 2.62 - 2.37 (m, 2H), 2.36 - 2.06 (m, 2H), 1.49 (d, J= 6.2 Hz, 2H), 1.32 (m, 4H). MS: 656.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyri - din-2-yl)-N-(3-fluorophenyl)-6-oxopiperidine-2-carboxamide(single enantiomer) - Compound 55
H NMR (400 MHz, CDC13): δ 8.59 (s, 1H), 8.28 (s, 1H), 7.72 (d, J= 7.2 Hz, 1H), 7.43 - 7.33 (m, 1H)„ 7.26 - 7.12 (m, 2H), 7.11 - 6.96 (m, 2H), 6.89 (dd, J= 8.3, 2.2 Hz, 1H), 6.46 (s, 1H), 6.27 (s, 1H), 5.00 (t, J= 4.6 Hz, 1H), 4.37-4.28 (m, 1H), 3.13 - 2.95 (m, 2H), 2.78 - 2.69 (m, 1H), 2.62 - 2.35 (m, 3H), 2.15 - 2.09 (m, 1H), 2.05 - 1.92 (m, 1H), 1.89 - 1.70 (m, 3H). MS: 596.2 (M+l)+.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyano - pyridin-2-yl)-N-(3-fluorophenyl)-6-oxopiperidine-2-carboxamide (single enantiomer) - Compound 75
H NMR (400 MHz, CDC13): δ 8.60 (s, 1H), 8.31 (s, 1H), 7.73-7.75 (m, 1H), 7.30 (m, 1H), 7.00- 7.17 (m, 5H), 6.87-6.91 (m, 1H), 6.45 (s, 1H), 5.50 (d, J= 7.0 Hz, 1H), 5.00-5.02 (m, 1H), 3.99 (m, 1H), 2.60 - 2.74 (m, 1H), 2.58-2.60 (m, 1H), 2.01 - 2.14 (m, 6H) , 1.83 - 1.92 (m, 4H), 1.42 - 1.46 (m, 3H). MS: 624.2 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-4-fluoro-N-(3-fluorophenyl)-5-oxopyrrolidine-2-carboxamide(single enantiomer) - Compound 151
Ή NMR (400 MHz, CDC13): δ 8.75 (s, 1H), 8.57 (s, 1H), 7.76 (s, 1H), 7.36 (m, 2H), 7.06 (m, 6H), 6.39 (s, 1H), 5.51 (d, J = 6.6 Hz, 1H), 5.12 (m, 1H), 4.82 (s, 1H), 3.91 (m, 1H), 2.69 - 2.26 (m, 2H), 2.05 (m, 6H), 1.53 - 1.38 (m, 2H). MS: 628.2 (M+l)+.
Example 9.Preparation of (2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2- oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3,5-dicyanophenyl)-5-oxopyrrolidine-2-carboxamide (racemic)- Compound 191
Step A:5-Nitroisophthaloyl dichloride.To a solution of 5-nitroisophthalic acid (2.3 g, 11 mmol) in SOCl
2 (6 mL) was added a drop of DMF and the mixture was stirred at reflux for 3hr.The resulting reaction mixture was concentrated to give the crude product which was used directly in the next step.
Step B:5-Nitroisophthalamide. 5-Nitroisophthaloyl dichloride (2.7 g, 9.7 mmol) was added portionwise to a cold solution of NH3 'H20 (40 mL) at 0°C. The reaction mixture was stirred overnight and a white precipitate formed. The mixture was then filtered, washed with excess of water, and dried at 110°C to give the crude product which was used directly in the next step. Step C:5-Aminoisophthalamide.To a solution of 5-nitroisophthalamide (2 g, 9.6 mmol) in MeOH (200 mL) was added Pd/C (200 mg). The reaction was stirred overnight under a hydrogen atmosphere. The suspension was filtered and the filtrate was concentrated to afford the desired product which was used directly in the next step.
StepD: 5-((2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl) pyridin-2-yl)-5-oxopyrrolidine-2-carboxamido)isophthalamide.A mixture of 2- chlorobenzaldehyde (1.0 mL, 7.3 mmol) and 5-aminoisophthalamide(1.3 g, 7.3 mmol) was stirred at room temperaturefor 30 min under N2, followed by addition of (S)-l-(4-cyanopyridin- 2-yl)-5-oxopyrrolidine-2-carboxylic acid (1.7 g, 7.3 mmol). After stirring for 10 min, 1 ,1 - difluoro-3-isocyanocyclobutane (854 mg, 7.3 mmol) was added. The mixture was then stirred overnight and filtered and purified by a standard method to give the title product.
Step E: (2S)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocychbutyl)amino)-2-oxoethyl)l-(4-cyano pyridin-2-yl)-N-(3,5-dicyanophenyl)-5-oxopyrrolidine-2-carboxamide. To a mixture of5-((2S)- N-(l -(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)l-(4-cyano pyridin-2-yl)-5- oxopyrrolidine-2-carboxamido)isophthalamide (850 mg,1.3 mmol) in pyridine (0.62 mL,7.8 mmol) and DCM (10 mL) was added TFAA (0.9 mL, 6.5 mmol). The reaction solution was stirred at room temperature overnight. The resulting mixture was concentrated and the residue was purified by a standard method to afford the titleproduct. H NMR (400 MHz, CDC13): δ 8.77 (s, 1H), 8.62 - 8.42 (m, 2H), 7.87 (s, 1H), 7.75 (s, 1H), 7.40 (d, J= 7.8 Hz, 1H), 7.31 (d, J= 4.2 Hz, 1H), 7.25 (d, J= 8.1 Hz, 1H), 7.10 (t, J= 7.3 Hz, 1H), 6.92 (d, J= 7.5 Hz, 1H), 6.47 (s, 1H), 6.11 (d, J= 6.6 Hz, 1H), 4.73 (dd, J = 9.4, 2.7 Hz, 1H), 4.35 (s, 1H), 3.14 - 2.82 (m, 3H), 2.68- 2.31 (m, 3H), 2.19 (m, 1H), 2.09-1.91 (m, 1H). MS : 614.1 (M+l)+.
The following analogs were synthesized via the procedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic(heteroaromatic) ring using the reagents and solvents set forth above, and purified via standard methods.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(3,5-dicyanophenyl)-5-oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 153
1H NMR (400 MHz, CDC13): δ 8.74 (s, 1H), 8.53 (m, 2H), 7.81 (m, 2H), 7.48 - 7.16 (m, 4H), 7.09 (t, J= 7.5 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.46 (s, 1H), 6.17 (d, J = 6.7 Hz, 1H), 4.72 (dd, J= 9.1, 2.3 Hz, 1H), 4.35 (s, 1H), 3.18 - 2.71 (m, 3H), 2.68 - 1.83 (m, 5H). MS : 614.1 (M+l)+.
Example 10.Preparation of(S)-tert-butyl 3-(((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)(3,5-difluorophenyl)carbamoyl)-5-oxopiperazine-l- carboxylate(single enantiomer) - Compound 97
Compound 97 was synthesized via the UGI reaction procedure set forth herein, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic(heteroaromatic) ring and purified viastandard methods.
1H NMR (400 MHz, CDC13): δ 8.75 - 8.44 (m, 2H), 7.81-7.41 (m, 1H), 7.46-7.35 (m, 2H), 7.24 (t, J= 7.2 Hz, 1H), 7.16 - 6.97 (m, 2H), 6.84-6.75 (m, 2H), 6.43 - 5.82 (m, 1H), 5.09-4.98 (m, 1H), 4.77-4.73 (m, 1H), 4.48 (d, J= 13.5 Hz, 1H), 4.27-4.07 (m, 2H), 3.45-2.76 (m, 4H), 1.54 (s, 9H). MS : 613.2 (M+l)+.
Example 11. Preparation of (3S)-tert-butyl 3-((l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl) amino)-2-oxoethyl)(3,5-difluorophenyl)carbamoyl)-4-(4-cyanopyrimidin-2-yl)-5- oxopiperazine- -carboxylate (racemic)- Compound 98
A mixture of (3S)-tert-butyl3-((l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl) (3,5-difluorophenyl)carbamoyl)-5-oxopiperazine-l-carboxylate(200 mg, 0.326 mmol), 2- bromopyrimidine-4-carbonitrile (0.489 mmol), Pd2(dba)3 (30.2 mg, 0.0323 mmol), XantPhos (19.1 mg, 0.03 mmol) and Cs2C03 (148.7 mg, 0.46 mmol) in 1 ,4-dioxane (10 mL) was stirred at 80°C for 3 hr under N2. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated and the residue was purified bya standard method to afford the desired product. 1H NMR (400 MHz, CDC13): δ 8.97 (d, J = 4.3 Hz, 1H), 7.85-7.55 (d, 1H), 7.51 -7.39 (m, 2H), 7.25 (t, J= 7.6 Hz, 1H), 7.13-6.26 (m, 6H), 5.91 (d, J= 7.6 Hz, 1H), 4.92-4.08 (m, 5H), 3.38 (t, J = 14.9 Hz, 1H), 3.02 (s, 2H), 2.83-2.22 (d, 2H), 1.61 (s, 9H). MS : 716.1 (M+l)+.
The following analogs were synthesized via the procedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocynide and halo-substitutedaromatic ring or heterocyclic(heteroaromatic) ring using the reagents and solvents set forth above, and purified viastandard methods.
(S)-tert-Butyl 3-(((S)-l-(2-chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)(3,5- difluorophenyl)carbamoyl)-4-(4-cyanopyrimidin-2-yl)-5-oxopiperazine-l-carboxylate (chiral)- Compound 93
¾ NMR (400 MHz, CDC1
3): δ 8.96 (d, J= A3 Hz, 1H), 7.83 (s, 1H), 7.43 (m, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.08 - 6.62 (m, 4H), 6.63 - 6.37 (m, 1H), 5.93 (m, 1H), 4.85 (t, J = 3.6 Hz, 1H), 4.63 - 4.23 (m, 2H), 4.16 (m, 1H), 3.93 (s, 1H), 3.43 (m, 1H), 2.24 - 1.91 (m, 5H), 1.79 (m, 3H), 1.60 (m, 1H). MS : 744.2 (M+l)
+.
(3S)-tert-Butyl 3-((l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)(3- fluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)-5-oxopiperazine-l-carboxylate(single enantiomer) - Compound 89
H NMR (400 MHz, DMSO-d6): δ 8.80-8.37 (m, 1H), 8.05-7.57 (m, 1H), 7.58-7.31 (m, 3H), 7.21 (s, 1H), 7.16-6.89 (m, 3H), 6.90-6.68 (m, 1H), 6.67-6.30 (m, 1H), 6.22 -5.84 (m, 1H), 5.09- 4.87(m, 1H), 5.83-4.57 (m, 1H), 4.50 (m, 1H), 4.25 (s, 1H), 4.08 (m, 1H), 3.50-2.70 (m, 4H), 2.60-2.10 (m, 1H), 1.70 (s, 2H), 1.54 (m, 1H). MS : 697.2 (M+l)+.
Example 12.Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyrimidin-2-yl)-N-(3,5-difluorophi oxopiperazine-2-carboxamide(single enantiomer) - Compound 99
TFA (0.3 mL) was added to a solution of (S)-tert-butyl 3-(((R)-l -(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)(3,5-difluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)-
5-oxopiperazine-l-carboxylate(60 mg, 0.08 mmol) in DCM (1.0 mL) at 0°C. The mixture was warmed toroom temperatureand stirred for lhr, and then concentrated. The residue was purified by a standard method to give the desired product. H NMR (400 MHz, CDC13): δ 8.94 (t, J= 4.6
Hz, 1H), 7.48-7.36 (m, 3H), 7.21 (m, J= 7.8, 1.5 Hz, 1H), 7.12-6.94 (m, 3H), 6.71-6.55 (m, 2H),
6.05 (d, J= 6.7 Hz, 1H), 4.73 (q, J= 4.0, 2.1 Hz, 1H), 4.36 (d, J = 6.7 Hz, 1H), 3.77-3.65 (m, 2H), 3.50-3.35 (m, 1H), 3.18 (m, 1H), 3.12 - 2.96 (m, 2H), 2.64-2.35 (m, 2H). MS : 616.1 (M+l)+.
The following compound was synthesized via the procedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic (heteroaromatic) ring using the reagents and solvents set forth above, and purified via standard methods.
(S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyano pyridin-2-yl)-N-(3,5-difluorophenyl)-6-oxopiperazine-2-carboxamide(single enantiomer) -
Compound 100
Ή NMR (400 MHz, CDC13): δ 8.68 - 8.28 (m, 1H), 7.61 - 7.28 (m, 2H), 7.20 (dd, J= 7.9, 1.3 Hz, OH), 7.02 - 6.90 (m, 1H), 6.66 (tt, J = 8.6, 2.3 Hz, 1H), 6.49 (d, J = 2.7 Hz, OH), 6.09 (m, 1H), 4.90 (dd, J = 3.8, 2.0 Hz, 1H), 4.42 - 4.16 (m, 1H), 3.71 (m, 1H), 3.50 - 3.23 (m, 1H), 3.18 - 2.78 (m, 2H), 2.63 - 2.13 (m, 2H). MS : 615.2 (M+l)+.
Example 13.(S)-4-Acetyl-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxo ethyl)-l-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-6-oxopiperazine-2-carboxamide(single enantiomer) - Compound 92
To a solution of (3S)-tert-butyl 3-((l -(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2- oxoethyl)(3-fluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)-5-oxopiperazine-l -carboxylate (100 mg, 0.14 mmol) in DCM (3 mL) was added TFA dropwise (1 mL) at 0°C. The mixture was stirred at room temperature for 2hr and then concentrated. The residue was dissolved in DCM
and cooled to 0°C. DIPEA (0.055 mL, 0.34 mmol) was added to the mixture followed by Ac20 (0.031 mL, 0.34 mmol) at 0°C. Then the mixture was stirred at room temperature for 2hr. The solution was concentrated and the residue was purified by a standard method to afford the desired product. 1H NMR (400 MHz, CDC13): δ 8.54 (s, 2H), 7.70-744 (m , 2H), 7.36 (m, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.14 - 6.99 (m, 2H), 6.94 (t, J = 7.4 Hz, 1H), 6.80 (s, 1H), 6.66 (d, J = 7.8 Hz, 1H), 6.58 - 6.42 (m, 1H), 5.09 (dt, J= 5.2, 3.1 Hz, 1H), 4.93 (m, 1H), 4.63 (m, 1H), 4.54 - 4.41 (m, 1H), 4.35-4.31 (m, 1H), 3.16 (s, 1H), 3.12 - 2.96 (m, 3H), 2.86 (s, 1H), 2.25 (s, 3H). MS: 639.2 (M+l)+.
Example 14. Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-4-cyclopropyl-N-(3,5- difluoropheny -6-oxopiperazine-2-carboxamide (single enantiomer) - Compound 106
TFA (0.3 mL) was added to a solution of (S)-tert-butyl 3-(((R)-l -(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)(3,5-difluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)- 5-oxopiperazine-l-carboxylate (60 mg, 0.084 mmol) in DCM (1.0 mL) at 0°C. The mixture was stirred at room temperaturefor lhr then concentrated. The residue was dissolved in MeOH (2 mL) followed by addition of (l-ethoxycyclopropoxy)trimethylsilane(88 mg, 0.50 mmol), AcOH (50 mg, 0.84 mmol) and NaBH3(CN)(27mg, 0.42 mmol). The resulting suspension was stirred at 80°C under N2 for 1.5hr. The reaction mixture was partitioned betweenEtOAc and H20. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated. The residue was purified by a standard method to afford the desired product. H NMR (400 MHz, CDC13): 57.64 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 5.3 Hz, 2H), 7.19 (s, 1H), 7.07 (s, 3H), 6.66 (s, 1H), 6.32 (s, 1H), 6.09 (m, 1H), 5.09 (s, 1H), 4.28 (s, 1H), 3.76 - 3.59 (m, 1H), 3.46 - 3.33 (m, 1H), 3.08-2.89 (m, 4H), 2.59 - 2.31 (m, 2H), 0.94 (s, 1H), 0.61-0.37 (m, 4H). MS: 655.2 (M+l)+.
Example 15. Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-4- methyl-6-oxopiperazine-2-carboxamide single enantiomer - Compound 101
TFA (0.6 mL) was added to a solution of (3 S)-tert-butyl 3-((l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)(3,5-difluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)- 5-oxopiperazine-l-carboxylate (30 mg, 0.042 mmol) in DCM (2 mL) at 0°C. The mixture was stirred at room temperature for lhr and then concentrated. The residue was dissolved in MeCN (4 mL) followed by addition of K2C03 (10 mg, 0.072 mmol) and iodomethane (2 mL). The resulting mixture was stirred at room temperaturefor 2hr and then concentrated. The residue was purified by a standard method to afford the desired product. H NMR (400 MHz, CDC13) : δ 8.60 (m, 2H), 7.80 (s, 1H), 7.45 (d, J = 7.9 Hz, lH), 7.33 (m, 1H), 7.07 (d, J= 4.3 Hz, 2H), 6.74 (t, J = 8.6 Hz, 1H), 6.48 - 5.91 (m, 3H), 4.92 (t, J = 4.7 Hz, 1H), 4.20 (m, lH), 3.61 - 3.40 (m, 1H), 3.14 (m, 1H), 3.02 - 2.77 (m, 3H), 2.71 (m, 1H), 2.42 - 2.26 (m, 5H), 2.04 (d, J= 9.0 Hz, 1H). MS: 629 (M+1) +.
Example 16. Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-4-(2- hydroxyethyl)-6-oxopiper azine-2-carboxamide(single enantiomer) - Compound 107
To a solution of (S)-tert-butyl 3-(((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl) amino)-2- oxoethyl)(3,5-difluorophenyl)carbamoyl)-4-(4-cyanopyridin-2-yl)-5-oxopiperazine-l -carboxyl ate (30 mg,0.04 mmol) in DCM (3 mL) was added TFA (1 mL) at 0°C.The mixture was stirred at
room temperature for 1 hr and concentratedin vacuo. The residue was dissolved in EtOH (3 mL) followed by addition of TBAI (16 mg, 0.04 mmol), Et3N (10 mg, 0.1 mol) and 2-bromoethanol (7 mg, 0.056 mmol). The resulting mixture was stirred at 85 °C for 3 hr and then filtered. The filtrate was concentrated and the residue was purified by a standard method to afford the desired product. Ή NMR (400 MHz, CDC13): δ 8.96 (t, J= 4.6 Hz, 1H), 7.53-7.36 (m, 3H), 7.23 (m, J = 7.8, 1.5 Hz, 1H), 7.14-6.94 (m, 3H), 6.68 (m, J = 8.6, 2.3 Hz, 1H), 6.60 (d, J= 3.1 Hz, 1H), 6.07 (d, J= 6.7 Hz, 1H), 4.75 (q, J = 4.0, 2.1 Hz, 1H), 4.38 (d, J= 6.7 Hz, 1H), 3.78-3.67 (m, 2H), 3.39 (m, 1H), 3.26-2.92 (m, 3H), 2.67-2.36 (m, 2H). MS : 659.2 (M+l)+.
The following compound was synthesized via the procedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic(heteroaromatic) ring using the reagents and solvents set forth above, and purified viastandard methods.
Compound 104
Ή NMR (400 MHz, CDC13): δ 8.60-8.56 (m, 2H), 7.47-7.28 (m, 3H), 7.22-7.01 (m, 4H), 6.72- 6.67 (m, 1H), 6.54-6.44 (m, 2H), 5.24 (m, 1H), 4.37-4.13 (m, 3H), 3.63-2.97 (m, 8H), 2.44-2.06 (m, 2H), 1.34-1.28 (m, 3H). MS : 701.2 (M+l)+.
Example 17. Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-l-(5-cyanooxa-zol-2-yl)-N-(3,5-difluorophenyl)-5- oxopyrrolidine-2-carboxamide (single enantiomer) - Compound 162
Step A : Oxazole-5-carboxamide.Eihy\ oxazole-5-carboxylate (2 g, 14.2 mmol) was dissolved in NH3 solution (7M in MeOH, 25 mL). The solution was stirred at room temperature for 2hr and
filtered. The solid was dried to give the desired product (1.5 g, 92% yield) as a whitepowder which was used directly in the next step.
Step B: 2-Iodooxazole-5-carboxamide.Oxazo\e-5-carboxamide (560 mg, 5.0 mmol) was dissolved in anhydrous THF (7.5 mL) and flushed with N2. The solution was cooled to -42 °C and treated with fresh LiHMDS (15 mL, 1M in THF). The solution became dark yellow wasstirred for 20 min and followed by the addition of a solution of ZnCl2 (30 mL, 0.5M in THF). The reaction was warmed to 0 °C for lhr. Aftersolid iodine (1.65g, 6.5mmol) was added, the reaction mixture was stirred at room temperaturefor another lhr and then poured into saturated Na2S203 solution containing 25% aq. NH3 solution. The resulting mixture was extracted with EtOAc (3 x 30 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated. The resulting residue was purified by a standard method to give the desired product.MS : 239.0 (M+l)+.
Step C: 2-((S)-2-(((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocycfabutyl)amino)-2-ox
difluorophenyl)carbamoyl)-5-oxopyrrolidin-l-yl)oxazole-5-carboxarmde. The product was prepared by the general procedure for the Buchwald reaction.1H NMR (400 MHz, CDC13): δ 7.59 (s, 1H), 7.53 (s, 1H), 7.37 (d, J = 7.9 Hz, 1H), 7.20 (t, J = 7.0 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.96 (d, J = 7.9 Hz, 2H), 6.68 (t, J = 8.7 Hz, 1H), 6.46 (s, 1H), 6.36 (d, J= 6.4 Hz, 1H), 5.68 (s, 1H), 4.82 (dd, J = 9.3, 2.6 Hz, 1H), 4.33 (s, 1H), 4.16 - 4.09 (m, 1H), 3.03-3.00 (m, 2H), 2.90 - 2.77 (m, 1H), 2.62 - 2.35 (m, 3H), 2.29-2.28 (m, 1H), 2.19 - 2.08 (m, 1H). MS: 608.1 (M+l)+.
Step D : (S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluoro(yclobufyl)amino)-2-oxoethyl)-l-(5- cyanooxazol-2-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxarmde.
2-((S)-2-(((S)-l -(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)(3,5-difluoro- phenyl)carbamoyl)-5-oxopyrrolidin-l-yl)oxazole-5-carboxamide (100 mg, 0.16 mmol) was dissolved in DCM (3 mL) and dry pyridine (0.8 mL). TFAA (0.1 mL) was addedand the reaction solution was stirred for 25 min at room temperature and then concentrated in vacuo. The residue was dissolved in EtOAc and washed with H20, saturated aq. NaHC03 and brine. The organic phase was separated, dried over anhydrous Na2S04, and concentrated. The residue was purified by a standard method to give the desired product. H NMR (400 MHz, CDC13): δ 7.63 (s, 1H),
7.55 (d, J = 7.0 Hz, 1H), 7.41 (d, J = 7.1 Hz, lH), 7.25 (td, J = 7.8, 1.5 Hz, 1H), 7.08 (t, J = 7.6
Hz, 1H), 6.98 - 6.91 (m, 1H), 6.80 (d, J = 6.7 Hz, 1H), 6.71 (dd, J = 9.7, 7.4 Hz, 1H), 6.49 (s, 1H), 5.97 (d, J = 6.8 Hz, 1H), 4.80 (dd, J = 9.3, 2.8 Hz, 1H), 4.36 (s, 1H), 3.06-3.03 (m, 2H), 2.92 - 2.79 (m, 1H), 2.62 - 2.29 (m, 4H), 2.18-2.12 (m, 1H). MS: 590.1 (M+l)+.
Example 18.Preparation of(2S,4R)-N-(l-(2-chlorophenyl)-2-((3,3- difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-cyano-phenyl)-l-(4-cyanopyridin-2-yl)-4- hydroxy-5-oxopyrrolidine-2-carboxamide (racemic) - Compound 170
Step A: (2S,4R)-l-tert-Butyl 2-methyl 4-((tert-butyldimethylsilyl)oxy)pyrrolidine-l,2- dicarboxylate. Imidazole (2.8 g, 40.8 mmol) was added to a solution of (2S,4R)-l-tert-butyl 2- methyl 4-hydroxypyrrolidine-l,2-dicarboxylate (5.0 g, 20.4 mmol) and TBSCl (4.6 g, 30.6 mmol) in anhydrous DMF (100 mL). The mixture was stirred at room temperatureovernight and then partitioned between EtOAc and H20. The organic layer was separated, washed with aq. LiCl (10%) and brine, dried over anhydrous Na2S04, and then concentrated. The residue was purified by column chromatography to afford the desired product as a colorless oil. MS: 360.2 (M+l)+. Step B : (2S,4R)-J -tert-Butyl 2-methyl 4-((tert-butyldimethylsilyl)oxy)-5-oxopyrrolidine-l,2- dicarboxylate. To a solution of NaI04 (7.5 g, 35.0 mmol) in water (80 mL) was added Ru02 (370 mg, 2.8 mmol) under the atmosphere of nitrogen. The resulting green-yellow solution was stirred for 5min followed by addition of (2S,4R)-l-tert-butyl-2-methyl4-((tert-butyldimethyl
silyl)oxy)pyrrolidine-l ,2-dicarboxylate (5.0 g, 14.0 mmol) in EtOAc(44 mL) in one portion. The
mixture was stirred at room temperatureovernight. The resulting mixture was then diluted with EtOAc and filtered through a pad of Celite. The organic layer was separated and washed with saturated aq. NaHS03, which resulted in precipitation of Ru black. The organic layer was then washed with brine and dried over anhydrous Na2S04. Evaporation of the solvent gave the desired productas a colorless oil. MS: 374.2 (M+l)+.
Step C: (2S,4R)-4-((tert-But ldimethylsilyl)oxy)-5-oxopyrrolidine-2-carboxylic acid. TFA (6 mL) was added to a solution of (2S,4R)-l -tert-butyl 2-methyl 4-((tert-butyldimethylsilyl)oxy)-5- oxopyrrolidine-l,2-dicarboxylate (2.5 g, 6.68 mmol) in DCM (18 mL) at 0°C. The mixture was stirred at room temperaturefor lh then concentrated. The residue was dissolved in
MeOH/THF(l OmL/1 OmL) followed by addition of a solution of LiOH (842 mg, 20.1 mmol) in water(5 mL). The resulting mixture was stirred at room temperature for lh and then partitioned between EtOAc and H20. The aqueous layer was separated and then adjusted to pH=6 with 1 N HC1 aq. and extracted with EtOAc (3 x 20 mL). Combined organic layers were washed with brine, dried over anhydrous Na2S04, and then concentrated to afford the desired product. 1H NMR (400 MHz, DMSO-d6): δ 12.87 (s, 1H), 8.17 (s, 1H), 4.21 (t, J= 8.0 Hz, 1H), 4.02 (d, J = 8.4 Hz, 1H), 2.39 - 2.23 (m, 1H), 2.09 (m, 1H), 0.84 (s, 9H), 0.07 (s, 6H). MS: 260.1 (M+l)+. Step D: The same as general procedure for UGI reaction set forth herein.
Step E: The same as general procedure for Buchwald reaction set forth herein.
Step F: (2S,4R)-N-(l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)andno)-2-oxoethyl)-N-(3 cyano^henyl)-l-(4-cyanopyridin-2-yl)^-hydroxy-5-oxopyrrolidine-2-carboxarmde. TBAF in THF(1N, 0.3 mL) was added to a solution of (2S,4R)-4-((tert-butyldimethylsilyl)oxy)-N-(l-(2- chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-N-(3-cyanophenyl)-l-(4-cyano pyridin-2-yl)-5-oxopyrrolidine-2-carboxamide (0.15 mmol) in THF at 0 °C and the reaction solution was stirred at this temperature for 20 min. The resulting mixture was concentrated and the residue was purified by a standard method to afford the desired product. H NMR (400 MHz, CDC13): δ 8.82 - 8.43 (m, 2H), 8.40 - 8.17 (m, 1H), 7.63 - 7.30 (m, 3H), 7.26 - 6.66 (m, 4H), 6.68 - 6.34 (m, 2H), 6.65 - 6.31 (m, 2H), 4.87 - 4.56 (m, 2H), 4.23 (m, 1H), 4.01 - 3.76 (m, 1H), 3.15 - 1.96 (m, 6H). MS: 605.1 (M+l)+.
The following analogs were synthesized via the procedure set forth herein, using the appropriate aldehyde, amine, carboxylic acid, isocynide and halo-substitutedaromatic ring or
heterocyclic(heteroaromatic) ring using the reagents and solvents set forth herein, and purified via various standard methods.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyano -pyridin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 113
H NMR (400 MHz, CDC13): δ 8.70 (m, 1H), 8.53 (s, 1H), 7.72 (d, J= 7.5 Hz, 1H), 7.32 (d, J = 4.9 Hz, 2H), 7.18 (d, J = 6.0 Hz, 1H), 7.09 - 6.85 (m, 4H), 6.43 (s, 1H), 6.20 (d, J= 5.3 Hz, 1H), 4.89 (s, 1H), 4.74 (t, J= 9.2 Hz, 1H), 4.37-4.32 (m, 1H), 3.40 (s, 1H), 3.11 - 2.87 (m, 2H), 2.77 - 2.14 (m, 3H), 2.03-1.91 (m, 1H). MS: 598.1 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide- Compound 120
Ή NMR (400 MHz, CDC13): δ 8.98 (d, J= 4.4 Hz, 1H), 7.70 (s, 1H), 7.39 (d, J= 4.9 Hz, 2H), 7.20 - 6.86 (m, 4H), 6.50 (s, 1H), 5.75 (s, 1H), 5.35 (s, 1H), 4.92 - 4.63 (m, 2H), 4.34 (s, 1H), 2.91 (m, 3H), 2.21 (m, 4H). MS: 599.1 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyano pyridin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 121
¾ NMR (400 MHz, CDC13): δ 8.78 (s, 1H), 8.54 (s, 1H), 7.77 (d, J= 8.1 Hz, 1H), 7.45 - 7.30 (m, 2H), 7.25 - 6.83 (m, 5H), 6.42 (s, 1H), 5.49 (d, J = 7.4 Hz, 1H), 4.83 (m„ 2H), 4.00 (s, 1H), 3.02 (s, 1H), 2.74 (m, 1H), 2.25 - 1.74 (m, 7H), 1.56 - 1.33 (m, 2H). MS : 626.2 (M+l)+.
(2S,4R)-N-((R)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 122
1H NMR (400 MHz, CDC13): δ 9.00 (d, J = 4.8 Hz, 1H), 7.83 (m, 1H), 7.42 (t, J = 6.6 Hz, 2H), 7.22 (m, 2H), 7.18 - 7.08 (m, 1H), 7.08 - 6.67 (m, 2H), 6.17 (m, 1H), 5.70 (d, J= 7.6 Hz, 1H), 4.93 - 4.66 (m, 2H), 3.88 (d, J= 7.7 Hz, 1H), 3.37 (s, 1H), 2.71 (m, 1H), 2.03 (m, 5H), 1.88 - 1.64 (m, 4H). MS : 627.2 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyano pyrimidin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide- Compound 123
1H NMR (400 MHz, CDC13): δ 8.99 (d, J= 4.4 Hz, 1H), 7.74 (d, J= 7.9 Hz, 1H), 7.47 - 7.29 3H), 7.08 (m, 6H), 6.51 (s, 1H), 5.61 (s, 1H), 4.81 (m, 2H), 4.02 (d, J = 7.2 Hz, 1H), 3.38 (s, 2.89 - 2.65 (m, 1H), 2.23 - 1.81 (m, 8H), 1.58 - 1.48 (m, 1H). MS : 627.2 (M+l)+.
(2S,4R)-N-((R)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cya nopyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 114
1H NMR (400 MHz, CDC13): δ 8.71 (d, J = 5.8 Hz, 1H), 8.64 - 8.50 (m, 1H), 7.94 - 7.56 (m, 1H), 7.47 - 7.31 (m, 2H), 7.29 (d, J= 2.2 Hz, 1H), 7.26 - 7.18 (m, 1H), 7.16 - 6.95 (m, 2H), 6.88 - 6.65 (m, 1H), 6.44 - 6.35 (m, 1H), 6.29 (s, 1H), 6.11 (d, J= 6.7 Hz, 1H), 4.77 (m, 2H), 4.40 - 4.08 (m, 1H), 3.27 (s, 1H), 3.09 - 2.58 (m, 3H), 2.54 - 2.12 (m, 2H), 2.10 - 1.95 (m, 1H). MS: 616 (M+1)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cya nopyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 115
1H NMR (400 MHz, MeOD): δ 8.65 - 8.50 (m, 2H), 7.54 (d, J= 9.5 Hz, 1H), 7.43 - 7.32 (m, 1H), 7.22 - 7.12 (m, 2H), 7.03 (m, 1H), 6.97 - 6.87 (m, 1H), 6.84 - 6.75 (m, 2H), 6.36 (d, J = 8.5 Hz, 1H), 4.89 (d, J= 8.6 Hz, 1H), 4.65 - 4.49 (m, 2H), 4.13 (m, 1H), 2.93 - 2.72 (m, 2H), 2.57 - 2.26 (m, 3H), 1.85 (m, 1H). MS: 616.1 (M+l)+.
(2S,4R)-N-((R)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 116
Ή NMR (400 MHz, CDC13): δ 8.98 (t, J = 5.0 Hz, 1H), 7.88 (s, 1H), 7.88 (s, 1H), 7.50 - 7.37 (m, 2H), 7.33 - 7.20 (m, 2H), 7.19 - 7.06 (m, 2H), 6.83 - 6.66 (m, 1H), 6.48 (m, 2H), 6.27 (s, 1H), 4.23 (s, 1H), 3.32 (s, 1H), 2.87 (m, 2H), 2.66 (m, 1H), 2.35 - 2.02 (m, 3H). MS: 617.1 (M+l)+. (2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide- Compound 117
Ή NMR (400 MHz, MeOD): δ 8.88 (d, J = 4.9 Hz, 1H), 7.56 (m, 2H), 7.34 (dd, J= 8.0, 1.1 Hz, lH), 7.16 (td, J = 7.8, 1.6 Hz, 1H), 7.09 - 7.00 (m, 1H), 6.98 - 6.85 (m, 2H), 6.81 (m, 1H), 6.42 (s, 1H), 4.87 (d, J = 8.8 Hz, 1H), 4.59 - 4.42 (m, 2H), 4.27 - 4.09 (m, 1H), 2.98 - 2.74 (m, 2H), 2.46 (m, 3H), 2.02 - 1.76 (m, 1H). MS: 617.1 (M+l)+.
(2S,4R)-N-((R)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 124
Ή NMR (400 MHz, CDC13): δ 8.71 (s, 1H), 8.64 (d, J= 5.0 Hz, 1H), 7.79 (s, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.35 (dd, J= 5.0, 1.0 Hz, 1H), 7.30 - 7.24 (m, 1H), 7.16 (d, J= 6.3 Hz, 1H), 7.14 - 7.05 (m, 1H), 6.79-6.68 (m, 2H), 6.27 (s, 1H), 5.87 (d, J = 7.5 Hz, 1H), 4.82 (t, J = 6.9 Hz, 1H),
4.74 (t, J= 9.2 Hz, 1H), 3.90 - 3.71 (m, 1H), 3.27 (s, 1H), 2.65 (m, 1H), 2.15 - 1.72 (m, 8H), 1.57-1.43 (m, 1H). MS: 644.2 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide (single enantiomer)- Compound 125
1H NMR (400 MHz, CDC13): δ 8.83 - 8.47 (m, 2H), 7.62 (d, J= 8.0 Hz, 1H), 7.38 (d, J= 8.0 Hz, 1H), 7.32(d, J= 5.0 Hz, 1H), 7.21 (t, J= 7.1 Hz, 1H), 7.05 (t, J= 7.5 Hz, 1H), 6.98 (d, J= 7.7 Hz, 1H), 6.85 (d, J = 7.7 Hz, 1H), 6.68 (t, J = 8.6 Hz, 1H), 6.40 (s, 1H), 5.62 (d, J= 7.7 Hz, 1H), 4.96 - 4.70 (m, 2H), 4.01 (d, J= 7.6 Hz, lH), 3.37 (s, 1H), 2.70 (m, 1H), 2.14 - 1.74 (m, 8H), 155-1.41(m, 1H). MS: 644.2 (M+l)+.
(2S,4R)-N-((R)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide (single enantiomer)- Compound 126
1H NMR (400 MHz, CDC13): δ 8.98 (dd, J = 4.7, 2.1 Hz, 1H), 7.63 (d, J = 7.3 Hz, 1H), 7.50 - 7.33 (m, 2H), 7.28 - 6.87 (m, 3H), 6.84 - 6.38 (m, 2H), 6.19 (s, 1H), 5.82 (d, J = 7.6 Hz, 1H), 4.94 - 4.65 (m, 2H), 3.86 (d, J= 7.5 Hz, 1H), 3.57-3.49 (m, 1H), 2.68 (m, 1H), 2.16 - 1.86 (m, 6H), 1.81-1.77 (m, 2H). MS: 645.2 (M+l)+.
(2S,4R)-N-((S)-l-(2-Chlorophenyl)-2-(4,4-difluorocyclohexylamino)-2-oxoethyl)-l-(4- cyanopyrimidin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 127
¾NMR (400 MHz, CDC13): δ 8.99 (d, J= 4.8 Hz, 1H), 7.62 (d,J= 8.7 Hz, 1H), 7.49 - 7.35 (m, 2H), 7.22 (td,J=7.8, 1.5 Hz, 1H), 7.07 (t,J=7.1 Hz, 1H), 6.98 (dd,J=7.8, 1.3 Hz, lH), 6.91 (d, J = 8.2 Hz, 1H), 6.72 (tt, J = 8.6, 2.2 Hz, 1H), 6.48 (s, 1H), 5.64 (d,J= 7.7 Hz, 1H), 4.94 - 4.69 (m, 2H), 4.11-3.91 (m, 1H), 3.46 (s, 1H), 2.79 (m, 1H), 2.19-1.85 (m, 7H), 1.61 - 1.40 (m, 2H). MS: 645.2 (M+l)+.
(2S,4R)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-N-(3-cyano-5- fluorophenyl)-l-(4-cyanopyridin-2-yl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide
(racemic)- Compound 169
H NMR (400 MHz, CDC13): δ 8.87-8.72 (m, 1H), 8.67 - 8.48 (m, 1H), 8.26 - 8.01 (m, 1H), 7.56-7.30 (m, 4H), 7.27-7.17 (m, 1H), 7.10 (m, 1H), 6.95 (t,J=7.3 Hz, 1H), 6.52-6.28 (m, 1H), 6.21 - 5.95 (m, 1H), 4.88 - 4.64 (m, 2H), 4.30 (m, 1H), 3.21 - 2.81 (m, 3H), 2.74 - 2.19 (m, 3H), 2.13 - 1.91 (m, 1H). MS: 623.1 (M+l)+.
(2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyano- pyri-din-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 118
Ή NMR (400 MHz, CD
3OD): δ 8.97 (d, J= 4.7 Hz, 1H), 7.81 - 7.62 (m, 2H), 7.41 - 7.35 (m, 2H), 7.26 - 6.96 (m, 5H), 6.46 (d, J = 12.0 Hz, lH), 4.81 - 4.75 (m, 1H), 4.37 - 4.28 (m, lH), 4.25 - 4.15 (m, 1H), 2.91 (s, 2H), 2.60 - 2.37 (m, 3H), 2.00 - 1.87 (m, 1H). MS: 598.1 (M+l)
+. (2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyano- pyrimidin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 119
1H NMR (400 MHz, CD3OD): δ 8.97 (d, J = 4.7 Hz, 1H), 7.81 - 7.62 (m, 2H), 7.41 - 7.35 (m, 2H), 7.26 - 6.96 (m, 5H), 6.46 (d, J = 12.0 Hz, lH), 4.81 - 4.75 (m, 1H), 4.37 - 4.28 (m, lH), 4.25 - 4.15 (m, 1H), 2.91 (s, 2H), 2.60 - 2.37 (m, 3H), 2.00 - 1.87 (m, 1H). MS: 599.1 (M+l)+. (2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyano -pyridin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 172
1H NMR (400 MHz, CDC13): δ 8.87 - 8.57 (m, 2H), 7.96 (s, 1H), 7.50 - 7.30 (m, 3H), 7.26 - 7.12 (m, 2H), 7.09 - 6.96 (m, 2H), 6.28 (s, 1H), 5.67 (d, J= 7.6 Hz, 1H), 4.74 (dd, J= 8.1, 4.6 Hz, 1H), 4.42-4.36 (m, 1H), 4.04 (s, 1H), 3.87 (d, J= 7.8 Hz, 1H), 2.54 - 2.41 (m, 1H), 2.22- 1.76 (m, 8H), 1.50-1.32(m,2H). MS: 626.2 (M+l) +.
(2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cya- nopyrimidin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-5-oxopyrrolidine-2-carboxamide (single enantiomer)- Compound 189
1HNMR (400 MHz, CDC13): δ 9.00 (d, J = 4.7 Hz, 1H), 7.76 (s, 1H), 7.47 - 7.30 (m, 2H), 7.24 - 6.88 (m, 6H), 6.47 (d, J = 6.7 Hz, 1H), 5.54 (s, lH), 4.74 (s, 1H), 4.35 (s, 1H), 3.99 (s, 1H), 3.72 (d,J=34.8Hz, 1H), 2.58 -2.18 (m,2H), 1.88 (m,4H), 1.56-1.42 (m, 2H). MS: 627.2 (M+l)+. (2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyano -pyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 171
1HNMR (400 MHz, CDC13): δ 8.68 (s, 1H), 8.52 (d, J = 5.0 Hz, 1H), 7.60 (d,J= 8.2 Hz, 1H), 7.45 - 7.17 (m, 4H), 7.15 - 6.91 (m, 2H), 6.84 (d,J= 8.7 Hz, 1H), 6.69 (t,J= 8.7 Hz, 1H), 6.54 - 6.36 (m, 2H), 4.87-4.60 (m, 1H), 4.31 (m, 2H), 3.99 - 3.77 (m, 1H), 3.15 - 2.78 (m, 2H), 2.62-2.26 (m, 3H), 2.26-2.08 (m, 1H). MS: 616.1 (M+l)+.
(2S,4S)-N-((S)-l-(2-Chlorophenyl)-2-((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3,5-difluorophenyl)-4-hydroxy-5-oxopyrrolidine-2- carboxamide(single enantiomer)- Compound 174
1HNMR (400 MHz, CDC13): δ 8.75 (s, 1H), 8.53 (d, J = 4.5 Hz, 1H), 7.62 (s, 1H), 7.44 - 7.18 (m, 3H), 7.09- 6.96 (m, 2H), 6.86 (s, 1H), 6.71 (t,J=8.7Hz, 1H), 6.38 (s, 1H), 5.58 (d,J= 7.6 Hz, 1H), 4.80 (dd,J= 8.0, 5.2 Hz, 1H), 4.37 (d,J= 5.6 Hz, 1H), 3.96 (s, 1H), 3.61 (d,J=7.7Hz, 1H), 2.62-2.29 (m, 1H), 2.13 (m, 6H), 1.48 (m, 2H). MS: 644.2 (M+l)+.
Example 19.Preparationof(2S)-N-((R)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)- 2-oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-4-hydroxy-4-methyl-5- oxopyrrolidine-2-carboxamide(single enantiomer)- Compound 183
Step A. (2S)-l-tert-Butyl 2-methyl 4-(tert-butyldimethylsilyloxy)-4-methyl-5-oxopyrrolidine-l,2- dica boxy late. LiHMDS (1 M in THF, 22.6 rtiL, 22.6 mmol) was added into a mixture of (2S,4R)-1 -tert-butyl 2-methyl 4-(tert-butyldimethylsilyloxy)-5-oxopyrrolidine-l ,2-dicar-boxylate (6.5 g, 17.4 mmol) in THF (60 mL) at -78°C under N2.The mixture was stirred at -78°C for 1 hr. A solution of iodomethane (2.7 g, 19.1 mmol) in THF (10 mL) was added dropwise to the above mixture over 30 min. Then the solution was stirred at -78°C for another 25 min. The resulting mixture was allowed to warm to room temperature and stirred overnight. The mixture was quenched with NH4C1 and extracted by ethyl acetate (60 mL x 3). The combined organic layers were dried and concentrated. The residue was purified by column chromatography to give the desired product. MS: 388 (M+l)+.
Step B. (2S,4S)-Methyl 4-((tert-butyldimethylsilyl)oxy)-5-oxopyrrolidine-2-carboxylate.A solution of(2S)-l -tert-butyl 2-methyl 4-(tert-butyldimethylsilyloxy)-4-methyl-5-oxopyrroli-dine- 1,2-dicarboxylate (960 mg, 25 mmol) in TFA/DCM (V:V = 1 : 3) was stirred at room
temperature for 1 h. The mixturewasthen concentrated to give the desired product which was used directly in the next step.MS: 288 (M+l)+.
Step C.(2S)-4-(tert-Butyldimethylsilyloxy)-4-methyl-5-oxopyrrolidine-2-carboxylic acid Yo a solution of (2S)-methyl 4-(tert-butyldimethylsilyloxy)-4-methyl-5-oxopyrrolidine-2-carbo-xylate (400 mg, 1.4 mmol) in MeOH/THF/H20 (V:V:V =2 : 2 : 1) was added LiOH (50 mg, 2.1 mmol). The mixture was stirred at room temperaturefor lhr and then concentrated. The residue was partitioned between ethyl acetate and water. The aqueous phase was separated and adjusted to
pH=3-4 with IN HC1 solution. The aqueous layer was then extracted with ethyl acetate (2x 20 mL). The combined organic layers were dried over anhydrous Na2S04and concentrated to give the desired product. MS: 274 (M+l)+.
Step D.(2S)-4-(tert-Butyldim thyhilyloxy)-N-(l-(2-chlorophenyl)-2-(^
ylamino)-2-oxoethyl)-N-(3-fluorophenyl)-4-methyl-5-oxopyrrolidine-2-carbo
of 3-fluoroaniline (83 mg, 0.75 mmol) and 2-chlorobenzaldehyde (105 mg, 0.75mmol) in MeOH (5 mL) was stirred for 30 min at room temperature, followed by addition of (2S)-4-(tert- butyldimethylsilyloxy)-4-methyl-5-oxopyrrolidine-2-carboxylic acid (205 mg, 0.75 mmol). The resulting mixture was stirred for lOmin and followed by the addition of l,l-difluoro-3- isocyanocyclobutane (105 mg, 0.9 mmol). The mixture was stirred at room
temperatureovemight and concentrated, and then the residue was purified by a standard method to give the desiredproduct. MS: 624 (M+l)+ .
Step E.(2S)^-(tert-Butyldimethylsilyloxy)-N-(l-(2-chlorophenyl)-2-(3,3-difluoro
amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-4-methyl-5-o
carboxamide. A mixture consisting of (2S)-4-(tert-butyldimethylsilyloxy)-N-(l-(2- chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-N-(3-fluorophenyl)-4-methyl-5- oxopyrrolidine-2-carboxamide (200 mg, 0.32 mmol), 2-bromoisonicotinonitrile (88 mg, 0.48 mmol), Cs2C03 (146 mg, 0.45 mmol), Pd2(dba)3 (29 mg, 0.032 mmol), Xant-Phos (19 mg, 0.032 mmol) and 1,4-dioxane (5 mL) was stirred under N2 at 80 °C overnight. After filtration, the filtrate was concentrated in vacuo and the residue was purified by a standard method to give desired product. MS: 726 (M+l)+ .
Step F.(2S)-N-((R)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluorophenyl)^-hydroxy-4-methyl-5-oxopyrrolidine-2- carboxamide.To a solution of (2S)-4-(tert-butyldimethylsilyloxy)-N-(l-(2-chlorophenyl)-2-(3,3- difluorocyclobutylamino)-2-oxoethyl)-l -(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-4-methyl-5- oxopyrrolidine-2-arboxamide (50 mg, 0.07 mmol) in THF (2 mL) was added TBAF (36 mg, 0.14 mmol) at 0 °C. The solution was stirred at 0 °C for 30 min and then partitioned between water and EtOAc. Combined organic layers were separated, dried, and concentratedin vacuo. The resulting residue was purified by a standard method to give the desired product. H NMR (400 MHz, CDC13): δ 8.57 (d, J = 5.0 Hz, 1H), 8.48 (d, J = 3.8 Hz, 1H), 7.54 - 7.17 (m, 5H), 6.98 -
6.84 (m, 3H), 6.67 (dd, J= 8.6 Hz, 1H), 6.33 (d, J= 5.2 Hz, 1H), 6.08 - 6.01 (m, 1H), 4.55 - 4.48 (m, 1H), 4.29 (s, 1H), 3.22 - 2.35 (m, 6H), 1.93 - 1.80 (m, 1H), 1.27 (s, 3H). MS: 612.2 (M+l)+.
Example 20. Preparation of (2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2- oxo-ethyl)-l-(4-cyanopyridin-2-yl)-N-(3-fluoro-5-sulfamoylphenyl)-5-oxopyrrolidine-2- carboxamide (racemic)- Compound 158
Step A. Benzyl(3-fluoro-5-nitrophenyl)sulfane. To a solution of 1 , 3-difluoro-5-nitrobenzene (15.9 g, 100 mmol) in DMF (160 mL) was added K2C03 (15.8 g, 110 mmol) and
phenylmethanethiol (12.4 g, 100 mmol) at 0 °C. The reaction was stirred at room temperaturefor 2 hr and then quenched with H20. The resulting mixture extracted with EtOAc (3x 100 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated in vacuo to afford the crude product as a yellow oil which was used in the next step without further purification. Step B. 3-Fluoro-5-nitrobenzene-l-sulfonyl chloride. To a solution of enzyl(3-fluoro-5- nitrophenyl)sulfane (3.0 g) in DCM (30 mL) was added deionized water (30 mL). Then chlorine was bubbled slowly into the mixture until the complete consumption of the starting material was observed (monitored by TLC). The organic layer was separated, washed with sat. aq.
Na2S203solution, dried and concentrated to afford the crude product which was used in the next step without further purification.
Step C. N-tert-butyl-3-fluoro-5-nitrobenzenesulfonamide. To a solution of 3-fluoro-5- nitrobenzene-l-sulfonyl chloride in dry dioxane (30 mL) was slowly added tert-butylamine (10 mL) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 2 hr. The mixture was then concentrated and the residue was purified by column chromatography to afford the desired product. 1H NMR (400 MHz, DMSO-d6): δ 8.43 (s, 1H), 8.40 - 8.32 (m, 1H), 8.10 - 8.05 (m, 1H), 7.99 (s, 1H), 1.12 (s, 9H).
Step D. 3-Amino-N-tert-butyl-5-fluorobenzenesulfonamide.N^ert-buty\-3- benzenesulfonamide (1.0 g, 3.6 mmol), iron powder (1.0 g, 18 mmol) and NH4C1 (1.0 g, 18 mmol) were mixed in EtOH (95%, 10 mL). The mixture was refluxed for 16 hr then filtered. The filtrate was concentrated and the residue was purified by a standard method to afford the desired product. 1H NMR (400 MHz, DMSO-d6): δ 7.45 (s, 1H), 6.88 - 6.85 (m, 1H), 6.66 - 6.62 (m, 1H), 6.48 - 6.42 (m, 1H), 5.89 (s, 2H), 1.11 (s, 9H).
Step E. The same as general procedures for UGI reaction set forth herein.
Step F. The same as general procedures for Buchwald reaction set forth herein.
Step G. (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocychbutylamino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-sulfamoylphenyl)-5-oxopyrrolidine-2-carboxandd To a solution of (2S)-N-(3 -(N-tert-butylsulfamoyl)-5 -fluorophenyl)-N-(l -(2-chlorophenyl)-2-(3 ,3 - difluorocyclo-butylamino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide (80 mg, 0.11 mmol) in DCM (1 mL) was added TFA (1 mL). Thereaction was stirred at room temperature for 16 hr and neutralized with saturated aq. NaHC03. The mixture was then extracted with EtOAc (3 x 10 mL). The combined organic layers were dried and concentrated. The residue was purified by a standard method to afford the target compound. 1H NMR (400 MHz, DMSO-d6): δ 8.90 - 8.84 (m, 1H), 8.67 - 8.62 (m, 1H), 8.55 (s, 1H), 8.19 (s, 1H), 7.87 - 7.76 (m, 1H), 7.65 - 7.60 (m, 2H), 7.45 - 7.40 (m, 3H), 7.21 (d, J = 7.0 Hz, 2H), 7.11 - 7.04 (m, 1H), 6.93 - 6.86 (m, 1H), 6.33 - 6.26 (m, 1H), 4.83 (m, 1H), 4.13 (s, 1H), 2.94 (m, 2H), 2.63 - 2.53 (m, 3H), 2.42 - 2.32 (m, 1H), 1.97 (s, 2H). MS: 661 (M+l)+.
Example 21.Preparation of (2S)-N-(lH-benzo[d]imidazol-7-yl)-N-(l-(2-chlorophenyl)-2- ((4,4-difluorocyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2- carboxamide (racemic)- Compound 141
Step A: 7-Nitro-lH-benzo[d]imidazole. A solution of 3 -nitrobenzene- 1,2-diamine (900 mg, 5.88 mmol) in AcOH (12 mL) was stirred at 100°C overnight. The mixture was neutralized with aq. NaHC03 to pH= 8 at 0°C and the precipitate was collected by filtration. The precipitate was dried in vacuo to afford the desired product.
Step B: 7-Nitro-l-((2-(Mmethylsilyl)ethoxy)methyl)-lH-benzofdJimidazole. NaH (331 mg, 8.28 mmol) was added to a solution of 7-nitro-lH-benzo[d]imidazole (900 mg, 5.52 mmol) in DMF (7 mL) at 0°C under N2. After stirring at 0 °C for lhr, SEMCl (1.38 g, 8.28 mmol) was added and the resulting mixture was stirred at room temperature for 2hr. The reaction mixture was quenched with H20 and extracted with EtOAc (3 x 30 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated. The residue was purified by column chromatography to afford the desired product as a yellow oil.
Step C: l-((2-(Trimethylsilyl)ethoxy)methyl)-lH-benzofdJimidazol-7-amine.To a solution of 7- nitro-l-((2-(trimethylsilyl)ethoxy)methyl)-lH-benzo[d]imidazole (600 mg, 2.05 mmol) in EtOH/EtOAc (10 mL/2mL) was added Pd/C (60 mg). After stirring under a hydrogen atmosphereat room temperature overnight, the reaction mixture was filtered and the filtrate was concentrated. The residue was purified by a standard method to afford the desired product. Step D: The same as general procedure for UGI reaction set forth herein.
Step E:(2S)-N-(lH-Benzo[d]imidazol-7-yl)-N-(l-(2-chlorophenyl)-2-((4,4-difluorocycloh amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide. TBAF (1M in
THF, 3 mL) was added to a solution of (2S)-N-(l -(2-chlorophenyl)-2-((4,4-difluoro - cyclohexyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxo-N-(l -((2-(trimethylsilyl)ethoxy) - methyl)-lH-benzo[d]imidazol-7-yl)pyrrolidine-2-carboxamide in THF (0.5 mL) at 0°C under N2. After stirring at room temperature for 7hr, the reaction was quenched with water at 0°C. The resulting mixture was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04and concentrated. Theresulting residue was purified by a standard method to afford the desired product. H NMR (400 MHz, CDC13): δ 13.08 (s, 1H), 8.92 - 8.39 (m, 2H), 8.19 (m, 1H), 7.82 (m, 1H), 7.51 - 7.31 (m, 2H), 7.25 (d, J = 5.2 Hz, 1H), 7.13 - 6.70 (m, 3H), 6.41 (m, 1H), 6.20 - 5.29 (m, 1H), 4.85 (m, 1H), 3.86 (s, 1H), 2.97 - 2.39 (m, 2H), 2.36 - 1.70 (m, 9H), 1.40 (m, 2H). MS: 632.2 (M+l)+.
Example 22. Preparation of (4S)-N-(l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)- 2-oxoethyl)-3-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-l-methyl-2-oxoimidazolidine-4- carboxamide (racemic) - Compou
Step A:(S)-3-(Benzyloxycarbonyl)-2-oxoimidazolidine-4-carbo -xylic acid. To a solution of 6.6 g of sodium hydroxide in 140 mL of waterat 0°C, 8.8 g of bromine was added dropwise, followed by addition of (S)-4-amino-2-(benzyloxycarbonylamino)-4-oxobutanoic acid (13.4 g, 50 mmol) portion wise over 3 min. The resulting yellow solution was heated to 50°C for lhr and thencooled to room temperature. After addition of sodium thiosulfate (2.0 g), the reaction mixture was washed with ether (2 x 30 mL). The aqueous layer was acidified to pHl-2 with 6N HC1. After the precipitate was formed, the suspension was filtered. The sticky material was collected and re-crystallized in MeOH to afford the desired product as a white solid.1H NMR (400 MHz, DMSO-d6): δ 13.29 (s, 1H), 7.57 (s, 1H), 7.40 - 7.27 (m, 4H), 5.27 - 5.04 (m, 2H), 4.66 (dd, J= 10.2, 3.2 Hz, 1H), 3.63 (t, J = 10.0 Hz, 1H), 3.20 (dd, J= 9.7, 3.2 Hz, 1H).
Step B:(S)-Dibenzyl 2-oxoimidazolidine- 1 , 5-dicarhoxylate. To a 500 mL-flask were added (S)- 3 -(benzyl oxycarbonyl)-2-oxoimidazolidine-4-carboxylic acid (5.3 g, 20 mmol), BnBr (2.8 mL, 23 mmol), K2C03 (8.28g, 60 mmol), and acetonitrile (250 mL). The reaction solution washeated to reflux for 6 hr, cooled and then filtered. The filtrate was concentrated invacuo and the residue was purified by column chromatography to afford the desired product as white solid. 1HNMR (400 MHz, CDC13): δ 7.41 - 7.25 (m, 10H), 6.36 (s, 1H), 5.30 - 5.05 (m, 4H), 4.80 (dd, J= 10.2, 3.6 Hz, 1H), 3.74 (t, J = 10.0 Hz, 1H), 3.41 (dd, J = 9.7, 3.7 Hz, 1H).
Step C: (S)-Dibenzyl 3-methyl-2-oxoimidazolidine-l,5-dicarboxylate. To a dry 1 OOmL-flask were added (S)-dibenzyl 2-oxoimidazolidine-l,5-dicarboxylate (1.5 g, 4.24 mmol), K2C03 (1.17 g,8.47 mmol), Mel (5.2 mL, 84.7mmol), and acetone (50 mL). The reaction solution was heated to reflux and stirred overnight. The resulting reaction mixture was cooled and filtered. The filtrate was concentrated invacuo and the residue was purified by column chromatography to afford the desired productas a white solid. 1H NMR (400 MHz, CDC13): δ 7.40 - 7.26 (m, 10H), 5.27 - 5.07 (m, 4H), 4.70 (dd, J = 10.2, 3.8 Hz, 1H), 3.63 (dd, J = 10.1, 9.7 Hz, 1H), 3.31 (dd, J = 9.6, 3.8 Hz, 1H), 2.84 (s, 3H). MS :369(M+1)+.
Step D: (S)-l-Methyl-2-oxoimidazolidine-4-carboxylic acid. To a dry 50 mL-flask were added (S)-dibenzyl 2-oxoimidazolidine-l,5-dicarboxylate (0.5 g, 1.36 mmol), Pd/C (10%, 100 mg) and MeOH (15 mL). The suspension was stirred overnight at room temperatureunder a hydrogen atmosphere. The resulting reaction mixture was filtered. The filtrate was concentrated invacuo to afford the desired product as an off-white solid. 1H NMR (400 MHz, CD3OD): 54.21 (dd, J= 9.9, 4.8 Hz, lH), 3.70 (t, J= 9.6 Hz, 1H), 3.55 (dd, J = 9.3, 4.8 Hz, 1H), 2.74 (s, 3H).
MS :145(M+1)+.
Step E:(4S)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl) -N-(3-fluoro- phenyl)-l-methyl-2-oxoimidazolidine-4-carboxamide.A mixture of 2-chlorobenzaldehyde (165 mg, 1.18 mmol) and 3-fluorobenzenamine (131 mg, 1.18 mmol) in MeOH (3 mL) was stirred at room temperature for 30 min. Then (S)-l -methyl -2-oxoimidazolidine-4-carboxylic acid (170 mg, 1.18 mmol)was added and the reaction mixture was stirred for another 15 min, followed by addition of l,l-difluoro-3-isocyanocyclobutane (138mg, 1.18 mmol). The reaction mixture was stirred overnight and concentrated in vacuo. The residue was purified by a standard method to give the desired product. MS :495(M+1)+.
Step F: The same as the Buchwald reaction procedure set forth herein. H NMR (400 MHz, CDCI3): δ 8.64 - 8.34 (m, 2H), 7.94 - 7.59 (m, 1H), 7.50 - 6.61 (m, 8H), 6.34 - 6.07 (m, 1H), 4.94 - 4.67 (m, 1H), 4.3-4.2 (m, 1H), 3.49 (m, 1H), 3.46 - 3.22 (m, 1H), 3.02-2.83 (m, 2H), 2.87 (s, 3H), 2.5-2.2 (m, 2H).MS :597(M+1)+.
Example 23.Preparation of (S)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)- 2-oxoethyl)-3-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-2-oxoimi dazolidine-4- carboxamide(single enantiomer)- Compound 80
Step A: (S)-3,4-Dibenzyl 1-tert-butyl 2-oxoimidazolidine-l,3,4-tricarboxylate. To a 25 mL-flask were added (S)-dibenzyl 2-oxoimidazolidine-l,5-dicarboxylate (40 mg, 0.11 mmol), (BOC)20 (26 mg,0.12 mmol), TEtOAc (0.06 mL,0.3 mmol), DMAP (cat.) and CH2C12 (2 mL). The mixture was stirred overnight. The solvent was then removed in vacuo and the residue was purified by column chromatography to give the desired product. H NMR (400 MHz, CDCI3): δ 7.39 - 7.27 (m, 10H), 5.24(s, 2H), 5.16(s, 2H), 4.67 (dd, J= 10.2, 3.5 Hz, lH), 3.94 (dd, J= 1 1.1, 10.3 Hz, 1H), 3.74 (dd, J = 11.2, 3.5 Hz, 1H), 1.51 (s, 9H).
Step B: (S)-l-(tert-Butoxycarbonyl)-2-oxoimidazolidine-4-carboxylic acid. To a dry 50 mL- flask were added (S)-3,4-dibenzyl 1-tert-butyl 2-oxoimidazolidine-l ,3,4-tricarboxylate (1.24 g, 2.73 mmol), Pd/ C(10%, 200 mg) and MeOH (30 mL). The suspension was stirred overnight at room temperatureunder a hydrogen atmosphere. The reaction mixture was filtered, and the filtrate was concentrated in vacuo to afford the desired product. H NMR (400 MHz, DMSO-d6): 56.06 (s, 2H), 4.31 (s, 1H), 4.25 - 3.94 (m, 2H), 1.52 (s, 9H).
Step C: (4S)-tert-Butyl4-((l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino) -2-oxoethyl)(3- fluorophenyl)carbamoyl)-2-oxoimidazolidine-l-carboxylate. A mixture of 2- chlorobenzaldehyde (122 mg, 0.87 mmol) and 3-fluorobenzenamine (97 mg, 0.87 mmol) in MeOH (2 mL) was stirred at room temperaturefor 30 min. Then (S)-l-(tert-butoxycarbonyl)-2- oxoimidazolidine-4-carboxylic acid (200 mg, 0.87 mmol)was added and the reaction mixture was stirred for another 15 minfollowed by addition of l,l-difluoro-3-isocyanocyclobutane (102 mg, 0.87 mmol). The reaction mixture was further stirred at room temperatureovemight and then concentrated in vacuo. The residue was purified by a standard method to give the desired product. ¾ NMR (400 MHz, CDC13):57.46 - 6.59 (m, 8H), 6.45 (s, 1H), 4.41 - 4.04 (m, 2H), 4.01 - 3.78 (m, 1H), 3.64 - 3.30 (m, 1H), 2.92 (m, 2H), 2.71 - 2.27 (m, 2H), 1.46 (s, 9H). MS : 581(M+1)+.
Step D: (4S)-tert-Butyl4-((l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino) -2-oxoethyl)(3- fluorophenyl)carbamoyl)-3-(4^yanopyridin-2-yl)-2-oxoinddazolidine-l-carboxylate. To a
25mL flask charged with 1 ,4-dioxane (4.5 mL)wereadded (4S)-tert-butyl 4-((l-(2-chlorophenyl)- 2-(3,3-difluoro cyclo butylamino)-2-oxoethyl)(3-fluoro -phenyl)carbamoyl)-2-oxoimidazolidine- 1 -carboxylate (250 mg, 0.43 mmol), 2-bromoisonicotinonitrile (122 mg, 0.65 mmol), CS2CO3 (281 mg,0.862 mmol), Xant-Phos (25 mg, 0.043 mmol) and Pd2(dba)3 (40 mg, 0.043 mmol). The mixture was degassed and refilled with nitrogen, and then heated to 100°C for 3hr. The resulting mixture was cooled and filtered. The filtrate was concentrated invacuo and the residue was purified by a standard methodto give both epimers. The epimers were further separated bya standard methodto give the desired product. ¾ NMR (400 MHz, CDC13): δ 8.58 (s, 1H), 8.48 (t, J= 5.9 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.37 - 7.16 (m, 4H), 7.15 - 6.76 (m, 4H), 6.56 - 6.31 (m, 2H), 4.95 - 4.75 (m, 1H), 4.31 (s, 1H), 3.86 (dd, J= 10.8, 5.1 Hz, 1H), 3.66 (m, 1H), 2.99 (m, 2H), 2.61 - 2.27 (m, 2H), 1.56 (s, 9H). MS : 683(M+1)+.
Step E: (S)-N-((S)-l-(2-Chlorophenyl)-2-(3,3-difluorocyclo butylamino)-2-oxoethyl)-3-(4- cyanopyridin-2-yl)-N-(3-fluorophenyl)-2-oxoimi dazolidine-4-carboxamide.To a solution of 2N HCl/MeOH (2 mL) at 0°C was added 50 mg of (S)-tert-butyl-4-(((S)-l-(2-chlorophenyl)-2-(3,3- difluorocyclobutylamino)-2-oxoethyl)(3 -fluorophenyl) carbamoyl)-3-(4-cyanopyridin-2-yl)-2- oxoimidazolidine-1 -carboxylate. The reaction mixture was warmed to room temperature and stirred for 5hr. The solvent was removed invacuo and the residue was purified by a standard
method to givethe desired product. Ή NMR (400 MHz, CD3OD): δ 8.50 (d, J = 4.5 Hz, 1H), 7.65 (d, J= 8.6 Hz, 1H), 7.50 - 6.81 (m, 8H), 6.47 (d, J= 11.6 Hz, 1H), 5.04 - 4.92 (m, 1H), 4.22 (m, 1H), 3.59 - 3.46 (m, 1H), 3.39 (dd, J = 9.9, 4.5 Hz, 1H), 2.91 (m, 2H), 2.63 - 2.36 (m, 2H). MS : 583(M+1)+.
Example 24.Preparation of(4S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutyl -amino)-2- oxoethyl)-3-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-l-(2-hydroxyeth-yl)-2-oxoimidazoli- dine-4-carboxamide (racemic)
Step A: (S)-Dibenzyl 3-(2-ethoxy-2-oxoethyl)-2-oxoimidazolidine- 1,5- dicarboxylate. To a dry
50mL-flask charged with DME (5 mL)were added (S)-dibenzyl 2-oxoimidazolidine-l,5- dicarboxylate (200 mg, 0.56 mmol), K2C03 (156 mg,1.13 mmol), and ethyl 2-bromoacetate (0.13 mL, 1.13 mmol). The mixture was heated to reflux for 3hr. The reaction mixture was cooled and filtered. The filtrate was concentrated invacuo and the residue was purified by column chromatography to afford the desired product as a colorless oil.1H NMR (400 MHz, CDC13): δ 7.45 - 7.25 (m, 10H), 5.41 - 5.05 (m, 4H), 4.80 (dd, J = 10.2, 3.5 Hz, 2H), 4.29 - 4.08 (m, 3H), 3.90 (dd, J = 12.2, 7.2 Hz, 2H), 3.45 (dd, J = 9.2, 3.5 Hz, 1H), 1.28 (td, J = 7.1, 2.1 Hz, 3H). Step B: (S)-l-(2-Ethoxy-2-oxoethyl)-2-oxoimidazolidine-4-carboxylic acid. To a dry 50 mL- flask were added (S)-dibenzyl 3 -(2-ethoxy-2-oxoethyl)-2-oxoimidazolidine-l , 5 -dicarboxylate (170 mg,0.386 mmol), Pd/C(10%, 35 mg) and MeOH (4 mL). The suspension was stirred at room temperature overnight under a hydrogen atmosphere. The reaction mixture was filtered, and the filtrate was concentrated invacuo to afford the desired product as an off-white solid.1H
NMR (400 MHz, CD3OD): δ 4.30 (dd, J= 10.0, 4.8 Hz, 1H), 4.20 (q, J= 7.1 Hz, 2H), 4.05 - 3.91 (m, 2H), 3.91 - 3.85 (m, 1H), 3.69 (dd, J= 9.0, 4.8 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H).
Step C: Ethyl 2-((4S)-4-((l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2 -oxoethyl)(3- fluorophenyl)carbamoyl)-2-oxoimidazolidin-l-yl)acetate. A mixture of 2-chlorobenzaldehyde (518 mg, 3.70 mmol) and 3-fluorobenzenamine (411 mg, 3.7 mmol) in MeOH (12 mL) was stirred at room temperature for 30 min. Then (S)-l-(2-ethoxy-2-oxoethyl) -2-oxoimidazolidine- 4-carboxylic acid (800 mg, 3.7 mmol)was added and the reaction mixture was stirred for another 30 min, followed by addition of l,l-difluoro-3-isocyanocyclobutane (600 mg, 3.7 mmol). The reaction mixture was stirred ovemightand concentrated in vacuo. The residue was purified by a standard method to give the desired product. MS : 567 : (M+l)+.
Step D: Ethyl 2-((4S)-4-((l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)(3- fluorophenyl)carbamoyl)-3-(4-cyanopyridin-2-yl)-2-oxoimMazolidin-l-yl)acetate- Compound
94. To a 25 mL-flask were addedethyl 2-((4S)-4-((l-(2-chlorophenyl)-2-(3,3- difluorocyclobutylamino)-2-oxoethyl) (3-fluoro -phenyl)carbamoyl)-2-oxoimidazolidin-l - yl)acetate (50 mg, 0.0882 mmol), 2-bromoisonicotinonitrile (21 mg, 0.115 mmol), Cs2C03 (58 mg,0.176 mmol), Xant-Phos (5.2 mg, 0.009mmol), Pd2(dba)3 (8.2 mg, 0.009 mmol) and 1,4- dioxane (1 mL). The mixture was degassed and refilled with nitrogen, and then heated to 100°C for 3hr. The resulting mixture was cooled and filtered and then the filtrate was concentrated in vacuo. The residue was purified by a standard method to give both epimers. XH NMR (400 MHz, CDC13): δ 8.63-8.57 (S, 1H), 8.55 - 8.38 (m, 1H), 7.63 (s, 1H), 7.46 - 6.84 (m, 8H), 6.45-6.37 (m, 1H), 6.22 - 5.94 (m,lH), 5.06 - 4.77 (m, 1H), 4.43-4.37 (m, 1H), 4.32-4.20 (m, 1H), 4.21 (q, J= 7.1 Hz, 2H), 3.82 - 3.46 (m, 3H), 3.12 - 2.82 (m, 2H), 2.66 - 2.25 (m, 2H), 1.29 (t, J = 7.1 Hz, 3H). MS : 669(M+1)+.
Step E: (4S)-N-(l-(2-Chlorophenyl)-2-(3,3-difluorocyclobutyl-amino)-2-oxoethyl)-3-(4- cyanopyridin-2-yl)-N-(3-fluorophenyl)-l-(2-hydroxyeth-yl)-2-oxoimidazolidine-4-carboxa - mide- Compound 112. To a solution of ethyl 2-((4S)-4-((l-(2-chlorophenyl)-2-(3,3- difluorocyclobutylamino)-2-oxoethyl)(3-fluorophenyl)carbamoyl)-2-oxo-3-(pyrimidin-2- yl)imidazolidin-l -yl)acetate (100 mg, 0.155 mmol)in DME (2 mL) at 0°C wasaddedLiBH4 (22 mg) in two portions. The mixture was stirred for 0.5hr, thenwarmed to room temperature. The resulting mixture was stirred for another 2hr and quenched with H20 (2 mL) at 0°C. The
resulting mixturewas extracted with EtOAc (2 x lOmL). The combined organic layers were combined, washed with brine, dried over anhydrous Na2S04, and concentrated invacuo. The residue was purified by a standard method to give the desired product. 1H NMR (400 MHz, CDCls): δ 8.62-8.55 (m, 2H), 7.63 (d, J = 8.1 Hz, 1H), 7.40 - 6.85 (m, 8H), 6.47 - 6.2 (m, 2H), 4.90 - 4.69 (m, 1H), 4.30 - 4.15 (m,lH), 3.87 - 3.72 (m, 2H), 3.71 - 3.19 (m, 5H), 3.08 - 2.85 (m, 2H), 2.63 - 2.35(m, 2H). MS : 603(M+1)+.
The following compound was synthesized via the procedure set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substitutedaromatic ring or heterocyclic(heteroaromatic) ring using the reagents and solvents set forth above, and purified via standard methods.
Ethyl 2-((4S)-4-((l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)(3-fluoro phenyl)carbamoyl)-3-(4-cyanopyrimidin-2-yl)-2-oxoimidazolidin-l-yl)acetate (racemic)- Compound 111
Ή NMR (400 MHz, CDC13): δ 8.90-8.82 (m, 1H), 7.62-7.57 (m, 1H), 7.46 - 6.82 (m, 8H), 6.52- 6.48(m, 1H), 6.15 - 5.85 (m, 2H), 4.88-4.82 (m, 1H), 4.45-4.35 (m, 1H), 4.32-4.13 (m, 2H), 3.86 - 3.46 (m, 3H), 3.05-2.85(m, 2H), 2.56-2.32 (m, 2H), 1.29 (t, J= 7.1 Hz, 3H). MS : 670(M+1)+.
Example 25. Preparation of Additional Compounds of Formula I
General procedures for the UGI reaction:
A mixture of aldehyde (3.5 mmol) and aniline (3.5 mmol) in MeOH (8 mL) was stirred at room temperature for 30 min. Then the acid (3.5 mmol) was added and the reaction mixture was stirred for another 30 min, followed by addition of the isocyanide (3.5 mmol). The resulting mixture was then stirred at room temperature overnight and quenched with H20. The resulting mixture was partitioned between EtOAc and H20. The organic layer was washed with brine, dried over
anhydrous Na2S04, and then concentrated. The resulting residue was purified by a standard method to afford the desired product.
General procedures for the Buchwald reaction:
A mixture of amine (0.30 mmol), aryl halide (0.30 mmol), CS2CO3 (129 mg, 0.39 mmol), Pd2(dba)3 (18 mg, 0.02 mmol) and Xant-Phos (9.4 mg, 0.02 mmol) in 1,4-dioxane (10 mL) was stirred under N2 at 80°C overnight. After filtration, the filtrate was concentrated under reduced pressure and the residue was purified by a standard method to give the desired products.
The following analogs were synthesized via the procedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substituted-aromatic ring or
heteroaromatic ring using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
2-(2-Chlorophenyl)-4-((S)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidin-2-yl)-N-(3,3- difluorocyclobutyl)-3-(lH-indazol-7-yl)-4-oxobutanamide - Compound 300
1H NMR (400 MHz, CDC13)5 13.18 (s, 1H), 8.79 (s, 0.5H), 8.70 - 8.63 (m, 1H), 8.53 (d, J= 5.0 Hz, 0.5H), 8.22 (s, 0.5H), 8.06 (s, 0.5H), 7.83 (d, J= 8.0 Hz, 0.5H), 7.61 (d, J= 8.0 Hz, 0.5H), 7.50 (d, J = 7.3 Hz, 0.5H), 7.38 (m, 3H), 7.13 (t, J = 7.7 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.98 (d, J= 7.3 Hz, 1H), 6.83 (t, J= 7.7 Hz, 0.5H), 6.67 (d, J= 7.2 Hz, 0.5H), 6.24 (d, J= 6.5 Hz, 0.5H), 6.02 (s, 0.5H), 5.79 (d, J = 6.3 Hz, 0.5H), 5.12 (dd, J = 9.3, 3.4 Hz, 0.5H), 4.95 (s, 0.5H), 4.61 (dd, J = 9.2, 3.0 Hz, 0.5H), 4.22 (d, J = 7.4 Hz, lH), 3.00 - 2.71 (m, 3H), 2.59 - 2.05 (m, 4H), 2.04 - 1.95 (m, 1H). MS: 603.2 (M+l)+.
(S)-3-(4-cyanopyridin-2-yl)-N-((R)-2-((3,3-difluorocyclobutyl)amino)-2-oxo-l-phenylethyl)- N-(5-fluoropyridin-3-yl)-2-oxo-lr5-oxazinane-4-carboxamide - Compound 301
1H NMR (400 MHz, CDC13): δ 8.54 - 8.21 (m, 3H), 8.08 - 7.99 (m, 1H), 7.31 - 7.27 (m, 2H), 7.26 - 7.20 (m, 2H), 7.09 - 6.87 (m, 2H), 6.38 - 6.17 (m, 1H), 5.89 - 5.60 (m, 1H), 4.93 - 4.70 (m, 1H), 4.62 - 4.46 (m, 1H), 4.42 - 4.21 (m, 2H), 3.12 - 2.91 (m, 2H), 2.60 - 2.10 (m, 4H). MS: 565.2 (M+l)+.
(S)-3-(4-cyanopyridin-2-yl)-N-((R)-2-(3,3-difluorocyclobutylamino)-l-(2-fluorophenyl)-2- oxoethyl)-N-(5-fluoropyridin-3-yl)-2-oxo-l,3-oxazinane-4-carboxamide - Compound 302
Ή NMR (400 MHz, CDC13): δ 8.95 - 7.77 (m, 5H), 7.21 (s, 1H), 7.15 (s, 1H), 7.00 - 6.71 (m, 3H), 6.45 (m, 2H), 4.81 (m, 1H), 4.38 (d, J = 7.8 Hz, 1H), 4.23 (m, 2H), 2.92 (s, 2H), 2.45 (s, 2H), 2.08 (s, 2H). MS: 583 (M+l)+.
2-(2-chlorophenyl)-4-((S)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidin-2-yl)-N-(3,3- difluorocyclobutyl)-3-(2-fluoropyridin-4-yl)-4-oxobutanamide - Compound 303
1HNMR (400 MHz, CDC13) δ 8.76 - 8.70 (m, 1H), 8.59 - 8.49 (m, 1H), 8.17 - 8.04 (m, 1H), 7.43 - 7.27 (m, 3H), 7.24 - 6.95 (m, 3H), 6.44 - 6.25 (m, 1H), 6.10 - 6.5.96 (m, 1H), 4.94 - 4.75 (m, 3.1 Hz, 1H), 4.34 - 4.18 (m, 1H), 3.03 (m, 1H), 2.93 - 2.81 (m, 2H), 2.65 - 2.09 (m, 5H). MS: 582.1 (M+l)+.
Compound 304
1H NMR (400 MHz, CDC13): δ 8.77 - 8.45 (m, 2H), 8.46 - 7.84 (m, 2H), 7.61 (m, 1H), 7.31 - 7.22 (m, 2H), 7.09 (m, 1H), 6.89 (d, J = 4.3 Hz, 2H), 6.46 (d, J = 5.7 Hz, 1H), 6.07 (m, 1H), 4.86 (m, 1H), 4.34 (d, J = 6.1 Hz, 1H), 3.19 - 2.76 (m, 3H), 2.63 - 2.08 (m, 4H), 1.93 (m, 1H). MS: 605.1 (M+l)+.
(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(7-fluoroquinolin-5-yl)-5-oxopyrrolidine-2-carboxamide - Compound
Ή NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 8.89 (s, 1H), 8.31 (m, 3H), 7.46 (s, 3H), 7.21 - 7.06 (m, 3H), 6.31 (s, 1H), 6.13 (s, 1H), 5.04 (d, J= 6.9 Hz, 1H), 4.86 (d, J= 6.5 Hz, 1H), 4.37 (s, 2H), 2.51 (m, 5H), 2.24 (s, 1H). MS: 633.2 (M+l)+.
Example 26.Preparation of (2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutyl amino)-2- oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(3-fluoro-5-(pyridin-4-yl)phenyl)-5-oxopyrrolidine-2- carboxamide - Compound 306
Compound306 was prepared according to the following scheme, using the following protocol.
Step A:(2S)-N-(3-bromo-5-fluorophenyl)-N-(l-(2-chlorophenyl)-2-(3,3-difluoro - cyclobutylamino)-2-oxoethyl)-l -(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxamide. 3 -
Bromo-5-fluoroaniline (189 mg, 1 mmol), 2-chlorobenzaldehyde (140 mg, 1 mmol), (S)-l-(4- cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxylic acid (231 mg, 1 mmol) and l,l-difluoro-3- isocyanocyclobutane (118 mg, 1 mmol) were used in the general UGI reaction to give the desired product. MS: 660.1 (M+l)+.
Step B:Compound306.A mixtureof (2S)-N-(3-bromo-5-fluorophenyl)-N-(l -(2-chlorophenyl)-2- (3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2- carboxamide (150 mg, 0.23 mmol), pyridin-4-ylboronic acid (42.4 mg, 0.35 mmol), Cs2C03 (224 mg, 0.69 mmol), Pd2(dba)3 (16.2 mg, 0.023 mmol) and Xant-Phos (27 mg, 0.046 mmol) in 1,4- dioxane (5 mL) was stirred under N2 at 100 °C overnight and then filtered. The filtrate was concentrated under reduced pressure and the residue was purified by a standard method to give the desired products. ¾ NMR (400 MHz, CDC13): δ 8.81 - 8.57 (m, 3H), 8.56 - 8.37 (m, 1H), 8.06 - 7.92 (m, 1H), 7.46 - 7.36 (m, 3H), 7.24 - 7.12 (m, 3H), 7.12 - 6.93 (m, 2H), 6.98 - 6.01 (m, 2H), 4.98 - 4.86 (m, 1H), 4.37 - 4.18 (m, 1H), 3.15 - 2.63 (m, 4H), 2.53 - 2.4 (m, 2H), 2.32 - 2.26 (m, 2H). MS: 659.1 (M+l)+.
The following analogs were synthesized via the procedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and boronic acid using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
(S)-N-((R)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(pyridin-4-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound307
1H NMR (400 MHz, CDC13): δ 8.76 - 8.58 (m, 3H), 8.47 (m, 1H), 8.08 (s, 1H), 7.79 (d, J= 8.7 Hz, 1H), 7.49 - 7.38 (m, 1H), 7.35 - 7.24 (m, 2H), 7.16 (m, 3H), 7.06 - 6.91 (m, 2H), 6.71 (m, 1H), 6.44 (m, 1H), 4.97 (dd, J= 9.2, 3.0 Hz, 1H), 4.32 (m, 1H), 3.14 - 2.75 (m, 3H), 2.36 (m, 4H), 2.15 - 1.66 (m, 3H). MS: 659.1 (M+l)+.
(2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyridin- -yl)-N-(3-fluoro-5-(pyridin-3-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 308
1H NMR (400 MHz, CDC13): δ 8.56 - 7.98 (m, 4H), 7.83 - 7.36 (m, 2H), 7.35 - 7.26 (m, 2H), 7.21 - 6.89 (m, 5H), 6.47 - 6.17 (m, 2H), 4.99 - 4.87 (m, 1H), 4.36 - 4.16 (m, 1H), 3.12 - 2.67 (m, 4H), 2.51 - 2.36 (m, 2H), 2.31 - 2.19 (m, 2H). MS: 659.1 (M+l)+.
(S)-N-((S)-l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxo-ethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(isoxazol-5-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 309
¾ NMR (400 MHz, CDC1
3): δ 8.77 - 8.32 (m, 4H), 7.79 (m, 1H), 7.59 (m, 2H), 7.42 (m, 3H), 7.25 - 6.92 (m, 3H), 6.71 (d, J= 6.7 Hz, 1H), 6.45 (m, 1H), 4.98 (dd, J = 9.1, 3.4 Hz, 1H), 4.35 (s, 1H), 3.10 - 2.74 (m, 3H), 2.64 - 2.19 (m, 4H), 2.17 - 2.00 (m, 1H), 1.91 - 1.73 (m, 1H). MS: 659.1 (M+l)
+.
(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-4-(pyridin-4-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 310
1H NMR (400 MHz, CDC13): δ 8.74 - 8.51 (m, 3H), 7.85 (s, 1H), 7.68 - 7.63 (m, 1H), 7.54 - 7.45 (m, 2H), 7.36 - 7.35 (m, 2H), 7.20 (m, 2H),7.03 - 6.98 (m, 2H), 6.45 (m, 1H), 6.17-6.16 (m, 1H), 4.94 - 4.95 (m, 1H), 4.34 (m, 1H), 3.0 - 2.84 (m, 3H), 2.6 - 2.26 (m, 4H), 2.07 (m, 1H). MS: 659.1 (M+l)+.
(S)-N-((R)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-4-(pyridin-3-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 311
Ή NMR (400 MHz, CDC13): δ 8.65 (m, 4H), 7.79 (d, J= 7.9 Hz, 1H), 7.36 (m, 5H), 7.25 - 7.03 (m, 3H), 6.46 (s, 1H), 6.29 (s, 1H), 4.93 (m, lH), 4.15 (s, 1H), 3.10 - 2.70 (m, 3H), 2.57 (m, 1H), 2.38 - 1.99 (m, 4H). MS: 659.1 (M+l)+.
(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-4-(pyridin-3-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 312
Ή NMR (400 MHz, CDC13): δ 8.80 - 8.46 (m, 4H), 7.96 - 7.73 (m, 2H), 7.35 (dd, J= 7.8, 4.9 Hz, 3H), 7.24 - 6.92 (m, 5H), 6.46 (s, 1H), 6.27 (d, J = 5.4 Hz, 1H), 4.98 (dd, J = 9.3, 3.2 Hz, 1H), 4.35 (m, 1H), 3.01 (m, 2H), 2.95 - 2.82 (m, 1H), 2.64 - 2.17 (m, 5H). MS: 659.1 (M+l)+.
Example 27. (S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxo -ethyl)- l-(4-cyanopyridin-2-yl)-N-(3-fluoro-4-(pyridin-2-yl)phenyl)-5-oxopyrrolidine-2- carboxamide - Compound 313
Compound 313 was prepared according to the following scheme, using the following protocol.
Step A:l-(4-Cyano-pyridin-2-yl)-5-oxo-pyrrolidine-2-carboxylic acid [(2-chloro- phenyl)-(3,3- difluoro-cyclobutylcarbamoyl)-methyl]-[3-fluoro^-(4,4,5,5 etramethyl-[l,3,2]dioxabo yl)-phenyl]-amide. To a solution of(S)-N-(4-bromo-3-fluoro -phenyl)-N-((S)-l-(2- chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4-cyanopyridin-2-yl)-5- oxopyrrolidine-2-carboxamide (500 mg, 0.758 mmol) in 1.4- dioxane (10 mL) was added
Pd(dppf)Cl2 (110.8 mg, 0.152 mmol), CH3COOK (185.6 mg, 1.90 mmol), and
bis(pinacolato)diboron (384.7 mg, 1.516 mmol). The mixture was stirred at 80 °C overnight and filtered. The filtrate was evaporated under reduced pressure and the residue was purified by a standard methodto give the desired product. MS: 708.2 (M+l)+.
Step B: Compound 313. To a solution of l-(4-cyano -pyridin-2-yl)-5-oxo-pyrrolidine-2- carboxylic acid [(2-chloro-phenyl)- (3,3-difluoro -cyclobutylcarbamoyl)-methyl]-[3-fluoro-4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-phenyl]-amide (650 mg, 0.919 mmol) and 2- bromo-pyridine (159.7 mg, 1.01 mmol) in 1.4-dioxane/H20 (7 mL/3 drops) was added
Pd(dppf)Cl2 (67.3 mg, 0.092 mmol), Cs2C03 (449.5 mg,1.38 mmol). The mixture was stirred at 95 °C for 3 hr and filtered. The filtrate was evaporated under reduced pressure and the residue was purified by a standard method to give the desired product. H NMR (400 MHz, CDC13): δ 8.71 (d, J = 11.6 Hz, 2H), 8.51 (s, 1H), 7.99 - 7.64 (m, 4H), 7.32 (s, 2H), 7.10 (m, 4H), 6.45 (m, 1H), 6.15 (m, 1H), 4.95 (d, J= 6.8 Hz, 1H), 4.34 (m, 1H), 3.09 - 2.81 (m, 3H), 2.61 - 2.20 (m, 4H), 2.12 - 2.00 (m, 1H). MS: 659.1 (M+l)+.
The following analog was synthesized via the procedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substituted-aromatic ring or heteroaromatic ring using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
(2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxoethyl)-l-(4-cyanopyridin- -yl)-N-(3-fluoro-5-(pyridin-2-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound314
Ή NMR (400 MHz, DMSO-d6): δ 8.84 - 8.64 (m, 3H), 8.65 - 8.24 (m, 1H), 8.03 - 7.60 (m, 3H), 7.53 - 7.31 (m, 3H), 7.26 - 6.95 (m, 3H), 6.80 (m, 1H), 6.43 (m, 1H), 5.12 - 4.85 (m, 1H), 4.28 (m, 1H), 2.85 (m, 3H), 2.67 - 1.98 (m, 5H). MS: 659 (M+l)+.
Example 28.(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxo -ethyl)- l-(4-cyanopyridin-2-yl)-N-(3-fluoro-5-(isoxazol-5-yl)phenyl)-5-oxopyrrolidine-2- carboxamide - Compound 315
Compound 315 was prepared according to the following scheme, using the following protocol.
Step A: 3-(dibenzylamino)-5-fluorobenzonitrile. A mixture of 3-amino-5-fluoro -benzonitrile (13.6 g, 0.1 mol), K2C03 (62.1 g,0.3 mol), BnBr (51.4 g, 0.3 mol) in CH3CN (150 mL) was stirred at 78 °C overnight. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by a standard method to give the desired product. MS: 317.1 (M+l)+.
Step B:l-(3-(dibenzylamino)-5-fluorophenyl)ethanone.To a mixture of 3-(dibenzyl -amino)-5- fluorobenzonitrile (16 g, 0.05 mol) in anhydrous THF (150 mL) at room temperature was dropwise added CH3MgBr (1 N solution in THF, 60 mL, 0.06 mol). The resulting mixture was stirred at 80 °C for 4 hr and then cooled down. The mixture was poured into 2N HC1 (68 mL), followed by addition of methanol (68 mL). The mixture was concentrated and the residue was extracted with EtOAc (3 x 100 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na2S04 and then concentrated in high vacuum. The residue was purified by a standard method to give the desired product. MS: 334.1 (M+l)+.
Step C:(E)-l-(3-(dibenzyl mino)-5-fluorophenyl)-3-(dimethylamino)prop-2-en -1-one. A mixture of l -(3-(dibenzylamino)-5-fluorophenyl)ethanone (2 g, 6 mmol), DMF-DMA (860 mg, 7.2 mmol) in toluene (30 mL) was stirred at 120 °C for 8 hr. The mixture was concentrated in high vacuum and the residue was purified by a standard method to give the desired product. MS: 389.2 (M+1 )+.
Step D: N,N-dibenzyl-3-fluoro-5-(isoxazol-5-yl)aniline. A mixture of (E)-l -(3-(di - benzylamino)-5-fluorophenyl)-3-(dimethylamino)prop-2-en- 1 -one (1.5 g, 3.86 mmol), hydroxylamine hydrochloride (534 mg, 7.73 mmol) and pyridine (611 mg, 7.73 mmol) in ethanol (20 mL) was stirred at 78 °C overnight. The resulting mixture was evaporated under reduced pressure and the residue was purified by a standard method to give the desired product. MS: 359.1 (M+l )+.
Step E:3-Fluoro-5-isoxazol-5-yl-phenylamine. 10% Pd/C (360 mg) was added to a solution of dibenzyl-(3-fluoro-5-isoxazol-5-yl-phenyl)-amine (200 mg, 0.559 mmol) in a mixed solvent composed by EtOAc (36 mL), MeOH (15 mL) and H20 (7.5 mL). 6 drops of aq. HC1 (6 N) was then added into the above suspension and the resulting reaction mixture was stirred at 25°C for 1 hr. The mixture was filtered through Celite. The filtrate was evaporated under reduced pressure and the residue was purified by a standard method to give the desired product. MS: 179.0 (M+l)+. Step F:(2S)-N-(l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)- N-(3- fluoro-5-(isoxazol-5-yl)phenyl)-5-oxopyrrolidine-2-carboxamide.2-Ch\orob^ (104 mg, 0.74 mmol), 3-fluoro-5-isoxazol-5-yl-phenylamine (132 mg, 0.74 mmol), (S)-5- oxopyrrolidine-2-carboxylic acid (95 mg, 0.74 mmol) and l ,l -difluoro-3-isocyanocyclobutane (87 mg, 0.74 mmol) were used in the general UGI reaction to afford the desired product. MS: 547.1 (M+l )+.
Step G: Compound 315. (2<Sr)-N-(l -(2-chloro -phenyl)-2-(3,3-difluorocyclobutylamino)-2- oxoethyl)-N-(3-fluoro-5-(isoxazol-5-yl)phenyl)-5-oxopyrrolidine-2-carboxamide (200 mg, 0.37 mmol), 2-bromopyrimidine (102 mg, 0.56 mmol), Cs2C03 (240 mg, 0.74 mmol), Pd2(dba)3 (37 mg, 0.04 mmol) and Xant-Phos (22 mg, 0.03 mmol) in 1 ,4-dioxane (15 mL) were stirred under N2 at 80 °C overnight and then filtered. The filtrate was concentrated under reduced pressure and the residue was purified by a standard method to give the desired product. ι~Ά NMR (400 MHz, CDC13): δ 8.69-8.17 (m, 3H), 7.80-7.28 (m, 3H), 7.25-6.93 (m, 5H), 6.63-6.30 (m, 3H), 4.96-
4.92 (m, 1H), 4.37-4.34 (m, 1H), 3.06-2.83 (m, 3H), 2.58-2.21 (m, 4H), 2.08-2.02 (m, 1H). MS: 649.1 (M+l)+.
Example 29.(2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-ox oethyl) -l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(oxazol-5-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 316
Compound?! 6 was prepared according to the following scheme, using the following protocol.
Step A: 5-(3-fluoro-5-nitrophenyl)oxazole. To a solution of 3-fluoro-5-nitrobenz -aldehyde (340 mg, 2.0 mmol) and 2-tosylacetonitrile (430 mg, 2.2 mmol) in MeOH (25 mL) and DME (25 mL) was added Amberlyst A26 OH-resin (3.7 g). The mixture was heated under reflux for 1 hr and cooled to r.t. The resin was filtered and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give the crude product which was used directly in the next step. MS: 209.1 (M+l)+.
Step B: 3-fluoro-5-(oxazol-5-yl)aniline. To a solution of 5-(3-fluoro-5-nitrophenyl) -oxazole (400 mg, 2.0 mmol) in TFA (10 mL) was added Zn powder (380 mg, 6.0 mmol). The mixture was stirred at room temperature for 2 hr and poured into ice. The resulting mixture was neutralized with NH3.H20 to pH= 10 and then extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2S04 and then concentrated in high vacuum. The residue was purified by a standard method to afford the desired product. MS: 179.0 (M+l)+.
Step C: Compound 316. 2-Chlorobenzaldehyde (104 mg, 0.74 mmol), 3-fluoro-5-(oxazol-5- yl)aniline(132 mg, 0.74 mmol), (S)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxylic acid (171 mg, 0.74 mmol) and 1,1-difluoro- 3-isocyanocyclobutane (87 mg, 0.74 mmol) were used in the general UGI reaction to afford the desired product. H NMR (400 MHz, CDC13): δ 8.63 (m, 2H), 8.12 - 7.82 (m, 1H), 7.78 - 7.30 (m, 2H), 7.26 (t, J= 7.4 Hz, 2H), 7.15 (m, 2H), 7.12 - 6.83 (m, 2H), 6.70 - 6.16 (m, 2H), 5.03 - 4.74 (m, 1H), 4.24 (m, 1H), 2.93 (m, 2H), 2.63 (m, 2H), 2.20 (m, 3H). MS: 649.1 (M+l)+.
The following analog was synthesized via the procedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substituted-aromatic ring or heteroaromatic ring using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(oxazol-5-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 317
XH NMR (400 MHz, CDC13): δ 8.68 (s, 1H), 8.60 - 8.45 (m, 1H), 8.06 -7.68 (m, 2H), 7.43 (s, 1H), 7.34 - 7.10 (m, 4H), 7.04 - 6.93 (m, 2H), 6.54 (d, J= 6.9 Hz, 1H), 6.43 (d, J= 11.5 Hz, 1H), 4.95 (t, J = 6.5 Hz, 1H), 4.35 (m, 1H), 3.1 1 - 2.74 (m, 3H), 2.66 - 2.14 (m, 4H), 2.09 - 1.97 (m, 1H). MS: 649.1 (M+l)+.
Example 30.(2S)-N-(l-(2-chlorophenyl)-2-(3,3-difluorocyclobutylamino)-2-oxo ethyl) -l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(oxazol-2-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 318
Compound318was prepared according to the following scheme, using the following protocol.
Step A: 2-(3-fluoro-5-nitrophenyl)oxazole. A mixture of 3-fluoro-5-nitrobenz -aldehyde (51 mg, 0.3 mmol) and 2,2-dimethoxyethanamine (32 mg, 0.3 mmol) was stirred at 110 °C for 2 hr and cooled to give a crude intermediate of (E)-N-(3-fluoro-5-nitrobenzylidene)-2,2- dimethoxyethanamine. A solution of 20 mg of the above intermediate in 0.5 mL of H2S04was added into a mixture of 18 mg of P205 in 0.1 mL of H2S04. The resulting mixture was heatedto 180 °C for 20 min, cooled down and neutralized with NH4OH to give the crude product which was used directly without further purification. MS: 209.1 (M+l)+.
Step B: 3-fluoro-5-(oxazol-2-yl)aniline.To a solution of 2-(3-fluoro-5-nitrophenyl) -oxazole (380 mg, 1.8 mmol) in TFA (10 mL) was added Zn powder (358 mg, 5.5 mmol). The mixture was stirred at r.t. for 2 hr and poured into ice. The resulting mixture was neutralized with
NH3.H20 to pH=10 and then extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2S04 and then concentrated in high vacuum. The residue was purified by a standard method to afford the desired product. MS: 179.0
(M+l)+.
Step C: Compound 31 #.2-Chlorobenzaldehyde (104 mg, 0.74 mmol), 3-fluoro-5-(oxazol-2- yl)aniline(132 mg, 0.74 mmol), (S)-l-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxylic acid (171 mg, 0.74 mmol) and l,l-difluoro-3-isocyano cyclobutane (87 mg, 0.74 mmol) were used in the general UGI reaction to afford the desired product. XH NMR (400 MHz, CDC13): δ 8.70 (m, 2H), 8.56 - 7.83 (m, 1H), 7.65 (m, 2H), 7.47 - 7.27 (m, 2H), 7.25 - 7.09 (m, 2H), 7.10 - 6.91 (m, 2H), 6.76 - 6.18 (m, 2H), 5.06 - 4.71 (m, 1H), 4.25 (m, 1H), 3.12 - 2.64 (m, 3H), 2.43 (m, 3H), 2.09 (m, 2H). MS: 649.1 (M+l)+.
The following analog was synthesized via the procedures set forth above, using the appropriate aldehyde, amine, carboxylic acid, isocyanide and halo-substituted-aromatic ring or heteroaromatic ring using the reagents and solvents set forth above or similar reagents and solvents thereof, and purified via standard methods.
(S)-N-((S)-l-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-l-(4- cyanopyridin-2-yl)-N-(3-fluoro-5-(oxazol-2-yl)phenyl)-5-oxopyrrolidine-2-carboxamide - Compound 319
1H NMR (400 MHz, CDC13): δ 8.82 - 8.26 (m, 2H), 7.94 - 7.46 (m, 3H), 7.38 - 6.81 (m, 6H), 6.44 (m, 1H), 6.20 (dd, J= 14.9, 6.9 Hz, 1H), 4.94 (m, 1H), 4.36 (m, 1H), 3.15 - 2.70 (m, 3H), 2.62 - 1.86 (m, 5H).
Example 31. (2S)-N-((lS)-l-(2-chlorophenyl)-2-(6,6-difluorospiro[3.3]heptan-2-yl amino)-2- oxoethyl)-l-(4-cyanopyridin-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2- carboxamide - Compound 320
Compound320was prepared according to the following scheme, using the following protocol.
Step A:N-(6,6-difluorospiro[3.3]heptan-2-yl)formamide. To a suspension of 6,6- difluorospiro[3.3]heptan-2-amine hydrochloride (500 mg, 2.73 mmol) in HCOOEt (5 mL) was added Et3N (552 mg, 5.47 mmol). The resulting mixture was stirred first at room temperature for
30 min in a sealed tank and then heated to 85 °C overnight. The mixture was concentrated and followed by addition of EtOAc (10 mL). The resulting suspension was stirred at room
temperaturefor 30 min and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by a standard method to afford the desired product. 1H NMR (400 MHz, CDC13): δ 8.06 (s, 1H), 5.71 (s, 1H), 4.42 (m, 1H), 2.74 - 2.45 (m, 8H).
Step B:2,2-difluoro-6-isocyanospiro[3.3]heptanes. A solution of N-(6,6- difluorospiro[3.3]heptan-2-yl)formamide (390 mg, 2.23 mmol), PPh3 (642 mg, 2.45 mmol), CC14 (339 mg, 2.23 mmol), and Et3N (225 mg, 2.23 mmol) in DCM (10 mL) was heated at 45 °C overnight. The mixture was concentrated under reduced pressure. The residue was suspended in Et20 (10 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by a standard method to afford the desired product. H NMR (400 MHz, CDC13): δ 4.01 - 3.85 (m, 1H), 2.80 - 2.37 (m, 8H).
Step C:(2S)-N-(l-(2-chlorophenyl)-2-(6, 6-difluorospiro[3.3]heptan-2-ylamino)-2- oxoethyl)-N- (5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide.2-Ch\orobenz -aldehyde (104 mg, 0.74 mmol), 5-fluoropyridin-3-amine (83 mg, 0.74 mmol), (S)-5-oxopyrrolidine-2-carboxylic acid (95 mg, 0.74 mmol) and 2,2-difluoro-6-isocyanospiro[3.3]heptane (116 mg, 0.74 mmol) were used in the general UGI reaction to afford the desired product.MS: 521 (M+l)+.
Step D:Cotnpound 320was synthesized via the general procedure for Buchwald reaction set forth above.1H NMR (400 MHz, CDC13): δ 8.97 (s, 0.5H), 8.70 (s, 1H), 8.48 (d, J= 4.6 Hz, 1H), 8.41 - 8.22 (m, 1.5H), 8.15 (d, J= 8.7 Hz, 1H), 7.31 (d, J= 7.1 Hz, 1H), 7.25 (d, J= 4.9 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.03 (t, J= 7.3 Hz, 1H), 6.91 (m, 1H), 6.42 (m, 1H), 6.05 (m, 1H), 4.83 (m, 1H), 4.38 (m, 1 1H), 2.97 - 2.75 (m, 1H), 2.68 - 2.11 (m, 8H), 2.10 - 1.82 (m, 3H). MS: 623.1 (M+l)+.
Example A: In Vitro Assays for IDHlm (R132H or R132C) Inhibitors
A test compound is prepared as 10 mM stock in DMSO and diluted to 50X final concentration in DMSO, for a 50 μΐ reaction mixture. IDH enzyme activity converting alpha- ketoglutarate to 2-hydroxyglutaric acid is measured using a NADPH depletion assay. In the assay the remaining cofactor is measured at the end of the reaction with the addition of a catalytic excess of diaphorase and resazurin, to generate a fluorescent signal in proportion to the
amount of NADPH remaining. IDH1 -R132 homodimer enzyme is diluted to 0.125 μg/ml in 40 μΐ of Assay Buffer(l 50 mM NaCl, 20 mM Tris-Cl pH 7.5, 10 mM MgCl2, 0.05% BSA, 2 mM b- mercaptoethanol); 1 μΐ of test compound dilution in DMSO is added and the mixture is incubated for 60 minutes at room temperature. The reaction is started with the addition of 10 μΐ of
Substrate Mix (20 μΐ NADPH, 5 mM alpha-ketoglutarate, in Assay Buffer) and the mixture is incubated for 90 minutes at room temperature. The reaction is terminated with the addition of 25 μΐ of Detection Buffer (36 μg/ml diaphorase, 30 mM resazurin, in IX Assay Buffer), and is incubated for 1 minute before reading on a SpectraMax platereader at Ex544/Em590.
Compounds are assayed for their activity against IDH1 R132C following the same assay as abovewith the following modifications: Assay Buffer is (50 mM potassium phosphate, pH 6.5; 40 mM sodium carbonate, 5 mM MgCl2, 10% glycerol, 2 mM b-mercaptoethanol, and0.03% BSA). The concentration of NADPH and alpha-ketoglutarate in the Substrate Buffer is 20 μΜ and 1 mM, respectively.
Representative compounds of formula I set forth in Table lwere tested in this assay or a similar assay and the results are set forth below in Table 3. As used in Table 3, "A" refers to an inhibitory activity against IDH1 R132H or IDH1 R132Cwith an IC50< 0.1 μΜ; "B" refers to an inhibitory activity against IDH1 R132H or IDH1 R132Cwith an IC50 between 0.1 μΜ and 0.5 μΜ; "C" refers to an inhibitory activity against IDH1 R132H or IDH1 R132Cwith an IC50 between 0.5 μΜ and 1 μΜ; "D" refers to an inhibitory activity against IDH1 R132H or IDH1 R132C with an IC50 between 1 μΜ and 2 μΜ.
Table 3. Inhibitory Activities of Representative Compounds of formula I
Example B: Cellular Assays for IDHlm (R132H or R132C) Inhibitors
Cells (HT1080 or U87MG) are grown in T125 flasks in DMEM containing 10% FBS, lx penicillin/streptomycin and 500ug/mL G418 (present in U87MG cells only). They areharvested by trypsin and seeded into 96 well white bottom plates at a density of 5000 cell/well in 100 ul/well in DMEM with 10% FBS. No cells are placed in columns 1 and 12. Cells areincubated overnight at 37°C in 5% C02. The next day test compounds aremade up at 2x the final concentration and l OOul areadded to each cell well. The final concentration of DMSO is 0.2% and the DMSO control wells are plated in row G. The plates are then placed in the incubator for 48 hours. At 48 hours, l OOul of media is removed from each well and analyzed by LC-MS for 2- HG concentrations. The cell plate is placed back in the incubator for another 24 hours. At 72 hours post compound addition, 10 mL/plate of Promega Cell Titer Glo reagent is thawed and mixed. The cell plate is removed from the incubator and allowed to equilibrate to
roomtemperature. Then lOOul of Promega Cell Titer Glo reagent is added to each well of media. The cell plate is then placed on an orbital shaker for 10 minutes and then allowed to sit at room temperature for 20 minutes. The plate is then read for luminescence with an integration time of 500ms.
The ICsofor inhibition of 2-HG production(concentration of test compound to reduce 2HG production by 50% compared to control) in these two cell lines for various compounds of formula I is set forth in Table 2 above. As used in Table 2, "A" refers to an IC5ofor inhibition of
2-HG production< 0.1 μΜ; "B" refers to an IC5ofor inhibition of 2-HG productionbetween 0.1 μΜ and 0.5 μΜ; "C" refers to an IC5ofor inhibition of 2-HG productionbetween 0.5μΜ and 1 μΜ; "D" refers to an IC5ofor inhibition of 2-HG productionbetween 1 μΜ and 2μΜ.
Example C: Metabolic Stabilities of Compounds of Formula I
Metabolic stabilities of compounds of formula I can be tested with the following assay and species specific liver microsomes (LM) extraction ratio (Eh) can be calculated:
1.Buffer A: 1.0 L of 0.1 M monobasic Potassium Phosphate buffer containing 1.0 mM
EDTA; Buffer B : 1.0 L of 0.1 M Dibasic Potassium Phosphate buffer containing 1.0 mM EDTA; Buffer C: 0.1 M Potassium Phosphate buffer, 1.0 mM EDTA, pH 7.4 by titrating 700 mL of buffer B with buffer A while monitoring with the pH meter.
2. Reference compounds (Ketanserin) and test compounds spiking solution:
500 μΜ spiking solution: add 10 μί of 10 mM DMSO stock solution into 190 ΐ, CAN;
1.5 μΜ spiking solution in microsomes (0.75 mg/mL): add 1.5 μΐ. of 500 μΜ spiking solution and 18.75 μϋ, of 20 mg/mL liver microsomes into 479.75 μί of Buffer C.
3. NADPH stock solution (6 mM) is prepared by dissolving NADPH into buffer C.
4. Dispense 30 μΐ. 1.5X compound/liver microsome solution in 96-well plate and immediately add 135 μΐ, ACN containing IS before adding 15uL Buffer C to prepare real 0 minute samples.
5. Add 15 μΐ. of NADPH stock solution (6 mM) to the wells designated as Time 30, and start timing.
6. At the end of incubation (0 min), add 135 μΐ. of ACN containing the internal standard
Osalmid) to all the wells (30 min, and 0 min). Then add 15 μΐ. of NADPH stock solution (6 mM) to the wells designated as Time 0.
7. After quenching, centrifuge the reaction mixtures at 3220g for 10 min.
8. Transfer 50 μΐ, of the supernatant from each well into a 96-well sample plate containing 50 μΐ, of ultra pure water (Millipore) for LC/MS analysis.