WO2015012348A1 - エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 - Google Patents
エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 Download PDFInfo
- Publication number
- WO2015012348A1 WO2015012348A1 PCT/JP2014/069550 JP2014069550W WO2015012348A1 WO 2015012348 A1 WO2015012348 A1 WO 2015012348A1 JP 2014069550 W JP2014069550 W JP 2014069550W WO 2015012348 A1 WO2015012348 A1 WO 2015012348A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- epoxy resin
- resin composition
- mass
- parts
- registered trademark
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/24—Di-epoxy compounds carbocyclic
- C08G59/245—Di-epoxy compounds carbocyclic aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3218—Carbocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/5006—Amines aliphatic
- C08G59/502—Polyalkylene polyamines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2400/00—Characterised by the use of unspecified polymers
- C08J2400/22—Thermoplastic resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2400/00—Characterised by the use of unspecified polymers
- C08J2400/26—Elastomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2453/00—Characterised by the use of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2477/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2481/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing sulfur with or without nitrogen, oxygen, or carbon only; Polysulfones; Derivatives of such polymers
- C08J2481/06—Polysulfones; Polyethersulfones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
- C08L2205/025—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group containing two or more polymers of the same hierarchy C08L, and differing only in parameters such as density, comonomer content, molecular weight, structure
Definitions
- the present invention relates to a fiber-reinforced composite material suitable for aerospace use, a prepreg for obtaining the same, and an epoxy resin composition suitably used as a matrix resin thereof.
- fiber reinforced composite materials using reinforced fibers such as carbon fibers and aramid fibers have utilized their high specific strength and specific elastic modulus to make sports such as aircraft and automobile structural materials, tennis rackets, golf shafts and fishing rods. It has been used for applications and general industrial applications.
- a prepreg that is a sheet-like intermediate material in which reinforcing fibers are impregnated with an uncured matrix resin is used, and a plurality of the prepregs are laminated and then heat-cured.
- a resin transfer molding method is used in which a liquid resin is poured into the arranged reinforcing fibers and then the resin is heated and cured.
- the method using a prepreg has an advantage that it is easy to obtain a high-performance fiber-reinforced composite material because the orientation of the reinforcing fibers can be strictly controlled and the design freedom of the laminated structure is high.
- a thermosetting resin is mainly used from the viewpoint of heat resistance and productivity.
- an epoxy resin is preferably used from the viewpoints of adhesive properties between the resin and the reinforcing fibers, dimensional stability, and mechanical properties such as strength and rigidity of the obtained composite material.
- amine-type epoxy resins that have a low epoxy equivalent and a high crosslink density can be used as a matrix resin for fiber-reinforced composite materials for aerospace applications that require excellent strength characteristics and durability stability.
- a cured resin product having a high elastic modulus and high heat resistance was obtained, but there was a tendency that the cured resin product had a small deformation ability and low toughness.
- Patent Document 3 discloses that toughness and heat resistance can be obtained by using an amine-type epoxy resin and a fluorene-type epoxy resin in combination.
- the elastic modulus of the cured resin and the strength characteristics as a fiber-reinforced composite material may still be insufficient.
- Patent Document 5 discloses that extremely high heat resistance and elastic modulus can be achieved by curing a fluorene type epoxy resin into which a condensed polycyclic group is introduced with a phenol novolac resin.
- the obtained resin cured product was low in elongation and brittle, and did not lead to a significant increase in toughness.
- nothing is mentioned about the compounding amount and the combination with other components, and it can be said that there is no knowledge about the physical properties of the epoxy resin composition obtained by using them.
- JP 2007-1514160 A International Publication No. 2010/035859 JP 2005-298815 A JP 2012-102228 A
- An object of the present invention is to provide an epoxy resin composition that provides a cured product having excellent heat resistance, toughness, and elastic modulus.
- the present invention is an epoxy resin composition
- an epoxy resin composition comprising the following components [A], [B] and [C]:
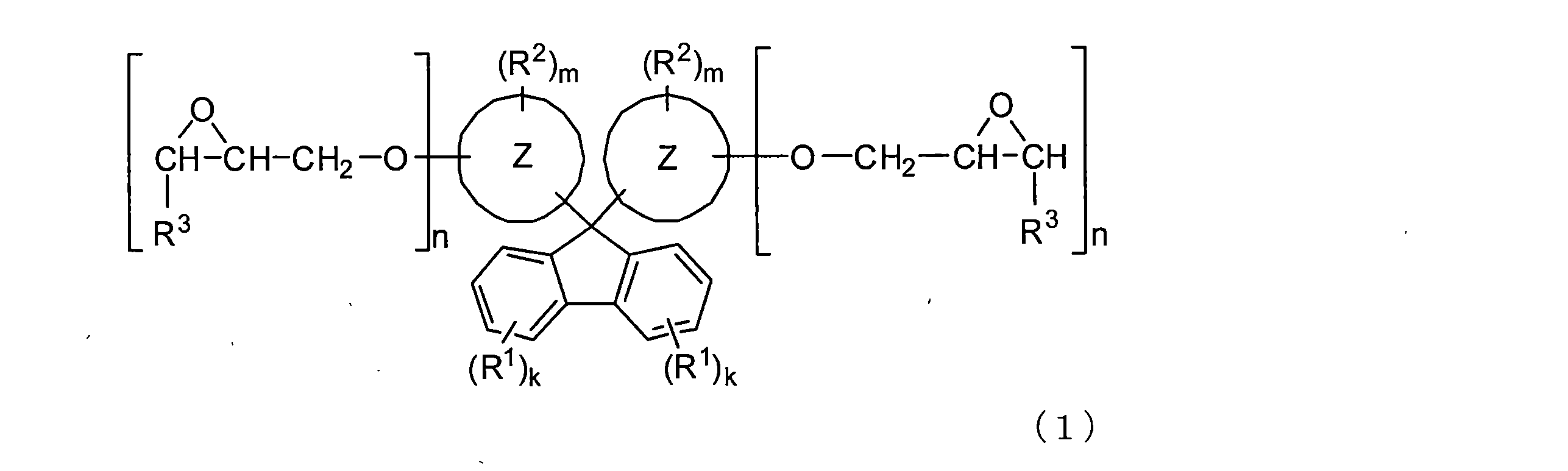
- ring Z is a condensed polycyclic aromatic hydrocarbon ring
- R 1 and R 2 are substituents
- R 3 is a hydrogen atom or a methyl group
- k is an integer of 0 to 4
- m is 0 or more.
- N is an integer of 1 or more;
- [B] At least one resin selected from the group consisting of the following [Bx], [By] and [Bz]; [Bx] bisphenol type epoxy resin; [By] amine-type epoxy resin; [Bz] thermoplastic resin; [C] Polyamine curing agent.
- a first preferred embodiment of the present invention is the above-mentioned epoxy resin composition, comprising the constituent elements [A], [Bx] and [C], and [A] in 100 parts by mass of the total amount of the epoxy resin.
- An epoxy resin composition comprising 10 to 70 parts by mass and 20 to 80 parts by mass of [Bx].
- the second preferred embodiment of the present invention is the epoxy resin composition described above, which is composed of the constituent elements [A], [By] and [C].
- a third preferred embodiment of the present invention is the epoxy resin composition described above, comprising the constituent elements [A], [Bz] and [C], and the glass transition temperature (Tg) of the thermoplastic resin [Bz].
- Tg glass transition temperature
- Another aspect of the present invention is a prepreg obtained by impregnating a reinforcing fiber with any of the epoxy resin compositions described above.
- Another aspect of the present invention is a fiber-reinforced composite material including reinforcing fibers and a cured product of the epoxy resin composition described above.
- an epoxy resin composition that provides a cured product having excellent heat resistance, toughness, and elastic modulus can be obtained.
- an epoxy resin composition that provides a cured product having further excellent heat and moisture resistance can be obtained.
- an epoxy resin composition giving a cured product exhibiting extremely high toughness can be obtained.
- the epoxy resin composition of the present invention comprises the following components [A], [B] and [C]: [A] an epoxy resin having a structure represented by the formula (1);
- ring Z is a condensed polycyclic aromatic hydrocarbon ring
- R 1 and R 2 are substituents
- R 3 is a hydrogen atom or a methyl group
- k is an integer of 0 to 4
- m is 0 or more.
- N is an integer of 1 or more;
- [B] At least one resin selected from the group consisting of the following [Bx], [By] and [Bz]; [Bx] bisphenol type epoxy resin; [By] amine-type epoxy resin; [Bz] thermoplastic resin; [C] Polyamine curing agent.
- an epoxy resin composition that gives a cured product having excellent heat resistance, toughness, and elastic modulus can be obtained.
- an epoxy resin composition that gives a cured product having excellent heat resistance, toughness, and elastic modulus can be obtained.
- a fiber-reinforced composite material having excellent compressive strength and interlayer toughness can be obtained.
- the epoxy resin composition contains the epoxy resin [A] having the structure represented by the formula (1), water absorption of the cured product is suppressed even in a high temperature and high humidity environment, and heat resistance and resin elastic modulus are maintained. it can. Thereby, the high compressive strength of the fiber-reinforced composite material obtained can be maintained.
- the condensed polycyclic aromatic hydrocarbon ring represented by ring Z is a condensed bicyclic hydrocarbon ring (preferably a C 8-20 condensed bicyclic ring such as an indene ring or a naphthalene ring). Hydrocarbon rings, more preferably C 10-16 condensed bicyclic hydrocarbon rings), condensed tricyclic hydrocarbon rings (preferably anthracene rings, phenanthrene rings, etc.), etc. Is mentioned.
- Preferred examples of the condensed polycyclic aromatic hydrocarbon ring include a naphthalene ring and an anthracene ring, and a naphthalene ring is particularly preferable.
- the two rings Z may be the same ring or different rings.
- the bonding position of the ring Z bonded to the 9-position of fluorene is not particularly limited.
- the ring Z is a naphthyl ring
- either 1-naphthyl or 2-naphthyl may be used.
- 2-naphthyl is particularly preferable.
- examples of the substituent R 1 include non-reactive substituents such as a cyano group, a halogen atom, and a hydrocarbon group. Of these, a halogen atom, a cyano group or an alkyl group is preferable, and an alkyl group is particularly preferable.
- halogen atom examples include a fluorine atom, a chlorine atom and a bromine atom.
- Examples of the hydrocarbon group include an alkyl group and an aryl group.
- the alkyl group a C 1-6 alkyl group is preferable, and a C 1-4 alkyl group is more preferable.
- Specific examples of the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, and a t-butyl group, and a methyl group is particularly preferable.
- As the aryl group a C 6-10 aryl group is preferable, and a phenyl group is particularly preferable.
- the groups R 1 may be different from each other or the same.
- the groups R 1 substituted on the two benzene rings constituting the fluorene may be the same or different.
- the bonding position (substitution position) of the group R 1 with respect to the benzene ring constituting the fluorene is not particularly limited.
- a preferred substitution number k is 0 to 1, and 0 is particularly preferred.
- the substitution number k may be the same as or different from each other.
- substituent R 2 examples include hydrocarbon groups such as alkyl groups, cycloalkyl groups, aryl groups, and aralkyl groups; groups represented by the general formula —OR 4 such as alkoxy groups, cycloalkoxy groups, and aryloxy groups ( R 4 represents the hydrocarbon group exemplified above); a group represented by the general formula —SR 4 such as an alkylthio group (R 4 is the same as above); an acyl group; an alkoxycarbonyl group; a halogen atom; a hydroxyl group; Nitro group; cyano group; substituted amino group and the like.
- hydrocarbon groups such as alkyl groups, cycloalkyl groups, aryl groups, and aralkyl groups
- groups represented by the general formula —OR 4 such as alkoxy groups, cycloalkoxy groups, and aryloxy groups
- R 4 represents the hydrocarbon group exemplified above
- SR 4 such as an alkylthio group (
- the alkyl group is preferably a C 1-12 alkyl group, more preferably a C 1-8 alkyl group, still more preferably a C 1-6 alkyl group.
- Specific examples of the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, and a butyl group.
- the cyclohexyl group is preferably a C 5-8 cycloalkyl group, more preferably a C 5-6 cycloalkyl group.
- the aryl group is preferably a C 6-14 aryl group, more preferably a C 6-10 aryl group, still more preferably a C 6-8 aryl group.
- a phenyl group, a tolyl group, a xylyl group and the like are preferable.
- the aralkyl group is preferably a C 6-10 aryl-C 1-4 alkyl group such as a benzyl group or a phenethyl group.
- alkoxy group a C 1-8 alkoxy group is preferable, and a C 1-6 alkoxy group is more preferable. Specific examples include a methoxy group.
- cycloalkoxy group a C 5-10 cycloalkyloxy group is preferable.
- aryloxy group a C 6-10 aryloxy group is preferable.
- alkylthio group a C 1-8 alkylthio group is preferable, and a C 1-6 alkylthio group is more preferable. Specific examples include a methylthio group.
- acyl group a C 1-6 acyl group is preferable. Specific examples include an acetyl group.
- alkoxycarbonyl group a C 1-4 alkoxy-carbonyl group is preferable. Specific examples include a methoxycarbonyl group.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- substituted amino groups include dialkylamino groups.
- alkyl include the above alkyl groups.
- a specific example is a dimethylamino group.
- the group R 2 includes a group selected from a hydrocarbon group, an alkoxy group, a cycloalkoxy group, an aryloxy group, an aralkyloxy group, an acyl group, a halogen atom, a nitro group, a cyano group, and a substituted amino group.
- a particularly preferred group R 2 is a group selected from a hydrocarbon group, an alkoxy group and a halogen atom.
- the groups R 2 may be different from each other or the same.
- the groups R 2 may be the same or different.
- the substitution number m is preferably 0 to 8, more preferably 0 to 6, more preferably 1 to 5, still more preferably 0 to 4, particularly preferably 0 to 2, and most preferably 0 to 1.
- the number of substitutions m may be the same or different from each other.
- the group R 3 is a hydrogen atom or a methyl group, preferably a hydrogen atom.
- the number of substitution n is 1 or more, preferably 1 to 4, more preferably 1 to 3, further preferably 1 to 2, particularly preferably 1.
- the number of substitutions n may be the same or different in each ring Z.
- the bonding position of the epoxy group with respect to ring Z is not particularly limited as long as it is bonded to an appropriate position of ring Z.
- the epoxy group is a hydrocarbon ring other than the hydrocarbon ring bonded to the 9th position of fluorene in the condensed polycyclic hydrocarbon ring constituting the ring Z (for example, the 5th and 6th positions of the naphthalene ring). It is preferable that it is couple
- Specific examples of the compound represented by the formula (1) include compounds in which n is 1 in the formula (1) such as 9,9-bis (glycidyloxynaphthyl) fluorene.
- Specific examples include 9,9-bis (6-glycidyloxy-2-naphthyl) fluorene, 9,9-bis (5-glycidyloxy-1-naphthyl) fluorene, and the like.
- the compound represented by the formula (1) is not particularly limited.
- the compound represented by the following formula (2) for example, 9,9-bis (hydroxynaphthyl) fluorene
- the following formula (3) It can manufacture by making it react with the compound represented. For example, it can be produced by the method described in JP2012-102228A.
- X represents a halogen atom.
- Z, R 1 , R 2 , k, m and n are the same as in the above formula (1).
- a halogen atom a chlorine atom, a bromine atom, an iodine atom, etc. are mentioned, A chlorine atom or a bromine atom is preferable, and a chlorine atom is especially preferable.
- Specific examples of the compound represented by the formula (3) include epihalohydrin (also referred to as halomethyloxirane), 1-halomethyl-2-methyloxirane, and the like.
- epihalohydrin examples include epichlorohydrin (chloromethyloxirane), epibromohydrin (bromomethyloxirane), and the like.
- 1-halomethyl-2-methyloxirane examples include 1-chloromethyl-2-methyloxirane.
- the epoxy resin composition contains the bisphenol type epoxy resin [Bx], the crosslink density of the cured product can be reduced while maintaining heat resistance, and a highly tough resin cured product can be obtained.
- the bisphenol type epoxy resin [Bx] is not particularly limited, and bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol AD type epoxy resin, bisphenol S type epoxy resin, or these bisphenol skeletons are halogen-substituted. , Alkyl-substituted, hydrogenated, etc. are used. Specific examples of such an epoxy resin include the following.
- bisphenol A type epoxy resins include “Epototo (registered trademark)” YD128 (manufactured by Nippon Steel & Sumikin Chemical Co., Ltd.), “jER (registered trademark)” 825, “jER (registered trademark)” 828, “ jER (registered trademark) 834, jER (registered trademark) 1001, jER (registered trademark) 1004, jER (registered trademark) 1007, jER (registered trademark) 1009, jER (registered trademark) "1010 (above, manufactured by Mitsubishi Chemical Corporation)”.
- Examples of commercially available bisphenol S-type epoxy resins include “Epiclon (registered trademark)” EXA-1514 (manufactured by DIC Corporation).
- the liquid bisphenol F type epoxy resin is particularly preferably used because it can increase the elastic modulus of the obtained cured product and can maintain heat resistance.
- the liquid bisphenol F type epoxy resin means a bisphenol F type epoxy resin that is liquid at room temperature (25 ° C.).
- the term “liquid” as used herein means that when a piece of metal having a specific gravity of 7 or more at the same temperature as the epoxy resin to be measured is placed on the epoxy resin and immediately buried under gravity, the epoxy resin is liquid. Define. Examples of the metal having a specific gravity of 7 or more include iron (steel), cast iron, copper, and the like.
- Examples of preferably used amine type epoxy resins [By] include tetraglycidyldiaminodiphenylmethane, tetraglycidyldiaminodiphenylsulfone, triglycidylaminophenol, triglycidylaminocresol, diglycidylaniline, diglycidyltoluidine, tetraglycidylxylylenediamine, These halogen-substituted products, alkyl-substituted products, hydrogenated products and the like can be mentioned.
- polyfunctional amine-type epoxy resins having three or more glycidyl groups are preferable, and tetraglycidyldiaminodiphenylmethane and triglycidylaminophenol are more preferable.
- Examples of commercially available tetraglycidyl diaminodiphenyl sulfone include TG3DAS (manufactured by Mitsui Chemicals Fine Co., Ltd.).
- triglycidylaminophenol or triglycidylaminocresol Commercially available products of triglycidylaminophenol or triglycidylaminocresol include “Sumiepoxy (registered trademark)” ELM100, “Sumiepoxy (registered trademark)” ELM120 (manufactured by Sumitomo Chemical Co., Ltd.), “Araldite (registered trademark)” “MY0500”, “Araldite (registered trademark)” MY0510, “Araldite (registered trademark)” MY0600 (manufactured by Huntsman Advanced Materials), “jER (registered trademark)” 630 (manufactured by Mitsubishi Chemical Corporation), etc. Is mentioned.
- Examples of commercially available diglycidyl aniline include GAN (manufactured by Nippon Kayaku Co., Ltd.), PxGAN (manufactured by Toray Fine Chemical Co., Ltd.), and the like.
- Examples of commercially available diglycidyl toluidine include GOT (manufactured by Nippon Kayaku Co., Ltd.).
- the epoxy resin may contain other epoxy resin components. These may be added in combination of not only one type but also a plurality of types. Specifically, phenol novolac type epoxy resin, cresol novolac type epoxy resin, resorcinol type epoxy resin, dicyclopentadiene type epoxy resin, epoxy resin having biphenyl skeleton, urethane and isocyanate modified epoxy resin, epoxy resin having fluorene skeleton, etc. Is mentioned.
- phenol novolac type epoxy resins include “jER (registered trademark)” 152, “jER (registered trademark)” 154 (manufactured by Mitsubishi Chemical Corporation), “Epicron (registered trademark)” N-740, “Epicron (registered trademark)” N-770, “Epicron (registered trademark)” N-775 (manufactured by DIC Corporation), and the like.
- cresol novolac type epoxy resins include “Epicron (registered trademark)” N-660, “Epicron (registered trademark)” N-665, “Epicron (registered trademark)” N-670, “Epicron (registered trademark)” “N-673”, “Epicron (registered trademark)” N-695 (above, manufactured by DIC Corporation), EOCN-1020, EOCN-102S, EOCN-104S (above, manufactured by Nippon Kayaku Co., Ltd.) It is done.
- resorcinol type epoxy resin examples include “Denacol (registered trademark)” EX-201 (manufactured by Nagase ChemteX Corporation).
- dicyclopentadiene type epoxy resins include “Epicron (registered trademark)” HP7200, “Epicron (registered trademark)” HP7200L, “Epicron (registered trademark)” HP7200H (manufactured by DIC Corporation), Tactix558 ( Huntsman Advanced Material), XD-1000-1L, XD-1000-2L (Nippon Kayaku Co., Ltd.).
- epoxy resins having a biphenyl skeleton include “jER (registered trademark)” YX4000H, “jER (registered trademark)” YX4000, “jER (registered trademark)” YL6616 (manufactured by Mitsubishi Chemical Corporation), NC -3000 (manufactured by Nippon Kayaku Co., Ltd.).
- Examples of commercially available urethane and isocyanate-modified epoxy resins include AER4152 (produced by Asahi Kasei E-Materials Co., Ltd.) and ACR1348 (produced by Asahi Denka Co., Ltd.) having an oxazolidone ring.
- epoxy resins having a fluorene skeleton include “ESF (registered trademark)” 300 (manufactured by Nippon Steel & Sumikin Chemical Co., Ltd.), “Oncoat (registered trademark)” EX-1010, and “Oncoat (registered trademark)”.
- thermoplastic resin [Bz] By mixing or dissolving the thermoplastic resin [Bz] in the epoxy resin composition, the brittleness of the epoxy resin is covered with the toughness of the thermoplastic resin [Bz], and the molding of the thermoplastic resin [Bz] is performed. The difficulty is covered with an epoxy resin, making it a well-balanced base resin.
- the thermoplastic resin [Bz] is generally a group consisting of a carbon-carbon bond, an amide bond, an imide bond, an ester bond, an ether bond, a carbonate bond, a urethane bond, a thioether bond, a sulfone bond, and a carbonyl bond in the main chain.
- a thermoplastic resin having a bond selected from Further, the thermoplastic resin [Bz] may have a partially crosslinked structure, and may be crystalline or amorphous.
- polyvinyl formal and polyethersulfone can be suitably used because of their excellent compatibility with epoxy resins.
- examples of commercially available polyvinyl formal include “Denka Formal (registered trademark)” (manufactured by Denki Kagaku Kogyo Co., Ltd.) and “Vinylec (registered trademark)” (manufactured by Chisso Corporation).
- polyethersulfone examples include “Sumika Excel (registered trademark)” PES5200P, “Sumika Excel (registered trademark)” PES4700P, “Sumika Excel (registered trademark)” PES3600P, “Sumika Excel (registered trademark)” PES5003P, “Sumika Excel (registered trademark)” PES5200P, “Sumika Excel (registered trademark)” PES7600P (manufactured by Sumitomo Chemical Co., Ltd.), “Ultrason (registered trademark)” E2020P SR, “Ultrason (registered trademark)” E2021SR ( As described above, BASF Co., Ltd.), “GAFONE (registered trademark)” 3600RP, “GAFONE (registered trademark)” 3000RP (above, Solvay Advanced Polymers Co., Ltd.) and the like can be mentioned.
- polyethersulfone and polyetherethersulfone copolymer oligomers as described in JP-T-2004-506789, and commercially available products of polyetherimide, “Ultem (registered trademark)” 1000, “Ultem ( Registered trademark) "1010,” Ultem (registered trademark) “1040 (above, Solvay Advanced Polymers Co., Ltd.) and the like.
- the oligomer refers to a polymer having a relatively low molecular weight in which about 10 to 100 finite number of monomers are bonded.
- thermoplastic resins soluble in epoxy resins and having hydrogen bonding functional groups include polyvinyl acetal resins such as Denkabutyral and Denka Formal (manufactured by Denki Kagaku Kogyo Co., Ltd.), “Vinylec (registered trademark)” ( Chisso Co., Ltd.), “UCAR (registered trademark)” PKHP (manufactured by Union Carbide) as phenoxy resin, “Macromelt (registered trademark)” (manufactured by Henkel Hakusui Co., Ltd.), “Amilan (registered trademark) as polyamide resin ) “CM4000 (manufactured by Toray Industries, Inc.),“ Ultem (registered trademark) ”(manufactured by General Electrics) as polyimide,“ Matrimid (registered trademark) ”5218 (manufactured by Ciba),“ Victrex (registered trademark) as polysulfone
- the glass transition temperature (Tg) of the thermoplastic resin [Bz] is preferably 150 ° C. or higher, and more preferably 170 ° C. or higher. If the glass transition temperature of the thermoplastic resin [Bz] is less than 150 ° C., it may be easily deformed by heat when used as a molded article.
- thermoplastic resin [Bz] it is also preferable to use a mixture of thermoplastic resin particles or dissolution. By blending the thermoplastic resin particles, the toughness of the matrix resin is improved, and the impact resistance is improved when a fiber-reinforced composite material is obtained.
- thermoplastic resin particles As the material for the thermoplastic resin particles, polyamide is most preferable. Among polyamides, nylon 12, nylon 6, nylon 11, nylon 6/12 copolymer, and epoxy compound described in Example 1 of JP-A-01-104624 are used. Nylon semi-IPN (polymer interpenetrating network structure) (semi-IPN nylon) gives particularly good adhesive strength with a thermosetting resin.
- the shape of the thermoplastic resin particles may be spherical particles, non-spherical particles, or porous particles, but the spherical shape is superior in viscoelasticity because it does not deteriorate the flow characteristics of the resin, and there is no origin of stress concentration. This is a preferred embodiment in terms of giving high impact resistance.
- polyamide particles include SP-500 (manufactured by Toray Industries, Inc.), “Trepearl (registered trademark)” TN (manufactured by Toray Industries, Inc.), and “Orgasol (registered trademark)” 1002D (manufactured by ATOCHEM). ), “Orgasol (registered trademark)” 2002 (manufactured by ATOCHEM), “Orgasol (registered trademark)” 3202 (manufactured by ATOCHEM), Trogamide T5000 and the like.
- the polyamine curing agent [C] is a curing agent for the epoxy resin contained in the epoxy resin composition, and is a compound having an active group capable of reacting with an epoxy group.
- the polyamine curing agent [C] include dicyandiamide, aromatic polyamine, imidazole derivative, aliphatic amine, tetramethylguanidine, thiourea-added amine, and the like.
- aromatic polyamines are preferable, and diaminodiphenyl sulfone or derivatives thereof, or various isomers thereof are more preferable.
- 3,3′-diaminodiphenylsulfone, 4,4′-diaminodiphenylsulfone, and combinations thereof are particularly preferably used because of excellent heat resistance and mechanical properties.
- a combination of dicyandiamide and a urea compound such as 3,4-dichlorophenyl-1,1-dimethylurea or imidazoles as a curing agent, high heat resistance and water resistance can be obtained while curing at a relatively low temperature.
- a latent product of these curing agents for example, a microencapsulated product, the storage stability of the prepreg, in particular, tackiness and draping properties hardly change even when left at room temperature.
- the optimum value of the addition amount of the polyamine curing agent [C] varies depending on the types of the epoxy resin and the curing agent, but the ratio of the active hydrogen amount of the polyamine curing agent [C] to the epoxy group amount of the epoxy resin is 0.6 to By setting the ratio to 1.2, and more preferably from 0.7 to 0.9, a higher elastic modulus resin may be obtained than when used in an equivalent amount.
- These curing agents may be used alone or in combination.
- aromatic polyamines include Seika Cure S (manufactured by Wakayama Seika Kogyo Co., Ltd.), MDA-220 (manufactured by Mitsui Chemicals), “jER Cure (registered trademark)” W (manufactured by Mitsubishi Chemical Corporation).
- these epoxy resin and polyamine curing agent [C], or a product obtained by pre-reacting a part thereof can be blended in the composition.
- This method may be effective for viscosity adjustment and storage stability improvement.
- the epoxy resin composition of this embodiment includes an epoxy resin [A] having a structure represented by the above formula (1), a bisphenol type epoxy resin [Bx], and a polyamine curing agent [C].
- the epoxy resin [A] having a structure represented by the formula (1) is preferably contained in an amount of 10 to 70 parts by mass, and more preferably 30 to 50 parts by mass in 100 parts by mass of the total amount of the epoxy resin. . Within this range, water absorption of the cured product can be suppressed even in a high temperature and high humidity environment, and heat resistance and resin modulus can be maintained. Thereby, the fiber-reinforced composite material obtained has a high compressive strength.
- the bisphenol-type epoxy resin [Bx] is preferably contained in an amount of 20 to 80 parts by mass, more preferably 40 to 60 parts by mass in 100 parts by mass of the total amount of epoxy resin. Within this range, the crosslink density of the cured product can be reduced while maintaining heat resistance, and a highly tough resin cured product can be obtained.
- an epoxy resin composition that gives a cured product having further excellent heat and moisture resistance.
- an epoxy resin composition a fiber-reinforced composite material having excellent compressive strength and interlayer toughness in a high temperature and high humidity environment can be obtained.
- the amine type epoxy resin [By] is further contained in an amount of 10 to 50 parts by mass in 100 parts by mass of the total amount of the epoxy resin.
- blending amine type epoxy resin [By] the elasticity modulus of hardened
- it is less than 10 parts by mass the effect of improving the elastic modulus of the cured product may be low.
- it exceeds 50 mass parts the crosslinked density and water absorption rate of hardened
- the epoxy resin composition is used by further mixing or dissolving the thermoplastic resin [Bz] to cover the brittleness of the epoxy resin with the toughness of the thermoplastic resin [Bz], and
- the molding difficulty of the thermoplastic resin [Bz] is covered with an epoxy resin, which is preferable because a balanced base resin is obtained.
- the blending amount of the thermoplastic resin [Bz] is preferably 2 to 30 parts by mass, more preferably 5 to 20 parts by mass with respect to 100 parts by mass of the total amount of the epoxy resin in terms of balance.
- the epoxy resin composition of this embodiment includes an epoxy resin [A] having a structure represented by the above formula (1), an amine type epoxy resin [By], and a polyamine curing agent [C].
- the elastic modulus of the cured product is improved, and the effect of improving the strength of the fiber reinforced composite material is exhibited.
- the epoxy resin composition which gives the hardened
- the compounding amount of the epoxy resin [A] having the structure represented by the formula (1) and the amine type epoxy resin [By] is good in the property balance of the cured resin and exhibits particularly excellent strength properties.
- the bisphenol type epoxy resin [Bx] it is also preferable that 20 to 50 parts by mass of bisphenol type epoxy resin [Bx] is further contained in 100 parts by mass of the total amount of epoxy resin.
- the bisphenol type epoxy resin [Bx] By blending the bisphenol type epoxy resin [Bx], the crosslink density of the cured product can be reduced while maintaining heat resistance, and a highly tough resin cured product can be obtained.
- the blending amount of [Bx] is less than 20 parts by mass, the toughness of the cured product may be lowered.
- the blending amount of [Bx] exceeds 50 parts by mass, the heat resistance of the cured product may be lowered.
- thermoplastic resin [Bz] it is also preferable to mix or dissolve the thermoplastic resin [Bz] in the above epoxy resin composition. Mixtures of epoxy resin and thermoplastic resin [Bz] give better results than when they are used alone. The brittleness of the epoxy resin is covered with the toughness of the thermoplastic resin [Bz], and the molding difficulty of the thermoplastic resin [Bz] is covered with the epoxy resin, thereby providing a well-balanced base resin.
- the blending amount of the thermoplastic resin [Bz] is preferably 2 to 30 parts by mass, more preferably 5 to 20 parts by mass with respect to 100 parts by mass of the total amount of the epoxy resin in terms of balance.
- the epoxy resin composition of this embodiment includes an epoxy resin [A] having a structure represented by the above formula (1), a thermoplastic resin [Bz], and a polyamine curing agent [C].
- the blending amount of the thermoplastic resin [Bz] is preferably 2 to 30 parts by mass, more preferably 5 to 20 parts by mass with respect to 100 parts by mass of the total amount of the epoxy resin in terms of balance. .
- the epoxy resin [A] having a structure represented by the formula (1) is preferably contained in an amount of 10 to 70 parts by mass, and more preferably 30 to 50 parts by mass in 100 parts by mass of the total amount of the epoxy resin. . Within this range, water absorption of the cured product can be suppressed even in a high temperature and high humidity environment, and heat resistance and resin modulus can be maintained. Thereby, the high compressive strength as a fiber reinforced composite material can be maintained.
- an elastomer [D] is further blended with the above epoxy resin composition.
- Such an elastomer [D] is blended for the purpose of forming a fine elastomer phase in the cured epoxy matrix phase.
- plane distortion that occurs when stress is applied to the cured resin can be eliminated by fracture voiding (cavitation) of the elastomer phase, and as a result of inducing plastic deformation of the epoxy matrix phase, large energy absorption is caused. It leads to the improvement of the interlayer toughness as a fiber reinforced composite material.
- Elastomer is a polymer material having a domain having a glass transition temperature lower than 20 ° C.
- examples thereof include a block copolymer.
- the elastomer [D] is preferably selected from block copolymers containing rubber having a glass transition temperature of 20 ° C. or less and rubber particles. This makes it possible to introduce a fine elastomer phase while minimizing the compatibility of the elastomer with the epoxy resin, thereby greatly improving the interlaminar toughness as a fiber-reinforced composite material while suppressing a decrease in heat resistance and elastic modulus. be able to.
- the rubber particles cross-linked rubber particles, and core-shell rubber particles obtained by graft polymerization of a different polymer on the surface of the cross-linked rubber particles are preferably used from the viewpoint of handleability and the like.
- the primary particle diameter of such rubber particles is preferably in the range of 50 to 300 ⁇ m, particularly preferably in the range of 80 to 200 ⁇ m.
- Such rubber particles preferably have good affinity with the epoxy resin used, and do not cause secondary aggregation during resin preparation or molding and curing.
- crosslinked rubber particles include FX501P (manufactured by Nippon Synthetic Rubber Industry Co., Ltd.) consisting of a crosslinked product of carboxyl-modified butadiene-acrylonitrile copolymer, and CX-MN series (Nippon Shokubai Co., Ltd.) consisting of fine acrylic rubber particles.
- YR-500 series manufactured by Nippon Steel & Sumikin Chemical Co., Ltd., etc. can be used.
- core-shell rubber particles include, for example, “Paraloid (registered trademark)” EXL-2655 (manufactured by Kureha Co., Ltd.) consisting of a butadiene / alkyl methacrylate / styrene copolymer, an acrylic ester / methacrylic ester copolymer "STAPHYLOID (registered trademark)” AC-3355 composed of coalescence, TR-2122 (manufactured by Takeda Pharmaceutical Co., Ltd.), “PARALOID (registered trademark)” EXL-2611 composed of butyl acrylate / methyl methacrylate copolymer EXL-3387 (manufactured by Rohm & Haas), “Kane Ace (registered trademark)” MX series (manufactured by Kaneka Corporation), and the like can be used.
- Paraloid (registered trademark)” EXL-2655 manufactured by Kureha Co., Ltd.
- the block copolymer containing a block having a glass transition temperature of 20 ° C. or lower is not particularly limited in chemical structure or molecular weight, but the block having a glass transition temperature of 20 ° C. or lower is incompatible with the epoxy resin. In addition, it is preferable that a block compatible with the epoxy resin is also included.
- the block copolymer containing a block having a glass transition temperature of 20 ° C. or lower includes at least one block copolymer selected from the group consisting of SBM type, BM type, and MBM type. It is also preferable to be a combination (hereinafter abbreviated as a block copolymer). Thereby, it is possible to greatly improve the interlaminar toughness while maintaining the excellent heat resistance as a fiber reinforced composite material and the mechanical strength in a severe use environment such as at low temperatures.
- Block M is a block made of a homopolymer of methyl methacrylate or a copolymer containing at least 50% by weight of methyl methacrylate.
- Introducing a monomer other than methyl methacrylate into the block M as a copolymerization component is preferably carried out from the viewpoint of compatibility with the epoxy resin and control of various properties of the cured product.
- Such a monomer copolymerization component is not particularly limited and can be appropriately selected. However, in order to obtain compatibility with an epoxy resin having a high SP value, a monomer having a higher SP value than methyl methacrylate, particularly a water-soluble component.
- Monomers are preferably used. Among these, acrylamide derivatives can be preferably used, and dimethylacrylamide can be particularly preferably used. A reactive monomer is also applicable.
- the reactive monomer means a monomer having a functional group capable of reacting with the oxirane group of the epoxy molecule or the functional group of the curing agent.
- a monomer having a reactive functional group such as an oxirane group, an amine group, or a carboxyl group can be exemplified, but the present invention is not limited thereto.
- (meth) acrylic acid in this specification, methacrylic acid and acrylic acid are abbreviated as “(meth) acrylic acid”), or (meth) acrylic acid can be hydrolyzed.
- Monomers that can be obtained can also be used.
- Use of a reactive monomer is preferred because compatibility with the epoxy resin and adhesion at the epoxy-block copolymer interface are improved.
- Examples of other monomers that can constitute the block M include glycidyl methacrylate or tert-butyl methacrylate, but the block M is preferably composed of syndiotactic PMMA (polymethyl methacrylate) at least 60%.
- Block B is a block that is incompatible with block M and has a glass transition temperature Tg (hereinafter sometimes referred to as Tg only) of 20 ° C. or lower.
- the glass transition temperature Tg of the block B is preferably 0 ° C. or lower, more preferably ⁇ 40 ° C. or lower.
- the glass transition temperature Tg is preferably as low as possible from the viewpoint of toughness, but if it is lower than ⁇ 100 ° C., there may be a problem in workability such as a roughened cutting surface when a fiber-reinforced composite material is obtained.
- the glass transition temperature Tg of the block B can be measured by the DMA method using RSAII (manufactured by Rheometrics) regardless of whether the epoxy resin composition or the block copolymer alone is used. That is, a 1 ⁇ 2.5 ⁇ 34 mm plate-like sample is measured by the DMA method with a traction period of 1 Hz at a temperature of ⁇ 60 to 250 ° C., and the tan ⁇ value is defined as the glass transition temperature Tg.
- the sample is manufactured as follows.
- the uncured resin composition is defoamed in vacuum, and then 130 mm in a mold set to a thickness of 1 mm by a 1 mm thick “Teflon (registered trademark)” spacer.
- a plate-like cured product having no voids can be obtained by curing at a temperature of 2 ° C. for 2 hours.
- a void-free plate can be similarly obtained by using a biaxial extruder. These plates can be evaluated by cutting them into the above size with a diamond cutter.
- the monomer constituting the block B is preferably a diene selected from butadiene, isoprene, 2,3-dimethyl-1,3-butadiene, 1,3-pentadiene and 2-phenyl-1,3-butadiene. It is particularly preferable from the viewpoint of toughness to select from polybutadiene, polyisoprene and their random copolymers or partially or fully hydrogenated polydienes.
- polybutadienes 1,2-polybutadiene (Tg: about 0 ° C.) and the like can be mentioned, but it is more preferable to use, for example, 1,4-polybutadiene (Tg: about ⁇ 90 ° C.) having the lowest glass transition temperature Tg. . This is because the use of the block B having a lower glass transition temperature Tg is advantageous from the viewpoint of impact resistance and toughness.
- Block B may be hydrogenated. This hydrogenation is carried out according to the usual methods.
- alkyl (meth) acrylate is also preferable. Specific examples include ethyl acrylate ( ⁇ 24 ° C.), butyl acrylate ( ⁇ 54 ° C.), 2-ethylhexyl acrylate ( ⁇ 85 ° C.), hydroxyethyl acrylate ( ⁇ 15 ° C.) and 2-ethylhexyl methacrylate ( ⁇ 10 ° C. ).
- the numerical value shown in parentheses after the name of each acrylate is the Tg of the block B obtained when each acrylate is used. Of these, butyl acrylate is preferably used.
- These acrylate monomers are incompatible with block M acrylates containing at least 50 parts by weight of methyl methacrylate.
- the B block is preferably a block made of a polymer selected from 1,4-polybutadiene, polybutyl acrylate and poly (2-ethylhexyl acrylate).
- Block S is incompatible with blocks B and M, and its glass transition temperature Tg is higher than that of block B.
- the Tg or melting point of the block S is preferably 23 ° C. or higher, and more preferably 50 ° C. or higher.
- the block S include aromatic vinyl compounds such as those obtained from styrene, ⁇ -methylstyrene or vinyltoluene.
- triblock copolymer SBM examples include styrene-butadiene-methyl methacrylate copolymers such as Nanostrength 123, Nanostrength 250, Nanostrength 012, Nanostrength E20, Nanostrength E40 manufactured by Arkema. It is done.
- Specific examples of the triblock copolymer MBM include, as a copolymer consisting of methyl methacrylate-butyl acrylate-methyl methacrylate, based on Nanostrength M22 manufactured by Arkema, and Nanostrength M22 manufactured by Arkema.
- Nanostrength M22N and Nanostrength SM4032XM10 copolymerized with a monomer having a high SP value are preferably used because they form a fine phase separation structure and give high toughness.
- the blending amount of the elastomer [D] is preferably 2 to 15 parts by mass, more preferably 3 to 15 parts by mass with respect to 100 parts by mass of the total amount of the epoxy resin, from the viewpoint of mechanical properties and compatibility with the composite production process. It is in the range of 10 parts by weight, more preferably 4-8 parts by weight.
- the blending amount is less than 2 parts by mass, the toughness and plastic deformation ability of the cured product are reduced, and the resulting fiber-reinforced composite material has low impact resistance.
- the blending amount exceeds 15 parts by mass, the elastic modulus of the cured product is lowered, the static strength characteristics of the resulting fiber reinforced composite material are lowered, and the resin flow at the molding temperature is lowered, resulting in the fiber reinforced composite obtained.
- the material is likely to contain voids.
- the epoxy resin composition of the present invention contains a coupling agent, thermosetting resin particles, or inorganic fillers such as silica gel, carbon black, clay, carbon nanotube, and metal powder as long as the effects of the present invention are not hindered. can do.
- components (components) other than the polyamine curing agent [C] are first heated and kneaded uniformly at a temperature of about 150 to 170 ° C, and then up to a temperature of about 80 ° C. After cooling, it is preferable to add and knead the polyamine curing agent [C], but the blending method of each component is not particularly limited to this method.
- the prepreg of the present invention is obtained by impregnating reinforcing fibers with the above-described epoxy resin composition.
- the fiber mass fraction in the prepreg is preferably 40 to 90% by mass, more preferably 50 to 80% by mass. If the fiber mass fraction is too low, the resulting composite material may have an excessive mass, which may impair the advantages of the fiber-reinforced composite material having excellent specific strength and specific modulus, and if the fiber mass fraction is too high. Insufficient impregnation of the resin composition occurs, and the resulting composite material tends to have a lot of voids, and its mechanical properties may be greatly deteriorated.
- the epoxy resin composition of the present invention preferably has a viscosity at 80 ° C. of 0.1 to 200 Pa ⁇ s, more preferably 0, from the viewpoint of processability such as tack and drape. 5 to 100 Pa ⁇ s, more preferably 1 to 50 Pa ⁇ s.
- the viscosity at 80 ° C. is 0.1 Pa ⁇ s or more, the shape of the prepreg can be maintained, the resin flow during molding hardly occurs, and variations in the reinforcing fiber content are reduced.
- the viscosity at 80 ° C. is 200 Pa ⁇ s or less, in the process of forming the epoxy resin composition into a film, fading is suppressed and the impregnation property to the reinforcing fibers is excellent.
- Viscosity here refers to, for example, a dynamic viscoelasticity measuring device such as ARES (manufactured by TA Instruments), using a parallel plate with a diameter of 40 mm, and depending on the measurement conditions of a heating rate of 2 ° C./min, a frequency of 0.5 Hz, and a gap of 1 mm. It refers to the obtained complex viscosity ⁇ * .
- the form of the reinforcing fibers is not particularly limited, and for example, long fibers, tows, woven fabrics, mats, knits, braids and the like that are aligned in one direction are used.
- an array in which reinforcing fibers are aligned in a single direction is most suitable. Arrangements are also suitable for the present invention.
- the reinforcing fiber examples include glass fiber, carbon fiber, graphite fiber, aramid fiber, boron fiber, alumina fiber, and silicon carbide fiber. Two or more kinds of these reinforcing fibers may be mixed and used, but in order to obtain a molded product that is lighter and more durable, it is preferable to use carbon fibers or graphite fibers. In particular, in applications where there is a high demand for reducing the weight and strength of materials, carbon fibers are preferably used because of their excellent specific modulus and specific strength.
- any type of carbon fiber can be used depending on the application, but a carbon fiber having a tensile elastic modulus of 400 GPa at the highest is preferable from the viewpoint of impact resistance.
- a carbon fiber having a tensile strength of preferably 4.4 to 6.5 GPa is preferably used because a composite material having high rigidity and mechanical strength can be obtained.
- the tensile elongation is an important factor, and it is preferable that the carbon fiber is a high strength and high elongation carbon fiber of 1.7 to 2.3%. Accordingly, carbon fibers having the characteristics that the tensile modulus is at least 230 GPa, the tensile strength is at least 4.4 GPa, and the tensile elongation is at least 1.7% are most suitable.
- Carbon fibers include “Torayca (registered trademark)” T800G-24K, “Torayca (registered trademark)” T800S-24K, “Torayca (registered trademark)” T700G-24K, and “Torayca (registered trademark)” T300- 3K, and “Torayca (registered trademark)” T700S-12K (manufactured by Toray Industries, Inc.).
- the form and arrangement of the carbon fibers can be appropriately selected from long fibers and woven fabrics arranged in one direction. However, in order to obtain a carbon fiber reinforced composite material that is lighter and more durable, It is preferably in the form of continuous fibers such as long fibers (fiber bundles) or woven fabrics arranged in one direction.
- the epoxy resin composition used as a matrix resin is dissolved in a solvent such as methyl ethyl ketone or methanol to lower the viscosity, and impregnated into reinforced fibers (wet method), and the matrix resin is heated.
- a solvent such as methyl ethyl ketone or methanol
- wet method impregnated into reinforced fibers
- examples thereof include a hot melt method (dry method) in which the viscosity is lowered and the reinforcing fibers are impregnated.
- the wet method is a method in which a reinforcing fiber is immersed in a solution of an epoxy resin composition that is a matrix resin, and then lifted and the solvent is evaporated using an oven or the like.
- the hot melt method (dry method) is a method of impregnating reinforcing fibers directly with an epoxy resin composition whose viscosity has been reduced by heating, or a film in which an epoxy resin composition is once coated on release paper or the like, Next, the reinforcing fiber is impregnated with resin by overlapping the film from both sides or one side of the reinforcing fiber and heating and pressing.
- the hot melt method is preferable because substantially no solvent remains in the prepreg.
- the fiber reinforced composite material of the present invention contains a reinforced fiber and a cured product of the epoxy resin composition of the present invention.
- a fiber-reinforced composite material is produced by a method of heat-curing the matrix resin while applying pressure to the laminate.
- a method of applying heat and pressure a press molding method, an autoclave molding method, a bagging molding method, a wrapping tape method, an internal pressure molding method, and the like are employed.
- the fiber reinforced composite material is a method in which an epoxy resin composition is directly impregnated into a reinforcing fiber without using a prepreg, followed by heat curing, such as a hand layup method, a filament winding method, a pultrusion method, a resin.
- -It can also be produced by a molding method such as an injection molding method and a resin transfer molding method. In these methods, it is preferable to prepare an epoxy resin composition by mixing two liquids of an epoxy resin main component and an epoxy resin curing agent immediately before use.
- the fiber reinforced composite material using the epoxy resin composition of the present invention as a matrix resin is suitably used for sports applications, aircraft applications and general industrial applications. More specifically, in aerospace applications, primary structural material applications such as main wings, tail wings and floor beams, secondary structural material applications such as flaps, ailerons, cowls, fairings and interior materials, rocket motor cases and satellites It is suitably used for structural material applications. Among such aerospace applications, impact resistance is required, and because it is exposed to low temperatures during high altitude flight, aircraft primary structure applications that require tensile strength at low temperatures, especially fuselage skins and main wing skins, Particularly preferably used.
- epoxy resin composition of the present invention will be described more specifically with reference to examples.
- the production methods and evaluation methods of the resin raw materials used in the examples are shown below.
- the organic layer was washed 5 times with 55 parts by weight of pure water, followed by drying through filter paper covered with 15 parts by weight of anhydrous magnesium sulfate (manufactured by Kanto Chemical Co., Ltd.), and then dried at 90 ° C. and 10 torr for 15 hours. As a result, 33.58 parts by weight of white powder (yield 62.8%) was obtained. As a result of analyzing the obtained white powder by HPLC and GPC, it was confirmed that it was a white powder containing a target product (9,9-bis (6-glycidyloxy-2-naphthyl) fluorene) with a purity of 85% or more. .
- a target product (9,9-bis (6-glycidyloxy-2-naphthyl) fluorene
- TPP Triphenylphosphine, curing accelerator, manufactured by Kay Kasei Co., Ltd.
- DCMU99 (3- (3,4-dichlorophenyl) -1,1-dimethylurea, curing accelerator, manufactured by Hodogaya Chemical Co., Ltd.)
- the initial precrack was introduced into the test piece by applying a razor blade cooled to liquid nitrogen temperature to the test piece and applying an impact to the razor with a hammer.
- the toughness of the cured resin refers to the critical stress intensity factor of deformation mode I (opening type).
- Viscosity measurement of epoxy resin composition The viscosity of the epoxy resin composition was measured using a dynamic viscoelasticity measuring device ARES (manufactured by TA Instruments), a parallel plate with a diameter of 40 mm, and a temperature increase rate of 2 ° C / min. The temperature was simply raised, and the value at 80 ° C. of the complex viscosity ( ⁇ * ) obtained under the measurement conditions of a frequency of 0.5 Hz and a gap of 1 mm was adopted.
- ARES dynamic viscoelasticity measuring device
- Example 1 In a kneading apparatus, 70 parts by mass of A-1 (epoxy resin [A] having a structure represented by the formula (1)), 20 parts by mass of “jER (registered trademark)” 806 (bisphenol type epoxy resin [Bx]) ) After kneading 10 parts by mass of “jER (registered trademark)” 152 (an epoxy resin other than [A], [Bx], [By]), 3,3′-DAS which is a polyamine curing agent [C] 20 parts by mass was kneaded to prepare an epoxy resin composition. Table 1 shows the composition and ratio (in Table 1, the numbers represent parts by mass). The properties of the obtained epoxy resin composition were measured as described above. The results are shown in Table 1.
- Examples 2 to 20, Comparative Examples 1 to 6 An epoxy resin composition was prepared in the same manner as in Example 1 except that the epoxy resin, thermoplastic resin, elastomer, other components, curing agent and blending amount were changed as shown in Tables 1 to 3. The properties of the obtained epoxy resin composition were measured as described above. The results are shown in Tables 1 to 3.
- an epoxy resin composition that provides a cured product having excellent heat resistance, toughness, and elastic modulus can be provided. Since the fiber reinforced composite material obtained by such an epoxy resin composition is excellent in compressive strength and interlayer toughness, it is particularly suitably used for a structural material.
- a structural material For example, for aerospace applications, primary aircraft structural materials such as main wing, tail and floor beams, secondary structural materials such as flaps, ailerons, cowls, fairings and interior materials, rocket motor cases and satellite structural materials Preferably used.
- structural materials for moving objects such as automobiles, ships, and railway vehicles, drive shafts, leaf springs, windmill blades, various turbines, pressure vessels, flywheels, paper rollers, roofing materials, cables, reinforcing bars
- civil engineering and building material applications such as repair and reinforcement materials.
- it is suitably used for golf shafts, fishing rods, tennis, badminton and squash rackets, hockey sticks, and ski pole applications.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Reinforced Plastic Materials (AREA)
- Epoxy Resins (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
[A]式(1)で表される構造を有するエポキシ樹脂;

[B]下記の[Bx]、[By]および[Bz]からなる群から選ばれる少なくとも1つの樹脂;
[Bx]ビスフェノール型エポキシ樹脂;
[By]アミン型エポキシ樹脂;
[Bz]熱可塑性樹脂;
[C]ポリアミン硬化剤。
[A]式(1)で表される構造を有するエポキシ樹脂;

[B]下記の[Bx]、[By]および[Bz]からなる群から選ばれる少なくとも1つの樹脂;
[Bx]ビスフェノール型エポキシ樹脂;
[By]アミン型エポキシ樹脂;
[Bz]熱可塑性樹脂;
[C]ポリアミン硬化剤。


<式(1)で表される構造を有するエポキシ樹脂[A]>
(A-1の製造方法:特開2012-102228号公報を参考とした。)
三方コックを装着した内容積300mlのセパラブルフラスコに9,9-ビス(6-ヒドロキシ-2-ナフチル)フルオレン(特開2007-99741号公報の実施例1に準じて合成したもの)45.1重量部(0.1mol)とエピクロルヒドリン(関東化学(株)製)92.0重量部(1.0mol)を仕込み、50℃まで昇温して溶解させた後、反応器内を窒素置換した。そして、反応器内に、フレークの水酸化ナトリウム10.0重量部(0.25mol)を、反応混合物の温度が60℃前後を保持するように、20分毎に4回に分けて添加し、さらに約7時間攪拌して反応させた。
・“jER(登録商標)”806(液状ビスフェノールF型エポキシ樹脂、三菱化学(株)製)
・“jER(登録商標)”828(液状ビスフェノールA型エポキシ樹脂、三菱化学(株)製)
・“jER(登録商標)”1001(固形ビスフェノールA型エポキシ樹脂、三菱化学(株)製)
<アミン型エポキシ樹脂[By]>
・ELM434(テトラグリシジルジアミノジフェニルメタン、住友化学(株)製)
・“jER(登録商標)”630(アミン型エポキシ樹脂、三菱化学(株)製)
・“アラルダイト(登録商標)”MY0600(トリグリシジルアミノフェノール、ハンツマン・アドバンスド・マテリアルズ社製)
・GAN(ジグリシジルアニリン、日本化薬(株)製)
<[A]、[Bx]、[By]以外のエポキシ樹脂>
・“jER(登録商標)”152(フェノールノボラック型エポキシ樹脂、三菱化学(株)製)
・“エピクロン(登録商標)”HP7200L(ジシクロペンタジエン型エポキシ樹脂、DIC(株)製)
・“オンコート(登録商標)”EX-1010(フルオレン型エポキシ樹脂、長瀬産業(株)製)
・“デコナート(登録商標)”EX-721(フタル酸エステル型エポキシ樹脂、長瀬産業(株)製)
・“jER(登録商標)”YX8000(水添ビスフェノールA型エポキシ樹脂、三菱化学(株)製)。
・3,3’-DAS(3,3’-ジアミノジフェニルスルホン、三井化学ファイン(株)製)
・DICY7(ジシアンジアミド、三菱化学(株))
<[C]以外の硬化剤>
・H-4(フェノールノボラック樹脂、明和化成(株)製)。
・“スミカエクセル(登録商標)”PES5003P(ポリエーテルスルホン、住友化学工業(株)製)
・“トレパール(登録商標)”TN(ポリアミド粒子、東レ(株)製、平均粒子径:13.0μm)。
・“ナノストレングス(Nanostrength)”M22N(Bがブチルアクリレート(Tg:-54℃)、Mがメタクリル酸メチルと極性アクリル系モノマーのランダム共重合鎖からなるM-B-M型のブロック共重合体、アルケマ(株)製)
・“カネエース(登録商標)”MX-416(スチレン-ブタジエン-メタクリル酸メチルからなるコアシェルゴム粒子、平均粒子径:100nm、カネカ(株)製)。テトラグリシジルジアミノジフェニルメタンをベースとする、濃度40質量部のマスターバッチ。表1~3にある実施例と比較例の組成表には、ゴム粒子としての配合部数を表記し、マスターバッチに含まれるテトラグリシジルジアミノジフェニルメタンはELM434に含めて表記した。
・TPP(トリフェニルホスフィン、硬化促進剤、ケイ・アイ化成(株)製)
・DCMU99(3-(3,4-ジクロロフェニル)-1,1-ジメチルウレア、硬化促進剤、保土ヶ谷化工業(株)製)。
ニーダー中に、エポキシ樹脂、ならびに、必要に応じて熱可塑性樹脂[Bz]およびエラストマー[D]を所定量加え、混練しつつ、160℃まで昇温し、160℃、1時間混練することで、透明な粘調液を得た。混練しつつ80℃まで降温させた後、ポリアミン硬化剤[C]を所定量加え、さらに混練し、エポキシ樹脂組成物を得た。
上記(1)で調製したエポキシ樹脂組成物を真空中で脱泡した後、2mm厚の“テフロン(登録商標)”製スペーサーにより厚み2mmになるように設定したモールド中に注入した。180℃の温度で2時間硬化させ、厚さ2mmの樹脂硬化物を得た。次に、得られた樹脂硬化物の板から、幅10mm、長さ60mmの試験片を切り出し、スパン間32mmの3点曲げを測定し、JIS K7171-1994に従い、曲げ弾性率と曲げ強度を求めた。湿熱時の曲げ弾性率は前述の方法により得られた樹脂硬化板を沸騰水中に48時間浸漬した後の曲げ弾性率を測定した。
上記(1)で調製したエポキシ樹脂組成物を真空中で脱泡した後、6mm厚の“テフロン(登録商標)”製スペーサーにより厚み6mmになるように設定したモールド中で、180℃の温度で2時間硬化させ、厚さ6mmの樹脂硬化物を得た。この樹脂硬化物を12.7×150mmのサイズにカットし、試験片を得た。インストロン万能試験機(インストロン社製)を用い、ASTM D5045(1999)に従って試験片の加工および靱性(KIC)の測定をおこなった。試験片への初期の予亀裂の導入は、液体窒素温度まで冷やした剃刀の刃を試験片にあてハンマーで剃刀に衝撃を加えることで行った。ここでいう、樹脂硬化物の靱性とは、変形モードI(開口型)の臨界応力拡大係数のことを指している。
上記(2)で作製した樹脂硬化物の板から、樹脂硬化物を7mg取り出し、TAインスツルメンツ社製DSC2910(型番)を用いて、30℃~350℃の温度範囲を昇温速度10℃/分にて、測定を行い、JIS K7121-1987に基づいて求めた中間点温度をガラス転移温度Tgとし、耐熱性を評価した。吸湿時のガラス転移温度は前述の方法により得られた樹脂硬化板を沸騰水中に48時間浸漬した後のガラス転移温度を測定した。
上記(2)で作製した樹脂硬化物の板を80℃、20時間で加熱乾燥した後の試料の重量と該樹脂硬化の板を沸騰水中に48時間浸漬した後の吸水した試料の重量の差から樹脂硬化物の吸水率を求めた。
エポキシ樹脂組成物の粘度は、動的粘弾性測定装置ARES(TAインスツルメンツ社製)を用い、直径40mmのパラレルプレートを用い、昇温速度2℃/minで単純昇温し、周波数0.5Hz、Gap1mmの測定条件で得られた、複素粘性率(η*)の80℃における値を採用した。
混練装置で、70質量部のA-1(式(1)で表される構造を有するエポキシ樹脂[A])、20質量部の“jER(登録商標)”806(ビスフェノール型エポキシ樹脂[Bx])、10質量部の“jER(登録商標)”152([A]、[Bx]、[By]以外のエポキシ樹脂)を混練した後、ポリアミン硬化剤[C]である3,3’-DASを20質量部混練して、エポキシ樹脂組成物を作製した。表1に、組成と割合を示す(表1中、数字は質量部を表す)。得られたエポキシ樹脂組成物の特性を、上記のように測定した。結果を表1に示す。
エポキシ樹脂、熱可塑性樹脂、エラストマー、その他の成分、硬化剤および配合量を、表1~3に示すように変更したこと以外は、実施例1と同様にしてエポキシ樹脂組成物を作製した。得られたエポキシ樹脂組成物の特性を上記のように測定した。結果を表1~3に示す。



Claims (14)
- 次の構成要素[A]、[B]および[C]からなるエポキシ樹脂組成物:
[A]式(1)で表される構造を有するエポキシ樹脂;
[B]下記の[Bx]、[By]および[Bz]からなる群から選ばれる少なくとも1つの樹脂;
[Bx]ビスフェノール型エポキシ樹脂;
[By]アミン型エポキシ樹脂;
[Bz]熱可塑性樹脂;
[C]ポリアミン硬化剤。 - 構成要素[A]、[Bx]および[C]からなるエポキシ樹脂組成物であって、エポキシ樹脂の総量100質量部中に[A]を10~70質量部、[Bx]を20~80質量部含む、請求項1に記載のエポキシ樹脂組成物。
- さらに熱可塑性樹脂[Bz]をエポキシ樹脂の総量100質量部に対して2~30質量部含む、請求項2に記載のエポキシ樹脂組成物。
- さらにアミン型エポキシ樹脂[By]をエポキシ樹脂の総量100質量部中に10~50質量部含む、請求項2または3に記載のエポキシ樹脂組成物。
- 構成要素[A]、[By]および[C]からなる、請求項1に記載のエポキシ樹脂組成物。
- エポキシ樹脂の総量100質量部中に[A]を10~50質量部、[By]を10~50質量部含む、請求項5に記載のエポキシ樹脂組成物。
- さらに熱可塑性樹脂[Bz]をエポキシ樹脂の総量100質量部に対して2~30質量部含む、請求項5または6に記載のエポキシ樹脂組成物。
- さらにビスフェノール型エポキシ樹脂[Bx]をエポキシ樹脂の総量100質量部中に20~50質量部含む、請求項5~7のいずれかに記載のエポキシ樹脂組成物。
- 構成要素[A]、[Bz]および[C]からなるエポキシ樹脂組成物であって、熱可塑性樹脂[Bz]のガラス転移温度(Tg)が150℃以上である、請求項1に記載のエポキシ樹脂組成物。
- さらにエラストマー[D]をエポキシ樹脂の総量100質量部に対して2~15質量部含む、請求項1~9のいずれかに記載のエポキシ樹脂組成物。
- 80℃における複素粘性率が0.1~200(Pa・s)の範囲にある、請求項1~10のいずれかに記載のエポキシ樹脂組成物。
- 請求項1~11のいずれかに記載のエポキシ樹脂組成物を強化繊維に含浸させてなるプリプレグ。
- 強化繊維が炭素繊維である、請求項12に記載のプリプレグ。
- 強化繊維およびエポキシ樹脂組成物の硬化物を含有する繊維強化複合材料であって、該エポキシ樹脂組成物が請求項1~11のいずれかに記載のエポキシ樹脂組成物である、繊維強化複合材料。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14830258.1A EP3026073B1 (en) | 2013-07-26 | 2014-07-24 | Epoxy resin composition, prepreg, and fiber-reinforced composite material |
| KR1020167003591A KR20160036566A (ko) | 2013-07-26 | 2014-07-24 | 에폭시 수지 조성물, 프리프레그 및 섬유 강화 복합 재료 |
| JP2014536024A JP5794391B2 (ja) | 2013-07-26 | 2014-07-24 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| US14/897,393 US9963589B2 (en) | 2013-07-26 | 2014-07-24 | Epoxy resin composition, prepreg, and fiber-reinforced composite material |
| CN201480041829.4A CN105408386B (zh) | 2013-07-26 | 2014-07-24 | 环氧树脂组合物、预浸料坯及纤维增强复合材料 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013155427 | 2013-07-26 | ||
| JP2013-155428 | 2013-07-26 | ||
| JP2013-155427 | 2013-07-26 | ||
| JP2013155428 | 2013-07-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015012348A1 true WO2015012348A1 (ja) | 2015-01-29 |
Family
ID=52393382
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/069550 Ceased WO2015012348A1 (ja) | 2013-07-26 | 2014-07-24 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9963589B2 (ja) |
| EP (1) | EP3026073B1 (ja) |
| JP (2) | JP5794391B2 (ja) |
| KR (1) | KR20160036566A (ja) |
| CN (1) | CN105408386B (ja) |
| WO (1) | WO2015012348A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016159147A1 (ja) * | 2015-03-31 | 2016-10-06 | 東邦テナックス株式会社 | エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料及びこれらの製造方法 |
| WO2016204173A1 (ja) * | 2015-06-19 | 2016-12-22 | 東レ株式会社 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2017122196A (ja) * | 2016-01-08 | 2017-07-13 | 大阪ガスケミカル株式会社 | エポキシ樹脂組成物及びその硬化物並びに新規ポリエーテルスルホン系樹脂 |
| JPWO2016147735A1 (ja) * | 2015-03-18 | 2018-01-25 | 三菱瓦斯化学株式会社 | 樹脂組成物、プリプレグ、金属箔張積層板、樹脂シート、及びプリント配線板 |
| US20180244880A1 (en) * | 2015-09-03 | 2018-08-30 | Toray Industries, Inc. | Epoxy resin composition, prepreg, and carbon fiber reinforced composite material |
| WO2021106588A1 (ja) * | 2019-11-27 | 2021-06-03 | Dic株式会社 | プリプレグ用樹脂組成物、プリプレグ及び成形品 |
| JP2022096595A (ja) * | 2020-12-17 | 2022-06-29 | 大阪ガスケミカル株式会社 | ポリアリーレンスルフィド系樹脂組成物およびその用途 |
| US12448510B2 (en) * | 2017-01-19 | 2025-10-21 | Toray Industries, Inc. | Prepreg, method for producing same, and slit tape prepreg |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106893257B (zh) * | 2015-12-17 | 2019-09-13 | 比亚迪股份有限公司 | 一种环氧预浸料复合材料及其制备方法 |
| WO2018057057A1 (en) * | 2016-09-26 | 2018-03-29 | Sabic Global Technologies B.V. | Homogeneous amorphous high heat epoxy blend composite compositions, articles, and uses thereof |
| US20200181391A1 (en) * | 2016-09-26 | 2020-06-11 | Sabic Global Technologies B.V. | High heat and high toughness epoxy compositions, articles, and uses thereof |
| CN106674898A (zh) * | 2016-12-21 | 2017-05-17 | 芜湖天道绿色新材料有限公司 | 核壳粒子环氧树脂预混物、碳纤维预浸料核壳粒子增韧环氧树脂组合物及其制备方法 |
| KR101884606B1 (ko) * | 2017-01-26 | 2018-08-02 | 주식회사 한국카본 | 고내충격성고강도 특성을 갖는 복합재료용 에폭시 수지 조성물 |
| EP3366584B1 (en) * | 2017-02-27 | 2019-04-17 | AIRBUS HELICOPTERS DEUTSCHLAND GmbH | Pitch control device for a ducted tail rotor of a rotorcraft |
| WO2018158792A1 (ja) * | 2017-02-28 | 2018-09-07 | 藤倉ゴム工業株式会社 | ゴルフクラブ、及び、ゴルフクラブシャフトとゴルフクラブヘッドの結合部材 |
| JP6555328B2 (ja) * | 2017-11-28 | 2019-08-07 | 横浜ゴム株式会社 | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| CN111040130A (zh) * | 2018-10-15 | 2020-04-21 | 沙特基础工业全球技术有限公司 | 可固化的高热环氧组合物、其固化的环氧组合物、其制造方法和包含其的制品 |
| US12404408B2 (en) | 2019-08-13 | 2025-09-02 | Exxon Mobil Technology and Engineering Company | Processes for functionalization and polymerization of polyaromatic feedstock |
| KR20260004589A (ko) * | 2019-08-13 | 2026-01-08 | 엑손모빌 테크놀로지 앤드 엔지니어링 컴퍼니 | 에폭사이드 작용화된 다환방향족 공급원료 및 이로부터 유도된 중합체 |
| JPWO2023089997A1 (ja) * | 2021-11-16 | 2023-05-25 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01132541A (ja) * | 1987-10-09 | 1989-05-25 | Shell Internatl Res Maatschappij Bv | 新規なジエポキシドおよびジフエノキシ化合物 |
| JP2004506789A (ja) | 2000-08-22 | 2004-03-04 | サイテク・テクノロジー・コーポレーシヨン | 分子鎖連結に適した組成物 |
| JP2005298815A (ja) | 2004-03-17 | 2005-10-27 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2007099741A (ja) | 2005-10-07 | 2007-04-19 | Osaka Gas Co Ltd | フルオレン骨格を有する化合物およびその製造方法 |
| JP2007154160A (ja) | 2005-11-14 | 2007-06-21 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| WO2010035859A1 (ja) | 2008-09-29 | 2010-04-01 | 東レ株式会社 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2012102228A (ja) | 2010-11-10 | 2012-05-31 | Osaka Gas Chem Kk | フルオレン骨格を有するエポキシ樹脂組成物およびその硬化物 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4882370A (en) * | 1988-01-27 | 1989-11-21 | Minnesota Mining And Manufacturing Company | Fiber reinforced composites with improved glass transition temperatures |
| WO1996017006A1 (en) * | 1994-12-02 | 1996-06-06 | Toray Industries, Inc. | Prepreg and fiber-reinforced composite material |
| JPH09194571A (ja) | 1996-01-18 | 1997-07-29 | Minnesota Mining & Mfg Co <3M> | エポキシ樹脂組成物、改質エポキシ樹脂組成物およびその製造方法 |
| JPH11302412A (ja) | 1998-04-16 | 1999-11-02 | Toray Ind Inc | プリプレグおよび繊維強化複合材料 |
| JP5249578B2 (ja) | 2007-12-26 | 2013-07-31 | 大阪瓦斯株式会社 | フルオレン骨格を有するエポキシ化合物 |
| CA2859629A1 (en) | 2011-12-27 | 2013-07-04 | Toray Industries, Inc. | Epoxy resin composition for fiber-reinforced composite materials, prepreg, and fiber-reinforced composite material |
| TWI633011B (zh) * | 2012-10-15 | 2018-08-21 | Ajinomoto Co., Inc. | Resin composition |
-
2014
- 2014-07-24 US US14/897,393 patent/US9963589B2/en not_active Expired - Fee Related
- 2014-07-24 EP EP14830258.1A patent/EP3026073B1/en not_active Not-in-force
- 2014-07-24 KR KR1020167003591A patent/KR20160036566A/ko not_active Withdrawn
- 2014-07-24 WO PCT/JP2014/069550 patent/WO2015012348A1/ja not_active Ceased
- 2014-07-24 CN CN201480041829.4A patent/CN105408386B/zh not_active Expired - Fee Related
- 2014-07-24 JP JP2014536024A patent/JP5794391B2/ja not_active Expired - Fee Related
-
2015
- 2015-04-23 JP JP2015088084A patent/JP5835513B2/ja not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01132541A (ja) * | 1987-10-09 | 1989-05-25 | Shell Internatl Res Maatschappij Bv | 新規なジエポキシドおよびジフエノキシ化合物 |
| JP2004506789A (ja) | 2000-08-22 | 2004-03-04 | サイテク・テクノロジー・コーポレーシヨン | 分子鎖連結に適した組成物 |
| JP2005298815A (ja) | 2004-03-17 | 2005-10-27 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2007099741A (ja) | 2005-10-07 | 2007-04-19 | Osaka Gas Co Ltd | フルオレン骨格を有する化合物およびその製造方法 |
| JP2007154160A (ja) | 2005-11-14 | 2007-06-21 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| WO2010035859A1 (ja) | 2008-09-29 | 2010-04-01 | 東レ株式会社 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2012102228A (ja) | 2010-11-10 | 2012-05-31 | Osaka Gas Chem Kk | フルオレン骨格を有するエポキシ樹脂組成物およびその硬化物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3026073A4 |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7046602B2 (ja) | 2015-03-18 | 2022-04-04 | 三菱瓦斯化学株式会社 | 樹脂組成物、プリプレグ、金属箔張積層板、樹脂シート、及びプリント配線板 |
| JPWO2016147735A1 (ja) * | 2015-03-18 | 2018-01-25 | 三菱瓦斯化学株式会社 | 樹脂組成物、プリプレグ、金属箔張積層板、樹脂シート、及びプリント配線板 |
| JPWO2016159147A1 (ja) * | 2015-03-31 | 2018-02-01 | 東邦テナックス株式会社 | エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料及びこれらの製造方法 |
| US10647849B2 (en) | 2015-03-31 | 2020-05-12 | Toho Tenax Co., Ltd. | Epoxy resin composition, prepreg, carbon fiber-reinforced composite material, and manufacturing methods therefor |
| WO2016159147A1 (ja) * | 2015-03-31 | 2016-10-06 | 東邦テナックス株式会社 | エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料及びこれらの製造方法 |
| WO2016204173A1 (ja) * | 2015-06-19 | 2016-12-22 | 東レ株式会社 | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| US20180244880A1 (en) * | 2015-09-03 | 2018-08-30 | Toray Industries, Inc. | Epoxy resin composition, prepreg, and carbon fiber reinforced composite material |
| US10851217B2 (en) * | 2015-09-03 | 2020-12-01 | Toray Industries, Inc. | Epoxy resin composition, prepreg, and carbon fiber reinforced composite material |
| JP2017122196A (ja) * | 2016-01-08 | 2017-07-13 | 大阪ガスケミカル株式会社 | エポキシ樹脂組成物及びその硬化物並びに新規ポリエーテルスルホン系樹脂 |
| US12448510B2 (en) * | 2017-01-19 | 2025-10-21 | Toray Industries, Inc. | Prepreg, method for producing same, and slit tape prepreg |
| WO2021106588A1 (ja) * | 2019-11-27 | 2021-06-03 | Dic株式会社 | プリプレグ用樹脂組成物、プリプレグ及び成形品 |
| JP2022096595A (ja) * | 2020-12-17 | 2022-06-29 | 大阪ガスケミカル株式会社 | ポリアリーレンスルフィド系樹脂組成物およびその用途 |
| JP7727469B2 (ja) | 2020-12-17 | 2025-08-21 | 大阪ガスケミカル株式会社 | ポリアリーレンスルフィド系樹脂組成物およびその用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20160036566A (ko) | 2016-04-04 |
| US9963589B2 (en) | 2018-05-08 |
| JPWO2015012348A1 (ja) | 2017-03-02 |
| EP3026073A1 (en) | 2016-06-01 |
| CN105408386A (zh) | 2016-03-16 |
| US20160130431A1 (en) | 2016-05-12 |
| JP5794391B2 (ja) | 2015-10-14 |
| CN105408386B (zh) | 2017-07-21 |
| EP3026073B1 (en) | 2017-12-13 |
| EP3026073A4 (en) | 2017-03-01 |
| JP5835513B2 (ja) | 2015-12-24 |
| JP2015157958A (ja) | 2015-09-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5835513B2 (ja) | プリプレグおよび繊維強化複合材料 | |
| US9738782B2 (en) | EPOXY resin composition, prepreg and fiber-reinforced composite materials | |
| KR101396031B1 (ko) | 에폭시 수지 조성물, 프리프레그, 섬유 강화 복합 재료 | |
| CN101945916B (zh) | 环氧树脂组合物、预浸料及纤维增强复合材料 | |
| JP5648679B2 (ja) | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP5564870B2 (ja) | エポキシ樹脂組成物、プリプレグ、および繊維強化複合材料 | |
| JP5747763B2 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| KR20140048340A (ko) | 에폭시 수지 조성물, 프리프레그 및 섬유 강화 복합 재료 | |
| KR20130118302A (ko) | 섬유 강화 복합 재료용 에폭시 수지 조성물, 프리프레그 및 섬유 강화 복합 재료 | |
| JP5747762B2 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| CN107949593B (zh) | 环氧树脂组合物、环氧树脂固化物、预浸料坯及纤维增强复合材料 | |
| JP5811883B2 (ja) | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP6079943B1 (ja) | エポキシ樹脂組成物、エポキシ樹脂硬化物、プリプレグおよび繊維強化複合材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480041829.4 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2014536024 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14830258 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014830258 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14897393 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20167003591 Country of ref document: KR Kind code of ref document: A |