WO2015016558A1 - 트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 - Google Patents
트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 Download PDFInfo
- Publication number
- WO2015016558A1 WO2015016558A1 PCT/KR2014/006894 KR2014006894W WO2015016558A1 WO 2015016558 A1 WO2015016558 A1 WO 2015016558A1 KR 2014006894 W KR2014006894 W KR 2014006894W WO 2015016558 A1 WO2015016558 A1 WO 2015016558A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- acid
- gram
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- Tricyclic benzoxaboro compound preparation method thereof and use [technical field]
- the present invention relates to a novel tricyclic benzoxabo derivative, a preparation method thereof and an antibiotic use containing the same as an active ingredient, and preferably relates to antibiotic use against Gram-negative bacteria. [Technique to become background of invention]
- MRSA Methicillin-resistant Staphylococcus aureus
- a new mechanism in 2010 reported the OBORT Oxaborole t-RNA trapping mechanism of Leucyl t-RNA synthetase. This is a novel mechanism by which oxabo compounds bind to the edi ting domain of the Leucyl t-RNA synthetase and covalently bind to the t-RNA terminus A76 to trap t-RNAs. Selectivity is due to differences in the structure of the edi ting domain of eukaryotes and bacteria. Therefore, Leucyl t-RNA synthetase inhibitor can be developed as an effective drug for Gram-negative bacteria.
- Benzoxaborole compounds are new synthetic antibiotics that are not fermentation products and derivatives of various structures are known. Compounds comprising boron, such as oxabolol, are described as useful antibiotics in US2006 / 0234981 and US2007 / 0155699. Benzoxabolic derivatives are also described in WO 2008/157726, WO 2009/140309, WO 2011/060196 and WO 2012/033858 and WO 2013/093615.
- W02013 / 093615 has a substituent at position 8 of the tricyclic benzoxabo Only the compound having and the compound in which the hydroxy methyl group was substituted at the 7-position are only specific synthesis examples.
- a novel benzoxaborole compound capable of selectively binding to Gram-negative bacteria and exhibiting functional activity and minimizing side effects, and a therapeutic agent for infectious diseases caused by Gram-negative bacteria, in particular, multidrug-resistant Gram-negative bacteria, which have recently become a great threat by using the same I really need it.
- the compounds of the present invention can minimize the side effects shown functional activity coming to selectively bind to gram-negative bacteria.
- a further object of the present invention relates to a method for the antibacterial, sterilization or sterilization of Gram-negative bacteria using the tricyclic benzoxabo compound according to the present invention.
- a further object of the present invention is to provide a method for preventing or treating an infection caused by Gram-negative bacteria, comprising administering a tricyclic benzoxabo according to the present invention to a subject in a therapeutically effective amount.
- Object of the invention is a tricyclic benzoxaborole according to the invention It relates to the use of antimicrobial, sterilization or sterilization of Gram-negative bacteria using a compound.
- a further object of the present invention is to provide a use for the prevention or treatment of infection by Gram-negative bacteria, comprising the tricyclic benzoxabo compound according to the present invention.
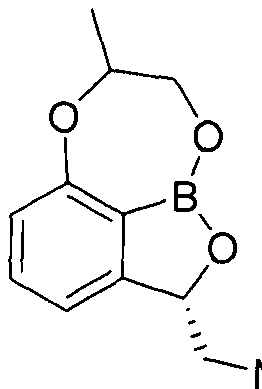
- the present invention discloses a tricyclic benzoxaborole compound represented by the following formula (1), an isomer thereof, or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present invention provides a pharmaceutical composition for antibiotics against Gram-negative bacteria comprising the compound of Formula 1, Isomer 1 thereof, or a pharmaceutically acceptable salt thereof as an active ingredient.
- the Gram-negative S. aureus is Acinetobacter baumannii, Citrobacter freundii, Escherichia coli, Enter obacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiel la oxytoca, Morganella morgan //, Pseudo onas aeruginosa Proteus isse vulgar naris Serratia marcescens 0
- the Gram-negative bacteria may be carbapenem resistant Gram-negative bacteria.
- the inventors of the present invention while studying benzoxabo having a therapeutic effect against bacterial infection, the equivalent in in vi t ro than conventionally known materials or It was confirmed that the compound having more antimicrobial effect and excellent antimicrobial effect against Gram-negative bacteria in vivo, and that it can be more usefully used as a therapeutic agent for Gram-negative bacteria infection was completed.
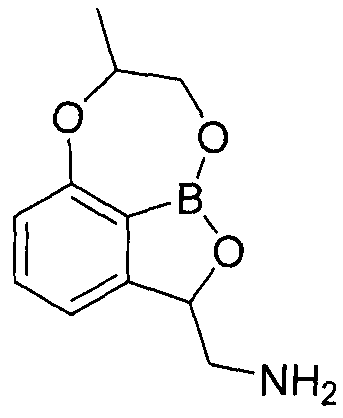
- the compounds of the present invention are conventionally known (8 -methyl-7,8-dihydro-2H-1,6,9-trioxa-9a-borabenzo [cd] azulen-2-yl) methanamine
- the compounds of the present invention are also known ((2S, 8R) -2- (aminomethyl) -7, 8-dihydro-2H-l, 6, 9-trioxa-9a—borabenzo [cd] azulene-8 -Compared with the 1) methanol hydrochloride, it showed strong in vitro antimicrobial activity against major pathogenic bacteria including Acinetobacter Bowmani, and also showed excellent bacterial infection treatment effect in the evaluation of in vivo drug efficacy model.
- the present inventors selectively bind to Gram-negative bacteria, exhibit a functional activity, and minimize the side effects, using the new benzoxaborole compound and antibiotics and / or bacterial infections caused by Gram-negative bacteria including Acinetobacter boumani using the same. To provide a therapeutic.
- the term "pharmaceutically ly acceptable salt” refers to a salt form of a compound that does not cause significant irritation to the organism to which the compound is administered and does not impair the biological activity and properties of the compound. Meaning, in the case of the present invention, it possesses the biological effectiveness and properties of the compound of the formula (I) equally, Any salt that is desirable in terms of pharmaceutical, biological or other properties may be generic.
- the pharmaceutically acceptable salts include, but are not limited to, acids that form non-toxic acid addition salts containing pharmaceutically acceptable anions, such as inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid, hydroiodic acid, and the like; Organic carboxylic acids such as tartaric acid, formic acid, citric acid, acetic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, benzoic acid, lactic acid, mandelic acid, fumaric acid, maleic acid, salicylic acid and the like; Or acid addition salts formed by sulfonic acids such as methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, P-luenesulfonic acid and the like.
- an acid addition salt of a compound of one embodiment can be obtained by reacting a compound in free base form with a stoichiometric amount
- the reaction may proceed in water, an organic solvent, or a combination thereof, and specifically, in a non-aqueous medium such as ether, ethyl acetate, ethane, isopropane, or acetonitrile.
- a non-aqueous medium such as ether, ethyl acetate, ethane, isopropane, or acetonitrile.
- the pharmaceutically acceptable salts include alkali metal salts or alkaline earth metal salts formed by lithium, sodium, potassium, calcium, magnesium, and the like; Amino acid salts such as lysine, arginine and guanidine; Or organic salts such as dicyclonuclear amine, N-methyl-D-glucamine, tris (hydroxymethyl) methylamine, diethanolamine, choline, triethylamine, and the like.
- the term "isomer 1" refers to a compound or salt thereof having the same chemical formula or molecular formula but which is optically or sterically different. Such isomers, salts thereof, and racemi c mixtures of isomers are also described herein. It is included in the range of.
- the tricyclic benzoxaborole compound according to the present invention may be a racemic, enantiomer, diastereoisomer, a mixture of enantiomers or a mixture of diastereomers of the compound.
- the compound represented by Formula 1 may have an asymmetric carbon center, may exist as each optical isomer, partial optical isomer or racemate when having an asymmetric carbon center, all forms of isomers, including these It may be included in the category of compounds according to one embodiment of the invention.
- the compounds of formula 1 or pharmaceutically acceptable salts according to the invention may exhibit po lymorphism and may exist as solvates (eg hydrates, etc.). And each of the compounds include the "individual stereoisomer or a common compound.
- the term "pharmaceutical effective amount (pharmaceutical i cal ly fect ive amount) 1" means the amount of active ingredient from which the desired pharmaceutical effect can be obtained, and in some cases, the desired pharmaceutical effect It may refer to the concentration or dosage of the active ingredient in the pharmaceutical composition for exercise.
- the isomer refers to a compound having the same chemical formula or molecular formula, but optically or stericly different, or a salt thereof.
- Such isomers, salts thereof, and racemic mixtures of isomers are also within the scope of the present invention.
- the isomer of the present invention may be the tricyclic benzoxaboro compound, the racemic, enantiomer, diastereoisomer, a mixture of enantiomers or a mixture of diastereomers of the compound.
- the isomer may be an optical isomer, stereoisomer, or a mixture (racemic mixture) of the compound of Formula 1.
- any asymmetric carbon atom on the compound may exist in any form of (R)-, (S)-or (R, S)-configuration, suitably in each separate form (R )-Or (S)-can be present in the configuration.
- At least one asymmetric carbon selected from the group consisting of carbon 2 and carbon 7 of the tricyclic benzoxabo ring may be an optical isomer, for example, (2S) isomer, (2R) isomer, (7S) It may be, but is not limited to, isomers, (7R) isomers, (2S, 7S) isomers, (2S, 7R) isomers, (2R, 7S) isomers, or (2R, 7R) isomers.
- the isomers of the invention may be the (2S) isomer represented by the formula:
- the isomer of the present invention may be the (2S, 7R) isomer represented by the following formula.
- the compound of Formula 1 or an isomer thereof may be selected from the group consisting of the following compounds:
- the tricyclic benzoxaborole compound of the present invention represented by Formula 1 may be used in the form of a pharmaceutically acceptable salt, and the salt may be added to a pharmaceutically acceptable free acid (free ac id). Acid addition salts formed by this are useful.
- the free acid examples include inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid, and hydroiodic acid; Organic carboxylic acids such as tartaric acid, formic acid, citric acid, acetic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, benzoic acid, lactic acid, manfelic acid, fumaric acid, maleic acid, salicylic acid, and the like; Or sulfonic acids such as methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, P-luenesulfonic acid, and the like, but are not limited thereto.
- the tricyclic benzoxaborole compound of the present invention may be a pharmaceutically acceptable acid addition salt hydrochloride.
- tricyclic benzoxabo compound represented by Chemical Formula 1 of the present invention includes not only pharmaceutically acceptable salts, but also all salts, hydrates, and solvates that can be prepared by conventional methods.
- the addition salts according to the invention can be prepared by conventional methods, for example by dissolving a compound of formula 1 in a water-soluble organic solvent such as acetone, methanol, ethane, or acetonitrile and adding an excess of an organic acid. It may be prepared by adding an acid aqueous solution of an inorganic acid and then precipitating or crystallizing it. This mixture can then be evaporated and dried to evaporate the solvent or excess acid to obtain additional salts or to precipitate prepared salts by suction filtration.
- a water-soluble organic solvent such as acetone, methanol, ethane, or acetonitrile
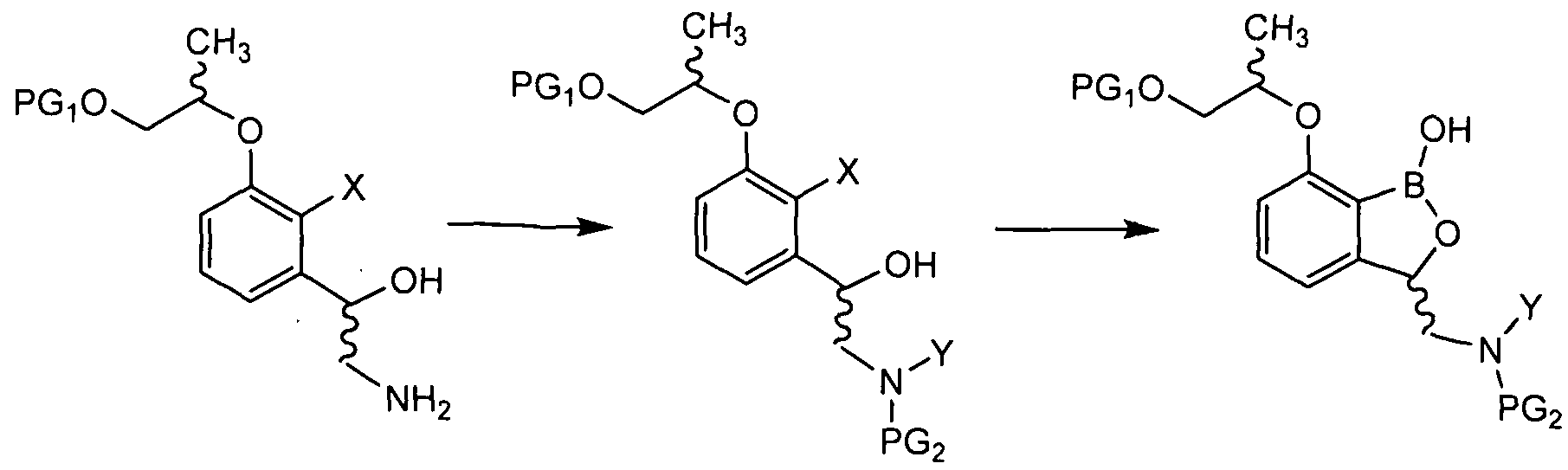
- the present invention also provides a method for preparing a tricyclic benzoxabo compound of formula (1).
- Tricyclic oxabolol derivatives of the present invention Depending on the type of stereoisomer may be prepared by a variety of methods, it may be prepared according to the method illustrated below. It is apparent that the preparation method presented below is merely illustrative and can be easily modified by those skilled in the art according to the desired compound, and therefore the method exemplified below limits the method of preparing the tricyclic benzoxabo compound according to the present invention. no.
- Deprotecting the amino group of the compound of Formula 11 (PG 2 ) may include preparing a compound of Formula 1.
- the method for preparing a compound of Formula 1 according to the present invention comprises the steps of coupling a compound of Formula 4 and a compound of Formula 5 to prepare a compound of Formula 6; [Formula 4] [Formula 5] [Formula 6]
- Deprotecting the amino group of the compound of Formula 11 may include preparing a compound of Formula 1.
- PGi and PG 2 are protecting groups that protect the active groups, each independently benzyl, t-butyl, Boc (tert-butyloxycarbonyl), pmb (4-methoxybenzyl), Fmoc (Fluorenylmethyloxycarbonyl), Ts (tosyl ate) , MOM (methoxymethyl), THP (tetrahydropyranyl), TBDMS (tert- butyldimethyl si lyl), or TBDPS (tert-butyldimethyl si lyl),
- LG is a leaving group which is eliminated from the condensation reaction, and is a halogen, a para-luenesulfonyl group, or a methanesulfonyl group.
- X is hydrogen, halogen or trifluoromethanesulfonyl.
- Y is hydrogen or PG 2 .
- the borylation reaction is bis (pinacolato) diboron or 2-isopropoxy ⁇ 4,4,5,5-tetramethyl-1,3,2-dioxaborolane It may be performed using but is not limited thereto.
- the pharmaceutical composition comprising at least one selected from the group consisting of the compound of Formula 1, an isomer thereof and a pharmaceutically acceptable salt thereof as an active ingredient, in particular a pharmaceutical composition for Gram-negative bacteria antibiotics To provide.
- Gram comprising the step of administering a tricyclic benzoxabo according to the invention in a therapeutically effective amount to a subject in need of prevention and / or treatment of a disease associated with infection with Gram-negative bacteria It is to provide a method for preventing or treating infection caused by negative bacteria. Identifying a patient in need of prevention and / or treatment of a disease associated with infection with Gram-negative bacteria may be further included prior to the administering step.
- the use of antibiotics of Gram-negative bacteria comprising one or more selected from the group consisting of the compound of Formula 1, an isomer thereof, and a pharmaceutically acceptable salt thereof as an active ingredient, or Gram-negative It relates to the use for preventing and / or treating bacterial infections.
- the novel tricyclic benzoxaborole compounds according to the invention have a broad antimicrobial spectrum against gram-negative bacteria, especially Gram-negative bacteria, for example acinetobacter bow, which exhibit multi-drug resistance (mu lti -drug resistance). Since it shows excellent antibacterial activity against Mani bacteria, it can be usefully used as a novel antibiotic.
- the Gram-negative bacterium is more preferably a carbapenem-resistant Gram-negative bacterium. Specific examples of Gram-negative bacteria are A. baumanni i, C. freundi i, E. coli, E. cloacae, E. aerogenes, K. pneumoniae, K. oxytoca, M.morgani i, P. aeruginosa P.
- P.wirabi 1 is N. gonorrhoeae or S can be .imrcescens °. Most preferably carbapenem-resistant acinetobacter boumani ⁇ . ⁇ ⁇ »37 77).
- the pharmaceutical composition containing the compound of Formula 1, an isomer thereof, or a pharmaceutically acceptable salt thereof as an active ingredient may be formulated and used in the form of a conventional pharmaceutical formulation.
- the pharmaceutical formulation may be prepared in various formulations for oral administration or parenteral administration, and the form of the formulation may be variously determined according to the method of use, administration method, administration purpose, and the like.
- Solid preparations for oral administration as tablets, pills, powders, granules, capsules The solid preparation may be one selected from the group consisting of the active ingredient and at least one excipient such as starch, calcium carbonate, sucrose, lactose, gelatin, and the like. The above can be mixed and manufactured. In addition to the simple excipients, lubricants such as magnesium styrate talc may also be used. In addition, liquid formulations for oral administration may include suspensions, solutions, emulsions, syrups and the like.
- water, and / or liquid paraffin which are conventionally used simple diluents, may be used, and optionally, in addition to various excipients, for example, a group consisting of wetting agents, sweeteners, fragrances, preservatives, and the like. One or more selected from may additionally be included.
- Parenteral administration may be carried out by the route of intravenous administration, intramuscular administration, subcutaneous administration, intraperitoneal administration, intranasal administration, transdermal administration and the like.
- Formulations for parenteral administration include sterile aqueous solutions, non-aqueous solutions, suspensions, emulsions, lyophilized preparations, suppositories, and the like.
- a non-aqueous solvent for preparing a non-aqueous solution, or a suspension for preparing a suspension propylene glycol, polyethylene glycol, injectable ester such as vegetable oil ethyl oleate such as olive oil, and the like can be used.
- injectable ester such as vegetable oil ethyl oleate such as olive oil, and the like can be used.
- As the base of the suppository witepsol, macrogol, tween 61, cacao butter, laurin butter, glycerogelatin and the like can be used.
- the content of at least one active ingredient selected from the group consisting of the compound of Formula 1, isomers thereof and pharmaceutically acceptable salts thereof in the pharmaceutical composition is, for example, 0.001 to 99.9% by weight, 0.01 to 90% by weight, or 0.1 to 50 weight 3 ⁇ 4>, but is not limited thereto and may be appropriately adjusted according to the type of formulation, administration method, administration purpose, and the like.
- a pharmaceutically effective amount of the pharmaceutical composition containing the compound of Formula 1, an isomer thereof, and / or a pharmaceutically acceptable salt thereof as an active ingredient of the present invention based on the amount of the active ingredient, from about 0.1 to about 0.1 to About l, 000 mg / l.
- the pharmaceutically effective amount may be administered or taken once or several times a day in consideration of the patient's weight, age, sex, health status, diet, administration time, administration method, excretion rate, severity of the disease, etc. And may be administered in various dosages and methods.
- the compound of formula 1 of the present invention or a pharmaceutically acceptable salt thereof may be contained in an amount of 1 to 95% by weight, preferably It may be contained 1 to 70% by weight.
- the patient may be a mammal, such as a primate, including a human, a rodent, including a mouse, a rat, or the like, specifically, a human.
- the patient may be a mammal, such as a human, in which a condition or disease is preventable, ameliorated and / or treated by the administration of a compound according to the invention.
- the tricyclic benzoxaborole compound according to the present invention has a broad antimicrobial spectrum against resistant bacteria, low toxicity, excellent antimicrobial activity against Gram-negative bacteria, and particularly Gram-negative bacteria, such as acinetobacter, which have antibiotic resistance.
- Microorganisms, such as Bowmani have a strong antimicrobial effect on human and various animal pathogens, and thus may be useful for the prevention, amelioration, and / or treatment of Gram-negative antibiotics or diseases related to their infection. [Specific contents to carry out invention]
- 1,2-propanediol (5 g, 65.7 ⁇ ol) and NaH (3.29 g, 82.0 ⁇ ol) were dissolved in ⁇ , ⁇ -dimethylformamide (70 mL), and then benzyl bromide (7.82 mL, 65.7 ⁇ 01) ( TC was added and stirred at room temperature for 2 hours, after completion of reaction, the mixture was extracted with water and ethyl acetate, the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, and the residue was purified by column chromatography to give the title compound (4.45). g, 41%).
- the reaction mixture was extracted with ice water (1 L) and heptane (500 ml twice), and then the organic layer was extracted with 0.02 N aqueous sodium hydroxide solution (200 ml, twice), 0.01 N aqueous hydrochloric acid solution (200 ml) and saturated aqueous sodium chloride solution (200 ml). After wiping, the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to dry the residue. The title compound (46g, 78%) was obtained.
- (LS) -2-amino-1- (3 ′ ((1 ′ (benzyloxy) propan-2-yl) oxy) phenyl) ethan-1-ol (22.2g, 73.6 ⁇ 0 1) prepared in step 4 Dissolve in all ethanol (245ml), add potassium carbonate (22.4g, 162 ⁇ ol) and stir for about 15 hours at phase silver. Methyl tertiary butyl ether (100ml) was added to the reaction mixture and the temperature was lowered to 0 ° C., followed by stirring for 30 minutes.
- step 4 (LS) -l- (3-((l- (benzyloxy) propan-2-yl) oxy) phenyl) -2- (dibenzylamino) ethan-1-ol hydrochloride prepared in step 4 (10 g> 19.3 ⁇ 0 1) are dissolved in anhydrous toluene (77 ml) in a nitrogen-filled flask (A). Unmelted The reaction mixture is kept in a nitrogen layered state, heated to 40-45 tons, and then normal butyllithium (2.5 M nucleic acid solution, 8.49 ml, 21.2 ⁇ 0 1) is slowly added over about 1 hour. After stirring for an hour, the reaction mixture is lowered to -30 ° C.
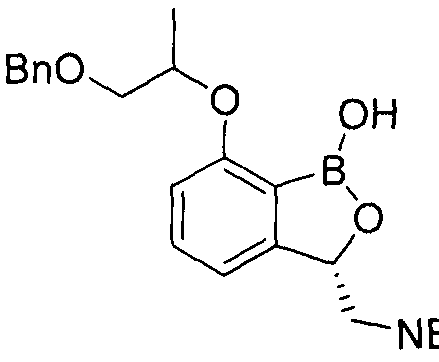
- the aqueous layer of the filtrate was extracted twice with ethyl acetate (100 ml), and then all organic layers were washed with saturated aqueous sodium chloride solution (100 ml), dried over anhydrous sodium sulfate, filtered and concentrated to obtain a residue by column chromatography. (6.03 g, 62%) was obtained.
- step 6 [c] [l, 2] oxabo of step 6 is -1 ( 3H)-(5.56g, 11.0 ⁇ ol) was dissolved in a mixture of 1N hydrochloric acid solution (13.2ml, 13.2 ⁇ 0 1) and methanol (110ml) at room temperature, and 5% palladium / activated carbon catalyst was added to the reaction product. Add. The reaction mixture is heated to 50 ° C and filled with hydrogen gas at a pressure of 50-60 psi while stirring.
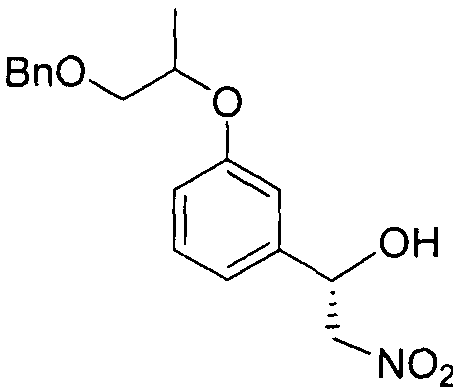
- Step 2 Synthesis of (R) -3-((l- (benzyloxy) propan-2-yl) oxy) benzaldehyde
- the starting material 3-hydroxy vanzaldehyde (26.5g, 217mmol) and potassium carbonate (36.0g, 261 ⁇ ol) are dissolved in dimethylformamide (540mL) and stirred at 0 ° C for 30 minutes.
- (S) -l- (benzyloxy) propane-2-ylmethanesulfonate (53 g, 217 ⁇ 01) prepared in Step 1 was slowly added to the reaction product, and the reaction product was stirred at 100 ° C. for 10 hours. Cool slowly to room temperature.
- the reaction was extracted with ice water (1 L) and heptane (500 ml, twice), and then the organic layer was extracted with 0.02 N aqueous sodium hydroxide solution (200 ml, twice), 0.01 N aqueous hydrochloric acid solution (200 ml), and saturated aqueous sodium chloride solution (200 ml). After drying, the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to dry the residue. The title compound (46g, 78%) was obtained.
- reaction mixture was extracted by adding 1N aqueous hydrochloric acid solution (300ml) and dichloromethane (300ml), and then the aqueous layer was further extracted with dichloromethane (80ml, twice).
- the organic layer was extracted with saturated brine—sodium aqueous solution (100 ml), washed, dried over anhydrous sodium sulfate, filtered and concentrated to give the residue, which was dried to give the title compound (25.3 g).
- the resulting solid precipitate was filtered using Celite, and then saturated hydrochloric acid solution (12.3ml, 147 ⁇ ol) was added to the filtrate and stirred for 30 minutes. After concentration to remove methyl tertiary butyl ether and excess hydrochloric acid isopropane (100ml) using azeotropic distillation to remove the remaining water. Repeat this azeotropic distillation two or three times to remove water, add isopropanol (45ml) to the remaining solid, and stir at 60 ° C for 2 hours to dissolve all solids.
- step 5 (S) -l- (3-(((R) -l- (benzyloxy) propan-2-yl) oxy) phenyl) -2- (dibenzylamino) ethan-1-ol hydrochloride prepared in step 5 (10g, 19.3 ⁇ 0 1) is dissolved in the anhydrous frame toluene (77ml) at (a) a nitrogen-filled flask. The reactant was dissolved in nitrogen and the temperature was raised to 40-45 ° C. Then normal butyllithium (2.5M nucleic acid solution, 8.49ml, 21.2 ⁇ 0 1) was added slowly over about 1 hour. .
- the aqueous layer of the filtrate was extracted twice with ethyl acetate (100 ml), and then all the organic layers were washed with saturated aqueous sodium chloride solution (100 ml), dried over anhydrous sodium sulfate, filtered and concentrated to obtain a residue by column chromatography. (6.03 g, 62%) was obtained.
- the filtered celite layer was washed with methane (10 ml) and the filtrate was concentrated. Isopropane (50ml) is added to the filtrate and azeotropic distillation to remove water. After adding 2-3 times of azeotropic distillation to remove water, Isopropanol (7ml) was added to the obtained solid, the mixture was stirred for at least 5 hours, and then the suspension was filtered. The solid is washed with isopropanol (3 ml). The filtered solid was collected and dried under reduced pressure to obtain the title compound (2.4 g, 86%).
- NCCLS National Committee for Clinical Laboratory Standards. 2000. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobical ly.Approved standard, Agar dilution using Mueller-Hinton agar according to NCCLS document M7-A5, 5 th ed, vol 20, no. 2. National Committee for Clinical Laboratory Standards, Wayne, PA.
- the test strain was a clinical isolate isolated from patients in the Korean general hospital from 2010 to 2013, and it was carbapenem-resistant acinetobacter bowmani.
- the title compound compound A a derivative having no substituents at positions 7 and 8 of tricyclic benzoxabo, may be obtained by using the method described in Example 24 of Patent Document WO 02013/093615. g was obtained.
- P.mirabi 1 is 8 8 8 16 0.0625
- the tricyclic benzoxaborole compounds according to the present invention are superior to or better than the compound A, compound B and compound C, which are the control group for a large number of Gram-negative bacteria. MIC results were shown. Similar to the compound of the present invention, the tricyclic benzoxabo compounds according to the present invention showed much better MIC results against a large number of Gram-negative bacteria compared to the compound of compound D having a hydroxymethyl group instead of methyl of carbon 7 .
- the compound of the present invention showed superior MIC not only to meropenem but also to Compound A, compound B and compound D, and even higher than that of compound C.
- Examples 2, 3 and 4 compounds showed a superior MIC compared to compound C. Since the current infection of carbapenem-resistant Acinetobacter boumani does not have an effective antimicrobial agent, thereby limiting the selection of a therapeutic agent, the tricyclic benzoxaborole compound of the present invention may be a very effective therapeutic agent for such bacterial infection.
- test strain used Acinetobacter Boumani BM-1605, which is resistant to carbapenem, and systemic infection was induced by intraperitoneal injection of mouse strains.
- the mice infected with the test strain were orally administered with Examples 1, 2, 3, and 5 compounds or controls of the present invention 1 hour after infection.
- the survival rate was then observed for 7 days, and the dose of the new compound (ED 50 ) required for 50% survival was calculated:
- the results for H acinetobacter Bowmani BAA-1605 are shown in Table 3.
- Example 1 exemplary ED 50 value of Example 2 as in Example 5.
- the compounds of the invention are shown, respectively 5.18, 0.69 6, 6.69 times the good ED 50 value of Compound C prepared.
- Example 3 compound of the present invention showed an ED 50 value that is 9.98 times superior to Compound C.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description














































Claims












Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ716154A NZ716154A (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method therefor, and use thereof |
| HK16106048.1A HK1218120A1 (zh) | 2013-07-30 | 2014-07-28 | 三环苯并氧杂硼化合物、其制备方法与应用 |
| AU2014297102A AU2014297102B2 (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method therefor, and use thereof |
| EP14831789.4A EP3029043B1 (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method therefor, and use thereof |
| JP2016531520A JP6130600B2 (ja) | 2013-07-30 | 2014-07-28 | トリサイクリックベンズオキサボロール化合物、その製造方法および用途 |
| CN201480043221.5A CN105452259B (zh) | 2013-07-30 | 2014-07-28 | 三环苯并氧杂硼化合物、其制备方法与应用 |
| BR112016001876-1A BR112016001876B1 (pt) | 2013-07-30 | 2014-07-28 | Composto de benzoxaborol tricíclico, método de preparação e composição farmacêutica do mesmo |
| ES14831789.4T ES2688597T3 (es) | 2013-07-30 | 2014-07-28 | Compuesto de benzoxaborol tricíclico, método de preparación y uso del mismo |
| CA2918249A CA2918249C (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method and use thereof |
| RU2016103919A RU2639153C2 (ru) | 2013-07-30 | 2014-07-28 | Трициклическое бензоксабороловое соединение, способ его получения и его применение |
| MX2016001138A MX363530B (es) | 2013-07-30 | 2014-07-28 | Compuesto de benzoxaborol tricíclico, método de preparación y uso del mismo. |
| SG11201600563UA SG11201600563UA (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method therefor, and use thereof |
| US14/908,006 US9676796B2 (en) | 2013-07-30 | 2014-07-28 | Tricyclic benzoxaborole compound, preparation method and use thereof |
| IL243699A IL243699A0 (en) | 2013-07-30 | 2016-01-20 | Benzoxaborole-type substances, a process for their preparation and preparations containing them |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2013-0090493 | 2013-07-30 | ||
| KR20130090493 | 2013-07-30 | ||
| KR10-2014-0093765 | 2014-07-24 | ||
| KR1020140093765A KR101636431B1 (ko) | 2013-07-30 | 2014-07-24 | 트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015016558A1 true WO2015016558A1 (ko) | 2015-02-05 |
Family
ID=52571626
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2014/006894 Ceased WO2015016558A1 (ko) | 2013-07-30 | 2014-07-28 | 트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US9676796B2 (ko) |
| EP (1) | EP3029043B1 (ko) |
| JP (1) | JP6130600B2 (ko) |
| KR (1) | KR101636431B1 (ko) |
| CN (1) | CN105452259B (ko) |
| AU (1) | AU2014297102B2 (ko) |
| BR (1) | BR112016001876B1 (ko) |
| CA (1) | CA2918249C (ko) |
| ES (1) | ES2688597T3 (ko) |
| HK (1) | HK1218120A1 (ko) |
| IL (1) | IL243699A0 (ko) |
| MX (1) | MX363530B (ko) |
| NZ (1) | NZ716154A (ko) |
| PT (1) | PT3029043T (ko) |
| RU (1) | RU2639153C2 (ko) |
| SG (1) | SG11201600563UA (ko) |
| WO (1) | WO2015016558A1 (ko) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016001834A1 (en) | 2014-07-01 | 2016-01-07 | Daiichi Sankyo Company, Limited | Tricyclic benzoxaboroles as antibacterial agents |
| WO2016128948A1 (en) | 2015-02-12 | 2016-08-18 | Glaxosmithkline Intellectual Property (No.2) Limited | 4 -substituted benzoxaborole compounds and uses thereof |
| US10308668B2 (en) | 2013-08-09 | 2019-06-04 | Glaxosmithkline Intellectual Property (No. 2) Limited | Tricyclic benzoxaborole compounds and uses thereof |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10070649B2 (en) | 2013-01-30 | 2018-09-11 | Agrofresh Inc. | Volatile applications against pathogens |
| US11039617B2 (en) | 2013-01-30 | 2021-06-22 | Agrofresh Inc. | Large scale methods of uniformly coating packaging surfaces with a volatile antimicrobial to preserve food freshness |
| RS64770B1 (sr) | 2016-03-07 | 2023-11-30 | Agrofresh Inc | Sinergistički postupci upotrebe jedinjenja benzoksaborola i gasova za prezerviranje kao antimikrobnih agenasa za useve |
| CN113214302B (zh) * | 2021-04-23 | 2022-06-24 | 四川大学 | 一种抑制金属β-内酰胺酶和/或丝氨酸β-内酰胺酶的3-取代五元环状硼酸酯衍生物 |
| CA3239096A1 (en) * | 2022-06-23 | 2023-12-28 | Shanghai Micurx Pharmaceutical Co., Ltd. | Methods and uses of boron compounds in the treatment of nontuberculous mycobacterium infections and pharmaceutical compositions for treatment of same |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060234981A1 (en) | 2005-02-16 | 2006-10-19 | Anacor Pharmaceuticals | Boron-containing small molecules |
| US20070155699A1 (en) | 2005-02-16 | 2007-07-05 | Anacor Pharmaceuticals | Boron-containing small molecules |
| KR20080110751A (ko) * | 2006-02-16 | 2008-12-19 | 아나코르 파마슈티칼스 인코포레이티드 | 항염증제로서 보론함유 소분자 |
| WO2008157726A1 (en) | 2007-06-20 | 2008-12-24 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| WO2009140309A2 (en) | 2008-05-12 | 2009-11-19 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| WO2011060196A1 (en) | 2009-11-11 | 2011-05-19 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| WO2012033858A2 (en) | 2010-09-07 | 2012-03-15 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US20130165411A1 (en) * | 2011-12-22 | 2013-06-27 | Micurx Pharmaceuticals, Inc. | Tricyclic boron compounds for antimicrobial therapy |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7390806B2 (en) | 2002-12-18 | 2008-06-24 | Anacor Pharmaceuticals, Inc. | Antibiotics containing borinic acid complexes and methods of use |
| RU2397986C2 (ru) * | 2002-12-18 | 2010-08-27 | Анакор Фармасьютикалз, Инк. | Антибиотики, содержащие комплексы бориновой кислоты, и способы их получения |
| KR100925333B1 (ko) | 2008-03-14 | 2009-11-04 | 엘지전자 주식회사 | 랜덤 액세스 과정에서 상향링크 동기화를 수행하는 방법 |
| ES2710105T3 (es) | 2010-04-07 | 2019-04-23 | Glaxosmithkline Llc | Proceso para preparar benzoxaboroles |
| US20160251380A1 (en) | 2013-08-09 | 2016-09-01 | Glaxosmithkline Intellectual Property (No.2) Limited | Tricyclic benzoxaborole compounds and uses thereof |
-
2014
- 2014-07-24 KR KR1020140093765A patent/KR101636431B1/ko not_active Expired - Fee Related
- 2014-07-28 ES ES14831789.4T patent/ES2688597T3/es active Active
- 2014-07-28 HK HK16106048.1A patent/HK1218120A1/zh unknown
- 2014-07-28 RU RU2016103919A patent/RU2639153C2/ru active
- 2014-07-28 US US14/908,006 patent/US9676796B2/en active Active
- 2014-07-28 CA CA2918249A patent/CA2918249C/en active Active
- 2014-07-28 NZ NZ716154A patent/NZ716154A/en unknown
- 2014-07-28 AU AU2014297102A patent/AU2014297102B2/en not_active Ceased
- 2014-07-28 SG SG11201600563UA patent/SG11201600563UA/en unknown
- 2014-07-28 MX MX2016001138A patent/MX363530B/es unknown
- 2014-07-28 BR BR112016001876-1A patent/BR112016001876B1/pt active IP Right Grant
- 2014-07-28 PT PT14831789T patent/PT3029043T/pt unknown
- 2014-07-28 WO PCT/KR2014/006894 patent/WO2015016558A1/ko not_active Ceased
- 2014-07-28 CN CN201480043221.5A patent/CN105452259B/zh active Active
- 2014-07-28 EP EP14831789.4A patent/EP3029043B1/en active Active
- 2014-07-28 JP JP2016531520A patent/JP6130600B2/ja not_active Expired - Fee Related
-
2016
- 2016-01-20 IL IL243699A patent/IL243699A0/en unknown
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060234981A1 (en) | 2005-02-16 | 2006-10-19 | Anacor Pharmaceuticals | Boron-containing small molecules |
| US20070155699A1 (en) | 2005-02-16 | 2007-07-05 | Anacor Pharmaceuticals | Boron-containing small molecules |
| KR20080110751A (ko) * | 2006-02-16 | 2008-12-19 | 아나코르 파마슈티칼스 인코포레이티드 | 항염증제로서 보론함유 소분자 |
| WO2008157726A1 (en) | 2007-06-20 | 2008-12-24 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| KR20100051615A (ko) * | 2007-06-20 | 2010-05-17 | 아나코르 파마슈티칼스 인코포레이티드 | 붕소를 함유하는 소분자 |
| WO2009140309A2 (en) | 2008-05-12 | 2009-11-19 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| WO2011060196A1 (en) | 2009-11-11 | 2011-05-19 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| WO2012033858A2 (en) | 2010-09-07 | 2012-03-15 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US20130165411A1 (en) * | 2011-12-22 | 2013-06-27 | Micurx Pharmaceuticals, Inc. | Tricyclic boron compounds for antimicrobial therapy |
| WO2013093615A1 (en) | 2011-12-22 | 2013-06-27 | Micurx Pharmaceuticals, Inc. | Tricyclic boron compounds for antimicrobial therapy |
Non-Patent Citations (4)
| Title |
|---|
| "Approved standard, NCCLS document M7-A5", vol. 20, 2000, NATIONAL COMMITTEE FOR CLINICAL LABORATORY STANDARDS, article "Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically" |
| S.CHOI ET AL., ANTIMICROB. AGENTS CHEMOTHER, vol. 56, no. 9, 2012, pages 4713 - 4717 |
| See also references of EP3029043A4 |
| VINCENT HERNANDEZ ET AL.: "Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria", ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 57, no. 3, March 2013 (2013-03-01), pages 1394 - 1403, XP002737653 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10308668B2 (en) | 2013-08-09 | 2019-06-04 | Glaxosmithkline Intellectual Property (No. 2) Limited | Tricyclic benzoxaborole compounds and uses thereof |
| US10526352B2 (en) | 2013-08-09 | 2020-01-07 | Glaxosmithkline Intellectual Property (No.2) Limited | Tricyclic benzoxaborole compounds and uses thereof |
| US10858376B2 (en) | 2013-08-09 | 2020-12-08 | Glaxosmithkline Intellectual Property (No.2) Limited | Tricyclic benzoxaborole compounds and uses thereof |
| WO2016001834A1 (en) | 2014-07-01 | 2016-01-07 | Daiichi Sankyo Company, Limited | Tricyclic benzoxaboroles as antibacterial agents |
| WO2016128948A1 (en) | 2015-02-12 | 2016-08-18 | Glaxosmithkline Intellectual Property (No.2) Limited | 4 -substituted benzoxaborole compounds and uses thereof |
| US10774096B2 (en) | 2015-02-12 | 2020-09-15 | Glaxosmithkline Intellectual Property (No.2) Limited | 4-substituted benzoxaborole compounds and uses thereof |
| US11214582B2 (en) | 2015-02-12 | 2022-01-04 | Glaxosmithkline Intellectual Property (No.2) Limited | 4-substituted benzoxaborole compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3029043A1 (en) | 2016-06-08 |
| IL243699A0 (en) | 2016-04-21 |
| RU2639153C2 (ru) | 2017-12-20 |
| KR20150014858A (ko) | 2015-02-09 |
| EP3029043A4 (en) | 2017-04-05 |
| MX363530B (es) | 2019-03-27 |
| MX2016001138A (es) | 2016-04-29 |
| EP3029043B1 (en) | 2018-08-29 |
| ES2688597T3 (es) | 2018-11-05 |
| SG11201600563UA (en) | 2016-02-26 |
| HK1218120A1 (zh) | 2017-02-03 |
| US20160168167A1 (en) | 2016-06-16 |
| RU2016103919A (ru) | 2017-08-31 |
| BR112016001876A2 (pt) | 2017-08-01 |
| CN105452259A (zh) | 2016-03-30 |
| JP2016530250A (ja) | 2016-09-29 |
| AU2014297102B2 (en) | 2017-02-23 |
| NZ716154A (en) | 2017-08-25 |
| BR112016001876B1 (pt) | 2022-02-22 |
| CA2918249A1 (en) | 2015-02-05 |
| JP6130600B2 (ja) | 2017-05-17 |
| PT3029043T (pt) | 2018-10-26 |
| AU2014297102A1 (en) | 2016-02-04 |
| KR101636431B1 (ko) | 2016-07-05 |
| CN105452259B (zh) | 2018-01-12 |
| CA2918249C (en) | 2018-05-08 |
| US9676796B2 (en) | 2017-06-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015016558A1 (ko) | 트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 | |
| JP6031122B2 (ja) | 抗微生物療法のための三環式ホウ素化合物 | |
| EP3662910B1 (en) | Pharmacophore for trail induction | |
| CA3062855A1 (en) | Aminopyridine compounds and methods for the preparation and use thereof | |
| TW201217373A (en) | Prodrug of substituted polycyclic carbamoyl pyridone derivative | |
| WO2015172732A1 (zh) | 作为二肽基肽酶-iv抑制剂的氨基四氢吡喃衍生物 | |
| WO2022227987A1 (zh) | 杂环类衍生物及其制备方法和用途 | |
| JP6824256B2 (ja) | ヒドロキシアルキルチアジアゾール誘導体 | |
| CN120303266A (zh) | 稠环化合物和包含该稠环化合物的药物 | |
| Yoshizawa et al. | S-3578, a new broad spectrum parenteral cephalosporin exhibiting potent activity against both methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa synthesis and structure-activity relationships | |
| EP3475260A1 (en) | Compounds for use as an anti-bacterial or anti-fungal agent and as a zinc sensor | |
| CN107531613B (zh) | 苯并脂肪环取代烷基胺类化合物及其用途 | |
| HUT71098A (en) | Pyrido[1,2,3-d,e][1,3,4]benzoxadiazine derivatives, process for preparing them and pharmaceutical compositions containing them | |
| CN111527093B (zh) | 抗菌杂环化合物及其合成 | |
| CA2971096A1 (en) | A process for preparing halogenated azaindole compounds using pybrop | |
| CN116239600B (zh) | 并环类化合物的制备方法及作为抗真菌剂的应用 | |
| CN119569728B (zh) | 吡啶并吡咯类衍生物及其在制备富亮氨酸重复激酶2抑制剂中的应用 | |
| JP2004203809A (ja) | 新規ビフェニル誘導体 | |
| HK40020077B (en) | Pharmacophore for trail induction | |
| WO2020225273A1 (en) | Novel compounds | |
| HK40020077A (en) | Pharmacophore for trail induction | |
| TW201840322A (zh) | 羥烷基噻二唑衍生物之n-膦醯基氧基甲基前驅藥 | |
| WO2017180248A9 (en) | Enantioenriched viridicatumtoxin b analogs | |
| HK1102562A (en) | Phosphate-bearing prodrugs of sulfonyl hydrazines as hypoxia-selective antineoplastic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480043221.5 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014831789 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2918249 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 243699 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2016/001138 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2016531520 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14908006 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2014297102 Country of ref document: AU Date of ref document: 20140728 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016001876 Country of ref document: BR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14831789 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016103919 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016001876 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160128 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 716154 Country of ref document: NZ |