WO2015024486A1 - Dgat1抑制剂及其制备方法和用途 - Google Patents
Dgat1抑制剂及其制备方法和用途 Download PDFInfo
- Publication number
- WO2015024486A1 WO2015024486A1 PCT/CN2014/084586 CN2014084586W WO2015024486A1 WO 2015024486 A1 WO2015024486 A1 WO 2015024486A1 CN 2014084586 W CN2014084586 W CN 2014084586W WO 2015024486 A1 WO2015024486 A1 WO 2015024486A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- dihydropyrimidine
- oxazepine
- oxo
- pyrazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1c(*)c(*)c(*)c(*)c1 Chemical compound *c1c(*)c(*)c(*)c(*)c1 0.000 description 11
- XVHJZYXOLKIDMQ-UHFFFAOYSA-N CC(C(C(C)=C1)C(C=C2)=CN(CC(F)(F)F)C2=O)C=C1N(CCOc1ncnc(N)c11)C1=O Chemical compound CC(C(C(C)=C1)C(C=C2)=CN(CC(F)(F)F)C2=O)C=C1N(CCOc1ncnc(N)c11)C1=O XVHJZYXOLKIDMQ-UHFFFAOYSA-N 0.000 description 1
- GKYRXEXUIXKPKF-UHFFFAOYSA-N CC(C[n]1ncc(-c(c(C)c2)c(C)cc2N(CCOc2c3c(N)ncn2)C3=O)c1C)(F)F Chemical compound CC(C[n]1ncc(-c(c(C)c2)c(C)cc2N(CCOc2c3c(N)ncn2)C3=O)c1C)(F)F GKYRXEXUIXKPKF-UHFFFAOYSA-N 0.000 description 1
- MAMDCMVYRQZSOP-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(CC3(C)OB(C(C=C4)=CN(CC(F)(F)F)C4=O)OC3(C)C)C(C)(C)O2)OC1(C)C Chemical compound CC1(C)OB(B2OC(C)(CC3(C)OB(C(C=C4)=CN(CC(F)(F)F)C4=O)OC3(C)C)C(C)(C)O2)OC1(C)C MAMDCMVYRQZSOP-UHFFFAOYSA-N 0.000 description 1
- VXGZBDIMTOHAKJ-UHFFFAOYSA-N CC1(C)OB(c2c[n](CC(F)(F)F)nc2C)OC1(C)C Chemical compound CC1(C)OB(c2c[n](CC(F)(F)F)nc2C)OC1(C)C VXGZBDIMTOHAKJ-UHFFFAOYSA-N 0.000 description 1
- NOUYQKUGPDXMAY-UHFFFAOYSA-N CC1(C[n]2nc(C)c(-c(c(C)c3)c(C)cc3N(CCOc3c4c(N)ncn3)C4=O)c2)COC1 Chemical compound CC1(C[n]2nc(C)c(-c(c(C)c3)c(C)cc3N(CCOc3c4c(N)ncn3)C4=O)c2)COC1 NOUYQKUGPDXMAY-UHFFFAOYSA-N 0.000 description 1
- HVSAAOKAHPMVQP-UHFFFAOYSA-N Cc(cc(cc1C)N(CCO)C(c(c(Cl)ncn2)c2Cl)=O)c1Br Chemical compound Cc(cc(cc1C)N(CCO)C(c(c(Cl)ncn2)c2Cl)=O)c1Br HVSAAOKAHPMVQP-UHFFFAOYSA-N 0.000 description 1
- DZZGIZGIAFKCAY-UHFFFAOYSA-N Cc1cc(N(CCOc2ncnc(Cl)c22)C2=O)cc(C)c1[Br]=C Chemical compound Cc1cc(N(CCOc2ncnc(Cl)c22)C2=O)cc(C)c1[Br]=C DZZGIZGIAFKCAY-UHFFFAOYSA-N 0.000 description 1
- ABDXGCYMBUOHCN-UHFFFAOYSA-N Cc1cc(N(CCOc2ncnc(N)c22)C2=O)cc(C)c1C(C=C1)=CN(CC(F)(F)F)C1=O Chemical compound Cc1cc(N(CCOc2ncnc(N)c22)C2=O)cc(C)c1C(C=C1)=CN(CC(F)(F)F)C1=O ABDXGCYMBUOHCN-UHFFFAOYSA-N 0.000 description 1
- WYJSWJFLKMGYKC-UHFFFAOYSA-N Cc1cc(N(CCOc2ncnc(N)c22)C2=O)cc(C)c1[Br]=C Chemical compound Cc1cc(N(CCOc2ncnc(N)c22)C2=O)cc(C)c1[Br]=C WYJSWJFLKMGYKC-UHFFFAOYSA-N 0.000 description 1
- OGBPXDJLNYCSJH-UHFFFAOYSA-N Cc1cc(N)cc(C)c1Br Chemical compound Cc1cc(N)cc(C)c1Br OGBPXDJLNYCSJH-UHFFFAOYSA-N 0.000 description 1
- GJLMELKPSAIDOM-UHFFFAOYSA-N Cc1n[n](CC(F)(F)F)cc1-c(c(C)c1)c(C)cc1N(CCOc1ncnc(N)c11)C1=O Chemical compound Cc1n[n](CC(F)(F)F)cc1-c(c(C)c1)c(C)cc1N(CCOc1ncnc(N)c11)C1=O GJLMELKPSAIDOM-UHFFFAOYSA-N 0.000 description 1
- SIPMCBGMOFFFQC-UHFFFAOYSA-N Nc1ncnc(OCCN2c3ccc(C4CN(CC(O)=O)CC4)cc3)c1C2=O Chemical compound Nc1ncnc(OCCN2c3ccc(C4CN(CC(O)=O)CC4)cc3)c1C2=O SIPMCBGMOFFFQC-UHFFFAOYSA-N 0.000 description 1
- BMZLLGYZNDRECU-UHFFFAOYSA-N O=C(C=C1)N(CC(F)(F)F)C=C1Br Chemical compound O=C(C=C1)N(CC(F)(F)F)C=C1Br BMZLLGYZNDRECU-UHFFFAOYSA-N 0.000 description 1
- NDMZZQRNZFWMEZ-UHFFFAOYSA-N Oc(cc1)ncc1Br Chemical compound Oc(cc1)ncc1Br NDMZZQRNZFWMEZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to a novel DGAT1 inhibitor, in particular a compound of the formula (I), a process for the preparation thereof, a pharmaceutical composition thereof and a FBS:) for preventing and treating familial sputum microparticle syndrome, obesity, high Use in drugs for lipoproteinemia or hypertriglyceridemia.
- a novel DGAT1 inhibitor in particular a compound of the formula (I), a process for the preparation thereof, a pharmaceutical composition thereof and a FBS:
- DGAT Diacylglycerol acyltransferase
- TAG Triacylglycerol acyltransferase
- DGAT plays a fundamental role in cellular glycerol metabolism and plays an important role in higher eukaryotic triglyceride metabolic pathways such as intestinal fat absorption, lipoprotein aggregation, lipogenesis and lactation.
- Triglyceride is essential for mammals to maintain normal physiological functions, but the storage of excess triglycerides (TAG) leads to obesity.
- DGAT1 diacylglycerol acyltransferase
- SCD1 sterol coenzyme A desaturase 1
- Diacylglycerol acyltransferase is a key enzyme that catalyzes the final step in the synthesis of triglycerides.
- the genetic defects and inhibition of DGAT1 prevent obesity caused by a high-fat diet and increase the body's sensitivity to insulin without side effects.
- Hypertriglyceridemia has been identified as a major independent risk for future cardiovascular disease Factors.
- diacylglycerol acyltransferase is a key enzyme in the final step of catalyzing the synthesis of triglycerides and is therefore identified as a potential therapeutic target against human hyperlipidemia and cardiovascular disease.
- DGAT1 is a major and fundamental factor for viral assembly, and it is also an indispensable factor in the transfer of HCV core from the endoplasmic reticulum membrane to lipid droplets to produce infectious particles.
- Selective inhibition or silencing of DGAT1, but not DGAT2 can greatly prevent the weakening of the production and secretion of infectious viruses, while the replication of HCV RNA is unaffected.
- Drugs for treating diabetes may reach $2 billion in 2017 from $1.1 billion at the end of 2009. By 2015, China's diabetes and obesity market could reach $6.7 billion.
- the number of obese people in China is 70 million, which is 14.6% of the total number of adults.
- the national weight loss market should have a capacity of 67.7 billion yuan, and the annual sales of the weight loss products market is less than 10 billion yuan.
- Novartis is developing Pradigastat (LCQ-908, Novartis), whose indications include familial sputum granulosis syndrome (FCS:), diabetes, obesity, hyperlipoproteinemia, hypertriglyceridemia, cardiovascular disease , renal dysfunction or liver damage, is currently in clinical trials.
- FCS familial sputum granulosis syndrome
- R la , R lb , R 2 , R 3a , R 3b , R 4a or R 4b each independently represent 11, alkyl
- c 3-10 ring ) c 3-10 c 3-10 cycloalkyl or (3 ⁇ 4. 1() cycloalkoxy is optionally substituted by halogen, OH, SH, dish 2 or ?1 ⁇ 4, the number of substituents is one or more;
- 1 6 represents an optionally substituted benzoheterocyclic ring, an optionally substituted monoheterocyclic ring, an optionally substituted cycloalkyl aldehyde group, and an optionally substituted aryl group which is optionally substituted with a benzoheterocyclic ring.
- the heterocyclic ring is saturated or unsaturated, the number of heteroatoms or heteroatoms is 1-8, respectively, independently selected from N, S, F, 0, SO and S0 2;
- the C 1-10 fluorenyl group, the C 1-10 decyloxy group, the C 3-10 cyclodecyl group, the C 3-10 cyclodecyloxy group, the —C 1-10 OH, —C 1-10 COOH , —OC 1-10 COOH , -C 1-10 OC 1-10 , -C 1-10 COO C 1-10 , -C 1-10 CN or -C 1-1Q CONH 2 is optionally halogen, OH, SH, NH 2 , PH 2 , CN, CF 3 , -OCF 3 , -OCH 3 , d. substituted by one or more of 6 alkyl, d. 6 cycloalkyl and d. 6 alkoxy;
- the carbon atom of the oxygen atom or the carbonyl group or the ipsilateral side of the -C ⁇ oO dK) or -d.wCOO C 1-10 is optionally bonded to form a ring;
- H of the N-position of the -CLK) amide is optionally substituted in whole or in part by one or two d. 6 alkyl groups;
- R 7 . 1Q independently represents du)alkyl, du)alkoxy, C 3 .1Q cycloalkyl, C 3 .1Q cycloalkoxy, H, halogen, OH, SH, ⁇ 2, PH 2 and CN, the C WQ alkyl group, the C WQ alkoxy group, the C 3 .1Q cycloalkyl group or the 3 (1) cycloalkoxy group is optionally halogen, OH, SH, NH 2 , PH 2 , CN , CH 3 or CF ⁇ substituted, the number of substituents is one or more; and
- the alkyl or C 3 . 1Q cycloalkoxy group is optionally substituted by halogen, OH, SH, ⁇ 2, PH 2 , CN, CF 3 , —OCF 3 or —OCH 3 , and the number of substituents is 1 to 3 .
- R la , R lb or R 2 is selected as ⁇ .
- R 3a , R 3b , ⁇ or ⁇ are each independently selected from H or methyl.
- the R 6 is selected from optionally substituted 9 to 10 membered benzo-heterocycle, optionally substituted heteromonocyclic -5 to 6, optionally substituted C 3. 1Q cycloalkyl group and an aldehyde group is connected An optionally substituted phenyl group of an optionally substituted 9 to 10 membered benzoheterocyclic ring, wherein
- the heterocyclic ring is saturated or unsaturated, the number of heteroatoms or heteroatoms is 1-8, respectively, independently selected from N, S, F, 0, SO and S0 2;
- the carbon atom of the oxygen atom or the carbonyl group or the ipsilateral side of the -C ⁇ oO dK) or -d.wCOO C 1-10 is optionally bonded to form a ring;
- H of the -CLK) amide N position is optionally substituted, in whole or in part, by one or two d. 6 alkyl groups.
- the above R6 is selected from
- R 14 and R 15 are each independently selected from H, d. 6 alkyl or d. 6 alkoxy, and the d. 6 alkyl or CL 6 alkoxy is optionally halogen, OH, SH, NH 2 Or substituted by PH 2 , the number of substituents is 1 to 3;
- R 16 . 4Q are each independently selected from the group consisting of H, halogen, OH, SH, NH 2 , PH 2 , d. 6 alkyl, d. 6 Alkoxy, -C 1-6 OH, -C 1-6 COOH, -OC 1-6 COOH, -C 1-6 0 C 1-6 , -C 1-6 COO C 1-6 , -C 1 -6 CN, -C 1-6 CONH 2 ,
- the C 6 alkyl group, the d. 6 alkoxy group, -C 1-6 OH, -C 1-6 COOH, -OC 1-6 COOH, -C 1-6 0 C 1-6 , -C 1 -6 COO C 1-6 , -C 1-6 CN, -C 1-6 CON3 ⁇ 4 are optionally halogen, OH, SH, ⁇ 2 , ? ! ⁇ or ⁇ cycloalkyl substituted, the number of substituents is 1 ⁇ 3;
- the carbon atom on either or both sides of the oxygen atom or the carbonyl group in the -CwO d. 6 or -C ⁇ COO d. 6 is optionally bonded to form a ring;
- H in the N position of the -C ⁇ amide is optionally substituted in whole or in part by one or two d. 6 alkyl groups.
- R 14 and R 15 are each independently selected from ⁇ , methyl and CF 3 .
- the above R 26 . 3Q is selected from H.
- R 16 . 25 , R 31 .4 Q are each independently selected from the group consisting of
- R 7 and R 8 described above are each independently selected from -CH 3 , -CH 2 CH 3 , -CF 3 , -OCH 3 , -OCF 3 , halogen, 011 or 11.
- R 9 and R 1Q described above are selected from H.
- R 1 W 3 are each independently selected from -OCH 3 , -OCF 3 , -OCH 2 CF 3 or -CF 3 .
- the compounds of the invention have the following preferred solutions:
- Another object of the present invention is to provide a process for the preparation of the above compound of the formula (I), wherein
- Xi, X 2 are independently selected from CF3, H, halogen, OH, SH, NH 2 , PH 2 , ⁇ and . ⁇ alkyl, the d. 6 alkyl group is optionally substituted by halogen, OH, SH, NH 2 or PH 2 , the number of substituents is one or more;
- R 6 ' represents R 6 protected by a protecting group
- R la , R lb , R 2 , R 3a , R 3b , R 4b , R 6-1Q are as defined in claims 1 to 10.
- Another object of the present invention is to provide another process for the preparation of the above compounds, wherein , including the following steps:
- Aj, A 2 and Y are each independently selected from the group consisting of CF 3 , halogen, OH, SH, NH 2 , PH 2 , NH 2 and ( ⁇ . 6 alkyl, the d. 6 alkyl optionally being halogen, OH, Substituted by SH, NH 2 or PH 2 , the number of substituents is one or more;
- R 6 " represents R 6 protected by a protecting group
- the protecting group described in the above preparation method is an amino protecting group or a hydroxy protecting group.
- the amino protecting group described in the above preparation method is selected from the group consisting of BOC, Cbz, Fmoc, Bn, PMB, Pht, Ac, Trt, CF 3 CO, Alloc, methyl ester, Troc or PMP; Base is selected from ethyl, TBS, TBDPS, TMS, Ac, Me, MOM, MEM, THP, Bn, PMB, MTM or Piv.
- Another object of the present invention is to provide a pharmaceutical composition
- a pharmaceutical composition comprising the above therapeutically effective amount of a compound of the formula ⁇ or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- an object of the present invention is to provide a compound of the above formula ⁇ or a pharmaceutically acceptable salt thereof or the above pharmaceutical composition for the preparation of a prophylactic or therapeutic FMS:), obesity, hyperlipoproteinemia Or the application of drugs with hypertriglyceridemia.
- the invention relates to the following definitions:
- Halogen includes fluorine, chlorine, bromine and iodine;
- CL 6 includes d, c 2 , c 3 , c 4 , c 5 and c 6 ;
- c 4-10 includes c 4 , c 5 , C 6 , C 7 , C 8 , C 9 and BC 10 ;
- C 1-10 include Ci, c 2 , c 3 , c 4 , c 5 , C 6 , C 7 , C 8 , C 9 and BC 10 ;
- 5-10 membered heterocyclic ring includes 5, 6 , 7, 8, 9 and 10 membered heterocycles.
- the compounds of the present invention may have asymmetric centers, and compounds containing asymmetrically substituted atoms in the present invention may be separated into optically active or racemic forms, and those skilled in the art know how to prepare optically active forms, such as by racemate resolution. Alternatively it is synthesized from an optically active starting material. Many geometric isomers of olefins, ON double bonds, and the like can also be present in the compounds of the present invention, all of which are part of the present invention. The invention also describes cis and trans geometric isomers which can be separated into a mixture of isomers or a single isomer form.
- the invention includes all chiral, diastereomeric, racemic and all geometric isomeric forms.
- Methods of preparing the compounds of the invention and intermediates thereof are part of the present invention. All tautomers of the compounds of the invention are also part of the invention.
- substituted means that any one or more hydrogen atoms on a particular atom are replaced by a substituent, including heavy gas and a variant of the atmosphere, as long as the valence of the particular atom is normal and the substituted compound is stable.
- Ketone substitution does not occur on the aryl group.
- a ring double bond is a double bond formed on two adjacent ring atoms, such as
- the invention includes all isotopes of atoms in the compounds of the invention.
- Isotopes include atoms with the same atomic number but different mass numbers.
- the isotopes of the atmosphere include strontium and barium
- the isotopes of carbon include 13 and 14.
- any variable e.g., R 6
- its definition in each case is independent.
- the group may optionally be substituted with up to two 16, and R6 in each case independently has the option.
- Combinations of this substituent and Z or variants thereof are permissible only if such a combination would result in a stable compound.
- substituents When a bond of a substituent can be cross-linked to two atoms on a ring, the substituent can be bonded to any atom on the ring.
- substituents do not indicate which atom is attached to a compound included in the chemical structural formula including but not specifically mentioned, such a substituent may be bonded through any atomic phase thereof. Combinations of substituents and Z or variants thereof are permissible only if such combinations result in stable compounds.
- the term "mercapto" itself or as part of another substituent means a straight, branched or cyclic hydrocarbon radical or a combination thereof, which may be fully saturated, unitary or polyunsaturated, A divalent or multivalent atomic group may be included having a specified number of carbon atoms (i.e., Crdo represents 1 to 10 carbons).
- the term “alkyl” refers to a straight or branched chain of atoms or a combination thereof, which may be fully saturated, mono- or polyunsaturated, and may include divalent and multivalent radicals.
- saturated hydrocarbon radicals include, but are not limited to, methyl, ethyl, n-propyl, Isopropyl, n-butyl', t-butyl, isobutyl, sec-butyl, isobutyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, and n-pentyl, n-hexyl, positive A homolog or isomer of an atomic group such as heptyl or n-octyl.
- the unsaturated fluorenyl group has one or more double or triple bonds, and examples thereof include, but are not limited to, a vinyl group, a 1-propenyl group, a T-alkenyl group, a crotonyl group, a 2-isopentenyl group, and a 2-butadienyl group. , 2, 4-pentadienyl, 3-(1,4-pentanyl), ethynyl, 1 and 3-propynyl, 3-butynyl, and higher homologs and isomers .
- decyloxy decyloxy
- alkylthio or thiodecyloxy
- heteroalkyl by itself or in conjunction with another term, denotes a stable straight-chain, branched or cyclic atomic group or a combination thereof, having a number of carbon atoms and at least one heteroatom.
- heteroalkyl by itself or in conjunction with another term denotes a stable straight chain, branched radical or combination thereof, having a number of carbon atoms and at least one heteroatom.
- the heteroatoms are selected from the group consisting of B, 0, N, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen heteroatoms are optionally quaternized.
- the heteroatoms B, 0, N and S may be located at any internal position of the heteroalkyl group or the sulfhydryl group is attached to the fluorene moiety of the molecule.
- Up to two heteroatoms may be consecutive, such as -CH 2 - Li -OCH 3.
- cycloalkyl and heterocycloalkyl mean cyclized “alkyl” and “heteroalkyl”, respectively.
- a hetero atom may occupy a position at which the heterocyclic ring is attached to the rest of the molecule.
- cycloalkyl groups include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
- heterocyclic groups include 1 (1,2,5,6-tetrazolopyridinyl), 1 1 pyridine, 2»piperidinyl, 3-cyclopentyl, 4 morpholinyl, 3 marinyl, tetrafuran-2-yl, Four-in-one furan noisy 3-based, four-individual thiazide- 2 ⁇ -based, four-in-one thiophene 3-yl, 1-pyridazinyl and 2-pyridazinyl.
- halo or halogen by itself or as part of another substituent denotes a fluorine, chlorine, bromine or iodine atom.
- haloalkyl is intended to include monohalofluorenyl and polyhaloalkyl.
- halo (C r C 4) alkyl is meant to include, but not limited. To, trifluoromethyl, 2, 2, 2-trifluoroethyl, 4-chlorobutyl and 34 ⁇ smell propyl Wait.
- aryl denotes a polyunsaturated, aromatic hydrocarbon substituent which may be monocyclic or polycyclic (preferably up to 3 rings) which are fused together or covalently bonded.
- heteroaryl refers to an aryl (or ring) containing one to four heteroatoms.
- the heteroatoms are selected from the group consisting of 13, N, 0, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom is optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom.
- Non-limiting examples of aryl or heteroaryl groups include phenyl, 1-tertyl, 2-naphthyl-, 4-biphenyl, ⁇ -[:, 2-pyryl, 3-pyridyl, 3- Pyrazolyl, 2-oxazolyl, 4-oxazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-iso Oxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-[pyrazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl 3-furanyl, 2-thiopanyl, 3 ⁇ , 2 ⁇ pyridyl, 3 ⁇ acridinyl, 4-pyridyl, 2 pyrimidinyl, 4 pyrimidinyl, 5-benzothiazolyl, fluorenyl, 2-benzoxazolyl, 5-noise , 1-iso
- aryl and heteroaryl ring systems are selected from those described below.
- the aryl group when used in combination with other terms includes ft The aryl and heteroaryl rings defined above.
- aralkyl is intended to include those radicals in which an aryl group is attached to an alkyl group (eg, a benzyl group, a phenethyl group, a pyridylmethyl group, etc.), including wherein a carbon atom (eg, a methylene group) has been
- alkyl groups replaced by oxygen atoms, such as phenoxymethyl, 2 pyridyloxymethyl 3 ⁇ -caioxy)propyl and the like.
- alkyl and heteroalkyl radicals including what are commonly referred to as alkylene, alkenyl, heteroalkyl, heteroalkenyl, styl, cycloalkyl, heterocycloalkyl, cyclopental and heterocycloalkenyl
- alkyl substituents which may be selected from, but are not limited to, one or more of the following groups:
- R', R", R"', R"" and R""' are each independently preferably an aryl group, a substituted or unbuffered heteroalkyl group, a substituted or unsubstituted aryl group (for example, 1 - ⁇ 3 halogen-substituted aryl), substituted or unsubstituted alkyl, alkoxy, thiomethoxy group or aryl fluorenyl.
- each R group is independently selected, as when more than one, R", R"', R"" and R""' are present. Each of these groups at the base S.
- R' and R" When R' and R" are attached to the same nitrogen atom, they may form a 5-, 6- or 7-membered ring with the nitrogen atom.
- NR'R is intended to include but is not limited to pyrrolidinyl and 4 ⁇ Loline.
- alkyl with is intended to include carbon atom is bonded to a non-hydrogen group consisting of groups, such as haloalkyl (e.g.
- each R group is independently selected, as when more than one R', R", R"', "" and R"'” groups are present. Each of these groups.
- Two substituents on adjacent atoms of the aryl or heteroaryl ring may be optionally substituted with a substituent of the formula -C(0)-(CRR')qU-, Wherein T and U are independently selected from -NR, 0, CRR' ⁇ or a single bond, and q is an integer from 0 to 3.
- two substituents on adjacent atoms of the aryl or heteroaryl ring may be optionally substituted with a substituent of the formula -A(CH2)r B-, wherein A and B are independently selected From -CRR'-, -0-, -NR -, -S -, -S(0)-, S(0)2-, -S(0)2NR'- or single bond, r is 1 ⁇ 4 Integer.
- a single bond on the new ring thus formed can be replaced with a double bond.
- two substituents on adjacent atoms of the aryl or heteroaryl ring may be optionally substituted with a substituent of the formula -A(CH2)r B-, wherein s and d are each independently An integer selected from 0 to 3, X is -0-, -NR', -S-, -S(0)-, -S(0) 2 - or -S(0) 2 NR substituent R, R' And R" and R'" are each independently preferably selected from hydrogen and substituted or unsubstituted alkyl.
- ring 5 denotes substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl or substituted or unrefluxed
- the so-called ring includes a fused ring.
- the number of atoms on the ring is usually defined as the number of atoms of the ring.
- “5 to 7-membered ring” means 5 to 7 atoms arranged around. Unless otherwise specified, the ring Optionally, it contains 1 to 3 hetero atoms.
- 5 to 7 membered ring includes, for example, phenylpyridine and piperidinyl; on the other hand, the term “5 to 7-membered hetero
- the cyclononyl ring “includes pyridinyl and I pyridine, but does not include phenyl.
- the term “ring” also includes ring systems containing at least one ring, each of which "ring” independently conforms to the above definition.
- heteroatom' as used herein includes atoms other than carbon (C) and hydrogen (H), including, for example, oxygen (0), nitrogen (N), sulfur (S), silicon (Si), germanium (Ge), Aluminum (A1) and code (B), etc.
- leaving group means a functional group or atom which may be substituted by another functional group or atom by a substitution reaction (e.g., an affinity substitution reaction).
- a substitution reaction e.g., an affinity substitution reaction
- representative leaving groups include triflate; chlorine, bromine, iodine; sulfonate groups such as mesylate, tosylate, p-bromobenzenesulfonate, p-toluene An acid ester or the like; an oxiranyl group such as an acetoxy group, a trifluoroacetoxy group or the like.
- R is a general abbreviation representing an alkyl group selected from substituted or unsubstituted, a heteroalkyl group substituted or unsubstituted, an aryl group substituted or unsubstituted, substituted or unsubstituted. a substituent such as a heteroaryl group, a substituted or unsubstituted cyclodecyl group, a substituted or unsubstituted heterocyclic fluorenyl group.
- ⁇ ективное amount of a drug, preparation or permeate is meant a sufficient amount of active agent to achieve the desired local or systemic effect.
- “Partially effective”, “cosmetically effective”, “pharmaceutically effective” or “clinically effective” amount refers to the amount of drug that is capable of achieving the desired therapeutic result.
- pharmaceutically acceptable salt refers to a salt of a compound of the invention prepared from a compound having a particular substituent found in the present invention and a relatively non-toxic acid or base.
- the base addition salt can be obtained by contacting a neutral amount of such a compound with a sufficient amount of a base in a neat solution or a suitable inert solvent.
- Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic ammonia or magnesium salts or similar salts.
- the acid addition salt can be obtained by contacting a sufficient amount of the acid with a neutral form of such a compound in a neat solution or a suitable inert solvent.
- pharmaceutically acceptable acid addition salts include inorganic acid salts including, for example, hydrochloric acid, gas bromic acid, 3 ⁇ 4 acid, carbonic acid, hydrogencarbonate, phosphoric acid, monosodium phosphate, dihydrogen phosphate, Sulfuric acid, hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and an organic acid salt, such as acetic acid, propionic acid, isobutyric acid, horse Acid, malonic acid, benzoic acid, crotonic acid, suberic acid, trans-butenedioic acid, lactic acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid and
- the salt is contacted with a base or acid in a conventional manner, and the parent compound is separated, thereby regenerating the neutral form of the compound.
- the parent form of the compound is different from the form of its various salts in that certain physical properties, such as solubility in polar solvents, are different.
- the compound blank provided by the present invention is in the form of a prodrug.
- Prodrugs of the compounds described herein are readily converted to chemical combinations of the invention by chemically changing the cluster under physiological conditions.
- prodrugs can be converted to the compounds of the invention by chemical or biochemical methods in an in vivo setting.
- Certain compounds of the invention may exist in unsolvated or solvated forms, including hydrated forms. In general, the solvated forms are equivalent to the unsolvated forms and are included within the scope of the invention. Certain compounds of the invention may exist in polycrystalline or amorphous form.
- Certain compounds of the invention have asymmetric carbon atoms (optical centers) or double bonds. Racemates, diastereomers, geometric isomers and individual isomers are included within the scope of the invention. Graphical representations of racemates, ambiscalemic and scalemic or enantiomerically pure compounds herein are from Maehr, J. Chem. Ed. 1985, 62: 114-120 ⁇ 1985, 62: 114-120. Unless otherwise stated Use the ⁇ and dashed keys to indicate the absolute ⁇ type of a solid center. When the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, they include ruthenium geometric isomers unless otherwise specified. Likewise, all tautomeric forms are included within the scope of the invention.
- the compounds of the invention may exist in specific geometric or stereoisomeric forms.
- the present invention contemplates Some such compounds include cis and trans isomers, (-)- and (+)-p-enantiomers, (R)- and (S)-enantiomers, diastereomers , () isomer, (L isomer, and its racemic mixture and other mixtures, such as enantiomeric or diastereomeric enriched mixtures, all of which are within the scope of the invention Additional asymmetric carbon atoms may be present in the substituents such as alkyl groups. All such isoindoles, as well as mixtures thereof, are included within the scope of the invention.
- optically active (R)- and (-isomer as well as Z) and iL isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If an enantiomer of a compound of the invention is desired, it can be prepared by asymmetric synthesis or by derivatization with a chiral auxiliary, wherein the resulting mixture of diastereomers is separated and the auxiliary groups are cleaved to provide pure The desired enantiomer.
- a salt of a diastereomer is formed with an appropriate optically active acid or base, followed by stepping as is known in the art.
- the diastereomeric resolution is carried out by crystallization or chromatography, and then the pure enantiomer is recovered. Separation of enantiomers and diastereomers is generally accomplished by the use of chromatographic methods employing a chiral stationary phase, optionally in combination with chemical derivatization (eg, from an amine) Carbamate).
- the compounds of the present invention may contain unnatural proportions of atomic isotopes on one or more of the atoms that make up the compound.
- radiolabeled compounds can be used, such as tritium (3 H), iodine -1 25 (i25 I) or C 14 (i4 C). All isotopic compositional changes of the compounds of the invention, whether radioactive or not, are included within the scope of the invention.
- pharmaceutically acceptable carrier refers to any formulation or carrier medium capable of delivering an effective amount of an active substance of the present invention, a biological activity that does not interfere with the active substance, and which has no toxic side effects to the host or patient, including water, oil. , vegetable and mineral rust, paste base, lotion base, ointment base, etc. These bases include suspending agents, tackifiers, transdermal enhancers, and the like.
- Their formulations are well known to those skilled in the art of veterinary or topical pharmaceuticals. For additional information on the vector, refer to Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), the contents of which are incorporated herein by reference.
- excipient generally refers to the carrier, diluent and/or vehicle required to formulate an effective pharmaceutical composition.
- the term "effective amount” or “therapeutically effective amount” with respect to a pharmaceutical or pharmacologically active agent refers to a sufficient amount of a drug or agent that is non-toxic but achieves the desired effect.
- the "effective amount" of an active substance in the composition means the amount required to achieve the desired effect in combination with another active substance in the composition.
- the determination of the effective amount will vary from person to person, depending on the age and general condition of the recipient, and also on the particular active enamel, and the appropriate effective amount in a case can be determined by one skilled in the art based on routine experimentation.
- active ingredient refers to a chemical entity that is effective in treating a target disorder, disease or condition.
- pharmaceutically acceptable is for those compounds, materials, compositions and/or dosage forms that are within the scope of sound medical judgment and are suitable for use in contact with human and animal tissues. Without excessive toxicity, irritability, allergic reactions or other problems or complications, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts are derivatives derived from the compounds of the present invention wherein the parent rust is modified by salt formation with an acid or with a hydrazine.
- pharmaceutically acceptable salts include, but are not limited to, inorganic or organic acid salts of bases such as amines, alkali metal or organic salts of acid groups such as carboxylic acids, and the like.
- Pharmaceutically acceptable salts include the conventional non-toxic salts or quaternary ammonium salts of the parent compound, for example salts formed from non-toxic inorganic or organic acids.
- salts include, but are not beta, those salts derived from inorganic acids and organic acids selected from the group consisting of 2-acetoxybenzoic acid, 2-hydroxyethanesulfonic acid, acetic acid, Ascorbic acid, benzenesulfonic acid, benzoic acid, carbonic acid gas, carbonic acid, citric acid, edetic acid, ethanedisulfonic acid, ethanesulfonic acid, fumaric acid, glucoheptose, gluconic acid, glutamic acid, ethanol Acid, glycollyarsanilic, hexyl-resorcinic hydrabamic, hydrobromic acid, Hydrochloric acid, gaseous iodate, hydroxyl, hydroxynaphthalene, isethionate, lactic acid, lactose, dodecylsulfonic acid, maleic acid, malic acid, mandelic acid, methanesulfonic acid, nitric acid
- the pharmaceutically acceptable salts of the present invention can be synthesized by a conventional chemical method from a parent compound containing an acid group or a base.
- such salts are prepared by reacting these compounds in a free acid or base form with a suitable amount of a suitable hydrazine or acid in water or an organic solvent or a mixture of the two.
- a nonaqueous medium such as ether, ethyl acetate, ethanol, isopropanol or acetonitrile is preferred.
- a list of suitable salts is disclosed in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, the disclosure of which is incorporated herein by reference.
- prodrugs can enhance many qualities of the drug (e.g., solubility, oyster availability, industrialization, etc.), the compounds of the present invention can be administered in the form of a prodrug. Accordingly, the invention is intended to cover prodrug forms of the compounds claimed herein, the mode of administration thereof, and the pharmaceutical compositions thereof. "Prodrug” is intended to include any covalently bonded carrier which, when administered to a mammalian receptor, will release the active parent drug of the present invention in vivo.
- the preparation of the prodrug of the present invention is carried out by modifying the functional group of the parent compound of the present invention, and the modified parent compound can be cleaved into the parent compound by a conventional operation or in an in vivo manner.
- the prodrug includes a compound of the present invention, wherein a group of a hydroxyl group, a nitrogen group or a thiol group is bonded to any functional group, and when the prodrug of the present invention is administered to a mammalian receptor, it will be separately cleaved to form a free hydroxyl group, free A gas based acid or a free sulfhydryl group.
- prodrugs include, but not beta, acetic acid, formic acid and benzoic acid derivatives of the alcohol or amine functional groups of the compounds of the invention.
- oral dosage form refers to administration of any pharmaceutical composition by buccal administration.
- Oral dosage forms include tablets, capsules, films, powders, sachets, granules, solutions, solids, suspensions, or a plurality of different dosage units (eg, granules, tablets containing different active ingredients) Agent and cockroach Or capsules are packaged together for administration, as well as other means known in the art.
- Oral dosage forms can be one, two, three, four, five or six units. When an oral dosage form has multiple units, all units are packaged in a single package (eg, bottles or other forms of packaging, such as Blister pack); When the oral dosage form is a separate unit, it may or may not be present in a single package.
- the oral dosage form is one, two or three units.
- the oral dosage form is a unit.
- Inhibition and blocking refer to the blockage of part or all of an enzyme such as a chloroglucose.
- leaving group means a functional group or atom which may be substituted by another functional group or atom by a substitution reaction (e.g., an affinity substitution reaction).
- a substitution reaction e.g., an affinity substitution reaction
- representative leaving groups include triflate; chlorine, bromine, bowl; sulfonate groups such as mesylate, tosylate, p-bromobenzenesulfonate, p-toluenesulfonic acid Ester and the like; an oxirane group such as an acetoxy group, a trifluoroacetoxy group or the like.
- amino protecting group refers to a protecting group suitable for preventing side reactions on the nitrogen nitrogen site.
- Representative amino protecting groups include, but are not limited to: formyl; a thiol group such as an alkanoyl group (ft!
- acetyl, trichloroacetyl or trifluoroethyl an alkoxycarbonyl group such as a T oxycarbonyl group (Boc); arylmethoxycarbonyl, such as ethoxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl, such as benzyl (Bn), trityl (Tr), 1 1-Zl- (4 f - methoxyphenyl) methyl; silyl groups embankment, such as a trimethylsilyl group (TMS) and tert-butyldimethylsilyl group embankment (TBS) and the like.
- TMS trimethylsilyl group
- TBS trimethylsilyl group
- TBS tert-butyldimethylsilyl group embankment
- hydroxy protecting group refers to a protecting group suitable for use in preventing hydroxy side reactions.
- Representative hydroxy protecting groups include, but are not limited to, alkyl groups such as methyl, ethyl and t-butyl groups; sulfhydryl groups such as alkanes Acyl (eg ethyl ketone); arylmethyl, m (Bn), p-methoxy (PMB:), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM :); silyl sulfhydryl groups such as trimethylsilyl thiol (TMS) and tert-butyldimethylsilyl thiol (TBS) and the like.
- alkyl groups such as methyl, ethyl and t-butyl groups
- sulfhydryl groups such as alkanes Acyl (eg ethyl ketone)
- haloalkyl groups include, but are not limited to: trifluoromethyl, trichloromethyl, pentafluoroethyl, and Pentachloroethyl.
- An "alkoxy" voltmeter is a sulfhydryl group having a specific number of g carbon atoms attached through an oxygen bridge.
- Ran 6 ⁇ oxy includes the alkoxy groups of ( ⁇ , C 2 , C frustrating C 4 , ( 5 and c 6 ).
- methoxy groups include, but are not limited to: methoxy, ethoxy, n-propoxy , isopropoxy, n-oxy, sec-butoxy, tert-butoxy, n-pentyloxy and s-pentyloxy.
- Cycloalkyl includes saturated cyclic groups such as cyclopropylcyclobutyl or Cyclopentyl. 3 7 cycloalkyl includes C 3 , C 4 , C 5 , ( 6 and €; 7 cycloalkyl.
- Alkenyl includes a hydrocarbon chain in a straight or branched configuration, wherein the chain One or more carbon-carbon double bonds, such as vinyl and propylene groups, are present at any stabilizing site.
- Alkenyl is meant to include c 2, c 3, c 4 , (5 and ⁇ ⁇ alkenyl.
- Alkynyl is intended to include a straight chain or branched chain configuration, hydrocarbon chain wherein any stable position on the chain One or more carbon "carbon triple bonds exist at the point, such as ethynyl and propynyl.
- C 2 ⁇ 6 blocks are meant to include C 2 , C 3 , C 4 , (: 5 and (: 6 alkynyl).
- halogen means fluorine, chlorine, O and iodine;
- counter ion is used to mean a small negatively charged enamel, such as chlorinated bromide oxidized acetate.
- sulfuric acid as used herein, "carbocyclic” or “carbocyclyl” means any stable 3, 4, 5, 6 or 7 membered single ring or ring or 7, 8, 9, 10 -, 11, 12 or 13-membered bicyclic or tricyclic, which may be saturated, partially unsaturated or unsaturated (aromatic).
- carbocyclic rings include, but are not limited to: cyclopropyl., cyclic T-based, cyclobutyl 'Alkenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptene, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctene, cyclooctadienyl, [3 , 3, 0]bicyclooctane, [4,3,0]bicyclic fluorene, [4, 4, 0]bicyclodecane, [2.2.2] Cyclooctyl, 3 ⁇ 4, phenyl, hydrazine, 2 , 3 indanyl, adamantyl and tetrachlorocainyl.
- the bridged ring is also included in the definition of a carbocyclic ring (eg [2, 2, 2]bicyclooctyl).
- a carbocyclic ring eg [2, 2, 2]bicyclooctyl
- one or more carbons Atom connects two non-adjacent carbons A bridged ring midnight.
- a bridge always converts a monocyclic ring into a tricyclic ring. Bridged ring substituents on the ring may also be present on the bridge.
- heterocycle or “heterocyclyl” as used herein means a stable 5 6 or 7 membered monocyclic or bicyclic ring. Or 7, 8, 9 or 10 membered bicyclic heterocycles which may be saturated, partially unsaturated or unsaturated (aromatic) which comprise a carbon atom and 1, 2, 3 or 4 independently selected from Ring heteroatoms of N, 0 and S, wherein any of the above heterocycles may be fused to a phenyl ring to form a bicyclic ring.
- the nitrogen and sulfur heteroatoms can be optionally oxidized (i.e., NO and S(0)p).
- the nitrogen atom can be substituted or unsubstituted (i.e., N or wherein R is H or other substituents as defined herein).
- the heterocyclic ring can be attached to the side groups of any hetero atom or carbon atom to form a stable structure. If the resulting compound is stable, the heterocycles described herein can undergo substitutions at the carbon or nitrogen sites.
- the nitrogen atom in the heterocycle is optionally quaternized. A preferred embodiment is that when the total number of S and 0 atoms in the heterocycle exceeds 1, these heteroatoms are not adjacent to each other.
- the total number of S and 0 atoms in the heterocycle does not exceed ⁇ ⁇
- aromatic heterocyclic group or "heteroaryl” means a stable 5, 6-, 7-membered single.
- the nitrogen atom can be substituted or unsubstituted (i.e., N or NR, wherein R is H or other substituents as already defined herein).
- the nitrogen and sulfur heteroatoms can be optionally oxidized (i.e., NO and SCp). It is worth noting that the total number of S and 0 atoms on the aromatic heterocycle does not exceed one.
- Bridged rings are also included in the definition of heterocycles.
- An anthracene ring is formed when one or more atoms (i.e., 0, N or S) join two non-adjacent carbon or nitrogen atoms.
- Preferred bridged rings include, but are not limited to: one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and one carbon group. It is worth noting that a bridge always converts a single ring into a three ring. In the bridge ring, the substituents on the ring can also appear on the bridge.
- heterocyclic compounds include, but are not limited to, acridinyl, P-octyl, benzoxazolyl, benzofuranyl, benzofuranylfuranyl, benzodecylphenyl, benzoxazolyl, benzene And oxazolidinyl, benzothiazolyl, benzotriazolyl, benzotetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimilin, carbazolyl, 4aH- ⁇ Azolyl, sulfonyl, benzodipyranyl, chromene, ⁇ Base ten quinoxaline, 2H, 6 , 5,2-dithiazyl, dihydrofuro[2 5 34]tetrahydrofuranyl, furyl, furazyl, oxazolidinyl, oxazolidinyl, favor Azolyl, carbazolyl, 0 decyl, alkeny
- “Stable compound” and “11-structure” refer to a compound which is capable of being separated from the reaction mixture and independently and stably present in a certain effective purity, and is formulated into an effective therapeutic drug.
- Substituted means that one or more nitrogen atoms on the "substituted” atom are replaced by a substituent (which has been defined herein) as long as the valence of the particular atom is normal and replaced The compound is stable.
- treatment is the treatment of a disease state, such as: (a) Prevention occurs in a mammalian condition, especially if the mammal is predisposed to the disease but has not yet been diagnosed with the disease; (b) inhibiting the state of the disease, ie preventing its development: and/or (e) Alleviating the disease, that is, causing the disease state to subside.
- Therapeutically effective amount means the amount required for the compound of the present invention to be effective when administered alone or in combination. “Therapeutically effective amount” also refers to the amount of the compound of the present invention which is required to work in combination.
- the combined use of the compounds of the invention is preferably used in a synergistic combination. As taught by Chou and Talalay, Adv. Enzyme egul. 1984, 22: 27-55, the combined effect of the drug is superior to the synergistic effect between the drugs when used alone. Under normal circumstances, suboptimal rolling compounds show the synergistic effects most clearly. The synergistic effect of the combination may be manifested in reducing cytotoxicity, increasing antithrombotic ability, or other beneficial effects as compared to administration alone.
- the compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments set forth below, combinations thereof with other chemical synthesis methods, and those well known to those skilled in the art. Equivalent alternatives, preferred embodiments include, but are not limited to, embodiments of the invention.
- NM displacement [delta] are given in units of 10- 6 (ppm) a.
- the NM was measured using a Bruker AVANCE-400 nuclear magnetic apparatus.
- the solvent was deuterated dimethyl sulfoxide (DMSO-i3 ⁇ 4, deuterated chloroform (CDC1 3 ), deuterated methanol (CD 3 OD), internal standard was tetramethyl).
- Silane TMS :).
- the HPLC was measured using a Shimadzu LC10AD high pressure liquid chromatograph (Xtimate C18 2.1*30m m column).
- the silica gel plate used in the spectral method (TLC) has a specification of 0.15 mm to 0.2 mm, and the thin layer chromatography separation and purification product has a specification of 0.4 mm to 0.5 mm.
- the known starting materials of the present invention may be synthesized by or according to methods known in the art, or may be purchased from ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, TCI, Alfa, ⁇ (Accela) ChemBio Inc), Beijing Coupling and other companies.
- reaction can be carried out under an argon atmosphere or a nitrogen atmosphere.
- An argon atmosphere or a nitrogen atmosphere means that the reaction flask is connected to an argon or nitrogen balloon having a volume of about 1 L.
- the hydrogen atmosphere means that the reaction flask is connected to a hydrogen balloon of about 1 L volume.
- the pressurized hydrogenation reaction was carried out using a Parr Model 3916EKX hydrogenation apparatus and a clear blue QL-500 type hydrogen generator or a HC2-SS type hydrogenation apparatus.
- the hydrogenation reaction is usually evacuated, charged with hydrogen, and operated three times.
- the microwave reaction was carried out using a CEM Discover-S Model 908860 or Biotage Initiator 60 microwave reactor.
- the solution means an aqueous solution.
- reaction temperature is room temperature and is 20 ° C to 30 ° C.
- the progress of the reaction in the examples was monitored by thin layer chromatography (TLC).
- TLC thin layer chromatography
- the system used for the reaction was: A: dichloromethane and methanol system, B: n-hexane and ethyl acetate system, C: petroleum ether And the ethyl acetate system, D: acetone, the volume ratio of the solvent is adjusted depending on the polarity of the compound.
- Purification compounds using column chromatography eluent systems and thin layer chromatography developers include: A: dichloromethane and methanol systems, B: petroleum ether and ethyl acetate systems, C: dichloromethane and acetone
- A dichloromethane and methanol systems
- B petroleum ether and ethyl acetate systems
- C dichloromethane and acetone
- the volume ratio of the solvent is adjusted depending on the polarity of the compound, and a small amount of an alkaline or acidic reagent such as triethylamine or acetic acid may be added for adjustment.
- aq stands for water
- HATU stands for 0-7-azabenzotriazole small base
- EDC stands for ⁇ G dimethyl propyl propyl) ⁇ ⁇ ' ⁇ ⁇ carbodiimide hydrochloride
- m-CPBA stands for 3 chloroperoxybenzoic acid: eq represents equivalent, equivalent
- CDI stands for carbonyl dicarbazole
- DCM stands for Dichloromethane
- PE stands for petroleum ether
- DIAD stands for diisopropyl azodicarboxylate
- DMF stands for N, N-dimethylformamide
- DMSO stands for dimethyl sulfoxide:
- EtOAc stands for ethyl acetate
- EtOH stands for ethanol MeOH stands for methanol
- CBz stands for oxycarbonyl group, is an amine protecting group
- BOC stands for t-
- the compound of the present invention is highly efficient, low in toxicity, and has great enthusiasm in terms of activity, half-life, solubility and pharmacokinetics, and is more suitable for pharmaceuticals. detailed description
- reaction solution was cooled to room temperature, 50 mL of water was added to the reaction solution, and 50 mL ⁇ 3 was extracted with ethyl acetate; and the organic phase was combined, and then washed with water (50 mL ⁇ 2;>, saturated sodium chloride solution 50 mL ⁇ 2; The aqueous solution was dried (MgSO4), filtered Ethyl silicon) oxo)ethyl)amino)-2-chlorobenzene)-2-oxoislimidin-1-yl)acetate EtOAc (550 mg, yellow oil), yield: 58.1%.
- the crude product is 2-(3-(4-(N-(2-((tert-butyldimethylsilyl)oxy)ethyl)- 4,6-dichloropyrimidine-5-amide)-2-fluorobenzene Ethyl)-2-oxoimidazolin-1-yl)acetate lk (1 g, 1.63 mmol) was dissolved in 10 mL of ethanol, concentrated hydrochloric acid (0.3 mL) was added, and the reaction was stirred for 1 hour.
- EtOAc EtOAc
- EtOAcjjjjjjj the crude title product 2-(3-(4-(4-amino-5-oxo-7,8-dihydropyrimidine[5,4-f][1,4]oxazepine-6 (5H) was obtained.
- Ethyl 4-(fluorophenyl)-2-oxoislimidin-1-yl)acetate ln (460 mg, yellow oil).
- the crude product is 2-(3-(4-(N-(2-((tert-butyldimethylsilyl)oxy)ethyl)- 4,6-dichloropyrimidin-5-amide)-2-toluene) - Ethyl 2- oxaimidazolin-1-yl)acetate lg (1.10 g, 18.01 mmol) was dissolved in 10 mL of ethanol, and concentrated hydrochloric acid (0.3 mL;) was added, and the reaction was stirred for 1 hour.
- the crude product is 2-(3-(4-(N-(2-((tert-butyldimethylsilyl)oxy)ethyl)- 4,6-dichloropyrimidine-5-amide)-2-ethyl- Ethyl 6-methylphenyl)-2-oxoislimidin-1-yl)acetate lg (662 mg, 1.04 mol) was dissolved in 10 mL of ethanol, and concentrated hydrochloric acid (0.3 mL) was added. 10 mL of water was added to the reaction mixture, and the mixture was extracted with ethyl acetate (10 mL ⁇ 3).
- reaction solution was cooled to room temperature, 50 mL of water was added to the reaction solution, and 50 mL ⁇ 3 was extracted with ethyl acetate; and the organic phase was combined and washed successively with water (50 mL ⁇ 2;>, saturated sodium chloride solution (50 mL ⁇ 2), with no The aqueous solution was dried (MgSO4).
- Silyl)oxo)ethyl)amino)-2-ethylphenyl)-2-oxoimidazoline-1-ethyl)acetic acid If (600 mg, yellow oil), yield: 59.4%.
- reaction solution was cooled to room temperature, 20 mL of water was added to the reaction solution, and 30 mL ⁇ 3 was extracted with ethyl acetate;>, the organic phase was combined, and then washed with water (30 mL ⁇ 2), saturated sodium chloride solution, 30 mL ⁇ 2; Drying over sodium sulfate, filtration, and EtOAcqqHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH
- Ethyl imidazoline-1-ethyl)acetate la (350 mg, 0.7 mmol) was dissolved in 10 mL dichloromethane, triethylamine (140 mg, 1.4 mmol) and 4,6-dichloropyrimidine-5- The carbonyl chloride (220 mg, 1.5 mmol) was stirred at room temperature for 12 hours. 20 mL of water was added to the reaction solution, and 20 mL of x3 was extracted with dichloromethane.
- reaction mixture was concentrated under reduced pressure, and then purified to purified crystals eluted elution , 2,2-trifluoroethyl)imidazolin-1 -yl)phenyl)-7,8-dihydropyrimidine [5,4-f][l,4]oxazepine-5(6H)- Ketone 1 (135 mg, white solid), Yield: 34.3%.
- N 1 - ⁇ bromo-2,6-dimethylphenyl)benzene-1,2-diamine Dissolve 4-bromo-2,6-dimethyl-N-(2-nitrophenyl)aniline lc (0.6 g, 1.87 mmol) in 10 mL of methanol and add nickel chloride (890 mg, 3.74 mmol) After 5 minutes, sodium borohydride (284 mg, 7.48 mmol) was added and the reaction was stirred for 10 minutes.
- reaction solution was quenched by adding 10 mL of saturated sodium chloride solution, and extracted with dichloromethane (30 mL ⁇ 3;), and the organic phases were combined, and then washed with water (50 mL ⁇ 2;>, saturated sodium chloride solution 50 mL ⁇ 2;) dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give the crude title product N 1 - ⁇ bromo-2,6-dimethylphenyl) benzene-1,2-diamine Id (570 mg, yellow solid), The product was directly subjected to the next reaction without purification.
- reaction solution was cooled to room temperature, 30 mL of water was added to the reaction mixture, and extracted with ethyl acetate (50 mL ⁇ 3). The organic phase was combined and washed with water (50 mL ⁇ 2) and saturated sodium chloride solution (50 mL ⁇ 2) The residue was dried over sodium sulfate, filtered, and evaporated. ))ethyl)amino)-2,6-dimethylphenyl)-3-(2,2,2-trifluoroethyl)-1H-benzo[d]imidazole-2(3H)-one lg (30 mg, yellow oil), Yield: 6.06 %.
- the crude phenyl 1-bromo-4-isocyanate lb (5.70 g, 28.79 mmol) was dissolved in 150 mL of tetrahydrofuran, and 2,2-dimethoxyethylamine (4.54 g, 42.80 mmol) was added. hour. Add 200 mL of water to the reaction solution, and extract with ethyl acetate (200 mL ⁇ 3;>, combine the organic phases, wash 100 mL ⁇ 2 with water (100 mL ⁇ 2), saturated sodium chloride solution;>, dry with anhydrous sodium sulfate, filter The filtrate was concentrated under reduced pressure.
- reaction solution was cooled to room temperature, 50 mL of water was added to the reaction solution, and 30 mL ⁇ 3 was extracted with dichloromethane; and the organic phase was combined, washed successively with water (30 mL ⁇ 2;), saturated sodium chloride solution (30 mL ⁇ 2), with no
- the aqueous solution was dried (MgSO4), filtered Ethyl silicon) oxo)ethyl)amino)phenyl)-2-oxo-2,3-dihydro-1H-imidazol-1-yl)acetate le (210 mg, yellow oil), yield : 15.5%.
- the crude product is 2-(3-(4-(N-(2-((tert-butyldimethylsilyl)oxy)ethyl)-4,6-dichloropyrimidine-5-amide)phenyl)-2- Oxy-2,3-dihydro-1H-imidazol-1-yl)acetate If (297 mg, 50 umol) was dissolved in 10 mL of ethanol, concentrated hydrochloric acid (0.3 mL:) was added, and the reaction was stirred for 1 hour.
- 4-bromo-3,5-dimethylpyrazole la (500 mg, 2.8 mmol) was dissolved in 8 mL of hydrazine, dimethyl-dimethylformamide, and 60% sodium hydrogen was slowly added (137). Mg, 5.7 mmol), after stirring for one hour, potassium iodide (450 mg, 2.7 mmol) and ethyl bromoacetate (524 mg, 3.1 mmol). Heat to 80 °C and stir for 12 hours. After the reaction temperature is lowered to room temperature, 20 mL of a saturated ammonium chloride solution is added dropwise to the reaction solution, and 20 mL ⁇ 3) is extracted with ethyl acetate.
- 3-Chloro-4-iodoaniline la (3.0 g, 11.84 mmol) was dissolved in anhydrous tetrahydrofuran (18 mL), and 60% sodium hydrogen (1.16 g, 29.06 mmol) was slowly added dropwise. After an hour, lb (3.48 g, 14.53 mmol) was added and stirred at 40 ° C for 12 hours. After the reaction was cooled to room temperature, 20 mL of a saturated solution of ammonium chloride was slowly added dropwise and water (50 mL) was used to quench excess sodium hydrogen.
- N-(2-; tert-butyldimethylsilyl:)oxy:)ethyl: )-4,6-dichloro-NP-chloro-4-iodobenzene:)pyrimidine-5-amide le (3.4 g, 5.79 mmol) was dissolved in a mixed solution of 40 mL of ethanol and 1.2 mL of hydrochloric acid, and stirred for 3 hours.
- 4-bromopyrazole la (2 g, 13.7 mmol) was dissolved in 40 mL of N,N-dimethylformamide under ice-cooling, 60% sodium hydrogen (657 mg, 16.4 mmol) was slowly added and stirred. After an hour, ethyl 2-bromopropionate pO g, 16.4 mmol) was added. The reaction was stirred for 12 hours. Add 200 mL of saturated ammonium chloride solution to the reaction solution, and extract 80 mL ⁇ 3) with ethyl acetate.
- Ethyl 2-(4-bromo-1H-pyrazol-1-yl)propanoate lb 50 mg, 0.18 mmol was dissolved in 2 mL of dioxane, and bis-n-n-nol borate ( 68 mg, 0.27 mmol), potassium acetate (53 mg, 0.54 mmol) and hydrazine, hydrazine-bis(diphenylphosphino)ferrocene palladium chloride (7.3 mg, 0.009 mmol), stirred at 110 ° C for 12 hours .
- Lithium hydroxide (30 mg, 63 umol) was added to the reaction liquid of the previous step, and the reaction was stirred for 3 hours. The residue was purified by EtOAc (EtOAc) elut elut 5,4-f][l,4]oxazepine-6(5H)-yl)-2-chlorobenzene)-3-methylpyrazol-1-yl)acetic acid 1 (5 mg, white solid) , Yield: 5.6%.
- EtOAc EtOAc
- 4-Bromopyrazole la (1.47 g, 10.00 mmol) was dissolved in 20 mL of N,N-dimethylformamide, and sodium hydrogen (480 mg, 12.00 mmol) was added under ice bath, and the reaction was stirred for one hour.
- Ethyl 3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazol-1-yl)propanoate Ethyl 3-(4-bromopyrazol-1-yl)propanoate lb (1.80 g, 7.28 mmol) was dissolved in 20 mL of N,N-dimethylformamide, and 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bis(1,3,2-dioxaoxypentane) (2.22 g, 8.74 mmol), potassium acetate (2.14 g, 21.84 mmol) and hydrazine, hydrazine-bis(diphenylphosphino)ferrocene palladium chloride (297 mg, 0.36 mmol), and stirred at 80 ° C for 12 hours.
- reaction solution was cooled to room temperature, and 20 mL of water was added to the reaction mixture, and the mixture was combined with ethyl acetate (20 mL ⁇ 3; >, the organic phase was combined, washed with a saturated sodium chloride solution, 20 mL ⁇ 2;), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give the crude title compound 6-(4-bromo-3-fluorophenyl)-4-chloro-7,8-dichloropyrimidine [5,4-f][l,4]oxazepine -5(6H)-one li (480 mg, yellow solid).
- Lithium hydroxide (30 mg, 63 umol) was added to the reaction liquid of the previous step, and the reaction was stirred for 3 hours. The residue was purified by EtOAc (EtOAc) elut elut 5,4-f][l,4]oxazepine-6(5H)-yl)-2-chlorobenzene)-3-methylpyrazol-1-yl)acetic acid 1 (12 mg, white solid) , Yield: 42.3%.
- reaction solution was cooled to room temperature, and 30 mL of water was added to the reaction mixture, and the mixture was extracted with ethyl acetate (30 mL ⁇ 3), and the organic phase was combined, and then washed with water (30 mL ⁇ 2) and saturated sodium chloride solution (30 mL ⁇ 2); The residue was dried over sodium sulfate, filtered, and evaporated, evaporated,3633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633633
- 3,5-Dimethylaniline la 80 g, 660 mmol was dissolved in 800 mL of acetonitrile under ice bath. A solution of N-bromosuccinimide (117 g, 660 mmol) in 400 ml of acetonitrile was slowly added dropwise, and stirred at room temperature for 16 hours. The reaction mixture was concentrated under reduced vacuolululululululululululululululululululululululululululululu %
- N-(4-bromo-3,5-dimethyl)-N-(2-(ethylhydroxy)-4,6-dichloro-5-pyrimidinecarboxamide N-(4-bromo-3, 5- Dimethyl)-N-(2-((tert-butyldimethyl)oxo)ethyl)-4,6-dichloro-5-pyrimidinecarboxamide ld (160 g, 299.6 mmol) dissolved in 1.5 L a mixture of ethanol and 45 mL of hydrochloric acid, stirred for 3 hours.
- Trifluoromethanesulfonic anhydride 34.39 g, 0.12 mol was dissolved in 20 mL of dichloromethane at -78 ° C, and 2,2-difluoroethanol la (10.00 g, 0.12 mol) and triethylamine were slowly added dropwise. (12 g, 0.12 mol) in dichloromethane (10 mL).
- 2,2-difluoroethane trifluoromethane decanoic acid lb (8.23 g, 0.38 mol), 3-methyl-4-(4,4,5,5-tetramethyl-1,3,2- Dioxaborolan-2-yl)-1H-pyrazole (500 mg, 1.92 mmol) and cesium carbonate (1.25 g, 3.84 mmol) dissolved in 10 mL of N, N-dimethylformamide, microwave 100 The reaction was carried out at ° C for 1 hour.
- the reaction solution was cooled to room temperature, 30 mL of water was added to the reaction solution, and 20 mL ⁇ 3 was extracted with ethyl acetate; and the organic phase was combined, and washed with water (10 mL ⁇ 2;>, saturated sodium chloride solution, 10 mL ⁇ 2;
- the aqueous solution was dried over Na2SO4HHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH
- 3-Bromo-2,2-dimethylpropanoic acid la (500 mg, 2.76 mmol) was dissolved in 5 mL of anhydrous ethanol, and 0.5 mL of concentrated sulfuric acid was added. Stir at 100 ° for 2 hours. The reaction mixture was cooled to room temperature, EtOAc EtOAc (EtOAc m. Mg, yellow liquid), Yield: 74.5%. The product was directly subjected to the next reaction without purification.
- reaction solution was cooled to room temperature, 10 mL of water was added to the reaction solution, and 100 mL ⁇ 3 was extracted with ethyl acetate; and the organic phase was combined, and 100 mL ⁇ 2 was washed successively with water (100 mL ⁇ 2) and saturated sodium chloride solution; The residue was dried over sodium sulfate, filtered, and evaporated, evaporated,36336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336336363363 , 3,2-Dioxaborolan-2
- reaction solution was cooled to room temperature, 50 mL of water was added to the reaction solution, and 50 mL ⁇ 3 was extracted with ethyl acetate;>, the organic phase was combined, and then washed with water (50 mL ⁇ 2;>, saturated sodium chloride solution 50 mL ⁇ 2; The aqueous solution was dried (MgSO4).
- -1H-pyrazole lc (652 mg, yellow liquid), Yield: 32.9%.
- reaction solution was cooled to room temperature, 20 mL of water was added to the reaction solution, and 20 mL ⁇ 3 was extracted with ethyl acetate; and the organic phase was combined and washed successively with water (20 mL ⁇ 2;>, saturated sodium chloride solution (20 mL ⁇ 2), with no
- the aqueous solution was dried over sodium sulfate, filtered, and the filtrate was evaporated, evaporated,jjjjjjjjjjjjjj )) propyl)-3-chloro-4-(1-isobutyl-1H-pyrazol-4-yl)aniline lf (1.00 g, yellow oil), yield: 83.3%.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Obesity (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
本发明公开了一种新的DGAT1抑制剂,尤其是式(I)所示化合物或其药学上可接受的盐,其制备方法、药物组合物及其在预防和治疗家族性糜微粒血症综合征(FCS)、肥胖病、高脂蛋白血症或高甘油三酯血症的药物中的用途。
Description
DGATl抑制剂及其制备方法和用途 本申请要求申请日为 2013年 8月 23日的中国专利申请
CN201310371069.0的优先权。 本申请引用上述中国专利申请的全文。 技术领域
本发明涉及一种新的 DGAT1抑制剂, 尤其是式 (I)所示化合物, 其制备 方法、 药物组合物及其在预防和治疗家族性糜微粒血症综合征FCS:)、 肥胖 病、 高脂蛋白血症或高甘油三酯血症的药物中的用途。 背景技术
二酰基甘油酰基转移酶 (DGAT)是一种微粒体酶, 与脂肪代谢、 脂类在 组织中的沉积有很大关系, 它的主要作用机制是使二酰甘油加上脂肪酸酰基 形成三酰甘油 (TAG:)。 DGAT在细胞甘油代谢中起根本性的作用, 并在高等 真核生物甘油三酯代谢途径如肠脂肪吸收、 脂蛋白集合、 脂肪形成和泌乳中 发挥着重要的功能。
甘油三脂 (TAG) 是哺乳动物维持正常生理功能的必须物质, 但是, 过 多甘油三脂 (TAG) 的存储则会导致肥胖遗传。
目前仅有一类新的药物 (多肽酶 -4抑制剂:)在 II型糖尿病患者血糖的控制 领域获得批准。 也出现了 CPDs 靶向脂质分区和脂质合成酶, 其中包括 DGAT1 (二酰甘油酰基转移酶) 和 SCD1 (固醇辅酶 A去饱和酶 1 ) 酶的抑 制剂。二酰甘油酰基转移酶是催化合成甘油三酯最后一步的关键酶。 DGAT1 的基因缺陷和抑制可以防止由高脂饮食引起的肥胖症以及在无副作用的前 提下提高机体对胰岛素的敏感性。
高甘油三酯血症已被确定为未来心血管疾病的一个主要的独立的危险
因素。 在甘油三酯的生化合成中, 二酰甘油酰基转移酶是催化合成甘油三酯 最后一步的关键酶, 因此被确定为对抗人类高脂血症与心血管疾病的潜在治 疗靶点。
关于丙型肝炎病毒, 对于病毒装配而言, DGAT1 是一个主要而又基本 的因素, 而且它在 HCV的核心从内质网膜转移到脂滴产生传染性粒子的过 程中也是不可缺少的因素。 选择性抑制或沉默 DGAT1 , 而不是 DGAT2, 可 以极大地阻止削弱传染病毒的生产和分泌, 而 HCV RNA的复制不受影响。
2011年全球糖尿病器材和药物市场达到 508亿美元,并且根据市场预期, 这一数值在 2018年将可达到 984亿美元, 从 2011 到 2018年增长率达到 9.9%。 中国糖尿病市场规模已达 150亿, 患者人数达 3980万, 每年的增长 率超过 40%。 治疗糖尿病直接人均医疗花费为 451美元, 治疗患有糖尿病并 发症的患者花费则高达 1694美元。 预计到 2025年, 中国的糖尿病患者总数 将达到 5930万左右。
治疗糖尿病的药物可能从 2009年底的 11亿美元达到 2017年的 20亿美 元。 而到 2015年中国的糖尿病和肥胖症市场可达到 67亿美元。 中国肥胖人 群达 7000万, 是成年人总数的 14.6%, 全国减肥市场容量应为 677亿元, 而目前减肥品市场年销售不到 100亿元。
诺华正在开发 Pradigastat (LCQ-908,诺华) ,其适应症包括家族性糜微粒 血症综合征 (FCS:)、 糖尿病、 肥胖病、 高脂蛋白血症、 高甘油三酯血症、 心 血管病、 肾功能损害或肝功能损害, 目前正处于临床试验阶段。

Pradigastat (LCQ-908,诺华)
辉瑞的国际专利申请 WO2009016462Al、WO2010086820Al公开了一类 化合物, 并在文献 "Discovery of PF-04620110: A Potent, Orally-Bioavailable Inhibitor of DGAT-l , ACS Med. Chem. Lett. 2011, 2, 407-412" 中着重描述了 以下结构的化合物:

PF-04620110 上述化合物在活性、半衰期和溶解度以及药代动力学等方面的表现有待 改i 发明内容
本发明的目的在于提供式①所示化合物或其药学上可接受的盐,

(I)
其中,
Rla、 Rlb、 R2、 R3a、 R3b、 R4a或 R4b分别独立地代表11、 烷基、
c3-10环:) c3-10
 c3-10 环烷基或(¾.1()环烷氧基任选地被卤素、 OH、 SH、 皿2或?1¾所取代, 取代 基的个数为 1个或多个;
c3-10 环烷基或(¾.1()环烷氧基任选地被卤素、 OH、 SH、 皿2或?1¾所取代, 取代 基的个数为 1个或多个;

1 6代表任选被取代的苯并杂环、任选被取代的单杂环、任选被取代的环 烷基醛基和连接任选被取代的苯并杂环的任选被取代的芳基, 其中,
所述杂环是饱和或者不饱和的,杂原子或杂原子团的数目为 1〜8,分别 独立地选自 N、 S、 F、 0、 SO和 S02;
取代基的数目为 1个或多个, 分别独立地选自卤素、 OH、 SH、 NH2、 P¾、 =0、 C1-10焼基、 C1-10焼氧基、。3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-10CONH2, 其中
所述 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-1QCONH2任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CF3、 -OCF3、 -OCH3、
d.6烷基、 d.6环烷基和 d.6烷氧基中的一个或多个所取代;
所述的 -C^oO d.K)或 -d.wCOO C1-10中氧原子或羰基两侧或同侧的碳原 子任选地连接成环;
所述 -CLK)酰胺 N位上的 H任选地全部或部分地被一个或两个 d.6烷基 所取代;
R7.1Q分别独立地代表 d.u)烷基、 d.u)烷氧基、 C3.1Q环烷基、 C3.1Q环烷氧 基、 H、 卤素、 OH、 SH、 丽 2、 PH2和 CN, 所述的 CWQ烷基、 CWQ烷氧基、 C3.1Q环烷基或 3.1()环烷氧基任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CH3 或 CF ^取代, 取代基的个数为 1个或多个; 和
别独立地代表 d.u)烷基、 d.u)烷氧基、 C3.1Q环烷基或 C3.1Q环烷 氧基, 所述 CLK)烷基、 d.u)烷氧基、 C3.1Q环烷基或 C3.1Q环烷氧基任选地被 卤素、 OH、 SH、 丽 2、 PH2、 CN、 CF3、 -OCF3或 -OCH3所取代, 取代基的 数目为 1〜3。
优选地, 上述 Rla、 Rlb或 R2选 § Η。
优选地, 上述 R3a、 R3b、 ^^或^^分别独立地选自 H或甲基。
优选地,上述 R6选自任选被取代的 9〜10元苯并杂环、任选被取代的 5〜 6单杂环、任选被取代的 C3.1Q环烷基醛基和连接任选被取代的 9〜10元苯并 杂环的任选被取代的苯基, 其中,
所述杂环是饱和或者不饱和的,杂原子或杂原子团的数目为 1〜8,分别 独立地选自 N、 S、 F、 0、 SO和 S02;
取代基的数目为 1〜3, 分别独立地选自卤素、 OH、 SH、 NH2、 PH2、 =0、 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH、 -OC1-10COOH、 -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN或 -C1-10CONH2, 其中 所述 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、
-C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-1QCONH2任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CF3、 -OCF3、 -OCH3、 d.6烷基、 d.6环烷基和 d.6烷氧基中的一个或多个所取代;
所述的 -C^oO d.K)或 -d.wCOO C1-10中氧原子或羰基两侧或同侧的碳原 子任选地连接成环; 和
所述 -CLK)酰胺 N位上的 H任选地全部或部分地被一个或两个 d.6烷基 所取代。
优选地, 上述 R6选自
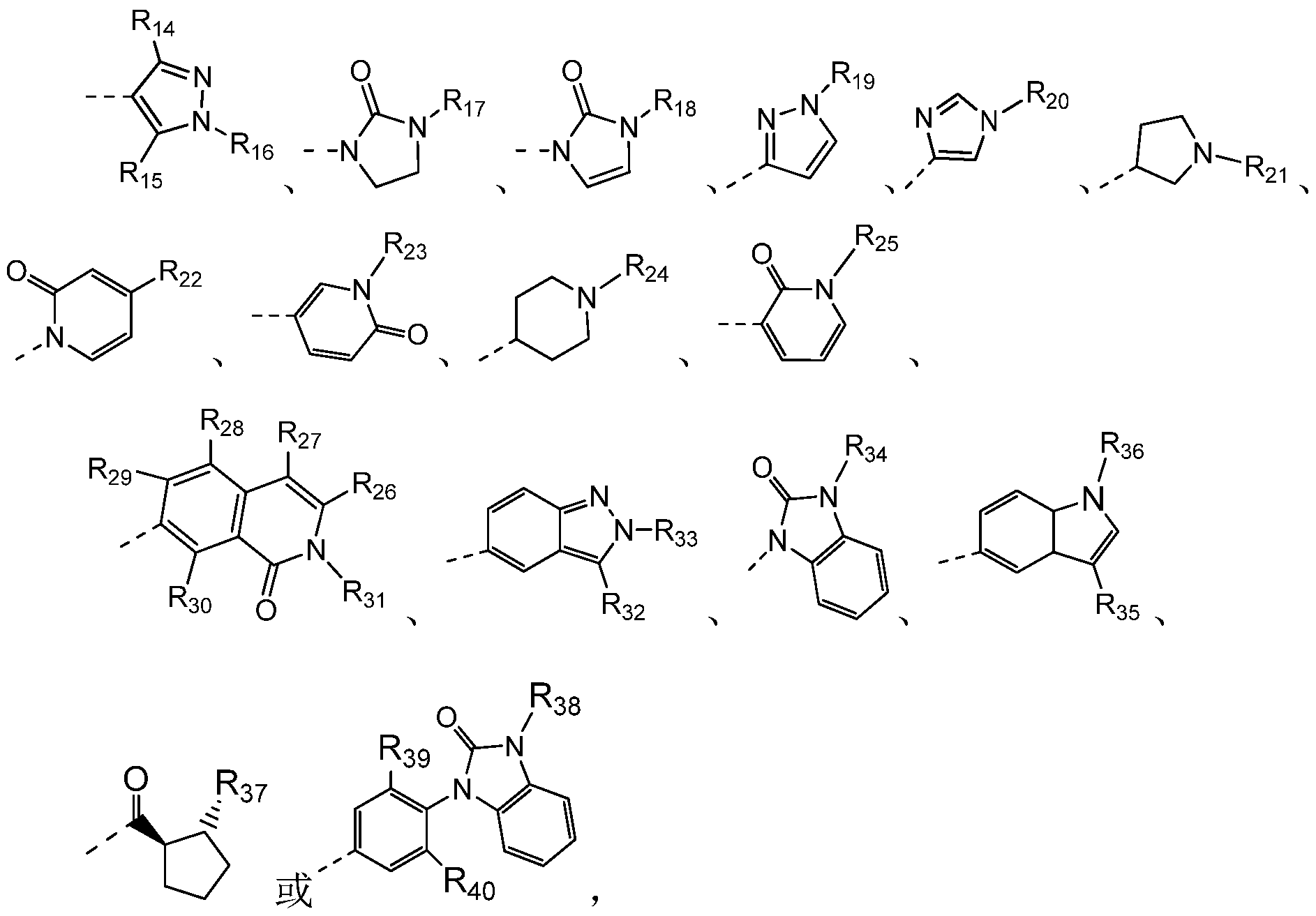
其中,
R14、 R15分别独立地选自 H、 d.6烷基或 d.6烷氧基, 所述 d.6烷基或 CL6烷氧基任选地被卤素、 OH、 SH、NH2或 PH2所取代,取代基的数目为 1〜 3; 和
R16.4Q分别独立地选自 H、 卤素、 OH、 SH、 NH2、 PH2、 d.6烷基、 d.6
烷氧基、 -C1-6OH、 -C1-6COOH、 -OC1-6COOH、 -C1-60 C1-6、 -C1-6COO C1-6、 -C1-6CN、 -C1-6CONH2,
其中,
所述的 C 6烷基、 d.6烷氧基、 -C1-6OH, -C1-6COOH, -OC1-6COOH, -C1-60 C1-6、 -C1-6COO C1-6, -C1-6CN、 -C1-6CON¾任选地被卤素、 OH、 SH、 丽 2、 ?!^或^^环烷基所取代, 取代基的数目为 1〜3;
所述的 -CwO d.6或 -C^COO d.6中氧原子或羰基两侧或同侧的碳原子 任选地连接成环; 和
所述 -C^酰胺 N位上的 H任选地全部或部分地被一个或两个 d.6烷基 所取代。
优选地, 上述 R14、 R15分别独立地选 βΗ、 甲基和 CF3。
优选地, 上述 R26.3Q选自 H。
优选地, 上述 R16.25、 R31.4Q分别独立地选自
1) 卤素;
2) -CH3、 -CH2CH (C¾) 2、 -C (C¾) 3、 -CH2CH (C¾) 2、

3 ) -CH2CF3、 -CH2C ( CH3 ) 2F、 -CH2C ( CH3 ) F2、 -CH2CHF2、 -OCH2CF3;
4) -CH2CH2OH, -CH2C (CH3) 2CH2OH、 -CH2C(CH3)2OH;
5) -COOH, -CH2COOH, -CH2CH2COOH, -CH2C(CH3)2COOH、 -CH2CH (CH3) COOH, -CH (CH3) COOH 、 -C (CH3) 2COOH、 -CH (CH2CH3)
COOH、 -C ( C ) 2COOH、 -CH2CH2COOH、 -CH ( C ) COOH、 -CH ( CH2C )
COOH,
 、 -OCH(CH3) COOH;
、 -OCH(CH3) COOH;

7 ) -CH2COOC2CH5;
8 ) -CH2CN; 和

优选地, 上述 R7 和 R8分别独立的选自 -CH3、 -CH2CH3、 -CF3、 -OCH3、 -OCF3、 卤素、 011或11。
优选地, 上述 R9 和 R1Q选自 H。
优选地, 上述 R1W3分别独立地选自 -OCH3、 -OCF3、 -OCH2CF3或 -CF3。 进一步, 本发明化合物具有以下优选方案:
1 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯苯) -2-氧代咪唑啉 -1-基)乙酸;
2 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氟苯) -2-氧代咪唑啉 -1-基)乙酸;
3 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 甲苯) -2-氧代咪唑啉 -1-基)乙酸;
4 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸;
5 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 乙基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
6 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯苯) -2-氧代咪唑啉 -1-基)丙酸;
7 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-
三氟甲氧基苯 )-2-氧代咪唑啉 - 1 -基)乙酸;
8 ) 4-氨基 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
9 ) 4-氨基 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
10) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
11 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代咪唑啉 -1-基)乙酸;
12) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代咪唑啉 -1-基)乙酸乙脂;
13 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代咪唑啉 -1-基)丙酸;
14) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧苯 )-2-氧代咪唑啉- 1 -基)乙酸;
15 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓- 6(5H)-基) 苯基 2-氧代咪唑啉 -1-基)丙酸;
16) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
17 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基) -三甲基丁酸;
18) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- ¾)-2,6-甲氧基苯基: )-2-氧代咪唑啉 - 1 -基:)乙酸;
19) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -
2,6-二甲基苯) -2-氧代咪唑啉 -1-基)乙酸;
20) 3-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)丙酸;
21 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2- (三氟甲氧基)苯基) -2-氧 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
22) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧基) -2,3-二氢 -1H-咪唑 -1-基)丙酸;
23 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基)丁酸;
24) 3-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基) -二甲基丙酸;
25 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) - 2- (三氟甲基)苯) -2-氧代咪唑啉- 1 -基)乙酸;
26) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
27 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
28) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 2- 氯苯) -2-氧代 -2,3-二氢 咪唑 -1-基)乙酸;
29) 4-氨基 -6-(3-氯 -4-(3-异丁基 -2-氧代咪唑啉 -1-基:)苯基: )-7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
30) 4-氨基-6-(3-氯-4-(3-(环丙基甲基)-2-氧代咪唑啉-1-基)苯基 7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
31 ) 4-氨基 -6-(3-氯 -4-(3-(2-氟 -2-二甲基丙基) -2-氧代咪唑啉 -1-基)苯
基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
32 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑 - 1 -基) -2-甲基丙酸;
33 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑 - 1 -基) -2-甲基丙酸;
34 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) 吡唑 -1-基)丙酸;
35 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) 吡唑 -1-基)丙酸;
36 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3-甲基 吡唑 -1-基)乙酸;
37 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) 吡唑 -1-基)丙酸;
38 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯基 )-3,5-二甲基 吡唑 -1-基)丙酸;
39 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -3- (三氟甲基) 吡唑 -1-基)乙酸;
40 ) 4-氨基-6-(3-氯-4-(3-甲基-1-(2,2,2-三氟乙基)-1 -吡唑-4-基)苯基)-7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
41 ) 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
42 ) 4-氨基 -6-(3-氯 -4-(1-(2-羟基 -2-甲基丙醇基) -1H-吡唑 -4-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-酮;
43 ) 4-氨基 -6-(4-(1-(2,2-二氟乙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基
苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
44) 4-氨基 -6-(3-氯 -4-(1-(3-羟基 -2,2-二甲基丙基) -1-H-吡唑 -4-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -5(6H)-酮;
45 ) (R)-4-氨基 -6-(3-氯 -4-(1-异丁基) -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
46) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -1H -吡唑 -1-基)乙酰胺;
47 ) 4-氨基 -6-(3,5-二甲基 -4-P-甲基 -1-((3-甲基氧杂环丁烷 -3-基)甲 基) -1H-吡唑 -4-基)苯基) - 7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
48) 4-氨基 -6-(4-(1-(2,2-二氟丙基) -5-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
49) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -1H-吡唑 -1-基)乙酸;
50) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -1H-吡唑 -1-基)丙酸;
51 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) -1H-吡咯 -1-基:)丁酸;
52) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) -1H-吡咯 -1-基:)丁酸;
53 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) -1H-吡咯 -1-基 )-3-甲基丁酸;
54) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) - 1 H-吡 P各- 1 -基: )-2-甲基丙酸;
55 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -
苯 )_3-甲基 -m -吡唑 -l-基)乙酸;
56) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑- 1 -基)乙酸;
57 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) - 1 H-吡唑- 1 -基)乙酸;
58) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基)丙酸;
59) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基) -3-甲基丁酸;
60) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基)丙酸;
61 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -1H-吡唑 -1-基)丁酸;
62 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-基) 苯基) -3- (三氟甲基) -1H-吡唑 -1-基)丙酸;
63 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基)
-2-甲基苯基) -m-吡唑小基) -2-甲基丙酸;
64) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) - 1H-吡唑- 1 -基) -丁酸;
65 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-氯苯基) -1H-吡唑 -1-基) -3-甲基丁酸;
66) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) - 1H-吡唑- 1 -基) -2-甲基丙酸;
67 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基)
-2,6-二甲基苯基) -m-吡唑 -1-基)乙酸;
68) 4-氨基-6-(3-氯-4-(1- (2,2,2-三氟乙基) -1氢 -吡唑 -4-基)苯基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
69) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧基苯基) -1H-吡唑 -1 -基)乙酸;
70) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) - 1H-吡唑- 1 -基)丙酸;
71 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基) -2- 氯苯) -1H-吡咯 -1-基) -2,2-二甲基丙酸;
72) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3,5-二甲基 -1H-吡唑 -1-基)乙酸;
73 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) -3 -甲基 - 1H-吡唑 - 1 -基)乙酸;
74) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧基苯) -1H-吡唑 -1-基)丙酸;
75 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) - 1 H-吡唑- 1 -基)乙酸;
76) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮杂卓 -6(5H)-基) -2-苯 基) -3,5-二甲基 -1H-吡唑 -1-基)丙酸;
77 ) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 苯甲基 )-3,5-二甲基 -1H-吡唑 -1-基)丙酸;
78) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3-甲基 -1H-吡唑 -1-基)丙酸;
79) 4-氨基 -6-(3-氯 -4-(1-乙基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]
氮氧杂卓 -5(6H)-酮;
80) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1Η-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
81 ) 4-氨基 -6-(3-氯 -4-(1-异丙基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
82) 4-氨基 -6-(3-氯 -4-(1-(2-羟基乙基) -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-酮;
83 ) 3-(4-(4-(4-氨基 5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) -3-甲基 -1H-吡唑 -1-基)丙酸;
84) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑- 1 -基)乙腈;
85 ) 4-氨基 -6-(3-氯 -4-(1-(2,2-二甲基 -3-氧 -3- (吡咯 -1-基)丙基) -1H-吡唑 -4- 基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
86) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1氢 -吡唑 -4-基)苯基) -8,8-二甲基 -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
87 ) 4-氨基 -6-(3-氯 -4-(1- (环丙基) -1氢 -吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
88) 4-氨基 -6-(3-氯 -4-(1-新戊基 -1氢 -吡唑 -4-基)苯基) - 7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
89) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基) -2- 氯苯基) -1H-吡唑 -1-基) -N,N,2,2-四甲基丙酰胺;
90) 4-氨基 -6-(3-乙基 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
91 ) 4-氨基 -6-(3-氯 -4-(1- (2-氟 -2-异丁基)-1氢 -吡唑 -4-基)苯基) -7,8-二氢
嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
92 ) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1氢 -吡唑 -4-基)苯基) -7,7-二甲基 -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
93 ) 4-氨基 -6-(4-(3,5-二甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基) -3,5-二甲 基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
94 ) 4-氨基 -6-(3-氯 -4-(1-(2-甲氧基 -2-甲基丙基) -1H-吡唑 -4-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
95 ) 4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-吡唑 -3-基)苯基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
96 ) (R)-4-氨基 -6-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -7-甲基 -7,8-二氢 嘧啶 [5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
97 ) 4-氨基 -6-(4-(1-(2-氟 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基 苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
98 ) 4-氨基 -6-(4-(1-(2-羟基 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲 基苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
99 ) 4-氨基 -6-(4-(1-(2-甲氧基 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二 甲基苯 )-7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
100) 4-氨基 -6-(4-(2-氧 -1-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)苯基) - 二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
101) 4-氨基 -6-(3,5-二甲基 -4-(6-氧 -1-(2,2,2-三氟乙基) -1,6-二氢吡啶 -5-基) 苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
102) 4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-咪唑 -4-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
103) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)基)苯
基)哌啶基)乙酸;
104) 2-(3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基)吡咯小基)乙酸;
105) 4-氨基 -6-(3,5-二甲基 -4-(1-氧代 -2-(2,2,2-三氟乙基) -1,2-二氢异喹啉 -7-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)- 酮;
106) (E)- 4-氨基 -6-(6-((2,2,2-三氟乙氧基)亚氨基 )-5,6,7,8-四氢萘 -2- 基) -7,8-二氢嘧啶 [5,4-f] [l,4]氧氮杂卓 -5(6H)-酮;
107) 4-氨基 -3,4-二氢萘 -1 ( 2H) -酮氧 -(2,2,2-三氟乙氧基) 肟 -7,8-二氢嘧 啶 [5,4-f][l,4] 咪唑啉 -5 ( 6H)-基) -酮;
108) 4-氨基 -6-(6-(2,2,2-三氟乙氧基) -5,6,7,8-四氢萘 -2-基) -7,8-二氢吡啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;和
109) 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -2-(2,2,2-三氟乙基) -2H-吲唑 -5-基) 苯基) -7,8-二氢嘧啶 [5,4-f][l ,4] 氧氮杂卓 -5(6H)-酮,
本发明将上述化合物依次命名为化合物 1〜109,其结构式和制备方法依 次详见实施例 1〜: 109。
本发明的另一目的在于提供上述式 (I)化合物的制备方法, 其中 代表

其中,
Xi、 X2分别独立地选自 CF3、
 H、卤素、 OH、 SH、 NH2、 PH2、 ^^和。^烷基、, 所述 d.6烷基任选地被卤素、 OH、 SH、 NH2或 PH2 所取代, 取代基的数目为 1个或多个;
H、卤素、 OH、 SH、 NH2、 PH2、 ^^和。^烷基、, 所述 d.6烷基任选地被卤素、 OH、 SH、 NH2或 PH2 所取代, 取代基的数目为 1个或多个;
R6 '代表 或者被保护基保护的 R6;
Rla、 Rlb、 R2、 R3a、 R3b、 R4b、 R6-1Q如权利要求 1〜10所定义。 本发明的另一目的在于提供上述式 化合物的另一制备方法, 其中
 , 包括如下步骤:
, 包括如下步骤:

其中,
Aj, A2和 Y分别独立地选自 CF3、 卤素、 OH、 SH、 NH2、 PH2、 NH2 和(^.6烷基, 所述 d.6烷基任选地被卤素、 OH、 SH、 NH2或 PH2 所取代, 取代基的数目为 1个或多个;
R6 "代表 或者被保护基保护的 R6;
Rla、 Rlb、 R2、 R3a、 R3b、 R4b、 R6-1Q如权利要求 1〜10所定义。 优选地, 上述制备方法中所述的保护基为氨基保护基或者羟基保护基。 优选地, 上述制备方法中所述的氨基保护基选自 BOC、 Cbz、 Fmoc、 Bn、 PMB、 Pht、 Ac、 Trt、 CF3CO、 Alloc, 甲基酯、 Troc或 PMP; 所述的 羟基保护基选自乙基、 TBS、 TBDPS、 TMS、 Ac、 Me、 MOM、 MEM, THP、
Bn、 PMB、 MTM或 Piv。
本发明的另一目的在于提供一种药物组合物, 其包括上述治疗有效量的 式 ω化合物或其药学上可接受的盐以及药学上可接受的载体。
进一步, 本发明的目的在于提供上述式 ω化合物或其药学上可接受的盐 或上述药物组合物在制备预防或治疗家族性糜微粒血症综合征FCS:)、 肥胖 病、 高脂蛋白血症或高甘油三酯血症的药物中的应用。
本发明可以通过不脱离其发明实质的其他具体方式实现, 包括上述本发 明优选方案的所有组合。本发明的所有具体实施方式可以与其他方式结合形 成更加优选的实施方式, 其中的特定技术特征可以与任何相关的其他技术特 征相结合形成新的实施方式;当然,也可以认为每个优选实施方式是独立的, 其中的特定技术特征仅限于本优选实施方式本身。
本发明涉及下述定义:
卤素包括氟、 氯、 溴和碘; CL6包括 d、 c2 、 c3、 c4 、 c5和 c6; c4-10 包括 c4、 c5 、 C6 、 C7、 C8 、 C9禾 B C10 ; C1-10 包括 Ci、 c2 、 c3 、 c4、 c5 、 C6 、 C7、 C8 、 C9禾 B C10 ; 5-10元杂环包括 5、 6、 7、 8、 9和 10元杂环。
本发明的化合物可能具有不对称中心,本发明中含有不对称取代原子的 化合物可以被分离成光学活性或者消旋体形式, 本领域技术人员知晓如何制 备光学活性形式, 比如通过消旋体拆分或者由光学活性的起始原料合成。 许 多几何异构体的烯烃、 ON双键等等也可以存在于本发明的化合物中, 所有 这些稳定的异构体都属于本发明的一部分。本发明也描述了顺式和反式几何 异抅体, 可以将它们分离成异构体的混合物或单一异构体形式。 除非特别指 明具体的立体化学或异构体形式, 本发明包括所有手性、 非对映异构体, 消 旋体和所有的几何异构体形式。制备本发明化合物的方法及其中间体属于本 发明的一部分。 本发明化合麴的所有互变异构体也属于是本发明的一部分。
术语"取代的 "是指特定原子上的任意一个或多个氢原子被取代基取代, 包括重氣和氛的变体, 只要特定原子的价态是正常的并且取代后的化合物是 稳定的。 当取代基为酮基 (即 =0) 时, 意味着两个氛原子被取代。 酮取代 不会发生在芳香基上。 环双键是指在两个相邻的环原子上形成的双键, 比如
OC、 ON和 N=Nc本发明一般如 N-卤素、 S(0)H以及 S02H这样的基团。
本发明包括本发明的化合物中原子的所有同位素。 同位素包括原子序数 相同、 但质量数不同的原子。 例如但不限于, 氛的同位素包括氚和氘, 碳的 同位素包括 13与 14。
当任何变量(例如 R6 )在化合物的组成或结构中出現一次以上时, 其在 每一种情况下的定义都是独立的。 因此, 例如, 如果一个基团被 0~2 个 R6 所取代, 则所述基团可以任选地至多被两个 1 6所取代, 并且每种情况下的 R6都有独立的选项。 此夕卜 取代基和 Z或其变体的组合只有在这样的组合会 产生稳定的化合物的情^下才是被允许的。
当一个取代基的键可以交叉连接到一个环上的两个原子时,这种取代基 可以与这个环上的任意原子相键合。当所列举的取代基中没有指明其通过哪 一个原子连接到化学结构通式中包括但未具体提及的化合物时, 这种取代基 可以通过其任何原子相键合。取代基和 Z或其变体的组合只有在这样的组合会 产生稳定的化合麴的情¾下才是被允许的。
除非另有规定, 术语"垸基"本身或者作为另一取代基的一部分表示直链 的、 支链的或环状的烃原子团或其组合, 可以是完全饱和的、 单元或多元不 饱和的, 可以包括二价或多价原子团, 具有指定数量的碳原子 (即 Crdo 表示 1至 10个碳)。 在一些实施例中, 术语"烷基"表示直链的或支链的原 子团或它们的组合, 可以是完全饱和的、 单元或多元不饱和的, 可以包括二 价和多价原子团。 饱和烃原子团的实例包括但不限于甲基、 乙基、 正丙基、
异丙基、 正丁 '基、 叔丁基、 异丁基、 仲丁基、 异丁基、 环己基、 (环己基) 甲基、 环丙基甲基, 以及正戊基、 正己基、 正庚基、 正辛基等原子团的同系 物或异构体。 不饱和垸基具有一个或多个双键或三键, 其实例包括但不限于 乙烯基、 1 ~丙烯基、 T烯基、 巴豆基、 2~异戊烯基、 2~ (丁二烯基)、 2 , 4- 戊二烯基、 3- ( 1, 4 戊二婦基)、 乙炔基、 1和 3-丙炔基, 3-丁炔基, 以及 更高级的同系物和异构体。
术语"垸氧基"、 "垸氯基"和"烷硫基" (或硫代垸氧基) 属于惯用表达, 是指分别通过一个氧原子、氣基或硫原子连接到分子的其余部分的那些垸基 基¾。
除非另有规定, 术语"杂烷基 "本身或者与另一术语联合表示稳定的直链 的、 支链的或环犹的经原子团或其组合, 有一定数目的碳原子和至少一个杂 原子组成。 在一些实施例中, 术语"杂烷基 "本身或者与另一术语联合表示稳 定的直链的、 支链的经原子团或其组合惣, 有一定数目的碳原子和至少一个 杂原子组成。 在一个典型实施例中, 杂原子选自 B、 0、 N和 S, 其中氮和 硫原子任选地被氧化, 氮杂原子任选地被季铵化。 杂原子 B、 0, N和 S可 以位于杂烷基的任何内部位置或者该垸基附着于分子其佘部分的位置。实例 包括但不限于 -CH2-CH2-0-CH3、 -CH2-CH2-丽 -CH3、 -CH2-CH2-N(CH3)-CH3、 -CH2-S-CH2-CH3、 -CH2-CH2、 -S(0)-CH3、 -CH2-CH2-S(0)2-CH3、 -CH=CH-0 -CH3、 -CH2-CH=N-OCH3和 -CH=CH-N(CH3)-CH3。 至多两个杂原子可以是 连续的, 例如 -CH2-丽 -OCH3。
除非另有规定, 术语"环烷基 "和"杂环烷基"本身或与其他术语联合分别 表示环化的"烷基"和"杂烷基"。 此外, 就杂环烷基而言, 杂原子可以占据该 杂环附着于分子其余部分的位置。 环垸基的实例包括但不限于环戊基、 环己 基、 1环己烯基、 3环己烯基、 环庚基等。 杂环基的非限制性实例包括 1
( 1,2,5,6-四氮吡啶基)、 1 1截啶基、 2»哌啶基, 3-喊锭基、 4吗啉基、 3』马琳 基、 四氛呋喃2-基、 四氛呋喃吲噪 3-基、 四氛噻玢 - 2~基、 四氛噻吩 3 基, 1~嚒嗪基和 2~嚒嗪基。
除非另有规定, 术语"卤代素 "或"卤素"本身或作为另一取代基的一部分 表示氟、 氯、 溴或碘原子。 此外, 术语"卤代烷基"意在包括单卤代垸基和 多卤代烷基。例如,术语"卤代(CrC4)烷基"意在包括但不仅限.于三氟甲基、 2 , 2 , 2-三氟乙基、 4~氯丁基和 34臭丙基等等。
除非另有规定, 术语"芳基"表示多不饱和的芳族烃取代基, 它可以是单 环或多环 (优选〗 至 3个环), 它们稠合在一起或共价连接。 术语"杂芳基" 是指含有一至四个杂原子的芳基 (或环)。 在一个示范性实例中, 杂原子选 自 13、 N、 0和 S, 其中氮和硫原子任选地被氧化, 氮原子任选地被季铵化。 杂芳基可通过杂原子连接到分子的其余部分。芳基或杂芳基的非限制性实施 例包括苯基、 1—蔡基、 2-萘基-、 4联苯基、 〗- [: 基、 2 比咯基、 3-吡 基、 3-吡唑基、 2-眯唑基、 4-睐唑基、 吡嗪基、 2恶唑基、 4-恶唑基、 2-苯基 4- 恶 唑基、 5 -恶唑基、 3 -异恶唑基、 4-异恶唑基、 5-异恶唑基、 2-〖塞唑基、 4 -噻唑 基、 5~噻唑基、 2~呋喃基 3呋喃基、 2-噻盼基、 3』塞玢基、 2~吡啶基、 3~ 匕 啶基、 4-吡啶基、 2嘧啶基、 4嘧啶基、 5-苯并噻唑基、 嘌呤基、 2-苯并味唑 基、 5 - 噪基、 1-异喹琳基、 5 -异喹啉基、 2~喹喔 基、 5喹喔啉基、 34奎啉 基和 6-喹啉基。上述任意一个芳基和杂芳基环系的取代基选自下文所述的可 为简便起见, 芳基在与其他术语联合使用时 (例如芳氧基、 芳硫基、 芳 垸基) 包括 ft!上定义的芳基和杂芳基环。 因此, 术语"芳烷基"意在包括芳基 跗着于烷基的那些原子团 (例如节基、 苯乙基、 吡啶基甲基等), 包括其中 碳原子 (如亚甲基) 己经被例如氧原子代替的那些烷基, 例如苯氧基甲基、
2吡啶氧甲基 3 α -蔡氧基) 丙基等。
每一个上述术语 (倒 ft! "烷基"、 "杂烷基"、 "芳基 "和 "杂芳基") 意在 被取代或未被取代的所述原子团。 下文会提供每种原子团所优选的取代基。
烷基和杂烷基原子团 (包括通常被称为亚烷基、 链烯基、 亚杂烷基、 杂 烯基、 块基、 环烷基., 杂环烷基、 环爆基和杂环烯基的那些基团) 的取代基 一般被称为"烷基取代基' 它们可以选自但不限于下列基团中的一个或多个:
-R,、 -OR,、 =0、 =NR,、 =N-OR,、- NR,R,,、 -SR,、卤素、 -SiR,R,,R,,,、 OC(0)R,、 -C(0) '、 -C02R'、 -CONR' "、 -OC(0)NR' "、 -NR"C(0) '、 NR' C(0)NR" "' 、 -NR"C(0)2R' 、 -NR"",-C(NR' " ,")=NR"" 、 NR"" C(NR,R,,)=NR,,,、 -S(0)R,、 -S(0)2R,、 -S(0)2NR,R,,、 NR,,S02R,、 -CN、 - N02、 -N3、 -CH(Ph)2和氟代 (d-C4)烷基, 取代基的数目为 0〜 (2m'+l ) ,其中 m' 是这类原子团中碳原子的总数。 R'、 R"、 R"'、 R""和 R""'各自独立地优选 氛、 被取代或未被取伏的杂烷基、 被取代或未被取代的芳基 (例如被 1 -〜 3 个卤素取代芳基)、 被取代或未被取代的烷基、 烷氧基、 硫代垸氧基基团或 芳垸基。 当本发明的化合物包括一个以上的 R基团时, 例如, 每一个 R基 团是独立地加以选择的, 如同当存在一个以上的 、 R"、 R"'、 R""和 R""' 基 S时的每个这些基团。 当 R'和 R"附着于同一个氮原子时, 它们可与该氮 原子结合形成 5-, 6-或 7-元环。 例如, NR'R"意在包括但不仅限于 吡咯烷 基和 4~吗琳基。 根据上述关于取代基的讨论中, 本领域技术人员可以理解, 术语"垸基"意在包括碳原子键合于非氢基团所构成的基团, 如卤代烷基 (例 如 CF3、 -CH2CF3 )和酰基(例如 - C(0)C¾、 - C(0)CF3、 - C(0)C OC¾等 λ 与烷基原子团所述取代基相似, 芳基和杂芳基取代基一般统称为"芳基 取代基",选自例如 -R'、 -OR'、 -NR'R"、 -SR'、 -卤素, -SiR'R"R"'、 OC(0)R'、 -C(0) '、 -C02 '、 -CONR'R"、 -OC(0)NR' "、 -NR"C(0) '、 NR'
C(0)NR" "' 、 -NR"C(0)2 ' 、 -N "",-C(N ' " ,")=NR"" 、 NR"" C(NR' ")=NR"\ -S(0)R,、 -S(0)2R,、 -S(0)2NR,R"、 NR"S02R,、 -CN、 - N02、 -N3、 -CH(Ph)2、 氟 (d-C )烷氧基和氟 (CrC )烷基等, 取代基的数量 为 0 到芳香环上开放化合价的总数之间; 其中 R'、 R"、 R"'、 ""和 R""' 独立地优选自氣、 被取代或未被取代的垸基、 被取代或未被取代的杂烷基、 被取代或未被取代的芳基和被取代或未被取代的杂芳基。当本发明的化合物 包括一个以上的 R基团时, 例如, 每个 R基团是独立地加以选择的, 如同 当存在一个以上 R'、 R"、 R"'、 ""和 R"' "基团时的每个这些基团。 芳基或 杂芳基环的相邻原子上的两个取代基可以任选地被通式为- C(0)-(CRR')q-U-的取代基所取代,其中 T和 U独立地选自- NR、 0、CRR'~ 或单键, q是 0到 3的整数。
作为替代选择, 芳基或杂芳基环的相邻原子上的两个取代基可以任选地 被通式为 -A (CH2)r B-的取代基所取代,其中 A和 B独立的选自 -CRR'-、-0-、 -NR -、 -S -、 -S(0)-、 S(0)2-、 -S(0)2NR'-或单键, r是 1〜4的整数。任选地, 由此形成的新环上的一个单键可以替换为双键。
作为替代选择, 芳基或杂芳基环的相邻原子上的两个取代基可以任选地 被通式为 -A (CH2)r B-的取代基所取代, 其中 s和 d分别独立的选自 0〜3的 整数, X是 -0-、 -NR'、 -S -、 -S(0)-、 -S(0)2-或 -S(0)2NR 取代基 R、 R'、 R"和 R' "分别独立地优选自氢和被取代或未被取代的 烷基。
本文所用的"环'5表示被取代或未被取代的环烷基、 被取代或未被取代的 杂环烷基、 被取代或未被取代的芳基或被取代或未被取伏的杂芳基。 所谓的 环包括稠环。 环上原子的数目通常被定义为环的元数, 例如, "5〜7 元环" 是指环绕排列 5〜7个原子。除非另有规定,该环任选地包含 1〜3个杂原子。 因此, "5〜7元环' '包括例如苯基吡啶和哌啶基; 另一方面, 术语" 5〜7元杂
环垸基环"包括吡锭基和 I 啶基, 但不包括苯基。 术语"环"还包括含有至少 一个环的环系, 其中的每一个"环"均独立地符合上述定义。
本文所用术语' '杂原子"包括碳(C )和氢(H ) 以外的原子, 例如包括氧 ( 0 ) , 氮 (N )、 硫 (S )、 硅 (Si)、 锗 ( Ge ),、 铝 (A1 ) 和碼 (B ) 等。
术语"离去基团 "是指可以被另一种官能团或原子通过取代反应(:例如亲 和取代反应)所取代的官能团或原子。 例 ftl, 代表性的离去基团包括三氟甲 磺酸酯; 氯、溴、碘; 磺酸酯基, 如甲磺酸酯、 甲苯磺酸酯、对溴苯磺酸酯、 对甲苯磺酸酯等; 轔氧基, 如乙酰氧基、 三氟乙酰氧基等等。
"R"是一个通用的缩写, 代表选自被取代或未被取代的烷基、 被取代或 未被取代的杂烷基、被取代或未被取代的芳基、被取代或未被取代的杂芳基、 被取代或未被取代的环垸基、 被取代或未被取代的杂环垸基等取代基。
所谓"有效"量的药麴、 制剂或渗透物是指能够获得所需的局部或全身性 的效果的足够量的活性剂。 "局部有效的"、 "美容上有效的"、 "药学上有效的" 或"临床上有效的"量是指能够实现所期望的治疗结果的药物用量。
术语"药学上可接受的盐"是指本发明化合物的盐, 由本发明发现的具有 特定取代基的化合物与相对无毒的酸或碱制备。 当本发明的化合麴中含有相 对酸性的功能团时, 可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱 与这类化合物的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成 盐包括钠、 钾、 钙、 铵、 有机氨或镁盐或类似的盐。 当本发明的化合物中含 有相对缄性的官能团时, 可以通过在纯的溶液或合适的惰性溶剂中用足够量 的酸与这类化合物的中性形式接触的方式莸得酸加成盐。药学上可接受的酸 加成盐的实例包括无机酸盐, 所述无机酸包括例^盐酸、 氣溴酸、 ¾酸、 碳 酸, 碳酸氢根, 磷酸、 磷酸一氛根、 磷酸二氫根 、 硫酸、 硫酸氢根、 氢碘 酸、 亚磷酸等; 以及有机酸盐, 所述有机酸包括如乙酸、 丙酸、 异丁酸、 马
来酸、 丙二酸、 苯甲酸、 璩珀酸、 辛二酸、 反丁 '烯二酸、 乳酸、 扁桃酸、 邻 苯二甲酸、 苯磺酸、 对甲苯磺酸、 柠檬酸、 酒石酸和甲磺酸等类似的酸; 坯 包括氨基酸(如精氨酸等)的盐, 以及如葡糖醛酸等有机酸的盐(参见 Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977》。 本发明的某些特定的化合惣含有碱性和酸性的官能团, 认而可以被转换成任 一减或酸加成盐。
优选地, 以常规方式使盐与碱或酸接触, 再分离母体化合物, 由此再生 化合物的中性形式。化合狻的母体形式与其各种盐的形式的不同之处在于某 些物理性质, 例如在极性溶剂中的溶解度不同。
除了盐的形式, 本发明所提供的化合物坯存在前药形式。 本文所描述的 化合物的前药容易地在生理条件下发生化学变化丛而转化成本发明的化合 麴。 此外, 前体药物可以在体内环境中通过化学或生化方法被转换到本发明 的化合物。
本发明的某些化合物可以以非溶剂化形式或者溶剂化形式存在, 包括水 合物形式。 一般而言, 溶剂化形式与非溶剂化的形式相当, 都包含在本发明 的范围之内。 本发明的某些化合物可以以多晶或无定形形式存在。
本发明的某些化合物具有不对称碳原子(光学中心)或双键。外消旋体、 非对映异构体、 几何异构体和单个的异构体都包括在本发明的范围之内。 本 文中消旋体、 ambiscalemic and scalemic或者对映体纯的化合物的图示法来自 Maehr, J. Chem. Ed. 1985, 62: 114- 120 ο 1985年, 62: 114-120. 除非另有说 明, 用禊形键和虚线键表示一个立体中心的绝对抅型。 当本文所述化合物含 有烯属双键或其它几何不对称中心, 除非另有规定, 它们包括 Ζ几何异 构体。 同样地, 所有的互变异构形式均包括在本发明的范围之内。
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所
有的这类化合惣,包括顺式和反式异构体、(-) - 和 (+)-对对映体., (R)- 和 (S)- 对映体、 非对映异构体、() 异构体、 (L 异构体, 及其外消旋混合物和其他 混合物, 例如对映异构体或非对映体富集的混合物, 所有这些混合物都属于 本发明的范围之内。 烷基等取代基中可存在另外的不对称碳原子。 所有这些 异抅体以及它们的混合物, 均包括在本发明的范围之内。
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的 (R)- 和 ( -异构体以及 Z)和 iL异构体。如果想得到本发明某化合物的一种对映体, 可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对 映体混合物分离, 并且辅助基团裂开以提供纯的所需对映异构体。 或者, 当分子中含有碱性官能团 (^氨基) 或酸性官能团 (如羧基) 时, 与适当的 光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的分步结 晶法或色谱法进行非对映异构体拆分, 然后回收得到纯的对映体。 此夕卜 对 映异构体和非对映异构体的分离通常是通过使用色谱法完成的, 所述色谱法 采用手性固定相,并任选地与化学衍生法相结合 (例如由胺生成氨基甲酸盐)。
本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然 比例的原子同位素。 例如, 可用放射性同位素标记化合物, 比如氚 (3H), 碘 -1 25 ( i25 I) 或 C 14 ( i4C)。 本发明的化合物的所有同位素组成的变换, 无论放射性与否, 都包括在本发明的范围之内。
术语"药学上可接受的载体 "是指能够递送本发明有效量活性物质、 不千 扰活性物质的生物活性并且对宿主或者患者无毒副作用的任何制剂或载体 介质代表性的载体包括水、 油、 蔬菜和矿锈质、 膏基、 洗剂基质、 软膏基质 等。 这些基质包括悬浮剂、 增粘剂、 透皮促迸剂等。 它们的制剂为化牧品领 域或局部药物领域的技术人员所周知。 关于载体的其他信息, 可以参考 Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott,
Williams & Wilkins (2005), 该文献的内容通过引用的方式并入本文。
术语"赋形剂 "通常是指配制有效的药物组合物所需要载体、 稀释剂和 / 或介质。
针对药物或药理学活性剂而言, 术语"有效量 "或"治疗有效量"是指无毒 的但能达到预期效果的药物或药剂的足够用量。 对于本发明中的口服剂型, 组合物中一种活性物质的 "有效量"是指与该组合物中另一种活性物质联用 时为了达到预期效果所需要的用量。 有效量的确定因人而异, 取决于受体的 年龄和一般情况, 也取决于具体的活性麴质, 个案中合适的有效量可以由本 领域技术人员根据常规试验确定。
术语"活性成分"、 "治疗剂", "活性 tJ质"或"活性剂"是指一种化学实体, 它可以有效地治疗目标紊乱、 疾病或病症。
这里所采用的术语"药学上可接受的", 是针对那些化合物、 材料、 组合 物和 /或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物 的组织接触使用, 而没有过多的毒性、 剌激性、 过敏性反应或其它问题或并 发症, 与合理的利益 /风险比相称。
本文所用的"药学上可接受的盐"属于本发明化合物的衍生称, 其中, 通 过与酸成盐或与緘成盐的方式修饰所述母体化合锈。药学上可接受的盐的实 例包括但不限于: 碱基比如胺的无机酸或有机酸盐、 酸根比如羧酸的碱金属 或有机盐等等。药学上可接受的盐包括常规的无毒性的盐或母体化合物的季 铵盐, 例如无毒的无机酸或有机酸所形成的盐。 常规的无毒性的盐包括但不 β艮于那些衍生自无机酸和有机酸的盐,所述的无机酸或有机酸选自 2-乙酰氧 基苯甲酸、 2-羟基乙磺酸、 乙酸、 抗坏血酸、 苯磺酸、 苯甲酸、 碳酸氣根、 碳酸、柠檬酸、依地酸、 乙垸二磺酸、 乙烷磺酸、富马酸、葡庚糖、葡糖酸、 谷氨酸、 乙醇酸、 glycollyarsanilic, hex基 resorcinic hydrabamic, 氢溴酸、
盐酸、 氣碘酸盐、 羟基、 羟萘、 羟乙磺酸、 乳酸、 乳糖、 十二垸基磺酸、 马 来酸、 苹果酸、扁桃酸、 甲烷磺酸、硝酸、草酸、双羟萘酸、泛酸、苯乙酸、 磷酸、 多聚半乳糖醛、丙酸、水杨酸、硬脂酸、亚乙酸、琥珀酸、氨基磺酸、 对氨基苯磺酸、 硫酸、 单宁、 酒石酸和对甲苯磺酸。
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常 规化学方法合成。 一般情况下, 这样的盐的制备方法是: 在水或有机溶剂或 两者的混合物中,经由游离酸或碱形式的这些化合物与化学什量的适当的緘 或酸反应来制备。 一般地, 优选醚、 乙酸乙酯、 乙醇、 异丙醇或乙腈等非水 介质。 Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445批露了合适的盐的列表, 其公开的内容在此引入本 文作为参考。
由于前药可以提高药狻的许多品质(如溶解度、生麴利用度、工业化等), 因此本发明的化合物可以以前药形式给药。 因此, 本发明意在覆盖本发明要 求保护的化合物的前药形式、 其给药方式及其药物组合麴。 "前药 "意在包括 任何共价键合的载体, 当将前药给予哺乳类受体时, 它将在体内释放本发明 的活性母体药狻。本发明前药的制备方法是通过修饰本发明母体化合物的官 能团实现的,修饰后的母体化合物可以通过常规操作的方式或者体内方式裂 解为母体化合物。 前药包括本发明的化合物, 其中、 羟基、 氮基或巯基的基 团键合到任何官能团, 当本发明的前体药物被给予哺乳类受体时, 它将分别 裂解形成游离的羟基、 游离氣基酸或游离的巯基。 前体药麴的实例包括但不 β艮于本发明化合物醇或胺官能团的乙酸、 甲酸及苯甲酸衍生物。
术语"口服剂型 "是指通过口腔给予受任意药物组合物。 口服剂型的实倒 包括片剂、 胶囊剂、 薄膜、 粉末、 小药囊、 颗粒剂、 溶液、 固体、 悬浮液, 或者将多个不同的药剂单元 (例如含有不同的活性成分的颗粒剂、 片剂和 Ζ
或胶囊)包装在一起联合给药, 以及本领域中己知的其他方式。 口服剂型可 以是一、 二、 三、 四、 五或六个单元, 当口服剂型有多个单元时, 所有的单 元都是包装在一一个包装中的 (如瓶子或其他形式的包装, 如泡罩包装); 当口服剂型是一个独立的单元时,它可能会也可能不会存在于在单一包装中。 在一个优选的实施方案中, 口服剂型是一个、 两个或三个单元。 在一个特别 优选的实施方案中, 口服剂型是一个单元。
"抑制 "和"阻挡 ",在本文中可互换使用, 是指对一种酶如丝氯酸蛋白酶 的部分或全部的阻断。
术语"离去基团"是指可以被另一种官能团或原子通过取代反应 (例如 亲和取代反应)所取代的官能团或原子。 例如, 代表性的离去基团包括三氟 甲磺酸酯; 氯、 溴、 碗; 磺酸酯基, 如甲磺酸酯、 甲苯磺酸酯、 对溴苯磺酸 酯、 对甲苯磺酸酯等; 轔氧基, 如乙酰氧基、 三氟乙酰氧基等等。
术语"氨基保护基"是指适合用于阻止氮基氮位上副反应的保护基团。 代表性的氨基保护基包括但不限于: 甲酰基; 酷基, 例如链烷酰基(ft!乙酰 基、 三氯乙酰基或三氟乙銑基); 烷氧基羰基, 如叙 T氧基羰基 (Boc); 芳基 甲氧羰基,如爷氧羰基 (Cbz)和 9~芴甲氧羰基 (Fmoc) ;芳基甲基,如苄基(Bn)、 三苯甲基 (Tr)、 1 ,1-Zl- ( 4f -甲氧基苯基) 甲基; 甲硅垸基, 如三甲基甲硅烷 基 (TMS ) 和叔丁基二甲基甲硅垸基 (TBS ) 等等。
术语"羟基保护基' '是指适合用于阻止羟基副反应的保护基。 代表性羟基 保护基包括但不限于: 烷基, 如甲基、 乙基和叔丁基; 轔基, 例如链烷酰基 (如乙酷基); 芳基甲基, m ( Bn), 对甲氧基 基 (PMB:)、 9-芴基甲 基 (Fm)和二苯基甲基(二苯甲基, DPM:);甲硅垸基,如三甲基甲硅垸基( TMS ) 和叔丁基二甲基甲硅垸基 (TBS ) 等等。
卤代烷基的实例包括但不仅限于: 三氟甲基、 三氯甲基、 五氟乙基, 和
五氯乙基。 "烷氧基"伏表通过氧桥连接的具有特定数 g碳原子的上述垸基。 „6垸氧基包括(^、 C2、 C„ C4、 (:5和 c6的烷氧基。 垸氧基的例子包括但 不限于: 甲氧基、 乙氧基、 正丙氧基、 异丙氧基、 正 T氧基、 仲丁氧基、 叔 丁氧基、 正戊氧基和 s~戊氧基。 "环烷基'包括饱和环基, 如环丙基 环丁基 或环戊基。 3 7环烷基包括 C3、 C4、 C5、 (6和€;7环烷基。 "链烯基"包括直链 或支链构型的烃链, 其中该链上任何的稳定位点上存在一个或多个碳 -碳双 键, 例如乙烯基和丙烯基。
链烯基是指包括 c2、 c3、 c4、 (5和 αύ的链烯基。 "炔基 "是指包括 直链或支链构型烃链, 其中该链上任何的稳定位点上存在一个或多个碳》碳 三键, 如乙炔基和丙炔基。 C2〜6块基是指包括 C2、 C3, C4, (:5和(:6的炔基。
本文所 ]¾的'¾"或"鹵素"是指氟、 氯、 澳和碘; "抗衡离子"是用来表示 一个小的 带负电荷的麴质, 如氯化 溴化物 氛氧化 乙酸盐和硫酸 如本文所用, "碳环 "或"碳环基 "是指任何稳定的 3、 4、 5、 6或 7元单环 或取环或 7、 8、 9、 10 -, 11、 12或 13元二环或三环, 它们可以是饱和的、 部分不饱和的或不饱和的 (芳族的)。 这样的碳环的例子包括但不限于: 环 丙基.,环 T基、环丁'烯基、环戊基、环戊烯基、环己基、环庚爆基、环庚基、 环庚烯基、 金刚烷基、 环辛基、 环辛烯、 环辛二烯基, [3, 3, 0]双环辛烷、 [4 , 3 , 0] 双环壬垸、 [4, 4, 0] 双环癸烷、 [2.2.2]取环辛垸、 ¾基、 苯基、 誦、 2,3二氢化茚基、 金刚烷基和四氯蔡基。 如上所示, 桥环也包含在碳 环的定义中 (例如 [2, 2, 2]双环辛垸)。 当一个或多个碳原子连接两个不相 邻的碳原子时形成桥环。 优选一个或两个碳原子的桥。 值得注意的是, 一个 桥总是将单环转换成三环。 桥环中, 环上的取代基也可以出现在桥上。
如本文所用,术语"杂环"或"杂环基"意指稳定的 5 6或 7元单环或双环
或 7、 8、 9或 10元双环杂环, 它们可以是饱和的、 部分不饱和的或不饱和 的 (芳族的), 它们包含碳原子和 1、 2、 3或 4个独立地选自 N、 0和 S的 环杂原子, 其中上述任意杂环可以稠合到一个苯环上形成双环。 氮和硫杂原 子可任选被氧化 (即 NO和 S (0) p ) 氮原子可以是被取代的或未取代的 (即 N或 其中 R是 H或本文已经定义过的其他取代基)。 该杂环可以附着 到任何杂原子或碳原子的侧基上从而形成稳定的结构。如果产生的化合物是 稳定的, 本文所述的杂环可以发生碳位或氮位上的取代。 杂环中的氮原子任 选地被季铵化。 一个优选方案是, 当杂环中 S及 0原子的总数超过 1时, 这 些杂原子彼此不相邻。另一个优选方案是,杂环中 S及 0原子的总数不超过 Ι ο 如本文所用, 术语"芳族杂环基团 "或"杂芳基 "意指稳定的 5、 6-, 7元单环 或双环或 7、 §、 9或 10元双环杂环基的芳香环, 它包含碳原子和 K 2、 3 或 4个独立地选自 N、 0和 S的环杂原子。 氮原子可以是被取代的或未取代 的 (即 N或 NR, 其中 R是 H或本文已经定义过的其他取代基)。 氮和硫杂 原子可任选被氧化 (即 NO和 S C p)。 值得注意的是, 芳香杂环上 S和 0 原子的总数不超过 1。
桥环也包含在杂环的定义中。 当一个或多个原子 (即 0、 N或 S ) 连接两个不相邻的碳原子或氮原子时形成挢环。 优选的桥环包括但不限于: 一个碳原子、 两个碳原子、 一个氮原子、 两个氮原子和一个碳 基。 值得 注意的是, 一个桥总是将单环转换成三环。 桥环中, 环上的取代基也可以出 現在桥上。
杂环化合物的实例包括但不限于: 吖啶基、 P丫辛因基、 苯并味唑基、 苯 并呋喃基、 苯并巯基呋喃基、 苯并巯基苯基、 苯并恶唑基、 苯并恶唑琳基、 苯并噻唑基、 苯并三唑基、 苯并四唑基、 苯并异恶唑基、 苯并异噻唑基、 苯 并咪睡琳基、 咔唑基、 4aH-咔唑基、 咔琳基、 苯并二氛吡喃基、 色烯、 嗜琳
基十氛喹唯基、 2H, 6 , 5,2-二噻嚷基、 二氢呋喃并 [2534]四氢呋喃基、 呋 喃基、 呋咱基、 味唑烷基、 眯唑琳基、 睐唑基、 吲唑基、 0引哚烯基、 二 氢嘲哚基、 中氮茚基、 ^哚基、 3/^ |垛基、 isatmo 基、 异苯并呋喃基、 Π比 喃、 异吲哚基、 异二氛吲哚基、 异 哚基、 吲哚基、 异喹啉基、 异噻唑基、 异恶唑基、 亚甲二氧基苯基、 吗啉基、 萘啶基, 八氢异喹啉基、 恶二唑基、 】 3-恶二哇基、 1 ,2,4-恶二唑基、 1 5恶二唑基、 1 ,3,4恶二唑基、 恶唑烷 基、 恶唑基、 异恶噔基、 羟嘲哚基、 嘧啶基、 菲啶基、 菲咯啉基、 盼嗪、 玢 噻嗪、 苯并黃嘌呤基、 酚恶嗪基、 駄嗉基、 喊嗉基、 哌啶基、 嚒啶酮基、 4~ 哌啶酮基、 胡椒基、 蝶啶基、 嘌呤基、 吡喃基、 吡嗪基、 吡唑垸基、 1¾唑琳 基、 Itt唑基、 ½嗪基、 吡啶并恶哇、 Π比啶并眯唑、 Itt啶并噻哇、 ¾啶基、 嘧 啶基、 吡咯垸基、 咯琳基、 2 /^吡咯基、 嗒基、 吡唑基、 喹噔啉基、 喹 I藝、 4 ~喹嗪基、 喹嗎輔;.、 奎宁环基、 四氯呋喃基、 四氯异喹琳基、 四 氛喹唯基、 四唑基, 5噻二嗪基., 1_ ,2,3-噻二唑基., 1 ,2,4-噻二唑基、 】 5 -噻二唑基、 1 ,3,4~噻二唑基、 噻蒽基、 噻哇基、 异壟唑基壟吩基、 麵 基、 噻玢并恶唑基、 噻吩并噻唑基、 噻吩并眯唑基、 噻玢基、 三嗪基、 〗,2,3- 三唑基、 1,2,4-三唑基 1 5~三哩基、 1,3,4~三锉基和咕吨基。 还包括稠环和 螺环化合, 例如上述杂环。
"稳定的化合物"和' 11定结构 "是指这样一种化合麴, 它能够脱离于反应 混合物并以一定的有效纯度独立、稳定地存在,并被配制成有效的治疗药物。
"被取代的"是指标识"被取代的"原子上的一个或多个氮原子被取代基 (本文已对这些取代基做过限定)取代, 只要特定原子的价态是正常的并且 取代后的化合物是稳定的。 当取代基为酮基 (即 = 0 ) 时, 意味着两个氛原 子被取代。
本文所用的"治疗"是对噹乳动麴特别是人的疾病状态的治疗,包括: ( a)
预防发生在哺乳动物的疾病状态,尤其是当该哺乳动物易患这种疾病但尚未 确诊其已经患上这种疾病; ( b ) 抑制疾病的状态, 即阻止了它的发展: 和, / 或 (e ) 缓解疾病犹态, 即造成疾病状态消退。
"治疗有效量 "是指本发明化合麴单独给药或联合给药时能够奏效所需 要的用量。 "治疗有效量 "也指本发明化合物组合使用时能够奏效所需要的用 量。本发明化合物的组合使用优选协同组合使用。正如 Chou and Talalay, Adv. Enzyme egul. 1984, 22: 27-55一文所教导的,组合使用药物所取得的效果优 于单独使用时药物之间发生协同效应。 在一般情^下, 次佳滚度的化合物最 能清楚地展示协同效应。 与单独用药相比, 联合用药的协同效应可以表现在 降低细胞毒性、 提高抗血栓能力或者其他的有益效果方面。
本发明化合物的制备方法:
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制 备, 包括下面列举的具体实施方式、 其与其他化学合成方法的结合所形成的 实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包 括但不限于本发明的实施例。
化合物的结构是通过核磁共振 (NMR)或 /和液相质谱 (LCMS)来确定的。 NM 位移 (δ)以 10—6 (ppm)的单位给出。 NM 的测定是用 Bruker AVANCE-400 核磁仪, 测定溶剂为氘代二甲基亚砜 (DMSO-i¾, 氘代氯仿 (CDC13), 氘代甲 醇 (CD3OD),内标为四甲基硅烷TMS:)。
液相质谱 LCMS的测定液相部分用安捷伦 1200(Xtimate C18 2.1*30mm 色谱柱:)和质谱部分用安捷伦 6110 (离子源: ESI
HPLC的测定使用岛津 LC10AD高压液相色谱仪 (Xtimate C18 2.1*30m m色谱柱)。
薄层层析硅胶板使用烟台黄海 HSGF254或青岛 GF254硅胶板, 薄层色
谱法 (TLC)使用的硅胶板采用的规格是 0.15 mm〜0.2 mm, 薄层层析分离纯 化产品采用的规格是 0.4 mm〜0.5 mm。
柱层析一般使用烟台黄海硅胶 200 300目硅胶为载体。
本发明的已知的起始原料可以采用或按照本领域已知的方法来合成,或 可购买自 ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, TCI, Alfa, 韶远化学科技 (Accela ChemBio Inc)、 北京偶合等公司。
实施例中无特殊说明, 反应能够均在氩气氛或氮气氛下进行。 氩气氛或 氮气氛是指反应瓶连接一个约 1L容积的氩气或氮气气球。
氢气氛是指反应瓶连接一个约 1L容积的氢气气球。
加压氢化反应使用 Parr 3916EKX型氢化仪和清蓝 QL-500型氢气发生器 或 HC2-SS型氢化仪。 氢化反应通常抽真空, 充入氢气, 反复操作 3次。
微波反应使用 CEM Discover-S 908860型或 Biotage Initiator 60微波反应 器。
实施例中无特殊说明, 溶液是指水溶液。
实施例中无特殊说明, 反应的温度为室温, 为 20°C〜30°C。
实施例中的反应进程的监测采用薄层色谱法 (TLC), 反应所使用的展开 剂的体系有: A: 二氯甲烷和甲醇体系, B: 正己烷和乙酸乙酯体系, C: 石 油醚和乙酸乙酯体系, D: 丙酮, 溶剂的体积比根据化合物的极性不同而进 行调节。
纯化化合物采用的柱层析的洗脱剂的体系和薄层色谱法的展开剂体系 包括: A: 二氯甲烷和甲醇体系, B: 石油醚和乙酸乙酯体系, C: 二氯甲烷 和丙酮体系, 溶剂的体积比根据化合物的极性不同而进行调节, 也可以加入 少量的三乙胺和醋酸等碱性或酸性试剂进行调节。
下面会通过实施例具体描述本发明,这些实施例并不意味着对本发明的
任何限制。
本发明所使用的所有溶剂是市售的, 无需迸一步纯化即可使用。
本发明采用下述缩略词: aq代表水; HATU代表 0-7 -氮杂苯并三唑小 基) Ν,Ν,Ν',Ν'四甲基脲六氟磷酸盐; EDC代表 Ν~ G二甲基氣基丙基) ~Ν'~Δ 基碳二亚胺盐酸盐; m-CPBA代表 3氯过氧苯甲酸: eq代表当量、 等量; CDI代表羰基二眯唑; DCM代表二氯甲垸; PE代表石油醚; DIAD代表偶 氮二羧酸二异丙酯; DMF 代表 N, N-二甲基甲酰胺; DMSO代表二甲亚砜: EtOAc代表乙酸乙酯; EtOH代表乙醇; MeOH代表甲醇; CBz代表节氧羰 基, 是一种胺保护基团; BOC 代表叔丁基羰基是一种胺保护基团; HOAc 代表乙酸; NaCNBH3代表氰基硼氫化钠; r.t.代表室温; 0/ N代表过夜; THF代表四氫呋喃; Boc20代表二 -叔丁基二碳酸酯; TFA代表三氟乙酸; DIPEA代表二异丙基乙基胺; S0C12代表氯化亚飒; CS2代表二硫化碳; TsOH 代表对甲苯磺酸; 代表 N-氟 (:苯磺酰基) 苯磺酰胺; NCS代表 1 - 氯比咯垸 2 二酮; n-Bu4NF 代表氟化四丁基铵: iPrOH代表 2-丙醇: mp 代表瑢点。
化合物经手工或者 ChemDraw®软件命名, 市售化合物采用供应商目录 名称。
与现有技术相比, 本发明化合麴高效、 低毒, 在活性、 半衰期、 溶解度 和药代动力学等方面均取得了巨大的迸歩, 更适合于制药。 具体实施方式
下面通过实施例对本发明进行详细描述,但并不意味着对本发明任何不 利限制。 本文已经详细地描述了本发明, 其中也公开了其具体实施例方式, 对本领域的技术人员而言, 在不脱离本发明精神和范围的情况下针对本发明 具体实施方式进行各种变化和改进将是显而易见的。
实施例 1
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯

4-溴 -2-氯 -1-苯基异氰酸酯
将 4-溴 -2-氯苯胺 la (1.00 g, 4.88 mmol)溶解于 20 mL四氢呋喃中, 加 入三光气 (478 mg, 1.63 mmol), 搅拌反应 3小时。 将反应液减压浓缩, 得到 粗品标题产物 4-溴 -2-氯 -1-苯基异氰酸酯 lb (1.12 g, 白色固体), 产物不经纯 化直接进行下一步反应。
第二步
1-(4-溴 -2-氯苯) -3-(2-氯乙基)脲
将粗品 4-溴 -2-氯 -1-苯基异氰酸酯 lb (1.12 g, 4.85 mmol)和三乙胺 (2.1 mL, 14.55 mmol)溶解于 100 mL四氢呋喃中,加入 2-氯乙胺盐酸盐 (844 mg, 7.27 mmol), 搅拌反应 2小时。 向反应液中加入 200 mL水, 用二氯甲烷萃 取 (200 mLx3),合并有机相,依次用水 (100 mLx2)、饱和氯化钠溶液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1-(4- 溴 -2-氯苯: )-3-(2-氯乙基:)脲 lb (1.12 g, 白色固体:), 产物不经纯化直接进行下 一步反应。
JH NM (400 MHz, CDC13) δ 8.03 (d, 1H), 7.36 (dd, 1H), 6.63 (d, 1H), 3.69-3.66 (m, 2H), 3.64-3.60 (m, 2H)
第三步
l-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-氯苯) -3-(2-氯乙基)脲 lc (1.12 g, 3.60 mmol)溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (433 mg, 10.80 mmol), 搅拌反应 12 小时。向反应液中加入 50 mL水,用乙酸乙酯萃取 50 mLx3;),合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;), 用无水硫酸钠干燥, 过滤,滤液减压浓缩,得到粗品标题产物 1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮 Id (986 mg, 白色固体:), 产物不经纯化直接进行下一步反应。
第四步
2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮 Id (986 mg, 3.60 mmol)溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (215 mg, 5.39 mmol), 搅拌反应 30 分钟。向反应液中加入溴乙酸乙酯 (596 mg, 5.37 mmol),搅拌反应 12小时。 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3;>, 合并有机相, 依次 用水 (50 mLx2;>、饱和氯化钠溶液洗涤 50 mLx2;>,用无水硫酸钠干燥,过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题 产物 2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 le (750 mg, 白色固体),
产率: 58.0%。
JH NM (400 MHz, CDC") δ 7.58 (d, 1H), 7.40 (dd, 1H), 7.25 (d, 1H), 4.20 (q, 2H), 4.04 (s, 2H), 3.84-3.80 (m, 2H), 3.67-3.62 (m, 2H), 1.28 (t, 3H) 第五步
2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氯苯) -2-氧代咪唑啉 -1- 基:)乙酸乙酯
将 2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 le (750 mg, 2.10 mol), 2- 叔丁基二甲基硅:)氧基)乙胺 (549 mg, 3.10 mmol, 采用公知的方法"专利 WO2010027500"制备而得)溶解于 15 mL甲苯中, 加入碳酸铯 (2.03 g, 6.22 mmol), 三 (二苯亚苄基丙酮)二钯 (50 mg, 0.05 mmol)和 4,5-双二苯基膦 -9,9- 二甲基氧杂蒽 (40 mg, 0.07 mmol), 100°C搅拌反应 12小时。 反应液冷却至 室温, 向反应液中加入 50 mL水,用乙酸乙酯萃取 50 mLx3;),合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得 到标题产物 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氯苯) -2-氧代咪 唑啉 -1-基)乙酸乙酯 lf (550 mg, 黄色油状物), 产率: 58.1%。
JH NM (400 MHz, CDC13) δ 7.05 (d, 1H), 6.58 (d, 1H), 6.43 (dd, 1H), 4.14 (q, 2H), 3.98 (s, 2H), 3.74-3.66 (m, 4H), 3.56-3.52 (m, 2H), 3.11 (q, 2H), 1.22 (t, 3H), 0.83 (s, 9H), 0.00 (s, 6H)
第六步
2-(3-(4-(N-(2- ;叔丁基二甲基硅)氧:)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-氯 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将 2-(3-(4-((2- ((叔丁基二甲基硅)氧)乙基)氨基) -2-氯苯) -2-氧代咪唑啉 -1- 基)乙酸乙酯 If (550 mg,1.20 mmol)溶解于 5 mL二氯甲烷中,加入三乙胺 (519 uL, 3.60 mmol)和 4,6-二氯 -5-酰氯嘧啶 (378 mg, 1.80 mmol), 搅拌反应 12 小时。向反应液中加入 10 mL水,用二氯甲烷萃取 10 mLx3;),合并有机相,
依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二甲基硅) 氧:)乙基: )-4,6-二氯嘧啶 -5-酰胺) -2-氯苯) -2-氧代咪唑啉 -1-基:)乙酸乙酯 lg (756 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第七步
2-(3-(2-氯 -4-(4,6-二氯 -N-(2-羟乙基:)嘧啶 -5-酰胺)苯基: )-2-氧代咪唑啉 -1- 基:)乙酸乙酯
将粗品 2-(3 -(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺:) -2-氯苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lg (756 mg, 1.20 mmol)溶解于 10 mL乙醇中, 加入浓盐酸 (0.3 mL;), 搅拌反应 1小时。 向反应液中加入 10 mL 水, 用乙酸乙酯萃取 (10 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和氯 化钠溶液洗涤 IO mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(2-氯 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉 _μ基)乙酸乙酯 ^^ !!^, 黄色固体), 产物不经纯化直接进行下一 步反应。
第八步
2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基)苯 基) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-氯 -4-(4,6-二氯 -Ν-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪 唑啉小基)乙酸乙酯 lh (618 mg, 1.20 mmol)溶解于 5 mL乙腈中, 加入三乙 胺 (519 uL, 3.60 mmol), 80°C搅拌反应 12小时。 反应液冷却至室温, 加入 20 mL水, 用乙酸乙酯萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2)、 饱和氯化钠溶液洗涤30 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (260 mg, 黄色固体), 产 物不经纯化直接进行下一步反应。
Ή NM (400 MHz, DMSO-d6) δ 8.83 (s, IH), 7.70 (d, IH), 7.52-7.46 (m, 2H), 4.75 (t, 2H), 4.21 (t, 2H), 4.15 (q, 2H), 4.03 (s, 2H), 3.80-3.76 (m, 2H), 3.62-3.58 (m, 2H), 1.23 (t, 3H)
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (260 mg, 543 umol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 加入二氯甲烷 30 mL, 依次用水 (30 mLx2), 饱和氯化钠溶液洗涤 (30 mLx2) , 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -6(5H)-基 )-2-氯 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lj (250 mg, 黄色固体), 产物不经纯化直接 进行下一步反应。
JH NMR (400 MHz, DMSO-d6) δ 8.18 (s, IH), 7.67 (d, IH), 7.66 (br. s, 2H) 7.47 (d, IH), 7.41 (dd, IH), 4.63 (t, 2H), 4.15 (q, 2H), 4.03 (s, 3H), 3.79-3.75 (m, 2H), 3.65 (s, 2H), 3.62-3.58 (m, 2H), 1.23 (t, 3H)
第十步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -2-氧代咪唑啉 -1-基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lj (250 mg, 543 umol)溶解于 8 mLl,4-二氧六环和水 (V/V = 3: 1)混合溶液中, 加入氢氧化锂 (68 mg, 1.60 mmol), 搅拌反应 12小时。滴加 1 M盐酸至反应液 pH为 5〜6, 过滤得到标 题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯苯) -2-氧代咪唑啉 -1-基;)乙酸 1 (74 mg, 白色固体:), 产率: 33.0%。
MS m/z (ESI): 433.1 [M+l]
JH NMR (400 MHz, DMSO-d6) δ 12.81 (s, IH), 8.17 (s, IH), 7.67-7.66 (m, 3H), 7.47-7.36 (m, 2H), 4.62 (t, 2H), 4.02 (t, 2H), 3.93 (s, 2H), 3.78-3.74 (m, 2H) 3.61-3.58 (m, 2H)
实施例 2
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟 苯) -2-氧代咪唑啉 -1-基)乙酸

第一步
4-溴 -2-氟 -1-苯基异氰酸酯
将 4-溴 -2-氟苯胺 la (5 g, 0.026 mol)溶解于 60 mL四氢呋喃中, 加入三 光气 (2.60 g, 8.77 mmol), 搅拌反应 3小时。 将反应液减压浓缩, 得到粗品
标题产物 4-溴 -2-氟 -1-苯基异氰酸酯 lb (5.68 g, 白色固体), 产物不经纯化直 接进行下一步反应。
MS m/z (ESI): 248.0 [M+l]
第二步
l-(4-溴 -2-氟苯) -3-(2-氯乙基)脲
将粗品 4-溴 -2-氟 -1-苯基异氰酸酯 lb (5.68 g, 2.6 mmol)和三乙胺 (7.98 g, 79 mmol)溶解于 100 mL四氢呋喃中,加入 2-氯乙胺盐酸盐 lc (4.54 g, 39.5 mmol), 搅拌反应 2小时。 向反应液中加入 200 mL水, 用二氯甲烷萃取 (200 mLx3),合并有机相,依次用水 (100 mLx2)、饱和氯化钠溶液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1-(4-溴 -2-氟 苯:) -3-(2-氯乙基:)脲 ld (7.67 g, 白色固体:), 产物不经纯化直接进行下一步反 应。
JH NM (400 MHz, CDC13)5 8.15-8.05 (m, 1H), 7.2-7.1 (m, 2H), 3.75-3.70 (m, 2H), 3.65-3.60 (m, 2H)
第三步
l-(4-溴 -2-氟苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-氟苯) -3-(2-氯乙基)脲 Id (2 g, 6.78 mmol)溶解于 60 mL
N, N-二甲基甲酰胺中, 加入钠氢 (410 mg, 10.2 mmol), 搅拌反应 12小时。 向反应液中加入 100 mL水, 用乙酸乙酯萃取 (200 mLx3;>, 合并有机相, 依 次用水 (300 mLx3;>、 饱和氯化钠溶液洗涤 (300 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮 le (2.20 g, 白色固体:), 产物不经纯化直接进行下一步反应。
MS m/z (ESI): 425.0 [M+l]
JH NMR (400 MHz, CDC13) δ 7.45 (t, 1H), 7.30-7.20 (m, 2H), 4.00-3.90 (m, 2H), 3.60-3.50 (m, 2H)
第四步
2-(3-(4-溴 -2-氟苯) -2-氧代咪唑啉 -1-乙基)乙酯
将粗品 1-(4-溴 -2-氟苯)氧代咪唑啉 -2-酮 le (1.96 g, 7.6 mmol)溶解于 40 mL N, N-二甲基甲酰胺中, 加入钠氢 (456 mg, 11.4 mmol), 搅拌反应 30分 钟。 向反应液中加入溴乙酸乙酯 If (1.90 g, 11.4 mmol), 室温搅拌反应 12小 时。 向反应中加入 50 mL水, 用乙酸乙酯萃取 100 mLx3;), 合并有机相, 依 次用水 (200 mLx3)、 饱和氯化钠溶液洗涤 (200 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得 到标题产物 2-(3-(4-溴 -2-氟苯) -2-氧代咪唑啉 -1-乙基)乙酯 lg (1.67 g, 白色固 体), 产率: 63.7 %。
MS m/z (ESI): 345.1 [M+l]
JH NM (400 MHz, CDC13) δ 7.50-7.40 (m, 1H), 7.30-7.20 (m, 2H), 4.22 (q, 2H), 4.06 (s, 2H), 3.9 (t, 2H), 3.65-3.55 (m, 2H), 1.30-1.25 (m, 3H)
第五步
2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氟苯) -2-氧代咪唑啉 -1- 基:)乙酸乙酯
将 2-(3-(4-溴 -2-氟苯) -2-氧代咪唑啉 -1-乙基)乙酯 lg (1.67 g, 7.84 mmol), 2-((叔丁基二甲基硅) -氧代) -乙胺 lh (1.72 g, 9.68 mmol)溶解于 30 mL甲苯中, 加入碳酸铯 (3.15 g, 9.68 mmol),三 (二苯亚苄基丙酮)二钯 (225.4 mg, 0.4 mmol) 和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (450 mg, 0.8 mmol), 100°C搅拌反应 12 小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3),合并有机相,依次用水 (50 mLx2;>、饱和氯化钠溶液洗涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨 基) -2-氟苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (1.10 g, 黄色油状物), 产率: 51.6 %。
JH NMR (400 MHz, CDC13) δ 7.19-7.15 (m, 1H), 6.39-6.33 (m, 2H),
4.22-4.18 (m, 2H), 4.05 (s, 2H), 3.8-3.75 (m, 4H), 3.63-3.57 (m, 2H), 3.17 (q, 2H), 1.27 (t, 3H), 0.90 (s, 9H)
第六步
2-(3-(4-(N-(2-((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-氟苯 基) -2-氧代咪唑啉 -1-基)乙酸乙酯
将 2-(3-(4-((2-((叔丁基二甲基硅)氧代)乙基)氨基) -2-氟苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (1.10 g, 2.5 mmol)溶解于 20 mL二氯甲烷中,加入三乙胺 (1 mL, 7.5 mmol)和 4,6-二氯 -5-酰氯嘧啶 lj (1.05 g, 5.0 mmol), 搅拌反应 12小 时。 向反应液中加入 50 mL水, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二甲基硅) 氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-氟苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lk (1.9 g, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.59 (s, 1Η), 7.05-7.03 (m, 2H), 4.51-3.91 (m 4H), 3.90-3.91 (m, 2H),2.6 (s, 2H), 1.61 (s, 6H,), 1.47-1.41 (m,3H)
第七步
2-(3-(2-氟 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉 -1- 基:)乙酸乙酯
将粗品 2-(3-(4-(N-(2- ((叔丁基二甲基硅) 氧代)乙基) -4,6-二氯嘧啶 -5-酰 胺) -2-氟苯基 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lk (1 g, 1.63 mmol)溶解于 10 mL 乙醇中,加入浓盐酸(0.3 mL),搅拌反应 1小时。向反应液中加入 10 mL水, 用饱和碳酸氢钠溶液调节 pH至 6~7,二氯甲烷萃取 (30 mLx3),合并有机相, 饱和氯化钠溶液洗涤 50 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-氟 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2- 氧代咪唑啉 -1-基)乙酸乙酯 11 (600 mg, 黄色油状), 产物不经纯化直接进行
第八步
2-(3-(2-氟 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-氟 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪 唑啉小基)乙酸乙酯 11 (600 mg,1.09 mmol)溶解于 20 mL乙腈中,加入三乙胺 (0.5 mL, 3.3 mmol), 80°C搅拌反应 12小时。 向反应液中加水 (30 mL)稀释, 用二氯甲烷萃取 50 mLx3;), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠 溶液洗涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品 标题产物 2-(3-(2-氟 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lm (490 mg, 黄色固体), 产物不经纯 化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.77 (s, IH), 7.75-7.65 (m, IH), 7.30-7.26 (m IH), 7.18-7.10 (m, IH), 4.75 (t, 2H), 4.24 (q, 2H), 4.13 (t, 2H), 4.10-4.02 (m, 2H) 3.95 (t, 2H), 3.66 (q, 2H), 1.30-1.20 (m, 3H)
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-氟 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lm (490 mg,1.05 mmol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 用二氯甲烷萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2)、 饱和氯 化钠溶液洗涤 PO mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -6(5H)-基) -2-氟苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 ln (460 mg, 黄色油状), 产 物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.30 (s, IH), 7.67 (t, IH), 7.10-7.02 (m, 2H),
4.69 (s, 2H), 4.23 (q, 2H), 4.07 (s, 2H), 4.0 (t, 2H), 3.98-3.92 (m, 2H), 3.70-3.60 (m, 2H), 1.30-1.20 (m, 3H)
第十步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟 苯) -2-氧代咪唑啉 -1-基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 In (230 mg, 0.5 mmol)溶解于 6 mLl,4-二氧六环和水 (V/V = 3: 1)混合溶液中, 加入氢氧化锂 (61 mg, 2.50 mmol), 搅拌反应 12小时。 滴加 1 M盐酸至反应液 pH为 5-6, 过滤得到标 题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氟苯) -2-氧代咪唑啉 -1-基)乙酸 1 (20 mg, 黄色固体), 产率: 9.7 %。
MS m/z (ESI): 417.1 [M+l]
JH NMR (400 MHz, CD3OD) δ 12.80 (s, IH), 8.13 (s, IH), 7.60 (s, 2H), 7.46 (t, IH), 7.40 (dd, IH), 7.23-7.21 (m, IH), 4.58 (t, 2H), 3.98-3.95 (m, 2H), 3.89 (s, 2H), 3.78 (t, 2H), 3.53 (t, 2H)
实施例 3
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-甲


4-溴 -2-甲基 -1-苯基异氰酸酯
将 4-溴 -2-甲基苯胺 la (10.00 g, 53.76 mmol)溶解于 200 mL四氢呋喃中, 加入三光气 (5.32 g, 17.92 mmol), 搅拌反应 3小时。 将反应液减压浓缩, 得到粗品标题产物 4-溴 -2-甲基 -1-苯基异氰酸酯 lb (11.40 g, 白色固体), 产 物不经纯化直接进行下一步反应。
第二步
1 -(4-溴 -2-甲苯) -3-(2-氯乙基)脲
将粗品 4-溴 -2-甲基 -1-苯基异氰酸酯 lb (11.40 g, 53.76 mmol)和三乙胺 (22.6 mL, 161.29 mmol)溶解于 350 mL四氢呋喃中, 加入 2-氯乙胺盐酸盐 (9.35 g, 80.65 mmol), 搅拌反应 2小时。 向反应液中加入 200 mL水, 用乙 酸乙酯萃取 (200 mLx3;), 合并有机相, 依次用水 (100 mLx2)、 饱和氯化钠溶 液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标 题产物 1-(4-溴 -2-甲苯: )-3-(2-氯乙基:)脲 lb (14.3 g, 白色固体 产物不经纯化 直接进行下一步反应。
Ή NM (400 MHz, CDC13) δ 7.37 (br.s., 1H), 7.33 (d, 1H), 7.25 (d, 1H), 6.08 (br, 1H), 5.07 (br.s., 1H), 3.64-3.61 (m, 2H), 3.57-3.54 (m, 2H), 2.25 (s, 3H)
第三步
l-(4-溴 -2-甲苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-甲苯) -3-(2-氯乙基)脲 lc (9.00 g, 30.87 mmol)溶解于 90 mL N, N-二甲基甲酰胺中, 冰浴下加入钠氢 (1.86 g, 46.55 mmol), 室温搅 拌反应 12小时。 向反应液中加入 100 mL饱和氯化铵溶液, 用乙酸乙酯萃取 (100 mLx3), 合并有机相, 依次用水 (100 mLx2)、 饱和氯化钠溶液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1-(4- 溴 -2-甲苯:)氧代咪唑啉 -2-酮 lc 6.3 g, 白色固体 产物不经纯化直接进行下 一步反应。
JH NMR (400 MHz, DMSO-d6) δ 7.44-7.43 (m, 1H), 7.36-7.33 (m, 1H), 7.14 (d, 1H), 6.70 (br.s., 1H), 3.69-3.65 (m, 2H), 3.40-3.36 (m, 2H), 2.17 (s, 3H)
第四步
2-(3-(4-溴 -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 1-(4-溴 -2-甲苯)氧代咪唑啉 -2-酮 Id (2.00 g, 7.84 mmol)溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (470 mg, 11.76 mmol), 搅拌反应 30 分钟。 向反应液中加入溴乙酸乙酯 (1.96 g, 11.76 mmol), 室温搅拌反应 12 小时。向反应液中加入 50 mL饱和氯化铵溶液,用乙酸乙酯萃取 50 mLx3), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤50 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 2-(3-(4-溴 -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 le ( 0.9 g, 白色固体), 产率: 33.7%。
JH NMR (400 MHz, CDC13) δ 7.39-7.38 (m, 1Η), 7.32-7.29 (m, 1H), 7.05 (d 1H), 4.20 (q, 1H), 4.03 (s, 2H), 3.64-3.60 (m, 2H), 2.26 (s, 3H), 1.28 (t, 1H)
2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-甲苯) -2-氧代咪唑啉 -1- 乙基)乙酸
将 2-(3-(4-溴 -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 le (900 mg, 2.64 mmol), 2- ((叔丁基二甲基硅)氧代)乙胺 (699 mg, 3.96 mmol)溶解于 15 mL甲 苯中, 加入碳酸铯 (2.58 g, 7.92 mmol), 三 (二苯亚苄基丙酮)二钯 (242 mg, 0.26 mmol)和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (253 mg, 0.53 mmol), 100°C 搅拌反应 12小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用乙酸 乙酯萃取 (50 mLx3;>, 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗 涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法 以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(3-(4-((2- ((叔丁基二甲基 硅)氧代)乙基)氨基) -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸 If (800 mg, 黄色油状 物), 产率: 70.2%。
JH NM (400 MHz, CDC13) δ 6.92 (d, 1H), 6.42-6.37 (m, 2H), 4.14 (q, 1H), 3.96 (s, 2H), 3.74-3.71 (m, 2H), 3.63-3.58 (m, 2H), 3.54-3.50 (m, 2H), 3.13-3.09 (m, 2H), 2.14 (s, 3H), 1.22 (t, 1H), 0.83 (s, 9H), 0.00 (s, 6H)
第六步
2-(3-(4-(N-(2- ;叔丁基二甲基硅)氧:)乙基: )-4,6-二氯嘧啶 -5-酰胺) -2-甲 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-甲苯) -2-氧代咪唑啉 -1- 乙基)乙酸 If (800 mg, 1.84 mmol)溶解于 10 mL二氯甲烷中,加入三乙胺 (558 mg, 5.52 mmol)和 4,6-二氯 -5-酰氯嘧啶 (446 mg, 2.20 mmol), 搅拌反应 12 小时。向反应液中加入 10 mL水,用二氯甲烷萃取 10 mLx3;),合并有机相, 依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二甲基硅) 氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lg (1.1 g, 黄色固体), 产物不经纯化直接进行下一步反应。
Ή NM (400 MHz, CDC") δ 8.52 (s, 1H), 7.26 (d, 1H), 7.16 (dd, 1H), 6.99 (d, 1H), 4.14 (q, 1H), 3.94 (s, 2H), 3.92-3.89 (m, 2H), 3.85-3.82 (m, 2H), 3.62-3.51 (m, 4H), 2.13 (s, 3H), 1.21 (t, 1H), 0.81 (s, 9H), 0.00 (s, 6H)
第七步
2-(3-(2-甲基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉
-1-基:)乙酸乙酯
将粗品 2-(3 -(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺) -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lg (1.10 g, 18.01 mmol)溶解于 10 mL乙醇中,加入浓盐酸 (0.3 mL;),搅拌反应 1小时。向反应液中加入 10 mL 水, 用乙酸乙酯萃取 (10 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和碳 酸氢钠溶液 IO mLx2;>、饱和氯化钠溶液洗涤 IO mLx2),用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-甲基 -4-(4,6-二氯 -N-(2-羟乙 基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lh (720 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.51 (s, 1H), 7.21 (d, 1H), 7.16-7.13 (m, 1H), 7.00 (d, 1H), 4.11 (q, 1H), 3.97-3.95 (m, 2H), 3.92 (s, 2H), 3.82-3.79 (m, 2H), 3.61-3.49 (m, 4H), 2.13 (s, 3H), 1.19 (t, 1H)
第八步
2-(3-(2-甲基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代咪唑啉- 1 -基)乙酸乙酯
将粗品 2-(3-(2-甲基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉 -1-基)乙酸乙酯 lh (720 mg, 1.45 mmol)溶解于 10 mL乙腈中,加入三 乙胺 (440 mg, 4.35 mmol), 80°C搅拌反应 12小时。 反应液冷却至室温, 向 反应液中加入 20 mL水, 用乙酸乙酯萃取 (20 mLx3;>, 合并有机相, 用饱和 氯化钠溶液洗涤20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(2-甲基 -4-(4-甲基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂
卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (450 mg, 黄色固体), 产 物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.75 (s, IH), 7.29 (d, IH), 7.28-7.24 (m, IH), 7.22- 7.19 (m, IH), 4.74-4.72 (m, 2H), 4.21 (q, IH), 4.05 (s, 2H), 4.02-4.00 (m, 2H), 3.78 -3.62 (m, 4H), 2.33 (s, 3H), 1.29 (t, 1H)。
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-甲 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-甲基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (450 mg, 0.98 mmol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 加入 30 mL 乙酸乙酯, 依次用水 (30 mLx2), 饱和氯化钠溶液洗涤 (30 mLx2) , 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -6(5H)-基 )-2-甲 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lj (230 mg, 黄色固体), 产物不经纯化直接 进行下一步反应。
第十步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-甲 苯) -2-氧代咪唑啉 -1-基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lj (230 mg, 0.52 mmol)溶解于 8 mLl,4-二氧六环和水 (V/V = 3: 1)混合溶液中, 加入氢氧化锂 (109 mg, 2.61 mmol), 搅拌反应 12小时。 滴加 1 M盐酸至反应液 pH为 5〜6, 减压浓缩, 残留物用制备高效液相色谱纯化得到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-甲苯) -2-氧代咪唑啉 -1-基)乙酸 1 (25 mg, 白色固体), 产率: 11.8%。
MS m/z (ESI): 413.1 [M+l]
JH NMR (400 MHz, DMSO-d6) δ 9.02 (br, s, 2H), 8.47 (s, 1H), 7.28-7.20 (m, 3H), 4.84-4.82 (m, 2H), 4.13-4.11 (m, 2H), 3.87 (s, 2H), 3.71-3.67 (m, 2H), 3.55-3.51 (m, 2H), 2.18 (s, 3H)
实施例 4
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-乙 基 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸

第一步
2-乙基 -6-甲基 -1-苯基异氰酸酯
将 4-溴 -2-乙基 -6-甲基苯胺 la (2.00 g, 9.40 mmol)溶解于 20 mL四氢呋 喃中, 加入三光气 (941 mg, 3.10 mmol), 搅拌反应 3小时。 将反应液减压浓
缩,得到粗品标题产物 2-乙基 -6-甲基 -1 -苯基异氰酸酯 lb (2.20 g,白色固体), 产物不经纯化直接进行下一步反应。
第二步
1-(4-溴 -2-乙基 -6-甲基苯 )-3-(2-氯乙基)脲
将粗品 2-乙基 -6-甲基 -1-苯基异氰酸酯 lb (2.20 g, 9.40 mol)和三乙胺 (4.0 mL, 28.20 mmol)溶解于 100 mL四氢呋喃中, 加入 2-氯乙胺盐酸盐 (1 .60 g, 14.10 mmol), 搅拌反应 2小时。 向反应液中加入 200 mL水, 用二氯甲烷萃 取 (200 mLx3),合并有机相,依次用水 (100 mLx2)、饱和氯化钠溶液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1 -(4- 溴 -2-乙基 -6-甲基苯 )-3-(2-氯乙基)脲 lb (2.87 g, 白色固体), 产物不经纯化直 接进行下一步反应。
JH NM (400 MHz, CDC13) δ 7.63 (s, 1Η), 7.23 (dd, 2H), 6.33 (br. s, 1H), 3.58 (t, 2H), 3.55 (s, 2H), 2.51 (s, 2H), 2.10 (s, 3H ), 1.05 (t, 3H)
第三步
l -(4-溴 -2-乙基 -6-甲基苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-乙基 -6-甲基苯 )-3-(2-氯乙基)脲 lc (2.86 g, 9.00 mmol) 溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (540 mg, 13.60 mmol), 搅 拌反应 12小时。 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3;), 合 并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;), 用无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1 -(4-溴 -2-乙基 -6-甲基 苯)氧代咪唑啉 -2-酮 Id (2.15 g, 白色固体),产物不经纯化直接进行下一步反 应。
JH NMR (400 MHz, CDC13) δ 7.25-7.24 (m, 2H), 3.70-3.58 (m, 4H), 2.69-2.48 (m, 2H), 2.24 (s, 3H), 1 .24-1.20 (m, 3H)
第四步
2-(3-(4-溴 -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 l-(4-溴 -2-乙基 -6-甲基苯)氧代咪唑啉 -2-酮 Id (1.00 g, 3.50 mol) 溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (210 mg, 5.20 mmol), 搅拌 反应 30分钟。 向反应液中加入溴乙酸乙酯 (871 mg, 5.20 mmol), 搅拌反应 12小时。 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3;), 合并有机 相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;>, 用无水硫酸钠干 燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(3-(4-溴 -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 le (650 mg, 白色固体), 产率: 50.0%。
JH NM (400 MHz, CDC13) δ 7.23 (s, 2H), 4.20 (q, 2H), 4.02 (s, 2H), 3.69-3.59 (m, 4H), 2.67-2.48 (m, 2H), 2.22 (s, 3H), 1.28 (t, 3H), 1.21 (t, 3H)
第五步
2-(3-(4-((2- ((叔丁基二甲基硅)氧)乙基)氨基) -2-乙基 -6-甲基) -2-氧代咪唑 啉 -1-基)乙酸乙酯
将 2-(3-(4-溴 -2-2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 le (650 mg, 1.77 mol), 2- ((叔丁基二甲基硅)氧代)乙胺 (467 mg, 2.66 mol)溶解于 15 mL甲苯中,加入碳酸铯 (1.73 g, 5.31 mmol),三 (二苯亚苄基丙酮)二钯 (50 mg, 0.05 mmol)和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (40 mg, 0.07 mmol), 100°C 搅拌反应 12小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用乙酸 乙酯萃取 (50 mLx3;>, 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗 涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法 以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(3-(4-((2- ((叔丁基二甲基 硅)氧)乙基)氨基) -2-乙基 -6-甲基) -2-氧代咪唑啉 -1-基)乙酸乙酯 If (480 mg, 黄色油状物), 产率: 58.0%。
JH NM (400 MHz, CDC13) δ 6.35 (dd, 1H), 4.19 (q, 2H), 4.03 (s, 2H), 3.78 (t, 2H), 3.61 (s, 4H), 3.18 (t, 2H), 2.61-2.42 (m, 2H), 2.16 (s, 3H), 1.28 (t, 3H), 1.19 (t, 3H), 0.89 (s, 9H), 0.06 (s, 6H)
2-(3_(4_(N_(2 叔丁基二甲基硅)氧)乙基) _4,6_二氯嘧啶 _5_酰胺) _2 -乙基
-6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯
将 2-(3-(4-((2- ((叔丁基二甲基硅)氧)乙基)氨基) -2-乙基 -6-甲基) -2-氧代咪 唑啉小基)乙酸乙酯 If (480 mg, 1.04 mol)溶解于 5 mL二氯甲烷中, 加入三 乙胺 (449 uL, 3.11 mmol)和 4,6-二氯 -5-酰氯嘧啶 (653 mg, 3.11 mmol), 搅拌 反应 12小时。 向反应液中加入 10 mL水, 用二氯甲烷萃取 (10 mLx3;), 合并 有机相, 依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 (10 mLx2;>, 用无水硫酸 钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二 甲基硅:)氧:)乙基: )-4,6-二氯嘧啶 -5-酰胺: )-2-乙基 -6-甲基苯 2-氧代咪唑啉 -1-基) 乙酸乙酯 lg (662 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第七步
2-(3-(2-乙基 -6-甲基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉小基)乙酸乙酯
将粗品 2-(3 -(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lg (662 mg, 1.04 mol)溶 解于 10 mL乙醇中, 加入浓盐酸0.3 mL:), 搅拌反应 1小时。 向反应液中加 入 10 mL水,用乙酸乙酯萃取 (10 mLx3),合并有机相,依次用水 (10 mL><2)、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-乙基 -6-甲基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lh (440 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第八步
2-(3-(2-乙基 -6-甲基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓
-6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3-(2-乙基 -6-甲基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯
基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lh (440 mg, 841 umol)溶解于 5 mL乙腈中, 加入三乙胺 (364 uL, 2.52 mmol), 80°C搅拌反应 12小时。 反应液冷却至室 温, 用乙酸乙酯萃取 (30 mLx3;), 合并有机相, 依次用水 (30 mLx2;)、 饱和氯 化钠溶液洗涤 PO mLx2) , 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)- 基) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 li (250 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, DMSO-d6) δ 8.80 (s, 1Η), 7.17 (d, 2H), 4.70 (t, 2H), 4.14-4.08 (m, 4H), 3.96 (s, 2H), 3.59 (s, 4H), 2.59-2.52 (m, 2H), 2.17 (s, 3H), 1.21-1.17 (m, 3H), 1.15-1.11 (m, 3H)
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-乙 基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 将粗品 2-(3-(4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)- 基) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 li (250 mg, 513 umol)溶 解于 10 mL 0.5 M氨气的 1 , 4-二氧六环溶液中, 搅拌反应 12小时。 反应液 减压浓缩, 用二氯甲烷萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2)、 饱和氯化钠溶液洗涤30 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lj (240 mg, 黄色 固体), 产物不经纯化直接进行下一步反应。
JH NMR (400 MHz,DMSO-^) δ 8.17 (s, 1Η), 7.63 (br. s, 2H), 7.14 (s, 2H), 4.60 (t, 2H), 4.12 (q, 2H), 4.03 (q, 2H), 3.98 (s, 2H), 3.61 (s, 4H), 2.59-2.52 (m, 2H), 2.19 (s, 3H), 1.24-1 .13 (m, 6H)
第十步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-乙
基 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)- 基) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lj (240 mg, 513 umol)溶 解于 8 mLl ,4-二氧六环和水 (V/V = 3 : 1)混合溶液中, 加入氢氧化锂 (64 mg, 1.52 mmol) , 搅拌反应 12小时。滴加 1 M盐酸至反应液 pH为 5〜6, 过滤得 到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)- 基) -2-乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸 1 (108 mg, 白色固体),产率: 47.0%。
MS m/z (ESI): 441 .1 [M+l]
JH NM (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 7.70 (br. s, 2H), 7.14 (s, 2H) 4.60 (s, 2H), 3.99 (s, 2H), 3.88 (s, 2H), 3.60 (s, 4H), 2.61-2.52 (m, 2H), 2.17 (s, 3H), 1 .14 (t, 3H)
实施例 5
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-乙 基


4-溴 -2-乙基 -1-苯基异氰酸酯
将 4—溴 _2—乙基苯胺 la (3.00 g, 14.99 mmol)溶解于 30 mL四氢呋喃 中,加入三光气 (1.48 g, 5.00 mmol),搅拌反应 3小时。将反应液减压浓缩, 得到粗品标题产物 4-溴 -2-乙基 -1-苯基异氰酸酯 lb (3.39 g, 白色固体), 产物 不经纯化直接进行下一步反应。
第二步
1 -(4-溴 -2-乙基苯 )-3-(2-氯乙基)脲
将粗品 4-溴 -2-乙基 -1-苯基异氰酸酯 lb (3.39 g, 14.99 mmol)和三乙胺 (6.3 mL, 45.00 mmol)溶解于 100 mL四氢呋喃中, 加入 2-氯乙胺盐酸盐 (2.61 g, 22.50 mmol), 搅拌反应 2小时。 向反应液中加入 200 mL水, 用乙酸乙 酯萃取 (200 mLx3;>, 合并有机相, 依次用水 (100 mLx2;>、 饱和氯化钠溶液洗 ¾100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产 物 1-(4-溴 -2-乙基苯 )-3-(2-氯乙基:)脲 lc (4 g, 白色固体:), 产物不经纯化直接
Ή NM (400 MHz, DMSO-d6) δ 7.84 (s, 1H), 7.75 (d, 1H), 7.28-7.23 (m, 2H), 6.91-6.88 (m, 1H), 3.64 (t, 2H), 3.40 (t, 2H), 2.53-2.49 (m, 2H), 1.09 (t, 3H)
第三步
l-(4-溴 -2-乙基苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-乙基苯 )-3-(2-氯乙基)脲 lc (3.39 g, 15.00 mmol)溶解于 30 mL N, N-二甲基甲酰胺中, 冰浴下加入钠氢 (792 mg, 19.80 mmol), 室温 搅拌反应 12小时。 向反应液中加入 120 mL饱和氯化铵溶液, 用乙酸乙酯萃 取 (100 mLx3),合并有机相,依次用水 (100 mLx2)、饱和氯化钠溶液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 1-(4- 溴 -2-乙基苯:)氧代咪唑啉 -2-酮 Id (2.2 g, 白色固体:), 产物不经纯化直接进行 下一步反应。
^ NM C^O MHz, DMSO-d6) :5 7.46 (d, 1H), 7.41-7.38 (m, 1H), 7.17 (d, 1H), 6.72 (s, 1H), 3.70 (t, 2H), 3.41 (t, 2H), 2.60-2.51 (m, 2H), 1.14 (t, 3H)
第四步
2-(3-(4-溴 -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 1-(4-溴 -2-乙基苯)氧代咪唑啉 -2-酮 ld (1.00 g, 3.72 mmol)溶解于 20 mL N, N-二甲基甲酰胺中, 加入钠氢 (223 mg, 5.58 mmol), 搅拌反应 30 分钟。 向反应液中加入溴乙酸乙酯 (802 mg, 4.84 mmol), 室温搅拌反应 12 小时。向反应液中加入 50 mL饱和氯化铵溶液,用乙酸乙酯萃取 50 mLx3), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤50 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 2-(3-(4-溴 -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 le ( 0.8 g, 白色固体), 产率: 60.6%。
JH NMR (400 MHz, CDC13) δ 7.43 (d, 1H), 7.35-7.33 (m, 1H), 7.07 (d, 1H), 4.25- 4.20 (q, 2H), 4.05 (s, 2H), 3.75-3.70 (m, 2H), 3.66-3.62 (m, 2H), 2.68-2.62 (q, 2H), 1.30 (t, 3H), 1.23 (t, 3H)
第五步
2-(3-(4-((2-((叔丁基二甲基硅)氧代)乙基)氨基) -2-乙基苯 )-2-氧代咪唑啉
-1-乙基)乙酸
将 2-(3-(4-溴 -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 le (800 mg, 2.25 mmol), 2- ((叔丁基二甲基硅)氧代)乙胺 (473 mg, 2.70 mmol)溶解于 15 mL甲 苯中,加入碳酸铯 (2.20 g, 6.75 mmol),三 (二苯亚苄基丙酮)二钯 (54 mg, 0.11 mmol)和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽(101 mg, 0.11 mmol), 100°C搅拌 反应 12小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用乙酸乙酯 萃取 50 mLx3;), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 C纯化所得残余物,得到标题产物 2-(3-(4-((2- ((叔丁基二甲基硅)氧代) 乙基)氨基) -2-乙基苯 )-2-氧代咪唑啉 -1-乙基)乙酸 If (600 mg, 黄色油状物), 产率: 59.4%。
JH NM (400 MHz, CDC13) δ 7.99 (d, 1H), 6.52 (d, 1H), 6.49-6.46 (dd, 1H) 4.24- 4.19 (q, 2H), 4.05 (s, 2H), 3.82-3.79 (m, 2H), 3.69-3.57 (m, 4H), 3.22 (t, 2H), 2.61-2.55 (q, 2H), 1.30 (t, 3H) , 1.21 (t, 3H), 0.91 (s, 9H) , 0.07 (s, 6H)
第六步
2-(3_(4_(N_(2 叔丁基二甲基硅)氧)乙基) _4,6_二氯嘧啶 _5_酰胺) _2 -乙基 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯
将 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-乙基苯 )-2-氧代咪唑 啉 -1-乙基)乙酸 lf (600 mg, 1.33 mmol)溶解于 10 mL二氯甲烷中, 加入三乙 胺 (0.56 mL, 3.99 mmol)和 4,6-二氯 -5-酰氯嘧啶 (423 mg, 2.00 mmol), 搅拌 反应 12小时。 向反应液中加入 10 mL水, 用二氯甲烷萃取 10 mLx3;), 合并 有机相, 依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 (10 mLx2;>, 用无水硫酸 钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二 甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙
酯 lg (800 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.58 (s, 1H), 7.33-7.27 (m, 2H), 7.08 (d, 1H), 4.24- 4.18 (q, 2H), 4.02-3.99 (m, 2H), 3.92-3.91 (m, 2H), 3.66-3.61 (m, 3H), 3.13-3.09 (m, 3H), 2.59- 2.54 (q, 2H), 1.29 (t, 3H), 1.11 (t, 3H), 0.89 (s, 9H) , 0.07 (s, 6H)
第七步
2-(3-(2-乙基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉
-1-基:)乙酸乙酯
将粗品 2-(3 -(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺) -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lg (800 mg, 1.28 mmol)溶解于 10 mL乙醇中,加入浓盐酸 (0.3 mL;),搅拌反应 1小时。向反应液中加入 10 mL 水, 用乙酸乙酯萃取 (10 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和碳 酸氢钠溶液 IO mLx2;>、饱和氯化钠溶液洗涤 IO mLx2),用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-乙基 -4-(4,6-二氯 -N-(2-羟乙 基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lh (600 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第八步
2-(3-(2-乙基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代咪唑啉- 1 -基)乙酸乙酯
将粗品 2-(3-(2-乙基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉 -1-基)乙酸乙酯 lh (600 mg, 1.18 mmol)溶解于 10 mL乙腈中,加入三 乙胺 (0.49 mL, 3.54 mmol), 80°C搅拌反应 12小时。 反应液冷却至室温, 向 反应液中加入 20 mL水, 用乙酸乙酯萃取 (20 mLx3;>, 合并有机相, 用饱和 氯化钠溶液洗涤20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(2-乙基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (500 mg, 黄色固体), 产物
不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.77 (s, 1H), 7.31-7.23 (m, 3H), 4.75 (t, 2H), 4.26- 4.21 (q, 2H), 4.07-4.04 (m, 2H), 3.79-3.75 (m, 2H), 3.69-3.65 (m, 3H), 2.75-2.69 (q, 3H), 1.33-1.25 (m, 6H)
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-乙 基苯) -2-氧代咪唑啉- 1 -基)乙酸乙酯
将粗品 2-(3-(2-乙基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 li (500 mg, 1.06 mmol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 加入 30 mL 乙酸乙酯, 依次用水 (30 mLx2), 饱和氯化钠溶液洗涤 (30 mLx2) , 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-乙基 苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lj (300 mg, 黄色固体), 产物不经纯化直接 进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.29 (s, 1Η), 8.16 (br.s., 1H), 7.30-7.13 (m, 3H), 5.66 (brs, 1H), 4.69 (t, 2H), 4.26- 4.20 (q, 2H), 4.07-4.01 (m, 2H), 3.79-3.71 (m, 2H), 3.68-3.64 (m, 3H), 2.74-2.68 (q, 3H), 1.33-1.24 (m, 6H)
第十步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-乙 基苯) -2-氧代咪唑啉- 1 -基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lj (300 mg, 0.66 mmol)溶解于 8 mLl,4-二氧六环和水 (V/V = 3: 1)混合溶液中, 加入氢氧化锂 (139 mg, 3.30 mmol), 搅拌反应 12小时。 滴加 1 M盐酸至反应液 pH为 5〜6, 减压浓缩, 残留物用制备高效液相色谱纯化得到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二
氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-乙基苯 )-2-氧代咪唑啉 -1-基)乙酸 1 (24.7 mg, 白色固体), 产率: 8.8%。
MS m/z (ESI): 427.2 [M+l]
JH NMR (400 MHz, DMSO-^) δ 8.37-8.18 (m, 2H), 7.31-7.23 (m, 2H), 4.73 (t, 2H), 4.08 (s, 3H), 3.89 (s, 3H), 3.71 (t, 2H), 3.45-3.43 (m, 4H), 2.59 (q, 2H), 1.16 (t, 3H)
实施例 6
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯

4-溴 -2-氯 -1 -苯基异氰酸酯
将 4-溴 -2-氯苯胺 la (7.00 g, 34 mmol)溶解于 250 mL四氢呋喃中, 加入 三光气 (3.30 g, 11.20 mmol) , 搅拌反应 3小时。 将反应液减压浓缩, 得到粗 品标题产物 4-溴 -2-氯 -1-苯基异氰酸酯 1 7.00 g, 白色固体:), 产物不经纯化 直接进行下一步反应。
第二步
1 -(4-溴 -2-氯苯) -3-(2-氯乙基)脲
将粗品 4-溴 -2-氯 -1-苯基异氰酸酯 lb (3.00 g, 12.90 mmol)和三乙胺 (1.96 g, 19.30 mmol)溶解于 100 mL四氢呋喃中, 加入 2-氯乙胺盐酸盐 (2.25 g, 19.30 mmol) , 搅拌反应 2小时。 向反应液中加入 200 mL水, 用二氯甲 烷萃取 (200 mLx3;>, 合并有机相, 依次用水 (100 mLx2;>、 饱和氯化钠溶液洗 涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产 物 1 -(4-溴 -2-氯苯: )-3-(2-氯乙基:)脲 lb (3.30 g, 白色固体:), 产物不经纯化直接 进行下一步反应。
第三步
1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮
将粗品 1-(4-溴 -2-氯苯) -3-(2-氯乙基)脲 lc (3.30 g, 10.80 mmol)溶解于 100 mL N, N-二甲基甲酰胺中, 加入钠氢 (634 mg, 15.80 mmol) , 搅拌反应 12 小时。向反应液中加入 70 mL水,用乙酸乙酯萃取70 mLx3;),合并有机相, 依次用水 (70 mLx2;>、 饱和氯化钠溶液洗涤70 mLx2;), 用无水硫酸钠干燥, 过滤,滤液减压浓缩,得到粗品标题产物 1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮 Id (3.10 g, 白色固体:), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, DMSO-^) δ 7.76 (d, 1Η), 7.54 (dd, 1H), 7.31-7.36 (m, 1H), 6.85 (br. s., 3H), 3.72 (t, 2H), 3.39 (t, 2H)
第四步
2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)丙酸乙酯 将粗品 1-(4-溴 -2-氯苯)氧代咪唑啉 -2-酮 Id (3.10 g, 7.80 mmol)溶解于 50 mL N, N-二甲基甲酰胺中, 加入钠氢 (472 mg, 11.80 mmol), 搅拌反应 30 分钟。向反应液中加入溴乙酸乙酯 (1.21 mL, 9.45 mmol),搅拌反应 12小时。 向反应液中加入 20 mL水, 用乙酸乙酯萃取20 mLx3;>, 合并有机相, 依次 用水 (20 mLx2;>、饱和氯化钠溶液洗涤20 mLx2;>,用无水硫酸钠干燥,过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题 产物 2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)丙酸乙酯 le (1.40 g, 白色固体), 产率: 47.0%。
JH NM (400 MHz, CDC13) δ 7.56 (d, 1Η), 7.40-7.37 (m, 1H), 7.24 (d, 1H), 4.15 (q, 2H), 3.75-3.71 (m, 2H), 3.59-3.52 (m, 4H), 2.60 (t, 2H), 1.26 (t, 3H)
第五步
2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氯苯) -2-氧代咪唑啉 -1- 基)丙酸乙酯
将 2-(3-(4-溴 -2-氯苯) -2-氧代咪唑啉 -1-基)丙酸乙酯 le (700 mg, 1.86 mol), 2-((叔丁基二甲基硅;)氧代)乙胺 (490 mg, 2.80 mmol)溶解于 15 mL甲苯中, 加入碳酸铯 (1.82 g, 5.59 mmol),三 (二苯亚苄基丙酮)二钯 (50 mg, 0.05 mmol) 和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (40 mg, 0.07 mmol), 100°C搅拌反应 12 小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3),合并有机相,依次用水 (50 mLx2;>、饱和氯化钠溶液洗涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨 基) -2-氯苯) -2-氧代咪唑啉 -1-基)丙酸乙酯 If (650 mg, 黄色油状物), 产率: 74.0%。
JH NM (400 MHz, CDC13) δ 7.01 (d, 1H), 6.58 (d, 1H), 6.43 (dd, 1H), 4.09 (q, 2H), 3.72 (d, 2H), 3.61-3.57 (m, 2H), 3.50 (t, 2H), 3.43-3.41 (m, 2H),
3.10 (q, 2H), 2.54 (t, 2H), 1.20 (t, 3H), 0.83 (s, 9H), 0.00 (s, 6H)
第六步
2-(3-(4-(N-(2- ;叔丁基二甲基硅)氧:)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-氯 苯) -2-氧代咪唑啉 -1-基)丙酸乙酯
将 2-(3-(4-((2- ((叔丁基二甲基硅)氧)乙基)氨基) -2-氯苯) -2-氧代咪唑啉 -1- 基)丙酸乙酯 If (650 mg, 1.38 mmol)溶解于 5 mL二氯甲烷中,加入三乙胺 (877 mg, 4.15 mmol)和 4,6-二氯 -5-酰氯嘧啶 (420 mg, 4.15 mmol), 搅拌反应 12 小时。向反应液中加入 10 mL水,用二氯甲烷萃取 10 mLx3;),合并有机相, 依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁基二甲基硅) 氧:)乙基: )-4,6-二氯嘧啶 -5-酰胺: )-2-氯苯: )-2-氧代咪唑啉 -1-基:)丙酸乙酯 lg (887 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第七步
2-(3-(2-氯 -4-(4,6-二氯 -N-(2-羟乙基:)嘧啶 -5-酰胺)苯基: )-2-氧代咪唑啉 -1- 基)丙酸乙酯
将粗品 2-(3 -(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺:) -2-氯苯: )-2-氧代咪唑啉 -1-基:)丙酸乙酯 lg (887 mg, 1.38 mmol)溶解于 10 mL乙醇中, 加入浓盐酸 (0.3 mL;), 搅拌反应 1小时。 向反应液中加入 10 mL 水, 用二氯甲烷萃取 (10 mLx3;), 合并有机相, 依次用水 (10 mLx2)、 饱和氯 化钠溶液洗涤 IO mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到 粗品标题产物 2-(3-(2-氯 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉 _μ基)丙酸乙酯 ^ !!^, 黄色固体), 产物不经纯化直接进行下一 步反应。
JH NM (400 MHz, CDC13) δ 8.63 (s, 1H), 7.52 (d, 1H), 7.33-7.29 (m, 2H), 4.15 (q, 2H), 4.03 (t, 2H), 3.91 (t, 2H), 3.71 (t, 2H), 3.59-3.51 (m, 4H), 2.59 (t, 2H), 1.26 (t, 3H)
第八步
2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -2-氧代咪唑啉 -1-基)丙酸乙酯
将粗品 2-(3-(2-氯 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代咪 唑啉 _1_基)丙酸乙酯 lh (732 mg, 1.38 mmol)溶解于 5 mL乙腈中, 加入三乙 胺 (600 uL, 4.14 mmol), 80°C搅拌反应 12小时。 反应液冷却至室温, 向反 应液中加入 20 mL水, 用乙酸乙酯萃取30 mLx3;>, 合并有机相, 依次用水 (30 mLx2), 饱和氯化钠溶液洗涤30 mLx2;>, 用无水硫酸钠干燥, 过滤, 滤 液减压浓缩, 得到粗品标题产物 2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)丙酸乙酯 li (240 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.76 (s, 1Η), 7.48-7.45 (m, 2H), 7.30 (dd, 1H), 4.74-4.72 (m, 2H), 4.16 (q, 2H), 4.03 (t, 2H), 3.79-3.76 (m, 2H), 3.61-3.55 (m, 4H), 2.62 (t, 2H), 1.27 (t, 3H)
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -2-氧代咪唑啉 -1-基)丙酸乙酯
将粗品 2-(3-(2-氯 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)丙酸乙酯 li (240 mg, 253 umol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 加入 30 mL 二氯甲烷, 依次用水 (30 mLx2), 饱和氯化钠溶液洗涤 (30 mLx2) , 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -6(5H)-基 )-2-氯 苯) -2-氧代咪唑啉 -1-基)丙酸乙酯 lj (170 mg, 黄色固体), 产物不经纯化直接 进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.28 (s, 1H), 7.45 (d, 1H), 7.38 (d, 1H), 7.30
(dd, IH), 4.68-4.66 (m, 2H), 4.16 (q, 2H), 3.99 (t, 2H), 3.77 (s, 2H), 3.69 (s, 2H) 3.61-3.55 (m, 4H), 2.62 (t, 2H), 1.27 (t, 3H)
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -2-氧代咪唑啉 -1-基)丙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l,4]氧氮杂卓 -6(5H)- 基:) -2-氯苯: )-2-氧代咪唑啉 -1-基:)丙酸乙酯 lj (170 mg, 358 umol)溶解于 8 mLl,4-二氧六环和水 (V/V = 3: 1)混合溶液中, 加入氢氧化锂 (45 mg, 1.07 mmol), 搅拌反应 12小时。滴加 1 M盐酸至反应液 pH为 5〜6, 过滤得到标 题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯苯 >-2-氧代咪唑啉 -1-基;)丙酸 1 (120 mg, 白色固体:), 产率: 75.0%。
MS m/z (ESI): 477.0 [M+l]
JH NMR (400 MHz, DMSO- ) δ 8.17 (s, IH), 7.65-7.64 {br. s, 3H), 7.45 (d IH), 7.39 (dd, IH), 4.63-4.60 (m, 2H), 4.01 (t, 2H), 3.72-3.68 (m, 2H), 3.52-3.48 (m, 2H), 3.40 (t, 2H), 2.49-2.47 (m, 2H)
实施例 7
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-三 氟甲氧基苯 )-2-氧代咪唑啉- 1 -基)乙酸


2-(3-(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-三氟 甲氧基苯) -2-氧代咪唑啉- 1 -基)乙酸乙酯
冰浴下,将 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-三氟甲氧基 苯) -2-氧代咪唑啉 -1-乙基)乙酸乙酯 la (350 mg, 0.7 mmol)溶解于 10 mL二氯 甲烷中, 加入三乙胺 (140 mg, 1.4 mmol)和 4,6-二氯嘧啶 -5-碳酰氯 (220 mg, 1.5 mmol), 室温搅拌反应 12小时。 向反应液中加入 20 mL水, 用二氯甲烷 萃取20 mLx3;>, 合并有机相, 用饱和氯化钠溶液洗涤 50 mL:), 无水硫酸钠 干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余 物, 得到标题产物 2-p- KN-(2- ;叔丁基二甲基硅:)氧:)乙基 )-4,6-二氯嘧啶 -5- 酰胺) -2-三氟甲氧基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 lb (170 mg,白色固体), 产率: 36.2%。
JH NM (400 MHz, CDC13) δ 8.56 (s, 1H), 7.48 (d, 1H), 7.31-7.33 (m, 2H), 4.16 (q, 2H), 3.97 (s, 2H), 3.92-3.94 (m, 2H), 3.85-3.87 (m, 2H), 3.76 (t, 2H), 3.55 (t, 2H), 1.21 (t, 3H), 0.81 (s, 9H), 0.00 (s, 6H)
第二步
2-(3-(2-三氟甲氧基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 咪唑啉小基)乙酸乙酯
将 2-(3-(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰胺) -2-三 氟甲氧基苯) -2-氧代咪唑啉 -1-基)乙酸乙酯 lb (170 mg, 0.28 mmol)溶解于 10
mL 乙醇中, 加入浓盐酸0.3 mL), 搅拌反应 2小时。 向反应液中加入饱和 碳酸氢钠溶液调节 pH大于 7, 用二氯甲烷萃取 G0 mLx3;>, 合并有机相, 依 次用水 (5 mLx2),饱和氯化钠溶液洗涤5 mLx2),用无水硫酸钠干燥,过滤, 滤液减压浓缩,得到粗品标题产物 2-(3-(2-三氟甲氧基 -4-(4,6-二氯 -N-(2-羟乙 基)嘧啶 -5-酰胺)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lc (140 mg, 黄色油状物), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.65 (s, 1Η), 7.57 (d, 1H), 7.37-7.42 (m, 2H), 4.22 (q, 2H), 4.06-4.09 (m, 2H), 4.04 (s, 2H), 3.93-3.96 (m, 2H), 3.85 (t, 2H), 3.61 (t, 2H), 1.27 (t, 3H)
第三步
2-(3-(2-三氟甲氧基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓
-6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯
将粗品 2-(3 -(2-三氟甲氧基 -4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯 基) -2-氧代咪唑啉 -1-基)乙酸乙酯 lc (140 mg, 0.25 mmol)溶解于 10 mL 乙腈 中, 加入三乙胺 (75 mg, 0.75 mmol), 80°C搅拌反应 12小时。 向反应液中加 入 40 mL水, 用乙酸乙酯萃取 (20 mLx3;>, 合并有机相, 饱和氯化钠溶液洗 涤 (100 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(2-三氟甲氧基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 Id (130 mg,黄色油状物), 产物不经 纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.79 (s, 1H), 7.67 (d, 1H), 7.31-7.39 (m, 2H), 4.76 (t, 2H), 4.25 (q, 2H), 4.04-4.21 (m, 5H), 3.91 (t, 2H), 3.68 (t, 2H), 1.27 (t, 3H)
第四步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-三 氟甲氧基苯) -2-氧代咪唑啉 - 1 -基)乙酸乙酯
将粗品 2-(3-(2-三氟甲氧基 -4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮 杂卓 -6(5H)-基)苯基) -2-氧代咪唑啉 -1-基)乙酸乙酯 Id (130 mg, 0.25 mmol)溶 解于 5 mL饱和氨气的 1,4-二氧六环中,搅拌反应 12小时。 向反应液中加入 20 mL水,用乙酸乙酯萃取20 mLx3),合并有机相,饱和氯化钠溶液洗涤 (100 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层层析以洗脱剂体系 A 纯化所得残余物, 得到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-三氟甲氧基苯 )-2-氧代咪唑啉 -1-基)乙酸乙 酯 le (80 mg, 黄色固体), 产率 64.0%。
JH NM (400 MHz, CD3OD) δ 8.15 (s, 1Η), 7.57 (d, 1H), 7.49 (s, 1H), 7.41 (dd, 1H), 4.70-4.72 (m, 2H), 4.22 (q, 2H), 4.07-4.09 (m, 2H), 3.86-3.91 (m, 2H), 3.69-3.71 (m, 2H), 3.30 (s, 2H), 1.28 (t, 3H)
第五步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-三 氟甲氧基苯 )-2-氧代咪唑啉- 1 -基)乙酸
将 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 三氟甲氧基苯 )-2-氧代咪唑啉 -1-基)乙酸乙酯 le (80 mg, 0.15 mmol)溶解于 4 mL 1,4-二氧六环和水中 (V/V=3/l), 加入氢氧化锂 (50 mg, 1.25 mmol), 搅拌 反应 12小时。 向反应液中加入 1M盐酸溶液调节 pH小于 5, 用乙酸乙酯萃 取 (10 mLx3),合并有机相,依次用水 (5 mLx2)、饱和氯化钠溶液洗涤 (5 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用 p-HPLC制备分离纯化所得残 余物,得到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-三氟甲氧基苯 )-2-氧代咪唑啉 -1-基)乙酸 1 (28 mg, 白色固体), 产率: 37.3%。
JH NMR (400 MHz, CD3OD) δ 8.55 (s, 2H), 8.34 (s, 1H), 7.40-7.55 (m, 3H), 4.75 (t, 2H), 4.10 (t, 2H), 3.89 (s, 2H), 3.72-3.77 (m, 4H)
实施例 8
4-氨基 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二氢 嘧啶

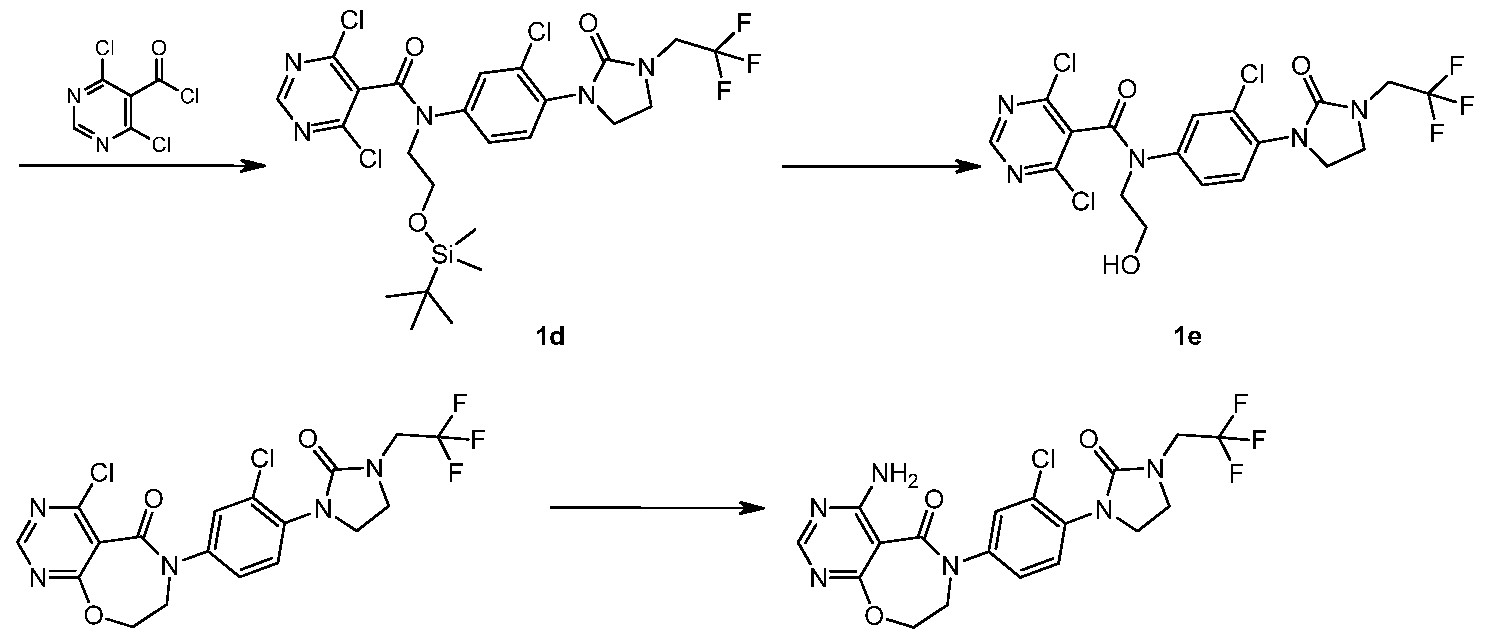
1-(4-溴 -2-氯苯) -3-(2,2,2-三氟乙基)咪唑啉 -2-酮
将 ι_(4-溴 -2-氯苯)咪唑啉 -2-酮 la (1.00 g, 3.63 mmol )溶解于 20 mL N,
N-二甲基甲酰胺中, 加入钠氢 (217 mg, 5.44 mmol ), 搅拌反应 30分钟。 向 反应液中加入 2,2,2-三氟乙基三氟甲磺酸脂 (1.26 g, 5.44 mmol ), 搅拌反应 12小时。 向反应液中加入 50 mL水, 用乙酸乙酯萃取 (20 mLx3;), 合并有机 相, 依次用水 (20 mLx2;>、 饱和氯化钠溶液洗涤 (20 mLx2;>, 用无水硫酸钠干 燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 1-(4-溴 -2-氯苯) -3-(2,2,2-三氟乙基)咪唑啉 -2-酮 lb (500 mg, 白 色固体), 产率: 38.5%。
Ή NM (400 MHz, CDC13): δ 7.60 (d, 1H), 7.42 (dd, 1H), 7.24 (d, 1H), 3.90-3.80 (m, 4H), 3.69-3.65 (m, 2H )
第二步
l-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氯苯) -3-(2,2,2-三氟乙基) 咪唑啉 -2-酮
将 1-(4-溴 -2-氯苯) -3-(2,2,2-三氟乙基)咪唑啉 -2-酮 lb (500 mg, 1.40 mol ),
2- ((叔丁基二甲基硅)氧代)乙胺 (367 mg, 2.10 mmol )溶解于 10 mL甲苯中, 加入碳酸铯 (1.30 g, 4.20 mmol ),三 (二苯亚苄基丙酮)二钯 (50 mg, 0.05 mmol ) 和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (40 mg, 0.07 mmol ), 100°C搅拌反应 12小时。反应液冷却至室温,向反应液中加入 30 mL水,用二氯甲烷萃取 (30 mLx3),合并有机相,依次用水 (30 mLx2;)、饱和氯化钠溶液洗涤 (30 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 1-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨 基) -2-氯苯) -3-(2,2,2-三氟乙基)咪唑啉 -2-酮 lc (420 mg,黄色油状物),产率: 66.4%。
JH NM (400 MHz, CDC13): δ 7.08 (d, 1H), 6.64 (d, 1H), 6.43 (dd, 1H), 3.84 (q, 7 = 9.2 Hz, 2H), 3.78 (t, 2H), 3.75-3.71 (m, 2H), 3.62-3.58 (m, 2H ), 3.16 (q, 2H ), 0.88 (s, 9H), 0.05 (s, 6H)
第三步
N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三 氟乙基)咪唑啉 -1-基)苯基)嘧啶 -5-酰胺
将 1-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2-氯苯) -3-(2,2,2-三氟乙 基) P米唑啉 -2-酮 lc (420 mg, 929 umol )溶解于 5 mL二氯甲烷中, 加入三乙 胺 (400 uL, 2.80 mmol )和 4,6-二氯 -5-酰氯嘧啶 (393 mg, 1.80 mmol ), 搅拌 反应 12小时。 向反应液中加入 20 mL水, 用二氯甲烷萃取 (20 mLx3;), 合并 有机相, 依次用水 (20 mLx2;)、 饱和氯化钠溶液洗涤20 mLx2;>, 用无水硫酸
钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 N-(2-((叔丁基二甲基硅) 氧)乙基) -4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基)嘧 啶 -5-酰胺 Id (582 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
第四步
4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -N-(2- 羟乙基)嘧啶 -5-酰胺
将粗品 N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基)嘧啶 -5-酰胺 Id (582 mg, 928 mmol )溶 解于 10 mL乙醇中, 加入浓盐酸0.3 mL:), 搅拌反应 1小时。 向反应液中加 入 10 mL水,用二氯甲烷萃取 10 mLx3;),合并有机相,依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1- 基)苯基) -N-(2-羟乙基)嘧啶 -5-酰胺 le (475 mg, 黄色固体), 产物不经纯化直 接进行下一步反应。
第五步
4-氯 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将粗品 4,6-二氯 -N-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯 基) -N-(2-羟乙基)嘧啶 -5-酰胺 le (475 mg, 926 umol )溶解于 5 mL乙腈中,加 入三乙胺 (400 uL, 2.80 mmol ), 80°C搅拌反应 12小时。反应液冷却至室温, 用二氯甲烷萃取 (10 mLx3), 合并有机相, 依次用水 (10 mLx2)、 饱和食盐水 洗涤 (10 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题 产物 4-氯 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 If (410 mg, 黄色固体), 产物不经纯化直接 进行下一步反应。
JH NM (400 MHz, CDC13): δ 8.77 (s, 1H), 7.51 (d, 1H), 7.48 (d, 1H), 7.32
(dd, J = 8.4 Hz, IH), 4.75 (t, 2H), 4.04 (t, 2H), 3.90-3.84 (m, 4H), 3.72-3.68 (m 2H)
4-氨基 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将粗品 4-氯 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 If (410 mg, 860 umol )溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。 反应液减压浓缩, 用 制备高效液相色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 4-氨基 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1 -基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (135 mg, 白色固体), 产率: 34.3%。
MS m/z (ESI): 456.9 [M+l]
JH NM (400 MHz, DMSO-d6): δ 8.59 (br, IH), 8.42 (br.s, IH), 8.36 (s, IH), 7.68 (d, IH), 7.49 (d, IH), 7.41 (dd, IH), 4.77 (t, 2H), 4.11 (t, 2H), 4.00 (q, 2H), 3.79-3.75 (m, 2H), 3.64-3.60 (m, 2H)
实施例 9
4-氨基 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -IH-苯并 [d] 咪唑 _1_基)苯基) _ 卓 -5(6H)-酮
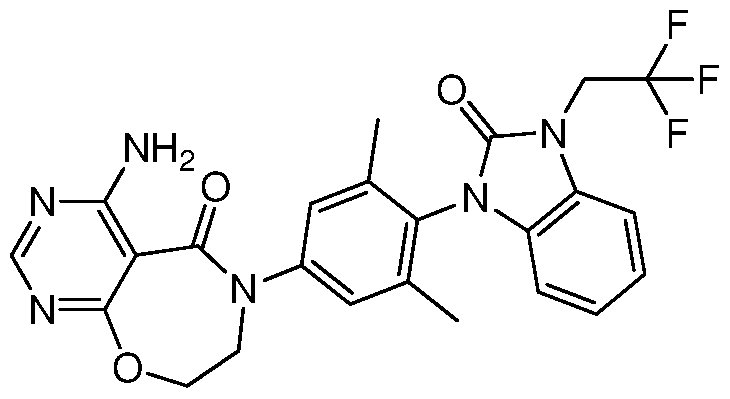


4-溴 -2,6-二甲基 -N-(2-硝基苯基:)苯胺
将 4一溴一 2,6 二甲基苯胺 la (5.0 g, 24.99 mmol)溶解于 50 mL二甲亚 砜中, 加入 1-氟 -2-硝基苯 lb (3.88 g, 27.49 mmol)和碳酸钾 (10.36 g, 74.97 mmol) o 120度下搅拌反应 12小时。反应液用 50 mL二氯甲烷和 50 mL水稀 释,用 l mol/L的稀盐酸将其 pH调至 6左右,溶液用二氯甲烷萃取 100 mLx3), 合并有机相, 依次用水 (300 mLx3;)、 饱和氯化钠溶液洗涤 (300 mLx 2), 用无 水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化 所得残余物, 得到标题产物 4-溴 -2,6-二甲基 -N-(2-硝基苯基:)苯胺 lc (640 mg, 黄色固体), 产率: 7.97 %。
JH NM (400 MHz, CDC13): δ 8.27 - 8.21 (m, IH), 7.41 - 7.41 (m, IH), 7.46 7.34 (m, 3H), 6.79 (d, 7=14.6 Hz, IH), 6.37 (d, 7=8.5 Hz, IH), 2.20 (s, 6H)。
第二步
N1-^溴 -2,6-二甲基苯基)苯 -1,2-二胺
将 4-溴 -2,6-二甲基 -N-(2-硝基苯基)苯胺 lc (0.6 g, 1.87 mmol)溶解于 10 mL甲醇中,加入氯化镍 (890 mg, 3.74 mmol), 5分钟后再加入硼氢化钠 (284 mg, 7.48 mmol), 搅拌反应 10分钟。 向反应液中加入 10 mL饱和氯化钠溶液 淬灭反应, 用二氯甲烷萃取30 mLx3;), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 N1-^溴 -2,6-二甲基苯基)苯 -1,2-二胺 Id (570 mg, 黄色固 体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13): δ 7.47 - 7.42 (m, 1Η), 7.26 - 7.23 (m, 2H), 7.15 (d, 7=7.5 Hz, 2H), 7.03 - 6.94 (m, 1H), 2.35 - 2.35 (m, 6H) 第三步
l-(4-溴 -2,6-二甲基苯基) -1H-苯并 [d]咪唑 -2(3H)-酮
将粗品 Nl-(4-溴 -2,6-二甲基苯基)苯 -1,2-二胺 Id (400 mg, 1.38 mmol)溶 解于 10 mL 四氢呋喃中, 加入三乙胺 (400 mg, 1.38 mmol)和羰基二咪唑 (894 mg, 5.52 mmol), 80度下搅拌反应 12小时。 向反应液中加入 30 mL水, 用 二氯甲烷萃取30 mLx3;), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶 液洗涤 (300 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法以洗脱剂体系 B纯化所得残余物得到标题产物 1-(4-溴 -2,6-二甲基苯 基) -1H-苯并 [d]咪唑 -2(3H)-酮 le (360 mg, 黄色固体)。
JH NMR (400 MHz, CDC13): δ 7.72 (d, J=2.0 Hz, 1H), 7.70 (m, 1H), 7.48 - 7.45 (m, 1H), 7.16-7.10(m, 2H), 7.06-7.03 (m, 1H), 6.70-6.07 (m, 1H)
第四步
l-(4-溴 -2,6-二甲基苯基) -3-(2,2,2-三氟乙基) -1H-苯并]咪唑 -2(3H)-酮 将 ι_(4-溴 -2,6-二甲基苯基) -1H-苯并 [d]咪唑 -2(3H)-酮 le (360 mg, l .lmmol)溶解于 5 mL N, N-二甲基甲酰胺中, 在零度下加入碳酸铯 (715 mg, 2.2 mmol), 2,2,2-三氟乙基三氟甲磺酸 (528.6 mg, 2.2 mmol), 搅拌反应 12小
时。 向反应液中加入 20 mL水, 用乙酸乙酯萃取20 mLx3;), 合并有机相, 依次用水 (60 mLx3;>、 饱和氯化钠溶液洗涤60 mLx2;>, 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层层析以洗脱剂体系 B纯化所得残余物, 得到标 题产物 1-(4-溴 -2,6-二甲基苯基) -3-(2,2,2-三氟乙基) -1H-苯并]咪唑 -2(3H)-酮 If (420 mg, 黄色固体), 产率: 95.9 %。
1H NM (400 MHz, CDC13): δ 7.39 (s, 2 Η), 7.14 7.24 (m, 4 H), 4.55 4.61 (m, 2 H), 2.05 - 2.08 (m, 6 H).
第五步
l-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基) -2,6-二甲基苯基) -3-(2,2,2-三 氟乙基) -1H-苯并 [d]咪唑 -2(3H)-酮
将 1-(4-溴 -2,6-二甲基苯基) -3-(2,2,2-三氟乙基) -1H-苯并]咪唑 -2(3H)-酮 If (0.4 g, l.Ol mmol), 2- ( (叔丁基二甲基硅) -氧代) -乙胺 (299 mg, 1.71 mmol ) 溶解于 10 mL甲苯中, 加入碳酸铯 (656.5 mg, 2.02 mmol), 三 (二苯亚苄基丙 酮)二钯 (15 mg)和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (15 mg) , 100°C搅拌反 应 12小时。 反应液冷却至室温, 向反应液中加入 30 mL水, 用乙酸乙酯萃 取 (50 mLx3), 合并有机相, 依次用水 (50 mLx2), 饱和氯化钠溶液洗涤 (50 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 C纯化所得残余物, 得到标题产物 1-(4-((2-((叔丁基二甲基硅)氧代) 乙基)氨基) -2,6-二甲基苯基) -3-(2,2,2-三氟乙基) -1H-苯并 [d]咪唑 -2(3H)-酮 lg (30 mg, 黄色油状物), 产率: 6.06 %。
MS m/z (ESI): 494.2 [M+l]
第六步
N-(2-((叔丁基二甲基硅)氧)乙基) -4,6-二氯 -N-(3,5-二甲基 -4-(2-氧 -3-(2,2,2-三氟乙基 2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基)嘧啶 -5-酰胺 将 1-(4-((2-((叔丁基二甲基硅)氧代)乙基)氨基) -2,6-二甲基苯基) -3-(2,2,2- 三氟乙基) -1H-苯并 [d]咪唑 -2(3H)-酮 lg (20 mg, 0.04 mmol)溶解于 2 mL二氯
甲烷中, 加入三乙胺 (8.08 mg, 0.08 mmol)和 4,6-二氯 -5-酰氯嘧啶 lh (17 mg, 0.08 mmol), 搅拌反应 3小时。 向反应液中加入 20 mL水, 用二氯甲烷萃取 (30 mLx3),合并有机相,依次用水 (30 mLx2)、饱和氯化钠溶液洗涤 (30 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 N-(2-((叔丁基 二甲基硅)氧)乙基) -4,6-二氯 -N-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3- 二氢 -1H-苯并 [d]咪唑 -1-基:)苯基:)嘧啶 -5-酰胺 li (40 mg, 黄色油状:),产物不经 纯化直接进行下一步反应。
JH NM (400 MHz, CDC13): δ 8.85 (s, 1 Η), 7.30 (s, 2 Η), 7.13 - 7.22 (m, 3 H), 7.07 (t, 1 H), 4.54 (q, 2 H), 4.10 - 4.18 (m, 2 H), 3.96 - 4.05 (m, 2 H), 2.01 (s, 6 H), 0.87 - 0.96 (m, 9 H), 0.08 - 0.08 (m, 1 H), 0.07 (s, 6 H).
第七步
4,6-氯 -N-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]
咪唑 -1 -基)苯基) -N-(2-羟乙基)嘧啶 -5-酰胺
将粗品 N-(2-((叔丁基二甲基硅:)氧:)乙基: )-4,6-二氯 -N-(3,5-二甲基 -4-(2-氧 代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基)嘧啶 -5-酰胺 li (40 mg, 0.06 mmol)溶解于 3 mL乙醇中, 加入浓盐酸(0.09 mL ), 搅拌反应 1小 时。 向反应液中加入 10 mL水, 用饱和碳酸氢钠溶液调节 pH至 6~7, 二氯 甲烷萃取 (20 mLx3), 合并有机相, 饱和氯化钠 (50 mLx2)洗涤, 无水硫酸钠 干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 4,6-氯 -N-(3,5-二甲基 -4-(2- 氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基) -N-(2-羟乙基) 嘧啶 -5-酰胺 lj (30 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NMR (400 MHz, CDC13): δ 8.87 - 8.83 (m, 1H), 8.68 (s, 1H), 7.30 (s, 5H), 7.22 7.02 (m, 1H), 4.58 4.49 (m, 2H), 4.16 4.10 (m, 2H), 4.02 3.97 (m, 2H), 2.01 (s, 6H).
第八步
4-氯 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪
唑 -1-基:)苯基 ,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮
将粗品 4,6-氯 -N-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H- 苯并 [d]咪唑 -1-基)苯基) -N-(2-羟乙基)嘧啶 -5-酰胺 lj (30 mg, 0.05 mmol)溶解 于 3 mL乙腈中, 加入三乙胺 (15 mg, 0.15 mmol), 80°C搅拌反应 12小时。 向 反应液中滴加饱和碳酸氢钠溶液至反应液 pH为 7〜8, 用二氯甲烷萃取 (20 mLx3), 合并有机相, 依次用水 (50 mLx2)、 饱和食盐水洗涤 (50 mLx2), 用 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 4-氯 -6-(3,5-二甲 基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 lk (30 mg,黄色固体),产物不经纯化直 接进行下一步反应。
MS m/z (ESI): 518.0 [M+l]
第九步
4-氨基 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -IH-苯并 [d] 咪唑 _1_基)苯基) _7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将粗品 4-氯 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯 并 [d]咪唑 -1-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 lk (30 mg, 0.058 mmo)溶解于 5 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小 时。反应液减压浓缩,用二氯甲烷萃取 (20 mLx3),合并有机相,依次用水 (30 mLx2), 饱和食盐水洗涤30 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓 缩, 所得物质用制备分离得到标题产物 4-氨基 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基 )-2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基 )-7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (2.30 mg, 白色固体), 产率: 7.9 %。
MS m/z (ESI): 499.2 [M+l]
JH NM (400 MHz, MeOD): δ 8.42 (s, IH), 7.40 (m, IH), 7.35 (s, 21H), 7.25 (m, IH), 7.15 (m, IH), 6.70 (m, IH), 5.15 (m, 2H), 4.70 (m, 2H), 4.28 (m, 2H), 2.13 (s, 6H).
实施例 10
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯
1 i
1-溴 -4-异氰酸苯酯
将 4-溴苯胺 la (5.00 g, 29.07 mmol )溶解于 200 mL四氢呋喃中, 加入 光气 (2.90 g, 9.69 mmol ), 搅拌反应 3小时。 将反应液减压浓缩, 得到粗 标题产物 1-溴 -4-异氰酸苯酯 lb 5.70 g, 白色固体:), 产物不经纯化直接进
l-(4-溴苯基 )-1Η-咪唑 -2(3H)-酮
将粗品 1-溴 -4-异氰酸苯酯 lb (5.70 g, 28.79 mmol )溶解于 150 mL四氢 呋喃中, 加入 2,2-二甲氧基乙胺 (4.54 g, 42.80 mmol ), 搅拌反应 12小时。 向反应液中加入 200 mL水, 用乙酸乙酯萃取 (200 mLx3;>, 合并有机相, 依 次用水 (100 mLx2)、 饱和氯化钠溶液洗涤 100 mLx2;>, 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得 到标题产物 1-(4-溴苯基) -1H-咪唑 -2(3H)-酮 lc (2.20 g, 白色固体), 产率: 31.9%。
JH NM (400 MHz, CDC13): δ 7.72-7.68 (m, 2Η), 7.59-7.55 (m, 2H), 6.98-6.96 (m, 1H), 6.60-6.58 (m, 1H)
第三步
2-(3-(4-溴苯基 )-2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯
将粗品 1-(4-溴苯基) -1H-咪唑 -2(3H)-酮 lc (1.10 g, 4.60 mmol )溶解于 25 mL N, N-二甲基甲酰胺中, 加入钠氢 (1.92 g, 6.90 mmol ), 搅拌反应 30分 钟。 向反应液中加入溴乙酸乙酯 (220 mg, 9.20 mmol ), 搅拌反应 12小时。 向反应液中加入 50 mL水, 用乙酸乙酯萃取 50 mLx3;>, 合并有机相, 依次 用水 (50 mLx2;>、饱和氯化钠溶液洗涤 50 mLx2;>,用无水硫酸钠干燥,过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题 产物 2-(3-(4-溴苯基 )-2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 Id (1.05 g, 白 色固体), 产率: 70.1%。
JH NM (400 MHz, CDC13): δ 7.54-7.48 (m, 4H), 6.59 (d, 1H), 6.42 (d, 1H) 4.43 (s, 2H), 4.24 (q, 2H), 1.29 (t, 3H)
第四步
2-(3-(4-((2-((叔丁基二甲基硅)氧代)乙基)氨基)苯基) -2-氧代 -2,3-二氢
_1H-咪唑 -1-基)乙酸乙酯
将 2_(3-(4-溴苯基 )-2-氧代 -2,3-二氢 -1Η-咪唑 -1-基)乙酸乙酯 Id (1.05 g, 3.23 mol ), 2- ((叔丁基二甲基硅)氧代)乙胺 (844 mg, 4.82 mmol )溶解于 15 mL甲苯中, 加入碳酸铯 (3.10 g, 9.60 mmol ), 三 (二苯亚苄基丙酮)二钯 (100 mg, 0.10 mmol )和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽(100 mg, 0.17 mmol ), 100°C搅拌反应 12小时。 反应液冷却至室温, 向反应液中加入 50 mL水, 用二氯甲烷萃取30 mLx3;), 合并有机相, 依次用水 (30 mLx2;)、 饱和氯化钠 溶液洗涤 (30 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱 色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(3-(4-((2- ((叔丁基 二甲基硅)氧代)乙基)氨基)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 le (210 mg, 黄色油状物), 产率: 15.5%。
JH NM (400 MHz, CDC13): δ 7.37-7.33 (m, 2Η), 6.69-6.65 (m, 2H), 6.53 (d, 1H), 6.38 (d, 1H), 4.47 (s, 2H), 4.26 (q, 2H), 3.84 (t, 2H), 3.70 (t, 1H), 3.38 (t, 1H), 1.32 (t, 3H), 0.93 (s, 9H), 0.09 (s, 6H)
第五步
2-(3_(4_(N_(2 叔丁基二甲基硅)氧)乙基) _4,6_二氯嘧啶 _5_酰胺)苯基 )-2- 氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯
将 2-(3-(4-((2- ((叔丁基二甲基硅)氧代)乙基)氨基)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 le (210 mg, 50 umol )溶解于 5 mL二氯甲烷中, 加 入三乙胺 (216 uL, 1.50 mmol )和 4,6-二氯 -5-酰氯嘧啶 (316 mg, 1.50 mmol ), 搅拌反应 12小时。 向反应液中加入 20 mL水, 用二氯甲烷萃取 (20 mLx3), 合并有机相, 依次用水 (20 mLx2;)、 饱和氯化钠溶液洗涤20 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(N-(2- ;叔丁 基二甲基硅:)氧)乙基: )-4,6-二氯嘧啶 -5-酰胺:)苯基: )-2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 If (297 mg,黄色固体),产物不经纯化直接进行下一步反应。
第六步
2-(3-(4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 -2,3-二氢 -1H-咪
唑 -1-基)乙酸乙酯
将粗品 2-(3-(4-(N-(2- ((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰胺) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 If (297 mg, 50 umol )溶解于 10 mL乙醇中, 加入浓盐酸 (0.3 mL:), 搅拌反应 1小时。 向反应液中加入 10 mL水, 用二氯甲烷萃取 IO mLx3;>, 合并有机相, 依次用水 (10 mLx2;>、 饱 和氯化钠溶液洗涤 IO mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 2-(3-(4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 lg (240 mg, 黄色固体), 产物不经纯化直 接进行下一步反应。
第七步
2-(3-(4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2- 氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯
将粗品 2-(3-(4-(4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺)苯基) -2-氧代 -2,3-二 氢 -1H-咪唑 -1-基)乙酸乙酯 lg (240 mg, 50 umol )溶解于 5 mL乙腈中, 加入 三乙胺 (216 uL, 1.50 mmol ), 80°C搅拌反应 12小时。 反应液冷却至室温, 用二氯甲烷萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2)、 饱和食盐水 洗涤 (30 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层色谱法 以展开剂体系 C纯化所得残余物, 得到标题产物 2-(3-(4-(4-氯 -5-氧代 -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙 酸乙酯 lh (40 mg, 黄色固体), 产率: 18.0%。
JH NM (400 MHz, CDC13): δ 8.76 (s, 1Η), 7.73-7.70 (m, 2H), 7.46-7.42 (m, 2H), 6.63 (d, 1H), 6.44 (d, 1H), 4.77-4.74 (m, 2H), 4.45 (s, 2H), 4.25 (q, 2H), 4.05 (t, 2H), 1.30 (t, 3H)
第八步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯
将粗品 2-(3-(4-(4-氯 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 lh (40 mg, 90 umol )溶解于
10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。反应液减压浓 缩, 加入 30 mL水, 用二氯甲烷萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2), 饱和食盐水洗涤30 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓 缩,得到粗品标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -6(5H)-基)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 li (38 mg, 黄色 固体), 产物不经纯化直接进行下一步反应。
第九步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸
将粗品 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基)苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸乙酯 li (38 mg, 89 umol )溶解于 8 mLl,4-二氧六环和水 (V/V = 3:1)混合溶液中, 加入氢氧化锂 (11 mg, 270 umol ), 搅拌反应 12小时。滴加 1 M盐酸至反应液 pH为 5〜6, 过滤得到标 题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸 l (22 mg, 白色固体),产率: 61.9%。
MS m/z (ESI): 397.2 [M+l]
JH NM (400 MHz, DMSO-d6): δ 8.15 (s, 1H), 7.76-7.73 (m, 2H), 7.61 (br.s, 2H), 7.45-7.41 (m, 2H), 7.06 (d, 1H), 6.75 (d, 1H), 4.60-4.58 (m, 2H), 4.33 (s, 2H), 3.98-3.96 (m, 2H) 参照实施例 1-10的制备方法还合成了以下化合物:






实施例 32
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯
-1-基) -2-甲基丙酸

2-(4-溴 -3,5_二甲基吡唑)乙酸乙酯
冰浴下, 将 4-溴 -3, 5-二甲基吡唑 la (500 mg, 2.8 mmol)溶解于 8 mL Ν,Ν- 二甲基甲酰胺中, 缓慢加入 60%的钠氢 (137 mg, 5.7 mmol), 搅拌一小 时后, 加入碘化钾 (450 mg, 2.7 mmol)和溴乙酸乙酯 (524 mg, 3.1 mmol)。 加热 至 80 °C, 搅拌 12小时。 待反应温度降至室温, 向反应液中滴加 20 mL的饱 和氯化铵溶液, 用乙酸乙酯萃取20 mLx3), 合并有机相, 用饱和氯化钠溶 液洗涤 (20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱 法以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(4-溴 -3,5-二甲基吡唑) 乙酸乙酯 lc ( 552 mg, 浅黄色液体), 产率: 74.0%。
MS m/z (ESI): 263 [M+l]
JH NM (400 MHz, CDC") δ 4.78 (s, 2H), 4.23 (q, 2 H), 2.21 (s, 6H), 1.28 (t, 3H)
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) - 苯) -3,5二甲基 -1H -吡唑 -1-基)乙酸
室温下, 将 2-(4-溴 -3,5-二甲基吡唑)乙酸乙酯 lc (55 mg, 0.21 mmol), 4-氨基 -6-(4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮 ld (80 mg, 0.21mmol, 采用公知的方法"专利 WO2011121350"制备而得), 碳酸钾 (57 mg, 0.42 mmol)和 [Ι,Γ-双 (二苯基膦) 二茂铁]二氯化钯 (lO mg)溶于 4 mLl,4-二氧六环中, 力 B入 1.5 mL水, 在 80°C 条件下, 反应 12小时。待反应温度降至室温, 反应液过滤, 滤液减压浓缩, 用制备高效液相色谱分离方法得到标题产物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -苯) -3,5二甲基 吡唑 -1-基)乙酸 1 ( 17 mg, 黄色固体), 产率: 20.4%。
MS m/z (ESI): 409.2 [M+l]
JH NM (400 MHz, CD3OD) δ 8.39 (s, 1H), 7.45 (m, 4H), 5.21 (s, 2H), 4.98 (m, 2H), 4.26 (m, 2H), 2.37 (s, 3H), 2.34 (s, 3H)
实施例 33
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 吡唑 -1-基) -2-甲基丙酸




N-(2-((叔丁基二甲基硅基)氧)乙基) -3-氯 -4-碘苯胺
冰浴下, 将 3-氯 -4-碘苯胺 la (3.0 g, 11.84 mmol)溶于无水四氢呋喃 (18 mL), 缓慢滴入 60%的钠氢 (1.16 g, 29.06 mmol), 室温搅拌一小时后, 加入 lb (3.48 g, 14.53 mmol), 在 40°C搅拌 12小时。 待反应冷却至室温, 缓慢 滴加氯化铵饱和溶液20 mL)和水 (50 mL)淬灭过量的钠氢。 反应液用乙酸乙 酯 (40 mLx3)萃取, 合并有机相, 用饱和氯化钠溶液 (30 mLx2)洗涤, 无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化所得残 余物,得到标题产物 Ν-Ρ- :叔丁基二甲基硅基:)氧:)乙基: )-3-氯 -4-碘苯胺 lc (3.5 g, 黄色固体), 产率: 58.5%。
MS m/z (ESI):412.0 [M+l]
JH NM (400 MHz, CDC13) δ 7.51 (d, 1H), 6.74 (d, 1H), 6.27 (dd, 1H),
4.12 (t, 2H), 3.17 (t, 2H), 0.90 (s, 9H), 0.07 (s, 6H)
第二步
N-(2-((叔丁基二甲基硅基:)氧:)乙基: )-4,6-二氯 -N-(3-氯 -4-碘苯:)嘧啶 -5-酰 胺
将 N-(2-((叔丁基二甲基硅基)氧:)乙基) -3-氯 -4-碘苯胺 lc (2.9 g, 7.03 mmol)溶于甲基四氢呋喃 (30 mL),加入 4,6-二氯 -5-嘧啶甲酰氯 Id (1.7 g, 8.06 mmol)和 Ν,Ν-二异丙基乙胺 (1.6 mL), 搅拌反应 12小时。 反应液用乙酸乙酯 (50 mL)和水 (50 mL)稀释,合并有机相,用饱和氯化钠溶液 (100 mLx2)洗涤, 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化 所得残余物, 得到标题产物 N-(2 叔丁基二甲基硅基:)氧)乙基: )-4,6-二氯 -N-P-氯 -4-碘苯)嘧啶 -5-酰胺 le (3.4 g, 黄色油状物), 产率 82.3 %
JH NM (400 MHz, CDC13) δ 8.89 (s, 1H), 7.92 (d, 1H), 6.65 (d, 1H), 7.07 (dd, 1H), 3.99 (t, 2H), 3.78 (t, 2H), 0.83 (s, 9H), 0.00 (s, 6H)
第三步
4,6-二氯 -N-P-氯 -4-碘苯) -N-(2-羟乙基)嘧啶 -5-酰胺
将 N-(2- ;叔丁基二甲基硅基:)氧:)乙基: )-4,6-二氯 -N-P-氯 -4-碘苯:)嘧啶 -5- 酰胺 le (3.4 g, 5.79 mmol)溶于 40 mL的乙醇和 1.2 mL盐酸的混合溶液, 搅 拌 3小时。 反应液用 50 mL水稀释, 用乙酸乙酯 (50 mLx3)萃取, 合并有机 相, 用饱和氯化钠溶液 lOO mL )洗涤, 无水硫酸钠干燥, 过滤, 滤液减压 浓缩得到标题产物 4,6-二氯 -N-P-氯 -4-碘苯:) -N-(2-羟乙基:)嘧啶 -5-酰胺 If (2.9 g, 浅黄色液体), 产率 105.8%。
JH NMR (400 MHz, CDC13) δ 8.67 (s, 1H), 7.76 (d, 1H), 7.56 (d, 1H), 7.03 (dd, 1H), 4.06 (t, 2H), 3.93 (t, 2H)
第四步
4-氯 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 室温下,将 4,6-二氯 -N-P-氯 -4-碘苯) -N-(2-羟乙基:)嘧啶 -5-酰胺 If (2.9 g,
6.14 mmol), 溶于 30 mL乙腈, 加入三乙胺 (2.4 mL), 在 80°C搅拌反应 12 小时。 反应液冷却至室温后, 加入 50 mL水稀释。 用二氯甲烷 50 mLx3;)萃 取, 合并有机相, 用饱和氯化钠溶液 (100 mLx2)洗涤, 无水硫酸钠干燥, 过 滤, 滤液减压浓缩得到较纯标题产物 4-氯 -6-P-氯 -4-碘苯: )-7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lg (l g, 浅黄色固体), 产率: 37.3%和粗品 (1.2 g,黄色液体)
第五步
4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将 4-氯 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lg (1 g, 2.29 mmol), 溶于 0.5摩尔氨气的 1,4-二氧六环溶液 (80 mL), 搅拌反应 12小时。将反应液过滤得到较纯标题产物 4-氨基 -6-P-氯 -4-碘苯: )-7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lh (300 mg, 黄色固体), 产率: 31.4%。
JH NM (400 MHz, DMS0- ) δ 8.14 (s, 1Η), 7.96 (d, 1H), 7.69 (d, 1H), 7.12 (dd, 1H), 4.57 (t, 2H), 3.96 (t, 2H)
第六步
2-(4-溴 吡唑 -1-基) -2-甲基丙酸乙酯
将 4-溴 吡唑 li (2 g, 13.7 mmol)溶解于 40 mL N,N-二甲基甲酰胺中, 加入钠氢 (657 mg, 16.4 mmol)和 2-溴 -2-甲基丙酸乙酯 (3.2 g, 16.4 mmol), 室 温搅拌反应 12小时。向反应液中加入 200 mL水,用乙酸乙酯萃取 (80 mLx3), 合并有机相, 用水溶液 100 mLx2;)、 饱和氯化钠溶液洗涤 100 mLx2;), 无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 2-(4-溴 吡唑 -1-基) -2-甲基丙酸乙酯 lj (3.4 g, 浅黄色液体), 产率: 95.5%。
JH NMR (400 MHz, CDC13) δ 7.58 (s, 1H), 7.50 (s, 1H), 4.17 (q, 2H), 1.83 (s, 6H), 1.22 (t, 3H)
第七步
2-甲基 -2-(4-(4,4,5,5-四甲基 -1,3,2-二氧杂 -2-基) 吡唑 -1-基)丙酸乙酯 将 2-(4-溴 吡唑 -1-基) -2-甲基丙酸乙酯 lj (3.4 g, 13 mmol)溶解于 50 mL N,N-二甲基甲酰胺中, 加入4,4,4',4',5,5,5',5'-八甲基-2,2'-双(1,3,2-二氧杂) (4 g, 15.6 mmol), 乙酸钾 (3.8 g, 39 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (1 g, 1.3 mmol), 90°C搅拌反应 12小时。 向反应液中加入 200 mL水, 用乙酸 乙酯萃取 (80 mLx3;), 合并有机相, 用水溶液 100 mLx2;)、饱和氯化钠溶液洗 涤 (100 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以 洗脱剂体系 C 纯化所得残余物, 得到标题产物 2-甲基 -2-(4-(4,4,5,5-四甲基 -1,3,2-二氧杂 -2-基:) 吡唑 -1-基:)丙酸乙酯 lk (1.7 g, 浅黄色液体 产率: 42.5%。
JH NM (400 MHz, CDC13) δ 7.87 (s, 1H), 7.86 (s, 1H), 4.14 (q, 2H), 1.83 (s, 6H), 1.30 (s, 12H), 1.18 (t, 3H)
第八步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) - 1 H-吡唑 - 1 -基) -2-甲基丙酸乙酯
将 4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lh (80 mg, 192 umol)和 2-甲基 -2-(4-(4,4,5,5-四甲基 -l,3,2-二杂氧戊硼烷-2- 基)-lH-吡唑-l-基)丙酸乙酯 lk (59 mg, 192 umol)溶解于 3 mLl,4-二氧六环 和水 (V/V = 2: l)混合溶液中, 加入碳酸钾 (80 mg, 577 umol)和 Ι,Γ-双 (二苯基 磷:)二茂铁氯化钯 (6 mg, 7.2 umol), 90°C搅拌反应 12小时。 反应液冷却至 室温后直接用于下一步反应。
第九步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) 吡唑 -1-基) -2-甲基丙酸
在上一步反应液中加入氢氧化锂 (30 mg, 63 umol), 搅拌反应 3小时。 减压浓缩, 用制备高效液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标
题产物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯苯) 吡唑 -1-基) -2-甲基丙酸 l (1.69 mg, 白色固体), 产率: 2.0%。
MS m/z (ESI): 443.1 [M+l]
JH NM (400 MHz, CD3OD) δ 8.45 (s, 1H), 8.26 (s, 1H), 7.95 (s, 1H), 7.70 (d, 1H), 7.60 (d, 1H), 7.36 (dd, 1H), 5.02 (t, 2H), 4.28 (t, 2H), 1.91 (s, 6H)
实施例 34
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) 吡唑 -1-基)丙酸

2-(4-溴 吡唑 -1-基:)丙酸乙酯
冰浴下, 将 4-溴 吡唑 la (2 g, 13.7 mmol)溶解于 40 mL N,N-二甲基 甲酰胺中, 缓慢加入 60%的钠氢 (657 mg, 16.4 mmol), 搅拌一小时后, 加入 2-溴丙酸乙酯 p.O g, 16.4 mmol)。反应搅拌 12小时。 向反应液中滴加 200 mL 的饱和氯化铵溶液, 用乙酸乙酯萃取 80 mLx3), 合并有机相, 用饱和氯化 钠溶液洗涤 (50 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱
色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 2-(4-溴 -1H-吡唑 -1- 基)丙酸乙酯 lb (1.4 g, 浅黄色液体), 产率: 42 %。
JH NM (400 MHz, CDC13) δ 7.56 (s, 1H), 7.48 (s, 1H), 5.03 (q, 1H), 4.19 (q, 2H), 1.76 (d, 3H), 1.25 (t, 3H)
第二步
2-(4-(4,4,5,5-四甲基 -1,3,2-二氧硼戊环 -2-基) -1H-吡唑 -1-基)丙酸乙酯 室温下,将 2-(4-溴 -1H-吡唑 -1-基)丙酸乙酯 lb (50 mg, 0.18 mmol)溶解于 2 mL二氧六环中,加入双联噸哪醇硼酸酯 (68 mg, 0.27 mmol),醋酸钾 (53 mg, 0.54 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (7.3 mg, 0.009 mmol), 110°C下 搅拌反应 12小时。 待反应温度降至室温, 减压浓缩, 向剩余物中加 10 mL 水, 用二氯甲烷萃取 (20 mLx3), 合并有机相, 无水硫酸钠干燥, 过滤, 滤 液减压浓缩, 用薄层层析以洗脱剂体系 C 纯化所得残余物, 得到标题产物 2-(4-(4,4,5,5-四甲基 -1,3,2-二氧硼戊环 -2-基) 吡唑 -1-基)丙酸乙酯 lc (90 mg, 白色固体), 产率: 100.0%。
第三步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -1H-吡唑 -1-基) 丙酸
将 4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (80 mg, 192 umol)和 2-(4-(4,4,5,5-四甲基 -1,3,2-二氧硼戊环 -2-基) 吡唑 -1-基) 丙酸乙酯 lc(59 mg, 192 umol)溶解于 3 mLl,4-二氧六环和水 (V/V = 2: 1)混合 溶液中, 加入碳酸钾 (80 mg, 577 umol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (6 mg, 7.2 umol), 90°C搅拌反应 12小时。 反应液冷却至室温, 减压浓缩, 用 制备高效液相色谱法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -6(5H)-基 )-2-氯 苯) _1H -吡唑 -1-基) 丙酸 (20 mg, 黄色固体), 产率: 25.0%。
MS m/z (ESI): 443.1 [M+l]
Ή NMR (400 MHz, CD3OD) δ 8.47 (s, IH), 8.30 (br.s, IH), 8.04 (br.s, 1H): 7.72 (d, IH), 7.63 (d, IH), 7.40 (dd, IH), 5.28 (br.s, IH), 5.04 (t, 2H), 4.30 (t 2H), 1.86 (d, 3H)
实施例 35
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯
-1-基)丙酸

1 第一步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) 吡唑 -1-基)丙酸乙酯
将 4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 la (80 mg, 192 umol)和 3-(4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) 吡唑 -1-基)丙酸乙酯 (56 mg, 192 umol)溶解于 3 mLl,4-二氧六环和水 (V/V = 2: 1) 混合溶液中, 加入碳酸钾 (80 mg, 577 umol)和 Ι,Γ-双 (二苯基磷:)二茂铁氯化 钯 (6 mg, 7.2 umol), 90°C搅拌反应 12小时。 反应液冷却至室温后直接用于
第二步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) 吡唑 -1-基)丙酸
在上一步反应液中加入氢氧化锂 (30 mg, 63 umol) , 搅拌反应 3小时。 减压浓缩, 用制备高效液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标 题产物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2- 氯苯) -1H-吡唑 -1-基)丙酸 l (l l mg, 白色固体), 产率: 14.3%。
MS m/z (ESI): 429.1 [M+l]
JH NMR (400 MHz, DMSO-^) 5 8.70 (br. s, 1H), 8.60 (br. s, 1H), 8.43 (d, 1H), 8.24 (s, 1H), 7.90 (s, 1H), 7.66 (d, 2H), 7.40 (dd, 1H), 7.36 (dd, 1H), 4.83 (s, 2H), 4.38 (t, 2H), 4.15 (s, 2H), 2.85 (t, 2H)
实施例 36
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-基) -2-氯

将 4-溴 -3-甲基 吡唑 la (500 mg, 3.1 mmol)溶解于 8 mL Ν,Ν-二甲基 甲酰胺中,加入 60%钠氢 (149 mg, 6.2 mmol)和 2-溴 -2-甲基乙酸乙酯 (570 mg, 3.4 mmol)和碘化钾 (489 mg, 2.9 mmol), 80°C搅拌反应 12小时。 向反应液 中加入 20 mL 水, 用乙酸乙酯萃取 (20 mLx3), 合并有机相, 用水溶液 (20 mLx2), 饱和氯化钠溶液洗涤20 mLx2;), 无水硫酸钠干燥, 过滤, 滤液减压 浓缩,用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物,得到标题产物 2-(4- 溴 -3-甲基 吡唑 -1-基) 乙酸乙酯 lb (522 mg,浅黄色液体),产率: 68.0%。
JH NM (400 MHz, CDC13) δ 7.44 (d, 1 H), 4.85 (s, 1H), 4.79 (s, 1H), 4.24 (q, 2 H), 2.23 (s, 3H), 1.29 (t, 3 H)
第二步
2·(3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 -1-基)乙 酸乙酯
将 2-(4-溴 -3-甲基 吡唑 -1-基)乙酸乙酯 lb (300 mg, 1.21 mmol)溶解于 10 mL N,N-二甲基甲酰胺中, 加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'-双 (1,3,2-二氧 杂) (369 mg, 1.46 mmol), 乙酸钾(178 g, 1.82 mmol)和 Ι,Γ-双 (二苯基磷)二茂 铁氯化钯 (10 mg, 130 umol), 90°C搅拌反应 12小时。 向反应液中加入 20 mL 水, 用乙酸乙酯萃取 (20 mLx3;), 合并有机相, 用水溶液 (20 mLx2;)、 饱和氯 化钠溶液洗涤 (20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶 柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 2-(3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) 吡唑 -1-基)乙酸乙酯 lc (160 mg, 浅黄色液体), 产率: 44.8%。
第三步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -3-甲基 吡唑 -1-基)乙酸乙酯
将 4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
(100 mg, 240 umol)和 2-(3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2- 基) 吡唑 -1-基)乙酸乙酯 (85 mg, 288 umol)溶解于 3 mLl,4-二氧六环和水 (V/V = 2:l)混合溶液中, 加入碳酸钾 (lOO mg, 821 umol)和 Ι,Γ-双 (二苯基磷) 二茂铁氯化钯 (12 mg, 15 umol), 90°C搅拌反应 12小时。反应液冷却至室温 后直接用于下一步反应。
第四步
2- (4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -3-甲基 吡唑 -1-基)乙酸
在上一步反应液中加入氢氧化锂 (30 mg, 63 umol), 搅拌反应 3小时。 减压浓缩, 用高效液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产 物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -3-甲基 吡唑 -1-基)乙酸 l (5 mg, 白色固体), 产率: 5.6%。
MS m/z (ESI): 429.0 [M+l]
JH NM (400 MHz, CD3OD) δ 8.41 (s, 1H), 7.82 (s, 0.4H), 7.62-7.60 (m, 1.5H), 7.48-7.42 (m, 1H), 7.38-7.35 (m, 1H), 5.00 (t, 4H), 4.26 (t, 2H), 2.23 (d, 3H)
实施例 37
3- (4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟
-1-基)丙酸


3-(4-溴 吡唑 -1-基:)丙酸乙酯
将 4-溴 吡唑 la (1.47 g, 10.00 mmol)溶解于 20 mL N, N-二甲基甲 酰胺中, 冰浴下加入钠氢 (480 mg, 12.00 mmol), 搅拌反应一个小时。 向反 应液中加入溴丙酸乙酯 (2.00 g, 11.05 mmol)和碘化钾 (1.74 g, 10.48 mmol), 70°C下搅拌反应 12小时。 向反应液中加入 100 mL饱和氯化铵溶液, 用乙 酸乙酯萃取 (100 mLx3;), 合并有机相, 依次用水 (100 mLx2;>、 饱和氯化钠溶 液洗涤 (100 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标 题产物 3-(4-溴 吡唑 -1-基)丙酸乙酯 lb (2.2 g,黄色液体),产率: 76.9%。
JH NM (400 MHz, CDC13) δ 7.46 (d, 2H), 4.39 (t, 2H), 4.14 (q, 2H), 2.86 (t, 2H), 1.24 (t, 3H)
第二步
3-(4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) 吡唑 -1-基)丙酸乙酯
将 3-(4-溴 吡唑 -1-基)丙酸乙酯 lb (1 .80 g, 7.28 mmol)溶解于 20 mL N, N-二甲基甲酰胺中, 加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'-二 (1 ,3,2-二杂氧 戊硼烷)(2.22 g, 8.74 mmol), 醋酸钾 (2.14 g, 21.84 mmol)和 Ι ,Γ-双 (二苯基磷) 二茂铁氯化钯 (297 mg, 0.36 mmol) , 80°C下搅拌反应 12小时。反应液冷却至 室温, 向反应液中加入 50 mL水,用乙酸乙酯萃取40 mLx3;),合并有机相, 用饱和氯化钠溶液洗涤30 mLx2;), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 3-(4-(4,4,5,5-四甲基 -1 ,3,2-二杂氧戊硼 -2-基) -1H-吡唑 -1-基)丙酸乙酯 lc (1.2 g, 无色液体:), 产率: 63.8%。
JH NM (400 MHz, CDC13) δ 7.75 (d, 2Η), 4.42 (t, 2H), 4.13 (q, 2H), 2.89 (t, 2H), 1 .31 (s, 12H), 1.26 (t, 3H)。
第三步
2-((4-溴 -3-氟苯)胺基)乙醇
将 4-溴 -3-氟苯胺 Id (2.00 g, 10.53 mmol)溶解于 20 mL N, N-二甲基甲 酰胺中, 加入碳酸钾 (7.26 g, 52.63 mmol), 100°C搅拌反应 12小时。 反应液 冷却至室温, 向反应液中加入 20 mL水, 用乙酸乙酯萃取 (20 mLx3;>, 合并 有机相, 依次用水 (100 mLx2)、 饱和氯化钠溶液洗涤 100 mLx2;>, 用无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得 残余物, 得到粗品标题产物 2-((4-溴 -3-氟苯)胺基)乙醇 le (630 mg,黄色油状 物), 产率: 25.6%。
JH NM (400 MHz, DMSO-^) δ 7.44-7.43 (m, 1H), 7.36-7.33 (m, 1H), 7.14 (d, 1H), 6.70 (br. s, 1H), 3.69-3.65 (m, 2H), 3.40-3.36 (m, 2H), 2.17 (s, 3H) 第四步
4-溴 -N-(2((叔丁基二甲基硅基)氧)乙基) -3-氟苯胺
将粗品 2-((4-溴 -3-氟苯)胺基)乙醇 le (630 mg, 2.96 mmol)溶解于 15 mL 二氯甲烷中, 加入叔丁基二甲基氯硅烷 (470 mg, 11.76 mmol) , 搅拌反应 30
分钟。 向反应液中加入溴乙酸乙酯 (1.96 g, 11.76 mmol), 室温搅拌反应 12 小时。向反应液中加入 50 mL饱和氯化铵溶液,用乙酸乙酯萃取 50 mLx3), 合并有机相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤50 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 4-溴 -N-(2((叔丁基二甲基硅)氧)乙基) -3-氟苯胺 If (530 mg, 无色液体), 产率: 49.6%。
JH NM (400 MHz, CDC13) δ 7.24-7.22 (m, IH), 6.40-6.37 (m, IH), 6.31-6.28 (m, IH), 4.19 {br. s, IH), 3.80 (t, 2H), 3.17 (t, 2H), 0.90 (s, 9H), 0.07 (s, 6H)。
第五步
N-(4-溴 -3-氟苯基) -N-(2-((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧啶 -5-酰 胺
将 4-溴 -N-(2((叔丁基二甲基硅)氧)乙基) -3-氟苯胺 If (510 mg, 1.41 mmol)溶解于 10 mL二氯甲烷中, 加入三乙胺 (445 mg, 4.41 mmol)和 4,6-二 氯 -5-酰氯嘧啶 (463 mg, 2.20 mmol), 搅拌反应 12小时。 向反应液中加入 10 mL水, 用二氯甲烷萃取 IO mLx3;>, 合并有机相, 依次用水 (10 mLx2;>、 饱 和氯化钠溶液洗涤 IO mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 N-(4-溴 -3-氟苯基) -N-(2-((叔丁基二甲基硅)氧)乙基) -4,6- 二氯嘧啶 -5-酰胺 lg (730 mg, 黄色固体), 产物不经纯化直接进行下一步反 应。
JH NM (400 MHz, CDC13) δ 8.86 (s, IH), 7.37-7.33 (m, IH), 7.27-7.19 (m, IH), 7.04-7.01 (m, IH), 3.94-3.91 (m, 2H), 3.87-3.85 (m, 2H), 0.81 (s, 9H), 0.00 (s, 6H)
第六步
N-(4-溴 -3-氟苯基 )-4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺
将粗品 N-(4-溴 -3-氟苯) -N-(2-((叔丁基二甲基硅)氧)乙基) -4,6-二氯嘧
啶 -5-酰胺 lg (730 mg, 1.39 mmol)溶解于 10 mL乙醇中,加入浓盐酸 (0.3 mL), 搅拌反应 1小时。 向反应液中加入 10 mL水, 用乙酸乙酯萃取 (10 mLx3;>, 合并有机相, 依次用水 (10 mLx2;>、 饱和碳酸氢钠溶液 (10 mLx2;>、 饱和氯化 钠溶液洗涤 (10 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗 品标题产物 N-(4-溴 -3-氟苯基 )-4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺 lh (560 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.65 (s, 1H), 7.48-7.44 (m, 1H), 7.29-7.24 (m, 1H), 7.13-7.10 (m, 1H), 4.06 (t, 2H), 3.93 (t, 2H)
第七步
6-(4-溴 -3-氟苯基 )-4-氯 -7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将粗品 N-(4-溴 -3-氟苯基 )-4,6-二氯 -N-(2-羟乙基)嘧啶 -5-酰胺 lh (560 mg, 1.37 mmol)溶解于 10 mL乙腈中, 加入三乙胺 (417 mg, 4.13 mmol), 80°C搅 拌反应 12小时。 反应液冷却至室温, 向反应液中加入 20 mL水, 用乙酸乙 酯萃取 (20 mLx3;>, 合并有机相, 用饱和氯化钠溶液洗涤20 mLx2;), 无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 6-(4-溴 -3-氟苯基 )-4-氯 -7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 li (480 mg, 黄色固体), 产物不 经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.79 (s, 1H), 7.67-7.63 (m, 1H), 7.27-7.24 (m, 1H), 7.11-7.08 (m, 1H), 4.76 (t, 2H), 4.06 (t, 2H)
第八步
4-氨基 -6-(4-溴 -3-氟苯基 )-7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将粗品 6-(4-溴 -3-氟苯基 )-4-氯 -7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)- 酮 li (480 mg, 1.29 mmol)溶解于 10 mL 0.5 M氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。 反应液减压浓缩, 加入 20 mL水, 用乙酸乙酯萃取 (20 mLx3),合并有机相,依次用水 (20 mLx2)、饱和氯化钠溶液洗涤 (20 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 4-氨基 -6-(4-
溴 -3-氟苯基 )-7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lj (340 mg, 黄色固 体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, DMSO-^) δ 8.18 (s, 1H), 7.77 (d, 1H), 7.64 (br, 2H), 7.58 (dd, 1H), 7.26 (dd, 1H), 4.62 (t, 2H), 4.01 (t, 2H)
第九步
3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟 苯) 吡唑 -1-基)丙酸乙酯
将 4-氨基 -6-(4-溴 -3-氟苯基 )-7,8-二氯嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lj (80 mg, 0.23 mmol)溶解于 5 mLl,4-二氧六环和水 (V/V=4: l)混合溶剂中, 加入 3-(4- (噸哪醇硼酸酯 -2-基) 吡唑 -1-基)丙酸乙酯 lc (64 mg, 0.23 mmol), 碳酸铯 (222 mg, 0.68 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (19 mg, 0.02 mmol), 微波 100°C下搅拌反应 0.5小时。 反应液冷却至室温, 过滤, 反应液 直接用于下一步。
第十步
3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟 苯) 吡唑 -1-基)丙酸
将粗品 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) 吡唑 -1-基)丙酸乙酯 lk (90 mg, 0.20 mmol)溶解于 8 mLl,4- 二氧六环和水 (V/V = 3: l)混合溶液中, 加入氢氧化锂 (34 mg, 0.82 mmol), 搅 拌反应 12小时。 滴加 1 M盐酸至反应液 pH为 5〜6, 减压浓缩, 残留物用 制备高效液相色谱纯化得到标题产物 -(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氟苯) 吡唑 -1-基)丙酸 1 (27 mg, 白色固 体), 产率: 33.2%。
MS m/z (ESI): 413.1 [M+l]
JH NM (400 MHz, DMSO-^) δ 8.55-8.43 (m, 3H), 8.16 (d, 1H), 7.92 (d, 1H), 7.74 (d, 1H), 7.41 (dd, 1H), 7.25 (dd, 1H), 4.77-4.75 (m, 2H), 4.34 (t, 2H),
4.10 (t, 2H), 2.81 (t, 2H)
实施例 38
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯基) -3,5-二甲基 吡唑 -1-基)丙酸

1
第一步
3-(3,5-二甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 -1-基) 丙酸乙酯
冰浴下, 将 3,5-二甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H- 吡唑 la (100 mg, 0.45 mmol)溶解于 1 mL Ν,Ν- 二甲基甲酰胺中, 加入碳酸 铯 (293 mg, 0.90 mmol)和溴丙酸乙酯 (151 mg, 0.90 mmol), 反应搅拌 12小 时。 向反应液中滴加 10 mL水, 用乙酸乙酯萃取 (10 mLx3;>, 合并有机相, 用饱和氯化钠溶液洗涤 10 mLx2;>, 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到标题产物 3-(3,5-二甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H- 吡唑 -1-基)丙酸乙酯 lc (200 mg, 浅黄色油状物)。
MS m/z (ESI): 309.1 [M+l]
第二步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯基) -3,5-二甲基 吡唑 -1-基)丙酸乙酯
室温下, 将 3-(3,5-二甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2- 基) -1H-吡唑 -1-基)丙酸乙酯 lc (63 mg, 0.19 mmol), 4-氨基 -6-(3-氯 -4-碘苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 ld (80 mg, 0.19 mmol), 碳酸 钾 (53.08 mg, 0.38 mmol)和 [Ι,Γ-双 (二苯基膦)二茂铁]二氯化钯 (5 mg)溶于 2 mLl,4-二氧六环中, 加入 0.6 mL水, 微波 100°C下搅拌反应 0.5小时。 反应 液冷却至室温, 过滤, 反应液直接用于下一步。
第三步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯基) -3,5-二甲基 吡唑 -1-基)丙酸
在上一步反应液中加入氢氧化锂 (30 mg, 63 umol), 搅拌反应 3小时。 减压浓缩, 用高效液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产 物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) -3-甲基 吡唑 -1-基)乙酸 l (12 mg, 白色固体), 产率: 42.3%。
MS m/z (ESI): 457.0 [M+l]
JH NM (400 MHz, CD3OD) δ 8.18 (s, 1H), 7.66-7.68 (m, 3H), 7.41 (dd, 1H), 7.32 (d, 1H), 4.64 (t, 2H), 4.19 (t, 2H), 4.05 (t, 2H), 2.80 (t, 2H), 2.12 (s, 3H), 2.01 (s, 3H)
实施例 39
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -3- (三氟甲基) 吡唑 -1-基)乙酸

2-(4-溴 -3- (三氟甲基) 吡唑 -1-基)乙酸乙酯
冰浴下,将 4-溴 -3-三氟甲基吡唑 la (500 mg,2.33 mmol)溶解于 10 mL N, N-二甲基甲酰胺中, 加入钠氢 (140 mg, 3.49 mmol), 搅拌反应 30分钟。 向 反应液中加入溴乙酸乙酯 (582 mg, 3.49 mmol), 室温搅拌反应 12小时。 向 反应液中加入 30 mL水, 用乙酸乙酯萃取 (20 mLx3;>, 合并有机相, 依次用 水 (10 mLx2;>、 饱和氯化钠溶液洗涤 (10 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题 产物 2-(4-溴 -3- (三氟甲基) 吡唑 -1-基)乙酸乙酯 lb (450 mg, 白色固体), 产率: 64.3%。
JH NM (400 MHz, CDC13) δ 7.60 (s, 1H), 4.90 (s, 2H), 4.25 (q, 2H), 1.29 (t, 3H)
第二步
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基) -3- (三氟甲基) 吡唑 -1-基)乙酸
将 2-(4-溴 -3- (三氟甲基) 吡唑 -1-基)乙酸乙酯 lb (50 mg, 170 umol) 和 4-氨基 -6-(4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基:)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (64 mg, 170 umol , 采用公知的方法"专利
WO2011121350"制备而得;)溶解于 5 mLl,4-二氧六环和水 (V/V = 4: 1)混合溶 液中,加入碳酸钾 (70 mg, 510 umol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (6.2 mg, 8.5 umol), 80°C反应 12小时。 反应液冷却至室温, 减压浓缩, 用制备高效 液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基) -3- (三氟甲基) 吡唑 -1-基)乙酸 l (16 mg, 白色固体), 产率: 21.5%。
MS m/z (ESI): 449.1 [M+l]
JH NM (400 MHz, DMSO-^) δ 8.20 (s, 1H), 8.18 (s, 1H), 7.71 {br. s, 2H), 7.47-7.41 (m, 4H), 5.10 (s, 2H), 4.62-4.60 (m, 2H), 4.03-4.01 (m, 2H)
实施例 40
4-氨基 -6-(3-氯 -4-(3-甲基 -l-(2,2,2-三氟乙基) 吡唑 -4-基)苯基) -7,8-二

4-溴 -3-甲基 -1-(2,2,2-三氟乙基) 吡唑
将 4-溴 -3-甲基吡唑 la (500 mg, 3.11 mmol)溶解于 10 mL N, N-二甲基甲
酰胺中,加入三氟甲磺酸三氟乙酯 (756 mg, 3.26 mmol)和碳酸铯 (2.02 g, 6.21 mmol), 微波 100°C搅拌反应 30分钟。 反应液冷却至室温, 向反应液中加入 30 mL水, 用乙酸乙酯萃取 (30 mLx3), 合并有机相, 依次用水 (30 mLx2)、 饱和氯化钠溶液洗涤30 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 4-溴 -3-甲 基 -1-(2,2,2-三氟乙基) -1H-吡唑 lb (516 mg, 白色固体), 产率: 68.4%。
JH NM (400 MHz, CDC13) δ 7.99 (s, 1H), 5.11-5.02 (m, 2H), 2.43 (s, 3H) 第二步
3·甲基 _4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -l-(2,2,2-三氟乙 基) -1H-吡唑
将 4-溴 -3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 lb (516 mg, 2.12 mmol)和 4,4,4',4',5,5,5',5'-八甲基 -2,2'-二 (1,3,2-二杂氧戊硼烷)(647 mg, 2.55 mmol)溶解 于 3 mLl, 4-二氧六环中, 加入乙酸钾 (625 mg, 6.37 mmol)和 Ι,Γ-双 (二苯基 磷:)二茂铁氯化钯 (86 mg, 106 umol), 90°C搅拌反应 12小时。 向反应液中加 入 20 mL水,用乙酸乙酯萃取 (20 mLx3),合并有机相,依次用水 (20 mL><2)、 饱和氯化钠溶液洗涤20 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1-(2,2,2-三氟乙基) -1H-吡唑 lc (200 mg, 白色固体), 产率: 32.5%。
第三步
4-氨基 -6-(3-氯 -4-(3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将 3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基 )-1-(2,2,2-三氟乙 基) -1H-吡唑 lc (41 mg, 144 umol)和 4-氨基 -6-(3-氯 -4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (60 mg, 144 umol)溶解于 3 mLl,4-二氧六环和 水 (V/V = 2: l)混合溶液中,加入碳酸钾 (60 mg, 432 umol)和 Ι,Γ-双 (二苯基磷)
二茂铁氯化钯6 mg, 7.2 umol),微波 90°C反应 30分钟。反应液冷却至室温, 减压浓缩, 用制备高效液相色谱法以洗脱剂体系 B纯化所得残余物, 得到标 题产物 4-氨基 -6-(3-氯 -4-(3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (28 mg, 白色固体), 产率: 43.5%。
MS m/z (ESI): 453.0 [M+l]
JH NM (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.97 (s, 1H), 7.71 (d, 1H), 7.48-7.41 (m, 2H), 5.18-5.07 (m, 2H), 4.75-4.73 (t, 2H), 4.12 (t, 2H), 2.27 (s, 3H)
实施例 41
4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -l-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯

4-溴 -3,5-二甲基苯胺
冰浴下,将 3,5-二甲基苯胺 la (80 g, 660 mmol)溶解于 800 mL乙腈中,
缓慢滴加 N-溴代丁二酰亚胺 (117 g, 660mmol)溶于 400 ml乙腈的溶液, 室 温搅拌 16小时后。 反应液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化 所得残余物,得到标题产物 4-溴 -3,5-二甲基苯胺 lb (90 g,黄色固体),产率: 68.2%
MS m/z (ESI): 200 [M+l]
第二步
4-溴 -N-(2- ((叔丁基二甲基)氧代)乙基) -3,5-二甲基苯胺
冰浴下, 将 4-溴 -3,5-二甲基苯胺 lb (100 g, 500 mmol)溶于无水四氢呋 喃 (1 L), 缓慢滴入 60%的钠氢 (40 g, 1000 mmol), 室温搅拌。 一小时后, 加 入 2-溴乙氧基-叔丁基二甲基硅 (179 g, 749 mmol), 在 40°C搅拌 16小时。 待 反应冷却至室温,缓慢滴加氯化铵饱和溶液50 mL)和水 (100 mL)淬灭过量的 钠氢。 反应液用乙酸乙酯 (500 mLx3)和水萃取 (500 mLx2), 合并有机相, 用 饱和氯化钠溶液洗涤300 mLx2;)洗涤, 无水硫酸钠干燥, 过滤, 滤液减压浓 缩, 硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 4-溴 -N-(2- ((叔丁基二甲基)氧代)乙基) -3,5-二甲基苯胺 lc (180 g, 黄色固体), 产 率: 100%
MS m/z (ESI):358 [M+l]
第三步
N-(4-溴 -3, 5-二甲基) -N-(2- ((叔丁基二甲基)氧代)乙基) -4,6-二氯 -5-嘧啶 甲酰胺
将 4-溴 -N-(2-((叔丁基二甲基)氧代)乙基) -3,5-二甲基苯胺 lc (180 g, 503 mmol)溶于无水二氯甲烷 (2 L),加入 4,6-二氯 -5-嘧啶甲酰氯 (138 g, 654 mmol) 和三乙胺 (210 mL),搅拌 16小时。反应液用二氯甲烷稀释 (1 Lx2)和水 (1 Lx2), 合并有机相, 用饱和氯化钠溶液洗涤 (300 mLx2)洗涤, 无水硫酸钠干燥, 过 滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标 题产物 N-(4-溴 -3, 5-二甲基) -N-(2- ((叔丁基二甲基)氧代)乙基) -4,6-二氯 -5-嘧
啶甲酰胺 ld(160 g, 黄色油状), 产率 59.7%
MS m/z (ESI):533.9 [M+l]
1H NM (400 MHz, CDC13): δ 8.61(s, 1H), 7.13 (s, 2H), 7.27 (s, 1H), 3.97 (t, 2H), 3.89 (t, 2H), 2.30 (s, 6H), 0.87 (s, 9H), 0.06 (s, 6H)
第四步
N-(4-溴 -3,5-二甲基) -N-(2- (乙羟基) -4,6-二氯 -5-嘧啶甲酰胺 将 N-(4-溴 -3, 5-二甲基) -N-(2- ((叔丁基二甲基)氧代)乙基) -4,6-二氯 -5-嘧 啶甲酰胺 ld(160 g, 299.6 mmol)溶于 1.5 L的乙醇和 45 mL盐酸的混合溶液, 搅拌 3小时。 反应液用乙酸乙酯 (500 mLx3)和水 (500 mLx2)萃取, 合并有机 相, 用用饱和氯化钠溶液洗涤300 mLx2;)洗涤, 无水硫酸钠干燥, 过滤, 滤 液减压浓缩得到标题产物 N-(4-溴 -3, 5-二甲基) -N-(2- (乙羟基) -4,6-二氯 -5-嘧 啶甲酰胺 le(125 g, 浅黄色固体), 产率 100%
MS m/z (ESI):419.9 [M+l]
1H NMR (400 MHz, CDC13): δ 8.62(s, 1H), 7.13 (s, 2H), 7.27 (s, 1H), 4.06 (t, 2H), 3.91 (t, 2H), 2.32 (s, 6H)
第五步
4-氯 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 室温下, 将 N-(4-溴 -3, 5-二甲基) -N-(2- (乙羟基) -4,6-二氯 -5-嘧啶甲酰胺 le(125 g, 298 mmol), 溶于 1.5 L乙腈, 加入三乙胺 (154.4 mL), 在 80°C搅 拌 16小时。 冷却至室温后, 反应液用二氯甲烷 (500 mLx3;)和水 (500 mLx2) 萃取, 合并有机相, 用饱和氯化钠溶液洗涤300 mLx2;)洗涤, 无水硫酸钠干 燥, 过滤, 滤液减压浓缩得到标题产物 4-氯 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lf(103 g, 浅黄色固体), 产率: 91%
MS m/z (ESI):383.9 [M+l]
JH NMR (400 MHz, CDC13): δ 8.76(s, 1H), 6.98 (s, 2H), 7.27 (s, 1H), 4.74 (t, 2H), 4.01 (t, 2H), 2.45 (s, 6H)
第六步
4-氨基 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)- 酮
将 4-氯 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)- 酮 If ( 103 g, 268.9 mmol),溶于 0.5摩尔氨气的 1,4-二氧六环溶液( 1.2 L), 搅拌 16小时。将反应液过滤得到 4-氨基 -6-(4-溴 -3,5-二甲基苯:) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lg (90 g, 白色固体) 产率: 92.4%
MS m/z (ESI): 364.9[M+1]
JH NM (400MHz, DMSO) δ 8.17 (s, 1H), 7.62 (br. s., 2H), 7.31 (s, 6H), 7.24 (s, 2H), 4.60 (t, 2H), 3.95 (t, 2H), 2.40 2.34 (s, 6H)
第七步
4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -l-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将 4-硼酸噸哪醇酯 -3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 lh (57 mg, 290 umol)和 4-氨基 -6-(4-溴 -3 ,5-二甲基苯) -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮 (60 mg, 165 umol)溶解于 3 mLl,4-二氧六环和水 (V/V = 2:1)混合溶 液中,加入碳酸钾 (68 mg, 495 umol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (6 mg, 7.2 umol), 微波 90°C反应 30分钟。 反应液冷却至室温, 减压浓缩, 用制备 高效液相色谱法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯基) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (30 mg, 白色固体), 产率: 40.7%。
MS m/z (ESI): 446.1 [M+l]
JH NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 7.68 (s, 1H), 7.16 (s, 1H), 5.07 (q, 2H), 4.75 (t, 2H), 4.09 (t, 2H), 2.01 (s, 6H), 1.94 (s, 3H)
实施例 42
4-氨基 -6-p-氯 -4-(l-(2-羟基 -2-甲基丙醇基) -1H-吡唑 -4-基)苯基) -7,8-二氢

第一步
2-(2-甲基 -2-羟基-丙醇基 1Η-吡唑 -5-硼酸噸哪醇酯
室温下, 将 4-吡唑硼酸噸哪醇酯 la (100 mg, 0.52 mmol) 溶解于 2 mL Ν,Ν二甲基甲酰胺中, 加入氧化异丁烯 lb (190 mg, 1.55 mmol) 和 碳酸铯 (251 mg, 0.77 mmol), 微波 110°C下反应 1 小时。 待反应温度降至室温, 向 反应液中加 20 mL水, 用乙酸乙酯萃取 (30 mLx3 ) , 合并有机相, 无水硫 酸钠干燥,过滤,滤液减压浓缩,得到标题产物 2-(2-甲基 -2-羟基-丙醇基 )-1Η- 吡唑 -5-硼酸噸哪醇酯 lc (90 mg, 白色固体:), 产率: 65.2 %。
MS m/z (ESI): 239.2 [M+l]
JH NM (400 MHz, CDC13) 5 1.15 (s, 6 H), 1.32 (s, 12 H), 4.07 (s, 2 H), 7.69 (s, 1 H), 7.82 (s, 1 H)
第二步
4-氨基 -6-p-氯 -4-(l-(2-羟基 -2-甲基丙醇) -1H-吡唑 -4-基)苯基) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-酮
室温下, 将 2-(2-甲基 -2-羟基 -丙醇基:) -1H-吡唑 -5-硼酸噸哪醇酯 lc (100 mg, 0.52 mmol), 4-氨基 -6-(3-氯 -4-碘苯基 )-7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -5(6H)-酮 Id (45 mg, 0.17 mmol), 碳酸铯 (110 mg, 0.34 mmol) 和 1,1'-双 (二苯基磷)二茂铁氯化钯 (5 mg, 0.006 mmol)溶解于 1.5 mL二氧六环中,加入 0.5 mL水,微波 110°C下反应 30分钟。待反应温度降至室温,反应液过滤,
滤液减压浓缩, 用制备分离方法得到标题产物 4-氨基 -6-(3-氯 -4-(1-(2-羟基 -2- 甲基丙醇) -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-酮 1 (4 mg,白色固体), 产率:8.3 %。
MS m/z (ESI): 429.1 [M+l]
JH NM (400 MHz, CD3OD) δ 1.25 (s, 6 H), 4.24 (s, 2 H), 4.27 - 4.33 (m, 2 H), 4.99 5.07 (m, 2 H), 7.40 (dd, 1 H), 7.64 (d, 1 H), 7.72 (d, 1 H), 8.09 (s, 1 H) 8.29 (s, 1 H) 8.46 (s, 1 H)
实施例 43
4-氨基 -6-(4-(l-(2,2-二氟乙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮

1
2,2-二氟乙烷三氟代甲烷璜酰氯
-78°C下,将三氟甲磺酸酐 (34.39 g,0.12 mol)溶解于 20 mL二氯甲烷中, 缓慢滴加 2,2-二氟乙醇 la (10.00 g, 0.12 mol)和三乙胺 (12 g, 0.12 mol)的二 氯甲烷 (10 mL)溶液。将反应液升至室温, 减压浓缩, 得到粗品标题产物 2,2- 二氟乙烷三氟代甲烷璜酸 lb (53.00 g,黄色油状物),产物不经纯化直接进行
l-(2,2-二氟乙基) -3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2- 基) -1H-吡唑
将 2,2-二氟乙烷三氟代甲烷璜酸 lb (8.23 g, 0.38 mol), 3-甲基 -4-(4,4,5,5- 四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 (500 mg, 1.92 mmol)和碳酸铯 (1.25 g,3.84 mmol)溶解于 10 mL N, N-二甲基甲酰胺中,微波 100°C反应 1小时。 反应液冷却至室温,向反应液中加入 30 mL水,用乙酸乙酯萃取20 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 1-(2,2-二氟乙基) -3-甲基 -4-(4,4,5,5-四甲基 -1,3,2- 二杂氧戊硼烷 -2-基) -1H-吡唑 lc (117 mg, 白色固体), 产率: 17.8%。
JH NM (400 MHz, CDC13) δ 7.63 (s, 1H), 6.23-5.89 (m, 1H), 4.41-4.31 (m 1H), 2.36 (s, 3H), 1.28 (s, 12H)
第三步
4-氨基 -6-(4-(l-(2,2-二氟乙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将 1-(2,2-二氟乙基) -3-甲基 -4-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2- 基) -1H-吡唑 lc (62 mg, 172 umol)和 4-氨基 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (117 mg, 342 umol)溶解于 2.5 mLl,4-二氧六 环和水 (V/V = 4: l)混合溶液中, 加入碳酸钾 (71 mg, 516 umol)和 Ι,Γ-双 (二苯 基磷)二茂铁氯化钯 (6.2 mg, 8.5 umol), 微波 100°C反应 40分钟。 反应液 冷却至室温, 减压浓缩, 用制备高效液相色谱法以洗脱剂体系 B纯化所得残 余物,得到标题产物 4-氨基 -6-(4-(1-(2,2-二氟乙基) -3-甲基 -1H-吡唑 -4-基) -3,5- 二甲基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 1 (30 mg, 白色固体), 产率: 40.9%。
MS m/z (ESI): 429.0 [M+l]
JH NM (400 MHz, DMSO-d6) δ 8.71 {br. s, IH), 8.58 {br. s, IH), 8.41 (s, IH), 7.58 (s, IH), 7.12 (s, IH), 6.50-6.20 (m, IH), 4.79 (t, 2H), 4.59-4.50 (m, 2H) 4.10 (t, 2H), 1.99 (s, 6H), 1.90 (s, 3H)
实施例 44
4-氨基 -6-(3-氯 -4-(l-(3-羟基 -2,2-二甲基丙基) -1-H-吡唑 -4-基)苯基) -7,8- -f][l,4] 咪唑啉 -5(6H)-酮

3-溴 -2,2-二甲基丙酸乙酯
将 3-溴 -2,2-二甲基丙酸 la ( 500 mg, 2.76 mmol )溶解于 5 mL无水乙 醇中, 加入 0.5 mL浓硫酸。 100°搅拌 2小时。 反应液冷却到室温, 用饱和 碳酸氢钠溶液调至中性, 乙酸乙酯萃取(30 mLx3 ), 合并有机相, 用无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 得到标题产物 lb (430 mg, 黄色液体), 产 率: 74.5%。 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) 4.18 (q , 2H), 3.5 (s, IH), 1.57 (s, 6H), 1.30 (q,
3H)
第二步
2,2-二甲基 -3-(4-(4,4,5,5-四甲基 -(1,3,2)-二杂氧戊硼烷 -2-基) -1Η-吡唑 -1- 基)丙酸乙酯
将 3-溴 -2,2-二甲基丙酸乙酯 lb (400 mg, 1.91 mmol) , 4-吡唑硼酸噸哪 醇酯 lc (330 mg, 1.73 mmol), Cs2C03 (1.2 g, 3.45 mmol) 混合于 10 mL DMF 中. 微波 100°C反应 25 分钟. 反应液冷却到室温, 用 30 mL乙酸乙酯 稀释, 乙酸乙酯层用水洗涤30 mLx3 无水硫酸钠干燥, 过滤, 滤液减压浓 缩得到标题产物 Id (170 mg, 黄色液体), 产率: 30.5%。 产物不经纯化直接 进行下一步反应。
JH NM (400 MHz, CDC13) 7.74 (s, 1H), 7.66 (s, 1H), 4.28 (s, 2H), 4.16 (q, 2H), 1.30 (s, 9H), 1.24 (t, 3H)
第三步
3-4_(4_(4_氨基 _5_氧代 _7,8-二氢嘧啶 [5,4-f][l,4]咪唑啉 -6(5H)-基) -2-氯苯 基) _1H-吡唑 -1-基)苯基) -2,2-二甲基丙酸乙酯 将 2,2-二甲基 -3-(4-(4,4,5,5-四甲基 -(1,3,2)-二杂氧戊硼烷 -2-基) -1H-吡唑 -1-基)丙酸乙酯 Id (94 mg, 0.29 mmol) , 4-氨基 -6- (3-氯 -4-溴苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]咪唑啉 -5(6Η)-酮 If (100 mg, 0.24mmol), 碳酸铯 (156 mg, 0.48 mmol) , Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (10 mg, .012 mmol) 混合于 8 mL 1, 4-二氧六环和水 (V/V = 3: 1)混合溶剂。 微波 100°C反应 30 分钟. 反应液 冷却到室温,用 30 mL乙酸乙酯稀释, 乙酸乙酯层用水洗涤 (30 mLx3), 无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层色谱法以展开剂体系 A纯化所得 残余物, 得到标题产物 3-4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]咪唑啉 -6(5H)-基) -2-氯苯基) -1H-吡唑 -1-基)苯基) -2,2-二甲基丙酸乙酯 lg (60 mg, 淡 黄色液体), 产率: 42.7 %。
JH NMR (400 MHz, CDC13) δ 8.30 (s, 1H), 7.83 (s, 1H), 7.76 (s, 1H), 7.52
(d, IH), 7.39 (d, IH), 7.21 (dd, IH), 4.74 4.69 (m, 2H), 4.34 (s, 2H), 4.22 4.14 (m, 2H), 4.05 - 4.01 (m, 2H), 1.57 (s, 9H)
第四步
4-氨基 -6-(3-氯 -4-(l-(3-羟基 -2,2-二甲基丙基) -IH-吡唑 -4-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]咪唑啉 -5(6H)-酮
冰浴下, 将 lg (80 mg, 0.16 mmol)溶解于 2 mL 四氢呋喃中, 依次加入 氯化钙 (36 mg, 0.32 mmol), 硼氢化钠 (31 mg, 0.82 mmol), 室温搅拌反应 12小时。 将反应液减压浓缩, 经 p-HPLC制备分离得到标题产物 lh (6 mg, 白色固体:), 产率: 8.1%。
MS m/z (ESI): 443.1 [M+l]
JH NM (400 MHz, CD3OD) δ 8.45 (s, IH), 8.20 (s, IH), 8.01 (s, IH), 7.70 (d, IH), 7.62 (d, IH), 7.39 (dd, IH), 5.00-5.07 (m, 2H), 4.26-4.33 (m, 2H), 4.18 (s, 2H), 0.97 (s, 6H)
实施例 45
(R)-4-氨基 -6-(3-氯 -4-(l-异丁基) -IH-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢嘧 啶 [5,4-f l,4]氧氮杂卓 -5(6H)-酮


1
1-异丁基 -(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 将 4-(4,4,5,5-四甲基 -1 ,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 la (5.00 g, 25.77 mmol)溶解于 50 mL N,N-二甲基甲酰胺中,加入碳酸铯 (16.79 g, 51.54 mmol) 和 1-溴 -2-甲基丙烷 (7.06 g, 51.54 mmol), 100°C下搅拌反应 2小时。 反应液 冷却至室温, 向反应液中加入 10 mL水, 用乙酸乙酯萃取 100 mLx3;), 合并 有机相, 依次用水 (100 mLx2)、 饱和氯化钠溶液洗涤 100 mLx2;>, 用无水硫 酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得 残余物, 得到标题产物 1-异丁基 -(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2- 基) -1H-吡唑 lb (2.05 g, 黄色液体), 产率: 31.8%。
JH NM (400 MHz, CDC13) δ 7.71 (s, 1H), 7.58 (s, 1H), 3.84 (d, 1H), 4.08-4.00 (m, 1H), 2.05-2.20 (m, 1H), 1.25 (s, 12H), 0.82 (d, 6H)
4-(4-溴 -2-氯苯基 )-1-异丁基 -m-吡唑
将 1-异丁基 -(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1H-吡唑 lb (2.05 g, 8.19 mmol), 4-溴 -2-氯 -1-碘苯 (2.00 g, 6.30 mmol), 碳酸钠(1.34 g, 12.60 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (257 mg, 0.31 mmol)溶解于 25 mL四 氢呋喃和水 (V/V = 4: 1)混合溶液中, 70°C下搅拌反应 12小时。 反应液冷却 至室温, 向反应液中加入 50 mL水, 用乙酸乙酯萃取50 mLx3;>, 合并有机 相, 依次用水 (50 mLx2;>、 饱和氯化钠溶液洗涤 50 mLx2;>, 用无水硫酸钠干 燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 4-(4-溴 -2-氯苯基 )-1-异丁基 -1H-吡唑 lc (652 mg, 黄色液体), 产率: 32.9%。
第三步
(R)-2- ((叔丁基二甲基硅)氧代)丙基 -1-胺
将 (R)-(-)-l-氨基 -2-丙醇 Id (19.00 g, 0.25 mol)溶解于 200 mL乙腈中, 冰浴下加入 1.8-二氮杂二环 [5.4.0] ^—烷 -7-烯 (42.30 g, 0.28 mmol))和叔丁基 二甲基氯硅烷 ( 42.20 g, 0.28 mol), 室温搅拌反应 12小时。 向反应液中加入 3 mL甲醇,减压浓缩。将残余物溶解于 100 mL乙酸乙酯中,用水 (50 mLx2), 饱和氯化钠溶液50 mL )洗涤, 无水硫酸钠干燥, 过滤, 滤液减压浓缩得 到粗品标题产物 (R)-2- ;叔丁基二甲基硅)氧代)丙基 -1-氨 le (40 g, 浅黄色液 体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 3.73-3.66 (m, 1Η), 2.59-2.48 (m, 2H), 1.03 (d, 6H), 0.82 (s, 9H), 0 (s, 6H)
第四步
(R)-2-N- ((叔丁基二甲基硅)氧代)丙基) -3-氯 -4-(l-异丁基 -1H-吡唑 -4-基) 苯胺
将 (R)-2- ((叔丁基二甲基硅)氧代)异丙基 -1-氨 le (900 mg, 2.87 mmol)溶 解于 10 mL甲苯中, 加入 4-(4-溴 -2-氯苯基 )小异丁基 -1H-吡唑 (652 mg, 3.44
mmol), 碳酸铯 (2.81 g, 8.61 mmol), 三 (二苯亚苄基丙酮)二钯 (265 mg, 0.29 mmol)和 4,5-双二苯基膦 -9,9-二甲基氧杂蒽(138 mg, 0.29 mmol), 100°C搅拌 反应 12小时。 反应液冷却至室温, 向反应液中加入 20 mL水, 用乙酸乙酯 萃取20 mLx3;), 合并有机相, 依次用水 (20 mLx2;>、 饱和氯化钠溶液洗涤 (20 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 C 纯化所得残余物, 得到标题产物 (R)-2-N- ((叔丁基二甲基硅)氧代) 丙基) -3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯胺 lf (1.00 g, 黄色油状物), 产率: 83.3%。
第五步
(R)-N-2- 叔丁基二甲基硅)氧代)丙基) -4,6-二氯 -N-(3-氯 -4-(1-异丁基
_1H-吡唑 -4-基:)苯基:)嘧啶 -5-酰胺
将 (R)-2-N-((叔丁基二甲基硅)氧代)丙基) -3-氯 -4-(1-异丁基 -1H-吡唑 -4-基) 苯胺 If (1.00 g, 2.37 mmol)溶解于 15 mL二氯甲烷中, 加入三乙胺 (1.0 mL, 7.11 mmol)和 4,6-二氯 -5-酰氯嘧啶 (1.00 g, 4.74 mmol), 搅拌反应 12小时。 向反应液中加入 30 mL水, 用二氯甲烷萃取 (20 mLx3;>, 合并有机相, 依次 用水 (20 mLx2;>、饱和氯化钠溶液洗涤20 mLx2;>,用无水硫酸钠干燥,过滤, 滤液减压浓缩,得到粗品标题产物 (R)-N-2- ;叔丁基二甲基硅)氧代)丙基 )-4,6- 二氯 -N-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基)嘧啶 -5-酰胺 lg (500 mg, 黄色 固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.63 (s, 1Η), 7.75 (d, 2H), 7.45 (d, 1H), 7.33 (d, 1H), 7.24-7.19 (m, 1H), 4.21 (d, 1H), 4.08-4.00 (m, 1H), 3.97-3.86 (m, 3H), 1.33 (d, 3H), 0.94 (d, 7H), 0.77 (s, 10H), 0.02 (s, 3H), -0.01 (s, 3H)
第六步
(R;>-4,6-二氯 -N-P-氯 -4-G-异丁基 -1H-吡唑 -4-基;)苯基; )-N-(2-羟丙基;)嘧啶 -5-酰胺
将粗品 (R)-N-2-((叔丁基二甲基硅)氧代)丙基) -4,6-二氯 -N-P-氯 -4-(1-异
丁基 -1H-吡唑 -4-基)苯基)嘧啶 -5-酰胺 lg (500 mg, 0.84 mmol)溶解于 10 mL 乙醇中,加入浓盐酸(0.3 mL),搅拌反应 1小时。向反应液中加入 10 mL水, 用乙酸乙酯萃取 10 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和碳酸氢 钠溶液 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过 滤, 滤液减压浓缩, 得到粗品标题产物 (R)-4,6-二氯 -N-(3-氯 -4-(1-异丁基 -1H- 吡唑 -4-基:)苯基:) -N-P-羟丙基:)嘧啶 -5-酰胺 lh 400 mg, 黄色固体), 产物不经 纯化直接进行下一步反应。
第七步
(R)-4-氯 -6-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将粗品 (R)-4,6-二氯 -N-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -N-(2-羟丙 基:)嘧啶 -5-酰胺 lh (100 mg, 0.21 mmol)溶解于 5 mL乙腈中, 加入碳酸钾 (86 mg, 0.62 mmol)和 30 mg分子筛, 80°C搅拌反应 12小时。 反应液冷却至室 温, 向反应液中加入 10 mL水, 用乙酸乙酯萃取 (10 mLx3), 合并有机相, 用饱和氯化钠溶液洗涤 10 mLx2;), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 (R)-4-氯 -6-P-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 li (40 mg, 黄色固体), 产物不经 纯化直接进行下一步反应。
第八步
(R)-4-氨基 -6-(3-氯 -4-(1-异丁基) -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将粗品 (R)-4-氯 -6-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 li (40 mg, 0.09 mmol)溶解于 5 mL 0.5 M 氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。 反应液减压浓缩, 用制备 高效液相色谱纯化所得残余物,得到粗品标题产物 (R 4-氯 -6-(3-氯 -4-(1-异丁 基 -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 1
(17.5 mg, 白色固体), 产率: 46.0%
MS m/z (ESI): 427.2 [M+l]
JH NM (400 MHz, CD30D) δ 8.42 (s, 1H), 8.12 (s, 1H), 7.90 (s, 1H), 7.68 (d, 1H), 7.59 (d, 1H), 7.38-7.35 (dd, 1H), 5.34-5.30 (m, 1H), 4.26-4.12 (m, 2H), 4.08 -4.05 (m, 2H), 2.28-2.19 (m, 1H), 0.96 (d, 6H)
实施例 46
2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯

4-硼酸噸哪醇酯 -2-乙酰基 -1H-吡唑
室温下, 将 4-吡唑硼酸噸哪醇酯 la (100 mg, 0.52 mmol) 溶解于 3 mL DMF中,在室温下加入 2-溴乙酰胺 lb (143 mg, 1.04 mmol), 碳酸铯 (253 mg, 0.78 mmol 100°C微波反应 35 分钟。 待温度降至室温, 加入 10 mL,乙酸 乙酯萃取 (10 mLx3;>, 合并有机相, 依次用水 p0 mLx3;)、 饱和氯化钠溶液洗 涤50 mL), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到标题产物 4-硼 酸噸哪醇酯 -2-乙酰基 -1H-吡唑 lc (70 mg, 黄色油状:),产物不需要纯化直接进 行下一步。
MS m/z (ESI): 252.2 [M+l]
JH NM (400 MHz, CDC13) δ 7.9 (d, 2 H), 2.05 (s, 2 H), 1.32 (s, 12 H)
第二步
2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯 苯) _1H -吡唑小基)乙酰胺
室温下, 将 2-(4-硼酸噸哪醇酯 -1H-吡唑 -1基)乙酰胺 lc (70 mg, 0.28 mmol), 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 氯碘苯 Id (80 mg, 0.19 mmol), 碳酸铯 (1.2 g, 0.38 mmol)和 Ι,Γ-双 (二苯基磷) 二茂铁氯化钯 (10 mg, 0.01 mmol) 溶解于 3 mL二氧六环中, 加入 1 mL水, 100°C微波反应 30分钟。 待反应温度降至室温, 过滤, 滤液减压浓缩, 用制 备分离方法纯化所得残余物, 得到标题产物 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2-氯苯) -1H-吡唑 -1-基)乙酰胺 1 (6 mg, 白 色固体), 产率: 7.6 %。
MS m/z (ESI): 414.0 [M+l]
JH NM (400 MHz, DMSO-d6) δ 8.38 (s, 1 H), 8.23 (s, 1 H), 7.91 (s, 1 H), 7.67-7.55 (m, 2 H), 7.60 (s, 1 H), 7.40 (q, 1 H), 7.30 (s, 1 H), 4.85-4.80 (m, 2 H), 4.14 (t, 2 H)
实施例 47
4-氨基 -6-(3,5-二甲基 -4-(3-甲基小((3-甲基氧杂环丁烷 -3-基)甲基) -1H-吡 唑 -4-基)苯基) - 7,8-二氢嘧啶 5,4-f][l,4]氧氮杂卓 -5(6H)-酮



4-溴 -3-甲基小((3-甲基氧杂环丁烷 -3-基)甲基) -1H-吡唑
将 4-溴 -3-甲基 -1H-吡唑 la (350 mgx4, 2.19 mmol) 溶解于 10 mL N,N- 二甲基甲酰胺中, 加入 3- (溴甲基: )-3甲基氧杂环丁烷 (400 mg, 2.44 mol) 和 碳酸铯 (1.44 g, 4.44 mmol), 在微波 100°C下搅拌反应 1.5小时。 反应液直接 过滤, 滤液浓缩得到标题产物(粗品) 4-溴 -3-甲基 -1-((3-甲基氧杂环丁烷 -3- 基)甲基) -1H-吡唑 lb (400 mg, 浅黄色固体), 产率: 74.9%。
第二步
3_甲基小 ((3_甲基氧杂环丁烷 _3_基)甲基) _4 噸哪醇硼酸酯 _2基) -1H-吡 唑
将 4-溴 -3-甲基 -1-((3-甲基氧杂环丁烷 -3-基)甲基) -1H-吡唑 lb (1.53 g, 6.25 mmol), 双联噸哪醇硼酸酯 (2.38 g, 9.38 mmol) 和乙酸钾 (1.23 g, 12.50 mmol) 溶解于 15 mL二氧六环中, 抽真空置换氮气后, 加入 Ι,Γ-双 (二苯基 磷:)二茂铁氯化钯 (229 mg, 0.32 mmol), 90°C下搅拌 16小时。反应液减压浓缩, 粗品直接用硅胶柱色谱法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 3_甲基小 ((3_甲基氧杂环丁烷 _3_基)甲基) _4 噸哪醇硼酸酯 _2基) -1H-吡唑 lc
(500 mg, 浅黄色油状胶状物), 产率: 28%
JH NM (400 MHz,CDCl3) δ 7.56 (s, 1H), 4.69-4.67 (d, 2H), 4.37-4.36 (d, 2H) ,4.23 (s, 2H), 1.30 (s, 6H), 1.26-1.23 (t, 12H)
4-氨基 -6-(3,5-二甲基 -4-(3-甲基小((3-甲基氧杂环丁烷 -3-基)甲基) -1Η-吡 唑 -4-基)苯基) - 7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 氮气保护条件下, 将 3-甲基 -1-((3-甲基氧杂环丁烷 -3-基)甲基: )-4-( (噸哪 醇硼酸酯 -2-基) -1H-吡唑 lc (80 mg, 0.27 mmol) 溶解于 3.6 mLl,4-二氧六环 和水 (V/V=5:l)混合溶剂中, 加入 4-氨基 -6-(3,5-二甲基苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (66 mg, 0.18 mmol), 碳酸钾 (54 mg, 0.55 mmol) 和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (7 mg, 0.01 mmol),微波 100°C下搅拌反应 12小时。反应液冷却至室温, 向反应液中加入 5 mL水, 用二氯甲烷萃取 (10 mLx3), 合并有机相, 用饱和氯化钠溶液洗涤 5 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用制备高相液相色谱纯化所得标题产物 4-氨基 -6-(3,5- 二甲基 -4-P-甲基 -1-((3-甲基氧杂环丁烷 -3-基)甲基) -1H-吡唑 -4-基:)苯基 )-7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (2.59 mg,棕色固体),产率: 2.1%。
MS m/z (ESI): 449.2 [M+l]
JH NM (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.62 (s, 2H), 7.56(s, 1H), 7.13 (s, 2H), 4.62 (s, 4H), 4.29 (s, 2H), 4.25 (d, 2H), 3.99 (s, 2H), 2.02 (s, 6H), 1.92 (s, 3H), 1.14 (d, 3H)
实施例 48
4-氨基 -6-(4-(l-(2,2-二氟丙基) -5-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯
基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮



1e 1f 第一步
l-(4-溴 -3-甲基 -m-吡唑 -1基)丙烷 -2-酮
将 4-溴 -3-甲基 -1H-吡唑 la (500 mg, 3.13 mmol)溶解于 3 mL N,N-二甲基 甲酰胺中, 加入 1-氯丙烷 2-酮 (862 mg, 9.38 mmol)和碳酸铯 (2.04 g, 6.25 mmol), 微波 90°C搅拌反应 1.5小时。 反应液冷却至室温, 向反应液中加入 15 mL水, 用二氯甲烷萃取30 mLx3;), 合并有机相, 依次用水 (15 mLx2;>、 饱和氯化钠溶液洗涤 15 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到粗品标题产物 1-(4- 溴 -3-甲基 -1H-吡唑 -1基:)丙烷 -2-酮 lb (700 mg,黄色油状物:)。
JH NM (400 MHz, CDC13) δ 7.50 (d, 1H), 4.86 (d, 2H), 2.31-2.15 (m, 6H) 第二步
4-溴 -l-(2,2-二氟丙基) -3-甲基 -1H-吡唑
-78 。c下,将粗品 1-(4-溴 -3-甲基 -1H-吡唑 -1基)丙烷 -2-酮 lb (700 mg, 3.24 mmol) 溶解于 10 mL二氯甲烷中。然后向反应中缓慢滴加 DAST (1.57 g, 9.72 mmol), 室温搅拌反应 12小时。 向反应液中加入 10 mL水, 用二氯甲烷萃 取20 mLx3;), 合并有机相, 依次用饱和碳酸氢钠 (10 mLx2;>, 饱和氯化钠溶 液洗涤 (10 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱 法以洗脱剂体系 B纯化所得残余物, 得到粗品标题产物 4-溴 -1-(2,2-二氟丙
基) -3-甲基 -1H-吡唑 lc (220 mg, 黄色油状物),产物不经进一步纯化直接进行 下一步操作。
第三步
1-(2,2-二氟丙基) -3-甲基 -4- (噸哪醇硼酸酯 -2-基) -1H-吡唑
氮气保护下, 将粗品 4-溴 -1-(2,2-二氟丙基) -3-甲基 -1H-吡唑 lc (200 mg, 0.84 mmol)和双联噸哪醇硼酸酯 (320 mg, 1.26 mmol)溶解于 10 mL 1, 4-二氧 六环中, 加入乙酸钾 (247 mg, 2.52 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (62 mg, 0.08 mmol), 90°C搅拌反应 12小时。 向反应液中加入 5 mL水, 用 乙酸乙酯萃取 (10 mLx3;), 合并有机相, 依次用水 (5 mLx2;>、饱和氯化钠溶液 洗涤 (5 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层层析以洗 脱剂体系 B纯化所得残余物得到标题产物 1-(2,2-二氟丙基 3-甲基 -4- (噸哪醇 硼酸酯 -2-基) -1H-吡唑 ld (80 mg, 黄色油状物), 产率: 31.0%。
JH NM (400 MHz, CDC13) δ 7.62 (d, 1H), 4.35-4.26 (m, 2H), 1.18 (d,
12H)
第四步
4-氨基 -6-(4-(l-(2,2-二氟丙基) -5-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯
基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
氮气保护下, 将 1-(2,2-二氟丙基) -3-甲基 -4- (噸哪醇硼酸酯 -2-基) -1H-吡 唑 Id (60 mg, 0.21 mmol)溶解于 3 mL 1,4-二氧六环和水 (V/V=5: l)混合溶剂中, 加入 4-氨基 -6-(4-溴 -3 ,5-二甲基苯基) -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮 (50 mg, 0.14 mmol), 碳酸钾 (38 mg, 0.28 mmol)和 1,1'-双 (二苯基磷) 二茂铁氯化钯 (5 mg, 0.007mmol), 90°C下搅拌反应 12小时。 反应液冷却至 室温, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (10 mLx3;>, 合并有机相, 用饱和氯化钠溶液洗涤 5 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用制备高相液相色谱纯化分离得到标题产物和 4-氨基 -6-(4-(1-(2,2-二氟丙 基) -5-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓
-5(6H)-酮 If (6.55 mg, 白色固体), 收率: 11.0%。
MS m/z (ESI): 443.2 [M+l]
JH NMR (400 MHz, DMSO-d6) δ 8.17 (s, IH), 7.34 (s, IH), 7.13 (s, 2H) 4.74-4.72 (t, 2H), 4.58-4.55 (t, 2H), 4.08-4.05 (t, 2H), 2.09-2.07 (d, 9H) 1.69-1.60 (t, 3H) 参照实施例 46〜48的制备方法还合成了以下 47个化合物:







7.60 2H), 4.30 1.18
(s, s., 1H), (dd, 1H), (m, 1H), (s, 3H)
8.30(s, 1H), (t, 2H),4.06 2H),0.43 (s, 2H)
δ 8.24 (s, (s, 1H), (d, 2H),4.04
8.28 7.51 1H), 4.70 3.01




实施例 100
4-氨基 -6-(4-(2-氧 -l-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)苯基) - 二氢嘧 啶 [ -f][l,4]氧氮杂卓 -5(6H)-酮
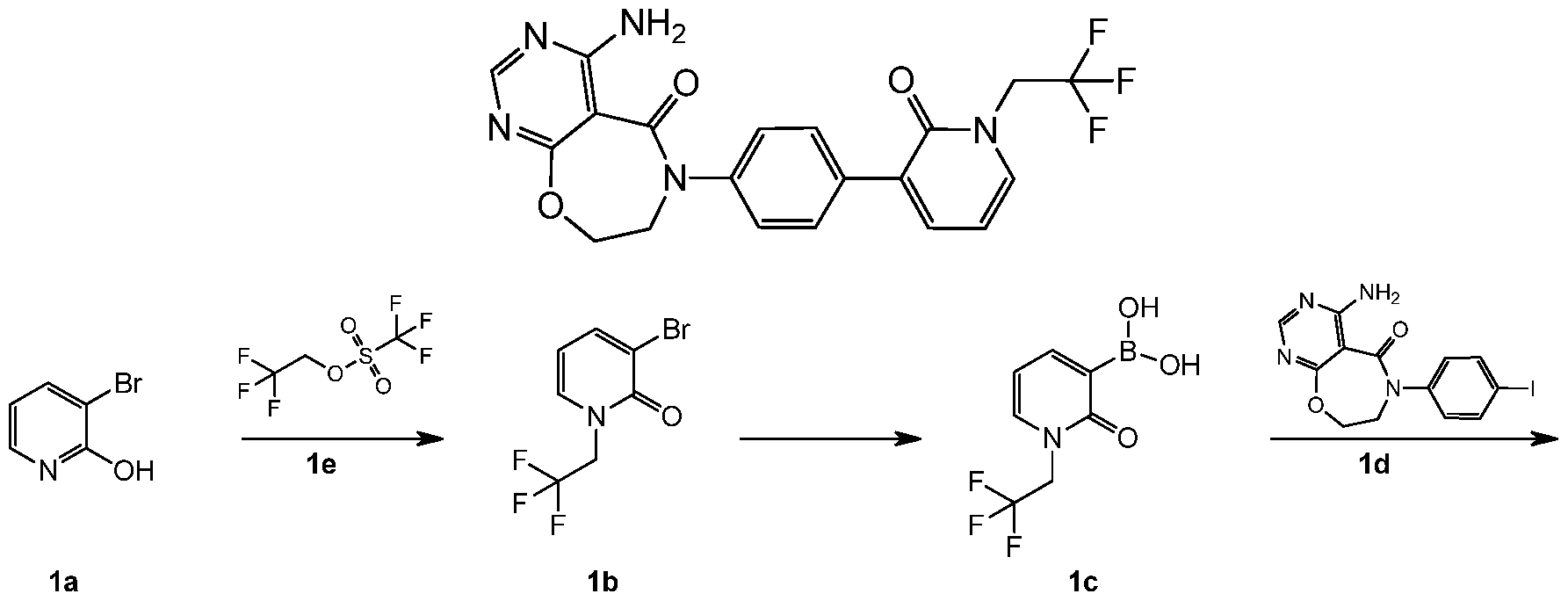

3-溴 -1-(2,2,2-三氟乙基)吡啶 -2(1Η)-酮
将 3-溴 -2-羟基吡啶 la (600 mg, 3.45 mmol)溶解于 10 mL Ν,Ν-二甲基甲 酰胺中, 加入碳酸钾 (950 mg, 6.9 mmol)和三氟甲磺酸三氟乙酯 (1.6 g, 6.9 mmol), 室温搅拌反应 12小时。 向反应液中加入 150 mL水, 用乙酸乙酯萃 取 (50 mLx3), 合并有机相, 用水溶液 (50 mLx2), 饱和氯化钠溶液洗涤 (50 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂 体系 C纯化所得残余物, 得到标题产物 3-溴 -1-(2,2,2-三氟乙基)吡啶 -2(1H)- 酮 lb (515 mg, 浅黄色液体), 产率: 58.6%。
JH NM (400 MHz, CDC13) δ 7.77 (d, 1H), 7.27 (d, 1H), 6.13 (t, 1H), 4.65(q, 2H)
第二步
(2-氧代 -l-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)硼酸
将 3-溴 -l-(2,2,2-三氟乙基)吡啶 -2(1H)-酮 lb (250 mg, 1 mmol)溶解于 20 mL 甲苯中,加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'-双 (1,3,2-二氧硼烷)(300 mg, 1.2 mmol), 乙酸钾 (300 mg, 3 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (40 mg, 0.05 mmol), 120°C微波反应 30分钟。 将反应液过滤, 滤液减压浓缩, 用薄 层色谱法以展开剂体系 C纯化所得残余物,得到标题产物 (2-氧 -1-(2,2,2-三氟 乙基) -1,2-二氢吡啶 -3-基)硼酸 lc (120 mg, 白色固体), 产率: 55.6%。
JH NM (400 MHz, DMSO-^6) δ 8.55 (s, 2Η), 7.98 (dd, 1H), 7.84 (d, 1H) 6.48 (t, 1H), 4.91 (q, 2H).
第三步
4-氨基 -6-(4-(2-氧代 -l-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)苯基) - 二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将 (2-氧 -1-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)硼酸 lc (50 mg, 0.13 mmol)溶解于 1.2 mL 二氧六环和水 (V/V=5/l)中,加入 4-氨基 -6-(4-碘苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 Id (50 mg, 0.13 mmol, 采用公知的方 法"专利 WO2009016462"制备而得), 碳酸钾 (40 mg, 0.26 mmol)和 Ι,Γ-双 (二 苯基磷)二茂铁氯化钯 (5 mg, 0.004 mmol), 100°C微波反应 30分钟。 向反应 液中加入 20 mL水, 用乙酸乙酯萃取 IO mLx3;>, 合并有机相, 饱和氯化钠 溶液洗涤 P0 mL:), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用 p-HPLC制备 分离纯化, 得到标题产物 4-氨基 -6-(4-(2-氧 -1-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)苯基) - 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 1 (18 mg, 白色固体), 产率: 26.9%。
JH NMR (400 MHz, CD3OD) δ 8.40 (s, 1H), 7.70-7.76 (m, 3H), 7.65 (d, 1H), 7.40 (d, 2H), 6.51 (t, 1H), 4.99 (s, 2H), 4.85-4.95 (m, 2H), 4.25 (s, 2H)
实施例 101
4-氨基 -6-(3,5-二甲基 -4-(6-氧 -l-(2,2,2-三氟乙基) -1,6-二氢吡啶 -5-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮

1
第一步
5-溴 -l-(2,2,2-三氟乙基)吡啶 -2(1H)-酮
室温下, 将 5-溴 -吲唑 la (300 mg, 1.72 mmol)溶解于 10 mL DMF中, 冰 浴下加入三氟甲磺酸三氟乙酯 lb (800 mg, 3.45 mmol), 碳酸铯 (476 mg, 3.45 mmol) o 室温搅拌反应 4 小时。反应液倒入冷水中,乙酸乙酯萃取 (30 mL><3), 合并有机相, 依次用水 (100 mLx3;>、 饱和氯化钠溶液洗涤 (100 mL:), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 5-溴 -1-(2,2,2-三氟乙基)吡啶 -2(1Η)-酮 lc (430 mg, 白色固体:), 产率: 36.4 %。
JH NM (400 MHz, CDC13) δ 7.90 (d, 1H), 7.64 (dd, 1H), 6.56 (d, 1H), 4.80 (q, 2H)
第二步
5-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基 )-l-(2,2,2-三氟乙基)吡啶
-2(1Η)-酮
室温下, 将 5-溴 -1-(2,2,2-三氟乙基)吡啶 -2(1H)-酮 lc (400 mg, 1.56 mmol 1)溶解于 5 mL二氧六环中,加入双联噸哪醇硼酸酯 Id (595 mg, 2.34 mmol), 醋酸钾 (458 mg, 4.63 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (63.60 mg, 0.08
mmol),微波 100°C下反应 30 分钟。 待反应温度降至室温, 减压浓缩, 向剩 余物中加 10 mL水,用二氯甲烷萃取 (20 mLx3), 合并有机相,无水硫酸钠干 燥, 过滤, 滤液减压浓缩, 用薄层层析以洗脱剂体系 C纯化所得残余物, 得 到标题产物 5-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基:) -1-(2,2,2-三氟乙基)吡 啶 -2(1H)-酮 le (800 mg, 白色固体), 产率: 100 %。
第三步
4-氨基 -6-(3,5-二甲基 -4-(6-氧 -1-(2,2,2-三氟乙基) -1,6-二氢吡啶 -5-基)苯 基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
室温下, 将 5-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -1-(2,2,2-三氟乙 基)吡啶 -2(1H)-酮 le (167 mg, 0.55 mmol), 4-氨基 -6-(4-溴 -3,5-二甲基苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-酮 If (100 mg, 0.28 mmol), 碳酸钾 (76 mg, 0.55 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (22 mg, 0.03 mmol) 溶解 于 1.5 mL 二氧六环中, 加入 0.5 mL水, 微波 100°C下反应 30 分钟。 待反 应温度降至室温, 加入二氯甲烷和水稀释, 用二氯甲烷萃取 (20 mLx3;), 合并 有机相, 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用制备分离方法纯化得到 标题产物 4-氨基 -6-(3,5-二甲基 -4-(6-氧 -1-(2,2,2-三氟乙基) -1,6-二氢吡啶 -5-基) 苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (37 mg,白色固体),产率: 28.2 %。
JH NM (400 MHz, CD3OD) δ 8.43 (s, 1Η), 7.50 (s, 1H), 7.41 (d, 1H), 7.20 7.09 (m, 2H), 6.76 6.65 (m, 1H), 5.00 (br,s, 2H), 4.93 4.88 (m, 2H), 4.23 (br,s, 2H), 2.18 (s, 6H)
实施例 102
4-氨基 -6-(3-氯 -4-(l-(2-氟 -2-甲基丙基) -1H-咪唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮

l-(4-溴 -m-咪唑 -1-基) -2-甲基丙烷 -2-醇
将 4-溴 -1H-咪唑 la(2g, 13.70mmol)溶解于 30mLN, N-二甲基甲酰胺 中,加入 20mL2,2-二甲基环氧乙烷和碳酸铯 (8g,24.62mmol), 90°C下搅拌 反应过夜。 反应液减压浓缩, 加入二氯甲烷 (30mL), 依次用水 (10mLx2)与 饱和氯化钠溶液 IO mL)洗涤, 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得 到粗品标题产物 1-(4-溴 -1H-咪唑 -1-基) -2-甲基丙烷 -2-醇 lb (2.7 g, 浅黄色固 体), 产率: 90.6%。
JH NM (400 MHz, CDC13) δ 7.33 (s, 1Η), 6.94 (s, 1H), 3.82 (s, 2H), 1.22 (s;
6H)
第二步
4-溴 -l-(2-氟 -2-甲基丙基) -1H-咪唑
在 -70°C, 氮气保护条件下, 将 1-(4-溴 -1H-咪唑 -1-基) -2-甲基丙烷 -2-醇 lb (2.70 g, 12.33 mmol)溶解于 30 mL二氯甲烷中, 逐滴滴加二乙氨基三氟化 (4.96 g, 30.83 mmol), 滴加完毕后, 缓慢升至室温下搅拌 12小时。 反应
液依次用水 G0 mLx2;>、饱和碳酸氢钠溶液5 mLx2;>、饱和氯化钠溶液洗涤 (:5 mLx2), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 B纯化所得残余物, 得到标题产物 4-溴 -1-(2-氟 -2-甲基丙基) -1H-咪唑 lc (1 g, 黄色油状物), 产率: 36.8%。
JH NM (400 MHz, CDC13) δ 7.37 (s, 1H), 6.95 (s, 1H), 4.00 (d, 2H), 1.33 (d, 6H)
第三步
4-氨基 -6-p-氯 -4- (噸哪醇硼酸酯 -2-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧 氮杂卓 -5(6H)-酮
氮气保护条件下, 将 4-氨基 -6-(4-溴 -3-氯苯基 )-7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 Id (100 mg, 0.27 mmol)溶解于 3 mLl,4-二氧六环中,加入 双联噸哪醇硼酸酯 (103 mg, 0.41 mmol), 醋酸钾 (79.38 mg, 0.81 mmol)和 Ι,Γ- 双 (二苯基磷)二茂铁氯化钯 (20 mg, 0.027 mmol), 80°C下搅拌 12小时。 反应 液减压浓缩, 用薄层色谱法以展开剂体系 B纯化所得残余物, 得到粗品标题 产物 4-氨基 -6-(3-氯 -4- (噸哪醇硼酸酯 -2-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧 氮杂卓 -5(6H)-酮 le (40 mg), 此产品与原料 Id的脱溴产品很难分离, 此粗产 品直接用于下一步反应。
第四步
4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-咪唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮
氮气保护条件下, 将 4-氨基 -6-P-氯 -4- (噸哪醇硼酸酯 -2-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 le粗品 (40 mg, 0.096 mmol)溶解于 1.2 mLl,4-二氧六环和水 (V/V=4: l)混合溶剂中, 加入 4-溴 -1-(2-氟 -2-甲基丙 基) -1H-咪唑 lc (21.22 mg, 0.096 mmol), 碳酸钾 (26.50 mg, 0.192 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (7 mg, 0.0096 mmol), 90°C下搅拌反应 12小 时。反应液冷却至室温,向反应液中加入 5 mL水,用乙酸乙酯萃取 5 mLx3),
合并有机相, 用饱和氯化钠溶液洗涤 5 mLx2;), 无水硫酸钠干燥, 过滤, 滤 液减压浓缩, 用制备高相液相色谱纯化所得残余物, 得到标题产物 4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-咪唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 1 (2.08 mg, 白色固体), 产率: 5.0%。
MS m/z (ESI): 431.1 [M+l]
JH NM (400 MHz, CD3OD) δ 9.15 (s, 1H), 8.40 (s, 1H), 8.02 (s, 1H), 7.76 (d, 2H), 7.55 (d, 1H), 4.98 (t, 2H), 4.52 (d, 2H), 4.29 (t, 2H), 1.42 (d, 6H)
实施例 103
2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)基)苯基)哌 啶基乙酸
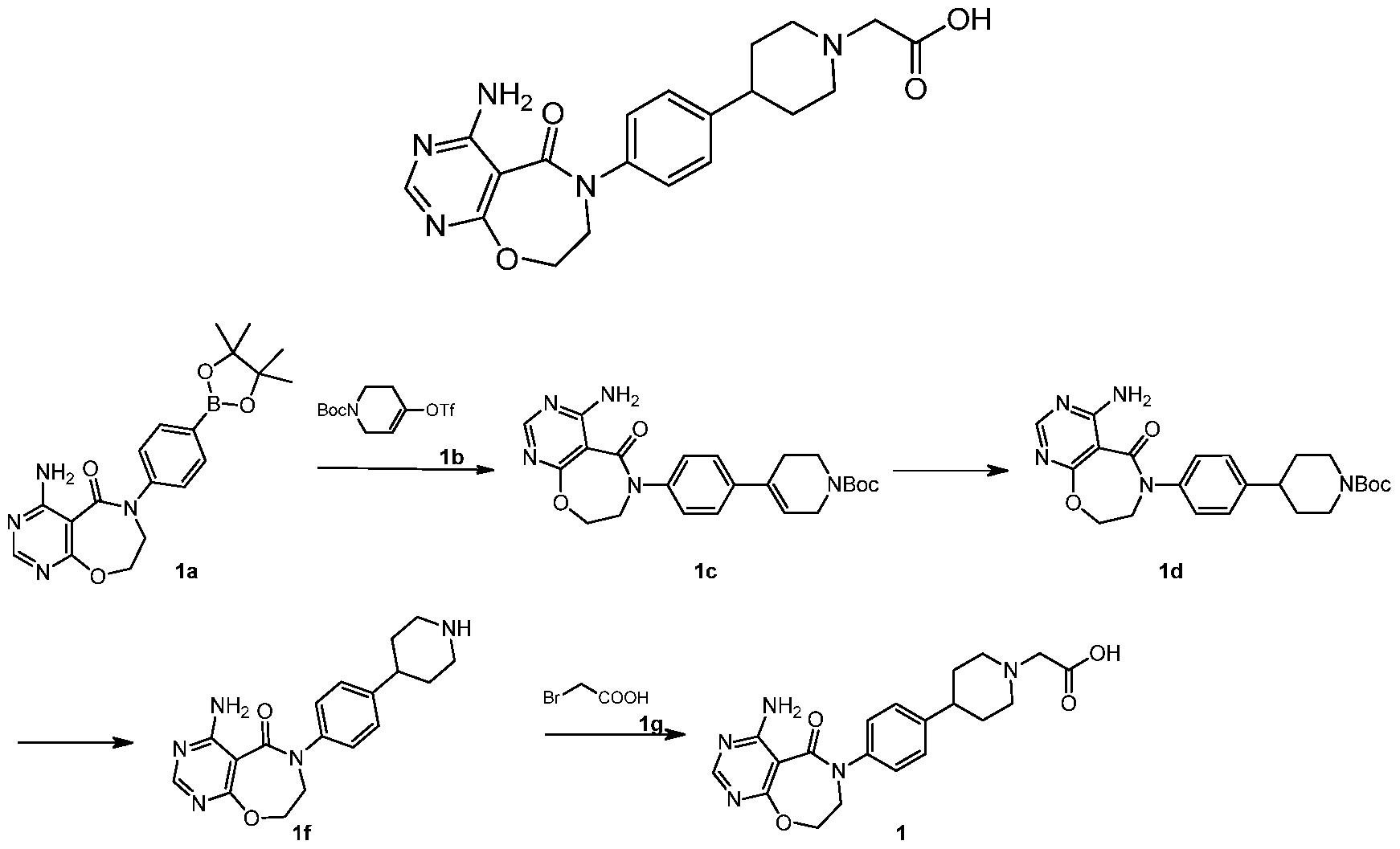
叔丁基 4_(4_(4_氨基 _5-氧代 -7,8-二氢嘧啶 -[5,4-f][l,4]氧氮杂卓 -6(5H)-基 苯基) -5,6-二羟吡啶 -1(2H)-羧酸酯
将 4-( ((三氟甲基)磺酰)氧基) -5,6-二氢 吡啶 -1(2H)-甲酸叔丁酯 1 (325 mg, 0.98 mmol, 采用公知的方法"专利 WO2005037826"制备而得), 1 (250 mg, 0.65 mmol, 采用公知的方法"专利 WO2011121350"制备而得), 碳酸钠
( 138 mg, 1.3 mmol), Ι,Γ-双 (二苯基磷)二茂铁氯化钯(24 mg, 0.033 mmol) 混合于 7.5 mL 四氢呋喃和水(V/V=2: l )混合溶剂中。 80°C 反应 12 小时。 向反应液中加入 10 mL水, 用乙酸乙酯萃取 (20 mLx3 ), 合并有机相, 用 饱和氯化钠溶液洗涤 IO mL), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用薄层层析以洗脱剂体系 C纯化所得残余物, 得到标题产物叔丁基 4-(4-(4- 氨基 -5-氧代 -7,8-二氢嘧啶 -[5,4-f][l,4]氧氮杂卓 -6(5H)-基苯基 )-5,6-二羟吡啶 -1(2H)-羧酸酯 3 (220 mg, 白色固体), 产率: 76.9%。
MS m/z (ESI): 438.3 [M+l]
JH NM (400 MHz, CDC13) δ 8.29 (s, 1H), 7.45 (d, 2H), 7.25 (d, 2H), 6.05 (s, 1H), 4.70 (t, 2H), 4.08 (m, 2H), 4.03 (t, 2H), 3.64 (t, 2H), 2.52 (m, 2H), 1.50 (s, 9H)
第二步
叔丁基 4_(4_(4_氨基 _5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基 苯基:)哌啶 -1-羧酸酯
将叔丁基 4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 -[5,4-f][l,4]氧氮杂卓 -6(5H)- 基苯基 )-5,6-二羟吡啶 -1(2H)- 羧酸酯 3 (220 mg, 0.50 mmol) 溶解于 10 mL 甲醇中, 加入乙酸钠 (410 mg, 5.0 mmol)和 10% 的湿钯碳 (lOO mg), 反应 液抽真空, 充入氢气, 反复操作 3次, 在氢气氛下室温搅拌反应 12小时。 将反应液过滤, 滤液减压浓缩, 得到粗品标题产物叔丁基 4-(4-(4-氨基 -5-氧 代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基苯基)哌啶 -1-羧酸酯 4 (200 mg, 白色固体:), 产物不经纯化直接进行下一步反应。
MS m/z (ESI): 440.3 [M+l]
第三步
4-氨基 -6-(4- (哌啶 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 冰浴下, 将叔丁基 4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -6(5H)-基苯基)哌啶 -1-羧酸酯 4 (200 mg, 0.45 mmol) 溶解于 4 mL 1,4-二
氧六环, 加入 1 mL 2 M氯化氢的 1, 4-二氧六环溶液。室温搅拌反应 1小时, 减压浓缩得到标题产物 4-氨基 -6-(4- (哌啶 -4-基:)苯基: )-7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 5 (140 mg, 淡黄色固体), 产率: 83.0%。
JH NM (400 MHz, DMSO-^) δ 8.32 (s, 1Η), 7.40-7.25 (m, 4H), 4.72 (t, 2H), 4.04 (t, 2H), 3.10-2.80 (m, 4H), 2.70-2.50 (m, 1H), 1.95-1.80 (m, 4H)
第四步
2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)基)苯基)哌 啶基)乙酸
将 4-氨基 -6-(4- (哌啶 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)- 酮 5 (80 mg, 0.21 mmol)溶解于 3 mL二氯甲烷中, 加入三乙胺 (0.15 mL, 1.06 mmol)和溴乙酸 6 (44 mg, 0.32 mmol), 室温搅拌反应 2小时。 将反应液减压 浓缩, 经 p-HPLC制备分离得到标题产物 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)基)苯基)哌啶基)乙酸 7 (13 mg, 白色固体),产率: 15.8%。
JH NMR (400 MHz, CD3OD) δ 8.43 (s, 1H), 7.45-7.30 (m, 4H), 5.00 (t, 2H), 4.23 (t, 2H), 3.79 (d, 1H), 3.51 (d, 1H), 3.30-3.10 (m, 2H), 3.05-2.90 (m, 1H), 2.18-1.90 (m, 4H)
实施例 104
2-(3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基)吡 咯 -1_基乙酸


3-0: (:三氟甲基:)磺酰:)氧 2,5-二氢 -m-吡咯 -1-羧酸叔丁酯
冰浴下,将 3-氧代吡咯烷 -1-羧酸叔丁酯 la (3 g, 16.2 mmol)溶解于 30 mL 四氢呋喃中, 加入六甲基二硅基氨基锂 (19.5 mL, 19.5 mmol)和 N-苯基双 (三 氟甲烷磺酰:)亚胺6.36 g, 18 mmol), 冰浴下搅拌反应 2小时。 向反应液中加 入 50 mL水, 用乙酸乙酯萃取30 mLx3;>, 合并有机相, 用饱和氯化钠溶液 洗涤 (50 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 3-(( (三氟甲基)磺酰)氧基) -2,5-二氢 -1H-吡咯 -1-羧酸叔丁酯 lb (9 g, 浅黄色油 状物), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 5.73 (d, 1H), 4.25-4.15 (m, 2H), 3.78-3.68 (m: 2H), 1.46 (s, 9H)
第二步
3-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -2,5-二氢 -1H-吡咯 -1-羧酸叔 丁酯
将粗品 3-(( (三氟甲基)磺酰)氧基) -2,5-二氢 -1H-吡咯 -1-羧酸叔丁酯 lb (9 g, 16.2 mmol)溶解于 100 mL二氧六环中, 加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'- 双 (1,3,2-二氧硼烷)(4. llg, 16.2 mmol), 乙酸钾 (4.72g, 48.6 mmol)和 Ι,Γ-双 (二 苯基磷)二茂铁氯化钯 (660 mg, 0.8 mmol), 90°C搅拌反应 12小时。 向反应液 中加入 lOO mL水, 用乙酸乙酯萃取 80 mLx3;), 合并有机相, 用饱和氯化钠 溶液洗涤 (lOO mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱
法以洗脱剂体系 C 纯化所得残余物, 得到粗品标题产物 3-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -2,5-二氢 -1H-吡咯 -1-羧酸叔丁酯 lc (4.5 g, 黄色油 状物)。
JH NM (400 MHz, CDC13) δ 6.50-6.40 (m, 1Η), 4.25-4.15 (m, 4H), 1.46 (s; 9H), 1.26 (s, 12H)
第三步
3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯
基:) -2,5-二氢 -1H-吡咯 -1-羧酸叔丁酯
将 3-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -2,5-二氢 -1H-吡咯 -1-羧酸 叔丁酯 lc (1.15 g, 0.78 mmol)溶解于 25 mL 二氧六环和水 (V/V=5/l)中,加入 4-氨基 -6-(4-碘苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (300 mg, 0.78 mmol, 采用公知的方法"专利 WO2009016462"制备而得), 碳酸钾 (216 mg, 1.57 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (64 mg, 0.08 mmol), 90°C搅拌 反应 12小时。 向反应液中加入 100 mL水, 用乙酸乙酯萃取 (60 mLx3;>, 合 并有机相, 饱和氯化钠溶液洗涤 lOO mL:), 无水硫酸钠干燥, 过滤, 滤液减 压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯基) -2,5-二 氢 -1H-吡咯 -1-羧酸叔丁酯 ld (160 mg, 白色固体), 产率: 48.19%。
JH NM (400 MHz, CDC13) δ 8.30 (s, 1H), 7.47 (d, 2H), 7.26(d, 2H), 6.17 (d, 1H), 4.71 (t, 2H), 4.31-4.52 (m, 4H), 4.04 (t, 2H), 1.53 (s, 9H)。
第四步
3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯基)吡咯 烷 -1-羧酸叔丁酯
将 3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基) -2,5-二氢 -1H-吡咯 -1-羧酸叔丁酯 Id (160 mg, 0.37 mmol)溶解于 5 mL 甲 醇中, 加入乙酸钠 (320 mg)和钯 /碳 (lOO mg), 反应液抽真空, 充入氢气, 反
复操作 3次, 在氢气氛下室温搅拌反应 1小时。 将反应液过滤, 滤液减压浓 缩, 得到粗品标题产物 3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6 5H;)-基:)苯基:)吡咯烷 -1-羧酸叔丁酯 le lOO mg, 白色固体:), 产物不经纯化 直接进行下一步反应。
JH NM (400 MHz, CDC13) δ 8.29 (s, IH), 8.18 (br. s., IH), 7.34 (d, 2H), 7.24 (d, 2H), 5.68 (br. s., IH), 4.70 (t, 2H), 4.01 (t, 2H), 3.80-3.70 (m, IH), 3.65-3.52 (m, IH), 3.50-3.25 (m, 3H), 2.35-2.20 (m, IH), 2.05-1.95 (m, IH), 1.49 (s, 9H)
第五步
4-氨基 -6-(4- (吡咯 -3-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 将 3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:)吡咯烷 -1-羧酸叔丁酯 le (100 mg, 0.23 mmol)溶解于 3 mL二氯甲烷中, 加入 1 mL氯化氢的 1,4-二氧六环 (V/V = 1:1)混合溶液, 搅拌反应 1小时。 将反应液减压浓缩, 得到粗品标题产物 4-氨基 -6-(4- (吡咯 -3-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H 酮 lf (50 mg, 黄色油状物), 产物不经纯化 直接进行下一步反应。
第六步
2-(3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基)吡 咯 -1_基)乙酸
将 4-氨基 -6-(4-(吡咯 -3-基)苯基 )-7,8-二氢嘧啶 [5,4-f] [1 ,4]氧氮杂卓 -5(6H)-酮 le (50 mg, 0.15 mmol)溶解于 3 mL二氯甲烷中,加入三乙胺 (78 mg, 0.77 mmol)和溴乙酸 (32 mg, 0.23 mmol), 搅拌反应 2小时。 将反应液减压浓 缩, 经 p-HPLC 制备分离得到标题产物 2-(3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯基)吡咯 -1-基)乙酸 1 (5 mg, 白色固体), 产 率: 8.52%。
JH NMR (400 MHz, CD3OD) δ 8.42 (s, IH), 7.44-7.52 (m, 2H), 7.39 (d,
2H), 4.99 (br. s., 2H), 4.28-4.35 (m, 2H), 4.23 (br. s., 2H), 3.32-4.14 (m, 5H), 2.43-2.61 (m, 1H), 2.07-2.38 (m, 1H)
实施例 105
4-氨基 -6-(3,5-二甲基 -4-(l-氧代 -2-(2,2,2-三氟乙基) -1,2-二氢异喹啉 -7-基)
-7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)- 酮

1
7-溴异喹啉 -1(2H)-酮
将 7-溴 -1-氯异喹啉 la (2.70 g, 11.10 mmol)溶解于 50 mL乙酸中,加入醋 酸铵 lb (8.50 g, 111.10 mmol), 在 100°C下搅拌反应 4小时。 将反应液减压 浓缩, 然后向剩余物中加水, 有固体物质析出, 过滤, 并用水洗, 得到标题 产物 7-溴异喹啉 -1(:2¾-酮 lc (2.7g, 白色固体:), 产物不经纯化直接进行下一 步反应。
MS m/z (ESI): 225.9 [M+l]
JH NM (400 MHz, CDC13): 8.55 (d, J = 1.8 Hz, 1H), 7.75 (dd, J = 2.1,
8.5 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 7.14 (d, J = 6.2 Hz, 1H), 6.52 (d, J = 7.3 Hz: 1H)
第二步
7-溴 -2-(2,2,2-三氟乙基)异喹啉 -1(2H)-酮
将粗品 7-溴异喹啉 -1(2H)-酮 lc (2.70 g, 12.05 mmol)溶解于 40 mL N, N- 二甲基甲酰胺中,加入碳酸铯 (7.80 g, 24.10 mmol)和三氟甲磺酸三氟乙酯 Id (3.35 g, 14.46 mmol), 搅拌反应 12小时。 反应液倒入 200 mL水, 有固体物 质析出, 过滤, 并用水洗固体物质, 将得到的固体物质溶于 200 mL乙酸乙 酯中, 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂 体系 C纯化所得残余物,得到标题产物 7-溴 -2-(2,2,2-三氟乙基)异喹啉 -1(2H)- 酮 le (2.5 g, 白色固体), 产率: 68.1 %。
JH NM (400 MHz, CDC13): 8.58 (d, J = 1.8 Hz, 1H), 7.78 (dd, J = 2.0, 8.4 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.09 (d, J = 7.3 Hz, 1H), 6.52 (d, J = 7.3 Hz, 1H), 4.67 (q, J = 8.6 Hz, 2H)
第三步
7-(4,4,5,5-四甲基 -1,3,2-二杂氧戊硼烷 -2-基) -2-(2,2,2-三氟乙基)异喹啉
-1(2H)-酮
将 7-溴 -2-(2,2,2-三氟乙基)异喹啉 -1(2H)-酮 le (2.50 g, 8.17 mmol )溶解 于 30 mL干燥的二氧六环中, 加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'-双 (1,3,2-二杂 氧戊硼烷) (3.10 g, 12.25 mmol), 醋酸钾 (2.40 g, 24.50 mmol) 和 Ι,Γ-双 (二苯 基磷)二茂铁氯化钯 (333 mg, 0.41 mmol), 在 100°C下反应 24小时。 待反应 温度降至室温,减压浓缩,向剩余物中加 40 mL水,用二氯甲烷萃取 (40 mLx3): 合并有机相, 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗 脱剂体系 C纯化所得残余物, 得到标题产物 7-(4,4,5,5-四甲基 -1,3,2-二杂氧 戊硼烷 -2-基) -2-(2,2,2-三氟乙基)异喹啉 -1(2氢) -酮 If (3.3 g,黄色油状),产率: 100 %。
Ή NM (400 MHz, CDC13): δ 8.92 (s, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.9 Hz, 1H), 7.09 (d, J = 7.5 Hz, 1H), 6.53 (d, J = 7.5 Hz, 1H), 4.68 (q, J = 8.7 Hz, 2H), 1.37 (s, 12H)。
第四步
4-氨基 -6-(3,5-二甲基 -4-(l-氧代 -2-(2,2,2-三氟乙基) -1 ,2-二氢异喹啉 -7-基) 苯基) -7,8-二氢嘧啶 [5,4-f] [l ,4] 氧氮杂卓 -5(6H)- 酮
将 7-(4,4,5,5-四甲基 -1 ,3,2-二杂氧戊硼烷 -2-基) -2-(2,2,2-三氟乙基)异喹啉 -1(2氢) -酮 If (3.12 g, 9.09 mmol) , 4-氨基 -6-(4-溴 -3,5-二甲基苯) -7,8-二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -6(5H)-酮 lg (3.0 g, 8.62 mmol) , 碳酸钾 (3.42 g, 24.78 mmol)和 Ι ,Γ-双 (二苯基磷)二茂铁氯化钯 (337 mg, 0.41 mmol) 溶解于 30 mL 二氧六环中, 加入 10 mL水, 100°C下搅拌反应 12小时。 待反应温度降至 室温, 反应减压浓缩, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C 纯化所得残余物, 用二氯甲烷重结晶所得粗产品, 得到标题产物 4-氨基 -6-(3,5-二甲基 -4-(1-氧代 -2-(2,2,2-三氟乙基) -1 ,2-二氢异喹啉 -7-基)苯基) -7,8- 二氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -5(6H)- 酮 1 (1.5 g, 白色固体),产率: 44.9 %。
MS m/z (ESI): 510.2 [M+l]
JH NMR (400 MHz, DMSO-d6): δ 8.16 (s, 1H), 7.95 (s, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.66 - 7.54 (m, 3H), 7.49 (d, J = 7.5 Hz, 1H), 7.18 (s, 2H), 6.78 (d, J = 7.5 Hz, 1H), 4.92 (q, J = 9.4 Hz, 2H), 4.61 (t, J = 4.5 Hz, 2H), 4.00 (t, J = 4.5 Hz, 2H), 1.97 (s, 6H)。
实施例 106
(E)- 4-氨基 -6-(6-((2,2,2-三氟乙氧基)亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8-二 氢嘧啶 [5,4-f] [l ,4]氧氮杂卓 -5(6H)-酮

第一步
6'-溴 -3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'-萘]
将 6-溴 2-四氢萘酮 la(10 g, 44.40 mmol)溶于 lOOmL的无水甲苯中, 室 温搅拌, 加入乙二醇 (2.7 mL, 48.30 mmol)和对甲苯磺酸 (3 mg:)。 反应物在 110°C下反应 6小时并且用分水器收集反应过程中生成的水。 待反应液冷却 至室温, 反应液分别用 4毫升 10%的氢氧化钠溶液, 20毫升的水洗涤。 有 机相用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 6'-溴 -3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] lb (10g, 黄色油状), 产率, 84.03%
MS m/z (ESI):270.9 [M+l]
JH NM (400 MHz, CDC13): δ 7.27 (s, 1H), 7.23 (d, 1H), 6.91 (d, 1H), 4.01-4.04 (m, 4H), 2.96 (t, 2H), 2.92 (s, 2H), 1.93 (t, 2H)
第二步
N-(2-((叔丁基二甲基硅基)氧代)乙基: )-3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊 环 -2,2,萘] -6,-胺
将 6'-溴 -3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] lb(10 g, 37.17 mmol), 2-((叔丁基二甲基硅基)氧代)乙胺 lc(7.8 g, 44.60 mmol)和碳酸铯 (36 g, 110.40 mmol)溶于 100 mL无水甲苯, 依次加入催化剂 Pd2(dba)3(700 mg) 和配 体 X-PhOS (700 mg;)。 反应物在 110°C下反应 18小时。 反应液冷却至室温, 加入二氯甲烷稀释, 并用水洗。 有机相用无水硫酸钠干燥, 过滤, 滤液减压 浓缩,硅胶柱色谱法以洗脱剂体系 C纯化所得残余物,得到标题产物 N-(2-((叔 丁基二甲基硅基)氧代)乙基) -3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-胺 Id (1.53克, 黄色油状), 产率, 11.3%
MS m/z (ESI):364.1 [M+l]
JH NM (400 MHz, CDC13): δ 6.7 (d, 1H), 6.3 (d, 1H), 6.26 (s, 1H)„ 3.87 (m, 4H), 2.77 (t, 2H), 2.72 (s, 2H), 1.77 (t, 2H) , 0.76 (s, 9) , 0.07 (s 6H)
第三步
N-(2-((叔丁基二甲基硅基)氧代)乙基: )-4,6二氯 -Ν-(3',4'-二氢 -ΓΗ-螺环
[[1,3]二氧戊环 -2,2'萘] -6'-基)嘧啶 -5-甲酰胺
将 Ν-(2- :叔丁基二甲基硅基:)氧代:)乙基: )-4,6二氯 -Ν-(3',4'-二氢 -ΓΗ-螺 环 [[1,3]二氧戊环 -2,2'萘] -6'-基)嘧啶 -5-甲酰胺 Id (1.53 g, 4.20 mmol)和 4,6- 二氯 -5-嘧啶甲酰氯 le (1.15 g, 5.40 mmol)溶于 15毫升无水二氯中, 缓慢滴 加三乙胺 1.6 mL:)。 反应物搅拌 18小时后, 反应液用二氯甲烷稀释, 并用水 洗涤。 有机相用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗 脱剂体系 C纯化所得残余物,得到标题产物 N-(2- ((叔丁基二甲基硅基)氧代) 乙基) -4,6二氯 -N-(3,,4,-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2,萘] -6,-基)嘧啶 -5-甲 酰胺 lf(2.1 g, 黄色油状), 产率 92.9%
MS m/z (ESI):538.0 [M+l]
第四步
4,6二氯 -N-(3,,4,-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2,萘] -6,-基) -N-(2-羟乙 基)嘧啶 _5_甲酰胺
将化合物 Ν-Ρ- :叔丁基二甲基硅基:)氧代:)乙基: )-4,6二氯 -Ν-ρ',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基)嘧啶 -5-甲酰胺 1 (1.7 g, 3.17 mmol)溶 于盐酸乙醇混合溶液(v:v =0.6mL:20mL)。 反应半小时后, 用饱和碳酸氢钠 溶液调节 pH至 8左右。 再用二氯甲烷稀释反应液, 用水洗涤。 有机相用无 水硫酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物,得到标题产物 4,6二氯 -Ν-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2' 萘] -6'-基;) -Ν-(2-羟乙基:)嘧啶 -5-甲酰胺 lg (450毫克, 黄色油状:), 产率 33%
MS m/z (ESI):424.0 [M+l]
第五步
4-氯 -6-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6Η)-酮
将 4,6二氯 -Ν-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -Ν-(2-羟 乙基)嘧啶 -5-甲酰胺 lg(450 mg, 1.06 mmol)和碳酸钾 (439 mg, 3.18 mmol)溶于 10毫升乙腈, 在 80°C下反应 16小时。待反应液冷却至室温后, 用二氯甲烷 稀释反应液, 用水洗涤。 有机相用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 硅胶柱色谱法以洗脱剂体系 C纯化所得残余物,得到标题产物 4-氯 -6-(3',4'- 二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -5(6H)-酮 lh (410 mg, 黄色固体), 产率 100%
MS m/z (ESI):388.0 [M+l]
JH NM (400 MHz, DMSO- ): δ 8.82 (s, 1H), 7.22 - 7.10 (m, 3H), 4.71 (t, 2H), 4.11 (t, 2H), 3.95 (s, 4H), 2.91 (br. s., 4H), 1.87 (t, H)
第六步
4-氨基 -6-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -7,8-二氢嘧 啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将 4-氨基 -6-(3',4'-二氢 -1Ή-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lh (410 mg, 1.11 mmol)溶于 0.5摩尔氨气 的 1,4-二氧六环溶液 (5 mL)。 搅拌 16小时后, 将反应液浓缩所得残余物, 得到标题产物 4-氨基 -6-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'- 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 li (370 mg, 黄色固体),产率: 100%
MS m/z (ESI):369[M+1]
第七步
4-氨基 -6-(6-氧代 -5,6,7,8-四氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓
-5(6H)-酮
将 4-氨基 -6-(3',4'-二氢 -ΓΗ-螺环 [[1,3]二氧戊环 -2,2'萘] -6'-基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 li (150 mg, 0.41 mmol)溶于三氟乙酸和二 氯甲烷的混合溶液 (V: v= l mL: 4mL),加入 0.12mL的水。搅拌 16小时后, 反应液用二氯甲烷稀释, 用水洗涤。 有机相用无水硫酸钠干燥, 过滤, 滤液 减压浓缩所得残余物, 得到标题产物 4-氨基 -6-(6-氧代 -5,6,7,8-四氢萘 -2- 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 lj(120mg, 黄色油状),产率: 90.9%
第八步
(E)- 4-氨基 -6-(6-((2,2,2-三氟乙氧基)亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮
将 4-氨基 -6-(6-氧代 -5,6,7,8-四氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂 卓 -5(6H)-酮 lj (90 mg, 0.278 mmol), 0-(2,2,2-三氟乙基)羟胺 lk (42 mg, 0.305 mmol)和醋酸钠 (68.3 mg, 0.833 mmol) 溶于乙醇和水的混合溶液 (v: v = 4mL : l mL), 在 80°C反应 3小时。 待反应温度降至室温, 反应液过滤, 滤液减压浓缩, 用制备高效液相色谱分离方法得到标题产物 ( - 4-氨基 -6-(6-((2,2,2-三氟乙氧基)亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6H)-酮 l (7.15 mg,黄色固体), 产率: 7.15%
MS m/z (ESI):422.1[M+l]
JH NMR (400 MHz, DMSO- ): δ 8.30 (s, IH), 7.34 - 7.17 (m, 3H), 4.74 - 4.58 (m, 4H), 4.03 (br. s., 2H), 3.79 (s, IH), 3.55 (s, IH), 2.86 (q, J = 6.0 Hz, 2H), 2.61 (m,lH), 2.54 (br. s., IH)
实施例 107
4-氨基 -3,4-二氢萘 -1 (2H) -酮氧 -(2,2,2-三氟乙氧基) 肟 -7,8-二氢嘧啶
5,4-f][l,4] 咪唑啉 -5 (6H)-基) -酮

1k
第一步
5-氧代 -5,6,7,8-四氢萘 -2-基三氟甲磺酸酯
将 6-羟基 -3,4-二氢萘 -1 (2H) -酮 (5 g, 30.8 mmol)和三乙胺 (3.1 g, 30.8 mmol)溶解于 50 mL无水二氯甲烷中, 在冰浴下, 加入三氟甲磺酸酐(8.7 g,
30.8 mmol)。室温搅拌 18小时。反应液依次用水, 饱和碳酸氢钠溶液洗涤, 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以展开剂体系 B 纯化所得残余物, 得到标题产物 lb 2.0 g, 无色液体:), 产率: 24.0%
JH NM (400 MHz, CDC13): 8.13 (d , 2Η), 7.20-7.15 (m, 2Η), 3.05-2.95 (m, 2H), 2.70-2.62 (m, 2H), 2.20-2.10 (m, 2H)。
第二步
(Z)-5-((2,2,2-三氟乙氧)亚氨基 )- 5,6,7,8-四氢萘 -2-基三氟甲磺酸酯 将 5-氧代 -5,6,7,8-四氢萘 -2-基三氟甲磺酸酯 1 (2.0 g, 6.8 mmol),氧 -(2,2,2- 三氟乙氧基)盐酸羟胺 3 (1.54 g, 10.2 mmol), 乙酸钠 (1.67 g, 20.4 mmol) 混 合于 25 mL 乙醇和水 (V:V=4:i;>的混合溶液中. 100°C回流反应 6小时. 反应 液冷却到室温, 用 50 mL乙酸乙酯稀释, 乙酸乙酯层用水洗涤 (30 mL), 无 水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以展开剂体系 B纯化 所得残余物, 得到标题产物 Id (1.6 g, 淡黄色液体), 产率: 61.0%。
JH NMR (400 MHz, CDC13): 8.02 (d , 2H), 7.10-7.05 (m, 2H), 4.58-4.50 (m, 2H), 2.70-2.65 (m, 4H), 1.90-1.82 (m, 2H)。
第三步
(Z)-6-((2- ((叔丁基二甲基硅基)乙基)氨) -3,4-二氢萘 -1 (2H) -酮氧 -(2,2,2- 三氟乙基) 肟
将 (Z)-5-((2,2,2-三氟乙氧)亚氨基) - 5,6,7,8-四氢萘 -2-基三氟甲磺酸酯 4 ( 1.6 g, 4.09 mmol) , 2- ( (叔丁基二甲基) 氧代) 乙胺 5 (1.07 g, 6.4 mmol), 碳酸铯 (2.66 g, 8.18 mmol) , Pd2(dba)3(100 mg), x-phos (100 mg) 混合于 15 mL 无水甲苯中。 110°C氮气保护下反应 18小时. 反应液冷却到室温, 减压浓 缩, 用硅胶柱色谱法以展开剂体系 B纯化所得残余物, 得到标题产物
(Z)-6-((2- ((叔丁基二甲基硅基)乙基)氨) -3,4-二氢萘 -1 (2H) -酮氧 -(2,2,2-三氟 乙基) 肟 If (440 mg, 淡黄色液体), 产率: 26.0 %。
JH NM (400 MHz, CDC13): 6.60 (q, 1H), 6.53 (d, 1H), 6.38 (q, 1H),
4.45-4.35 (d, 2H), 3.70 (t, 2H), 3.15-3.11 (m, 2H), 2.70 - 2.55 (m, 4H), 1.80-1.70 (m, 2H), 0.86 (s, 6H), 0.82 (s, 9H)。
第四步
(Z)-氮 -(2((叔丁基二甲基硅基)氧代)乙基) -4,6-二氯-氮 -(5-((2,2,2-三氟乙 氧基) 亚氨基 )-5,6,7,8-四氢萘 -2-基)嘧啶 -5-甲酰胺 将 (Z)氮 -(2((叔丁基二甲基硅基)氧代)乙基) -4,6-二氯-氮 -(5-((2,2,2-三氟 乙氧基) 亚氨基 )-5,6,7,8-四氢萘 -2-基)嘧啶 -5-甲酰胺 If (440 mg, 1.06 mmol) 和三乙胺 (440 umL, 1.38 mmol) 溶解于 6 mL二氯甲烷中, 在冰浴下, 加入 4,6-二氯嘧啶 -5-甲酰胺 lg (336 mg, 1.59 mmol)。 室温反应 1小时。 将反应液 减压浓缩, 用硅胶柱色谱法以展开剂体系 B纯化所得残余物, 得到标题产物 lh (320 mg, 淡黄色固体), 产率: 51.0 %。
JH NM (400 MHz, CDC13): 58.59 (s, 1H), 8.20 (s, 1H), 7.25-7.17 (m, 2H), 4.51 (q, 2H), 4.05-3.90 (m, 4H), 2.72-2.62 (m, 4H), 1.80-1.70 (m, 2H), 0.87 (s, 9H), 0.06 (s, 6H)。
第五步
(Z)-4,6-二氯-氮 -(2-羟基乙基) -氮 -(5-((2,2,2-三氟乙氧基) 亚氨
基:) -5,6,7,8-四氢萘 -2-基:)嘧啶 -5-甲酰胺
将 (Z)-4,6-二氯-氮 -(2-羟基乙基) -氮 -(5-((2,2,2-三氟乙氧基) 亚氨 基) -5,6,7,8-四氢萘 -2-基)嘧啶 -5-甲酰胺 lh (270 mg, 0.46 mmol) 溶解于 5 mL 乙醇中, 加入盐酸 (0.15 mL)。 室温反应 1.5 小时。 用 30 mL乙酸乙酯稀释, 乙酸乙酯层用饱和碳酸氢钠溶液洗涤, 无水硫酸钠干燥, 过滤, 滤液减压浓 缩得到标题产物 li (150 mg, 淡黄色固体), 产率: 69.0%。
JH NM (400 MHz, CDC13): 58.61 (s, 1H), 7.81-7.92 (m, 1H), 7.21 (s, 2H), 4.51 (q, 2H), 4.06-4.13 (m, 2H), 3.88-3.97 (m, 2H), 2.57-2.83 (m, 4H), 2.11-2.23 (m, 1H), 1.72-1.89 (m, 2H)。
第六步
(Z)-4-氯 -6-(5-((2,2,2-三氟乙氧基) 亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8-二氢 嘧啶 [5,4-f][l,4] 咪唑啉 -5(6H)-基) -酮
将 (Z)-4,6-二氯-氮 -(2-羟基乙基) -氮 -(5-((2,2,2-三氟乙氧基) 亚氨 基) -5,6,7,8-四氢萘 -2-基) 嘧啶 -5-甲酰胺 lj (100 mg, 0.20 mmol)和三乙胺
(0.084 mL , 0.60 mmol) 溶解于 4 mL 乙睛中, 80°C反应 18 小时。 反应 液冷却到室温, 用 30 mL乙酸乙酯稀释, 乙酸乙酯层用水洗涤, 无水硫酸钠 干燥, 过滤, 滤液减压浓缩得到标题产物 lk (80 mg, 淡黄色固体), 产率: 91.0%。
MS m/z (ESI): 441.1 [M+l]
第七步
4-氨基 -3,4-二氢萘 -1(2H)-酮氧 -(2,2,2-三氟乙氧基) 肟 -7,8-二氢嘧啶
[5,4-f][l,4] 咪唑啉 -5(6H)-基) -酮
将 (Z)-4-氯 -6-(5-((2,2,2-三氟乙氧基) 亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8-二 氢嘧啶 [5,4-f][l,4] 咪唑啉 -5(6H)-基) -酮 lk (80 mg, 0.18 mmol)和 5 mL 0.5M 氨气的 1, 4-二氧六环溶液混合, 室温反应 18 小时。 反应液减压浓缩, 用薄 层色谱法以展开剂体系 C纯化所得残余物, 得到标题产物得到标题产物 (Z)-4-氯 -6-(5-((2,2,2-三氟乙氧基) 亚氨基 )-5,6,7,8-四氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -5(6H)-基) -酮 1 (58.94 mg, 白色固体), 产率: 78.0%。
JH NM (400 MHz, MeOD): 58.19 (s, 1H), 8.03 (d, 1H), 7.24 (d, 2H), 4.74 (q, 2H), 4.65 (q, 2H), 4.07-4.12 (m, 2H), 2.83 (t, 4H), 1.90 (q, 2H)
MS m/z (ESI): 422.0 [M+l]
实施例 108
4-氨基 -6-(6-(2,2,2-三氟乙氧基) -5,6,7,8-四氢萘 -2-基) -7,8-二氢吡啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮

1f 19
第一步
7-溴 -3-(2,2,2-三氟乙氧基) -1,2-二氢萘
将 6-溴 -3 ,4-二氢萘 -2-醇 la (2.0 g, 8.9 mmol )溶解于 30 mL N, N-二甲基 甲酰胺中, 加入三氟甲磺酸三氟乙酯 (792 mg, 19.8 mmol ) )和碳酸铯 (5.80 g, 17.8 mmol ), 室温搅拌反应 12小时。 向反应液中加入 90 mL水, 用乙酸乙 酯萃取 (40 mLx3;), 合并有机相, 次用水 (20 mLx2;)、 饱和氯化钠溶液洗涤 (20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 7-溴 -3-(2,2,2-三氟乙氧基) -1,2-二氢萘 lb (2.7 g,棕色固体),产物不经纯化直接进 行下一步反应。
Ή NM (400 MHz, CDC13): δ 7.23-7.19 (m, 2H), 6.79-6.77 (dd, 2H), 5.45 (s, 1H), 4.22-4.15 (m, 2H), 2.89-2.84 (m, 2H), 2.47-2.41 (m, 2H)。
第二步
N-(2- ((叔丁基二甲基硅)氧代)乙基) -6-氯 -(2,2,2-三氟乙氧基) -7,8-二氢萘
-2-酰胺
将 7-溴 -3-(2,2,2-三氟乙氧基) -1,2-二氢萘 lb (2.7 g, 8.9 mmol)溶解于 30 mL甲苯中, 加入 2- ((叔丁基二甲基硅)氧代)乙胺 (1.87 g, 10.7 mmol), 碳酸 铯 (5.80 g, 17.8 mmol),三 (二苯亚苄基丙酮)二钯 (412 mg, 0.45 mmol)和 4,5- 双二苯基膦 -9,9-二甲基氧杂蒽 (212 mg, 0.45 mmol), 90°C搅拌反应 12小时。 反应液冷却至室温,向反应液中加入 30 mL水,用乙酸乙酯萃取20 mLx3;), 合并有机相, 依次用水 (20 mLx2;)、 饱和氯化钠溶液洗涤20 mLx2;), 用无水 硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所 得残余物, 得到标题产物 N-(2- ((叔丁基二甲基硅)氧代)乙基) -6-氯 -(2,2,2-三 氟乙氧基) -7,8-二氢萘 -2-酰胺 lc (800 mg, 黄色油状物), 产率: 22.4%。
第三步
N-(2- ((叔丁基二甲基硅)氧代)乙基) -4,6-二氯 -N-(6-(2,2,2-三氟乙氧
基 )-7,8_二氢萘 _2_基)嘧啶 _5_酰胺
将 N-(2- ((叔丁基二甲基硅)氧代)乙基) -6-氯 -(2,2,2-三氟乙氧基) -7,8-二氢 萘 -2-酰胺 lc (800 mg, 1.99 mmol)溶解于 15 mL二氯甲烷中,加入三乙胺 (0.83 mL, 6.0 mmol )和 4,6-二氯 -5-酰氯嘧啶 (506 mg, 2.39 mmol), 搅拌反应 12 小时。向反应液中加入 30 mL水,用二氯甲烷萃取20 mLx3;),合并有机相, 依次用水 (20 mLx2;)、 饱和氯化钠溶液洗涤20 mLx2;), 用无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 N-(2-((叔丁基二甲基硅)氧代)乙 基) -4,6-二氯 -N-(6-(2,2,2-三氟乙氧基) - 7,8-二氢萘 -2-基)嘧啶 -5-酰胺 Id (570 mg, 黄色固体), 产物不经纯化直接进行下一步反应。
JH NM (400 MHz, CDC13): δ 8.51 (s, 6Η), 7.07-7.03 (m, 2H), 6.70 (d, 1H),
5.36 (s, 6H), 4.16-4.09 (q, 2H), 3.92 (t, 2H), 3.92 (t, 2H), 2.74 (t, 2H), 2.35 (t, 2H), 0.81 (s, 9H) , 0.00 (s, 6H)。
第四步
4,6-二氯 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) -7,8-二氢嘧啶
[5,4-f][l,4] 氧氮杂卓 -5(6氢) -酰胺
将粗品 N-(2- ((叔丁基二甲基硅)氧代)乙基) -4,6-二氯 -N-(6-(2,2,2-三氟乙 氧基) - 7,8-二氢萘 -2-基)嘧啶 -5-酰胺 Id (570 mg, 0.99 mmol)溶解于 10 mL乙 醇中, 加入浓盐酸 (0.3 mL:), 搅拌反应 1小时。 向反应液中加入 10 mL水, 用乙酸乙酯萃取 10 mLx3;), 合并有机相, 依次用水 (10 mLx2;>、 饱和碳酸氢 钠溶液 (10 mLx2;>、 饱和氯化钠溶液洗涤 10 mLx2;), 用无水硫酸钠干燥, 过 滤, 滤液减压浓缩, 得到粗品标题产物 4,6-二氯 -6-(6-(2,2,2-三氟乙氧基) -7,8- 二氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6氢) -酰胺 le (460 mg, 黄 色固体), 产物不经纯化直接进行下一步反应。
第五步
4-氯 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6氢:) -酮
将粗品 4,6-二氯 -N-(2-羟乙基) -N-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) 嘧啶 -5-酰胺 le (460 mg, 1.00 mmol)溶解于 10 mL乙腈中, 加入三乙胺 (0.42 mL, 3.02 mmol), 80°C搅拌反应 12小时。 反应液冷却至室温, 向反应液中 加入 20 mL水, 用乙酸乙酯萃取 (20 mLx3), 合并有机相, 用饱和食盐水洗 涤 (20 mLx2), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到粗品标题产物 4-氯 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮 杂卓 -5(6氢) -酮 If (350 mg,黄色固体),产物不经纯化直接进行下一步反应。
第六步
4-氨基 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6氢:) -酮
将粗品 4-氯 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基 )-7,8-二氢嘧啶
[5,4-f][l,4] 氧氮杂卓 -5(6氢) -酮 If (350 mg, 0.82 mmol)溶解于 10 mL 0.5 M 氨气的 1, 4-二氧六环溶液中, 搅拌反应 12小时。 反应液减压浓缩, 得到粗 品标题产物 4-氨基 -6-(6-(2,2,2-三氟乙氧基) -7,8-二氢萘 -2-基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮 lg (300 mg,黄色固体),产物不经纯化直接进 行下一步反应。
第七步
4-氨基 -6-(6-(2,2,2-三氟乙氧基) -5,6,7,8-四氢萘 -2-基) -7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮
将 4-氨基 -6-(6-(2,2,2-三氟乙氧基 )-7,8-二氢萘 -2-基 )-7,8-二氢嘧啶
[5,4-f][l,4] 氧氮杂卓 -5(6氢) -酮 lg (300 mg, 0.74 mmol) 溶解于 10 mL甲醇中, 先在空气中加入 10%湿钯碳 (60 mg),通入氢气三次,排出空气,加压到 50 psi, 70°C下搅拌反应 12小时。 反应液冷却至室温, 过滤, 滤液减压浓缩, 用制 备高相液相色谱纯化所得残余物,通过手性分离得到标题产物的两个同分异 构体 4-氨基 -6-(6-(2,2,2-三氟乙氧基 )-5,6,7,8-四氢萘 -2-基 )-7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮 1 前峰 (26.71 mg,白色固体)和后峰 (52.85 mg, 白色固体:), 产率: 26.3%。
MS m/z (ESI): 409.1 [M+l]
JH NM (400 MHz, Methanol-d4) δ 8.16 (s, 1H), 7.18-7.07 (m, 3H), 4.71-4.69 (m, 2H), 4.07-3.96 (m, 5H), 3.31-2.77 (m, 4H), 2.10-1.88 (m, 2H)。
JH NM (400 MHz, Methanol-d4) δ 8.16 (s, 1H), 7.19-7.07 (m, 3H), 4.71-4.69 (m, 2H), 4.07-3.96 (m, 5H), 3.31-2.77 (m, 4H), 2.08-1.89 (m, 2H)。
实施例 109
4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -2-(2,2,2-三氟乙基) -2H-吲唑 -5-基)苯
基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮


(E)-(l-(5-溴 -2-氟苯基) 亚乙基)肼
将 1-(5-溴 -2-氟苯基)乙酮 la (2 g, 9.2 mmol)和水合肼 (7.8 g, 16 mmol) 的混合物在 100°C下搅拌反应 12小时。向反应液中加入水 (150 mL:),用乙酸 乙酯萃取 (50 mLx3;>, 合并有机相, 用水溶液50 mL:)、 饱和氯化钠溶液洗漆 (50 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 得到标题产物 (1-(5- 溴 -2-氟苯基) 亚乙基)肼 lb (2.48 g, 白色固体), 产率: 100%。
第二步
5-溴 -3-甲基 -1H-吲唑
将 (E)-(l-(5-溴 -2-氟苯基) 亚乙基)肼 lb (2.48 g, 10.8 mmol)溶解于乙二醇 (24 πι 中, 165°C微波反应 90分钟。 向反应液中加入水 (100 mL:), 用乙酸 乙酯萃取 (50 mLx3;>, 合并有机相, 用水溶液50 mL:)、 饱和氯化钠溶液洗漆 (50 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 将反应液过滤, 滤液减 压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 5-溴 -3-甲基 -1H-吲唑 lc (1.26 g, 白色固体), 产率: 55.50%。
Ή NM (400 MHz, CDC13): 57.82(d,7= 1.6 Hz, 1H), 7.44(dd, J}=S.S Hz, J2= 1.6 Ηζ,ΙΗ), 7.32 (d,7=8.4 Hz, 1H), 4.73 (br, s, 1H), 2.55(s, 3H).
第三步
5-溴 -3-甲基 -2-(2,2,2-三氟甲基) -2H-吲唑
将 5-溴 -3-甲基 -1H-吲唑 lc (1.26 g, 6 mmol)溶解于 10 mLN,N,-二甲基甲 酰胺中,加入碳酸铯 (3.9 g, 12 mmol), 2,2,2-三氟乙基-三氟甲磺酸酯 (2.1 g, 9 mmol), 室温反应 12h。 向反应液中加入水 (100 mL), 用乙酸乙酯萃取 (50 mLx3), 合并有机相, 用水溶液 (50 mL), 饱和氯化钠溶液洗涤 (50 mL), 无 水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化 所得残余物, 得到标题产物 5-溴 -3-甲基 -2-(2,2,2-三氟甲基) -2H-吲唑 Id (260 mg, 白色固体), 产率: 14.80%。
JH NMR (400 MHz, CDC13): δ 7.73 (d, 7 = 1.2 Hz, 1H), 7.53 (d, J = 9.2 Hz, 1H), 7.35 (dd, J}= 9.2 Hz, J2= 2.0 Hz, 1H), 4.93 (q, J}= 16.4 Hz, J2= 8.4 Hz, 2H), 2.63 (s, 3H).
第四步
3-甲基 -5-(4,4,5,5-八甲基 -1,3,2-二氧硼烷 -2-基) -2-(2,2,2-三氟乙基) -2H-吲 唑
将 5-溴 -3-甲基 -2-(2,2,2-三氟甲基) -2H-吲唑 Id (260 mg, 0.9 mmol) 溶于 1,4-二氧六环 (5 mL) 中,加入 4,4,4',4',5,5,5',5'-八甲基 -2,2'-双 (1,3,2-二氧硼烷) (250 mg, 0.98 mmol),乙酸钾 (260 mg, 2.7 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯 化钯 (36 mg, 0.04 mmol), 90°C 下搅拌 12h。 向反应液中加入水 (30 mL), 用 乙酸乙酯萃取 (20 mLx3;>, 合并有机相, 用水溶液 PO mL:)、 饱和氯化钠溶液 洗涤 (30 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以 洗脱剂体系 A纯化所得残余物,得到标题产物 3-甲基 -5-(4,4,5,5-八甲基 -1,3,2- 二氧硼烷 -2-基) -2-(2,2,2-三氟乙基) -2H-吲唑 le (200 mg, 白色固体), 产率: 66.67%。
Ή NM (400 MHz, CDC13): δ 8.15 (s, 1H), 7.65 (d, J = 8.8 Hz, 1H), 7.59 (d, 7=8.8 Hz, 1H), 4.93 (q, J} = 16.8 Hz, J2 = 8.4 Hz, 2H), 2.67 (s, 3H), 1.36 (s, 12H).
第五步
4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -2-(2,2,2-三氟乙基) -2H-吲唑 -5-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 将 3-甲基 -5-(4,4,5,5-八甲基 -1,3,2-二氧硼烷 -2-基) -2-(2,2,2-三氟乙基) -2H- 吲唑 le (94 mg, 0.27 mmol)溶解于 2.4 mL二氧六环和水 (V/V=5/l)中, 加入 4-氨基 -6-(3,5-二甲基 -4-溴苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮 (80 mg, 0.22 mmol),碳酸钾 (61 mg, 0.44 mmol)和 Ι,Γ-双 (二苯基磷)二茂铁氯化钯 (9 mg, 0.011 mmol), 100°C微波反应 40分钟。 向反应液中加入 20 mL水, 用乙酸乙酯萃取 10 mLx3;), 合并有机相, 饱和氯化钠溶液洗涤 PO mL:), 无 水硫酸钠干燥, 过滤, 滤液减压浓缩, 用 p-HPLC制备分离纯化, 得到标题 产物 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -2-(2,2,2-三氟乙基) -2H-吲唑 -5-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮 1 (52 mg, 白色固体),产率: 47.70%。
JH NMR (400 MHz, DMSO-^): δ 8.48 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.46 (s, 1H), 7.15 (s, 2H), 7.00 (d, J = 9.6 Hz, 1H), 5.41 (q, J} = 18.0 Hz, J2 = 8.8 Hz, 2H), 4.86 (s, 2H), 4.15 (s, 2H), 2.65 (s, 3H), 2.00 (s, 6H).
实验例 1 : 体外评价
通过测定 IC50值来评价受试化合麴对人 /鼠 DGAH 的挿制能力, 通过 IC50 值可以计算抑制常数 Ki ( Cheng-Prusoff equation: IQ = IC50/ (1 +
[S]/Km) )
( 1 ) 人 DGAT1 IC50(nM)实验
供试样品: 从实施例 1〜109所制备的产物中选取一部分结构有代表性 的化合物进行人 DGAT1 IC50(nM)实验。
实验方法: 采用表达纯化的人 DAGAT1酶, 以二酰基甘油和 14C标记 的油酰基辅酶 A为底物, 选用 PE公司的 Flashplate检测板, 构建人 DGAT1 酶学反应体系。 14C标记的油酰基辅酶 A在酶学反应后与二酰基甘油共价结 合, 产生 14C标记的产物三酰基甘油。 由于脂溶性差异, 14C标记的底物油 酰基辅酶 A溶于反应液, 产物 14C标记的三酰基甘油与 Flashplate板底的磷 脂层结合, 从而有效分离标记底物和产物。 通过测定与磷脂层结合的 14C标 记的三酰基甘油的放射性, 计算化合物对产物生成的抑制性 (IC50) 。
实验结果见表 1:
表 1 人 DGAT1 IC50(nM)测试结果

化合物 90 61.98 57.99 59.99
化合物 91 12.46 27.49 19.98
化合物 93 64.51 74.20 69.36
化合物 94 63.06 55.12 59.09
化合物 95 32.18 52.85 42.52
化合物 96 69.35 71.72 70.54
化合物 97 30.28 23.41 26.85
化合物 99 104.30 79.10 91.70
化合物 105 27.05 36.43 31.74
化合物 108 (前峰) 103.20 43.32 73.26
化合物 108 (后峰) 56.38 48.78 52.58 注: hDGATl IC50<100 nM即表明具有较高的体外生物活性; 阳性化合 物 PF-04620110采用公知的方法" Discovery of PF-04620110: A Potent,
Orally-Bioavailable Inhibitor of DGAT-1, ACS Med. Chem. Lett. 2011, 2, 407- 412"制备而得; 阳性化合物 Pradigastat采用公知的方法"专利 WO2007126957" 制备而得。
结论: 与现有技术相比, 本发明化合惣表现出较强或相当的人 DGAH 的抑制能力。
(2) 其他体外活性实验
供试样品: 在上述人 DGATl IC50(nM;)实验的基础上, 选择其中一些高
活性、 结构有代表性的化合物开展进一步实验。
实验方法:
鼠 DGATl IC50测定方法:除了应用表达纯化的鼠 DGAT1,与人 DGAT1 IC50实验方法相同。
细胞HEK) DGAT1抑制检测:
以稳定同位素 13C标记的油酸分子处理 HEK293细胞, 细胞内源性表达 的 DGAT-1催化 13C标记的油酸分子整合到甘油三脂中; 分离萃取细胞悬液 中的甘油三脂成分; 通过液相 -质谱联用法(LC-MS/MS )检测产中甘油三酯 中 13C含量, 确定 DGAT1催化反应速率, 进而确定化合物对 DGAT1抑制 作用 (IC50 ) 。
实验结果见表 2:

结论: 与 Pradigas 相比, 本发明化合物的 cLogP明显更接近理想值, 溶解度方面具有明显优势 (3~8 倍 对鼠 DGAT1 酶的抑制活性相当: 与 PF-04620110相比, 化合物 105有更好的细胞活性。
( 3 ) 体外药代动力学性质研究
供试样品: 在上述实验的基础上, 选择其中一些高活性、 结构有代表性 的化合物开展进一步实验。
实验方法: 该研究的目的是为了测定该化合麴药代动力学参数 并计
算其在雄性 SD大鼠中的口服生物利用度。 该项目使用四只雄性 SD大鼠, 两只大鼠迸行静脉注射给药, 给药剂量为 2 mg/kg, 收集 O h (给药前) 和 给药后 0.0833, 0.25, 0.5, 1, 2, 4, 7, 24h的血浆样品, 另外两只大鼠口 服灌胃给药, 给药剂量为 0 mg/kg, 收集 O h (给药前) 和给药后 0.5, 1, 2, 3, 4, 6, 24 h的血浆样品, 然后对收集的样品进行 LCZMS/MS分析并采 集数据, 采集的分析数据用 Phoemx WinNonlio 6.2.1软件计算相关药代动力


实验例 2: 体内研究
在上述体外评价的基础上, 选择其中一些高活性、 结构有代表性的化合 物开展体内研究。
( 1 ) 小鼠脂耐受性急性体内药效实验
供试样品: 在上述实验的基础上, 选择其中一些高活性、 结构有代表性 的化合物开展进一步实验。
实验方法: 实验动物选用 CD1 品系小鼠, 体重为 30g左右。 给予 12h 光照 /12h黑暗, 保持温度 20-25°C, 湿度 40-70%的饲养环境, 并保证动物的 自由饮食摄水, 实验开始前 16h所有动物禁食, 实验当天动物按组别给予相 对应的药物, 并在给药后 lh按 6ml/kg剂量灌胃给予玉米油。 在动物摄取玉 米油的 -0.5, 0.5, 1, 2, 4h时间点对其进行尾尖采血, 采血量为 20ul, 并立即将 血液按 7000rpm离心 10min, 分装出血浆供测试 TG用。 TG的测定采用甘 油三脂试剂盒检测法。 实验数据通过 SPSS统计分析软件分析其显著性。
实验结果见表 4:
表 4小鼠脂耐受体内药效实验结果

实验结论: 在体内药效方面, 本发明化合物较 Pradigastat具有显著地甚 至意料不到的进步。
(2 ) DIO鼠上的七天体重及食物摄取变化
供试样品: 在上述实验的基础上, 选择其中一些高活性、 结构有代表性 的化合物开展进一步实验。
实验方法:
DIO小鼠模型的建立:购入 6周龄雄性 C57W/6J 小鼠,保持 12h光照 /12h 黑暗, 温度 20-25°C, 湿度 40-70%的饲养环境, 动物适应 1-2周后开始食用 高脂饲料 D12492i (Research Diets, Inc), 自由饮水。 每天监测其体重及耗食 量变化, 12-14周后, 体重达 40-50g的动物视为造模成功。
实验方案: 选取造模成功的动物单笼饲养, 对其灌胃给予相对应的溶媒 2周左右, 待体重稳定后进行分组。 分组后的动物按组别分别给予相应的受 试药物, 每天 2次, 连续 7天, 并每天记录动物体重及耗食量的变化, 绘制 动物累积体重及累积耗食量曲线图。 实验数据通过 SPSS统计分析软件分析 其显著性。
实验结果见表 5和表 6:
表 5 DIO鼠上的七天体重变化
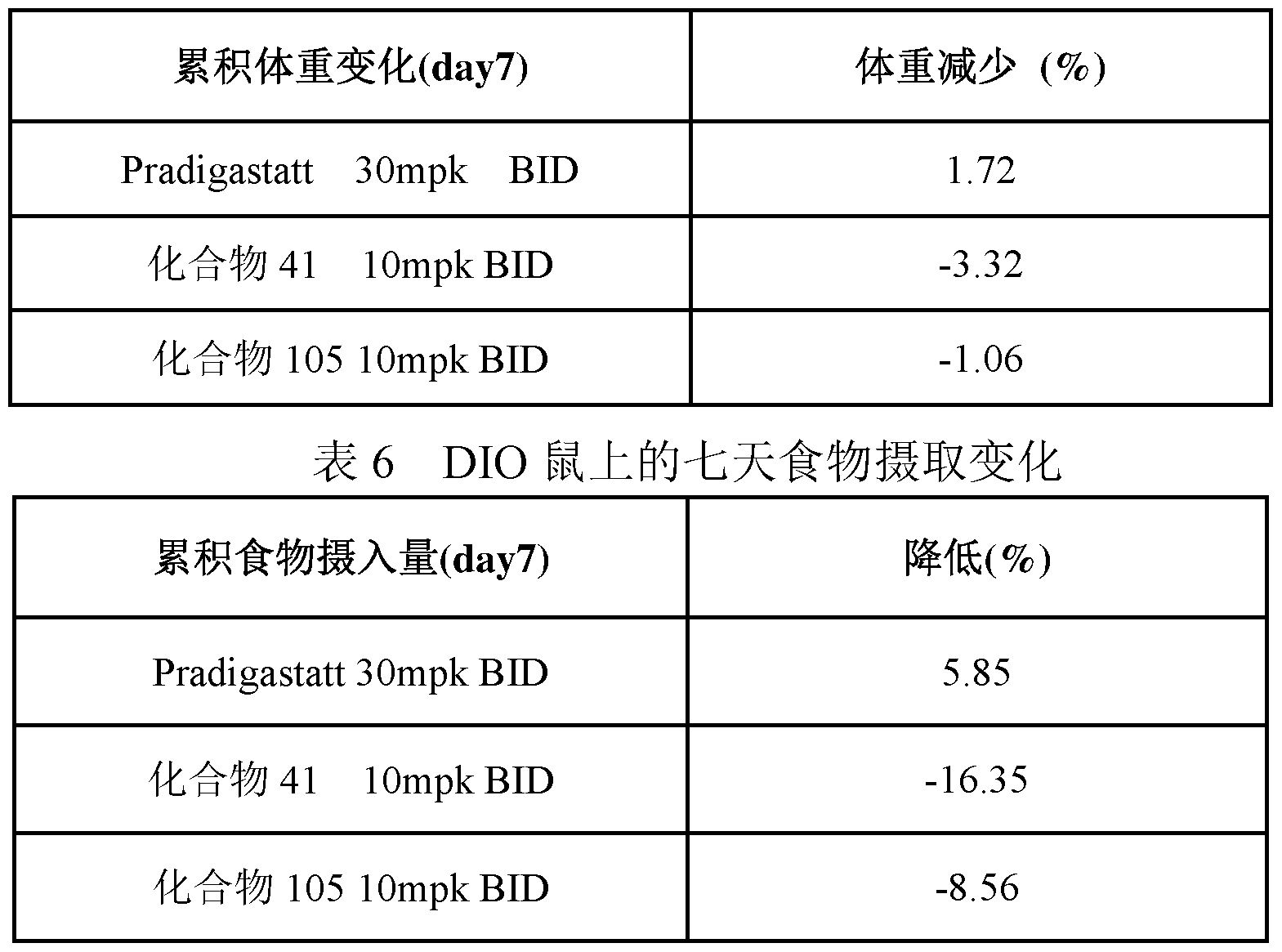
实验结论:本发明化合物给药 lOmpk BID都能有效降低体重和较大地降
低食物摄取; 而 Pradigastatt即使给药 30mpk BID,体重和食物摄取依然略增 加。
虽然以上描述了本发明的具体实施方式,但是本领域的技术人员应当理 解, 这些仅是举例说明, 在不背离本发明的原理和实质的前提下, 可以对这 些实施方式做出多种变更或修改。 因此, 本发明的保护范围由所附权利要求 书限定。
Claims
1. 式 (I)所示化合物或其药学上可接受的盐,

(I)
其中,
Rla、 Rlb、 R2、 R3a、 R3b、 R4a或 R4b分别独立地代表11、 烷基、
c3-10环:) c3-10
 c3-10 环烷基或(¾.1()环烷氧基任选地被卤素、 OH、 SH、 皿2或?1¾所取代, 取代 基的个数为 1个或多个;
c3-10 环烷基或(¾.1()环烷氧基任选地被卤素、 OH、 SH、 皿2或?1¾所取代, 取代 基的个数为 1个或多个;

1 6代表任选被取代的苯并杂环、任选被取代的单杂环、任选被取代的环 烷基醛基和连接任选被取代的苯并杂环的任选被取代的芳基, 其中,
所述杂环是饱和或者不饱和的,杂原子或杂原子团的数目为 1〜8,分别 独立地选自 N、 S、 F、 0、 SO和 S02;
取代基的数目为 1个或多个, 分别独立地选自卤素、 OH、 SH、 NH2、 P¾、 =0、 C1-10焼基、 C1-10焼氧基、。3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-10CONH2, 其中
所述 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、
-C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-1QCONH2任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CF3、 -OCF3、 -OCH3、 d.6烷基、 d.6环烷基和 d.6烷氧基中的一个或多个所取代;
所述的 -C^oO d.K)或 -d.wCOO C1-10中氧原子或羰基两侧或同侧的碳原 子任选地连接成环;
所述 -CLK) CONH2的 N位上的 H任选地全部或部分地被一个或两个 d.6 烷基所取代;
R7.1Q分别独立地代表 d.u)烷基、 d.u)烷氧基、 C3.1Q环烷基、 C3.1Q环烷氧 基、 H、 卤素、 OH、 SH、 丽 2、 PH2和 CN, 所述的 CWQ烷基、 CWQ烷氧基、 C3.1Q环烷基或 3.1()环烷氧基任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CH3 或 CF ^取代, 取代基的个数为 1个或多个; 和
别独立地代表 d.u)烷基、 d.u)烷氧基、 C3.1Q环烷基或 C3.1Q环烷 氧基, 所述 CLK)烷基、 d.u)烷氧基、 C3.1Q环烷基或 C3.1Q环烷氧基任选地被 卤素、 OH、 SH、 丽 2、 PH2、 CN、 CF3、 -OCF3或 -OCH3所取代, 取代基的 数目为 1〜3。
2. 根据权利要求 1所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中所述的 RLA、 RLB或 选 § Η。
3. 根据权利要求 1所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中所述的 R3A、 R3B、 1 4£1或1 41)分别独立地选自 H或甲基。
4. 根据权利要求 1所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中 R6选自任选被取代的 9〜10元苯并杂环、 任选被取代的 5〜6单 杂环、任选被取代的 C3.1Q环烷基醛基和连接任选被取代的 9〜10元苯并杂环 的任选被取代的苯基, 其中,
所述杂环是饱和或者不饱和的,杂原子或杂原子团的数目为 1〜8,分别
独立地选自 N、 S、 F、 0、 SO和 S02 ;
取代基的数目为 1〜3, 分别独立地选自卤素、 OH、 SH、 NH2、 PH2、 =0、 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH、 -OC1-10COOH、 -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN或 -C1-10CONH2, 其中 所述 C1-10焼基、 C1-10焼氧基、 C3-10环焼基、 C3-10环焼氧基、 -C1-10OH、 -C1-10COOH , -OC1-10COOH , -C1-10O C1-10、 -C1-10COO C1-10、 -C1-10CN 或 -C1-1QCONH2任选地被卤素、 OH、 SH、 NH2、 PH2、 CN、 CF3、 -OCF3、 -OCH3、 d.6烷基、 d.6环烷基和 d.6烷氧基中的一个或多个所取代;
所述的 -C^oO d.K)或 -d.wCOO C1-10中氧原子或羰基两侧或同侧的碳原 子任选地连接成环; 和
所述 -CLK) CONH2的 N位上的 H任选地全部或部分地被一个或两个 d.6 烷基所取代。
5. 根据权利要求 4所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中 R6选自


其中,
R14、 R15分别独立地选自 H、 d.6烷基或 d.6烷氧基, 所述 d.6烷基或 CL6烷氧基任选地被卤素、 OH、 811、皿2或?¾所取代,取代基的数目为 1〜 3; 和
R16.4Q分别独立地选自 H、 卤素、 OH、 SH、 NH2、 PH2、 d.6烷基、 d.6 烷氧基、 -C1-6OH、 -C1-6COOH、 -OC1-6COOH、 -C1-60 C1-6、 -C1-6COO C1-6、 -C1-6CN、 -C1-6 CONH2,
其中,
所述的 C 6烷基、 d.6烷氧基、 -C1-6OH, -C1-6COOH, -OC1-6COOH, -C1-60 C1-6、 -C1-6COO C1-6, -C1-6CN、 -C1-6 CON¾任选地被卤素、 OH、 SH、 丽 2、 ?!^或^^环烷基所取代, 取代基的数目为 1〜3;
所述的 -CwO d.6或 -C^COO d.6中氧原子或羰基两侧或同侧的碳原子 任选地连接成环; 和
所述 -CL6NH2的 N位上的 H任选地全部或部分地被一个或两个 d.6烷基 所取代。
6. 根据权利要求 5所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中 R14、 R15分别独立地选自11、 甲基和 CF3。
7. 根据权利要求 5所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中 R26.3Q选自 Η。
8. 根据权利要求 5所述的式①所示化合物或其药学上可接受的盐,其特 征是, 其中 R16.25、 R31.4Q分别独立地选自
1) 卤素;
2) -CH3、 -CH2CH (C¾) 2、 -C (C¾) 3、 -CH2CH (C¾)

3 ) -CH2CF3、 -CH2C ( CH3 ) 2F、 -CH2C ( CH3 ) F2、 -CH2CHF2、 -OCH2CF3;
4) -CH2CH2OH, -CH2C (CH3) 2CH2OH、 -CH2C(CH3)2OH;
5) -COOH, -CH2COOH, -CH2CH2COOH, -CH2C(CH3)2COOH、 -CH2CH (CH3) COOH, -CH (CH3) COOH 、 -C (CH3) 2COOH、 -CH (CH2CH3)
COOH、 -C ( C ) 2COOH、 -CH2CH2COOH、 -CH ( C ) COOH、 -CH ( CH2C )
COOH,
 -OCH(CH3) COOH;
-OCH(CH3) COOH;

7) -CH2COOC2CH5;
8) -CH2CN; 和

9. 根据权利要求 1所述的式①所示化合物或其药学上可接受的盐,其特 征是,其中 R7 和 R8分别独立的选自 -CH3、 -CH2CH3、 -CF3、 -OCH3、 -OCF3、 卤素、 011或11。
10. 根据权利要求 1所述的式 ω所示化合物或其药学上可接受的盐, 其 特征是, 其中 R9 和 R1Q选自 H。
11. 根据权利要求 1所述的式 ω所示化合物或其药学上可接受的盐, 其
特征是, 其中 R1W3分别独立地选自 -OCH3、 -OCF3、 -OCH2CF3或 -CF3。
12. 根据权利要求 1所述的式①所示化合物或其药学上可接受的盐, 其 特征是, 其中该化合物选自:
1 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 氯苯) -2-氧代咪唑啉 -1-基)乙酸;
2 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 氟苯) -2-氧代咪唑啉 -1-基)乙酸;
3 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 甲苯) -2-氧代咪唑啉 -1-基)乙酸;
4 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 乙基 -6-甲基苯 )-2-氧代咪唑啉 -1-基)乙酸;
5 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 乙基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
6 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 氯苯) -2-氧代咪唑啉 -1-基)丙酸;
7 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5Η)-基) -2- 三氟甲氧基苯 )-2-氧代咪唑啉 - 1 -基)乙酸;
8 ) 4-氨基 -6-(3-氯 -4-(2-氧代 -3-(2,2,2-三氟乙基)咪唑啉 -1-基)苯基) -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
9 ) 4-氨基 -6-(3,5-二甲基 -4-(2-氧代 -3-(2,2,2-三氟乙基) -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
10) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
11 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)
苯) -2-氧代咪唑啉 -1-基)乙酸;
12) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代咪唑啉 -1-基)乙酸乙脂;
13 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代咪唑啉 -1-基)丙酸;
14) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧苯 )-2-氧代咪唑啉- 1 -基)乙酸;
15 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓- 6(5H)-基) 苯基 2-氧代咪唑啉 -1-基)丙酸;
16) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
17 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基) -三甲基丁酸;
18) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- ¾)-2,6-甲氧基苯基: )-2-氧代咪唑啉 - 1 -基:)乙酸;
19) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) - 2,6-二甲基苯) -2-氧代咪唑啉 -1-基)乙酸;
20) 3-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧代 -2,3-二氢 -1H-咪唑 -1-基)丙酸;
21 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2- (三氟甲氧基)苯基) -2-氧 -2,3-二氢 -1H-咪唑 -1-基)乙酸;
22) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -2-氧基) -2,3-二氢 -1H-咪唑 -1-基)丙酸;
23 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-
基) -2-氟苯) -2-氧代咪唑啉 -1-基)丁酸;
24 ) 3-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) -2-氧代咪唑啉 -1-基) -二甲基丙酸;
25 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) - 2- (三氟甲基)苯) -2-氧代咪唑啉- 1 -基)乙酸;
26 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
27 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯 -6-甲基苯 )-2-氧代咪唑啉- 1 -基)乙酸;
28 ) 2-(3-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 2- 氯苯) -2-氧代 -2,3-二氢 咪唑 -1-基)乙酸;
29 ) 4-氨基 -6-(3-氯 -4-(3-异丁基 -2-氧代咪唑啉 -1-基:)苯基: )-7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
30 ) 4-氨基-6-(3-氯-4-(3-(环丙基甲基)-2-氧代咪唑啉-1-基)苯基 7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
31 ) 4-氨基 -6-(3-氯 -4-(3-(2-氟 -2-二甲基丙基) -2-氧代咪唑啉 -1-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
32 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑 - 1 -基) -2-甲基丙酸;
33 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑 - 1 -基) -2-甲基丙酸;
34 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) 吡唑 -1-基)丙酸;
35 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-
基) -2-氯苯) 吡唑 -1-基)丙酸;
36) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3-甲基 吡唑 -1-基)乙酸;
37 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) 吡唑 -1-基)丙酸;
38) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯基 )-3,5-二甲基 吡唑 -1-基)丙酸;
39) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -3- (三氟甲基) 吡唑 -1-基)乙酸;
40) 4-氨基-6-(3-氯-4-(3-甲基-1-(2,2,2-三氟乙基)-1 -吡唑-4-基)苯基)-7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
41 ) 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
42) 4-氨基 -6-(3-氯 -4-(1-(2-羟基 -2-甲基丙醇基) -1H-吡唑 -4-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-酮;
43 ) 4-氨基 -6-(4-(1-(2,2-二氟乙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基 苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
44) 4-氨基 -6-(3-氯 -4-(1-(3-羟基 -2,2-二甲基丙基) -1-H-吡唑 -4-基)苯 基) -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -5(6H)-酮;
45 ) (R)-4-氨基 -6-(3-氯 -4-(1-异丁基) -1H-吡唑 -4-基)苯基) -8-甲基 -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
46) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -1H -吡唑 -1-基)乙酰胺;
47 ) 4-氨基 -6-(3,5-二甲基 -4-P-甲基 -1-((3-甲基氧杂环丁烷 -3-基)甲
基) -1Η-吡唑 -4-基)苯基) - 7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
48) 4-氨基 -6-(4-(1-(2,2-二氟丙基) -5-甲基 -1H-吡唑 -4-基) -3,5-二甲基苯 基:) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
49) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -1H-吡唑 -1-基)乙酸;
50) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -1H-吡唑 -1-基)丙酸;
51 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基) -1H-吡咯 -1-基:)丁酸;
52 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基 )-1Η-吡咯 -1-基:)丁酸;
53 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) - 1H-吡咯 - 1 -基:) -3 -甲基丁酸;
54) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基)苯 基:) -1H-吡咯 -1-基) -2-甲基丙酸;
55 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) - 苯) -3-甲基 -1H -吡唑 -1-基)乙酸;
56) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑 - 1 -基)乙酸;
57 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 ( 5H)-基) -2-甲基苯基) - 1H-吡唑 - 1 -基)乙酸;
58) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基)丙酸;
59) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基)
-2-甲基苯基) -m-吡唑 -1-基) -3-甲基丁酸;
60) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基)丙酸;
61 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -1H-吡唑小基)丁酸;
62) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) 苯基) -3- (三氟甲基) -1H-吡唑 -1-基)丙酸;
63 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基) -2-甲基丙酸;
64) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-甲基苯基) -1H-吡唑 -1-基) -丁酸;
65 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 (5H)-基) -2-氯苯基) -1H-吡唑 -1-基) -3-甲基丁酸;
66) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) -1H-吡唑 -1-基) -2-甲基丙酸;
67 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4] 咪唑啉 -6 ( 5H)-基) -2,6-二甲基苯基) -1H-吡唑 -1-基)乙酸;
68) 4-氨基-6-(3-氯-4-(1- (2,2,2-三氟乙基) -1氢 -吡唑 -4-基)苯基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
69) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧基苯基) - 1H-吡唑 - 1 -基)乙酸;
70) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基:) -2,6-二甲基苯 )- 1 H-吡唑- 1 -基)丙酸;
71 ) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基) -2-
氯苯 )- m-吡咯 - 1 -基:) -2,2-二甲基丙酸;
72 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3,5-二甲基 -1H-吡唑 -1-基)乙酸;
73 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2,6-二甲基苯) -3 -甲基 - 1H-吡唑 - 1 -基)乙酸;
74 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-甲氧基苯) -1H-吡唑 -1-基)丙酸;
75 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氟苯) - 1 H-吡唑- 1 -基)乙酸;
76 ) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮杂卓 -6(5H)-基) -2-苯 基) -3,5-二甲基 -1H-吡唑 -1-基)丙酸;
77 ) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基) -2- 苯甲基 )-3,5-二甲基 -1H-吡唑 -1-基)丙酸;
78 ) 3-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) -3-甲基 -1H-吡唑 -1-基)丙酸;
79 ) 4-氨基 -6-(3-氯 -4-(1-乙基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氮氧杂卓 -5(6H)-酮;
80 ) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
81 ) 4-氨基 -6-(3-氯 -4-(1-异丙基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
82 ) 4-氨基 -6-(3-氯 -4-(1-(2-羟基乙基) -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -6(5H)-酮;
83 ) 3-(4-(4-(4-氨基 5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-
基) -2,6-二甲基苯) -3-甲基 -1H-吡唑 -1-基)丙酸;
84 ) 2-(4-(4-(4-氨基 -5-氧代 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)- 基) -2-氯苯) - 1 H-吡唑- 1 -基)乙腈;
85 ) 4-氨基 -6-(3-氯 -4-(1-(2,2-二甲基 -3-氧 -3- (吡咯 -1-基)丙基) -1H-吡唑 -4- 基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
86 ) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1氢 -吡唑 -4-基)苯基) -8,8-二甲基 -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
87 ) 4-氨基 -6-(3-氯 -4-(1- (环丙基) -1氢 -吡唑 -4-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
88 ) 4-氨基 -6-(3-氯 -4-(1-新戊基 -1氢 -吡唑 -4-基)苯基) - 7,8-二氢嘧啶
[5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
89 ) 3-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氮氧杂卓 -6(5H)-基) -2- 氯苯基) -1H-吡唑 -1-基) -N,N,2,2-四甲基丙酰胺;
90 ) 4-氨基 -6-(3-乙基 -4-(1-异丁基 -1H-吡唑 -4-基)苯基) -7,8-二氢嘧啶
[5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
91 ) 4-氨基 -6-(3-氯 -4-(1- (2-氟 -2-异丁基)-1氢 -吡唑 -4-基)苯基) -7,8-二氢 嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;
92 ) 4-氨基 -6-(3-氯 -4-(1-异丁基 -1氢 -吡唑 -4-基)苯基) -7,7-二甲基 -7,8-二 氢嘧啶 [5,4-f] [ 1 ,4]氧氮杂卓 -5(6H)-酮;
93 ) 4-氨基 -6-(4-(3,5-二甲基 -1-(2,2,2-三氟乙基) -1H-吡唑 -4-基) -3,5-二甲 基苯) -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
94 ) 4-氨基 -6-(3-氯 -4-(1-(2-甲氧基 -2-甲基丙基) -1H-吡唑 -4-基)苯基) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
95 ) 4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-吡唑 -3-基)苯基) -7,8-二氢
嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
96 ) (R)-4-氨基 -6-(3-氯 -4-(1-异丁基 -1Η-吡唑 -4-基)苯基) -7-甲基 -7,8-二氢 嘧啶 [5,4-f][l,4]氮氧杂卓 -5(6H)-酮;
97 ) 4-氨基 -6-(4-(1-(2-氟 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲基 苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
98 ) 4-氨基 -6-(4-(1-(2-羟基 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二甲 基苯) -7,8- 二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
99 ) 4-氨基 -6-(4-(1-(2-甲氧基 -2-甲基丙基) -3-甲基 -1H-吡唑 -4-基) -3,5-二 甲基苯 )-7,8- 二氢嘧啶 [5,4-f] [l,4]氧氮杂卓 -5(6H)-酮;
100) 4-氨基 -6-(4-(2-氧 -1-(2,2,2-三氟乙基) -1,2-二氢吡啶 -3-基)苯基) - 二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
101) 4-氨基 -6-(3,5-二甲基 -4-(6-氧 -1-(2,2,2-三氟乙基) -1,6-二氢吡啶 -5-基) 苯基) -7,8-二氢嘧啶 [5,4-f][l ,4]氧氮杂卓 -5(6H)-酮;
102) 4-氨基 -6-(3-氯 -4-(1-(2-氟 -2-甲基丙基) -1H-咪唑 -4-基)苯基) -7,8-二 氢嘧啶 [5,4-f][l,4]氧氮杂卓 -5(6H)-酮;
103) 2-(4-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)基)苯 基)哌啶基)乙酸;
104) 2-(3-(4-(4-氨基 -5-氧 -7,8-二氢嘧啶 [5,4-f][l,4]氧氮杂卓 -6(5H)-基)苯 基)吡咯小基)乙酸;
105) 4-氨基 -6-(3,5-二甲基 -4-(1-氧代 -2-(2,2,2-三氟乙基) -1,2-二氢异喹啉 -7-基)苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)- 酮;
106) (E)- 4-氨基 -6-(6-((2,2,2-三氟乙氧基)亚氨基 )-5,6,7,8-四氢萘 -2- 基) -7,8-二氢嘧啶 [5,4-f] [l,4]氧氮杂卓 -5(6H)-酮;
107) 4-氨基 -3,4-二氢萘 -1 ( 2H) -酮氧 -(2,2,2-三氟乙氧基) 肟 -7,8-二氢嘧
啶 [5,4-f][l,4] 咪唑啉 -5 (6H)-基) -酮;
108) 4-氨基 -6-(6-(2,2,2-三氟乙氧基) -5,6,7,8-四氢萘 -2-基) -7,8-二氢吡啶 [5,4-f][l,4]氧氮杂卓 -5(6氢) -酮;或
109) 4-氨基 -6-(3,5-二甲基 -4-(3-甲基 -2-(2,2,2-三氟乙基) -2H-吲唑 -5-基) 苯基) -7,8-二氢嘧啶 [5,4-f][l,4] 氧氮杂卓 -5(6H)-酮。
13. 权利要求 1〜10或 12任意一项所述的式 (X)化合物的制备方法,其中
R5

PH2和 d.6烷基,所述 d.6烷基任选地被卤素、 OH、 SH、 NH2或 PH2所取代, 取代基的数目为 1个或多个;
R6 '代表 或者被保护基保护的 R6;
Rla、 Rlb、 R2、 R3a、 R3b、 R4b、 R6.1Q如权利要求 1〜10所定义。
14. 权利要求 1〜10或 12任意一项所述的式 (X)化合物的制备方法,其中
R5
 , 包括如下步骤:
, 包括如下步骤:

其中,
Aj, A2和 Y分别独立地选自 CF3、 卤素、 OH、 SH、 NH2、 PH2和 C1-6 烷基, 所述 CL6烷基任选地被卤素、 OH、 SH、 NH2或 PH2 所取代, 取代基 的数目为 1个或多个;
R6 "代表 或者被保护基保护的 R6;
R2、 R3a、 b、 R4a、 R4b、 R6-1Q如权利要求 1〜10所定义。
15. 根据权利要求 13或 14所述的制备方法, 其特征是, 其中所述的保 护基为氨基保护基或者羟基保护基。
16. 根据权利要求 15所述的制备方法,其特征是,其中所述的氨基保护 基选自 BOC、 Cbz、 Fmoc、 Bn、 PMB、 Pht、 Ac、 Trt、 CF3CO、 Alloc、 甲 基酯、 Troc或 PMP; 所述的羟基保护基选自乙基、 TBS、 TBDPS、 TMS、 Ac、 Me、 MOM、 MEM, THP、 Bn、 PMB、 MTM或 Piv。
17. 一种药物组合物, 其包括权利要求 1〜12任意一项所述的治疗有效 量的式①化合物或其药学上可接受的盐以及药学上可接受的载体。
18. 权利要求 1〜12任意一项所述的式 (X)化合物或其药学上可接受的盐, 或根据权利要求 17所述的药物组合物在制备预防或治疗家族性糜微粒血症 综合征、 肥胖病、 高脂蛋白血症或高甘油三酯血症的药物中的应用。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14837191.7A EP3042907B1 (en) | 2013-08-23 | 2014-08-18 | Dgat1 inhibitor and preparation method and use thereof |
| US14/912,428 US9809604B2 (en) | 2013-08-23 | 2014-08-18 | DGAT1 inhibitor and preparation method and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310371069.0 | 2013-08-23 | ||
| CN201310371069.0A CN104418866B (zh) | 2013-08-23 | 2013-08-23 | Dgat1抑制剂及其制备方法和用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015024486A1 true WO2015024486A1 (zh) | 2015-02-26 |
Family
ID=52483075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/084586 Ceased WO2015024486A1 (zh) | 2013-08-23 | 2014-08-18 | Dgat1抑制剂及其制备方法和用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9809604B2 (zh) |
| EP (1) | EP3042907B1 (zh) |
| CN (1) | CN104418866B (zh) |
| TW (1) | TWI636055B (zh) |
| WO (1) | WO2015024486A1 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109970763B (zh) * | 2017-12-27 | 2021-10-08 | 青岛黄海制药有限责任公司 | 一种dgat1抑制剂的制备方法 |
| EP3819305A4 (en) | 2018-07-04 | 2022-04-06 | Mitsubishi Tanabe Pharma Corporation | AMIDE COMPOUND WITH BET PROTEOLYSIS-INDUCING ACTIVITY AND MEDICAL USE THEREOF |
| AU2020285269B2 (en) * | 2019-05-31 | 2022-11-17 | Medshine Discovery Inc. | Bicyclic compound as RIP-1 kinase inhibitor and application thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005037826A1 (en) | 2003-10-17 | 2005-04-28 | Incyte Corporation | Substituted cyclic hydroxamates as inhibitors of matrix metalloproteinases |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| WO2009016462A2 (en) | 2007-08-02 | 2009-02-05 | Pfizer Products Inc. | Substituted bicyclolactam compounds |
| WO2010086820A1 (en) | 2009-02-02 | 2010-08-05 | Pfizer Inc. | 4-amino-5-oxo-7, 8-dihydropyrimido [5,4-f] [1,4] oxazepin-6 (5h) -yl) phenyl derivatives, pharmaceutical compositions and uses thereof |
| WO2011121250A2 (fr) | 2010-04-02 | 2011-10-06 | Libragen | Composition cosmetique et pharmaceutique comprenant du n-acetyl-glucosamine-6-phosphate |
| WO2011121350A1 (en) | 2010-04-01 | 2011-10-06 | Astrazeneca Ab | 4 -amino -7,8- dihydropyrimido [5, 4 - f] [1, 4] oxazepin- 5 ( 6h) - one based dgat1 inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2805951B1 (en) | 2009-03-20 | 2018-03-14 | Metabasis Therapeutics, Inc. | Inhibitors of diacylglycerol o-acyltransferase 1 (DGAT-1) and uses thereof |
-
2013
- 2013-08-23 CN CN201310371069.0A patent/CN104418866B/zh active Active
-
2014
- 2014-08-14 TW TW103127847A patent/TWI636055B/zh active
- 2014-08-18 WO PCT/CN2014/084586 patent/WO2015024486A1/zh not_active Ceased
- 2014-08-18 EP EP14837191.7A patent/EP3042907B1/en active Active
- 2014-08-18 US US14/912,428 patent/US9809604B2/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005037826A1 (en) | 2003-10-17 | 2005-04-28 | Incyte Corporation | Substituted cyclic hydroxamates as inhibitors of matrix metalloproteinases |
| WO2007126957A2 (en) | 2006-03-31 | 2007-11-08 | Novartis Ag | New compounds |
| WO2009016462A2 (en) | 2007-08-02 | 2009-02-05 | Pfizer Products Inc. | Substituted bicyclolactam compounds |
| CN101772504A (zh) * | 2007-08-02 | 2010-07-07 | 辉瑞产品公司 | 经取代的双环内酰胺化合物 |
| WO2010086820A1 (en) | 2009-02-02 | 2010-08-05 | Pfizer Inc. | 4-amino-5-oxo-7, 8-dihydropyrimido [5,4-f] [1,4] oxazepin-6 (5h) -yl) phenyl derivatives, pharmaceutical compositions and uses thereof |
| WO2011121350A1 (en) | 2010-04-01 | 2011-10-06 | Astrazeneca Ab | 4 -amino -7,8- dihydropyrimido [5, 4 - f] [1, 4] oxazepin- 5 ( 6h) - one based dgat1 inhibitors |
| WO2011121250A2 (fr) | 2010-04-02 | 2011-10-06 | Libragen | Composition cosmetique et pharmaceutique comprenant du n-acetyl-glucosamine-6-phosphate |
Non-Patent Citations (7)
| Title |
|---|
| "Discovery ofPF-04620110: A Potent, Orally-Bioavailable Inhibitor of DGAT-1", ACS MED. CHEM. LETT., vol. 2, 2011, pages 407 - 412 |
| "Remington: The Science and Practice of Pharmacy", 2005, LIPPINCOTT, WILLIAMS & WILKINS |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING COMPANY, pages: 1445 |
| ACS MED. CHEM. LETT., vol. 2, 2011, pages 407 - 412 |
| BERGE ET AL.: "Pharmaceutical Salts", JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 1 - 19 |
| CHOU; TALALAY, ADV. ENZYME REGUL, vol. 22, 1984, pages 27 - 55 |
| MAEHR, J. CHEM. ED., vol. 62, 1985, pages 114 - 120 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104418866B (zh) | 2018-10-16 |
| US9809604B2 (en) | 2017-11-07 |
| EP3042907A4 (en) | 2017-06-07 |
| TW201536794A (zh) | 2015-10-01 |
| TWI636055B (zh) | 2018-09-21 |
| EP3042907A1 (en) | 2016-07-13 |
| EP3042907B1 (en) | 2021-01-13 |
| CN104418866A (zh) | 2015-03-18 |
| US20160272651A1 (en) | 2016-09-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3555070B1 (en) | Amine-substituted heterocyclic compounds as ehmt2 inhibitors and methods of use thereof | |
| US8338437B2 (en) | Amines as small molecule inhibitors | |
| JP7797486B2 (ja) | 新規なrho関連タンパク質キナーゼ阻害剤の調製方法およびその調製方法における中間体 | |
| CN111944012B (zh) | 一种芳香胺类靶向ar和bet的蛋白降解嵌合体化合物及用途 | |
| CN121013839A (zh) | 作为ras-pi3k调节剂用于治疗例如癌症的1-(([1, 1',-联苯基]-2-基)磺酰基)-4-氟-n-(3-(甲基磺酰基)烯丙基)哌啶-4-甲酰胺衍生物 | |
| JP2020537645A (ja) | Ehmt2阻害剤としてのアミン置換複素環化合物及びその誘導体 | |
| CN115991706A (zh) | Pim激酶抑制剂 | |
| US20170291890A1 (en) | Heterocycle substituted amino-pyridine compounds and methods of use thereof | |
| WO2018064557A1 (en) | Substituted fused bi- or tri- heterocyclic compounds as ehmt2 inhibitors | |
| CN114105950A (zh) | 吡唑类化合物及其应用 | |
| WO2018090982A1 (zh) | 苯并二环烷烃衍生物、其制法及其用途 | |
| CN121586705A (zh) | 小分子前列腺素f受体拮抗剂 | |
| CN107709323B (zh) | 羟基嘌呤类化合物及其应用 | |
| WO2015024486A1 (zh) | Dgat1抑制剂及其制备方法和用途 | |
| AU2017239295A1 (en) | Compound having mutant IDH inhibitory activity, preparation method and use thereof | |
| KR101998011B1 (ko) | 디아자-벤조플루오란텐류 화합물 | |
| CN120381447A (zh) | 作为乙醇酸氧化酶抑制剂的杂环羧酸酯化合物 | |
| JP2024544529A (ja) | Btk阻害剤 | |
| CN114853812A (zh) | 含氧化膦基团的化合物、其制备方法及其在医药上的应用 | |
| JP2024521741A (ja) | アルギニンメチルトランスフェラーゼ阻害剤およびその用途 | |
| WO2026055780A1 (en) | Compounds and compositions for treating mc4r related conditions | |
| WO2026040870A1 (zh) | 一种靶向磷酸二酯酶亚型pde4b的化合物及其用途 | |
| JP2024542036A (ja) | Gpr39タンパク質のアンタゴニスト | |
| WO2022067063A1 (en) | Mutant selective egfr inhibitors and methods of use thereof | |
| HK40085073A (zh) | 新型rho相关蛋白激酶抑制剂的制备方法和此制备方法中的中间体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14837191 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14912428 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014837191 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014837191 Country of ref document: EP |