WO2015048250A1 - Preparation of radioiodinated 3-fluoropropyl-nor-beta-cit - Google Patents
Preparation of radioiodinated 3-fluoropropyl-nor-beta-cit Download PDFInfo
- Publication number
- WO2015048250A1 WO2015048250A1 PCT/US2014/057404 US2014057404W WO2015048250A1 WO 2015048250 A1 WO2015048250 A1 WO 2015048250A1 US 2014057404 W US2014057404 W US 2014057404W WO 2015048250 A1 WO2015048250 A1 WO 2015048250A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- reaction mixture
- reaction
- fluoropropyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- XMSZNCPTMJREEB-SSDOTTSWSA-N CN([C@@H](C=C1)C=C2)C1=C2C(OC)=O Chemical compound CN([C@@H](C=C1)C=C2)C1=C2C(OC)=O XMSZNCPTMJREEB-SSDOTTSWSA-N 0.000 description 1
- FOAZXCIJRVJHOR-UHFFFAOYSA-N C[n]1c2ccc1CC(c(cc1)ccc1I)=C2C(OC)=O Chemical compound C[n]1c2ccc1CC(c(cc1)ccc1I)=C2C(OC)=O FOAZXCIJRVJHOR-UHFFFAOYSA-N 0.000 description 1
- WJPSQSYMPDVAAR-UHFFFAOYSA-N C[n]1c2ccc1CC(c1ccccc1)=C2C(OC)=O Chemical compound C[n]1c2ccc1CC(c1ccccc1)=C2C(OC)=O WJPSQSYMPDVAAR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0446—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K51/0448—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil tropane or nortropane groups, e.g. cocaine
Definitions
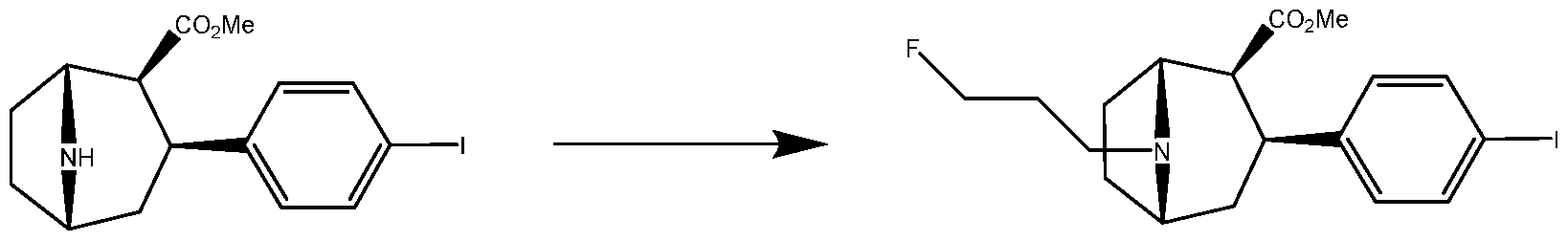
- the present invention relates to improved processes for the preparation of radioiodinated 3-fluoropropyl-nor-3-CIT.
- radioiodinated 3-fluoropropyl-nor-3-CIT (i.e., compound I) is a diagnostic agent useful for diagnosing and monitoring movement disorders and dementia, and has specific use in the diagnosis and monitoring of Parkinson's disease.
- 1-123 radioiodinated 3-fluoropropyl-nor-3-CIT has the following structure:
- This compound can be prepared from anhydroecgonine methyl ester using a six step process, which relies on the conversion of an arylstannane precursor to the 1-123 labeled compound.
- a six step process is generally disclosed in, for example, Swahn, et al., Journal of Labelled Compounds and Radiopharmaceuticals, 1996, Vol.
- the present invention is directed to a process for preparation of compound (I) comprising the following reaction scheme:
- the present invention is directed to a process for the preparation of a N-(3-fluoropropyl) compound (III), the process comprising contacting a nortropane compound (II) with 3-fluoropropanal to form compound (III):
- R 1 is selected from the group consisting of halogen, ⁇ - ⁇ Si(CH 3 ) 3 , and ⁇ - ⁇ Sn(CH 3 ) 3 .
- the present invention relates to the preparation of 1-123 radioiodinated 3-fluoropropyl-nor-3-CIT using an arylsilane intermediate.
- the process avoids the use of hexamethylditin, and has a reduced number of steps as compared to previously known processes for the preparation of radioiodinated 3- fluoropropyl-nor-3-CIT from anhydroecgonine methyl ester (compound 1-1).
- the present invention also relates to the alkylation of a nortropane to the corresponding N-(3-fluoropropyl) analogue using 3-fluoropropanal. The process avoids the use of the ozone depleting compound 3-fluoro-l-bromopropane.
- the present invention provides a process for the preparation of compound (I) comprising the following reaction scheme:
- the present invention is directed to a process for the preparation of a N-(3-fluoropropyl) compound (III), the process comprising contacting a nortropane compound (II) with 3-fluoropropanal to form compound (III):
- R 1 is selected from the group consisting of halogen, ⁇ - ⁇ Si(CH 3 ) 3 , and ⁇ - ⁇ Sn(CH 3 ) 3 .
- the present invention relates to the preparation of I- 123 radioiodinated 3-fluoropropyl-nor-3-CIT (compound I) using an arylsilane intermediate.
- the process avoids the use of hexamethylditin, and reduces the number of steps previously required for the preparation of radioiodinated 3-fluoropropyl-nor-3-CIT from anhydroecgonine methyl ester (compound 1-1).
- Step A conversion of compound 1-1 to compound 2-1
- Step A of the process involves or comprises the formation of an arylsilane precursor by reacting anhydroecogonine methyl ester (compound 1-1) with a halogen-substituted phenyltrimethylsilane in a Grignard addition reaction:
- X is a halogen, and preferably is I or Br.
- a Grignard reagent may be prepared by contacting the halogen- substituted phenyltrimethylsilane with magnesium in the presence of a solvent.
- Suitable solvents for use in formation of the Grignard reagent include ethereal solvents, such as tetrahydrofuran, 2-methyltetrahydrofuran, tert-butyl methyl ether, diethyl ether, and the like.
- the halogen-substituted phenyltrimethylsilane is typically present in the solvent in at a molar concentration of from about 0.2 M to about 1.0 M.
- the reaction mixture may optionally further include or comprise a compound for initiating formation of the Grignard reagent.
- Suitable compounds include an iodine crystal or ethylene dibromide.
- the compound is preferably included in the reaction mixture in an amount sufficient to catalyze formation of the Grignard reagent.
- the reaction may be conducted at a variety of temperatures, for example, from about 30°C to about 90°C, and preferably is conducted at reflux.
- the reaction is allowed to proceed until substantially complete, and typically for at least 30 minutes, and more typically from about 30 minutes to about 180 minutes, or from about 45 minutes to about 180 minutes.
- the Grignard reagent forming reaction may be conducted under an inert atmosphere, such as argon or nitrogen.
- the solution comprising the Grignard reagent is contacted with compound 1-1 to form an arylsilane precursor (compound 2-1).
- the Grignard reagent is preferably present in the reaction mixture in an amount of from about 2.2 to about 2.4 equivalents relative to compound 1-1 (e.g., about a 2.2:1 to about a 2.4:1 molar equivalent).
- an additional solvent may be added to the reaction mixture.
- suitable solvents include methylene chloride, tetrahydrofuran, diethyl ether, 2-methyltrethydrofuran, tert-butyl methyl ether, and the like.
- the solvent comprises methylene chloride and diethyl ether (e.g., diethyl ether added during preparation of the Grignard reagent) in a volume:volume ratio of about 1.2:1.
- compound 1-1 is present in the reaction mixture in a concentration of from about 0.1 M to about 0.3 M, and preferably is at a concentration of 0.17 M.
- the reaction may be conducted at a variety of temperatures, for example, from about -40°C to about -90°C, including from about -60°C to about - 85°C, and from about -75°C to about -80°C.
- the reaction is conducted at a temperature of -78°C and allowed to warm to 0°C. The reaction is allowed to proceed until completed, typically for at least 20 minutes.
- the temperature of the reaction mixture may be further lowered following reaction, for example, to a temperature of from about - 75°C to about -80°C, and preferably to a temperature of -78°C.
- the reaction is quenched with an acid, such as trifluoroacetic acid.
- the reaction mixture is allowed to warm following addition of compound 1-1, for example, to from about 0°C to about 22°C, and then cooled again to a temperature of from about -40°C to about -90°C, and preferably to a temperature of -78°C. Following this second cooling, the reaction may be quenched with an acid, such as trifluoroacetic acid to obtain the arylsilane precursor (compound 2-1). Typically, the acid is added to the reaction mixture in a 1:1 molar ratio with the Grignard reagent.
- an acid such as trifluoroacetic acid
- Compound 2-1 may be isolated from the reaction mixture using any suitable technique known in the art, including, for example, filtration, ether extraction, rotary evaporation of solvent, chromatography, or combinations thereof.
- Step B conversion of compound 2-1 to compound 3-1
- Step B of the process involves or comprises demethylating the arylsilane precursor (compound 2-1) to form compound 3-1:
- Suitable demethylation reagents include 1- chloroethylchloroformate, vinyl chloroformate, 2,2,2-triethylchloroformate, and the like.
- the demethylation reagent is preferably present in the reaction mixture in an amount of from about 1 equivalent to about 10 equivalents, and more preferably in an amount of about 7 equivalents, relative to compound 2-1.
- the reaction mixture may further include or comprise one or more aprotic solvent.
- suitable aprotic solvents include 1,2- dichloroethane, ethyl acetate, toluene, chloroform, and combinations thereof.
- the solvent is 1,2-dichloroethane or toluene.
- the solvent may be present in the reaction mixture in an amount of from about 1 mL to about 100 mL per gram of compound 2-1, and preferably in an amount of about 20 mL per gram of compound 2-1.
- the reaction mixture may optionally further include or comprise an inorganic base.
- suitable inorganic bases include sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, and combinations thereof.
- the reaction mixture typically comprises the inorganic base in a molar ratio of compound 2-1 to inorganic base of from about 1:1 to about 1:6.
- the demethylation reaction may result in formation of a carbamate intermediate compound.
- the mixture is typically concentrated and heated in methanol, to decompose the intermediate carbamate.
- the reaction mixture may optionally further comprise one or more reagents to assist in the decomposition of the intermediate carbamate. Suitable reagents include combinations of zinc and acetic acid.
- the reaction may be conducted at a variety of temperatures, for example, from about 60°C to about 115°C, including from about 75°C to about 115°C, and preferably is conducted at about 80°C.
- the reaction is allowed to proceed typically for at least about 1 hour, and preferably proceeds for about 3 hours.
- a base such as ⁇ , ⁇ -diisopropylethylamine may be added to the reaction mixture after about 3 hours of reaction.
- the base is typically added in an amount of about 1 equivalent, relative to compound 2-1.
- the reaction mixture may then be reheated to a temperature of from about 60°C to about 115°C, including from about 75°C to about 115°C, and preferably about 80°C, and the reaction allowed to proceed for an additional 3 hours.
- the demethylation reagent is 1- chloroethylchloroformate
- the reaction mixture contains about 7 equivalents of 1-chloroethylchloroformate relative to compound 2-1.
- the reaction mixture is heated to 80°C for 3 hours, followed by addition of N,N- diisopropylethylamine, and heating to 80°C for an additional 3 hours.
- Compound 3-1 may be isolated from the reaction mixture using any suitable technique known in the art, including, for example, filtration, chromatography, or combinations thereof.
- Step C conversion of compound 3-1 to compound 4-1
- Step C of the process involves or comprises the N-alkylation of compound 3-1 to form compound 4-1:
- compound 3-1 is contacted with a suitable alkylating agent, such as 3-fluoro-l-bromopropane, 3-fluoro-l-iodopropane, or 3- fluoropropanal.
- a suitable alkylating agent such as 3-fluoro-l-bromopropane, 3-fluoro-l-iodopropane, or 3- fluoropropanal.
- the alkylating agent is 3-fluoropropanal.
- 3-fluoropropanal is commercially available, or may be prepared by oxidation of 3- fluoropan-l-ol using Dess- Martin periodinane or trichloroisocyanuric acid and TEMPO (i.e., 2,2,6,6-tetramethyl-l-piperidinyloxy).
- the reaction mixture will comprise from about 1 to about 3 equivalents of alkylating agent, relative to compound 3-1.
- the reaction mixture may further comprise a solvent.
- suitable solvents include ethyl acetate, 1,2-dichloroethane, acetonitrile, dichloromethane, chloroform, and combinations thereof.
- the reaction mixture may include a reducing agent.
- suitable reducing agents include sodium triacetoxyborohydride, sodium cyanoborohydride, and formic acid.
- the reducing agent is present in the reaction mixture in an amount of from about 2 equivalents to about 15 equivalents, relative to compound 3-1.
- the reaction may be conducted at a variety of temperatures, for example, from about -20°C to about 50°C, and typically is conducted at about 20°C.
- the reaction is allowed to proceed until substantially complete, and typically for from about 15 minutes to about 24 hours, and preferably for about 2 hours. Reaction completion may be determined using any suitable technique, such as H PLC.
- Compound 4-1 may be isolated from the reaction mixture using any suitable technique known in the art, including, for example, solvent evaporation, filtration, chromatography, or combinations thereof.
- Step D conversion of compound 4-1 to compound (I)
- Step D of the process is a radioiododesilation reaction, in which the trimethylsilane moiety of compound 4-1 is replaced with 1-123 to form compound (I):
- reaction mixture may further comprise a buffer, such as acetate.
- Non-limiting examples of suitable solvents include methanol, acetic acid, trifluoroacetic acid, and acetic acid in ethanol.
- Non-limiting examples of suitable sources of 1-123 include sodium iodide (i.e., [ 123 l] Nal) and iodinemonochloride (i.e., [ 123 I]ICI).
- Non-limiting examples of suitable oxidizing agents include Chloramine-T (i.e., (N-chloro-p-toluenesulfonamido)sodium), tert-butyl hypochlorite, peracetic acid, and combinations thereof.
- the reaction mixture may comprise a reagent, such as silver tetrafluoroborate, which increases the electrophilicity of the 1-123 source.
- the reaction mixture will comprise from about 0.6 to about 10 micrograms of compound 4-1 per mCi of 1-123 from the 1-123 source.
- the reaction mixture may comprise, per mCi of 1-123, from about 0.6 to about 10 micrograms of compound 4-1, about 3 microliters of trifluoroacetic acid, about 1.5 microliters of Chloramine-T, and about 0.25 microliters of methanokacetic acid (99:1).
- the reaction mixture comprises [ 123 l]Nal, chloramine-T, and trifluoroacetic acid.
- the reaction mixture comprises [ 123 I]ICI and silver tetrafluoroborate.
- the reaction mixture comprises [ 123 l]Nal and tert-butyl hypochlorite.
- the reaction mixture comprises [ 123 l]Nal and peracetic acid in acetate buffer.
- the reaction is typically conducted at a temperature of from about 21°C to about 25°C, and allowed to proceed for at least about 15 minutes, and typically from about 15 minutes to about 2 hours.
- reaction may be quenched by addition of a base, such as N H 4 OH, and compound (I) may be isolated using any suitable technique known in the art, such as solvent evaporation, chromatography, and combinations thereof.
- a base such as N H 4 OH
- suitable reaction conditions for Step D are described in, for example, Musachio, et al., Appl. Radiat. Isoi, 1996, Vol. 47, No. 1, p. 79-81.
- the present invention relates to the alkylation of a nortropane to the corresponding N-(3-fluoropropyl) analogue using 3- fluoropropanal.
- the process advantageously avoids the use of the ozone depleting compound 3-fluoro-l-bromopropane.
- R 1 is selected from the group consisting of halogen, ⁇ - ⁇ Si(CH 3 ) 3 , and ⁇ - ⁇ Sn(CH 3 ) 3 .
- the halogen is I.
- R 1 is I or ⁇ - ⁇ Si(CH 3 ) 3 .
- 3-fluoropropanal is commercially available, or may be prepared by oxidation of 3-fluoropan-l-ol using Dess-Martin periodinane or trichloroisocyanuric acid and TEMPO (i.e., 2,2,6,6-tetramethyl-l-piperidinyloxy) according to the following reaction:
- the reaction mixture may further comprise a solvent.
- suitable solvents include ethyl acetate, 1,2-dichoroethane, acetonitrile, dichloromethane, chloroform, and combinations thereof.
- R 1 is as defined above.
- a reducing agent in the reaction mixture to facilitate reduction of the iminium intermediate compound 1-2 to compound (III).
- suitable reducing agents include sodium triacetoxyborohydride, sodium cyanoborohydride, and formic acid.
- the reducing agent is present in the reaction mixture in an amount of from about 2 equivalents to about 15 equivalents, relative to compound (II).
- the reducing agent may be added to the reaction mixture prior to or subsequent to reaction of compound (II) with the 3-fluoropropanal.
- Suitable reaction conditions are described a bove for Step C of Reaction Scheme 1.
- Compound (III) may be isolated from the reaction mixture using any suitable technique known in the art, including, for example, solvent evaporation, filtration, chromatography, or combinations thereof.
- the Grignard reagent was prepared as follows: The reaction was initiated by adding about 0.6 mL (4 mmol) of (4-bromophenyl)trimethylsilane to a suspension of magnesium (0.286 g, 12 mmol) and an iodine crystal in 7 mL of ether and heating at reflux in an oil bath. After the reaction initiated, the mixture was diluted with 7 mL of ether and the remaining 1.7 mL (8 mmol) of (4- bromophenyl)trimethylsilane was added. The mixture was then heated at reflux for 1.5 hr.
- reaction was worked up by concentrating in vacuo, adding 2 mL of 1:1 waterxoncentrated ammonium hydroxide. The mixture was extracted with three 1 mL portions of dichloromethane. The combined organic layers of each extraction were dried over sodium sulfate. A sample of the organic extracts was analyzed by gas chromatography, which showed 80% compound 3-1.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description



















Claims


Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2014326601A AU2014326601B2 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodinated 3-fluoropropyl-nor-beta-CIT |
| MX2016003035A MX382629B (en) | 2013-09-25 | 2014-09-25 | PREPARATION OF RADIOIODINATED 3-FLUOROPROPYL-NOR-BETA-CIT. |
| EP14849233.3A EP3049378B1 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodinated 3-fluoropropyl-nor-beta-cit |
| CA2922723A CA2922723C (en) | 2013-09-25 | 2014-09-25 | Improved process for the preparation of radioiodinated 3-fluoropropyl-nor-b-cit |
| JP2016545225A JP6224256B2 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodine labeled 3-fluoropropyl-NOR-β-CIT |
| BR112016006129A BR112016006129A2 (en) | 2013-09-25 | 2014-09-25 | preparation of radioiodinated 3-fluoropropyl-nor-beta-cit |
| PL14849233T PL3049378T3 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodinated 3-fluoropropyl-nor-beta-cit |
| ES14849233T ES2711122T3 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodinated 3-fluoropropyl-nor-beta-CIT |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361882039P | 2013-09-25 | 2013-09-25 | |
| US61/882,039 | 2013-09-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015048250A1 true WO2015048250A1 (en) | 2015-04-02 |
Family
ID=52691501
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/057404 Ceased WO2015048250A1 (en) | 2013-09-25 | 2014-09-25 | Preparation of radioiodinated 3-fluoropropyl-nor-beta-cit |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9339565B2 (en) |
| EP (1) | EP3049378B1 (en) |
| JP (2) | JP6224256B2 (en) |
| AU (1) | AU2014326601B2 (en) |
| BR (1) | BR112016006129A2 (en) |
| CA (1) | CA2922723C (en) |
| ES (1) | ES2711122T3 (en) |
| MX (1) | MX382629B (en) |
| PL (1) | PL3049378T3 (en) |
| TR (1) | TR201901758T4 (en) |
| WO (1) | WO2015048250A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016191397A1 (en) | 2015-05-22 | 2016-12-01 | Td2 Inc. | Benzamide and active compound compositions and methods of use |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2569230T3 (en) | 2010-05-12 | 2016-09-19 | Nestec Sa | Capsule, system and method for preparing a drink at the spin |
| US9513907B2 (en) * | 2013-08-06 | 2016-12-06 | Intel Corporation | Methods, apparatus, instructions and logic to provide vector population count functionality |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4190601A (en) * | 1978-05-31 | 1980-02-26 | Union Carbide Corporation | Production of tertiary amines by reductive alkylation |
| US6123917A (en) * | 1990-08-09 | 2000-09-26 | Research Triangle Institute | Method for making cocaine receptor binding ligands and intermediates therefor |
| US6447747B1 (en) * | 1996-10-29 | 2002-09-10 | Guilford Pharmaceuticals Inc. | Process for the preparation of radiopharmaceuticals |
| US6548041B1 (en) * | 1999-05-12 | 2003-04-15 | Organix, Inc. | Dopamine transporter imaging agents |
| US20030109708A1 (en) * | 2000-02-02 | 2003-06-12 | Nihon Medi-Physics Co., Ltd. | Process for synthesizing anhydroecgonine derivative |
| US20100210843A1 (en) * | 2009-02-17 | 2010-08-19 | Mallinckrodt Inc. | Process for the Reductive Alkylation of Normorphinans |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5698179A (en) * | 1992-02-25 | 1997-12-16 | Neuro Imaging Technologies, Llc | Iodinated neuroprobe for mapping monoamine reuptake sites |
| FI104048B (en) * | 1997-06-16 | 1999-11-15 | Map Medical Technologies Oy | Process for the preparation of radioiodinated receptor substances for in vivo use |
| EP1682545B1 (en) * | 2003-10-03 | 2007-12-12 | Pfizer Limited | Imidazopyridine substituted tropane derivatives with ccr5 receptor antagonist activity for the treatment of hiv and inflammation |
| KR100789847B1 (en) * | 2004-12-15 | 2007-12-28 | (주)퓨쳐켐 | Process for preparing organofluoro compound in alcohol solvent |
| RU2474435C2 (en) * | 2007-11-07 | 2013-02-10 | Джи-И Хелткер БВ | Stabilisation of radiopharmaceutical compositions |
| GB0922023D0 (en) * | 2009-12-17 | 2010-02-03 | Ge Healthcare Ltd | Preparation of n-monofluoroalkyl compounds |
-
2014
- 2014-09-25 PL PL14849233T patent/PL3049378T3/en unknown
- 2014-09-25 EP EP14849233.3A patent/EP3049378B1/en active Active
- 2014-09-25 BR BR112016006129A patent/BR112016006129A2/en not_active Application Discontinuation
- 2014-09-25 AU AU2014326601A patent/AU2014326601B2/en active Active
- 2014-09-25 MX MX2016003035A patent/MX382629B/en unknown
- 2014-09-25 CA CA2922723A patent/CA2922723C/en active Active
- 2014-09-25 WO PCT/US2014/057404 patent/WO2015048250A1/en not_active Ceased
- 2014-09-25 JP JP2016545225A patent/JP6224256B2/en active Active
- 2014-09-25 ES ES14849233T patent/ES2711122T3/en active Active
- 2014-09-25 TR TR2019/01758T patent/TR201901758T4/en unknown
- 2014-09-25 US US14/496,376 patent/US9339565B2/en active Active
-
2017
- 2017-09-22 JP JP2017182197A patent/JP6423936B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4190601A (en) * | 1978-05-31 | 1980-02-26 | Union Carbide Corporation | Production of tertiary amines by reductive alkylation |
| US6123917A (en) * | 1990-08-09 | 2000-09-26 | Research Triangle Institute | Method for making cocaine receptor binding ligands and intermediates therefor |
| US6447747B1 (en) * | 1996-10-29 | 2002-09-10 | Guilford Pharmaceuticals Inc. | Process for the preparation of radiopharmaceuticals |
| US6548041B1 (en) * | 1999-05-12 | 2003-04-15 | Organix, Inc. | Dopamine transporter imaging agents |
| US20030109708A1 (en) * | 2000-02-02 | 2003-06-12 | Nihon Medi-Physics Co., Ltd. | Process for synthesizing anhydroecgonine derivative |
| US20100210843A1 (en) * | 2009-02-17 | 2010-08-19 | Mallinckrodt Inc. | Process for the Reductive Alkylation of Normorphinans |
Non-Patent Citations (3)
| Title |
|---|
| BOIS, F ET AL.: "Synthesis, radiolabeling, and baboon SPECT imaging of 2beta-carbomethoxy- 3beta-(3'-[1231]iodophenyl)tropane ([1231]YP256) as a serotonin transporter radiotracer.([1231] YP256) a potential serotonin transporter radiotracer", NUCL MED BIOL., vol. 35, no. 1, 2008, pages 1 - 15, XP022460551, Retrieved from the Internet <URL:http://www.nr-bi.nlm.nih.gov/pmc/articles/PMC2276982> * |
| See also references of EP3049378A4 * |
| STEHOUWER, JS ET AL.: "Synthesis, Radiosynthesis, and Biological Evaluation of Fluorine-18 Labeled 2beta-Carbo(fluoroalkoxy)-3beta-(3'-((Z)-2-haloethenyl phenyl) nortropanes: Candidate Radioligands for In Vivo Imaging of the Serotonin Transporter with Positron Emission Tomography", J MED CHEM., vol. 51, no. 24, 2008, pages 7788 - 7799, XP055335440, Retrieved from the Internet <URL:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668213/pdf/nihms-86624.pdf> [retrieved on 20141124] * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016191397A1 (en) | 2015-05-22 | 2016-12-01 | Td2 Inc. | Benzamide and active compound compositions and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2711122T3 (en) | 2019-04-30 |
| JP2016533390A (en) | 2016-10-27 |
| JP6224256B2 (en) | 2017-11-01 |
| JP6423936B2 (en) | 2018-11-14 |
| US20150087838A1 (en) | 2015-03-26 |
| EP3049378A4 (en) | 2017-04-19 |
| US9339565B2 (en) | 2016-05-17 |
| MX2016003035A (en) | 2016-06-10 |
| JP2017218460A (en) | 2017-12-14 |
| EP3049378B1 (en) | 2018-12-05 |
| EP3049378A1 (en) | 2016-08-03 |
| BR112016006129A2 (en) | 2017-08-01 |
| MX382629B (en) | 2025-03-13 |
| CA2922723A1 (en) | 2015-04-02 |
| PL3049378T3 (en) | 2019-06-28 |
| CA2922723C (en) | 2022-04-19 |
| AU2014326601A1 (en) | 2016-03-17 |
| TR201901758T4 (en) | 2019-03-21 |
| AU2014326601B2 (en) | 2018-01-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4500454B2 (en) | 1,4-diazabicyclo [3.2.2] nonane-4-carboxylate and carboxamide derivatives, their preparation and use in therapy | |
| EP1161434B1 (en) | Pyridopyranoazepine derivatives, preparation and therapeutic use | |
| AU742090B2 (en) | Dopamine transporter imaging ligand | |
| EP0905135B1 (en) | Cocaine receptor binding ligands | |
| CA2658165A1 (en) | Process for the preparation of tiotropium bromide | |
| JP6423936B2 (en) | Preparation of radioiodine labeled 3-fluoropropyl-NOR-β-CIT | |
| US7217822B2 (en) | Process for making cabergoline | |
| CN112876461A (en) | Process for the preparation of nicotine and intermediates therefor | |
| CN101805339B (en) | Entecavir compound preparation method | |
| JP5452165B2 (en) | Process for producing quinuclidine | |
| HRP20050893A2 (en) | Process for the preparation of a cyano-isobenzofuran | |
| JP4480802B2 (en) | Brominating agent | |
| WO2013050929A1 (en) | Manufacturing process for tiotropium bromide | |
| EP2699570B1 (en) | 1,4-disubstituted 1,2,3-triazoles, their preparation processes and their diagnostic and therapeutic uses | |
| JPS5980681A (en) | Manufacture of vincamine derivative | |
| ITMI961918A1 (en) | PROCEDURE FOR THE PREPARATION OF CEPHALOSPORANIC DERIVATIVES | |
| JP2012158524A (en) | Novel carbasugar precursor and method for producing the same, and method for producing bioactive carbasugar amine derivative using them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14849233 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2922723 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016545225 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2016/003035 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2014326601 Country of ref document: AU Date of ref document: 20140925 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014849233 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014849233 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016006129 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112016006129 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160321 |