WO2015077866A1 - Protein kinase inhibitors - Google Patents
Protein kinase inhibitors Download PDFInfo
- Publication number
- WO2015077866A1 WO2015077866A1 PCT/CA2014/000848 CA2014000848W WO2015077866A1 WO 2015077866 A1 WO2015077866 A1 WO 2015077866A1 CA 2014000848 W CA2014000848 W CA 2014000848W WO 2015077866 A1 WO2015077866 A1 WO 2015077866A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mmol
- inhibitors
- treatment
- agents
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*OCc1cnc(C2CC2)nc1 Chemical compound C*OCc1cnc(C2CC2)nc1 0.000 description 3
- IVSZLXZYQVIEFR-UHFFFAOYSA-N Cc1cc(C)ccc1 Chemical compound Cc1cc(C)ccc1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- YSKMVOKQVKWXIY-RUZDIDTESA-N CC(C)(C)OC(N(CCC1)C[C@@H]1[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc2cc(OCc3cnc(C)nc3)cc(F)c2)c1)=O Chemical compound CC(C)(C)OC(N(CCC1)C[C@@H]1[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc2cc(OCc3cnc(C)nc3)cc(F)c2)c1)=O YSKMVOKQVKWXIY-RUZDIDTESA-N 0.000 description 1
- XVJPHCMTROBMCA-UHFFFAOYSA-N CC(C)[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc(cc2)cc(OCc3cnc(C)nc3)c2F)c1 Chemical compound CC(C)[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc(cc2)cc(OCc3cnc(C)nc3)c2F)c1 XVJPHCMTROBMCA-UHFFFAOYSA-N 0.000 description 1
- HVHZPWHNJSXHGP-UHFFFAOYSA-N CC(C)[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc2cccc(OCc3cnc(C)nc3)c2F)c1 Chemical compound CC(C)[n]1c2ncnc(N)c2c(-c(cc2)ccc2Oc2cccc(OCc3cnc(C)nc3)c2F)c1 HVHZPWHNJSXHGP-UHFFFAOYSA-N 0.000 description 1
- PXZMBUKDGVRCBA-UHFFFAOYSA-N CC(C)c1ncc(CO)cn1 Chemical compound CC(C)c1ncc(CO)cn1 PXZMBUKDGVRCBA-UHFFFAOYSA-N 0.000 description 1
- KRJLBBTVAZUERA-UHFFFAOYSA-N CC(C)c1ncc(COC)cn1 Chemical compound CC(C)c1ncc(COC)cn1 KRJLBBTVAZUERA-UHFFFAOYSA-N 0.000 description 1
- HHFZLXYKHWLAPS-UHFFFAOYSA-N CC(C)c1ncc(COc2cc(Br)cc(F)c2)cn1 Chemical compound CC(C)c1ncc(COc2cc(Br)cc(F)c2)cn1 HHFZLXYKHWLAPS-UHFFFAOYSA-N 0.000 description 1
- JEXVKNFMKLKJMM-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(CCC3CC3)C(C)(C)O2)OC1(C)C Chemical compound CC1(C)OB(B2OC(C)(CCC3CC3)C(C)(C)O2)OC1(C)C JEXVKNFMKLKJMM-UHFFFAOYSA-N 0.000 description 1
- GPCBHDGADSSHKM-UHFFFAOYSA-N CCCCN(C)CC(O)=O Chemical compound CCCCN(C)CC(O)=O GPCBHDGADSSHKM-UHFFFAOYSA-N 0.000 description 1
- AJOKFCAKYWTSGP-UHFFFAOYSA-N CCCc1ncc(COC)cn1 Chemical compound CCCc1ncc(COC)cn1 AJOKFCAKYWTSGP-UHFFFAOYSA-N 0.000 description 1
- HYFLWBNQFMXCPA-UHFFFAOYSA-N CCc1ccccc1C Chemical compound CCc1ccccc1C HYFLWBNQFMXCPA-UHFFFAOYSA-N 0.000 description 1
- FFDGPVCHZBVARC-UHFFFAOYSA-N CN(C)CC(O)=O Chemical compound CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 description 1
- GRXPOIRCBRHSCW-UHFFFAOYSA-N COCc1cnc(CO)cc1 Chemical compound COCc1cnc(CO)cc1 GRXPOIRCBRHSCW-UHFFFAOYSA-N 0.000 description 1
- TWCXNDFLOHYYHZ-UHFFFAOYSA-N COCc1cnc(CO)nc1 Chemical compound COCc1cnc(CO)nc1 TWCXNDFLOHYYHZ-UHFFFAOYSA-N 0.000 description 1
- RKHKHCHHOHWWKY-UHFFFAOYSA-N COCc1cncnc1 Chemical compound COCc1cncnc1 RKHKHCHHOHWWKY-UHFFFAOYSA-N 0.000 description 1
- RZXMPPFPUUCRFN-UHFFFAOYSA-N Cc(cc1)ccc1N Chemical compound Cc(cc1)ccc1N RZXMPPFPUUCRFN-UHFFFAOYSA-N 0.000 description 1
- JJYPMNFTHPTTDI-UHFFFAOYSA-N Cc1cc(N)ccc1 Chemical compound Cc1cc(N)ccc1 JJYPMNFTHPTTDI-UHFFFAOYSA-N 0.000 description 1
- YSFBAGSOKZIXIZ-UHFFFAOYSA-N Cc1ccc(COC)cn1 Chemical compound Cc1ccc(COC)cn1 YSFBAGSOKZIXIZ-UHFFFAOYSA-N 0.000 description 1
- HOBMGFDJYBEWLS-UHFFFAOYSA-N Cc1ncc(CO)cn1 Chemical compound Cc1ncc(CO)cn1 HOBMGFDJYBEWLS-UHFFFAOYSA-N 0.000 description 1
- RTSYMKPGWBDUCB-UHFFFAOYSA-N Cc1ncc(COC)[s]1 Chemical compound Cc1ncc(COC)[s]1 RTSYMKPGWBDUCB-UHFFFAOYSA-N 0.000 description 1
- BKCJLEABVDUETF-UHFFFAOYSA-N Cc1ncc(COC)cn1 Chemical compound Cc1ncc(COC)cn1 BKCJLEABVDUETF-UHFFFAOYSA-N 0.000 description 1
- QLHIUVWJBJUAOF-UHFFFAOYSA-N Cc1ncc(COc2cc(F)cc(Oc(cc3)ccc3-c3c[n](C4CNCCC4)c4ncnc(N)c34)c2)cn1 Chemical compound Cc1ncc(COc2cc(F)cc(Oc(cc3)ccc3-c3c[n](C4CNCCC4)c4ncnc(N)c34)c2)cn1 QLHIUVWJBJUAOF-UHFFFAOYSA-N 0.000 description 1
- OQUKJSVQMVBUBY-UHFFFAOYSA-N Fc1cc(I)cc(Br)c1 Chemical compound Fc1cc(I)cc(Br)c1 OQUKJSVQMVBUBY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/10—Organic compounds
- A61K49/101—Organic compounds the carrier being a complex-forming compound able to form MRI-active complexes with paramagnetic metals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/58—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/58—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances
- G01N33/582—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances with fluorescent label
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to a novel family of protein kinase inhibitors, to the processes for preparation of these compounds, to pharmaceutical compositions comprising them, and to their use in the treatment of proliferative, inflammatory, autoimmune, or infectious diseases, disorders, or conditions associated with kinase function.
- Protein kinases are a large group of intracellular and transmembrane signaling proteins in eukaryotic cells (Manning G. et al, (2002) Science, 298: 1912-1934). These enzymes are responsible for transfer of the terminal (gamma) phosphate from ATP to specific amino acid residues of target proteins. Phosphorylation of specific amino acid residues in target proteins can modulate their activity leading to profound changes in cellular signaling and metabolism. Protein kinases can be found in the cell membrane, cytosol and organelles such as the nucleus and are responsible for mediating multiple cellular functions including metabolism, cellular growth and differentiation, cellular signaling, modulation of immune responses, and cell death.
- Serine kinases specifically phosphorylate serine or threonine residues in target proteins.
- tyrosine kinases including tyrosine receptor kinases, phosphorylate tyrosine residues in target proteins.
- Tyrosine kinase families include: Tec, Src, Abl, Jak, Csk, Fak, Syk, Fer, Ack and the receptor tyrosine kinase subfamilies including EGFR, FGFR, VEGFR, RET and Eph.
- Kinases exert control on key biological processes related to health and disease. Furthermore, aberrant activation or excessive expression of various protein kinases are implicated in the mechanism of multiple diseases and disorders characterized by benign and malignant proliferation, as well as diseases resulting from inappropriate activation of the immune system (Kyttaris V.C., Drug Des. Devel. Ther. 2012, 6:245-50 and Fabbro D. et al. Methods Mol. Biol., 2012, 795:1-34).
- inhibitors of select kinases or kinase families are expected to be useful in the treatment of cancer, vascular disease, autoimmune diseases, and inflammatory conditions including, but not limited to: solid tumors, hematological malignancies, thrombus, arthritis, graft versus host disease, lupus erythematosus, psoriasis, colitis, illeitis, multiple sclerosis, uveitis, coronary artery vasculopathy, systemic sclerosis, atherosclerosis, asthma, transplant rejection, allergy, dermatomyositis, pemphigus, and the like.
- Tec kinases are a family of non-receptor tyrosine kinases predominantly, but not exclusively, expressed in cells of hematopoietic origin (Bradshaw J.M. Cell Signal. ,2010 ,22:1175-84).
- the Tec family includes Tec, Bruton's tyrosine kinase (Btk), inducible T- cell kinase (Itk), resting lymphocyte kinase (Rlk Txk), and bone marrow-expressed kinase (Bmx/Etk).
- Btk is important in B-cell receptor signaling and regulation of B-cell development and activation (W.N. Khan et al.
- Btk is activated by Src-family kinases and phosphorylates PLC gamma leading to effects on B-cell function and survival. Additionally, Btk is important in signal transduction in response to immune complex recognition by macrophage, mast cells and neutrophils. Btk inhibition is also important in survival of lymphoma cells (Herman SEM. Blood, 2011 , 117:6287-6289) suggesting that inhibition of Btk may be useful in the treatment of lymphomas. As such, inhibitors of Btk and related kinases are of great interest as anti-inflammatory as well as anticancer agents. Btk is also important for platelet function and thrombus formation suggesting that Btk-selective inhibitors may prove to be useful antithrombotic agents (Liu J. Blood, 2006,108:2596-603).
- Bmx another Tec family member which has roles in inflammation, cardiovascular disease, and cancer (Cenni B. et al. Int Rev. Immunol. 2012, 31 : 166-173) is also important for self-renewal and tumerogenic potential of glioblastoma stem cells (Guryanova OA et al. Cancer Cell Cancer Cell 2011 ,19:498-511). As such, Bmx inhibitors are expected to be useful in the treatment of various diseases including cancer, cardiovascular disease and inflammation.
- the SRC family of tyrosine kinases includes cSRC, Lyn, Fyn, Lck, Hck, Fgr, Blk, Syk, Yrk and Yes.
- cSRC is critically involved in signaling pathways involved in cancer and is often over-expressed in human malignancies (Kim L.C. et al. (2009) Nat. Rev. Clin. Oncol. 6:587-9). cSRC is involved in signaling downstream of growth factor receptor tyrosine kinases and regulates cell cycle progression suggesting that cSRC inhibition would impact cancer cell proliferation. Furthermore, Src inhibitors or downregulation of Hck sensitize tumor cells to immunotoxins (Lui X.F., Mol. Cancer Ther. 2013, Oct. 21).
- SRC family members may be useful in treatments designed to modulate immune function.
- SRC family members including Lck, regulate T-cell receptor signal transduction which leads to gene regulation events resulting in cytokine release, survival and proliferation.
- inhibitors of Lck may be useful immunosuppressive agents with potential application in graft rejection and T-cell mediated autoimmune disease (Martin et al. Expert Opin. Ther. Pat. 2010, 20:1573-93).
- the Src family member HCK is implicated in regulation of cytokine production suggesting that inhibition of this kinase may be useful in treatment of inflammatory disease (Smolinska M.J. et al. J. Immunol. 2011 ;187:6043-51).
- Src family kinase Fgr is critical for activation of mast cells and IgE-mediated anaphylaxis suggesting that this kinase is a potential therapeutic target for allergic diseases (Lee J.H. et al. J. Immunol. 2011 ;187:1807-15)
- the present invention relates to a novel family of kinase inhibitors.
- Compounds of this class have been found to have inhibitory activity against members of the Tec or Scr protein kinase families.
- One aspect of the present invention is directed to a compound of Formula I:
- R is selected from the group consisting of:
- heteroaryl wherein the alkyl, heteroalkyl, carbocyclyl, heterocyclyl, aryl, or heteroaryl are optionally substituted;
- R is selected from the group consisting of:
- E is oxygen
- R 2 is substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl; wherein Y-E-Z-W is
- X 1 and X 2 are independently hydrogen or halogen; m is an integer from 0 to 4,
- n' is an integer from 0 to 4.
- Another embodiment includes compounds of Formula I, wherein Z is selected from the group consisting of:
- Another embodiment of the present invention includes compounds of Formula I, wherein Y is
- Preferred embodiment includes compounds of Formula I, wherein R 1 is hydrogen.
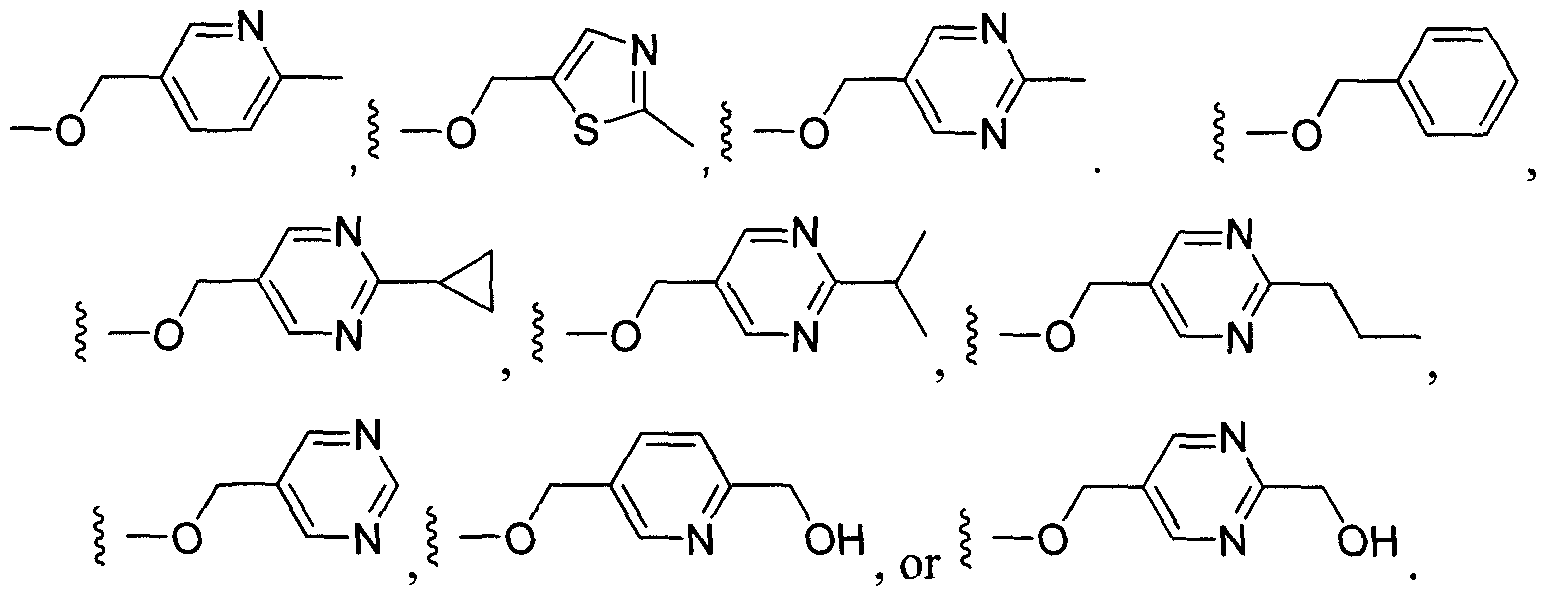
- Another embodiment of the present invention includes compounds of Formula I, wherein R is selected from the group consisting of:
- R is selected from the group consisting of:
- heteroaryl wherein the alkyl, heteroalkyi, carbocyclyl, heterocyclyl, aryl, or heteroaryl are optionally substituted;
- R 2 is substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl.
- Another embodiment of the present invention includes compounds of Formula II, wherein W is selected from the group consisting of:
- Another embodiment of the present invention includes compounds of Formula II, wherein R is selected from the group consisting of:
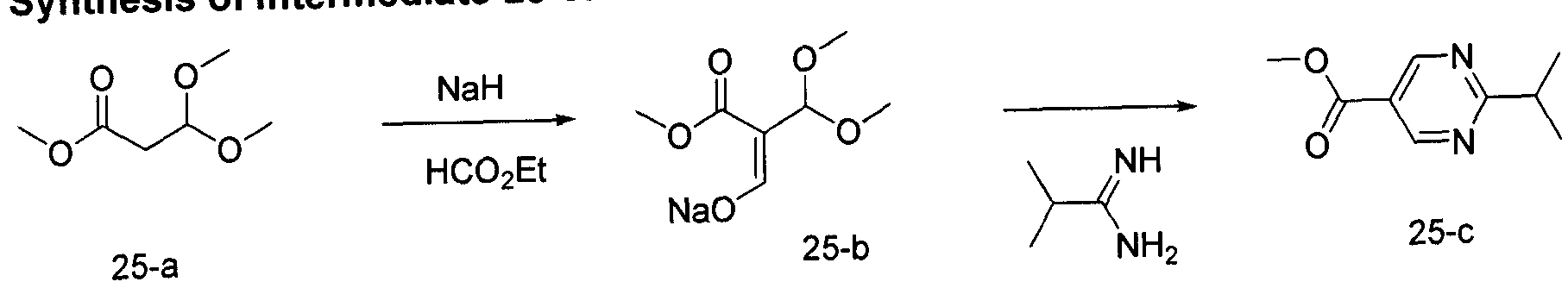
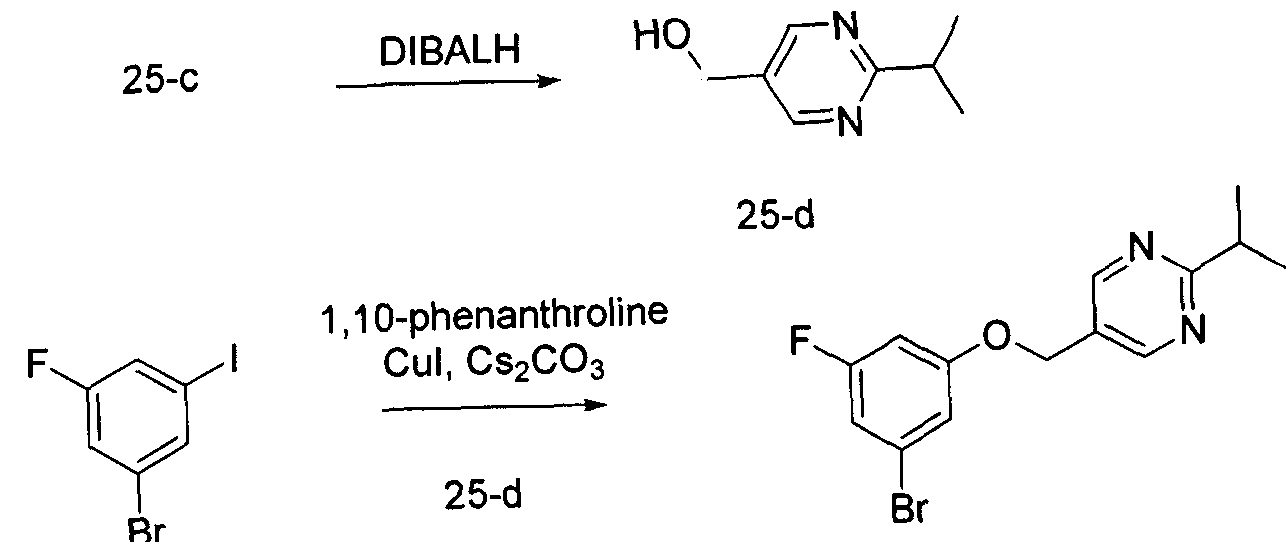
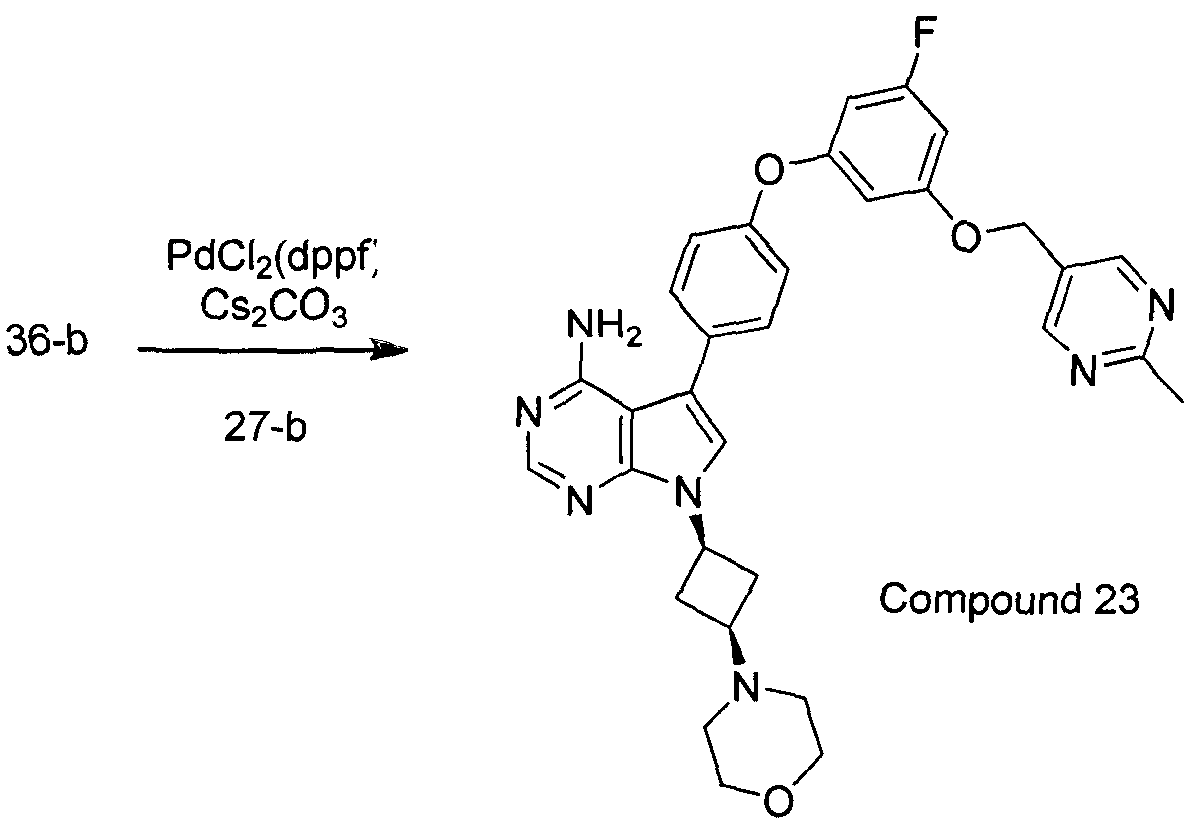
- Another aspect of the present invention provides intermediates and their synthesis related to a process of production of compounds of the invention as defined herein, or a pharmaceutically acceptable salt, or solvate, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, or a pharmaceutical composition as defined herein.
- the present invention relates to a process for preparing a compound of Formula I or Formula II, wherein the process comprises:
- Another aspect of the present invention provides the process for preparing a compound of Formula I or Formula II, wherein the process comprises:
- Another aspect of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I, or Formula II, or a pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, and at least one pharmaceutically acceptable carrier, diluents, or excipient.
- the present invention relates to a compound of the invention as defined herein, or a pharmaceutically acceptable salt, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, or a pharmaceutical composition as defined herein, for use in therapy.
- the present invention relates to a compound of the invention as defined herein, or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition as defined herein, for use in the treatment of subjects suffering from a protein kinase mediated diseases or conditions.
- Another aspect of the present invention provides a use of the compound of Formula I, or Formula II, or a pharmaceutically acceptable salt, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, as an inhibitor of protein kinase, more particularly, as an inhibitor of members of the Tec family of kinases.
- a further aspect of the present invention provides a use of the compound of Formula I, or Formula II, or a pharmaceutically acceptable salt, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, as an inhibitor of protein kinase, more particularly, as an inhibitor of members of the Src family of kinases.
- Another aspect of the present invention provides a use of the compound of Formula I, or Formula II, as an inhibitor of protein kinase, more particularly, as an inhibitor wherein the diseas is a protein kinase mediated disease, disorder, or condition in which Btk kinase activity is implicated.
- the present invention relates to the use of a compound of the invention as defined herein, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for use in the treatment of subjects suffering from a protein kinase mediated diseases or conditions.
- a further aspect of the present invention provides a pharmaceutically acceptable salt, or solvate thereof, for use in manufacturing of a pharmaceutical composition, for use in treatment of proliferative, inflammatory, infectious, or autoimmune diseases.
- Another aspect of the present invention provides a compound, or pharmaceutically acceptable salts, or solvates thereof, or a pharmaceutical composition, as defined in present invention, for use in the treatment of a proliferative disorder, inflammatory, or autoimmune disease.
- the proliferative disorder, inflammatory, or autoimmune disease is cancer. More particular, is a human cancer.
- a further aspect of the present invention provides the use of a compound, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for use in the treatment of a proliferative disorder, such as cancer.
- Another aspect of the present invention provides a compound of Formula I, or Formula II, or a pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes, or biologically active metabolites thereof, for use in the treatment of a proliferative, inflammatory, or autoimmune diseases, or disorder state in combination v/ith an agent selected from: an estrogen receptor modulator; an androgen receptor modulator; a retinoid receptor modulator; a cytotoxic agent; an anti-proliferative agent comprises adriamycin, dexamethasone, vincristine, cyclophosphamide, fluorouracil, topotecan, taxol, interferons, or platinum derivatives; an anti-inflammatory agent comprises corticosteroids, TNF blockers, IL-1 RA, azathioprine, cyclophosphamide, or sulfasalazine; a
- the medicament is for the treatment of a proliferative disorder or disease state in combination with a death receptor agonist.
- Another aspect of the present invention provides a compound, or pharmaceutically acceptable salts, or solvates thereof, or a pharmaceutical composition as defined in present invention, for use in the treatment of diseases or disorders selected from: cancer, myeloproliferative disorders, lung fibrosis, hepatic fibrosis, cardiovascular diseases: cardiac hypertrophy, cardiomyopathy, restenosis; thrombosis, heart attacks or stroke; alopecia, emphysema; atherosclerosis, psoriasis or dermatological disorders, lupus, multiple sclerosis, macular degeneration, asthma, reactive synoviotides, viral disorders; CNS disorders; auto-immune disorders: glomerulonephritis or rheumatoid arthritis; hormone-related diseases, metabolic disorders; inflammatory diseases; infectious or fungal diseases, malaria or parasitic disorders.
- diseases or disorders selected from: cancer, myeloproliferative disorders, lung fibrosis, hepatic fibrosis, cardiovascular diseases: cardiac hypertrophy,
- Another aspect of the present invention provides a compound, or pharmaceutically acceptable salts, or solvates thereof, or a pharmaceutical composition, as defined in present invention, for use in the manufacture of a medicament for the treatment of: arthritis, tenosynovial giant cell tumour, pigmented villonodular synovitis, and other reactive synoviotides, bone metastases formation and progression, acute myeloid leukemia, or human cancer, or select subsets of cancer, for example breast tumours and gastric cancer by inhibition of kinase activity.
- the present invention relates to a method of treating a disease or condition associated with protein kinase activity, said method comprising administering to a subject a therapeutically effective amount of a compound of the invention as defined herein, or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition as defined herein.
- the present invention provides a method of treating a proliferative disorder, said method comprising administering to a subject a therapeutically effective amount of a compound, or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition as defined herein.
- the proliferative disorder is a cancer.
- Another aspect of the present invention provides a method of modulating kinase function, the method comprising contacting a cell with a compound of the present invention in an amount sufficient to modulate the enzymatic activity of a given kinase, or kinases from Tec family kinases, thereby modulating the kinase function.
- a further aspect of the present invention provides a method of modulating kinase function, the method comprising contacting a cell with a compound of the present invention in an amount sufficient to modulate the enzymatic activity of a given kinase, or kinases from Src family, thereby modulating the kinase function.
- Another aspect of the present invention provides a method of inhibiting cell proliferation or survival in vitro or in vivo, said method comprising contacting a cell with an effective amount of a compound as defined herein, or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of producing a protein kinase inhibitory effect in a cell or tissue, said method comprising contacting the cell or tissue with an effective amount of a compound, or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of producing a protein kinase inhibitory effect in vivo, said method comprising administering to a subject an effective amount of a compound, or a pharmaceutically acceptable salt or solvate thereof.
- the administration may be by any suitable route of administration, such including parenteral or oral administration.
- the dosage amount may be any suitable amount, for example, the dosage unit for parenteral or oral administration may contain from about 50 mg to about 5000 mg of a compound of Formula I, or Formula II, or a pharmaceutical acceptable salt, or solvate thereof.
- the compound of the present invention may be administered 1 to 4 times a day. A dosage of between 0.01-100 mg/kg body weight/day of the compound of the present invention can be administered to a patient receiving these compositions.
- the compounds of the present invention may be used alone or in combination with one or more other therapeutic agents.
- the combination may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of treatment.
- Such combination products employ the compounds of this invention within the dose range described hereinbefore and the other pharmaceutically active agent within its approved dose range.
- Another aspect of the present invention provides a method of modulating the target kinase function.
- the method comprising:
- the present invention further provides a method of synthesis a compound, or a pharmaceutically acceptable salt or solvate thereof, as defined herein.
- probe comprising a compound of Formula I, or Formula II, labeled with a detectable label or an affinity tag.
- the probe comprises a residue of a compound of Formula I, or Formula II, covalently conjugated to a detectable label.
- detectable labels include, but are not limited to, a fluorescent moiety, a chemiluminescent moiety, a paramagnetic contrast agent, a metal chelate, a radioactive isotope-containing moiety, or biotin.
- the present invention relates to novel kinase inhibitors. These compounds are found to have activity as inhibitors of protein kinases, including members of the Src or Tec kinase families.
- Compounds of the present invention may be formulated into a pharmaceutical composition, which comprises ⁇ effective amount of a compound of the present invention, with at least one pharmaceutically acceptable diluent, carrier, or excipient.
- pharmaceutically effective amount refers to any amount of the composition for the prevention and treatment of subjects that is effective in treating a disease, disorder, or condition associated with protein kinase activity.
- a pharmaceutical composition which comprises a compound of Formula I, Formula II, combinations thereof, or a pharmaceutically acceptable salt, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, or mixtures of the compounds of the present invention, in association with at least one pharmaceutically acceptable excipient, diluent, or carrier.
- the pharmaceutical compositions may be in a conventional pharmaceutical form suitable for oral administration (e.g., tablet, capsule, granules, powder, liquid solution, suspension or syrup); for parenteral administration (e.g., cutaneous, subcutaneous, intramuscular, intraperitoneal, intravenous, intra-arterial, intra-cerebral, intraocular injection, or infusion); suppository, rectal or vaginal; bronchial, nasal, topical, buccal, sub-lingual, transdermal, or drop infusion preparations, inhalation or insufflations, eye lotion or liquid aerosol.
- the compounds may be formulated into pharmaceutically acceptable dosage forms by conventional methods known to those skilled in the art.
- the choice of the core excipients is extremely important.
- Several aspects of the finished dosage form must be considered such as the nature of the active pharmaceutical ingredient (API), the intended delivery method of the API (immediate release, modified, sustained, extended, delayed release etc), and the manufacturing process.
- API active pharmaceutical ingredient
- compositions comprising a compound of Formula I, or Formula II (or combinations of the inventive compounds), according to the present invention, and at least one pharmaceutically acceptable excipient, such as a binder, a disintegrating agent, a lubricant, a diluent, a solubilizing agent, an emulsifier, a coating agent, a cyclodextrin or buffer, for use in formulation of suitable release dosage forms: "prolonged release”, “extended release”, “modified release”, “delayed release”, “sustained release”, or “immediate release”, "orally disintegrating tablets", or "sustained release parenteral depot” pharmaceutical compositions.
- a pharmaceutically acceptable excipient such as a binder, a disintegrating agent, a lubricant, a diluent, a solubilizing agent, an emulsifier, a coating agent, a cyclodextrin or buffer
- controlled release pharmaceutical compositions there are different dosage forms with plurality of "controlled release” pharmaceutical compositions, particularly “prolonged release”, “extended release”, “modified release”, “delayed release”, or “sustained release” compositions.
- Examples for controlled release pharmaceutical compositions are immediate release pharmaceutical compositions, enteric coated pharmaceutical compositions, pulsed release pharmaceutical compositions, or sustained release pharmaceutical compositions.
- An oral "controlled release pharmaceutical composition” means a pharmaceutical composition including at least one active pharmaceutical ingredient which is formulated with at least one pharmaceutically acceptable film forming polymer, and optionally with at least one pharmaceutically acceptable excipient, where the pharmaceutical composition shows a pH-dependent. or a pH-independent reproducible release profile.
- oral controlled release pharmaceutical composition is defined to mean oral pharmaceutical compositions which when administered releases the active ingredient at a relatively constant rate, and provide plasma concentrations of the active ingredient that remain substantially invariant with time within the therapeutic range of the active ingredient over a 24-hour period, and encompasses "prolonged release", “extended release”, “modified release”, “delayed release” or “sustained release” compositions.
- modified release means that the escape of the drug from the tablet has been modified in some way. Usually, this is to slow the release of the drug so that the medicine doesn't have to be taken too often, and therefore improves compliance.
- the other benefit from modifying release is that the drug release is controlled, and there are smaller peaks, and troughs in blood levels therefore reducing the chance of peak effects, and increasing the likelihood of therapeutic effectiveness for longer periods of time.
- continuous release means that a term applied to a drug that is designed to deliver a dose of a medication over an extended period.
- the most common device for this purpose is a soft, soluble capsule containing minute pellets of the drug for release at different rates in the Gl tract, depending on the thickness and nature of the oil, fat, wax, or resin coating on the pellets.
- Another system consists of a porous plastic carrier, impregnated with the drug, and a surfactant to facilitate the entry of Gl fluids that slowly leach out of the drug. Ion exchange resins that bind to drugs and liquids containing suspensions of slow-release drug granules, are also used to provide medication over an extended period.
- pulsatile release means that a drug is delivered in one, or more doses that fluctuate between a maximum and minimum dose, over a predetermined time intervals. This can be represented by a dose release profile having one or more distinct peaks, or valleys. However, two or more pulsed releases may produce an overlapping, overall, or composite release profile that appears, or effectively is constant.
- the need for pulsatile release may include the desire to avoid drug degradation in the stomach, or first pass metabolism.
- Pulsatile release can be achieved via coating of multiparticulates with pH dependent, and/or barrier membrane coating systems, followed by blending of the multiparticulates to achieve desired release profiles.
- the term “delayed” release” refers to the onset of release in relationship to administration of the drug. “Delayed”, means that the release of drug is postponed, and begins, or is triggered some period of time after administration (e.g., the lag time), typically a relatively long period of time, e.g. more than one hour.
- immediate release means that oral pharmaceutical compositions, which when administered release the active ingredient within a small period of time, typically less than 45 minutes after admin stration.
- Oral formulations for immediate release drug delivery system is a conventional type of drug delivery system that designed to disintegrate, and release their pharmaceutically active ingredient with no rate controlling features, such as special coatings and other techniques.
- Orally Disintegrating Tablets refers to the tablet that have a disintegration time less than 60 seconds, with good mouth feel and friability that did not exceed 1%. Orally Disintegrating Tablet (ODT) allows to improve patient compliance, in particular with pediatric, geriatric, and institutionalized patients, or patients with chemotherapy-induced nausea.
- Oral dosage forms which may be employed with the present invention include: tablets, granules, spheroids, or pellets in a capsule, or in any other suitable solid form.
- a “depot formulation” may be formulated to provide slow absorption of the molecules of Formula I, or Formula II, or combinations thereof, or pharmaceutically acceptable salts, derivatives, isomers, polymorphs, solvates, hydrates, analogues, enantiomers, tautomeric forms, or mixtures thereof from the site of administration, often keeping therapeutic levels of the molecule, or an active metabolite in the patient's system for days or weeks at a time.
- a depot formulation may provide convenience for a patient in need of chronic medication. By delivering molecules of the present invention without exposure to the Gl tract.
- a depot formulation may provide better compliance due to the infrequent dosing regimen and convenience.
- a depot formulation that will enhance patient compliance are good local tolerance at the injection site and ease of administration.
- the dosage form will vary depending on the symptoms, age, and body weight of the patient, the nature and severity of the disorder to be treated or prevented, the route of administration, and the form of the drug. In general a daily dosage form 0.01 to 2000 mg of the compound is recommended for an adult human patient, and this may be administered in a single dose, or in divided doses.
- the amount of active ingredient, which can be combined with at least one carrier material, to produce a single dosage form will generally be that amount of the compound which produces a therapeutic effect.
- time of administration or amount of the composition that will yield the most effective results in terms of efficacy of treatment, in a given patient will depend upon the activity, pharmacokinetics, and bioavailability of a particular compound, physiological condition of the patient (including age, sex, disease type, and stage, general physical condition, responsiveness to a given dosage form, and type of medication), route of administration, etc.
- phrases "pharmaceutically acceptable” is employed herein to refer to those ligands, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable carrier means a pharmaceutically acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material.
- a pharmaceutically acceptable material such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material.
- Each carrier must be acceptable in the sense of being compatible with the other ingredients of the formulation, including the active ingredient, and not injurious or harmful to the patient.
- materials which can serve as pharmaceutically acceptable carriers include:
- sugars such as lactose, glucose, or sucrose
- starches such as corn starch, potato starch, and substituted or unsubstituted ⁇ -cyclodextrin
- cellulose, and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose, or cellulose acetate
- excipients such as cocoa butter or suppository waxes
- oils such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil, or soybean oil
- glycols such as propylene glycol
- polyols such as glycerin, sorbitol, mannitol, or polyethylene glycol
- esters such as ethyl oleate, or ethyl laurate
- agar such as agar
- buffering agents such as magnesium hydroxide or aluminum hydroxide
- alginic acid such as glycerin, sorbitol, mannitol, or polyethylene glycol
- esters such as ethyl oleate, or ethyl laurate
- agar such as agar
- buffering agents such as magnesium hydroxide or aluminum hydroxide
- alginic acid such as pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; and (21 ) other non-toxic compatible substances employed in pharmaceutical formulations.
- pharmaceutically acceptable salt refers to the relatively non-toxic, inorganic and organic acid addition salts of the compound(s). These salts can be prepared in situ during the final isolation and purification of the compound(s), or by separately reacting a purified compound(s) in its free base form with a suitable organic or inorganic acid, and isolating the salt thus formed.
- Representative salts include the hydrobromide, hydrochloride, sulfate, bisulfate, phosphate, nitrate, acetate, valerate, oleate, palmitate, stearate, laurate, benzoate, lactate, phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate, naphthylate, mesylate, glucoheptonate, lactobionate, laurylsulphonate salts, and amino acid salts, and the like (See, for example, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66: 1-19).
- halo or halogen refers to chlorine, bromine, fluorine, or iodine. Fluorine is a preferred halogen.
- compositions of the present invention may be obtained by conventional procedures using conventional pharmaceutical excipients, well known in the art.
- the compounds of the present invention may contain one or more acidic functional groups and, thus, are capable of forming pharmaceutically acceptable salts with pharmaceutically acceptable bases, such as the hydroxide, carbonate, or bicarbonate of a pharmaceutically acceptable metal cation, with ammonia, or with a pharmaceutically acceptable organic primary, secondary, or tertiary amine.
- pharmaceutically acceptable bases such as the hydroxide, carbonate, or bicarbonate of a pharmaceutically acceptable metal cation, with ammonia, or with a pharmaceutically acceptable organic primary, secondary, or tertiary amine.
- Representative alkali or alkaline earth salts include the lithium, sodium, potassium, calcium, magnesium, and aluminum salts, and the like.
- Representative organic amines useful for the formation of base addition salts include ethylamine, diethylamine, ethylenediamine, ethanolamine, diethanolamine, piperazine, and the like (see, for example, Berge et al.).
- affinity tag means a ligand or group
- alkyl refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups, including haloalkyl groups such as trifluoromethyl and 2,2,2-trifluoroethyl, etc.
- Representative alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, (cyclohexyl)methyl, cyclopropylmethyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- alkenyl and alkynyl refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- Representative alkenyl groups include vinyl, propen-2-yl, crotyl, isopenten-2-yl, 1 ,3-butadien-2-yl), 2,4- pentadienyl, and 1 ,4-pentadien-3-yl.
- Representative alkynyl groups include ethynyl, 1- and 3-propynyl, and 3-butynyl.
- alkyl substituents are lower alkyl groups, e.g., having from 1 to 6 carbon atoms.
- alkenyl and alkynyl preferably refer to lower alkenyl and alkynyl groups, e.g., having from 2 to 6 carbon atoms.
- alkylene refers to an alkyl group with two open valencies (rather than a single valency), such as -(CH 2 )i-io- and substituted variants thereof.
- alkoxy refers to an alkyl group having an oxygen attached thereto. Representative alkoxy groups include methoxy, ethoxy, propoxy, tert-butoxy and the like.
- An "ether” is two hydrocarbons covalently linked by an oxygen. Accordingly, the substituent of an alkyl that renders that alkyl an ether is or resembles an alkoxy.
- alkoxyalkyi refers to an alkyl group substituted with an alkoxy group, thereby forming an ether.
- amide and “amido”, are art-recognized as an amino-substituted carbonyl and includes a moiety that can be represented by the general formula:
- R 9 , R 10 are as defined above.
- Preferred embodiments of the amide will not include imides, which may be unstable.
- amine and “amino”, are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by the general formulae:
- R 9 , R 10 and R 10 each independently represent a hydrogen, an alkyl, an alkenyl, -(CH 2 ) -R 8 , or R 9 and R 10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure;
- R 8 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocyclyl, or a polycyclyl; and
- p is zero or an integer from 1 to 8.
- only one of R 9 or R 10 can be a carbonyl, e.g., R 9 , R 10 , and the nitrogen together do not form an imide.
- R 9 and R 10 each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH 2 ) P -R 8 .
- the amino group is basic, meaning the protonated form has a pK a > 7.00.
- aralkyl refers to an alkyl group substituted with an aryl group, for example -(CH 2 ) P -Ar.
- heteroaryl refers to an alkyl group substituted with a heteroaryl group, for example -(CH 2 ) P -Het.
- aryl as used herein includes 5-, 6-, or 7-membered substituted, or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon.
- aryl also includes polycyclic ring systems, having two or more cyclic rings, in which two or more carbons are common to two adjoining rings, wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, or heterocyclyls.
- Aryl groups include benzene, naphthalene, phenanthrene, phenol, aniline, anthracene, or phenanthrene.
- Carbocycle and “carbocyclyl”, as used herein, refer to a non-aromatic substituted or unsubstituted ring in which each atom of the ring is carbon.
- the terms “carbocycle” and “carbocyclyl” also include polycyclic ring systems having two or more cyclic rings, in which two or more carbons are common to two adjoining rings, wherein at least one of the rings is carbocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, or heterocyclyls.
- Representative carbocyclic groups include cyclopentyl, cyclohexyl, 1-cyclohexenyl, or 3- cyclohexen-1- yl, cycloheptyl.
- carbonyl is art-recognized and includes such moieties as can be represented by the general formula:
- X is a bond or represents an oxygen, or a sulfur
- R 1 represents a hydrogen, an alkyl, an alkenyl, -(CH 2 ) P -R 8 or a pharmaceutically acceptable salt.
- X is oxygen and R 11 is not hydrogen, the formula represents an "ester”.
- X is oxygen, and R 11 is hydrogen, the formula represents a "carboxylic acid”.
- heteroaryl includes substituted or unsubstituted aromatic 5- to 7-membered ring structures, more preferably 5- to 6-membered rings, whose ring structures include one to four heteroatoms.
- heteroaryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, isoxazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine, or pyrimidine, and the like.
- heteroatom as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, or sulfur.
- heterocyclyl or “heterocyclic group” refer to substituted or unsubstituted non- aromatic 3- to 10-membered ring structures, more preferably 3- to 7-membered rings, whose ring structures include one to four heteroatoms.
- heterocyclyl or “heterocyclic group” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heterocyclyl groups include, for example, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, pyrrolidine, morpholine, lactones, or lactams.
- Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocycle, alkyl, alkenyl, alkynyl, or combinations thereof.
- polycyclyl or “polycyclic”, refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings".
- rings e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls
- Each of the rings of the polycycle can be substituted or unsubstituted.
- the term "probe” means a compound of the invention which is labeled with either a detectable label, or an affinity tag, and which is capable of binding, either covalently or non-covalently, to a protein kinase domain.
- the probe When, for example, the probe is non-covalently bound, it may be displaced by a test compound.
- the probe When, for example, the probe is bound covalently, it may be used to form cross-linked adducts, which may be quantified and inhibited by a test compound.
- substituted refers to moieties having substituents replacing a hydrogen on one or more atoms of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with T/CA2014/000848 permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same, or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein, which satisfy the valences of the heteroatoms.
- Substituents can include, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiet/. It will be
- Compounds of the invention also include all isotopes of atoms present in the intermediates or final compounds.
- Isotopes include those atoms having the same atomic number, but different mass numbers.
- isotopes of hydrogen include deuterium and tritium.
- the compounds of the present invention are inhibitors of protein kinase activity.
- An aspect of the present invention provides a method of inhibiting protein kinase activity in a cell, the method comprising administering to said cell compound of Formula I, or Formula II, as defined herein, combinations thereof, or a pharmaceutically acceptable salt, or solvate thereof.
- the present invention provides a method of inhibiting protein kinase in vitro or in vivo, said method comprising contacting a cell with an effective amount of a compound, or a pharmaceutically acceptable salt, or solvate thereof, as defined herein.
- a further aspect of the preser.t invention provides a method of inhibiting protein kinase activity in a human or an.mal subject, the method comprising administering to said subject an effective amount of a compound of Formula I, or Formula II, combinations thereof, as defined herein, or a pharmaceutically acceptable salt or solvate thereof.
- the protein kinase is selected from the following group: Tec, Src, Abl, Jak, Csk, Fak, Syk, Fer, Ack kinases, or receptor protein kinases.
- the protein kinases are from Tec or Src kinase family.
- the protein kinase is Bruton's tyrosine kinase (Btk).
- the compounds of the present invention are suitable for the treatment of diseases or conditions, in which one or more of the protein kinase targets are implicated.
- the compounds are suitable for inhibition of a proliferative disorder mediated by one of the aforementioned protein kinase targets.
- the compounds are suitable for inhibition of a proliferative disorder mediated by Tec kinase targets.
- the compounds are suitable for inhibition of a proliferative disorder mediated by Src kinase targets.
- proliferative disorder is used herein in a broad sense to include any disorder that requires control of deleterious cell proliferation, for example cancers and other disorders associated with uncontrolled cellular proliferation, such as dermatological disorders such as psoriasis, certain viral disorders, certain cardiovascular diseases such as restenosis, or cardiomyopathy, certain CNS disorders, auto-immune disorders such as glomerulonephritis or rheumatoid arthritis, hormone-related diseases, metabolic disorders, stroke, alopecia, emphysema, inflammatory diseases, or infectious diseases such fungal diseases, or parasitic disorders such as malaria.
- the compounds of the present invention may induce apoptosis, or maintain stasis within the desired cells as required.
- protein kinase mediated disease is used herein associated with abnormal cellular responses triggered by protein kinase-mediated events. Furthermore, aberrant activation or excessive expression of various protein kinases are implicated in the mechanism of multiple diseases and disorders characterized by benign and malignant proliferation. These diseases include, but are not limited to allergies and asthma, Alzheimer's disease, autoimmune diseases, bone diseases, cancer, cardiovascular diseases, inflammatory diseases, hormone-related diseases, metabolic diseases, neurological and neurodegenerative diseases.
- inhibitors of kinase families are expected to be suitable in the treatment of cancer, vascular disease, autoimmune diseases, and inflammatory conditions including, but not limited to: solid tumors, hematological malignancies, thrombus, arthritis, graft versus host disease, lupus erythematosus, psoriasis, colitis, illeitis, multiple sclerosis, uveitis, coronary artery vasculopathy, systemic sclerosis, atherosclerosis, asthma, transplant rejection, allergy and dermatomyositis.
- the compound of Formula I, Formula II, combinations thereof, or pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes, or biologically active metabolites thereof is acting by inhibiting one or more of the host cell kinases involved in cell proliferation, cell survival, viral replication, cardiovascular disorders, neurodegeneration, autoimmunity, a metabolic disorder, stroke, alopecia, an inflammatory disease, or an infectious disease.
- the proliferative disorder is cancer.
- the cancer may be selected from the group consisting of chronic lymphocytic leukaemia (CLL), lymphoma, leukaemia, breast cancer, lung cancer, prostate cancer, colon cancer, melanoma, pancreatic cancer, ovarian cancer, squamous carcinoma, carcinoma of head or neck, endometrial cancer, or oesophageal carcinoma.
- CLL chronic lymphocytic leukaemia
- the infectious disease includes diseases that are caused by protozoal infestations in humans or animals.
- Such veterinary and human pathogenic protozoas are preferably intracellular active parasites of the phylum Apicomplexa, or Sarcomastigophora, especially Trypanosoma, Plasmodia, Leishmania, Babesia, or Theileria, Cryptosporidia, Sacrocystida, Amoebia, Coccidia, or Trichomonadia.
- the compounds of the present invention are particularly suitable for the treatment of Malaria tropica caused by Plasmodium falciparum, Malaria tertiana caused by Plasmodium vivax, or Plasmodium ovale, or for the treatment of Malaria quartana caused by Plasmodium malariae.
- Toxoplasmosis caused by Toxoplasma gondii Coccidiosis caused for instance by Isospora belli, intestinal Sarcosporidiosis caused by Sarcocystis suihominis, dysentery caused by Entamoeba histolytica, Cryptosporidiosis caused by Cryptosporidium parvum, Chagas disease caused by Trypanosoma cruzi, sleeping sickness caused by Trypanosoma brucei, rhodesiense or gambiense, the cutaneous or visceral, as well as other forms of Leishmaniosis.
- the present invention is also suitable for the treatment of animals infected by veterinary pathogenic Protozoa, like Theileria parva, the pathogen causing bovine East coast fever, Trypanosoma congolense or Trypanosoma vivax, Trypanosoma brucei, pathogens causing Nagana cattle disease in Africa, Trypanosoma brucei evansi causing Surra, Babesia bigemina, the pathogen causing Texas fever in cattle and buffalos, Babesia bovis, the pathogen causing European bovine Babesiosis, as well as Babesiosis in dogs, cats or sheep, Sarcocystis ovicanis or Sarcocystis ovifelis pathogens causing Sarcocystiosis in sheep, cattle or pigs, Cryptosporidia, pathogens causing Cryptosporidioses in cattle and birds, Eimeria or Isospora species, pathogens causing Coccidiosis in rabbits,
- the compounds of the present invention is particularly preferred for use in the treatment of Coccidiosis or Malaria infections, or for the preparation of a drug, or feed stuff for the trea t ment of these diseases. These treatments can be prophylactic or curative.
- the protein kinase inhibitor as defined above may be combined with other anti-malaria agents.
- the present compound described may further be used for viral infections, or other infections caused by Pneumocystis carinii. These compounds may be used alone, or in combination with one, or more agents for the efficient therapy.
- Tec kinases is a family of non-receptor tyrosine kinases predominantly, but not exclusively, expressed in cells of hematopoietic origin.
- the Tec family comprises: Tec, Bruton's tyrosine kinase (Btk), inducible T-cell kinase (Itk), resting lymphocyte kinase (Rlk/Txk), or bone marrow-expressed kinase (Bmx/Etk).
- Btk Bruton's tyrosine kinase
- Itk inducible T-cell kinase
- Rlk/Txk resting lymphocyte kinase
- Bmx/Etk bone marrow-expressed kinase
- Btk is activated by Src-family kinases and phosphorylates PLC gamma leading to effects on B-cell function and survival. Additionally, Btk is important in signal transduction in response to immune complex recognition by macrophage, mast cells and neutrophils. Btk inhibition is also important in survival of lymphoma cells (Herman SEM. Blood, 2011 , 117:6287-6289) suggesting that inhibition of Btk may be useful in the treatment of lymphomas. Bmx, another Tec family member are expected to be suitable in the treatment of various diseases including cancer, cardiovascular disease and inflammation. These compounds may be used alone, or in combination with one or more agents for the therapy.
- the compound of Formula I, Formula II, combinations thereof, or pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes, or biologically active metabolites thereof is acting as inhibitor of cell kinases, as anti-inflammatory, anticancer, or as antithrombotic agents.
- These compounds may be used alone, or in combination with one or more agents, for the treatment of cancer, inflammatory or infectious diseases, or thrombi.
- the compounds of the present invention can be used in combination with at least one chemotherapeutic agent for use particularly in treatment of cancer, neoplasms, or other proliferative diseases or disorder.
- the compounds of Formula I, Formula II, combinations thereof, or pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, can be used in combination with, but not limiting to:
- Anti-proliferative agents selected from the group of: adriamycin, dexamethasone, vincristine, cyclophosphamide, fluorouracil, topotecan, taxol, interferons, platinum derivatives; anti-inflammatory agents comprising corticosteroids, TNF blockers, IL-1 RA, azathioprine, cyclophosphamide, or sulfasalazine;
- Angiogensis inhibitors comprising: sorafenib, sunitinib, pazopanib, or everolimus;
- Immunomodulatory or immunosuppressive agents selected from the group comprising: cyclosporin, tacrolimus, rapamycin, mycophenolate mofetil, interferons, corticosteroids, cyclophophamide, azathioprine, or sulfasalazine; PPAR- ⁇ agonists such as thiazolidinediones;
- Agents for the treatment of anemia comprising erythropoiesis, stimulating agents, vitamins, or iron supplements;
- Anti-emetic agents including: 5-HT3 receptor antagonists, dopamine antagonists, NK1 receptor antagonists, H1 histamine receptor antagonists, cannabinoids, benzodiazepines, anticholinergic agents, or steroids;
- Modulators of the immune system including: interferon-alpha, Bacillus Calmette- Guerin (BCG), or ionizing radition (UVB) that can induce the release of cytokines, such as the interleukins, TNF, or induce release of death receptor ligands such as TRAIL;
- BCG Bacillus Calmette- Guerin
- UVB ionizing radition
- Modulators of death receptors TRAIL or TRAIL- agonists including humanized antibodies HGS-ETR1 , or HGS-ETR in combination, or sequentially with radiation therapy;
- Neurotrophic factors comprising: acetylcholinesterase inhibitors, MAO inhibitors, interferons, anti-convulsants, ion channel blockers, or riluzole; 19.
- Anti-Parkinsonian agents comprising: anticholinergic agents, dopaminergic agents, including dopaminergic precursors, monoamine oxidase B inhibitors, COMT inhibitors, or dopamine receptor agonists;
- Agents for treating cardiovascular disease comprising: beta-blockers, ACE inhibitors, diuretics, nitrates, calcium channel blockers, or statins;
- Agents for treating liver disease comprising: corticosteroids, cholestyramine, or interferons;
- Anti-viral agents including: nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, protease inhibitors, integrase inhibitors, fusion inhibitors, chemokine receptor antagonists, polymerase inhibitors, viral proteins synthesis inhibitors, viral protein modification inhibitors, neuraminidase inhibitors, fusion or entry Inhibitors;
- Agents for treating blood disorders including: corticosteroids, anti-leukemic agents, or growth factors;
- Agents for treating immunodeficiency disorders comprising: gamma globulin, adalimumab, etarnecept, or infliximab; or
- HMG-CoA reductase inhibitors comprising: torvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, simvastatin, or pravastatin.
- an effect against a proliferative disorder mediated by a kinase within the scope of the present invention may be demonstrated by the ability to inhibit a purified kinase in vitro or to inhibit cell proliferation or survival in an in vitro cell assay, for example in Btk Kinase Inhibition Assay and Splenic Cell Proliferation Assay.
- the present invention includes the transdermal, rectal, parenteral, or oral administration of compounds of Formula I, or Formula II (or combinations thereof) to a human or animal subject.
- the dosage unit may contain any suitable amount a compound of Formula I, Formula II, combinations thereof (or a pharmaceutical acceptable salt or solvate thereof, or combinations thereof), for example from about 10 mg to about 5000 mg.
- the dosage unit for oral administration may contain from 50mg to 500mg, per human individual condition. 2014/000848
- the compound of the present invention may be administered 1 to 4 times a day.
- a dosage may be between 0.01-100 mg/kg body weight/day of the compound of the present invention may be administered to a patient receiving these compositions.
- the dose can vary within wide limits and is to be suited to the individual conditions in each individual case. For the above uses the appropriate dosage will vary depending on the mode of administration, the particular condition to be treated and the effect desired. Preferably a dose of 1 to 50 mg/kg body weight/day may be used.
- suitable dosage rates for larger mammals are of the order of from about 10 mg to 3 g/day, administered orally once, or divided doses, such as 2 to 4 times a day, or in sustained release form.
- suitable dosage rates for topical delivery depending on the permeability of the skin, the type and the severity of the disease and dependent on the type of formulation and frequency of application, different concentrations of active compounds within the medicament can be sufficient to elicit a therapeutic effect by topical application.
- the concentration of an active compound pharmaceutically acceptable salts, solvates, solvates of salts, stereoisomers, tautomers, isotopes, prodrugs, complexes or biologically active metabolites thereof, within a medicament according to the present invention is in the range of between 1 mol/L and 100 mmol/L.
- Fluorescence polarization-based kinase assays were performed in 384 well-plate format using histidine tagged recombinant human full-length Bruton Agammaglobulinemia Tyrosine Kinase (Btk) and a modified protocol of the KinEASETM FP Fluorescein Green Assay supplied from Millipore®. Kinase reaction were performed at room temperature for 60 minutes in presence of 250 ⁇ substrate, 10 ⁇ ATP and variable test article concentrations. The reaction was stopped with EDTA/kinease detection reagents. Phosphorylation of the substrate; peptide was detected by fluorescence polarization measured with a Tecan 500 instrument.
- Btk histidine tagged recombinant human full-length Bruton Agammaglobulinemia Tyrosine Kinase
- Kinase reaction were performed at room temperature for 60 minutes in presence of 250 ⁇ substrate, 10 ⁇ ATP and variable test article concentrations. The reaction was stopped with EDTA/kin
- the IC 50 was calculated using Graph Pad Prisms® using a non linear fit curve.
- the Km for ATP on each enzyme was experimentally determined and the Ki values calculated using the Cheng-Prusoff equation (see: Cheng Y, Prusoff W.H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (l 50 ) of an enzymatic reaction”. Biochem Pharmacol 22 (23): 3099-108).
- hBTK kinase is diluted in buffer and all compounds were prepared to 50x final assay concentration in 100% DMSO. Tiis working stock of the compound was added to the assay well as the first component in the reaction, followed by the remaining components as detailed in the assay protocol listed above. The reaction was initiated by the addition of the MgATP mix. The kinase reaction was performed at room temperature for 40 P T/CA2014/000848 minutes in presence of 250 ⁇ substrate, 10 mM MgAcetate, [ ⁇ -33 ⁇ - ⁇ ] (specific activity approx. 500 cpm/pmol, concentration as required) and variable test article concentrations. The ATP concentrations in the assays were with 5 ⁇ of the apparent. The reaction was stopped by the addition of 3% phosphoric acid solution.
- a - EC 50 ⁇ 100 nM; b - 100 nM ⁇ EC 50 ⁇ 1000 nM, c - EC 50 >1000 nM.
- Splenocytes were obtained from (5 week old male CD1 mice (Charles River Laboratories Inc.). Mouse spleens were manually disrupted in PBS and filtered using a 70um cell strainer followed by ammonium chloride red blood cell lysis. Cells were washed, resuspended in Splenocyte Medium (HyClone RPMI supplemented with 10% heat- inactivated FBS, 0.5X non-essential amino acids, 10 mM HEPES, 50 uM beta mercaptoethanol) and incubated at 37°C, 5% C0 2 for 2h to remove adherent cells.
- Splenocyte Medium HyClone RPMI supplemented with 10% heat- inactivated FBS, 0.5X non-essential amino acids, 10 mM HEPES, 50 uM beta mercaptoethanol
- Suspension cells were seeded in 96 well plates at 50,000 cells per well and incubated at 37°C, 5% C0 2 for 1h.
- Splenocytes were pre-treated in triplicate with 10,000 nM curves of Formula I compounds for 1h, followed by stimulation of cell proliferation with 2.5ug/ml anti-IgM F(ab') 2 (Jackson ImmuncResearch) for 72h.
- Cell proliferation was measured by Cell Titer-Glo Luminescent Assay (Promega).
- EC 50 values (50% proliferation in the presence of compound as compared to vehicle treated controls) were calculated from dose response compound curves using GraphPad Prism Software.
- a - EC 50 ⁇ 100 nM; b - 100 nM ⁇ EC 50 ⁇ 1000 nM, c - EC 50 >1000 nM,
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- General Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Pathology (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Radiology & Medical Imaging (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Optics & Photonics (AREA)
- Transplantation (AREA)
- Vascular Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
Abstract
Description






























































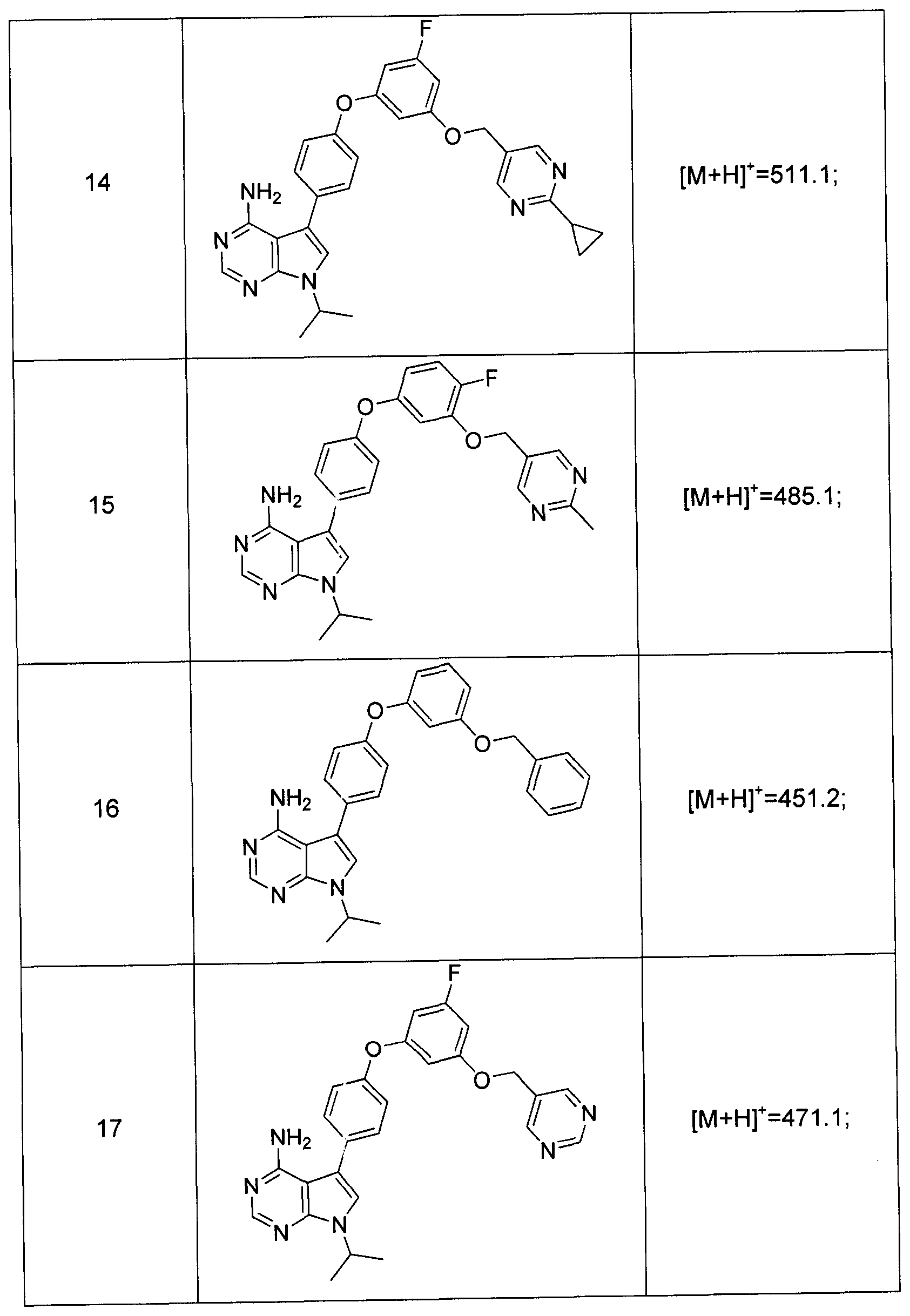






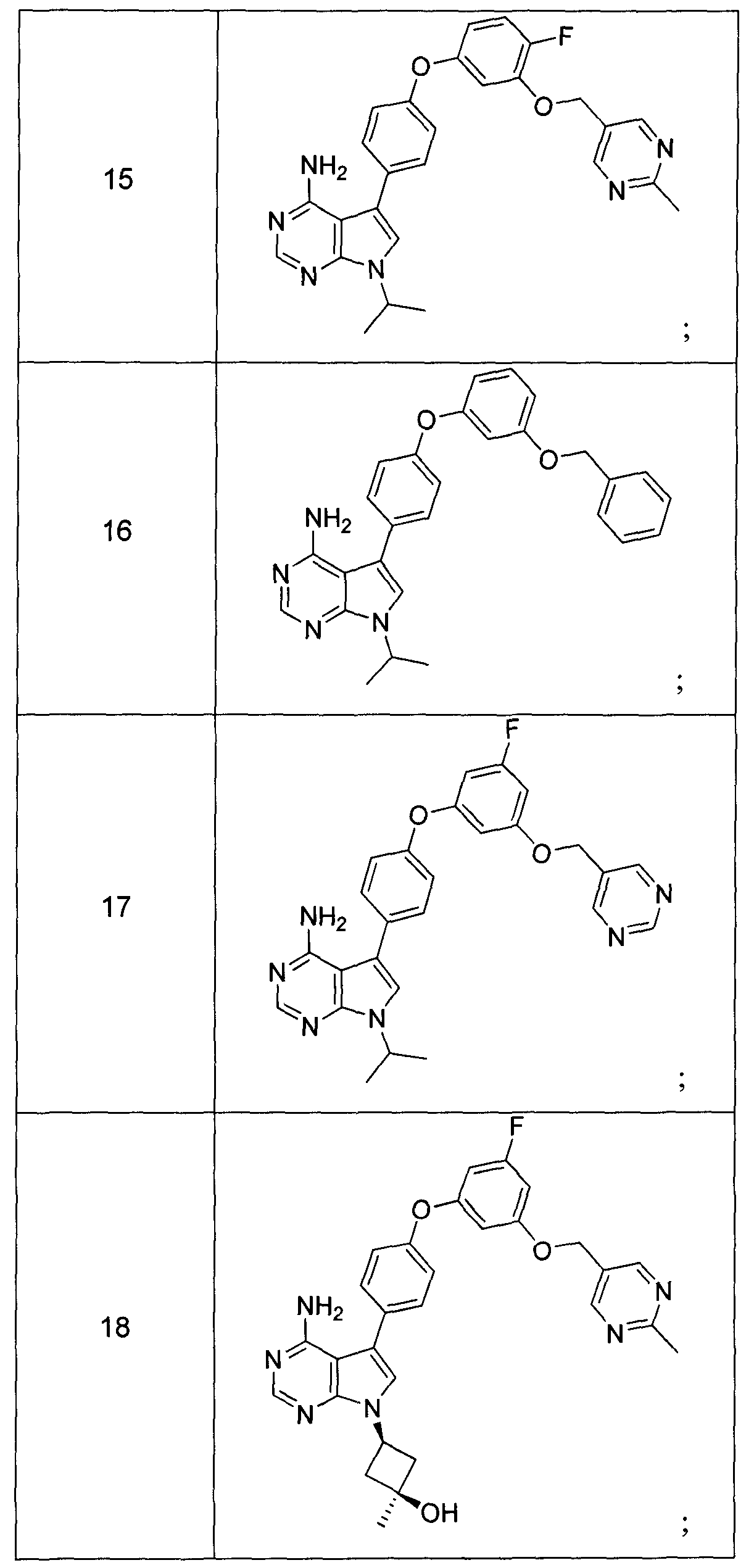
Claims



















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/039,127 US20170158697A1 (en) | 2013-11-26 | 2014-11-26 | Protein Kinase Inhibitors |
| CA2929889A CA2929889A1 (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitors |
| JP2016554766A JP2017503006A (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitor |
| CN201480064253.3A CN105764906A (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitors |
| KR1020167014057A KR20160089378A (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitors |
| RU2016124366A RU2016124366A (en) | 2013-11-26 | 2014-11-26 | PROTEINKINASE INHIBITORS |
| EP14865858.6A EP3074401A4 (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2834528A CA2834528A1 (en) | 2013-11-26 | 2013-11-26 | Protein kinase inhibitors |
| CA2,834,528 | 2013-11-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015077866A1 true WO2015077866A1 (en) | 2015-06-04 |
Family
ID=53198134
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2014/000848 Ceased WO2015077866A1 (en) | 2013-11-26 | 2014-11-26 | Protein kinase inhibitors |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20170158697A1 (en) |
| EP (1) | EP3074401A4 (en) |
| JP (1) | JP2017503006A (en) |
| KR (1) | KR20160089378A (en) |
| CN (1) | CN105764906A (en) |
| CA (2) | CA2834528A1 (en) |
| RU (1) | RU2016124366A (en) |
| TW (1) | TW201602112A (en) |
| WO (1) | WO2015077866A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3025813A1 (en) * | 2015-05-27 | 2016-12-01 | Gb005, Inc. | Inhibitors of the tec kinase enzyme family |
| CN108794483B (en) * | 2018-04-27 | 2021-04-23 | 四川大学华西医院 | A 7-deazapurine derivative and its six-membered ring supramolecular structure |
| KR20230064355A (en) | 2021-11-03 | 2023-05-10 | 고려대학교 산학협력단 | Pharmaceutical composition for preventing or treating pain comprising N-Acetyl-L-tryptophan 3,5-bis(trifluoromethyl)benzyl ester compound as an active ingredient |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000017203A1 (en) * | 1998-09-18 | 2000-03-30 | Basf Aktiengesellschaft | Pyrrolopyrimidines as protein kinase inhibitors |
| WO2001072751A1 (en) * | 2000-03-29 | 2001-10-04 | Knoll Gesellschaft Mit Beschraenkter Haftung | Pyrrolopyrimidines as tyrosine kinase inhibitors |
| US20030153752A1 (en) * | 1998-09-18 | 2003-08-14 | Hirst Gavin C. | Pyrrolopyrimidines as therapeutic agents |
| US20030187001A1 (en) * | 1997-03-19 | 2003-10-02 | David Calderwood | 4-aminopyrrolopyrimidines as kinase inhibitors |
| CA2779184A1 (en) * | 2012-05-31 | 2013-11-30 | Pharmascience Inc. | Protein kinase inhibitors |
| CA2782774A1 (en) * | 2012-07-06 | 2014-01-06 | Pharmascience Inc. | Protein kinase inhibitors |
| CA2813299A1 (en) * | 2013-04-17 | 2014-10-17 | Pharmascience Inc. | Protein kinase inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2253930T3 (en) * | 1998-09-18 | 2006-06-01 | ABBOTT GMBH & CO. KG | 4-AMINOPIRROLOPIRIMIDINAS AS QUINASA INHIBITORS. |
| WO2013177668A1 (en) * | 2012-05-31 | 2013-12-05 | Pharmascience, Inc. | Protein kinase inhibitors |
-
2013
- 2013-11-26 CA CA2834528A patent/CA2834528A1/en not_active Abandoned
-
2014
- 2014-11-26 KR KR1020167014057A patent/KR20160089378A/en not_active Withdrawn
- 2014-11-26 EP EP14865858.6A patent/EP3074401A4/en not_active Withdrawn
- 2014-11-26 WO PCT/CA2014/000848 patent/WO2015077866A1/en not_active Ceased
- 2014-11-26 CA CA2929889A patent/CA2929889A1/en not_active Abandoned
- 2014-11-26 RU RU2016124366A patent/RU2016124366A/en unknown
- 2014-11-26 TW TW103141063A patent/TW201602112A/en unknown
- 2014-11-26 CN CN201480064253.3A patent/CN105764906A/en active Pending
- 2014-11-26 US US15/039,127 patent/US20170158697A1/en not_active Abandoned
- 2014-11-26 JP JP2016554766A patent/JP2017503006A/en not_active Withdrawn
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030187001A1 (en) * | 1997-03-19 | 2003-10-02 | David Calderwood | 4-aminopyrrolopyrimidines as kinase inhibitors |
| WO2000017203A1 (en) * | 1998-09-18 | 2000-03-30 | Basf Aktiengesellschaft | Pyrrolopyrimidines as protein kinase inhibitors |
| US20030153752A1 (en) * | 1998-09-18 | 2003-08-14 | Hirst Gavin C. | Pyrrolopyrimidines as therapeutic agents |
| WO2001072751A1 (en) * | 2000-03-29 | 2001-10-04 | Knoll Gesellschaft Mit Beschraenkter Haftung | Pyrrolopyrimidines as tyrosine kinase inhibitors |
| CA2779184A1 (en) * | 2012-05-31 | 2013-11-30 | Pharmascience Inc. | Protein kinase inhibitors |
| CA2782774A1 (en) * | 2012-07-06 | 2014-01-06 | Pharmascience Inc. | Protein kinase inhibitors |
| CA2813299A1 (en) * | 2013-04-17 | 2014-10-17 | Pharmascience Inc. | Protein kinase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3074401A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105764906A (en) | 2016-07-13 |
| US20170158697A1 (en) | 2017-06-08 |
| CA2834528A1 (en) | 2015-05-26 |
| EP3074401A4 (en) | 2017-05-10 |
| RU2016124366A (en) | 2018-01-09 |
| EP3074401A1 (en) | 2016-10-05 |
| JP2017503006A (en) | 2017-01-26 |
| TW201602112A (en) | 2016-01-16 |
| CA2929889A1 (en) | 2015-06-04 |
| KR20160089378A (en) | 2016-07-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2487875C2 (en) | IMIDAZO[1,2-b]PYRIDAZINE AND PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AND USE THEREOF AS PROTEIN KINASE INHIBITOR | |
| US8026246B2 (en) | Aurora kinase inhibitors for inhibiting mitotic progression | |
| US12551563B2 (en) | ERK5 degraders as therapeutics in cancer and inflammatory diseases | |
| US20180179210A1 (en) | Inhibitors of the TEC Kinase Enzyme Family | |
| US20080207632A1 (en) | Protein kinase inhibitors | |
| US12440494B2 (en) | Compounds useful for inhibiting RAF dimers | |
| US11897883B1 (en) | Pyrrolo[3,2-c][1,6]naphthyridine-2-carboxylic acid compounds as CK2 inhibitors | |
| CN103172641B (en) | Heterocyclic amino and alkoxy-replaced quinazoline derivative and application thereof | |
| EP3071572A1 (en) | Protein kinase inhibitors | |
| EP3074401A1 (en) | Protein kinase inhibitors | |
| US9822120B2 (en) | Protein kinase inhibitors | |
| US11987581B1 (en) | Imidazo[1,2-c]pyrido[3,4-e]pyrimidine-2-carboxylic acids as CK2 inhibitors | |
| US11891394B1 (en) | Pyrrolo[2,3-c][1,6]naphthyridine-8-carboxylic acid compounds as CK2 inhibitors | |
| CN119343346A (en) | Benzenesulfonamide derivative, preparation method thereof and pharmaceutical composition containing the same as active ingredient for preventing or treating cancer | |
| US11945823B1 (en) | Substituted pyrrolo[2,3-d]pyrimidines as CK2 inhibitors | |
| US12286436B2 (en) | Substituted pyrimido[4,5-b]indoles as CK2 inhibitors | |
| US12173001B1 (en) | Pyrido[3,4-e][1,2,4]triazolo[4,3-c]pyrimidines as CK2 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14865858 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2929889 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016554766 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15039127 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20167014057 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014865858 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014865858 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016011865 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2016124366 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016011865 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160524 |