WO2015094970A1 - Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof - Google Patents
Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof Download PDFInfo
- Publication number
- WO2015094970A1 WO2015094970A1 PCT/US2014/070021 US2014070021W WO2015094970A1 WO 2015094970 A1 WO2015094970 A1 WO 2015094970A1 US 2014070021 W US2014070021 W US 2014070021W WO 2015094970 A1 WO2015094970 A1 WO 2015094970A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- tetrahydrofuran
- fdm
- process according
- mono
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C(CC1C2C(*[C@](C3)C[C@]4*3C(C3)C3CC4)CC1C2)C1*C1 Chemical compound C(CC1C2C(*[C@](C3)C[C@]4*3C(C3)C3CC4)CC1C2)C1*C1 0.000 description 6
- YAIFIJXVZLUCFH-UHFFFAOYSA-N CCCCCCCCCCCCCCCCCCCC1CC(CC(C)(CC2)C3(CC(C)CC3)C2(C2)C2C2(C3)C(CC4)C4C3C(C)C2)CC1 Chemical compound CCCCCCCCCCCCCCCCCCCC1CC(CC(C)(CC2)C3(CC(C)CC3)C2(C2)C2C2(C3)C(CC4)C4C3C(C)C2)CC1 YAIFIJXVZLUCFH-UHFFFAOYSA-N 0.000 description 1
- IHMXVSZXHFTOFN-ZCFIWIBFSA-N CC[C@H]1OCCC1 Chemical compound CC[C@H]1OCCC1 IHMXVSZXHFTOFN-ZCFIWIBFSA-N 0.000 description 1
- DSLRVRBSNLHVBH-UHFFFAOYSA-N OCc1ccc(CO)[o]1 Chemical compound OCc1ccc(CO)[o]1 DSLRVRBSNLHVBH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/14—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/40—Radicals substituted by oxygen atoms
- C07D307/42—Singly bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/52—Radicals substituted by nitrogen atoms not forming part of a nitro radical
Definitions
- the present disclosure relates to certain cyclic bi-functional materials that are useful as monomers in polymer synthesis, as well as intermediate chemical compounds.
- the present invention pertains to ethers of furan-2,5-dimethanol (FDM) and/or (tetrahydrofuran-2,5- diyl)dimethanol (bHMTHF), methods for their preparation, and derivative chemical compounds thereof.
- HMF 5- (hydroxymethyl)furfural
- Figure 1 a salient dehydration product of the abundant, inexpensive monosaccharide, fructose.
- HMF is a versatile chemical antecedent to various furanic ring-based derivatives that are known intermediates for a multitude of chemical syntheses, and as plausible surrogates for aromatic hydrocarbons that derive from petroleum resources. Due to HMF's diverse functionalities, some have proposed that HMF be used to produce a wide range of commodities such as polymers, solvents, surfactants, pharmaceuticals, and plant protection agents. As alternates, derivatives of HMF are comparable to benzene -based aromatic compounds or to other compounds containing a furan or tetrahydrofuran (THF). HMF and 2,5-disubstituted furans and THF analogs, therefore, have great potential in the field of intermediate chemicals from renewable agricultural resources.
- THF tetrahydrofuran
- HMF itself, however, is rather unsuitable as a chemical intermediate substrate, given its propensity to decompose under thermo-oxidative conditions.
- FDM furan-2,5-dimethanol
- Scheme 1 is produced from partial hydrogenation (aldehyde reduction) of HMF.
- bHMTHF 2,5-bis(hydroxymethyl)tetrahydrofuran
- B 2,5-bis(hydroxymethyl)tetrahydrofuran
- C diastereometic ratio when both the ring and aldehyde moieties of HMF are reduce completely
- These materials can be of value as a molecular antecedent, for example, to polyesters, polyurethane foams, FDCA, plasticizers, additives, lubricants, and amphiphiles.
- the present disclosure describes, in part, linear mono- and di-alkyl ethers of furan-2,5- dimethanol (FDM) and/or 2,5-bis(hydroxymethyl)tetrahydrofuran (bHMTHF), and a process for synthesis.
- FDM furan-2,5- dimethanol
- bHMTHF 2,5-bis(hydroxymethyl)tetrahydrofuran
- the process includes contacting either FDM or bHMTHF in a polar aprotic organic solvent having a permittivity ( ⁇ ) >8, at a temperature ranging from about -25°C to about 100°C, with either a) an unhindered Bronsted base having a difference in pKa (ApKa) >15 relative to the pKa of a hydroxyl group of either FDM or bHMTHF or b) a hindered Bronsted base and a nucleophile.
- a polar aprotic organic solvent having a permittivity ( ⁇ ) >8, at a temperature ranging from about -25°C to about 100°C with either a) an unhindered Bronsted base having a difference in pKa (ApKa) >15 relative to the pKa of a hydroxyl group of either FDM or bHMTHF or b) a hindered Bronsted base and a nucleophile.
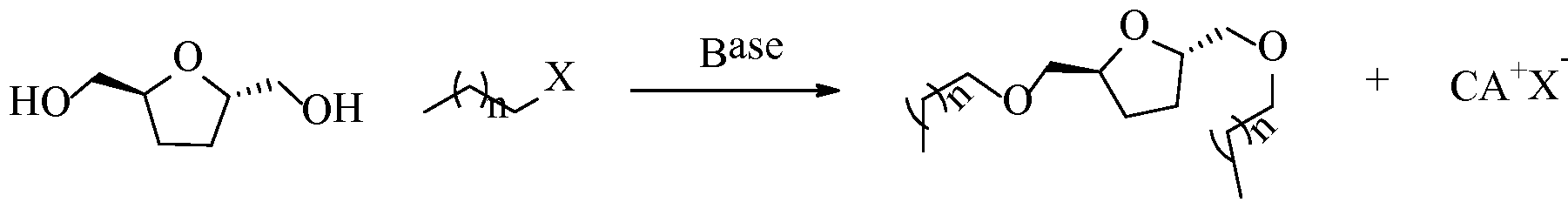
- the present disclosure provides a method of preparing a mono- ether involving: contacting FDM with a Bronsted base and one or less molar equivalents of an alkyl-X species according to the following: wherein: "X” is the leaving group (nucleofuge), "n” is an integer from 5 to 25, and "CA” is a conjugate acid.
- the resultant mono-ether of FDM can be, for example, at least one of the following compounds:
- the method involves: contacting FDM with a Bronsted base and a minimum of 2 molar equivalents of an alkyl-X species according to the foll
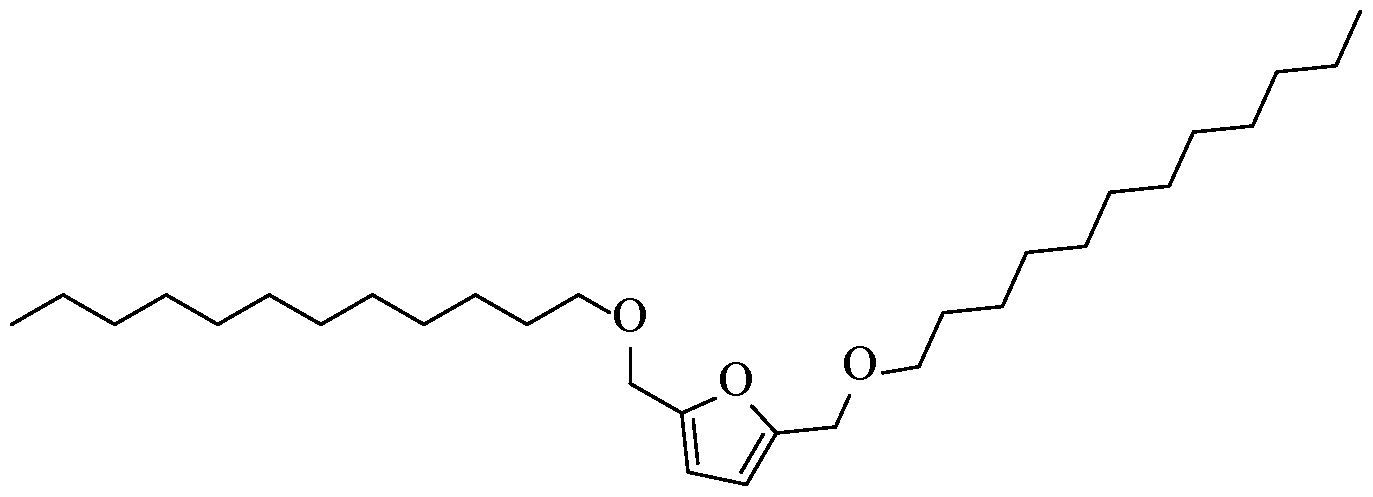
- the resultant di-ether of FDM can be, for instance, at least one of the following compounds: a. -bis((hexyloxy)methyl)furan
- the present disclosure provides a method of preparing a mono- ether involving: contacting bHMTHFs with a Bronsted base and 1 or less molar equivalents of an alkyl-X species according to the following:
- the resultant mono-ether of bHMTHF can be, for example, at least one of the following compounds:
- the method involves: contacting bHMTHFs with a Bronsted base and a minimum of two molar equivalents of an alkyl-X species according to the following:
- X is the nucleofuge
- n is an integer from 5 to 25
- CA is a conjugate acid.
- the resultant di-ethers of bHMTHF can be, for instance, at least one of the following compounds:
- the present disclosure pertains to derivative compounds from the linear mono-ethers of FDM and bHMTHF described above and methods for making the derivatives.
- These derivative compounds are amphiphilic variants of the mono-ethers and are valued as precursors or plausible bio-based surfactants, dispersants, and/or hydrophiles.
- the present synthetic processes opens a pathway for direct preparation of linear alkyl ethers from the glycols FDM and/or bHMTHF, molecules that arise from the reduction of fructose derived 5 -(hydroxymethyl) furfural (HMF) under mild conditions, and their derivative chemical compounds.
- the process may also include either first partially reducing HMF to FDM or fully reducing HMF to bHMTHFs in hydrogenation steps prior to selective etherification according to the present reaction process described herein.
- the alkyl ethers are valuable precursors with bio-based amphiphilic properties that can be used in surfactants, dispersants, and plasticizers.
- the process for generating alkyl ethers can be implemented in a single reaction step, in which the FDM or bHMTHF glycol is reacted with either one or two equivalents of a halogenated or sulfonated (leaving group) alkane, depending respectively on whether a mono- or di- ether product is desired.
- a hindered Bronsted base with a minimum pKa of about 10, preferably about 16, or an unhindered Bronsted base having a difference in pKa (ApKa) of >15 relative to the pKa of a hydroxyl group of either the FDM or bHMTHF is used to deprotonate the -OH moieties of the glycols, enhancing their nucleophilicities by several orders of magnitude towards nucleofuge displacement.
- the Bronsted base should have a limited propensity to react with an alkyl halide or sulfonate in a nucleophilic substitution and/or elimination.
- a polar aprotic organic solvent with a dielectric constant of >10, preferably > 30, is employed to augment the basicity of the Bronsted base via charge separation capacities.
- the reaction is conducted at a temperature in a range from about -20°C to about 100°C, over a period of about 2 or 3 hours. In some other iterations the time may involve about 4 or 8 hours up to about 12 or 24 hours, as conditions may dictate.
- the Bronsted base in the reaction serves to deprotonate the -OH moieties of the glycols. This helps to enhance the corresponding nucleophilicity of the glycols FDM and bHMTHF by about at least 6 or more orders of magnitude (e.g., 8-10-12) and drives halide/sulfonate displacement on the alkyl reagent.
- the relative strength of a Bronsted base used in the reaction is of essence in furnishing high conversions of the glycols to, in particular, mono-alkyl ethers.
- Bronsted bases that have a pKa of at least 10 to about 15
- the synthesis reaction usually requires the addition of heat to proceed; hence, reaction temperatures of about 45°C-50°C or greater.
- This can increase the risk of generating side -products (e.g., product of Bronsted base-nucleophilic substitution with the alkyl halide/sulfonate and/or alkenes formed from Bronsted base- mediated elimination of the alkyl halide/sulfonate) and reducing the overall yield of the desired synthesis.
- side -products e.g., product of Bronsted base-nucleophilic substitution with the alkyl halide/sulfonate and/or alkenes formed from Bronsted base- mediated elimination of the alkyl halide/sulfonate
- a Bronsted base that has a pKa of at least -16, typically >20 is favored according to certain embodiments of the present process.
- Bronsted bases with a greater pKa more easily reacts with the -OH moieties of the glycols. This is an advantage that helps one operate effectively the reaction at about ambient room temperatures (e.g., ⁇ 18°C -22°C) or lower temperatures.
- Some suitable Bronsted bases may include, for example, hydroxides (e.g., methoxide, ethoxide, i-butoxide, and benzyl oxide).
- Bronsted bases having pKa's > 30 are used, as the equilibrium for deprotonation favors generation of the desired products, such as illustrated in the examples in Scheme 3.
- Certain favored Bronsted bases of this type may include, for example, metallic hydrides (e.g., lithium, potassium, or sodium hydrides); metal amides (e.g., potassium or sodium amides); lithium diisopropylamide (LDA);
- metallic hydrides e.g., lithium, potassium, or sodium hydrides
- metal amides e.g., potassium or sodium amides
- organometalic compounds e.g., alkyl lithium (e.g., methyl-lithium, n-butyl-lithium, or phenyl- lithium), alkyl magnesium, or alkyl cuprate) and Grignard reagents (e.g., ethylmagnesium bromide, phenylmagnesium bromide).
- certain disfavored Bronsted bases may include, for example, nitrogen-centered bases (e.g., tertiary amines, aryl amine), because of low-pKa- favoring reactants and nucleophilic propensities.
- Reaction (a) shows when using a Bronsted base having a pKa -16, the reaction tends to be at equilibrium between product and reactants.
- Reaction (b) when using a Bronsted base having a pKa -20, the reaction tend to favor the product more, whereas in Reaction (c) when using a Bronsted base having pKa >30, the reaction is driven completely towards product formation.
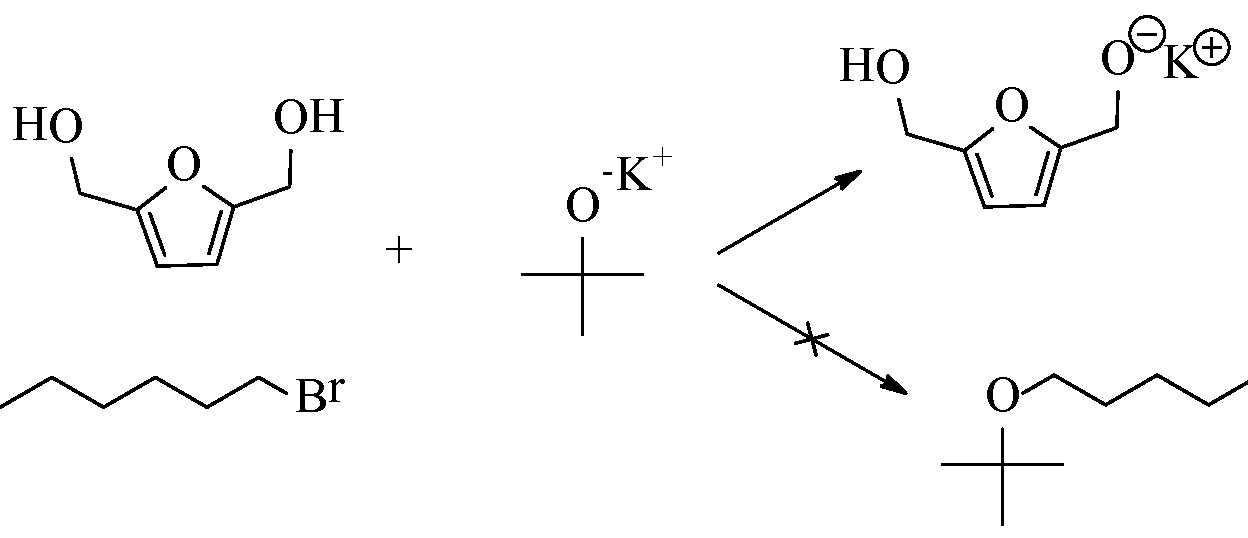
- Another factor according to an embodiment of the present invention is to employ a Bronsted base that has molecular bulk.
- the bulky Bronsted base impedes undesired nucleophilic substitutions of the Bronsted base with the alkyl halide/sulfonate.
- a more sterically hindered Bronsted base enhances more effectively the reaction to produce predominantly the ether product.
- Scheme 4 illustrates this feature.
- reaction (a) using an unhindered Bronsted base tends to make a mixed product of both straight-chain and FDM ethers.
- reaction (b) with a more bulky, hindered Bronsted base generates the FDM ethers alone.
- the etherification reaction of the present description can be characterized as a base-mediated, second order substitution reaction between a glycol and activated alkane.
- the leaving group affixed to the alkane should exhibit favorable nucleofugal properties.
- Some species in this context can be, for example, halides (e.g., CI, Br, I) and sulfonates (e.g., -OTf, -OTs,-OMs).
- halides e.g., CI, Br, I
- sulfonates e.g., -OTf, -OTs,-OMs
- the alkyl chain lengths may range from about 5 or 8 to about 16 or 18 carbons, or about 6 or 10 to about 20 or 22 carbons (e.g., Cs-Cis; C5-C15; C6-C12), or any iteration therein between.
- tosylate p-toluenesulfonate
- CH3C6H4SO2O- [ ⁇ 0 R ] esylate
- ethanesulfonate ethanesulfonate
- halides such as bromides
- alcohols are more economically accessible commercial alkane sources, they may be favored for larger scale, industrial uses according to some embodiments.
- an alkyl halide is unavailable or prohibitively expensive, but the corresponding alcohol available, one may substitute the alcohol for the corresponding sulfonate through a simple sulfonation reaction.
- the sulfonate is preferably a triflate because it is a powerful leaving group.
- This reaction exhibits relatively fast kinetics and generates an activated triflic complex.
- the reaction is usually conducted at a low temperature, less than 0°C (e.g., typically about -10°C or - ⁇ 2°C to about -20°C or -25°C), to control the reaction kinetics more easily.
- This reaction is essentially irreversible, as the liberated triflate is entirely non-nucleophilic.
- the triflic complex then reacts readily with the FDM or bHMTHF, forming respectively a FDM or bHMTHF-triflate with concomitant release and protonation of a nucleophilic base (e.g., pyrimidine, dimethyl-aminopyridine, imidazole, pyrrolidine, and morpho line).
- a nucleophilic base e.g., pyrimidine, dimethyl-aminopyridine, imidazole, pyrrolidine, and morpho line.
- the tosylate, mesylate, brosylate, benzenesulfonate, ethylsulfonate or other sulfonate species can be as effective as triflate in imparting leaving groups, and manifesting overall yields that were commensurate with that achieved with triflate. But, these other sulfonates tend to react more slowly in comparison to the triflate. To compensate for this, operations at higher temperatures are typically needed for better yields when using these other species.
- aprotic solvents are used, as they contain no functionality labile to covalent modifications with the glycol, alkyl halide/sulfonate and Bronsted base of the title reaction, and thus do not interfere with the Sn2-driven process.
- polar aprotic solvents i.e., solvents with a permanent dipole moment but without the ability to act as hydrogen bond donors
- Polar aprotic solvents adequately dissolve the glycols and the alkyl halide/sulfonate, a feature for an efficient reaction to occur.
- polar aprotic solvents tend not to react with the alkyl halide/sulfonate (cf., Scheme 6, ethanol, a polar protic solvent, which can generate undesired side products).
- a greater dielectric constant can help prevent the solvent from reacting with the primary reagents, hence minimizing formation of side -products.
- the reactions of the present synthesis process are conducted in solvents with a relative permittivity > 8 r 25, typically about 30 or 35.
- DMSO and DMF exhibit relatively high dielectric constants (e.g., -30 or 32).
- Other solvents with high boiling points and dielectric constants, such as NMP and DMA, are effective in cyanide for sulfonate displacement reactions.
- the reaction to derivatize FDM or bHMTHF with a sulfonate is performed in a solution of solvent having a boiling point > 110°C.
- dimethylformamide (DMF), dimethylsulfoxide (DMSO), dimethylacetamide (DMA), N- methylpyrrolidone (NMP), hexamethylphosphoramide (HMPA), acetone, acetonitrile (ACN), nitromethane, sulfolane, tetrahydrofuran (THF), 1,4-dioxane, and ethyl acetate.
- a further consideration when using polar aprotic solvent in the etherification process is to amply charge separate the Bronsted base so that the glycol -OH moieties can be deprotonated.
- a reflection of the power to charge separate is the permittivity of dielectric constant, represented by ⁇ (no units), with the larger number signifying a greater capacity to sequester the ions.
- ⁇ dielectric constant
- the preferred ⁇ is >30.
- reaction temperatures can span between about -25°C or -20°C to about 80°C or 100°C.
- reaction temperature is in a range from about -12°C or -7°C to about 65°C or 70°C, more typically from about -10°C or -5°C to about 40°C or 50°C.
- preferred temperatures may range from about -10°C or -8°C to about 25°C or 30°C, or about -3°C or 0°C to about 32°C or 35°C, inclusive.
- the reaction can be performed at or below ambient room temperatures (e.g., ⁇ about 22°C or 25°C). Because of a potential or tendency to generate olefins from base-mediated elimination of an alkyl halide/sulfonate at elevated temperatures, and potential slow reaction kinetics when uses certain Bronsted bases (Scheme 7), temperature control for the present selective etherification is an important factor.
- various amphiphilic compounds can be synthesized from FDM or bHMTHF ethers as a starting or precursor material.
- Such derivative materials can be useful as substitutes for existing compounds or new chemical building blocks in surfactant, dispersant, plasticizer or a component in other applications.
- the derivative amphiphilic compounds can be prepared according to various chemical reactions available for organic synthesis. Preparations of some representative derivative compounds are further described in the accompanying examples below.
- the methods may include: reacting either a mono-ether of bHMTHF or FDM with: a) chlorosulfonic acid to generate a sulfate, or b) trifluoromethanesulfonic anhydride to generate a trifluoromethanesulfonate, respectively, of each glycol species.
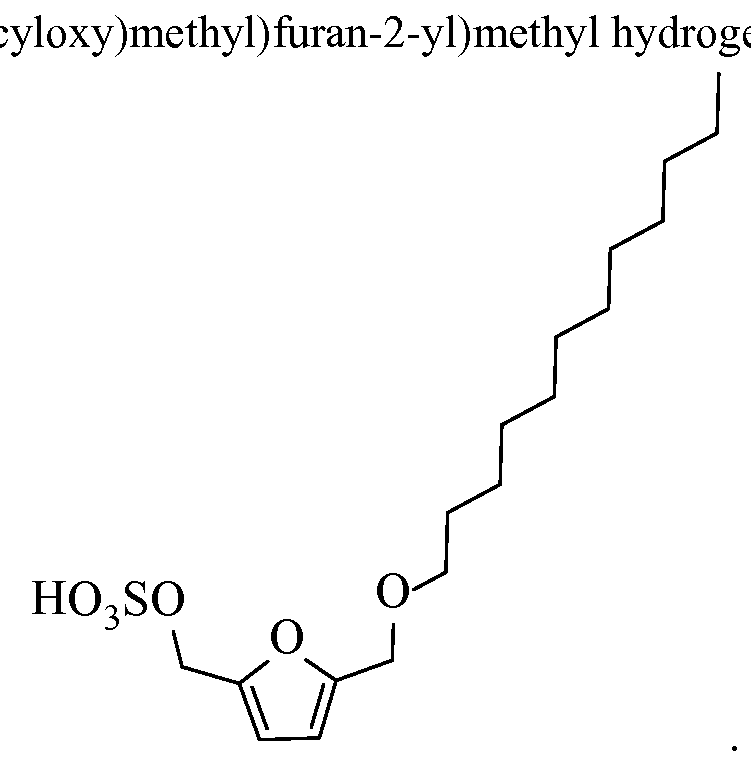
- a sulfate product can be, for example, at least one of the following compounds:
- a trifluoromethanesulfonated mono-ether generated from the bHMTHF mono-ether can be, for example, at least one of the following compounds:
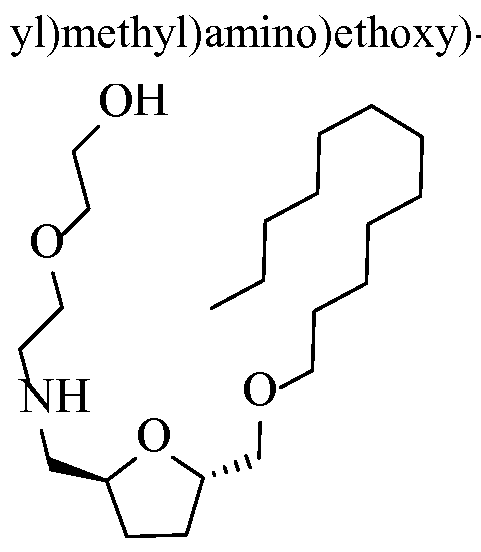
- the process may further involve generating an ethoxyethanolamine derivative of the bHMTHF mono-ether sulfonate compound by substitution of a sulfonate group with an ethanolamine.
- the resultant ethoxyethanolamine prepared can be, for instance, at least one of the following compounds:
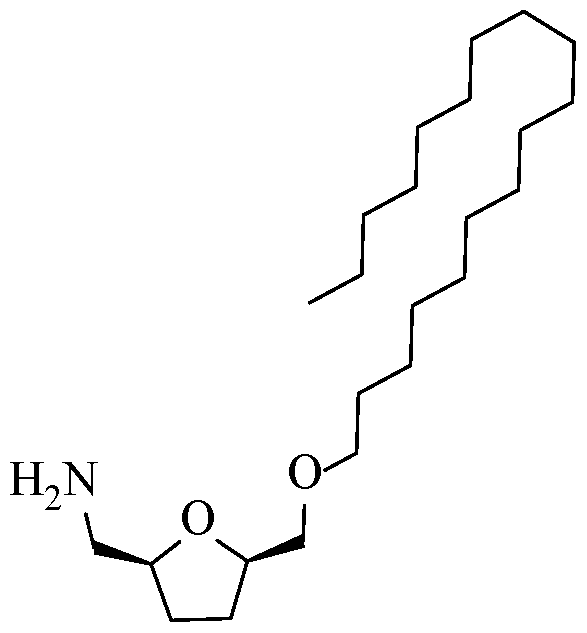
- the process may further include generating a primary amine of a bHMTHF monoether by substitution of a trifluoromethanesulfonate group to form a benzyl-amine, such as one of the following: a) N-benzyl-l-(5-( e
- the resultant primary amine can be, for instance, at least one of the following compounds:
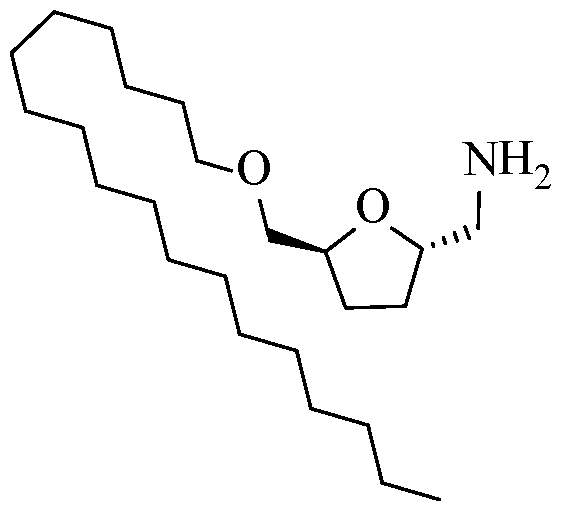
- the process may further include preparing a primary ammonium salt of the bHMTHF monoether by substitution of a trifluoromethanesulfonate group followed by catalytic debenzylation and protonation by a Bronsted acid having a pKa ⁇ 0 (e.g., HC1, HBr, HI).
- the resultant primary ammonium group can be, for example, at least one of the following compounds:
- the salt version of the primary amine renders the molecule more amphiphilic with a polar head for cationic surfactants.
- the resultant sulfate product can be for example:
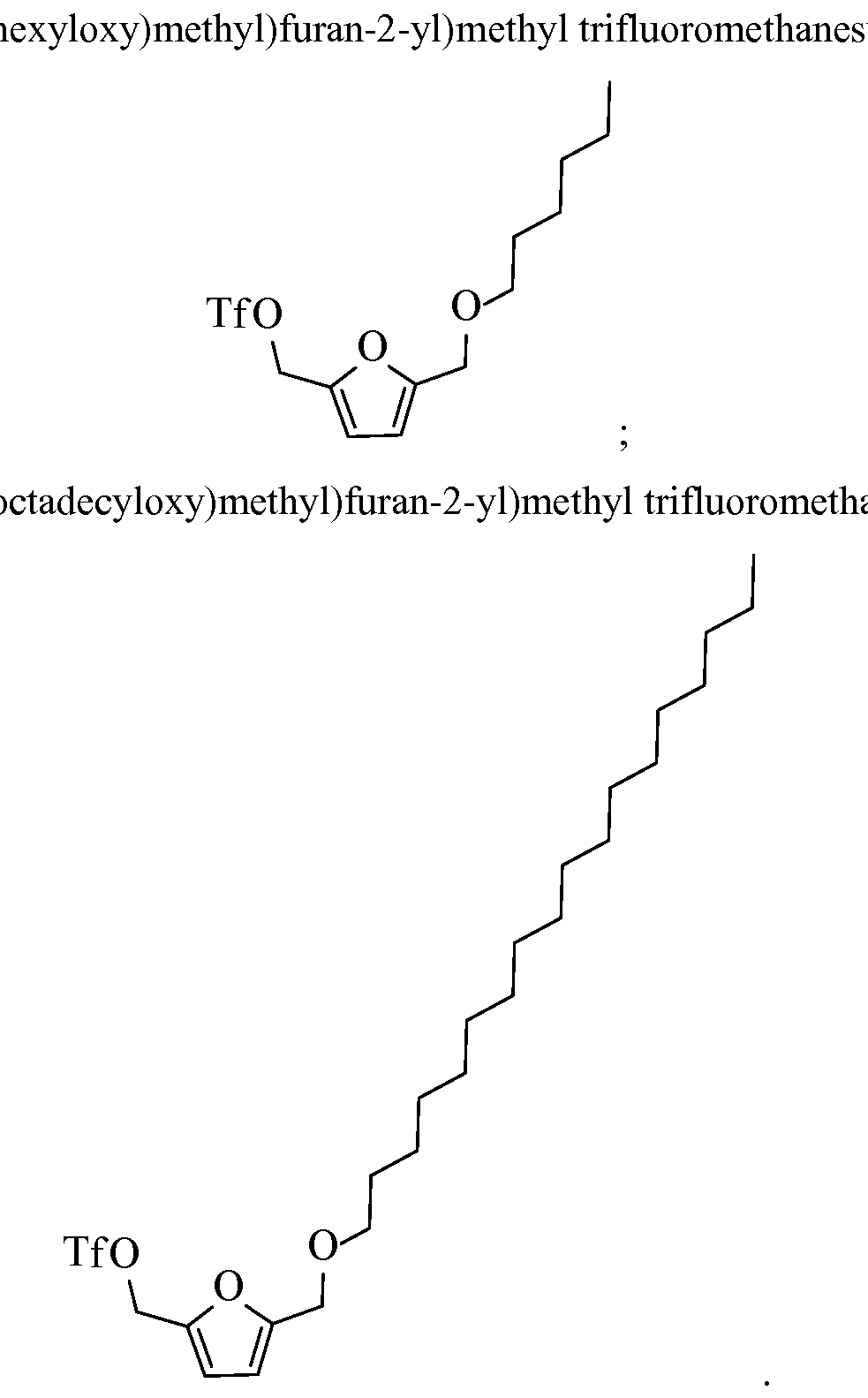
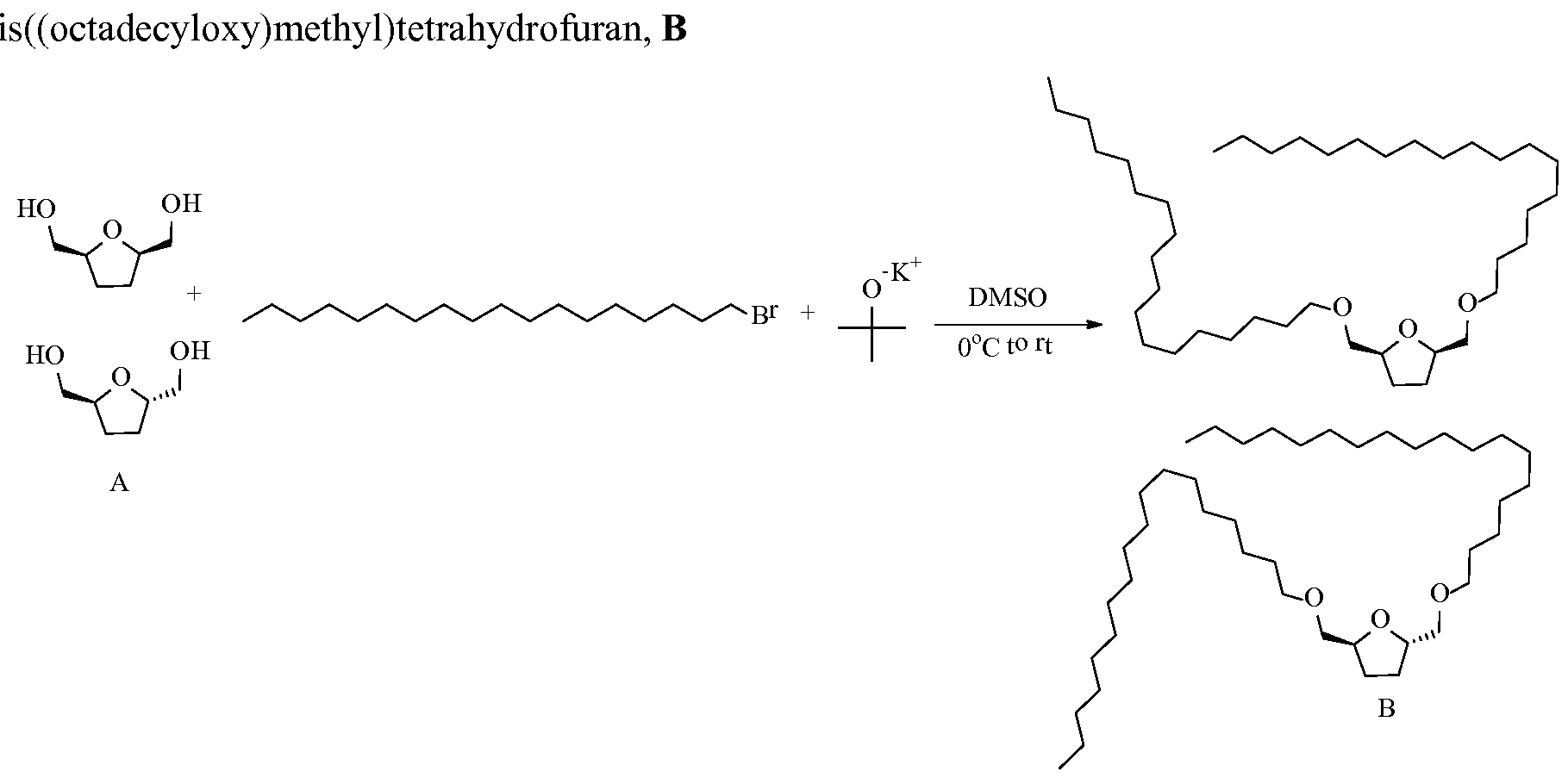
- the resultant trifluoromethanesulfonate from FDM mono-ether can be, for example, at least one of the following structures:
- the process for preparing a primary ammonium group using FDM mono-ethers also involves substitution of a trifluoromethanesulfonate group followed by catalytic debenzylation and protonation by a Bronsted acid having a pKa ⁇ 0.
- the resultant aminoethylethanolamine can be, for example, the following:
- a primary amine derivative that is prepared using FDM mono- ether as the starting material can be, for example, the following: (5-((hexyloxy)methyl)furan-2- yl)methanamine
- quaternary trimethylammonium salt such as: l-(5- ((hexyloxy)methyl)furan-2-yl)-N,N,N de
- the present synthesis system is further illustrated in the following examples for making: A) bHMTHF di-ethers; B) bHMTHF mono-ethers; C) derivatives of bHMTHF mono-ethers; D) FDM di- ethers; E) FDM mono-ethers; and F) amphiphilic derivatives of FDM mono-ethers .
- the mixture was diluted with 5 mL of water and 5 mL of methylene chloride and partitioned and the aqueous layer extracted with 3-5 mL volumes of methylene chloride.
- the organic phases were combined, dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum.
- the oily residue was dissolved in a minimum amount of methylene chloride and added to 20 g of silica gel, which was then dried under vacuum, furnishing product adsorbed silica gel.
- This material was added to a pre-fabricated silica gel column, where flash chromatography with hexanes to 10% ethyl acetate in hexanes afforded 64 mg of a B as light yellow oil after inspissation (56% of theoretical).
- the mixture was diluted with 5 mL of water and 5 mL of methylene chloride and partitioned and the aqueous layer extracted with 3-5 mL volumes of methylene chloride.
- the organic phases were combined, dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum.
- the oily residue was dissolved in a minimum amount of methylene chloride and added to 20 g of silica gel, which was then dried under vacuum, furnishing product adsorbed silica gel.
- This material was added to a pre-fabricated silica gel column, where flash chromatography with hexanes to 7% ethyl acetate in hexanes afforded 118 mg of a B as a beige solid after concentration (65% of theoretical).
- the mixture was diluted with 5 mL of water and 5 mL of methylene chloride and partitioned and the aqueous layer extracted with 3-5 mL volumes of methylene chloride.
- the organic phases were combined, dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum.
- the oily residue was dissolved in a minimum amount of methylene chloride and added to 20 g of silica gel, which was then dried under vacuum, furnishing product adsorbed silica gel.
- This material was added to a pre-fabricated silica gel column, where flash chromatography with hexanes to 5% ethyl acetate in hexanes afforded 132 mg of a B as an off-white solid after concentration (55% of theoretical).
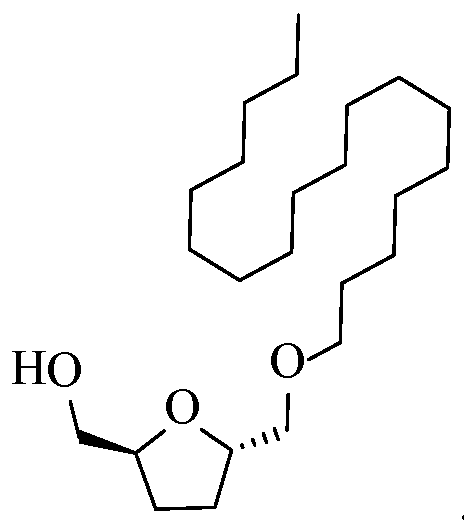
- Example 4 Synthesis of ((2S,5R)-5-((hexyloxy)methyl)tetrahydrofuran-2-yl)methanol, ((2S,5S)-5- ((hexyloxy)methyl)tetrahydrofuran-2-yl)methanol, and ((2S,5S)-5- (
- Example 6 Synthesis of ((2S,5R)-5-((octadecyloxy)methyl)tetrahydrofuran-2-yl)methanol, ((2S,5S)- 5-((octadecyloxy)methyl)tetrahydrofuran-2-yl)methanol, ((2S,5S)- 5(
- a reflux condenser was outfitted to the flask, and while stirring, the solution was heated to 50°C, 4 hours. After this time, an aliquot was extracted and analyzed by TLC (cerium molybdate visualization), demonstrating that B had entirely disappeared.
- a single neck, 50 mL round bottomed flask equipped with a 1 cm PTFE coated magnetic stir bar was charged with 175 mg of B (0.339 mmol), 65 of Hunig's base (0.373 mmol), 37 of benzylamine and 10 mL of ethanol.
- the neck was capped with a reflux condenser, and while vigorously stirring, the mixture was heated to 50°C for 2hrs. After this time, TLC (UV and cerium molybdate visualization) indicated a single band and full consumption of both reagents.
- the mixture was then diluted with 10 mL of water and 10 mL of methylene chloride and layers partitioned by liquid-liquid extraction.
- aqueous layer was extracted with 5 mL volumes of methylene chloride (x2), organic layers combined and dried, affording a pale yellow waxy solid.
- This material was charged to a 25 mL round bottomed flask equipped with a 0.5" PTFE coated magnetic stir bar, along with 100 mg of 10%> Pd/C and 10 mL of absolute ethanol.
- the neck was capped with a rubber septum and a balloon filled with 3 ⁇ 4 was inserted via a 9 inch, 16" needle; the mixture was stirred vigorously and monitored by TLC (UV-vis visualization). After 2 h, the reaction was deemed complete; catalyst filtered through a pad of Celite and filtrate concentrated under vacuum overnight, affording 74 mg of C (52%) as light yellow, loose oil. This material was used in the supervening step without further purification.
- a single neck, 10 mL round bottomed flask equipped with a 0.5" octagonal PTFE coated magnetic stir bar was charged with 50 mg of C (0.130 mmol) and 2 mL of a IN ethanolic HC1 solution. The mixture was stirred for 15 minutes, after which time excess solvent was removed first with a rotary evaporator (50°C, 30 mmHg) then under high vacuum ( ⁇ 1 torr) for 1 week. After this time, a yellow semi-solid corresponding to D was observed, weighing 49 mg (88%).
- the mixture was diluted with 5 mL of water and 5 mL of methylene chloride and partitioned and the aqueous layer extracted with 3-5 mL volumes of methylene chloride.
- the organic phases were combined, dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum.
- the oily residue was dissolved in a minimum amount of methylene chloride and added to 20 g of silica gel, which was then dried under vacuum, furnishing product adsorbed silica gel.
- the mixture was diluted with 5 mL of water and 5 mL of methylene chloride and partitioned and the aqueous layer extracted with 3-5 mL volumes of methylene chloride.
- the organic phases were combined, dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum.
- the oily residue was dissolved in a minimum amount of methylene chloride and added to 20 g of silica gel, which was then dried under vacuum, furnishing product adsorbed silica gel.
- This material was added to a pre-fabricated silica gel column, where flash chromatography with hexanes to 9% ethyl acetate in hexanes afforded 139 mg of a B as a beige solid after concentration (39%> of theoretical).
- various derivative species can also be made from FDM-monoethers, and the preparation of the FDM derivatives employ the same or similar reaction protocols, mutatis mutandis, as that used to synthesize the derivatives from bHMTHF as a starting material, such as described in the foregoing examples.
- reaction protocols mutatis mutandis
- the following examples are of alternative compounds that illustrate certain variance in synthesis.
- Each of the compounds in these variant examples is expected to parallel that of a derivative bHMTHF mono-ether (e.g., non- hydrolyzable amphiphiles with potential applications as surfactants, dispersants, plasticizers, etc). fate, B.
- a single neck, 25 mL round bottomed flask equipped with a PTFE coated magnetic stir bar was charged with 50 mg of (5-((hexyloxy)methyl)furan-2-yl)methanamine C (0.237 mmol) and 5 mL of anhydrous DMF.
- the flask was capped with a rubber septum affixed to an argon inlet and immersed in a saturated brine/ice bath mixture ( ⁇ 0°C). While vigorously stirring and under argon, 74 ⁇ ⁇ of methyl iodide (167 mg, 1.18 mmol) the mixture was added dropwise over 10 minutes. Upon complete addition, the ice bath was withdrawn and the mixture stirred at room temperature overnight.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description























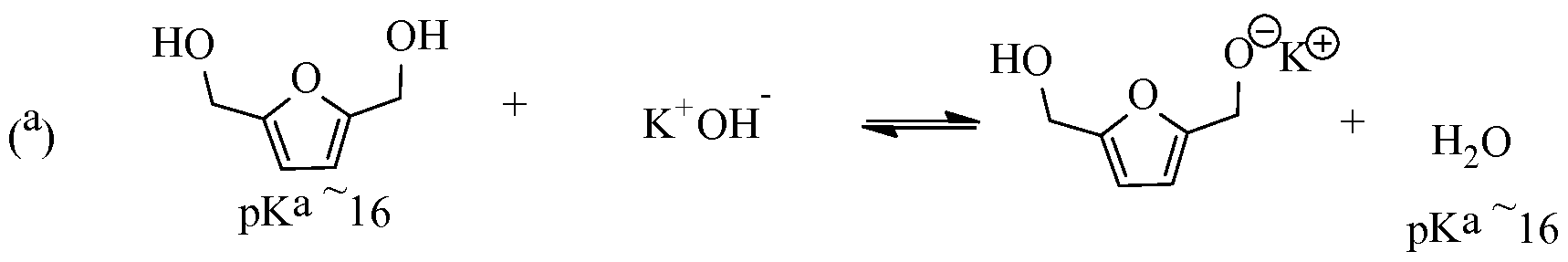









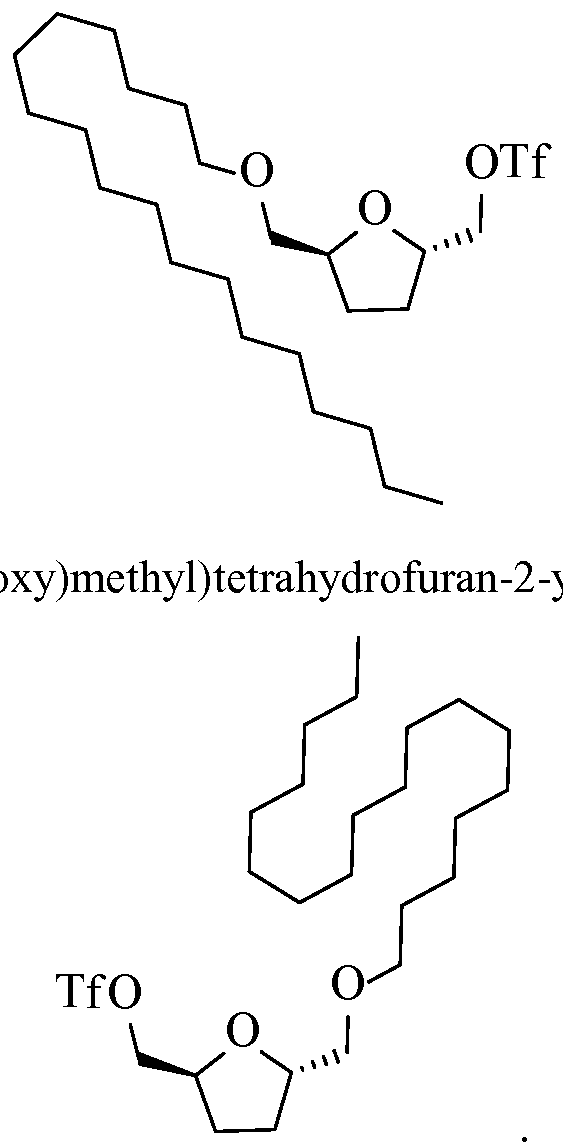


























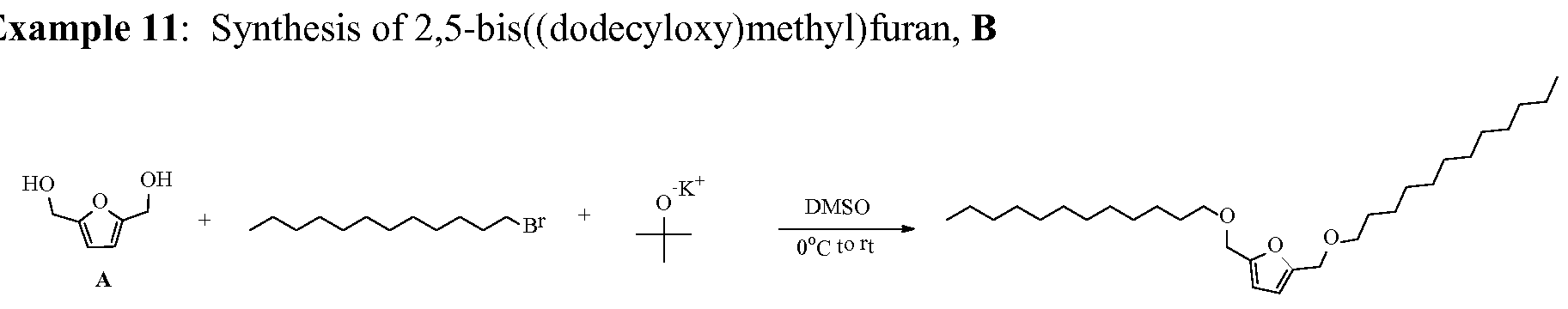







Claims














































Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/038,066 US9670174B2 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| MX2016007952A MX2016007952A (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof. |
| CA2931554A CA2931554C (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| JP2016530946A JP6396462B2 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetrahydrofuran-2,5-diyl) dimethanol and their amphiphilic derivatives |
| AU2014366334A AU2014366334B2 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| CN201480067612.0A CN105814030A (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| KR1020167018816A KR102100539B1 (en) | 2013-12-19 | 2014-12-12 | Mono- and Dialkyl Ethers of Furan-2,5-Dimethanol and (Tetra-hydrofuran-2,5-diyl)dimethanol and Amphiphilic Derivatives Thereof |
| EP14871772.1A EP3083576A4 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| US15/581,613 US20170233358A1 (en) | 2013-12-19 | 2017-04-28 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361918239P | 2013-12-19 | 2013-12-19 | |
| US61/918,239 | 2013-12-19 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/038,066 A-371-Of-International US9670174B2 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
| US15/581,613 Division US20170233358A1 (en) | 2013-12-19 | 2017-04-28 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015094970A1 true WO2015094970A1 (en) | 2015-06-25 |
Family
ID=53403552
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/070021 Ceased WO2015094970A1 (en) | 2013-12-19 | 2014-12-12 | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US9670174B2 (en) |
| EP (1) | EP3083576A4 (en) |
| JP (1) | JP6396462B2 (en) |
| KR (1) | KR102100539B1 (en) |
| CN (1) | CN105814030A (en) |
| AU (1) | AU2014366334B2 (en) |
| CA (1) | CA2931554C (en) |
| HK (1) | HK1226391A1 (en) |
| MX (1) | MX2016007952A (en) |
| WO (1) | WO2015094970A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102016009800A1 (en) | 2016-08-12 | 2018-02-15 | Henkel Ag & Co. Kgaa | Detergents and cleaning agents with anionic surfactants from renewable raw materials |
| WO2018029202A1 (en) * | 2016-08-12 | 2018-02-15 | Henkel Ag & Co. Kgaa | New anionic surfactants and detergents and cleaning agents containing same |
| WO2019042804A1 (en) * | 2017-08-28 | 2019-03-07 | Henkel Ag & Co. Kgaa | NEW ANIONIC SURFACTANTS AND WASHING AND CLEANING AGENTS CONTAINING THEM |
| US10538499B2 (en) | 2015-04-14 | 2020-01-21 | Dupont Industrial Biosciences Usa, Llc | Processes for producing 2,5-furandicarboxylic acid and derivatives thereof and polymers made therefrom |
| KR20200042942A (en) * | 2017-08-28 | 2020-04-24 | 헨켈 아게 운트 코. 카게아아 | New anionic surfactants and detergents and cleaning agents containing them |
| WO2020229158A1 (en) | 2019-05-10 | 2020-11-19 | Unilever Plc | Compound and detergent composition |
| WO2022008150A1 (en) | 2020-07-06 | 2022-01-13 | Unilever Ip Holdings B.V. | Irritation mitigating surfactants |
| WO2026002783A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Cleansing composition |
| WO2026002784A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Furan-based cleansing surfactant |
| WO2026002781A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Cleansing composition |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2903281C (en) | 2013-03-08 | 2022-11-29 | The University Of British Columbia | Substituted organofluoroborates as imaging agents |
| CA2931552A1 (en) * | 2013-12-19 | 2015-06-25 | Archer Daniels Midland Company | Sulfonates of furan-2,5-dimethanol and (tetrahydrofuran-2,5-diyl)dimethanol and derivatives thereof |
| DE102017008072A1 (en) * | 2017-08-28 | 2019-02-28 | Henkel Ag & Co. Kgaa | New anionic surfactants and detergents and cleaners containing them |
| AU2019407851B8 (en) | 2018-12-18 | 2025-10-09 | Provincial Health Services Authority | Dual mode 18f-labelled theranostic compounds and uses thereof |
| CN114163407B (en) * | 2020-09-11 | 2023-10-24 | 中国科学院宁波材料技术与工程研究所 | Furan ring ingredients that inhibit or kill microorganisms, products containing them and their applications |
| KR102665866B1 (en) | 2021-11-08 | 2024-05-14 | 한국생산기술연구원 | Method of preparing, separating and purifying tetrahydrofuran-2,5-diester using tetrahydrofuran-2,5-dimethanol |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208352A (en) * | 1992-04-02 | 1993-05-04 | Sandoz Ltd. | Process for preparing the R- and S-isomers of 2-hydroxy-methyl-2-octadecyloxymethyl-tetrahydrofuran and their use in preparing stereoisomers of pharmacologically active compounds |
| US20130303791A1 (en) * | 2010-12-16 | 2013-11-14 | Archer Daniels Midland Company | Preparation of aminomethyl furans and alkoxymethyl furan derivatives from carbohydrates |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0178261A3 (en) * | 1984-10-10 | 1986-12-30 | Sandoz Ag | Substituted 2-furanyl or 5-oxo-2-furanyl-alkoxy phosphoryl alkyl cyclimmonium salts |
| US4619917A (en) * | 1985-07-18 | 1986-10-28 | Sandoz Pharm. Corp. | Substituted 2-furanyl- or 5-oxo-2-furanyl methoxy phosphoryl alkyl cyclimmonium salts |
| ES2010568A6 (en) * | 1988-08-04 | 1989-11-16 | Uriach & Cia Sa J | New 2,5-disubstituted tetrahydrofuran derivatives. |
| JP4419761B2 (en) * | 2004-09-06 | 2010-02-24 | ダイソー株式会社 | A chiral phase transfer catalyst having a spiro skeleton, a process for producing the same, and an asymmetric catalytic reaction using the same. |
| DE102005023588A1 (en) * | 2005-05-18 | 2006-11-23 | Grünenthal GmbH | Salts of substituted allophanate esters and their use in medicaments |
| JO3240B1 (en) * | 2007-10-17 | 2018-03-08 | Janssen Pharmaceutica Nv | Inhibitors of c-fms Kinase |
-
2014
- 2014-12-12 AU AU2014366334A patent/AU2014366334B2/en active Active
- 2014-12-12 KR KR1020167018816A patent/KR102100539B1/en active Active
- 2014-12-12 JP JP2016530946A patent/JP6396462B2/en active Active
- 2014-12-12 HK HK16114626.5A patent/HK1226391A1/en unknown
- 2014-12-12 CA CA2931554A patent/CA2931554C/en active Active
- 2014-12-12 MX MX2016007952A patent/MX2016007952A/en unknown
- 2014-12-12 US US15/038,066 patent/US9670174B2/en active Active
- 2014-12-12 CN CN201480067612.0A patent/CN105814030A/en active Pending
- 2014-12-12 WO PCT/US2014/070021 patent/WO2015094970A1/en not_active Ceased
- 2014-12-12 EP EP14871772.1A patent/EP3083576A4/en not_active Withdrawn
-
2017
- 2017-04-28 US US15/581,613 patent/US20170233358A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208352A (en) * | 1992-04-02 | 1993-05-04 | Sandoz Ltd. | Process for preparing the R- and S-isomers of 2-hydroxy-methyl-2-octadecyloxymethyl-tetrahydrofuran and their use in preparing stereoisomers of pharmacologically active compounds |
| US20130303791A1 (en) * | 2010-12-16 | 2013-11-14 | Archer Daniels Midland Company | Preparation of aminomethyl furans and alkoxymethyl furan derivatives from carbohydrates |
Non-Patent Citations (4)
| Title |
|---|
| DATABASE PUBCHEM [online] 26 October 2006 (2006-10-26), "C30H56O3, SCHEMBL16853025", XP055352025, accession no. NCBI Database accession no. CID 10917601 * |
| DATABASE PUBCHEM. [online] 1 December 2012 (2012-12-01), "SCHEMBL10865666 | C24H48O3", XP055352027, accession no. NCBI Database accession no. CID 70458981 * |
| DATABASE PUBCHEM. [online] 9 February 2007 (2007-02-09), "SCHEMBL2672596 , C22H44O3", XP055352029, accession no. NCBI Database accession no. 14785823 * |
| See also references of EP3083576A4 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11028063B2 (en) | 2015-04-14 | 2021-06-08 | Dupont Industrial Biosciences Usa, Llc | Processes for producing 2,5-furandicarboxylic acid and derivatives thereof and polymers made therefrom |
| US10538499B2 (en) | 2015-04-14 | 2020-01-21 | Dupont Industrial Biosciences Usa, Llc | Processes for producing 2,5-furandicarboxylic acid and derivatives thereof and polymers made therefrom |
| US10745369B2 (en) | 2015-04-14 | 2020-08-18 | Dupont Industrial Biosciences Usa, Llc | Processes for producing 2,5-furandicarboxylic acid and derivatives thereof and polymers made therefrom |
| WO2018029201A1 (en) | 2016-08-12 | 2018-02-15 | Henkel Ag & Co. Kgaa | Detergents and cleaning agents having anionic surfactants consisting of renewable raw materials |
| WO2018029202A1 (en) * | 2016-08-12 | 2018-02-15 | Henkel Ag & Co. Kgaa | New anionic surfactants and detergents and cleaning agents containing same |
| US11193089B2 (en) | 2016-08-12 | 2021-12-07 | Henkel Ag & Co. Kgaa | Detergents and cleaning agents having anionic surfactants consisting of renewable raw materials |
| DE102016009800A1 (en) | 2016-08-12 | 2018-02-15 | Henkel Ag & Co. Kgaa | Detergents and cleaning agents with anionic surfactants from renewable raw materials |
| US11174451B2 (en) | 2016-08-12 | 2021-11-16 | Henkel Ag & Co. Kgaa | Anionic surfactants and detergents and cleaning agents containing same |
| KR20200042940A (en) * | 2017-08-28 | 2020-04-24 | 헨켈 아게 운트 코. 카게아아 | New anionic surfactants and detergents and cleaning agents containing them |
| KR102647146B1 (en) | 2017-08-28 | 2024-03-14 | 헨켈 아게 운트 코. 카게아아 | Novel anionic surfactants and detergents and cleaning agents containing them |
| JP2020531665A (en) * | 2017-08-28 | 2020-11-05 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェンHenkel AG & Co. KGaA | New anionic surfactants, as well as detergents and cleaning agents containing them |
| KR20200042942A (en) * | 2017-08-28 | 2020-04-24 | 헨켈 아게 운트 코. 카게아아 | New anionic surfactants and detergents and cleaning agents containing them |
| KR102635150B1 (en) | 2017-08-28 | 2024-02-13 | 헨켈 아게 운트 코. 카게아아 | Novel anionic surfactants and detergents and cleaning agents containing them |
| WO2019042804A1 (en) * | 2017-08-28 | 2019-03-07 | Henkel Ag & Co. Kgaa | NEW ANIONIC SURFACTANTS AND WASHING AND CLEANING AGENTS CONTAINING THEM |
| JP7220705B2 (en) | 2017-08-28 | 2023-02-10 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェン | Novel anionic surfactants and detergents and cleaners containing them |
| US11535816B2 (en) | 2017-08-28 | 2022-12-27 | Henkel Ag & Co. Kgaa | Surfactants with tetrahydrofuranyl system |
| CN113710785A (en) * | 2019-05-10 | 2021-11-26 | 联合利华知识产权控股有限公司 | Compound and detergent composition |
| WO2020229158A1 (en) | 2019-05-10 | 2020-11-19 | Unilever Plc | Compound and detergent composition |
| WO2022008150A1 (en) | 2020-07-06 | 2022-01-13 | Unilever Ip Holdings B.V. | Irritation mitigating surfactants |
| WO2026002783A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Cleansing composition |
| WO2026002784A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Furan-based cleansing surfactant |
| WO2026002781A1 (en) | 2024-06-24 | 2026-01-02 | Unilever Ip Holdings B.V. | Cleansing composition |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2014366334A1 (en) | 2016-06-02 |
| JP2017501123A (en) | 2017-01-12 |
| AU2014366334B2 (en) | 2018-01-18 |
| US20170233358A1 (en) | 2017-08-17 |
| US20160297785A1 (en) | 2016-10-13 |
| JP6396462B2 (en) | 2018-09-26 |
| US9670174B2 (en) | 2017-06-06 |
| HK1226391A1 (en) | 2017-09-29 |
| CN105814030A (en) | 2016-07-27 |
| KR20160098382A (en) | 2016-08-18 |
| KR102100539B1 (en) | 2020-04-13 |
| EP3083576A4 (en) | 2017-08-02 |
| EP3083576A1 (en) | 2016-10-26 |
| MX2016007952A (en) | 2016-09-09 |
| CA2931554A1 (en) | 2015-06-25 |
| CA2931554C (en) | 2020-10-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2931554C (en) | Mono- and dialkyl ethers of furan-2,5-dimethanol and (tetra-hydrofuran-2,5-diyl)dimethanol and amphiphilic derivatives thereof | |
| Jadhav et al. | Efficient selective dehydration of fructose and sucrose into 5-hydroxymethylfurfural (HMF) using dicationic room temperature ionic liquids as a catalyst | |
| Yang et al. | Lewis basic ionic liquids-catalyzed synthesis of 5-aryl-2-oxazolidinones from aziridines and CO 2 under solvent-free conditions | |
| JP5766717B2 (en) | Highly efficient metathesis catalyst for ROMP and RCM reaction | |
| EP2307431B1 (en) | Process for preparing amines from alcohols and ammonia | |
| Jadhav et al. | Short oligo (ethylene glycol) functionalized imidazolium dicationic room temperature ionic liquids: Synthesis, properties, and catalytic activity in azidation | |
| AU2014366329A1 (en) | Sulfonates of furan-2,5-dimethanol and (tetrahydrofuran-2,5-diyl)dimethanol and derivatives thereof | |
| Yang et al. | Efficient and sustainable transformation of gamma-valerolactone into nylon monomers | |
| EP3008049B1 (en) | Process for the production of furanic compounds comprising at least one amine function | |
| CA2909059A1 (en) | Synthesis of dicarbamates from reduction products of 2-hydroxymethyl-5-furufural (hmf) and derivatives thereof | |
| Song et al. | Efficient conversion of glucose and cellulose to 5-hydroxymethylfurfural in DBU-based ionic liquids | |
| AU2014260269A1 (en) | 5-(hydroxymethyl) furan-2-carbaldehyde (HMF) sulfonates and process for synthesis thereof | |
| Lee et al. | Cycloaddition of carbon dioxide to epichlorohydrin using ionic liquid as a catalyst | |
| AU2014334822A1 (en) | Synthesis of diacids, dialdehydes, or diamines from THF-diols | |
| Wu et al. | Synthesis of dicationic alkyl imidazolium peroxopolyoxotungsten-based phase transfer catalyst and its catalytic activity for olefin epoxidation | |
| Olszewska et al. | Asymmetric synthesis of optically active vinyltetrahydrofurans via palladium-catalysed cyclisation of bis (hydroxymethyl) allylic carbonates | |
| Yu et al. | Cobalt complex with a tetradentate aminopyridine ligand: a single-component and efficient catalytic system for the cycloaddition reactions of CO 2 and epoxides | |
| JP7475638B2 (en) | Optically active compound and its manufacturing method, coordination compound, cyclic compound, and intermediate compound containing optically active compound | |
| Avola | II; MECHANISTIC INTERPRETATIONS OF THE NEGISHIALKYL-ALKYL CROSS COUPLING PROTOCOL USING NMR SPECTROSCOPY |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14871772 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016530946 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15038066 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2931554 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2014366334 Country of ref document: AU Date of ref document: 20141212 Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014871772 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2016/007952 Country of ref document: MX Ref document number: 2014871772 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016014118 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20167018816 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016014118 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160616 |