WO2015105174A1 - 5-ヒドロキシ-1h-イミダゾール-4-カルボキサミドの有効投与量または感受性の予測方法および予測装置、キサントシン一リン酸の量の測定方法ならびに骨髄異形成症候群の処置剤および処置方法 - Google Patents
5-ヒドロキシ-1h-イミダゾール-4-カルボキサミドの有効投与量または感受性の予測方法および予測装置、キサントシン一リン酸の量の測定方法ならびに骨髄異形成症候群の処置剤および処置方法 Download PDFInfo
- Publication number
- WO2015105174A1 WO2015105174A1 PCT/JP2015/050478 JP2015050478W WO2015105174A1 WO 2015105174 A1 WO2015105174 A1 WO 2015105174A1 JP 2015050478 W JP2015050478 W JP 2015050478W WO 2015105174 A1 WO2015105174 A1 WO 2015105174A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salt
- hydrate
- imidazole
- hydroxy
- carboxamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images








Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/483—Physical analysis of biological material
- G01N33/487—Physical analysis of biological material of liquid biological material
- G01N33/49—Blood
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/62—Detectors specially adapted therefor
- G01N30/72—Mass spectrometers
- G01N30/7233—Mass spectrometers interfaced to liquid or supercritical fluid chromatograph
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/5308—Immunoassay; Biospecific binding assay; Materials therefor for analytes not provided for elsewhere, e.g. nucleic acids, uric acid, worms, mites
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/5758—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumours, cancers or neoplasias, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides or metabolites
- G01N33/57595—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumours, cancers or neoplasias, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides or metabolites involving intracellular compounds
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6848—Methods of protein analysis involving mass spectrometry
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N2030/022—Column chromatography characterised by the kind of separation mechanism
- G01N2030/027—Liquid chromatography
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
- G01N2030/8809—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86 analysis specially adapted for the sample
- G01N2030/8813—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86 analysis specially adapted for the sample biological materials
- G01N2030/8822—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86 analysis specially adapted for the sample biological materials involving blood
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/22—Haematology
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/52—Predicting or monitoring the response to treatment, e.g. for selection of therapy based on assay results in personalised medicine; Prognosis
Definitions
- the present invention relates to a method for predicting an effective dose of 5-hydroxy-1H-imidazole-4-carboxamide or a salt thereof or a hydrate thereof for a patient with myelodysplastic syndrome.
- the invention further relates to a method for predicting whether a patient with myelodysplastic syndrome is susceptible to treatment with 5-hydroxy-1H-imidazole-4-carboxamide or a salt or hydrate thereof.
- the present invention further relates to a method for measuring the amount of xanthosine monophosphate contained in blood.
- the present invention further provides a device for predicting an effective dosage of 5-hydroxy-1H-imidazole-4-carboxamide for a patient with myelodysplastic syndrome, and a patient with myelodysplastic syndrome, wherein 5-hydroxy-1H-imidazole It relates to a device for predicting whether it is sensitive to treatment with 4-carboxamide or a salt thereof or a hydrate thereof.
- the present invention further provides a therapeutic agent for use in the treatment of patients with myelodysplastic syndrome predicted to be sensitive to treatment with 5-hydroxy-1H-imidazole-4-carboxamide or a salt thereof or a hydrate thereof. And a treatment method.
- Compound A 5-Hydroxy-1H-imidazole-4-carboxamide
- MDS myelodysplastic syndrome
- Compound A or a salt thereof or a hydrate thereof is converted in vivo by adenine phosphoribosyltransferase (APRT) to ribosyl monophosphate (RMP) and inhibits inosine-5'-monophosphate dehydrogenase (IMPDH).
- APRT adenine phosphoribosyltransferase
- RMP ribosyl monophosphate
- IMPDH inosine-5'-monophosphate dehydrogenase
- IMPDH is an enzyme that produces xanthosine monophosphate (XMP) from inosine monophosphate (IMP).
- IMPDH inhibitors As IMPDH inhibitors, immunosuppressive agents such as mycophenolic acid and mycophenolic acid mofetil are known. About these immunosuppressive agents, IMP is added in vitro to cell lysates of peripheral blood mononuclear cells (PBMC), which is a fraction containing lymphocytes obtained from blood, and the enzyme reaction by IMPDH is performed. A method for evaluating the degree of inhibition by the IMPDH inhibitor by measuring the amount of XMP was reported (Non-patent Document 2).
- PBMC peripheral blood mononuclear cells
- Non-patent Document 3 For analysis of nucleotides, methods using hydrophilic interaction chromatography-mass spectrometry (HILIC-MS) and aqueous normal phase chromatography-mass spectrometry have been reported (Non-patent Document 3). For XMP analysis, methods using reverse phase chromatography and ion exchange chromatography using an ion pair reagent have been reported.
- XMP produced by IMPDH is further converted into guanosine monophosphate (GMP), guanosine diphosphate (GDP), and guanosine triphosphate (GTP) by an in vivo enzyme reaction, and becomes a material for nucleic acid synthesis (non-patented) Reference 4).
- GMP guanosine monophosphate
- GDP guanosine diphosphate
- GTP guanosine triphosphate
- MDS is a disease in which dysplasia of blood cell morphology and cytopenia are observed, and the therapeutic effect of therapeutic agents is mainly evaluated by hematological remission and improvement.
- the method described in Non-Patent Document 2 requires complicated operation and time for fractionation of PBMC.
- this method is a method for evaluating by adding non-endogenous IMP, and does not accurately reflect the degree of IMPDH inhibition in vivo.
- nucleotides are hydrophilic small molecules, they are difficult to retain in reversed-phase chromatography usually used for analysis. Moreover, since it has a phosphate group, an adsorption phenomenon and / or tailing is likely to occur during separation analysis. For this reason, quantification of endogenous nucleotides is difficult. Numerous nucleotide metabolites are similar in mass and structure. Since these often approximate the behavior and retention time on liquid chromatography (LC), it is difficult to distinguish and quantify them by liquid chromatography-mass spectrometry (LC-MS).
- LC-MS liquid chromatography-mass spectrometry
- This invention made it the subject which should be solved to provide the method of estimating the effective dose of the compound A or its salt, or its hydrate with respect to a patient with MDS in a short time by simple operation.
- the present invention further provides a method for predicting whether or not a patient with MDS is susceptible to treatment with Compound A or a salt or hydrate thereof in a short time with a simple operation. It was a problem to be solved.
- Another object of the present invention is to provide a method for accurately measuring a trace amount of XMP contained in blood.
- the present invention further provides a device for predicting an effective dose of Compound A or a salt thereof or a hydrate thereof to a patient with MDS and a patient with MDS in a short time in a simple operation. It was a problem to be solved to provide a device for predicting whether or not it is sensitive to treatment using the hydrate.
- the present invention further solves the problem of providing a therapeutic agent and method for use in the treatment of patients with MDS predicted to be sensitive to treatment with Compound A or a salt or hydrate thereof. It was a problem to be solved.
- the present inventors have found that an effective dose of Compound A or a salt or hydrate thereof and a treatment using Compound A or a salt or hydrate thereof for patients with MDS It was found that the susceptibility of patients with MDS to can be predicted by measuring variations in the amount of XMP in the blood. Furthermore, the present inventors have found that the amount of trace XMP contained in blood can be accurately measured by setting measurement conditions in the MS method. The present invention has been completed based on these findings.
- a method for predicting an effective dose of Compound A or a salt thereof or a hydrate thereof for patients with MDS (A) (i) the amount of XMP contained in blood collected from a patient prior to administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof
- the amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Measuring the amount of XMP contained in the obtained blood;
- [2] The method according to [1], wherein the blood is peripheral blood.
- [3] The method according to [1] or [2], wherein the step of measuring the amount of XMP is a step carried out under a condition in which contaminants contained in blood and XMP can be distinguished and measured.
- [4] The method according to any one of [1] to [3], wherein the step of measuring the amount of XMP is a step of measuring by a mass spectrometry (MS) method.
- MS mass spectrometry
- the step of measuring the amount of XMP comprises (A1) measuring XMP contained in blood under two different measurement conditions by MS method; (A2) A step of obtaining an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the step (a1); (A3) The method according to [4], including a step of confirming that an ionic strength ratio of XMP contained in blood obtained in the step (a2) is within a predetermined range.
- the method according to [5], wherein the step (a1) is a step of measuring XMP contained in blood under two different measurement conditions by an LC-MS method.
- the method according to [6] wherein the mobile phase used for liquid chromatography (LC) has a pH value of 7 to 11.
- Step (c) (C1) expressing the relationship between the time after administration or contact time of Compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in step (b) as a function; (C2) A step of predicting an effective dose of compound A or a salt thereof or a hydrate thereof from the function obtained in step (c1), wherein any one of [1] to [11] The method described.
- Step (c2) predicts that the dose of Compound A or a salt thereof or a hydrate thereof such that the variation rate of XMP is not less than the predetermined variation rate during the predetermined treatment period is an effective dose.
- the dose of Compound A or a salt thereof or a hydrate thereof is such that the variation rate of XMP is not less than a predetermined variation rate in a period not less than a predetermined ratio during a predetermined treatment period.
- the method according to [12] which comprises a step of predicting an effective dose.
- a method for predicting whether a patient with MDS is sensitive to treatment with Compound A or a salt or hydrate thereof comprising: (A) (i) the amount of XMP contained in blood collected from a patient prior to administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof The amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Measuring the amount of XMP contained in the obtained blood; (B) a step of obtaining a variation rate of XMP from the amount of XMP obtained in step (a); (C) predicting whether the patient is sensitive to treatment with Compound A or a salt or hydrate thereof from the variation rate of XMP obtained in step (b). .
- the step of measuring the amount of XMP comprises (A1) measuring XMP contained in blood under two different measurement conditions by MS method; (A2) A step of obtaining an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the step (a1); (A3) The method according to [18], comprising a step of confirming that the ionic strength ratio of XMP contained in blood obtained in the step (a2) is within a predetermined range.
- the method according to [19], wherein the step (a1) is a step of measuring XMP contained in blood under two different measurement conditions by an LC-MS method.
- the method according to [20] wherein the mobile phase used for LC has a pH value of 7 to 11.
- Step is (C1) expressing the relationship between the time after administration or contact time of Compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in step (b) as a function; (C2) predicting whether or not the patient is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof from the function obtained in Step (c1). [15] The method according to any one of [25].
- step (c2) when the variation rate of XMP is not less than the predetermined variation rate in the predetermined treatment period, the patient is sensitive to the treatment using Compound A or a salt thereof or a hydrate thereof.
- step (c2) when the variation rate of XMP is not less than a predetermined variation rate in a period that is not less than a predetermined rate during a predetermined treatment period, the patient has a compound A or a salt thereof or a hydrate thereof.
- the method comprises a step of predicting that the treatment is sensitive to the treatment using the method.
- a method for measuring the amount of XMP contained in blood (A1) measuring XMP contained in blood under two different measurement conditions by MS method; (A2) A step of obtaining an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the step (a1); (A3) a step of confirming that the ionic strength ratio of XMP contained in the blood obtained in the step (a2) is within a predetermined range.
- the step (a1) is a step of measuring XMP contained in blood under two different measurement conditions by an LC-MS method.
- a device for predicting an effective dose of Compound A or a salt thereof or a hydrate thereof for a patient with MDS (A) (i) the amount of XMP contained in blood collected from a patient before administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof The amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Means for measuring the amount of XMP contained in the obtained blood; (B) a means for determining the variation rate of XMP from the amount of XMP obtained by the means described in (A); (C) A device comprising: a means for predicting an effective dose of Compound A or a salt thereof or a hydrate thereof from the variation rate of XMP obtained by the means described in (B). [37] The device according to [36], wherein the means for measuring the amount of
- a device for predicting whether a patient with MDS is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof (A) (i) the amount of XMP contained in blood collected from a patient before administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof The amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Means for measuring the amount of XMP contained in the obtained blood; (B) a means for determining the variation rate of XMP from the amount of XMP obtained by the means described in (A); (C) a means for predicting whether or not a patient is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof from the variation rate of XMP obtained by the means described in (B); Including the device. [40] The device according to [39
- An active ingredient for use in the treatment of a patient with MDS comprising the step of predicting whether the patient with MDS is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof.
- a therapeutic agent that is Compound A wherein the predicting step comprises: (A) (i) the amount of XMP contained in blood collected from a patient prior to administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof The amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Measuring the amount of XMP contained in the obtained blood; (B) a step of obtaining a variation rate of XMP from the amount of XMP obtained in step (a); (C) predicting whether the patient is sensitive to treatment with Compound A or a salt or hydrate thereof from the variation rate of XMP obtained in Step
- An active ingredient for use in the treatment of a patient with MDS comprising predicting whether the patient with MDS is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof.
- a method of treating MDS, which is Compound A wherein the predicting step comprises: (A) (i) the amount of XMP contained in blood collected from a patient prior to administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof The amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof Measuring the amount of XMP contained in the obtained blood; (B) a step of obtaining a variation rate of XMP from the amount of XMP obtained in step (a); (C) predicting whether the patient is sensitive to treatment with Compound A or a salt or hydrate thereof from the variation rate of XMP obtained
- the prediction method of the present invention is a method using blood as a sample, whereby the effective dose of Compound A or a salt thereof or a hydrate thereof can be predicted in a short time by a simple operation. In addition, it can be predicted in a short time by a simple operation whether or not a patient with MDS is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof.
- the measurement method of the present invention can accurately measure the amount of trace XMP contained in blood.
- the prediction device of the present invention is a device that uses blood as a sample, and is used for predicting the effective dose of Compound A or a salt thereof or a hydrate thereof to a patient with MDS in a short time with a simple operation. Can do. Moreover, it can be used for predicting in a short time by a simple operation whether or not a patient with MDS is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof.
- the treatment agent and the treatment method of the present invention are used for the treatment of patients with MDS predicted to be sensitive to treatment with Compound A or a salt thereof or a hydrate thereof, Compound A or a salt thereof or a salt thereof Unwanted administration to patients who are not sensitive to hydrates can be avoided.
- the method and treatment agent of the present invention can be applied to a kit for treatment of MDS patients according to a conventional method.
- FIG. 1 shows the antitumor effect of Compound A.
- FIG. 2 shows the result of PK / PD analysis (PK parameter: C max ).
- FIG. 3 shows the results of PK / PD analysis (PK parameter: Time above IC 50 ).
- FIG. 4 shows the result of PK / PD analysis (PK parameter: AUC 0-48 ).
- FIG. 5 shows the measurement results of XMP contained in the XMP standard substance and human blood (sample) (measurement conditions: A and B).
- FIG. 6 shows the measurement results of XMP contained in the XMP standard substance and human blood (sample) (measurement conditions: C, D).
- FIG. 7 shows the measurement results of XMP contained in the XMP standard substance and human blood (sample) (measurement conditions: A ′, B ′).
- FIG. 8 shows the measurement results of XMP contained in human blood (LC conditions: a, b).
- Treatment means prevention or treatment for diseases.
- a therapeutic agent means a substance that is provided for the purpose of prevention or treatment of a disease.
- the treatment period means a period during which treatment is performed.
- the ion intensity includes, for example, the intensity of the corresponding ion on the peak area or MS spectrum obtained from the chromatogram in the LC-MS method.
- the XMP standard substance means a standard used when measuring XMP contained in blood. XMP standard substances include commercially available sodium salts of xanthosine monophosphate.
- a method for predicting an effective dose of Compound A or a salt thereof or a hydrate thereof to a patient with MDS, and whether a patient with MDS is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof in the method according to the present invention, as the step (a), as the step (a), (i) the amount of XMP contained in blood collected from a patient before administering Compound A or a salt thereof or a hydrate thereof, (Ii) The amount of XMP contained in blood collected from a patient after administration of Compound A or a salt thereof or a hydrate thereof, or collected from a patient before administration of Compound A or a salt thereof or a hydrate thereof The amount of XMP contained in blood obtained by contacting blood with compound A or a salt thereof or a hydrate thereof is measured.
- Examples of the salt of compound A include salts of a commonly known basic group or acidic group.
- Examples of basic group salts include salts with mineral acids such as hydrochloric acid, hydrogen bromide, phosphoric acid and sulfuric acid; tartaric acid, formic acid, acetic acid, fumaric acid, maleic acid, citric acid, trichloroacetic acid and trifluoroacetic acid.
- salts with organic carboxylic acids and salts with sulfonic acids such as methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, mesitylenesulfonic acid and naphthalenesulfonic acid.
- salts with alkali metals such as sodium and potassium
- salts with alkaline earth metals such as calcium and magnesium
- Preferred salts include pharmacologically acceptable salts.
- Compound A or a salt thereof or a hydrate thereof is, for example, JP-A-53-32124, JP-A-58-24569, International Publication No. 2009/035168, International Publication No. 2013/047758, or the like. It can be produced by the method described in 1.
- One of the features of the present invention is that the amount of XMP contained in blood is used as an index.
- the frequency is limited due to the great mental load. Therefore, the bone marrow cells necessary for evaluation of the IMPDH inhibitory effect are not usually available.
- the IMPDH inhibitory effect of compound A or a salt thereof or a hydrate thereof is equivalent between bone marrow cells and peripheral blood cells.
- the blood used in the present invention is not particularly limited, and may be, for example, peripheral blood, blood pooled in bone marrow, spleen, or liver, lymph fluid, tissue fluid, or umbilical cord blood, preferably peripheral blood. Collection of blood from a patient can be performed by conventional methods well known to those skilled in the art.
- the blood used in the present invention is preferably whole blood from the viewpoint of performing prediction in a short time with a simple operation.
- the step (a) in the present invention can be performed in either of the following two modes.
- the amount of XMP contained in blood collected from a patient before administration of Compound A or a salt thereof or a hydrate thereof and from a patient after administration of Compound A or a salt thereof or a hydrate thereof In this embodiment, the amount of XMP contained in the collected blood is measured.
- the amount of XMP contained in blood collected from a patient before administration of Compound A or a salt thereof or a hydrate thereof and the patient before administration of Compound A or a salt thereof or a hydrate thereof are used.
- the amount of XMP contained in blood obtained by contacting compound A or a salt or hydrate thereof with the collected blood is measured.
- Compound A or a salt thereof or a hydrate thereof administered to a patient exerts its action by contacting blood cells in the patient.
- compound A or a salt thereof or a hydrate thereof is contacted in vitro with blood collected from a patient prior to administration of compound A or a salt thereof or a hydrate thereof. Or its salt or its hydrate exerts an action on blood cells.
- the amount of XMP can be measured, for example, by the MS method.
- a blood sample Prior to measurement by the MS method, a blood sample may be subjected to rough separation of the sample such as protein removal, or separation of components in the sample by LC or the like.
- proteins, peptides and lipids contained in blood are typical interfering components of MS measurement
- these interfering components can be removed in advance by rough separation of the sample.
- fractionation cell fractionation by centrifugation, removal of liquid components and / or erythrocytes by hemolysis
- filtration protein removal treatment
- liquid phase extraction and / or solid phase extraction, etc. It can be performed.
- the rough separation of the sample can be performed on-line by pre-column or column switching together with the separation and detection operation of components described later.
- LC liquid crystal display
- MS reverse phase chromatography
- chromatography using an ion pair reagent ion exchange chromatography, or the like
- the mode and measurement conditions are not particularly limited as long as necessary separation and performance can be obtained and elution can be performed with a mobile phase composition that can be introduced into MS.
- a separation method such as capillary electrophoresis or gas chromatography can also be used.
- Contaminants that can be detected in XMP measurement include compounds that approximate mass with XMP, compounds that generate a compound that approximates mass with XMP by decomposition, and / or compounds that approximate the elution time of XMP and LC, etc. Is mentioned.
- Examples of the compound whose mass approximates that of XMP include an isotope of GMP.
- Examples of the compound that generates a compound having a mass approximate to that of XMP by decomposition or the like include an isotope of GDP and an isotope of GTP.
- Examples of the compounds having similar XMP and LC elution times include reduced nicotinamide adenine dinucleotide phosphate (NADPH).
- the step of measuring the amount of XMP by the MS method is preferably performed under conditions that allow measurement by distinguishing contaminants contained in blood and XMP.
- impurities that can be detected in the measurement of XMP include GMP (mass 363.06) isotopes (mass 364.06 and mass 364.06), GDP, It was found that the isotopes of GTP in-source decomposition products (decomposed at the ionization part to produce GMP) and divalent ions derived from NADPH were included.
- the step of measuring the amount of XMP comprises (A1) measuring XMP contained in blood under two different measurement conditions by MS method; (A2) A step of obtaining an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the step (a1); (A3) a step of confirming that the ionic strength ratio of XMP contained in blood obtained in the step (a2) is within a predetermined range; It is a process including.
- Quantification of XMP can be performed by a known quantitative method such as a calibration curve method (external standard method, standard addition method or internal standard method) or isotope dilution method, and is not particularly limited.
- step (b) a step of obtaining the variation rate of XMP from the amount of XMP obtained in the step (a) is performed.
- the method for obtaining the variation rate is not particularly limited, but can be obtained by the following equation, for example.
- Fluctuation rate (%) (1- (Amount of XMP after administration of Compound A or its salt or hydrate thereof / Amount of XMP before administration of Compound A or its salt or its hydrate)) ⁇ 100
- Fluctuation (%) (1- (Amount of XMP after contacting Compound A or a salt thereof or hydrate thereof / Amount of XMP before contact with Compound A or a salt or hydrate thereof)) ⁇ 100
- the value per unit blood volume can also be used for the quantity of XMP, it is preferable to use the value per unit blood cell number.
- step (c) the effective dose of compound A or a salt thereof or a hydrate thereof is predicted from the variation rate of XMP obtained in step (b), Alternatively, it is predicted from the rate of variation of XMP obtained in step (b) whether the patient is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof.
- step (c1) a step of expressing the relationship between the time after administration of the compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in the step (b) as a function; ) (C1) The step of predicting the effective dose of compound A or a salt thereof or a hydrate thereof from the function obtained in step (c1).
- the step (c) includes (c1) a step of expressing the relationship between the time after administration of the compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in the step (b) as a function; ) (C1) can be performed by predicting whether the patient is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof from the function obtained in Step (c1).
- the step (c) includes (c1) a step of expressing the relationship between the contact time of the compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in the step (b) as a function; (C1) The step of predicting the effective dose of Compound A or a salt thereof or a hydrate thereof from the function obtained in the step can be performed.
- the step (c) includes (c1) a step of expressing the relationship between the contact time of the compound A or a salt thereof or a hydrate thereof and the variation rate of XMP obtained in the step (b) as a function; (C1) It can be performed by predicting whether or not the patient is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof from the function obtained in Step (c1).
- the effective dose is the dose of Compound A or a salt thereof or a hydrate thereof such that the variation rate of XMP is not less than the predetermined variation rate (decrease rate) during the predetermined treatment period. Predict that there is. Further, in the step (c2), when the XMP fluctuation rate is equal to or higher than the predetermined fluctuation rate (decrease rate) in the predetermined treatment period, the patient uses Compound A, a salt thereof or a hydrate thereof. Predicting sensitivity to treatment.
- the predetermined treatment period can be appropriately selected according to the species, but may be, for example, one day to two months, or a period of a predetermined ratio or more during the predetermined treatment period. Good.
- the period equal to or higher than the predetermined ratio in the predetermined treatment period can be appropriately selected according to the species, and can be, for example, a period of 40% or more in the predetermined treatment period.
- the predetermined variation rate can be appropriately selected according to the species, and can be, for example, 45% or more (preferably 50% or more, more preferably 55% or more).
- the variation rate (reduction rate) of XMP is 45% or more (preferably 50% or more, more preferably 55% or more).
- Such a dose can be predicted as an effective dose.
- the effective dose of Compound A or a salt thereof or a hydrate thereof obtained here may be administered to a patient with MDS.
- the variation rate (decrease rate) of XMP is 45% or more (preferably 50% or more, more preferably 55% If this is the case, it can be predicted that the patient is sensitive to treatment with Compound A or a salt or hydrate thereof.
- Compound A or a salt thereof or a hydrate thereof may be administered to a patient who is predicted to be susceptible.
- the method for measuring the amount of XMP contained in blood comprises: (A1) measuring XMP contained in blood under two different measurement conditions by MS method; (A2) A step of obtaining an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the step (a1); (A3) a step of confirming that the ionic strength ratio of XMP contained in blood obtained in the step (a2) is within a predetermined range; It is a method including.
- step (a1) XMP is detected by the MS method.
- A1 The measurement is performed under two different measurement conditions in the step, (a2) the ionic strength ratio of XMP contained in blood under the two different measurement conditions is obtained in the step (a3), By confirming that it is within the range, XMP can be measured separately from impurities.
- step (a1) it is preferable to separate a sample by LC and detect XMP by MS method (LC-MS method).
- the method for measuring the amount of XMP contained in the blood of the present invention comprises: (A11) a step of measuring XMP standard substance and XMP contained in blood under two different measurement conditions by MS method; (A12) a step of obtaining an ionic strength ratio of the XMP standard substance and an ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in the steps (a11); (A13) comparing the ionic strength ratio of the XMP standard substance obtained in the step (a12) with the ionic strength ratio of XMP contained in blood; Can be used.
- the ionic strength ratio of the XMP standard substance and the ionic strength ratio of XMP contained in the blood under two different measurement conditions obtained in the step (a11) are obtained.
- the ionic strength ratio of the XMP standard substance obtained in the step is compared with the ionic strength ratio of XMP contained in blood.
- the ionic strength ratio of the blood sample and the ionic strength ratio of the XMP standard substance are within a predetermined range, it can be determined that XMP in blood can be detected with high specificity.
- LC HILIC
- RP chromatography using an ion pair reagent, ion exchange chromatography or the like
- HILIC is preferable.
- a mobile phase A in an aqueous solution which is preferably basic, and a mobile phase B containing an organic solvent can be used.
- the pH value of the mobile phase A is preferably 7 to 11, more preferably 8 to 11, further preferably 8.4 to 10.4, and particularly preferably 9.0 to 9.8. . What is necessary is just to set the pH value of the mobile phase A according to the kind of column to be used.
- the salt concentration of the mobile phase used for LC is preferably 5 to 300 mmol / L, more preferably 20 to 200 mmol / L.
- the salt concentration is usually low (for example, 10 mmol / L, etc.) in order to obtain high sensitivity, but in the present invention, by adopting a salt concentration higher than usual, XMP and impurities It became possible to isolate.
- the mobile phase A used for LC preferably contains an aqueous ammonium bicarbonate solution.
- organic solvent used for the mobile phase B examples include acetonitrile, methanol, 2-propanol and ethanol, preferably acetonitrile or methanol, more preferably acetonitrile.
- a HILIC column for example, SeQuant ZIC-pHILIC, Merck
- 50 mmol / L ammonium bicarbonate aqueous solution (adjusted to pH 9.4 with 25% by mass ammonia aqueous solution) and acetonitrile as mobile phases.
- a gradient method in which the ratio of mobile phase A and mobile phase B is changed over time or an isocratic method in which the ratio of mobile phase A and mobile phase B is fixed.
- the concentration of acetonitrile in the mobile phase is preferably 20 to 90% by volume, more preferably 35 to 70% by volume, and further preferably 65% by volume.
- the elution may be performed by gradient elution in which the ratio of mobile phase A and mobile phase B is changed over time. However, the elution can be carried out in that the separation from impurities is improved and the analysis can be performed under the condition where the injection amount is increased. An isocratic method in which the ratio of the phase A and the mobile phase B is not changed is preferable.
- XMP contained in blood is measured under two different measurement conditions by the MS method.
- the MS method components in a sample are ionized by an MS apparatus, and ions derived from XMP are selected and detected.
- ions derived from XMP are selected and detected.
- the method of the present invention is characterized in that separation conditions and MS conditions (detection ions and / or ionization conditions) are set so that high selectivity can be achieved.
- Whether the selectivity is sufficient can be evaluated by the methods shown in the steps (a1), (a2) and (a3).
- the amount of XMP contained in blood can be accurately measured even when a low-resolution MS apparatus commonly used in pharmacokinetics is used. .
- MS / MS measurement such as MRM (Multiple Reaction Monitoring) mode while ensuring specificity. It is preferable to measure under a plurality of conditions, such as detecting a plurality of MS / MS product ions generated from the measurement target. Conditions with good specificity can be selected at the time of analysis, and specificity can be verified as described later.
- MS / MS measurement or MS measurement using a high-resolution apparatus such as an Orbitrap type MS (for example, Q-Exactive, Thermo) or a TOF type MS (for example, 6550i Funnel Q-TOF, Agilent, SYNAPT, Waters) It can be measured with higher specificity. It is preferable that the resolution of the apparatus is high, but it is preferable to use an apparatus that can obtain the sensitivity and dynamic range necessary for the quantitative analysis of XMP.
- XMP contained in blood is measured under two different measurement conditions by the MS method. That is, XMP-derived ions are measured under two different MS conditions.
- the two different measurement conditions are preferably two measurement conditions having different detection ions and / or ionization conditions in the MS method. Specifically, measurement of multiple MS / MS product ions generated in MS / MS measurement, measurement under different ionization conditions (such as ion source temperature (turbo heater gas temperature) or ion introduction part (orifice plate) voltage)) And measurements in the posi mode and the negative mode.
- a product ion of 97.0 (m / z) when 365.0 (m / z) is used as a precursor ion and 365.0 (m / z) Combination with 153.0 (m / z) product ion when the precursor ion is a precursor ion, and 97.0 (m / z) product ion when the precursor ion is 365.0 (m / z)
- a combination of product ions of 213.0 (m / z) when 365.0 (m / z) is used as a precursor ion can be used.
- the product ion of 97.0 (m / z) when 365.0 (m / z) is used as the precursor ion and the precursor of 365.0 (m / z) are used. It is preferable to use a combination with a product ion of 213.0 (m / z) as an ion.
- 97.0 (m / z) product ion when 365.0 (m / z) is used as a precursor ion, and 365.0 (m / z) as precursor ion It is preferable to use a combination with a product ion of 153.0 (m / z).
- step (a2) of the present invention the ionic strength ratio of XMP contained in blood under two different measurement conditions obtained in step (a1) is obtained, and in step (a3), in step (a2) It is confirmed that the ionic strength ratio of XMP contained in the obtained blood is within a predetermined range.
- the ionic strength ratio can be obtained by the following equation, for example.
- Ion intensity ratio (Ion intensity under other measurement conditions) / (Intensity of 97.0 (m / z) product ion when 365.0 (m / z) is used as a precursor ion)
- the ion intensity under other conditions for example, the intensity of the product ion of 153.0 (m / z) when 365.0 (m / z) is used as the precursor ion or 365.0 (m / z)
- the ionic strength ratio of the blood sample when the ionic strength ratio of the blood sample is within a predetermined range, it can be determined that XMP in the blood can be detected with high specificity. On the other hand, if the ionic strength ratio of the blood sample is outside the predetermined range, it is determined that the selectivity is insufficient under either or both of the conditions where the ratio is taken. “Within a predetermined range” refers to the case where the ionic strength ratio is less than 1 when the ionic strength ratio is determined by the above formula. When the ion intensity under other conditions is the intensity of a product ion of 213.0 (m / z) when 365.0 (m / z) is used as a precursor ion, the ion intensity ratio is 0.1 to 0.00.
- the inventors have found that the detection of contaminants can be suppressed by changing the MS / MS product ion to be detected or decreasing the ion source temperature (turbo heater gas temperature) and the voltage of the ion introduction part (orifice plate). Found by examination.
- the predetermined range when comparing the ionic strength ratio of the XMP standard substance and the ionic strength ratio of the XMP contained in the blood means that the ionic strength ratio of the XMP contained in the blood is the ionic strength ratio of the XMP standard substance. Of 0.5 to 1.5.
- XMP can be quantified from ions detected by MS measurement with sufficient specificity.
- a device for predicting an effective dose of compound A or a salt thereof or a hydrate thereof and a device for predicting sensitivity to compound A or a salt thereof or a hydrate thereof
- the present invention further includes MDS Apparatus for predicting the effective dose of Compound A or a salt or hydrate thereof for patients with MDS and whether the patient with MDS is sensitive to treatment with Compound A or a salt or hydrate thereof An apparatus for predicting this is provided.
- the device of the present invention (A) (i) the amount of XMP contained in blood collected from a patient before administration of Compound A or a salt or hydrate thereof, and (ii) administration of Compound A or a salt or hydrate thereof
- the amount of XMP contained in blood collected from a later patient, or blood collected from a patient before administration of Compound A or a salt or hydrate thereof is contacted with Compound A or a salt or hydrate thereof
- Means for measuring the amount of XMP contained in the obtained blood (B) a means for determining the variation rate of XMP from the amount of XMP obtained by the means described in (A);
- the means described in (A) is not particularly limited as long as it is a means capable of measuring the amount of XMP contained in blood.
- an MS apparatus or the like can be used, and an apparatus that combines a separation apparatus and an MS apparatus can be used. It can also be used.
- the separation device a high performance liquid chromatography device, capillary electrophoresis, gas chromatography device or the like can be used.
- An LC-MS device is preferable.
- the MS apparatus usually has a sample introduction unit, an ion source, an analysis unit, an ion detection unit, and a data processing unit.
- the sample introduction part is a part for introducing a sample into the MS apparatus.
- the MS apparatus can be connected to a high performance liquid chromatography apparatus, capillary electrophoresis or gas chromatography apparatus, and a sample can be introduced from these apparatuses into the MS apparatus.
- the ion source is a part that charges the sample. Electron ionization method (EI method), chemical ionization method (CI method), field desorption method (FD method), fast atom bombardment method (FAB method), matrix support Laser desorption ionization (MALDI), atmospheric pressure photoionization (APPI), atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI) are known.
- EI method Electron ionization method
- CI method chemical ionization method
- FD method field desorption method
- FAB method fast atom bombardment method
- MALDI matrix support Laser desorption ionization
- APPI atmospheric pressure photoionization
- APCI atmospheric pressure chemical ionization
- ESI electrospray ionization
- an ion source used as the LC-MS method an APPI method, an APCI method, or an ESI method can be used, and the ESI method is preferable.
- the analysis unit is a part that separates the ionized sample.
- a magnetic field deflection type, a quadrupole type, an ion trap type, a time-of-flight type, a Fourier transform ion cyclotron resonance type, a tandem type, and the like are known.
- the detection unit is a site that detects and sensitizes ions selected by the analysis unit with an electron multiplier or a microchannel plate.
- the data processing unit is a part for creating a mass spectrum from the obtained data.
- any means can be used as long as it can obtain the fluctuation rate from the amount of XMP obtained and the above formula.
- an electronic computer having calculation software or a program can be used.
- the means for predicting whether or not it is sensitive to the treatment using hydrate is not particularly limited as long as it is a means capable of predicting an effective dose or sensitivity based on a predetermined standard from the variation rate of XMP. For example, it can be performed using general-purpose calculation software.
- means for expressing the relationship between the time after administration or contact time of Compound A or a salt thereof or a hydrate thereof and the variation rate of XMP as a function, and from the obtained function, Compound A or a salt thereof or water thereof An electronic computer (computer) having a program that includes means for predicting the effective dose of the sum or the sensitivity to treatment with Compound A or a salt or hydrate thereof can be used.
- a patient with MDS further uses Compound A or a salt thereof or a hydrate thereof.
- a therapeutic agent for MDS in which the active ingredient is Compound A and a method for treating MDS in which the active ingredient is Compound A, comprising the step of predicting whether or not the treatment is sensitive.
- the process of predicting whether a patient with MDS of the present invention is sensitive to treatment with Compound A or a salt thereof or a hydrate thereof is as described above.
- a therapeutic agent for MDS whose active ingredient is Compound A is used to treat the patient.
- formulation adjuvants such as excipients, carriers and diluents usually used for formulation may be appropriately mixed.
- excipients such as excipients, carriers and diluents usually used for formulation
- formulation adjuvants such as excipients, carriers and diluents usually used for formulation may be appropriately mixed.
- excipients such as excipients, carriers and diluents usually used for formulation
- these are tablets, capsules, powders, syrups, granules, pills, suspensions, emulsions, solutions, powder formulations, suppositories, eye drops, nasal drops, ear drops, patches in accordance with conventional methods. It can be administered orally or parenterally in the form of an agent, ointment or injection.
- the administration method, the dose, and the number of administrations can be appropriately selected according to the age, weight and symptoms of the patient, but the variation rate (decrease rate) of XMP is 45 in a period of 40% or more of the treatment period. It is preferable to select a dose such that it is at least% (preferably at least 50%, more preferably at least 55%).
- UltrafreeMC-PLHCC 250 / pk for Metabolome Analysis (Human Metabolome Technologies) was used as the ultrafiltration tube.
- miVac Duo HV (Genevac) was used as a centrifugal evaporator.
- PBS, pH 7.4 (Thermo Fisher Scientific) was used as the PBS solution.
- Example 1 Cell Growth Inhibitory Action of Compound A 3/4 hydrate of Compound A was used as a test compound.
- SKM-1 Independent Administrative Institution JCRB Cell Bank
- SKM-1 Independent Administrative Institution JCRB Cell Bank
- the human myeloid leukemia cell line SKM-1 was seeded in a 96-well plate at 90 ⁇ L (5000 cells). 10 ⁇ L of a test compound in PBS (0, 1, 3, 10, 30, 100, 300, 1000, 3000 or 10000 ⁇ mol / L as compound A) was added to the plate and incubated at 37 ° C., 5% CO 2 for 72 hours. . CellTiter-Glo (Promega) was added in an amount of 100 ⁇ L to prepare a cell disruption solution. The relative value of the chemiluminescence intensity was determined with a plate reader.
- the IC 50 value of Compound A was 21.5 ⁇ mol / L (Compound A: 2.73 ⁇ g / mL). Compound A exhibited excellent growth inhibitory activity.
- Example 2 Prediction of drug concentration by PK / PD analysis (Pharmacokinetics / Pharmacodynamics analysis) (1) Antitumor effect of Compound A 3/4 hydrate of Compound A was used as a test compound. .
- SKM-1 cells were used as human myeloid leukemia cells. Human myeloid leukemia cells 5.0 ⁇ 10 6 cells were transplanted subcutaneously into the right flank of female nude mice (BALB / cAJcl-nu / nu) to form subcutaneous tumors. On the 9th day after transplantation, the groups were divided into 7 groups of 10 animals per group. Table 1 shows the group structure.
- Inhibition rate (%) (1 ⁇ (tumor volume 39 days after transplantation) / (tumor volume 39 days after transplantation in group 1)) ⁇ 100
- the tumor volume 39 days after transplantation is shown in FIG. Group 4, Group 5, Group 6 and Group 7 showed excellent antitumor effects.
- test compound A (10, 40, 80 and 160 mg / kg as Compound A) was orally administered to a non-fasting female mouse (BALB / cAJcl). After heparin blood was collected after 5 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, 5 hours, 10 hours or 24 hours, the concentration of compound A in plasma was measured using LC / MS / MS, and PK parameters ( tmax , Cmax , t1 / 2 and AUC 0-24 ) were calculated.
- tumor volume was statistically significantly suppressed in the group with Time above IC 50 of 40% or more. From this, it was predicted that it is important to maintain the drug concentration in human plasma at 2.73 ⁇ g / mL or more as Compound A in a period of 40% or more in the treatment period.
- Example 3 Quantification of XMP (1-1) 450 ⁇ L of blood collected from a healthy person by heparinized blood collection and 50 ⁇ L of PBS solution were added to a tube and stirred for 8 hours in a 37 ° C. incubator.
- 400 ⁇ L of methanol was added to 100 ⁇ L of the sample obtained in (1-1).
- 400 ⁇ L of chloroform was added, and after stirring with a vortex mixer, 120 ⁇ L of ultrapure water was added.
- the mixture was centrifuged at 4 ° C. and 10,000 ⁇ g for 15 minutes.
- the peak area ratio (B / A) of the sample was 1.2. Since the peak area ratio of the sample was 1 or more, it was determined that either one or both of the measurement conditions A and B had insufficient specificity (contamination was detected by overlapping).
- the peak area ratio (D / C) of the sample was 0.4. Since the peak area ratio of the sample was less than 1, the specificity was determined to be good. From this result, it was determined that the amount of XMP could be measured under measurement conditions C and D.
- the peak area ratio (B ′ / A ′) of the sample was 0.2. Since the peak area ratio of the sample was in the range of 0.1 to 0.3, the specificity was judged to be very good. From this result, it was determined that the amount of XMP could be measured under the measurement conditions A ′ and B ′.
- Example 4 XMP in human myeloid leukemia cells and XMP in blood cells of healthy humans
- a test compound 3/4 hydrate of Compound A was used.
- K562 RIKEN BiResource Center
- (1) 2 ⁇ 10 7 human myeloid leukemia cells were suspended in a culture flask containing 18 mL of RPMI1640 medium (Life Technologies).
- 2 mL of a test compound in PBS (0, 10, 100, or 1000 ⁇ g / mL as Compound A) was added to the flask and incubated at 37 ° C., 5% CO 2 for 24 hours. After centrifuging at 20 ° C.
- the supernatant was removed and suspended in 10 mL of PBS solution. Then, after centrifuging at 4 ° C. and 300 ⁇ g for 5 minutes, the supernatant was removed. After adding 1000 ⁇ L of methanol and stirring, 500 ⁇ L of ultrapure water was added and further stirred. Each was divided into two tubes of 600 ⁇ L, and 400 ⁇ L of chloroform was added to each tube and stirred, followed by centrifugation at 4 ° C. and 10,000 ⁇ g for 15 minutes. 400 ⁇ L of the aqueous phase was recovered, added to an ultrafiltration tube, and centrifuged at 9200 ⁇ g for 2 hours at 12 ° C. The filtrates were combined and dried under reduced pressure at 40 ° C. for 2 hours using a centrifugal evaporator. After drying, it was stored in a -80 ° C freezer.
- Example A and sample B Blood (sample A and sample B) collected from 2 healthy individuals by heparinized blood collection is added to each 1.5 mL tube at 450 ⁇ L, and the test compound in PBS (compound A, 0, 10, 30, 100, 300 or 1000 ⁇ g / 50 mL of each concentration was added to the tube. The mixture was stirred at 37 ° C. in an incubator for 8 hours. Thereafter, BD Pharm Lyse (555899, Becton, Dickinson and Company) was added to the blood (2 mL of BD Pharm Lyse was added to 0.2 mL of blood). After stirring with a vortex mixer, it was shielded from light and allowed to stand at room temperature for 15 minutes. After centrifuging at 20 ° C.
- the supernatant was removed and suspended in 10 mL of PBS solution. After centrifuging at 4 ° C. and 1000 ⁇ g for 5 minutes, the supernatant was removed, 1000 ⁇ L of methanol was added to each and stirred, and then 500 ⁇ L of ultrapure water was added and further stirred. Each was divided into two tubes of 600 ⁇ L, 400 ⁇ L of chloroform was added to each tube, stirred, and then centrifuged at 4 ° C. and 10,000 ⁇ g for 15 minutes. 400 ⁇ L of the aqueous phase was recovered, added to an ultrafiltration tube, and centrifuged at 9200 ⁇ g for 2 hours at 12 ° C. The filtrates were combined and dried under reduced pressure at 40 ° C. for 2 hours using a centrifugal evaporator. After drying, it was stored in a -80 ° C freezer.
- Example 5 XMP in blood cells after hemolysis and XMP in whole blood As a test compound, 3/4 hydrate of Compound A was used.
- Blood serum (sample A and sample B) collected from two healthy individuals by heparinized blood collection is added to each tube by 450 ⁇ L, and the test compound in PBS (compound A, 0, 10, 30, 100, 300, or 1000 ⁇ g / mL) was added to each tube at a concentration of 50 ⁇ L. The mixture was stirred at 37 ° C. in an incubator for 8 hours.
- BD Pharm Lyse (555899, Becton, Dickinson and Company) was added to the blood sample obtained in (1) (2 mL of BD Pharm Lyse was added to 0.2 mL of blood). After stirring with a vortex mixer, it was shielded from light and allowed to stand at room temperature for 15 minutes. After centrifugation at 1000 ° C. for 5 minutes at 20 ° C., the supernatant was removed and suspended in 10 mL of PBS. After centrifugation at 1000 ° C. for 5 minutes at 20 ° C., the supernatant was removed and suspended in 10 mL of PBS. After centrifuging at 4 ° C.
- the amount of XMP in the blood cells after hemolysis was almost equal to the amount of XMP in whole blood.
- the amount of XMP in whole blood reflected the amount of XMP in blood cells.
- Example 6 XMP in blood cells and XMP in bone marrow cells As a test compound, 3/4 hydrate of Compound A was used.
- Female mice (BALB / cAJcl) were orally administered 0.5% MC or 0.5% MC of the test compound (100 mg / kg as Compound A).
- Example 4 From the results of Example 4, Example 5 and Example 6, it was shown that the change in XMP in peripheral blood whole blood reflects the change in XMP in blasts of bone marrow of MDS patients.
- Example 7 XMP in human myeloid leukemia cells As a test compound, 3/4 hydrate of Compound A was used. SKM-1 was used as human myeloid leukemia cells.
- XMP amount (%) ((XMP peak area) / ((2′-fluoro-2′-deoxycytidine monophosphate peak area) ⁇ (cell count))) / ((XMP of untreated group) Peak area) / ((peak area of 2′-fluoro-2′-deoxycytidine monophosphate in the untreated group) ⁇ (number of cells in the untreated group))) ⁇ 100
- the XMP reduction rate (%) was calculated using the following formula.
- XMP reduction rate (%) 100-XMP amount (%) The results are shown in Table 14.
- the IC 50 value of Compound A in SKM-1 cells is 2.73 ⁇ g / mL (Example 1). Therefore, the XMP decrease rate around the IC 50 value was 43 to 55%.
- Example 7 From the results of Example 2 and Example 7, it can be predicted that the dose is effective when the XMP reduction rate is 45% or more in a period of 40% or more of the treatment period.
- Example 8 XMP, GMP and GTP in whole blood As a test compound, 3/4 hydrate of Compound A was used. (1) 450 ⁇ L of blood collected from a healthy person by heparinized blood collection was added to each tube, and a PBS solution of the test compound (0, 1, 10 or 100 ⁇ g / mL as compound A) was added to each tube at a concentration of 50 ⁇ L. The mixture was stirred at 37 ° C. in an incubator for 8 hours.
- the amount of XMP in whole blood decreased as the concentration of Compound A increased.
- the amount of GMP and GTP in the whole blood did not change much even when the concentration of Compound A was increased. From these results, it was shown that the therapeutic effect of Compound A could not be evaluated with metabolites downstream of XMP such as GMP or GTP, and the therapeutic effect of Compound A could be evaluated with XMP.
- Example 9 LC Gradient Conditions Samples were prepared in the same manner as described in Example 3 (1-1) and (1-2). The sample was dissolved in water and centrifuged, and then the supernatant was recovered and used as a measurement sample. The measurement sample and XMP standard were subjected to LC-MS / MS. The apparatus configuration and measurement conditions are shown below.
- the prediction method of the present invention can predict the effective dose of Compound A or a salt thereof or a hydrate thereof to a patient with MDS in a short time with a simple operation. In addition, it can be predicted in a short time by a simple operation whether or not a patient with MDS is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof.
- the measurement method of the present invention can accurately measure the amount of trace XMP contained in blood.
- the prediction apparatus of the present invention can be used for predicting the effective dose of Compound A or a salt thereof or a hydrate thereof to a patient with MDS in a short time with a simple operation. Moreover, it can be used for predicting in a short time by a simple operation whether or not a patient with MDS is sensitive to a treatment using Compound A or a salt thereof or a hydrate thereof.
- the treatment agent and the treatment method of the present invention are used for the treatment of patients with MDS predicted to be sensitive to treatment with Compound A or a salt thereof or a hydrate thereof, Compound A or a salt thereof or a salt thereof Unwanted administration to patients who are not sensitive to hydrates can be avoided.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Urology & Nephrology (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Biophysics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Bioinformatics & Computational Biology (AREA)
- Epidemiology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Ecology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
本発明はさらに、簡便な操作で、短時間に、MDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する方法を提供することを解決すべき課題とした。
本発明はさらに、簡便な操作で、短時間に、MDSの患者に対する化合物Aもしくはその塩またはその水和物の有効投与量を予測するための装置およびMDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測するための装置を提供することを解決すべき課題とした。
本発明はさらに、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測されたMDSの患者の処置に用いるための、処置剤および処置方法を提供することを解決すべき課題とした。
[1] MDSの患者に対する化合物Aもしくはその塩またはその水和物の有効投与量を予測する方法であって、
(a)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する工程と、
(b)(a)工程で得られるXMPの量からXMPの変動率を求める工程と、
(c)(b)工程で得られるXMPの変動率から化合物Aもしくはその塩またはその水和物の有効投与量を予測する工程と、を含む方法。
[3] XMPの量を測定する工程が、血液中に含まれる夾雑物とXMPとを区別して測定できる条件下で行う工程である、[1]または[2]に記載の方法。
[4] XMPの量を測定する工程が、マススペクトロメトリー(MS)法により測定する工程である、[1]から[3]のいずれか一に記載の方法。
(a1)MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるXMPのイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるXMPのイオン強度比が所定の範囲内にあることを確認する工程と、を含む工程である、[4]に記載の方法。
[6] (a1)工程が、LC-MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程である、[5]に記載の方法。
[7] 液体クロマトグラフィー(LC)に用いる移動相のpH値が、7~11である、[6]に記載の方法。
[8] LCに用いる移動相の塩濃度が、5~300mmol/Lである、[6]または[7]に記載の方法。
[9] LCに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、[6]から[8]のいずれか一に記載の方法。
[11] 異なる2つの測定条件が、MS法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、[5]から[10]のいずれか一に記載の方法。
[12] (c)工程が、
(c1)化合物Aもしくはその塩またはその水和物の投与後の時間または接触の時間と(b)工程で得られるXMPの変動率との関係を関数で表す工程と、
(c2)(c1)工程で得られる関数から化合物Aもしくはその塩またはその水和物の有効投与量を予測する工程と、を含む工程である、[1]から[11]のいずれか一に記載の方法。
[13] (c2)工程が、XMPの変動率が所定の処置期間において所定の変動率以上となるような化合物Aもしくはその塩またはその水和物の投与量が、有効投与量であると予測する工程を含む工程である、[12]に記載の方法。
[14] (c2)工程が、XMPの変動率が所定の処置期間中の所定の割合以上の期間において所定の変動率以上となるような化合物Aもしくはその塩またはその水和物の投与量が、有効投与量であると予測する工程を含む工程である、[12]に記載の方法。
(a)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する工程と、
(b)(a)工程で得られるXMPの量からXMPの変動率を求める工程と、
(c)(b)工程で得られるXMPの変動率から、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む方法。
[17] XMPの量を測定する工程が、血液中に含まれる夾雑物とXMPとを区別して測定できる条件下で行う工程である、[15]または[16]に記載の方法。
[18] XMPの量を測定する工程が、MS法により測定する工程である、[15]から[17]のいずれか一に記載の方法。
(a1)MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるXMPのイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるXMPのイオン強度比が所定の範囲内にあることを確認する工程と、を含む工程である、[18]に記載の方法。
[20] (a1)工程が、LC-MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程である、[19]に記載の方法。
[21] LCに用いる移動相のpH値が、7~11である、[20]に記載の方法。
[22] LCに用いる移動相の塩濃度が、5~300mmol/Lである、[20]または[21]に記載の方法。
[23] LCに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、[20]から[22]のいずれか一に記載の方法。
[25] 異なる2つの測定条件が、MS法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、[19]から[24]のいずれか一に記載の方法。
[26] (c)工程が、
(c1)化合物Aもしくはその塩またはその水和物の投与後の時間または接触の時間と(b)工程で得られるXMPの変動率との関係を関数で表す工程と、
(c2)(c1)工程で得られる関数から、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程である、[15]から[25]のいずれか一に記載の方法。
[27] (c2)工程が、XMPの変動率が所定の処置期間において所定の変動率以上である場合に、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測する工程を含む工程である、[26]に記載の方法。
[28] (c2)工程が、XMPの変動率が所定の処置期間中の所定の割合以上の期間において所定の変動率以上である場合に、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測する工程を含む工程である、[26]に記載の方法。
(a1)MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるXMPのイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるXMPのイオン強度比が所定の範囲内にあることを確認する工程と、を含む方法。
[30] (a1)工程が、LC-MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程である、[29]に記載の方法。
[32] LCに用いる移動相の塩濃度が、5~300mmol/Lである、[30]または[31]に記載の方法。
[33] LCに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、[30]から[32]のいずれか一に記載の方法。
[34] LCが、HILICである、[30]から[33]のいずれか一に記載の方法。
[35] 異なる2つの測定条件が、MS法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、[29]から[34]のいずれか一に記載の方法。
(A)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する手段と、
(B)(A)に記載の手段により得られるXMPの量からXMPの変動率を求める手段と、
(C)(B)に記載の手段により得られるXMPの変動率から化合物Aもしくはその塩またはその水和物の有効投与量を予測する手段と、を含む装置。
[37] XMPの量を測定する手段が、MS装置を用いる手段である、[36]に記載の装置。
[38] MS装置が、LC装置と連結されたMS装置(LC-MS装置)である、[37]に記載の装置。
(A)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する手段と、
(B)(A)に記載の手段により得られるXMPの量からXMPの変動率を求める手段と、
(C)(B)に記載の手段により得られるXMPの変動率から、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する手段と、を含む装置。
[40] (A)に記載の手段が、MS装置を用いる手段である、[39]に記載の装置。
[41] MS装置が、LC装置と連結されたMS装置(LC-MS装置)である、[40]に記載の装置。
(a)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する工程と、
(b)(a)工程で得られるXMPの量からXMPの変動率を求める工程と、
(c)(b)工程で得られるXMPの変動率から、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程であって、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し感受性であると予測された患者である、処置剤。
(a)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する工程と、
(b)(a)工程で得られるXMPの量からXMPの変動率を求める工程と、
(c)(b)工程で得られるXMPの変動率から、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程であって、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し感受性であると予測された患者である、処置方法。
本発明の方法および処置剤は、常法に従って、MDSの患者の処置用のキットに応用することができる。
処置とは、疾患に対する予防または治療などを意味する。
処置剤とは、疾患に対して予防または治療などの目的で供せられる物質を意味する。
処置期間とは、処置を施す期間を意味する。
イオン強度には、例えば、LC-MS法におけるクロマトグラムから得られるピークエリアまたはMSスペクトル上の該当するイオンの強度が含まれる。
XMP標準物質とは、血液に含まれるXMPを測定するときに用いられる標品を意味する。XMP標準物質としては、市販のキサントシン一リン酸のナトリウム塩などが挙げられる。
本発明による方法においては、(a)工程として、(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られる血液に含まれるXMPの量とを測定する。
第二の態様は、化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量および化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量を測定する態様である。
試料中の成分の分離方法としては、HILIC、逆相クロマトグラフィー(RP)、イオンペア試薬を用いたクロマトグラフィーまたはイオン交換クロマトグラフィーなどを使用することができる。必要な分離と性能が得られ、MSへの導入が可能な移動相組成で溶出できる限り、モードおよび測定条件は特に限定されない。また、キャピラリー電気泳動またはガスクロマトグラフィーなどの分離法を用いることもできる。
(a1)MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるXMPのイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるXMPのイオン強度比が所定の範囲内にあることを確認する工程と、
を含む工程である。
変動率(%)=(1-(化合物Aもしくはその塩またはその水和物の投与後のXMPの量/化合物Aもしくはその塩またはその水和物の投与前のXMPの量))×100
または
変動率(%)=(1-(化合物Aもしくはその塩またはその水和物を接触させた後のXMPの量/化合物Aもしくはその塩またはその水和物の接触前のXMPの量))×100
なお、XMPの量は、単位血液量あたりの値を用いることもできるが、単位血液細胞数あたりの値を用いることが好ましい。
また、(c2)工程において、XMPの変動率が所定の処置期間において、XMPが所定の変動率(減少率)以上となる場合に、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測する。
所定の処置期間中の所定の割合以上の期間とは、生物種に応じて適宜選択することができるが、例えば、所定の処置期間中の40%以上の期間とすることができる。
所定の変動率とは、生物種に応じて適宜選択することができるが、例えば、45%以上(好ましくは50%以上、より好ましくは55%以上)とすることができる。
また、例えば、化合物Aもしくはその塩またはその水和物を用いる処置期間の40%以上の期間において、XMPの変動率(減少率)が45%以上(好ましくは50%以上、より好ましくは55%以上)となる場合に、患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測することができる。感受性であると予測された患者に対して、化合物Aもしくはその塩またはその水和物を投与すればよい。
本発明による血液に含まれるXMPの量を測定する方法は、
(a1)MS法により、異なる2つの測定条件で血液に含まれるXMPを測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるXMPのイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるXMPのイオン強度比が所定の範囲内にあることを確認する工程と、
を含む方法である。
(a1)工程においては、LCにより試料の分離を行い、MS法によりXMPを検出すること(LC-MS法)が好ましい。
(a11)MS法により、異なる2つの測定条件でXMP標準物質および血液に含まれるXMPを測定する工程と、
(a12)(a11)工程で得られる異なる2つの測定条件でのXMP標準物質のイオン強度比および血液に含まれるXMPのイオン強度比を求める工程と、
(a13)(a12)工程で得られるXMP標準物質のイオン強度比と血液に含まれるXMPのイオン強度比とを比較する工程と、
を含む方法を用いることができる。
(a12)工程においては、(a11)工程で得られる異なる2つの測定条件でのXMP標準物質のイオン強度比および血液に含まれるXMPのイオン強度比を求め、(a13)工程においては、(a12)工程で得られるXMP標準物質のイオン強度比と血液に含まれるXMPのイオン強度比とを比較する。
ここで、血液試料のイオン強度比とXMP標準物質のイオン強度比とが、所定の範囲内である場合には、血液中のXMPを高い特異性で検出できていると判断することができる。反対に、血液試料のイオン強度比とXMP標準物質のイオン強度比とが所定の範囲外である場合には、比を取った条件のどちらかまたは両方で選択性が不十分であったと判断する。
移動相AのpH値は、好ましくは7~11であり、より好ましくは8~11であり、さらに好ましくは8.4~10.4であり、特に好ましくは9.0~9.8である。
移動相AのpH値は、使用するカラムの種類に応じて設定すればよい。
LCに用いる移動相の塩濃度は、好ましくは5~300mmol/Lであり、より好ましくは20~200mmol/Lである。
LC-MS測定では、通常、高い感度を出すために塩濃度を低く(例えば、10mmol/Lなど)するが、本発明においては、通常より高い塩濃度を採用することによって、XMPと夾雑物とを分離することが可能になった。
LCに用いる移動相Aとしては、重炭酸アンモニウム水溶液を含むことが好ましい。
イオン強度比=(他の測定条件のイオンの強度)/(365.0(m/z)をプリカーサーイオンとした際の97.0(m/z)のプロダクトイオンの強度)
ここで、他の条件のイオンの強度として、例えば、365.0(m/z)をプリカーサーイオンとした際の153.0(m/z)のプロダクトイオンの強度または365.0(m/z)をプリカーサーイオンとした際の213.0(m/z)のプロダクトイオンの強度を用いることができる。
また、XMP標準物質のイオン強度比と血液に含まれるXMPのイオン強度比とを比較する場合の所定の範囲内とは、血液に含まれるXMPのイオン強度比が、XMP標準物質のイオン強度比の0.5~1.5である場合である。
本発明は、さらに、MDSの患者に対する化合物Aもしくはその塩またはその水和物の有効投与量を予測するための装置およびMDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測するための装置を提供する。本発明の装置は、
(A)(i)化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるXMPの量と、(ii)化合物Aもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるXMPの量、または化合物Aもしくはその塩またはその水和物を投与する前の患者から採取した血液に化合物Aもしくはその塩またはその水和物を接触させて得られた血液に含まれるXMPの量とを測定する手段と、
(B)(A)に記載の手段により得られるXMPの量からXMPの変動率を求める手段と、
(C)(B)に記載の手段により得られるXMPの変動率から化合物Aもしくはその塩またはその水和物の有効投与量を予測する手段または患者が化合物Aもしくはその塩またはその水和物を用いる処置に対し感受性であるか否かを予測する手段と、
を含む装置である。
試料導入部は、試料をMS装置内に導入する部位である。例えば、MS装置を、高速液体クロマトグラフィー装置、キャピラリー電気泳動またはガスクロマトグラフィー装置に連結し、これらの装置から試料をMS装置に導入することもできる。
データ処理部では、得られたデータからマススペクトルを作製する部位である。
本発明は、さらに、MDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程を含む、有効成分が化合物AであるMDSの処置剤、および有効成分が化合物AであるMDSの処置方法を提供する。
本発明のMDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程は、上記のとおりである。
MDSの患者が、化合物Aもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測された場合、有効成分が化合物AであるMDSの処置剤が患者の処置に用いられる。
限外ろ過チューブは、UltrafreeMC-PLHCC 250/pk for Metabolome Analysis(Human Metabolome Technologies社)を用いた。
遠心エバポレーターは、miVac Duo HV(Genevac社)を用いた。
PBS溶液は、PBS,pH7.4(サーモフィッシャーサイエンティフィック社)を用いた。
試験化合物として、化合物Aの3/4水和物を用いた。
ヒト骨髄性白血病細胞株として、SKM-1(独立行政法人医薬基盤研究所JCRB細胞バンク)を用いた。
プレートリーダーにより化学発光強度の相対値を求めた。
(1)化合物Aの抗腫瘍効果
試験化合物として、化合物Aの3/4水和物を用いた。
ヒト骨髄性白血病細胞として、SKM-1細胞を用いた。
ヒト骨髄性白血病細胞5.0×106個を雌性ヌードマウス(BALB/cAJcl-nu/nu)の右側腹部皮下に移植し、皮下腫瘍を形成した。移植後9日目に群分けを行い、1群10匹として7群を作成した。群構成を表1に示す。移植後10日目から対照溶媒(0.5w/v%メチルセルロース400水溶液、以下、「0.5%MC」とも称する。)または試験化合物を間欠経口投与(2日間投与した後4日間休薬を1クールとして、合計5クール)し、移植後39日目の腫瘍体積を測定し、抗腫瘍効果を評価した。抑制率は以下の式で算出した。
抑制率(%)=(1-(移植後39日目の腫瘍体積)/(群1の移植後39日目の腫瘍体積))×100

群4、群5、群6および群7は、優れた抗腫瘍効果を示した。
非絶食下の雌性マウス(BALB/cAJcl)に試験化合物(化合物Aとして、10、40、80および160mg/kg)を単回経口投与した。5分間、15分間、30分間、1時間、2時間、5時間、10時間または24時間後にヘパリン採血後、血漿中の化合物Aの濃度をLC/MS/MSを用いて測定し、PKパラメータ(tmax、Cmax、t1/2およびAUC0-24)を算出した。
化合物Aを単回経口投与したときの血漿中濃度推移より、two-compartment modelの薬物動態パラメータであるV1/F、K01、K10、K12およびK21を算出した。これらのパラメータを用いて、様々な用法用量における反復投与時の化合物Aの血漿中濃度推移を予測した。なお、10mg/kgから80mg/kgまでは線形性が認められたため、10mg/kg投与のパラメータを用いて20、40および80mg/kgの反復投与時の予測を行った。120および240mg/kgについては160mg/kgのパラメータを用いて予測を行った。得られた化合物Aの血漿中濃度推移から、Cmax(ng/mL)、AUC0-48 (ng・時間/mL)およびTime above IC50(%、IC50値:21.5μmol/L(化合物Aとして、2.73μg/mL))を算出した。PKパラメータ(Cmax、Time above IC50およびAUC0-48)をX軸にプロットし、腫瘍体積の抑制率をY軸にプロットした。結果を図2、3および4に示す。
(1-1)ヘパリン添加採血により健常人から採取した血液450μLおよびPBS溶液50μLをチューブに加え、37℃のインキュベーターで8時間攪拌した。
(1-2)(1-1)で得られた試料100μLにメタノール400μLを添加した。ボルテックスミキサーで攪拌後、クロロホルム400μLを添加し、ボルテックスミキサーで攪拌後、超純水120μLを添加した。ボルテックスミキサーで攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を併せ、遠心エバポレーターを用いて40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(1-3)試料を水に溶解し、遠心分離した後、上清を回収して測定試料とした。測定試料およびXMP標準物質をLC-MS/MSに供した。XMP標準物質は、XMP(Xanthosine-5'-monophosphate, Sodium salt)(ジェナバイオサイエンス社)を用いた。装置構成および測定条件を以下に示す。
LC:UFLC(島津製作所)
MS:QTRAP 5500(エービー・サイエックス社)
(LC)
カラム:SeQuant ZIC-pHILIC 5μm, polymeric PEEK 150×2.1 mm metal-free HPLC column (メルク社)
カラムオーブン温度:40℃
移動相A:50mmol/L 重炭酸アンモニウム(25質量%アンモニア水溶液でpH9.4に調整)
移動相B:アセトニトリル
送液条件:0.3mL/分
グラジエントサイクルを表2に示す。表2中の%は体積%を示す。
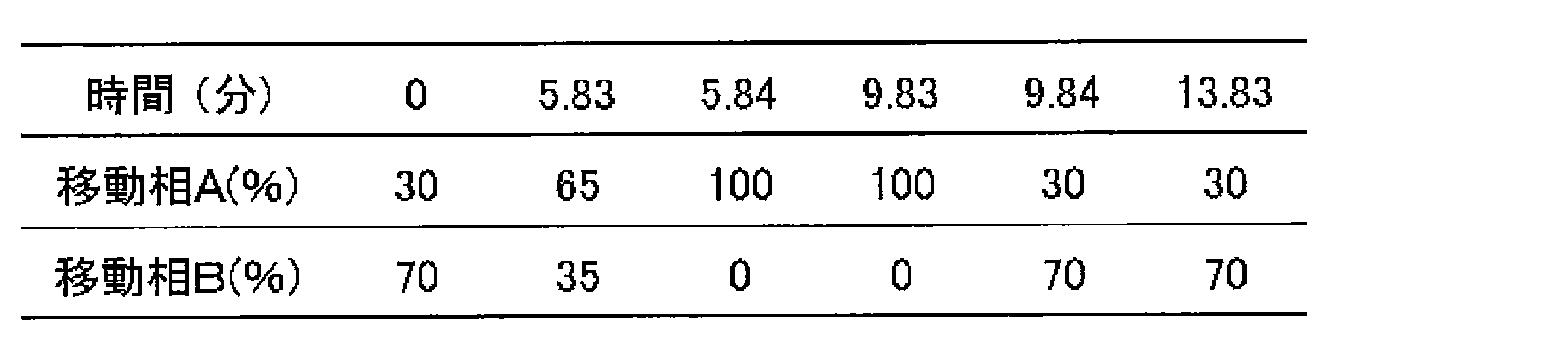
イオン化:ESI positive(Electrospray ionization positive)
スキャンモード:MRM (Multiple Reaction Monitoring)
測定条件を表3に示す。

試料のクロマトグラムでは複数の近接ピークが見られた。XMP標準物質のピークから試料のXMPのピークを推定し、面積を算出した。特異性を検証するため、測定条件Aにおけるピークエリアおよび測定条件Bにおけるピークエリアの比(B/A)を算出した。結果を表4に示す。

測定条件CおよびDで、XMP標準物質および(3)で得られた試料を測定した。測定条件CおよびDを表5に示す。XMP標準物質および試料の測定結果を図6および表6に示す。


この結果から、測定条件CおよびDにより、XMPの量を測定できると判定した。

特異性を検証するため、測定条件A’におけるピークエリアおよび測定条件B’におけるピークエリアの比(B’/A’)を算出した。結果を表8に示す。

この結果から、測定条件A’およびB’により、XMPの量を測定できると判定した。
試験化合物として、化合物Aの3/4水和物を用いた。
ヒト骨髄性白血病細胞として、K562(独立行政法人理化学研究所バイリソースセンター)を用いた。
(1)
ヒト骨髄性白血病細胞2×107個をRPMI1640培地(Life Technologies社)18mLが入った培養フラスコに懸濁した。試験化合物のPBS溶液(化合物Aとして、0、10、100または1000μg/mL)2mLをフラスコに加え、37℃、5%CO2条件下で24時間インキュベートした。20℃、300×gで3分間遠心分離した後、上清を除去し、PBS溶液10mLに懸濁した。その後、4℃、300×gで5分間遠心分離した後、上清を除去した。メタノール1000μLを添加して攪拌した後、超純水500μLを添加し、さらに攪拌した。600μLずつ2つのチューブに分割し、それぞれにクロロホルム400μLを添加して攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を併せ、遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
ヘパリン添加採血により健常人2人から採取した血液(検体Aおよび検体B)を450μLずつ1.5mLチューブに加え、試験化合物のPBS溶液(化合物Aとして、0、10、30、100、300または1000μg/mL)を各濃度それぞれ50μLずつチューブに加えた。37℃でインキュベーターで8時間攪拌した。その後、血液にBD Pharm Lyse(555899、Becton, Dickinson and Company社)を加えた(血液0.2mLに対し、BD Pharm Lyse2mLを添加した)。ボルテックスミキサーで攪拌後、遮光し、室温で15分静置した。20℃、1000×gで5分間遠心分離した後、上清を除去し、PBS溶液10mLに懸濁した。4℃、1000×gで5分間遠心分離した後、上清を除去し、それぞれにメタノール1000μLを添加し、攪拌した後、超純水500μLを添加し、さらに攪拌した。600μLずつ2つのチューブに分割し、それぞれにクロロホルム400μLを添加し、攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を併せ、遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(1)および(2)で保存した試料を室温に戻し、それぞれに超純水40μLを加えて攪拌し、4℃、21500×gで5分間遠心分離した。上清35μLをバイアルに移し、4℃で保存後、実施例3(3)に記載したLC-MS/MS測定装置、LC条件およびMS/MS条件で、試料中のXMPを測定した。
化合物A無処理群のXMPの値(ピークエリア値)を100%とし、XMPの量(%)を算出した。
結果を表9に示す。

試験化合物として、化合物Aの3/4水和物を用いた。
(1)
ヘパリン添加採血により健常人2人から採取した血液(検体Aおよび検体B)を450μLずつチューブに加え、試験化合物のPBS溶液(化合物Aとして、0、10、30、100、300または1000μg/mL)を各濃度それぞれ50μLずつチューブに加えた。37℃でインキュベーターで8時間攪拌した。
(1)で得られた血液サンプルにBD Pharm Lyse(555899、Becton, Dickinson and Company社)を加えた(血液0.2mLに対し、BD Pharm Lyse2mLを添加した。)。ボルテックスミキサーで攪拌後、遮光し、室温で15分静置した。20℃、1000×gで5分間遠心分離した後、上清を除去し、PBS10mLに懸濁した。20℃、1000×gで5分間遠心分離した後、上清を除去し、PBS10mLに懸濁した。4℃、1000×gで5分間遠心分離した後、上清を除去し、それぞれにメタノール1000μLを添加し、攪拌した後、超純水500μLを添加し、さらに攪拌した。600μLずつ2つのチューブに分割し、それぞれにクロロホルム400μLを添加し、攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブ(UltrafreeMC-PLHCC 250/pk for Metabolome Analysis(Human Metabolome Technologies社)))に添加し、12℃、9200×gで2時間遠心分離した。ろ液を併せ、遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(1)で得られた血液サンプル100μLに400μLのメタノールを添加し、攪拌した後、400μLのクロロホルムを添加し、さらに攪拌した。120μLの超純水を添加し、攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(2-1)および(2-2)で得られた試料を室温に戻し、それぞれに超純水40μL加えて攪拌し、4℃、21500×gで5分間遠心分離した。上清35μLをバイアルに移し、4℃で保存後、実施例3(3)に記載したLC-MS/MS測定装置、LC条件およびMS/MS条件で、試料中のXMPを測定した。
化合物A無処理群でのXMPの量を100%とし、XMPの量(%)を算出した。
結果を表10に示す。

試験化合物として、化合物Aの3/4水和物を用いた。
(1)
雌性マウス(BALB/cAJcl)に0.5%MCまたは試験化合物の0.5%MC(化合物Aとして100mg/kg)を経口投与した。
投与後から1、4、8または24時間後にイソフルラン吸入による麻酔下で後大静脈からエチレンジアミン四酢酸ジナトリウム塩で処理したポリプロピレン製注射筒および25ゲージの注射針を用いて全採血した。血液にBD Pharm Lyse(555899、Becton, Dickinson and Company社)を加えた(0.2mLの血液に対し、2mLのBD Pharm Lyseを添加した。)。ボルテックスミキサーで攪拌後、遮光し、室温で15分静置した。20℃、1000×gで5分間遠心後、上清を除去し、PBS溶液10mLに懸濁した。4℃、1000×gで5分間遠心分離した後、上清を除去し、それぞれに1000μLのメタノールを添加して攪拌した後、500μLの超純水を添加し、さらに攪拌した。600μLずつ2チューブに分割し、それぞれに400μLのクロロホルムを添加して攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心した。ろ液を併せ、遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
投与後から1、4、8または24時間後にイソフルラン吸入による麻酔下で左右大腿骨を採取、骨端を切断し、4℃、3000rpmで1分間遠心分離し、骨髄細胞を回収した。1mLのPBSに懸濁し、細胞数を計数した。4℃、1000×gで5分間遠心分離した後、上清を除去し、それぞれに1000μLのメタノールを添加して攪拌した後、500μLの超純水を添加し、さらに攪拌した。600μLずつ2つのチューブに分割しそれぞれに400μLのクロロホルムを添加して攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心した。ろ液を併せ、遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(2-1)および(2-2)で得られた試料を室温に戻し、超純水を溶血サンプルに40μL、骨髄サンプルに150μL加えた。攪拌後、4℃、21500×gで5分間遠心した。溶血サンプルは上清35μLを、骨髄サンプルは上清140μLをバイアルに移し、4℃で保存後、実施例3(3)に記載したLC-MS/MS測定装置、LC条件およびMS/MS条件で、試料中のXMPを測定した。
化合物A無処理群でのXMPの量を100%とし、XMPの量(%)を算出した。
結果を表11に示す。

試験化合物として、化合物Aの3/4水和物を用いた。
ヒト骨髄性白血病細胞として、SKM-1を用いた。
ヒト骨髄性白血病細胞2×107個をRPMI1640培地(Life Technologies社)18mLが入った培養フラスコに懸濁した。試験化合物のPBS溶液(化合物Aとして、0、10、30、100、300または1000μg/mL)2mLをフラスコに加え、37℃、5%CO2条件下で24時間インキュベートした。20℃、300×gで3分間遠心分離した後、上清を除去し、PBS溶液10mLに懸濁した。10 μLを分取し、細胞数をTC10(BioRad社)を用いて計測した。300×gで5分間遠心分離した後、上清を除去した。メタノール1000μLを添加して攪拌した後、超純水500μLを添加し、さらに攪拌した。600μLずつ2つのチューブに分割し、それぞれに400μLのクロロホルムを添加して攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を1つのチューブにまとめ、遠心エバポレーターを用いて、40℃で約2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(1)で得られた試料を室温に戻し、それぞれに、4ng/μL2'-フルオロ-2'-デオキシシチジン1リン酸を含有した5mmol/Lギ酸アンモニウム50μLを加えて攪拌した。上清12.5μLをバイアルに移し、以下の測定条件で試料中のXMPを測定した。
LC:UFLC(島津製作所)
MS:QTRAP3200(エービー・サイエックス社)
(LC)
カラム:SeQuant ZIC-pHILIC 5μm, polymeric PEEK 150×2.1mm metal-free HPLC column (メルク社)
カラムオーブン温度:40℃
移動相A:10mmol/L重炭酸アンモニウム(25質量%アンモニア水溶液でpH9.4に調整)
移動相B:アセトニトリル
送液条件:0.5mL/分
グラジエントサイクルを表12に示す。表12中の%は体積%を示す。

イオン化:ESI positive
スキャンモード:MRM
測定条件を表13に示す。

化合物A無処理群のXMPの値を100%とし、XMP量(%)を以下の式で算出した。
XMPの量(%)=((XMPのピークエリア)/((2’-フルオロ-2’-デオキシシチジン1リン酸のピーク面積)×(細胞数)))/((無処理群のXMPのピークエリア)/((無処理群の2’-フルオロ-2’-デオキシシチジン1リン酸のピークエリア)×(無処理群の細胞数)))×100
以下の式でXMPの減少率(%)を算出した。
XMPの減少率(%)=100-XMPの量(%)
結果を表14に示す。
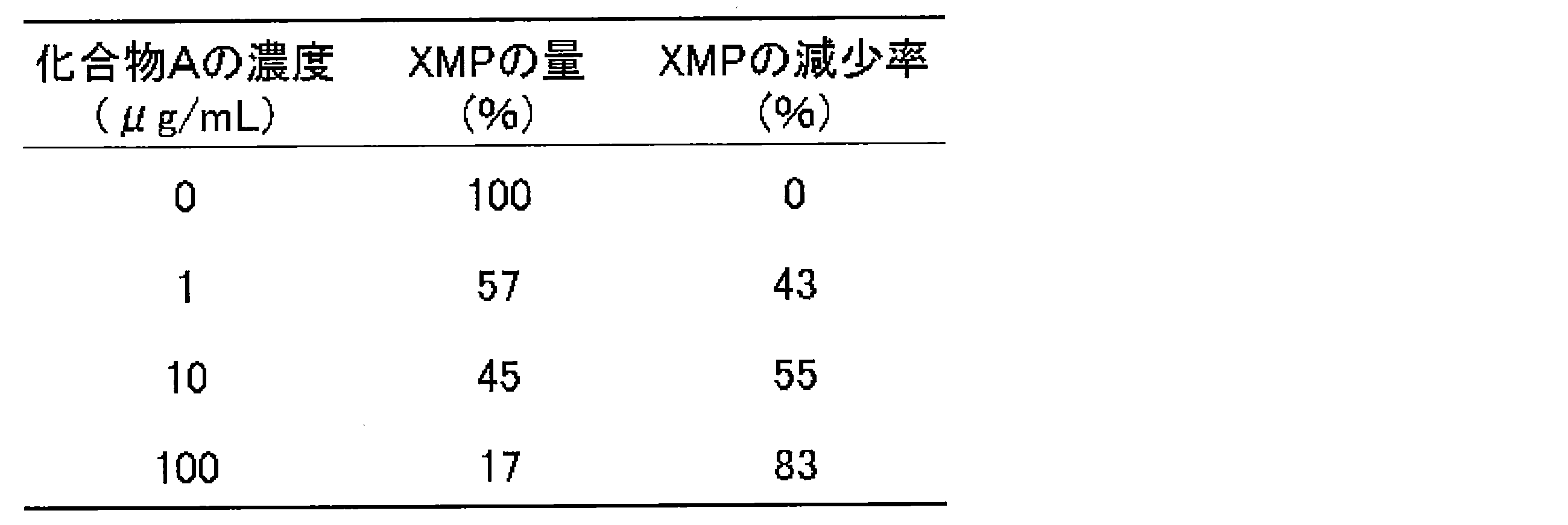
試験化合物として、化合物Aの3/4水和物を用いた。
(1)
ヘパリン添加採血により健常人から採取した血液を450μLずつチューブに加え、試験化合物のPBS溶液(化合物Aとして、0、1、10または100μg/mL)を各濃度それぞれ50μLずつチューブに加えた。37℃でインキュベーターで8時間攪拌した。
(1)で得られた血液サンプル100μLに400μLのメタノールを添加し、攪拌した後、400μLのクロロホルムを添加し、さらに攪拌した。120μLの超純水を添加し、攪拌した後、4℃、10000×gで15分間遠心分離した。水相400μLを回収し、限外ろ過チューブに添加し、12℃、9200×gで2時間遠心分離した。ろ液を遠心エバポレーターを用いて、40℃で2時間減圧乾燥した。乾燥後、-80℃のフリーザで保存した。
(2)で得られた試料を室温に戻し、それぞれに超純水40μL加えて攪拌し、4℃、21500×gで5分間遠心分離した。上清35μLをバイアルに移し、4℃で保存後、実施例3(3)に記載したLC-MS/MS測定装置およびLC条件ならびに表15に示すMS/MS条件で、試料中のXMP、GMPおよびGTPを測定した。

化合物A無処理群のXMP、GMPおよびGTPの値(Peak Area値)を100%とし、XMP、GMPおよびGTPの量(%)を算出した。結果を表16に示す。

実施例3(1-1)および(1-2)に記載の方法と同様にして試料を作製した。試料を水に溶解し、遠心分離した後、上清を回収して測定試料とした。測定試料およびXMP標準物質をLC-MS/MSに供した。装置構成および測定条件を以下に示す。
LC:UFLC(島津製作所)
MS:QTRAP 5500(エービー・サイエックス社)
(LC)
カラム:SeQuant ZIC-pHILIC 5μm, polymeric PEEK 150×2.1 mm metal-free HPLC column (メルク社)
カラムオーブン温度:40℃
移動相A:50mmol/L 重炭酸アンモニウム(25質量%アンモニア水溶液でpH9.4に調整)
移動相B:アセトニトリル
送液条件:0.3mL/分
注入量:2μL(条件a)、5μL(条件b)
条件aおよびbのグラジエントサイクルを表17および18に示す。表17及び表18中の%は体積%を示す。

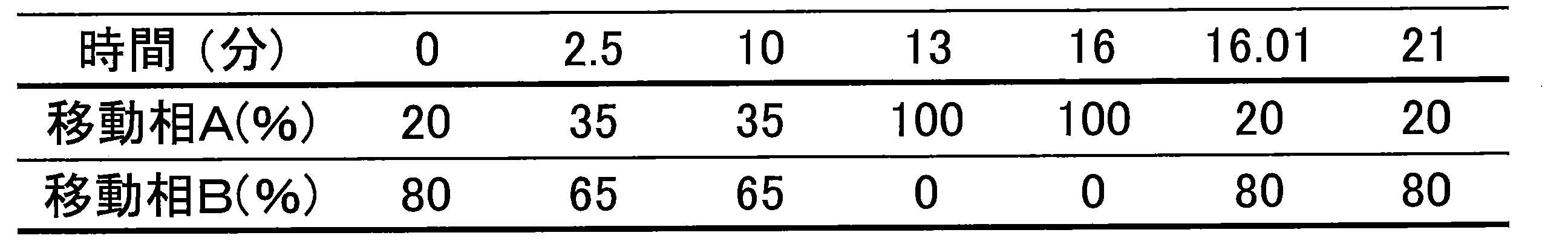
イオン化:ESI positive
スキャンモード:MRM
測定条件を表19に示す。

LC条件bのXMP溶出でアイソクラッティック法を用いた場合には、LC条件aのグラジエント法を用いた場合に比べ、試料中のXMPと夾雑物との分離度が向上し、試料中のXMPをより高い特異性で検出できることが確認された。さらに、LC条件bでは、サンプル注入量をLC条件aの2.5倍まで増加させることができた。
Claims (43)
- 骨髄異形成症候群の患者に対する5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の有効投与量を予測する方法であって、
(a)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する工程と、
(b)(a)工程で得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める工程と、
(c)(b)工程で得られるキサントシン一リン酸の変動率から5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の有効投与量を予測する工程と、を含む方法。 - 血液が、末梢血である、請求項1に記載の方法。
- キサントシン一リン酸の量を測定する工程が、血液に含まれる夾雑物とキサントシン一リン酸とを区別して測定できる条件下で行う工程である、請求項1または2に記載の方法。
- キサントシン一リン酸の量を測定する工程が、マススペクトロメトリー法により測定する工程である、請求項1から3のいずれか一項に記載の方法。
- キサントシン一リン酸の量を測定する工程が、
(a1)マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるキサントシン一リン酸のイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるキサントシン一リン酸のイオン強度比が所定の範囲内にあることを確認する工程と、を含む工程である、請求項4に記載の方法。 - (a1)工程が、液体クロマトグラフィー-マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程である、請求項5に記載の方法。
- 液体クロマトグラフィーに用いる移動相のpH値が、7~11である、請求項6に記載の方法。
- 液体クロマトグラフィーに用いる移動相の塩濃度が、5~300mmol/Lである、請求項6または7に記載の方法。
- 液体クロマトグラフィーに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、請求項6から8のいずれか一項に記載の方法。
- 液体クロマトグラフィーが、親水性相互作用クロマトグラフィーである、請求項6から9のいずれか一項に記載の方法。
- 異なる2つの測定条件が、マススペクトロメトリー法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、請求項5から10のいずれか一項に記載の方法。
- (c)工程が、
(c1)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の投与後の時間または接触の時間と(b)工程で得られるキサントシン一リン酸の変動率との関係を関数で表す工程と、
(c2)(c1)工程で得られる関数から5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の有効投与量を予測する工程と、を含む工程である、請求項1から11のいずれか一項に記載の方法。 - (c2)工程が、キサントシン一リン酸の変動率が所定の処置期間において所定の変動率以上となるような5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の投与量が、有効投与量であると予測する工程を含む工程である、請求項12に記載の方法。
- (c2)工程が、キサントシン一リン酸の変動率が所定の処置期間中の所定の割合以上の期間において所定の変動率以上となるような5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の投与量が、有効投与量であると予測する工程を含む工程である、請求項12に記載の方法。
- 骨髄異形成症候群の患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する方法であって、
(a)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する工程と、
(b)(a)工程で得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める工程と、
(c)(b)工程で得られるキサントシン一リン酸の変動率から、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む方法。 - 血液が、末梢血である、請求項15に記載の方法。
- キサントシン一リン酸の量を測定する工程が、血液中に含まれる夾雑物とキサントシン一リン酸とを区別して測定できる条件下で行う工程である、請求項15または16に記載の方法。
- キサントシン一リン酸の量を測定する工程が、マススペクトロメトリー法により測定する工程である、請求項15から17のいずれか一項に記載の方法。
- キサントシン一リン酸の量を測定する工程が、
(a1)マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるキサントシン一リン酸のイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるキサントシン一リン酸のイオン強度比が所定の範囲内にあることを確認する工程と、を含む工程である、請求項18に記載の方法。 - (a1)工程が、液体クロマトグラフィー-マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程である、請求項19に記載の方法。
- 液体クロマトグラフィーに用いる移動相のpH値が、7~11である、請求項20に記載の方法。
- 液体クロマトグラフィーに用いる移動相の塩濃度が、5~300mmol/Lである、請求項20または21に記載の方法。
- 液体クロマトグラフィーに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、請求項20から22のいずれか一項に記載の方法。
- 液体クロマトグラフィーが、親水性相互作用クロマトグラフィーである、請求項20から23のいずれか一項に記載の方法。
- 異なる2つの測定条件が、マススペクトロメトリー法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、請求項19から24のいずれか一項に記載の方法。
- (c)工程が、
(c1)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の投与後の時間または接触の時間と(b)工程で得られるキサントシン一リン酸の変動率との関係を関数で表す工程と、
(c2)(c1)工程で得られる関数から、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程である、請求項15から25のいずれか一項に記載の方法。 - (c2)工程が、キサントシン一リン酸の変動率が所定の処置期間において所定の変動率以上である場合に、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測する工程を含む工程である、請求項26に記載の方法。
- (c2)工程が、キサントシン一リン酸の変動率が所定の処置期間中の所定の割合以上の期間において所定の変動率以上である場合に、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であると予測する工程を含む工程である、請求項26に記載の方法。
- 血液に含まれるキサントシン一リン酸の量を測定する方法であって、
(a1)マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程と、
(a2)(a1)工程で得られる異なる2つの測定条件での血液に含まれるキサントシン一リン酸のイオン強度比を求める工程と、
(a3)(a2)工程で得られる血液に含まれるキサントシン一リン酸のイオン強度比が所定の範囲内にあることを確認する工程と、を含む方法。 - (a1)工程が、液体クロマトグラフィー-マススペクトロメトリー法により、異なる2つの測定条件で血液に含まれるキサントシン一リン酸を測定する工程である、請求項29に記載の方法。
- 液体クロマトグラフィーに用いる移動相のpH値が、7~11である、請求項30に記載の方法。
- 液体クロマトグラフィーに用いる移動相の塩濃度が、5~300mmol/Lである、請求項30または31に記載の方法。
- 液体クロマトグラフィーに用いる移動相が、重炭酸アンモニウム水溶液を含む移動相である、請求項30から32のいずれか一項に記載の方法。
- 液体クロマトグラフィーが、親水性相互作用クロマトグラフィーである、請求項30から33のいずれか一項に記載の方法。
- 異なる2つの測定条件が、マススペクトロメトリー法における検出イオンおよび/またはイオン化条件が異なる2つの測定条件である、請求項29から34のいずれか一項に記載の方法。
- 骨髄異形成症候群の患者に対する5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の有効投与量を予測するための装置であって、
(A)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する手段と、
(B)(A)に記載の手段により得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める手段と、
(C)(B)に記載の手段により得られるキサントシン一リン酸の変動率から5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物の有効投与量を予測する手段と、を含む装置。 - (A)に記載の手段が、マススペクトロメトリー装置を用いる手段である、請求項36に記載の装置。
- マススペクトロメトリー装置が、液体クロマトグラフィー装置と連結されたマススペクトロメトリー装置である、請求項37に記載の装置。
- 骨髄異形成症候群の患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測するための装置であって、
(A)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する手段と、
(B)(A)に記載の手段により得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める手段と、
(C)(B)に記載の手段により得られるキサントシン一リン酸の変動率から、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する手段と、を含む装置。 - (A)に記載の手段が、マススペクトロメトリー装置を用いる手段である、請求項39に記載の装置。
- マススペクトロメトリー装置が、液体クロマトグラフィー装置と連結されたマススペクトロメトリー装置である、請求項40に記載の装置。
- 骨髄異形成症候群の患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程を含む、
骨髄異形成症候群の患者の処置に用いるための、有効成分が5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドである骨髄異形成症候群の処置剤であって、
予測する工程が、
(a)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する工程と、
(b)(a)工程で得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める工程と、
(c)(b)工程で得られるキサントシン一リン酸の変動率から、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程であって、
患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し感受性であると予測された患者である、処置剤。 - 骨髄異形成症候群の患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程を含む、
骨髄異形成症候群の患者の処置に用いるための、有効成分が5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドである骨髄異形成症候群の処置方法であって、
予測する工程が、
(a)(i)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に含まれるキサントシン一リン酸の量と、(ii)5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与した後の患者から採取した血液に含まれるキサントシン一リン酸の量、または5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を投与する前の患者から採取した血液に5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を接触させて得られた血液に含まれるキサントシン一リン酸の量とを測定する工程と、
(b)(a)工程で得られるキサントシン一リン酸の量からキサントシン一リン酸の変動率を求める工程と、
(c)(b)工程で得られるキサントシン一リン酸の変動率から、患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し、感受性であるか否かを予測する工程と、を含む工程であって、
患者が、5-ヒドロキシ-1H-イミダゾール-4-カルボキサミドもしくはその塩またはその水和物を用いる処置に対し感受性であると予測された患者である、処置方法。
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2015205151A AU2015205151B2 (en) | 2014-01-10 | 2015-01-09 | 5-hydroxy-1H-imidazole-4-carboxamide effective-dose/sensitivity prediction method and prediction device, xanthosine-monophosphate-amount measurement method, and myelodysplastic-syndrome treatment agent and treatment method |
| CN201580004057.1A CN105899951B (zh) | 2014-01-10 | 2015-01-09 | 5-羟基-1h-咪唑-4-甲酰胺的有效给予量的预测装置、黄苷一磷酸的量的测定方法 |
| HK16110606.7A HK1222453A1 (zh) | 2014-01-10 | 2015-01-09 | 5-羟基-1h-咪唑-4-甲醯胺的有效给予量或感受性的预测方法及预测装置、黄苷一磷酸的量的测定方法、以及骨髓增生异常综合征的处置剂及处置方法 |
| JP2015556844A JP6294357B2 (ja) | 2014-01-10 | 2015-01-09 | 5−ヒドロキシ−1h−イミダゾール−4−カルボキサミドの有効投与量または感受性の予測方法および予測装置、キサントシン一リン酸の量の測定方法ならびに骨髄異形成症候群の処置剤および処置方法 |
| EP15735185.9A EP3093662B1 (en) | 2014-01-10 | 2015-01-09 | 5-hydroxy-1h-imidazole-4-carboxamide effective-dose/sensitivity prediction method and prediction device, xanthosine-monophosphate-amount measurement method, and myelodysplastic-syndrome treatment agent and treatment method |
| RU2016127534A RU2672872C2 (ru) | 2014-01-10 | 2015-01-09 | Способ и устройство для прогнозирования эффективной дозы или восприимчивости к 5-гидрокси-1н-имидазол-4-карбоксамиду, способ определения количества ксантозинмонофосфата, а также средство и способ для лечения миелодиспластического синдрома |
| CA2935905A CA2935905C (en) | 2014-01-10 | 2015-01-09 | Method and apparatus for predicting effective dose or sensitivity of 5-hydroxy-1h-imidazole-4-carboxamide, method for determining amount of xanthosine monophosphate, and treatmentagent and method for treating myelodysplastic syndrome |
| KR1020167018364A KR101856889B1 (ko) | 2014-01-10 | 2015-01-09 | 5-하이드록시-1h-이미다졸-4-카복사마이드의 유효 투여량 또는 감수성의 예측 방법 및 예측 장치, 잔토신 1인산의 양의 측정 방법 그리고 골수 이형성 증후군의 처치제 및 처치 방법 |
| US15/205,444 US20160320368A1 (en) | 2014-01-10 | 2016-07-08 | Method and apparatus for predicting effective dose or sensitivity of 5-hydroxy-1h-imidazole-4-carboxamide, method for determining amount of xanthosine monophosphate, and treatment agent and method for treating myelodysplastic syndrome |
| AU2018202975A AU2018202975A1 (en) | 2014-01-10 | 2018-04-30 | 5-hydroxy-1H-imidazole-4-carboxamide effective-dose/sensitivity prediction method and prediction device, xanthosine-monophosphate-amount measurement method, and myelodysplastic-syndrome treatment agent and treatment method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-003611 | 2014-01-10 | ||
| JP2014003611 | 2014-01-10 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/205,444 Continuation US20160320368A1 (en) | 2014-01-10 | 2016-07-08 | Method and apparatus for predicting effective dose or sensitivity of 5-hydroxy-1h-imidazole-4-carboxamide, method for determining amount of xanthosine monophosphate, and treatment agent and method for treating myelodysplastic syndrome |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015105174A1 true WO2015105174A1 (ja) | 2015-07-16 |
Family
ID=53523998
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/050478 Ceased WO2015105174A1 (ja) | 2014-01-10 | 2015-01-09 | 5-ヒドロキシ-1h-イミダゾール-4-カルボキサミドの有効投与量または感受性の予測方法および予測装置、キサントシン一リン酸の量の測定方法ならびに骨髄異形成症候群の処置剤および処置方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20160320368A1 (ja) |
| EP (1) | EP3093662B1 (ja) |
| JP (1) | JP6294357B2 (ja) |
| KR (1) | KR101856889B1 (ja) |
| CN (1) | CN105899951B (ja) |
| AU (2) | AU2015205151B2 (ja) |
| CA (1) | CA2935905C (ja) |
| HK (1) | HK1222453A1 (ja) |
| RU (1) | RU2672872C2 (ja) |
| WO (1) | WO2015105174A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3513791A4 (en) * | 2016-09-13 | 2019-08-28 | Fujifilm Corporation | MEDICAMENT, METHOD, USE AND COMPOSITION FOR PREVENTING OR TREATING MLL-CONTAINED LEUKEMIA |
| WO2020049786A1 (ja) * | 2018-09-03 | 2020-03-12 | 富士フイルム株式会社 | 組み合わせ医薬、ピリミジン代謝拮抗剤の耐性化の予防または抑制薬、および疾患の処置方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5332124A (en) | 1976-09-07 | 1978-03-27 | Sumitomo Chem Co Ltd | Carcinostatic agent |
| JPS5824569A (ja) | 1981-08-05 | 1983-02-14 | Sumitomo Chem Co Ltd | イミダゾ−ル誘導体の精製方法 |
| JPH1151885A (ja) * | 1997-08-04 | 1999-02-26 | Nippon Telegr & Teleph Corp <Ntt> | 二次イオン質量分析法 |
| WO2008053530A1 (en) * | 2006-10-31 | 2008-05-08 | Shimadzu Corporation | Sample quantification method |
| WO2009035168A1 (ja) | 2007-09-14 | 2009-03-19 | Mbr Co., Ltd. | 4-カルバモイル-5-ヒドロキシ-イミダゾール誘導体のスルホン酸塩化合物 |
| WO2009123297A1 (ja) * | 2008-04-03 | 2009-10-08 | 株式会社日立ハイテクノロジーズ | 質量分析装置を用いた定量分析方法 |
| JP2012127771A (ja) * | 2010-12-15 | 2012-07-05 | Hitachi High-Technologies Corp | 質量分析装置 |
| WO2013047758A1 (ja) | 2011-09-28 | 2013-04-04 | 富士フイルム株式会社 | 5-ヒドロキシ-1h-イミダゾール-4-カルボキサミド水和物の結晶及び5-ヒドロキシ-1h-イミダゾール-4-カルボキサミド・3/4水和物の結晶及びその製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4753935A (en) * | 1987-01-30 | 1988-06-28 | Syntex (U.S.A.) Inc. | Morpholinoethylesters of mycophenolic acid and pharmaceutical compositions |
| DE19811313A1 (de) * | 1998-03-16 | 1999-09-23 | Merckle Gmbh | Verfahren zur Bestimmung der Aktivität von Inosin 5'-monophosphat dehydrogenase |
| CN102373261A (zh) * | 2011-09-26 | 2012-03-14 | 上海交通大学 | 一种二氧化钛纳米颗粒的细胞生物安全性评价方法 |
-
2015
- 2015-01-09 WO PCT/JP2015/050478 patent/WO2015105174A1/ja not_active Ceased
- 2015-01-09 KR KR1020167018364A patent/KR101856889B1/ko not_active Expired - Fee Related
- 2015-01-09 RU RU2016127534A patent/RU2672872C2/ru not_active IP Right Cessation
- 2015-01-09 HK HK16110606.7A patent/HK1222453A1/zh unknown
- 2015-01-09 EP EP15735185.9A patent/EP3093662B1/en active Active
- 2015-01-09 JP JP2015556844A patent/JP6294357B2/ja not_active Expired - Fee Related
- 2015-01-09 CN CN201580004057.1A patent/CN105899951B/zh not_active Expired - Fee Related
- 2015-01-09 AU AU2015205151A patent/AU2015205151B2/en not_active Ceased
- 2015-01-09 CA CA2935905A patent/CA2935905C/en not_active Expired - Fee Related
-
2016
- 2016-07-08 US US15/205,444 patent/US20160320368A1/en not_active Abandoned
-
2018
- 2018-04-30 AU AU2018202975A patent/AU2018202975A1/en not_active Abandoned
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5332124A (en) | 1976-09-07 | 1978-03-27 | Sumitomo Chem Co Ltd | Carcinostatic agent |
| JPS5824569A (ja) | 1981-08-05 | 1983-02-14 | Sumitomo Chem Co Ltd | イミダゾ−ル誘導体の精製方法 |
| JPH1151885A (ja) * | 1997-08-04 | 1999-02-26 | Nippon Telegr & Teleph Corp <Ntt> | 二次イオン質量分析法 |
| WO2008053530A1 (en) * | 2006-10-31 | 2008-05-08 | Shimadzu Corporation | Sample quantification method |
| WO2009035168A1 (ja) | 2007-09-14 | 2009-03-19 | Mbr Co., Ltd. | 4-カルバモイル-5-ヒドロキシ-イミダゾール誘導体のスルホン酸塩化合物 |
| WO2009123297A1 (ja) * | 2008-04-03 | 2009-10-08 | 株式会社日立ハイテクノロジーズ | 質量分析装置を用いた定量分析方法 |
| JP2012127771A (ja) * | 2010-12-15 | 2012-07-05 | Hitachi High-Technologies Corp | 質量分析装置 |
| WO2013047758A1 (ja) | 2011-09-28 | 2013-04-04 | 富士フイルム株式会社 | 5-ヒドロキシ-1h-イミダゾール-4-カルボキサミド水和物の結晶及び5-ヒドロキシ-1h-イミダゾール-4-カルボキサミド・3/4水和物の結晶及びその製造方法 |
Non-Patent Citations (9)
| Title |
|---|
| "Stryer Biochemistry", 2013, pages: 689 - 693 |
| ANALYTICAL CHEMISTRY, vol. 84, 2012, pages 1994 - 2001 |
| ANALYTICAL CHEMISTRY, vol. 84, 2012, pages 216 - 223 |
| CANCER AND CHEMOTHERAPY, vol. 16, no. 1, 1989, pages 123 - 130 |
| DAISUKE TESHIMA: "TDM for Second-Line Medicine (Mycophenolate Mofetil) in the Immunosuppresive Medication", JAPANESE JOURNAL OF THERAPEUTIC DRUG MONITORING, vol. 23, no. 2, 2006, pages 61 - 62, XP008182445 * |
| KIYOJI KIMURA ET AL.: "Zoketsuki Shuyo ni Taisuru SM-108(4-Carbamoylimidazolium-5-olate) no Phase II study", JAPANESE JOURNAL OF CANCER AND CHEMOTHERAPY, vol. 16, no. 1, 1989, pages 123 - 130, XP008182427 * |
| LAVERDIERE ISABELLE ET AL.: "Liquid Chromatography-Coupled Tandem Mass Spectrometry Based Assay to Evaluate Inosine-5'- monophosphate Dehydrogenase Activity in Peripheral Blood Mononuclear Cells from Stem Cell Transplant Recipients", ANAL CHEM, vol. 84, no. 1, 2012, pages 216 - 223, XP055295182 * |
| See also references of EP3093662A4 |
| ZHANG TONG ET AL.: "Evaluation of Coupling Reversed Phase, Aqueous Normal Phase, and Hydrophilic Interaction Liquid Chromatography with Orbitrap Mass Spectrometry for Metabolomic Studies of Human Urine", ANAL CHEM, vol. 84, no. 4, 2012, pages 1994 - 2001, XP055295184 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3513791A4 (en) * | 2016-09-13 | 2019-08-28 | Fujifilm Corporation | MEDICAMENT, METHOD, USE AND COMPOSITION FOR PREVENTING OR TREATING MLL-CONTAINED LEUKEMIA |
| WO2020049786A1 (ja) * | 2018-09-03 | 2020-03-12 | 富士フイルム株式会社 | 組み合わせ医薬、ピリミジン代謝拮抗剤の耐性化の予防または抑制薬、および疾患の処置方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2015105174A1 (ja) | 2017-03-23 |
| AU2015205151B2 (en) | 2018-02-01 |
| HK1222453A1 (zh) | 2017-06-30 |
| CA2935905A1 (en) | 2015-07-16 |
| EP3093662A4 (en) | 2017-01-11 |
| CN105899951A (zh) | 2016-08-24 |
| KR101856889B1 (ko) | 2018-05-10 |
| CA2935905C (en) | 2019-02-05 |
| AU2018202975A1 (en) | 2018-05-17 |
| EP3093662A1 (en) | 2016-11-16 |
| JP6294357B2 (ja) | 2018-03-14 |
| RU2672872C2 (ru) | 2018-11-20 |
| KR20160095142A (ko) | 2016-08-10 |
| RU2016127534A (ru) | 2018-02-16 |
| US20160320368A1 (en) | 2016-11-03 |
| EP3093662B1 (en) | 2019-05-15 |
| CN105899951B (zh) | 2019-07-12 |
| AU2015205151A1 (en) | 2016-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhang et al. | Exploratory urinary metabolic biomarkers and pathways using UPLC-Q-TOF-HDMS coupled with pattern recognition approach | |
| Cala et al. | Urinary metabolite and lipid alterations in Colombian Hispanic women with breast cancer: A pilot study | |
| US20130330419A1 (en) | Compositions and methods for personal tumor profiling treatment | |
| Hung et al. | Simultaneous determination of pyrazinamide, rifampicin, ethambutol, isoniazid and Acetyl Isoniazid in human plasma by LC-MS/MS method | |
| EP3891508A1 (en) | Methods of analysis using in-sample calibration curve by multiple isotopologue reaction monitoring | |
| Ye et al. | Quantification of sorafenib, lenvatinib, and apatinib in human plasma for therapeutic drug monitoring by UPLC-MS/MS | |
| Su et al. | Network‐based biomarkers for cold coagulation blood stasis syndrome and the therapeutic effects of Shaofu Zhuyu decoction in rats | |
| CN110045048B (zh) | 一种测定人血浆中两种抗肿瘤药物浓度的hplc-msms方法 | |
| Yang et al. | Development of UPLC-MS/MS method for studying the pharmacokinetic interaction between dasatinib and posaconazole in rats | |
| JP6294357B2 (ja) | 5−ヒドロキシ−1h−イミダゾール−4−カルボキサミドの有効投与量または感受性の予測方法および予測装置、キサントシン一リン酸の量の測定方法ならびに骨髄異形成症候群の処置剤および処置方法 | |
| Parmar et al. | Recent method development by analytical techniques of new FDA-approved rugs in 2021 | |
| Zuo et al. | Pharmacokinetics and tissue distribution study of prucalopride in rats by ultra high performance liquid chromatography with tandem mass spectrometry | |
| Zhou et al. | Independent or integrative processing approach of metabolite datasets from different biospecimens potentially affects metabolic pathway recognition in metabolomics | |
| Gupta et al. | Selective and rapid determination of raltegravir in human plasma by liquid chromatography–tandem mass spectrometry in the negative ionization mode | |
| WO2025002914A1 (en) | Antagonists of the imidazoline-1 receptor for use in the prevention and/or treatment of an autoinflammatory or autoimmune disease | |
| Kobayashi et al. | Liquid chromatography-mass spectrometry measurements of blood diphosphoinositol pentakisphosphate levels | |
| KR101703132B1 (ko) | 이매티닙에 대한 약물 반응 예측용 마커 및 이를 이용한 약물 반응 예측 방법 | |
| Zhou et al. | Metabolomics investigation of cutaneous T cell lymphoma based on UHPLC-QTOF/MS | |
| Yu et al. | Simultaneous determination of eight antiepileptic drugs and two metabolites in human plasma by liquid chromatography/tandem mass spectrometry | |
| Yan et al. | Development and optimization of a high-throughput HPLC-MS/MS method for the simultaneous determination of Cedazuridine, Gemcitabine and its metabolite in mouse plasma | |
| Macioszek et al. | A multiplatform metabolomics approach for comprehensive analysis of GIST xenografts with various KIT mutations | |
| Arvonio | DEVELOPMENT OF ANALYTICAL METHODS IN | |
| Berardi | Untargeted metabolomics and proteomics application of mass spectrometry: from biomarker identification within cells to their spatial localization on tissues | |
| Dan et al. | Pharmacokinetic Characterization of ZT55, A Novel Indole Derivative Isolated from Radix Isatidis, using Liquid Chromatography/Tandem Mass and Q-TOF/Tandem Mass Spectrometry | |
| Kunati | Development of Bioanalytical Methods for Quantitative Measurement of Anticancer Agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15735185 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2935905 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2015556844 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015735185 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015735185 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167018364 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2015205151 Country of ref document: AU Date of ref document: 20150109 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016016128 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2016127534 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112016016128 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160711 |