WO2015107888A1 - 幹細胞における腫瘍化原因細胞の新たな標識法と治療法 - Google Patents
幹細胞における腫瘍化原因細胞の新たな標識法と治療法 Download PDFInfo
- Publication number
- WO2015107888A1 WO2015107888A1 PCT/JP2015/000138 JP2015000138W WO2015107888A1 WO 2015107888 A1 WO2015107888 A1 WO 2015107888A1 JP 2015000138 W JP2015000138 W JP 2015000138W WO 2015107888 A1 WO2015107888 A1 WO 2015107888A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- cells
- undifferentiated
- gene
- promoter
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6897—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids involving reporter genes operably linked to promoters
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5011—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing antineoplastic activity
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5073—Stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/90—Fusion polypeptide containing a motif for post-translational modification
- C07K2319/92—Fusion polypeptide containing a motif for post-translational modification containing an intein ("protein splicing")domain
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/15011—Lentivirus, not HIV, e.g. FIV, SIV
- C12N2740/15041—Use of virus, viral particle or viral elements as a vector
- C12N2740/15043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/008—Vector systems having a special element relevant for transcription cell type or tissue specific enhancer/promoter combination
Definitions
- the present invention relates to a method for searching for a promoter for use in labeling and removing tumorigenic cells remaining after differentiation of stem cells.
- the present invention also relates to a method for labeling and removing tumorigenic cells remaining after differentiation of stem cells, and a viral vector for use in the method.
- Human embryonic stem cells (ES cells) and human induced pluripotent stem cells (iPS cells) are (1) human cells used for drug toxicity testing in drug development, and (2) elucidation of development and pathology in vitro. It is expected as a basic tool for medical / pharmaceutical creation such as disease model cells used for this purpose, and (3) cells for transplantation of regenerative medicine. Regarding the most promising regenerative medicine, clinical trials in human patients using human ES cells have already been started for some diseases in the United States. In Japan, clinical studies using human iPS cells for eye diseases have been started.
- Non-patent Document 1 The biggest problem at present in clinical application of human ES / iPS cells is a mixture of undifferentiated cells remaining after differentiation into target cells. It has been reported that residual undifferentiated cells form teratomas after transplantation of cells subjected to differentiation induction treatment (Non-patent Document 1). Teratomas are benign tumors but can have a variety of side effects, such as fatal arrhythmias, such as when the heart is transplanted with cells other than cardiomyocytes .
- Non-patent Document 2 discloses a high incidence of not only teratomas but also malignant tumors.
- c-Myc which is also known as a proto-oncogene, activates cells, and retroviruses in the establishment of iPS cells.
- Non-patent Documents 3 and 4 the method of establishing iPS cells with 3 factors excluding c-Myc (Non-patent Documents 3 and 4) and the suppression of chromosome damage by establishing iPS cells using plasmids instead of viral vectors Methods (Non-Patent Documents 5 and 6) have been reported.
- Non-Patent Documents 5 and 6 the suppression of chromosome damage by establishing iPS cells using plasmids instead of viral vectors Methods.
- these can all be expected to have a certain effect of suppressing cell "tumorization", they do not prevent the remaining of undifferentiated cells, and for example, they cannot remove undifferentiated cells that have undergone reverse differentiation after transplantation. There was a problem.
- Patent Document 1 As another approach, there has been reported a method of selecting and using a clone that is unlikely to become a tumor even when differentiated (Patent Document 1). However, even in this case, it did not prevent the remaining of undifferentiated cells, and it was not possible to completely suppress tumorigenesis.
- a method for killing the remaining undifferentiated cells as a target has been studied.
- a lentiviral vector having a herpes simplex virus thymidine kinase (HSV-tk) gene downstream of the Nanog promoter is introduced into mouse ES / iPS cells or human ES / iPS cells, and these cells are rendered ganciclovir (GCV) sensitive. It has been tried. It has been reported that tumors shrink by administration of GCV after transplanting the cells subcutaneously to immunodeficient mice to form tumors (Non-patent Documents 7 and 8).
- HSV-tk herpes simplex virus thymidine kinase
- Such a suicide gene therapy method using a combination of HSV-tk and GCV is a stage in which a part of the above research has been announced as a technique for removing stem cell tumorigenic cells in regenerative medicine such as the present invention. There have been no clinical applications in stem cell regenerative medicine.
- HSV-tk / GCV has a long history of research as an application to gene therapy for cancer, and HSV-tk / GCV gene therapy (a typical method is to introduce and express HSV-tk gene).
- a large number of clinical trials have already been conducted for cancer patients as injecting GCV directly into a patient's tumor and then injecting GCV into the patient.
- HSV-tk When a gene derived from HSV-tk, which is not a human-derived virus, is introduced into cells, it is suggested that immunity against heterologous proteins may be induced.
- cancer treatment removal of cancer cells into which HSV-tk gene has been introduced by immunity induction centered on cellular immunity leads to amplification of the therapeutic effect. It is done.
- the transplanted cell may be removed by immunity induction against a heterologous protein. In this respect, it is more preferable to use a human-derived gene. Presumed to be desirable.
- Patent Document 2 a technique has been reported in which p53 and p16 genes that act to suppress the cell growth cycle are induced by doxycycline.
- this technique is not able to kill all tumorigenic cells because tumor suppressor genes do not kill tumor cells actively or directly in the first place.
- the possibility that differentiated cells will start to proliferate after the release of doxycycline induction, and because the so-called doxycycline gene expression induction system has low specificity, these genes are expressed non-specifically to induce differentiation of stem cells and biological dynamics, etc.
- SP1 Specificity Protein 1
- a protein contained in a Kahal body a protein contained in a nuclear lamina
- a protein contained in a paranuclear compartment a protein contained in a paranuclear compartment
- PML Transformed cells contained in the cell population can be identified by identifying the presence or absence of expression of hTERT gene or IPAS gene mRNA (Patent Document 3) by identifying the amount of protein contained in the body in the cell nucleus, etc.
- a method of detecting (Patent Document 4) and a method of identifying cells expressing podocalyxin-like protein (PODXL) on the surface Patent Document 5 are disclosed.
- all of these methods require complicated operations. For example, residual undifferentiated cells in a tissue transplanted in a living body cannot be easily identified.
- Patent Document 6 Attempts have been made to identify undifferentiated cells by their promoter activity, and a method using the Stm1 promoter sequence has been disclosed.
- Patent Document 7 a method for selectively killing remaining undifferentiated cells using a multifactor cancer cell-specific growth control virus. While this method is very useful for solving this problem from the point of directly and aggressively killing tumorigenic cells and undifferentiated cells, and the effect of killing tumorigenic cells into which no gene has been introduced. Since the target gene has not been introduced into all stem cells in advance, if a tumorigenic cell emerges from a stem cell that has not reached the multifactor cancer cell-specific growth-control virus, the virus is transferred to this virus in vivo. It was not possible to guarantee that all immunity induction could be eliminated.
- an object of the present invention is to provide various methods that can be used to achieve more efficient labeling and removal of remaining undifferentiated cells.
- an object of the present invention is to provide a technique (vector) capable of efficiently introducing a foreign gene into all undifferentiated cells without inhibiting a gene that maintains the normal function of the cell.
- Another object of the present invention is to provide a method for searching for the best promoter for reliably targeting tumorigenic cells remaining after differentiation of stem cells.
- Another object of the present invention is to find a gene capable of efficiently killing / removing cells that cause tumorigenesis of stem cells.
- an object of the present invention is to provide a vector capable of both simpler labeling and removal of tumorigenic cells.
- a viral vector having an expression cassette in which a marker gene and a killing gene are linked downstream of a possible promoter portion via a sequence capable of simultaneously expressing the two genes by a single promoter was found.
- the marker gene and the killing gene are linked via a sequence that allows the two genes to be expressed simultaneously by a single promoter. The function of can be provided.
- this vector can be easily incorporated with any promoter by a recombination system, it is appropriately used depending on, for example, the cell to be differentiated, the timing of cell removal (before / after transplantation), and the like. It is possible to select a promoter. Moreover, it can be used for the purpose of simply analyzing whether or not the promoter has activity specific to undifferentiated cells or tumorigenic cells. Furthermore, by adopting a drug-dependent suicide gene as a killing gene in the vector of the present invention, in addition to the cell selectivity of a tumor-specific / undifferentiated cell-specific promoter, a cell killing function is exhibited if there is no induction by a drug. It can be set as the structure which does not.
- such a vector of the present invention does not kill those cells only by introducing them into stem cells, and therefore can be introduced into all stem cells before differentiation induction treatment. It can be prevented from occurring. Therefore, double safety can be ensured by the cell selectivity of the tumor-specific / undifferentiated cell-specific promoter and the drug dependency. Further, by using a killing gene that provides a (drug-dependent) proliferating cell-specific killing mechanism as a killing gene in the vector of the present invention, cell selectivity of a tumor-specific / undifferentiated cell-specific promoter, In addition to drug dependence, it can impart proliferative cell selectivity of the killing mechanism, ensuring triple specificity and safety.
- the present inventors have found that the problem of immunogenicity can be solved by using human-derived enzymes human-tmpk and iCaspase9 as drug-dependent suicide genes carried by the vectors of the present invention.
- the present inventors have searched for a promoter specifically active in undifferentiated cells or tumorigenic cells, which is carried by the vector of the present invention.
- tumors cancers
- the survivin promoter known as a specific promoter, was unexpectedly found to be highly active in normal, undifferentiated human ES / iPS cells.
- the present inventors have used the vector of the present invention to efficiently and comprehensively label / remove undifferentiated cells remaining after differentiation induction treatment, and the promoter is an undifferentiated cell or a cause of tumorigenesis.
- the present inventors have found a method for simply analyzing whether or not a cell-specific activity is present.
- the present invention relates to the following inventions.
- a viral vector having a nucleic acid sequence in which a marker gene and a killing gene are linked via a sequence capable of simultaneously expressing the two genes by a single promoter, and a recombination cassette including a promoter region A viral vector in which the promoter region is linked so that the marker gene and the killing gene can be expressed.
- the marker gene is a fluorescent protein.
- the viral vector according to (5), wherein the fluorescent protein is a red or green fluorescent protein.
- the viral vector according to (5), wherein the fluorescent protein is mKate2 or green fluorescent protein.
- the viral vector according to any one of (1) to (7), wherein the sequence capable of simultaneously expressing the two genes by one promoter is a 2A sequence or an IRES sequence.
- the viral vector according to any one of (1) to (8) which is a lentiviral vector.
- a screening method for an undifferentiated cell-specific promoter comprising: Here, the rate at which the expression of the labeled gene is detected in cells identified as undifferentiated cells is high, and the rate at which the expression of the labeled gene is detected in cells identified as differentiated cells is low In the case, the method is carried out by determining that the test promoter is specific to undifferentiated cells.
- the level of the expression product of the marker gene indicates the amount (or ratio) of undifferentiated cells.
- (22) A) Infecting a target cell with the viral vector according to any one of (10) to (12), B) detecting an expression product of the marker gene in the cell after differentiation treatment; and C) A method for monitoring undifferentiated cells remaining or generated after differentiation treatment, comprising a step of determining that undifferentiated cells remain or have been generated when the expression product of the marker gene is detected.
- (23) A) detecting the expression product of the marker gene in a cell into which the viral vector according to any one of (10) to (12) has been introduced and subjected to differentiation induction; and B) A method for monitoring undifferentiated cells after differentiation processing of stem cells, comprising a step of determining that undifferentiated cells remain or are generated when the expression product of the marker gene is detected.
- a method for killing undifferentiated cells comprising the step of administering a drug capable of exerting the above.
- An undifferentiated cell labeling agent comprising the viral vector according to any one of (10) to (12).
- An undifferentiated cell killing agent comprising the viral vector according to any one of (10) to (12).
- the cell according to (24), wherein the stem cell is an ES cell, iPS cell, neural stem cell, hematopoietic stem cell, mesenchymal stem cell, hepatic stem cell, pancreatic stem cell, skin stem cell, muscle stem cell, or germline stem cell.
- stem cell means a cell having pluripotency and self-renewal ability.
- pluripotency is synonymous with pluripotency, and means a state of a cell that can differentiate into cells of a plurality of lineages by differentiation.
- pluripotency refers to a state that can be differentiated into all types of cells constituting a living body (totipotency), and a state that can be differentiated into all types of cells other than extraembryonic tissues (differentiation).
- Pluripotency a state that can differentiate into cells belonging to some cell lineages (multipotency), and a state that can differentiate into one type of cell (unipotency) Including.
- stem cells in the present specification include stem cells, ES cells, iPS cells, neural stem cells, hematopoietic stem cells, mesenchymal stem cells, hepatic stem cells, pancreatic stem cells, skin stem cells, muscle stem cells, or germline stem cells.
- the “stem cell” in the present specification is a cell having pluripotency, more preferably an embryonic stem cell (ES cell) and an induced pluripotent stem cell (iPS cell). Whether a certain cell is a stem cell is determined by, for example, a cell that forms an embryoid body in an in vitro culture system, or a cell that differentiates into a desired cell after culturing under differentiation-inducing conditions (differentiation treatment).
- stem cell It can be confirmed as a stem cell. Or, whether it is a stem cell is determined by using a living body, transplanting it to an immunodeficient mouse, a cell forming a teratoma, a cell forming a chimeric embryo by injection into a blastocyst, or a living tissue It can be confirmed that cells proliferating by transplantation or injection into ascites are stem cells.
- a stem cell is a cell positive for alkaline phosphatase staining, SSEA3 staining, SSEA4 staining, TRA-1-60 staining, and / or TRA-1-81 staining; oct3 / 4, nanog, sox2, clipto Cells expressing the, dax1, eras, fgf4, esg1, rex1, zfp296, utf1, gdf3, all4, tbx3, tcf3, dnmt3l, and / or dnmt3b genes; miR-290 and / or miR-302 It can also be confirmed that the cell is a stem cell. Alternatively, whether or not a certain cell is a stem cell can be determined, for example, as a cell having a high expression level of telomerase reverse transcriptase or survivin as a stem cell.
- differentiation refers to a phenomenon in which daughter cells having specific functional or morphological characteristics are generated by the division of pluripotent cells.
- “differentiation treatment” and “differentiation induction treatment” are synonymous and mean treatment for inducing stem cells into differentiated cells. It has been reported that cell differentiation is induced by various methods. Pluripotent cells can be differentiated by differentiation-inducing treatment using a differentiation-inducing substance or the like according to the type of cells to be differentiated.
- a “differentiated cell” means a daughter cell that has a specific functional or morphological characteristic resulting from differentiation. Differentiated cells are usually stable, their ability to proliferate is low, and differentiation into other types of cells occurs only exceptionally.
- Undifferentiated cell means an undifferentiated cell, an undifferentiated cell in the middle of differentiation, or a cell that is incompletely differentiated.
- undifferentiated cell means a cell that has not been differentiated even though it has undergone differentiation induction treatment.
- the undifferentiated cells in the present specification do not necessarily need to have the above-mentioned stem cell properties completely, and have a high (self) proliferation ability compared to differentiated cells (for example, the proliferation rate is twice or more, (3 times or more, 5 times or more, 10 times or more) means a cell.
- the “undifferentiated cell” is a “tumor causing cell”.
- Whether a cell is an undifferentiated cell is determined by, for example, determining whether a cell having c-Myc activation or a cell having a high expression level of telomerase reverse transcriptase and / or survivin is a cell having tumorigenic potential. Can be determined.
- tumor-causing cell means an undifferentiated cell that has not differentiated even though the stem cell has been subjected to differentiation treatment, and can generate a tumor cell.
- Tumor-causing cells are not only cells that maintain the same properties as stem cells before differentiation, but also cells that have different properties from stem cells before differentiation but have not become differentiated cells, such as small numbers of cells. It also includes functional cells (cells that can be differentiated into only a few types of cells), cells that have tumor-forming ability (for example, cancer stem cells, etc.), and the like.
- cancer stem cells for example, cancer stem cells, etc.
- Whether or not a certain cell has tumorigenicity can be determined, for example, by c-Myc-activated cells or cells having high expression levels of telomerase reverse transcriptase and / or survivin as cells having tumorigenicity. Can be determined.
- differentiation into undifferentiated cells particularly stem cells expressed by “undifferentiated cells remaining after differentiation treatment”, “residual undifferentiated cells”, “undifferentiated cells in cells after differentiation treatment”, etc.
- the undifferentiated cells remaining after the induction treatment may be “tumor-causing cells”.
- the “labeling gene” means a gene encoding a labeling substance capable of detecting or measuring a cell into which the gene has been introduced.
- the marker gene include green fluorescent protein (GFP), EGFP (enhanced GFP) and Venus whose labeling ability is further improved by changing the amino acid sequence of GFP, red fluorescent protein (RFP) exhibiting other fluorescent wavelengths, blue Fluorescent protein (BFP), yellow fluorescent protein (YFP), red, green, blue, or yellow fluorescent proteins such as mKate2; mentions genes encoding ⁇ -glucuronidase, ⁇ -galactosidase, luciferase, dihydrofolate reductase Preferably, mKate2 or Venus.
- labeling refers to identifying the presence or position of a target cell temporarily or for a long period of time depending on its purpose of use, and measuring the number of target cells (may be an amount or a ratio). Or, it means that the target cell can be distinguished from other cells.
- identify means to temporarily or long-term specify the presence or position of a target cell, and “identify” means to temporarily or long-term target cell. It means to distinguish cells from other cells and tissues.
- the label in the present specification is not particularly limited as long as the presence or position of the target cell can be specified or the target cell can be distinguished from other cells, and cells other than the target cell are completely stained. It does not require that they are not equal.
- the undifferentiated cell can be distinguished from the differentiated cell in the cell. It only needs to be stained and does not prevent (weakly) staining of differentiated cells.
- the “killing gene” means a gene having the ability to finally kill the introduced cell, for example, any apoptosis-inducing gene (Bax, p53, DP5, PL-3, reaper, hidden). ), A gene that induces cell death, or a gene that suppresses tumor cells. Further, a gene that promotes differentiation induction may be used instead of the “killing gene”.
- the “killing gene” depends on a drug-dependent suicide gene, induction (light, heat, temperature, RNA, compound, radiation, ultrasound, etc.) Sex gene expression system or inducible killing gene.
- “Drug-dependent suicide gene” is a gene encoding a protein that converts a non-toxic prodrug (drug) into a toxic substance, or a non-toxic protein that is converted into a toxic substance by adding a drug Means the gene encoding
- “a drug capable of exerting toxicity by a drug-dependent suicide gene” means a drug that causes toxicity by interacting with the drug-dependent suicide gene.
- the drug-dependent suicide gene of the present invention may be a gene encoding a metabolic enzyme protein (prodrug converting enzyme) that converts a non-toxic prodrug (drug) into a toxic substance.
- a drug capable of exerting toxicity by a drug-dependent suicide gene means a non-toxic prodrug that is converted into a toxic substance by the drug-dependent suicide gene (metabolic enzyme).
- Induction (light, heat, temperature, RNA, compound, radiation, ultrasound, etc.)-Dependent gene expression system, or inducible killing gene means light, heat, temperature, RNA, compound, radiation, ultrasound stimulation It means a gene expression system whose expression is induced, or a killed gene whose expression is induced by stimulation.
- drug-dependent suicide gene may be read as “inducible killing gene” unless such interpretation is incompatible.
- Combinations of drug-dependent suicide genes and prodrugs include, for example, herpes simplex virus thymidine kinase, ganciclovir or acyclovir, Varicella Zoster virus thymidine kinase, and 6 methoxypurine arabinonucleoside (Huber et al., 1999 , Proc . ... Natl Acad Sci USA 88 : 8039), E. coli and cytosine deaminase, fluorouracil (Mullen et al., 1992, Proc Natl Acad Sci USA 89: doi 33), E.
- thymidine kinase such as HSV-tk
- GCV ganciclovir
- HSV-tk when thymidine kinase such as HSV-tk is used as a drug-dependent suicide gene and ganciclovir (GCV) is used as a drug, GCV is metabolized by HSV-tk to generate gancyclovir triphosphate which is a toxic substance. it can.
- human-tmpk is used as a drug-dependent suicide gene and azidothymidine (AZT) is used as a non-toxic prodrug
- AZT when human-tmpk is used as a drug-dependent suicide gene and azidothymidine (AZT) is used as a non-toxic prodrug, AZT generates AZT triphosphate, which is a toxic substance metabolized by human-tmpk. be able to.
- CD cytosine deaminase
- coli is used as a drug-dependent suicide gene and 5-fluorotoxin (5-FC) is used as a non-toxic prodrug, 5-FC is metabolized by CD. It can give rise to the toxic substance 5-fluouricil (5-FU).
- 5-FU 5-fluouricil
- Ara-M 6-methoxypurine arabinonucleoside
- Ara-M is metabolized by varicella virus thymidine kinase and becomes a toxic substance.
- Certain 6-methoxypurine arabinonucleosides can be generated.
- the drug-dependent suicide gene of the present invention may be a gene encoding a protein that converts a non-toxic protein into a toxic substance by the addition of a drug.
- the drug-dependent suicide gene of the present invention may encode a protein that does not exhibit toxicity with a monomer but exhibits toxicity by becoming a multimer (eg, dimer).
- a drug capable of exerting toxicity by a drug-dependent suicide gene means a substance that converts the drug-dependent suicide gene into a toxic substance.
- caspases such as Caspase 9 and Caspase 3 are used as drug-dependent suicide genes and Dimerizer is used as a drug
- the caspase-9 dimer that is an toxic substance by dimerizing caspase by Dimerizer And can induce apoptosis.
- the drug-dependent suicide gene that exhibits toxicity by becoming such a multimer include Fas receptor and FADD.
- a sequence capable of simultaneously expressing two genes by one promoter means a sequence capable of translating two types of proteins or peptides when activated by one promoter. means.
- Such a sequence is a sequence having a self-cleavage activity, and is expressed as two proteins or peptides linked through the sequence by expressing two genes together by activation of one promoter,
- the two proteins or peptides linked via the sequence may be a sequence in which the two separated proteins or peptides are translated by self-cleavage in the region of the sequence.
- a sequence that allows two genes to be expressed simultaneously by one promoter means that a ribosome is attracted to initiate the second translation from the middle of the mRNA, and two different genes from one mRNA are bicistronic. It may be a sequence to be translated.
- 2A sequence can be mentioned, and as the latter, for example, “IRES (internal ribosome entry site) sequence” can be mentioned.
- 2A sequences include 2A sequences derived from foot-and-mouth disease virus and 2A sequences derived from equine rhinitis A virus (Furler, S et al. (2001) Gene Ther. 8, 864-73; and Hasegawa et al. ( 2007) See Stem Cells 25, 1707-12).
- the vector of the present invention has a recombination cassette containing a promoter region.
- the recombination cassette means a region where a gene can be recombined by recombination (recombinase), and includes a nucleic acid sequence recognized by the recombinase and a foreign gene insertion site sandwiched between the nucleic acid sequences.
- Examples of the recombination cassette include LR recombination (bacteriophage ⁇ integrase and excinase, and E.
- coli integration host factor protein in which recombination occurs between an attL-terminal DNA fragment and an attR-containing donor vector
- ⁇ phage recombination such as BP recombination (bacteriophage ⁇ integrase and E. coli integration host factor protein), where recombination occurs between the attB-terminal DNA fragment and the attP-containing donor vector.
- an undifferentiated cell-specific promoter can be used as the promoter.
- the “undifferentiated cell-specific promoter” refers to a marker gene and / or a drug-dependent suicide gene that is specifically and downstream bound to an undifferentiated cell and / or a tumorigenic cell. It means a promoter exhibiting an activity that can be expressed to a sufficient degree to exhibit its function. Examples of such promoters include telomerase reverse transcriptase (tert) promoter (see Takakura, M. et al., Cancer Res., 59: 551-557, 1999), survivin promoter (Li, F.
- operably linked to means (in an in vitro transcription / translation system or in the host cell if the vector is introduced into the host cell). This means that the nucleic acid sequence of the target gene is bound to a control sequence (promoter sequence) in a state that enables expression of the nucleic acid sequence of the gene.
- the viral vector is not particularly limited as long as it is a viral vector that can be used for the purpose of the present invention.
- lentivirus adenovirus, retrovirus, adeno-associated virus, herpes virus, herpes simplex virus, vaccinia Virus vectors such as viruses, box viruses, polioviruses, Sindbis viruses, Sendai viruses and the like are included.
- the present invention also relates to a cell into which the above-described viral vector has been introduced.
- the present cell may be a stem cell or a cell obtained by inducing differentiation of a stem cell (for example, a differentiated cell).
- the marker gene and the killing gene are linked by a single promoter through a sequence capable of simultaneously expressing the two genes. Both killing functions can be given.
- this vector can be easily incorporated with any promoter by a recombination system, for example, depending on the purpose of differentiation, the timing of cell removal (before / after transplantation), etc. It is possible to efficiently and quickly prepare various viral vectors by selecting a plurality of candidate promoters suitable for the purpose. Moreover, it can be used for the purpose of simply analyzing whether or not the promoter has activity specific to undifferentiated cells or tumorigenic cells.
- the cell killing function is not exhibited unless induced by the drug. It has become. Therefore, even if it is introduced into all stem cells before differentiation induction treatment, cells will not die unless a drug is added. Therefore, a gene should be introduced before differentiation induction, and only the stem cell into which the gene has been introduced should be used for differentiation induction. Therefore, leakage of gene transfer can be prevented. Moreover, since the drug-dependent suicide gene carried by the vector of the present invention is a human-derived enzyme, the problem of immunogenicity can be solved.
- the viral vector having an undifferentiated cell-specific promoter of the present invention is specifically and comprehensively labeled / killed after the differentiation treatment by introducing it into all stem cells before the differentiation treatment. Therefore, the problem of tumorigenesis due to remaining undifferentiated cells in regenerative medicine can be solved.
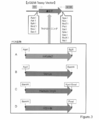
- Promoter recombination was performed using Gateway® cloning, the promoter (“Promoter” cassette) subcloned into Shuttle vector plasmid was inserted into the RC portion of pLenti6-RC-fluorescent protein-2A-suicide gene, and plasmid “pLenti6 -Promoter-fluorescent protein-2A-suicide gene "was completed. It is the schematic of pLenti6-RC structure and Promoter insertion.
- A. The structure of Lentiviral vector plasmid (pLenti6-RC) including RC is shown.
- B. Figure 5 shows the insertion of a promoter cassette using LR coronase.
- pLenti6-RC and the shuttle vector plasmid for gene recombination contain a homologous att sequence.
- a shuttle vector plasmid, pLenti6-RC, in which the target Promoter is cloned, is reacted with LR clone, which is a recombinant enzyme, and a promoter sequence is incorporated into the pLenti vector.
- LR clone which is a recombinant enzyme
- FIG. 3 is a schematic diagram showing the cloning of a fluorescent protein gene into the pFlag-CMV-2A vector.
- mKate2 (A) and Venus (B) subcloned into pGEM-Teasy were excised with restriction enzymes and inserted between CMV and 2A of pFlag-CMV-2A cleaved with NotI / BglII.
- FIG. 3 is a schematic diagram showing insertion of a “fluorescent protein + suicide gene” expression cassette into pLenti6-RC plasmid. Two types of “fluorescent protein + suicide gene” expression cassettes (B) shown in FIG.
- FIG. 3 is a schematic diagram showing insertion of a “fluorescent protein + suicide gene” expression cassette into pLenti6-RC plasmid.
- Two types of “fluorescent protein + suicide gene” expression cassettes (B) shown in FIG. 5 were inserted into the AgeI and KpnI sites (C) in pLenti6-RC (A) containing RC shown in FIG.
- FIG. 2 is a schematic diagram showing the preparation of pLenti6-RC having an “iCaspase + fluorescent protein” expression cassette.
- PCR was performed using pLenti6-RC as a vector and designing primers with 15 PB homologous sequences at both ends so that iCaspase 9-2A and a fluorescent protein gene could be inserted downstream of RC. These were combined and cloned by reacting with IN-FUSION enzyme. The photograph which confirmed the expression of the subcloned fluorescent protein is shown. Expression of mKate2 (A) and Venus (B) inserted into pFlag-CMV-2A was confirmed. It is the photograph which introduce
- the plasmid shown in FIG. 2B was introduced into HEK293 cells, and GCV was distributed and administered at concentrations of 0, 0.01, 0.1, 1, 10, and 100 mg / mL for 3 days from 48 hours later.
- Cells into which the plasmid having HSV-tk was introduced were observed with a fluorescence microscope, and the fluorescence expression of mKate2 (red) and Venus (green) was confirmed over time after GCV administration.
- the result of having counted the cell survival number after GCV administration about the cell of FIG. 10A is shown.
- the numerical value on the vertical axis represents the percentage (%) when the cell survival number of the drug 0 mg / mL is 100.
- the photograph which confirmed the expression of the "fluorescent protein + suicide gene” cassette is shown.
- the plasmid shown in FIG. 2B was introduced into HEK293 cells, and AZT was distributed and administered at concentrations of 0, 0.1, 1, 10, 100, and 1000 nm for 3 days from 48 hours later.
- Cells into which a plasmid having human-tmpk was introduced were observed with a fluorescence microscope, and the fluorescence expression of mKate2 (red) and Venus (green) was confirmed over time after AZT administration.
- the result of having counted the cell survival number after AZT administration about the cell of FIG. 10C is shown.
- the numerical value on the vertical axis represents the percentage (%) when the number of viable cells at 0 nm of the drug is 100.
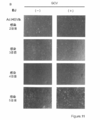
- FIG. 2 is a photograph showing GCV sensitivity after introduction of HSV-tk in KhES1 cells.
- FIG. 2 is a photograph showing AZT sensitivity after introduction of human-tmpk in KhES1 cells.
- PHR'-cPPT-EF-Tmpk F105Y
- AZT was administered at 0, 30, 100, and 300 mm after 24 hours, and the cytotoxic effect after 3 and 4 days was confirmed.
- It is a photograph which shows the AZT sensitivity after human-tmpk introduction
- PHR'-cPPT-EF-Tmpk (F105Y) was introduced into BJ cells, and AZT was administered at 0, 30, 100, and 300 mm after 24 hours, and the cytotoxic effect after 3 days and 4 days was confirmed.
- AZT was administered at 0, 0.1, 1, 10, 100, and 1000 mm, respectively.
- the state of the cell after 3 days of AZT treatment in Jurkat is shown.
- the left is a tmpk constant expression cell group, and the right is an NC group.
- It is a photograph showing the results of confirming drug sensitivity in human-tmpk constant expression MKN cells.
- Sorted human-tmpk constitutively expressing cells (tmpk) and non-infected cells (NC) were seeded at 1 ⁇ 10 5 cells per well in a 24 well plate, and AZT was set to 0 for both tmpk and NC groups for 3 days from the next day. , 0.1, 1, 10, 100, and 1000 mm, respectively.
- the mode of the cell after 3 days of AZT treatment in MKN is shown.
- the left is a tmpk constant expression cell group, and the right is an NC group.
- It is a graph which shows the result of having measured the viable cell number of the Jurkat cell 3 days after administration of the experiment of FIG. 14A by cell count. The numerical value is shown as a percentage (%) when the cell survival number of the drug 0 mg / mL is 100.
- the numerical value is shown as a percentage (%) when the cell survival number of the drug 0 mg / mL is 100.
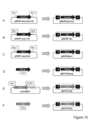
- A The survivin promoter part, which is a candidate for an undifferentiated specific promoter, was excised from pGEMT-easy-survivin that had already been prepared with NotI, subjected to blunt end treatment, subcloned into pENTR-vector, and pENTR-survivin Built.
- B The tert promoter part, which is a candidate for an undifferentiated specific promoter, was excised from pGEMT-EASY-tert that had already been prepared with MluI / BglII, blunt-ended, subcloned into pENTR-vector, and pENTR -Constructed Tert.
- C The Rex1 promoter part, which is a candidate for an undifferentiated specific promoter, was excised from pGEMT-easy-Rex1 that had already been prepared with MunI / NheI, blunt-ended, subcloned into pENTR-vector, and pENTR -Rex1 was constructed.
- D Nanog promoter which is a candidate for undifferentiated specific promoter is a material extracted from genomic DNA from mouse ES cells (mES), and CACC is used as a protruding end for pENTR-vector cloning on the 5 'side of Nanog promoter region.
- CAG promoter part which is a candidate for constitutive promoter was excised from pCX-EGFP which had already been prepared with SalI / EcoRI, blunted, and subcloned into pENTR-vector to construct pENTR-CAG did.
- a PGK promoter which is a candidate for a constitutive promoter is prepared by PCR using a primer added with CACC as a protruding end for pENTR-vector cloning on the 5 ′ side and a primer added with a PstI site on the 3 ′ side. After amplification, pENTR-PGK was constructed by cloning into pENTR-vector. Among the plasmids shown in FIG. 7, various promoters were inserted into the RC part of pLenti6-RC-Venus-2A-HSV-tk by recombination.
- tert, Rex1, and Nanog were inserted as undifferentiated specific promoters, and constitutive promoters CAG and PGK were inserted as controls.
- various promoters were inserted into the RC part of pLenti6-RC-Venus-2A-human-tmpk by recombination.
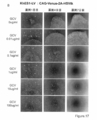
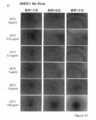
- Survivin, tert, Rex1, and Nanog were inserted as undifferentiated specific promoters, and constitutive promoters CAG and PGK were inserted as controls. It is the photograph which confirmed the expression of Venus in the cell which infected the lentiviral vector which has survivin-Venus-2A-HSV-tk to the KhES1 cell.
- the state of the cells after 1, 4, and 7 days from GCV treatment is shown from the left. It is the photograph which confirmed the expression of Venus in the cell which infected the lentiviral vector which has CAG-Venus-2A-HSV-tk to the KhES1 cell. The state of the cells after 1, 4, and 7 days from GCV treatment is shown from the left. It is the photograph which confirmed the expression of Venus in the cell which infected the lentiviral vector which has survivin-Venus-2A-puromycin with no HSV-tk to KhES1 cell. The state of the cells after 1, 4, and 7 days from GCV treatment is shown from the left. It is the photograph of the cell which infected the lentiviral vector non-infected KhES1 cell (No virus).
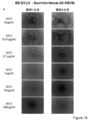
- the state of the cells after 1, 4, and 7 days from GCV treatment is shown from the left. It is the photograph which confirmed the expression of Venus in the cell which infected the lentiviral vector which has survivin-Venus-2A-HSV-tk to the D3 cell. From the left, the state of the cells 1 to 4 days after GCV treatment is shown. It is the photograph which confirmed the expression of Venus in the cell which infected the lentiviral vector which has CAG-Venus-2A-HSV-tk to the D3 cell. From the left, the state of the cells 1 to 4 days after GCV treatment is shown.
- FIG. 18 is a result of measuring the cell viability of each cell by WST-8 Assay 7 days after GCV treatment of the cells shown in FIGS. 18A-D.
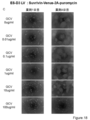
- Each group shows the survival rate after administration of survivin-Venus-2A-HSV-tk, CAG-Venus-2A-HSV-tk, survivin-Venus-2A-puromycin and uninfected D3 cells from the left. It is the photograph which infected the lentiviral vector which has survivin-Venus-2A-HSV-tk to KhES1 cell, and confirmed the expression of Venus after differentiation induction processing. From the left, the state of the cells 1 to 4 days after GCV treatment is shown. It is the photograph which infected the lentiviral vector which has CAG-Venus-2A-HSV-tk to KhES1 cell, and confirmed the expression of Venus after differentiation induction processing.
- FIG. 18 is a result of measuring the cell viability of each cell by WST-8 Assay 7 days after GCV treatment of the cells shown in FIGS. 18A-D.
- Each group shows the survival rate after administration of survivin-Venus-2A-HSV-tk, CAG-Venus-2A-HSV-tk, survivin-Venus-2A-puromycin and uninfected KhES1 cells from the left.
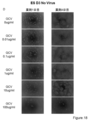
- the cells were subcutaneously transplanted into an immunodeficient mouse (NOD-Scid mouse) and a teratoma formation experiment was conducted. went. The hematoxylin-eosin (HE) dyeing
- HE hematoxylin-eosin
- the vector of the present invention comprises a nucleic acid sequence in which a marker gene and a killing gene are linked via a sequence capable of simultaneously expressing the two genes by one promoter, and a promoter region.
- the viral vector of the present invention can incorporate an arbitrary test promoter region or a target promoter region using a recombination method.
- the gene transfer by recombination can be carried out by a method well known to those skilled in the art by using a shuttle vector and a recombinase corresponding to the gene sequence used for the recombination cassette in the vector.
- the viral vector of the present invention specifically labels undifferentiated cells remaining after differentiation treatment, and / or Can be used for killing methods.
- the “target cell” may be a stem cell (for example, a stem cell before differentiation treatment, a stem cell under differentiation treatment, etc.) or a cell after differentiation treatment of the stem cell.
- the “target cell” that is infected with the viral vector of the present invention in the method of the present invention means a target cell that is infected with the vector of the present invention, and when it remains or develops as an undifferentiated cell after differentiation induction treatment.
- any cell that is intended to be removed or detected may be before or after differentiation induction.
- a differentiation treatment step for inducing differentiation of cells infected with the virus vector of the present invention may be included.
- “differentiated cells” or “differentiated cells” mean cells obtained by subjecting stem cells to differentiation treatment.
- the “undifferentiated cell-specific marker / killing gene expression vector” means the vector of the present invention described above, and specifically, the marker gene and the killing gene are one.
- Undifferentiated cell-specific labeling method for example, the viral vector of the present invention having an undifferentiated cell-specific promoter region is used for specifically labeling undifferentiated cells remaining or generated after differentiation of stem cells. Can be used.
- the present invention relates to a method for determining whether or not undifferentiated cells remain in cells after differentiation treatment by specifically labeling undifferentiated cells.
- the present invention includes A) a step of infecting a target cell with an undifferentiated cell-specific labeling / killing gene expression vector, and B) detecting an expression product of the marker gene in the cell after differentiation treatment.
- the method does not include an infection step by using a cell in which a stem cell into which the vector of the present invention has already been introduced has been induced to differentiate or a cell in which a stem cell has been induced to differentiate and into which the vector of the present invention has been introduced.
- the present invention comprises: A) detecting an expression product of the marker gene in a cell after differentiation treatment into which an undifferentiated cell-specific marker / killing gene expression vector has been introduced; and B) expression of the marker gene When a product is detected, there may be a method for determining the presence or absence of undifferentiated cells in a cell after differentiation treatment, comprising a step of determining that undifferentiated cells are present.
- the method of the present invention is a method for determining the content or content ratio of undifferentiated cells using as an index the level (amount) of the marker gene contained in the cell population, instead of simply determining the presence or absence of undifferentiated cells.
- the method of the present invention comprises A) a step of infecting a target cell with an undifferentiated cell-specific labeling / killing gene expression vector, and B) an expression product of the marker gene in the cell after differentiation treatment.
- a method for determining an amount (or percentage) comprising:
- the present invention relates to a method in which the level of the expression product of the marker gene indicates the amount (or ratio) of undifferentiated cells.
- the method does not include an infection step by using a cell in which a stem cell into which the vector of the present invention has already been introduced has been induced to differentiate or a cell in which a stem cell has been induced to differentiate and into which the vector of the present invention has been introduced. May be.
- the present invention includes A) a step of measuring the level of the expression product of the marker gene in a cell after differentiation treatment into which an undifferentiated cell-specific marker / killing gene expression vector has been introduced, and B) the measured A method for determining the amount (or ratio) of undifferentiated cells in a cell after differentiation treatment, comprising the step of determining the amount (or ratio) of undifferentiated cells from the level of an expression product of a marker gene,
- the level of the expression product of the marker gene may indicate the amount (or ratio) of undifferentiated cells.
- “level” usually indicates the amount of the expression product of the marker gene, but other values (for example, measurement) that serve as an indicator of the amount. Fluorescent intensity etc.) may be used.
- “determining the amount (or ratio) of undifferentiated cells from the measured level of the expression product of the labeled gene” means, for example, the level of the expression product of the labeled gene and the cell in a standard cell prepared in advance. It may mean that the specific number of assumed cells is calculated from a calibration curve with the number, or the amount (or ratio) of undifferentiated cells in comparison with other samples or measured values at different processing elapsed times.
- differentiated cells and undifferentiated cells can be distinguished at the cell level.
- a method for identifying an undifferentiated cell (or a position of an undifferentiated cell) included in a cell group subjected to differentiation treatment comprises: A) a step of infecting a target cell with an undifferentiated cell-specific labeling / killing gene expression vector, and B) an expression product of the marker gene in the cell after differentiation treatment.
- the present invention relates to a method for identifying undifferentiated cells (or positions of undifferentiated cells) in treated cells.
- the present invention relates to an undifferentiated cell (or an undifferentiated cell) in a cell in which a stem cell into which the vector of the present invention has already been introduced has been induced to differentiate, or in a cell in which a stem cell has been induced to differentiate and introduced into the vector of the present invention.
- the present invention includes A) a step of measuring an expression product of the marker gene in a cell after differentiation treatment into which an undifferentiated cell-specific marker / killing gene expression vector has been introduced, and B) expression of the marker gene.
- the present invention relates to a method for identifying a position.
- the measurement of the expression product of the marker gene is performed by a method that can identify the cell expressing the expression product or the location of the cell. .
- the present invention since differentiated cells and undifferentiated cells can be distinguished at the cell level, the undifferentiated cells contained in the cell population after differentiation induction can also be counted as the number of cells. Therefore, the present invention provides A) a step of infecting a target cell with an undifferentiated cell-specific labeling / killing gene expression vector, and B) counting the number of cells expressing the marker gene in the cells after differentiation treatment. And C) a method of determining the number of undifferentiated cells in a cell after differentiation treatment, comprising the step of determining that the number of cells expressing the marker gene is the number of undifferentiated cells.
- the present invention is a method for determining the number of undifferentiated cells in a cell in which a stem cell into which the vector of the present invention has already been introduced has been induced to differentiate, or in a cell into which a stem cell has been induced to undergo differentiation induction and the vector of the present invention has been introduced.
- the present invention includes A) a step of counting the number of cells that have been infected with an undifferentiated cell-specific labeling / killing gene expression vector and in which the marker gene is expressed in the cells after differentiation treatment.
- the method for determining the number of undifferentiated cells contained in the cell population after differentiation is induced by setting a predetermined number of cells, a predetermined culture area, or a predetermined culture amount as a target to be counted.
- the number of undifferentiated cells per number, the number of undifferentiated cells per predetermined culture region, or the number of undifferentiated cells per predetermined culture amount may be determined.
- the above-described method for determining the presence, abundance or number of cells, or identification method of undifferentiated cells can also be used as a method for monitoring cell quality by performing over time.
- the present invention A) A differentiation-treated cell or a differentiation-treated cell, into which an undifferentiated cell-specific labeling / killing gene expression vector has been introduced and the differentiation-expressed cell expression product Detecting step, and B) A method for monitoring undifferentiated cells after differentiation of stem cells, comprising a step of determining that undifferentiated cells remain or have been generated when an expression product of the marker gene is detected.
- the monitoring method of the present invention can be carried out at two or more points in time and compared with past values to confirm increase / decrease in undifferentiated cells or change over time in development.
- the marker gene of the present invention can be detected noninvasively, the monitoring can be performed in real time while culturing the cell or after transplanting the cell into the living body. Therefore, the method of the present invention provides A) a differentiation-treated cell or a differentiation-treated cell, in which the cell is introduced with an undifferentiated cell-specific labeling / killing gene expression vector.
- the present invention further includes a method for monitoring undifferentiated cells generated after transplantation of differentiated cells into the body.
- the present invention provides A) transplantation of a cell that has undergone differentiation treatment, and into which the cell has been introduced with an undifferentiated cell-specific marker / killing gene expression vector (hereinafter referred to as “transplanted cell”). Detecting the expression product of the marker gene, measuring the level, identifying or measuring the level of the marker gene in the treated patient, and B) detecting or increasing the expression product of the marker gene based on the result determined by the step.
- the present invention relates to a method for monitoring the presence or absence of undifferentiated cells in transplanted cells, comprising the step of determining that undifferentiated cells are generated or increasing.
- whether or not “the determined result means that the expression product of the marker gene is detected or increased” is determined by the above-described determination of the presence, abundance or number of cells of undifferentiated cells. It can be determined according to the method or the identification method.
- the viral vector of the present invention having a promoter region specific to undifferentiated cells is used in a method for specifically killing undifferentiated cells remaining after differentiation treatment. Can be used.
- the present invention comprises a step of infecting a target cell with an undifferentiated cell-specific label / killing gene expression vector, and a step of killing undifferentiated cells remaining after differentiation treatment with a killing gene. It may be a method of killing or removing undifferentiated cells remaining or generated after the treatment.
- the killing gene is not a drug-dependent or inducible killing gene
- the target cell is a cell after differentiation induction.
- the method of the present invention can administer a drug that induces the expression of the drug-dependent killing gene. Includes steps. Therefore, the present invention provides A) a nucleic acid sequence in which a marker gene and a drug-dependent killing gene are linked via a sequence capable of simultaneously expressing the two genes by a single promoter, and undifferentiated cell-specific A viral vector having a recombination cassette containing a typical promoter, wherein the promoter region is linked to express the marker gene and the drug-dependent killing gene (hereinafter referred to as “undifferentiated” in the present specification).
- a step of killing remaining undifferentiated cells with a drug-dependent killing gene may be a method of killing or removed after treatment.
- the target cell when using a drug-dependent killing gene as a killing gene, the target cell may be a stem cell before or during differentiation, or may be a cell after differentiation.
- a method for specifically killing undifferentiated cells remaining after differentiation treatment is performed on cells after differentiation induction treatment into which the virus vector of the present invention has already been introduced. It can also be carried out by administering a drug that specifically kills undifferentiated cells. That is, the present invention is a differentiation-treated cell, and the drug-dependent killing gene can exert toxicity on a cell into which the undifferentiated cell-specific label / drug-dependent killing gene expression vector has been introduced. It may be a method of removing undifferentiated cells remaining or generated after differentiation treatment, comprising a step of killing remaining undifferentiated cells by a killing gene by administering a drug.
- the method of the present invention may be a method of removing undifferentiated cells remaining or generated in cells transplanted into a patient's body after differentiation induction treatment. That is, the method of the present invention comprises: A) a cell differentiated from a stem cell, into which the cell has been introduced with an undifferentiated cell-specific label / drug-dependent killing gene expression vector (hereinafter referred to as “transplanted cell”).
- a method of killing or removing undifferentiated cells that remain in the transplanted cells or that have occurred in the transplanted cells, comprising administering a drug capable of exerting toxicity to the drug-dependent killing gene to a patient transplanted with It may be.
- the dose of a drug capable of exhibiting toxicity by a drug-dependent killing gene can be appropriately selected according to the type of each drug-dependent killing gene.
- the drug-dependent killing gene can exhibit toxicity. More preferably, it is an amount capable of killing cells in which the drug-dependent killing gene is expressed.
- the method of the present invention includes an administration step of a drug that induces the expression of the inducible killing gene. Therefore, the present invention provides A) a nucleic acid sequence in which a marker gene and an inducible killing gene are linked via a sequence capable of simultaneously expressing the two genes by a single promoter, and undifferentiated cell-specific A viral vector having a recombination cassette containing a promoter, wherein the promoter region is linked to express the marker gene and the drug-dependent killing gene (hereinafter referred to as “undifferentiated cells” in the present specification).
- a specific label / inducible killing gene expression vector ”), infecting the target cell; and B) remaining by adding a stimulus capable of exerting toxicity to the inducible killing gene to the differentiated cell.
- Remaining after stem cell differentiation treatment comprising the step of killing undifferentiated cells to be killed by an inducible killing gene
- That or undifferentiated cells generated may be a method of killing or removal.
- the target cell when using an inducible killing gene as a killing gene, the target cell may be a stem cell before or during differentiation, or may be a cell after differentiation.
- a method for specifically killing undifferentiated cells remaining after differentiation treatment is a method for the cells after differentiation induction treatment into which the virus vector of the present invention has already been introduced. It can also be performed by administering a drug that specifically kills undifferentiated cells. That is, the present invention provides a drug capable of exerting toxicity to the inducible killing gene in a cell that has been subjected to differentiation treatment, and into which the cell has been introduced with an undifferentiated cell-specific label / inducible killing gene expression vector. It may be a method of removing undifferentiated cells remaining or generated after differentiation treatment, comprising a step of killing undifferentiated cells remaining by administration with a killing gene.
- each of the above methods is a step of infecting if necessary (or a step of differentiation treatment if necessary) Thereafter, a step of transplanting the cells into a necessary organ or tissue of a patient may be included, or all the steps without including the transplanting step may be performed in vitro. Therefore, in the above description, “differentiated cells” or “differentiated cells” may be before transplantation or after transplantation.
- a drug is administered after transplantation, it can be performed in accordance with the drug administration method in ordinary pharmaceuticals, and can be administered, for example, in an oral dosage form or a parenteral dosage form such as an injection or infusion. .
- a drug When a drug is administered to a mammal or the like, it may be administered orally as a tablet, powder, granule, syrup or the like, or may be administered parenterally as an injection or infusion. Dosage depends on the number of cells transplanted, the tissue or site, the time since transplantation, the amount of undifferentiated cells remaining or generated, the age, weight and sex of the patient, the route of administration, the mode of administration, the drug selected It can be set as appropriate depending on the suicide gene and the type of drug. For example, it is usually possible to administer 50 to 500 mg per adult in 1 to several divided doses.
- the present invention can be used in a method for measuring the activity of a test promoter in undifferentiated cells using the viral vector of the present invention.
- the method of the present invention comprises: A) recombining the test promoter in the promoter region of the undifferentiated cell-specific labeling / killing gene expression vector to produce a viral vector having the test promoter; B) Infecting a subject cell with a viral vector having a test promoter; C) detecting the expression of the marker gene in the cell after differentiation induction treatment; D) identifying whether the cell after differentiation induction treatment is an undifferentiated cell or a differentiated cell, and E) determining whether the test promoter is specific to the undifferentiated cell.
- a screening method for an undifferentiated cell-specific promoter comprising:
- the determination has a high rate at which the expression of the labeled gene is detected in cells identified as undifferentiated cells, and the rate at which the expression of the labeled gene is detected in cells identified as differentiated cells. If low, it relates to a method performed by determining that the test promoter is specific for undifferentiated cells.
- the screening method may be, for example, the following method: A) recombining the test promoter with the promoter part of the undifferentiated cell-specific labeling / killing gene expression vector to produce a viral vector having the test promoter; B) Infecting undifferentiated cells (eg, ES cells or iPS cells) with a viral vector having a test promoter, C) differentiating the undifferentiated cells; D) detecting the expression of the marker gene in each cell after differentiation induction treatment; E) identifying whether each cell after differentiation induction treatment is an undifferentiated cell or a differentiated cell, and F) determining whether the test promoter is specific to the undifferentiated cell.
- a screening method for an undifferentiated cell-specific promoter comprising:
- the determination has a high rate at which the expression of the labeled gene is detected in cells identified as undifferentiated cells, and the rate at which the expression of the labeled gene is detected in cells identified as differentiated cells. If low, it relates to a method performed by determining that the test promoter is specific for undifferentiated cells.
- the step of discriminating whether each cell after differentiation induction treatment is an undifferentiated cell or a differentiated cell includes alkaline phosphatase staining, SSEA3 staining, SSEA4 staining, Tra-1-60 staining, and / or Cells positive for Tra-1-81 staining; Oct3 / 4, Nanog, Sox2, Cripto, Dax1, ERas, Fgf4, Esg1, Rex1, zfp296, UTF1, GDF3, Sall4, Tbx3, Tcf3, DNMT3MT, and / or DMT3L
- a cell expressing a gene a cell expressing miR-290 and / or miR-302 as an undifferentiated cell, and a cell negative for the staining or a cell not expressing the gene as a differentiated cell This can be done by identifying.
- the expression of the marker gene is detected in the cells identified as undifferentiated cells, and the expression of the marker gene is detected in the cells identified as differentiated cells.
- Low means that the expression level of the early marker gene in the differentiated and undifferentiated cells is compared, and the expression level of the early marker gene in the undifferentiated cell is higher than the expression level of the early marker gene in the differentiated cell. To do.
- the ratio is low in which the expression of the marker gene is detected in the cells identified as the “high ratio” differentiated cells in which the expression is detected.
- the viral vector of the present invention is a stem cell before differentiation treatment or during differentiation treatment (for example, pluripotent stem cells such as ES cells or iPS cells), or after differentiation treatment. Can infect cells.
- the cytotoxicity of the vector of the present invention is drug-dependent, even if the cell exhibits promoter activity, the vector of the present invention does not exhibit cytotoxicity under the condition that no drug is added. Therefore, the context of the differentiation step and the infection step is not limited because of the damage to the stem cells before the differentiation treatment due to the cytotoxicity of the introduced vector.
- the infection is performed in vitro before the differentiation process.
- the viral vector of the present invention may be infected after stem cell differentiation treatment.
- the viral vector of the present invention is infected before differentiation treatment.
- the infection is performed prior to transplantation to ensure infection of all subject cells. Infection can be performed using methods well known to those skilled in the art by appropriately selecting the number of viral vectors and the duration of infection according to the type and nature of the viral vector used. For example, infection can be performed in the range of MOI 1-1000, preferably in the range of MOI 10-100.
- the method of the present invention is a virus having a recombination cassette comprising a nucleic acid sequence in which a marker gene and a drug-dependent suicide gene are bound via a 2A sequence, and an undifferentiated cell-specific promoter region.
- a viral vector in which the promoter region is linked to express the marker gene and the drug-dependent suicide gene in an undifferentiated cell for example, pluripotent stem cell such as ES cell or iPS cell
- a step (differentiation treatment step) of inducing the undifferentiated cells into differentiated cells may be further included before, after, or simultaneously with the infection step performed by the infection.
- the cell differentiation induction treatment can be performed by bringing a differentiation inducer for differentiation induction into contact with an undifferentiated cell and culturing under an appropriate culture condition.
- Various culture conditions and culture methods for differentiation induction treatment have been reported, and can be appropriately selected according to the type of cells to be used and the cells to be differentiated.
- Detection, identification, and measurement of marker gene The method for detecting, identifying, or measuring the level of the expression product of the marker gene can be selected as appropriate according to the marker gene used. For example, when a fluorescent gene is used, the expression product can be detected using a fluorescence detection device, flow cytometry, a real-time fluorescence detection device, or the like.
- Example 1 Preparation of lentiviral vector plasmid (1) Design of lentiviral vector plasmid A schematic diagram of a vector to be prepared is shown in FIG. A lentiviral vector was used as the vector base, and the following recombination cassette (RC) was used for the promoter portion. This utilizes a site-specific recombination system involved in the entry of ⁇ phage into the E. coli chromosome, and exchanges DNA sequences sandwiched between att sequences modified between vectors (James, L. et al. (2000). ) Genome Res. 10: 1788-1795).
- RC recombination cassette
- the RC portion has a DNA sequence (att R sequence) that specifically interacts at the time of recombination at both ends, and has a chloramphenicol resistance gene (CMR) and ccdB, which is an E. coli suicide gene, between them.
- CMR chloramphenicol resistance gene
- ccdB chloramphenicol resistance gene
- a shuttle vector plasmid for gene recombination was prepared by cloning a lentiviral vector in which RC was inserted into the promoter site and various promoter sequences between att L sequences corresponding to the att R sequence of RC (FIGS. 1 and 2A).
- mKate2 As fluorescent proteins, the luminous efficiency of mKate2, which has a fluorescence intensity about 100 times that of TAGFP635, which is a near-infrared fluorescent protein, and Green Fluorescent Protein (GFP), which is a green fluorescent protein derived from a luminescent jellyfish, are improved. These two types were used so that they could be used properly according to the purpose.
- HSV-tk HSV-tk is sensitive to GCV and has been reported to act as a suicide gene (Yasuhiro TERAZAKI et al. (2003) Hepatology 37: 155-63).
- GCV is phosphorylated by HSV-tk, and further phosphorylated by an endogenous kinase in human cells, and finally has a highly toxic gancyclovir triphosphate (GCV).
- GCV highly toxic gancyclovir triphosphate
- GCV-3P inhibits DNA synthesis, and thus the HSV-tk gene-introduced cell is killed.
- HSV-tk it is well known for its powerful tumor therapeutic effect, which is a bi-standard effect on tumor cells, in which many of the surrounding tumor cells into which the HSV-tk gene has not been introduced are also killed. ing. Therefore, the combination of HSV-tk and GCV can strongly cause killing action selectively on cells with vigorous cell division (such as tumor cells and stem cell undifferentiated cells).
- human-tmpk / AZT HSV-tk has a very high killing effect, but since it is derived from herpes simplex, an immune response occurs in the human body, and the problem of eliminating genes introduced into cells is also inferred. Therefore, a method of combining the human-derived enzyme Thymylate Kinase (tmpk) with 3′-azido-3′-deoxythymidine (AZT) was also examined.
- AZT a prodrug, is phosphorylated and converted to AZT triphosphate (AZT-3P).
- AZT-3P inhibits human immunodeficiency virus (HIV) replication and eukaryotic DNA synthesis (Fei CHEN et al.
- the rate-limiting enzyme in the conversion of AZT to toxic AZT-3P is a cellular Thymylate Kinase (tmpk) that catalyzes the conversion of AZT monophosphate (AZT-MP) to AZT diphosphate (AZT-DP).
- tmpk Thymylate Kinase
- tmpk has low enzyme efficiency
- tmpk F105Y
- tmpk seems to retain all the advantages described in the above HSV-tk as a suicide gene. Further advantages include killing effects of higher tumor cells and undifferentiated cells, and immune induction from heterologous proteins. It is possible that there is no. However, there have been no reports of applying tmpk to stem cells, not limited to the vector system of the present invention.
- iCaspase9 / Dimeriser Unlike the two genes described above, it is a gene that induces cell death by inducing apoptosis.
- Caspase constitutes apoptotic signaling.
- Caspase-9 which is involved in the early stage of the caspase pathway from mitochondria, is usually in a state of being phosphorylated and inactivated by AKT or the like. Inactive caspase-9 is activated by stimulation from mitochondria, and apoptosis is caused by activating caspase-3 and caspase-7.
- a drug-induced Caspase-9 activation system was used.
- AP20187 which is a cell membrane-permeable low molecular weight compound that binds to FKBP after introducing and expressing iCaspase9 (iCasp9), which is a fusion protein of the Caspase-9 gene and the human FK506 binding protein (FKBP) protein gene, in a target cell.
- iCasp9 iCaspase9
- FKBP human FK506 binding protein
- Plasmid construction was performed in two steps as shown in detail below. That is, a lentiviral vector inserted with a recombination cassette (RC) (FIG. 1A) and a fluorescent protein-2A-suicide gene cassette (FIG. 1B) were prepared and finally combined into a lentiviral vector. The plasmid was used (FIG. 1C).
- mKate2 was designed with primers with AgeI on the 5 ′ side and BGI II site added on the 3 ′ side (Table 1), amplified by PCR method, and then PCR product cloning vector pGEM-Teasy vector (Invitrogen) To construct pGEM-Teasy-mKate2 (FIG. 3A). After cloning, sequence analysis was performed using 5'-side T7 and 3'-side M13R from the primer sequences at both ends of the cloning site in pGEM-Teasy vector.
- pFlag-CMV-2A in which a synthetic double-stranded 2A sequence DNA oligo having a protruding end of a BamHI site on the 5 ′ side and a BamHI site on the 3 ′ side is inserted into pFlag-CMV-2 (SIGMA) cleaved with BglII / BamHI
- SIGMA pFlag-CMV-2
- This pFlag-CMV-2A was similarly cleaved with Not I / BglII, and mKate2 was inserted between the CMV promoter and 2A sequence to construct pFlag-CMV-mKate2-2A (FIG. 4A).
- a primer with NotI on the 5 ′ side and a BamHI site on the 3 ′ side was designed for Venus (Table 1), amplified by PCR, cloned into pGEM-Teasy vector, and pGEM-Teasy-Venus was cloned. It was constructed. After cloning, sequence analysis was performed using the primers. Next, Venus was cut out from pGEM-Teasy-Venus at the NotI / BamHI site (FIG. 4B) and inserted into pFlag-CMV-2A in the same manner as mKate2 to construct pFlag-CMV-Venus-2A (FIG. 4B). .
- HSV-tk Suicide gene (HSV-tk, human-tmpk)
- primers were prepared (Table 1), amplified by PCR, and then cloned into a cloning vector pGEM-Teasy vector to construct pGEM-Teasy-HSV-tk (FIG. 3).
- sequence analysis was performed with the above primers in the same manner as mKate2 and Venus.
- pGEM-Teasy-HSVtk was cleaved with SPEI on the 3 ′ side of HSV-tk, blunt-ended, and cleaved with BamHI on the 5 ′ side of HSV-tk to obtain HSV-tk (FIG. 5A). ).
- primers were prepared in the same manner as HSV-tk (Table 1), amplified by PCR, and then cloned into pGEM-Teasy vector to construct pGEM-Teasy-human-tmpk (FIG. 3D). . After cloning, sequence analysis was performed using the above primers in the same manner as mKate2-Venus. Next, human-tmpk was obtained by cutting from pGEM-Teasy-human-tmpk with 5′-side BamHI and 3′-side SmaI (blunt end) (FIG. 5A).
- the gene fragment obtained in (B) was inserted into the plasmid prepared in (A) to prepare a vector plasmid having an expression cassette for fluorescent protein-2A-suicide gene.
- pFlag-CMV-mKate2-2A and pFlag-CMV-Venus-2A were cleaved with 3′-side BstXI, blunt-ended, and cut out with BamHI on the 5 ′ side (FIG. 5B).
- the HSV-tk or human-tmpk obtained in (B) was inserted into the 3 ′ side of 2A, and four types of plasmids, pFlag-CMV-mKate2-2A-HSVtk, pFlag-CMV-mKate2-2A-tmpk, pFlag -CMV-Venus-2A-HSVtk and pFlag-CMV-Venus-2A-tmpk were constructed.
- the 3 ′ side was cleaved with KpnI, blunt-ended, and the 5 ′ side was cleaved with AgeI (FIG. 7A).
- pFlag-CMV-mKate2-2A-tmpk and pFlag-CMV-Venus-2A-tmpk are cut on the 5 ′ side with AgeI and on the 3 ′ side with KpnI, respectively, and mKate2-2A-tmpk and Venus ⁇ The 2A-tmpk portion was cut out (FIG. 6B).
- HSVtk has a KpnI cleavage site inside, pFlag-CMV-mKate2-2A-HSVtk and pFlag-CMV-Venus-2A-HSVtk are cut with AgeI on the 5 'side and PmeI (blunt end) on the 3' side. Then, mKate2-2A-HSVtk and Venus2-2A-HSVtk were cut out, respectively (FIG. 7B).
- pLenti-RC-mKate2-2A-tmpk By ligating these in combination, pLenti-RC-mKate2-2A-tmpk, pLenti-RC-Venus-2A-tmpk, pLenti-RC-mKate2-2A-HSVtk, and pLenti-RC-Venus-2A- HSVtk was prepared.
- Plasmids were constructed using In-Fusion (registered trademark) HD Cloning Kit (TAKARA BIO), which is a DNA fragment prepared by PCR or a linearized vector containing 15 bases homologous to the ends of each DNA fragment.
- plasmid having the expression cassette of fluorescent protein-2A-suicide gene was introduced into HEK293 cells and evaluated at each stage of preparation, thereby confirming the expression of fluorescence.
- HSK293 cells were seeded on a 6-well plate at 6 ⁇ 10 5 cells / well, and the next day, the plasmid was introduced at 2 mg / well, using a Polyfect Transfection Reagent Kit (QIAGEN) according to the protocol provided by the manufacturer. The expression of fluorescence was confirmed with a fluorescence microscope 48 hours after gene introduction.
- Example 2 Verification of cytotoxic effect of suicide gene in cultured human cells
- three types of suicide genes HSV-tk, human-tmpk, and iCaspase9 were expressed. After the cells were infected with the virus vector, the corresponding drugs were added to compare the toxic effects on each cell.
- HSV-tk used an adenovirus vector that had already been prepared (Yasuhiro Terazaki et al. (2003) Hepatology 37; 155-63).
- human-tmpk and iCaspase9 were assigned to the plasmids pHR′-cPPT-EF-Tmpk (F105Y) (Takeya Sato et al.
- the plasmid iCasp9-2A-DCD19RRF (Carlos A RAMOS et al. (2010) STEM Cells 28; 1107-15) was used. These plasmids were introduced directly into cells or lentiviral vectors were produced and used.
- iPS cells 201B7 derived from human adult fibroblasts (HDF) and introduced with 4 factors of OCT3 / 4, KLF4, SOX2 and c-Myc, and HDF derived c- 253G1 established by introducing three factors other than Myc was used.
- D3 cells were used as mouse ES cells.
- KhES1 assigned from Kyoto University
- normal human fibroblasts (BJ) and human cancer cells human T lymphoma cell line Jurkat, human gastric cancer cell MKN45
- Human iPS / ES cells were co-cultured on mouse fetal fibroblasts (MEF) treated with mitomycin C.
- Mouse ES cells were cultured on gelatin-coated dishes.
- the medium used for culturing each cell is as follows: (Human iPS / ES cells) 20% KSR (Invitrogen), 200 mM L-Glutamine, 0.1 mM Non Essential Amino Acid (Sigma), 0.1 mM 2-Mercaptoethanol (Sigma), 5 ng / mL Basic-Fgfin (Wakopyl / Wakopyl) (P / S) (Nacalai Tesque) -containing DMEM-F12 (Sigma) (Mouse ES cells) 20% Fetal Bovine Serum (FBS) (GIBCO), 0.1 mM Non Essential Amino Acid, 1 mM Sodium Pyruvate (Gibco), 70 ⁇ M 2-Mercaptoethanol, 1 ⁇ 10 3 unit / mL / S-containing DMEM (SIGMA) (Jurkat cells and MKN45 cells) RPMI-1640 (SIGMA
- CM Conditional Medium
- MEF medium of MEF (1.5 ⁇ 10 6 cells / 10 CM dish) treated with mitomycin C was replaced with a medium for human ES cells (BFGF-). After overnight culture in an incubator at 0 ° C., the supernatant was used.
- KhES1 was used for virus infection experiments by carrying out monodisperse culture under a feeder-free condition using a ROCK inhibitor (Y27632, WAKO).
- HEK293T cells were seeded at 6 ⁇ 10 5 cells / well in a 6-well plate coated with POLY-L-LYSINE (SIGMA).
- SIGMA POLY-L-LYSINE
- 0.5 mg of each lentivector plasmid was mixed with PLP1, PLP2, and PLP-VSVG, 50 mL of OPTI-MEM was added, 6 mL of FUGENE HD (PROMEGA) was added, and vortexed, and then at room temperature for 15 minutes. Left to stand.
- each cell was seeded in a 24-well plate at the following cell number: KhES1 cells 4 ⁇ 10 4 cells / well Jurkat cells 1.25 ⁇ 10 5 cells / well MKN45 cells 2 ⁇ 10 4 cells / well HEK293T cells 5 ⁇ 10 4 cells / well
- medium containing 4 mg / mL polybrene And 20 mL of each lentiviral vector was added to infect for 24 hours. All cell media were changed after 24 hours and continued to culture.
- Cells infected with an EGFP-expressing lentiviral vector were evaluated with a flow cytometer (Cyan Adp Analyzer, Beckman Coulter) 72 hours after infection, and the titer of the produced viral vector was calculated.
- Alexa Fluor 488 anti-Human SSEA4 (BIOLEGEND) and anti-Human CD19-APC (EBIOSCIENCE), which are undifferentiated cell-labeled antibodies, were added to a cell suspension (100 mL (1 ⁇ 10 6 cells)) on ice. The cells were labeled by allowing them to stand for 30 minutes while suspending the cells every 10 minutes. After washing once with PBS containing 10 mm Y27632, the suspension was suspended in a medium for ES containing 10 mm Y27632, and KhES1 labeled with both antibodies was evaluated as undifferentiated CD19-positive cells using a flow cytometer.
- HSV-tk HSV-tk adenovirus (Ad.CAG-HSV-tk) already prepared was used.
- human-tmpk and iCasp9 introduced the plasmid into the cells.
- plasmid As for human-tmpk and iCasp9, 0.5 mg of each plasmid was added to 25 mL of OPTI-MEM, 2 mL of FUGENE HD (QIAGEN) was added, and the whole volume was administered after standing for 15 minutes. After changing the medium 24 hours later, AZT was administered to each well at 0, 30, 100, and 300 mm for 3 days for the human-tmpk-introduced cells. For iCasp3, Dimerizer was administered at 0, 0.03, 0.1, 0.3 mm to each well for 3 days. After drug administration, the cytotoxic effect was measured by measuring the number of viable cells. On the measurement day, KhES1 was treated with CTK and separated into single cells, and 10 mL was collected.
- FUGENE HD QIAGEN
- a constant expression strain was prepared and further analyzed for human-tmpk, whose effect on drug sensitivity was observed by transient expression with a plasmid.
- a lentiviral vector expressing human-tmpk and CD19 was produced from PHR-CPPT-EF-tmpk (F105Y), and the cells were infected, and CD19 positive was collected as an indicator.
- KhES1, Jurkat, and MKN45 cells were collected, proliferated, and analyzed again by a flow cytometer using an anti-CD19 antibody, it was confirmed that CD19 positive cells were present at a higher rate than before the sorting. (FIGS. 14A-C).
- Example 3 Verification of cytotoxic effect of suicide gene in human cultured cells using the prepared lentiviral vector plasmid
- RC prepared in Example 1 was first used.
- Various promoters were inserted into the contained lentiviral vector plasmid by recombination to complete the plasmid.
- Lentiviral vectors were produced from these plasmids and the cells were infected, and then the corresponding drugs were added to compare the toxic effects on each cell.
- pENTR-vector contains an attL sequence and is used as a shuttle vector to incorporate the promoter into RC by reacting with pLenti6-RC in the LR clone.
- promoters for subcloning survivin, tert, Rex1, and Nanog were used as candidates for undifferentiation-specific promoters, and constitutive promoters CAG and PGK were used as controls, and inserted into pENTR-vector by the following methods.
- CAG promoter The CAG promoter portion was excised from pCX-EGFP that had already been prepared by SalI / EcoRI (blunted) and cloned into pENTR-vector to construct pENTR-CAG (FIG. 15E).
- F PGK promoter A primer with CACC added as a protruding end for pENTR-vector cloning on the 5 ′ side of the PGK promoter region and a PstI site added on the 3 ′ side (Table 3) is amplified by PCR. Was then cloned into pENTR-vector to construct pENTR-PGK (FIG. 15F).
- tert, Rex1, and Nanog were inserted as undifferentiated specific promoters, and the constitutive promoters CAG and PGK were inserted as controls (FIG. 16A). Also, among the plasmids shown in FIG. 7, the various promoters shown in FIG. 15 were inserted into the RC part of pLenti6-RC-Venus-2A-human-tmpk by recombination (pLenti6-Promoter-Venus-2A-human). -Tmpk). Survivin, tert, Rex1, and Nanog were inserted as undifferentiated specific promoters, and constitutive promoters CAG and PGK were inserted as controls (FIG. 16B).
- Alexa Fluor 488 anti-Human SSEA4 (BIOLEGEND), an undifferentiated cell-labeled antibody, was added at 5 mL per 100 mL of cell suspension (1 ⁇ 10 6 cells) and allowed to stand for 30 minutes while suspending the cells every 10 minutes on ice. The cells were labeled. After washing once with 10 mm Y27632-containing PBS, the suspension was suspended in a 10 mm Y27632-containing ES medium, and antibody-labeled GFP-positive cells were evaluated as undifferentiated lentivirus-infected KhES1 using a flow cytometer. D3 cells were trypsinized and the cell mass was dispersed into single cells by pipetting.
- the number of cells was counted, the required amount of D3 cells was centrifuged (1000 rpm, 5 minutes), resuspended in the medium, and GFP positive cells were evaluated as lentivirus-infected D3 cells using a flow cytometer. After confirming GFP positive cells with a flow cytometer and increasing the number of cells, GFP positive cells were sorted by BD FACSARIA II. The sorted cells were immediately centrifuged (1000 rpm, 5 minutes), resuspended in fresh medium, KhES1 cells were co-cultured with MEF cells, and D3 cells were seeded in gelatin-coated dishes.
- KhES1 cells were seeded at 1 ⁇ 10 5 cells per well in a 96-well plate and survivin-Venus-2A-HSV-tk (A), CAG-Venus-2A-HSV-tk (B) for 7 days from the next day.
- D3 cells were seeded at 1 ⁇ 10 5 cells per well in 96-well plates, and survivin-Venus-2A-HSV-tk (A), CAG-Venus-2A-HSV-tk (B) for 4 days from the next day.
- the cytotoxic effect after 4 days of administration was measured using WST-8 assay, which is a modified method of MTT assay.
- the reagent SF (Nacalai) used was a tetrazolium salt that produces a highly sensitive water-soluble formazan
- WST-8 [2- (20methoxy-4-nitrophenyl) -3- (nitrophenyl) -5- (2,4-disulphenphenyl)- 2H-tetrazolinium monosodium salt] is used as a chromogenic substrate, which enables measurement with higher sensitivity than conventional MTT, and WST-8 is reduced by intracellular dehydrogenase to produce water-soluble formazan.
- the number of viable cells was measured by directly measuring the absorbance of this formazan at 450 nm (E). The results are shown in FIG.
- Differentiated cells were replated at 1 ⁇ 10 5 cells per well in a 96-well plate, and survivin-Venus-2A-HSV-tk (A), CAG-Venus-2A-HSV-tk (A) for 4 days from the next day.
- B survivin-Venus-2A-puromycin
- C survivin-Venus-2A-puromycin
- NC non-infected cells
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Cell Biology (AREA)
- General Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Food Science & Technology (AREA)
- Pathology (AREA)
- Toxicology (AREA)
- General Physics & Mathematics (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Plant Pathology (AREA)
- Virology (AREA)
- Developmental Biology & Embryology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
Abstract
Description
(1) 標識遺伝子と殺傷遺伝子とが、一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列を介して結合した核酸配列、及び、プロモーター領域を含むリコンビネーションカセットを有するウイルスベクターであって、該プロモーター領域が、該標識遺伝子及び該殺傷遺伝子を発現可能に連結されているウイルスベクター。
(2) 殺傷遺伝子が、自殺遺伝子である、(1)に記載のウイルスベクター。
(3) 自殺遺伝子が、薬剤依存性自殺遺伝子である、(2)に記載のウイルスベクター。
(4) 薬剤依存性自殺遺伝子が、HSV-tk、human-tmpk、シトシンデアミナーゼ(CD)遺伝子、水痘ウイルスチミジンキナーゼ、又はCaspaseである、(3)に記載のウイルスベクター。
(5) 標識遺伝子が蛍光タンパク質である、(1)~(4)のいずれか1項に記載のウイルスベクター。
(6) 蛍光タンパク質が、赤色又は緑色の蛍光蛋白質である、(5)に記載のウイルスベクター。
(7) 蛍光タンパク質が、mKate2又は緑色蛍光蛋白質である、(5)に記載のウイルスベクター。
(8) 一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列が、2A配列又はIRES配列である、(1)~(7)のいずれか1項に記載のウイルスベクター。
(9) レンチウイルスベクターである、(1)~(8)のいずれか1項に記載のウイルスベクター。
(10) プロモーター領域が未分化細胞特異的プロモーター領域である、(1)~(9)のいずれか1項に記載のウイルスベクター。
(11) 未分化細胞特異的プロモーターが、survivinプロモーター又はtertプロモーターである、(10)に記載のウイルスベクター。
(12) 未分化細胞特異的プロモーターが、survivinプロモーターである、(10)に記載のウイルスベクター。
(13)A)(1)~(9)のいずれか1項に記載のウイルスベクターのプロモーター部分に被験プロモーターを組み替えて被験プロモーターを有するウイルスベクターを作製するステップ、
B)被験プロモーターを有するウイルスベクターを対象の細胞に感染させるステップ、
C)分化誘導処理後の前記細胞における前記標識遺伝子の発現を検出するステップ、
D)分化誘導処理後の前記細胞が未分化細胞であるか、分化細胞であるかを識別するステップ、及び
E)被験プロモーターが未分化細胞に特異的であるか否かを判定するステップを備える、未分化細胞特異的プロモーターのスクリーニング方法であって、
ここで、該判定が未分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が高く、かつ、分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が低い場合、該被験プロモーターは未分化細胞に特異的であると判定することにより行われる方法。
(14) A)(10)~(12)のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中の前記標識遺伝子の発現産物を検出するステップ、及び、
C)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が存在すると判定するステップを備える、分化処理後の細胞における未分化細胞の有無を判定する方法。
(15) A)(10)~(12)のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中の前記標識遺伝子の発現産物を検出するステップ、及び、
B)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が存在すると判定するステップを備える、分化処理後の細胞における未分化細胞の有無を判定する方法。
(16) A)(10)~(12)のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における前記標識遺伝子の発現産物を同定するステップ、及び、
C)前記標識遺伝子が発現している細胞が未分化細胞であると同定するステップを備える、分化処理後の細胞における未分化細胞を同定する方法。
(17) A)(10)~(12)のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における前記標識遺伝子の発現産物を同定するステップ、及び、
B)前記標識遺伝子が発現している細胞が未分化細胞であると同定するステップを備える、分化処理後の細胞における未分化細胞を同定する方法。
(18) A)(10)~(12)のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における、前記標識遺伝子の発現産物のレベルを測定するステップ、及び、
C)測定された前記標識遺伝子の発現産物のレベルから、未分化細胞の量(又は割合)を決定するステップを備える、分化処理後の細胞における未分化細胞の量(又は割合)を決定する方法であって、
ここで、前記標識遺伝子の発現産物のレベルが、未分化細胞の量(又は割合)を示す方法。
(19) A)(10)~(12)のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における、前記標識遺伝子の発現産物のレベルを測定するステップ、及び、
B)測定された前記標識遺伝子の発現産物のレベルから、未分化細胞の量(又は割合)を決定するステップを備える、分化処理後の細胞における未分化細胞の量(又は割合)を決定する方法であって、
ここで、前記標識遺伝子の発現産物のレベルが、未分化細胞の量(又は割合)を示す方法。
(20) A)(10)~(12)のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における、前記標識遺伝子が発現している細胞数を計数するステップ、及び、
C)前記標識遺伝子が発現している細胞数が未分化細胞の数であると決定するステップを備える、分化処理後の細胞中における未分化細胞数を決定する方法。
(21) A)(10)~(12)のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における、前記標識遺伝子が発現している細胞数を計数するステップ、及び、
B)前記標識遺伝子が発現している細胞数が未分化細胞の数であると決定するステップを備える、分化処理後の細胞中における未分化細胞の数を決定する方法。
(22) A)(10)~(12)のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の細胞中における、前記標識遺伝子の発現産物を検出するステップ、及び、
C)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が残存し又は発生したと判定するステップを備える、分化処理後に残存又は発生する未分化細胞のモニタリング方法。
(23) A)(10)~(12)のいずれか1項に記載のウイルスベクターが導入され、分化誘導処理された細胞における、前記標識遺伝子の発現産物を検出するステップ、及び、
B)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が残存又は発生していると判定するステップを備える、幹細胞の分化処理後の未分化細胞のモニタリング方法。
(24) A)(10)~(12)のいずれか1項に記載のウイルスベクターであって、殺傷遺伝子が薬剤依存性殺傷遺伝子であるウイルスベクターを対象の細胞に感染させるステップ、及び、
B)前記細胞に前記薬剤依存性殺傷遺伝子が毒性を発揮可能な薬剤を投与するステップを備える、未分化細胞を殺傷する方法。
(25) (10)~(12)のいずれか1項に記載のウイルスベクターであって、殺傷遺伝子が薬剤依存性殺傷遺伝子であるウイルスベクターが導入された細胞に前記薬剤依存性殺傷遺伝子が毒性を発揮可能な薬剤を投与するステップを備える、未分化細胞を殺傷する方法。
(26) (10)~(12)のいずれか1項に記載のウイルスベクターを含む、未分化細胞標識剤。
(27) (10)~(12)のいずれか1項に記載のウイルスベクターを含む、未分化細胞殺傷剤。
(23) (1)~(12)のいずれか1項に記載のウイルスベクターが導入された細胞。
(24) 幹細胞である(23)に記載の細胞。
(25) 幹細胞が、ES細胞、iPS細胞、神経幹細胞、造血幹細胞、間葉系幹細胞、肝幹細胞、膵幹細胞、皮膚幹細胞、筋幹細胞、又は生殖幹細胞である、(24)に記載の細胞。
(26) 幹細胞を分化誘導処理することにより得られた細胞である、(23)に記載の細胞。
本発明のベクターは、標識遺伝子と殺傷遺伝子とが、一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列を介して結合した核酸配列、及び、プロモーター領域を含むリコンビネーションカセットを有するウイルスベクターであって、該プロモーター領域が、該標識遺伝子及び該殺傷遺伝子を発現可能に連結されているウイルスベクターを当業者周知の遺伝子組み換え技術を用いて作製することにより得ることができる。
本発明のウイルスベクターは、リコンビネーション法を用いて任意の被験プロモーター領域、又は目的のプロモーター領域を組み込むことができる。リコンビネーションによる遺伝子導入は、ベクター中のリコンビネーションカセットに使用した遺伝子配列に対応するシャトルベクター及びリコンビナーゼを用いることにより、当業者周知の方法を用いて実施することができる。
一態様において、未分化細胞特異的なプロモーター領域を用いることによって本発明のウイルスベクターは、分化処理後に残存する未分化細胞を特異的に標識し、かつ/又は殺傷する方法に使用することができる。なお、以下の全ての方法において、「対象の細胞」とは、幹細胞(例えば、分化処理前の幹細胞、分化処理中の幹細胞など)、又は幹細胞を分化処理した後の細胞であってもよい。例えば、本発明の方法において本発明のウイルスベクターを感染させる「対象の細胞」とは、本発明のベクターを感染させる標的細胞を意味し、分化誘導処理後に未分化細胞として残存又は発生した場合に除去又は検出することを意図する細胞であれば分化誘導の前後を問わない。また、対象の細胞が、分化処理前の幹細胞又は分化処理中の幹細胞の場合、本発明のウイルスベクターが感染した細胞の分化を誘導する分化処理ステップを含んでいてもよい。また、以下の全ての方法において、「分化処理された細胞」又は「分化処理後の細胞」とは、幹細胞に分化処理を施すことにより得られた細胞を意味する。また、以下の全ての方法において、「未分化細胞特異的標識/殺傷遺伝子発現ベクター」とは、上述の本発明のベクターを意味し、具体的には、標識遺伝子と殺傷遺伝子とが、一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列を介して結合した核酸配列、及び、未分化細胞特異的プロモーターを含むリコンビネーションカセットを有するウイルスベクターであって、該プロモーター領域が、該標識遺伝子及び該殺傷遺伝子を発現可能に連結されているウイルスベクターを意味する。
例えば、未分化細胞特異的なプロモーター領域を有する本発明のウイルスベクターは、幹細胞を分化処理した後に残存する又は発生した未分化細胞を特異的に標識する目的で用いることができる。
ここで、前記標識遺伝子の発現産物のレベルが、未分化細胞の量(又は割合)を示す方法に関する。
A)分化処理された細胞又は分化処理中の細胞であって、該細胞が未分化細胞特異的標識/殺傷遺伝子発現ベクターが導入され、分化誘導処理された細胞における、前記標識遺伝子の発現産物を検出するステップ、及び、
B)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が残存又は発生していると判定するステップを備える、幹細胞の分化処理後の未分化細胞のモニタリング方法を含む。
別の態様において、未分化細胞特異的なプロモーター領域を有する本発明のウイルスベクターは、分化処理後に残存する未分化細胞を特異的に殺傷する方法に用いることができる。
一態様において、本発明は、本発明のウイルスベクターを用いた、被験プロモーターの未分化細胞における活性を測定する方法に使用することができる。
A)未分化細胞特異的標識/殺傷遺伝子発現ベクターのプロモーター領域に被験プロモーターを組み替えて被験プロモーターを有するウイルスベクターを作製するステップ、
B)被験プロモーターを有するウイルスベクターを対象の細胞に感染させるステップ、
C)分化誘導処理後の前記細胞における前記標識遺伝子の発現を検出するステップ、
D)分化誘導処理後の前記細胞が未分化細胞であるか、分化細胞であるかを識別するステップ、及び
E)被験プロモーターが未分化細胞に特異的であるか否かを判定するステップを備える、未分化細胞特異的プロモーターのスクリーニング方法であって、
ここで、該判定が、未分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が高く、かつ、分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が低い場合、該被験プロモーターは未分化細胞に特異的であると判定することにより行われる方法に関する。
A)未分化細胞特異的標識/殺傷遺伝子発現ベクターのプロモーター部分に被験プロモーターを組み替えて被験プロモーターを有するウイルスベクターを作製するステップ、
B)被験プロモーターを有するウイルスベクターを未分化細胞(例えば、ES細胞又はiPS細胞)に感染させるステップ、
C)前記未分化細胞を分化させるステップ、
D)分化誘導処理後の各細胞における前記標識遺伝子の発現を検出するステップ、
E)分化誘導処理後の各細胞が未分化細胞であるか、分化細胞であるかを識別するステップ、及び
F)被験プロモーターが未分化細胞に特異的であるか否かを判定するステップを備える、未分化細胞特異的プロモーターのスクリーニング方法であって、
ここで、該判定が、未分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が高く、かつ、分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が低い場合、該被験プロモーターは未分化細胞に特異的であると判定することにより行われる方法に関する。
上述の本発明の方法において、本発明のウイルスベクターは分化処理前又は分化処理中の幹細胞(例えば、ES細胞又はiPS細胞等の多能性幹細胞)、あるいは分化処理後の細胞に感染させることができる。本発明のベクターの細胞毒性が薬剤依存性である場合、プロモーター活性を発揮する細胞であっても薬剤不添加の条件下では本発明のベクターが細胞毒性を発揮することはない。そのため、導入ベクターの細胞毒性による分化処理前の幹細胞への障害を理由として分化処理ステップと感染ステップの前後関係が制限されることない。よって、本発明のベクターのこのような特徴は、全ての対象の細胞への感染を確実とするために最適なタイミングで感染の時期を選択することを可能とする。例えば、感染は、in vitroで分化処理の前に行われる。あるいは、本発明のウイルスベクターは、幹細胞の分化処理後に感染させてもよい。好ましくは、本発明のウイルスベクターは、分化処理前に感染させる。また、本発明の方法が移植ステップを含む場合、全ての対象の細胞への感染を確実とするため、感染は移植の前に行われる。感染は、使用するウイルスベクターの種類及び性質に応じて、適宜ウイルスベクター数や感染期間を選択し、当業者周知の方法を用いて実施することができる。例えば、感染はMOI1~1000の範囲で行うことができ、好ましくは、MOI10~100の範囲で行うことができる。
よって、例えば、本発明の方法は、標識遺伝子と薬剤依存性自殺遺伝子とが2A配列を介して結合した核酸配列、及び、未分化細胞特異的プロモーター領域を含むリコンビネーションカセットを有するウイルスベクターであって、該プロモーター領域が、該標識遺伝子及び該薬剤依存性自殺遺伝子を発現可能に連結されているウイルスベクターを未分化細胞(例えば、ES細胞又はiPS細胞等の多能性幹細胞)に感染させることにより行われる感染ステップの前、後、又は該感染ステップと同時に、更に、前記未分化細胞を分化細胞へと誘導させるステップ(分化処理ステップ)を含んでいてもよい。細胞の分化誘導処理は、未分化細胞に分化誘導のための分化誘導物質を接触させ、適切な培養条件により培養することにより行うことができる。分化誘導処理のための各種培養条件、及び培養方法が報告されており、使用する細胞及び分化させようとする細胞の種類に応じて適宜選択することができる。
前記標識遺伝子の発現産物の検出、同定、又はレベルの測定方法は、使用する標識遺伝子に応じて適宜選択することができる。例えば、蛍光遺伝子を使用する場合、発現産物の検出は、蛍光検出装置、フローサイトメトリー、又はリアルタイム蛍光検出装置等を用いて実施することができる。
(1) レンチウイルスベクタープラスミドの設計
作成するベクターの概略図を図1に示す。ベクターの基盤としてレンチウイルスベクターを使用し、プロモーター部分に以下のようなリコンビネーションカセット(RC)を用いた。これはΛファージが大腸菌染色体へ侵入する際に関与する部位特異的組換えシステムを利用し、ベクター間で改変したatt配列に挟まれたDNA配列の交換反応を行う(James,L.ら (2000)Genome Res.10: 1788-1795)。RC部分は組換え時に特異的に相互作用するDNA配列(att R配列)が両端にあり、その間にクロラムフェニコール耐性遺伝子(CMR)と大腸菌自殺遺伝子であるccdBを有する。RCをプロモーター部位に挿入したレンチウイルスベクターと、様々なプロモーター配列をRCのatt R配列に対応するatt L配列間にクローニングした遺伝子組み換え用シャトルベクタープラスミドを作製した(図1及び図2A)。レンチウイルスベクターのatt R配列とシャトルベクターのatt L配列とを反応させるLR clonaseを使用することにより、レンチウイルスベクターにプロモーター配列を組換えることを可能とした(図2B)。これにより、様々なプロモーターを簡便に入れ替えることができ、各種プロモーターの未分化細胞/腫瘍化細胞における活性について検証を行うことを可能とした。
プロモーター部分の下流に目的細胞への遺伝子導入を可視化するための蛍光タンパク質(図3及び8)と、遺伝子が導入された細胞を薬剤により選択的に殺傷可能とするための自殺遺伝子(図5及び8)とを組み合わせた遺伝子カセットを挿入した(図1及び7-9)。
(I)HSV-tk
HSV-tkはGCVに感受性があり、自殺遺伝子として働くことが報告されている(Yasuhiro TERAZAKIら(2003) Hepatology 37:155-63)。HSV-tk発現細胞をGCVを含む培地で培養すると、GCVはHSV-tkによりリン酸化され、さらにヒト細胞内の内在性キナーゼによりリン酸化され、最終的に強い毒性を有するガンシクロビル三リン酸(GCV-3P)となり、GCV-3PはDNA合成を阻害するためHSV-tk遺伝子導入細胞が死滅する。一方、HSV-tkを発現しない細胞ではGCVはGCV-3Pに変換されないため毒性を示さない。さらにこのシステムの長所として、上記のようにGCV-3Pの細胞殺傷作用はDNA合成阻害であるため、多くの正常細胞は体内では細胞分裂はしていないか限定的であるため、例え正常細胞にHSV-tkが導入されて発現しても細胞障害は無いか軽微であり、異常に恒常的な細胞分裂が起こっている腫瘍細胞を優位に特異的に殺傷していくという、高度な腫瘍選択制が上げられる。よってHSV-tk遺伝子を腫瘍特異的プロモーター、あるいは未分化特異的プロモーターで発現させた場合、そのプロモーターの特異性とHSV-tk/GCV自体の特異性という二重の特異性により、非常に高度な腫瘍細胞や幹細胞の未分化細胞に対する特異性を保持させることが期待できる。またHSV-tkのもう一つの利点として腫瘍細胞に対してはバイスタンダード効果という、HSV-tk遺伝子が導入されていない周辺の腫瘍細胞の多くも殺傷されるという強力な腫瘍治療効果がよく知られている。従って、HSV-tkとGCVとの組み合わせにより、細胞分裂が旺盛な細胞(腫瘍細胞や幹細胞の未分化細胞など)選択的に殺傷作用を強力に引き起こすことができる。
HSV-tkは非常に高い殺傷効果を有するが、単純ヘルペス由来であるためヒトの体内において免疫応答が起こり、細胞内に導入された遺伝子を排除するという問題も推察される。そこで、ヒト由来の酵素Thymidylate Kinase(tmpk)と3’-azido-3’-deoxythymidine(AZT)とを組み合わせる方法についても検討を行った。プロドラッグであるAZTは、リン酸化されてAZT三リン酸(AZT-3P)に変換される。AZT-3Pはヒト免疫不全ウイルス(HIV)の複製や、真核生物のDNAの合成を阻害する(Fei CHENら (2013) Biomaterials 34:1701-11)。AZTから毒性を持つAZT-3Pへの転換における律速酵素は、AZT一リン酸(AZT-MP)からAZT二リン酸(AZT-DP)への変換を触媒する細胞性Thymidylate Kinase(tmpk)である。tmpkの酵素効率は低いが、最小限に遺伝子改変を加えてAZT-MPへの作用が200倍であるtmpk(F105Y)が報告されている(13;14)。本実験においては、この遺伝子改良型tmpkを使用した。またtmpkは自殺遺伝子としての上記のHSV-tkに記載した長所は全て保持していると思われ、さらなる長所として、さらに高い腫瘍細胞や未分化細胞の殺傷効果と、異種蛋白由来の免疫誘導がない、ということが考えられる。但しtmpkを幹細胞に応用した報告は、当該発明のベクターシステムに限らずこれまで全くない。
上述の2遺伝子と異なり、アポトーシスを誘導することで細胞死を引き起こす遺伝子である。Caspaseはアポトーシスのシグナル伝達を構成している。ミトコンドリアからのカスパーゼ経路の初期に関わるCaspase-9は、通常AKTなどによりリン酸化され不活性化された状態にある。ミトコンドリアからの刺激により不活性型のCaspase-9が活性化され、Caspase-3やCaspase-7を活性化することでアポトーシスを引き起こす。Caspase-9の活性化を任意のタイミングで制御するため、薬剤誘導性のCaspase-9の活性化システムを用いた。これはCaspase-9遺伝子とヒトFK506結合蛋白(FKBP)蛋白遺伝子の融合タンパク質であるiCaspase9(iCasp9)を目的細胞に遺伝子導入・発現させた後、FKBPと結合する細胞膜透過性低分子化合物であるAP20187(クロンテック社)を培地に添加して、Caspase-9二量体を形成させ、活性化させることによりアポトーシスを誘導し、細胞死を引き起こすことができる(Carlos A Ramosら(2010)Stem Cells28:1107-15)。
プラスミドの構築は、以下に詳細を示すように2段階に分けて行った。即ち、リコンビネーションカセット(RC)を挿入したレンチウイルスベクターの作製(図1A)と、蛍光タンパク質-2A-自殺遺伝子カセットの作製(図1B)とを行い、これを最終的に組み合わせてレンチウイルスベクタープラスミドとした(図1C)。
レンチウイルスベクタープラスミドであるpLenti6-V5-RC-LacZ(Invitrogen社)をMluIサイトで切断し、V5-LacZ領域を除去したものを基本のベクタープラスミドとした。制限酵素で切断した後に平滑末端化を行い、Gateway(登録商標)プラスミド(Invitrogen社)からリコンビネーションカセット部分を挿入して、pLenti6-RC-BIsを構築した(図2)。このカセット部位にリコンビネーション反応を用いて種々のプロモーター配列を挿入することができる。
上述において作製したpLenti6-RC-BIsのRCカセットの下流に挿入するための遺伝子カセットを以下の方法により作製した。遺伝子カセットは、蛍光タンパク質と自殺遺伝子が2A配列を介して結合されたものとした。
赤色蛍光タンパク質mKate2及び緑色蛍光タンパク質Venusの2種類を使用した。mKate2は、5’側にAgeI、3’側にBGI IIサイトを付加したプライマーを設計し(表1)、PCR法により増幅した後、PCR産物クローニング用ベクターであるpGEM-Teasy vector(Invitrogen社)にクローニングしてpGEM-Teasy-mKate2を構築した(図3A)。クローニング後、pGEM-Teasy vector内のクローニング部位の両端にあるプライマー配列より、5’側のT7及び3’側のM13Rを用いてシークエンス解析を行った。次に、pGEM-Teasy-mKate2から、NotI/BglIIでmKate2を切り出した(図4A)。5’側にBglII、3’側にBamHIサイトの突出末端を持つ合成二本鎖2A配列DNAオリゴをBglII/BamHIで切断したpFlag-CMV-2(SIGMA)へ挿入したpFlag-CMV-2Aを構築し、このpFlag-CMV-2Aも同様にNot I/BglIIで切断し、CMVプロモーターと2A配列の間にmKate2を挿入してpFlag-CMV-mKate2-2Aを構築した(図4A)。
HSV-tkは、プライマーを作製して(表1)、PCR法により増幅した後、クローニング用ベクターであるpGEM-Teasy vectorにクローニングしてpGEM-Teasy-HSV-tkを構築した(図3)。クローニング後、mKate2、Venusと同様に前記プライマーでシークエンス解析を行った。次に、pGEM-Teasy-HSVtkを、HSV-tkの3’側のSPEIで切断した後、平滑末端化し、HSV-tkの5’側のBamHIで切断してHSV-tkを得た(図5A)。

(B)で得られた遺伝子断片を(A)で作製したプラスミドに挿入し、蛍光タンパク質-2A-自殺遺伝子の発現カセットを持つベクタープラスミドの作製を行った。
pFlag-CMV-mKate2-2A及びpFlag-CMV-Venus-2Aは、3’側BstXIで切断した後、平滑末端化し、5’側をBamHIで切断して切り出した(図5B)。(B)で得られたHSV-tk又はhuman-tmpkを2Aの3’側へ挿入し、4種類のプラスミド、pFlag-CMV-mKate2-2A-HSVtk、pFlag-CMV-mKate2-2A-tmpk、pFlag-CMV-Venus-2A-HSVtk、及びpFlag-CMV-Venus-2A-tmpkを構築した。
上述のとおり作製したRCカセットを持つpLenti6-RC-BIsに、上述のとおり作製した蛍光タンパク質-2A-自殺胃遺伝子の発現カセットを挿入し、モニターベクタープラスミドを完成させた。
pLenti6-RC-BIsを3’側KpnIで切断した。human-tmpkを持つplasmidとのライゲーション用に、5’側をAgeIで切断した(図6A)。また、HSV-tkを有する2種類のプラスミドとのライゲーションに使用するために、3’側をKpnIで切断後、平滑末端化を行い、5’側をAgeIで切断した(図7A)。
また、pFlag-CMV-mKate2-2A-tmpk、及びpFlag-CMV-Venus-2A-tmpkは、5’側をAgeI、3’側をKpnIで切断し、それぞれ、mKate2-2A-tmpk、及びVenus-2A-tmpk部分を切り出した(図6B)。HSVtkは内部にKpnI切断サイトを持つため、pFlag-CMV-mKate2-2A-HSVtk、及びpFlag-CMV-Venus-2A-HSVtkは5’側をAgeI、3’側をPmeI(ブラントエンド)で切断し、それぞれ、mKate2-2A-HSVtk、及びVenus2-2A-HSVtkを切り出した(図7B)。
これらをそれぞれ組み合わせてライゲーションすることにより、pLenti-RC-mKate2-2A-tmpk、pLenti-RC-Venus-2A-tmpk、pLenti-RC-mKate2-2A-HSVtk、及び、pLenti-RC-Venus-2A-HSVtkを作製した。
induced-Caspase9(iCasp9-2Aの配列を持つプラスミド(Brenner博士から譲渡)から、iCasp9-2Aの領域をサブクローニングし、In-Fusion(登録商標)HD Cloning KIT(TAKARA BIO社)を用いてプラスミドを構築した。本KITはPCRで調整したDNA断片や線状化したベクターを、各DNA断片末端の相同な15塩基を正確に認識してクローニングすることができ、複数のDNA断片をあらゆるベクターへ迅速にクローニングできる(Zhu Bら (2007)Biotechniques. 43(3):354-9)。そこで、記述のpLenti6-RC-BISのRCの下流にiCasp9-2A-Venus又はiCasp9-2A-mKate2を挿入するため、iCasp9-2Aの前後、及びVenusとmKate2の前後にそれぞれお150bpのプライマーを設計し、PCRを行った(図8、表2)。
その後、pLenti6-RC-BIs、iCasp9-2AのDNA配列と、Venus又はmKate2を混合して酵素反応させることにより、2種類のプラスミド、pLenti6-RC-iCasp9-2A-Venus、及びpLenti6-RC-iCasp9-2A-mKate2を完成させた。
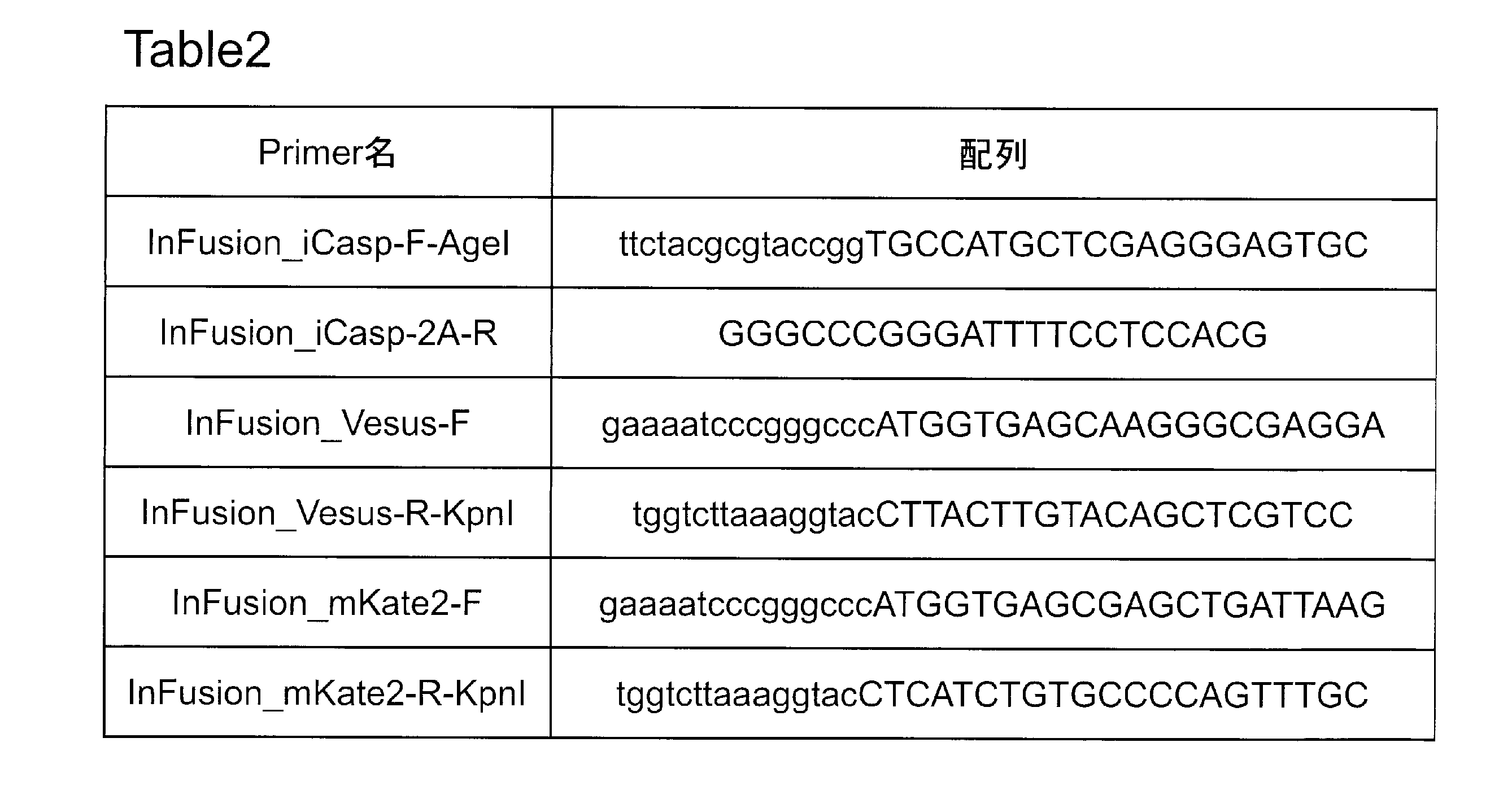
蛍光タンパク質-2A-自殺遺伝子の発現カセットを持つプラスミドは、作成の各段階でHEK293細胞に導入して評価することにより、蛍光の発現確認を行った。HSK293細胞は6ウェルプレートに6×105細胞/ウェルで播種し、翌日にプラスミドを2mg/ウェルで、Polyfect Transfection Reagent Kit(QIAGEN社)を用いて製造者提供のプロトコルに従って遺伝子導入を行った。遺伝子導入の48時間後に蛍光顕微鏡で蛍光の発現を確認した。
pFlag-CMV-mKate2-2, pFlag-CMV-Venus-2AをHEK293細胞に導入し、蛍光タンパク質の発現について評価を行った結果、恒常性プロモーターであるCMVの制御下で、mKate2及びVenusが正常に発現することが確認された(図9)。また、pFlag-CMV-mKate2-2A-HSVtk、pFlag-CMV-Venus-2A-HSVtk、pFlag-CMV-mKate2-2A-Human tmpk、及びpFlag-CMV-Venus-2A-Human tmpkについても同様にHEK293細胞に導入し、蛍光タンパク質及び自殺遺伝子の発現について評価を行った結果、mKate2及びVenusが正常に発現することが確認された(図10A及び図10C)。また、自殺遺伝子は各遺伝子に対応する薬剤を加えたところ、薬剤の濃度依存的に細胞傷害効果がみられた(図10A~D)。
ヒト培養細胞における自殺遺伝子の細胞毒性効果を検証するため、HSV-tk、human-tmpk、及びiCaspase9の3種類の自殺遺伝子を発現するウイルスベクターを細胞に感染させた後、それぞれに対応する薬剤を加えて各細胞に対する毒性効果を比較した。HSV-tkは既に作製済みのアデノウイルスベクターを使用した(Yasuhiro Terazakiら(2003)Hepatology 37;155-63)。human-tmpk及びiCaspase9は、それぞれ、Medin博士に譲渡頂いたプラスミドpHR’-cPPT-EF-Tmpk(F105Y)(Takeya Satoら(2007)Mol Ther 15:962-70)、及びbrenner博士に譲渡頂いたプラスミドiCasp9-2A-DCD19RRF(CARLOS A RAMOSら(2010)STEM Cells 28;1107-15)を使用した。これらのプラスミドを直接細胞に導入し、又はレンチウイルスベクターを産生して使用した。
ヒトiPS細胞として、ヒト成人繊維芽細胞(HDF)由来で、OCT3/4、KLF4、SOX2及びc-Mycの4因子を導入して樹立された201B7、及びHDF由来でc-Myc以外の3因子を導入して樹立された253G1を使用した。また、マウスES細胞としてD3細胞を使用した。ヒトES細胞は、KhES1(京都大学より譲渡)を使用した。また、ヒト正常繊維芽細胞(BJ)、ヒト癌細胞(ヒトTリンパ腫細胞株Jurkat、ヒト胃癌細胞MKN45)を使用した。ヒトiPS/ES細胞はマイトマイシンC処理したマウス胎仔繊維芽細胞(MEF)上で共培養した。マウスES細胞はゼラチンコートしたディッシュ上で培養した。 各細胞の培養に使用した培地は以下のとおりである:
(ヒトiPS/ES細胞)
20% KSR(Invitrogen社)、200mM L-Glutamine、0.1mM Non Essential Amino Acid(Sigma社)、0.1mM 2-Mercaptoethanol(Sigma社)、5ng/mL Basic-Fgf(Wako社)、Penicillin/Streptomycin(P/S)(Nacalai Tesque)含有 DMEM-F12(Sigma社)
(マウスES細胞)
20% Fetal Bovine Serum(FBS)(GIBCO社)、0.1mM Non Essential Amino Acid、1mM Sodium Pyruvate(Gibco社)、70μM 2-Mercaptoethanol、1×103unit/mL LIF mouse recombinant(ナカライ社)、P/S含有 DMEM(SIGMA社)
(Jurkat細胞及び MKN45細胞)
10% FBS(Bio West社)及びP/S含有 RPMI-1640(SIGMA社)
(BJ細胞)
10% FCS、200mM L-Glutamine(Gibco社)、0.1mM Non Essential Amino Acid、1mM Sodium Pyruvate、P/S含有 Minimum Essential Medium Eagle(Sigma社)
KhES1は、ROCK阻害剤(Y27632、WAKO社)を用いて、単一分散培養を無フィーダー下で行い、ウイルス感染実験に用いた。24ウェルプレートに無血清培地(DMEM-F12)で40倍に希釈したgrowth Factor Reduced BD Matrigel(BD Bioscience社)を加え、37℃のCO2インキュベーター内で1時間以上静置し、マトリゲルコート処理を行った。6CMディッシュのKhES1の培地を新しい培地2mLに交換し、最終濃度が10mmとなるようにY27632を加え、37℃のCO2インキュベーターにて1時間以上培養した。Y27632処理ヒトES細胞を2mLのPBSで洗浄し、ヒトES細胞用解離液(CTK)を0.4mL加え、37℃で5分静置した後、10mm Y27632含有ヒトES細胞用培地を2mL加えてピぺッティングにより細胞塊を単一細胞に分散させた。細胞数をカウントし、必要量のKhES1を遠心(1000rpm、5分)して、CMに懸濁させ、マトリゲルコート処理したプレートに播種した。
レンチウイルスベクターとして、PHR-CPPT-EF-tmpk(human-tmpk発現用)、iCasp9.2A.DCD19.RRF(iCasp9発現用)、及びCMV-EGFPを使用した。PHR-CPPT-EF-tmpk、及びiCasp9.2A.DCD19.RRFは自殺遺伝子と同時にCD19を発現することから、各自殺遺伝子の発現は抗CD19抗体により確認することができる。CMV-EGFPは、レンチウイルスベクターの力価確認のために用いた。
POLY-L-LYSINE(SIGMA社)でコートした6ウェルプレートにHEK293T細胞を6×105細胞/ウェルで播種した。翌日、各レンチベクタープラスミド0.5mgにPLP1,PLP2、及びPLP-VSVGを混合し、50mLのOPTI-MEMを添加した後、FUGENE HD(PROMEGA社)を6mL加え、voltexした後、室温で15分静置した。この全量を1ウェルに添加してトランスフェクションし、24時間後に培地を10%FCS/DMEM,P/S(-)と交換し、48及び72時間後の2回、上清を回収した(6ウェルプレート各2mL×4ウェルで2回回収、合計16mL)。これをLenti-X Concentrator Kit(Clonetech社)を用いて製造者提供のプロトコルに従い、100倍に濃縮した。
感染前日に、各細胞を以下の細胞数で24ウェルプレートに播種した:
KhES1細胞 4×104細胞/ウェル
Jurkat細胞 1.25×105細胞/ウェル
MKN45細胞 2×104細胞/ウェル
HEK293T細胞 5×104細胞/ウェル
ウイルス感染当日、4mg/mLのポリブレンを含む培地に交換し、各レンチウイルスベクターを20mL添加して24時間感染させた。全ての細胞の培地を24時間後に交換し、更に培養を続けた。EGFP発現レンチウイルスベクターを感染させた細胞は、感染72時間後にフローサイトメーター(Cyan Adp Analyzer、ベックマンコールター社)で評価し、産生したウイルスベクターの力価を計算した。
human-tmpk発現レンチウイルスベクターを感染させた細胞について、FACS ARIA II(BD社)を使用して感染細胞(CD19陽性細胞)を分取した。
KhES1は、10mm Y27632を含む培地で一晩培養したKhES1をCTKで処理した後、単一細胞へと分散させた。細胞数をカウントした後、必要量の細胞を遠心(1000rpm、5分)し、10mm Y27632含有PBSに再懸濁させた。未分化細胞標識抗体である、Alexa Fluor 488 anti-Human SSEA4(BIOLEGEND社)及びanti-Human CD19-APC(EBIOSCIENCE社)を、細胞懸濁液100mL(1×106細胞)あたり5mL加え、氷上で10分毎に細胞を懸濁しながら30分間静置して細胞のラベリングを行った。10mm Y27632含有PBSで1回洗浄後、10mm Y27632含有ES用培地に懸濁させ、両抗体に標識されたKhES1を未分化なCD19陽性細胞としてフローサイトメーターで評価した。
Jurkat、MKN45については、細胞数のカウント後、必要量の細胞を遠心(1000rpm、5分)し、10%FCS含有PBSに再懸濁させた。細胞懸濁液100mL(1×106細胞)あたり5mLのanti-Human CD19-APC(EBIOSCIENCE社)を加え、氷上で10分毎に細胞を懸濁しながら30分間静置して細胞のラベリングを行った。10%FCS含有PBSで1回洗浄後、10%FCS含有PBSに懸濁し、標識されたJurkat、及びMKN45をCD19陽性細胞としてフローサイトメーターで評価した。
フローサイトメーターでCD19陽性細胞を確認後、細胞を増やし、再び上記プロトコルに従って抗体で標識した後、BD FACSARIA IIにより陽性細胞の分取を行った。KhES1は、SSEA4及びCD19両陽性な細胞を、Jurkat及びMKN45はCD19陽性細胞を分取した。分取した細胞は直ちに遠心(1000rpm、5分)し、新しい培地に再懸濁させ、KhES1についてはMEF細胞と共培養を行った。Jurkat、MKN45については、通常培養を行った。
各細胞における自殺遺伝子の細胞毒性を評価するため、HSV-tk、human-tmpk、及びiCaspase9を細胞に導入し、薬剤を加えて細胞の殺傷効果を比較検討した。HSV-tkについては、既に作製済みのHSV-tkアデノウイルス(Ad.CAG-HSV-tk)を用いた。human-tmpk、及びiCasp9は、プラスミドを細胞に導入した。各細胞を以下の細胞数で24ウェルプレートに播種した:
KhES1細胞 4×104細胞/ウェル
MKN45細胞 2×104細胞/ウェル(tmpk、iCasp9のみ)
BJ細胞 1×104細胞/ウェル
翌日、Ad.CAG-HSV-tkをMOI=10で感染させ、24時間後に培地交換した後、GCV(10mg/mL)を10mL、4日間投与した。human-tmpk、及びiCasp9は、各プラスミド0.5mgをOPTI-MEM25mLに加え、FUGENE HD(QIAGEN社)2mLを添加して、voltexした後、15分静置したものを全量投与した。24時間後に培地交換した後、human-tmpk導入細胞にはAZTを0,30,100,300mmで各ウェルに3日間投与した。iCasp3にはDimerizerを0,0.03,0.1,0.3mmで各ウェルに3日間投与した。薬剤投与後、細胞毒性効果の測定は生細胞数の測定により行った。測定当日、KhES1はCTK処理し、単一細胞へと分離した後、10mLを採取した。Jurkatは細胞懸濁後10mLを採取し、MKN45はトリプシンで処理し、細胞懸濁後10mLを採取した。採取した細胞懸濁液のそれぞれにトリパンブルー10mLを加え、生細胞数をカウントした。
Jurkat-tmpk/NC 1×105細胞/ウェル
MKN45細胞-tmpk/NC 2×104細胞/ウェル
翌日よりAZTを0,0.1,1,10,100,1000mmで各ウェルに3日間投与し、上述の方法で生細胞数をカウントした。
コントロールであるAd.CAG-HSV-tk感染KhES1細胞は、GCV投与により傷害を受けた一方、GCV非投与ではそのような傷害は受けていなかった
(図11A)。BJでは、GCV非投与で障害がみられず、GCV投与による細胞障害はKhES1と比較してわずかであった(図11B)。
pHR-CPPT-EF-tmpk(F105Y)導入KhES1では、低濃度のAZT投与でも細胞傷害効果がみられ、アデノウイルスベクターで導入したHSV-tk(図11A)とほぼ同様の殺傷効果を示した(図12A)。AZT高濃度投与ではhuman-tmpk非導入細胞でも細胞傷害が観察された。BJでは、human-tmpk導入/非導入細胞間で細胞傷害に差は見られなかった(図12B)。MKN45では、未分化細胞同様、AZT投与により低濃度でも細胞傷害効果がみられ、高濃度ではhuman-tmpk非導入細胞でも細胞傷害が確認された(図12C)。しかし、iCasp9.2A.DCD19.RRFを導入したKhES1、BJ、及びMKN45については、いずれもほとんど変化が見られなかった。
ヒト培養細胞における自殺遺伝子の細胞毒性効果を検証するため、まず実施例1で作成したRCを含むレンチウイルスベクタープラスミドに、リコンビネーションにより各種プロモーターを挿入してプラスミドを完成させた。これらのプラスミドよりレンチウイルスベクターを産生し、細胞に感染させた後、それぞれに対応する薬剤を加えて各細胞に対する毒性効果を比較した。
各種プロモーターをpENTR-vector(LIFE TECHNOLOGIES社)に挿入したものを作製した(図15)。pENTR-vectorはattL配列を含んでおり、LR clonaseでpLenti6-RCと反応させることにより、プロモーターをRC内に組み込むためのシャトルベクターとして使用する。サブクローニングずるプロモーターには、未分化特異的プロモーターの候補として、survivin,tert,Rex1,Nanogを、またコントロールとして恒常性プロモーターのCAG,PGKを使用し、それぞれ以下の方法でpENTR-vectorに挿入した。
(A)survivinプロモーター
既に作製済みであったpGEMT-easy-survivinより、survivinプロモーター部分をNotIにより切り出し、pENTR-vectorにクローニングしてpENTR-survivinを構築した(図15A)
(B)tertプロモーター
既に作製済みであったpGEMT-easy-tertより、tertプロモーター部分をMluI/BglIIにより切り出し、pENTR-vectorにクローニングしてpENTR-Tertを構築した(図15B)
(C)Rex1プロモーター
既に作製済みであったpGEMT-easy-Rex1より、Rex1プロモーター部分をMunI/Nhe Iにより切り出し、pENTR-vectorにクローニングしてpENTR-Rex1を構築した(図15C)
(D)Nanogプロモーター
Nanogプロモーター領域の3’側、及び5’側にpENTR-vectorクローニング用の突出末端としてCACCを付加したプライマーを作製して(表3)、PCR法により増幅した後、pENTR-vectorにクローニングしてpENTR-Nanogを構築した(図15D)
(E)CAGプロモーター
既に作製済みであったpCX-EGFPよりCAGプロモーター部分をSalI/EcoRI(平滑末端化)により切り出し、pENTR-vectorにクローニングしてpENTR-CAGを構築した(図15E)
(F)PGKプロモーター
PGKプロモーター領域の5’側にpENTR-vectorクローニング用の突出末端としてCACCを付加し、3’側にPstIサイトを付加したプライマーを作製して(表3)、PCR法により増幅した後、pENTR-vectorにクローニングしてpENTR-PGKを構築した(図15F)

図7で示したプラスミドのうち、pLenti6-RC-Venus-2A-HSV-tkのRC部分に、図15で示した各種プロモーターをリコンビネーションにより挿入した(pLenti6-Promoter-Venus-2A-HSV-tk)。RCを含むレンチウイルスベクタープラスミドと、各プロモーターを含むpENTR vectorプラスミドをそれぞれ合わせて、LRクロナーゼを反応させることでプロモーターをRC部分に挿入した。未分化特異的プロモーターとしてsurvivin,tert,Rex1,Nanogを、またコントロールとして恒常性プロモーターのCAG,PGKをそれぞれ挿入した(図16A)。また,図7で示したプラスミドのうち、pLenti6-RC-Venus-2A-human-tmpkのRC部分に、図15で示した各種プロモーターをリコンビネーションにより挿入した(pLenti6-Promoter-Venus-2A-human-tmpk)。未分化特異的プロモーターとしてsurvivin,tert,Rex1,Nanogを、またコントロールとして恒常性プロモーターのCAG,PGKをそれぞれ挿入したものを作製した(図16B)。
pLenti6-Promoter-Venus-2A-HSV-tkのうち、survivinおよびCAGプロモーターのプラスミドよりレンチウイルスベクターを産生し、感染させた細胞についてFACS ARIA II(BD社)を使用して感染細胞(GFP陽性細胞)を分取した。
KhES1は、10mm Y27632を含む培地で一晩培養したKhES1をCTKで処理した後、単一細胞へと分散させた。細胞数をカウントした後、必要量の細胞を遠心(1000rpm、5分)し、10mm Y27632含有PBSに再懸濁させた。未分化細胞標識抗体である、Alexa Fluor 488 anti-Human SSEA4(BIOLEGEND社)を、細胞懸濁液100mL(1×106細胞)あたり5mL加え、氷上で10分毎に細胞を懸濁しながら30分間静置して細胞のラベリングを行った。10mm Y27632含有PBSで1回洗浄後、10mm Y27632含有ES用培地に懸濁させ、抗体に標識されたGFP陽性細胞を未分化なレンチウイルス感染KhES1としてフローサイトメーターで評価した。
D3細胞はトリプシン処理し、ピぺッティングにより細胞塊を単一細胞に分散させた。細胞数をカウントし、必要量のD3細胞を遠心(1000rpm、5分)して、培地で再懸濁させ、GFP陽性細胞をレンチウイルス感染D3細胞としてフローサイトメーターで評価した。
フローサイトメーターでGFP陽性細胞を確認し、細胞を増やした後、BD FACSARIA IIによりGFP陽性細胞の分取を行った。分取した細胞は直ちに遠心(1000rpm、5分)し、新しい培地に再懸濁させ、KhES1細胞はMEF細胞と共培養を行い、D3細胞はゼラチンコートしたディッシュに播種した。
上記の分取したKhES1細胞およびD3細胞を用いて、GCV投与後の細胞毒性効果について解析を行った。KhES1細胞は、1wellあたり1×105個の細胞を96well plateに播種し、翌日より7日間、survivin-Venus-2A-HSV-tk(A),CAG-Venus-2A-HSV-tk(B)、およびHSV-tkを持たないsurvivin-Venus-2A-puromycin(C)、及び非感染細胞(NC)(D)の各群にGCVを0,0.01,0.1,1,10,100mg/mLでそれぞれ毎日、7日間投与した。結果を図17に示す。D3細胞は、1wellあたり1×105個の細胞を96well plateに播種し、翌日より4日間、survivin-Venus-2A-HSV-tk(A),CAG-Venus-2A-HSV-tk(B)、およびHSV-tkを持たないsurvivin-Venus-2A-puromycin(C)、及び非感染細胞(NC)(D)の各群にGCVを0,0.01,0.1,1,10,100mg/mLでそれぞれ毎日、4日間投与した。4日間投与後の細胞毒性効果の測定は、MTT assayの変法であるWST-8 assayを用いて行った。使用した試薬SF(ナカライ社)は高感度水溶性ホルマザンを生成するテトラゾリウム塩;WST-8 [2-(20methoxy-4-nitrophenyl)-3-(nitrophenyl)-5-(2,4-disulgophenyl)-2H-tetrazolium monosodium salt]を発色基質として使用することでこれまでのMTTよりも高感度測定を可能としており、WST-8が細胞内脱水素酵素により還元され、水溶性のホルマザンを生成する。このホルマザンの450nmの吸光度を直接測定することにより生細胞数を測定した(E)。結果を図18に示す。
LV.survivin-Venus-2A-HSV-tk感染KhES1細胞は、コントロールであるLV.CAGと共に、GCV1mg/mLと低用量で十分な細胞毒性効果を示し、細胞がほぼ全滅していた(図17A,B)。一方で、HSV-tkを持たないLV.survivin-Venus-2A-puromycin感染細胞、および非感染細胞(NC)細胞では同様量で細胞毒性は見られず(図17C,D)、GCV1mg/mL投与時にHSV-tk依存性の特異的な細胞傷害効果であることが示された。またGCVが10μg/mL以上の高濃度になるとHSV-tk非特異的な細胞毒性が見られた。また、D3細胞でも同等の結果が得られ、特にLV.survivin-Venus-2A-HSV-tk感染D3細胞では、同ウイルス感染KhES1細胞よりも感受性の高い細胞毒性効果を示した(図18A-E)
上記の分取したKhES1細胞を用いて、分化誘導後にGCV投与を行った際の安全性を確認するための解析を行った。分化誘導は、細胞を1wellあたり3×103個の細胞を96well 低接着plateにES培地(LIFを除く)で播種し、1週間培養して胚葉体を形成させた。その後、ゼラチンコートしたディッシュに継代を続け、28日以上培養して十分に分化させて作製した。分化誘導した細胞を1wellあたり1×105個の細胞を96well plateに再播種し、翌日より4日間、survivin-Venus-2A-HSV-tk(A),CAG-Venus-2A-HSV-tk(B)、およびHSV-tkを持たないsurvivin-Venus-2A-puromycin(C)、及び非感染細胞(NC)(D)の各群にGCVを0,0.01,0.1,1,10,100mg/mLでそれぞれ毎日、4日間投与した。結果を図19に示す。
LV.survivin-Venus-2A-HSV-tk感染KhES1細胞は、コントロールであるLV.CAGと共に、未分化細胞では毒性効果を示していたが、上述の方法で分化させた細胞ではほとんど障害がみられず、未分化細胞特異的な殺傷効果を示した(図19A,B)。HSV-tkを持たないLV.survivin-Venus-2A-puromycin感染細胞、および非感染細胞(NC)細胞では未分化細胞と同じく細胞毒性は見られず(図19C,D)、GCV10mg/mL以上投与時でも未分化細胞に比べて非特異的な殺傷が見られないことから、HSV-tk存在下で、かつ未分化な細胞でのみGCVの毒性が現れることが示された。(図19A-E)
上記の分取したKhES1細胞を用いて、レンチウイルス感染後の分化多能性に対する影響がないことを確認するための検証を行った。免疫不全マウスに4×106個の細胞を皮下移植して、テラトーマを形成させた。テラトーマが直径5mm以上になったものをそれぞれ取り出し、ホルマリン固定後、パラフィン包埋切片を作製した。各切片は、脱パラフィンした後、HE染色を行った。結果を図20に示す。
LV.survivin-Venus-2A-HSV-tk、及びLV.CA-Venus-2A-HSV-tk感染KhES1細胞のいずれにおいても、三胚葉由来の各細胞が見られ(図20A-F)、非感染細胞と同等に多能性を維持していることが示された。この結果より、以前の研究で多数報告のあるとおり、レンチウイルス感染が、多能性幹細胞が有する分化多能性に影響しないことが確認された。
Claims (31)
- 標識遺伝子と殺傷遺伝子とが、一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列を介して結合した核酸配列、及び、プロモーター領域を含むリコンビネーションカセットを有するウイルスベクターであって、該プロモーター領域が、該標識遺伝子及び該殺傷遺伝子を発現可能に連結されているウイルスベクター。
- 殺傷遺伝子が、自殺遺伝子である、請求項1に記載のウイルスベクター。
- 自殺遺伝子が、薬剤依存性自殺遺伝子である、請求項2に記載のウイルスベクター。
- 薬剤依存性自殺遺伝子が、HSV-tk、human-tmpk、シトシンデアミナーゼ請求項CD遺伝子、水痘ウイルスチミジンキナーゼ、又はCaspaseである、請求項3に記載のウイルスベクター。
- 標識遺伝子が蛍光タンパク質である、請求項1~請求項4のいずれか1項に記載のウイルスベクター。
- 蛍光タンパク質が、赤色又は緑色の蛍光蛋白質である、請求項5に記載のウイルスベクター。
- 蛍光タンパク質が、mKate2又は緑色蛍光蛋白質である、請求項5に記載のウイルスベクター。
- 一つのプロモーターにより前記2つの遺伝子を同時に発現させることが可能な配列が、2A配列又はIRES配列である、請求項1~請求項7のいずれか1項に記載のウイルスベクター。
- レンチウイルスベクターである、請求項1~請求項8のいずれか1項に記載のウイルスベクター。
- プロモーター領域が未分化細胞特異的プロモーター領域である、請求項1~請求項9のいずれか1項に記載のウイルスベクター。
- 未分化細胞特異的プロモーターが、survivinプロモーター又はtertプロモーターである、請求項10に記載のウイルスベクター。
- 未分化細胞特異的プロモーターが、survivinプロモーターである、請求項10に記載のウイルスベクター。
- A)請求項1~請求項9のいずれか1項に記載のウイルスベクターのプロモーター部分に被験プロモーターを組み替えて被験プロモーターを有するウイルスベクターを作製するステップ、
B)被験プロモーターを有するウイルスベクターを対象の細胞に感染させるステップ、
C)分化誘導処理後の前記細胞における前記標識遺伝子の発現を検出するステップ、
D)分化誘導処理後の前記細胞が未分化細胞であるか、分化細胞であるかを識別するステップ、及び
E)被験プロモーターが未分化細胞に特異的であるか否かを判定するステップを備える、未分化細胞特異的プロモーターのスクリーニング方法であって、
ここで、該判定が未分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が高く、かつ、分化細胞として識別された細胞において前記標識遺伝子の発現が検出される割合が低い場合、該被験プロモーターは未分化細胞に特異的であると判定することにより行われる方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中の前記標識遺伝子の発現産物を検出するステップ、及び、
C)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が存在すると判定するステップを備える、分化処理後の細胞における未分化細胞の有無を判定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中の前記標識遺伝子の発現産物を検出するステップ、及び、
B)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が存在すると判定するステップを備える、分化処理後の細胞における未分化細胞の有無を判定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における前記標識遺伝子の発現産物を同定するステップ、及び、
C)前記標識遺伝子が発現している細胞が未分化細胞であると同定するステップを備える、分化処理後の細胞における未分化細胞を同定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における前記標識遺伝子の発現産物を同定するステップ、及び、
B)前記標識遺伝子が発現している細胞が未分化細胞であると同定するステップを備える、分化処理後の細胞における未分化細胞を同定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における、前記標識遺伝子の発現産物のレベルを測定するステップ、及び、
C)測定された前記標識遺伝子の発現産物のレベルから、未分化細胞の量請求項又は割合を決定するステップを備える、分化処理後の細胞における未分化細胞の量請求項又は割合を決定する方法であって、
ここで、前記標識遺伝子の発現産物のレベルが、未分化細胞の量請求項又は割合を示す方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における、前記標識遺伝子の発現産物のレベルを測定するステップ、及び、
B)測定された前記標識遺伝子の発現産物のレベルから、未分化細胞の量請求項又は割合を決定するステップを備える、分化処理後の細胞における未分化細胞の量請求項又は割合を決定する方法であって、
ここで、前記標識遺伝子の発現産物のレベルが、未分化細胞の量請求項又は割合を示す方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の前記細胞中における、前記標識遺伝子が発現している細胞数を計数するステップ、及び、
C)前記標識遺伝子が発現している細胞数が未分化細胞の数であると決定するステップを備える、分化処理後の細胞中における未分化細胞数を決定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターが導入された分化処理後の細胞中における、前記標識遺伝子が発現している細胞数を計数するステップ、及び、
B)前記標識遺伝子が発現している細胞数が未分化細胞の数であると決定するステップを備える、分化処理後の細胞中における未分化細胞の数を決定する方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターを対象の細胞に感染させるステップ、
B)分化処理後の細胞中における、前記標識遺伝子の発現産物を検出するステップ、及び、
C)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が残存し又は発生したと判定するステップを備える、分化処理後に残存又は発生する未分化細胞のモニタリング方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターが導入され、分化誘導処理された細胞における、前記標識遺伝子の発現産物を検出するステップ、及び、
B)前記標識遺伝子の発現産物が検出された場合には、未分化細胞が残存又は発生していると判定するステップを備える、幹細胞の分化処理後の未分化細胞のモニタリング方法。 - A)請求項10~請求項12のいずれか1項に記載のウイルスベクターであって、殺傷遺伝子が薬剤依存性殺傷遺伝子であるウイルスベクターを対象の細胞に感染させるステップ、及び、
B)前記細胞に前記薬剤依存性殺傷遺伝子が毒性を発揮可能な薬剤を投与するステップを備える、未分化細胞を殺傷する方法。 - 請求項10~請求項12のいずれか1項に記載のウイルスベクターであって、殺傷遺伝子が薬剤依存性殺傷遺伝子であるウイルスベクターが導入された細胞に前記薬剤依存性殺傷遺伝子が毒性を発揮可能な薬剤を投与するステップを備える、未分化細胞を殺傷する方法。
- 請求項10~請求項12のいずれか1項に記載のウイルスベクターを含む、未分化細胞標識剤。
- 請求項10~請求項12のいずれか1項に記載のウイルスベクターを含む、未分化細胞殺傷剤。
- 請求項1~請求項12のいずれか1項に記載のウイルスベクターが導入された細胞。
- 幹細胞である請求項28に記載の細胞。
- 幹細胞が、ES細胞、iPS細胞、神経幹細胞、造血幹細胞、間葉系幹細胞、肝幹細胞、膵幹細胞、皮膚幹細胞、筋幹細胞、又は生殖幹細胞である、請求項29に記載の細胞。
- 幹細胞を分化誘導処理することにより得られた細胞である、請求項28に記載の細胞。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20159605.3A EP3690055B1 (en) | 2014-01-14 | 2015-01-14 | Method for labelling cancerization-causing cell in stem cells |
| US15/111,084 US20170051306A1 (en) | 2014-01-14 | 2015-01-14 | Novel method for labelling cancerization-causing cell in stem cells, and therapy method |
| EP15737214.5A EP3095864B1 (en) | 2014-01-14 | 2015-01-14 | Method for labelling cancerization-causing cell in stem cells |
| JP2015557768A JP6358713B2 (ja) | 2014-01-14 | 2015-01-14 | 幹細胞における腫瘍化原因細胞の新たな標識法と治療法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014004262 | 2014-01-14 | ||
| JP2014-004262 | 2014-01-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015107888A1 true WO2015107888A1 (ja) | 2015-07-23 |
Family
ID=53542785
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/000138 Ceased WO2015107888A1 (ja) | 2014-01-14 | 2015-01-14 | 幹細胞における腫瘍化原因細胞の新たな標識法と治療法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20170051306A1 (ja) |
| EP (2) | EP3690055B1 (ja) |
| JP (1) | JP6358713B2 (ja) |
| WO (1) | WO2015107888A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018122932A1 (ja) * | 2016-12-26 | 2018-07-05 | オリンパス株式会社 | 細胞の選別方法、純化された細胞集団の製造方法、及び発光イメージングシステム |
| JPWO2020171222A1 (ja) * | 2019-02-22 | 2020-08-27 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110603045A (zh) * | 2017-05-09 | 2019-12-20 | 学校法人庆应义塾 | 脑肿瘤治疗用细胞制剂 |
| GB201721069D0 (en) * | 2017-12-15 | 2018-01-31 | Glaxosmithkline Biologicals Sa | Hepatitis B Immunisation regimen and compositions |
| ES2957688T3 (es) * | 2018-11-07 | 2024-01-24 | Consejo Superior Investigacion | Construcción génica suicida doble e inducible y su uso en terapia génica |
| CN111909986B (zh) * | 2019-05-10 | 2024-03-26 | 潘雨堃 | 一种基因治疗安全评估与毒副作用预防的方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005095027A (ja) | 2003-09-22 | 2005-04-14 | Reprocell Inc | 細胞の未分化状態マーカープロモーターおよびその利用 |
| JP2008541768A (ja) * | 2005-06-01 | 2008-11-27 | カリフォルニア インスティテュート オブ テクノロジー | ウィルスベクターを使用した標的化遺伝子送達方法 |
| JP2009528838A (ja) | 2006-03-06 | 2009-08-13 | エージェンシー フォー サイエンス,テクノロジー アンド リサーチ | ヒト胚性幹細胞の方法及びpodxlの発現 |
| JP2012072063A (ja) | 2010-09-27 | 2012-04-12 | Japan Health Science Foundation | 細胞の造腫瘍性試験方法及び腫瘍マーカー |
| WO2012115270A1 (ja) | 2011-02-25 | 2012-08-30 | 学校法人慶應義塾 | iPS細胞クローンの選択方法、及びその選択方法に用いる遺伝子の選択方法 |
| WO2012133674A1 (ja) | 2011-03-29 | 2012-10-04 | 国立大学法人 鹿児島大学 | 幹細胞における腫瘍化原因細胞の除去方法 |
| JP2012223104A (ja) | 2011-04-15 | 2012-11-15 | Olympus Corp | 誘導多能性幹細胞の識別方法 |
| JP2013009742A (ja) | 2011-06-28 | 2013-01-17 | Nagoya City Univ | 多能性幹細胞の増殖を抑制するための組成物及び方法 |
| JP2013530699A (ja) * | 2010-06-15 | 2013-08-01 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 生物学的応答の調査のための既製の幹細胞モデルの概要 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060288431A1 (en) * | 2003-02-13 | 2006-12-21 | Norio Nakatsuji | Marker for undifferentiated state of cell and composition and method for separation and preparation of stem cells |
| EP2251423B1 (en) * | 2008-03-07 | 2016-01-13 | National University Corporation University Of Toyama | Homologous recombination method, cloning method, and kit |
| EP2438159B1 (en) * | 2009-05-29 | 2018-10-03 | Kyoto University | Method for selecting clone of induced pluripotent stem cells |
| JP2011201813A (ja) * | 2010-03-25 | 2011-10-13 | Kagoshima Univ | サービビンプロモーターを含む増殖制御型ウイルスベクターによる造血器腫瘍の遺伝子治療 |
| CA2875858C (en) * | 2011-06-21 | 2022-05-03 | Georgia Tech Research Corporation | Adhesive signature-based methods for the isolation of stem cells and cells derived therefrom |
| WO2014052458A1 (en) * | 2012-09-25 | 2014-04-03 | Yale University | Differentiation of human ips cells to human alveolar type ii via definitive endoderm |
| AU2014248167B2 (en) * | 2013-04-03 | 2019-10-10 | FUJIFILM Cellular Dynamics, Inc. | Methods and compositions for culturing endoderm progenitor cells in suspension |
-
2015
- 2015-01-14 WO PCT/JP2015/000138 patent/WO2015107888A1/ja not_active Ceased
- 2015-01-14 EP EP20159605.3A patent/EP3690055B1/en active Active
- 2015-01-14 EP EP15737214.5A patent/EP3095864B1/en active Active
- 2015-01-14 US US15/111,084 patent/US20170051306A1/en not_active Abandoned
- 2015-01-14 JP JP2015557768A patent/JP6358713B2/ja active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005095027A (ja) | 2003-09-22 | 2005-04-14 | Reprocell Inc | 細胞の未分化状態マーカープロモーターおよびその利用 |
| JP2008541768A (ja) * | 2005-06-01 | 2008-11-27 | カリフォルニア インスティテュート オブ テクノロジー | ウィルスベクターを使用した標的化遺伝子送達方法 |
| JP2009528838A (ja) | 2006-03-06 | 2009-08-13 | エージェンシー フォー サイエンス,テクノロジー アンド リサーチ | ヒト胚性幹細胞の方法及びpodxlの発現 |
| JP2013530699A (ja) * | 2010-06-15 | 2013-08-01 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 生物学的応答の調査のための既製の幹細胞モデルの概要 |
| JP2012072063A (ja) | 2010-09-27 | 2012-04-12 | Japan Health Science Foundation | 細胞の造腫瘍性試験方法及び腫瘍マーカー |
| WO2012115270A1 (ja) | 2011-02-25 | 2012-08-30 | 学校法人慶應義塾 | iPS細胞クローンの選択方法、及びその選択方法に用いる遺伝子の選択方法 |
| WO2012133674A1 (ja) | 2011-03-29 | 2012-10-04 | 国立大学法人 鹿児島大学 | 幹細胞における腫瘍化原因細胞の除去方法 |
| JP2012223104A (ja) | 2011-04-15 | 2012-11-15 | Olympus Corp | 誘導多能性幹細胞の識別方法 |
| JP2013009742A (ja) | 2011-06-28 | 2013-01-17 | Nagoya City Univ | 多能性幹細胞の増殖を抑制するための組成物及び方法 |
Non-Patent Citations (40)
| Title |
|---|
| ANTONIW ET AL., BRIT J. CANCER, vol. 62, 1990, pages 909 |
| BESHARD ET AL., MOL. CELL BIOL., vol. 7, 1987, pages 4139 |
| BETBEDER ET AL., NUCLEICACIDS RES, vol. 17, 1989, pages 4217 |
| BOSSLET ET AL., CANCER RES., vol. 54, 1994, pages 2151 |
| CARLOS A RAMOS ET AL., STEM CELLS, vol. 28, 2010, pages 1107 - 15 |
| FEI CHEN ET AL., BIOMATERIALS, vol. 34, 2013, pages 1701 - 11 |
| FEI, CHEN ET AL., BIOMATERIALS, vol. 34, 2013, pages 1701 - 11 |
| FURLER, S. ET AL., GENE THER., vol. 8, 2001, pages 864 - 73 |
| FUYI, CHENG ET AL., BIOMATERIALS, vol. 33, 2012, pages 3195 - 204 |
| HAEBERLIN ET AL., PHARMACEUTICAL RES., vol. 10, 1993, pages 1553 |
| HAENSELER ET AL., BIOCHEMISTRY, vol. 31, 1992, pages 891 |
| HASEGAWA ET AL., STEM CELLS, vol. 25, 2007, pages 1707 - 12 |
| HOF ET AL., IMMUNITAT UNDINFEKTION, vol. 16, 1988, pages 220 |
| HUBER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 8039 |
| JAMES, L. ET AL., GENOME RES., vol. 10, 2000, pages 1788 - 1795 |
| KATZ ET AL., J. ANTIBIOTICS, vol. 43, 1990, pages 231 |
| KOSZALKA; KRENITSKY, J. BIOL CHEM, vol. 254, 1979, pages 8185 |
| LANDY, A, ANN. REV. BIOCHEM., vol. 58, 1989, pages 913 - 949 |
| LI, F. ET AL., BIOCHEM. J., vol. 344, 1999, pages 305 - 311 |
| LI, F. ET AL., CANCER RES., vol. 59, 1999, pages 3143 - 3151 |
| MEYER ET AL., CANCERRES., vol. 53, 1993, pages 3956 |
| MIURA, K. ET AL., NATURE BIOTECHNOLOGY, vol. 27, 2009, pages 743 - 745 |
| MULLEN ET AL., PROC. NATL. ACAD. SCI. USA., vol. 89, 1992, pages 33 |
| NAKAGAWA, M. ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 101 - 6 |
| OKITA, K. ET AL., SCIENCE, vol. 7, 2008, pages 949 - 53 |
| PTASHNE, M: "A Genetech Switch: Phage (Lambda) And Higher Organisms", 1992, CAMBRIDGE, MA: CELL PRESS |
| See also references of EP3095864A4 |
| SENTER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 4842 |
| SONG-TAO YU ET AL.: "Noninvasive and real-time monitoring of the therapeutic response of tumors in vivo with an optimized hTERT promoter", CANCER, vol. 118, no. ISSUE., 25 August 2011 (2011-08-25), pages 1884 - 1893, XP055213274, Retrieved from the Internet <URL:http://onlinelibrary.wiley.com/doi/10.1002/cncr.26476/full> * |
| SORSCHER ET AL., GENETHERAPV, vol. 1, 1994, pages 233 |
| TAKAHASHI, K. ET AL., CELL, vol. 131, 2007, pages 861 - 72 |
| TAKAKURA, M. ET AL., CANCER RES., vol. 59, 1999, pages 551 - 557 |
| TAKEYA SATO ET AL., MOL THER, vol. 15, 2007, pages 962 - 70 |
| UPCROFT ET AL., INT. J. PARASITOLO, vol. 20, 1990, pages 489 |
| VRADHULA ET AL., BIOCONJUGATE CHEMISTRY, vol. 4, 1993, pages 334 |
| WALTON ET AL., BIOCHEM. PHARMACOL., vol. 44, 1992, pages 251 |
| WERNIG, M. ET AL., CELL STEM CELL, vol. 2, 2008, pages 10 - 2 |
| YASUHIRO TERAZAKI ET AL., HEPATOLOGY, vol. 37, 2003, pages 155 - 63 |
| YU, J. ET AL., SCIENCE, vol. 8, 2009, pages 797 - 801 |
| ZHU B. ET AL., BIOTECHNIQUES, vol. 43, no. 3, 2007, pages 354 - 9 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018122932A1 (ja) * | 2016-12-26 | 2018-07-05 | オリンパス株式会社 | 細胞の選別方法、純化された細胞集団の製造方法、及び発光イメージングシステム |
| JPWO2020171222A1 (ja) * | 2019-02-22 | 2020-08-27 | ||
| WO2020171222A1 (ja) * | 2019-02-22 | 2020-08-27 | 国立大学法人 鹿児島大学 | ヒト多能性幹細胞の安全領域に長い外来遺伝子を組み込み正常機能させる方法 |
| JP7491589B2 (ja) | 2019-02-22 | 2024-05-28 | 国立大学法人 鹿児島大学 | ヒト多能性幹細胞の安全領域に長い外来遺伝子を組み込み正常機能させる方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6358713B2 (ja) | 2018-07-18 |
| EP3095864B1 (en) | 2020-04-15 |
| US20170051306A1 (en) | 2017-02-23 |
| EP3690055B1 (en) | 2024-12-11 |
| EP3095864A1 (en) | 2016-11-23 |
| EP3095864A4 (en) | 2017-09-20 |
| EP3690055A1 (en) | 2020-08-05 |
| JPWO2015107888A1 (ja) | 2017-03-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230282445A1 (en) | Method of nuclear reprogramming | |
| JP7377309B2 (ja) | 体細胞を再プログラミングすることにおけるwnt経路の刺激 | |
| JP6358713B2 (ja) | 幹細胞における腫瘍化原因細胞の新たな標識法と治療法 | |
| JP2021151254A (ja) | 遺伝的プログラミングによる多系統造血前駆細胞の作製 | |
| EP2732029B1 (en) | Methods for cell reprogramming and genome engineering | |
| CA2697621C (en) | Method of efficiently establishing induced pluripotent stem cells | |
| CN103562376B (zh) | 复壮细胞的方法 | |
| JP2018531020A6 (ja) | 遺伝的プログラミングによる多系統造血前駆細胞の作製 | |
| US20110003365A1 (en) | Method of preparing induced pluripotent stem cells deprived of reprogramming gene | |
| US8709805B2 (en) | Canine iPS cells and method of producing same | |
| Lee et al. | Efficient exogenous DNA-free reprogramming with suicide gene vectors | |
| Malecki | ‘Above all, do no harm’: safeguarding pluripotent stem cell therapy against iatrogenic tumorigenesis | |
| Schlaeger | Nonintegrating human somatic cell reprogramming methods | |
| JP7491589B2 (ja) | ヒト多能性幹細胞の安全領域に長い外来遺伝子を組み込み正常機能させる方法 | |
| Kramer et al. | Full biological characterization of human pluripotent stem cells will open the door to translational research | |
| US11078496B2 (en) | System for F-box hormone receptor regulated protein expression in mammalian cells | |
| US11542524B2 (en) | Elimination of proliferating cells from stem cell-derived grafts | |
| Kubarakos | A CRISPR/Cas9-mediated FACS screen reveals novel regulators of Wnt/β-catenin signalling in human embryonic stem cells | |
| Chakraborty | Synthetic Biology-Based Approaches to Enhance Transgene Attributes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15737214 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015557768 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015737214 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015737214 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15111084 Country of ref document: US |