WO2015124559A1 - Polymerisierbares dentalmaterial - Google Patents
Polymerisierbares dentalmaterial Download PDFInfo
- Publication number
- WO2015124559A1 WO2015124559A1 PCT/EP2015/053303 EP2015053303W WO2015124559A1 WO 2015124559 A1 WO2015124559 A1 WO 2015124559A1 EP 2015053303 W EP2015053303 W EP 2015053303W WO 2015124559 A1 WO2015124559 A1 WO 2015124559A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- methyl
- dental material
- material according
- ethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/60—Preparations for dentistry comprising organic or organo-metallic additives
- A61K6/62—Photochemical radical initiators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/80—Preparations for artificial teeth, for filling teeth or for capping teeth
- A61K6/884—Preparations for artificial teeth, for filling teeth or for capping teeth comprising natural or synthetic resins
- A61K6/887—Compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
Definitions
- the invention relates to a polymerizable dental material according to the preamble of claim 1.
- Object of the invention It is ⁇ further provides the use of a tertiary aromatic amine according to claim 13 as co-initiator in a po- lymerisierbaren dental material.
- the invention is also a co-initiator with enhanced solubility in water and a radically polymerizable dental restorers ⁇ tion material containing the co-initiator whose adhesive strength to the tooth, in relativelyster application improved.
- the radically polymerizable dental material is preferably a flowable, self-etching, self-adhesive composite for direct fillings.
- Dental restoration materials are subject to special requirements. This is particularly true for direct fuel ⁇ lung materials, fixing cements or adhesives, which contain a polymerizable acid as an adhesive and / or caustic component, intra administered orally and lichtgehär ⁇ tet.
- the dental restorative materials must be biologically and toxicologically harmless, the highest possible depth of cure (DHT), low polymerization shrinkage, high storage stability (2 or more years, preferably at room temperature) and low sensitivity to ambient light and in the cured state the desired Lichtopazmaschine, color stability, radio-opacity, a ge ⁇ rings water absorption (WA) and water solubility (WS), and have the highest possible bending strength (BF).
- DHT depth of cure
- WA ge ⁇ rings water absorption
- WS water solubility
- BF bending strength
- the polymerized restoration should have the highest possible shear bond strength (SBS) on dentin.
- a proportion of hydrophilic monomers (water ⁇ solubility> 50 g / 1) is preferably included, containing no acid group. Preference is given to those hydrophilic monomers which are miscible with water in any ratio, in particular 2-hydroxyethyl methacrylate (HEMA).
- HEMA 2-hydroxyethyl methacrylate
- the polymerization shrinkage and the strength of dental restorative materials can be improved by adding known crosslinkers and inorganic fillers.
- crosslinking agents mixtures of di (meth) ac triacrylate are preferably based on bisphenol-A and a low molecular weight crosslinkers such as triethylene glycol dimethacrylate (TEDMA) was ⁇ sets.
- TEDMA triethylene glycol dimethacrylate
- Suitable inorganic fillers have particle sizes of less than 30 ⁇ m and a hydrophobized surface.
- X-ray opaque silanized glasses are preferably used as inorganic fillers.
- camphorquinone and / or phosphine oxide compounds as photoinitiators and alkyl (4-dialkylaminobenzoic acid) esters or butoxyethyl (4-dialkylaminobenzoic) esters as coinitiators.
- alkyl (4-dialkylaminobenzoic acid) esters or butoxyethyl (4-dialkylaminobenzoic) esters as coinitiators.
- Such materials have been described many times, examples being the following documents WO2011139936, EP2286784, EP2153811, WO02007100569,
- WO 00/49064 describes a cationically curable dental restorative which comprises ⁇ -ethylpoly (oxyethylene) -4- [bis (co (2-hydroxyethyl) poly (oxyethylene)] amino] benzoate (PEG-25).
- PBA ⁇ -ethylpoly (oxyethylene) -4- [bis (co (2-hydroxyethyl) poly (oxyethylene)] amino] benzoate
- PBA a coinitiator.
- This co-initiator is described there, however, adversely esters as the alkyl (4-Dialkylaminobenzo ⁇ eklare) opposite, since it has only a low reactivity.
- Object of the present invention is to provide an improvedfitzu ⁇ dental material for direct dental restoration having a high DHT and a high SBS and BF of the cured Restaurativs.
- the simultaneous compliance or improvement of properties such as biological and toxicological safety, storage stability, insensitivity to ambient light and polymerization shrinkage and color stability, low water absorption (WA) and water solubility (WS) in casualhär ⁇ ended state is preferred.
- the invention solves this problem by the features of the independent patent claims. Advantageous embodiments are given in the dependent claims.
- the invention provides a dental material insbeson ⁇ particular a light-curing self-adhesive and self-etching radically polymerizable Dentalrestaurativ containing: a. at least one monomer containing at least one radically polymerizable ethylenically unges2011 ⁇ preferential group and at least one acid group, b.
- the invention provides a dental material, preferably dental adhesive, with the properties mentioned in the task, which can be applied simply and safely with a few treatment steps.
- the adhesive surface (the dentin) must not be previously treated with etchants and Be prepared for adhesion agents.
- the term polymerizable dental material designates any dental materials whose curing takes place, at least essentially, by (preferably free-radical) polymerization and which can be used for or in the case of a dental restoration.
- a dental material according to the invention at least one further be ⁇ vorzugt two, more preferably all three of the following characteristics on egg ⁇ : light-curing, self-adhesive andsruc- zend. It can be a restoration material or an adhesive.
- a Applicant does not binding attempt to explain the effect of the invention is that the improved adhesion to dentin and the good mechanical properties of the inventive dental material (high reactivity) are based on increased amphiphilicity of the co-initiator which a ⁇ hand a very good solubility in a aqueous hybrid ⁇ layer of the dentin as well as in the dental material itself ⁇ acts, the dental material itself is relatively hydrophobic.
- the invention has recognized that high proportions of polymerizable Barer acids and HEMA according to the prior art in a dental restorative material, various disadvantages ha ⁇ ben, in particular, they can use the hardening depth (DHT) comparable wrestlers and water absorption (WA) and water solubility (WS ) so that the restorations are no longer biologically and toxicologically safe. Furthermore, it is difficult with such compositions to achieve the desired flexural strength of the restorative material.
- DHT hardening depth
- WA water absorption
- WS water solubility
- the preferred radically polymerizable ethylenically unsaturated groups are the acrylate, the methacrylate, the acrylamide and methacrylamide groups.
- (meth) acrylic always means both - acrylic and methacrylic.
- Monomers containing at least one free-radically polymerizable ethylenically unsaturated group and at least one acid group are known to the person skilled in the art.
- Preferred acid groups are carboxylic acid, phosphorus ⁇ acid, phosphonic acid, bisphosphonic acid and sulfonic acid group. Particularly preferred are the phosphorus-containing acid groups.
- the polymerizable acid is (a) before ⁇ Trains t to 1 to 20 wt -.%, More preferably from about 1 to 15 -.%, More preferably from 5 to 15 wt .-%, based on the total mass of the dental material contained monomers containing ⁇ at least one free-radically polymerizable ethylenically unsaturated group.
- the photoinitiator (c) is preferably from 0.05 to 10, preferably from 0.05 to 5, most preferably from 0.1 to 2 wt .-%, based on the total mass of the monomers contained in the dental material containing at least one free radical polymerizable ethylenically unsaturated
- Preferred photoinitiators (c) absorb more light in the wavelength range between 390 and 560 nm, more preferably between 420 and 480 nm.
- the photoinitiator is preferably a dental material-soluble aliphatic 1,2-diketone, such as camphorquinone (CQ) or propyl-phenyl-diketone (PPD), particularly preferably camphorquinone and / or a camphorquinone derivative, for example having a carboxyl, carboxylate, sulfonic acid or sulfonate group.
- CQ camphorquinone
- PPD propyl-phenyl-diketone
- camphorquinone and / or a camphorquinone derivative for example having a carboxyl, carboxylate, sulfonic acid or sulfonate group.
- the tertiary aromatic amine is preferably (co-initiator according to feature group (d)) selected from the group ⁇ best starting from:
- R 1 and R 2 are independently of one another) C 1 -C 5 -alkyl, preferably methyl,
- R3 is alkyl, preferably methyl
- R 4 is H or C 1 -C 5 -alkyl, preferably methyl
- R 5 and R 6 are, independently of one another, C 1 -C 5 -alkyl, preferably methyl,
- R 7 and R 8 are independently selected from H, alkyl, alkoxyalkyl and polyalkylene oxide, preferably polyalkylene oxide having at least two oxyethylene and / or oxypropylene units, x ⁇ 0 and y> 1.
- the sum of x and y is preferably 3 to 20, more preferably 5 to 17.
- x ⁇ y is preferably 3 to 20, more preferably 5 to 17.
- the polyalkylene oxide may be formed as block copolymers or as statis ⁇ diagram copolymers.
- a polyalkylene oxide chain preferably has a molar mass between 130 and 1200 g / mol, more preferably between 220 and 1200 g / mol, more preferably between 300 and 800 g / mol.
- An example is 4-dimethylaminobenzoic acid poly (ethylene glycol-350) monomethyl ether ester.
- Suitable coinitiators (d) of the formulas III and IV are 4-dimethylaminobenzamide; 4-dimethylamino-N-propylbenzamide; 3-dimethylamino-N-propylbenzamide; 4-dimethylamino-N- (hydroxyethyl) benzamide; 4-dimethylamino-N- (hydroxypropyl) benzamide; 4-dimethylamino-N- (hydroxybutyl) benzamide; 4-dimethylamino-N- (2-methoxyethyl) benzamide; 3-dimethylamino-N- (2-methoxyethyl) benzamide; 4-Dimethylamino-N- (2- (2-hydroxyethyl) ethyl) benzamide.
- coinitiators (d) of formulas III and IV are 4-dimethylamino-N- (2- (2- (2-methoxyethoxy) ethoxy) ethyl) benzamide; 4-dimethylamino-N-poly (ethylene oxide) methyl ether benzamide; 4-dimethylamino-N-poly (propylene oxide-jblocif-ethylene oxide) methyl ether-benzamide; 4-dimethylamino-N-poly (ethylene oxide-propylene oxide-stat) me ⁇ methyl ether-benzamide.
- the polyalkylene oxide chains can be used as block copolymers or as random copolymers.
- a polyalkylene oxide chain particularly preferably has a molar mass between 130 and 1200 g / mol.
- the coinitiators without amide group according to the invention have a water solubility greater than 2 g / l, more preferably greater than 50 g / l, more preferably greater than 100 g / l, more preferably greater than 500 g / l.
- the coinitiators with amide group according to the invention have a water solubility greater than 0.4 g / l, preferably> 2 g / l, more preferably> 50 g / l, more preferably> 100 g / l, even more preferably> 500 g / l.
- the coinitiator (d) is from 0.05 to 10, preferably from 0.1 to 10, most preferably from 0.5 to 5 Wt .-%, based on the total mass of the monomers contained in the dental material containing at least one radically polymerizable ethylenically unsaturated group enthal ⁇ th.
- Suitable further monomers (b), ie monomers containing at least one free-radically polymerizable ethylenically unsaturated group which have no acid group are known to the person skilled in the art.
- the at least one monomer that no acid groups and at least one radically polymerizable ethylenically unsaturated group contains (feature b) of claim 1.
- R 1 is H, methyl or ethyl, preferably H or methyl;
- R 8 is H, methyl or ethyl, preferably H or methyl;
- HG is OH, (O-CH 2 -CH 2 ) m -OR 7 ; preferably OH;
- R 7 is H or methyl;
- n 3-20, preferably 6-20;
- v 0 or 1; preferably 0.
- R 8 is H, methyl or ethyl, preferably H or methyl
- HG is (O-CH 2 -CH 2 ) m ;
- n 4-20, preferably 6-20;
- v 0 or 1; preferably 0.
- Preferred monomers bl) as described above are known to the person skilled in the art. Examples include hydroxyethyl methacrylate, hy- roxypropylmethacrylat, hydroxyethylacrylamide, Hydroxypropy- lacrylamid, 2, 3-dihydroxypropyl, 1, 3-dihydroxy-2-propanylmethacrylate, methoxy (polyethylene glycol-350) me ⁇ methacrylate or methoxy (polyethylene glycol 500) methacrylate. Particularly preferred is hydroxyethyl methacrylate (HEMA).
- HEMA hydroxyethyl methacrylate
- Preferred monomers b2) as described above are known to the person skilled in the art. Examples are glycerol dimethacrylate and N, '- (1, 2-dihydroxyethylene) bisacrylamide. Further preferred monomers are described in EP2133368 (Sekiguchi). Preferred monomers b3) as described above are known to the skilled man. Examples are polyethylene glycol 200
- Suitable monomers b1) to b3) preferably have at least one water solubility of 50 g / l of water.
- the dental material according to the invention may preferably zusharm ⁇ Lich comprise at least one monomer b4) containing no acid groups and at least two radically polymerizable ethylenically unsaturated groups, said we ⁇ tendonss a monomer b4) is not selected from the group bl), b2) and b3) is selected.
- Preferred monomers b4) as described above are known to the skilled man.
- Examples are allyl (meth) acrylate; 1,3-propanediol di (meth) acrylate; 1,3-butanediol di (meth) acrylate; 1,4-butanediol di (meth) acrylate; 1,5-pentanediol di (meth) acrylate; Neopentyl glycol di (meth) acrylate; 1, 6-hexanediol dimethacrylate; 1, 9-nonanediol di (meth) acrylate; 1, 10-decanediol di (meth) ac ⁇ triacrylate; 1,12-dodecanediol di (meth) acrylate; Ethylene glycol di (meth) acrylate; Diethylene glycol di (meth) acrylate;
- the water solubility of the monomer b4) is preferably less than 50 g / l.
- mixtures of at least one monomer b1) to b3) and at least one monomer or crosslinker of group b4) are particularly preferred.
- the dental material radically polymerizable by irradiation with visible light furthermore preferably contains 0.1 to 90% by weight, preferably 45 to 70% by weight, of organic and / or inorganic fillers.
- Suitable fillers are known to the person skilled in the art. Surface-treated inorganic fillers ge ⁇ geninate the acids a resin matrix are substantially preferred are inert, such as in the dental material, that is, the resin matrix are un- soluble, wherein the free-radically poly- merisierbaren monomers and dissolved additives therein are understood resin matrix.
- Suitable fillers are disclosed, for example, in EP 1 720 506 (Neffgen et al).
- the radically polymerizable by irradiation with visible light dental material further preferably contains 0.01 to 20, preferably 0.01 to 10 wt .-% dental ad ⁇ additives.
- dental additives include polymerization inhibitors such as BHT, UV stabilizers, rheology modifiers, dyes and radiopaque.
- the means of irradiation with visible light radically polymerizable dental material may also contain non-polymerizable organic solvent, for example, volatile solvents such as ethanol or acetone, as well as contain What ⁇ ser.
- non-polymerizable organic solvent for example, volatile solvents such as ethanol or acetone
- the invention furthermore relates to a composition according to the invention for use as a light-curing self-adhesive and self-etching radically polymerizable dental restorative or the use of such a composition as or in the production of a light-curing self-adhesive and self-etching radically polymerizable dental restorative.
- Another object of the invention is the use of a tertiary aromatic amine containing a benzene ring comprises the at least one dialkylamine and min ⁇ least one further group are directly bonded, the further group is selected from: i. Carbonklareester weakness units containing at least one polyoxyalkylene group having Minim ⁇ least 2 oxyethylene and / or Oxypropylenein-, and
- sulfur dioxide is developed from 63 ga 2 S0 3 and 100 ml of a 6 MH 2 SO 4 within one hour and passed through a capillary into the filtrier ⁇ th approach.
- the red-brown mixture stirs after completion of gas introduction overnight. Thereafter, the above-described gas evolution and introduction are repeated.
- the red-brown-black mixture is filtered through Celite 545 and the filter residue washed with deionized water. From the approach, about 130 ml of water are distilled off at 100 mbar.
- the distillation residue is neutralized with solid BaCC> 3.
- the thick reddish-brown mass is filtered through Celite 545 and the filter residue washed with about 150 ml of deionized water. A clear, yellow solution is obtained.
- the Lö ⁇ solution is concentrated on a rotary evaporator until about 80 ml of liquid remain. A white None ⁇ Derschlag which is filtered is produced.
- the crude product (about 10.9 g) is dissolved in 100 ml of methanol.
- the batch is stirred for about 70 h with exclusion of moisture and light at room temperature. Here ent ⁇ is little rainfall.
- the solution is decanted from the precipitate.
- the precipitate is washed with Et ⁇ THF.
- the organic phases are combined and the solvent is removed on a rotary evaporator. The residue is taken up in 50 ml of dichloromethane.
- the organic phase is shaken out with 2 ⁇ 10 ml of 0.5 N HCl solution, 1 ⁇ 10 ml of saturated NaHCO 3 solution and 1 ⁇ 10 ml of saturated NaCl solution. Subsequently, the organic phase is dried over Na 2 SC> 4.
- the batch is filtered.
- the filter residue is washed with a little THF.
- the solvent is removed on Rotati ⁇ onsverdampfer and the residue evaporated in high vacuum of volatile constituents.
- the crude product is poured into a mixture of 80 ml deionized water and 0.8 ml 2N HCl.
- the resulting emulsion is diluted once more with 100 ml of deionized water and filtered.
- To the filtered solution 0.5 ml of saturated sodium bisulfite solution is added with a 2 N NaOH to a pH value of 7.5 is ⁇ represents.
- the aqueous phase is extracted with 3x60 ml of dichloromethane.
- the batch was filtered through Celite 545 and the filter residue was washed with 20 ml of THF.
- THF was removed on a rotary evaporator and the residue in 100 Added dichloromethane.
- the organic phase was extracted with 2x20 ml of 0.5N HCl, 1x20 ml of saturated NaHCO 3 and 1x20 ml of saturated NaCl solution.
- the organic phase was dried over a 2 SÜ 4 and the solvent removed on a rotary evaporator.
- the aqueous washing phases were combined and water was removed on a rotary evaporator.
- the residue was hiert ⁇ extracting with 100 ml of acetone and 50 ml of ethanol.
- the mixture is extracted with 20 ml of a 2N HCl solution.
- a seeded saturated NaHC03 ⁇ ⁇ solution is then extracted with 20 ml.
- the organic phase is dried over Na 2 SC> 4, filtered and the solvent removed on a rotary evaporator.
- the crude product is 3.04 g of a slightly greenish-yellow, highly viscous liquid.
- the crude product is further purified by column chromatography on ⁇ . Stationary phase: AI 2 O 3 , neutral.
- a mixture of 10 ml of dichloromethane and 1.64 ml (20 mmol) of n-propylamine is prepared.
- the flask is cooled with an ice / salt mixture.
- the mixture from the dropping funnel is slowly added dropwise to the reaction mixture within 15 min. 10 minutes after addition, the cooling bath is removed and the An ⁇ set 72 h stirred at room temperature.
- the mixture is about 50 g Al 2 O 3 added to neutral and the solution ⁇ medium removed on a rotary evaporator. Further drying takes place in vacuo at room temperature.
- An MPLC system from Buchi was used. It consisted of a pump module C-601, a pump manager C-615 and a fraction collector C-660. The samples to be chromatographed were dissolved in the eluent and applied to a sample loop (up to 20 ml) via a 4-way injection valve. Glass columns type C-690 were used. The exact dimensions are described in the syntheses.
- the measurements were carried out on a Gemini 300 MHz NMR spectrometer from Varian.
- the calibration of the spectra was based on the residual proton signal of the deuterated solvent used.
- DHT Depth of cure
- Bovine teeth without pulp were embedded in a cold polymerizing resin (Viscovoss GTS with MEKP MEH Hardener, Voss Chemie). Immediately before the experiment ⁇ passage 10 bovine teeth were wet to the desired surface (enamel or dentin) per test series
- the adhesives were triturated s using a disposable brush 20 on the tooth top ⁇ surface and then exposed for 20 s with an LED lamp (Mini LED, Acteon, blue light, 182 mW / cm 2). Subsequently, a two-part Teflon mold (height 3 mm) with a bore (diameter 3 mm) was placed flat on the prepared area and fixed with metal clamps. The bore was filled with the adhesive without air bubbles. Then it was irradiated for 40 s with a LED lamp (Mini LED, Acteon). After curing, the Teflon mold was removed and excess residues of the cured adhesive were removed with a scalpel.
- an LED lamp Mini LED, Acteon, blue light, 182 mW / cm 2
- test specimens were then stored for 1 day at 37 ° C in a water bath. Immediately prior to measurement of shear adhesive force was allowed to cool the water in ⁇ nergur one hour at 23 ° C.
- the test specimens were clamped in a shearing apparatus according to ISO 10477: 2004 for measuring the shear adhesion force and the SBS was measured with a universal testing machine (type Z 010 / TN2A, Zwick) at a feed rate of 0.5 mm / min.
- the resulting ⁇ nisse were given in the form of a mean value with standard deviation.
- Test specimens whose bond has already dissolved before the measurement or whose bonding was not better than the pre-load of 0.01 N set, are included in the averaging with a shear adhesion force of 0 MPa. Determination of water solubility
- the water solubility was determined in deionized water by UV / Vis spectroscopy.
- the spectra were with a UV / Vis spectrometer (Evolution 600, Thermo Scientific) recorded between 200-900 nm. The measurement was carried out in
- Quartz cuvettes (layer thickness 10 mm).
- ⁇ the pure solvent deionized water was used. From the spectra of the coinitiators, the background spectrum was subtracted.
- the absorption of the different coinitiators was measured in the absorption maximum (between 290-310 nm) and a linear relationship between the known concentration and the measured absorption was determined.
- resin compositions were prepared from the components shown in the tables, respectively. These resin compositions served as a matrix for the fillers added to the paste preparation. To prepare the composite pastes, 60 parts of glass powder and 3 parts of silica were suspended in 37 parts of resin matrix. The preparation was otherwise as described in the methods.
- hydrophilic photoinitiator and hydrophobic coinitiator had the lowest shear adhesion to both enamel and dentin.
- Co-initiator (C9) provided a similar shear strength to enamel as Cll. Shear adhesion to dentine was significantly higher than Cll.
- the combination of hydrophilic photoinitiator and hydrophilic co-initiator (CIO) delivered compared to C9 and Cll a slightly clotting ⁇ gere shear bond strength to enamel.
- the shear strength on dentine was also significantly higher than in Cll. From this series of tests, it can be seen that a hydrophilic or water-soluble, aromatic, tertiary amine as a coinitiator allows a significant improvement in the adhesion to dentin.
- the flexural strength can be further improved.
Landscapes
- Health & Medical Sciences (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Plastic & Reconstructive Surgery (AREA)
- Biophysics (AREA)
- Dental Preparations (AREA)
Abstract
Die Erfindung betrifft ein Dentalmaterial, das wenigstens ein Monomer enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe und mindestens eine Säuregruppe; wenigstens ein Monomer, das keine Säuregruppen und mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe enthält; wenigstens einen Photoinitiator; sowie mindestens ein tertiäres aromatisches Amin als Coinitiator, das einen Benzolring aufweist, an den mindestens eine Dialkylamingruppe und mindestens eine weitere Gruppe direkt gebunden sind, enthält. Erfindungsgemäß ist vorgesehen, dass die weitere Gruppe ausgewählt ist aus Carbonsäureestergruppen enthaltend mindestens eine Polyoxyalkylengruppe mit mindestens 2 Oxyethylen- und/oder Oxypropyleneinheiten; und Amidgruppen.
Description
Polymerisierbares Dentalmaterial
Die Erfindung betrifft ein polymerisierbares Dentalmaterial gemäß dem Oberbegriff des Anspruchs 1. Gegenstand der Er¬ findung ist ferner die Verwendung eines tertiären aromatisches Amins gemäß Anspruch 13 als Coinitiator in einem po- lymerisierbaren Dentalmaterial. Gegenstand der Erfindung ist auch ein Coinitiator mit verbesserter Wasserlöslichkeit sowie ein radikalisch polymerisierbares dentales Restaura¬ tionsmaterial enthaltend den Coinitiator, dessen Haftkraft am Zahn, bei einfachster Anwendung, verbessert wird. Das radikalisch polymerisierbare Dentalmaterial ist bevorzugt ein fließfähiges, selbstätzendes, selbsthaftendes Komposit für direkte Füllungen.
An dentale Restaurationsmaterialien werden besondere Anfor- derungen gestellt. Dies gilt insbesondere für direkte Fül¬ lungsmaterialien, Befestigungszemente oder Adhäsive, die eine polymerisierbare Säure als adhäsive und/oder ätzende Komponente enthalten, intraoral appliziert und lichtgehär¬ tet werden. Die dentalen Restaurationsmaterialien müssen biologisch und toxikologisch unbedenklich sein, eine möglichst hohe Durch- härtetiefe (DHT) , einen geringen Polymerisationsschrumpf, eine hohe Lagerstabilität (2 oder mehr Jahre, bevorzugt bei Raumtemperatur) und geringe Empfindlichkeit gegenüber Umge- bungslicht sowie im ausgehärteten Zustand die gewünschte Lichtopazität, Farbstabilität, Röntgenopazität , eine ge¬ ringe Wasseraufnahme (WA) und Wasserlöslichkeit (WS) sowie
eine möglichst hohe Biegefestigkeit (BF) aufweisen. Die po- lymerisierte Restauration soll insbesondere eine möglichst hohe Scherhaftkraft (SBS) auf Dentin aufweisen.
Insbesondere um die SBS von Restaurationsmaterialien auf Dentin zu steigern, ist es bekannt, dentalen Restaurations¬ materialien höhere Anteile an polymerisierbaren Säuren zuzufügen .
Bevorzugt ist auch ein Anteil hydrophiler Monomere (Wasser¬ löslichkeit >50 g/1) enthalten, die keine Säuregruppe ent- halten. Bevorzugt sind solche hydrophile Monomere, die in jedem Verhältnis mit Wasser mischbar sind, zu nennen wäre hier insbesondere 2-Hydroxethylmethacrylat (HEMA) .
Der Polymerisationsschrumpf und die Festigkeit dentaler Restaurationsmaterialien kann durch Zugabe von bekannten Vernetzern und anorganischen Füllstoffen verbessert werden. Als Vernetzer werden bevorzugt Mischungen von Di (meth) ac- rylat auf Basis von Bisphenol-A und einem niedermolekularen Vernetzer wie Triethylenglycoldimethacrylat (TEDMA) einge¬ setzt. Geeignete anorganische Füllstoffe weisen Teilchen- großen unter 30 ym und eine hydrophobisierte Oberflächene auf. Als anorganische Füllstoffe werden bevorzugt röntgen- opake silanisierte Gläser eingesetzt.
Bekannte selbsthaftende und selbstätzende radikalisch poly- merisierbare dentale Restaurationsmaterialien, insbesondere fließfähige Komposite für die direkte Füllung, weisen Ver¬ besserungsbedarf bezüglich oben genannter Eigenschaften auf. Insbesondere besteht weiterhin Bedarf die SBS fließfä¬ higer Komposite an Dentin zu verbessern, ohne dass dieses zuvor mit separaten Ätzmitteln und/oder separaten Haftver- mittlem vorbereitet wurde.
Bekannte lichthärtende selbsthaftende und selbstätzende ra¬ dikalisch polymerisierbare Dentalrestaurative enthalten ei¬ nen Photoinitiator und ein tertiäres aromatisches Amin als Coinitiator. Besonders bevorzugt wird eine Kombination aus Campherchinon und/oder Phosphinoxid-Verbindungen als Photoinitiatoren sowie Alkyl- (4-Dialkylaminobenzoesäure) -ester oder Butoxyethyl- (4-Dialkylaminobenzoesäure) -ester als Coinitiatoren . Solche Materialien sind vielfach beschrieben, beispielhaft seien hier folgende Dokumente genannt WO2011139936, EP2286784, EP2153811, W02007100569 ,
EP1784155, EP1387657 und EP0136186.
In der WO 00/49064 wird ein kationisch härtbares dentales Restaurativ beschrieben, welches ω-Ethylpoly (oxyethylen) - 4- [bis ( co- (2-hydroxy-ethyl) poly (oxyethylen) ) -amino] benzoat (PEG-25-PBA) als Coinitiator enthalten soll. Dieser Coinitiator wird dort jedoch als den Alkyl- (4-Dialkylaminobenzo¬ esäure) -estern gegenüber nachteilig beschrieben, da er eine nur geringe Reaktivität aufweist.
Aufgabe der vorliegenden Erfindung ist es, ein verbessertes Dentalmaterial für die direkte Zahnrestauration bereitzu¬ stellen, welches eine hohe DHT aufweist sowie eine hohe SBS und BF des gehärteten Restaurativs . Bevorzugt ist die gleichzeitige Einhaltung oder Verbesserung von Eigenschaften wie biologischer und toxikologischer Unbedenklichkeit, Lagerstabilität, Unempfindlichkeit gegenüber Umgebungslicht und Polymerisationsschrumpf sowie Farbstabilität, geringe Wasseraufnahme (WA) und Wasserlöslichkeit (WS) im ausgehär¬ teten Zustand.
Die Erfindung löst diese Aufgabe durch die Merkmale der un- abhängigen Patentansprüche. Vorteilhafte Ausführungsformen sind in den abhängigen Patentansprüchen angegeben.
Gegenstand der Erfindung ist ein Dentalmaterial, insbeson¬ dere ein lichthärtendes selbsthaftendes und selbstätzendes radikalisch polymerisierbares Dentalrestaurativ, das enthält : a. wenigstens ein Monomer enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesät¬ tigte Gruppe und mindestens eine Säuregruppe, b. wenigstens ein Monomer, das keine Säuregruppen und mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe enthält, wenigstens einen Photoinitiator, mindestens ein tertiäres aromatisches Amin als Co-Initiator, das einen Benzolring aufweist, an den mindestens eine Dialkylamingruppe und mindes tens eine weitere Gruppe direkt gebunden sind, dadurch gekennzeichnet, dass die weitere Gruppe ausgewählt ist aus: i. Carbonsäureestergruppen enthaltend mindes- tens eine Polyoxyalkylengruppe mit mindes¬ tens 2 Oxyethylen- und/oder Oxypropylenein- heiten, und ii. Amidgruppen.
Die Erfindung stellt ein Dentalmaterial, bevorzugt Dental- adhäsiv, mit den in der Aufgabenstellung genannten Eigenschaften zur Verfügung, welches einfach und sicher mit wenigen Behandlungsschritten anzuwenden ist. Bevorzugt muss die Haftfläche (das Dentin) zuvor nicht mit Ätzmitteln und
Haftvermittlern vorbereitet werden. Der Begriff polymeri- sierbares Dentalmaterial bezeichnet im Rahmen der Erfindung jegliche Dentalmaterialien, deren Aushärtung mindestens in wesentlichen Teilen durch (bevorzugt radikalische) Poly- merisation erfolgt und die für eine oder bei einer dentalen Restauration verwendet werden können. Bevorzugt weist ein erfindungsgemäße Dentalmaterial wenigstens eine, weiter be¬ vorzugt zwei, weiter bevorzugt alle drei der folgenden Ei¬ genschaften auf: lichthärtend, selbsthaftend und selbstät- zend. Es kann sich um ein Restaurationsmaterial oder um ein Adhäsiv handeln.
Ein die Anmelderin nicht bindender Erklärungsversuch der Wirkung der Erfindung ist, dass die verbesserte Haftung auf Dentin und die guten mechanischen Eigenschaften des erfin- dungsgemäßen Dentalmaterials (bei hoher Reaktivität) auf erhöhter Amphiphilie des Coinitiators beruhen, die einer¬ seits eine sehr gute Löslichkeit in einer wässrigen Hybrid¬ schicht des Dentins als auch im Dentalmaterial selbst be¬ wirkt, wobei das Dentalmaterial selbst vergleichsweise hyd- rophob ist.
Die Erfindung hat erkannt, dass hohe Anteile polymerisier- barer Säuren und HEMA gemäß dem Stand der Technik in einem dentalen Restaurationsmaterial verschiedene Nachteile ha¬ ben, insbesondere können sie die Durchhärtetiefe (DHT) ver- ringern und die Wasseraufnahme (WA) und Wasserlöslichkeit (WS) soweit erhöhen, sodass die Restaurationen nicht mehr biologisch und toxikologisch unbedenklich sind. Weiterhin ist es mit solchen Zusammensetzungen schwierig, die gewünschte Biegefestigkeit des Restaurationsmaterials zu er- reichen.
Die bevorzugten radikalisch polymerisierbaren ethylenisch ungesättigten Gruppen sind die Acrylat-, die Methacrylat- ,
die Acrylamid- und die Methacrylamidgruppe . (Meth) acryl steht im Folgenden stets für beides - Acryl und Methacryl .
Geeignete polymerisierbare Säuren gemäß Merkmalsgruppe (a) , d.h. Monomere enthaltend mindestens eine radikalisch poly- merisierbare ethylenisch ungesättigte Gruppe und mindestens eine Säuregruppe sind dem Fachmann bekannt.
Bevorzugte Säuregruppen sind die Carbonsäure-, Phosphor¬ säure-, Phosphonsäure- , Bisphosphonsäure- und Sulfonsäure- gruppe . Besonders bevorzugt sind die phosphorhaltigen Säu- regruppen.
Exemplarisch seien genannt: 4- (Meth) acryloyloxyethyl-tri- mellitsäure ; Bis- [2- (meth) acryloyloxyethyl] -phosphat; 10- (Meth) acryloyl-oxydecyl-dihydrogenphosphat (MDP) oder 2-Ac- rylamido-2 -methylpropansulfonsäure . Im Dentalmaterial ist die polymerisierbare Säure (a) bevor¬ zugt zu 1 bis 20 Gew. -%, weiter bevorzugt zu 1 bis 15 Gew. - %, besonders bevorzugt zu 5 bis 15 Gew.-%, bezogen auf die Gesamtmasse der im Dentalmaterial enthaltenen Monomere ent¬ haltend mindestens eine radikalisch polymerisierbare ethyl- enisch ungesättigte Gruppe, enthalten.
Im Dentalmaterial ist der Photoinitiator (c) bevorzugt zu 0,05 bis 10, bevorzugt zu 0,05 bis 5, am bevorzugtesten zu 0,1 bis 2 Gew.-% , bezogen auf die Gesamtmasse der im Dentalmaterial enthaltenen Monomere enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte
Gruppe, enthalten.
Bevorzugte Photoinitiatoren (c) absorbieren verstärkt Licht im Wellenlängenbereich zwischen 390 und 560 nm, besonders bevorzugt zwischen 420 und 480 nm. Der Photoinitiator ist bevorzugt ein im Dentalmaterial lösliches aliphatisches
1, 2-Diketon, wie Campherchinon (CQ) oder Propyl-phenyl-dik- eton (PPD) , besonders bevorzugt jedoch Campherchinon und/oder ein Campherchinonderivat bspw. eine Carboxyl-, Car- boxylat-, Sulfonsäure- oder Sulfonatgruppe aufweisend. Die Lichtabsorption im angegebenen Bereich löst einen initiierenden Übergang aus, der zur Radikalbildung im Photoinitiatorsystem führt.
Bevorzugt ist das tertiäre aromatische Amin (Coinitiator gemäß Merkmalsgruppe (d) ) ausgewählt aus der Gruppe beste¬ hend aus :
a) Formel I


c) Formel III


wobei in den Formeln bedeuten:
- Ri und R2 sind unabhängig voneinander) C1-C5- Alkyl, bevorzugt Methyl,
- R3 ist Alkyl, bevorzugt Methyl,
- R4 ist H oder Cl-C5-Alkyl, bevorzugt Methyl, - R5 und R6 sind unabhängig voneinander C1-C5-A1- kyl, bevorzugt Methyl,
- R7 und R8 sind unabhängig voneinander ausgewählt aus H, Alkyl, Alkoxyalkyl und Polyalkylen- oxid, bevorzugt Polyalkylenoxid mit mindestens zwei Oxyethylen- und/oder Oxypropyleneinheiten, x ^0 und y >1. Die Summe aus x und y beträgt bevorzugt 3 bis 20, weiter vorzugsweise 5 bis 17. Bevorzugt ist x < y.
Für die Coinitiatoren (d) der Formeln I und II können die Polyalkylenoxidketten als Blockcopolymere oder als statis¬ tische Copolymere ausgebildet sein. Eine Polyalkylen- oxidkette hat bevorzugt eine molare Masse zwischen 130 und
1200 g/mol, weiter bevorzugt zwischen 220 und 1200 g/mol, weiter bevorzugt zwischen 300 und 800 g/mol. Ein Beispiel ist 4 -Dirnethylamino-benzoesäure-poly (ethylenglykol-350 ) mo- nomethylether-ester . Geeignete Coinitiatoren (d) der Formeln III und IV sind 4- Dimethylaminobenzamid; 4-Dimethylamino-N-propylbenzamid; 3- Dimethylamino-N-propylbenzamid; 4-Dimethylamino-N- (hydro- xyethyl ) benzamid; 4-Dimethylamino-N- (hydroxypro- pyl ) benzamid; 4-Dimethylamino-N- (hydroxybutyl) benzamid; 4- Dimethylamino-N- (2-methoxyethyl) benzamid; 3-Dimethylamino- N- (2-methoxyethyl) benzamid; 4-Dimethylamino-N- (2- (2-hydro- xyethoxy) ethyl ) benzamid . Besonders geeignete Coinitiatoren (d) der Formeln III und IV sind 4-Dimethylamino-N- (2- (2- (2- methoxyethoxy) ethoxy) ethyl) benzamid; 4-Dimethylamino-N- poly (ethylenoxid) methylether-benzamid; 4-Dimethylamino-N- poly (propylenoxid-jblocif-ethylenoxid) methylether-benzamid; 4-Dimethylamino-N-poly (ethylenoxid-stat-propylenoxid) me¬ thylether-benzamid.
Die Polyalkylenoxidketten können als Blockcopolymere oder auch als statistische Copolymere verwendet werden. Eine Po- lyalkylenoxidkette hat besonders bevorzugt eine molare Masse zwischen 130 und 1200 g/mol.
Die erfindungsgemäßen Coinitiatoren ohne Amidgruppe weisen eine Wasserlöslichkeit größer > 2 g/1, weiter bevorzugt > 50 g/1, weiter bevorzugt > 100 g/1, weiter bevorzugt > 500 g/1 auf.
Die erfindungsgemäßen Coinitiatoren mit Amidgruppe weisen eine Wasserlöslichkeit größer 0,4 g/1, bevorzugt > 2 g/1, weiter bevorzugt > 50 g/1, weiter bevorzugt > 100 g/1, wei- ter bevorzugt > 500 g/1 auf.
Im Dentalmaterial ist der Coinitiator (d) zu 0,05 bis 10, bevorzugt zu 0,1 bis 10, am bevorzugtesten zu 0,5 bis 5
Gew.-%, bezogen auf die Gesamtmasse der im Dentalmaterial enthaltenen Monomere enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe, enthal¬ ten . Geeignete weitere Monomere (b) , d.h. Monomere enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe, die keine Säuregruppe aufweisen sind dem Fachmann bekannt.
Bevorzugt ist das wenigstens eine Monomer, das keine Säure- gruppen und mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe enthält (Merkmal b) des An¬ spruchs 1) ausgewählt aus der Gruppe bestehend aus bl), b2) und b3 ) , mit : bl)
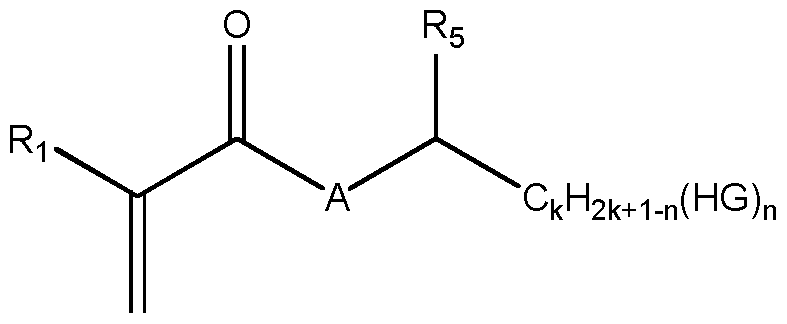
A ist 0 oder NR3, mit R3 = H, Methyl, Ethyl oder Propyl, bevorzugt R3 = H; HG ist eine hydrophile Gruppe ausgewählt aus OH und (O-CH2- CH2)m-OR4; bevorzugt ist HG = OH;
R5 ist H, Methyl, Ethyl oder CkH2 k+i-n(HG)n; bevorzugt H; R4 ist H oder Methyl;
k, m, n sind natürliche Zahlen; k = 1-5; bevorzugt 1-2; m= 3-20; bevorzugt 5-20; n= 1-5; n < k; jedes Kohlenstoffatom trägt höchstens eine hydrophile Gruppe HG;
b2) (Q A Ev) r CtH(2t+2)-(r+s)) (HG) : mit Q


R8 ist H, Methyl oder Ethyl, bevorzugt H oder Methyl; A ist O oder NR3, mit R3 = H, Methyl, Ethyl oder Propyl, bevorzugt R3 = H;
HG ist OH, (0-CH2-CH2)m-OR7; bevorzugt OH; R7 ist H oder Methyl;
m, r, s, t, v sind natürliche Zahlen; m= 3-20, bevorzugt 6-20;
r= 2-4,
s= 1-4
t= 2-8 bevorzugt
v= 0 oder 1; bevorzugt 0.
b3) Q-A-EV-C2H2-HG-EV-A-Q mit Q
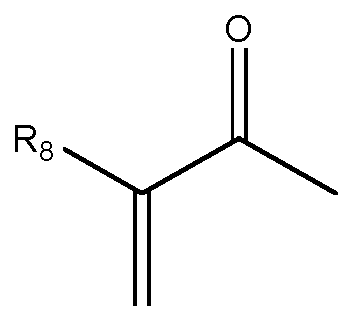

oder OH

wobei in den Formeln bedeuten
R8 ist H, Methyl oder Ethyl, bevorzugt H oder Methyl;
A ist 0 oder NR3, mit R3 = H, Methyl, Ethyl oder Propyl, bevorzugt R3 = H;
HG ist (0-CH2-CH2)m;
m= 4-20, bevorzugt 6-20;
v= 0 oder 1; bevorzugt 0.
Bevorzugte Monomere bl) wie oben beschrieben sind dem Fach- mann bekannt. Beispiele sind Hydroxyethylmethacrylat , Hyd- roxypropylmethacrylat , Hydroxyethylacrylamid, Hydroxypropy- lacrylamid, 2 , 3-Dihydroxypropylmethacrylat , 1 , 3-Dihydroxy- 2-propanylmethacrylate, Methoxy (polyethylenglycol-350 ) me¬ thacrylat oder Methoxy (polyethylenglycol-500) methacrylat . Besonders bevorzugt ist Hydroxyethylmethacrylat (HEMA) .
Bevorzugte Monomere b2) wie oben beschrieben sind dem Fachmann bekannt. Beispiele sind Glyceroldimethacrylat und N, ' - ( 1 , 2-Dihydroxyethylen) bisacrylamid . Weitere bevorzugte Monomere sind in der EP2133368 (Sekiguchi) beschrieben. Bevorzugte Monomere b3) wie oben beschrieben sind dem Fach¬ mann bekannt. Beispiele sind Polyethylenglykol 200
Di (meth) acrylat ; Polyethylenglykol 300 Di (meth) acrylat ; Po¬ lyethylenglykol 400 Di (meth) acrylat ; Polyethylenglykol 600 Di (meth) acrylat ; PPGDMA, Polypropylenglykoldimethacrylat . Geeignete Monomere bl) bis b3) weisen bevorzugt mindestens eine Wasserlöslichkeit von 50 g/1 Wasser auf.
Das erfindungsgemäße Dentalmaterial kann bevorzugt zusätz¬ lich wenigstens ein Monomer b4), das keine Säuregruppen und mindestens zwei radikalisch polymerisierbare ethylenisch ungesättigte Gruppen enthält, aufweisen, wobei dieses we¬ nigstens eine Monomer b4) nicht aus der Gruppe bl), b2) und b3) ausgewählt ist.
Bevorzugte Monomere b4) wie oben beschrieben sind dem Fach¬ mann bekannt. Beispiele sind Allyl (meth) acrylat ; 1,3-Pro- pandioldi (meth) acrylat ; 1 , 3-Butandioldi (meth) acrylat ; 1,4- Butandioldi (meth) acrylat; 1, 5-Pentandioldi (meth) acrylat; Neopentylglykoldi (meth) acrylat ; 1 , 6-Hexandioldimethacrylat ; 1, 9-Nonandioldi (meth) acrylat; 1, 10-Decandioldi (meth) ac¬ rylat; 1, 12-Dodecandioldi (meth) acrylat; Ethylengly- koldi (meth) acrylat; Diethylenglykoldi (meth) acrylat;
Triethylenglykoldi (meth) acrylat; Tetraethylengly- koldi (meth) acrylat ; Propoxyliertes (2) Neopentylgly¬ koldi (meth) acrylat ; Bisphenol-A-di (meth) acrylat ; Bisphenol- A-glyceroldi (meth) acrylat (BisGMA) ; Ethoxyliertes Bi- sphenol-A-Di (meth) acrylat ; Propoxyliertes Bisphenol-A- Di (meth) acrylat ; Diurethandi (meth) acrylat (UDMA) ;
Tricyclo [ 5.2.1.0 ] decan-dimethanoldi (meth) acrylate ; Ν,Ν'- Ethylenbisacrylamid; N, N ' -Diethyl (1, 3-propylen)bisac- rylamid; N, ' - (2,2, 4-Trimethyl-hexamethylen) -bismethac- rylamid; Bis [2- (2-methylacryl-amino) -ethoxycarbonyl ] -hexa- methylendiamin; Trimethylolpropan-tri (meth) acrylat ; Di-Tri- methylolpropan-tetra (meth) acrylat ; Di-Pentaerythritol- penta (meth) -acrylat; Di-Pentaerythritolhexa (meth) acrylat .
Die Wasserlöslichkeit des Monomers b4) beträgt bevorzugt weniger als 50 g/1.
Besonders bevorzugt sind Mischungen von mindestens einem Monomeren bl) bis b3) und mindestens einem Monomeren bzw. Vernetzer der Gruppe b4) .
Das mittels Bestrahlung mit sichtbarem Licht radikalisch polymerisierbare Dentalmaterial enthält weiterhin bevorzugt 0,1 bis 90 Gew.-%, bevorzugt 45 bis 70 Gew.-% organische und/oder anorganische Füllstoffe.
Geeignete Füllstoffe sind dem Fachmann bekannt. Bevorzugt sind oberflächenbehandelte anorganische Füllstoffe, die ge¬ genüber den Säuren einer Harzmatrix im Wesentlichen inert sind, z.B. die im Dentalmaterial, d.h. der Harzmatrix un- löslich sind, wobei unter Harzmatrix die radikalisch poly- merisierbaren Monomere und darin gelöste Zusätze verstanden werden. Geeignete Füllstoffe werden beispielsweise in EP 1 720 506 (Neffgen et al) offenbart.
Das mittels Bestrahlung mit sichtbarem Licht radikalisch polymerisierbare Dentalmaterial enthält weiterhin bevorzugt 0,01 bis 20, bevorzugt 0,01 bis 10 Gew.-% dentalübliche Ad¬ ditive .
Die Auswahl der Additive nimmt der Fachmann in Abhängigkeit von der vorgesehenen Verwendung des Dentalmaterials vor. Übliche dentale Additive sind Polymerisationsinhibitoren, wie BHT, UV-Stabilisatoren, Rheologiemodifizierer, Farbstoffe und Röntgenopaker .
Das mittels Bestrahlung mit sichtbarem Licht radikalisch polymerisierbare Dentalmaterial kann weiterhin nicht poly- merisierbare organische Lösungsmittel, beispielweise leicht flüchtige Lösungsmittel wie Ethanol oder Aceton, sowie Was¬ ser enthalten.
Gegenstand der Erfindung ist ferner eine erfindungsgemäße Zusammensetzung zur Verwendung als ein lichthärtendes selbsthaftendes und selbstätzendes radikalisch polymeri- sierbares Dentalrestaurativ bzw. die Verwendung einer solchen Zusammensetzung als oder bei der Herstellung eines lichthärtenden selbsthaftenden und selbstätzenden radikalisch polymerisierbaren Dentalrestaurativs . Ein weiterer Gegenstand der Erfindung ist die Verwendung eines tertiären aromatisches Amins, das einen Benzolring
aufweist, an den mindestens eine Dialkylamingruppe und min¬ destens eine weitere Gruppe direkt gebunden sind, wobei die weitere Gruppe ausgewählt ist aus: i. Carbonsäureestergruppen enthaltend mindestens eine Polyoxyalkylengruppe mit mindes¬ tens 2 Oxyethylen- und/oder Oxypropylenein- heiten, und
11 Amidgruppen als Coinitiator in einem lichthärtenden selbsthaftenden und selbstätzenden radikalisch polymerisierbaren Dentalrestaurativ.
Ausführungsbeispiele der Erfindung werden nachfolgend er¬ läutert .
Verwendete Materialien:
1 O-Methacryloyl-oxydecyl-dihydrogenphos-
PCM Products phat (MDP)
2- (2-Aminoethoxy) ethanol Alfa Aeser
2, 6-Di-tert-butyl-4-methylphenol (BHT) Merck
2-Hydroxyethylmethacrylat (HEMA) Melrob (Evonik)
2-Methoxyethylamin Merck
3-Dirnethylaminobenzoesäure Merck
3-Trimethoxysilylpropylmethacrylat
Evonik
(MEMO)
4 -Dirnethylaminobenzoesäure Alfa Aeser
4 -Dirnethylaminobenzoesäure-2 -ethylhe-
Rahn
xylester (EHA)
4-Dimethylaminobenzoesäure-ethylester Sigma Aldrich
4 -Dirnethylaminobenzoesäure-2 -n- butoxyethylester
4-Dimethylaminobenzonitril Alfa Aeser
4-Dirnethylaminobenzoylchlorid Alfa Aeser
Aktivkohle Merck
A1203 DC-Karten (Polygram Alox N/UV 254) Macherey-Nagel
AI2O3 für Säulenchromatographie
Merck
(Aluminiumoxid 90 aktiv, neutral)
BaC03 Sigma Aldrich
Bisphenol-A-diglycidyldimethacrylat
CCP Composites (BisGMA)
CaCl2, wasserfrei Sigma Aldrich
Campher- 10-sulfonsäure Merck
Celite 545 Merck
CuS04 x 5 H20 Merck
D, L-Campherquinon (CQ) Rahn
Dibenzo-18-krone-6 Sigma Aldrich
Dichlormethan Merck
Diethylether Carl Roth
Dirnethy1sulfoxid Merck
Dowex 50 WX 8 Fluka
Essigsäure Merck
Ethanol Walter CMP
Ethylacetat Merck
Ethyldiisopropylamin Acros
Glaspulver BAF GM 27884 0,7 ym, mit 3- Trimethoxysilylpropylmethacrylat silani- Schott
siert
H2O2, 35 % Sigma Aldrich
HCl Merck
Hydrophobe, hochdisperse Kieselsäure
Wacker
(HDKH 2000)
Jeffamin M 1000 Huntsman
K2CO3 J.T. Baker
K3PO4 Alfa Aeser
Methanol Sigma Aldrich
Na2S205 Sigma Aldrich
Na2S03 Merck
Na2S04 Merck
NaBH4 Merck
NaCl Merck
NaHC03 J.T. Baker
NaN3 Merck
NaOH Merck
Natriumethoxid-Lösung, 21% in Ethanol Sigma Aldrich n-Heptan Merck
n-Propylamin Sigma Aldrich
0- (Benzotriazol-l-yl) -Ν,Ν,Ν' ,Ν' -tetrame- Iris Biotech thyluronium tetrafluoroborat (TBTU)
Polyethylenglykolmonomethylether 350 Sigma Aldrich p-Toluolsulfonsäurechlorid Fluka
Schwefelsäure Sigma Aldrich
Selendioxid Merck
Si02 DC-Karten (Polygram Sil G/UV 254) Macherey-Nagel
S1O2 für Mitteldruckchromatographie
Merck
(Kieselgel 60, 0,04-0,063 mm)
S1O2 für Säulenchromatographie
Merck
(Kieselgel 60, 0,063-0,200 mm)
Tetrahydrofuran (THF) Sigma Aldrich
Tetrahydrofuran (THF) , wasserfrei Sigma Aldrich
Toluol Merck
Triethylenglykoldimethacrylat (TEDMA) Lehmann & Voss
Triethylenglykolmonomethylether Merck
VE-Wasser DMG
Synthesen
Campherchinon-1 Q-sulfonsäure
In einem Einhalskolben mit Magnetrührfisch werden 3,15 g (28,4 mmol) Selendioxid in 20 ml VE-Wasser gelöst. Dazu werden 10,6 g (45,6 mmol) Campher-10-sulfonsäure gegeben und der Kolben mit einem Rückflusskühler ausgestattet. Das Reaktionsgemisch wird auf 110 °C unter Rückflusskühlung erhitzt. Im Abstand von 3 h werden drei weitere Portionen von jeweils 3,15 g Selendioxid zum Ansatz gegeben. Nach der
letzten Zugabe wird für 4,5 h bei 110 °C unter Rückfluss¬ kühlung erhitzt. Nach Abkühlung auf Raumtemperatur wird der Ansatz über Celite 545 filtriert und der Filterrückstand mit 2x50 ml VE-Wasser gewaschen. Die Lösung wird mit weite- ren 50 ml VE-Wasser verdünnt, sowie mit 2 ml einer 20 %-i- gen Schwefelsäure-Lösung versetzt und in einen Zweihalskolben überführt.
In einer Gasentwicklungsapparatur wird aus 63 g a2S03 und 100 ml einer 6 M H2SO4 innerhalb von einer Stunde Schwefel- dioxid entwickelt und durch eine Kapillare in den filtrier¬ ten Ansatz geleitet. Der rotbraune Ansatz rührt nach beendeter Gaseinleitung über Nacht. Danach wird die oben beschriebene Gasentwicklung und Einleitung wiederholt. Der rotbraun-schwarze Ansatz wird über Celite 545 filtriert und der Filterrückstand mit VE-Wasser gewaschen. Vom Ansatz werden bei 100 mbar ca. 130 ml Wasser abdestilliert. Der Destillationsrückstand wird mit festem BaCC>3 neutralisiert. Die dickflüssige rötlich-braune Masse wird über Celite 545 filtriert und der Filterrückstand mit ca. 150 ml VE-Wasser gewaschen. Eine klare, gelbe Lösung wird erhalten. Die Lö¬ sung wird am Rotationsverdampfer eingeengt, bis ca. 80 ml Flüssigkeit übrig bleiben. Dabei entsteht ein weißer Nie¬ derschlag, der abfiltriert wird. Die klare, gelbe Lösung wird über eine Säule (h = 12 cm, d = 2 cm) mit Ionenaustau- scher (Dowex 50WX8, H-Form) gegeben. Nachdem die Lösung vollständig in die Säule eingezogen ist, wird die Säule mit VE-Wasser gespült, bis die austretende Flüssigkeit aus der Säule farblos ist. Die gelbe Lösung wird am Rotationsver¬ dampfer bis zur Trockene eingedampft und der Rückstand im Vakuum bei Raumtemperatur getrocknet. Das Rohprodukt (ca. 10,9 g) wird in 100 ml Methanol gelöst. Zu der Lösung wer¬ den ca. 2 g Aktivkohle gegeben und die Suspension wird zwei Stunden unter Rückflusskühlung erhitzt. Nach Abkühlung auf
Raumtemperatur wird über Celite 545 filtriert und der Fil¬ terrückstand mit 3x20 ml Methanol gewaschen. Man erhält eine klare, hellgelbe Lösung. Methanol wird am Rotations¬ verdampfer entfernt. Nach Trocknung im Vakuum verbleibt eine hochviskose, hellgelbe Flüssigkeit, die über Nacht im Kühlschrank auskristallisiert. Der gelbe Feststoff wird aus Ethylacetat/n-Heptan umkristallisiert. Ausbeute: 6,02 g. 1H-NMR (d6-DMSO) ppm: 0,82 (s, 3H) , 0,11 (s, 3H) , 1,50 (m, 1H) , 1,65 (m, 1H) , 2,13 (m, 1H) , 2,59 (d, 1H) , 2,65 (d, 1H) , 2,75 (dt, 1H) , 2,95 (d, 1H) , Schmelzpunkt: 155 °C. Als Verunreinigung sind ca. 10 % des Edukts Campher-10-sulfon- säure vorhanden
Natrium-Campherchinon-1 Q-sulfonat
1,01 g (4,1 mmol) der Campherchinon-10-sulfonsäure werden in 0,5 ml VE-Wasser gelöst. Die Lösung wird in einem Eisbad gekühlt. Mit einer Eppendorf-Pipette werden 2 ml einer 2N NaOH-Lösung langsam zugetropft. Das Eisbad wird entfernt und die Lösung wird drei Stunden bei Raumtemperatur gerührt. Am Rotationsverdampfer wird das Wasser abdestil- liert. Der Rückstand wird im Vakuum über CaCl2 getrocknet. Das Produkt fällt als gelber Feststoff an. Ausbeute: 0,99 g, Schmelzpunkt: 250 °C (Zersetzung).
4-Dimethylaminobenzoesäure-poly (ethylenglykol ) -monomethyl- ether-esters
In einem Zweihalskolben mit Magnetrührfisch werden 35 g Po- lyethylenglykolmonomethylether (100 mmol) und 29 g (150 mmol) 4-Dimethylaminobenzoesäureethylester gegeben. Dazu werden 2 ml einer Natriumethoxid-Lösung (21 Gew. % in Etha- nol (5 mmol)) zugefügt. Der Kolben wird mit einem Hahn und Stopfen verschlossen und der Inhalt unter Vakuum (750 mbar) gesetzt. Der Kolben wird in ein vortemperiertes Ölbad (100
°C) gehängt und das Vakuum wird schrittweise auf 40 mbar runtergeregelt. Nach zwei Stunden wird das Vakuum gebrochen und der Ansatz auf ca. 50 °C abgekühlt. Dann werden erneut 2 ml der Natriumethoxid-Lösung (5 mmol in Ethanol) zuge- fügt. Es wird wieder ein leichtes Vakuum von 750 mbar ange¬ legt und der Kolben in ein 100 °C warmes Ölbad überführt. Das Vakuum wird dann schrittweise auf 40 mbar reduziert und der Ansatz drei Stunden bei diesen Bedingungen im Ölbad belassen. Danach wird der Ansatz auf Raumtemperatur gebracht und das Vakuum gebrochen. Zur Aufarbeitung wird der Ansatz in eine Mischung aus 500 ml VE-Wasser und 5 ml einer 2N HCl-Lösung gegeben. Es entsteht ein weißer Niederschlag. Der Feststoff wird abfiltriert. Zum Filtrat wird 1 ml einer gesättigten Natriumbisulfit-Lösung gegeben und mit einer 2N NaOH-Lösung der pH-Wert auf ca. 7,5 eingestellt. Die gelb¬ lich-grüne Emulsion wird mit 6,3 g Aktivkohle versehen und eine Stunde auf einer Rüttelplatte geschüttelt. Die Suspen¬ sion wird über Celite 545 filtriert. Die wässrige Lösung wird mit 3x150 ml Dichlormethan extrahiert. Die organische Phase wird über Na2SC>4 getrocknet. Das Trocknungsmittel wird abgetrennt und das Lösungsmittel am Rotationsverdamp¬ fer entfernt. Nach Trocknung im Vakuum fällt das Produkt als schwach gelbliches Öl an. Ausbeute 28,2 g, 1H-NMR
(CDCL3) ppm: 3,00 (s, 6H) , 3,35 (s, 3H) , 3,52 (t, 2H) , 3, 56-3, 75 (br, ca. 30H) , 3,79 (t, 2H) , 4,39 (t, 2H) , 6,61 (d, 2H) , 7,89 (d, 2H)
4-Dimethylaminobenzoesäure-tri (ethylenglykol) monomethyl- ether-esters
In einem Einhalskolben mit Magnetrührfisch, Tropftrichter und CaCl2-Trockenrohr werden bei Raumtemperatur 0,92 g (5 mmol) 4-Dimethylaminobenzoylchlorid, 20 ml wasserfreies THF und 1,16 ml (7 mmol) Diisopropylethylamin vorgelegt. Die Lösung wird im Eisbad auf 0 °C gekühlt. Im Tropftrichter
wird eine Lösung aus 5 ml wasserfreiem THF und 0,78 ml (4,9 mmol) Triethylenglykolmonomethylether zubereitet. Diese Lösung wird langsam unter Eiskühlung zum Reaktionsansatz getropft. Nach Zugabe lässt man den Ansatz auf Raumtemperatur kommen. Der Ansatz wird ca. 70 h unter Feuchtigkeits- und Lichtausschluss bei Raumtemperatur gerührt. Hierbei ent¬ steht etwas Niederschlag. Zur Aufarbeitung wird die Lösung vom Niederschlag dekantiert. Der Niederschlag wird mit et¬ was THF gewaschen. Die organischen Phasen werden vereinigt und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wird in 50 ml Dichlormethan aufgenommen. Die organische Phase wird mit 2x10 ml 0,5 N HCl-Lösung, 1x10 ml gesättigter NaHC03-Lösung und 1x10 ml gesättigter NaCl-Lö- sung ausgeschüttelt. Anschließend wird die organische Phase über Na2SC>4 getrocknet. Nach Filtration wird das Lösungs¬ mittel am Rotationsverdampfer entfernt und der Rückstand im Hochvakuum getrocknet. Es fallen 1,47 g Rohprodukt an. Das Rohprodukt wird durch Säulenchromatographie weiter aufge¬ reinigt. Stationäre Phase: Si02- Laufmittel: Ethylacetat/n- Heptan 1:1. Säule: h = 35 cm, d = 4 cm. Das Produkt hat ei¬ nen Rf = 0,17. Das Produkt fällt als klares, blassgelbli¬ ches Öl an. Ausbeute: 0,75 g. 1H-NMR (CDCL3) ppm: 3,00 (s, 6H) , 3,34 (s, 3H) , 3,50 (m, 2H) , 3, 60-3, 70 (br, ca. 6H) , 3,78 (t, 2H) , 4,39 (t, 2H) , 6,60 (d, 2H) , 7,89 (d, 2H)
4-Dimethylaminobenzamid
In einem Zweihalskolben mit Magnetrührfisch, Tropftrichter und Rückflusskühler werden 2,31 g (15,8 mmol) umkristalli¬ siertes 4-Dimethylbenzonitril in 10 ml DMSO gelöst. Zu der hellbraunen Lösung werden 0,32 g (2,3 mmol) K2CO3 gegeben. Innerhalb von 5 min werden 1,9 ml einer 35 %-igen H2O2-LÖ- sung (18,6 mmol in Wasser) bei Raumtemperatur langsam zugetropft. Dabei fällt ein weißer Feststoff aus. Nach Zugabe
werden weitere 45 min bei Raumtemperatur unter Rückflusskühlung gerührt. Während dieser Phase steigt kurzzeitig die Temperatur an. Es bildet sich so viel weißer Feststoff, dass der Magnetrührfisch sich nicht mehr bewegt. Zur Aufar- beitung werden 50 ml VE-Wasser zum Ansatz gegeben und mit dem Spatel wird der Brei homogenisiert. Der Feststoff wird über eine Glasfritte abgetrennt und mit 3x20 ml VE-Wasser gewaschen. Im Exsikkator wird der Feststoff im Vakuum bei Raumtemperatur über CaCl2 getrocknet. Das Produkt fällt als weißer Feststoff an. Ausbeute: 2,42 g. 1H-NMR (d6-DMSO) ppm: 2,95 (s, 6H) , 6,67 (d, 2H) , 6,89 (br. s, NH) , 7,60 (br. s, NH) , 7,73 (d, 2H)
4-Dimethylamino-N-poly (propylenoxid-felocic-ethylenoxid) - methylether-benzamids In einem Dreihalskolben mit Magnetrührfisch, Tropftrichter, Rückflusskühler und Hahn werden 4,25 g (20 mmol) gemörser- tes, wasserfreies K3PO4 vorgelegt. Der Kolben wird mit Stickstoff gespült und die gesamte Apparatur mit einer Heißluftpistole ausgeheizt. Im N2-Strom wird auf Raumtempe- ratur abgekühlt. Dann werden 80 ml wasserfreies THF zum
K3PO4 gegeben. Zur Suspension werden 2,94 g (16 mmol) 4-Di- methylamino-benzoylchlorid und 0,04 g (0,1 mmol) Dibenzo- 18-krone-6 zugefügt. Eine Lösung aus 8,04 g Jeffamin M 1000 in 12 ml wasserfreiem THF wird zubereitet und in den Tropf- trichter überführt. Unter Ausschluss von Feuchtigkeit wird die Jeffamin M 1000-Lösung bei Raumtemperatur innerhalb von 15 min zum Ansatz gegeben. Dann wird ein Ballon, befüllt mit N2, auf die Apparatur gesteckt. Tagsüber wird der An¬ satz unter Schutzgas und Rückflusskühlung für 5 bis 8 h im Ölbad auf 50 °C erwärmt. Die sonstige Zeit wird bei Raum¬ temperatur gerührt. Insgesamt wird der Ansatz für 44 h bei 50 °C und für 148 h bei Raumtemperatur gerührt. Zur Aufarbeitung wird der Ansatz filtriert. Der Filterrückstand wird
mit etwas THF gewaschen. Das Lösungsmittel wird am Rotati¬ onsverdampfer entfernt und der Rückstand im Hochvakuum von flüchtigen Bestandteilen befreit. Das Rohprodukt wird in eine Mischung aus 80 ml VE-Wasser und 0,8 ml 2 N HCl gege- ben. Die sich bildende Emulsion wird nochmals mit 100 ml VE-Wasser verdünnt und filtriert. Zur filtrierten Lösung werden 0,5 ml gesättigte Natriumbisulfit-Lösung zugegeben und mit einer 2 N NaOH auf einen pH-Wert von 7,5 einge¬ stellt. In einem Scheidetrichter wird die wässrige Phase mit 3x60 ml Dichlormethan extrahiert. Die vereinigten orga¬ nischen Phasen werden über Na2SÜ4 getrocknet. Zur extrahierten wässrigen Phase wird festes NaCl gegeben, woraufhin sich eine organische Phase absetzt. Diese wird abgetrennt und ebenfalls über Na2SÜ4 getrocknet. Alle getrockneten or- ganischen Phasen werden vereinigt und das Lösungsmittel im Vakuum am Rotationsverdampfer entfernt. Das Produkt fällt als leicht beiger, wachsartiger Feststoff an. Ausbeute: 5,94 g. !H-NMR (CDC13) ppm: 1,04 (m, ca. 5H) , 1,17 (m, ca. 3H) , 1,76 (m, ca. 1H) , 2,93 (s, 6H) , 3,29 (s, ca. 3H) , 3,31 - 3,51 (br, ca. 10H) , 3,56 (s, ca. 78H) , 6,34 (d, NH) , 6,58 (d, ca. 2H) , 7,67 (m, ca. 2H)
4-Dimethylamino-N- (2- (2-hydroxyethoxy) ethyl) -benzamids
In einem Einhalskolben mit Magnetrührfisch und Stopfen wurden 6, 61 g (40 mmol) 4-Dimethylaminobenzoesäure in 200 ml Dichlormethan suspendiert und durch Zugabe von 13,22 ml (80 mmol) Ethyldiisopropylamin gelöst. Zu der klaren Lösung wurden 12,85 g (40 mmol) TBTU in Portionen bei Raumtempera¬ tur gegeben. Nach beendeter Zugabe wurde der Kolben mit einem Stopfen verschlossen und 14 h bei Raumtemperatur ge- rührt. Es entstand eine gelbe, leicht trübe Lösung. In ei¬ nem 500 ml Vierhalskolben mit zwei Stopfen, Innenthermometer, Magnetrührfisch und Tropftrichter mit CaCl2-Trocken- rohr wurden 4 ml (40 mmol) 2- (2-Aminoethoxy) -ethanol in 100
ml Dichlormethan gelöst. Durch ein Eisbad wurde die Lösung auf 0 °C gekühlt. Das Gemisch aus 4-Dimethylaminobenzoe- säure, Ethyldiisopropylamin und TBTU in Dichlormethan wurde in den Tropftrichter überführt und langsam, innerhalb von 90 min zur 2- (2-Aminoethoxy) ethanol-Lösung zugegeben, wobei die Innentemperatur nicht über 5 °C anstieg. Nach beendeter Zugabe wurde das Eisbad entfernt und das Reaktionsgemisch 48 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch in einen Scheidetrichter überführt und mit 4x25 ml 0,5 N Essigsäure-, 2x25 ml gesättigter aHC03~ und 1x25 ml gesättigter NaCl-Lösung extrahiert. Die organische Phase wurde über a2S04 getrocknet. Das Lösungsmittel wurde am Rotationsverdampfer entfernt. Zum Rückstand wurden 180 ml Diethylether gegeben. Dabei löste sich nicht alles. Vom Feststoff wurde die klare Diethylether-Phase abdekantiert. Das Lösungsmittel wurde am Rotationsverdampfer entfernt. Ausbeute Rohprodukt: 12,12 g. Zur weiteren Aufreinigung wurde das Rohprodukt in 70 ml einer Mischung aus Ethyl- acetat/Ethanol 10:2 gelöst. Dazu wurden 65 g AI2O3 gegeben und das Lösungsmittelgemisch am Rotationsverdampfer entfernt. Das auf A1203 aufgezogene Rohprodukt wurde auf eine mit 100 g AI2O3 befüllte Säule (d = 3 cm) gegeben und das Rohprodukt durch Säulenfiltration aufgereinigt . Laufmittel: Ethylacetat/Ethanol 10:2. Das Laufmittel wurde am Rotati- onsverdampfer entfernt und der Rückstand im Vakuum von flüchtigen Bestandteilen befreit. Ausbeute aufgereinigtes Rohprodukt: 9,24 g.
2- (2- (2-Methoxyethoxy) ethoxy) ethyl-tosylat
18,73 g (114 mmol) Methoxytriethylenglykol wurden in einem Dreihalskolben vorgelegt und in 60 ml THF gelöst. Mittels eines Eisbades wurde die Lösung auf ca. 0 °C gekühlt. Zu der Lösung wurden 35 ml einer 6 N NaOH-Lösung zugegeben.
Dabei erwärmte sich die Mischung auf 8 °C. Nachdem die In¬ nentemperatur wieder bei ca. 0 °C lag, wurde eine Lösung von 40,4 g (212 mmol) p-Toluolsulfonyl-chlorid in 70 ml THF zur Lösung unter starkem Rühren getropft, wobei die Innen- temperatur unter 5 °C gehalten wurde. Nach beendeter Zugabe wurde 30 min bei ca. 0 °C gerührt. Danach wurde das Eisbad entfernt und der Ansatz wurde 45 min bei Raumtemperatur gerührt. Zur Aufarbeitung wurde der Ansatz auf 300 ml Diet- hylether gegeben. Dazu wurden 50 ml einer 1 NaOH-Lösung ge- geben und extrahiert. Die organische Phase wurde abgetrennt und nochmals mit 50 ml einer 1 N NaOH-Lösung extrahiert. Nach erneuter Abtrennung der organischen Phase wurde diese über Na2SC>4 getrocknet und das Lösungsmittel am Rotations¬ verdampfer entfernt. Ausbeute Rohprodukt: 44,91 g. Das Rohprodukt wurde durch Säulenchromatographie weiter aufgereinigt . Dazu wurden ca. 5 g auf eine Kieselgelsäule aufgebracht (Säule: d = 3 cm, stationäre Phase 75 g Si02) · Zunächst wurde mit dem Laufmittelgemisch n-Heptan/Ethyl- acetat 10:1 eluiert (ca. 1,2 1), bis durch Dünnschichtchro- matographie kein Edukt (p-Toluol-sulfonylchlorid) mehr nachgewiesen konnte. Dann wurde das Laufmittelgemisch umgestellt auf n-Heptan/Ethylacetat 1:1 und das Produkt elu¬ iert. Das Lösungsmittel wurde am Rotationsverdampfer ent¬ fernt und der Rückstand im Vakuum von flüchtigen Bestand- teilen befreit. Ausbeute: 4,21 g. 1H-NMR (CDC13) ppm: 2,42 (s, 3H) , 3,35 (s, 3H) , 3,51 (m, 2H) , 3,57-3,60 (m, 6H) , 3, 64-3, 67 (t, 2H) , 4,11-4,15 (t, 2H) , 7,32 (d, 2H) , 7,77 (d, 2H)
2- (2- (2-Methoxyethoxy) ethoxy) ethyl-azid
In einem verschraubbaren Reagenzglas mit Magnetrührfisch wurden 0,45 g (6,9 mmol) a 3 in 1 ml VE-Wasser suspendiert. Dazu wurde eine Lösung von 1,50 g (4,7 mmol) 2- (2- (2-Methoxyethoxy) -ethoxy) ethyl-tosylat in 0,5 ml THF gege- ben. Der Ansatz wurde verschlossen und 7,5 h unter starkem Rühren im Ölbad auf 70 ° C erhitzt. Der Ansatz wurde zur Aufarbeitung auf 15 ml VE-Wasser gegeben und mit 4x25 ml Diethylether extrahiert. Die organische Phase wurde über Na2SC>4 getrocknet und anschließend wurde das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wurde im Va¬ kuum von flüchtigen Bestandteilen befreit. Ausbeute: 0,71 g. 1H-NMR (CDC13) ppm: 3,31-3,34 (m, 5H) , 3, 47-3, 50 (m, 2H) , 3, 58-3, 63 (m, 8H)
2- (2- (2-Methoxyethoxy) ethoxy) ethyl-amin
In einem Kolben wurden 1,54 g (6,17 mmol) CU S C X 5 H2O in 180 ml Methanol gelöst. Die Lösung wurde in einem Eisbad auf 0 ° C gekühlt. Unter Rühren wurden 0,69 g NaBH4 zur Lösung gegeben. Eine starke Gasentwicklung und eine leichte Erwärmung setzten ein und es entstand eine schwarze Suspen¬ sion. 11,68 g (61,7 mol) 2- (2- (2-Methoxyethoxy) ethoxy) - ethyl-azid gelöst in 90 ml Methanol wurden zur schwarzen Suspension gegeben. Bei 0 ° C wurden innerhalb von 3,75 h 6x412 mg (6 x 10,9 mmol) NaBH4 zum Ansatz gegeben. Nach der letzten Zugabe wurde 30 min bei 0 ° C nachgerührt. Zur Auf¬ arbeitung wurde der Ansatz mit 2 N NaOH-Lösung auf pH = 12 eingestellt. Dabei entstand ein Niederschlag, der über eine Fritte mit einer dünnen Schicht Celite 545 abfiltriert wurde. Der Rückstand wurde mit etwas Methanol gewaschen. Die Methanol-Lösung wurde über Nacht stehen gelassen, es bildete sich wieder etwas Niederschlag. Methanol wurde am Rotationsverdampfer entfernt und der Rückstand in 800 ml Dichlormethan aufgenommen. Die organische Phase wurde über
Na2SC>4 getrocknet. Das Lösungsmittel wurde am Rotationsver¬ dampfer entfernt und der Rückstand im Vakuum von flüchtigen Bestandteilen entfernt. Der Rückstand wurde in 30 ml Toluol aufgenommen, filtriert und mit weiteren 200 ml Toluol ver- setzt. Ca. 70 ml Toluol wurden abdestilliert, um das Roh¬ produkt azeotrop zu trocknen. Restliches Toluol wurde am Rotationsverdampfer entfernt und der Rückstand im Vakuum von flüchtigen Bestandteilen befreit. Das Rohprodukt (8,12 g) wurde durch eine Destillation im Kugelrohr weiter aufge- reinigt. Das Produkt ging bei 5 mbar zwischen 160 und 200 °C als farbloses Öl über. Ausbeute: 6,58 g. 1H-NMR (CDC13) ppm: 2,82 (t, 2H) , 3,33 (s, 3H) , 3,47-3,51 (m, 4H) , 3,58- 3,61 (m, 6H)
4-Dimethylamino-N- (2- (2- (2-methoxyethoxy) ethoxy) ethyl) -ben- zamids
5,43 g (25 mmol) fein gemorstes K3PO4 wurde in einem Zwei¬ halskolben mit Tropftrichter, Hahn und Magnetrührfisch in einem schwachen 2-Strom mit einem Heizgebläse ausgeheizt. Unter 2 wurde auf Raumtemperatur abgekühlt und anschlie¬ ßend 100 ml wasserfreies THF zugegeben. In einem Wägeschäl- chen wurden 1,84 g (10 mmol) 4-Dimethylaminobenzoylchlorid und 26 mg (0,07 mmol) Dibenzo-18-krone-6 abgewogen und zu dem Ansatz gegeben. Im Tropftrichter wurden 25 ml THF was- serfrei mit 1,63 g (10 mmol) 2- (2- (2-Me- thoxyethoxy) ethoxy) ethyl-amin vermischt. Unter 2 wurde diese Lösung bei Raumtemperatur langsam zum Ansatz getropft. Nach beendeter Zugabe wurde ein CaCl2-Trockenrohr auf den Ansatz gesetzt. Das Reaktionsgemisch wurde 24 h bei Raumtemperatur und anschließend 2,5 h bei 60 °C gerührt.
Zur Aufarbeitung wurde der Ansatz über Celite 545 filtriert und der Filterrückstand mit 20 ml THF gewaschen. THF wurde am Rotationsverdampfer entfernt und der Rückstand in 100
Dichlormethan aufgenommen. Die organische Phase wurde mit 2x20 ml 0,5 N HCl-, 1x20 ml gesättigte NaHC03- und 1x20 ml gesättigte NaCl-Lösung extrahiert. Die organische Phase wurde über a2SÜ4 getrocknet und das Lösungsmittel am Rota- tionsverdampfer entfernt. Die wässrigen Waschphasen wurden vereint und Wasser am Rotationsverdampfer entfernt. Der Rückstand wurde mit 100 ml Aceton und 50 ml Ethanol extra¬ hiert. Das Aceton- und Ethanol-Extrakt wurde vereint und das Lösungsmittel am Rotationsverdampfer entfernt. Dünn- schichtchromatograpie (S1O2, Laufmittel Ethylacetat/Ethanol 95:5) der Rohprodukte aus der Dichlormethan-Phase und der wässrigen Phase zeigten beide einen Produktfleck bei Rf = 0,22. Daraufhin wurden beide Rohprodukte in Dichlormethan gelöst, vereint und das Lösungsmittel am Rotationsverdamp- fer entfernt. Ausbeute Rohprodukt: 2,53 g. Weitere Aufrei- nigung erfolgte durch Mitteldruckchromatographie. Statio¬ näre Phase: Si02- Laufmittel: Ethylacetat/Ethanol 95:5.
Flussgeschwindigkeit: 40 ml/min. Säule: h = 46 cm, d = 2,6 cm. Ausbeute: 0,8 g. 1H-NMR (CDC13) ppm: 2,98 (s, 6h), 3,32 (s, 3H) , 3, 49-3, 52 (m, 2H) , 3,61-3,63 (m, 10H) , 6,63 (d, 2H) , 7,68 (d, 2H)
Synthese des 4-Dimethylamino-N-propylbenzamids
In einem Zweihalskolben mit Magnetrührfisch, Stopfen, Rückflusskühler und CaCl2-Trockenrohr werden 1,66 g (10 mmol) 4-Dimethylaminobenzoesäure in 100 ml Dichlormethan suspendiert. Zu der Suspension werden 3,37 ml (20 mmol) Diisopropylethylamin gegeben, woraufhin eine klare Lösung entsteht. Der Lösung werden portionsweise 3,22 g (10 mmol) TBTU (2- (lH-Benzotriazol-l-yl) -tetramethyluronium tetraflu- oroborat) zugefügt. Es entsteht eine leicht trübe, gelbe Lösung. Diese Mischung wird eine Stunde bei Raumtemperatur
gerührt. Unter Kühlung werden dann 1,64 ml (20 mmol) n-Pro- pylamin zugetropft. Der Ansatz erwärmt sich leicht dabei und die Farbe der Lösung hellt sich auf. Das Kühlwasser wird nach beendeter Zugabe ausgeschaltet und der Ansatz über Nacht bei Raumtemperatur gerührt. Es entsteht dabei eine leicht beige, klare Lösung.
Zur Aufarbeitung wird der Ansatz mit 20 ml einer 2N HCl-Lö- sung extrahiert. Anschließend wird mit 20 ml einer gesät¬ tigten NaHC03~Lösung extrahiert. Die organische Phase wird über Na2SC>4 getrocknet, filtriert und das Lösungsmittel am Rotationsverdampfer entfernt. Als Rohprodukt fallen 3,04 g einer leicht grünlich-gelben, hochviskosen Flüssigkeit an. Das Rohprodukt wird durch Säulenchromatographie weiter auf¬ gereinigt. Stationäre Phase: AI2O3, neutral. Laufmittel: Ethylacetat/n-Heptan 1:1. Säule: h = 50 cm, d = 4 cm. Das Produkt hat einen Rf = 0,3. Die reinen Fraktionen werden gesammelt, das Lösungsmittel am Rotationsverdampfer ent¬ fernt und der Rückstand im Vakuum bei Raumtemperatur getrocknet. Das Produkt fällt als weißer Feststoff an. Aus- beute: 0,64 g. 1H-NMR (CDC13) ppm: 0,98 (t, 3H) , 1,63
(sext., 2H) , 3,02 (s, 6H) , 3,42 (m, 2H) , 6,03 (s, 1H) , 6,72 (d, 2H) , 7,69 (d, 2H)
Synthese des 3-Dimethylamino-N-propyl-benzamids In einem Dreihalskolben mit Magnetrührfisch, Stopfen mit
Innenthermometer, Tropftrichter, Rückflusskühler und CaCl2~ Trockenrohr werden 1,66 g (10 mmol) 3-Dimethylamino-benzoe- säure in 50 ml Dichlormethan suspendiert. Zu der Suspension werden 3,37 ml (20 mmol) Diisopropylethylamin gegeben, wo- raufhin eine hellbraune, klare Lösung entsteht. Der Lösung werden portionsweise 3,21 g (10 mmol) TBTU (2- ( lH-Benzotri- azol-l-yl) -tetramethyluronium tetrafluoroborat ) zugefügt.
Es entsteht eine leicht trübe, gelbbraune Lösung. Diese Mi¬ schung wird eine Stunde bei Raumtemperatur gerührt. Im Tropftrichter wird eine Mischung aus 10 ml Dichlormethan und 1,64 ml (20 mmol) n-Propylamin zubereitet. Der Kolben wird mit einer Eis/Kochsalz-Mischung gekühlt. Bei ca. 0 °C Innentemperatur wird die Mischung aus dem Tropftrichter langsam innerhalb von 15 min zum Reaktionsgemisch getropft. 10 min nach Zugabe wird das Kältebad entfernt und der An¬ satz 72 h bei Raumtemperatur gerührt. Zur Aufarbeitung wird dem Ansatz ca. 50 g AI2O3 neutral zugefügt und das Lösungs¬ mittel am Rotationsverdampfer entfernt. Weitere Trocknung erfolgt im Vakuum bei Raumtemperatur. Das AI2O3 wird in eine Säule mit Fritte überführt und mit 800 ml einer Mi¬ schung aus Ethylacetat/n-Heptan 1:1 eluiert. Das Lösungs- mittel wird am Rotationsverdampfer entfernt und der Rückstand im Vakuum bei Raumtemperatur getrocknet. Es fallen 3,31 g einer gelben, hochviskosen Flüssigkeit als Rohpro¬ dukt an. Durch Säulenchromatographie wird das Rohprodukt weiter aufgereinigt . Stationäre Phase: Si02- Laufmittel: Ethylacetat/n-Heptan 1:1. Säule: h = 50 cm, d = 3 cm. Das Produkt hat einen Rf = 0,19. Die reinen Fraktionen werden gesammelt, das Lösungsmittel am Rotationsverdampfer ent¬ fernt und der Rückstand im Vakuum bei Raumtemperatur getrocknet. Das Produkt fällt als klare, leicht gelbliche, hochviskose Flüssigkeit an, die über Nacht im Kühlschrank zu einem leicht gelblichen Feststoff auskristallisiert. Ausbeute: 1,84 g. 1H-NMR (CDC13) ppm: 0,98 (t, 3H) , 1,63 (sext., 2H) , 3,00 (s, 6H) , 3,42 (m, 2H) , 6,24 (s, 1H) , 6,87 (dd, 1H) , 7,00 (d, 1H) , 7,24-7,30 (br, 2H)
Synthese des 4-Dimethylamino-N- (2-methoxyethyl) -benzamids
In einem Dreihalskolben mit Magnetrührfisch, Stopfen mit Innenthermometer, Tropftrichter, Rückflusskühler und CaCl2~
Trockenrohr werden 1,66 g (10 mmol) 4-Dimethylamino-benzoe- säure in 90 ml Dichlormethan suspendiert. Zu der Suspension werden 3,37 ml (20 mmol) Diisopropylethylamin gegeben, woraufhin eine helle Lösung entsteht. Der Lösung werden por- tionsweise 3,22 g (10 mmol) TBTU (2- (lH-Benzotriazol-l-yl) - tetramethyluronium tetrafluoroborat ) zugefügt. Es entsteht eine klare, gelbe Lösung. Diese Mischung wird eine Stunde bei Raumtemperatur gerührt. Im Tropf-trichter wird eine Mischung aus 10 ml Dichlormethan und 1,72 ml (20 mmol) 2-Me- thoxyethylamin zubereitet. Der Kolben wird mit einer
Eis/Kochsalz-Mischung gekühlt. Bei ca. 0 °C Innentemperatur wird die Mischung aus dem Tropftrichter langsam innerhalb von 15 min zum Reaktionsgemisch getropft. Es wird weitere 20 min gekühlt und dann das Kältebad entfernt. Eine klare Lösung mit einem weißen Feststoff ist entstanden. Bei Erwärmung auf Raumtemperatur löst sich der Feststoff wieder auf und die Lösung bleibt leicht trübe. Es wird 72 Stunden bei Raumtemperatur gerührt. Zur Aufarbeitung wird dem Ansatz ca. 40 g AI2O3 neutral zugefügt und das Lösungsmittel am Rotationsverdampfer entfernt. Weitere Trocknung erfolgt im Vakuum bei Raumtemperatur. Das AI2O3 wird in eine Säule mit Fritte überführt und mit einer Mischung aus Ethyl- acetat/n-Heptan 1:1 eluiert. Das Lösungsmittel wird am Ro¬ tationsverdampfer entfernt. Es werden 2,90 g Rohprodukt isoliert. Weitere Aufreinigung erfolgt durch zweimaliges Umkristallisieren aus Ethylacetat. Das Produkt fällt als weißer Feststoff an. Ausbeute: 1,74 g. 1H-NMR (CDC13) ppm: 3,02 (s, 6H) , 3,38 (s, 3H) , 3,54 (t, 2H) , 3,63 (m, 2H) , 6,44 (s, 1H) , 6,73 (d, 2H) , 7,71 (m, 2H)
Synthese des 3-Dimethylamino-N- (2-methoxyethyl) -benzamids einem Dreihalskolben mit Magnetrührfisch, Innenthermome , Rückflusskühler mit CaCl2-Trockenrohr, sowie einem
Tropftrichter mit Stopfen wurden 3,31 g (20mmol) 3-Dime- thylaminobenzoesäure in 125 ml Dichlormethan suspendiert. Dazu wurden 6,62 ml (40 mmol) Ethyldiisopropylamin gegeben. Es entstand eine leicht bräunliche, klare Lösung. In einem Wägeschiffchen wurden 6,43 g (20 mmol) TBTU abgewogen und portionsweise bei Raumtemperatur zum Ansatz zugegeben. Es entstand eine gelbbraune Lösung. Die Lösung wurde 1 h bei Raumtemperatur gerührt. Anschließend wurde der Ansatz in einem Eisbad auf 0 °C gekühlt. Im Tropftrichter wurde eine Mischung aus 3,45 ml (40 mmol) 2-Methoxyethylamin in 25 ml Dichlormethan zubereitet. Diese Lösung wurde innerhalb von 35 min zum Ansatz getropft, sodass die Innentemperatur nicht über 3 °C anstieg. Die Farbe des Reaktionsgemisches hellte sich auf und es entstand ein Feststoff. Nach beende- ter Zugabe wurde das Eisbad entfernt und der Ansatz wurde
48 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch in einen Scheidetrichter überführt und mit 2x50 ml 0,5 N HCl-, 1x50 ml gesättigter NaHC03- und 1x50 ml gesättigter NaCl-Lösung extrahiert. Die organsiche Phase wird über a2SÜ4 getrocknet und das Lösungsmittel am Rota¬ tionsverdampfer entfernt. Ausbeute Rohprodukt: 2,85 g.
Methoden
Alle Untersuchungen und Synthesen wurden bei 23±2°C und normalem Luftdruck durchgeführt, wenn nicht ausdrücklich etwas anderes angegeben ist.
Für die Herstellung von Pasten wurde ein Speedmixer DAC 150 FV der Firma Hauschild verwendet. Zur Homogenisierung der Pasten wurde ein Dreiwalzwerk Exakt 50 der Firma Exakt verwendet. Die Pasten wurden im Vakuum entgast.
Kleine Substanzmengen wurden mit dem Glasofen B 585 Kugelrohr der Firma Büchi destilliert.
Mitteldruckchromatographie
Es wurde ein MPLC-System der Firma Büchi verwendet. Es be- stand aus einem Pumpenmodul C-601, einem Pumpen-Manager C- 615 und einem Fraktionssammler C-660. Die zu chromatogra- phierenden Proben wurden im Laufmittel gelöst und über ein 4-Wege-Inj ektionsventil auf eine Probenschlaufe (bis 20ml) aufgebracht. Es wurden Glassäulen vom Typ C-690 verwendet. Die genauen Dimensionen sind bei den Synthesen beschrieben.
1H-NMR Spektroskopie
Die Messungen erfolgten auf einem Gemini 300 MHz NMR- Spektrometer der Firma Varian. Die Kalibrierung der Spektren erfolgte anhand des Restprotonen-Signals des verwende- ten deuterierten Lösungsmittels.
Durchhärtetiefe (DHT)
Die Durchhärtetiefe wurde entsprechend der Prüfung „Poly¬ merisationstiefe" nach der ISO 4049:2000 bestimmt. Hierzu wurden die Pasten in zylindrische Teflonformen (5 mm Durch- messer, 10 mm Höhe) gefüllt und mit einer Halogenlampe
(Translux EC, Heraeus Kulzer) oder einer LED-Lampe (Mini LED, Acteon) 5, 10 oder 20 s von oben belichtet. Direkt nach der Belichtung wurden die ausgehärteten Prüfkörper entnommen und mit einem Messer von nicht ausgehärtetem Ma- terial befreit. Die Höhe des ausgehärteten Bereiches wurde mit einer Schieblehre ermittelt und als Durchhärtetiefe (DHT) in mm angegeben.
Biegefestigkeit
Mit Hilfe einer Form aus Kunststoff wurden Prüfkörper mit den Abmessungen (40±2) mm x (2±0,1) mm x (2±0,1) mm herge¬ stellt. Hierzu wurden die jeweiligen Pasten im Lichtofen (HiLite Power, Heraeus Kulzer) lichtgehärtet. Die Pasten wurden in der Form zunächst 90 s belichtet, anschließend gewendet und nochmals 90 s belichtet. Nach Entformung wur¬ den die Prüfkörper in VE-Wasser für 23 h bei 37 °C und dann 1 h bei 23 °C gelagert. Anschließend wurde mit Hilfe einer Universalprüfmaschine (Typ Z 010/TN2A, Zwick) die Biegefes- tigkeit (BF) gemäß ISO 4049:2000 gemessen. Die Messung er¬ folgte bei konstanter Vorschubgeschwindigkeit von 0,8 mm/min. Die Berechnung der BF [MPa] erfolgte nach der Formel :
 F = maximale auf den Prüfkörper ausgeübte Kraft in Newton; 1 = Abstand zwischen den Auflagen mit einer Genauigkeit von ± 0,01 mmm; b = Breite der Prüfkörper unmittelbar vor der Prüfung, gemessen in mm; h = Höhe der Prüfkörper unmittelbar vor der Prüfung gemessen in mm. Scherhaftkraft (SBS) auf Rinderschmelz und Rinderdentin
F = maximale auf den Prüfkörper ausgeübte Kraft in Newton; 1 = Abstand zwischen den Auflagen mit einer Genauigkeit von ± 0,01 mmm; b = Breite der Prüfkörper unmittelbar vor der Prüfung, gemessen in mm; h = Höhe der Prüfkörper unmittelbar vor der Prüfung gemessen in mm. Scherhaftkraft (SBS) auf Rinderschmelz und Rinderdentin
Rinderzähne ohne Pulpen (Rocholl) wurden in ein kalt poly- merisierendes Harz (Viscovoss GTS mit MEKP MEH-Härter, Voss Chemie) eingebettet. Unmittelbar vor der Versuchs¬ durchführung wurden pro Versuchsserie 10 Rinderzähne auf den gewünschten Untergrund (Schmelz oder Dentin) nass mit
Schleifpapier 120 Grit abgeschliffen. Danach fand ein Feinschliff mit Schleifpapier der Körnung 500 Grit statt. Das Zahnmaterial hatte eine plane und glatte Oberfläche. Die geschliffenen Zähne wurden bis zur weiteren Behandlung in VE-Wasser aufbewahrt. In Etiketten wurden Löcher mit einem Durchmesser von 3 mm gestanzt. Der zu präparierende Zahn
wurde unmittelbar vor der Behandlung aus dem Wasser genommen und die Feuchtigkeit mit ölfreier Druckluft entfernt, wobei ein übertrocknen vermieden wurde. Das gelochte Eti¬ kett wurde so mittig auf die Zahnoberfläche geklebt, dass das Loch für das Kleben zur Verfügung stand. Die Adhäsive wurden mit Hilfe eines Einmalpinsels 20 s auf der Zahnober¬ fläche verrieben und anschließend 20 s mit einer LED-Lampe (Mini LED, Acteon, blaues Licht, 182 mW/cm2) belichtet. Anschließend wurde eine zweiteilige Teflonform (Höhe 3 mm) mit Bohrung (Durchmesser 3 mm) auf die präparierte Stelle plan aufgelegt und mit Metallklammern fixiert. Die Bohrung wurde mit dem Adhäsiv luftblasenfrei befüllt. Dann wurde 40 s mit einer LED-Lampe (Mini LED, Acteon) bestrahlt. Nach der Aushärtung wurde die Teflonform entfernt und überste- hende Reste des ausgehärteten Adhäsivs mit einem Skalpell entfernt. Die so hergestellten Prüfkörper wurden danach 1 Tag bei 37 °C in einem Wasserbad gelagert. Unmittelbar vor der Messung der Scherhaftkraft ließ man das Wasserbad in¬ nerhalb einer Stunde auf 23 °C abkühlen. Die Prüfkörper wurden in eine Abschervorrichtung gemäß ISO 10477:2004 zur Messung der Scherhaftkraft eingespannt und mit einer Universalprüfmaschine (Typ Z 010/TN2A, Zwick) bei einem Vorschub von 0,5 mm/min die SBS gemessen. Die Ergeb¬ nisse wurden in Form eines Mittelwerts mit Standardabwei- chung angegeben. Prüfkörper, deren Verklebung sich vor der Messung schon aufgelöst hat oder deren Verklebung nicht besser war als die eingestellte Vorkraft von 0,01 N, gehen mit einer Scherhaftkraft von 0 MPa in die Mittelwertbildung ein . Bestimmung der Wasserlöslichkeit
Die Wasserlöslichkeit wurde in VE-Wasser durch UV/Vis- Spektroskopie bestimmt. Die Spektren wurden mit einem
UV/Vis-Spektrometer (Evolution 600, Thermo Scientific) zwischen 200-900 nm aufgenommen. Die Messung erfolgte in
Quarzküvetten (Schichtdicke 10 mm) . Für das Hintergrund¬ spektrum wurde das reine Lösungsmittel VE-Wasser verwendet. Von den Spektren der Coinitiatoren wurde das Hintergrundspektrum subtrahiert.
Verdünnte Lösungen der unterschiedlichen Coinitiatoren mit bekannter Konzentration wurden in VE-Wasser hergestellt.
Die Absorption der unterschiedlichen Coinitiatoren wurde im Absorptionsmaximum (zwischen 290-310 nm) gemessen und eine lineare Beziehung zwischen der bekannten Konzentration und der gemessenen Absorption ermittelt.
Gesättigte, wässrige Lösungen der Coinitiatoren wurden soweit verdünnt, dass die gemessene Absorption in den oben gefundenen linearen Bereich fällt. Über das Lambert-Beer- Gesetz wird die Konzentration der Coinitiatoren in der gesättigten, wässrigen Lösung ermittelt.
Folgende Löslichkeiten in Wasser wurden ermittelt:
Löslichkeit
Coinitiator
[mg/1]
4-Dimethylaminobenzoesäure-ethylester1' 36
4-Dimethylaminobenzoesäure-2-n-
35 butoxyethylester1'
EHA 4-Dimethylaminobenzoesäure-2-ethylhe-
26 xylester1'
4-Dimethylaminobenzonitril1' 278
PEGA 4 -Dirnethylaminobenzoesäure-poly (ethylen-
>766300 glykol) monomethylether-ester
4-Dimethylaminobenzamid 427
4-Dimethylamino-N-propyl-benzamid 680
4-Dimethylamino-N- (2-methoxyethyl) -ben-
2670 zamid
1 Coinitiator gemäß Stand der Technik
Beispielzusammensetzungen Beispiel 1
Es wurden einteilige radikalisch polymerisierbare licht¬ härtbare adhäsive Dentalkomposite hergestellt und Ihre Haftkraft (SBS) , Durchhärtungstiefe (DHT) und Biegefestig- keit (BF) gemessen.
Zunächst wurden aus den in den Tabellen jeweils angegebenen Komponenten Harzzusammensetzungen hergestellt. Diese Harzzusammensetzungen dienten als Matrix für die zur Pastenher- Stellung zugefügten Füllstoffe. Zur Herstellung der Kompositpasten wurden in 37 Teilen Harzmatrix 60 Teile Glaspulver und 3 Teile Kieselsäure suspendiert. Die Herstellung erfolgte ansonsten wie bei den Methoden beschrieben.
Erfindungs- Erfindungs-
Vergleich 1 Vergleich 2 Vergleich 3 gemäß gemäß
Composite 3 Composite 11 Composite 4 Composite 9 Composite 6
Harz
64,15 64,38 79,1 64,04 78,7 55/45*
HEMA 24,95 24,76 11,99 24,63 11,93
MPD 9,98 9,91 8,04 9,86 7,95
BHT 0,05 0,05 - 0,05 -
CQ 0,2 0,2 0,2 0,2 0,2
EHA 0,67 0,7 0,67 - -
PEGA - - - 1,22 1,22
BF 131 - 148 - 136
DHT (5s) 7,7 - 9,1 - 8,7
DHT (10
9,1 - 9,9 - 9,8 s)
DHT
10 - - - - (20s)
SBS
5,95 5,12
Schmelz - - - (2,15) (1,5)
(SD)
SBS Den2,34
- - 5 (2,2) - tin (SD) (1,4)
* (Bis-GMA 54,92; TEGDMA 44,93; BHT 0,15%)
Für den Photoinitiator CQ erkennt man, dass beim äquimola- ren Austausch eines Coinitiators des Standes der Technik
(EHA, Composite 11) durch einen erfindungsgemäßen Coinitia- tor (PEGA, Composite 9) , der eine höherer Wasserlöslichkeit aufweist, nahezu eine Verdopplung der Haftkraft auf Dentin erzielt wird. Gleichzeitig werden gute Werte für die DHT und BF erreicht.
Beispiel 2

* (Bis-GMA 54,92; TEGDMA 44,93; BHT 0,15%) Bei äquimolarem Austausch des Photoinitiators CQ durch seine hydrophileren Derivate wurde überraschend gefunden,
dass auch mit diesen durch einen erfindungsgemäßen Coinitiator mit höherer Wasserlöslichkeit (PEGA, Composite 10) eine sprunghafte Verbesserung der Haftkraft auf Dentin er¬ zielt wird, jedoch sind die absoluten Werte nicht weiter verbessert. Die Kombination von CQ-10-sulfonat und EHA führt andererseits zu einer signifikanten Verschlechterung der Haftung.
Die Kombination aus hydrophilem Photoinitiator und hydro- phobem Coinitiator (C12) hatte die geringsten Scherhaftkräfte, sowohl auf Schmelz als auch auf Dentin. Die Kombi¬ nation aus hydrophobem Photoinitiator und hydrophilem
Coinitiator (C9) lieferte auf Schmelz eine vergleichbare Scherhaftkraft wie Cll. Die Scherhaftung auf Dentin war da- gegen signifikant höher als bei Cll. Die Kombination aus hydrophilem Photoinitiator und hydrophilem Coinitiator (CIO) lieferte im Vergleich zu C9 und Cll eine etwas gerin¬ gere Scherhaftkraft auf Schmelz. Die Scherhaftkraft auf Dentin war dagegen ebenfalls signifikant höher als bei Cll. Aus dieser Testreihe ist ersichtlich, dass ein hydrophiles bzw. wasserlösliches, aromatisches, tertiäres Amin als Coinitiator eine deutliche Verbesserung der Haftung auf Dentin ermöglicht.
Beispiel 3

* (Bis-GMA 54,92; TEGDMA 44,93; BHT 0,15%)
Durch Erhöhung des Anteils an erfindungsgemäßem Coinitiator in den erfindungsgemäßen Kompositen lässt sich insbesondere die Biegefestigkeit weiter verbessern.
Das molare Verhältnis von Photoinitiator zu Coinitiator ist in folgender Tabelle aufgeführt:
Composites
4 5 6 7 8 molares Verhältnis
Photoinitiator zu 1:2,03 1:1,04 1:2,04 1:3,06 1:4, 07 Coinitiator
Claims
1. Dentalmaterial, das enthält: a) wenigstens ein Monomer enthaltend mindestens eine radikalisch polymerisierbare ethylenisch ungesät¬ tigte Gruppe und mindestens eine Säuregruppe, b) wenigstens ein Monomer, das keine Säuregruppen und mindestens eine radikalisch polymerisierbare ethylenisch ungesättigte Gruppe enthält, c) wenigstens einen Photoinitiator, d) mindestens ein tertiäres aromatisches Amin als Coinitiator, das einen Benzolring aufweist, an den mindestens eine Dialkylamingruppe und mindes¬ tens eine weitere Gruppe direkt gebunden sind, dadurch gekennzeichnet, dass die weitere Gruppe ausgewählt ist aus: i. Carbonsäureestergruppen enthaltend mindestens eine Polyoxyalkylengruppe mit mindes¬ tens 2 Oxyethylen- und/oder Oxypropylenein- heiten, und ii. Amidgruppen.
2. Dentalmaterial nach Anspruch 1, dadurch gekennzeichnet, dass die mindestens eine weitere Gruppe mindes¬ tens eine Polyoxyalkylengruppe mit 3 bis 20, Vorzugs-
weise 5 bis 17 Oxyethylen- und/oder Oxypropyleneinhei ten enthält.
Dentalmaterial nach Anspruch 1 oder 2, dadurch gekenn zeichnet, dass das tertiäre aromatische Amin ausge¬ wählt ist aus der Gruppe bestehend aus: a) Formel I




- Ri und R2 sind unabhängig voneinander) C1-C5- Alkyl, bevorzugt Methyl,
- R3 ist Alkyl, bevorzugt Methyl,
- R4 ist H oder Cl-C5-Alkyl, bevorzugt Methyl,
- R5 und R6 sind unabhängig voneinander C1-C5-A1- kyl, bevorzugt Methyl,
- R7 und R8 sind unabhängig voneinander ausgewählt aus H, Alkyl, Alkoxyalkyl und Polyalkylenoxid, bevorzugt Polyalkylenoxid mit mindestens zwei Oxyethylen- und/oder Oxypropyleneinheiten, x ^0 und y >1.
Dentalmaterial nach Anspruch 3, dadurch gekennzeichnet, dass die Summe aus x und y 3 bis 20, vorzugsweise 5 bis 17 beträgt.
Dentalmaterial nach Anspruch 3 oder 4, dadurch gekennzeichnet, dass x kleiner als y ist.
Dentalmaterial nach einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, dass die Wasserlöslichkeit ei¬ nes Coinitiators enthaltend wenigstens eine Amidgruppe wenigstens 0,4 g/1, vorzugsweise wenigstens 2 g/1, weiter vorzugsweise wenigstens 50 g/1 beträgt; und dass die Wasserlöslichkeit eines Co-Initiators enthal¬ tend keine Amidgruppe wenigstens 2 g/1, weiter vor¬ zugsweise wenigstens 50 g/1 beträgt.
Dentalmaterial nach einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, dass der Gehalt des Co-Initia¬ tors 0,1 bis 10 Gew.-%, vorzugsweise 0,5 bis 5 Gew.-% beträgt .
Dentalmaterial nach einem der Ansprüche 1 bis 7, dadurch gekennzeichnet, dass das wenigstens eine Mono¬ mer, das keine Säuregruppen und mindestens eine radika lisch polymerisierbare ethylenisch ungesättigte Gruppe enthält, ausgewählt ist aus der Gruppe bestehend aus: bl)
 wobei in der Formel bedeutet:
wobei in der Formel bedeutet:
R1 ist H, Methyl oder Ethyl, bevorzugt H oder Methyl;
A ist O oder NR3, mit R3 = H, Methyl, Ethyl oder Pro- pyl, bevorzugt R3 = H;
HG ist eine hydrophile Gruppe ausgewählt aus OH und (0 CH2-CH2)m-OR4; bevorzugt ist HG = OH;
ist H, Methyl, Ethyl oder CkH2k+i-n (HG) n; bevorzugt
R4 ist H oder Methyl; k, m, n sind natürliche Zahlen; k = 1-5; bevorzugt 1-2; m= 3-20; bevorzugt 5-20; n= 1-5; n < k; jedes Kohlenstoffatom trägt höchstens eine hydrophile Gruppe HG; b2) (Q A Ev)r CtH2t+2-(r+S) (HG) mit Q


R8 ist H, Methyl oder Ethyl, bevorzugt H oder Methyl;
A ist 0 oder NR3, mit R3 = H, Methyl, Ethyl oder Pro- pyl, bevorzugt R3 = H;
HG ist OH, (0-CH2-CH2)m-OR7; bevorzugt OH;
R7 ist H oder Methyl;
m, r, s, t, v sind natürliche Zahlen;
m= 3-20, bevorzugt 6-20;
r= 2-4,
s= 1-4
t= 2-8 bevorzugt
oder 1; bevorzugt 0; und b3) Q-A-EV-C2H2-HG-EV-A-Q mit Q


oder OH

wobei in den Formeln bedeuten:
A ist O oder NR3, mit R3 = H, Methyl, Ethyl oder Pro- pyl, bevorzugt R3 = H;
HG ist (0-CH2-CH2)m;
m= 4-20, bevorzugt 6-20;
v= 0 oder 1; bevorzugt 0.
Dentalmaterial nach einem der Ansprüche 1 bis 8, dadurch gekennzeichnet, dass es zusätzlich wenigstens ein Monomer b4), das keine Säuregruppen und mindestens zwei radikalisch polymerisierbare ethylenisch ungesät¬ tigte Gruppen enthält, aufweist, wobei dieses wenigs¬ tens eine Monomer b4) nicht aus der Gruppe bl), b2) und b3) ausgewählt ist.
Dentalmaterial nach einem der Ansprüche 8 bis 9, dadurch gekennzeichnet, dass das wenigstens eine Mono mer bl) bis b3) eine Wasserlöslichkeit von wenigstens 50 g/1 aufweist.
Dentalmaterial nach Anspruch 9 oder 10, dadurch gekennzeichnet, dass das wenigstens eine Monomer b4)) eine Wasserlöslichkeit von weniger als 50 g/1 auf¬ weist.
Dentalmaterial nach einem der Ansprüche 1 bis 11 zur Verwendung als lichthärtendes selbsthaftendes und selbstätzendes radikalisch polymerisierbares Dental- restaurativ .
Verwendung eines tertiären aromatisches Amins, das ei nen Benzolring aufweist, an den mindestens eine Dial- kylamingruppe und mindestens eine weitere Gruppe di¬ rekt gebunden sind, wobei die weitere Gruppe ausge¬ wählt ist aus :
i. Carbonsäureestergruppen enthaltend mindestens eine Polyoxyalkylengruppe mit mindes¬ tens 2 Oxyethylen- und/oder Oxypropylenein- heiten, und
5
ii. Amidgruppen als Coinitiator in einem polymerisierbaren Dentalmaterial .
10
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15704566.7A EP3107520B1 (de) | 2014-02-21 | 2015-02-17 | Polymerisierbares dentalmaterial |
| US15/120,372 US10058487B2 (en) | 2014-02-21 | 2015-02-17 | Polymerizable dental material |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102014203166.4A DE102014203166A1 (de) | 2014-02-21 | 2014-02-21 | Polymerisierbares Dentalmaterial |
| DE102014203166.4 | 2014-02-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015124559A1 true WO2015124559A1 (de) | 2015-08-27 |
Family
ID=52472336
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2015/053303 Ceased WO2015124559A1 (de) | 2014-02-21 | 2015-02-17 | Polymerisierbares dentalmaterial |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10058487B2 (de) |
| EP (1) | EP3107520B1 (de) |
| DE (1) | DE102014203166A1 (de) |
| WO (1) | WO2015124559A1 (de) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102015217418A1 (de) | 2015-09-11 | 2017-03-16 | Mühlbauer Technology Gmbh | Radikalisch polymerisierbares Dentalmaterial |
| EP3308765A1 (de) | 2016-10-17 | 2018-04-18 | Mühlbauer Technology GmbH | Radikalisch polymerisierbare verbindung |
| EP3653194A1 (de) | 2018-11-15 | 2020-05-20 | Dentsply DeTrey GmbH | Bifunktionelle und polyfunktionelle co-initiatoren in zahnärztlichen zusammensetzungen |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3677241B1 (de) * | 2017-08-28 | 2023-02-15 | Kuraray Noritake Dental Inc. | Lösungsmittelfreie klebstoffzusammensetzung für zahnärztliche zwecke |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19848886A1 (de) * | 1998-10-23 | 2000-05-04 | Heraeus Kulzer Gmbh & Co Kg | Lichtpolymerisierbares Einkomponenten-Dentalmaterial |
| US6084004A (en) * | 1997-08-21 | 2000-07-04 | Espe Dental Ag | Compositions which undergo light-induced cationic curing and their use |
| WO2000049064A1 (de) * | 1999-02-17 | 2000-08-24 | Lsp Dental Chemistry Ag | Epoxidverbindungen für zahnmedizinische und/oder zahntechnische anwendungen |
| US20060052470A1 (en) * | 2003-07-16 | 2006-03-09 | Eric Grech | Photopolymerizable composition based on an epoxyvinylester resin and on a urethane acrylate resin and use thereof for making dental prosthesis preforms and or models |
| EP2153811A2 (de) * | 2008-08-13 | 2010-02-17 | Kerr Corporation | N.a. |
| WO2011139936A2 (en) * | 2010-05-03 | 2011-11-10 | Levin, Leana | Dental compositions |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3541068A (en) * | 1968-07-12 | 1970-11-17 | Minnesota Mining & Mfg | Dental restorative compositions having enhanced stability |
| US4548689A (en) | 1983-09-28 | 1985-10-22 | Mitsui Petrochemical Industries, Ltd. | Photocurable resin composition |
| GB9507808D0 (en) * | 1995-04-18 | 1995-05-31 | Lambson Fine Chemicals Limited | Novel amine compounds |
| JP4535309B2 (ja) * | 2000-11-10 | 2010-09-01 | デュランド テクノロジー リミテッド | 光学記録材料 |
| DE10124028B4 (de) | 2001-05-16 | 2010-02-18 | 3M Espe Ag | Selbstadhäsive Dentalmaterialien |
| EP1570831A1 (de) | 2004-03-02 | 2005-09-07 | Ernst Mühlbauer GmbH & Co.KG | Gefülltes, polymerisierbares Dentalmaterial und Verfahren zu dessen Herstellung |
| CA2575976A1 (en) | 2004-08-11 | 2006-02-23 | 3M Innovative Properties Company | Self-adhesive compositions including a plurality of acidic compounds |
| US7906564B2 (en) | 2006-02-23 | 2011-03-15 | Pentron Clinical Technologies, Llc | Self etch all purpose dental cement composition, method of manufacture, and method of use thereof |
| JP5416581B2 (ja) | 2007-03-20 | 2014-02-12 | クラレノリタケデンタル株式会社 | 重合性単量体、重合性組成物及び歯科用材料 |
| EP2226061B2 (de) * | 2009-03-06 | 2017-01-25 | Ernst Mühlbauer GmbH & Co.KG | Infiltrant für die Dentalapplikation |
| JP2011042609A (ja) | 2009-08-20 | 2011-03-03 | Gc Corp | 歯質接着性組成物 |
-
2014
- 2014-02-21 DE DE102014203166.4A patent/DE102014203166A1/de not_active Withdrawn
-
2015
- 2015-02-17 US US15/120,372 patent/US10058487B2/en active Active
- 2015-02-17 EP EP15704566.7A patent/EP3107520B1/de active Active
- 2015-02-17 WO PCT/EP2015/053303 patent/WO2015124559A1/de not_active Ceased
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6084004A (en) * | 1997-08-21 | 2000-07-04 | Espe Dental Ag | Compositions which undergo light-induced cationic curing and their use |
| DE19848886A1 (de) * | 1998-10-23 | 2000-05-04 | Heraeus Kulzer Gmbh & Co Kg | Lichtpolymerisierbares Einkomponenten-Dentalmaterial |
| WO2000049064A1 (de) * | 1999-02-17 | 2000-08-24 | Lsp Dental Chemistry Ag | Epoxidverbindungen für zahnmedizinische und/oder zahntechnische anwendungen |
| US6620864B2 (en) * | 1999-02-17 | 2003-09-16 | Lsp Dental Chemistry Ag | Epoxy compounds for use in dental medicine and/or dentistry |
| US20060052470A1 (en) * | 2003-07-16 | 2006-03-09 | Eric Grech | Photopolymerizable composition based on an epoxyvinylester resin and on a urethane acrylate resin and use thereof for making dental prosthesis preforms and or models |
| EP2153811A2 (de) * | 2008-08-13 | 2010-02-17 | Kerr Corporation | N.a. |
| WO2011139936A2 (en) * | 2010-05-03 | 2011-11-10 | Levin, Leana | Dental compositions |
| US20110275035A1 (en) * | 2010-05-03 | 2011-11-10 | Dentsply International Inc. | Dental compositions |
Non-Patent Citations (1)
| Title |
|---|
| N.S. ALLEN ET AL.: "Photochemistry and photo-induced co-synergistic polymerisation activities of novel N,N,-dimethylaminobenzoates and benzamides", 4 December 2000 (2000-12-04), XP055186056, Retrieved from the Internet <URL:http://www.sciencedirect.com/science/article/pii/S1010603000003646> [retrieved on 20150427] * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102015217418A1 (de) | 2015-09-11 | 2017-03-16 | Mühlbauer Technology Gmbh | Radikalisch polymerisierbares Dentalmaterial |
| EP3308765A1 (de) | 2016-10-17 | 2018-04-18 | Mühlbauer Technology GmbH | Radikalisch polymerisierbare verbindung |
| US10633491B2 (en) | 2016-10-17 | 2020-04-28 | Muhlbauer Technology Gmbh | Radically polymerizable compound |
| EP3653194A1 (de) | 2018-11-15 | 2020-05-20 | Dentsply DeTrey GmbH | Bifunktionelle und polyfunktionelle co-initiatoren in zahnärztlichen zusammensetzungen |
| WO2020099518A1 (en) | 2018-11-15 | 2020-05-22 | Dentsply Detrey Gmbh | Bifunctional and polyfunctional coinitiators in dental compositions |
| AU2019379295B2 (en) * | 2018-11-15 | 2023-05-25 | Dentsply Detrey Gmbh | Bifunctional and polyfunctional coinitiators in dental compositions |
Also Published As
| Publication number | Publication date |
|---|---|
| US10058487B2 (en) | 2018-08-28 |
| EP3107520B1 (de) | 2019-10-30 |
| US20170065496A1 (en) | 2017-03-09 |
| EP3107520A1 (de) | 2016-12-28 |
| DE102014203166A1 (de) | 2015-08-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3150641B1 (de) | Acylgermanium-photoinitiatoren und verfahren zu deren herstellung | |
| EP1374829B1 (de) | Dentalmaterialien auf der Basis von Acrylesterphosphonsäuren | |
| EP2753291B1 (de) | Dentalmaterialien auf basis von verbindungen mit debonding-on-demand eigenschaften | |
| EP3166569B1 (de) | Komposite mit gesteuerter netzwerkstruktur | |
| EP2910236B1 (de) | Dentalmaterialien auf der Basis von geruchsarmen Thiolen | |
| DE3516256A1 (de) | (meth)-acrylsaeureester und ihre verwendung | |
| EP2755624B1 (de) | Dentalmaterialien auf basis von stark aciden polymerisierbaren bisphosphonsäuren | |
| EP2823801B1 (de) | Dentalmaterialien auf der Basis von harnstoffgruppenhaltigen Monomeren | |
| EP3166568B1 (de) | Dentalmaterialien mit debonding-on-demand eigenschaften | |
| JP5688933B2 (ja) | アシルホスフィンオキシド化合物及びそれを含む重合性組成物 | |
| EP3107520B1 (de) | Polymerisierbares dentalmaterial | |
| WO2017064042A1 (de) | Polymerisierbare zusammensetzungen auf basis von thermisch spaltbaren verbindungen | |
| EP2816049B1 (de) | ß-Ketophosphonsäuren und Dentalmateriallen auf deren Basis | |
| EP0798286A1 (de) | Multifunktionelle Vinylcy-clopropan-Derivate | |
| JP5688939B2 (ja) | ビスアシルホスフィンオキサイド化合物及びそれを含む重合性組成物 | |
| EP1825843B1 (de) | Dentalmaterialien auf der Basis von multicyclischen Allylsulfiden | |
| DE10234326B3 (de) | Dentalmaterialien auf der Basis von Acrylesterphosphonsäuren | |
| EP1027377B1 (de) | Polymerisierbare zusammensetzung und ihre verwendung als haftvermittler | |
| EP3328871B1 (de) | Vernetzende monomere mit mindestens einem schwefelatom | |
| EP3346969B1 (de) | Radikalisch polymerisierbares dentalmaterial |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15704566 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15120372 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015704566 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015704566 Country of ref document: EP |