WO2015131299A1 - 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 - Google Patents
手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 Download PDFInfo
- Publication number
- WO2015131299A1 WO2015131299A1 PCT/CN2014/000197 CN2014000197W WO2015131299A1 WO 2015131299 A1 WO2015131299 A1 WO 2015131299A1 CN 2014000197 W CN2014000197 W CN 2014000197W WO 2015131299 A1 WO2015131299 A1 WO 2015131299A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxypiperidine
- benzyl
- formula
- substituted
- butoxycarbonyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/42—Oxygen atoms attached in position 3 or 5
Definitions
- the present invention relates to the synthesis of a compound, in particular to the preparation of a chiral 1-tert-butoxycarbonyl-3-hydroxypiperidine and a chiral turning
- the present invention provides a process for the preparation of chiral 1-tert-butoxycarbonyl-3-hydroxypiperidine, which comprises the steps of:
- the resolving agent for the resolution reaction in the step 1) is chiral d-camphorsulfonic acid D-CSA or L-CSA of L-camphoric acid, the chiral dextrose camphorsulfonic acid D-CSA or L-camphor
- the molar ratio of the acid L-CSA to the starting material N-benzyl-3-hydroxypiperidine is 0 to 0.6: 1, preferably: 0.3 to 0.5: 1.
- the solvent system of the resolution reaction in the step 1) is: an organic alcohol solvent such as methanol, ethanol or isopropanol; an organic ester solvent such as ethyl acetate or isopropyl acetate; such as 2-butanone, An organic ketone solvent of acetone; an organic ether solvent such as THF, diisopropyl ether, methyl tert-butyl ether; or any combination thereof.
- an organic alcohol solvent such as methanol, ethanol or isopropanol
- an organic ester solvent such as ethyl acetate or isopropyl acetate
- 2-butanone An organic ketone solvent of acetone
- an organic ether solvent such as THF, diisopropyl ether, methyl tert-butyl ether; or any combination thereof.
- the base of the step 2) is sodium hydroxide, lithium hydroxide, potassium hydroxide, sodium carbonate, sodium hydrogencarbonate, potassium carbonate or potassium hydrogencarbonate, wherein the base and the raw material N-benzyl-3-hydroxypiperidin
- the molar ratio of pyridine is from 1 to 10: 1, preferably from 1 to 1.5:1.
- the solvent system of the step 3) reaction is: an organic alcohol solvent such as methanol, ethanol or isopropanol; an organic ester solvent such as ethyl acetate or isopropyl acetate; such as THF, isopropyl ether, methyl uncle An organic ether solvent of butyl ether; or any combination thereof.
- step 3 the weight of palladium on carbon and (S)-1-benzyl-3-hydroxypiperidine of formula III s or (R)-l-benzyl-3-hydroxypiperidine of formula III r
- the ratio is 0.01 ⁇ 0.5:1; the reaction temperature is between 0 °C and 80 °C; the reaction pressure is latm ⁇ 30 atm ; wherein the tert-butoxycarbonyl anhydride and the substrate of formula III s are (S) -1-
- the molar ratio of benzyl-3-hydroxypiperidine or (R)-l-benzyl-3-hydroxypiperidine of formula III r is from 1 to 2:1.
- the present invention also provides a chiral inversion method for the above chiral 1-tert-butoxycarbonyl-3-hydroxypiperidine, which comprises the steps of:
- Step 4) The (R)-1-substituted-3-hydroxypiperidinyl sulfonate of the formula VI r or the (S)-1-substituted-3-hydroxypiperidine sulfonate of VI s is substituted Substituted by a carboxylate, the structure is inverted to give (S)-1-substituted-3-hydroxypiperidinecarboxylate of formula VII s or (R)-1-substituted-3-hydroxypiperidinecarboxylate of formula VII r;
- R is benzyl, benzyloxycarbonyl or t-butoxycarbonyl
- Ri is a lower hydrazine of 1 to 3, substituted phenyl
- the chiral-1-substituted-3-hydroxypiperidine is (S)-1-benzyl-3-hydroxypiperidine of the formula III s according to the step 2) of claim 1, or the formula III r (R)-l-benzyl-3-hydroxypiperidine; or (S)-l-tert-butoxycarbonyl-3-hydroxypiperidine of formula IVs as described in step 3 of claim 1 or formula IV r (R)-1-tert-Butoxycarbonyl-3-hydroxypiperidine.
- the sulfonyl chloride described in the step 4) is methylsulfonyl chloride, p-methylbenzenesulfonyl chloride, p-nitrobenzenesulfonyl chloride or p-bromobenzenesulfonyl chloride;
- the solvent system of the acylation reaction is: ethyl acetate,
- Organic ester solvents of isopropyl acetate organic chlorinated solvents such as methylene chloride, trichloromethane, dichloroacetamidine, chlorobenzene: organic such as isopropyl ether, methyl tert-butyl ether, tetrahydrofuran Ether solvent.
- the carboxylate described in the step 5) is sodium acetate or sodium benzoate
- the solvent system for replacing the inversion reaction is: an organic amide solvent such as DMF or formamide; an organic ester such as ethyl acetate or isopropyl acetate; Solvents; organic chlorinated solvents such as methylene chloride, trichloromethane, dichloroacetamidine, chlorobenzene; organic ether solvents such as isopropyl ether, methyl tert-butyl ether, tetrahydrofuran; Organic alcohol solvent of ethanol: or a mixed solvent of one or more types of the above solvents and a mixed solvent system with water.
- the base of the hydrolysis reaction in the step 6) is sodium hydroxide, lithium hydroxide or potassium hydroxide; sodium carbonate, potassium carbonate or ammonia water; wherein the above base and the raw material N-benzyl-3- described in the step 4)
- the molar ratio of hydroxypiperidine is from 1 to 100:1.
- the solvent for the hydrolysis reaction is water; organic such as methanol or ethanol An alcohol solvent; an organic ether solvent such as tetrahydrofuran; or a mixed solvent system thereof.
- the above method of the present invention solves the problem that it is difficult to industrialize in the chemical resolution method because it is difficult to purify due to the water solubility of 3-hydroxypiperidine.
- the utilization of chiral impurities during the splitting process is solved.
- the synthetic route has not been reported in the literature, and the reaction is clean and suitable for industrial large-scale production.
- Step 6) Base hydrolysis to give (S) or (R) -l-substituted -3-hydroxypiperidine
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法。主要包含步骤:以N-苄基-3-羟基哌啶为原料,拆分得(S)或(R)-1-苄基-3-羟基哌啶樟脑磺酸盐;经碱游离得(S)或(R)-1-苄基-3-羟基哌啶;经钯炭氢化脱苄/叔丁氧羰基保护得(S)或(R)-1-叔丁氧羰基-3-羟基哌啶。以(R)或(S)-1-取代-3-羟基哌啶为原料,取代磺酰氯酰化得(R)或(S)-1-取代-3-羟基哌啶磺酸酯;被取代羧酸盐取代,得(S)或(R)-1-取代-3-羟基哌啶羧酸酯;经碱水解得(S)或(R)-1-取代-3-羟基哌啶。上述合成路线反应条件温和,适合工业化大生产。
Description
手性 -1 -叔丁氧羰基 -3-羟基哌啶的制备以及手性翻转的方法 技术领域
本发明涉及一种化合物的合成, 具体涉及一种手性 -1-叔丁氧羰基 -3- 羟基哌啶的制备以及手性翻 背景技术
(S)-l-叔丁氧羰基 -3-羟基哌啶是一种重要的医药中间体。 现今文 献中报道的其合成方法主要如下:
方法一 (不对称合成):
,、、ΟΗ

主要文献: Tetrahedron, 63, 331 -336; 2007。 方
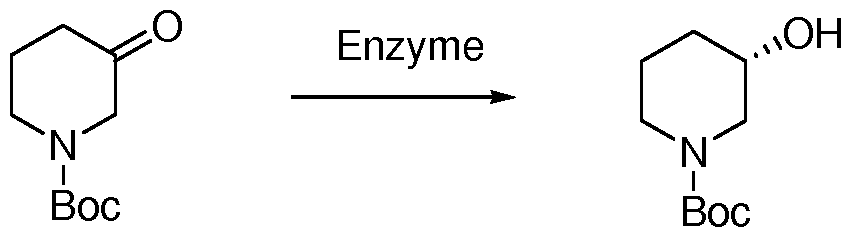
主要文献: Organic Letters, 11 (6), 1245- 1248 ; 2009。 方法三 (化学拆分法):

主要文献: WO2004064730 ; WO2004072086。 化学拆分法中, 由于 3-羟基哌啶的水溶性而不易纯化获得, 较难工业 化的问题。 同时拆分过程中容易存在手性杂质。
发明内容 本发明的目的是提供一种新的手性 1-叔丁氧羰基 -3-羟基哌啶的制备 方法。 该合成路线未见文献报道, 反应条件温和, 适合工业化大生产。
具体地, 本发明提供一种手性 -1-叔丁氧羰基 -3-羟基哌啶的制备方法, 主要包含步骤:
1 ) 以式 I 的 N-苄基 -3-羟基哌啶为原料, 经手性樟脑磺酸 CSA的作 用下拆分得式 Π s的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐或式 II r 的 (R)-l-节 基 -3-羟基哌啶樟脑磺酸盐;
2)步骤 1 )所述的式 II s的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐或式 II r 的 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐经碱游离,得式 III s的 (S) -1-苄基 -3- 羟基哌啶或式 Ill r的 (R)-l-苄基 -3-羟基哌啶;
3 )步骤 2)所述的式 III s的 (S) -1-苄基 -3-羟基哌啶或式 Ill r的 (R)-l- 苄基 -3-羟基哌啶经钯炭催化氢化脱苄, 同时用叔丁氧酸酐羰基保护得式 IVs的 (S)-l-叔丁氧羰基 -3-羟基哌啶或式 IV r的 (R) -1-叔丁氧羰基 -3-羟基 哌啶

III IV
其中,步骤 1 )中的拆分反应的拆分剂为手性右旋樟脑磺酸 D-CSA或 左旋樟脑磺酸 L-CSA, 所述手性右旋樟脑磺酸 D-CSA 或左旋樟脑磺酸 L-CSA与原料 N-苄基 -3-羟基哌啶的摩尔比为 0〜0.6: 1, 优选: 0.3〜0.5:
1。
其中, 步骤 1 ) 中的拆分反应的溶剂体系为: 如甲醇、 乙醇、 异丙醇 的有机醇类溶剂、如醋酸乙酯、醋酸异丙酯的有机酯类溶剂; 如 2—丁酮、 丙酮的有机酮类溶剂; 如 THF、 异丙醚, 甲基叔丁基醚的有机醚类溶剂; 或其中的任意组合。
其中, 步骤 2 )所述的碱为氢氧化钠、 氢氧化锂、 氢氧化钾、 碳酸钠、 碳酸氢钠、 碳酸钾或碳酸氢钾, 其中上述碱与原料 N-苄基 -3-羟基哌啶的 摩尔比为 1〜10: 1, 优选 1〜1.5: 1。
其中, 步骤 3 ) 反应的溶剂体系为: 如甲醇、 乙醇、 异丙醇的有机醇 类溶剂; 如醋酸乙酯、 醋酸异丙酯的有机酯类溶剂; 如 THF、 异丙醚, 甲 基叔丁基醚的有机醚类溶剂; 或其中的任意组合。
其中,步骤 3 )中钯炭与底物式 III s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶的重量比为 0.01〜0.5: 1 ;反应温度在 0°C〜80°C 之间; 所述的反应压力为 latm〜30atm; 其中叔丁氧羰基酸酐与底物式 III s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶的摩尔比为 1〜2: 1。
本发明还提供上述的手性 -1-叔丁氧羰基 -3-羟基哌啶的手性翻转方 法, 主要包含步骤:
4 ) 以式 V r的 (R) -1-取代 -3-羟基哌啶或式 V s的 (S)-l-取代 -3-羟基哌 啶 (V) 为原料, 在取代磺酰氯的作用下酰化得式 VI r的 (R) -l-取代 -3-羟 基哌啶磺酸酯或 VI s的 (S) -1-取代 -3-羟基哌啶磺酸酯;
5 ) 步骤 4 ) 所述的式 VI r的 (R) -1-取代 -3-羟基哌啶磺酸酯或 VI s的 (S) -1-取代 -3-羟基哌啶磺酸酯被取代羧酸盐取代, 结构翻转得式 VII s的 (S) -1-取代 -3-羟基哌啶羧酸酯或式 VII r的 (R) -1-取代 -3-羟基哌啶羧酸酯;
6 )步骤 5 )所述的式 VII s的 (S) -1-取代 -3-羟基哌啶羧酸酯或式 VII r 的 (R) -1-取代 -3-羟基哌啶羧酸酯经碱水解得式 VIII s 的 (S) -1-取代 -3-羟基 哌啶或式 VIII r 的 (R) -l-取代 -3-羟基哌啶;

其中, R是苄基、 苄氧羰基或叔丁氧羰基;
Ri, 是( 1~ 3的低级垸烃, 取代苯基;
其中, 所述的手性 -1-取代 -3-羟基哌啶为权利要求 1的步骤 2)所述的 式 III s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶;或权 利要求 1步骤 3 ) 所述的式 IVs的 (S)-l-叔丁氧羰基 -3-羟基哌啶或式 IV r 的 (R) -1-叔丁氧羰基 -3-羟基哌啶。
其中, 步骤 4) 中所述的磺酰氯为甲基磺酰氯、 对甲基苯磺酰氯、 对 硝基苯磺酰氯或对溴苯磺酰氯; 酰化反应的溶剂体系为: 如醋酸乙酯、 醋 酸异丙酯的有机酯类溶剂: 如二氯甲垸、 三氯甲垸、 二氯乙垸、 氯苯的有 机氯代类溶剂: 如异丙醚, 甲基叔丁基醚、 四氢呋喃的有机醚类溶剂。
其中, 步骤 5 ) 中所述的羧酸盐为醋酸钠或苯甲酸钠; 取代翻转反应 的溶剂体系为: 如 DMF、 甲酰胺的有机酰胺类溶剂; 如醋酸乙酯、 醋酸 异丙酯的有机酯类溶剂; 如二氯甲垸、 三氯甲垸、 二氯乙垸、 氯苯的有机 氯代类溶剂; 如异丙醚、 甲基叔丁基醚、 四氢呋喃的有机醚类溶剂; 如甲 醇、 乙醇的有机醇类溶剂: 或包括上述溶剂的一类或几类混合溶剂以及和 水的混合溶剂体系。
其中, 步骤 6) 中的水解反应的碱为氢氧化钠、氢氧化锂、氢氧化钾; 碳酸钠、碳酸钾或氨水; 其中上述碱与步骤 4)所述的原料 N-苄基 -3-羟基 哌啶的摩尔比为 1〜100: 1。 水解反应的溶剂为水; 如甲醇、 乙醇的有机
醇类溶剂; 如四氢呋喃的有机醚类溶剂; 或包括其混合溶剂体系。
本发明的上述方法解决了化学拆分法中, 由于 3-羟基哌啶的水溶性而 不易纯化获得,较难工业化的问题。同时解决拆分过程中手性杂质的利用。 该合成路线未见文献报道, 反应清洁, 适合工业化大生产。
为让本发明的上述和其它目的、 特征和优点能更明显易懂, 下文特举 较佳实施例, 作详细说明如下。 具体实施方式 以下是本发明的具体实施例, 对本发明的技术方案作进一步的描述, 但本发明并不限于此实施例。
步骤 1 ) (S)或 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成
实施例 1 : (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成
( 1 ) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (一)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 异 丙醇 478 ml滴加 L-CSA (58 g, 0.25 mol) 的 116 ml 异丙醇溶液, 室温 搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 冷异丙醇 30 ml洗 涤,干燥得 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 75 g。(ee: 95% ) (理论: 105.9 g)。 95 %ee值的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐以 3倍量的异丙醇重结 晶, 可以得到 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 (99%ee)。
(2) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (二)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 乙 酸乙酯 478ml滴加 L-CSA (58 g, 0.25 mol) 的 290 ml 乙酸乙酯溶液, 室温搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 乙酸乙酯 30 ml 洗涤, 干燥得 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 95 g。 (ee: 88% ) (理论: 105.9 g)。
( 3 ) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (三)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 2-
丁酮 478 ml滴加 L-CSA (58 g, 0.25 mol) 的 290 ml 2-丁酮溶液, 室温 搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 2-丁酮 30 ml洗涤, 干燥得 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 88g。 (ee: 93% ) (理论: 105.9 g)。
(4) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (四)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 2- 丁酮 478 ml滴加 L-CSA (69.6 g, 0.3 mol) 的 290 ml 2-丁酮溶液, 室温 搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 2-丁酮 30 ml洗涤, 干燥得 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 108 g。 (ee: 75% ) (理论: 105.9 g)。
(5 ) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (五)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 2- 丁酮 478 ml滴加 L-CSA (52.2 g, 0.225 mol) 的 290 ml 2-丁酮溶液, 室 温搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 2-丁酮 30 ml洗 涤,干燥得 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 83 g。 (ee: 95% ) (理论: 95.3 g)。
(6) (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成 (六)
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , THF 478 ml, 滴加 L-CSA (58 g, 0.25 mol) 的 290 ml THF溶液, 室温搅拌 1 小时,析出固体出现, 0°C保温 2小时,过滤, THF 30 ml洗涤,干燥得 (S)-l- 苄基 -3-羟基哌啶樟脑磺酸盐 83g。 (ee: 92% ) (理论: 105.9 g)。
实施例 2: (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐的合成
1000 ml三口瓶中, 加入 N-苄基 -3-羟基哌啶 (95.6 g, 0.5 mol) , 异 丙醇 478 ml滴加 D-CSA (58 g, 0.25 mol) 的 116 ml异丙醇溶液, 室温 搅拌 1小时, 析出固体出现, 0°C保温 2小时, 过滤, 冷异丙醇 30 ml洗 涤,干燥得 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐 75g。 (ee: 95% ) (理论: 105.9 g)。 95 %ee值的 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐以 3倍量的异丙醇重结 晶, 可以得到 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐 (99%ee)。
步骤 2) (S)或 (R)-l-苄基 -3-羟基哌啶的合成
实施例 3: (S)-l-苄基 -3-羟基哌啶的合成
(1) (S)-l-苄基 -3-羟基哌啶的合成 (一)
1000ml三口瓶中, 加入 实施例 1中 (1)、 (2)、 (3) 中任一制备的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐 (99%ee , 84.7 g, 0.2 mol), 二氯甲垸 423 ml, 滴加 1N氢氧化钠水溶液(210 ml), 室温搅拌 1小时, 分去水相, 有机相无水硫酸钠干燥, 浓缩得 (S)-l-苄基 -3-羟基哌啶 38g。 (ee: 99%; LC-MS: m/e = 191.3) (理论: 38.25 g)。
(2) (S)-l-苄基 -3-羟基哌啶的合成 (二)
1000 ml三口瓶中,加入实施例 1中(4)、(5)、(6)中任一制备的(S)-l- 苄基-3-羟基哌啶樟脑磺酸盐(99%ee , 84.7 g, 0.2 mol), 乙酸乙酯 423ml, 滴加 IN碳酸钾水溶液(150 ml), 室温搅拌 1小时, 分去水相, 有机相无 水硫酸钠干燥, 浓缩得 (S)-l-苄基 -3-羟基哌啶 36g。 (ee: 99%; LC-MS: m/e = 191.3) (理论: 38.25 g)。
实施例 4: (R)-l-苄基 -3-羟基哌啶的合成
1000 ml三口瓶中, 加入实施例 2中制备的 (R)-l-苄基 -3-羟基哌啶樟 脑磺酸盐(99%ee , 84.7 g, 0.2 mol), 二氯甲垸 423 ml, 滴加 IN氢氧化 钠水溶液 (210 ml), 室温搅拌 1小时, 分去水相, 有机相无水硫酸钠干 燥, 浓缩得 (R)-l-苄基 -3-羟基哌啶 38g。 (ee: 99%; LC-MS: m/e = 191.3) (理论: 38.25 g)。
步骤 3): (S)或 (R)-l-叔丁氧幾基 -3-羟基哌啶的合成
实施例 5: (S)-l-叔丁氧羰基 -3-羟基哌啶的合成
(1) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (一)
lOOOmL高压反应釜中, 加入实施例 3中 (1) 制备的 (S)-l-苄基 -3-羟 基哌啶 (38.25 g, 0.2 mol), 甲醇 200 ml, 加入 10%钯炭 (3.8 g), 以及 叔丁氧羰基酸酐(43.6g, 0.2 mol),氢气氛围,控制压力〜 5 atm, 〜40°C,
反应 15小时, HPLC原料((S)-l-苄基 -3-羟基哌啶) < 0.2%; 反应液冷却 至室温, 停止反应, 过滤, 回收钯炭, 滤液减压浓缩至干, 加乙酸乙酯 / 正己垸重结晶,析出固体,过滤;固体以少量正己垸洗涤;干燥得产品 (S)-l- 叔丁氧羰基 -3-羟基哌啶〜 36 g。 (理论: 40.2 g) (HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 )。
(2) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (二)
1000 mL高压反应釜中, 加入实施例 3中 (2) 制备 (S)-l-苄基 -3-羟基 哌啶 (38.25 g, 0.2 mol), 乙酸乙酯 200 ml, 加入 10%钯炭 (1.9 g), 以 及叔丁氧羰基酸酐 (48 g, 0.22 mol) , 氢气氛围, 控制压力〜 10 atm, 〜 50°C, 反应 15小时, HPLC原料((S)-l-苄基 -3-羟基哌啶) < 0.2%; 反应 液冷却至室温, 停止反应, 过滤, 回收钯炭, 滤液减压浓缩至 1/2, 加正 己垸重结晶, 析出固体, 过滤; 固体以少量正己垸洗涤; 干燥得产品 (S)-l- 叔丁氧羰基 -3-羟基哌啶〜 37 g, (理论: 40.2 g) (HPLC含量 〜99 %; ee: 99%; LC-MS: m/e = 201.3 )。
(3 ) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (三)
1000 mL高压反应釜中, 加入实施例 3中 (2) 制备 (S)-l-苄基 -3-羟基 哌啶 (38.25g, 0.2mol), THF 200ml, 加入 10%钯炭 (0.95g), 以及叔丁 氧羰基酸酐 (54.5g, 0.25mol), 氢气氛围, 控制压力〜 20atm, 〜40°C, 反应 15小时, HPLC原料((S)-l-苄基 -3-羟基哌啶) <0.2%; 反应液冷却 至室温, 停止反应, 过滤, 回收钯炭, 滤液减压浓缩至干, 加乙酸乙酯 / 正己垸重结晶,析出固体,过滤;固体以少量正己垸洗涤;干燥得产品 (S)-l- 叔丁氧羰基 -3-羟基哌啶〜 38g, (理论: 40.2g) (HPLC含量〜 99% ; ee: 99%; LC-MS: m/e=201.3 )。
实施例 6: (R)-l-叔丁氧羰基 -3-羟基哌啶的合成
1000 mL高压反应釜中, 加入实施例 4中制备的 (R)-l-苄基 -3-羟基哌 啶 (38.25 g, 0.2 mol), 甲醇 200 ml, 加入 10%钯炭 (0.95 g), 以及叔丁 氧羰基酸酐 (43.6 g, 0.2 mol), 氢气氛围, 控制压力〜 5 atm, 〜60°C, 反应 15小时, HPLC原料 ((R)-l-苄基 -3-羟基哌啶) < 0.2%; 反应液冷
却至室温, 停止反应, 过滤, 回收钯炭, 滤液减压浓缩至干, 加乙酸乙酯
/正己垸重结晶,析出固体,过滤;固体以少量正己垸洗涤;干燥得产品 (R)- 1 - 叔丁氧羰基 -3-羟基哌啶〜 36 g。 (理论: 40.2 g) (HPLC含量〜 99% ; ee: 99%; LC-MS : m/e = 201.3 )。
步骤 4) (R)或 (S) -1-取代 -3-羟基哌啶磺酸酯的合成
实施例 7:
( 1 ) (R)-l-叔丁氧羰基 -3-甲磺酸基哌啶的合成 (一)
500 ml三口瓶中, 加入实施例 6中制备的 (R)-l-叔丁氧羰基 -3-羟基哌 啶 (30.2 g, 0.15 mol) , 二氯甲垸 180 ml, 三乙胺 ( 18.2 g; 0.18 mol) , 控 制 0〜10°C, 滴加甲磺酰氯 ( 18.9 g, 0.165 mol) , 室温搅拌 1小时, 加水 淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓缩得 (R)-l-叔丁氧羰 基 -3-甲磺酸基哌啶 39.8g。 (yield: 95%; LC-MS : m/e = 279.1 ) (理论: 41.9 g
(2) (R)-l-叔丁氧羰基 -3-甲磺酸基哌啶的合成 (二)
500 ml三口瓶中, 加入实施例 6中制备的 (R)-l-叔丁氧羰基 -3-羟基哌 啶 (30.2 g, 0.15 mol) , 醋酸异丙酯 180 ml, 三乙胺 ( 18.2 g; 0.18 mol) , 控制 0〜10°C, 滴加甲磺酰氯 (18.9g, 0.165mol), 室温搅拌 1 小时, 加 水淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓缩得 (R)-l-叔丁氧 羰基 -3-甲磺酸基哌啶 39.8g。 (yield: 95%; LC-MS : m/e = 279.1 ) (理论: 41.9 g
实施例 8: (R)-l-苄基 -3-甲磺酸基哌啶的合成
500 ml 三口瓶中, 加入实施例 4 中制备的 (R)-l-苄基 -3-羟基哌啶 (28.7g, 0.15mol), 二氯甲垸 180 ml, 三乙胺(18.2g; 0.18mol),控制 0〜 10°C, 滴加甲磺酰氯 (18.9g, 0.165mol), 室温搅拌 1小时, 加水淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓缩得 (R)-l-苄基 -3-甲磺酸基 哌啶 37.2g。 (yield: 92%; LC-MS: m/e=269.4) (理论: 40.4g)。
实施例 9: (R)-l-叔丁氧羰基 -3-对硝基苯磺酸基哌啶的合成
500 ml三口瓶中, 加入实施例 6中制备的 (R)-l-叔丁氧羰基 -3-羟基哌 啶 (30.2 g, 0.15 mol), 二氯甲垸 180 ml, 三乙胺 (18.2 g; 0.18 mol), 控 制 0〜10°C, 滴加对硝基苯磺酰氯 (36.6 g, 0.165 mol) 的二氯甲垸溶液, 室温搅拌过夜, 加水淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓 缩得 (R)-l-叔丁氧羰基 -3-对硝基苯磺酸基哌啶 54.5 go(yield:94%;LC-MS: m/e = 386.4) (理论: 58.0 g)。
实施例 10: (R)-l-叔丁氧羰基 -3-对甲基苯磺酸基哌啶的合成
500 ml 三口瓶中, 加入实施例 6中制备的 (R)-l-叔丁氧羰基 -3-羟基 哌啶 (30.2 g, 0.15 mol), 二氯乙垸 180 ml, 三乙胺 (18.2 g; 0.18 mol), 室温滴加对甲基苯磺酰氯(31.4 g, 0.165 mol)的二氯乙垸溶液, 50〜60°C 反应过夜, 加水淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓缩得 (R)-l-叔丁氧羰基 -3-对甲基苯磺酸基哌啶 48.0 g。 (yield: 90%; LC-MS: m/e = 355.5) (理论: 53.3 g)。
实施例 11: (S)-l-叔丁氧羰基 -3-甲磺酸基哌啶的合成
500 ml三口瓶中, 加入实施例 5 (1) (2) (3) 中任一制备的 (S)-l-叔 丁氧羰基 -3-羟基哌啶(30.2 g, 0.15 mol), 二氯甲垸 180ml, 三乙胺(18.2 g; 0.18 mol), 控制 0〜10°C, 滴加甲磺酰氯 (18.9 g, 0.165 mol), 室温 搅拌 1小时, 加水淬灭, 分去水相, 有机相水洗, 无水硫酸钠干燥, 浓缩 得 (S)-l-叔丁氧羰基 -3-甲磺酸基哌啶 39.8g。 (yield: 95%; LC-MS: m/e = 279.1) (理论: 41.9 g)。
步骤 5)翻转得 (S)或 (R) -1-取代 -3-羟基哌啶羧酸酯
实施例 12: (S)-l-叔丁氧羰基 -3-乙酰氧基哌啶的合成
500 ml三口瓶中, 加入实施例 7中 (1) 或 (2) 制备的 (R)-l-叔丁氧 羰基 -3-甲磺酸基哌啶 (27.9 g, 0.1 mol), DMFlOOml, 醋酸钠 (16.4 g; 0.2 mol), 60°C反应过夜, 加水淬灭, 乙酸乙酯提取, 有机相水洗, 浓缩
得 (S)-l-叔丁氧羰基 -3-乙酰氧基哌啶粗品。 (yield: 〜100%; LC-MS: m/e = 243.3 ) (理论: 24.3 g) o
实施例 13:
( 1 ) (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶的合成 (一)
500 ml三口瓶中, 加入实施例 7中 (1 ) 或 (2) 制备的 (R)-l-叔丁氧 羰基 -3-甲磺酸基哌啶 (27.9 g, O.l mol) , DMF lOO ml, 苯甲酸钠 (28.8 g; 0.2 mol), 80°C反应过夜, 反应液冷却, 加水淬灭, 二氯甲垸提取, 有机 相水洗,浓缩得 (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶粗品。(yield:〜100%; LC-MS: m/e = 305.4) (理论: 30.5 g)。
(2) (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶的合成 (二)
500 ml三口瓶中, 加入实施例 9中制备的 (R)-l-叔丁氧羰基 -3-对硝基 苯磺酸基哌啶(38.6 g, 0.1 mol), DMF 100 ml,苯甲酸钠(28.8 g; 0.2 mol), 80°C反应过夜, 反应液冷却, 加水淬灭, 二氯甲垸提取, 有机相水洗, 浓 缩得 (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶粗品。 (yield: 〜100%; LC-MS: m/e = 305.4) (理论: 30.5 g)。
(3 ) (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶的合成 (三)
500 ml 三口瓶中, 加入实施例 10中制备的 (R)-l-叔丁氧羰基 -3-对甲 基苯磺酸基哌啶(35.5g, O.lmol), DMF lOO ml,苯甲酸钠(28.8g; 0.2mol), 90°C反应过夜, 反应液冷却, 加水淬灭, 二氯甲垸提取, 有机相水洗, 浓 缩得 (S)-l-叔丁氧羰基 -3-苯甲酰氧基哌啶粗品。 (yield: 〜100%; LC-MS: m/e = 305.4) (理论: 30.5g)。
实施例 14: (S)-l-苄基 -3-苯甲酰氧基哌啶的合成
500 ml 三口瓶中, 加入实施例 8中制备的 (R)-l-苄基 -3-甲磺酸基哌啶 (26.9 g, O.l mol), DMF lOO ml, 苯甲酸钠 (28.8 g; 0.2 mol), 90°C反 应过夜,反应液冷却,加水淬灭,二氯甲垸提取,有机相水洗,浓缩得 (S)-l- 苄基 -3-苯甲酰氧基哌啶粗品。 (yield: -100%; LC-MS: m/e = 295.4) (理 论: 29.5 g)。
实施例 15: (R)-l-叔丁氧羰基 -3-乙酰氧基哌啶的合成
500 ml 三口瓶中, 加入实施例 11 中制备的 (S)-l-叔丁氧羰基 -3-甲磺 酸基哌啶 (26.9 g, O.l mol) , DMF 100 ml, 乙酸钠 ( 16.4 g; 0.2 mol), 90°C反应过夜, 反应液冷却, 加水淬灭, 二氯甲垸提取, 有机相水洗, 浓 缩得 (R)-l-叔丁氧羰基 -3-乙酰氧基哌啶粗品。 (yield: 〜100%; LC-MS: m/e = 233.3 ) (理论: 23.3 g)。
步骤 6): 碱水解得 (S)或 (R) -l-取代 -3-羟基哌啶
实施例 16: (S)-l-苄基 -3-羟基哌啶的合成
500 ml三口瓶中, 加入实施例 14中制备的 (S)-l-苄基 -3-苯甲酰氧基 哌啶粗品(O.lmol), 甲醇 50ml; 水 50ml, 再加入氢氧化钠 6g (0.15mol), 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 得 产品 (S)-l-苄基 -3-羟基哌啶〜 18g。(HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 191.3 ) (理论: 19.1 g)。
实施例 17: (S)-l-叔丁氧羰基 -3-羟基哌啶的合成
( 1 ) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (四)
500 ml 三口瓶中, 加入实施例 12制备的 (S)-l-叔丁氧羰基 -3-乙酰氧 哌啶粗品(O.lmol), 甲醇 50ml; 水 50ml, 再加入氢氧化钠 6g (0.15mol), 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 乙 酸乙酯 /正己垸重结晶, 干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 16g。
(HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。
(2) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (五)
500 ml 三口瓶中, 加入实施例 12制备的 (S)-l-叔丁氧羰基 -3-乙酰氧 哌啶粗品 (0.1 mol),甲醇 50 ml;水 50 ml,再加入氢氧化钠 6 g (0.15 mol) , 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 乙 酸乙酯 /正己垸重结晶, 干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 16 g o (HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。
(3 ) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (六)
500 ml 三口瓶中, 加入实施例 12制备的 (S)-l-叔丁氧羰基 -3-乙酰氧 哌啶粗品(O.l mol), 甲醇 50 ml; 水 50 ml, 再加入氢氧化钾 9.1g (〜0.15 mol), 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓 缩, 乙酸乙酯 /正己垸重结晶, 干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 15 g。 (HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。
(4) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (七)
500 ml 三口瓶中, 加入实施例 12制备的 (S)-l-叔丁氧羰基 -3-乙酰氧 哌啶粗品(0.1 mol), THF 50 ml;水 50 ml,再加入氢氧化钠 6g (0.15 mol), 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 乙 酸乙酯 /正己垸重结晶, 干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 16 g o (HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。
(5 ) (S)-l-叔丁氧羰基 -3-羟基哌啶的合成 (八)
500 ml 三口瓶中, 加入实施例 12制备的 (S)-l-叔丁氧羰基 -3-乙酰氧 哌啶粗品 (O.lmol), 乙醇 100ml; 浓氨水 10ml, 室温反应过夜, 反应液 减压浓缩, 加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 乙酸乙酯 /正己 垸重结晶,干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 16g。 (HPLC含量〜 99%; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。
实施例 18: (R)-l-叔丁氧羰基 -3-羟基哌啶的合成
500 ml 三口瓶中, 加入实施例 15中制备的 (R)-l-叔丁氧羰基 -3-乙酰 氧基哌啶粗品(O.lmol),甲醇 50ml;水 50ml,再加入氢氧化钠 6g(0.15mol), 室温反应过夜, 反应液加水稀释, 二氯甲垸提取, 有机相水洗, 浓缩, 乙 酸乙酯 /正己垸重结晶, 干燥得产品 (R)-l-叔丁氧羰基 -3-羟基哌啶〜 16g。
(HPLC含量〜 99% ; ee: 99%; LC-MS: m/e = 201.3 ) (理论: 20.1 g)。 虽然本发明已以较佳实施例披露如上, 然其并非用以限定本发明, 任 何所属技术领域的技术人员, 在不脱离本发明之精神和范围内, 当可作些 许之更动与改进, 因此本发明之保护范围当视权利要求所界定者为准。
Claims
1. 一种手性 -1-叔丁氧羰基 -3-羟基哌啶的制备方法, 其特征在于, 包 含步骤:
1 ) 以式 I 的 N-苄基 -3-羟基哌啶为原料, 经手性樟脑磺酸 CSA的作 用下拆分得式 Π s的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐或式 II r 的 (R)-l-节 基 -3-羟基哌啶樟脑磺酸盐;
2)步骤 1 )所述的式 II s的 (S)-l-苄基 -3-羟基哌啶樟脑磺酸盐或式 II r 的 (R)-l-苄基 -3-羟基哌啶樟脑磺酸盐经碱游离,得式 III s的 (S) -1-苄基 -3- 羟基哌啶或式 Ill r的 (R)-l-苄基 -3-羟基哌啶;
3 )步骤 2)所述的式 III s的 (S) -1-苄基 -3-羟基哌啶或式 Ill r的 (R)-l- 苄基 -3-羟基哌啶经钯炭催化氢化脱苄, 同时用叔丁氧酸酐羰基保护得式 IVs的 (S)-l-叔丁氧羰基 -3-羟基哌啶或式 IV r的 (R) -1-叔丁氧羰基 -3-羟基 哌啶

III IV
2. 根据权利要求 1所述的方法, 其特征在于, 步骤 1 ) 中的拆分反应 的拆分剂为手性右旋樟脑磺酸 D-CSA或左旋樟脑磺酸 L-CSA, 所述手性 右旋樟脑磺酸 D-CSA或左旋樟脑磺酸 L-CSA与原料 N-苄基 -3-羟基哌啶 的摩尔比为 0〜0.6: 1。
3. 根据权利要求 1所述的方法, 其特征在于, 步骤 1 ) 中的拆分反应 的溶剂体系为: 如甲醇、 乙醇、 异丙醇的有机醇类溶剂、 如醋酸乙酯、 醋 酸异丙酯的有机酯类溶剂; 如 2—丁酮、丙酮的有机酮类溶剂; 如异丙醚, 甲基叔丁基醚的有机醚类溶剂; 或其中的任意组合。
4. 根据权利要求 1所述的方法, 其特征在于, 步骤 2 )所述的碱为氢 氧化钠、 氢氧化锂、 氢氧化钾、 碳酸钠、 碳酸氢钠、 碳酸钾或碳酸氢钾, 其中上述碱与原料 N-苄基 -3-羟基哌啶的摩尔比为 1〜10: 1。
5. 根据权利要求 1所述的方法, 其特征在于, 步骤 3 )反应的溶剂体 系为: 如甲醇、 乙醇、 异丙醇的有机醇类溶剂; 如醋酸乙酯、 醋酸异丙酯 的有机酯类溶剂; 如异丙醚, 甲基叔丁基醚的有机醚类溶剂; 或其中的任 意组合。
6. 根据权利要求 1所述的方法, 其特征在于, 步骤 3 ) 中钯炭与底物 式 III s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶的重量 比为 0.01〜0.5: 1;反应温度在 0°C〜80°C之间;所述的反应压力为 latm〜 30atm; 其中叔丁氧羰基酸酐与底物式 Ill s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶的摩尔比为 1〜2: 1。
7. 一种手性 -1-取代 -3-羟基哌啶的手性翻转方法, 其特征在于, 包含 步骤:
4 ) 以式 V r的 (R) -1-取代 -3-羟基哌啶或式 V s的 (S)-l-取代 -3-羟基哌 啶 (V) 为原料, 在取代磺酰氯的作用下酰化得式 VI r的 (R) -l-取代 -3-羟 基哌啶磺酸酯或 VI s的 (S) -1-取代 -3-羟基哌啶磺酸酯;
5 ) 步骤 4 ) 所述的式 VI r的 (R) -1-取代 -3-羟基哌啶磺酸酯或 VI s的 (S) -1-取代 -3-羟基哌啶磺酸酯被取代羧酸盐取代, 结构翻转得式 VII s的 (S) -1-取代 -3-羟基哌啶羧酸酯或式 VII r的 (R) -1-取代 -3-羟基哌啶羧酸酯;
6 )步骤 5 )所述的式 VII s的 (S) -1-取代 -3-羟基哌啶羧酸酯或式 VII r 的 (R) -1-取代 -3-羟基哌啶羧酸酯经碱水解得式 VIII s 的 (S) -1-取代 -3-羟基 哌啶或式 VIII r 的 (R) -l-取代 -3-羟基哌啶;

其中, R是苄基、 苄氧羰基或叔丁氧羰基;
Ri, 是( 1~ 3的低级垸烃, 取代苯基;
其中, 所述的手性 -1-取代 -3-羟基哌啶为权利要求 1的步骤 2)所述的 式 III s的 (S) -1-苄基 -3-羟基哌啶或式 III r的 (R)-l-苄基 -3-羟基哌啶;或权 利要求 1步骤 3 ) 所述的式 IVs的 (S)-l-叔丁氧羰基 -3-羟基哌啶或式 IV r 的 (R) -1-叔丁氧羰基 -3-羟基哌啶。
8. 根据权利要求 7所述的方法, 其特征在于, 步骤 4) 中所述的磺酰 氯为甲基磺酰氯、 对甲基苯磺酰氯、 对硝基苯磺酰氯或对溴苯磺酰氯; 酰 化反应的溶剂体系为: 如醋酸乙酯、 醋酸异丙酯的有机酯类溶剂: 如二氯 甲垸、 三氯甲垸、 二氯乙垸、 氯苯的有机氯代类溶剂: 如异丙醚, 甲基叔 丁基醚、 四氢呋喃的有机醚类溶剂。
9. 根据权利要求 7所述的方法, 其特征在于, 步骤 5 ) 中所述的羧酸 盐为醋酸钠或苯甲酸钠; 取代翻转反应的溶剂体系为: 如 DMF、 甲酰胺 的有机酰胺类溶剂; 如醋酸乙酯、 醋酸异丙酯的有机酯类溶剂; 如二氯甲 烧、 三氯甲垸、 二氯乙垸、 氯苯的有机氯代类溶剂; 如异丙醚、 甲基叔丁 基醚、 四氢呋喃的有机醚类溶剂; 如甲醇、 乙醇的有机醇类溶剂: 或包括 上述溶剂的一类或几类混合溶剂以及和水的混合溶剂体系。
10. 根据权利要求 7所述的方法, 其特征在于, 步骤 6) 中的水解反 应的碱为氢氧化钠、 氢氧化锂、 氢氧化钾; 碳酸钠、 碳酸钾或氨水; 其中 上述碱与步骤 4) 所述的原料的摩尔比为 1〜100: 1; 水解反应的溶剂为
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14884887.2A EP3115358B1 (en) | 2014-03-04 | 2014-03-04 | Method of preparation and chiral inversion of chiral-1-t-butoxycarbonyl-3-hydroxy piperidine |
| PCT/CN2014/000197 WO2015131299A1 (zh) | 2014-03-04 | 2014-03-04 | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/000197 WO2015131299A1 (zh) | 2014-03-04 | 2014-03-04 | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015131299A1 true WO2015131299A1 (zh) | 2015-09-11 |
Family
ID=54054316
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/000197 Ceased WO2015131299A1 (zh) | 2014-03-04 | 2014-03-04 | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP3115358B1 (zh) |
| WO (1) | WO2015131299A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104693108B (zh) * | 2013-12-06 | 2017-05-31 | 重庆博腾制药科技股份有限公司 | 一种布鲁顿酪氨酸激酶(Btk)中间体的制备方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104140390B (zh) * | 2014-07-16 | 2016-06-08 | 雅本化学股份有限公司 | 一种手性1-苄基-3-羟基哌啶的消旋回收方法 |
| CN113461598B (zh) * | 2021-07-28 | 2023-04-18 | 山东华素制药有限公司 | 哌啶醇化合物的制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4501748A (en) * | 1983-06-03 | 1985-02-26 | Kyowa Hakko Kogyo Co., Ltd. | 1,4-Dihydropyridine derivatives |
| CN1738793A (zh) * | 2003-01-14 | 2006-02-22 | 赛特凯恩蒂克公司 | 化合物、组合物及方法 |
| CN1809576A (zh) * | 2003-02-14 | 2006-07-26 | 辉瑞大药厂 | 抗寄生物萜类生物碱 |
| WO2012138678A1 (en) * | 2011-04-04 | 2012-10-11 | Merck Sharp & Dohme Corp. | Gamma secretase inhibitors |
| CN103864673A (zh) * | 2014-03-04 | 2014-06-18 | 雅本化学股份有限公司 | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE552236T1 (de) * | 2003-01-14 | 2012-04-15 | Cytokinetics Inc | Verbindungen, zusammensetzungen und verfahren zur behandlung von herzinsuffizienz |
-
2014
- 2014-03-04 WO PCT/CN2014/000197 patent/WO2015131299A1/zh not_active Ceased
- 2014-03-04 EP EP14884887.2A patent/EP3115358B1/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4501748A (en) * | 1983-06-03 | 1985-02-26 | Kyowa Hakko Kogyo Co., Ltd. | 1,4-Dihydropyridine derivatives |
| CN1738793A (zh) * | 2003-01-14 | 2006-02-22 | 赛特凯恩蒂克公司 | 化合物、组合物及方法 |
| CN1809576A (zh) * | 2003-02-14 | 2006-07-26 | 辉瑞大药厂 | 抗寄生物萜类生物碱 |
| WO2012138678A1 (en) * | 2011-04-04 | 2012-10-11 | Merck Sharp & Dohme Corp. | Gamma secretase inhibitors |
| CN103864673A (zh) * | 2014-03-04 | 2014-06-18 | 雅本化学股份有限公司 | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104693108B (zh) * | 2013-12-06 | 2017-05-31 | 重庆博腾制药科技股份有限公司 | 一种布鲁顿酪氨酸激酶(Btk)中间体的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3115358B1 (en) | 2019-09-11 |
| EP3115358A1 (en) | 2017-01-11 |
| EP3115358A4 (en) | 2018-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7100125B2 (ja) | リボシクリブおよびその塩の改善された調製のためのプロセス | |
| CN103880903B (zh) | 一种泰乐菌素类大环内酯及其衍生物的制备方法 | |
| CN109956870A (zh) | 一种罗沙司他的合成方法及其中间体化合物 | |
| CN112174798A (zh) | 一种沙库巴曲缬沙坦钠lcz696的合成方法 | |
| CN104910231B (zh) | 一种25-羟基-7-脱氢胆固醇的合成方法 | |
| CN114478690B (zh) | 一种6,6-二甲基-3-氮杂双环[3.1.0]己烷衍生物的制备方法 | |
| WO2015131299A1 (zh) | 手性-1-叔丁氧羰基-3-羟基哌啶的制备以及手性翻转的方法 | |
| CN107011404A (zh) | 一种以胆酸为原料合成石胆酸的方法 | |
| EP3413891B1 (en) | Processes for the preparation of highly pure prucalopride succinate | |
| CN111072660B (zh) | 一种瑞来巴坦的简便制备方法 | |
| JP5585822B2 (ja) | 光学活性ニペコチン酸誘導体の製造方法 | |
| NZ580271A (en) | Process for making galantamine | |
| CN110845512A (zh) | 杂萜类天然产物(+)-Arisugacins F/G的全合成方法 | |
| US20240336603A1 (en) | Process for the preparation of a cyp11a1 inhibitor and intermediates thereof | |
| CN116396290B (zh) | 一种制备莫西沙星中间体(s,s)-2,8-二氮杂双环[4,3,0]壬烷的方法 | |
| CN108440349B (zh) | 一种手性光学纯对甲苯亚磺酰胺的制备方法 | |
| CN107935909B (zh) | 一种尼达尼布(nintedanib)及其中间体的合成方法 | |
| CN113004245B (zh) | 一种地氯雷他定制备方法 | |
| CN104892491B (zh) | 一种合成帕罗西汀手性中间体的方法 | |
| WO2011161646A2 (en) | Process for the preparation of alvimopan or its pharmaceutically acceptable salt or solvate thereof | |
| JPH01190655A (ja) | 飽和脂肪族α,ω−ジアミノモノカルボン酸のN,ωトリフルオロアセチル化方法 | |
| CN103012264A (zh) | 3-取代氨基-六氢-1h-氮杂环庚烷的拆分方法 | |
| CN104876872A (zh) | 一种制备1-叔丁氧基羰基-3-羟甲基吲唑的方法及应用 | |
| CN103483286B (zh) | 樟脑衍生的噁唑烷-2-苯亚胺及其制备方法和用途 | |
| CN120623077A (zh) | 一种司帕生坦的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14884887 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014884887 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014884887 Country of ref document: EP |