WO2015166745A1 - 電着塗料組成物、電着塗料組成物用触媒 - Google Patents
電着塗料組成物、電着塗料組成物用触媒 Download PDFInfo
- Publication number
- WO2015166745A1 WO2015166745A1 PCT/JP2015/058532 JP2015058532W WO2015166745A1 WO 2015166745 A1 WO2015166745 A1 WO 2015166745A1 JP 2015058532 W JP2015058532 W JP 2015058532W WO 2015166745 A1 WO2015166745 A1 WO 2015166745A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- electrodeposition coating
- compound
- chemical formula
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/44—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2226—Anionic ligands, i.e. the overall ligand carries at least one formal negative charge
- B01J31/223—At least two oxygen atoms present in one at least bidentate or bridging ligand
- B01J31/2234—Beta-dicarbonyl ligands, e.g. acetylacetonates
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/44—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications
- C09D5/4488—Cathodic paints
- C09D5/4496—Cathodic paints characterised by the nature of the curing agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/18—Arsenic, antimony or bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2213—At least two complexing oxygen atoms present in an at least bidentate or bridging ligand
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D201/00—Coating compositions based on unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/08—Anti-corrosive paints
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/44—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications
- C09D5/4419—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications with polymers obtained otherwise than by polymerisation reactions only involving carbon-to-carbon unsaturated bonds
- C09D5/4465—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/44—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications
- C09D5/448—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes for electrophoretic applications characterised by the additives used
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/41—Organic pigments; Organic dyes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
- C09D7/63—Additives non-macromolecular organic
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/005—General concepts, e.g. reviews, relating to methods of using catalyst systems, the concept being defined by a common method or theory, e.g. microwave heating or multiple stereoselectivity
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/50—Complexes comprising metals of Group V (VA or VB) as the central metal
- B01J2531/54—Bismuth
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0025—Crosslinking or vulcanising agents; including accelerators
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0091—Complexes with metal-heteroatom-bonds
Definitions
- the present invention includes an organic tin-free electrodeposition coating composition that does not contain an organic tin compound and can ensure good coating curability under the same baking conditions as the present, and is contained in this composition.
- the present invention relates to a catalyst that promotes a crosslinking reaction.
- Electrodeposition coating is a primer coating for parts with a bag structure such as automobiles, electrical appliances, etc., because it has better throwing power and less environmental pollution than air spray coating and electrostatic spray coating. As a result, it has been widely put into practical use.
- cationic electrodeposition coating can be applied continuously, and is therefore widely used as a method for undercoating a large article such as an automobile body that requires high corrosion resistance.
- Cationic electrodeposition coating generally uses a coating component as a cathode in a cationic electrodeposition coating composition in which a binder component containing a cationic resin and a curing agent is dispersed in an aqueous medium containing a neutralizing agent such as an organic acid. It is performed by immersing and applying a voltage.
- an electrodeposition coating film is deposited on the surface of the cathode (substrate) due to an electrochemical reaction. Since the electrodeposition coating film thus formed contains a curing agent together with a cationic resin, the coating film is cured by baking the coating film after completion of electrodeposition coating, and a desired cured coating film is formed. Is done.
- cationic resin used in the cationic electrodeposition coating composition from the viewpoint of corrosion resistance, an amine-modified epoxy resin is used, and as the curing agent, a polyisocyanate blocked with a blocking agent such as alcohol is used. It has been.
- organotin compounds can cause deodorization catalyst poisoning in baking furnaces in painting lines, and future use may be restricted due to recent environmental regulations for organotin compounds.
- Development of a cationic electrodeposition coating composition that uses an alternative catalyst has been strongly desired.
- Cationic electrodeposition coating compositions using zinc borate, quaternary ammonium organic acid salts, zinc compounds and the like have been proposed as alternative catalysts for the organic tin compounds.
- Patent Documents 1 to 3 Cationic electrodeposition coating compositions containing metal chelate compounds, tetravalent organic titanium / zirconium / hafnium complexes having oxygen-containing ligands, and fluoro metal ions such as zirconium / titanium have been proposed.
- Patent Documents 4 to 6 However, these compounds alone need to be used in combination with an organic tin compound because of insufficient film curability.
- Patent Document 7 a bismuth acetylacetone complex has also been proposed (Patent Document 7).
- Patent Document 7 since the storage stability in the dispersion paste is poor, the catalyst curing performance is greatly reduced when stored for a certain period of time, or the gel is thickened during storage. There was a problem that caused.
- a cationic electrodeposition coating composition that does not contain an organic tin compound and that can ensure good coating curability under the same baking conditions as the current one.
- JP 7-331130 A Japanese Patent Laid-Open No. 11-152432 JP 2000-336287 A JP-A-2-265974 Special table 2011-513525 gazette JP 2006-257268 A JP 2002-129100 A
- the present invention has been made in view of the above circumstances, and does not contain an organic tin compound, and an organic tin-free cationic electrodeposition coating composition that can ensure good coating curability under the same baking conditions as the current one. And a catalyst for the composition.
- a catalyst for an electrodeposition coating composition containing a bismuth compound (A),
- the catalyst for an electrodeposition coating composition wherein the bismuth compound is a compound having a ligand prepared from a ⁇ -diketone represented by the following chemical formula (1), is provided.
- Chemical formula (1) (In the formula, R 1 s are the same or different from each other and represent a hydrocarbon group, and the total number of carbon atoms of the two R 1 s is 4 or more.)
- the present inventors have evaluated the catalytic performance of many substances. As a result, the present inventors have found that the bismuth compound (A) has very excellent characteristics, and have completed the present invention. .
- the present inventors produced bismuth compounds (A) from various ⁇ diketones in which the total number of carbon atoms of two R 1 in the chemical formula (1) is 4 or more, and their catalytic activity. As a result, excellent results were obtained. Therefore, in order to investigate the stability of the bismuth compound (A) in an aqueous medium, the catalyst paste was stored at 35 ° C. for 30 days and again electrodeposited to confirm the catalytic activity. As shown in Table 3, the catalyst activity was excellent. It turns out that a result is obtained.
- the bismuth acetylacetone complex of the prior art is a compound obtained from acetylacetone, which is a ⁇ -diketone in which the total number of carbon atoms of two R 1 in chemical formula (1) is 2.
- Comparative Examples 1 to 4 when the catalytic activity was investigated by performing electrodeposition coating using a catalyst paste in which the catalyst paste was stored at 35 ° C. for 30 days because of poor stability in the aqueous medium, It did not show good results.
- a cationic electrodeposition coating composition excellent in curability, anticorrosiveness, and finishability equivalent to or higher than that in the case where it is blended without using an organic tin compound, and a catalyst for this composition Can be provided.
- the electrodeposition coating composition of the present invention contains a catalyst for an electrodeposition coating composition containing a bismuth compound (A) and a base resin (B).
- the catalyst for an electrodeposition coating composition of the present invention contains a bismuth compound (A).
- the bismuth compound (A) is a compound having a ligand prepared from a ⁇ diketone represented by the following chemical formula (1).
- R 1 s are the same or different from each other and represent a hydrocarbon group, and the total carbon number of R 1 is 4 or more.
- Examples of the hydrocarbon group represented by R 1 include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, a t-butyl group, a pentyl group, a hexyl group, a cyclohexyl group, a heptyl group, and an octyl group.
- Saturated hydrocarbon groups such as dodecanyl group and octadecanyl group, unsaturated groups such as vinyl group, allyl group, prenyl group, crotyl group, cyclopentadienyl group, phenyl group, tolyl group, xylyl group, and substituted aryl group
- a hydrocarbon group is mentioned.
- a hydrocarbon group having 1 to 12 carbon atoms is preferable, and a methyl group, a phenyl group, and an aryl group having a substituent are particularly preferable.
- the combination of R 1 in which the total carbon number of two R 1 is 4 or more includes a combination of a methyl group and a hydrocarbon group having 3 or more carbon atoms, a hydrocarbon group having 2 or more carbon atoms and a carbon number of 2 or more.
- Examples include a combination of two or more hydrocarbon groups. Specifically, for example, ethyl group and ethyl group, methyl group and t-butyl group, methyl group and phenyl group, phenyl group and phenyl group, 4-t-butyl group Examples thereof include a phenyl group and a 4-methoxyphenyl group.
- the bismuth compound (A) is preferably at least one bismuth compound having a group represented by the following chemical formula (4).
- the bismuth compound (A) is more preferably at least one bismuth compound represented by the following chemical formula (5).
- the bismuth compound (A) is more preferably a kind of bismuth compound represented by the following chemical formula (2) or chemical formula (3).
- Chemical formula (2) (Wherein R 1 is the same as in chemical formula (1), R 2 is a hydrogen atom, an alkyl group, an acyl group, —Bi— (O—R 2 ) 2 , or two R 2 is substituted with a Bi atom to connect two oxygen atoms, and a represents an integer of 1, 2 or 3.
- Chemical formula (3) (In the formula, R 1 is the same as chemical formula (1). R 2 is the same as chemical formula (2). B and c represent an integer of 1 or more.)
- Examples of the alkyl group represented by R 2 or R 3 include a methyl group, an ethyl group, a butyl group, an octyl group, a 2-ethylhexyl group, and an octadecanyl group, and those having 1 to 18 carbon atoms are preferable.
- Examples of the acyl group represented by R 2 or R 3 include, for example, an acetyl group, a glucoroyl group, a propionyl group, a lactoyl group, a butyryl group, a benzoyl group, a substituted aromatic acyl group, an octanoyl group, and 2-ethylhexanoyl.
- Bi atom stearoyl group and the like, and those having 1 to 18 carbon atoms are preferable. Moreover, it may couple
- dibasic acid acyl groups such as a malonyl group, a succinyl group, and an adipoloyl group
- the -Bi- (O-R 2) 2 represented by R 2 for example, via a bismuth atom, a hydroxyl group, an alkoxy group include those having an acyloxy group.
- two R 2 may be substituted with a Bi atom and bridged or cyclized by connecting two oxygen atoms.
- the number of bismuth atoms in the bismuth compound (A) is not particularly limited, but is, for example, 1 to 1000, preferably 1 to 200, and more preferably 1 to 50.
- the method for producing the bismuth compound (A) is not particularly limited.
- the bismuth compound (A) can be produced by reacting the ⁇ diketone represented by the chemical formula (1) with bismuth alkoxide or bismuth oxide.
- the bismuth compound (A) can be produced by the following method.
- bismuth trialkoxide and the ⁇ -diketone are reacted in a molar ratio of 0.1 to 10, preferably 0.2 to 3.3.
- the bismuth trialkoxide include bismuth trimethoxide, bismuth triethoxide, bismuth triisopropoxide, bismuth tributoxide, and the like.
- the reaction solvent is not particularly limited.
- alcohol solvents such as methanol, ethanol, isopropanol and butanol
- hydrocarbon solvents such as hexane, heptane, cyclohexane, toluene and xylene
- ethers such as diethyl ether, dibutyl ether and tetrahydrofuran
- Solvents such as solvents, ketone solvents such as acetone, methyl ethyl ketone, and methyl isobutyl ketone
- ester solvents such as ethyl acetate and butyl acetate
- amide solvents such as DMF
- sulfoxide solvents such as DMSO
- mixed solvents thereof can be mentioned.
- the reaction temperature is not particularly limited, but the reaction is usually carried out in the range of 0 to 200 ° C. Alcohol produced in the reaction system may be distilled out of the reaction system. Further, water or carboxylic acid may be added to the reaction solution. The reaction solution thus obtained can be concentrated to obtain the bismuth compound (A). If necessary, it can be purified by distillation, sublimation, washing with a solvent, reprecipitation, recrystallization and the like.
- the bismuth compound (A) may be a substantially single bismuth compound or a mixture of a plurality of bismuth compounds depending on the reaction conditions. A specific compound may be isolated from such a mixture and used as a catalyst, or the mixture may be used as a catalyst as it is. In either case, it functions as a catalyst.
- the content of the bismuth compound (A) in the electrodeposition coating composition of the present invention is not particularly limited, but usually the total solid content of the base resin (B) and the curing agent (C) in the electrodeposition coating composition is 100.
- the amount is 0.2 to 10 parts by mass, preferably 0.4 to 4.0 parts by mass with respect to parts by mass. Even if the addition amount is out of the above range, there is no particular problem in the paint performance. However, if it is within the range of 0.2 to 10 parts by mass, the curability, corrosion resistance, stability of the electrodeposition paint, etc. Practical balance is improved.
- a curing agent (C), a metal compound (D), a neutralizing agent (E) can be blended.
- Base resin (B) any resin such as epoxy, acrylic, polybutadiene, alkyd, and polyester can be used by introducing a cationic group. Of these, an epoxy-modified resin having a cationic group is preferable.
- the epoxy-modified resin includes an epoxy ring of an epoxy resin as a starting material, an amine such as a primary amine and a secondary amine, a quaternary ammonium salt which is a reaction product of a tertiary amine and an acid, a sulfide and an acid. Ring-opened by reaction with a mixture of
- the “cationic group” in the present specification means one that itself is a cation and one that becomes a cation by adding an acid.
- the polyepoxide compound used in the production of the epoxy-modified resin is a compound having at least two epoxy groups in one molecule, and is generally at least 200, preferably 400 to 4000, more preferably 800 to 3000. Those having a number average molecular weight are suitable, and those obtained by reaction of a polyphenol compound and epichlorohydrin are particularly preferred.
- Examples of the polyphenol compound that can be used for forming the polyepoxide compound include 2,2-bis (4-hydroxyphenyl) propane, 1,1-bis (4-hydroxyphenyl) ethane, and 1,1-bis (4 -Hydroxyphenyl) isobutane, 2,2-bis (4-hydroxy-t-butylphenyl) propane, 4,4-dihydroxybenzophenone, bis (2,4-dihydroxyphenyl) methane, bis (2-hydroxynaphthyl) methane, Examples thereof include 1,5-dihydroxynaphthalene, 4,4-dihydroxydiphenyl sulfone, phenol novolak, cresol novolak and the like.
- the polyepoxide compound may be partially reacted with polyol, polyether polyol, polyester polyol, polyamidoamine, polycarboxylic acid, polyisocyanate compound, or the like. Further, the polyepoxide compound may be further obtained by graft polymerization of ⁇ -caprolactone, an acrylic monomer or the like.
- Examples of amines used for opening an epoxy ring and introducing an amino group include primary amines such as butylamine, octylamine, monoethanolamine, 2- (2-aminoethoxy) ethanol, diethylamine, and diamine. Secondary amines such as butylamine, methylbutylamine, diethanolamine, N-methylethanolamine, polyamines such as ethylenediamine, diethylenetriamine, ethylaminoethylamine, methylaminopropylamine, N, N-dimethylaminopropylamine, aminoethylethanolamine methylisobutylketimine Ketimine block primary amino group-containing secondary amines such as can also be used.
- primary amines such as butylamine, octylamine, monoethanolamine, 2- (2-aminoethoxy) ethanol, diethylamine, and diamine.
- Secondary amines such as butylamine, methylbutylamine, diethanol
- quaternary ammonium salt which is a reaction product of a tertiary amine and an acid such as N, N-dimethylethanolamine, N-methyldiethanolamine, triethanolamine, or triethylamine can be used for opening the epoxy ring.
- Examples of opening an epoxy ring by reaction with a mixture of sulfide and acid include diethyl sulfide, dipropyl sulfide, dibutyl sulfide, diphenyl sulfide, ethylphenyl sulfide, tetramethylene sulfide, thiodiethanol, thiodipropanol, thiodibutanol Examples include 1- (2-hydroxyethylthio) -2-propanol, 1- (2-hydroxyethylthio) -2,3-propanediol, and 1- (2-hydroxyethylthio) -2-butanol.
- Examples of the acid used above include formic acid, acetic acid, propionic acid, lactic acid, dimethylolpropionic acid, sulfamic acid and the like.
- the base resin (B) may be of any type of external crosslinking type and internal (or self) crosslinking type. Since the cross-linking reaction requires a cross-linked part and an active hydrogen-containing part (eg, amino group, hydroxyl group) that reacts with the cross-linked part, both the cross-linked part and the active hydrogen-containing part are included in the base resin (B). When the base resin (B) contains only one of them, it becomes the external cross-linking type.
- an active hydrogen-containing part eg, amino group, hydroxyl group
- Examples of the internal cross-linking type include those in which a blocked isocyanate group or the like is introduced into the molecule of the base resin (B).
- a method for introducing the blocked isocyanate group into the base resin (B) a known method can be used.
- a free isocyanate group in the partially blocked polyisocyanate compound and an active hydrogen-containing part in the base resin are used. It can be introduced by reacting.
- the curing agent (C) used in combination is a crosslinking agent having a crosslinking part (eg, a block polyisocyanate compound) or a compound having an active hydrogen-containing part ( Examples: Resins containing amino groups, hydroxyl groups, etc.). More specifically, when the base resin (B) contains an active hydrogen-containing part, it is preferable to use a crosslinking agent as the curing agent, and when the base resin (B) contains a cross-linking part. It is preferable to use a compound having an active hydrogen-containing part as a curing agent.
- the block polyisocyanate compound can be obtained by addition reaction of a theoretical amount of a polyisocyanate compound and an isocyanate blocking agent.
- polyisocyanate compound examples include aromatics such as tolylene diisocyanate, xylylene diisocyanate, phenylene diisocyanate, bis (isocyanate methyl) cyclohexane, tetramethylene diisocyanate, hexamethylene diisocyanate, methylene diisocyanate, isophorone diisocyanate, and polymethylene polyphenyl polyisocyanate.
- terminal polyisocyanate compounds obtained by reacting low molecular weight active hydrogen-containing compounds such as ethylene glycol, propylene glycol, trimethylolpropane, hexanetriol, castor oil with an excess amount of these aliphatic isocyanate compounds A compound can be mentioned.
- the isocyanate blocking agent it is blocked by adding to the isocyanate group of the polyisocyanate compound, and the blocked polyisocyanate compound produced by the addition is stable at room temperature and when heated to about 100 to 200 ° C., It is desirable to be able to dissociate and regenerate free isocyanate groups.
- the blocking agent examples include halogenated hydrocarbons such as 1-chloro-2-propanol and ethylene chlorohydrin, heterocyclic alcohols such as furfuryl alcohol and alkyl group-substituted furfuryl alcohol, phenol, m-cresol, phenols such as p-nitrophenol, p-chlorophenol, nonylphenol, oximes such as methyl ethyl ketoxime, methyl isobutyl ketone oxime, acetone oxime, cyclohexanone oxime, active methylene compounds such as acetylacetone, ethyl acetoacetate, diethyl malonate, Lactams such as ⁇ -caprolactam, aliphatic alcohols such as methanol, ethanol, n-propanol, isopropanol and 2-ethylhexanol, and aromatic alcohols such as benzyl alcohol , Ethylene glycol monomethyl ether,
- the blocking agent dissociation temperature of alcohols and glycol ethers is higher than the dissociation temperature of oximes, active methylene compounds, and lactams.
- alcohols and glycol ethers are cheaper than other blocking agents, they are generally used in fields requiring large economy such as automobile bodies.
- the solid content mass ratio of the base resin (B) / curing agent (C) is preferably 20/80 to 90/10, more preferably 30/70 to 80/20.
- Metal compound (D) examples include at least one compound selected from the group consisting of titanium, zinc, iron, magnesium, aluminum, and calcium.
- a titanium compound the compound as described in WO2013 / 125562, WO2013 / 137174 is mentioned, for example.
- the titanium compound may be added alone to the electrodeposition coating composition of the present invention, or a composite catalyst of the bismuth compound (A) and the titanium compound is prepared by the method described in the above patent, and the composite catalyst is used for the electrolysis.
- a coating composition may be prepared.
- Examples of the zinc compound include unsubstituted aliphatic and aromatic zinc carboxylates having various substituents such as zinc acetate, zinc lactate, zinc dimethylolpropionate and zinc benzoate, 2,4-pentanedione, 1 -Zinc chelate complexes having ligands prepared from 1,3-dicarbonyl compounds such as phenyl-1,3-butanedione and 1,3-diphenyl-1,3-propanedione.
- iron compound examples include carboxylates such as tris (2-ethylhexanoic acid) iron (III), 2,4-pentanedione, 1-phenyl-1,3-butanedione, 1,3-diphenyl-1, And iron chelate complexes having ligands prepared from 1,3-dicarbonyl compounds such as 3-propanedione.
- magnesium compound examples include carboxylates such as magnesium acetate and magnesium lactate, 1,4-pentanedione, 1-phenyl-1,3-butanedione, 1,3-diphenyl-1,3-propanedione and the like.
- a magnesium chelate complex having a ligand prepared from a 3-dicarbonyl compound examples include 1,3 such as phosphates such as aluminum polyphosphate, 2,4-pentanedione, 1-phenyl-1,3-butanedione, 1,3-diphenyl-1,3-propanedione, and the like.
- the calcium compound include carboxylates such as calcium acetate, 1,3-pentanedione, 1-phenyl-1,3-butanedione, 1,3-diphenyl-1,3-propanedione, and the like.
- Examples include calcium chelate complexes having a ligand prepared from a dicarbonyl compound.
- the content of the metal compound (D) in the electrodeposition coating composition of the present invention is not particularly limited, but is usually 5 to 300 parts by mass, preferably 10 to 100 parts per 100 parts by mass of the bismuth compound (A). Part by mass. When it is in the above range, the effect of improving the physical properties of the electrodeposition paint can be obtained.
- the electrodeposition coating composition of the present invention may further include a neutralizing agent (E) for dispersing the above components in water.
- a neutralizing agent (E) for dispersing the above components in water.
- the neutralizing agent (E) include aliphatic carboxylic acids such as formic acid, acetic acid, propionic acid, and lactic acid, sulfamic acid, and the like.
- the amount of the neutralizing agent (E) varies depending on the amount of amino groups in the base resin (B), and may be any amount that can be dispersed in water.
- the pH of the electrodeposition paint is 3.0 to 9. What is necessary is just to be an amount kept in the range of 0.
- the equivalent number of the neutralizing agent (E) necessary for neutralizing the amino group contained in the base resin (B) is 0.25 to 1.5, preferably 0.5 to 1.25. It is. When it is in the above range, the effect of improving the finish, throwing power, low temperature curability and the like of the composition can be obtained.
- the electrodeposition coating composition of the present invention can be produced by mixing the above components all at once, but can also be produced by the following method. For example, first, the base resin (B) and the curing agent (C) are mixed, and the neutralizing agent (E) is added. An emulsion is produced by dispersing the mixture of the base resin (B), the curing agent (C), and the neutralizing agent (E) in an aqueous medium that is water alone or a mixture of water and a hydrophilic organic solvent.
- the base resin (B) and the curing agent (C) are mixed, and the base is added to an aqueous solution to which the neutralizing agent (E) is added or a mixed solution of water to which the neutralizing agent (E) is added and a hydrophilic organic solvent.
- An emulsion is produced by dispersing a mixture of the resin (B) and the curing agent (C).
- a predetermined amount of the above bismuth compound (A), metal compound (D), other additives, pigment, pigment dispersant, etc. is added to the previously prepared base resin (B) solution for cationic pigment catalyst dispersion and mixed.
- a pigment catalyst dispersion paste is produced by dispersing well until the solid in the mixture becomes a certain particle size or less using a normal dispersing device such as a ball mill or a sand mill. Finally, the emulsion and a predetermined amount of the pigment catalyst dispersion paste are mixed well to produce an electrodeposition coating composition.
- the electrodeposition coating composition of the present invention can be applied to a desired substrate surface by electrodeposition coating.
- electrodeposition coating is diluted with deionized water or the like so that the solid content concentration is about 5 to 40% by mass, and the pH is adjusted within the range of 3.0 to 9.0.
- An electrodeposition bath composed of a coating composition can be usually adjusted to a bath temperature of 15 to 45 ° C. and under a load voltage of 100 to 400V.
- the film thickness of the electrodeposition coating film that can be formed using the electrodeposition coating composition of the present invention is not particularly limited, but is generally 5 to 40 ⁇ m, particularly 10 to 10 ⁇ m based on the cured coating film. Within the range of 30 ⁇ m is preferable.
- the baking temperature of the coating film is generally in the range of 100 to 200 ° C., preferably 140 to 180 ° C. on the surface of the object to be coated, and the baking time is 5 to 60 minutes, preferably about 10 to 30 minutes. It is preferable that the surface of the object to be coated is held.
- Parts and % indicate “parts by mass” and “% by mass”.
- the reaction solution was transferred to a 1 L eggplant flask and concentrated under reduced pressure while heating in a 50 ° C. hot water bath to obtain 62 g of a concentrated solution. While stirring, 40 g of heptane was added dropwise to the flask containing the concentrated solution over 10 minutes, followed by stirring for 10 minutes. The obtained slurry solution was filtered by suction to obtain a pale yellow wet solid, dried under reduced pressure (decompression degree 10-20 mmHg) for 4 hours while heating in a hot water bath at 60 ° C., and 18.2 g of a dried pale yellow solid was obtained. . Bi content analysis value by EDTA titration of the obtained pale yellow solid bismuth compound (A) was 65.2%.
- Ion-exchanged water 3.6 g (0.20 mol) was added dropwise over 5 minutes at an internal temperature range of 20 to 30 ° C., and the mixture was stirred for 1 hour at the same temperature range.
- the reaction solution was transferred to a 1 L eggplant flask and concentrated under reduced pressure while heating in a 50 ° C. hot water bath to obtain 57 g of a concentrated solution.
- a 3 L 4-neck round bottom flask equipped with a stirrer, a thermometer and a condenser was charged with 700 g of heptane, and the above concentrated solution was added dropwise over 30 minutes at an internal temperature of 20 to 30 ° C. with stirring. I washed it in. The mixture was stirred for 1 hour in the same temperature range.
- the obtained slurry solution was subjected to suction filtration to obtain a pale yellow wet solid, which was dried under reduced pressure (decompression degree 10 to 20 mmHg) for 4 hours while heating in a 60 ° C. hot water bath to obtain 36.0 g of a dried pale yellow solid.
- Bi content analysis value by EDTA titration of the obtained pale yellow solid bismuth compound (A) was 56.3%.
- the concentrated solution was transferred to a 500 mL eggplant flask and concentrated under reduced pressure while heating in a 60 ° C. hot water bath to obtain 51 g of a concentrated solution, 50 g of heptane was added to precipitate a yellow solid, and concentrated under reduced pressure while heating in a 60 ° C. hot water bath.
- the solid was dried (reduced pressure: 10 to 20 mmHg) for 6 hours to obtain 43.7 g of a dried yellow solid.
- the Bi content analysis value of the obtained yellow solid bismuth compound (A) by EDTA titration was 23.8%.
- the liquid temperature was cooled to 20 ° C. while stirring, and 4.5 g (0.25 mol) of ion-exchanged water was added dropwise over 5 minutes within the range of the internal temperature of 20-30 ° C., and stirring was continued for 1 hour at the same temperature range. did.
- the reaction solution was transferred to a 500 mL eggplant flask and concentrated under reduced pressure while heating in a 60 ° C. hot water bath to obtain 60 g of a concentrated solution.
- a 3 L 4-neck round bottom flask equipped with a stirrer, a thermometer and a condenser was charged with 700 g of heptane, and the above concentrated solution was added dropwise over 30 minutes at an internal temperature of 20 to 30 ° C. with stirring.
- the obtained yellow solid bismuth compound (A) had a Bi content analysis value by EDTA titration of 52.5%.
- Production Example 8 A bismuth compound (A) was obtained in the same manner as in Production Example 7, except that the amount of 1,3-diphenyl-1,3-propanedione and water charged and the reaction time were changed. The results are shown in Table 1.
- the solid was transferred to a 300 mL eggplant flask, 100 g of heptane was added and stirred for 15 minutes, and the slurry was suction filtered to obtain a yellow wet solid, which was dried under reduced pressure (decompression degree 10-20 mmHg) for 4 hours while heating in a 60 ° C. hot water bath. 13.8 g of a dry pale yellow solid was obtained.
- the Bi content analysis value of the obtained yellow solid bismuth compound (A) by EDTA titration was 73.5%.
- the mixture was heated in an oil bath with stirring, and the solvent was distilled off at normal pressure until the internal temperature reached 100 ° C. While stirring, the liquid temperature was cooled to 20 ° C., and 2.0 g (0.11 mol) of ion-exchanged water was added dropwise over 1 minute in the range of 20-30 ° C., and stirring was continued for 3 hours in the same temperature range. did.
- the reaction solution was transferred to a 500 mL eggplant flask and concentrated to dryness under reduced pressure (pressure reduction 10-20 mmHg) for 8 hours while heating in a 60 ° C. hot water bath to obtain 26.5 g of a dried yellow solid.
- the yellow solid bismuth compound (A) obtained had an Bi content analysis value by EDTA titration of 40.1%.
- the reaction solution was transferred to a 500 mL eggplant flask and concentrated under reduced pressure while heating in a 50 ° C. hot water bath to obtain a concentrated solution 41 g.
- Heptane: 50 g was added, and the solution was concentrated under reduced pressure while heating in a 60 ° C. hot water bath. 10 to 20 mmHg) for 6 hours to obtain 24.2 g of a dry pale yellow solid.
- the Bi content analysis value of the obtained yellow solid bismuth compound (A) by EDTA titration was 43.0%.
- Ion-exchanged water 5.4 g (0.30 mol) was added dropwise over 5 minutes at an internal temperature of 20 to 30 ° C., and the mixture was stirred for 1 hour at the same temperature range.
- the reaction solution was transferred to a 1 L eggplant flask and concentrated under reduced pressure while heating in a 50 ° C. hot water bath to obtain 62 g of a concentrated solution.
- 300 g of heptane was added dropwise over 30 minutes with stirring, and the mixture was stirred for 10 minutes.
- the obtained slurry solution was subjected to suction filtration to obtain a pale yellow wet solid, which was dried under reduced pressure (decompression degree: 10 to 20 mmHg) for 4 hours while being heated in a hot water bath at 60 ° C. to obtain 29.6 g of a dried pale yellow solid. .
- the Bi content analysis value of the obtained pale yellow solid bismuth compound (A) by EDTA titration was 70.0%.
- the hydroxyl value of the resin solid content obtained by measuring the hydroxyl value of B-1 and subtracting and correcting the hydroxyl value of the solvent butyl cellosolve was 199 mgKOH / g (OH group conversion: 3.55 mmol / g).
- the calculated amine content was 0.63 mmol / g.
- Quaternary ammonium salt type for pigment catalyst dispersion >> Tolylene diisocyanate (hereinafter referred to as TDI): 420.6 g (2.41 mol), methyl isobutyl ketone (hereinafter referred to as MIBK) in a 2 L four-necked flask equipped with a stirrer, a thermometer and a condenser in a nitrogen atmosphere. ): 82 g was charged and heated with stirring to raise the internal temperature to 50 ° C. Butyl cellosolve: 285.4 g (2.41 mol) was added dropwise over 4 hours while maintaining the internal temperature at 50 to 55 ° C. The mixture was reacted at 50 ° C.
- TDI Tolylene diisocyanate
- MIBK methyl isobutyl ketone
- butyl cellosolv half-blocked TDI 788 g.
- a 1 L four-necked flask equipped with a stirrer, a thermometer, and a condenser was charged with 106.0 g (1.19 mol) of dimethylethanolamine, and the above-mentioned butyl cellosolv half-blocked TDI: 394 g (with stirring) 1.21 mol) was added dropwise over a period of 2 hours at an internal temperature of 25 to 50 ° C. Then, it heated up to 80 degreeC and stirred for 1 hour.
- aqueous lactic acid solution 142 g (1.19 mol) was added dropwise in the range of 75 to 85 ° C. over 1 hour, butyl cellosolve: 88 g was added, and the mixture was stirred at 65 to 70 ° C. for 3 hours. To obtain 730 g of a quaternizing agent.
- Epoxy resin “jER1001AF” (manufactured by Mitsubishi Chemical Corporation, epoxy equivalent 468 g / eq, average molecular weight of about 900): 378 g (epoxy group equivalent 0) in a 3 L four-necked flask equipped with a stirrer, thermometer, and cooler under nitrogen atmosphere .81 mol) was charged and heated and stirred in an oil bath at 120 ° C. to melt the resin. While stirring, 394 g of the butyl cellosolv half-blocked TDI was added dropwise over 2 hours at an internal temperature range of 117 to 123 ° C. After reacting for 2 hours in the same temperature range, it was cooled to 90 ° C.
- an epoxy resin “jER1001AF” manufactured by Mitsubishi Chemical Corporation, epoxy equivalent 468 g / eq, average molecular weight of about 900): 234.5 g (epoxy group) 0.50 mol
- jER1001AF manufactured by Mitsubishi Chemical Corporation, epoxy equivalent 468 g / eq, average molecular weight of about 900
- 234.5 g (epoxy group) 0.50 mol was charged, and the resin was melted by heating and stirring in an oil bath at 140 ° C.
- 190 g (0.50 mol) of the butyl cellosolv half-blocked IPDI was added dropwise over 1 hour at an internal temperature range of 137 to 143 ° C.
- 100 g of butyl cellosolve was added and cooled to 70 ° C.
- Titanium Compound D1 >> A titanium compound D1 having a ligand prepared from 1-phenyl-1,3-butanedione by the method described in Production Example 3 of WO2013 / 125562 was obtained.
- Titanium Compound D2 >> A titanium compound D2 having a ligand prepared from 1,3-diphenyl-1,3-propanedione was obtained by the method described in Production Example 5 of WO2013 / 125562.
- Pigment catalyst dispersion quaternary ammonium salt resin solution B-2 obtained in Production Example 12 (solid content 60%): 250 g, ion-exchanged water: 594 g, “Nonion K-220” (manufactured by NOF Corporation, surfactant) ): 6 g was mixed well to prepare 850 g (solid content 17.6%) of pigment catalyst dispersion paste solution B-2: Pigment catalyst-dispersed tertiary sulfonium salt resin solution B-3 obtained in Production Example 13 (solid content 60%): 250 g, ion-exchanged water: 594 g, “Nonion K-220” (manufactured by NOF Corporation, surfactant) ): 6 g was mixed well to prepare 850 g of pigment catalyst dispersion paste solution B-3 (solid content 17.6%).
- Production Example P1 >> The pigment dispersion paste solution B-2: 28.4 g and the bismuth compound (A) B 1: 5.0 g obtained in Production Example 1 were charged into a 100 ml flask equipped with a stirrer and stirred for 10 minutes. Thereto, glass beads (particle diameter: 2.5 mm to 3.5 mm): 60 g was added, and the mixture was stirred as it was for 2 hours, and the glass beads were separated by filtration to obtain pigment catalyst dispersed paste P1.
- Comparative Production Examples RP2 to RP8 >> The types and amounts of the pigment catalyst dispersion paste solution, the bismuth compound synthesized in the comparative production example, the comparative bismuth compound, and the comparative organotin compound were changed as shown in Table 2, and the pigment catalyst dispersion was performed in the same manner as in Production Example P1. Pastes RP2 to RP8 were obtained.
- bismuth compound content% means the mass of the bismuth compound (A) and the comparative bismuth compound relative to the total solid content of the base resin (B) and the curing agent (C) blocked isocyanate in the electrodeposition coating composition. %. Moreover, “organotin compound content%” shows weight% of an organotin compound with respect to the total solid of base resin (B) and hardening
- Electrodeposition coating, curability confirmation test> A 0.8x70x150mm cold-rolled steel plate (standard test plate manufactured by Japan Test Panel Co., Ltd., certified by Japan Anticorrosion Technology Association) treated with "Palbond L3080" (manufactured by Nihon Parkerizing Co., Ltd., zinc phosphate treatment agent) Weighed in advance and dipped in the electrodeposition coating compositions obtained in Examples 1 to 23 and Comparative Examples 1 to 8, and this was used as a cathode, and electrodeposition coating was performed on each of four test plates.
- the electrodeposition conditions were a voltage of 300 V, energization for 15 seconds, and a paint temperature in the electrodeposition tank of 20 to 30 ° C.
- the electrodeposition coated film was washed with ion exchange water and air-dried for 6 hours. Thereafter, the test plate was heated and baked in a gear oven (manufactured by ESPEC, model GPHH-202). The baking conditions were 170 ° C./20 minutes, 160 ° C./20 minutes, and each condition was performed on two test plates. Each test plate was weighed to calculate the weight of the electrodeposition-coated cured coating film. Thereafter, each test plate was immersed in an acetone bath at 20 ° C. for 16 hours, air-dried, and then heat-dried at 100 ° C. for 1 hour. Each test plate was weighed, and the weight of the remaining dry coating film after immersion in acetone was calculated.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Inorganic Chemistry (AREA)
- Paints Or Removers (AREA)
Abstract
Description
また、金属キレート化合物、酸素含有配位子を有する四価の有機チタン・ジルコニウム・ハフニウム錯体、ジルコニウム・チタン等のフルオロ金属イオンを含有するカチオン性電着塗料組成物が提案されている。(特許文献4~6)しかし、これらの化合物だけでは、塗膜硬化性が不十分なため有機錫化合物を併用する必要があった。
さらに、ビスマスアセチルアセトン錯体も提案されているが(特許文献7)、分散ペースト中での貯蔵安定性が悪いため、一定期間貯蔵すると触媒硬化性能が大幅に低下したり、貯蔵中に増粘ゲル化を起こしたりする問題があった。
このように、有機錫化合物を含まず、現行と同等の焼き付け条件にて良好な塗膜の硬化性を確保することができるカチオン電着塗料組成物はこれまでになかった。
前記ビスマス化合物は、下記化学式(1)で表されるβジケトンから調製した配位子を有する化合物である、電着塗料組成物用触媒が提供される。
化学式(1):

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、2つのR1の炭素数合計は4以上である。)
本発明の電着塗料組成物は、ビスマス化合物(A)を含有する電着塗料組成物用触媒と、基体樹脂(B)を含有する。
本発明の電着塗料組成物用触媒は、ビスマス化合物(A)を含有する。
ビスマス化合物(A)は、下記化学式(1)で表されるβジケトンから調製した配位子を有する化合物である。

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、R1の炭素数合計は4以上である。)
また、2つのR1の炭素数合計が4以上となるR1の組合せとしては、メチル基と炭素数が3以上の炭化水素基の組み合わせ、炭素数が2以上の炭化水素基と炭素数が2以上の炭化水素基の組み合わせが挙げられ、具体的には、例えば、エチル基とエチル基、メチル基とt-ブチル基、メチル基とフェニル基、フェニル基とフェニル基、4-t-ブチルフェニル基と4-メトキシフェニル基などが挙げられる。
化学式(4):
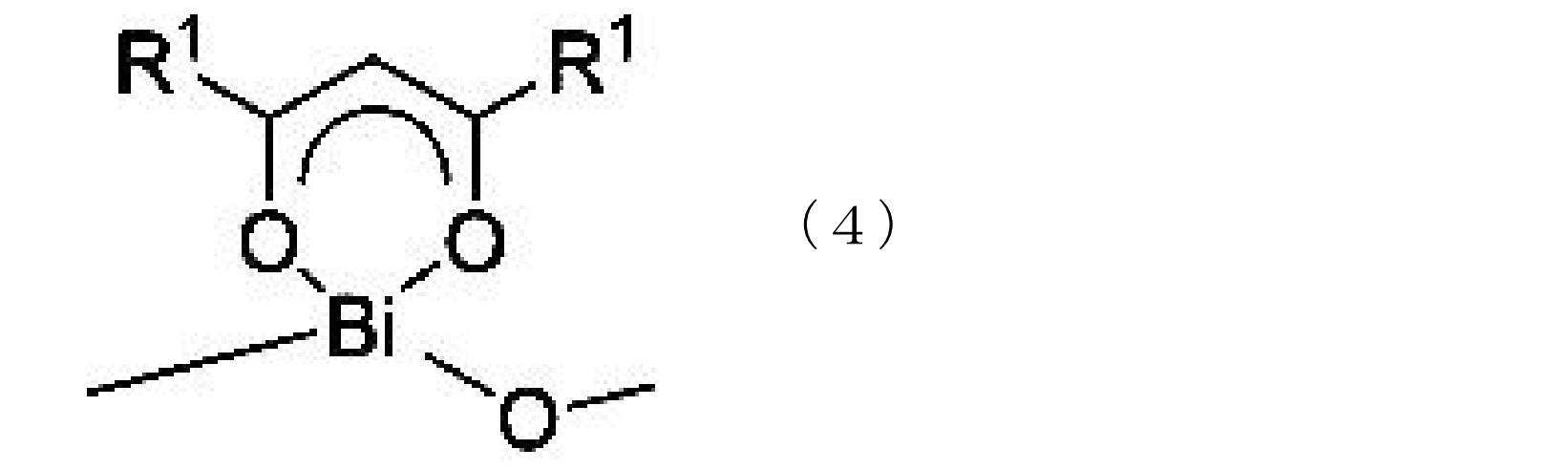
(式中、R1は、化学式(1)と同様である。)
化学式(5):

(式中、Xは、-OR3、又は前記化学式(1)のβジケトンに由来する1,3-ジカルボニレートであり、複数個のXのうちの少なくとも1つが前記1,3-ジカルボニレートであり、R3は、水素原子、アルキル基、アシル基、-Bi-X2の何れかであるか、又は2つのR3がBi原子で置換されて2つの酸素原子を連結する。)

(式中、R1は、化学式(1)と同様である。R2は、水素原子、アルキル基、アシル基、-Bi-(O-R2)2の何れかであるか、又は2つのR2がBi原子で置換されて2つの酸素原子を連結する。aは、1、2又は3の整数を表す。)

(式中、R1は、化学式(1)と同様である。R2は、化学式(2)と同様である。bとcは、1以上の整数を表す。)
R2又はR3で示されるアシル基としては、例えば、アセチル基、グルコロイル基、プロピオニル基、ラクトイル基、ブチリル基、ベンゾイル基、置換基を有する芳香族アシル基、オクタノイル基、2-エチルヘキサノイル基、ステアロイル基などが挙げられ、炭素数1~18のものが好ましい。また、マロニル基、スクシニル基、アジポロイル基等の二塩基酸アシル基として同じBi原子に結合していても良く、異なるBi原子間に結合していても良い。
反応溶媒は特に制限がないが、例えば、メタノール、エタノール、イソプロパノール、ブタノール等のアルコール系溶媒、ヘキサン、ヘプタン、シクロヘキサン、トルエン、キシレン等の炭化水素系溶媒、ジエチルエーテル、ジブチルエーテル、テトラヒドロフラン等のエーテル系溶媒、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン系溶媒、酢酸エチル、酢酸ブチル等のエステル系溶媒、DMF等のアミド系溶媒、DMSO等のスルホキシド系溶媒等の単独溶媒、それらの混合溶媒が挙げられる。反応温度は特に制限がないが、通常、0~200℃の範囲で反応させる。反応系で生成するアルコールを反応系外に留出させても良い。また、反応液に水やカルボン酸を添加しても良い。このようにして得られた反応液を濃縮してビスマス化合物(A)を得ることができる。必要に応じて、蒸留、昇華、溶媒による洗浄、再沈殿、再結晶等により精製することもできる。ビスマス化合物(A)は、反応条件等によって実質的に単一のビスマス化合物になる場合もあれば、複数のビスマス化合物の混合物になる場合もある。このような混合物から特定の化合物を単離して触媒として用いてもよく、混合物をそのまま触媒として用いてもよい。どちらの場合であっても触媒として機能する。
基体樹脂(B)としては、エポキシ系、アクリル系、ポリブタジエン系、アルキド系、ポリエステル系などのいずれの樹脂でもカチオン性基を導入することにより使用することができる。なかでも、カチオン性基を有するエポキシ変性樹脂が好ましい。
前記基体樹脂(B)が外部架橋型の樹脂の場合、併用される硬化剤(C)としては、架橋部を有する架橋剤(例:ブロックポリイソシアネート化合物)や、活性水素含有部を有する化合物(例:アミノ基、水酸基等を含有する樹脂)が挙げられる。より具体的には、基体樹脂(B)に活性水素含有部が含まれている場合には、硬化剤として架橋剤を用いることが好ましく、基体樹脂(B)に架橋部が含まれている場合には、硬化剤として活性水素含有部を有する化合物を用いることが好ましい。
金属化合物(D)としては、例えば、チタン、亜鉛、鉄、マグネシウム、アルミニウム、カルシウムからなる群から選ばれた少なくとも1種の化合物が挙げられる。
チタン化合物としては、例えば、WO2013/125562,WO2013/137174に記載の化合物が挙げられる。チタン化合物は、単独で本発明の電着塗料組成物に添加しても良いし、前記特許記載の方法で、ビスマス化合物(A)とチタン化合物の複合触媒を調製して、該複合触媒から電着塗料組成物を調製しても良い。
亜鉛化合物としては、例えば、酢酸亜鉛、乳酸亜鉛、ジメチロールプロピオン酸亜鉛、安息香酸亜鉛等の無置換及び種々の置換基を有する脂肪族及び芳香族カルボン酸亜鉛、2,4-ペンタンジオン、1-フェニル-1,3-ブタンジオン、1,3-ジフェニル-1,3-プロパンジオン等の1,3-ジカルボニル化合物から調製した配位子を有する亜鉛キレート錯体などが挙げられる。
鉄化合物としては、例えば、トリス(2-エチルヘキサン酸)鉄(III)等のカルボン酸塩、2,4-ペンタンジオン、1-フェニル-1,3-ブタンジオン、1,3-ジフェニル-1,3-プロパンジオン等の1,3-ジカルボニル化合物から調製した配位子を有する鉄キレート錯体などが挙げられる。
マグネシウム化合物としては、例えば、酢酸マグネシウム、乳酸マグネシウム等のカルボン酸塩、2,4-ペンタンジオン、1-フェニル-1,3-ブタンジオン、1,3-ジフェニル-1,3-プロパンジオン等の1,3-ジカルボニル化合物から調製した配位子を有するマグネシウムキレート錯体などが挙げられる。
アルミニウム化合物としては、例えば、ポリリン酸アルミニウム等のリン酸塩、2,4-ペンタンジオン、1-フェニル-1,3-ブタンジオン、1,3-ジフェニル-1,3-プロパンジオン等の1,3-ジカルボニル化合物から調製した配位子を有するアルミニウムキレート錯体などが挙げられる。
カルシウム化合物としては、例えば、酢酸カルシウム等のカルボン酸塩、2,4-ペンタンジオン、1-フェニル-1,3-ブタンジオン、1,3-ジフェニル-1,3-プロパンジオン等の1,3-ジカルボニル化合物から調製した配位子を有するカルシウムキレート錯体などが挙げられる。
本発明の電着塗料組成物中における金属化合物(D)の含有量は、特に制限されないが、通常、ビスマス化合物(A)100質量部に対して、5~300質量部、好ましくは10~100質量部である。前記範囲にある場合、電着塗料物性の向上の効果が得られる。
本発明の電着塗料組成物は、前記成分を水分散するための中和剤(E)をさらに含むことができる。中和剤(E)としては、例えば、ギ酸、酢酸、プロピオン酸、乳酸などの脂肪族カルボン酸、スルファミン酸等が挙げることができる。この中和剤(E)の量は、上記基体樹脂(B)中のアミノ基の量によって異なるものであり、水分散できる量であればよく、電着塗料のpHを3.0~9.0の範囲に保つ量であればよい。本発明では前記基体樹脂(B)に含まれるアミノ基を中和するのに必要な中和剤(E)の当量数は、0.25~1.5、好ましくは0.5~1.25である。前記範囲にある場合、組成物の仕上り性、つきまわり性、低温硬化性などの向上の効果が得られる。
本発明の電着塗料組成物には、さらに必要に応じて、着色顔料、体質顔料、有機溶剤、顔料分散剤、塗面調整剤、界面活性剤、酸化防止剤、紫外線吸収剤などの慣用の塗料添加物を配合することができる。
本発明の電着塗料組成物は、上記成分を一括して混合することにより製造することができるが、以下のような方法でも製造することができる。
例えば、まず、基体樹脂(B)と硬化剤(C)を混合し、中和剤(E)を加える。水単独又は水と親水性有機溶剤の混合物である水性媒体中に、前記基体樹脂(B)・硬化剤(C)・中和剤(E)の混合物を分散させてエマルションを製造する。あるいは、基体樹脂(B)と硬化剤(C)を混合し、中和剤(E)を添加した水溶液又は中和剤(E)を添加した水と親水性有機溶剤の混合溶液に、前記基体樹脂(B)・硬化剤(C)の混合物を分散させてエマルションを製造する。
次いで、予め調製したカチオン性の顔料触媒分散用の基体樹脂(B)溶液に、上記ビスマス化合物(A)、金属化合物(D)、その他添加物、顔料、顔料分散剤等を所定量加えて混合した後、必要に応じて、ボールミルやサンドミルなどの通常の分散装置を用いて混合物中の固体が一定の粒径以下になるまで良く分散させて顔料触媒分散ペーストを製造する。
最後に、前記エマルションと所定量の上記顔料触媒分散ペーストを良く混合し電着塗料組成物を製造する。
本発明の電着塗料組成物は、電着塗装によって所望の基材表面に塗装することができる。
電着塗装は、一般には、固形分濃度が約5~40質量%となるように脱イオン水などで希釈し、さらにpHを3.0~9.0の範囲内に調整した本発明の電着塗料組成物からなる電着浴を、通常、浴温15~45℃に調整し、負荷電圧100~400Vの条件で行うことができる。
製造例1~10及び比較製造例1~2に従って、ビスマス化合物(A)を製造した。これらの製造例及び比較製造例の内容を表1にまとめた。

窒素雰囲気下、攪拌装置、温度計、冷却器を備えた10L4つ口丸底フラスコに、塩化ビスマス(和光純薬工業社製):500g(1.58mol)、エタノール:365g、トルエン:2175gを加え攪拌し、加熱昇温して30分間還流させた。20%ナトリウムエトキシドエタノール溶液(和光純薬工業社製):1620g(4.76mol)を加熱還流下4時間かけて滴下し、その後3時間加熱還流した。その後、攪拌しながら20℃まで冷却し、不溶物を窒素雰囲気下、吸引濾過し、ビスマストリエトキシド溶液:4340gを得た。EDTA滴定によるBi濃度:0.336mmol/g、Bi基準収率92.3%

窒素雰囲気下、攪拌装置、温度計、冷却器を備えた1L4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液300g(0.10mol)を仕込み、化学式(7)で表される52,52-ジメチル-23,45-ヘキサジオン14.2g(0.10mol)を内温20~30℃の範囲で15分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。イオン交換水5.4g(0.30mol)を内温20~30℃の範囲で5分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。反応液を1Lナスフラスコに移し、50℃の湯浴で加熱しながら減圧濃縮して濃縮液68gを得た。濃縮液の入ったフラスコにヘプタン:20gを攪拌しながら10分間かけて滴下し、そのまま10分間攪拌した。得られたスラリー溶液を吸引濾過し、淡黄色湿固体を得、60℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間し、乾燥した淡黄色固体11.8gを得た。得られた淡黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は64.0%であった。

窒素雰囲気下、攪拌装置、温度計、冷却器を備えた1L4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:300g(0.10mol)を仕込み、化学式(8)で表される1-フェニル-1,3-ブタンジオン:19.4g(0.12mol)をトルエン:50gに溶解した溶液を内温20~30℃の範囲で30分間かけて滴下し、同温度範囲でそのまま2時間攪拌した。イオン交換水:3.6g(0.20mol)を内温20~30℃の範囲で5分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。反応液を1Lナスフラスコに移し、50℃の湯浴で加熱しながら減圧濃縮して濃縮液57gを得た。攪拌装置、温度計、冷却器を備えた3L4つ口丸底フラスコにヘプタン:700gを仕込み、攪拌しながら上記濃縮液を内温20~30℃の範囲で30分間かけて滴下し、トルエン:10gで洗いこんだ。同温度範囲でそのまま1時間攪拌した。得られたスラリー溶液を吸引濾過し、淡黄色湿固体を得、60℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間し、乾燥した淡黄色固体36.0gを得た。得られた淡黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は56.3%であった。

1-フェニル-1,3-ブタンジオン及び水の仕込み量を変更し、製造例3と同様の方法でビスマス化合物(A)を得た。結果を表1に示す。
窒素雰囲気下、攪拌装置、温度計、蒸留ヘッド、冷却器を備えた1L4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:150g(0.050mol)を仕込み、化学式(9)で表される1,3-ジフェニル-1,3-プロパンジオン:33.7g(0.15mol)をトルエン:180gに溶解した溶液を内温20~30℃の範囲で30分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。攪拌しながら油浴で加熱して液内温が100℃になるまで溶媒を常圧で留去した。濃縮液を500mLナスフラスコに移し、60℃湯浴で加熱しながら減圧濃縮して濃縮液51gを得、ヘプタン:50gを添加し黄色固体を析出させ、60℃の湯浴で加熱しながら減圧濃縮乾固(減圧度10~20mmHg)6時間し、乾燥した黄色固体43.7gを得た。得られた黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は23.8%であった。

窒素雰囲気下、攪拌装置、温度計、蒸留ヘッド、冷却器を備えた1L4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:300g(0.10mol)を仕込み、1,3-ジフェニル-1,3-プロパンジオン:18.0g(0.080mol)をトルエン:100gに溶解した溶液を内温20~30℃の範囲で30分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。攪拌しながら油浴で加熱して液内温が100℃になるまで溶媒を常圧で留去した。攪拌しながら液内温を20℃まで冷却し、イオン交換水:4.5g(0.25mol)を内温20~30℃の範囲で5分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。反応液を500mLナスフラスコに移し、60℃の湯浴で加熱しながら減圧濃縮して濃縮液60gを得た。攪拌装置、温度計、冷却器を備えた3L4つ口丸底フラスコにヘプタン:700gを仕込み、攪拌しながら上記濃縮液を内温20~30℃の範囲で30分間かけて滴下し、トルエン:10gで洗いこんだ。同温度範囲でそのまま1時間攪拌した。得られたスラリー溶液を吸引濾過し、淡黄色湿固体を得、60℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間し、乾燥した淡黄色固体37.3gを得た。得られた黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は52.5%であった。
1,3-ジフェニル-1,3-プロパンジオン及び水の仕込み量、反応時間を変更し、製造例7と同様の方法でビスマス化合物(A)を得た。結果を表1に示す。
攪拌装置、温度計、冷却器を備えた300mL4つ口丸底フラスコに、酸化ビスマスBi2O3:11.7g(0.025mol)、1,3-ジフェニル-1,3-プロパンジオン:33.6g(0.15mol)、イオン交換水:50gを仕込み攪拌した。攪拌しながら100℃まで昇温し同温度で5時間反応した。25℃まで冷却しトルエン:40gを添加しスラリーを吸引濾過し黄色湿固体を得た。固体を300mLナスフラスコに移しヘプタン:100gを添加し15分間攪拌しスラリーを吸引濾過し黄色湿固体を得、60℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間し、乾燥した淡黄色固体13.8gを得た。得られた黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は73.5%であった。
窒素雰囲気下、攪拌装置、温度計、蒸留ヘッド、冷却器を備えた500mL4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:150g(0.050mol)を仕込み、化学式(10)で表される1-(4-t-ブチルフェニル)-3-(4-メトキシフェニル)-1,3-プロパンジオン:15.5g(0.050mol)をトルエン:90gに溶解した溶液を内温20~30℃の範囲で30分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。攪拌しながら油浴で加熱して液内温が100℃になるまで溶媒を常圧で留去した。攪拌しながら液内温を20℃まで冷却し、イオン交換水:2.0g(0.11mol)を内温20~30℃の範囲で1分間かけて滴下し、同温度範囲でそのまま3時間攪拌した。反応液を500mLナスフラスコに移し、60℃の湯浴で加熱しながら減圧濃縮乾固(減圧度10~20mmHg)8時間し、乾燥した黄色固体26.5gを得た。得られた黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は40.1%であった。

窒素雰囲気下、攪拌装置、温度計、冷却器を備えた500mL4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:150g(0.050mol)を仕込み、化学式(11)で表されるアセチルアセトン:15.0g(0.15mol)を内温20~30℃の範囲で30分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。反応液を500mLナスフラスコに移し、50℃湯浴で加熱しながら減圧濃縮して濃縮液41gを得、ヘプタン:50gを添加し、60℃の湯浴で加熱しながら減圧濃縮乾固(減圧度10~20mmHg)6時間し、乾燥した淡黄色固体24.2gを得た。得られた黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は43.0%であった。

窒素雰囲気下、攪拌装置、温度計、冷却器を備えた1L4つ口丸底フラスコに、製造例1で合成したビスマストリエトキシド溶液:300g(0.10mol)を仕込み、アセチルアセトン:10.0g(0.10mol)を内温20~30℃の範囲で15分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。イオン交換水:5.4g(0.30mol)を内温20~30℃の範囲で5分間かけて滴下し、同温度範囲でそのまま1時間攪拌した。反応液を1Lナスフラスコに移し、50℃の湯浴で加熱しながら減圧濃縮して濃縮液62gを得た。濃縮液の入ったフラスコにヘプタン:300gを攪拌しながら30分間かけて滴下し、そのまま10分間攪拌した。得られたスラリー溶液を吸引濾過し、淡黄色湿固体を得、60℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間し、乾燥した淡黄色固体29.6gを得た。得られた淡黄色固体ビスマス化合物(A)のEDTA滴定によるBi含量分析値は70.0%であった。
<<製造例11 メインバインダー用>>
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた3L4つ口フラスコに、エポキシ樹脂「jER1004AF」(三菱化学社製、エポキシ当量896g/eq、平均分子量 約1650):1425g(エポキシ基換算1.59mol)、エチレングリコールモノブチルエーテル(以下、ブチルセルソルブと表記):406gを仕込み、120℃油浴で加熱し攪拌して樹脂を溶解させた。ジエタノールアミン:175.5g(1.67mol)を滴下漏斗で内温95~115℃の範囲で1時間かけて滴下し、滴下漏斗をブチルセルソルブ:64gで洗いこんだ。そのまま、内温115~120℃の範囲で16時間加熱攪拌した。その後、攪拌しながら、ブチルセルソルブ:597gを30分かけて滴下し、そのまま50℃まで攪拌しながら放冷し、ジエタノールアミン付加エポキシ樹脂ブチルセルソルブ溶液B-1(固形分60%):2667gを得た。B-1の水酸基価を測定し溶媒ブチルセルソルブの水酸基価を差し引き補正した樹脂固形分の水酸基価は、199mgKOH/g(OH基換算3.55mmol/g)であった。計算によるアミン含量は、0.63mmol/gであった。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた2L4つ口フラスコに、トリレンジイソシアネート(以下、TDIと表記):420.6g(2.41mol)、メチルイソブチルケトン(以下、MIBKと表記):82gを仕込み、攪拌しながら加熱して内温50℃まで昇温した。ブチルセルソルブ:285.4g(2.41mol)を内温50~55℃に保ち4時間かけて滴下した。50℃で4時間反応しブチルセルソルブハーフブロック化TDI:788gを得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた1L4つ口フラスコに、ジメチルエタノールアミン:106.0g(1.19mol)に仕込み、攪拌しながら上記ブチルセルソルブハーフブロック化TDI:394g(1.21mol)を内温25~50℃の範囲で2時間かけて滴下した。その後、80℃まで昇温し1時間攪拌した。その後、攪拌しながら、75%乳酸水溶液:142g(1.19mol)を75~85℃の範囲で1時間かけて滴下し、ブチルセルソルブ:88gを添加し、65~70℃で3時間攪拌して4級化剤:730gを得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた3L4つ口フラスコに、エポキシ樹脂「jER1001AF」(三菱化学社製、エポキシ当量468g/eq、平均分子量 約900):378g(エポキシ基換算0.81mol)を仕込み、120℃の油浴で加熱攪拌して樹脂を溶融させた。攪拌しながら、上記ブチルセルソルブハーフブロック化TDI:394gを内温117~123℃の範囲で2時間かけて滴下した。同温度範囲で2時間反応した後、90℃まで冷却した。攪拌しながら、上記4級化剤:495g(0.81mol)を85~90℃の範囲で1時間かけて滴下した。ブチルセルソルブ:136gを添加し75~85℃の範囲で16時間反応させ、ブチルセルソルブ:500g添加して希釈し攪拌しながら50℃まで放冷し顔料触媒分散用4級アンモニウム塩型樹脂溶液B-2(固形分60%):1903gを得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた1L4つ口フラスコに、イソホロンジイソシアネート(以下、IPDIと表記):222.3g(1.00mol)、MIBK:39.5gを仕込み、攪拌しながら加熱して内温50℃まで昇温した。ブチルセルソルブ:118.2g(1.00mol)を内温47~53℃に保ち2時間かけて滴下した。同温度範囲で10時間反応しブチルセルソルブハーフブロック化IPDI:380gを得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた500mL4つ口フラスコに、2-メルカプトエタノール:75.3g(0.96mol)、MIBK:154gを仕込み攪拌して均一溶液とした。
ジメチルベンジルアミン:0.39g(2.9mmol)を添加し、攪拌しながら内温50℃まで昇温した。攪拌しながら、グリシドール:78.6g(1.06mol)を47~53℃の範囲で2時間かけて滴下し、同温度範囲で5時間反応した。反応液を1Lナスフラスコに移し、50℃の湯浴で減圧濃縮(減圧度10~20mmHg)3時間し淡黄色粘張液体1-(2-ヒドロキシエチルチオ)-2,3-プロパンジオール:163gを得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた3L4つ口フラスコに、エポキシ樹脂「jER1001AF」(三菱化学社製、エポキシ当量468g/eq、平均分子量 約900):234.5g(エポキシ基換算0.50mol)を仕込み、140℃の油浴で加熱攪拌して樹脂を溶融させた。
攪拌しながら、上記ブチルセルソルブハーフブロック化IPDI:190g(0.50mol)を、内温137~143℃の範囲で1時間かけて滴下した。同温度範囲で2時間反応した後、ブチルセルソルブ:100g添加し70℃まで冷却した。攪拌しながら上記1-(2-ヒドロキシエチルチオ)-2,3-プロパンジオール:83g、ジメチロールプロピオン酸:59g(0.5mol)、イオン交換水:60gを添加した。内温67~73℃の範囲で6時間攪拌後、ブチルセルソルブ:50gを添加し67~73℃の範囲で12時間反応させた。ブチルセルソルブ:134.5g添加して希釈し攪拌しながら50℃まで放冷し顔料触媒分散用3級スルホニウム塩型樹脂溶液B-3(固形分60%):910gを得た。
<<製造例14>>
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた3L4つ口フラスコに、ポリメチレンポリフェニルポリイソシアネート「スミジュール44V20」(住化バイエルウレタン社製、イソシアネート基含有率31.5%):798g(イソシアネート基換算6.0mol)を仕込み、攪拌しながら内温95℃まで加熱した。加熱を停止し、攪拌しながら、ブチルセルソルブ:1063g(9.0mol)を内温95~120℃の範囲で2時間かけて滴下した。その後、加熱して内温115~120℃の範囲で5時間攪拌した。その後、加熱を停止し、サンプリングしてIRスペクトルでイソシアネート基の吸収(2241cm-1)の消失を確認した。攪拌しながら、ブチルセルソルブ:290gを15分間かけて滴下し、そのまま50℃まで攪拌しながら放冷し、ブチルセルソルブでブロック化したポリメチレンポリフェニルポリイソシアネートブチルセルソルブ溶液(固形分70%):2150gを得た。計算による本溶液中のブロックイソシアネート基含量は、2.79mmol/gであった。
<<製造例15>>
製造例11で得たジエタノールアミン付加エポキシ樹脂ブチルセルソルブ溶液B―1(固形分60%):100g、製造例14で得たポリメチレンポリフェニルポリイソシアネートブチルセルソルブ溶液(固形分70%):128g、ブチルセルソルブ:21gを良く混合し、基体樹脂(B)及び硬化剤(C)の固形分60%ブチルセルソルブ溶液を調製した。
TKホモミキサーMARKII 2.5型(プライミクス社製)を備えた3Lビーカーに、イオン交換水:880g、酢酸:3.6g、ブチルセルソルブ:13gを仕込み、1000rpmで攪拌混合した。ホモミキサー回転数を12000rpmとし、内温を15~20℃に保ちながら、上記基体樹脂(B)及び硬化剤(C)のブチルセルソルブ溶液を6時間かけて滴下し、そのまま、同温度範囲で6時間攪拌し、固形分13%のエマルション溶液:1145gを得た。
<<製造例16:チタン化合物D1>>
WO2013/125562の製造例3記載の方法で1-フェニル-1,3-ブタンジオンから調製した配位子を有するチタン化合物D1を得た。
WO2013/125562の製造例5記載の方法で1,3-ジフェニル-1,3-プロパンジオンから調製した配位子を有するチタン化合物D2を得た。
窒素雰囲気下、攪拌装置、温度計、冷却器を備えた300mL4つ口丸底フラスコに、1-フェニル-1,3-ブタンジオン:23.8g(0.15mol)を仕込み、THF45gを添加し攪拌して溶解した。予め調製した水酸化ナトリウム:5.9g(0.15mol)をメタノール50gに溶解した溶液を、内温15~20℃の範囲で30分かけて滴下し、同温度範囲でそのまま30分間攪拌した。その後、予め調製した塩化亜鉛:10.0g(0.07mol)をイオン交換水25gに溶解した溶液の約1/2を20~25℃の範囲で5分間かけて滴下した。次いで、メタノール25g、THF10gを添加し、残りの塩化亜鉛水溶液を同温度範囲で5分間かけて滴下し、そのまま1時間攪拌した。得られたスラリー溶液を吸引濾過し、濾過物をイオン交換水100gで3回洗浄し湿固体を得、80℃の湯浴で加熱しながら減圧乾燥(減圧度10~20mmHg)4時間することにより、1-フェニル-1,3-ブタンジオンから調製した配位子を有する亜鉛化合物D3:24.7gを得た。
製造例12で得た顔料触媒分散用4級アンモニウム塩型樹脂溶液B-2(固形分60%):250g、イオン交換水:594g、「ノニオンK-220」(日油社製、界面活性剤):6gを良く混合し顔料触媒分散ペースト用溶液B-2:850g(固形分17.6%)を調製した。
製造例13で得た顔料触媒分散用3級スルホニウム塩型樹脂溶液B-3(固形分60%):250g、イオン交換水:594g、「ノニオンK-220」(日油社製、界面活性剤):6gを良く混合し顔料触媒分散ペースト用溶液B-3:850g(固形分17.6%)を調製した。

攪拌機を備えた100mlフラスコに、前記顔料分散ペースト用溶液B-2:28.4g、製造例1で得たビスマス化合物(A)B1:5.0gを仕込み、10分間攪拌混合した。そこへガラスビーズ(粒子径2.5mm~3.5mm):60gを加え、そのまま2時間攪拌し、ガラスビーズを濾別し、顔料触媒分散ペーストP1を得た。
前記顔料触媒分散ペースト用溶液、ビスマス化合物(A)、及び金属化合物(D)を表2に示すように配合し、製造例P1と同様の方法で顔料触媒分散ペーストP2~P23を得た。
本発明のビスマス化合物の代わりに、比較製造例1で合成したビスマス化合物RB1:5.0gを使用して製造例P1と同様の方法で顔料触媒分散ペーストRP1を得た。
前記顔料触媒分散ペースト用溶液、比較製造例で合成したビスマス化合物、比較ビスマス化合物、比較有機スズ化合物の種類及び量を表2に示すように変更し、製造例P1と同様の方法で顔料触媒分散ペーストRP2~RP8を得た。
製造例P1~P23、比較製造例RP1~RP8で得た顔料触媒分散ペーストをガラス製サンプル瓶に入れ密栓し35℃30日間恒温槽で保存した。保存ペーストを撹拌し再分散可能か確認し、電着塗料組成物の製造、硬化試験に供した。
<<実施例1~23及び比較例1~8>>
表3に示すエマルション溶液、顔料触媒分散ペーストを、表3に示す割合(質量部)で配合し、混合分散することにより電着塗料組成物を製造した。

「パルボンドL3080」(日本パーカライジング社製、リン酸亜鉛処理剤)で化成処理した0.8x70x150mmの冷間圧延鋼板(日本テストパネル社製標準試験板、(社)日本防錆技術協会認定品)を予め秤量し、実施例1~23及び比較例1~8で得られた電着塗料組成物中に浸漬し、これを陰極として電着塗装を各々4枚の試験板で行った。電着条件は、電圧300V、15秒通電、電着槽内塗料温度20~30℃で実施した。電着塗装した塗膜はイオン交換水で水洗し6時間風乾した。その後、該試験板をギヤーオーブン(エスペック製、GPHH-202型)にて加熱焼き付けを行った。焼き付け条件は、170℃/20分、160℃/20分、各条件該試験板2枚ずつで実施した。各試験板を秤量して電着塗装硬化塗膜重量を算出した。その後、各試験板を20℃、16時間アセトン浴に浸漬し、風乾後、100℃1時間加熱乾燥した。各試験板を秤量しアセトン浸漬後の残存乾燥塗膜の重量を算出した。下式に従い、ゲル分率を算出し、以下の基準で塗膜の硬化性を評価した。その結果を表3に示す。
ゲル分率(%)=100×(アセトン浸漬後の残存塗膜重量(g))/(アセトン浸漬前の塗膜重量(g))
○:80%以上90%未満
X:80%未満
以上の結果から、本発明のビスマス化合物(A)が優れた触媒活性及び水系媒体中の安定性を示すことが実証された。
Claims (6)
- ビスマス化合物(A)を含有する電着塗料組成物用触媒であって、
前記ビスマス化合物は、下記化学式(1)で表されるβジケトンから調製した配位子を有する化合物である、電着塗料組成物用触媒。
化学式(1):

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、2つのR1の炭素数合計は4以上である。) - 前記ビスマス化合物(A)は、下記化学式(4)で表される基を有する少なくとも一種のビスマス化合物である、請求項1に記載の電着塗料組成物用触媒。
化学式(4):

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、2つのR1の炭素数合計は4以上である。) - 前記ビスマス化合物(A)は、下記化学式(5)で表される少なくとも一種のビスマス化合物である、請求項1又は請求項2に記載の電着塗料組成物用触媒。
化学式(5):

(式中、Xは、-OR3、又は前記化学式(1)のβジケトンに由来する1,3-ジカルボニレートであり、複数個のXのうちの少なくとも1つが前記1,3-ジカルボニレートであり、R3は、水素原子、アルキル基、アシル基、-Bi-X2の何れかであるか、又は2つのR3がBi原子で置換されて2つの酸素原子を連結する。) - 前記ビスマス化合物(A)は、下記化学式(2)又は下記化学式(3)で表される少なくとも一種のビスマス化合物である、請求項1~請求項3の何れか1つに記載の電着塗料組成物用触媒。
化学式(2):

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、2つのR1の炭素数合計は4以上である。R2は、水素原子、アルキル基、アシル基、-Bi-(O-R2)2の何れかであるか、又は2つのR2がBi原子で置換されて2つの酸素原子を連結する。aは1,2又は3の整数を表す。)
化学式(3):

(式中、R1は、互いに同一又は異なって、炭化水素基を表し、かつ、2つのR1の炭素数合計は4以上である。R2は、水素原子、アルキル基、アシル基、-Bi-(O-R2)2の何れかであるか、又は2つのR2がBi原子で置換されて2つの酸素原子を連結する。bとcは、1以上の整数を表す。添字bが付された基と、添字cが付された基の並び順は任意である。) - 請求項1~請求項4の何れか1つに記載の電着塗料組成物用触媒と基体樹脂(B)とを含有する電着塗料組成物。
- 前記基体樹脂(B)がブロックイソシアネート基を含有するか、前記電着塗料組成物がブロックポリイソシアネート化合物からなる硬化剤(C)を含有する、請求項5に記載の電着塗料組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/306,463 US9822263B2 (en) | 2014-04-28 | 2015-03-20 | Electrodeposition coating material composition and catalyst for electrodeposition coating material |
| KR1020167033198A KR101939571B1 (ko) | 2014-04-28 | 2015-03-20 | 전착 도료 조성물, 전착 도료 조성물용 촉매 |
| CN201580022435.9A CN106232750B (zh) | 2014-04-28 | 2015-03-20 | 电沉积涂料组合物、电沉积涂料组合物用催化剂 |
| JP2015517540A JP6019223B2 (ja) | 2014-04-28 | 2015-03-20 | 電着塗料組成物、電着塗料組成物用触媒 |
| EP15786511.4A EP3138885A4 (en) | 2014-04-28 | 2015-03-20 | Electrodeposition coating material composition and catalyst for electrodeposition coating material |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014092739 | 2014-04-28 | ||
| JP2014-092739 | 2014-04-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015166745A1 true WO2015166745A1 (ja) | 2015-11-05 |
Family
ID=54358484
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/058532 Ceased WO2015166745A1 (ja) | 2014-04-28 | 2015-03-20 | 電着塗料組成物、電着塗料組成物用触媒 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9822263B2 (ja) |
| EP (1) | EP3138885A4 (ja) |
| JP (1) | JP6019223B2 (ja) |
| KR (1) | KR101939571B1 (ja) |
| CN (1) | CN106232750B (ja) |
| WO (1) | WO2015166745A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017187900A1 (ja) * | 2016-04-25 | 2017-11-02 | 日東化成株式会社 | 電着塗料組成物及びその製造方法 |
| JP2020147703A (ja) * | 2019-03-14 | 2020-09-17 | 日東化成株式会社 | 粉体塗料組成物、該組成物用触媒 |
| WO2025204073A1 (ja) * | 2024-03-25 | 2025-10-02 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物およびその製造方法 |
| WO2025243636A1 (ja) * | 2024-05-23 | 2025-11-27 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物および硬化電着塗膜の形成方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11339260B2 (en) | 2019-08-01 | 2022-05-24 | Covestro Llc | Pultrusion processes for producing fiber reinforced polyurethane compositions and polyurethane-forming reaction mixtures suitable for use in such processes |
| KR102940527B1 (ko) | 2020-06-08 | 2026-03-18 | 주식회사 케이씨씨 | 양이온성 우레탄 경화 첨가제 |
| CN114507331A (zh) * | 2022-01-28 | 2022-05-17 | 重庆毂运科技有限公司 | 一种高柔韧性电泳树脂及其制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002129100A (ja) * | 2000-08-18 | 2002-05-09 | Nippon Paint Co Ltd | カチオン電着塗料組成物 |
| JP2006057025A (ja) * | 2004-08-20 | 2006-03-02 | Sanyo Chem Ind Ltd | 活性エネルギー線硬化型ウレタン(メタ)アクリレート組成物 |
| JP2007084727A (ja) * | 2005-09-22 | 2007-04-05 | Dainippon Printing Co Ltd | 光学シート形成用紫外線硬化型接着剤および光学シート |
| JP2009508985A (ja) * | 2005-09-15 | 2009-03-05 | モーメンティブ・パフォーマンス・マテリアルズ・インク | 有機ビスマス触媒を用いたアミノシラン末端含有ポリマーの調製方法、及びスズ触媒を用いずにそれにより得られる硬化ポリマー |
| JP2011501774A (ja) * | 2007-10-17 | 2011-01-13 | ビーエーエスエフ ソシエタス・ヨーロピア | 有機金属化合物を主成分とする光潜伏性触媒 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5464533A (en) * | 1977-11-02 | 1979-05-24 | Kansai Paint Co Ltd | Aqueous corrosion-resistant coating composition |
| US4321304A (en) * | 1980-10-02 | 1982-03-23 | Ppg Industries, Inc. | Beta-diketone-epoxy resin reaction products blended with monomeric or polymeric phosphonium salts useful for providing corrosion resistance |
| JP2815386B2 (ja) | 1989-04-05 | 1998-10-27 | 関西ペイント株式会社 | 電着塗料組成物 |
| DE4331673A1 (de) * | 1993-09-17 | 1995-05-11 | Herberts Gmbh | Verfahren zur Herstellung von Mehrschichtlackierungen |
| JP3375736B2 (ja) | 1994-06-02 | 2003-02-10 | 神東塗料株式会社 | カチオン性電着塗料用樹脂組成物 |
| US5908912A (en) * | 1996-09-06 | 1999-06-01 | Ppg Industries Ohio, Inc. | Electrodepositable coating composition containing bismuth and amino acid materials and electrodeposition method |
| JPH11152432A (ja) | 1997-11-19 | 1999-06-08 | Shinto Paint Co Ltd | カチオン性電着塗料組成物 |
| CA2292483A1 (en) * | 1998-12-17 | 2000-06-17 | Sinzi Hirato | Electrodeposition paint composition |
| JP2000336287A (ja) | 1999-05-26 | 2000-12-05 | Nippon Paint Co Ltd | 低光沢鉛フリーカチオン電着塗料組成物、塗膜形成方法および塗装物 |
| JP2001192611A (ja) * | 2000-01-07 | 2001-07-17 | Nippon Paint Co Ltd | カチオン電着塗料組成物 |
| JP3846545B2 (ja) * | 2000-06-08 | 2006-11-15 | 信越化学工業株式会社 | コーティング剤組成物、コーティング方法及び被覆物品 |
| TWI303533B (en) * | 2001-06-15 | 2008-11-21 | Oled T Ltd | Electroluminescent devices |
| US6811667B2 (en) * | 2002-03-04 | 2004-11-02 | E. I. Du Pont De Nemours And Company | Cathodic electrodeposition coating agents containing bismuth complexes, preparation and use thereof |
| US6969754B2 (en) * | 2003-12-11 | 2005-11-29 | Valspar Sourcing, Inc. | Blocked isocyanate crosslinker solution |
| JP4473755B2 (ja) | 2005-03-17 | 2010-06-02 | 関西ペイント株式会社 | 電着塗料及び塗装方法と塗装物品 |
| PA8785001A1 (es) * | 2007-06-18 | 2008-06-17 | Johnson Matthey Plc | Compuestos estables en agua, catalizadores y reacciones catalizadas novedosos |
| JP2009084727A (ja) | 2007-09-28 | 2009-04-23 | Bridgestone Corp | ゴム−スチールコード複合体、その製造方法およびそれを用いた空気入りタイヤ |
| DE102008012085A1 (de) | 2008-02-29 | 2009-09-10 | Basf Coatings Ag | Kathodischer Elektrotauchlack enthaltend metallorganische Verbindung |
| JP5894919B2 (ja) * | 2009-09-15 | 2016-03-30 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 光潜在性チタンキレート触媒 |
| US8795836B2 (en) * | 2010-03-03 | 2014-08-05 | Ppg Industries Ohio, Inc | Electrodepositable coating composition comprising a bismuth salt and a stabilizing compound |
| WO2013125562A1 (ja) * | 2012-02-21 | 2013-08-29 | 日東化成株式会社 | 電着塗料組成物、電着塗料組成物用触媒 |
| JP2015108029A (ja) * | 2012-03-12 | 2015-06-11 | 日東化成株式会社 | 電着塗料組成物、電着塗料組成物用触媒 |
| MX390670B (es) * | 2013-11-18 | 2025-03-20 | Basf Coatings Gmbh | Metodo de dos etapas para el recubrimiento por inmersion de sustratos electricamente conductores utilizando una composicion que contiene bi(iii). |
-
2015
- 2015-03-20 EP EP15786511.4A patent/EP3138885A4/en not_active Withdrawn
- 2015-03-20 JP JP2015517540A patent/JP6019223B2/ja active Active
- 2015-03-20 CN CN201580022435.9A patent/CN106232750B/zh not_active Expired - Fee Related
- 2015-03-20 US US15/306,463 patent/US9822263B2/en not_active Expired - Fee Related
- 2015-03-20 WO PCT/JP2015/058532 patent/WO2015166745A1/ja not_active Ceased
- 2015-03-20 KR KR1020167033198A patent/KR101939571B1/ko not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002129100A (ja) * | 2000-08-18 | 2002-05-09 | Nippon Paint Co Ltd | カチオン電着塗料組成物 |
| JP2006057025A (ja) * | 2004-08-20 | 2006-03-02 | Sanyo Chem Ind Ltd | 活性エネルギー線硬化型ウレタン(メタ)アクリレート組成物 |
| JP2009508985A (ja) * | 2005-09-15 | 2009-03-05 | モーメンティブ・パフォーマンス・マテリアルズ・インク | 有機ビスマス触媒を用いたアミノシラン末端含有ポリマーの調製方法、及びスズ触媒を用いずにそれにより得られる硬化ポリマー |
| JP2007084727A (ja) * | 2005-09-22 | 2007-04-05 | Dainippon Printing Co Ltd | 光学シート形成用紫外線硬化型接着剤および光学シート |
| JP2011501774A (ja) * | 2007-10-17 | 2011-01-13 | ビーエーエスエフ ソシエタス・ヨーロピア | 有機金属化合物を主成分とする光潜伏性触媒 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3138885A4 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017187900A1 (ja) * | 2016-04-25 | 2017-11-02 | 日東化成株式会社 | 電着塗料組成物及びその製造方法 |
| JPWO2017187900A1 (ja) * | 2016-04-25 | 2019-02-28 | 日東化成株式会社 | 電着塗料組成物及びその製造方法 |
| JP2020147703A (ja) * | 2019-03-14 | 2020-09-17 | 日東化成株式会社 | 粉体塗料組成物、該組成物用触媒 |
| WO2020184505A1 (ja) * | 2019-03-14 | 2020-09-17 | 日東化成株式会社 | 粉体塗料組成物、該組成物用触媒 |
| JP7292708B2 (ja) | 2019-03-14 | 2023-06-19 | 日東化成株式会社 | 粉体塗料組成物 |
| WO2025204073A1 (ja) * | 2024-03-25 | 2025-10-02 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物およびその製造方法 |
| WO2025243636A1 (ja) * | 2024-05-23 | 2025-11-27 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物および硬化電着塗膜の形成方法 |
| JP2025177425A (ja) * | 2024-05-23 | 2025-12-05 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物および硬化電着塗膜の形成方法 |
| JP7804720B2 (ja) | 2024-05-23 | 2026-01-22 | 日本ペイント・オートモーティブコーティングス株式会社 | カチオン電着塗料組成物および硬化電着塗膜の形成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3138885A4 (en) | 2017-12-27 |
| JPWO2015166745A1 (ja) | 2017-04-20 |
| US20170051160A1 (en) | 2017-02-23 |
| CN106232750A (zh) | 2016-12-14 |
| US9822263B2 (en) | 2017-11-21 |
| KR20170003940A (ko) | 2017-01-10 |
| EP3138885A1 (en) | 2017-03-08 |
| KR101939571B1 (ko) | 2019-01-17 |
| CN106232750B (zh) | 2018-05-22 |
| JP6019223B2 (ja) | 2016-11-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6019223B2 (ja) | 電着塗料組成物、電着塗料組成物用触媒 | |
| EP0509437B1 (en) | Electrocoating composition | |
| CN111670226A (zh) | 可电沉积的涂料组合物和由其产生的导电涂层 | |
| WO1999031187A1 (en) | Cationic electrodeposition paint composition | |
| WO1999006493A1 (en) | Cationic electrodeposition coating composition | |
| JP2000290542A (ja) | カチオン電着塗料組成物および塗膜 | |
| JP4825841B2 (ja) | 皮膜形成方法 | |
| JP4060620B2 (ja) | 無鉛性カチオン電着塗料を用いる電着塗装方法 | |
| JP2008195925A (ja) | 電着塗膜の形成方法 | |
| EP3034656A1 (en) | Multi-layered coating film formation method | |
| WO2013099750A1 (ja) | 電着塗料組成物、電着塗料組成物用解離触媒 | |
| JP5918845B2 (ja) | 電着塗料組成物、電着塗料組成物用触媒 | |
| WO2014054549A1 (ja) | カチオン電着塗料組成物 | |
| JP6406848B2 (ja) | 電着塗料組成物 | |
| JPH05239386A (ja) | 電着塗料用組成物 | |
| WO2012147478A1 (ja) | カチオン電着塗料組成物及び塗装物品 | |
| JP3685297B2 (ja) | 鉛フリーカチオン電着塗膜の暴露耐食性を向上させる方法 | |
| WO2017187900A1 (ja) | 電着塗料組成物及びその製造方法 | |
| JP3874386B2 (ja) | カチオン電着塗料組成物 | |
| WO2016167147A1 (ja) | 電着塗料組成物、電着塗料組成物用触媒 | |
| JP3124088B2 (ja) | 電着塗料組成物 | |
| JP2015108029A (ja) | 電着塗料組成物、電着塗料組成物用触媒 | |
| WO2016158319A1 (ja) | 電着塗料組成物、電着塗料組成物用触媒 | |
| WO2013099749A1 (ja) | 電着塗料組成物、電着塗料組成物用解離触媒 | |
| JP3910698B2 (ja) | カチオン電着塗料組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2015517540 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15786511 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15306463 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015786511 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015786511 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167033198 Country of ref document: KR Kind code of ref document: A |