WO2015173970A1 - 抗悪性腫瘍剤組成物 - Google Patents
抗悪性腫瘍剤組成物 Download PDFInfo
- Publication number
- WO2015173970A1 WO2015173970A1 PCT/JP2014/065163 JP2014065163W WO2015173970A1 WO 2015173970 A1 WO2015173970 A1 WO 2015173970A1 JP 2014065163 W JP2014065163 W JP 2014065163W WO 2015173970 A1 WO2015173970 A1 WO 2015173970A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- compound
- lat1
- cell
- inhibitor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images










































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/235—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group
- A61K31/24—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group having an amino or nitro group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/243—Platinum; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/50—Hydrolases (3) acting on carbon-nitrogen bonds, other than peptide bonds (3.5), e.g. asparaginase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/01—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in linear amides (3.5.1)
- C12Y305/01001—Asparaginase (3.5.1.1)
Definitions
- the present invention relates to a novel antineoplastic agent composition, for example, a novel pharmaceutical composition for treating T cell acute lymphoblastic leukemia / lymphoma, a pharmaceutical composition for treating gastric cancer, a pharmaceutical composition for treating pancreatic cancer, or the large intestine.
- a novel antineoplastic agent composition for example, a novel pharmaceutical composition for treating T cell acute lymphoblastic leukemia / lymphoma, a pharmaceutical composition for treating gastric cancer, a pharmaceutical composition for treating pancreatic cancer, or the large intestine.
- the present invention relates to a pharmaceutical composition for cancer treatment.
- LAT1 System L amino acid transporter 1
- cancer types include lung cancer, stomach cancer, pancreatic cancer, esophageal cancer, breast cancer, colon cancer, bladder cancer, prostate cancer, blood cancer, etc. Taking cancer, stomach cancer, pancreatic cancer, and colon cancer as examples.
- Blood cancer includes leukemia, lymphoma, myeloma and the like.
- Leukemia is classified into 4 types (Acute myelocytic leukemia, Chronic myelogenous leukemia, Acute lymphoblastic leukemia (ALL), Chronic lymphocytic leukemia).
- Acute leukemia is a disease in which tumorigenesis occurs in hematopoietic stem cells or hematopoietic progenitor cells, and only specific cells proliferate as leukemia cells.
- Lymphoma is a tumor derived from the cells constituting the lymphoid tissue, and is a hematopoietic tumor mainly caused by lymphocyte oncogenesis. Malignant lymphoma is divided into Hodgkin's disease and non-Hodgkin's lymphoma.
- Lymphocytic leukemia and lymphoma are tumors of the main constituent cells of lymphocytes, and can be divided into B cell tumors and T cell tumors.
- T-cell tumors include T-cell acute lymphoblastic leukemia (T-ALL), T-cell lymphoblastic lymphoma (T-LL), T-cell chronic lymphocytic leukemia (T-CLL), adult T Cellular leukemia / lymphoma (ATL) and the like are included.
- Lymphoblastic lymphoma is a cancer of lymphocytes that are more immaturely differentiated compared to general lymphoma, and the essence of cancer cells is the same as acute lymphocytic leukemia It is thought that. Therefore, in the WHO classification, it is classified as a group of diseases as “Precursor T or B Lymphoblastic leukemia / lymphoma” (Precursor T-or B-Lymphoblastic Leukemia / Lymphoma).
- T-cell lymphoblastic lymphoma T-LL
- T-ALL T-cell acute lymphoblastic leukemia
- Non-patent Document 3 Treatment with invasive chemotherapy in T-ALL patients has improved the cure rate to more than 40% in adults and more than 80% in pediatric patients (2), but refractory recurrence has It is well known that it occurs in all available medications (Non-Patent Document 3). Consequently, we have identified more novel molecular and molecular treatments involved in the pathogenesis of T-cell malignancies that identify novel and / or combination treatments to enhance these cure rates. I think it is very important to do.
- Tumorized cells are a hallmark property that allows them to continue to proliferate, avoid elimination by apoptosis, spread and resist treatment (Non-Patent Document 4).
- Cancer cells are reprogrammed for their metabolism (Non-Patent Document 5), which produces new proteins, lipids, and DNA for cancer cells to grow quickly and actively divide. Their properties are enhanced properties so as to occupy the nutrients that are needed.
- T-ALL / T-LL cells are very heterogeneous at the molecular level because they contain a variety of genetic disorders such as chromosomal translocations that promote tumorigenic growth and disrupt differentiation. Yes (Non-Patent Document 1).
- Non-Patent Document 6 PI3K / Akt is suppressed by a tumor suppressor gene PTEN (phosphotase and tensin homolog deleted on chromosome 10) that is frequently inactivated in human cancer.
- PTEN phosphotase and tensin homolog deleted on chromosome 10.
- PTEN lipid phosphatase activity is achieved by converting phosphatidylinositol (3,4,5) -triphosphate (PIP3) to phosphatidylinositol 4,5-bisphosphate (PIP2) which prevents Akt activation and downstream proliferation events. Counteracts the action of PI3K, which is clearly involved in cell survival (ie growth, proliferation, intracellular transport). In the absence of PTEN, Akt is sufficiently active and stimulates cells towards cancerous transformation (8, 9).
- mice exhibit conditional T cell-specific PTEN deletion (PTENf / f x lck-Cre)
- PTENf / f x lck-Cre conditional T cell-specific PTEN deletion
- Non-Patent Document 10 The resulting offspring (tPTEN ⁇ / ⁇ mice) immediately develop high grade, aggressive T cell lymphoma and die within 6-20 weeks.
- a transcriptome (expression profiling) analysis was performed that identified SLC7A5 gene upregulation.
- SLC7A5 encodes system L amino acid transporter 1 (LAT1).
- Non-Patent Document 11 On-Patent Document 11
- Non-Patent Document 12 On-Patent Document 13
- Non-Patent Document 14 On-Patent Document 15
- Non-Patent Document 16 activities to develop potent and selective inhibitors of LAT1 are ongoing (Non-Patent Document 17).
- LAT proteins ie LAT1-4
- LAT1 and LAT3 are the two isoforms that are most frequently overexpressed in tumor cells (18).
- LAT protein imports large neutral essential amino acids (LNEAA) such as leucine, valine, isoleucine, phenylalanine and tyrosine in exchange for glutamine.
- LAT1 requires a disulfide bond linked to a single transmembrane protein 4F2 cell surface antigen heavy chain (encoded by CD98hc / 4F2hc; SLC3A2 gene) so that delivery is functional (Non-patent Document 11). . It is also known to interact with ⁇ integrins (Non-patent Document 19).
- Non-Patent Document 18 the potential pharmacological role for LAT1 targeting by utilizing COMPOUND-JP, a potent and selective LAT1 inhibitor Therefore, it was to conduct research (Non-Patent Document 18); (Non-Patent Document 17); (Non-Patent Document 20).
- Kanai Expression of L-type amino acid transporter 1 (LAT1) and 4F2 heavy chain (4F2hc) in liver tumor lesions of rat models surgical 265 (Dec, 2001).
- K. Nakanishi et al. LAT1 expression in normal lung and in atypical adenomatous hyperplasia and adenocarcinoma of the lung. Virchows Archiv: an international journal of pathology 448, 142 (Feb, 2006).
- an object of the present invention is to provide a satisfactory antineoplastic agent composition that reliably suppresses the growth of cancer (malignant tumor) and has few side effects, and includes, for example, T-LL and T- It is an object of the present invention to provide a novel pharmaceutical composition, a pharmaceutical composition for gastric cancer treatment, a pharmaceutical composition for pancreatic cancer treatment, or a pharmaceutical composition for colorectal cancer treatment for enhancing the cure rate of ALL patients.
- L amino acid transporter 1 SLC7A5
- T-ALL human T cell acute lymphoblastic leukemia
- T-LL lymphoma
- COMPOUND-JP O- (5-amino-2-phenylbenzoxazol-7-yl) methyl-3,5-dichloro-L- Tyrosine or a pharmacologically acceptable salt thereof, hereinafter referred to as “COMPUND-JP” ⁇ was used.
- COMPOUND-JP decreases the viability and proliferation of leukemic cells and induces transient autophagy, followed by apoptosis, and COMPOUND-JP is a heterologous dose administered in nude mice. In vivo growth of transplanted luciferase-expressing tPTEN-/-cells could be altered.
- COMPOUND-JP was nontoxic to normal mouse thymocytes and human peripheral blood lymphocytes.
- COMPOUND-JP inhibited constitutive activation of mTORC1 and Akt.
- COMPOUND-JP caused an unfolded protein response (UPR) mediated by CHOP transcription factors associated with cell death.
- URR unfolded protein response
- the inventors also generated a CHOP-induced deficient COMPOUND-JP resistant tPTEN ⁇ / ⁇ clone.
- COMPOUND-JP is synergistic with rapamycin, dexamethasone, doxorubicin, velcade, and L-asparaginase and can alter the viability of leukemic cells.
- the results demonstrate that targeting of essential amino acid influx mediated by LAT1 can be an efficient broad-assist approach for treating fatal T cell malignancies.
- LAT1 and LAT2 genes expressed in cultured cells derived from various human cancers LAT2 was found to be low only in 5 types of cells, but 46 types of cells. LAT1 mRNA is expressed in all, and LAT1 can be said to be a cancer type transporter. On the other hand, LAT2 is understood to be a normal type with zero or extremely low expression in cancer cells.
- both human Skills gastric cancer-derived 44As3-11 cells and human pancreatic cancer-derived Panc-1 cells clearly showed a reduction in the dose-dependent proliferation activity of COM-JP. Many other LAT1-expressing cancer cells are expected to have a COM-JP growth-inhibitory effect according to their LAT1 expression level.
- the present inventors have found that the LAT1 inhibitor and the combined use of the LAT1 inhibitor and other agents are effective as the anti-malignant tumor agent composition, and have completed the present invention.
- the present invention (1) includes a LAT1 inhibitor, an alkylating agent, a platinum preparation, an antimetabolite, a topoisomerase inhibitor, a microtubule polymerization inhibitor, an endocrine therapy agent, a microtubule depolymerization inhibitor, an antitumor antibiotic
- An antineoplastic agent composition comprising, as an active ingredient, one or more agents selected from the group consisting of substances and molecular target drugs.
- the anti-neoplastic agent composition is a pharmaceutical composition for treating T cell acute lymphoblastic leukemia / lymphoma, a pharmaceutical composition for treating gastric cancer, a pharmaceutical composition for treating pancreatic cancer, or a treatment for colorectal cancer. It is a composition as described in the said invention (1) which is a pharmaceutical composition.
- the LAT1 inhibitor is O- (5-amino-2-phenylbenzoxazol-7-yl) methyl-3,5-dichloro-L-tyrosine or a pharmacologically acceptable salt thereof.
- the agent is an anthracycline antibiotic such as cisplatin, 5-fluorouracil (5-FU), gemcitabine, L-asparaginase (leinase), paclitaxel, dexamethasone, doxorubicin, velcade (bortezomib)
- anthracycline antibiotic such as cisplatin, 5-fluorouracil (5-FU), gemcitabine, L-asparaginase (leinase), paclitaxel, dexamethasone, doxorubicin, velcade (bortezomib)
- an anthracycline antibiotic such as cisplatin, 5-fluorouracil (5-FU), gemcitabine, L-asparaginase (leinase), paclitaxel, dexamethasone, doxorubicin, velcade (bortezomib)
- the present invention (5) is an antineoplastic agent composition
- an LAT1 inhibitor and one or more agents selected from an mTOR inhibitor, a PI3K inhibitor and an Akt inhibitor as active ingredients.
- the antineoplastic agent composition is a pharmaceutical composition for treating T cell acute lymphoblastic leukemia / lymphoma, a pharmaceutical composition for treating gastric cancer, a pharmaceutical composition for treating pancreatic cancer, or a treatment for colorectal cancer. It is a composition as described in said invention (5) which is a pharmaceutical composition.
- the LAT1 inhibitor is O- (5-amino-2-phenylbenzoxazol-7-yl) methyl-3,5-dichloro-L-tyrosine or a pharmacologically acceptable salt thereof.
- the present invention (8) is the composition according to any one of the inventions (5) to (7), wherein the agent is one or more agents selected from rapamycin, KU0063794 and PI-103.
- the “LAT1 inhibitor” is not particularly limited as long as it is an agent that selectively inhibits the system L amino acid transporter 1 (LAT1) and the LAT1 gene, and includes compounds and salts thereof, anti-LAT1 antibodies, aptamers, and nucleic acid drugs. .
- the LAT1 inhibitor includes a single agent or a combination of two or more agents.
- alkylating drugs is not particularly limited as long as it is a drug that alkylates DNA.
- the alkylating agent includes a single agent or a combination of two or more agents.
- Platinum preparation is not particularly limited as long as it is an antineoplastic agent containing platinum.
- any one or more existing platinum preparations approved for manufacture and sale as pharmaceuticals by government agencies, or currently clinical trials Alternatively, it refers to one or more of any platinum preparations that are in preclinical trials or that will be used in future clinical trials and that will be approved for future marketing by government agencies after the trial.
- the platinum preparation includes a single agent or a combination of two or more agents.
- the “antimetabolite” is not particularly limited as long as it is an agent that inhibits the metabolism of cancer cells and suppresses the growth.
- an antimetabolite or any one or more that are currently in clinical trials or preclinical trials, or will be used in future clinical trials and will be approved for future marketing by government agencies after the trial Of antimetabolite.
- An antimetabolite includes a single agent or a combination of two or more agents.
- topoisomerase inhibitor is not particularly limited as long as it is an agent that interferes with the action of an enzyme called topoisomerase. Or any one or more topoisomerase inhibitors that are currently in clinical trials or pre-clinical trials or will be used in future clinical trials and will be approved for future marketing by government agencies after the trial. means.
- the topoisomerase inhibitor includes a single agent or a combination of two or more agents.
- microtubule polymerization inhibitor is not particularly limited as long as it is an agent that prevents the formation of microtubules.
- any one or a plurality of existing microtubule polymerization inhibitors that have been approved for manufacture and sale as pharmaceuticals by government agencies.
- the microtubule polymerization inhibitor includes a single agent or a combination of two or more agents.
- the “endocrine therapeutic agent” is not particularly limited as long as it is an agent that suppresses secretion or action of endocrine (hormone), and for example, any one or more existing endocrine approved for manufacture and sale as a pharmaceutical product by a government agency Therapeutic agent or any one or more endocrine that is currently in clinical trials or preclinical trials or will be used in future clinical trials and will be approved for marketing as a pharmaceutical product by government agencies in the future after the trial Means therapeutic agent.
- the endocrine therapy agent includes a single agent or a combination of two or more agents.
- microtubule depolymerization inhibitor is not particularly limited as long as it is an agent that stabilizes microtubule polymerization.
- the microtubule depolymerization inhibitor includes a single agent or a combination of two or more agents.
- antineoplastic antibiotics The “antitumor antibiotic” is not particularly limited as long as it is a substance derived from a product of a microorganism that functions as an antitumor agent. Multiple optional anti-tumor antibiotics, or currently in clinical trials or preclinical trials, or used in future clinical trials, and will be approved for future marketing by government agencies after that trial By one or more optional anti-tumor antibiotics. Antitumor antibiotics include a single agent or a combination of two or more agents.
- the “molecular target drug” is not particularly limited as long as it is a drug that targets a molecule involved in tumor cell proliferation, invasion, metastasis, etc.
- an existing 1 Any one or more, or any one or more that are currently in clinical trials or preclinical trials, or will be used in future clinical trials and will be approved for future marketing by government agencies after the trial This means a molecular target drug.
- the molecular target drug includes a single agent or a combination of two or more agents.
- Antineoplastic agents include, for example, alkylating agents, platinum preparations, antimetabolites, plant alkaloids (topoisomerase inhibitors, microtubule polymerization inhibitors, microtubule depolymerization inhibitors, etc.), endocrine therapy agents, antitumors Antibiotics, molecular targeted drugs, biological response modifiers, and other drugs, for example, any one or more existing antineoplastic agents that have been approved for manufacture and sale as pharmaceuticals by government agencies Or any one or more antineoplastic tumors that are currently in clinical trials or preclinical trials or that will be used in future clinical trials and that will be approved for future marketing by government agencies after the trial. Means an agent.
- the antineoplastic agent includes a single agent or a combination of two or more agents.
- Analog means a molecule that is substantially similar in structure and function to O- (5-amino-2-phenylbenzoxazol-7-yl) methyl-3,5-dichloro-L-tyrosine.
- Cisplatin means cisplatin (CDDP), a platinum preparation, and includes cisplatin and all analogs, derivatives and congeners that have the same pharmacological properties as cisplatin.
- 5-Fluorouracil (5-FU) “5-Fluorouracil (5-FU)” means 5-fluorouracil (5-FU), which is an antimetabolite, and includes 5-fluorouracil (5-FU) and 5-fluorouracil (5-FU). Includes all analogs, derivatives and congeners with the same pharmacological properties.
- Gamcitabine refers to the antimetabolite gemcitabine and includes gemcitabine and all analogs, derivatives and congeners that have the same pharmacological properties as gemcitabine.
- L-asparaginase (leonase) “L-asparaginase (leonase)” means L-asparaginase (leonase) which is an antineoplastic agent, and has the same pharmacological properties as L-asparaginase (leonase) and L-asparaginase (leonase). , Including all analogs, derivatives and congeners.
- “Paclitaxel” refers to paclitaxel (taxol), a microtubule depolymerization inhibitor, and includes paclitaxel and all analogs, derivatives and congeners that have the same pharmacological properties as paclitaxel.
- Dexamethasone means dexamethasone, a drug with anti-inflammatory and immunosuppressive effects that contains corticosteroids as the main component, and has the same pharmacological properties as dexamethasone and dexamethasone , Including all analogs, derivatives and congeners.
- Anthracycline antibiotics including doxorubicin are not particularly limited as long as they are a group of drugs used as antitumor antibiotics derived from microorganisms belonging to the genus Streptomyces. Any existing one or more, or currently in clinical trials or preclinical trials, or used in future clinical trials, and will be approved for future marketing and marketing by government agencies in the future 1 Or a plurality of any anthracycline antibiotics.
- the anthracycline antibiotics include a single agent or a combination of two or more agents.
- Doxorubicin means doxorubicin, an anthracycline antibiotic, and includes doxorubicin and all analogs, derivatives and congeners that have the same pharmacological properties as doxorubicin.
- Velcade (bortezomib) “Velcade (Bortezomib)” means the molecular targeted drug Velcade (Bortezomib), and all analogs, derivatives and congeners that have the same pharmacological properties as Velcade (Bortezomib) and Velcade (Bortezomib) Including the body.
- Japanese Patent Application Laida “Rapamycin” means rapamycin, a molecular targeted drug, and includes rapamycin and all analogs, derivatives and congeners that have the same pharmacological properties as rapamycin.
- KU0063794 means KU0063794, a molecular targeted drug, and includes all analogs, derivatives and congeners that have the same pharmacological properties as KU0063794 and KU0063794.
- PI-103 means PI-103, a molecular targeted drug, and includes PI-103 and all analogs, derivatives and congeners that have the same pharmacological properties as PI-103.
- the “mTOR inhibitor” is not particularly limited as long as it is an agent that directly or indirectly targets mTOR (mammalian target of rapamycin) or reduces or inhibits the activity / function of mTOR. Any one or more existing mTOR inhibitors that are approved for manufacture and sale as pharmaceuticals from, or are currently in clinical trials or preclinical trials or are used in future clinical trials and are Means any one or more mTOR inhibitors that will be approved for marketing.
- the mTOR inhibitor includes a single agent or a combination of two or more agents.
- P13K inhibitor is not particularly limited as long as it is an agent that directly or indirectly targets P13K (PI3 kinase), or reduces or inhibits the activity / function of P13K. Any one or more existing P13K inhibitors approved for manufacture and sale as, or currently in clinical trials or in preclinical trials or used in future clinical trials, after which By any one or more P13K inhibitors that will be approved for marketing.
- the P13K inhibitor includes a single agent or a combination of two or more agents.
- Akt inhibitor is not particularly limited as long as it is a drug that directly or indirectly targets Akt, or reduces or inhibits Akt activity / function.
- it is manufactured and sold as a pharmaceutical from a government agency.
- the Akt inhibitor includes a single agent or a combination of two or more agents.
- FIG. 1 is a diagram for tPTEN ⁇ / ⁇ tumors, T-ALL / T-LL cell lines, and primary samples with elevated levels of CD98 expression.
- FIGS. 1B-E show flow cytometric analysis of surface CD98 levels after anti-mouse CD98 rat mAb staining followed by development with secondary anti-rat FITC antibody, specific MFI / non-specific MFI ratio.
- FIG. 1C shows CD98 expression levels between quiescent or PMA + ionomycin activated (24 hours) normal mouse thymocytes and KO99L cells.
- FIG. 1C shows CD98 expression levels between quiescent or PMA + ionomycin activated (24 hours) normal mouse thymocytes and KO99L cells.
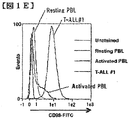
- FIG. 1E shows the Facs profile for CD98 expression between quiescent or 24-hour activated human PBL and T-ALL sample # 1.
- FIG. 2 is a diagram for the functional effects of LAT-1 inhibition in tPTEN ⁇ / ⁇ and T-ALL / T-LL cell models.
- FIG. 2A shows an analysis of metabolic activity using the WST-1 cell viability assay in the presence of amino acid uptake disruptors. Cells were incubated with COMPOUND-JP, BCH, or D-leucine for 48 hours. COMPOUND-JP: average of triplicate 7 independent experiments +/ ⁇ SEM; p-value was calculated using a two-sided student t test; BCH, D-Leu: triplicate one experiment.
- FIG. 1E shows the Facs profile for CD98 expression between quiescent or 24-hour activated human PBL and T-ALL sample # 1.
- FIG. 2 is a diagram for the functional effects of LAT-1 inhibition in tPTEN ⁇ / ⁇ and T-ALL / T
- FIG. 2B shows proliferation assessed by BrdU incorporation Elisa. Data are shown as the average of two independent experiments performed in quadruplicate (mean +/ ⁇ SEM; student's t test).
- FIG. 2C is a diagram in which cells are treated with COMPOUND-JP or not and supplemented with nonessential (N) or essential (E) amino acids, if indicated, in the media. Cell viability was measured after 48 hours (mean of triplicate independent experiments +/ ⁇ SEM; Student's t test).
- FIG. 2D shows cell death analyzed by flow cytometry after 48 hours of treatment and after DAPI staining (mean of triplicate independent experiments +/ ⁇ SEM, Student's t test).
- FIG. 2E-G nude mice were inoculated subcutaneously with KO99L-LUC cells and treated daily with COMPOUND-JP (1.5 mg / mouse / day) 48 hours later. Tumor measurements were taken after 18 days; mean +/ ⁇ SEM. Bioluminescence was recorded with a BioImager after intraperitoneal injection of D-luciferin.
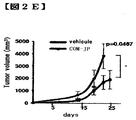
- FIG. 2E shows tumor volume; Student's t test.
- FIG. 2F shows whole body mouse imaging.
- the pseudo-color scale shows the relative bioluminescence change over time (ph / s / r: photon / second / radiance).
- FIG. 2G shows the bioluminescence quantification of the tumor. The p value was calculated using a two-sided Mann-Whitney test.
- FIG. 2H shows a measurement of cell viability of Jurkat human T-ALL cells in the presence of the indicated doses of COMPOUND-JP.
- COMPOUND-JP affects the T-ALL / T-LL model.
- FIG. 2I shows the measurement of the same cell proliferation ability (average of three independent experiments +/ ⁇ SEM; Student's t test).
- FIG. 3 shows that COMPOUND-JP induces caspase activation, apoptotic cell death, and autophagy.
- FIG. 3 shows that COMPOUND-JP induces caspase activation, apoptotic cell death, and autophagy.
- FIG. 3A shows a representative cytometry plot of KO99L cells after a 48 hour incubation period with effectors followed by staining with Annexin-V-FITC and DAPI. The percentage of cells in each state is shown in the quadrant. Data are representative of 5 independent experiments.
- FIG. 3B is a graph showing the mitochondrial potential difference measured by Facs after TMRE staining after stimulation of KO99L cells with COMPOUND-JP for 48 hours. B1, Facs profile; B2, quantification of results. Representative of 2 independent experiments.
- FIG. 3D shows the Facs profile for caspase 3 activation after 48 hours using D1, Red-DEVD-fmk fluorescent substrate. D2, quantification of results representative of 2 independent experiments.
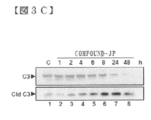
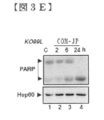
- FIG. 3E shows a time course Western blotting analysis of PARP cleavage by caspase 3 in KO99L cells.
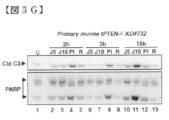
- FIG. 3G shows Western blotting analysis of caspase 3 activation and PARP cleavage in primary tPTEN ⁇ / ⁇ tumor cells.
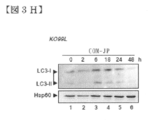
- FIG. 3H shows Western blotting analysis of LC3 processing induced by COMPOUND-JP (10.0 ⁇ M) in KO99L cells.
- FIG. 3I shows the time course effect of caspase inhibition on COMPOUND-JP-induced LC3 processing and JNK activation in KO99L cells. QVD-OH is used at 20.0 ⁇ M. All Western blot data are representative of at least 3 independent experiments.
- FIG. 4 illustrates that targeting of LAT1 function interferes with mTORC1 activation in vitro and in vivo.
- FIG. 4 illustrates that targeting of LAT1 function interferes with mTORC1 activation in vitro and in vivo.
- FIG. 4A shows a time course experiment on KO99L cells with the indicated doses of COMPOUND-JP. Total cell lysates were analyzed by Western blotting with specific antibodies. Data are representative of at least 3 independent experiments.
- FIG. Isotype controls are shown as gray lines (mean +/ ⁇ SEM). Untreated or treated sample curves were normalized to peak height. An event (10 4 ) was obtained for each sample. The conditions were run in duplicate.
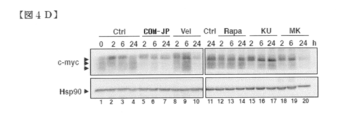
- FIG. 4D shows a Western blotting analysis of c-myc levels following stimulation of KO99L cells with the indicated inhibitors.
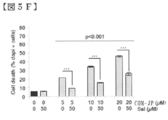
- FIG. 5 is a diagram regarding the involvement of an induced stress response to the action of COMPOUND-JP.
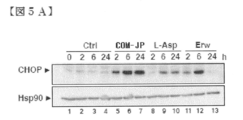
- FIG. 5A is a diagram showing analysis of CHOP protein levels by Western blotting in KO99L cells.
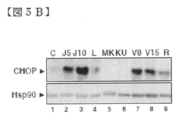
- FIG. 5B is a diagram showing analysis of CHOP protein levels by Western blotting in KO99L cells.
- COMPOUND-JP 5.0 ⁇ M (J5), 10.0 ⁇ M (J10); Velcade 8.0 nM (V8), 15.0 nM (J15), rapamycin 10.0 nM (R).
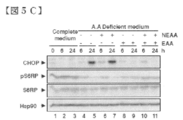
- FIG. 5C shows the effect of amino acid depletion / complementation on the signal pathway. The medium was diluted to 10% with HBSS and supplemented with nonessential (NEAA) (100 ⁇ ) or essential (EAA) amino acids (50 ⁇ ) prior to recovery and Western blotting analysis.
- NEAA nonessential
- EAA essential amino acids
- FIG. 5E cells were treated with salubrinal (50 ⁇ M) for 30 minutes prior to the addition of COMPOUND-JP (10 ⁇ M).
- FIG. 5F is a diagram showing the reactivity of the COMPOUND-JP effect on salubrinal. Cell death visualized by DAPI staining (data are representative of 3 independent experiments performed in triplicate; mean +/ ⁇ SEM; student t test). Growth is BrdU Elisa
- FIG. 5G is a diagram showing the reactivity of the COMPOUND-JP effect on salubrinal. (One experiment performed in triplicate; mean +/ ⁇ SEM; Student t test) or cell count.
- FIG. 5G is a diagram showing the reactivity of the COMPOUND-JP effect on salubrinal. (One experiment performed in triplicate; mean +/ ⁇ SEM; Student t test) or cell count.
- FIG. 5H is a diagram showing the reactivity of the COMPOUND-JP effect on salubrinal. (Data are representative of 3 independent experiments performed in triplicate; mean +/ ⁇ SEM; Student's t test).
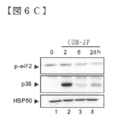
- FIG. 6 is a diagram for molecular characterization of the inducible stress response mobilized by COMPOUND-JP.
- FIGS. 6A-C show time-course western blotting analysis of ISR components upon stimulation of COMPOUND-JP (10 ⁇ M) in KO99L cells.
- FIG. 6D is a diagram showing a time course western blotting analysis of Bcl2 family members upon stimulation of COMPOUND-JP (10 ⁇ M) of KO99L cells.
- FIG. 6E shows the effect of salubrinal on COMPOUND-JP-induced Bcl2 family members. Hsp90 was used as a loading control. All data are representative of at least 3 independent experiments.
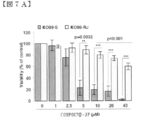
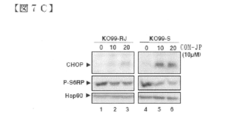
- FIG. 7 is a diagram for characterization of COMPOUND-JP resistant mutants. The parent cells of KO99L cells (KO99-S) sensitive to COMPOUND-JP were incubated for several months with increasing drug concentration to obtain viable resistant cells (KO99-RJ). It was.
- FIG. 7A shows an analysis of cell viability by WST-1 assay after 48 hours of culture with increasing concentrations of COMPOUND-JP (average of 5 independent experiments performed in quadruplicate). +/- SEM; Student's t test).
- FIG. 7A shows an analysis of cell viability by WST-1 assay after 48 hours of culture with increasing concentrations of COMPOUND-JP (average of 5 independent experiments performed in quadruplicate). +/- SEM; Student's t test).
- FIG. 7B shows analysis of cell death by PI staining 48 hours after culture with increasing concentrations of COMPOUND-JP (average of at least 3 independent experiments performed in quadruplicate; average + / -SEM, Student's t test).
- FIG. 7C shows Western blotting analysis of CHOP induction and S6RP phosphorylation in resistant mutants compared to parental cells. Data are representative of at least 3 independent experiments.
- FIG. 8 is a diagram for a combination study between COMPOUND-JP and a chemotherapeutic agent.
- FIG. 8A is a diagram showing a heat map for the expression of the combination index (CI). Synergy between drugs was analyzed using the Chou-Talalay method.
- FIG. 8B shows an isobologram for the combination effect.
- FIGS. 9-1 and 2 are diagrams showing a reaction scheme of the compound according to the present invention.
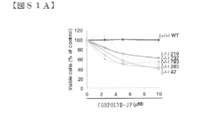
- FIG. S1A shows that COMPOUND-JP interferes with the survival of primary tPTEN ⁇ / ⁇ cells but not normal thymocytes. Fresh primary cells from tPTEN KO tumors (-/-) or normal thymocytes (+ / +) were incubated with the indicated doses of COMPOUND-JP prior to WST-1 analysis for cell viability.
- FIGS. S1B-D show that COMPOUND-JP is not toxic to normal human blood cells.
- STS staurosporine (1.0 ⁇ M) is a positive inducer of cell death.
- FIG. S1D shows the viability of normal human cord blood cells in the presence of COMPOUND-JP (48 hours) analyzed after DAPI staining and Facs quantification of live cells.
- S2 shows the effect of a signal transduction inhibitor.
- Total cell lysates separated by SDS-PAGE before analysis by Western blotting with specific antibodies are shown.
- Time course for KO99L cells with indicated doses of MK2206 (5 ⁇ M), KU0063794 (5 ⁇ M), rapamycin (10 nM), Erwinase (2.5 UI / ml), L-asparaginase (2.5 UI / ml), Velcade (15 nM) Experiment. Data are representative of 3 independent experiments.
- FIGS. S3-1 and S2-1 show a combination study.
- FIG. S4 is a diagram showing molecular events for LAT1 targeting by COMPOUND-JP.
- FIG. 10 is a diagram showing the results of quantification of neutral amino acid transporters, LAT1 and LAT2 genes expressed in cultured cells derived from various human cancers. The amount of mRNA per unit RNA is indicated on the vertical axis, the name of the cancer cell used is indicated on the horizontal axis, and the cancer type name is indicated in the table. The upper row shows LAT1 and the lower row shows LAT2.
- FIG. 11 is a graph showing the inhibitory effect of the LAT1 selective inhibitor, COM-JP, on the proliferation of human skill gastric cancer-derived 44As3-11 cells and human pancreatic cancer-derived Panc-1 cells. Each value was expressed as an average value of 4 cases under the same conditions as a relative value with the activity in the absence of COM-JP as 100%. Mean values, standard errors, and p-values for significant difference tests are shown in the figure.
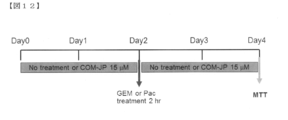
- FIG. 12 is a diagram showing an experimental procedure showing the action of LAT1 selective inhibitor, COM-JP, existing drug gemcitabine (GEM) and paclitaxel (Pac) alone and in combination.
- FIG. 12 is a diagram showing an experimental procedure showing the action of LAT1 selective inhibitor, COM-JP, existing drug gemcitabine (GEM) and paclitaxel (Pac) alone and in combination.
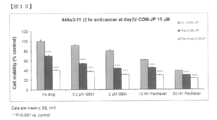
- FIG. 13 is a graph showing the combined effect of COM-JP and other agents on the proliferation of 44 As3-11 cells derived from human Skills gastric cancer.
- the effect of Gemstabine and Paclitaxel acting on the growth of 44 As3-11 cells derived from human Skills gastric cancer alone for 2 hours corresponds to the difference in the value of the bar graph on the left of each group column.
- FIG. 14 shows the combined effect of COM-JP and other agents on the proliferation of human pancreatic cancer-derived Panc-1 cells.
- FIG. 15 is a diagram showing the combined effect of COM-JP and 5-FU on tumor growth in a human colon cancer-derived HT-29 cell seeded nude mouse model.
- FIG. 16 shows the synergistic effect of the combined use of COM-JP and CDDP on tumor growth in an orthotopic seeded nude mouse model (SOI model) of human colon cancer-derived HT-29 cells.
- SOI model orthotopic seeded nude mouse model
- Group 16 is a bar graph showing the size of the tumor in each treatment group.
- Group 1 is the control group, 2 to 4 are COM-JP, 3.1, 12.5 and 50 mg / Kg / day, each body weight is administered intraperitoneally for 4 weeks, and Group 5 is CDDP 2.5 mg / Kg / day.
- 0, 6 and 13 3 intraperitoneal single administrations; group 6-8: combined administration of 2-4 and 5; group 9: COM-JP 300 mg / Kg / day for 4 weeks Oral administration was performed every day.
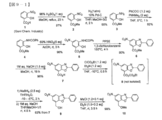
- the LAT1 inhibitor according to the present invention contains an aromatic amino acid derivative described in the following compounds 1 to 10 or a pharmacologically acceptable salt thereof as an active ingredient.
- Compound 1 is Formula (I): [Wherein X is a halogen atom, an alkyl group or an alkoxyl group, Y is O or NH; l is 0 or 1, m is 0, 1 or 2; n is an integer from 0 to 5, R 1 is a hydrogen atom or an amino protecting group, R 2 is a hydrogen atom or an alkyl, aralkyl or aryl group, R 3 represents (1) a halogen atom; (2) an aroylamino group in which the amino moiety may be substituted with lower alkyl; (3) A phenyl group substituted with phenyl, phenoxy, pyridyl, pyrimidinyl or quinolyl (wherein each substituent in the phenyl group is a halogen atom, cyano, hydroxy, carboxy, lower alkoxy, lower alkoxycarbonyl, phenyl, di Optionally substituted with lower alkylamino or thiomorpholinyl);
- Compound 2 is Formula (I): [Wherein X is a chlorine or iodine atom or an alkyl or alkoxyl group; Y is O or NH; l is 0 or 1, m is 0 or 2, n is an integer from 0 to 5, R 1 is a hydrogen atom or a trifluoroacetyl or t-butoxycarbonyl group, R 2 is a hydrogen atom or an alkyl group, R 3 represents (1) a bromine atom; (2) an aroylamino group in which the amino moiety may be substituted with lower alkyl; (3) a phenyl group substituted with a phenyl, phenoxy, pyridyl, pyrimidinyl or quinolyl group (wherein each substituent in the phenyl group is a halogen atom or cyano, hydroxy, carboxy, lower alkoxy, lower alkoxycarbonyl, phenyl, Optionally further substituted with di-lower alky
- Compound 3 is The aromatic according to Compound 2, wherein the unsaturated monocyclic heterocyclic group in (3) of R 3 is a 5-membered or 6-membered heterocyclic group containing N, N and O, or N and S. An amino acid derivative or a pharmacologically acceptable salt thereof.
- Compound 4 is The aromatic amino acid derivative or the pharmaceutically acceptable salt thereof according to compound 2 or 3, wherein the unsaturated monocyclic heterocyclic group in (3) of R 3 is pyridyl, oxazolyl or thiazolyl.
- Compound 5 is The aromatic amino acid derivative or a pharmaceutically acceptable salt thereof according to any one of compounds 2 to 4, wherein R 1 is a hydrogen atom.
- Compound 6 is An aromatic amino acid derivative or a pharmaceutically acceptable salt thereof according to any one of compounds 2 to 4, wherein R 1 is trifluoroacetyl or t-butoxycarbonyl group.
- Compound 7 is R 3 is (3) a phenyl group substituted with a phenyl, phenoxy, pyridyl, pyrimidinyl or quinolyl group (wherein each substituent in the phenyl group is further substituted with a halogen atom or a hydroxy or di-lower alkylamino group)
- X is a chlorine atom
- (4) a naphthyl group optionally substituted with hydroxy where X is a chlorine atom and m is 2 when naphthyl is substituted with hydroxy
- a phenyl or naphthyl group A substituted N and O-containing unsaturated monocyclic heterocyclic group wherein each substituent in the monocyclic heterocyclic group may be further substituted with a hydroxy group, wherein X is chlorine Atoms, m is 2 and l is 1); Is, The aromatic amino acid derivative described in Compound 2 or a pharmacologically acceptable salt thereof.
- Compound 8 is R 3 is (3) a phenyl group substituted with a phenyl or pyridyl group (wherein each substituent in the phenyl group may be further substituted with a halogen atom or a di-lower alkylamino group, X is a chlorine atom); or (4) a naphthyl group optionally substituted with a hydroxy group (wherein when naphthyl is substituted with hydroxy, X is a chlorine atom and m is 2) Is, The aromatic amino acid derivative described in Compound 2 or a pharmacologically acceptable salt thereof.
- Compound 9 is R 3 is (3) a phenyl group substituted with a pyridyl substituted with a di-lower alkylamino group (wherein X is a chlorine atom in this case); (4) a naphthyl group (where n is 0 or 2); or (5) an N and O-containing unsaturated monocyclic heterocyclic group substituted with a naphthyl group,
- Compound 10 is The aromatic amino acid derivative or the pharmaceutically acceptable salt thereof according to any one of compounds 2 to 9, wherein X is a chlorine or iodine atom.
- COMPOUND-JP which is a LAT1 inhibitor according to the present invention is O- (5-amino-2-phenylbenzoxazol-7-yl) methyl-3,5-dichloro-L represented by the following formula (II).
- Tyrosine or a pharmacologically acceptable salt thereof is particularly suitable as the antineoplastic agent composition according to the present invention.
- “pharmacologically acceptable salt” means, for example, alkali metal salts (sodium salt, potassium salt, etc.), alkaline earth metal salts (calcium salt, magnesium salt, etc.), ammonium salts, organic bases (trimethylamine).
- organic acids acetic acid, benzoic acid, succinic acid, fumaric acid, maleic acid, lactic acid, citric acid, tartaric acid, gluconic acid, methanesulfonic acid, Salts with benzen
- the compound and its salt may be in the form of a hydrate or a solvate such as ethanolate.
- the compound or a salt thereof is preferably an inclusion body of cyclodextrin (for example, ⁇ -cyclodextrin).
- cyclodextrin mass ratio
- the compound or a salt thereof: cyclodextrin (mass ratio) is preferably 1:10 to 70, more preferably 1:20 to 50, and particularly preferably 1:25 to 35.
- the LAT1 inhibitor according to the present invention includes an anti-LAT1 antibody.
- the anti-LAT1 antibody is not particularly limited as long as it is an antibody that can specifically recognize LAT1, and examples thereof include an anti-hLAT1 monoclonal antibody, an anti-mouse LAT1 monoclonal antibody, an anti-hLAT1 polyclonal antibody, and an anti-mouse LAT1 polyclonal antibody.
- the anti-LAT1 antibody is not particularly limited as long as LAT1 is used as an antigen and binds to the antigen, and a mouse antibody, a rat antibody, a rabbit antibody, a sheep antibody, or the like can be appropriately used.
- a hybridoma producing a monoclonal antibody can be basically produced using a known technique as follows. That is, a desired antigen or a cell expressing the desired antigen is used as a sensitizing antigen and immunized according to a normal immunization method, and the resulting immune cell is fused with a known parent cell by a normal cell fusion method. And can be prepared by screening monoclonal antibody-producing cells (hybridomas) by a normal screening method.
- the hybridoma can be prepared, for example, according to the method of Milstein et al. (Kohler. G. and Milstein, C., Methods Enzymol. (1981) 73: 3-46).
- LAT1 or a fragment of the protein When producing an anti-LAT1 monoclonal antibody, LAT1 or a fragment of the protein can be used as an antigen, and cells expressing LAT1 or the protein fragment can be used as an antigen.
- LAT1 or a fragment of the protein is in accordance with, for example, the method described in Molecular Cloning: A Laboratory Manual 2nd edition Vol. 1-3 brook Sambrook, J. et al., Cold Spring Harber Laboratory Press publication New York ⁇ 1989. Can be obtained.
- cells expressing LAT1 or a fragment of the protein can also be obtained by the method described in Molecular Cloning: A Laboratory Manual 2nd edition Vol. 1-3 brook Sambrook, J. et al., Cold Spring Harber Laboratory Press publishing New York 1989. Can be obtained according to the above.
- Polyclonal antibodies are obtained by administering an antigen subcutaneously or intraperitoneally about 2 to 4 times every 2 to 3 weeks together with a commercially available adjuvant (for example, complete or incomplete Freund's adjuvant). Can be obtained by collecting whole blood about 3 to 10 days after the final immunization and purifying the antiserum.
- adjuvant for example, complete or incomplete Freund's adjuvant
- animals to which the antigen is administered include mammals such as rats, mice, rabbits, goats, guinea pigs, and hamsters.
- antineoplastic agent composition according to the present invention can be administered, for example, orally, transdermally, or by injection.
- Tablets, granules and capsules for oral administration are conventional additives such as binders (eg syrup, gum arabic, gelatin, sorbitol, tragacanth or polyvinylpyrrolidone); fillers (eg lactose, sugar, corn starch, calcium phosphate) Sorbitol or glycine); lubricants (eg magnesium stearate, talc, polyethylene glycol or silica); disintegrants (eg potato starch) or wetting agents (eg sodium lauryl sulfate). Tablets, granules and capsules may be formed into a film by a method known in the field of ordinary preparations.
- binders eg syrup, gum arabic, gelatin, sorbitol, tragacanth or polyvinylpyrrolidone
- fillers eg lactose, sugar, corn starch, calcium phosphate) Sorbitol or glycine
- lubricants eg magnesium
- Liquid formulations for oral administration may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or as lyophilized formulations that dissolve in water or other suitable solvent prior to use. Good.
- Liquid formulations are conventional additives such as suspending agents (eg sorbitol, syrup, methylcellulose, glucose syrup, gelatin water edible fat); emulsifiers (eg lecithin, sorbitan monooleate or gum arabic); non-aqueous excipients (Eg almond oil, fractionated coconut oil or oily esters such as glycerin, propylene glycol or ethyl alcohol); including preservatives (eg methyl or propyl p-hydroxybenzoate or sorbic acid) and flavoring or coloring agents You may go out.
- suspending agents eg sorbitol, syrup, methylcellulose, glucose syrup, gelatin water edible fat
- emulsifiers eg lecithin, sorbitan monooleate or gum arabic
- non-aqueous excipients Eg almond oil, fractionated coconut oil or oily esters such as glycerin, propylene glycol or ethyl alcohol
- the active ingredient When administered transdermally, the active ingredient may take the form of a cream, lotion or ointment. Cream or ointment preparations that can be used as drugs can be prepared by methods well known in the art.
- An injection can be produced by suspending or dissolving the compound or a salt thereof in an appropriate medium.
- the injection may contain adjuvants such as local anesthetics, preservatives and buffering agents.
- the dosage of the antineoplastic agent composition according to the present invention should be appropriately adjusted according to various factors including the age, weight, general health, sex, administration time, administration route, and severity of disease of the patient. Although not limited, for example, it is usually appropriate to administer about 10 to 5000 mg, preferably about 100 to 3000 mg per day for an adult in 1 to 5 divided doses.
- LAT1 inhibitor according to the present invention, alkylating agent, platinum preparation, antimetabolite, topoisomerase inhibitor, microtubule polymerization inhibitor, endocrine therapy agent, microtubule depolymerization inhibitor, antitumor antibiotic and molecular target drug
- the dose (mass ratio) of one or more agents selected from the group consisting of various factors including the patient's age, weight, general health, sex, administration time, route of administration, severity of disease Although it is not limited, for example, if the normal dose per day for an adult for each single use is 1, usually an LAT1 inhibitor, an alkylating agent, a platinum preparation,
- the reaction solution was diluted with methylene chloride (200 ml), washed successively with water and saturated brine, and dried, and the solvent was evaporated under reduced pressure.
- the methyl ester (2.37 g, 73%) was obtained as pale yellow crystals. m. p.
- Lithium aluminum hydride 53 mg, 1.42 mmol was added to a solution of [2- (4-pyridyl) -benzoxazol-7-yl] carboxylic acid methyl ester (360 mg, 1.42 mmol) in tetrahydrofuran (15 ml) under ice-cooling and stirring. ) was added over 10 minutes and stirred at the same temperature for 0.5 hour.
- the catalyst was removed by filtration, the solvent of the filtrate was concentrated to about 5 ml, and the precipitated crystals were collected by filtration.
- Ethyl acetate (50 ml) -water (30 ml) was added to the collected crystals, and the mixture was adjusted to pH 8-9 with saturated aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate.
- the extracted organic layer is washed with saturated brine and dried, and the solvent is distilled off under reduced pressure to obtain 1,2,3,4-tetrahydronaphthalene [1,2-a] benzimidazole-7-carboxylic acid methyl ester. (96 mg, 20%) was obtained as slightly yellow crystals. m. p.
- 2-phenylbenzimidazole-4-carboxylic acid methyl ester (3.89 g, 64%) as pale yellow crystals.
- Tri-n-butyltin chloride (2.5 ml) was then added dropwise, and the mixture was stirred at the same temperature for 0.5 hours and at room temperature for 1 hour. did.
- a 10% aqueous potassium fluoride solution (50 ml) and ethyl acetate (50 ml) were sequentially added to the reaction solution, and the mixture was stirred at room temperature for 0.5 hour, and then the organic layer was separated. The organic layer was washed sequentially with water and saturated brine, and after drying, the solvent was distilled off under reduced pressure.
- reaction mixture was cooled to ⁇ 10 ° C., ethyl chlorocarbonate (10.68 ml, 0.112 mol) was added, and the mixture was stirred at the same temperature for 30 min.
- Sodium borohydride (8.84 g, 233.7 mmol) was added and stirred for 30 minutes, and then THF / water (317 ml / 64 ml) was added dropwise at ⁇ 10 to ⁇ 6 ° C. over 3 hours.
- the reaction solution was further stirred for 2 hours, after which ice water was poured, and the precipitated solid was collected by filtration and washed with water.
- the reaction solution was stirred at the same temperature for 1 hour, then gradually warmed up and stirred at room temperature for 20 hours.
- Diisopropyl ether was added and the solid was collected by filtration and washed with diisopropyl ether.
- the obtained solid was divided into 3 lots and dissolved in ethanol (2300 ml in total), and insoluble matter was filtered off.
- the filtrate was concentrated to about 1000 ml under reduced pressure at room temperature, and the resulting solution was stirred at room temperature.
- the produced crystal was filtered and dried to obtain 66.11 g (81%) of a compound salt (17) according to the present invention as a pale yellow solid.
- the filtrate was placed in a sterile 5 L container and diluted with the SBE-CD solution prepared in (1) to adjust the final volume to 2800 ml.
- (5) After further stirring for 5 minutes, the pH was measured and confirmed to be ⁇ 6.5. Using a sterile dispenser, each 15 ml was dispensed into 30 ml vials. The final compound concentration was 10 mg / ml.
- Each vial was processed in a lyophilizer (Lyostar3, FTS Systems SP Scientific) for 1 week.
- the vial after freeze-drying was sealed with a rubber stopper, metal creased, and stored in a refrigerator.
- (7) For re-solutionization, 14.1 ml of distilled water for injection was added, and the solution was turned into a 15 ml light yellow solution by a voltex mixer for 5-10 minutes.
- Pten-deficient mouse (Pten-deficient mouse (tPTEN-/-)) Animal experiments were performed in accordance with the Declaration of Helsinki, and the protocol was approved by the institutional ethics committee (2011-73) and CIEPAL (NCE / 2011-33). Mice were maintained in a C3M animal room facility under specific pathogen removal conditions. Pten-deficient mice (tPTEN ⁇ / ⁇ ) were generated by crossing mice with two Pten flox alleles with proximal lck promoter-cre transgenic mice (reference 53). Animals were characterized by PCR as previously described (ref. 53).
- mice were sacrificed by lethal pentobarbital injection. Tumors were dissected and homogenized in PBS on a 70 ⁇ m cell strainer (BD Biosciences) and total RNA was isolated and transcribed into cDNA as previously described (reference 54). Four unrelated tumors were analyzed and thymocytes from two wild type mice were also processed.
- cDNA is hybridized to an Affymetrix DNA chip (Affymetrix-MoGene-1_0-st-v1), gene expression data is normalized (GSE 39591), and a 25% false discovery rate is used to supervised SAM. (Significance Analysis of Microarrays) Analysis was performed. Then, a linear model (LIMMA) for microarray analysis, p value ⁇ 10 4, was used to identify the probes that are differentially expressed. This results in gene selection with abs (log Fold Change)> 0.5. After removing duplicates with smaller p-values between phenotype and non-annotated sequences, the combination of analyzes resulted in 498 up-regulated genes and 279 down-regulated genes in the tumor. Identified.
- Affymetrix DNA chip Affymetrix-MoGene-1_0-st-v1
- GSE 39591 Gene expression data is normalized
- a 25% false discovery rate is used to supervised SAM. (Significance Analysis of Microarrays) Analysis
- Cell culture In addition to mouse cell lines designated as KO99L, KO1081L, and KO631L established from irrelevant tPTEN-/-tumors, human Ke37, DND41, Sil-ALL, Peer, Molt-16, Jurkat (T-ALL), And SupT1 (T-LL), 10 or 20% (Sil-ALL and mouse cell lines) fetal bovine serum, and 50 units / ml penicillin, 50 mg / ml streptomycin, and 1.0 mM sodium pyruvate (Sigma-Aldrich) ) In RPMI 1640 medium (Invitrogen) supplemented. Cell cultures were maintained at 37 ° C. under 5% CO 2 .
- TMB peroxidase
- Cell extracts were washed with cell lysis buffer (50 mM HEPES supplemented with a cocktail of protease inhibitors, pH 7.4, 150 mM NaCl, 20 mM EDTA, 100 mM NaF, 10 mM Na 3 VO 4 , 0.5% nonylphenoxypolyethoxyl ethanol (NP40 )).
- cell lysis buffer 50 mM HEPES supplemented with a cocktail of protease inhibitors, pH 7.4, 150 mM NaCl, 20 mM EDTA, 100 mM NaF, 10 mM Na 3 VO 4 , 0.5% nonylphenoxypolyethoxyl ethanol (NP40 )).
- Annexin V / PI staining assay Apoptosis assessment was performed by measuring phosphatidyl-serine membrane redistribution with Annexin-V-FITC (Invitrogen). Cells (5.0 ⁇ 0.1 ⁇ 10 5 per well; 2.0 mL) are incubated with effector, resuspended in staining solution (110 ⁇ L; Annexin-V (1/10)), and in the dark (15 minutes) , RT).
- TMRE tetramethylrhodamine ethyl ester perchlorate assay
- mice in each experimental group were injected subcutaneously with 99KOL-LUC T lymphoma cells (200 ⁇ L; 5.0 ⁇ 0.1 ⁇ 10 6 ) stably expressing the reporter luciferase gene.
- COMPOUND-JP prepared by dissolving and mixing the drug powder in saline (NaCl 0.9%) (50 mg / kg / day / 5 per week). Days).
- mice received an intraperitoneal injection (150 ⁇ L, 30 mg / mL) of D-luciferin (Caliper Life Science) and anesthetized with inhaled isoflurane (2.0%).
- the bioluminescent signal was monitored using a Photon imager (Biospace Lab) equipped with a sensitive cooled CCD camera, and all images were collected 10 minutes after D-luciferin injection. Data were analyzed using total photon flux emission (photons / second) focused on the image area of interest (ROI) covering the tumor area.
- ROI image area of interest
- Sections (4 ⁇ m) in dissected tumor paraffin blocks were labeled with primary rabbit phospho S6RP antibody (# 4858, Cell Signaling Technology).
- a secondary anti-rabbit antibody conjugated with peroxidase (HRP) (SignalStain®, # 8114, Cell Signaling Technology), as recommended by the manufacturer, prior to development with diaminobenzidine (DAB), Added to the section.
- Sections were stained with hematoxylin / eosin / saffron (HES) for assessment of necrosis. Images were analyzed and necrotic area was quantified using ImageJ software (NIH).
- the culture was continued for 5 days in the absence of COM-JP and in the presence of different concentrations of COM-JP in the culture solution of each cancer cell.
- the proliferation activity of the cells over the course of each day was measured by the MTT method.
- HT-29 cells were seeded subcutaneously in mice, and the tumor mass size changed with COM-JP and 5-FU alone or in combination from the state where the size of the tumor grew to a certain size or more. Measured daily.
- FIG. 16 is a bar graph showing the size of the tumor in each treatment group.
- Group 1 is the control group
- 2 to 4 are COM-JP, 3.1, 12.5 and 50 mg / Kg / day, each body weight is administered intraperitoneally for 4 weeks
- Group 5 is CDDP 2.5 mg / Kg / day.
- 0, 6 and 13 3 intraperitoneal single administrations
- group 6-8 combined administration of 2-4 and 5
- group 9 COM-JP 300 mg / Kg / day for 4 weeks Oral administration was performed every day.
- TPTEN-/-tumors human T-ALL / T-LL cell lines, and primary cells have enhanced LAT1 / CD98hc (slc7a5 / slc3a2) expression
- Mice tPTEN-/- develop high-grade, invasive lymphoma.
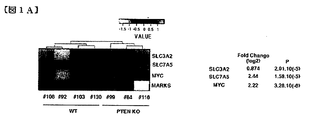
- tPTEN-/-tumor cells showed 498 up-regulated genes.
- SLC7A gene encoding LAT1 was upregulated in tPTEN ⁇ / ⁇ compared to normal cells (log2: 2.44 times).
- SLC3A2 encoding CD98hc was only slightly enhanced (0.874-fold) in tumors.
- c-myc gene (2.22 fold), which is also highly expressed in tPTEN ⁇ / ⁇ cells, Some marks were not significantly different between the two categories.
- the three primary human T-ALL samples showed higher CD98 levels compared to quiescent and activated peripheral blood lymphocytes (PBL) from healthy donors (FIGS. 1D-E). Generally speaking, these results indicate that CD98 expression was enhanced in activated mouse and human T cells and reached a higher level in transformed lymphocytes.
- BCH is a classic system L inhibitor, but lacks LAT1 selectivity (Non-patent Document 18). As shown in FIG. 2A, BCH and D-leucine maintain high cell viability at high dose concentrations, with cell viability being 30% and 60% at 10.0 and 30.0 mM, respectively. FIG. 2A).
- COMPOUND-JP decreased cell cycle (proliferation) by 75% after 48 hours (FIG. 2B).
- COMPOUND-JP affected the cell viability of freshly isolated tPTEN-/-tumor cells but was non-toxic in normal mouse thymocytes (Supplementary Figure S1A) .
- COMPOUND-JP inhibits essential amino acid influx
- COMPOUND-JP alters human T-ALL / T-LL cell lines and patient primary cells
- COMPOUND-JP alters human T-ALL / T-LL cell lines and primary tumor cells.
- the human Jurkat T-ALL strain was utilized to demonstrate that COMPOUND-JP can alter cell survival (FIG. 2H) and proliferation (FIG. 2I).
- primary T-ALL cells isolated from 4 different patients also showed COMPOUND-JP sensitivity (cell viability) in a concentration-dependent manner (FIG. 2J). Although very different, COMPOUND-JP did not induce cell death in normal quiescent or activated human PBL cells (FIG. S2B).
- COMPOUND-JP has PBL cell proliferation (1.0-50.0 ⁇ M; Neither S2C) nor normal cord blood mononuclear cells (5.0 and 10.0 ⁇ M; FIG. S2D) were significantly altered. Consistent with its known LAT1 / LAT2 inhibitor selectivity, these experimental results show that COMPOUND-JP is more leukemic (LAT1 expression) compared to normal (healthy, LAT2 expression) T cells. On the other hand, it demonstrates that it acts preferentially.
- COMPOUND-JP causes apoptosis and autophagy
- Annexin-V + and Annexin-V + / DAPI + cell analysis (FIG. 3A) after 48 hours compared to the control (no COMPOUND-JP) is 10.0 and 20.0 ⁇ M, 2.8 and 3. It showed 8 times higher level of apoptosis.
- Mitochondrial outer membrane permeabilization is an early apoptotic cell death event that distorts mitochondrial transmembrane potential ( ⁇ m) (Ref. 23).
- TMRE tetramethylrhodamine ethyl ester perchlorate staining assay
- COMPOUND-JP 5.0 and 10.0 ⁇ M was shown to cause MOMP in KO99L cells (FIG. 3B1).
- the mitochondrial depolarization observed in KO99L cells was found to be COMPOUND-JP concentration dependent (2.5-20.0 ⁇ M; 48 hours), reaching 70% at the highest concentration tested (FIG. 3B2). .
- LC3-II accumulation peaked at 6.0 hours (FIG. 3H, lane 3) and then decreased (lanes 4-6).
- the presence of QvD-OPH (20.0 ⁇ M) was able to counter apoptosis and was able to maintain LC3-II for up to 18.0 hours (lane 7 and Lane 5) compared.
- Autophagosome formation-related Atg5 levels increased progressively to a maximum at 6.0 hours (lane 6) and returned to baseline at 18.0 hours (lane 7).
- pJNK stress pathway activation was detected when apoptosis was blocked (5 compared to lane 7).
- COMPOUND-JP In KO99L cells, COMPOUND-JP also interfered with Akt activation as measured by serine 473 phosphorylation (FIG. 4A). In contrast, COMPOUND-JP has no effect on 4E-BP1 phosphorylation, demonstrating that COMPOUND-JP only partially perturbs mTORC1 activation.
- the Akt inhibitor MK-2206 (5.0 ⁇ M) and the Akt / mTOR dual inhibitor KU0063794 (5.0 ⁇ M) completely block S6RP, 4EBP1, and Akt phosphorylation as early as 2.0 hours (FIG. S2).
- Rapamycin (10.0 nM) was similar to the action observed with COMPOUND-JP (10 ⁇ M), but rapamycin is an mTORC1 inhibitor rather than mTORC2 (reference 25), which is not 4EBP1, S6RP phosphorylation was completely inhibited (FIG. S2). Rapamycin also affected Akt phosphorylation / activation at a later time point compared to COMPOUND-JP (FIG. S2). Since LAT1 has been established to be an extracellular EAA exchanger with intracellular glutamine (Ref. 26), we have determined that various levels of glutamine contribute to EAA influx and mTORC1 activation. I tried to find out if it could interfere.
- L-asparaginase L-Asp
- Erw Erwinase
- c-myc protein has been shown to be upregulated in human lymphomas and tPTEN-/-tumors (ref. 28).
- tPTEN ⁇ / ⁇ cells c-myc was observed to migrate in triplicate around 60 kDa in SDS-PAGE (FIG. 4D; lanes 1-4).
- COMPOUND-JP COMPOUND-JP (10.0 ⁇ M)
- the total c-myc was already reduced at 2.0 hours (lane 5) and maintained until 24 hours (lane 7), in particular c-myc. Two lower forms of movement were significantly reduced. Similar observations were made when incubated with Velcade (8 nM; 24 hours; lane 10).
- Akt / mTOR inhibition by KU0063794 (10.0 ⁇ M) also slightly affected the two lower bands (lanes 15-17).
- MK-2206 (10.0 ⁇ M) -mediated Akt inhibition resulted in a dramatic disappearance of all three c-myc isoforms within 24 hours (lanes 18-20).
- rapamycin 100 nM did not affect c-myc levels (lanes 12-14) and consequently lacked apoptosis induction.
- Hsp90 level fluctuations cannot be clearly observed, so that the various observed drug-induced results show significant changes in overall cellular protein content. Help to confirm that it was not the result.
- COMPOUND-JP As assessed by immunoblotting in KO99L cells, COMPOUND-JP (10.0 ⁇ M) rapidly induced CHOP expression by 2.0 hours (FIG. 5A, lane 5), which was 6.0 hours (lane 6). ) And remained stable at 24 hours (lane 7). Although less in volume compared to COMPOUND-JP, induction of CHOP by L-Asp (2.5 units / ml) was evident at 6.0 hours (lane 9) and 24 hours (lane 10). . Erw (2.5 units / ml) also elicited a transient response that was maximal at 6.0 hours (lane 12) and returned to basal levels by 24 hours (lane 13). As demonstrated in FIG.
- CHOP showed a prominent, COMPOUND-JP dose-dependent induction (5.0 and 10.0 ⁇ M; lanes 2 and 3, respectively), and CHOP induction was also induced by Velcade. (8.0 and 15.0 ⁇ M; lanes 7 and 8, respectively) and to a lesser extent L-Asp (2.5 units / ml) and rapamycin (10.0 nM), also by lanes 4 and 9, respectively. Strongly triggered.
- Akt and Akt / mTOR inhibitors MK-2206 (10.0 ⁇ M; lane 5) and KU0063794 (10.0 ⁇ M; lane 6) did not result in CHOP induction, indicating that this pathway is PI3K Suggests that it is not caused by the / Akt / mTOR pathway.
- UPR Denatured protein response
- IRE1 ⁇ inositol-requiring enzyme 1 alpha
- PKR-like ER kinase PKR-like ER kinase
- ATF6 activated transcription factor
- Activated PKR-like ER kinase is Phosphorylates eIF2 ⁇ (serine-51A), leading to the induction of translation of ATF4 (ref. 34)
- ATF6 is cleaved during ER stress, and its cytosolic domain [ATF6 (N)] is located in the nucleus.
- Migrate and regulate transcription (Ref. 35)
- XBP-1, ATF4, and ATF6 are three transcription factors known to induce chop during ER stress (Ref.
- COMPOUND-JP regulates pro- and anti-apoptotic Bcl2 family members
- COMPOUND-JP To molecularly define the induction of apoptosis by COMPOUND-JP, we analyzed Bcl2 family members and we are known to control the initiation of apoptosis at the mitochondrial and ER membrane levels. Attempted to measure members who are. Starting at 6.0 hours (FIG. 6D), COMPOUND-JP (10.0 ⁇ M) is expressed in four pro-apoptotic members: Bak (FIG. 6D; lanes 2-4), Bax (lane 4), PUMA (lane 2). ⁇ 4), and Bim (lanes 3 and 4) protein levels were increased. Induction of Bax, Bak, PUMA, and Bim was sensitive to inhibition of CHOP by salubrinal (FIG. 6E, lanes 3 and 4), but total Hsp90 protein levels were not affected.
- COMPOUND-JP enhances chemotherapeutic and signaling inhibitors
- COMPOUND-JP was tested (in vitro) in combination with various chemotherapeutic or signaling inhibitors, as shown in FIG.
- the calculated combination index (CI) value is shown in FIG. 8A.
- COMPOUND-JP appears to be strongly synergistic with rapamycin, reducing overall metabolic activity / cell survival.
- COMPOUND-JP was synergistic with dexamethasone and doxorubicin.
- LAT1 inhibitors could enhance the effects of velcade and L-asparaginase, but appeared to be less efficient with PI103 and KU-0063794.
- the isobologram derived from WST1 further shows the possibilities for the various combinations tested (FIG. 8B).
- the data show that COMPOUND-JP can have a positive effect when combined with other molecules that interfere with T-ALL cell survival.
- each value is expressed as an average value of four cases under the same conditions as a relative value with the activity in the absence of COM-JP as 100%. Mean values, standard errors, and p-values for significant difference tests are shown in the figure. Although there was a difference depending on the type of cancer cell, a dose-dependent decrease in the growth activity of COM-JP was clearly observed in both cells.
- the combined effect of the person who was present for the entire period was stronger than the result of treatment with COM-JP (15 ⁇ M) for the previous two days (the bar graph in the middle of each group).
- the inhibitory effect of the combined use is a result of adding the actions of each individual, and shows a so-called additive effect.
- EAA EAA
- COMPOUND-JP resulted in a significant decrease in tumor volume.
- COMPOUND-JP has already been reported to reduce the growth of HT-29 colon cancer cells transplanted into nude mice (ref. 43), but this study is a LAT1 inhibitor both in vitro and in vivo. Further, the utility of COMPOUND-JP as will be described.
- COMPOUND-JP does not induce cell death in normal quiescent or activated human PBL cells, and a LAT1-selective inhibitor is a normal (LAT2 expressing) cell.
- LAT1 expression the idea of having a preferential effect on leukemia (LAT1 expression) was supported.
- COMPOUND-JP could partially interfere with mTORC1 activation.
- COMPOUND-JP blocked S6RP phosphorylation but did not affect 4EBP1 phosphorylation.
- Xenograft tPTEN-/-tumors in mice treated with COMPOUND-JP also showed a significant decrease in phosphoS6RP.
- COMPOUND-JP An important specificity of COMPOUND-JP is its ability to induce CHOP transcription factors in the overall stress response (ISR) / denatured protein response (UPR). This response, triggered by misfolded protein accumulation or AA deficiency in the endoplasmic reticulum (ER), allows cells to restore protein homeostasis in the ER (ref. 44). As they fade, cellular apoptotic clearance begins. Furthermore, changing the AA content in the medium resulted in a clear time-dependent induction of CHOP. CHOP was also strongly induced by velcade proteasome inhibitors, but also by L-asparaginase and Erwinase, which affect asparagine and glutamine levels, and by rapamycin, although less intense.
- Inhibitors of PI3K / Akt / mTOR did not induce CHOP.
- COMPOUND-JP-induced CHOP was modulated by the addition of extracellular EAA, demonstrating that COMPOUND-JP acts through inhibition of EAA uptake.
- salubrinal (ref. 32), an established compound that prevents eIF2 ⁇ dephosphorylation and induces cytoprotection against ER stress, counteracts the CHOP induction caused by COMPOUND-JP, KO99L
- UPR can be caused by at least three pathways by ATF6, IRE-1 and PERK (Ref. 44).
- COMPOUND-JP could mobilize three pathways through ATF6, IRE-1, and PERK, suggesting that it could interfere with common upstream events.
- COMPOUND-JP induced the expression of four pro-apoptotic Bcl-2 family members: Bax, Bak, Bim, and Puma associated with the involvement of UPR and CHOP.
- the BH3-only protein Puma is induced by tunicamycin by Puma ⁇ / ⁇ cells that are resistant to ER stress-induced apoptosis (ref. 45).
- CHOP interacts with AP-1 within the Puma promoter region (Ref. 46) and directly regulates the Bim promoter (Ref. 47).
- Bax and Bak can form a protein complex with the cytosolic domain of IRE1 ⁇ , which is essential for IRE1 ⁇ activation (Ref. 48).
- COMPOUND-JP showed a dose-dependent mitochondrial depolarization in tPTEN-/-cells that promotes caspase activation. Apoptosis was found to follow an early autophagy response, possibly resulting from the inhibitory effect of COMPOUND-JP on mTORC1.
- COMPOUND-JP a COMPOUND-JP resistant cell line designated as KO99-RJ.
- COMPOUND-JP not only failed to induce cell death in KO99-RJ, but also CHOP induction could not be observed, demonstrating the importance of CHOP in COMPOUND-JP-induced cell death did.
- a tentative model of the COMPOUND-JP mode of action is shown in supplementary figure S4.
- COMPOUND-JP did not affect overall protein synthesis, but COMPOUND-JP is very short half-life (15 minutes) that makes c-myc highly sensitive to reductions in EAA availability Changed the expression of c-myc with, preventing its continued resynthesis. This reduction in c-myc will have a dramatic outcome on lymphoma cell proliferation and survival.
- LAT1 / SLC7A5 is under the control of the T cell antigen receptor (TCR), and its induction properly mounts the immune response and its reprogramming of T cell metabolism. Critical to reprogramming (ref. 28). T cells having defects in SLC7A5 are unable to stimulate mTORC1 upon TCR triggering and show incomplete c-myc expression.
- LAT1 and 4F2 / CD98hc are the light chain and heavy chain of the heterodimeric CD98 complex, respectively.
- LAT1 carries large EAA
- CD98hc interacts with ⁇ integrin (Non-patent Document 19). It is not yet clear whether the two characteristics are functionally related. Disruption of CD98hc in ES cells interfered with integrin function and disrupted cell diffusion and migration rather than adhesion (Non-patent Document 19). Null cell reconstitution with a CD98hc mutant that interacts with integrin but not light chain restored integrin signaling, but surprisingly was not necessarily required for Akt activation and protection from apoptosis (Non-patent document 19).
- COMPOUND-JP in vitro experiments in combination with various chemotherapeutic agents or signal transduction inhibitors to calculate combination index (CI) values.
- CI combination index
- COMPOUND-JP is a stronger inducer of CHOP compared to rapamycin and can inhibit Akt activation.
- COMPOUND-JP can also enhance the anti-leukemic effects of dexamethasone, doxorubicin, and L-asparaginase. It has been reported that the toxic effect of the proteasome inhibitor Velcade on cancer cells is due to depletion of the intracellular AA pool (Ref. 27). This is likely to be amplified by COMPOUND-JP and may explain the observed additive effect of the two molecules on cell viability.
- COMPOUND-JP also slightly increased the effects of two inhibitors, KU0063794 and PI-103, acting on the PI3K / Akt / mTOR pathway.
- both Gemstabine and Paclitaxel decreased the cell proliferation activity in a dose-dependent manner.
- the inhibitory effect of Paclitaxel was stronger than that of Gemstabine.
- the combined effect of the group (the bar graph on the right side of each group) was more powerful.
- the inhibitory effect of the combined use is a result of adding the actions of each individual, and shows a so-called additive effect.
- both Gemstabine and Paclitaxel decreased the cell proliferation activity in a dose-dependent manner.
- the inhibitory effect of Paclitaxel was stronger than that of Gemstabine.
- the effect of Gemstabine alone was extremely small and was clearly weaker than the effect on the proliferation of Skills gastric cancer-derived 44As3-11 cells.
- the increase in tumor showed a significant decrease in each administration of both drugs compared to the untreated control group.
- the combined effect of COM-JP and 5-FU on tumor growth in a human colorectal cancer-derived HT-29 cell seeded nude mouse model the combined increase in the residual mass of each single group was further reduced. This combined effect was additive and was found to be due to the different mechanism of action of both drugs.
- a medicinal effect that can further enhance the therapeutic effect of each individual by such a combination is sufficiently expected in clinical treatment of colorectal cancer.
- the LAT1 inhibitor according to the present invention alkylating agents, platinum preparations, antimetabolites
- a malignant tumor agent composition is effective.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Emergency Medicine (AREA)
- Inorganic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
「LAT1阻害剤」とは、システムLアミノ酸トランスポーター1(LAT1)及びLAT1遺伝子を選択的に阻害する剤である限り特に限定されず、化合物及びその塩、抗LAT1抗体、アプタマー、核酸医薬を含む。当該LAT1阻害剤は、単独の剤であっても2種以上の剤の併用をも含む。
「アルキル化薬」とは、DNAをアルキル化する薬剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意のアルキル化薬、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のアルキル化薬を意味する。アルキル化薬は、単独の剤であっても2種以上の剤の併用をも含む。
「白金製剤」とは、白金を含む抗悪性腫瘍剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の白金製剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の白金製剤を意味する。白金製剤は、単独の剤であっても2種以上の剤の併用をも含む。
「代謝拮抗剤」とは、癌細胞の代謝を阻害して、増殖を抑制する剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の代謝拮抗剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の代謝拮抗剤を意味する。代謝拮抗剤は、単独の剤であっても2種以上の剤の併用をも含む。
「トポイソメラーゼ阻害薬」とは、トポイソメラーゼと呼ばれる酵素の働きを妨げる剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意のトポイソメラーゼ阻害薬、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のトポイソメラーゼ阻害薬を意味する。トポイソメラーゼ阻害薬は、単独の剤であっても2種以上の剤の併用をも含む。
「微小管重合阻害薬」とは、微小管の形成を妨げる剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の微小管重合阻害薬、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の微小管重合阻害薬を意味する。微小管重合阻害薬は、単独の剤であっても2種以上の剤の併用をも含む。
「内分泌療法剤」とは、内分泌(ホルモン)の分泌又は作用を抑制する剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の内分泌療法剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の内分泌療法剤を意味する。内分泌療法剤は、単独の剤であっても2種以上の剤の併用をも含む。
「微小管脱重合阻害薬」とは、微小管重合を安定化させる剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の微小管脱重合阻害薬、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の微小管脱重合阻害薬を意味する。微小管脱重合阻害薬は、単独の剤であっても2種以上の剤の併用をも含む。
「抗腫瘍性抗生物質」とは、抗腫瘍剤として機能する、微生物の産生物に由来する物質であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の抗腫瘍性抗生物質、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の抗腫瘍性抗生物質を意味する。抗腫瘍性抗生物質は、単独の剤であっても2種以上の剤の併用をも含む。
「分子標的薬」とは、腫瘍細胞の増殖、浸潤、転移等に関わる分子を標的とする薬剤であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の分子標的薬を意味する。当該分子標的薬は、単独の剤であっても2種以上の剤の併用をも含む。
「抗悪性腫瘍剤」とは、例えば、アルキル化薬、白金製剤、代謝拮抗剤、植物アルカロイド(トポイソメラーゼ阻害薬、微小管重合阻害薬、微小管脱重合阻害薬等)、内分泌療法剤、抗腫瘍性抗生物質、分子標的薬、生物学的応答調節剤及びその他の薬剤等を挙げることができ、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の抗悪性腫瘍剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意の抗悪性腫瘍剤を意味する。当該抗悪性腫瘍剤は、単独の剤であっても2種以上の剤の併用をも含む。
「シスプラチン」とは、白金製剤であるシスプラチン(CDDP)を意味し、シスプラチン、ならびに、シスプラチンと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「5-フルオロウラシル(5-FU)」とは、代謝拮抗剤である5-フルオロウラシル(5-FU)を意味し、5-フルオロウラシル(5-FU)、ならびに、5-フルオロウラシル(5-FU)と同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「ゲムシタビン」とは、代謝拮抗剤であるゲムシタビンを意味し、ゲムシタビン、ならびに、ゲムシタビンと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「L-アスパラギナーゼ(ロイナーゼ)」とは、抗悪性腫瘍剤であるL-アスパラギナーゼ(ロイナーゼ)を意味し、L-アスパラギナーゼ(ロイナーゼ)、ならびに、L-アスパラギナーゼ(ロイナーゼ)と同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「パクリタキセル」とは、微小管脱重合阻害薬であるパクリタキセル(タキソール)を意味し、パクリタキセル、ならびに、パクリタキセルと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「デキサメタゾン」とは、主な成分として副腎皮質ステロイドが含まれている抗炎症作用や免疫抑制作用などをもつ薬物であるデキサメタゾンを意味し、デキサメタゾンン、ならびに、デキサメタゾンと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「アントラサイクリン系抗生物質」とは、ストレプトマイセス属微生物に由来する抗腫瘍性抗生物質として用いられる薬剤の一群であれば特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意の、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のアントラサイクリン系抗生物質を意味する。当該アントラサイクリン系抗生物質は、単独の剤であっても2種以上の剤の併用をも含む。「ドキソルビシン」とは、アントラサイクリン系抗生物質であるドキソルビシンを意味し、ドキソルビシン、ならびに、ドキソルビシンと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「ベルケイド(ボルテゾミブ)」とは、分子標的薬であるベルケイド(ボルテゾミブ)を意味し、ベルケイド(ボルテゾミブ)、ならびに、ベルケイド(ボルテゾミブ)と同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「ラパマイシン」とは、分子標的薬であるラパマイシンを意味し、ラパマイシン、ならびに、ラパマイシンと同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「KU0063794」とは、分子標的薬であるKU0063794を意味し、KU0063794、ならびに、KU0063794と同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「PI-103」とは、分子標的薬であるPI-103を意味し、PI-103、ならびに、PI-103と同じ薬理学的特性を持つ、全ての類似体、誘導体および同属体を含む。
「mTOR阻害剤」とは、mTOR(mammalian target of rapamycin)を直接的又は間接的に標的にする、若しくはmTORの活性/機能を低減又は阻害する薬剤である限り特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意のmTOR阻害剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のmTOR阻害剤を意味する。当該mTOR阻害剤は、単独の剤であっても2種以上の剤の併用をも含む。
「P13K阻害剤」とは、P13K(PI3キナーゼ)を直接的又は間接的に標的にする、若しくはP13Kの活性/機能を低減又は阻害する薬剤である限り特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意のP13K阻害剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のP13K阻害剤を意味する。当該P13K阻害剤は、単独の剤であっても2種以上の剤の併用をも含む。
「Akt阻害剤」とは、Aktを直接的又は間接的に標的にする、若しくはAktの活性/機能を低減又は阻害する薬剤である限り特に限定されず、例えば、政府機関から医薬品としての製造販売承認された既存の1または複数の任意のAkt阻害剤、または現在臨床試験若しくは前臨床試験中であるかまたは将来臨床試験において使用され、当該試験の後に将来政府機関から医薬品としての製造販売承認がされるであろう1または複数の任意のAkt阻害剤を意味する。当該Akt阻害剤は、単独の剤であっても2種以上の剤の併用をも含む。
式(I):
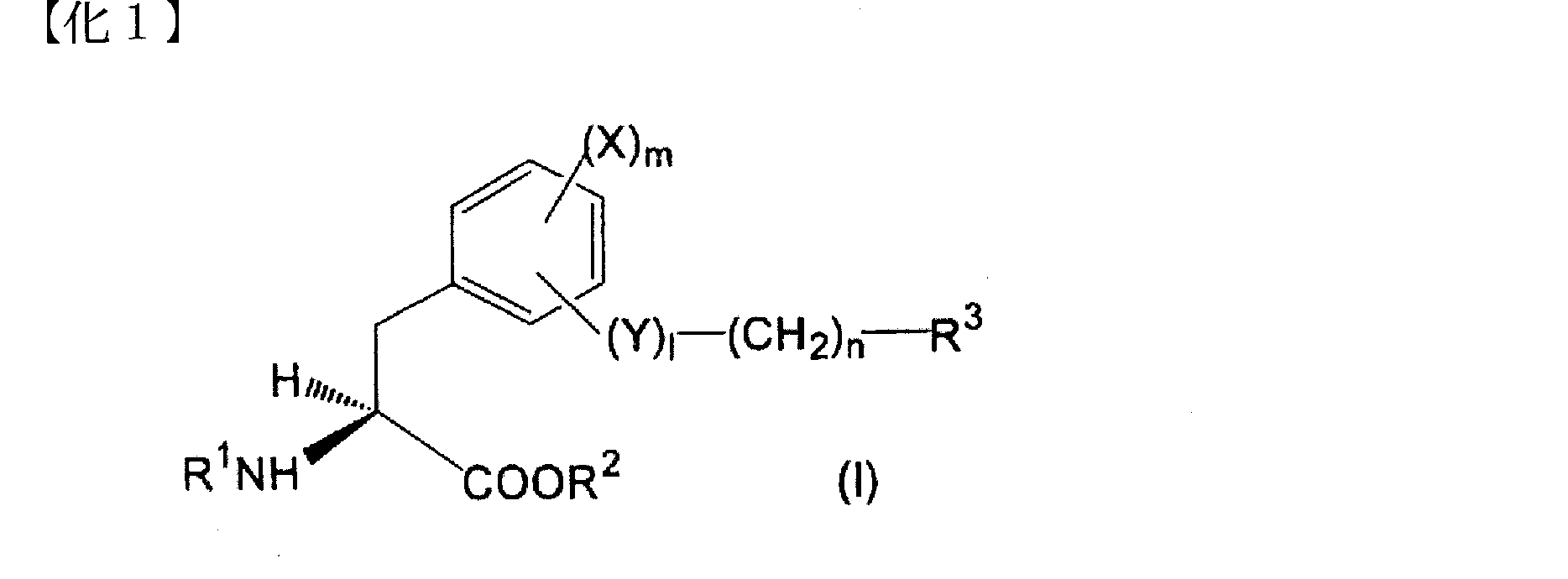
YはO又はNHであり、
lは0又は1であり、
mは0、1又は2であり、
nは0~5の整数であり、
R1は、水素原子又はアミノ保護基であり、
R2は、水素原子もしくはアルキル、アラルキル又はアリール基であり、
R3は、(1)ハロゲン原子;
(2)アミノ部分が低級アルキルで置換されていてもよいアロイルアミノ基;
(3)フェニル、フェノキシ、ピリジル、ピリミジニルもしくはキノリルで置換されたフェニル基(ここで、該フェニル基における各置換基は、ハロゲン原子、シアノ、ヒドロキシ、カルボキシ、低級アルコキシ、低級アルコキシカルボニル、フェニル、ジ低級アルキルアミノもしくはチオモルホリニルでさらに置換されていてもよい);
(4)ナフチル基もしくはテトラヒドロナフチル基(ここで、該ナフチル基はヒドロキシ、低級アルコキシもしくはジ低級アルキルアミノで置換されていてもよく、ジ低級アルキルアミノはハロゲン原子もしくはヒドロキシでさらに置換されていてもよく、ナフチル基が置換されていないとき、nは0または2であり、
ナフチル基がヒドロキシで置換されているとき、Xはクロル原子であり、mは0でない);
(5)低級アルキル、フェニル、ナフチルもしくはテトラヒドロキノリルで置換されたN、O及び/又はS含有の、不飽和の単環複素環式基(ここで、該単環複素環式基における各置換基はハロゲン原子、ヒドロキシもしくはフェニルでさらに置換されていてもよく、この場合、mは1又は2であり、lは1である);または
(6)オキソ、カルボキシ、アミノ、低級アルキル、低級アルコキシ、シクロアルキル、ジ低級アルキルアミノ、低級アルコキシカルボニル、ジ低級アルキルカルバモイル、フェニル又はN、O及び/又はS含有の、飽和もしくは不飽和の単環複素環式基で置換されていてもよいN、O及び/又はS含有の、不飽和又は部分的に飽和されていてもよい縮合複素環式基(ここで、該縮合複素環式基における各置換基は、ハロゲン原子、ヒドロキシ、低級アルキル、低級アルコキシ、フェニル、ジ低級アルキルアミノ、低級アルカノイルオキシ、ビス〔ハロ(低級)アルキル〕アミノもしくはN-低級アルキル-N-ヒドロキシ(低級)アルキルアミノでさらに置換されていてもよく、この場合、mは1又は2である)
である]
で表される芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
式(I):
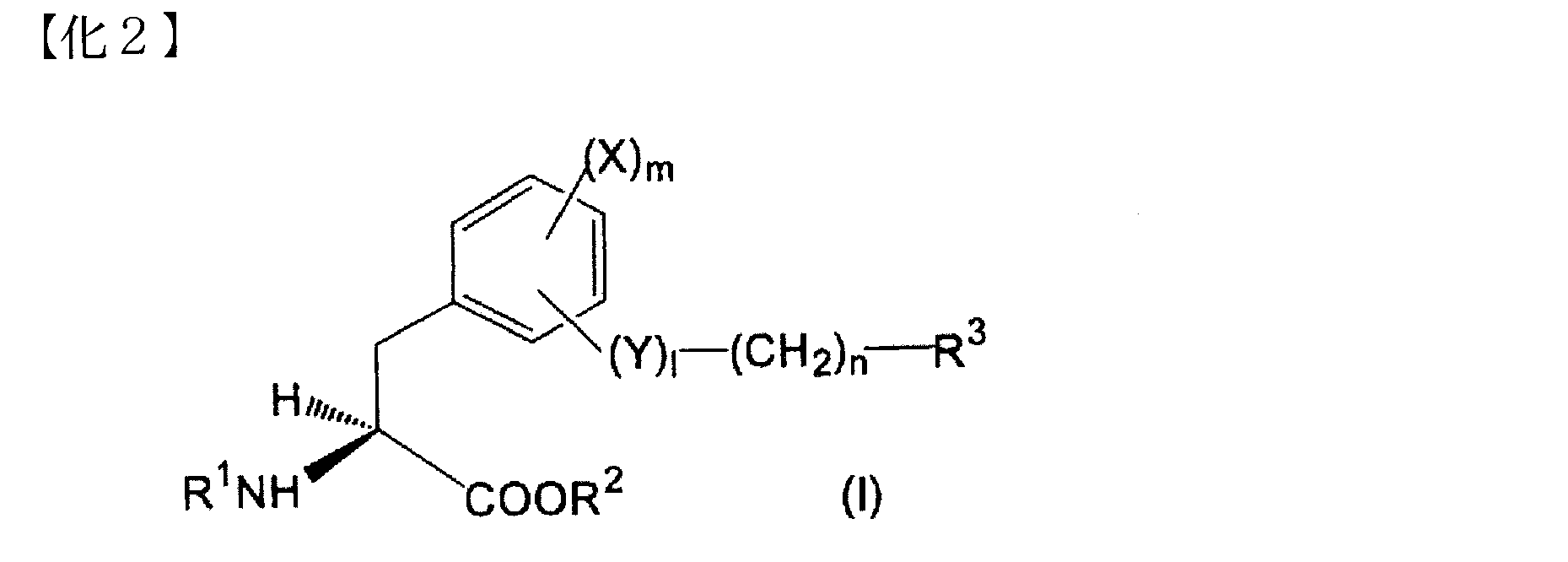
YはO又はNHであり、
lは0又は1であり、
mは0又は2であり、
nは0~5の整数であり、
R1は、水素原子又はトリフルオロアセチルもしくはt-ブトキシカルボニル基であり、
R2は、水素原子もしくはアルキル基であり、
R3は、(1)臭素原子;
(2)アミノ部分が低級アルキルで置換されていてもよいアロイルアミノ基;
(3)フェニル、フェノキシ、ピリジル、ピリミジニルもしくはキノリル基で置換されたフェニル基(ここで、該フェニル基における各置換基は、ハロゲン原子又はシアノ、ヒドロキシ、カルボキシ、低級アルコキシ、低級アルコキシカルボニル、フェニル、ジ低級アルキルアミノもしくはチオモルホリニルでさらに置換されていてもよく、この場合、Xは塩素原子である);
(4)ナフチル基もしくはテトラヒドロナフチル基(ここで、該ナフチル基はヒドロキシ、ジ(2-ヒドロキシエチル)アミノもしくはジ(2-クロロエチル)アミノ基で置換されていてもよく、
ナフチル基が置換されていないとき、nは0または2であり、
ナフチルがヒドロキシで置換されているとき、Xは塩素原子であり、mは2である);または、
(5)フェニル、ナフチルもしくはテトラヒドロキノリルで置換されたN、O及び/又はS含有の、不飽和の単環複素環式基(ここで、該単環複素環式基における各置換基はハロゲン原子又はヒドロキシもしくはフェニルでさらに置換されていてもよく、この場合、Xは塩素原子であり、mは2であり、lは1である)
である]
で表される芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R3の(5)における不飽和の単環複素環式基が、N含有、N及びO含有又はN及びS含有の5員環又は6員複素環式基である化合物2に記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R3の(5)における不飽和の単環複素環式基が、ピリジル、オキサゾリル又はチアゾリルである化合物2または3に記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R1が水素原子である化合物2~4のいずれか1つに記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R1がトリフルオロアセチル又はt-ブトキシカルボニル基である化合物2~4のいずれか1つに記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R3が、(3)フェニル、フェノキシ、ピリジル、ピリミジニルもしくはキノリル基で置換されたフェニル基(ここで、該フェニル基における各置換基は、ハロゲン原子又はヒドロキシもしくはジ低級アルキルアミノ基でさらに置換されていてもよく、この場合、Xは塩素原子である);
(4)ヒドロキシで置換されていてもよいナフチル基(ここで、ナフチルがヒドロキシで置換されているとき、Xは塩素原子であり、mは2である);または
(5)フェニルもしくはナフチル基で置換されたN及びO含有の不飽和の単環複素環式基(ここで、該単環複素環式基における各置換基はヒドロキシ基でさらに置換されていてもよく、この場合、Xは塩素原子であり、mは2であり、lは1である);
である、
化合物2に記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R3が、(3)フェニルもしくはピリジル基で置換されたフェニル基(ここで、該フェニル基における各置換基は、ハロゲン原子もしくはジ低級アルキルアミノ基でさらに置換されていてもよく、この場合、Xは塩素原子である);または
(4)ヒドロキシ基で置換されていてもよいナフチル基(ここで、ナフチルがヒドロキシで置換されているとき、Xは塩素原子であり、mは2である)
である、
化合物2に記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
R3が、(3)ジ低級アルキルアミノ基で置換されたピリジルで置換されたフェニル基(ここで、この場合、Xは塩素原子である);
(4)ナフチル基(ここで、nは0または2である);または
(5)ナフチル基で置換されたN及びO含有の不飽和単環複素環式基
である、
化合物2に記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。
Xが塩素又はヨウ素原子である化合物2~9のいずれか1つに記載の芳香族アミノ酸誘導体又はその薬理学的に許容しうる塩である。

本発明に係る抗悪性腫瘍剤組成物は、例えば、経口、経皮又は注射により投与することができる。
本発明に係る抗悪性腫瘍剤組成物の投与量は、患者の年齢、体重、全身的な健康度、性別、投与時間、投与経路、疾患の重篤度を含む種々の要因によって適宜調整することができ、限定されないが、例えば、通常、成人1日あたり10~5000mg程度、好ましくは100~3000mg程度を、1~5回に分けて投与することが適当である。
(製造例1)
1)28%アンモニア水(20ml)とテトラヒドロフラン(30ml)の混液中に、氷冷攪拌下、2-ナフトイルクロリド(5.70g,29.9mmol)のテトラヒドロフラン(60ml)溶液を滴下し、室温で4時間攪拌した。反応液から溶媒を減圧留去して、残渣に水を加えて、析出物をろ取し、乾燥して2-カルバモイルナフタレン(2.68g,52%)を無色固体として得た。
IR(Nujol)3378,3194,1685,1655cm-1、APCI-MS m/z:172[M+H]+
2)2-カルバモイルナフタレン(2.64g,15.4mmol)と4-クロロアセト酢酸エチル(2.06g,12.5mmol)の混合物を160℃で1時間攪拌後、室温にて酢酸エチル(200ml)で希釈して、飽和炭酸水素ナトリウム水溶液、飽和食塩水で順に洗浄後、乾燥した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=8~4)で精製して、[2-(2-ナフチル)-オキサゾール-4-イル]酢酸エチルエステル(459mg,13%)を黄色結晶として得た。
m.p.:61.5~64℃、IR(Nujol)1737,1591cm-1、1H NMR(CDCl3):δ 1.31(3H,t,J=7.1Hz),3.73(2H,d,J=1.1Hz),4.24(2H,q,J=7.1Hz),7.49-7.57(2H,m),7.76(1H,t,J=1.1Hz),7.82-7.95(3H,m),8.11(1H,dd,J=1.7,8.7Hz),8.58(1H,br d,J=1.1Hz)、APCI-MS m/z:282[M+H]+
3)リチウムアルミニウムハイドライド(66mg,1.74mmol)のテトラヒドロフラン(15ml)懸濁液に、氷冷攪拌下、[2-(2-ナフチル)-オキサゾール-4-イル]酢酸エチルエステル(434mg,1.54mmol)のテトラヒドロフラン(20ml)溶液を滴下し、同温度で2.5時間攪拌した。反応液に同温度で水(0.1ml)、15%水酸化ナトリウム水溶液(0.1ml)、水(0.3ml)、硫酸ナトリウム(3g)を順に加え、不溶物をろ去し、ろ液から溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=2~1)で精製して、2-[2-(2-ナフチル)-オキサゾール-4-イル]エタノール(323mg,88%)を淡黄色結晶として得た。
m.p.:96~98℃、IR(Nujol)3466,1591,1543cm-1、1H NMR(CDCl3):δ 2.85-2.90(2H,m),2.93(1H,t,J=6.0Hz),3.99(2H,q,J=6.0Hz),7.50-7.57(2H,m),7.58(1H,t,J=1.0Hz),7.82-7.96(3H,m),8.10(1H,dd,J=1.7,8.6Hz),8.52(1H,br)、APCI-MS m/z:240[M+H]+
1)3-アミノ-2-ヒドロキシ安息香酸メチルエステル塩酸塩(2.0g,12.0mmol)、N,N’-ジメチルアニリン(3.04ml,24.0mmol)、及びテトラヒドロフラン(20ml)の混合物に氷冷攪拌下、イソニコチン酸クロリド塩酸塩(2.34g,13.2mmol)、トリエチルアミン(1.83ml,13.2mmol)、及びテトラヒドロフラン(10ml)の混合物を滴下し、同温度で2時間攪拌した。反応液を塩化メチレン(200ml)で希釈し、水、飽和食塩水で順に洗浄後、乾燥して、溶媒を減圧留去した。残渣をアミンシリカゲルカラムクロマトグラフィー(クロマトレックス(登録商標)NH)(n-ヘキサン/酢酸エチル=4及びクロロホルム/酢酸エチル=1)で精製して、2-ヒドロキシ-3-イソニコチノイルアミノ安息香酸メチルエステル(2.37g,73%)を淡黄色結晶として得た。
m.p.:157~158℃、IR(Nujol)3423,1669,1555,15
42cm-1、APCI-MS m/z:273[M+H]+
2)2-ヒドロキシ-3-イソニコチノイルアミノ安息香酸メチルエステル(500mg,1.84mmol)、p-トルエンスルホン酸一水和物(349mg,1.84mmol)及びキシレン(20ml)の混合物を14時間加熱還流した後、p-トルエンスルホン酸一水和物(349mg,1.84mmol)を追加して、2時間加熱還流した。反応液を氷冷し、酢酸エチル、10%炭酸カリウム水溶液を加え、分液し、有機相を水、飽和食塩水で順に洗浄後、乾燥した。溶媒を減圧留去して、残渣をn-ヘキサン/ジイソプロピルエーテル(4/1)混液で粉砕し、ろ取後乾燥して、[2-(4-ピリジル)-ベンズオキサゾール-7-イル]カルボン酸メチルエステル(382mg,82%)を淡黄色結晶として得た。
m.p.:149~151℃、IR(Nujol)1725cm-1、APCI-MS
m/z:255[M+H]+
3)[2-(4-ピリジル)-ベンズオキサゾール-7-イル]カルボン酸メチルエステル(360mg,1.42mmol)のテトラヒドロフラン(15ml)溶液に氷冷攪拌下、リチウムアルミニウムハイドライド(53mg,1.42mmol)を10分間かけ加え、同温度で0.5時間攪拌した。反応液に同温度で10%含水テトラヒドロフラン(2ml)、30%水酸化ナトリウム水溶液(0.5ml)を順に滴下し室温で2時間攪拌した。生じた不溶物をろ去し、ろ液の溶媒を減圧留去した。残渣を酢酸エチルで希釈し、水、飽和食塩水で順に洗浄後乾燥し、溶媒を減圧留去した。得られた結晶をn-ヘキサン/ジイソプロピルエーテル(1/1)混液で粉砕し、ろ取後乾燥して、[2-(4-ピリジル)-ベンズオキサゾール-7-イル]メタノール(216mg,67%)を無色結晶として得た。
m.p.:146~147℃、IR(Nujol)3221,1595,1541cm-1、1H NMR(CDCl3):δ 2.35(1H,t,J=5.9Hz),5.08(2H,d,J=5.9Hz),7.38-7.51(2H,m),7.76(1H,dd,J=1.4,7.8Hz),8.07-8.09(2H,m),8.78-8.81(2H,m)、APCI-MS m/z:227[M+H]+
1)3-ヒドロキシベンズアルデヒド(1.72g,14.1mmol)、4-メトキシフェニルボロン酸(3.16g,20.8mmol)、モレキュラーシーブス4A粉末(1.95g)及び酢酸第二銅(2.96g,16.3mmol)の塩化メチレン(100ml)懸濁液に室温でトリエチルアミン(7.1ml,51mmol)を滴下し、27.5時間攪拌した。反応液を酢酸エチル(200ml)で希釈し、不溶物をろ去し、酢酸エチルで洗浄した。ろ洗液を10%塩酸、飽和食塩水で順に洗浄後、乾燥して、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=7)で精製して、3-[(4-メトキシ)フェノキシ]ベンズアルデヒド(596mg,19%)を淡褐色油状物として得た。
IR(Neat)2835,1699,1584,1504cm-1、GCEI-MS m/z:228(M+)
2)3-[(4-メトキシ)フェノキシ]ベンズアルデヒド(573mg,2.51mmol)のエタノール(10ml)溶液に氷冷攪拌下、水素化ほう素ナトリウム(158mg,4.18mmol)を加え、室温で25分間攪拌した。反応液の溶媒を減圧留去し、残渣を酢酸エチルで希釈し、水、飽和食塩水で順に洗浄後乾燥した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/酢酸エチル=20)で精製して、3-[(4-メトキシ)フェノキシ]ベンジルアルコール(469mg,81%)を無色油状物として得た。
IR(Neat)3356,1611,1586,1504cm-1、1H NMR(CDCl3):δ 1.67(1H,t,J=5.3Hz),3.81(3H,s),4.65(2H,br d,J=4.6Hz),6.84-6.91(3H,m),6.93-7.06(4H,m),7.28(1H,t,J=7.9Hz)、GCEI-MS m/z:230(M+)
製造例3で得た3-[(4-メトキシ)フェノキシ]ベンジルアルコールを常法により脱メチル化して、3-[(4-ヒドロキシ)フェノキシ]ベンジルアルコールを淡黄色油状物として得た。
IR(Neat)3350,1603,1585,1505cm-1、1H NMR(CDCl3+DMSO-d6):δ 3.57(1H,t,J=5.9Hz),4.60(2H,d,J=5.5Hz),6.79-6.90(5H,m),6.92-6.96(1H,m),6.98-7.04(1H,m),7.24(1H,t,J=7.8Hz),8.49(1H,s)、ESI-MS m/z:215[M-H]-
2-ブロモ-6-メトキシナフタレン(3.0g,12.7mmol)のテトラヒドロフラン(45ml)溶液に、アルゴン雰囲気下-60℃でn-ブチルリチウムのヘキサン溶液(1.5M;8.9ml,13.4mmol)を滴下し、同温度で70分間攪拌後、トリn-ブチルボレート(5.2ml,19.0mmol)を加え同温度で1時間、5℃で1.5時間攪拌した。反応液に5℃で20%塩酸(13ml)を滴下後、水を加え、室温にて酢酸エチルで抽出した。有機層を水、飽和食塩水で順に洗浄後乾燥し、溶媒を減圧留去した。残渣を酢酸エチル及びジエチルエーテルで粉砕後、ろ取し乾燥して、(2-メトキシ)-6-ナフタレンボロン酸(1.50g,59%)を無色固体として得た。
m.p.:301~311℃、IR(Nujol)3290,1625cm-1、1H NMR(DMSO-d6):δ 3.87(3H,s),7.14(1H,dd,J=2.6,9.0Hz),7.28(1H,d,J=2.6Hz),7.74(1H,d,J=8.2Hz),7.79-7.84(2H,m),8.06(2H,s),8.28(1H,s).
[2-(3-メトキシフェニル)-ベンズオキサゾール-7-イル]メタノール(457mg,1.79mmol)の塩化メチレン(10ml)懸濁液に-78℃で三臭化ほう素の塩化メチレン溶液(1.0M;7ml,7mmol)を滴下し、同温度で1時間攪拌後、冷却浴をはずして、室温で2.5時間攪拌した。反応液を氷水(50ml)中に注ぎ込み、有機層を分取後、水層を酢酸エチルで抽出した。有機層を合わせ乾燥後、溶媒を減圧留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=3~1)で精製して、7-ブロモメチル-2-(3-ヒドロキシフェニル)-ベンズオキサゾール(535mg,98%)を褐色固体として得た。
m.p.:225~228℃、IR(Nujol)3147,1602,1559cm-1、1H NMR(DMSO-d6):δ 5.02(2H,s),7.04(1H,ddd,J=0.9,2.4,8.0Hz),7.36-7.54(3H,m),7.62-7.71(2H,m),7.78(1H,dd,J=1.2,8.0Hz),10.0(1H,s)、APCI-MS m/z:304/306[M+H]+
5-ブロモベンゾ[b]フラン(1.0g,5.0mmol)のテトラヒドロフラン(10ml)溶液に、アルゴン雰囲気下、-60℃でn-ブチルリチウムのヘキサン溶液(1.5M;3.72ml,5.6mmol)を滴下し、同温度で30分間攪拌後、トリメチルボレート(0.69ml,6.0mmol)を加え4時間かけて室温まで昇温させた。反応液に5℃で水(5ml)を加え、テトラヒドロフランを減圧留去し、残渣に1N塩酸を加えpH1として、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄後乾燥し、溶媒を減圧留去した。残渣をn-ヘキサン/ジエチルエーテル混液で粉砕後、ろ取し、乾燥して、5-ベンゾ[b]フランボロン酸(551mg)を淡褐色固体として得た。
1)2-ブロモベンゾチアゾール-7-カルボン酸エチルエステル(572mg,2.00mmol)、ピペリジン(5ml)及びエタノール(1ml)の混合物を70~75℃で2.5時間攪拌した。反応液を室温としてジエチルエーテル(15ml)を加え析出物をろ去し、ジエチルエーテルで洗浄した。ろ液と洗液を合わせて溶媒を減圧留去して、残さをシリカゲルカラムクロマトグラフィー(クロロホルム)で精製して2-ピペリジノベンゾチアゾール-7-カルボン酸エチルエステル(526mg,90%)を微橙色結晶として得た。
m.p.:88~89℃、IR(Nujol)1791,1595,1541cm-1、APCI-MS m/z:291[M+H]+
2)2-ピペリジノベンゾチアゾール-7-カルボン酸エチルエステル(475mg,1.64mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-ピペリジノベンゾチアゾール-7-メタノール(375mg,91%)を微黄色結晶として得た。
m.p.:107~108℃、IR(Nujol)3261,3200,1595,1541cm-1、APCI-MS m/z:249[M+H]+
1)2-ブロモベンゾチアゾール-7-カルボン酸エチルエステル(715mg,2.50mmol)及びフェニルボロン酸(341mg,2.8mmol)の脱気したジメトキシエタン(20ml)溶液にテトラキス(トリフェニルホスフィン)パラジウム(144mg,0.125mmol)、炭酸水素ナトリウム(630mg,7.50mmol)の脱気水溶液(10ml)を順に加え50℃で10分間攪拌後、室温としてヨウ化第一銅(24mg,0.125mmol)を加え4時間加熱還流した。ジメトキシエタンを減圧留去して酢酸エチルで抽出し、有機層を飽和食塩水で洗浄後乾燥し、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=10)で精製し2-フェニルベンゾチアゾール-7-カルボン酸エチルエステル(500mg,70%)を無色結晶として得た。
m.p.:86.5~87.5℃、IR(Nujol)1716cm-1、APCI-MS m/z:284[M+H]+
2)2-フェニルベンゾチアゾール-7-カルボン酸エチルエステル(425mg,1.50mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-フェニルベンゾチアゾール-7-メタノール(340mg,94%)を無色結晶として得た。
m.p.:101~102℃、IR(Nujol)3241,1572,1507cm-1、APCI-MS m/z:242[M+H]+
1)5-クロロ-3-ニトロサリチル酸メチルエステル(2.32g,10.0mmol)の塩化メチレン(25ml)溶液に氷冷攪拌下2,6-ルチジン(1.83ml,15.7mmol)、無水トリフルオロメタンスルホン酸(3.67g,13mmol)を順に滴下し、氷冷で0.5時間攪拌した。反応液に塩化メチレン(20ml)-氷水(30ml)を加え分液し、水層を塩化メチレンで抽出した。有機層を合わせて水洗後乾燥して溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=10)で精製して5-クロロ-2-トリフロロメタンスルホニルオキシ-3-ニトロ安息香酸メチルエステル(3.33g,91%)を淡黄色油状物質として得た。
IR(Neat)3088,2961,1742,1603,1552cm-1、APCI-MS m/z:381[M+NH4]+
2)5-クロロ-2-トリフロロメタンスルホニルオキシ-3-ニトロ安息香酸メチルエステル(820mg,2.25mmol)のジメチルスルホキシド(5ml)溶液に、トリエチルアミン(0.33ml,2.37mmol)及びイソインドリン(0.27ml,2.38mmol)を順に加え、室温で1時間攪拌した。反応液を氷冷して氷水(50ml)を加え攪拌後、析出物をろ取し水洗して乾燥した。得られた黄色結晶をシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=15)で精製して5-クロロ-2-(1,2,3,4-テトラヒドロイソキノリニオ)-3-ニトロ安息香酸メチルエステル(608mg,81%)を黄色結晶として得た。
m.p.:114~115℃、IR(Nujol)1743,1705,1603,1589,1529,1503cm-1、APCI-MS m/z:333[M+H]+
3)5-クロロ-2-(1,2,3,4-テトラヒドロイソキノリニオ)-3-ニトロ安息香酸メチルエステル(590mg,1.77mmol)のメタノール(6ml)-テトラヒドロフラン(9ml)溶液に、10%パラジウム炭素(水分51.7%,190mg)を加え水素雰囲気下、室温で24時間攪拌した。触媒をろ去し、ろ液の溶媒を約5mlまで濃縮して、析出結晶をろ取した。ろ取した結晶に酢酸エチル(50ml)-水(30ml)を加え飽和炭酸水素ナトリウム水でpH8~9として、酢酸エチルで抽出した。抽出した有機層は、飽和食塩水で洗浄後乾燥して、溶媒を減圧留去することにより1,2,3,4-テトラヒドロナフタレン[1,2-a]ベンズイミダゾール-7-カルボン酸メチルエステル(96mg,20%)を微黄色結晶として得た。
m.p.:152~153℃、IR(Nujol)1710,1627,1611,1589,1579,1551cm-1、APCI-MS m/z:265[M+H]+
4)1,2,3,4-テトラヒドロナフタレン[1,2-a]ベンズイミダゾール-7-カルボン酸メチルエステル(80mg,0.30mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、1,2,3,4-テトラヒドロナフタレン[1,2-a]ベンズイミダゾール-7-メタノール(67mg,crude 94%)を淡灰白色固体として得た。
IR(Nujol)3170,1615,1547cm-1、APCI-MS m/z:237[M+H]+
1)2-クロロニコチン酸エチルエステル(928mg,5.00mmol)の脱気した1,4-ジオキサン(25ml)溶液に及び3-ビフェニルフェニルボロン酸(1089mg,5.50mmol)、テトラキス(トリフェニルホスフィン)パラジウム(578mg,0.50mmol)、炭酸カリウム(2.07g,15.0mmol)を順に加え18時間加熱還流した。水を加え酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄後乾燥し、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=5)で精製し、2-(3-ビフェニル)ニコチン酸エチルエステル(1.411g,93%)を無色油状物質として得た。
IR(Neat)2981,1722,1716cm-1、APCI-MS m/z:304[M+H]+
2)2-(3-ビフェニル)ニコチン酸エチルエステル(1.38g,4.55mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-(3-ビフェニル)ピリジン-3-メタノール(1.115g,94%)を無色結晶として得た。
m.p:91~93℃、IR(Nujol)3265,1579,1566cm-1、APCI-MS m/z:262[M+H]+
1)2-クロロニコチン酸エチルエステル(557mg,3.0mmol)のテトラヒドロフラン(17ml)溶液に1,2,3,4-テトラヒドロイソキノリン(0,49ml,3.9mmol)及びトリエチルアミン(0.47ml,3.34mmol)を順に加え、18時間加熱還流した。反応液を室温として、析出物をろ去してろ液の溶媒を減圧留去した。残さを酢酸エチルに溶解し、水、飽和食塩水で順に洗浄後乾燥して溶媒を減圧留去した。残さをアミンシリカゲルカラムクロマトグラフィー(クロマトレックス(登録商標)NH)(n-ヘキサン/酢酸エチル=19)で精製して2-(1,2,3,4-テトラヒドロイソキノリニオ)ニコチン酸エチルエステル(529mg,62%)を無色油状物質として得た。
IR(Neat)2979,1711,1584,1555cm-1、APCI-MS m/z:283[M+H]+
2)2-(1,2,3,4-テトラヒドロイソキノリニオ)ニコチン酸エチルエステル(511mg,1.81mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-(1,2,3,4-テトラヒドロイソキノリニオ)ピリジン-3-メタノール(386mg,89%)を無色結晶として得た。
m.p.:95~97℃、IR(Nujol)3184,1595,1575cm-1、APCI-MS m/z:241[M+H]+
1)3-アミノサリチル酸メチルエステル塩酸塩及び2-メチルチオピリミジン-5-カルボン酸クロリド塩酸塩を原料にして製造例2に準じて、2-(2-メチルチオピリミジン-5-イル)ベンズオキサゾール-7-カルボン酸メチルエステルを微黄色結晶として得た。
m.p.:190~191℃、IR(Nujol)1719cm-1、APCI-MS m/z:302[M+H]+
2)2-(2-メチルチオピリミジン-5-イル)ベンズオキサゾール-7-カルボン酸メチルエステル(400mg,1.33mmol)のテトラヒドロフラン(10ml)の懸濁液に氷冷下77%メタクロル過安息香酸(476mg,2.12mmol)を加え15分間攪拌後、さらに室温で3時間攪拌した。本反応液に室温で50%ジメチルアミン水溶液を滴下し、室温で1時間攪拌した。反応液を氷冷し、水(25ml)を加え15分間攪拌して酢酸エチルで抽出した。有機層を水、飽和食塩水で順に洗浄後、乾燥して、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(クロロホルム/酢酸エチル=4)で精製して2-(2-ジメチルアミノピリミジン-5-イル)ベンズオキサゾール-7-カルボン酸メチルエステル(328mg,83%)を無色結晶として得た。
m.p.:199~201℃、IR(Nujol)1719,1628,1605cm-1、APCI-MS m/z:299[M+H]+
3)2-(2-ジメチルアミノピリミジン-5-イル)ベンズオキサゾール-7-カルボン酸メチルエステル(480mg,1.61mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-(2-ジメチルアミノピリミジン-5-イル)ベンズオキサゾール-7-メタノール(234mg,54%)を黄色結晶として得た。
m.p.:212~215℃、IR(Nujol)3283,1617cm-1、APCI-MS m/z:271[M+H]+
1)2-(2-メチルチオピリミジン-5-イル)ベンズオキサゾール-7-カルボン酸メチルエステル(400mg,1.33mmol)を原料にして製造例13の2)に準じて、2-[2-[N-2-(ヒドロキシ)エチル-N-メチル]アミノピリミジン-5-イル]ベンズオキサゾール-7-カルボン酸メチルエステル(322mg,74%)を無色結晶として合成した。
m.p.:157~159℃、IR(Nujol)3529,1713,1626,1601cm-1、APCI-MS m/z:329[M+H]+
2)2-[2-[N-2-(ヒドロキシ)エチル-N-メチル]アミノピリミジン-5-イル]ベンズオキサゾール-7-カルボン酸メチルエステル(300mg,0.914mmol)のヒドロキシ基を常法によりテトラヒドロピラニル化して、2-[2-[N-2-(テトラヒドロピラン-2-イルオキシ)エチル-N-メチル]アミノピリミジン-5-イル]ベンズオキサゾール-7-カルボン酸メチルエステル(286mg,69%)を無色結晶として得た。
m.p.:126~128℃、IR(Nujol)1717,1625,1601cm-1、APCI-MS m/z:413[M+H]+
3)2-[2-[N-2-(テトラヒドロピラン-2-イルオキシ)エチル-N-メチル]アミノピリミジン-5-イル]ベンズオキサゾール-7-カルボン酸メチルエステル(275mg,0.667mmol)を製造例2の3)と同様にリチウムアルミニウムハイドライドで還元して、2-[2-[N-2-(テトラヒドロピラン-2-イルオキシ)エチル-N-メチル]アミノピリミジン-5-イル]ベンズオキサゾール-7-メタノール(128mg,50%)を無色結晶として得た。
m.p.:110~112℃、IR(Nujol)3287,1622,1603cm-1、APCI-MS m/z:385[M+H]+
1)水素化ホウ素ナトリウム(422mg,11.14mmol)のテトラヒドロフラン(30ml)懸濁液に、三フッ化ホウ素エーテル錯体(1.83ml,14.86mmol)を氷冷攪拌下10分間かけて滴下し、本反応液に(±)-2-フェニル-1,4-ベンズオキサジン-3-オン-8-カルボン酸(500mg,1.86mmol)のテトラヒドロフラン(6ml)溶液を加え同温で5分間、室温で1.5時間攪拌した。反応液を氷冷して水(20ml)を滴下し、飽和炭酸水素ナトリウム水溶液で中和して酢酸エチルで抽出した。抽出した有機層は、水、飽和食塩水で順に洗浄後乾燥して、溶媒を減圧留去することにより(±)-2-フェニル-1,4-ベンズオキサジン-8-メタノール(397mg,89%)を微橙色油状物質として得た。
IR(Neat)3375,1607cm-1
2)(±)-2-フェニル-1,4-ベンズオキサジン-8-メタノール(600mg,2.49mmol)のジエチルエーテル(22ml)溶液に、氷冷攪拌下炭酸ナトリウム(2.90g,27.35mmol)を加えた後、無水トリフルオロ酢酸(3.86ml,27.35mmol)を滴下し、同温で15分間、室温で15分間攪拌した。反応液を氷冷して氷水中に注ぎ込み酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄後乾燥して、溶媒を減圧留去した。残さにジイソプロピルエーテルを加えて粉砕し、ろ取して乾燥することにより(±)-4-トリフロロアセチル-8-トリフロロアセトキシメチル-2-フェニル-1,4-ベンズオキサジン(973mg,90%)を無色結晶として得た。
m.p.:91~92℃、IR(Nujol)1781,1705cm-1
3)(±)-4-トリフロロアセチル-8-トリフロロアセトキシメチル-2-フェニル-1,4-ベンズオキサジン(953mg,2.20mmol)のメタノール(19ml)溶液に、室温でグリシン緩衝液(pH10,6.33ml)を滴下し、30分間攪拌した。本反応液に水(70ml)を加え室温で20分間攪拌後、酢酸エチルで抽出した。抽出した有機層は、水、飽和食塩水で順に洗浄後乾燥して、溶媒を減圧留去することにより(±)-4-トリフロロアセチル-2-フェニル-1,4-ベンズオキサジン-8-メタノール(793mg,収率定量的)を微橙色油状物質として得た。
IR(Neat)3400,1704cm-1
(5-メトキシ-2-フェニル)ベンゾ[b]フラン-7-カルボン酸を常法によりリチウムアルミニウムハイドライドで還元して、(5-メトキシ-2-フェニル)ベンゾ[b]フラン-7-メタノールを合成した。
m.p.:150~151℃
3-ヒドロキシメチルフラボン-8-カルボン酸を常法によりヒドロキシ基をアセチル化後カルボキシル基を還元して、3-アセトキシメチルフラボン-8-メタノールを合成した。
m.p.:204~206℃、IR(Nujol)1739,1616cm-1、ESI-MS m/z:337[M-H]-.
3-ボロモメチルフラボン-8-カルボン酸エチルエステルに常法によりジメチルアミンを反応させた後還元して、3-ジメチルアミノメチルフラボン-8-メタノールを合成した。
m.p.:149.5~150.5℃、IR(Nujol)3448,1627cm-1、APCI-MS m/z:310[M+H]+
1)製造例17で合成した3-アセトキシメチルフラボン-8-メタノールのヒドロキシ基を常法によりメトキシメチル化しアセチル基を除去後生成したアルコールを酸化して8-メトキシメトキシメチルフラボン-3-カルボン酸を合成した。
m.p.:142~143℃、IR(Nujol)1731,1621,1606cm-1、ESI-MS m/z:339[M-H]-
2)8-メトキシメトキシメチルフラボン-3-カルボン酸にアジ化ジフェニルホスホリル、t-ブタノールを順に反応させた後、塩酸水-ジオキサンで加水分解して3-アミノフラボン-8-メタノールを合成した。
m.p.:191.5~192.5℃、IR(Nujol)3391,3302,1606cm-1、APCI-MS m/z:268[M+H]+
(5-アミノ-2-フェニル)ベンゾ[b]フラン-7-カルボン酸メチルエステルを常法によりアミノ基をジメチル化後エステル基をリチウムアルミニウムハイドライドで還元して、(5-ジメチルアミノ-2-フェニル)ベンゾ[b]フラン-7-メタノールを合成した。
m.p.:115~116℃、IR(Nujol)3243,1734,1703cm-1、APCI-MS m/z:268[M+H]+
1)2-アセトアミノ-3-ニトロ安息香酸メチルエステル(1.444g,6.06mmol)及び6N塩酸(30ml)の混合物を15分間加熱還流した。反応液を氷冷し、10%炭酸カリウム水溶液でpH8として酢酸エチルで抽出した。抽出した有機層は水、飽和食塩水で順に洗浄後、乾燥して溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=1)で精製し、2-アミノ-3-ニトロ安息香酸メチルエステル(987mg,83%)を黄色結晶として得た。
m.p.:94~96℃、APCI-MS m/z:197[M+H]+
2)2-アミノ-3-ニトロ安息香酸メチルエステル(980mg,5.00mmol)のメタノール(20ml)-テトラヒドロフラン(10ml)溶液に10%パラジウム炭素(水分51.7%,250mg)を加え、水素気流下室温で3時間攪拌した。触媒をろ去し、テトラヒドロフランで洗浄した。ろ液、洗液を合わせて溶媒を減圧留去することにより2,3-ジアミノ安息香酸メチルエステル(814mg,98%)を黄緑色固体として得た。
m.p.:65~67℃、IR(Nujol)3451,3314,1701,1619cm-1、APCI-MS m/z:167[M+H]+
3)2,3-ジアミノ安息香酸メチルエステル(4.00g,24.07mmol)及びベンズアルデヒド(2.56g,24.07mmol)のニトロベンゼン(60ml)溶液を155~160℃で3時間加熱攪拌した。反応液を室温としてそのままシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=20及び4)で精製し、2-フェニルベンズイミダゾール-4-カルボン酸メチルエステル(3.89g,64%)を淡黄色結晶として得た。
m.p.:125~127℃、IR(Nujol)3364,1709cm-1、APCI-MS m/z:253[M+H]+
4)60%水素化ナトリウム(32mg,0.793mmol)をn-ヘキサンで洗浄後、2-フェニルベンズイミダゾール-4-カルボン酸メチルエステル(200mg,0.793mmol)のDMF(1.2ml)溶液を室温で滴下し、1.5時間室温攪拌した。本反応液に2-(トリメチルシリル)エトキシメチルクロリド(0.15ml,0.841mmol)を滴下し、室温で2時間攪拌後、氷冷して水を加え酢酸エチルで抽出した。抽出した有機層は水、飽和食塩水で順に洗浄後、乾燥して溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=7及び2.5)で精製し、(溶出した順に)1-[2-(トリメチルシリル)エトキシメチル]-2-フェニルベンズイミダゾール-4又は7-カルボン酸メチルエステル(113mg,37%)及び1-[2-(トリメチルシリル)エトキシメチル]-2-フェニルベンズイミダゾール-4又は7-カルボン酸メチルエステル(133mg,44%)をそれぞれ無色油状物質として得た。溶出した化合物の順に物性を示す。
IR(Neat)2952,1722cm-1、APCI-MS m/z:383[M+H]+、IR(Neat)2951,1723cm-1、APCI-MS m/z:383[M+H]+
5)1-[2-(トリメチルシリル)エトキシメチル]-2-フェニルベンズイミダゾール-4又は7-カルボン酸メチルエステル(120mg,0.314mmol)(後から溶出した化合物)をリチウムアルミニウムハイドライドで還元して、1-[2-(トリメチルシリル)エトキシメチル]-2-フェニルベンズイミダゾール-4又は7-メタノール(94mg,85%)を無色結晶として得た。
m.p.:96~100℃、IR(Nujol)3181cm-1、APCI-MS m/z:355[M+H]+
製造例19の1)で得た8-メトキシメトキシメチルフラボン-3-カルボン酸を常法によりアミド化し、塩酸水-メタノールでメトキシメチル基を除去した後、塩化チオニルで処理して8-クロロメチル-3-ジメチルカルバモイルフラボンを合成した。
m.p.:202~203℃、IR(Nujol)1638,1630,1616cm-1、APCI-MS m/z:324[M+H]+
製造例19の1)で得た8-メトキシメトキシメチルフラボン-3-カルボン酸を常法によりメチルエステル化し、塩酸水-メタノールでメトキシメチル基を除去した後、塩化チオニルで処理して8-クロロメチル-3-メトキシカルボニルフラボンを合成した。
m.p.:149.5~150.5℃、IR(Nujol)1732,1637cm-1、APCI-MS m/z:311[M+H]+
製造例19の1)で得た8-メトキシメトキシメチルフラボン-3-カルボン酸を常法によりメトキシメチル基を除去し、カルボキシル基をジフェニルメチルエステルに変換した後、塩化チオニルで処理して8-クロロメチル-3-ジフェニルメトキシカルボニルフラボンを合成した。
m.p.:185.5~186℃、IR(Nujol)1731,1629cm-1、ESI-MS m/z:485[M+Na]+
8-ヒドロキシメチル-3-メチルフラボンを塩化チオニルで処理して8-クロロメチル-3-メチルフラボンを合成した。
m.p.:112.5~113.5℃、IR(Nujol)1622,1601cm-1、APCI-MS m/z:285[M+H]+
1)2,6-ジブロモナフタレン(2.20g,7.70mmol)を原料にして、パラジウム触媒を用いるアミノ化反応で2-[ビス-2-(ベンジルオキシ)エチル]アミノ-6-ブロモナフタレン(2.29g,61%)を黄色油状物質として得た。
IR(Neat)1625,1585cm-1、APCI-MS m/z:490/492[M+H]+
2)2-[ビス-2-(ベンジルオキシ)エチル]アミノ-6-ブロモナフタレン(205mg,0.42mmol)を原料にして、製造例5に準じて2-[ビス-2-(ベンジルオキシ)エチル]アミノ-ナフタレン-6-ボロン酸(124mg,65%)を黄色油状物質として得た。
1)2-アミノ-5-ブロモピリジン(2.0g,11.56mmol)のメタノール(465ml)溶液に、室温で37%ホルムアルデヒド水溶液(13.55ml,180.3mmol)を滴下後、塩化亜鉛(3.94g,28.90mmol)及びシアノ水素化ホウ素ナトリウム(3.63g,57.80mmol)のメタノール(155ml)溶液を滴下して室温で4時間攪拌した。反応液に5°Cで氷水(300ml)を加えた後、メタノールを減圧留去して酢酸エチル-テトラヒドロフラン(1/1)で抽出した。有機層を水、飽和食塩水で順に洗浄後乾燥し、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=24及び5)で精製して5-ブロモ-2-ジメチルアミノ-ピリジン(1.00g,43%)を無色結晶として得た。
m.p.:39~41℃、IR(Nujol)1588cm-1、APCI-MS m/z:201/203[M+H]+
2)5-ブロモ-2-ジメチルアミノピリジン(402mg,2.00mmol)を原料にして、製造例5に準じて2-ジメチルアミノピリジン-5-ボロン酸(321mg,crude 97%)を微褐色粉末として得た。
1)5-ブロモ-2-クロロピリジン(600mg,3.12mmol)及びチオモルホリン(1.60g,15.59mmol)の混合物を100°Cで16時間攪拌した。反応液に飽和炭酸水素ナトリウムを加え酢酸エチルで抽出した。有機層を水、飽和食塩水で順に洗浄後乾燥し、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=50)で精製して5-ブロモ-2-チオモルホリノピリジン(465mg,58%)を無色油状物質として得た。
IR(Neat)1581,1481cm-1、APCI-MS m/z:259/261[M+H]+
2)5-ブロモ-2-チオモルホリノピリジン(706mg,2.72mmol)の脱気したトルエン(30ml)-1,4-ジオキサン(30ml)溶液に室温でトリエチルアミン(30ml)、ビス(トリブチルスズ)(3.05ml,6.04mmol)の及びテトラキス(トリフェニルホスフィン)パラジウム(315mg,0.272mmol)を順に加え脱気してアルゴン置換後、95-100℃で14時間加熱攪拌した。反応液を室温とし溶媒を減圧留去した。残さをアミンシリカゲルカラムクロマトグラフィー(クロマトレックス(登録商標)NH)(n-ヘキサン/酢酸エチル=100)で精製し5-トリ-n-ブチルスタニル-2-チオモルホリノピリジン(467mg,37%)を無色油状物質として得た。
IR(Neat)1575,1535,1483cm-1、APCI-MS m/z:467/469/471[M+H]+
1)5-ブロモ-2-クロロピリミジン(1.79g,10mmol)に50%ジメチルアミン水溶液(30ml)を加え、アルゴン雰囲気下、室温で5時間攪拌した。反応液に飽和炭酸水素ナトリウム水溶液(15ml)を加え、酢酸エチルで抽出した。抽出層は、飽和食塩水で洗浄して乾燥後溶媒を減圧留去した。残さを40℃で2時間減圧乾燥して5-ブロモ-2-ジメチルアミノピリミジン(1.83g,90%)を無色結晶として得た。
m.p.:81~82℃、IR(Nujol)1586,1527cm-1、APCIMS m/z:202/204[M+H]+
2)5-ブロモ-2-ジメチルアミノピリミジン(1.75g,8.66mmol)のテトラヒドロフラン(18ml)溶液にアルゴン雰囲気下、-78℃でn-ブチルリチウム(1.5M n-ヘキサン溶液;6.06ml,9.09mmol)を15分間かけて滴下し、同温で2時間攪拌後、塩化トリ-n-ブチルスズ(2.5ml)を滴下し、同温で0.5時間、室温で1時間攪拌した。反応液に10%フッ化カリウム水溶液(50ml)、酢酸エチル(50ml)を順に加え、室温で0.5時間攪拌後、有機層を分取した。有機層は、水、飽和食塩水で順に洗浄して、乾燥後溶媒を減圧留去した。残さをアミンシリカゲルカラムクロマトグラフィー(クロマトレックス(登録商標)NH)(n-ヘキサン)で精製し、5-トリ-n-ブチルスタニル-2-ジメチルアミノピリミジン(2.65g,74%)を無色油状物質として得た。
IR(Neat)2955,2925,2870,2853,1569,1519cm-1、APCI-MS m/z:410/412/414[M+H]+
N-t-ブトキシカルボニル-3,5-ジヨード-L-チロシン メチルエステル(700mg,1.28mmol)の脱気したN-メチルピロリドン(2.1ml)溶液にアルゴン雰囲気下、トリス(ジベンジリデンアセトン)ジパラジウム(37mg,0.04mmol)、トリフェニルホスフィン(69mg,0.261mmol)を順に加え50℃で10分間加熱攪拌後、室温としてヨウ化第一銅(24mg,0.125mmol)を加えた。反応液を50℃で10分間加熱攪拌後、室温としてテトラメチルスズ(0.39ml,2.82mmol)を滴下して封管中65℃で18時間攪拌した。反応液を氷冷して、水(10ml)、飽和フッ化ナトリウム水溶液を順に加え、酢酸エチルで抽出した。抽出層は、水、飽和食塩水で順に洗浄後、乾燥して溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=5)で精製してN-t-ブトキシカルボニル-3,5-ジメチル-L-チロシン メチルエステル(230mg,56%)を褐色油状物質として得た。
IR(Neat)3389,1739,1695cm-1、APCI-MS m/z:324[M+H]+
4-アミノ-N-トリフルオロアセチル-L-フェニルアラニン エチルエステル(9.30g,30.6mmol)のジメチルホルムアミド(305ml)溶液にN-クロロコハク酸イミド(9.79g,73.32mmol)を加え、アルゴン雰囲気下、55℃で2.5時間攪拌した。反応液に氷冷下、酢酸エチル及び水を加え飽和炭酸水素ナトリウム水溶液でpH8として、酢酸エチルで抽出した。抽出層は、水、飽和食塩水で順に洗浄後、乾燥して、溶媒を減圧留去した。残さをシリカゲルカラムクロマトグラフィー(n-ヘキサン/酢酸エチル=6)で精製して4-アミノ-3,5-ジクロロ-N-トリフルオロアセチル-L-フェニルアラニン エチルエステル(8.49g,74%)を無色結晶として得た。
m.p.:124~125℃、IR(Nujol)3300,1742,1707cm-1、ESI-MS m/z:371/373[M-H]-
図9-1、2の反応スキームに従って、本発明に係るO-(5-アミノ-2-フェニルベンズオキサゾール-7-イル)メチル-3,5-ジクロロ-L-チロシン・二塩酸塩のβ-シクロデキストリン包接体を調製した。当該塩は、淡黄色結晶又は結晶性の粉末であり、融点が180~200℃(分解)であった。
(2)の合成
3-ニトロサリチル酸(1)(201g、1.09mol)のメタノール(1600ml)溶液に室温で96%硫酸(60.6ml)を滴下し、その後23時間還流した。反応液を約1/3になるまで減圧濃縮した後氷浴にて冷却した。生成した固体を濾取、冷メタノール(600ml)、冷水(600ml)にて洗浄、減圧下40℃にて乾燥することにより3-ニトロサリチル酸メチル(2)を209.7g(97%)黄色固体として得た。
(3)の合成
3-ニトロサリチル酸メチル(2)(108.2g、0.549mol)のTHF(1140ml)/MeOH(810ml)溶液に10%Pd-C(15.2g)を加え、水素1気圧下室温にて2時間接触還元した。触媒を濾別し、THFで洗浄した後溶媒留去した。残渣をジイソプロピルエーテルにて洗浄することにより3-アミノサリチル酸メチル(3)を166.7g(95%)褐色固体として得た。
(4)の合成
3-アミノサリチル酸メチル(3)(74.8g、0.447mol)のTHF(1130ml)溶液に5℃でN,N-ジメチルアニリン(108.7g、0.897mol)を40分で滴下した。反応液を15分間撹拌した後ベンゾイルクロリド(75.7g、0.538mol)のTHF(570ml)溶液を1時間かけて滴下した。同温で1時間撹拌した後、水(100ml)を加え、THFを留去した。残渣に水(900ml)を加え、酢酸エチルにて抽出し、有機層を10%塩酸、水、飽和食塩水で洗浄、亡硝にて乾燥し、溶媒を留去した。得られた粗結晶とアミノ体(3)(90.16g)より同様の操作で得られた結晶を合わせ酢酸エチルより再結晶することにより3-ベンゾイルアミノサリチル酸メチル(4)を247.1g(92%)無色固体として得た。
(5)の合成
3-ベンゾイルアミノサリチル酸メチル(4)(83.2g、0.296mol)の酢酸(1480ml)溶液に内温15℃で69%硝酸(d=1.42)(95ml、1.458mol)を滴下した。反応液を室温で3時間撹拌した後、氷水(300ml)に注ぎ10分間撹拌した。生成した固体を濾過し、水(3回)、エーテルで洗浄した後風乾することにより3-ベンゾイルアミノ-5-ニトロサリチル酸メチル(5)を93.4g(96%)黄色固体として得た。
(6)の合成
3-ベンゾイルアミノ-5-ニトロサリチル酸メチル(5)(72.2g、0.260mol)とPPSEの1,2-ジクロロベンゼン溶液(780ml)の混合物を150℃で4時間撹拌した。反応液を氷冷し、析出した結晶を濾取、n-ヘキサン、冷メタノールで洗浄、減圧で乾燥することにより、5-ニトロ-2-フェニルベンゾオキサゾール-7-カルボン酸メチルエステル(6)を61.2g(90%)淡黄色固体として得た。
(7)の合成
5-ニトロ-2-フェニルベンゾオキサゾール-7-カルボン酸メチルエステル(6)(59.2g、0.198mol)のメタノール(990ml)懸濁液に1M水酸化ナトリウム水溶液(297ml、0.297mol)を滴下し、その後室温にて19時間撹拌した。氷冷下に10%塩酸にて反応液を酸性とし数分間撹拌した。生成した固体を濾取、水、メタノールで洗浄した後減圧乾燥することにより5-ニトロ-2-フェニルベンゾオキサゾール-7-カルボン酸(7)を54.2g(96%)無色固体として得た。
(9)の合成
5-ニトロ-2-フェニルベンゾオキサゾール-7-カルボン酸(7)(26.4g、0.0931mol)のTHF(470ml)懸濁液にトリエチルアミン(15.57ml、0.112mol)を加え室温で30分間撹拌した。反応液を-10℃に冷却し、クロロ炭酸エチル(10.68ml、0.112mol)を加え同温で30分撹拌した。水素化ホウ素ナトリウム(8.84g、233.7mmol)を加え30分撹拌した後、THF/水(317ml/64ml)を3時間かけて-10~-6℃にて滴下した。反応液をさらに2時間撹拌し、その後に氷水を注ぎ、析出した固体を濾取、水で洗浄した。5-ニトロ-2-フェニルベンゾオキサゾール-7-カルボン酸(7)(14.6g)より同様の操作により得られた固体を合わせ、THF(600ml)/メタノール(600ml)に懸濁し、1M水酸化ナトリウム水溶液(115ml)を加えた後室温で4.5時間撹拌した。反応液を氷水にあけ、析出した固体を濾過した後風乾することにより7-ヒドロキシメチル-5-ニトロ-2-フェニルベンゾオキサゾール(9)を24.4g(63%)ほぼ無色の固体として得た。
(10)の合成
7-ヒドロキシメチル-5-ニトロ-2-フェニルベンゾオキサゾール(9)(46.2g、0.171mol)のTHF(1150ml)懸濁液に氷冷下トリエチルアミン(34.9ml、0.250mol)を加えた。メタンスルホニルクロリド(23.1g、0.200mol)を滴下し、氷冷下で30分その後室温にて3時間撹拌した。トリエチルアミン(4.55ml、0.033mol)、メタンスルホニルクロリド(1.29ml、0.017mol)を追加しさらに30分撹拌した。反応液を氷水に注ぎ析出した固体を濾過した。得られた固体を水、ジイソプロピルエーテルで洗浄した後40℃で減圧乾燥することにより5-ニトロ-2-フェニルベンゾオキサゾール-7-イルメチルメタンスルホネート(10)を59.1g(99%)淡黄色固体として得た。
2)ジクロロチロシン誘導体の合成
(12)の合成
(S)-チロシンメチルエステル塩酸塩(11)(60.15g、0.26mol)の酢酸(294ml)懸濁液に室温にてスルフリルクロリド(52.22ml、0.65mol)を滴下し、その後3.5時間撹拌した。反応液を濃縮し、残渣をエーテルにて充分に洗浄、乾燥することにより(S)-ジクロロチロシンメチルエステル塩酸塩(12)を72.70g(93%)無色固体として得た。
(13)の合成
(S)-ジクロロチロシンメチルエステル塩酸塩(12)(72.42g、0.24mol)の水(1.4l)懸濁液に氷冷下で2M炭酸カリウム水溶液(120.5ml)を滴下した。ジ-t-ブチルジカーボネート(58.1ml、0.25mol)のジクロロメタン(1.4l)溶液を加え氷冷下で1.5時間、室温にて40時間撹拌した。反応液をセライトを用いて濾過した後、濾液をクロロホルムで抽出した。有機層を水、飽和食塩水で洗浄、亡硝乾燥し、溶媒を留去した。残渣をn-ヘキサンで洗浄することにより(S)-N-Boc-ジクロロチロシンメチルエステル(13)を76.95g(88%)無色固体として得た。
3)本発明に係る化合物塩の合成
(14)の合成
5-ニトロ-2-フェニルベンゾオキサゾール-7-イルメチルメタンスルホネート(10)(29.11g、83.6mmol)をTHF(820ml)に溶解し、アセトン(820ml)を加えた。(S)-N-Boc-ジクロロチロシンメチルエステル(13)(36.54g、100mmol)、ヨウ化ナトリウム(12.53g、83.6mmol)、炭酸カリウム(17.37g、125mmol)を加え、室温にて21時間撹拌した。反応液を濾過し、濾液を約1/2まで濃縮した。酢酸エチル、水を加え分液した後、有機層を水、5%チオ硫酸ナトリウム水溶液、飽和食塩水で洗浄し、亡硝乾燥溶媒留去した。5-ニトロ-2-フェニルベンゾオキサゾール-7-イルメチルメタンスルホネート(10)(30.06g)、(S)-N-Boc-ジクロロチロシンメチルエステル(13)(37.66g)より同様の操作によって得られた生成物を加え、計116.2gの粗生成物をジイソプロピルエーテルにて洗浄した後乾燥することによりニトロ体(14)を98.43g(94%)淡黄色固体として得た。
(15)の合成
ニトロ体(14)(57.04g、92.5mmol)をTHF(1340ml)に溶解し、イソプロパノール(1340ml)を加え内温60℃に加熱した。5M塩化アンモニウム水溶液(278ml、1.39mol)、続いて鉄粉(103.32g、1.85mol)を加え、60℃にて3時間撹拌した。反応液をセライトを用いて濾過し、濾液を約1/3まで濃縮した。セライト上の不要物は熱酢酸エチルにて3回洗浄し、濾液と合わせて水、飽和食塩水で洗浄した後亡硝にて乾燥した。溶媒を留去し、得られた固体をジイソプロピルエーテルにて洗浄、乾燥することによりアミノ体(15)を53.03g(98%)黄色固体として得た。
(16)の合成
アミノ体(15)(86.91g、0.148mol)をTHF(1430ml)に溶解し、内温が約10℃になるように氷浴で冷却しつつ0.5M水酸化リチウム水溶液(444ml、0.222mol)を滴下し、さらに氷浴中で1時間撹拌した。反応液を約1/2になるまで濃縮し、水(600ml)を加え、さらに10%クエン酸水溶液にてpHを約4とした後酢酸エチル-THF(10:1)にて抽出した。抽出液を水、飽和食塩水で洗浄し、亡硝乾燥、溶媒留去した。得られた残渣を酢酸エチル-THF(10:1)より再結晶し、ほぼ無色の固体(72.94g、粗収率86%)を得た。この固体(10.03g)をクロロホルム-メタノール(10:1)より再結晶し、カルボン酸(16)を8.63g(回収率86%)ほぼ無色の固体として得た。
(17)本発明に係る化合物塩の合成
氷冷下カルボン酸(16)(85.23g、0.149mol)のTHF(750ml)溶液に5~10℃で4M塩化水素-ジオキサン(1117ml、4.47mol)を2時間かけて滴下した。反応液を同温で1時間撹拌した後、徐々に昇温し室温にて20時間撹拌した。ジイソプロピルエーテルを加え固体を濾取し、ジイソプロピルエーテルにて洗浄した。得られた固体を3ロットに分けてエタノール(計2300ml)に溶解し、不溶物を濾別した。濾液を室温、減圧にて約1000mlにまで濃縮し、得られた溶液を室温にて撹拌した。生成した結晶を濾過し、乾燥することにより本発明に係る化合物塩(17)を66.11g(81%)淡黄色固体として得た。
4)サイクロデキストリン包接体の合成
(1)Sulfobutyl ether-β-cyclodextrin (SBE-CDと略す:商品名Captisol)を1000 g秤量し、細胞培養用の無菌フード内で、5 Lの容器に取り、HPLC用グレードの純水1800 mlを加えて、一晩撹拌して澄んだ溶液を得た(約36 重量%となる)。
(2)翌朝、水槽中で32-36℃に加温し、この温度を保持して、本発明に係る化合物のHCl塩を45.5 g; free baseとして35.5 g相当に100 mlのエタノール、200 mlの純水、そして300 mlの上記SBE-CD溶液を添加し、化合物溶液を得た。この溶液のpHが3~4であることを確認した。
(3)3.4 gの苛性ソーダを200 mlの純水に溶解し、上記溶液を撹拌しながらこの苛性ソーダ水を加えて、pHを6.5に調製した。
(4)この暖かい状態で、溶液を真空ポンプを用いて、0.4 μmのフィルターで滅菌ろ過した。ろ液は滅菌済の5 Lの容器に取り、(1)で調製したSBE-CD溶液で希釈し、最終容量を2800 mlに調整した。
(5)更に5分間撹拌後に、pHを測定し、~6.5であることを確認した。滅菌分注器を用いて、30 mlバイアル瓶に各15 mlづつ分注した。最終化合物濃度は10 mg/mlとなった。各バイアルは1週間にわたり凍結乾燥機(Lyostar3, FTS Systems SP Scientific)で処理された。
(6)凍結乾燥後のバイアル瓶はゴム栓で密封し、金属ヒダ留めをし、冷蔵庫で保存した。
(7)再溶液化には14.1 mlの注射用蒸留水を加えて5-10 分間のvoltex mixerにより15 mlの淡黄色の溶液になった。
以下の材料および方法により、試験を行った。
動物実験は、ヘルシンキ宣言に従って行い、また、プロトコールは、施設内の倫理委員会(2011~73)およびCIEPAL(NCE/2011~33)によって承認された。マウスは、C3Mの動物飼育室施設で、特定病原体除去条件下で維持した。Pten欠損マウス(tPTEN-/-)は、2つのPten flox対立遺伝子を持つマウスを、近位lckプロモーター-creトランスジェニックマウスと交雑させることによって生成した(参考文献53)。動物は、先に記載されるように(参考文献53)、PCRによって特徴付けた。
リンパ腫形成の臨床徴候が観察された場合(たとえば毛の逆立ち(fur ruffling)、丸まった姿勢、顔面浮腫、および/または表在呼吸)、マウスは、致命的なペントバルビタール注射により犠牲死させた。腫瘍を解剖し、70μm細胞ストレーナ(BD Biosciences)上でPBS中でホモジナイズし、全RNAを単離し、先に記載されるように(参考文献54)、cDNAに転写した。無関係な4つの腫瘍を分析し、2つの野生型マウス由来の胸腺細胞もまた、処理した。cDNAを、AffymetrixDNAチップ(Affymetrix-MoGene-1_0-st-v1)にハイブリダイズさせ、遺伝子発現データを標準化し(GSE 39591)、25%の偽発見率を使用して、監視下の(supervised)SAM(マイクロアレイの有意性分析(Significance Analysis of Microarrays))分析にかけた。次いで、マイクロアレイ分析についての線形モデル(LIMMA)を、p値<104で、異なって発現されるプローブを同定するために使用した。これにより、abs(log倍数変化(log Fold Change))>0.5を有する遺伝子選択がもたらされる。フェノタイプおよびアノテートされていない配列の間でより小さなp値を有する複写物を除去した後、分析の組み合わせにより、腫瘍において、498のアップレギュレートされた遺伝子および279のダウンレギュレートされた遺伝子が同定された。
無関係なtPTEN-/-腫瘍から確立されたKO99L、KO1081L、およびKO631Lとして示されたマウス細胞株に加えて、ヒトKe37、DND41、Sil-ALL、Peer、Molt-16、Jurkat(T-ALL)、およびSupT1(T-LL)を、10または20%(Sil-ALLおよびマウス細胞株)ウシ胎児血清、ならびに50ユニット/mlペニシリン、50mg/mlストレプトマイシン、および1.0mMピルビン酸ナトリウム(Sigma-Aldrich))を補足したRPMI 1640培地(Invitrogen)において増殖させた。細胞培養物は、5%CO2下で37℃で維持した。
本研究において使用した化学化合物:2-アミノビシクロ-(2,2,1)-ヘプタン-2-カルボン酸(BCH)、デキサメタゾン、塩酸ドキソルビシン、必須アミノ酸(EAA;50×溶液)、非必須アミノ酸(NEAA;100×溶液)、D-ロイシン、およびワートマニン(Sigma-Aldrich);KU0063794(rel-5-[2-[(2R,6S)-2,6-ジメチル-4-モルホリニル]-4-(4-モルホリニル)ピリド[2,3-d]ピリミジン-7-イル]-2-メトキシベンゼンメタノール)、PI-103塩酸塩(3-[4-(4-モルホリニルピリド[3’,2’:4,5]フロ[3,2-d]ピリミジン-2-イル]フェノール塩酸塩)、およびラパマイシン(Tocris Bioscience);L-アスパラギナーゼ、Erwinase(登録商標)(クリサンタスパーゼ)(EUSA-PHARMA)。Salubrinal(N-(2,2,2-トリクロロ-1-(3-(キノリン-8-イル)チオウレイド)エチル)シンナムアミド)(Enzo Life Science);Q-VD-OHP(MPBiomedicals)。
インフォームドコンセントは、施設内のガイドラインに従って得た。ヘパリン処理した血液は、3人のT-ALL患者および2人の健康なドナーから得た。患者リンパ芽球は、ベンダー推奨の手順に従って、Ficoll-Paque PLUS密度勾配を使用して、収集後24時間以内に単離した(GE Healthcare Life Sciences)。
2つの異なるアッセイを用いた:i)96ウェルプレートフォーマットにおいて、細胞(ウェル当たり4.0±0.1×104;100μL)を、エフェクターと共にインキュベートし(48時間、37℃;実験当たりn=4)、次いで、WST-1試薬(10μL;Roche Diagnostics)を、96ウェルプレートのそれぞれのウェルに追加し、インキュベートし、ホルマザン生成を490nmで測定した;およびii)細胞(ウェル当たり2.0±0.1×104;100μL)を、エフェクターと共にインキュベートし(48時間、37℃;実験当たりn=4)、次いで、ブロモデオキシウリジン試薬(10μL/ウェル;Roche Diagnostics)を、それぞれのウェルに追加し、インキュベートした(8時間、37℃)。次いで、細胞を、固定し、変性させ、ペルオキシダーゼ(POD)抗体とコンジュゲートされた抗BrdUとインキュベートした(100μL/ウェル)。比色定量基質(TMB:テトラメチル-ベンジジン;100μL/ウェル)を追加し、BrdUの取込みを、DNA合成および増殖細胞に対応する光学濃度測定(450nm)によって定量化した。
細胞抽出物を、細胞溶解バッファー(プロテアーゼ阻害剤のカクテルを補足した50mM HEPES、pH7.4、150mM NaCl、20mM EDTA、100mM NaF、10mM Na3VO4、0.5%ノニルフェノキシポリエトキシルエタノール(NP40))を使用して調製した。SDS-PAGEによる分離およびニトロセルロースへの転写の後に、イムノブロッティングを、以下の抗体を使用して実行した:Akt(Cell Signaling Technologies、#9272)、CHOP(#2895)、切断されたカスパーゼ3(#9664)、c-Myc(#9402)、eIF2α(#9721)、4EBP1(#9644)、LC3A/B(#12741)、PARP(#9542)、pJnk(T183/Y185)(#9251)、pSer235-236-S6RP(#4856)、pSer473-Akt(#9271)、pSer65-4EBP1(#9451)、およびS6RP(#2217);Bax(Sigma-Aldrich、B8429);PUMA(Abcam、ab54288);Bak(Millipore、06536);ATF4(Santa Cruz Biotechnologies、sc200)、GADD34(sc8327)、Hsp60(sc-1722)、Hsp90(sc-13119)、およびXBP1(sc-7160);ATF6(Imgenex、IMG-273)。
アポトーシス評価は、アネキシン-V-FITC(Invitrogen)によってホスファチジル-セリン膜再分布を測定することによって実行した。細胞(ウェル当たり5.0±0.1×105;2.0mL)を、エフェクターと共にインキュベートし、染色溶液(110μL;アネキシン-V(1/10))中に再懸濁し、暗中(15分間、RT)でインキュベートした。細胞をPBSにより洗浄し、プロピジウムヨージド染色溶液(1.0μg/ml;2.0mL)中に再懸濁し、MacsQuant Analyser(Miltenyi Biotech)でフローサイトメトリによって直ちに分析した。基底のアポトーシスおよび壊死は、コントロール未処理細胞に対して決定した。
細胞を収集し、PBSにより洗浄した。回転させた後に、細胞(106;PBS、EDTA 5.0mM、0.1%BSA中1.0mL)を、暗中で、4℃で、30分間、ラット抗CD98抗体(H202-141、5.0μg/ml、BDPharmingen)と共にインキュベートした。細胞を、洗浄し、4℃で、30分間、ヤギ抗ラットAlexaFluor(登録商標)-488二次抗体(A11006、2μg/ml、Life Technologies)と共にインキュベートした。細胞を、遠心沈殿し、2回洗浄し、MacsQuant Analyser(Miltenyi Biotech)での分析まで1%PFAにより固定した。
アポトーシスのイベントと関連したミトコンドリア膜内外電位差(ψm)の変化を、テトラメチルローダミンエチルエステル過塩素酸塩アッセイ(TMRE;Molecular Probes)を使用して測定した。マウスまたはヒト細胞(ウェル当たり5.0±0.1×105;2.0mL)を、24時間まで、様々な濃度のCOMPOUND-JPに曝露し、次いで、TMREと共にインキュベートした(1.0μM;30分間、37℃)。その後、細胞を、PBSにより2回洗浄し、次いで、DAPI(Sigma-Aldrich、1.0μg/ml)により染色し、MACSQuant Analyser(Miltenyi Biotech)により検査し、データを、FlowJoソフトウェアによって評価した。
細胞(ウェル当たり1.0±0.1×106;1.0mL)を、カスパーゼ3阻害剤Qvd-OH(20μM)ありまたはなしで、様々なCOMPOUND-JP用量により刺激した。次いで、細胞は、カスパーゼ3阻害剤コンジュゲート(RED-DEVD-FMK;Biovision)により染色し、インキュベートした(1.0時間、5%CO2で37℃)。細胞を、洗浄バッファーにより洗浄し、次いで、遠心分離し(3000rpm、5.0分間)、30分間、アネキシンV-FITC(10μL/ml;Miltenyi Biotech)により染色し、DAPI(1.0μg/ml)を、MacsQuant Analyserによる検査の前に追加した。
細胞(ウェル当たり1.0±0.1×106;1mL)を、様々なエフェクター(24時間、37℃)と共にインキュベートし、次いで、Cytofix/Cytopermバッファー(100μL、30分間、4℃)により透過処理し、次いで、PermWash(BD Biosciences)により洗浄し、ホスホ-Ser235/236-S6RP抗体により染色した(30分間、4℃)。さらなるPermWashの後に、細胞を、次いで、二次抗ウサギalexaF 488(20μg/ml;Life Technologies)により染色した(30分間、4℃)。次いで、細胞を、PermWash中で洗浄し、染色バッファー(PBS+0.1%ウシ血清アルブミン+1.0%パラホルムアルデヒド)中に再懸濁し、MacsQuant Analyser(Miltenyi Biotech)でフローサイトメトリによって分析した。
それぞれの実験群におけるマウスすべてに、レポータールシフェラーゼ遺伝子を安定して発現する99KOL-LUC Tリンパ腫細胞(200μL;5.0±0.1×106)を皮下注射した。注射の2日後に、マウスに、食塩水(NaCl 0.9%)に薬粉を溶解し、混合することによって調製したCOMPOUND-JPを腹腔内投与した(50mg/kg/日/1週間当たり5日間)。体重および腫瘍体積を、5日ごとに測定した。腫瘍体積は、数式に従って計算した:V=(L*W2)/2(L:長さ;W:幅)。生体発光イメージングを行うために、マウスに、D-ルシフェリン(Caliper Life Science)の腹腔内注射(150μL、30mg/mL)を受けさせ、吸入イソフルラン(2.0%)によって麻酔をかけた。生体発光シグナルは、高感度冷却CCDカメラを装備したPhoton imager(Biospace Lab)を使用してモニターし、画像はすべて、D-ルシフェリン注射の10分後に収集した。データは、腫瘍領域を覆う注目画像領域(ROI)に集中して、全光子束放射(total photon flux emission)(光子/秒(photons/second))を使用して分析した。
解剖した腫瘍のパラフィンブロック中の切片(4μm)を、一次ウサギホスホS6RP抗体(#4858、Cell Signaling Technology)により標識した。ペルオキシダーゼ(HRP)とコンジュゲートされた二次抗ウサギ抗体(SignalStain(登録商標)、#8114、Cell Signaling Technology)を、メーカーによって推奨されるように、ジアミノベンジジン(DAB)による顕色の前に、切片に追加した。壊死の評価のために、切片を、ヘマトキシリン/エオシン/サフラン(HES)により染色した。画像を分析し、壊死面積は、ImageJソフトウェア(NIH)を使用して定量化した。
インビトロ実験についての統計分析は、95%の信頼水準で、両側スチューデントのt試験を使用して実行した、またはANOVAを使用し、その後、Dunnettの多重比較検定を続けることによって計算した。組み合わせについては、実験群の間の有意性を決定するために、ANOVA、その後に続くTukey多重比較検定によって分析した。薬剤の間の相乗作用は、Chou-Talalay法を使用して分析した。インビボ実験については、統計分析は、両側Mann-Whitney順位和検定を使用して実行した。P値<0.05は、統計的に有意であるとして許容された(*:p<0.05;**:p<0.01;***:p<0.001)。分析は、GraphPAd Prism 3.0ソフトウェアを使用して実行した。
各細胞からRNAを抽出し、LAT1及びLAT2特異プライマーを用いて各mRNAを定量PCR法で測定した。
各がん細胞の培養液中にCOM-JPの非存在並びに異なる濃度のCOM-JPの存在下に5日間培養を継続した。各日の経過における細胞の増殖活性をMTT法で測定した。
ヒトスキルス胃がん由来44As3-11細胞とヒト膵がん由来Panc-1細胞の2種のがん細胞をCOM-JPの存在、非存在下に培養し、2日目にGEM及びPacを添加して2時間培養継続の後に、培地を交換し、COM-JPの存在、非存在下にさらに2日間培養を行った。最後に細胞を収集して、細胞の生存活性をMTT法により定量した。各実験は同一条件の群を夫々3例測定し、平均値と標準誤差並びに有意差検定のp値を図中に示した。
HT-29細胞をマウスの皮下に播種し、腫瘤の大きさが或る一定の大きさ以上に成長した状態から、COM-JP及び5-FUの単独並びに併用による腫瘍塊の大きさの推移を経日的に測定した。
ヒト大腸がん由来HT-29細胞に海ほたるルシフェリンを発現させて、マウスの大腸壁に移植したメタマウスを作出した。これにより腫瘤の大きさを発光強度で経日的に定量できる。図16には各処置グループでの腫瘤の大きさを棒グラフに示した。グループ1は対照群、2~4はCOM-JPを夫々3.1,12.5,50mg/Kg/日体重を4週間連日腹腔内単独投与、グループ5はCDDPを2.5mg/Kg/日、0,6,13日目の3回腹腔内単独投与、グループ6~8には同2~4と5の組み合わせでの併用投与、グループ9はCOM-JPを300mg/Kg/日を4週間連日経口投与を行った。
以上の試験の結果を以下に示す。
マウス(tPTEN-/-)は、高悪性度で侵襲性のリンパ腫を発症する。正常胸腺細胞およびtPTEN-/-胸腺腫瘍細胞の全体的な遺伝子発現は、全ゲノム(pan-genomic)AffymetrixマウスDNAチップを使用して確立した。データの標準化の後に、野生型胸腺細胞(n=3)およびtPTEN-/-腫瘍細胞(n=4)由来の遺伝子発現プロファイル(GEP)を評価した。野生型と比較して、tPTEN-/-腫瘍細胞は、498のアップレギュレートされた遺伝子を示した。図1Aにおいて示されるように、本発明者らは、LAT1をコードするSLC7A遺伝子が、正常細胞と比較して、tPTEN-/-においてアップレギュレートされたことを観察した(log2:2.44倍)。対照的に、CD98hcをコードするSLC3A2の発現は、腫瘍においてほんのわずかに増強された(0.874倍)。そのうえ、レファレンスとして取り扱うために、本発明者らはまた、これもまたtPTEN-/-細胞において高度に発現されたc-myc遺伝子(2.22倍)の結果をも示すが、ハウスキーピング遺伝子であるmarksは、2つのカテゴリーの間で有意に異なっていなかった。定量的PCR分析により、悪性細胞において、SLC3A2ではなく、LAT1およびc-mycの発現の増強が観察されるのを確認することができた。高度なLAT1発現は、2つの初代tPTEN-/-腫瘍(KOマウス#99、#631)において観察され、それぞれ、KO99LおよびKO631Lと示される細胞株を確立するために使用した。序論において先に論じられたように、LAT1は、運搬活性となるために、ジスルフィド結合によって連結されたCD98hcを必要とする。そのため、本発明者らはまた、抗CD98hc抗体を使用して、フローサイトメトリ分析を実行することによって、その表面発現をも分析した。正常(WT)胸腺細胞と比較して(図1B)、5つのtPTEN-/-腫瘍は、より高度なCD98発現を示した。細胞活性化:(PMA+イオノマイシン)は、腫瘍胸腺細胞における17倍と比較して、2.8倍の発現の増加をもたらした(図1D)。
本発明者らは、様々な濃度のBCH、D-ロイシン、およびCOMPOUND-JPの存在下において、細胞生存能力を調べるために、KO99Lと示される、確立されたtPTEN-/-腫瘍細胞株を使用した。BCHは、古典的なシステムL阻害剤であるが、LAT1選択性を欠く(非特許文献18)。図2Aにおいて示されるように、BCHおよびD-ロイシンは、高用量濃度で高度な細胞生存能力を維持し、細胞生存能力は、それぞれ、10.0および30.0mMで30%および60%であった;図2A)。対照的に、LAT1選択的阻害剤COMPOUND-JPは、はるかに大きな程度までKO99L細胞生存能力に影響を及ぼし、半分の細胞を死滅させるのに、より低い薬剤濃度を示し、IC50=2.4μMであった(図2A)。10.0μMで、COMPOUND-JPは、48時間後に75%、細胞周期(増殖)を減少させた(図2B)。濃度依存性の方式で、COMPOUND-JPは、新たに単離されたtPTEN-/-腫瘍細胞の細胞生存能力に影響を及ぼしたが、正常マウス胸腺細胞において無毒であった(補足資料図S1A)。
LAT1が、大きな中性アミノ酸の取込みを媒介することは、十分に確証されている(参考文献21);(参考文献22)。図2Cにおいて示されるように、COMPOUND-JP(2.5~10.0μM)とのKO99L細胞のインキュベートは、細胞生存能力の用量依存的な減少をもたらし、これは、必須アミノ酸(EAA)を培養培地に補足することによっていくぶんか逆転され得たのに対して、非必須アミノ酸(NEAA)は、救出効果を有していなかった。並行して、図2Dにおいて要約されるように、COMPOUND-JPにより誘発された細胞死は、NEAAではなく、EAAの追加に際して、2.5倍減少した。これらのデータは、COMPOUND-JPが、EAAの取込みを変化させることによって機能することをさらに実証する。
ルシフェラーゼ発現ベクターを安定してトランスフェクトしたKO99L細胞を、ヌードマウスに皮下注射し、その後(2日目)、動物を、COMPOUND-JP(50mg/kg/日=1.5mg/マウス/日)により治療した。腫瘍関連性の生体発光および腫瘍体積データを5日ごとに収集した。毎日のCOMPOUND-JP治療は、ビヒクル治療群と比較して、18日目に、平均腫瘍体積を45%(p<0.05)減少させた(図2E)。腫瘍発光(図2F)もまた、COMPOUND-JP治療によって統計的に有意な減少(59%;p<0.05)を示した(図2G)。腫瘍の免疫組織学分析は、JPH206治療が、腫瘍内で壊死性の応答をもたらしたことを示した(10.5+/-0.5%対4.6+/-0.5%;図4C)。
本発明者らは、COMPOUND-JPによるLAT1のターゲティングもまた、T-ALL/T-LL細胞株および初代腫瘍細胞に影響を与え得るかどうかを確証しようと試みた。ヒトJurkat T-ALL株は、COMPOUND-JPが、細胞生存(図2H)および増殖(図2I)を変化させ得ることを実証するために利用した。そのうえ、4人の異なる患者から単離された初代T-ALL細胞もまた、濃度依存性の様式で、COMPOUND-JP感受性(細胞生存能力)を示した(図2J)。大きく異なるが、COMPOUND-JPは、正常な静止または活性化ヒトPBL細胞において、細胞死を誘発しなかった(図S2B)。さらに、細胞増殖に強く影響を与えた、アポトーシス誘発剤であるスタウロスポリン(STS:1.0μM)とは対照的に、COMPOUND-JPは、PBL細胞増殖(1.0~50.0μM;図S2C)も、正常な臍帯血単核細胞(5.0および10.0μM;図S2D)も有意に変化させなかった。その知られているLAT1/LAT2阻害剤選択性と一致して、これらの実験結果は、COMPOUND-JPが、正常(健康、LAT2発現)T細胞と比較して、白血病性細胞(LAT1発現)に対して、優先的に作用することを実証する。
KO99L細胞に対するCOMPOUND-JPの曝露は、アポトーシスを誘発した。コントロール(COMPOUND-JPなし)と比較して、48時間後のアネキシン-V+およびアネキシン-V+/DAPI+細胞分析(図3A)は、それぞれ、10.0および20.0μMで、2.8および3.8倍高いレベルのアポトーシスを示した。COMPOUND-JPが、アポトーシスの細胞の量を著しく減少させた、カスパーゼ活性化を妨げる阻害剤であるQvD-OPH(20.0μM)と同時投与された場合に、これらの結果は、さらに確認されるものとなった。ミトコンドリア外膜透過性化(MOMP)は、ミトコンドリア膜内外電位差(△ψm)を歪める初期のアポトーシス細胞死イベントである(参考文献23)。テトラメチルローダミンエチルエステル過塩素酸塩染色アッセイ(TMRE)を使用すると、COMPOUND-JP(5.0および10.0μM)は、KO99L細胞においてMOMPを引き起こすことが示された(図3B1)。KO99L細胞において観察されたミトコンドリア脱分極は、COMPOUND-JP濃度依存性であることが分かり(2.5~20.0μM;48時間)、試験した最も高い濃度で70%に達した(図3B2)。本発明者らは、ウェスタンブロッティング法によって48時間の期間にわたってカスパーゼ3切断を検査することにより、カスパーゼ活性化をモニターした(図3C)。COMPOUND-JP追加(10.0μM)の4~6時間後、カスパーゼ3切断を観察することができた(図3C、レーン4および5)。48時間(レーン8)までに、本質的にすべての細胞カスパーゼ3が、プロセシングされた。そのうえ、本発明者らはまた、よく知られているカスパーゼ基質であるRed-DEVD-fmkによって、細胞関連性の蛍光を増加させる、COMPOUND-JPの能力について調べ、結果は、QvD-OPH(20.0μM;図3D1)とのプレインキュベーションに際してベースラインに戻った。図3D2において示されるように、COMPOUND-JPの結果は、用量および時間依存的であり、QvD-OPHとの同時投与によって打ち消すことができた(図3D1、2)。カスパーゼ3基質であるPARPもまた、COMPOUND-JP(10.0μM;24時間)の存在下において、完全に切断されることが観察され、細胞のHsp60レベルにおける観察可能な変動はなかった(図3E、レーン4)。COMPOUND-JPはまた、初代tPTEN-/-細胞に対しても有効であり、用量依存的な(2.5~20.0μM)様式で、細胞死を誘発した(図3F;データは、5つの初代tPTEN-/-腫瘍の平均である)。初代KO#732細胞(図3G)を使用して、アポトーシスを分子的に確認した。COMPOUND-JPは、カスパーゼ3プロセシングおよびPARP切断を誘発した(18.0時間;レーン10、11)。ラパマイシンは、効果を有していなかった(レーン5、9、13)が、PI103は、初期(2.0時間、レーン4)のおよび効率的な細胞死誘発因子(レーン8、12)のように思われた。栄養素の不足の間に起こり得る保護応答として、自食作用が、アポトーシスの前に起こり得る。図3Hにおいて示されるように、COMPOUND-JP(10.0μM)の曝露は、LC3-IIの蓄積を刺激したが、LC3-IIは、LC3の自食形態であり、自食作用の第1のステップと関連する(参考文献24)。LC3-IIの蓄積は、6.0時間でピークに達し(図3H、レーン3)、その後、減少した(レーン4~6)。図3Iにおいて示されるように、QvD-OPH(20.0μM)の存在は、アポトーシスに対抗することができ、18.0時間までの間、LC3-IIを維持することができた(レーン7と比較したレーン5)。オートファゴソーム形成関連性のAtg5レベルは、次第に増加して、6.0時間で最大になり(レーン6)、18.0時間でベースラインに戻った(レーン7)。さらに、pJNKストレス経路活性化は、アポトーシスがブロックされた場合に検出された(レーン7と比較した5)。全体として、これらの実験結果は、COMPOUND-JPが、最初に、自食応答を引き起こし、アポトーシスが速やかにそれに続いたという考えを支持する。
KO99L細胞におけるpS6RP低下によって確立されるように(図4A)、本発明者らは、COMPOUND-JP(10.0μM)が、早くも2.0時間でmTORC1活性化に影響を与えることができ(レーン5)、6.0時間(レーン6)および24時間(レーン7)で、維持されたことを観察した。COMPOUND-JPのmTOR阻害効果はまた、2つの初代T-ALLサンプルに対するホスホフローサイトメトリによっても観察された(図4B)。さらに、pS6RP染色は、COMPOUND-JP治療マウス由来の腫瘍において劇的に減少し、化合物のインビボにおける阻害効果を実証した(図4C)。KO99L細胞において、COMPOUND-JPはまた、セリン473リン酸化によって測定されるように、Akt活性化にも干渉した(図4A)。対照的に、COMPOUND-JPは、4E-BP1リン酸化に対して影響を示さず、COMPOUND-JPが、mTORC1活性化を部分的にのみ乱すことを実証する。Akt阻害剤であるMK-2206(5.0μM)およびAkt/mTOR2重阻害剤であるKU0063794(5.0μM)は、早くも2.0時間で、S6RP、4EBP1、およびAktリン酸化を完全にブロックすることが示された(図S2)。ラパマイシン(10.0nM)は、COMPOUND-JP(10μM)で観察された作用に類似していたが、ラパマイシンは、mTORC2ではなくmTORC1阻害剤であり(参考文献25)、これは、4EBP1ではなく、S6RPリン酸化を完全に阻害した(図S2)。ラパマイシンはまた、COMPOUND-JP(図S2)と比較して、より後の時点で、Aktリン酸化/活性化に影響を与えた。LAT1が、細胞内グルタミンとの細胞外EAA交換体であることが確立されているので(参考文献26)、本発明者らは、様々なグルタミンレベルが、どのようにEAA流入およびmTORC1活性化に干渉し得るのかについて調べようと試みた。このために、本発明者らは、細胞外アスパラギンを分解することができ、かつグルタミナーゼ活性を持つ2つの酵素、L-アスパラギナーゼ(L-Asp)およびErwinase(Erw)を使用した。図S2において示されるように、L-AspおよびErw(2.5ユニット/ml)の両方が、S6RPリン酸化を変化させたが、Erwがより高いグルタミナーゼ活性を有するので、それはL-Aspよりも強力であった。コントロール(図S2;レーン15~17)と比較して、Erw(レーン18~20)およびL-Asp(レーン21~23)はまた、4EBP1リン酸化をも変化させたのに対して、それらは、Aktリン酸化に有意に影響を及ぼさなかった。プロテアソーム活性の阻害は、アミノ酸ホメオスタシスを妨害することが報告されている(参考文献27)。本発明者らのモデルにおいて、プロテアソーム阻害剤であるベルケイド(Vel;図S2)は、初期の時点(2.0および6.0時間;それぞれレーン24および25)ではなく、24時間(レーン26)で、S6RP、4EBP1、およびAktリン酸化を完全に消失させた。これらのすべての実験において、S6RP、4EBP1、Akt、およびHsp 90の全体のレベルは、様々な薬剤によって有意に変化せず、リン酸化に対して観察された影響が、タンパク質の量における差異によるものではなかったことを実証する。
c-mycタンパク質は、ヒトリンパ腫およびtPTEN-/-腫瘍においてアップレギュレートされることが示された(参考文献28)。tPTEN-/-細胞において、c-mycは、SDS-PAGE(図4D;レーン1~4)において60kDaのあたりで三つ組として移動するように観察された。COMPOUND-JP(10.0μM)との細胞のインキュベーションに際して、全c-mycは、2.0時間で既に減少し(レーン5)、24時間まで維持され(レーン7)、特に、c-mycの2つの下位の移動形態は、有意に減少した。ベルケイドとインキュベートした場合に、類似した観察がなされた(8nM;24時間;レーン10)。KU0063794(10.0μM)によるAkt/mTOR阻害もまた、2つの下位のバンドにわずかに影響を及ぼした(レーン15~17)。MK-2206(10.0μM)媒介性のAkt阻害は、24時間以内に3つのc-mycアイソフォームすべての劇的な消失をもたらした(レーン18~20)。COMPOUND-JPと著しく異なって、ラパマイシン(100 nM)は、c-mycレベルに影響を与えず(レーン12~14)、結果としてアポトーシス誘発を欠いた。これらの様々な実験を通じて、Hsp90レベル変動は、明確に観察することができず、それによって、様々な観察された、薬剤により引き起こされた結果が、全体的な細胞タンパク質含有量における有意な変化の結果ではなかったということを確証するのを支援する。さらに、本発明者らは、全体的なタンパク質合成に対するCOMPOUND-JPのいかなる干渉をも観察しなかった。まとめると、実験データは、COMPOUND-JPによってもたらされるc-mycダウンレギュレーションが、その抗白血病性の効果の重要な特質であるかもしれないという考えと一致している。
タンパク質および/またはアミノ酸ホメオスタシス異常は、自食作用および細胞死に至り得る小胞体/総合的ストレス応答(ISR)/変性タンパク質応答(UPR)を引き起こすことが知られている(参考文献29)。「C/EBP相同タンパク質」(CHOP)として知られている転写因子の発現は、ERストレスの間に誘発性になり、ER媒介性のアポトーシスに参加する(参考文献30)。無秩序なCHOP活性は、細胞生存能力を損ない、CHOPを欠く細胞は、ERストレスの致命的な転帰から有意に保護される(参考文献31)。KO99L細胞においてイムノブロッティングによって評価すると、COMPOUND-JP(10.0μM)は、2.0時間(図5A、レーン5)までにCHOP発現を速やかに誘発し、これは、6.0時間(レーン6)で最大となり、24時間(レーン7)で安定したままであった。COMPOUND-JPと比較して量が少なかったが、L-Asp(2.5ユニット/ml)によるCHOPの誘発は、6.0時間(レーン9)および24時間(レーン10)で明らかであった。Erw(2.5ユニット/ml)もまた、一過性の応答を誘発し、これは、6.0時間(レーン12)で最大となり、24時間(レーン13)までに基本レベルに戻った。図5Bにおいて実証されるように、CHOPは、著明な、COMPOUND-JP用量依存的な誘発を示し(5.0および10.0μM;それぞれレーン2および3)、CHOP誘発はまた、ベルケイドによっても(8.0および15.0μM;それぞれレーン7および8)、また、程度はより小さいが、L-Asp(2.5ユニット/ml)およびラパマイシン(10.0nM)、それぞれレーン4および9によっても強く誘発された。対照的に、AktおよびAkt/mTOR阻害剤であるMK-2206(10.0μM;レーン5)およびKU0063794(10.0μM;レーン6)は、CHOP誘発をもたらさず、これは、この経路が、PI3K/Akt/mTOR経路によって引き起こされないことを示唆する。
本発明者らは、次に、tPTEN-/-細胞が、培地アミノ酸組成の修飾に対してどのように応答するかについて調査した。完全培地をHBSSにより10倍に希釈すると(図5C;6.0および24時間、レーン4および5)、明らかな時間依存的なCHOP誘発が、観察された。NEAAを栄養欠損培地に補足しても(図5C;レーン6および7)、CHOP誘発を修飾しなかったが、EAA(レーン8および9)またはNEAA+EAA(レーン10および11)の組み合わせの追加は、CHOP出現を妨げた。S6RPリン酸化がCHOP発現と逆相関したことは、興味深く、培地におけるEAAの欠乏は、mTORC1ダウンレギュレーションに至る。そのうえ、本発明者らは、COMPOUND-JP媒介性のCHOPの誘発(図5D)が、細胞外アミノ酸含有量によって調整されることを観察した。実際に、EAAの補足は、CHOP誘発を明確に妨げた(レーン8および9)。CHOPタンパク質が、6.0時間(レーン6)と比較して、24時間(レーン7)でほとんどを検出することができなかったので、NEAAは、応答を短くし、これに反して、EAA+NEAAの組み合わせは、中間の応答を示した(レーン10および11)。この実験結果は、COMPOUND-JPによって引き起こされるストレス応答が、細胞外EAA濃度を増加させることによって相殺され得ることを示し、COMPOUND-JPの作用が、EAA取込みの阻害によるものであることをさらに実証する。
COMPOUND-JP(LAT1阻害)によって観察されるISR/UPRが、続くアポトーシスの原因となったかどうかについて検査するために、本発明者らは、eIF2α脱リン酸化を妨げ、ERストレスに対して細胞保護を誘発した阻害剤として確立されたsalubrinal化合物を使用した(参考文献32)。図5Eにおいて示されるように、salubrinal(50.0μM)は、COMPOUND-JP(10.0μM)によるCHOPの誘発を相殺することができた(レーン6および7と比較した8および9)。そのうえ、これらの結果には、カスパーゼ3切断の減少が伴った(中央のパネル)が、Hsp60レベルは、変わらなかった(下位のパネル)。さらに、サルブリナル(salubrinal)(50.0μM)は、COMPOUND-JPによって誘発された細胞死からKO99L細胞を救出し(図5F)、かつ、BrdU取込み(図5G)または細胞計数(図5H)によって評価される細胞増殖を回復させた。これらの結果は、COMPOUND-JPが、細胞死応答の間に、CHOP誘発を引き起こすことができることを実証する。
変性タンパク質応答(UPR)は、i)イノシトール要求性酵素1アルファ(IRE1α);ii)PKR様ERキナーゼ;およびiii)活性化転写因子(ATF6)((参考文献33)において概説される)を含む、複数のERタンパク質の調和的な活性化によって特徴付けられる。活性化されたIRE1αは、chopの発現を誘発する転写因子XBP-1のアイソフォームをコードするmRNAの非通常性のスプライシングを開始する((参考文献30)。活性化されたPKR様ERキナーゼは、eIF2α(セリン-51A)をリン酸化し、ATF4の翻訳の誘発をもたらす(参考文献34)。ATF6は、ERストレスの間に切断され、そのサイトゾルドメイン[ATF6(N)]は、核に移動し、転写を調節するであろう(参考文献35)。XBP-1、ATF4、およびATF6は、ERストレスの間にchopを誘発することが知られている3つの転写因子である(参考文献30)。そのため、本発明者らは、COMPOUND-JP(10.0μM)との細胞のインキュベーションの後のATF4、ATF6、およびXBP-1のステータスについて検査した。本発明者らは、ATF4(6.0時間)およびATF6(18.0時間)が誘発され、一方、完全長XBP1が、徐々に消失し(図6A;レーン3および4)、CHOPおよびGADD34が、同時に発現した(図6B;レーン3および4)のを観察した。eIF2αの脱リン酸化は、GADD34ホスファターゼの平行した発現と共に24時間で観察された(図6C)。p38ストレス経路は、2.0時間で強く活性化された(図6C;レーン2)。
COMPOUND-JPによるアポトーシス誘発を分子的に定義するために、本発明者らは、Bcl2ファミリーメンバーを分析し、本発明者らは、ミトコンドリアおよびER膜レベルでアポトーシスの開始をコントロールすることが知られているメンバーを測定しようと試みた。6.0時間から開始すると(図6D)、COMPOUND-JP(10.0μM)は、4つのアポトーシス促進性メンバー:Bak(図6D;レーン2~4)、Bax(レーン4)、PUMA(レーン2~4)、ならびにBim(レーン3および4)のタンパク質レベルを増加させた。Bax、Bak、PUMA、およびBimの誘発は、salubrinalによるCHOPの阻害に対して感受性であった(図6E、レーン3および4)が、全Hsp90タンパク質レベルは、影響を与えられなかった。
COMPOUND-JPの濃度を増加させながらKO99L細胞を定期的に培養することによって、本発明者らは、COMPOUND-JPの抗増殖およびアポトーシス効果に抵抗する、細胞の変異体を得ることができた。およそ4か月後に、本発明者らは、COMPOUND-JP(20.0μM)の存在下においてなお増殖することができるKO99-RJとして示される細胞株を得た。図7Aにおいて要約されるように、細胞生存能力における減少は、COMPOUND-JP用量依存的であり、変異細胞株(つまりKO99-RJ)と比較して、親の株(つまりKO99-S)においてはるかに有力であり、たとえば、40μM COMPOUND-JPで、細胞生存能力は、KO99-Sについて本質的に95~100%であったのに対して、KO99-RJにおいておよそ40%減少した。図7Bにおいて示されるように、COMPOUND-JPは、KO990-Sにおいて濃度依存性の細胞死を示したが、KO99-RJにおいてはるかに効率的でなかった。さらに、CHOP誘発の非存在によって確立されたように(図7C)、COMPOUND-JPが、KO99-RJ細胞において、総合的ストレス応答を引き起こさなかったことを観察することは興味深かった。これらのデータは、COMPOUND-JPによって誘発されるアポトーシス応答を引き起こすこの経路の重要な役割をさらに強調するものである。最後に、S6RPリン酸化における減少を観察することができなかったので(図7C)、COMPOUND-JPはまた、KO99-RJにおいて、mTORをも阻害できず、KO99-RJは、親の細胞と同等のレベルでCD98を発現する。
図8において示されるように、COMPOUND-JPを、様々な化学療法剤またはシグナル伝達阻害剤と組み合わせて試験した(インビトロにおいて)。本発明者らは、白血病治療の導入期の間に臨床的に使用される2つの化学療法剤であるデキサメタゾンおよびドキソルビシンについて、試験した(参考文献36)。これらの実験において、化合物はすべて、最適以下の濃度で使用し、これらの結果は、補足の資料の図S3-1、2において示す。算出された組み合わせインデックス(CI)値は、図8Aにおいて示す。COMPOUND-JPは、ラパマイシンと強く相乗作用を示すように思われ、全体的な代謝活性/細胞生存を減少させる。相乗効果または相加効果は、すべての化合物で観察され、特に、COMPOUND-JPは、デキサメタゾンおよびドキソルビシンと相乗作用を示した。LAT1阻害剤は、ベルケイドおよびL-アスパラギナーゼの効果を強化することができるが、PI103およびKU-0063794とはそれほど効率的でないように思われた。WST1から導き出したアイソボログラムは、試験した様々な組み合わせについての可能性をさらに示す(図8B)。全体として、データは、T-ALL細胞生存に干渉する他の分子と組み合わせた場合、COMPOUND-JPがポジティブな効果をもたらすことができることを示す。
図10において、単位RNA当りのmRNA量を縦軸に、使用したがん細胞名を横軸に、がん種別名を表中に記した。5つの例外を除けば46種類の細胞全部にLAT1 mRNAは発現しており、がんタイプのトランスポーターと言うことができる。他方、LAT2はがん細胞での発現はゼロか極端に低値であり、正常タイプである、と理解できる。
図11において、各値は同一条件での4例の平均値をCOM-JPの非存在での活性を100%とした相対値として表示した。平均値と標準誤差並びに有意差検定のp値を図中に示した。がん細胞の種類による差異は認められるものの、両細胞共にCOM-JPの用量依存的な増殖活性の低下が明らかに認められた。
ヒトスキルス胃がん由来44As3-11細胞の増殖に及ぼすGemstabine並びにPaclitaxelを単独で2時間作用させた効果は各群カラムの左の棒グラフの値の差に相当する。各実験は同一条件の群を夫々3例測定し、平均値と標準誤差並びに有意差検定のp値を図中に示した。両者共に用量依存性に細胞の増殖活性を低下させた。GemstabineよりはPaclitaxelの抑制効果が強力であった。COM-JP(15μM)を前2日間処置した結果(各群の真ん中の棒グラフ)よりも全期間存在させた方(各群の右側の棒グラフ)の併用効果がより強力であった。併用による抑制効果は各単独の作用を加算した結果であり、所謂相加効果を示した。
ヒト膵がん由来Panc-1細胞の増殖に及ぼすGemstabine並びにPaclitaxelを単独で2時間作用させた効果は各群カラムの左の棒グラフの値の差に相当する。各実験は同一条件の群を夫々3例測定し、平均値と標準誤差並びに有意差検定のp値を図中に示した。両者共に用量依存性に細胞の増殖活性を低下させた。GemstabineよりはPaclitaxelの抑制効果が強力であった。しかし、Gemstabineの単独効果は極めて小さく、スキルス胃がん由来44As3-11細胞の増殖への作用よりも明らかに弱い作用であった。COM-JP(15μM)を前2日間処置した結果(各群の真ん中の棒グラフ)よりも全期間存在させた方(各群の右側の棒グラフ)の併用効果は僅かに抑制が見られたものの、その程度はスキルス胃がん由来44As3-11細胞の増殖に比して明らかに低い抑制であった。Panc-1細胞における各併用による抑制効果は各単独の作用を加算した結果であり、44As3-11細胞の場合と同様に所謂相加効果であった。
両薬物の単独投与では無処置の対照群に比して腫瘍の増大は夫々有意な減少を示した。両者の併用により、各単独群の残存腫瘤増大は更に減少した。この併用効果は相加的であり、両薬物の作用機序が異なることに起因することが明らかにされた。
図16のグループ8と9では対照群に比して腫瘤の増大は有意に低下した。この結果より、COM-JPとCDDPの単独投与では認められなかった各薬効は両者の併用で明らかな薬効を認められた。この併用による薬効は両者の相乗効果と結論された。
上記の試験結果により、以下が明らかとなった。
(参考文献21)H. Uchino et al., Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): insights into the mechanisms of substrate recognition. Molecular pharmacology 61, 729 (Apr, 2002).
(参考文献22)E. Morimoto et al., Establishment and characterization of mammalian cell lines stably expressing human L-type amino acid transporters. Journal of pharmacological sciences 108, 505 (Dec, 2008).
(参考文献23)L. Galluzzi, N. Larochette, N. Zamzami, G. Kroemer, Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene 25, 4812 (Aug 7, 2006).
(参考文献24)A. Puissant, G. Robert, P. Auberger, Targeting autophagy to fight hematopoietic malignancies. Cell Cycle 9, 3470 (Sep 1, 2010).
(参考文献25)M. R. Janes et al., Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 16, 205 (Feb, 2010).
(参考文献26)P. Nicklin et al., Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521 (Feb 6, 2009).
(参考文献27)A. Suraweera, C. Munch, A. Hanssum, A. Bertolotti, Failure of amino acid homeostasis causes cell death following proteasome inhibition. Molecular cell 48, 242 (Oct 26, 2012).
(参考文献28)L. V. Sinclair et al., Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nature immunology 14, 500 (May, 2013).
(参考文献29)I. Tabas, D. Ron, Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13, 184 (Mar, 2011).
(参考文献30)S. Oyadomari, M. Mori, Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation 11, 381 (Apr, 2004).
(参考文献31)H. Zinszner et al., CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & development 12, 982 (Apr 1, 1998).
(参考文献32)M. Boyce et al., A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935 (Feb 11, 2005).
(参考文献33)P. Walter, D. Ron, The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081 (Nov 25, 2011).
(参考文献34)H. P. Harding et al., An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell 11, 619 (Mar, 2003).
(参考文献35)J. Ye et al., ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Molecular cell 6, 1355 (Dec, 2000).
(参考文献36)C. H. Pui, W. E. Evans, Treatment of acute lymphoblastic leukemia. N Engl J Med 354, 166 (Jan 12, 2006).
(参考文献37)N. Yanagisawa et al., High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J Clin Pathol 65, 1019 (Nov, 2012).
(参考文献38)H. Nawashiro et al., L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int J Cancer 119, 484 (2006).
(参考文献39)M. Furuya, J. Horiguchi, H. Nakajima, Y. Kanai, T. Oyama, Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci 103, 382 (Feb, 2012).
(参考文献40)M. Ichinoe et al., High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: comparison with non-cancerous lesions. Pathol Int 61, 281 (May, 2011).
(参考文献41)K. Kaira et al., l-type amino acid transporter 1 and CD98 expression in primary and metastatic sites of human neoplasms. Cancer Sci 99, 2380 (Dec, 2008).
(参考文献42)K. Yamauchi et al., System L amino acid transporter inhibitor enhances anti-tumor activity of cisplatin in a head and neck squamous cell carcinoma cell line. Cancer Lett 276, 95 (Apr 8, 2009).
(参考文献43)K. Oda et al., L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci 101, (2010).
(参考文献44)Y. P. Vandewynckel et al., The paradox of the unfolded protein response in cancer. Anticancer Res 33, 4683 (Nov, 2013).
(参考文献45)C. Reimertz, D. Kogel, A. Rami, T. Chittenden, J. H. Prehn, Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. The Journal of cell biology 162, 587 (Aug 18, 2003).
(参考文献46)S. C. Cazanave et al., CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. American journal of physiology. Gastrointestinal and liver physiology 299, G236 (Jul, 2010).
(参考文献47)H. Puthalakath et al., ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337 (Jun 29, 2007).
(参考文献48)C. Hetz et al., Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312, 572 (Apr 28, 2006).
(参考文献49)K. Hayashi, P. Jutabha, H. Endou, H. Sagara, N. Anzai, LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol 191, 4080 (Oct 15, 2013).
(参考文献50)N. Bulus, C. Feral, A. Pozzi, R. Zent, CD98 increases renal epithelial cell proliferation by activating MAPKs. PLoS One 7, e40026 (2012).
(参考文献51)E. Boulter et al., CD98hc (SLC3A2) regulation of skin homeostasis wanes with age. J Exp Med 210, 173 (Jan 14, 2013).
(参考文献52)C. Rosilio et al., The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett 336, 114 (Aug 9, 2013).
(参考文献53)T. Hagenbeek et al., The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. J Exp Med 200, 883 (2004).
(参考文献54)N. Lounnas et al., Pharmacological inhibition of carbonic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas. Cancer Lett 333, 76 (Jun 1, 2013).
Claims (8)
- LAT1阻害剤と、アルキル化薬、白金製剤、代謝拮抗剤、トポイソメラーゼ阻害薬、微小管重合阻害薬、内分泌療法剤、微小管脱重合阻害薬、抗腫瘍性抗生物質及び分子標的薬からなる群より選択される一以上の剤と、を有効成分として含む抗悪性腫瘍剤組成物。
- 前記抗悪性腫瘍剤組成物が、T細胞急性リンパ性白血病/リンパ腫治療用医薬組成物、胃がん治療用医薬組成物、膵がん治療用医薬組成物又は大腸がん治療用医薬組成物である、請求項1に記載の組成物。
- 前記LAT1阻害剤が、O-(5-アミノ-2-フェニルベンズオキサゾール-7-イル)メチル-3,5-ジクロロ-L-チロシン、その類似体又はそれらの薬理学的に許容しうる塩である、請求項1又は2に記載の組成物。
- 前記剤が、シスプラチン、5-フルオロウラシル(5-FU)、ゲムシタビン、L-アスパラギナーゼ(ロイナーゼ)、パクリタキセル、デキサメサゾン、ドキソルビシンをはじめとするアントラサイクリン系抗生物質、ベルケイド(ボルテゾミブ)、ラパマイシン、KU0063794及びPI-103から選択される一以上の剤である、請求項1~3のいずれかに記載の組成物。
- LAT1阻害剤と、mTOR阻害剤、PI3K阻害剤及びAkt阻害剤から選択される一以上の剤と、を有効成分として含む抗悪性腫瘍剤組成物。
- 前記抗悪性腫瘍剤組成物が、T細胞急性リンパ性白血病/リンパ腫治療用医薬組成物、胃がん治療用医薬組成物、膵がん治療用医薬組成物又は大腸がん治療用医薬組成物である、請求項5に記載の組成物。
- 前記LAT1阻害剤が、O-(5-アミノ-2-フェニルベンズオキサゾール-7-イル)メチル-3,5-ジクロロ-L-チロシン、その類似体又はそれらの薬理学的に許容しうる塩である、請求項5又は6に記載の組成物。
- 前記剤が、ラパマイシン、KU0063794及びPI-103から選択される一以上の剤である、請求項5~7のいずれかに記載の組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14809268.7A EP2959918B1 (en) | 2014-05-15 | 2014-06-07 | Anti-malignant tumor agent composition |
| KR1020147034643A KR20170001962A (ko) | 2014-05-15 | 2014-06-07 | 항 악성 종양제 조성물 |
| CN201480001519.XA CN105392498A (zh) | 2014-05-15 | 2014-06-07 | 抗恶性肿瘤剂组合物 |
| US14/409,371 US10172835B2 (en) | 2014-05-15 | 2014-06-07 | Anticancer agent composition |
| JP2014557910A JP6402393B2 (ja) | 2014-05-15 | 2014-06-07 | 抗悪性腫瘍剤組成物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1454362 | 2014-05-15 | ||
| FR1454362 | 2014-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015173970A1 true WO2015173970A1 (ja) | 2015-11-19 |
Family
ID=54479535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/065163 Ceased WO2015173970A1 (ja) | 2014-05-15 | 2014-06-07 | 抗悪性腫瘍剤組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10172835B2 (ja) |
| EP (1) | EP2959918B1 (ja) |
| JP (1) | JP6402393B2 (ja) |
| KR (1) | KR20170001962A (ja) |
| CN (1) | CN105392498A (ja) |
| WO (1) | WO2015173970A1 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017155023A (ja) * | 2016-03-04 | 2017-09-07 | ジェイファーマ株式会社 | Lat1阻害活性を有する芳香族アミノ酸誘導体を含有する注射剤 |
| WO2019130637A1 (ja) * | 2017-12-28 | 2019-07-04 | ジェイファーマ株式会社 | がん治療薬 |
| WO2020004043A1 (ja) | 2018-06-28 | 2020-01-02 | 株式会社トクヤマ | α-アジドアニリン誘導体又はα,α'-ジアジド誘導体の製造方法 |
| KR20200009001A (ko) | 2017-05-17 | 2020-01-29 | 가부시끼가이샤 도꾸야마 | 디아미노벤젠 화합물의 제조 방법 |
| WO2020096041A1 (ja) * | 2018-11-09 | 2020-05-14 | ジェイファーマ株式会社 | (S)-2-アミノ-3-{4-[(5-アミノ-2-フェニルベンゾ[d]オキサゾール-7-イル)メトキシ]-3,5-ジクロロフェニル}プロパン酸・1塩酸塩、その1水和物、およびその結晶とその製造方法 |
| CN111253271A (zh) * | 2020-02-11 | 2020-06-09 | 浙江聚贤医药科技有限公司 | 一种制备2-氨基-3-硝基苯甲酸甲酯的方法 |
| JPWO2019093383A1 (ja) * | 2017-11-07 | 2020-09-24 | ジェイファーマ株式会社 | 抗pd−1抗体若しくは抗pd−l1抗体療法の奏効性を予測する方法、がんの悪性度を評価する方法、及び抗pd−1抗体若しくは抗pd−l1抗体療法の奏効性を上昇させる方法 |
| WO2022102687A1 (ja) * | 2020-11-11 | 2022-05-19 | ジェイファーマ株式会社 | がん治療剤 |
| US20220288035A1 (en) * | 2019-08-30 | 2022-09-15 | J-Pharma Co., Ltd. | A pharmaceutical composition for treating cancer used for a patient having specific genetic marker |
| WO2023286823A1 (ja) * | 2021-07-15 | 2023-01-19 | 国立大学法人大阪大学 | 医薬組成物 |
| CN115772130A (zh) * | 2021-09-08 | 2023-03-10 | 四川大学华西医院 | 一种微管解聚剂及其制备方法和用途 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4067368A1 (en) | 2016-06-01 | 2022-10-05 | Athira Pharma, Inc. | Compounds |
| CN112972391B (zh) * | 2021-03-10 | 2022-06-21 | 温州医科大学附属第二医院(温州医科大学附属育英儿童医院) | 一种胆红素-jph203纳米颗粒及其制备和应用 |
| CN114652718B (zh) * | 2022-03-08 | 2024-03-12 | 浙江大学 | Lat1抑制剂和奥沙利铂在制备肾细胞癌治疗药物中的应用 |
| US20250262190A1 (en) * | 2022-04-25 | 2025-08-21 | Georgetown University | Use of lat1 inhibitors to treat obesity |
| WO2024105699A1 (en) * | 2022-11-18 | 2024-05-23 | Council Of Scientific And Industrial Research | Composition for treating paclitaxel-resistant breast cancer and method for preparation thereof |
| CN115814090B (zh) * | 2022-11-22 | 2024-05-24 | 广州医科大学附属第一医院 | 烟草引发炎症的药物靶点 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008081537A1 (ja) * | 2006-12-28 | 2008-07-10 | Human Cell Systems, Inc. | Lat1阻害活性を有する芳香族アミノ酸誘導体、それを含有するlat1阻害活性剤及びその製造方法 |
| WO2013183786A1 (ja) * | 2012-06-08 | 2013-12-12 | 学校法人近畿大学 | トランスポーターに対する抗体およびその用途 |
-
2014
- 2014-06-07 JP JP2014557910A patent/JP6402393B2/ja not_active Expired - Fee Related
- 2014-06-07 WO PCT/JP2014/065163 patent/WO2015173970A1/ja not_active Ceased
- 2014-06-07 US US14/409,371 patent/US10172835B2/en not_active Expired - Fee Related
- 2014-06-07 CN CN201480001519.XA patent/CN105392498A/zh active Pending
- 2014-06-07 EP EP14809268.7A patent/EP2959918B1/en not_active Not-in-force
- 2014-06-07 KR KR1020147034643A patent/KR20170001962A/ko not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008081537A1 (ja) * | 2006-12-28 | 2008-07-10 | Human Cell Systems, Inc. | Lat1阻害活性を有する芳香族アミノ酸誘導体、それを含有するlat1阻害活性剤及びその製造方法 |
| WO2013183786A1 (ja) * | 2012-06-08 | 2013-12-12 | 学校法人近畿大学 | トランスポーターに対する抗体およびその用途 |
Non-Patent Citations (75)
| Title |
|---|
| 1. SANSAL; W. SELLERS: "The biology and clinical relevance of the PTEN tumor suppressor pathway", J CLIN ONCOL, vol. 22, 2004, pages 2954, XP003011859, DOI: doi:10.1200/JCO.2004.02.141 |
| A. LEVINE; A. PUZIO-KUTER: "The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes", SCIENCE, vol. 330, 2010, pages 1340 |
| A. PUISSANT; G. ROBERT; P. AUBERGER: "Targeting autophag y to fight hematopoietic malignancies", CELL CYCLE, vol. 9, 1 September 2010 (2010-09-01), pages 3470 |
| A. SURAWEERA; C. MUNCH; A. HANSSUM; A. BERTOLOTTI: "Fail ure of amino acid homeostasis causes cell death following proteasome inhibition", MOLECULAR CELL, vol. 48, 26 October 2012 (2012-10-26), pages 242 |
| AYRAL-KALOUSTIAN S. ET AL.: "Hybrid inhibitors of phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR): design, synthesis, and superior antitumor activity of novel wortmannin-rapamycin conjugates.", JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 1, 2010, pages 452 - 459, ISSN: 0022-2623 * |
| B. D. CHESON: "Novel therapies for peripheral T-cell non-Hodgkin's lymphomas", CURR OPIN HEMATOL, vol. 16, July 2009 (2009-07-01), pages 299, XP008172491, DOI: doi:10.1097/MOH.0b013e32832ad69a |
| BRESSANIN D. ET AL., ONCOTARGET, vol. 3, no. 8, 2012, pages 811 - 823, ISSN: 1949-2553 * |
| C. C. FERAL ET AL.: "CD98hc (SLC3A2) mediates integrin signaling", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 102, 11 January 2005 (2005-01-11), pages 355, XP002667312, DOI: doi:10.1073/PNAS.0404852102 |
| C. H. PUI; W. E. EVANS: "Treatment of acute lymphoblasti c leukemia", N ENGL J MED, vol. 354, 12 January 2006 (2006-01-12), pages 166 |
| C. HETZ ET AL.: "Proapoptotic BAX and BAK modulate the un folded protein response by a direct interaction with IRElalpha", SCIE NCE, vol. 312, 28 April 2006 (2006-04-28), pages 572 |
| C. PUI; M. RELLING; D. PHARM; J. DOWNING: "Acute lymphoblastic leukemia", N ENGL J MED, vol. 350, 2004, pages 1535 |
| C. REIMERTZ; D. KOGEL; A. RAMI; T. CHITTENDEN; J. H. PRE HN: "Gene expression during ER stress-induced apoptosis in neurons: i nduction of the BH3-only protein Bbc3/PUMA and activation of the mito chondrial apoptosis pathway", THE JOURNAL OF CELL BIOLOGY, vol. 162, 18 August 2003 (2003-08-18), pages 587 |
| C. ROSILIO ET AL.: "The metabolic perturbators metformi n, phenformin and AICAR interfere with the growth and survival of mur ine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cell s", CANCER LETT, vol. 336, 9 August 2013 (2013-08-09), pages 114 |
| CHAIROUNGDUA A. ET AL.: "Amino acid transporters in malignant tumors as a candidate target for anti-cancer therapy.", JOURNAL OF JAPANESE BIOCHEMICAL SOCIETY, vol. 75, no. 8, 2003, pages 744, ISSN: 0037-1017 * |
| D. HANAHAN; R. WEINBERG: "Hallmarks of cancer : the next generation", CELL, vol. 144, 2011, pages 646 |
| E. BOULTER ET AL.: "CD98hc (SLC3A2) regulation of skin h omeostasis wanes with age", J EXP MED, vol. 210, 14 January 2013 (2013-01-14), pages 173 |
| E. MORIMOTO ET AL.: "Establishment and characterization of mammalian cell lines stably expressing human L-type amino acid tra nsporters", JOURNAL OF PHARMACOLOGICAL SCIENCES, vol. 108, December 2008 (2008-12-01), pages 505 |
| FUKUMOTO S. ET AL.: "A new treatment for human malignant melanoma targeting L-type amino acid transporter 1 (LAT1): a pilot study in a canine model.", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 439, no. 1, 2013, pages 103 - 108, XP028715152, ISSN: 0006-291X * |
| FUKUMOTO S. ET AL.: "L-type amino acid transporter 1 (LAT1): a new therapeutic target for canine mammary gland tumour.", VETERINARY JOURNAL, vol. 198, no. 1, 2013, pages 164 - 169, XP055237372, ISSN: 1090-0233 * |
| H. KOBAYASHI; Y. ISHII; T. TAKAYAMA: "Expression of L-type amino acid transporter 1 (LAT1) in esophageal carcinoma", JOURNAL OF SURGICAL ONCOLOGY, vol. 90, 15 June 2005 (2005-06-15), pages 233 |
| H. NAWASHIRO ET AL.: "L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors", TNT J CANCER, vol. 119, 2006, pages 484 |
| H. OHKAME; H. MASUDA; Y. ISHII; Y. KANAI: "Expression of L-type amino acid transporter 1 (LAT1) and 4F2 heavy chain (4F2hc) in liver tumor lesions of rat models", JOURNAL OF SURGICAL ONCOLOGY, vol. 78, December 2001 (2001-12-01), pages 265 |
| H. P. HARDING ET AL.: "An integrated stress response reg ulates amino acid metabolism and resistance to oxidative stress", MOL ECULAR CELL, vol. 11, March 2003 (2003-03-01), pages 619 |
| H. PUTHALAKATH ET AL.: "ER stress triggers apoptosis by activating BH3-only protein Bim", CELL, vol. 129, 29 June 2007 (2007-06-29), pages 1337 |
| H. UCHINO ET AL.: "Transport of amino acid-related compo unds mediated by L-type amino acid transporter 1 (LAT1): insights int o the mechanisms of substrate recognition", MOLECULAR PHARMACOLOGY, vol. 6 1, April 2002 (2002-04-01), pages 729 |
| H. ZINSZNER ET AL.: "CHOP is implicated in programmed ce 11 death in response to impaired function of the endoplasmic reticulu m", GENES & DEVELOPMENT, vol. 12, 1 April 1998 (1998-04-01), pages 982 |
| HAYASHI K. ET AL.: "Role of c-Myc for regulation of LAT1 in cancer cells.", JOURNAL OF PHARMACOLOGICAL SCIENCES, vol. 118, no. 1, 2012, pages 120P, ISSN: 1347-8613 * |
| HISAO IMAI ET AL.: "L-gata Amino-san Transporter 1 (LAT1) Sogai ni yoru Hi Shosaibo Haigan Saibokabu deno Ko Shuyo Koka no Kento", JAPANESE JOURNAL OF LUNG CANCER, vol. 51, no. 5, 2011, pages 551, ISSN: 0386-9628 * |
| HUO H.Z. ET AL., BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 443, no. 2, January 2014 (2014-01-01), pages 406 - 412, ISSN: 0006-291X * |
| I. AIFANTIS; E. RAETZ; S. BUONAMICI: "Molecular pathogenesis of T-cell leukeamia and lymphoma", NATURE REVIEWS IMMUNOLOGY, vol. 8, 2008, pages 380 |
| I. TABAS; D. RON: "Integrating the mechanisms of apoptos is induced by endoplasmic reticulum stress", NAT CELL BIOL, vol. 13, March 2011 (2011-03-01), pages 184 |
| J. TOYOSHIMA; H. KUSUHARA; M. F. WEMPE; H. ENDOU; Y. SUGIYAMA: "Investigation of the role of transporters on the hepatic elimination of an LAT1 selective inhibitor JPH203", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 102, September 2013 (2013-09-01), pages 3228 |
| J. YE ET AL.: "ER stress induces cleavage of membrane-bo und ATF6 by the same proteases that process SREBPs", MOLECULAR CELL, vol. 6, December 2000 (2000-12-01), pages 1355 |
| K. HAYASHI; P. JUTABHA; H. ENDOU; H. SAGARA; N. ANZAI: "L AT1 is a critical transporter of essential amino acids for immune rea ctions in activated human T cells", J IMMUNOL, vol. 191, 15 October 2013 (2013-10-15), pages 4080 |
| K. KAIRA ET AL.: "1-type amino acid transporter 1 and CD9 8 expression in primary and metastatic sites of human neoplasms", CANC ER SCI, vol. 99, December 2008 (2008-12-01), pages 2380, XP055073515, DOI: doi:10.1111/j.1349-7006.2008.00969.x |
| K. KAIRA ET AL.: "Fluorine-18-alpha-methyltyrosine positron emission tomography for diagnosis and staging of lung cancer: a clinicopathologic study", CLINICAL CANCER RESEARCH : AN OFFICIAL JOURNAL OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, vol. 13, 1 November 2007 (2007-11-01), pages 6369, XP002636302, DOI: doi:10.1158/1078-0432.CCR-07-1294 |
| K. NAKANISHI ET AL.: "LAT1 expression in normal lung and in atypical adenomatous hyperplasia and adenocarcinoma of the lung", VIRCHOWS ARCHIV : AN INTERNATIONAL JOURNAL OF PATHOLOGY, vol. 448, February 2006 (2006-02-01), pages 142, XP019344892, DOI: doi:10.1007/s00428-005-0063-7 |
| K. ODA ET AL.: "L-type amino acid transporter 1 inhibito rs inhibit tumor cell growth", CANCER SCI, 2010, pages 101 |
| K. ODA ET AL.: "L-type amino acid transporter 1 inhibitors inhibit tumor cell growth", CANCER SCI, vol. 101, January 2010 (2010-01-01), pages 173 |
| K. YAMAUCHI ET AL.: "System L amino acid transporter inh ibitor enhances anti-tumor activity of cisplatin in a head and neck s quamous cell carcinoma cell line", CANCER LETT, vol. 276, 8 April 2009 (2009-04-08), pages 95 |
| KANAI Y. ET AL., JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 273, 1998, pages 23629 |
| KHARAS M.G. ET AL.: "Ablation of PI3K blocks BCR -ABL leukemogenesis in mice, and a dual PI3K/ mTOR inhibitor prevents expansion of human BCR- ABL+ leukemia cells.", JOURNAL OF CLINICAL INVESTIGATION, vol. 118, no. 9, 2008, pages 3038 - 3050, XP009123405, ISSN: 0021-9738, DOI: doi:10.1172/JCI33337 * |
| KOHLER, G.; MILSTEIN, C., METHODS ENZYMOL., vol. 73, 1981, pages 3 - 46 |
| L. GALLUZZI; N. LAROCHETTE; N. ZAMZAMI; G. KROEMER: "Mit ochondria as therapeutic targets for cancer chemotherapy", ONCOGENE, vol. 2 5, 7 August 2006 (2006-08-07), pages 4812 |
| L. SALMENA; A. CARRACEDO; P. PANDOLFI: "Tenets of PTEN tumor suppression", CELL, vol. 133, 2008, pages 403, XP055332237, DOI: doi:10.1016/j.cell.2008.04.013 |
| L. V. SINCLAIR ET AL.: "Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essenti al for T cell differentiation", NATURE IMMUNOLOGY, vol. 14, May 2013 (2013-05-01), pages 500 |
| M. BOYCE ET AL.: "A selective inhibitor of eIF2alpha dep hosphorylation protects cells from ER stress", SCIENCE, vol. 307, 25 November 2011 (2011-11-25), pages 935 |
| M. F. WEMPE ET AL.: "Metabolism and pharmacokinetic studies of JPH203, an L-amino acid transporter 1 (LAT1) selective compound", DRUG METABOLISM AND PHARMACOKINETICS, vol. 27, 2012, pages 155, XP055394226, DOI: doi:10.2133/dmpk.DMPK-11-RG-091 |
| M. FURUYA; J. HORIGUCHI; H. NAKAJIMA; Y. KANAI; T. OYAM A: "Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis", CANCER SCI, vol. 103, February 2012 (2012-02-01), pages 382 |
| M. ICHINOE ET AL.: "High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: comparison with non-can cerous lesions", PATHOL INT, vol. 61, May 2011 (2011-05-01), pages 281 |
| M. R. JANES ET AL.: "Effective and selective targeting o f leukemia cells using a TORC1/2 kinase inhibitor", NAT MED, vol. 16, February 2010 (2010-02-01), pages 205, XP002673719, DOI: doi:10.1038/nm.2091 |
| N. BULUS; C. FERAL; A. POZZI; R. ZENT: "CD98 increases re nal epithelial cell proliferation by activating MAPKs", PLOS ONE, vol. 7, 2012, pages E4 0026 |
| N. LOUNNAS ET AL.: "Pharmacological inhibition of carbon ic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas", CANCER LETT, vol. 333, 1 June 2013 (2013-06-01), pages 76 |
| N. YANAGISAWA ET AL.: "High expression of L-type amino a cid transporter 1 (LAT1) predicts poor prognosis in pancreatic ducta 1 adenocarcinomas", J CLIN PATHOL, vol. 65, November 2012 (2012-11-01), pages 1019 |
| ODA K. ET AL.: "L-type amino acid transporter 1 inhibitors inhibit tumor cell growth.", CANCER SCIENCE, vol. 101, no. 1, 2010, pages 173 - 179, ISSN: 1347-9032 * |
| P. NICKLIN ET AL.: "Bidirectional transport of amino aci ds regulates mTOR and autophagy", CELL, vol. 136, 6 February 2009 (2009-02-06), pages 521 |
| P. STECK ET AL.: "Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers", NAT GENET, vol. 15, 1997, pages 356, XP002066156, DOI: doi:10.1038/ng0497-356 |
| P. WALTER; D. RON: "The unfolded protein response: from stress pathway to homeostatic regulation", SCIENCE, vol. 334, 25 November 2011 (2011-11-25), pages 1081 |
| S. C. CAZANAVE ET AL.: "CHOP and AP-1 cooperatively medi ate PUMA expression during lipoapoptosis. American journal of physio logy", GASTROINTESTINAL AND LIVER PHYSIOLOGY, vol. 299, July 2010 (2010-07-01), pages G236 |
| S. OYADOMARI; M. MORI: "Roles of CHOP/GADD153 in endopla smic reticulum stress", CELL DEATH AND DIFFERENTIATION, vol. 11, April 2004 (2004-04-01), pages 381 |
| SAMBROOK, J. ET AL.: "Molecular Cloning: A Laboratory Manual, 2 d. Ed.,", vol. 1-3, 1989, COLD SPRING HARBOR LABORATORY PRESS |
| See also references of EP2959918A4 |
| SHI L. ET AL.: "Downregulation of L-type amino acid transporter 1 expression inhibits the growth, migration and invasion of gastric cancer cells.", ONCOLOGY LETTERS, vol. 6, no. 1, 2013, pages 106 - 112, XP055237375, ISSN: 1792-1074 * |
| SUGURU HAYASE ET AL.: "Igaku Yogo Kaisetsu", LAT1, G.I. RESEARCH, vol. 17, no. 3, 2009, pages 268 - 270, ISSN: 0918-9408 * |
| T. HAGENBEEK ET AL.: "The loss of PTEN allows TCR alphabe ta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling", J EXP MED, vol. 200, 2004, pages 883 |
| T. HAGENBEEK; H. SPITS: "T-cell lymphomas in T-cell specific Pten-deficient mice originate in the thymus", LEUKEMIA, vol. 22, 2007, pages 608 |
| T. SAKATA ET AL.: "L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer", PATHOL INT, vol. 59, January 2009 (2009-01-01), pages 7, XP055311922, DOI: doi:10.1111/j.1440-1827.2008.02319.x |
| VAAGE J. ET AL.: "Therapy of a xenografted human colonic carcinoma using cisplatin or doxorubicin encapsulated in long-circulating pegylated stealth liposomes.", INTERNATIONAL JOURNAL OF CANCER, vol. 80, no. 1, 1999, pages 134 - 137, XP000979545, ISSN: 0020-7136, DOI: doi:10.1002/(SICI)1097-0215(19990105)80:1<134::AID-IJC24>3.0.CO;2-Q * |
| W. L. ZHAO: "Targeted therapy in T-cell malignancies: dysregulation of the cellular signaling pathways", LEUKEMIA, vol. 24, January 2010 (2010-01-01), pages 13 |
| Y. P. VANDEWYNCKEL ET AL.: "The paradox of the unfolded p rotein response in cancer", ANTICANCER RES, vol. 33, November 2013 (2013-11-01), pages 4683 |
| YAMAUCHI K. ET AL.: "System L amino acid transporter inhibitor enhances anti-tumor activity of cisplatin in a head and neck squamous cell carcinoma cell line.", CANCER LETTERS, vol. 276, no. 1, 2009, pages 95 - 101, XP025953194, ISSN: 0304-3835 * |
| YANAGIDA ET AL.: "Human L-type amino acid transporter 1 (LAT1) : characterization of function and expression in tumor cell lines", BIOCHEM BIOPHYS ACTA, vol. 1514, 1 October 2001 (2001-10-01), pages 291, XP004319633, DOI: doi:10.1016/S0005-2736(01)00384-4 |
| YANAGIDA O. ET AL.: "Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines.", BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1514, no. 2, 2001, pages 291 - 302, XP004319633, ISSN: 0006-3002, DOI: doi:10.1016/S0005-2736(01)00384-4 * |
| YOSHIKATSU KANAI: "Cancer drug delivery targeting amino acid transporters", DRUG DELIVERY SYSTEM, vol. 27, no. 5, 2012, pages 342 - 349, ISSN: 0913-5006 * |
| YUN D.W. ET AL.: "JPH203, an L-type amino acid transporter 1-selective compound, induces apoptosis of YD-38 human oral cancer cells.", JOURNAL OF PHARMACOLOGICAL SCIENCES, vol. 124, no. 2, February 2014 (2014-02-01), pages 208 - 217, ISSN: 1347-8613 * |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017155023A (ja) * | 2016-03-04 | 2017-09-07 | ジェイファーマ株式会社 | Lat1阻害活性を有する芳香族アミノ酸誘導体を含有する注射剤 |
| KR20200009001A (ko) | 2017-05-17 | 2020-01-29 | 가부시끼가이샤 도꾸야마 | 디아미노벤젠 화합물의 제조 방법 |
| JPWO2019093383A1 (ja) * | 2017-11-07 | 2020-09-24 | ジェイファーマ株式会社 | 抗pd−1抗体若しくは抗pd−l1抗体療法の奏効性を予測する方法、がんの悪性度を評価する方法、及び抗pd−1抗体若しくは抗pd−l1抗体療法の奏効性を上昇させる方法 |
| US11899019B2 (en) | 2017-11-07 | 2024-02-13 | J-Pharma Co., Ltd. | Method for predicting efficacy of anti-PD-1 antibody or anti-PD-L1 antibody therapy, method for evaluating cancer grade, and method for enhancing efficacy of anti-PD-1 antibody or anti-PD-L1 antibody therapy |
| JP7369378B2 (ja) | 2017-11-07 | 2023-10-26 | ジェイファーマ株式会社 | 抗がん剤及びがんを治療するための医薬組成物 |
| JP7298827B2 (ja) | 2017-11-07 | 2023-06-27 | ジェイファーマ株式会社 | 抗pd-1抗体若しくは抗pd-l1抗体療法の奏効性を予測する方法、がんの悪性度を評価する方法、及び抗pd-1抗体若しくは抗pd-l1抗体療法の奏効性を上昇させる方法 |
| JP2022025113A (ja) * | 2017-11-07 | 2022-02-09 | ジェイファーマ株式会社 | 抗pd-1抗体若しくは抗pd-l1抗体療法の奏効性を予測する方法、がんの悪性度を評価する方法、及び抗pd-1抗体若しくは抗pd-l1抗体療法の奏効性を上昇させる方法 |
| JP2023101705A (ja) * | 2017-12-28 | 2023-07-21 | ジェイファーマ株式会社 | がん治療薬 |
| JP2020073586A (ja) * | 2017-12-28 | 2020-05-14 | ジェイファーマ株式会社 | がん治療薬 |
| US12458628B2 (en) | 2017-12-28 | 2025-11-04 | J-Pharma Co., Ltd. | Cancer therapeutic |
| JP7541400B2 (ja) | 2017-12-28 | 2024-08-28 | ジェイファーマ株式会社 | がん治療薬 |
| WO2019130637A1 (ja) * | 2017-12-28 | 2019-07-04 | ジェイファーマ株式会社 | がん治療薬 |
| JP6546367B1 (ja) * | 2017-12-28 | 2019-07-17 | ジェイファーマ株式会社 | がん治療薬 |
| JP2019151672A (ja) * | 2017-12-28 | 2019-09-12 | ジェイファーマ株式会社 | がん治療薬 |
| WO2020004043A1 (ja) | 2018-06-28 | 2020-01-02 | 株式会社トクヤマ | α-アジドアニリン誘導体又はα,α'-ジアジド誘導体の製造方法 |
| KR20210025517A (ko) | 2018-06-28 | 2021-03-09 | 가부시끼가이샤 도꾸야마 | α-아지도아닐린 유도체 또는 α,α'-디아지드 유도체의 제조 방법 |
| WO2020096041A1 (ja) * | 2018-11-09 | 2020-05-14 | ジェイファーマ株式会社 | (S)-2-アミノ-3-{4-[(5-アミノ-2-フェニルベンゾ[d]オキサゾール-7-イル)メトキシ]-3,5-ジクロロフェニル}プロパン酸・1塩酸塩、その1水和物、およびその結晶とその製造方法 |
| JP7622986B2 (ja) | 2018-11-09 | 2025-01-28 | ジェイファーマ株式会社 | (S)-2-アミノ-3-{4-[(5-アミノ-2-フェニルベンゾ[d]オキサゾール-7-イル)メトキシ]-3,5-ジクロロフェニル}プロパン酸・1塩酸塩、その1水和物、およびその結晶とその製造方法 |
| JPWO2020096041A1 (ja) * | 2018-11-09 | 2021-09-24 | ジェイファーマ株式会社 | (S)−2−アミノ−3−{4−[(5−アミノ−2−フェニルベンゾ[d]オキサゾール−7−イル)メトキシ]−3,5−ジクロロフェニル}プロパン酸・1塩酸塩、その1水和物、およびその結晶とその製造方法 |
| US20220288035A1 (en) * | 2019-08-30 | 2022-09-15 | J-Pharma Co., Ltd. | A pharmaceutical composition for treating cancer used for a patient having specific genetic marker |
| CN111253271A (zh) * | 2020-02-11 | 2020-06-09 | 浙江聚贤医药科技有限公司 | 一种制备2-氨基-3-硝基苯甲酸甲酯的方法 |
| WO2022102687A1 (ja) * | 2020-11-11 | 2022-05-19 | ジェイファーマ株式会社 | がん治療剤 |
| WO2023286823A1 (ja) * | 2021-07-15 | 2023-01-19 | 国立大学法人大阪大学 | 医薬組成物 |
| CN115772130A (zh) * | 2021-09-08 | 2023-03-10 | 四川大学华西医院 | 一种微管解聚剂及其制备方法和用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20160279103A1 (en) | 2016-09-29 |
| CN105392498A (zh) | 2016-03-09 |
| EP2959918B1 (en) | 2019-05-22 |
| KR20170001962A (ko) | 2017-01-06 |
| JPWO2015173970A1 (ja) | 2017-04-20 |
| JP6402393B2 (ja) | 2018-10-10 |
| EP2959918A4 (en) | 2016-03-09 |
| EP2959918A1 (en) | 2015-12-30 |
| US10172835B2 (en) | 2019-01-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6402393B2 (ja) | 抗悪性腫瘍剤組成物 | |
| JP7337133B2 (ja) | TAMおよびMETキナーゼの阻害薬としてのピラゾロ[3,4-b]ピリジン化合物 | |
| US20220411389A1 (en) | Compounds and methods for treating cancer | |
| KR102772574B1 (ko) | 키랄 디아릴 매크로사이클 및 이것의 용도 | |
| JP2020530858A (ja) | 肝疾患を治療する方法 | |
| CN108368110A (zh) | 用于激酶调节的化合物和方法以及其适应症 | |
| UA123964C2 (uk) | Сполука піридину | |
| CN107683139A (zh) | 用于治疗b细胞恶性肿瘤的赛度替尼 | |
| US9469625B2 (en) | Organic compounds | |
| US12318393B2 (en) | Modulators of orphan nuclear receptors for NASH and other metabolic disorders | |
| EP3242661A1 (en) | Myc g-quadruplex stabilizing small molecules and their use | |
| JPWO2018101329A1 (ja) | 新規エステル体化合物並びにそれを用いたPin1阻害剤、炎症性疾患治療剤及び大腸癌治療剤 | |
| JP6034880B2 (ja) | オーロラおよびflt3キナーゼモジュレーター | |
| JP2019526617A (ja) | 治療用化合物 | |
| US20200325140A1 (en) | Fused bicyclic compounds for the treatment of disease | |
| US20240238274A1 (en) | Inhibitors of ubiquitin specific peptidase 22 (usp22) and uses thereof for treating disease and disorders | |
| WO2016098042A1 (en) | Use of ceritinib (ldk-378) in the treatment of fes or fer mediated disorders, in particular proliferative disorders | |
| US20230339942A1 (en) | Inhibitors of ubiquitin specific peptidase 22 (usp22) and uses thereof for treating diseases and disorders | |
| WO2025255213A1 (en) | Niclosamide prodrugs for treating cancers | |
| CN117561255A (zh) | 作为mek抑制剂的3,4-二氢-2,7-萘啶-1,6(2h,7h)-二酮化合物 | |
| CN121108109A (zh) | 一种靶向alk蛋白的protac化合物及其抗肿瘤应用 | |
| HK1254606B (zh) | 手性二芳基大环及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480001519.X Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2014557910 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147034643 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014809268 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14409371 Country of ref document: US |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14809268 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |