СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ, НАРУШАЮЩИЕ ВЗАИМОДЕЙСТВИЕ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ И FRS2, ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ
РАКА И ДРУГИХ ЗАБОЛЕВАНИЙ
Описание
Краткое описание изобретения
Изобретение относится к медицине и включает использование ингибиторов, которые избирательно нарушают взаимодействие рецептора фактора роста фибробластов с FRS2, а также взаимодействие с другими компонентами комплекса FRS2. Применение селективных ингибиторов, описанных в настоящем изобретении, приводит к достоверному повышению противоопухолевой эффективности по сравнению с другими ингибиторами и моноклональными антителами, снижению токсичности проводимого лечения, возможности использовать низкую концентрацию агента для полного блокирования, проводить длительное лечение. Преимущество изобретения заключается в разработке нового класса лекарственных препаратов для лечения злокачественных новообразований и других болезней.
Полное описание изобретения
Известно, что в основе развития злокачественных новообразований лежит избыточная пролиферация клеток, а также образование кровеносных сосудов в опухоли, через которые происходит ее питание (ангиогенез) (J. Folkman et al. Nature; 339, 58 (1989). Образование новых кровеносных сосудов происходит из уже существующего эндотелия и является важным компонентом многих заболеваний и нарушений, в том числе таких, как рост и метастазирование опухолей, ревматоидный артрит, псориаз, атеросклероз, диабетическая ретинопатия, ретролентальная фиброплазия, неоваскулярная глаукома, гемангиомы, иммунное отторжение трансплантированной роговицы и других тканей, а также хронические воспаления.
Пролиферация клеток опухоли, равно как и эндотелиальных клеток, может быть вызвана различными факторами, которые естественным образом встречаются в природе. Данные факторы связываются с рецепторами на поверхности опухолевых, эндотелиальных и других клеток, что приводит к активации рецепторов и проведению сигнала внутрь клетки с последующим делением.
По многочисленным данным, рецепторы с фактора роста фибробластов (ФРФР) часто экспрессированы на клетках опухолей, что приводит к пролиферации клеток самой
опухоли и эндотелиоцитов, способствует опухолевой прогрессии. Зачастую активирующие мутации ФРФР также изменяют течения болезни и чувствительность к проводимой терапии.
Семейство ФРФР участвует в физиологических процессах, таких как ангиогенез, миграция, пролиферация, дифференцировка и выживаемость различных клеток.
Семейство насчитывает 18 факторов, которые условно можно разделить на две группы: классические (ФРФ1-10, 16-18 и 20) и гормоноподобные (ФРФ19,21 и 23) [Beenken et al. 2009]. Фактор роста фибробластов (ФРФ) связывается специфически с ФРФР, представленными пятью типами. ФРФР 1-4 типов являются типичными рецепторами с тирозинкиназной активностью, то есть внутриклеточная часть представлена тирозинкиназой. В основе процессов активации тирозинкиназы и дальнейшей передачи сигнала внутри клетки лежат процессы фосфорилирования, присущие для всех подобных рецепторов. Экстрацеллюлярная часть ФРФР состоит из 3 иммуноглобулин-подобных доменов; II и III домены содержат лиганд-связывающий регион. Благодаря альтернативному сплайсингу, домен III изменяет форму и находится в двух состояниях - ШЬ и Шс. Это может влиять на силу связывания с лигандом и активность терапевтических агентов, направленных против рецептора [Eswarakumar et al. 2005; Brooks et al. 2012]. Более того, существуют сведения о различной пролиферативной активности клеток опухоли при связывании ФРФ с различными субъединицами домена Шс - 1а или 1β [Tomlinson et al. 2010]. ФРФ относятся к группе гепаринсвязывающих белков. Для того чтобы передать сигнал в клетку и активировать внутриклеточные процессы, ФРФ должен связаться с рецептором и гепаран сульфатом (гепарином) на поверхности клетки [Abuharbeid et al. 2006]. Это отличает механизм активации рецептора ФРФ от других факторов, в частности VEGF, для которого гепаран сульфат не нужен. Гормоноподобные ФРФ имеют низкую аффинность к гепаран сульфату. При связывании ФРФ с рецептором происходит активация основных внутриклеточных путей RAS-RAF-MAPK и PI3K-AKT- mTOR с распространением сигнала в ядро [Turner et al. 2010]. Главным компонентом передачи сигнала с ФРФР к внутриклеточным киназам является белок FRS2 [Ong et al. 2000; Kouhara et al. 1997]. FRS2 - якорный белок ("docking protein"), активирующийся при связывании ФРФР с лигандом и формирующий комплекс с Shp2 и Grb2, в связи с чем активация FRS2 приводит к активации обоих сигнальных путей RAS-RAF-MAPK и PI3K- AKT-mTOR.
В настоящее время создано большое количество таргетных препаратов, блокирующих/ингибирующих активность различных типов ФРФР. К ним относятся ингибиторы тирозинкиназы, а также моноклональные антитела. Существующие
ингибиторы тирозинкиназы снижают активность внутриклеточной тирозинкиназы путем блокирования различных активных частей одного или, как правило, нескольких рецепторов. Неизбирательное блокирование сопряжено со снижением эффективности воздействия на опухолевые клетки, клетки сосудов, а также другие структуры, экспрессирующие соответствующий тип рецептора. Известно, что эффективность терапевтических моноклональных антител, направленных против ФРФР, может существенно снижаться, если домен III рецептора ФРФ меняет форму (ШЬ и Шс) [Wesche et al. 201 1 ; Tsimafeyeu et al. 2012]. Тоже самое происходит, если ФРФР существует на клетке в различных изоформах [Dienstmann et al. 2013]. Существующие низкомолекулярные ингибиторы тирозинкиназы не обладают селективным действием в отношении одного типа рецептора ФРФ, блокируют несколько типов ФРФ, а также другие рецепторы. Так, бриваниб, доватиниб ингибируют ФРФР и рецепторы фактора роста эндотелия сосудов [Ayers et al. 2007; Sarker et al. 2008], понатиниб, BGJ398, AZD4547, J J-42756493, BAY 1163877 ингибируют несколько типов ФРФР и, хоть в меньшей степени, но также рецепторы фактора роста эндотелия сосудов и некоторые другие киназы [Gozgit et al. 2012; Arai et al. 2014; Gavine et al. 2012; Perera et al. 2014; Heroult et al. 2014]. При этом показано, что клетки определенной опухоли экспрессируют свой тип ФРФР (например, рак почки - ФРФР1 [Tsimafeyeu et al. 2011], люминальный тип рака молочной железы - ФРФР1 [Tenhagen et al. 2012], трижды-негативный рак молочной железы - ФРФР2 [Tannheimer et al. 2000], рак желудка - ФРФР2 [Xie et al. 2013], рак мочевого пузыря - ФРФРЗ [Gust et al. 2013] и т.п.). Следовательно, одновременное блокирование нескольких типов ФРФР не требуется и может приводить не только к повышению токсичности, но и к снижению эффективности. Во многих работах показано, что существующие низкомолекулярные ингибиторы тирозинкиназы в наномолярных концентрациях подавляют активность внтуриклеточной тирозинкиназы ФРФР, при этом слабо влияют на фосфорилирование (активность) FRS2. Другими словами, взаимодействие между ФРФР и FRS2 даже при подавлении активности внутриклеточной тирозинкиназы ФРФР представляется важным, а его отсутствие может быть фактором резистентности. Так, например, сведений о действии бриваниба на FRS2 нет (Huynh et al. 2008); довитиниб влияет на фосфорилирование ФРФР 1/3 в наномолярной концентрации, и в меньшей степени подавляет активность FRS2 (Zhang et al. 2014); понатиниб, как наиболее селективный ингибитор ФРФР, ингибирует фосфорилирование ФРФР с IC50 в пределах 3-18 нмоль/л, a FRS2 - только 33-40 нмоль/л (Gozgit et al. 2012); высокоселективный ингибитор AZD4547, подавляя активность ФРФР2 с IC50 в пределах 2 нмоль/л, угнетает активность FRS2 при использовании концентрации 100 нмоль/л
(Gavine et al. 2012). To есть существующие ингибиторы имеют слабый опосредованный ингибирующий эффект на FRS2, тем более, не вызывают нарушение взаимодействия ФРФР с FRS2.
В настоящем изобретении описываются ингибиторы, механизм действия которых включает все из перечисленного:
1) нарушение взаимодействия ФРФР и FRS2, что в результате приводит к снижению фосфорилирования и потери активности FRS2, а также, возможно, но необязательно, нарушение взаимодействия с другими компонентами комплекса FRS2;
2) отсутствие влияния на внутриклеточную тирозинкиназу ФРФР и на фосфорилирование самого рецептора;
3) отсутствие воздействия на внутриклеточную тирозинкиназу других рецепторов с тирозинкиназной активностью и на их фосфорилирование;
4) не связываются с активным центром ФРФР и с активным центром других рецепторов;
5) избирательное нарушение взаимодействия FRS2 преимущественно с одним типом ФРФР, или в некоторых случаях - несколькими типами ФРФР;
Также подразумевается отсутствие ингибитор-обусловленной выраженной токсичности как результата селективного действия.
Описываемые в изобретении ингибиторы нарушают взаимодействие ФРФР с FRS2, что существенно влияет на белок FRS2, активность которого снижается (Пример 1). Важнейшим в механизме действия описываемых ингибиторов является отсутствие влияния на активность общего ФРФР, а также внутриклеточной части рецептора, представленной тирозинкиназой, и отсутствие влияния на активность других рецепторов с тирозинкиназной активностью (Пример 2). Это характеризует высокоселективное действие ингибиторов и подчеркивает значение нарушения взаимодействия ΦPΦP-FRS2 без необходимости блокировать киназную активность рецептора и другие механизмы. Важно отметить, что описываемые ингибиторы не взаимодействуют с активным центром ФРФР и не препятствуют связи ФРФР с лигандом (Пример 3).
Снижение активности FRS2 - основного проводника сигнала от рецептора к внутриклеточным белкам, приводит к подавлению сигнальной трансдукции внутри клетки. При этом описываемые ингибиторы могут не влиять непосредственно на другие составляющие сигнальных путей RAS-RAF-MAP и PDK-AKT-mTOR, что характеризует их избирательное действие только в отношении взаимодействия ФРФР и FRS2 (Примеры 4, 5). Для некоторых типов описываемых ингибиторов действие против одного или нескольких компонентов сигнальных путей исключить нельзя (Пример 6). Однако, это может быть как независимым влиянием на структуру компонента сигнального пути, так и
следствием блокирования OPOP-FRS2. Также описываемые ингибиторы могут нарушать комплекс FRS2 с Shp2 и/или Grb2, что может привести к усилению действия, но не является обязательным, так как главным в ограничении проведения сигнала является блокирование взаимодействия OPOP-FRS2 (Пример 7). Описываемые ингибиторы могут ингибировать только один тип ФРФР, как описано в Примере 8 для ФРФР2, или несколько (Пример 6). Зависимость активности ингибитора от изоформы определенного типа ФРФР в большинстве случаев должна исключаться (Пример 9).
Такое селективное нарушение взаимодействия ФРФР и FRS2 приводит к необходимым терапевтическим эффектам описываемых ингибиторов, например, к подавлению пролиферации и миграции эндотелиальных клеток, формирования тубулярных структур сосуда, роста зрелых сосудов (Примеры 10 и 11), к угнетению пролиферации опухолевых клеток (Пример 12), к торможению роста опухоли (Пример 13), к развитию некроза в опухоли (Пример 14), к торможению развития других заболеваний (Пример 15). Также селективное действие описываемых ингибиторов и отсутствие влияния на другие мишени приводит к снижению выраженной токсичности, что является крайне важным при использовании терапевтических средств (Пример 16).
Основным объектом настоящего изобретения является использование ингибиторов, которые избирательно нарушают взаимодействие рецептора фактора роста фибробластов с FRS2, а также взаимодействие с другими компонентами комплекса FRS2, с описанными свойствами выше. Примером такого ингибитора, нарушающего взаимодействие ФРФР 2 типа и FRS2, может быть вещество, определенное химической Формулой I.
Фо мула I
о Kg
RI, R2, R3, R-ь R5, R6, R7S Rs, 9, Rio могут быть одинаковыми или различными и могут включать независимо группы -NH2, -N02, -СН3, -CH2NH2, -F, -CI, -Br, -I, -CF3, -OCH3, -
C2H5, -H, замещенные или незамещенные первичные, вторичные или третичные алкильные группы, замещенные или незамещенные арильные группы, замещенные или незамещенные алкенил- группы, замещенные или незамещенные алкинил- группы, замещенные или незамещенные гетероциклические группы, замещенные или
незамещенные гетероциклилалкильные группы, замещенные или незамещенные алкоксиалкильные группы, замещенные или незамещенные арилоксиалкильные группы, замещенные или незамещенные гетероциклилоксиалкильные группы.
X может быть представлен CHjN или СН или N.
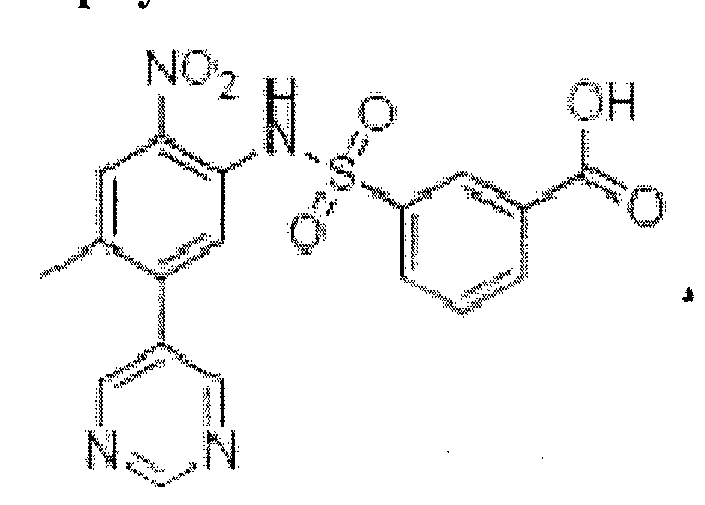
Как пример ингибитора, избирательно нарушающего взаимодействие рецептора фактора роста фибробластов с FRS2, описываемого общей формулой I, в настоящем изобретении, можно представить химическое вещество RPT835, продемонстрированное Формулой II и относящееся к 3 -(Ы-(4-метил-2-нитро-5 -(пиридин-3 -ил)фенил)сульфамоил)бензойная кислота.
Формула II
■мог
Кроме того, похожий ингибитор может относится к 3-(]ЧГ-(4-метил-2-нитро-5-(пиримидин- 5-ил)фенил)сульфамоил)бензойная кислота и быть представлен Формулой III
Формула III
или относится к 3-(М-(5-(фуран-3-ил)-4-метил-2-нитрофенил)сульфамоил)бензойная кислота и быть представлен Формулой IV,
Формула IV
а может относится к 3-(Т -(2-амино-4-метил-5-(пиримидин-5- ил)фенил)сульфамоил)бензойная кислота и быть описан по Формуле V.
Формула V
Также описываемый в изобретении ингибитор может сочетать в себе несколько компонентов, выбранных из Формул I, II, III, IV, V. Так, химическая формула такой комбинированной субстанции может быть, к примеру, такой (Формула VI).
Формула VI
Ингибитор, нарушающий взаимодействие рецептора фактора роста фибробластов и FRS2, может быть выбран из любого класса веществ, описанных Формулой VII.
Ri, R2, R3, R4, R5, R6, R7, R8, R9 могут быть одинаковыми или различными и могут включать независимо группы -NH2, -CH2NH2, -N02, -СН3, -F, -CI, -Br, -I, -CF3, -OCH3, - C2H5, -H, замещенные или незамещенные первичные, вторичные или третичные алкильные группы, замещенные или незамещенные арильные группы, замещенные или незамещенные алкенил- группы, замещенные или незамещенные алкинил- группы, замещенные или незамещенные гетероциклические группы, замещенные или незамещенные гетероциклилалкильные группы, замещенные или незамещенные алкоксиалкильные группы, замещенные или незамещенные арилоксиалкильные группы, замещенные или незамещенные гетероциклилоксиалкильные группы.
Самым простым примером химических веществ, относящихся к классу по Формуле VII, могут быть 3-(3-оксо-1,2,3,4-тетрагидрохиноксалин-1-карбонил)бензойная кислота (Формула VIII) и 3-(2-метил-3-оксо-1,2,3,4-тетрагидрохиноксалин-1-карбонил)бензойная кислота (Формула IX).
Формула VIII Формула IX
Ингибитор, нарушающий взаимодействие рецептора фактора роста фибробластов и FRS2, также может быть выбран из любого класса веществ, описанных Формулой X.
Ri, R2, R3, R4, R5, Яб, R7, R8, R9, Rio, Ri i могут быть одинаковыми или различными и могут включать независимо группы -NH2, -CH2NH2, -N02, -СН3, -F, -CI, -Br, -I, -CF3, -OCH3, - C2H5, -H, замещенные или незамещенные первичные, вторичные или третичные алкильные группы, замещенные или незамещенные арильные группы, замещенные или незамещенные алкенил- группы, замещенные или незамещенные алкинил- группы, замещенные или незамещенные гетероциклические группы, замещенные или незамещенные гетероциклилалкильные группы, замещенные или незамещенные алкоксиалкильные группы, замещенные или незамещенные арилоксиалкильные группы, замещенные или незамещенные гетероциклилоксиалкильные группы.
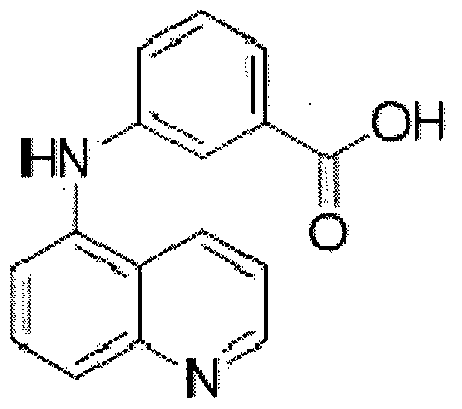
Простейшим примером химического вещества, относящегося к этому классу, может быть 3-(хинолин-5-иламино)бензойная кислота с Формулой XI
Фо мула XI
Также ингибитор может включать в свой состав все структуры описанные в формулах I- XI и быть представлять комбинированное вещество.
Химические вещества по формулам I, II, III, IV, V, VI, VII, VIII, IX, X, XI могут быть представлены фармацевтически приемлемой солью, таутомером, фармацевтически приемлемой солью таутомера или их комбинацией.
Терапевтическое использование ингибиторов
Для использования ингибиторов, описанных в настоящем изобретении, в терапевтической практике требуется их введение млекопитающему, предпочтительно человеку, в фармацевтически приемлемой форме, включая введение внутривенно, а также
следующими путями: пероральным, внутримышечным, подкожным, интраперитонеальным, интрацереброспинальным, внутрисуставным, внутрисиновиальным, внутриоболочечным, локальным или ингаляционным. Ингибиторы также могут вводиться внутриопухолевым, околоопухолевым, внутриочаговым и околоочаговым путями для обеспечения локального действия наряду с системным терапевтическим действием.
Подобные формы введения включают фармацевтически приемлемые носители, которые по своей природе не обладают ни токсическим, ни терапевтическим действием. Вспомогательные вещества, которые используются для приготовления лекарственного препарата на основе описываемого в изобретении ингибитора, в зависимости от влияния на физико-химические характеристики и фармакокинетику лекарственных форм можно разделить на следующие группы:
• формообразующие
• стабилизирующие
• пролонгирующие
• солюбилизирующие
• корригирующие
Могут использоваться носители. Примерами таких носителей являются ионообменные вещества, квасцы, стеарат алюминия, лецитин, белки плазмы (такие, как белок плазмы человека), буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, частичные глицеридные смеси насыщенных овощных жирных кислот, вода, соли или электролиты, такие как сульфат протамина, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидная окись кремния, трисиликат магния, поливинилпирролидон, вещества с целлюлозной основой и полиэтиленгликоль.
Носители для локальной или основанной на геле форм антагонистов включают полисахариды, такие как натриевая соль карбоксиметилцеллюлозы или метилцеллюлозы, поливинилпирролидон, полиакрилаты, полимеры полиоксиэтиленполиоксипропиленового блока, полиэтиленгликоль и спирты. Для введения во всех случаях используются обычные лекарственные формы, получаемые со складов. К таким формам относятся, например, микрокапсулы, нанокапсулы, липосомы, пластыри, ингаляционные препараты, аэрозоли, подъязычные таблетки и препараты с постоянным высвобождением вещества.
Подходящие примеры препаратов с постоянным высвобождением вещества включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащих ингибитор; подобные матрицы имеют определенную форму, например это могут быть пленки или микрокапсулы. К примерам матриц с постоянным высвобождением относятся полиэфиры,
гидрогели [например, поли(2-гидроксиэтил-метакрилат)] , описанные Лангером и др. (J. Biomed. Mater. Res. 15, 167 (1981) и Лангером (Chem. Tech. 12 (1982), или поли(винилалкоголь), полилактиды (Патент США N 3773919), сополимеры L- глутаминовой кислоты и гаммаэтил-Ь-глутамата, описанные Сидман и др. (Biopolymers 22, 547 (1983), недеградируемый этиленвинилацетат (Лангер и др. см. выше), деградируемые сополимеры молочной и гликолевой кислот, такие как Lupron Depot™ (инъецируемые микросферы, состоящие из полимеров молочной и гликолевой кислот и ацетата лейпролида), и поли-О-(-)- 3-гидроксибутировой кислоты. В то время, как такие полимеры, как этиленвинилацетат и сополимер молочной и гликолевой кислот, способны к постоянному высвобождению молекул в течение более 100 дней, определенные гидрогели высвобождают белки за более короткие периоды времени. С целью стабилизации могут быть разработаны разумные стратегии, в зависимости от действующего механизма. Например, если обнаружен механизм агрегации, выражающийся в формировании межмолекулярной S-S-связи посредством тиодисульфидного обмена, стабилизация может быть достигнута путем модификации сульфгидрильных остатков, лиофилизации с целью удаления кислых растворов, контролирования влажности, использования соответствующих добавок и разработки специфических полимерных матричных составов.
Составы с постоянным высвобождением ингибитора включают также заключение в липосомы. Липосомы, содержащие ингибиторы, могут быть получены известными в данной области методами, например, описанными Эпстейном и др. (Proc. Nat. Acad. Sci. 82, 3688 (1985); Хуанг и др. (Procc. Nat. Acad. Sci. 77, 4030 (1980); Патент США N 4485045 и Патент США N 4544545. Липосомы, как правило, имеют небольшую величину (величиной около 200-800 ангстрем) и принадлежат к однослойному типу, в котором содержание липидов выше, чем 30 мол.% холестерина; выбранное соотношение может изменяться для подбора оптимальных условий терапии. Липосомы с продолжительным сроком циркуляции покрываются Патентом США N 5013556,
Еще одним путем использования данного изобретения является инкорпорирование ингибитора внутрь изделий, имеющих определенную форму. Такие изделия могут быть использованы для модулирования роста клеток эндотелия и ангиогенеза. Кроме того, такие изделия могут быть использованы для модулирования инвазии опухолей и метастазов.
Возможна конъюгация описываемого ингибитора и другого лечебного средства. Описываемый ингибитор может быть использован в различных режимах в комбинации с другими терапевтически активными и/или адъювантными препаратами для усиления
терапевтического действия, а также с целью уменьшения частоты побочных эффектов и нежелательных явлений.
При профилактике или лечении заболевания необходимая доза описываемого ингибитора будет зависеть от типа заболевания, от его степени серьезности и протекания, от того, вводится ли ингибитор с профилактической или терапевтической целью, от предыдущей терапии, от истории болезни пациента и его реакции на ингибитор и от указаний лечащего врача. Ингибитор может вводиться пациенту различными способами, единовременно или в качестве серии назначений, на протяжении времени, когда терапия ингибитором будет считаться эффективной по стандартным критериям эффективности, использующимся в практике на момент назначения ингибитора.
Описываемые в изобретении ингибиторы могут быть использованы для лечения различных неопластических и ненеопластических заболеваний и нарушений. Опухоли и близкие состояния, которые поддаются такому лечению, например, включают рак молочной железы, немелкоклеточный и мелкоклеточный рак легкого, опухоли трахеи, рак желудка, рак пищевода, колоректальный рак, рак печени, рак поджелудочной железы, опухоли желчного пузыря и желчевыводящих путей, гастроинтестинальные опухоли, рак яичников, рак шейки матки, рак тела матки, гиперплазию эндометрия, эндометриоз, саркомы, опухоли головы и шеи, гепатобластому, меланому, рак кожи, гемангиому, кавернозную гемангиому, гемангиобластому, рак надпочечников, ретинобластомы, астроцитому, глиобластому, шванному, олигодендроглиому, медуллобластому, нейробластому, рабдомиосаркому, лейомиосаркому, почечноклеточный рак, рак мочевого пузыря и уротелиальный рак, рак полового члена, рак предстательной железы, опухоль Вилмса, герминогенные опухоли, нейроэндокринные опухоли, опухоли невыявленной первичной локализации, лейкозы и лимфомы, аномальную пролиферацию сосудов, связанную с факоматозами.
Возможно применение описываемых ингибиторов при неонкологических заболеваниях, которые поддаются лечению, включая такие как ревматические болезни, сердечно- сосудистые болезни, в т.ч. атеросклероз, болезни органов дыхания, болезни органов пищеварения, заболевания почек, включая гломерулонефрит, кожные болезни, в т.ч. псориаз, диабетические и другие ретинопатии, фиброплазии, неоваскулярную глаукому, эндокринные заболевания и болезни обмена, трансплантацию роговицы и других тканей, хронические воспаления, асцит, преэклампсию, перикардиальНый выпот (например, связанный с перикардитом) и плевральный выпот, неврологические болезни.
В зависимости от типа заболевания и от степени его серьезности первоначальная доза для введения пациенту будет составлять от 0,001 мг/кг до 200 мг/кг, или более
предпочтительно от 1 мг/кг до 100 мг/кг, или еще боле предпочтительно от 10 мг/кг до 50 мг/кг, и может вводиться путем одного или многих отдельных назначений/введений или путем постоянного вливания. Обычная дневная доза может варьировать примерно от 1 мг/кг до 100 мг/кг и более, в зависимости от вышеупомянутых факторов. Для повторного назначения в течение нескольких дней и более, в зависимости от условий, лечение повторяется, пока не будет достигнуто желаемое подавление симптомов болезни. Однако могут использоваться и другие режимы дозировки. Успех лечения легко определяется обычными методами и анализами, например методами рентгено- визуализации опухолей. В соответствии с другим применением изобретения эффективность ингибитора в предотвращении или лечении болезней может быть улучшена путем введения ингибитора серийно или же в комбинации с другим веществом, эффективным для данной цели, таким, например, как фактор некроза опухоли, интерфероны, интерлейкины; моноклональные антитела, "ловушки" факторов роста (traps) и другие ингибиторы, способные нейтрализовать или ингибировать активность фактора роста эндотелия сосудов и/или его рецепторов и/или фактора роста гепатоцитов и/или его рецепторов и/или эпидермального фактора роста и/или его рецепторов и/или фактора роста плаценты и/или его рецепторов и/или инсулиноподобного фактора роста и/или его рецепторов и/или фактора роста фибробластов и/или его рецепторов и/или mTOR и/или других внутриклеточных киназ, или одно или более обычных терапевтических веществ, таких как, например, алкилирующие соединения, антибиотики, антиметаболиты, антрациклины, винкаалкалоиды, эпиподофиллотоксины, другие цитостатики. Также возможно сочетание ингибиторов, описываемых в изобретении, с противовоспалительными препаратами, препаратами для лечения сердечно-сосудистой патологии. Подобные вещества могут присутствовать во вводимом составе или могут вводиться отдельно. Кроме того, ингибитор по способу может вводиться серийно или же в комбинации с радиологическим лечением.
Один ингибитор или более вводятся пациенту с опухолью в терапевтически эффективных дозах, определенных, например, при наблюдении некроза опухоли или ее метастатических фокусов, если они имеются. Такая терапия продолжается до тех пор, пока перестает наблюдаться дальнейшее улучшение, или клиническое обследование показывает, что опухоль или ее метастазы исчезли. При прогрессировании болезни вводится одно или несколько описанных выше веществ(о) или используются другие методы лечения. Поскольку эффективность дополнительных веществ будет варьировать, желательно сравнить их влияние на опухоль путем стандартного скрининга. Производится повторное введение ингибитора и дополнительного агента, пока не будет достигнут желаемый
клинический эффект. В альтернативном случае ингибитор(ы) вводятся совместно и, при желании, вместе с дополнительными веществами. При лечении ингибиторами других заболеваний, например, ревматоидных болезней, используются стандартные шкалы оценки терапевтического эффекта.
Ингибиторы, описываемые в изобретении, могут быть использованы с препаратами поддерживающей и сопроводительной терапии, например, с эритропоэтинами, препаратами, стимулирующими лейкопоэз или повышающими количество тромбоцитов и/или нейтрофилов, макрофагов, с нутритивной поддержкой, нестероидными противовоспалительными средствами, компонентами крови, кортикостероидами, золедроновой кислотой, антителами против RANKL, противорвотными препаратами и другими.
Ниже в качестве примеров представлены некоторые доказательства целесообразности использования ингибиторов, описываемых в изобретении. Нижеследующие примеры предлагаются только в качестве иллюстрации и не должны восприниматься как в чем- либо ограничивающие настоящее изобретение.
Пример 1 (Результаты исследования): нарушение взаимодействия ФРФР2 с FRS2 при добавлении ингибитора RPT835
Для оценки эффекта ингибитора RPT835, охарактеризованного в настоящем изобретении формулой II, на фосфорилирование FRS2 были использованы клетки рака желудка КАТО III, которые экспрессируют ФРФР2. Для стимуляции ФРФР к клеткам добавляли ФРФ-2 в концентрации 1 мг/мл и гепарин 10 мг/мл. RPT835 добавляли в различных концентрациях. Часть клеток осталась без добавления ингибитора, а часть клеток - без добавления как ингибитора, так и стимулирующих агентов (контрольные группы).
Для оценки уровня фосфорилирования ФРФР и FRS2 были использованы 2 стандартных методики:
- Вестерн-блоттинг с использованием ручной работы;
- автоматический капиллярный электрофорез и иммунодетекция с использованием автоматической системы Wes™ (ProteinSimple; SantaClara,CA).
Стимуляция клеток КАТО III фактором роста фибробластов приводила к достоверному повышению активности (фосфорилирования) ФРФР (в 4,3 раза), FRS2a (в 11,4 раза), р44/42(МАРК, в 12 раз) и АКТ (в 4 раза) по сравнению с нестимулированными клетками. Фосфорилирования белка mTOR и киназы p70S6 наблюдалось в меньшей степени (Фигура 1).
При добавлении к клеткам ингибитора RPT835 наблюдалось нарушение взаимодействия между ФРФР и FRS2, проявляющееся в значительном снижении фосфорилирования FRS2 (Фигура 2) без влияния на фосфорилирование общего ФРФР (Фигура 3). Более того, концентрация полумаксимального ингибирования (IC50) FRS2 при использовании ингибитора RPT835 составила менее 10 пМ.
Основываясь на полученных данных, можно сделать вывод, что ФРФ стимулирует весь сигнальный путь от рецептора ФРФ до внутриклеточных киназ, включая взаимодействие ФРФР и FRS2. Ингибитор RPT835 не влияет на активность ФРФР, но приводит к нарушению взаимодействия ФРФР и FRS2, что в свою очередь сказывается на подавлении активности FRS2 в наномолярной концентрации.
Пример 2 (Результаты исследования): ингибитор RPT835 не влияет на тирозинкиназную активность ФРФР
В предыдущем Примере уже было показано отсутствие влияния ингибитора RPT835 на фосфорилирование общего ФРФР. В настоящем исследовании, для того, чтобы опровергнуть механизм действия RPT835 как ингибитора тирозинкиназы, был оценен уровень общего ФРФР и фосфо-ФРФР до и после добавления RPT835.
Как и в Примере 1 , были использованы клетки рака желудка КАТО III, сильно экспрессирующие ФРФР2. Часть клеток стимулировали ФРФ-2 в концентрации 1 мг/мл и гепарин 10 мг/мл, другую часть клеток оставляли без стимуляции. К клеткам добавляли ингибитор RPT835 в дозе 1, 10, 100, 1000 пМ. Группа контроля оставалась без добавления RPT835.
Уровень фосфорилирования ФРФР оценивали с помощью стандартного Вестерн- блоттинга и автоматического капиллярного электрофореза с иммунодетекцией и использованием автоматической системы Wes™ (ProteinSimple; SantaClara,CA).
Ингибитор RPT835 не оказывал никакого действия на фосфорилирование ФРФР2, стимулированное ФРФ-2 (Фигура 4), то есть не проявлял ингибирующей активности на тирозинкиназу даже в максимальной концентрации.
Пример 3 (Результаты исследования): ингибитор RPT835 не препятствует связыванию ФРФ-2 с ФРФР2
Для того, чтобы оценить влияет ли RPT835 на связывание ФРФР2 и лиганда (ФРФ-
2), был проведен нерадиоактивный иммуноферментный анализ (non-radioactive enzyme linked FGF2 binding assay). В анализе для связывания ФРФ-2 использовался рекомбинантный химерный белок ФРФР2-Рс, который состоял из человеческого
экстрацеллюлярного домена ΦΡΦΡ2α (Шс) и Fc-фрагмента человеческого IgG). ΦΡΦΡ2α содержал все 3 иммуноглобулин-подобные домены и представлял собой домен Шс ФРФР2 с высокой аффинностью к ФРФ-2. Белком ФРФР2-Рс покрывали 96-луночный планшет, затем в лунки добавляли:
1) только ФРФ-2
2) ФРФ-2 + гепарин
3) ингибитор RPT835 в различных концентрациях + ФРФ-2 + гепарин
4) часть лунок оставалась без стимуляторов (ФРФ-2 + гепарин) и RPT835 (негативный контроль).
После инкубации был проведен иммуноферментный анализ с использованием соответствующих антител и последующим определением OD650 (оптическая плотность при длине волны 650 нм).
В первой группе без добавления ингибитора и гепарина было показано, что ФРФ-2 хорошо связывается с белком ФРФР2-Рс. Связывание ФРФ-2 с рецептором было дозо-зависимым. Во второй группе добавление гепарина улучшало связывание ФРФ-2 с рецептором. При этом для достижения той же силы связывания ФРФ-2 можно было использовать в меньших дозах, чем без гепарина. В группе негативного контроля сигнал был отрицательный (менее 1% от позитивного контроля). Результаты представлены на Фигуре 5. Они подтверждают значимость данного метода: ФРФ-2 связывается с ФРФР2-Рс в присутствии гепарина или без него.
Следующей задачей было оценить влияния ингибитора RPT835 на связывание ФРФ-2 и ФРФР2-Рс. RPT835 в концентрациях от 1 до 1000 пМ не влиял на связывания лиганда с рецептором (Фигура 6А). Чтобы исключить неспецифическое действие RPT835 на связывание, был проведен еще один иммуноферментный анализ, в котором лунки были покрыты иммобилизованным анти-ФРФ-2 антителом. К лункам добавлялся ФРФ-2 ± RPT835 в различных концентрациях. RPT835 в концентрациях от 1 до 1000 пМ не влиял на связывание ФРФ-2 с анти-ФРФ-2 антителом (Фигура 6В).
Обобщенные результаты исследования доказывают, что ингибитор RPT835, приведенный как пример в настоящем изобретении, не препятствует связыванию лиганда (то есть ФРФ-2) с его рецептором (то есть с ФРФР2).
Пример 4 (Результаты исследования): нарушение взаимодействия ФРФР с FRS2 при использовании ингибиторов, определенных формулами V, IX, XI
Ингибиторы V, IX, XI, определенные в настоящем изобретении соответствующими формулами V, IX, XI, также нарушают передачу сигнала от ФРФР к FRS2. Путем химического синтеза были получены указанные соединения. Клетки рака желудка Snul6, экспрессирующие ФРФР2, были культивированы по стандартной методике. Часть клеток была оставлена в качестве негативного контроля, к другой части был добавлен ФРФ-2 (позитивный контроль). Терапевтические группы включали: добавление ингибитора по формуле V и последующую стимуляцию ФРФ-2, добавление ингибитора по формуле IX и последующую стимуляцию ФРФ-2, добавление ингибитора по формуле XI и последующую стимуляцию ФРФ-2. Для оценки уровня фосфорилирования различных киназ использовали метод Вестерн-блоттинга.
Результаты: в клетках без стимуляции и без лечения уровень фосфорилирования ФРФР2, FRS2 были низкие (отсутствие фосфорилирования); клетки после стимуляции ФРФ-2 имели высокий уровень активности ФРФР2 и FRS2 (в 14 и 17 раз выше, чем в негативном контроле); в летках, к которым были добавлены ингибиторы, уровень активности ФРФР2 не менялся по сравнению со стимулированным ФРФ-2 контролем, при этом фосфорилирования FRS2 отмечено не было (во всех случаях ингибирующая концентрация IC50 менее 10 пМ). Кроме того, было показано, что ингибиторы не влияют на другие внутриклеточные киназы, в том числе Akt, Erk 1/2.
Также было подсчитано количество клеток. Клетки без добавления ингибиторов и ФРФ-2 пролиферировали слабо, в отличие от клеток, к которым был добавлен ФРФ-2. Количество клеток при добавлении ингибиторов было сравнимым с группой негативного контроля (р=0,5) и достоверно меньше, чем в группе со стимуляцией ФРФ-2 и без добавления ингибиторов (р<0,0001).
Таким образом, вещества, описанные в формулах V, IX, XI, являются селективными ингибиторами взаимодействия ФРФР2 и FRS2, что влияет на пролиферативную активность клеток опухоли, экспрессирующих ФРФР.
Пример 5 (Результаты исследования): оценка влияния ингибиторов, описываемых в изобретении, на различные внутриклеточные киназы
Для того, чтобы доказать избирательное действие ингибиторов только на передачу сигнала от ФРФР к FRS2 без влияния на внутриклеточные киназы, был оценен уровень фосфорилирования внутриклеточных киназ, а именно р44/42(МАРК), Akt, mTOR, p70S6.
Для оценки эффекта ингибитора RPT835, охарактеризованного в настоящем изобретении формулой II, на фосфорилирование перечисленных киназ были использованы клетки рака желудка КАТО III, которые экспрессируют ФРФР2. Для стимуляции ФРФР к клеткам
добавляли ФРФ-2 в концентрации 1 мг/мл и гепарин 10 мг/мл. RPT835 добавляли в различных концентрациях. Часть клеток осталась без добавления ингибитора, а часть клеток - без добавления как ингибитора, так и стимулирующих агентов (контрольные группы).
Для оценки уровня фосфорилирования были использованы 2 стандартных методики:
- Вестерн-блоттинг с использованием ручной работы;
- автоматический капиллярный электрофорез и иммунодетекция с использованием автоматической системы Wes™ (ProteinSimple; SantaClara,CA).
Стимуляция клеток КАТО III фактором роста фибробластов приводила к достоверному повышению активности (фосфорилирования) р44/42(МАРК, в 12 раз), АКТ (в 4 раза), по сравнению с нестимулированными клетками. Фосфорилирования белка mTOR и киназы p70S6 наблюдалось в меньшей степени.
При добавление ингибитора RPT835 активность перечисленных киназ не изменилась по сравнению с клетками, только стимулированными ФРФ-2 (без добавления RPT835). Уровни фосфорилирования представлены на Фигуре 7.
Следовательно, RPT835 не оказывает влияния на фосфорилирование других внутриклеточных киназ.
Пример 6 (Результаты исследования): оценка влияния ингибитора, определенного формулой VIII, на ФРФР1, ФРФР2, ФРФРЗ, VEGFR1 и внутриклеточные киназы
По методике, описанной выше, методом Вестерн-блоттинга, были проанализированы уровни фософорилирования рецепторов фактора роста фибробластов 1-3 типа, FRS2, а также уровни фософорилирования VEGFR1 и внутриклеточных киназ PDK, р44/42(МАРК), Akt, mTOR.
В исследовании использовались эндотелиальные клетки HUVEC, клетки рака человеческого почечноклеточного рака типа Caki-1, клетки рака желудка КАТО III. Все перечисленные клетки экспрессировали различные виды перечисленных рецепторов. Как и в Примере 5, часть клеток стимулировали ФРФ-2 и/или VEGF-A, часть клеток культивировалась с ингибитором, описанным формулой VIII, часть клеток оставалось без стимуляции и лечения.
ФРФ-2 сильно стимулировал фосфорилирование всех трех типов ФРФР, а также PI3K,
Akt и FRS2 по сравнению с нестимулированными клетками (Р<0,0001 для всех сравнений). VEGF-A не оказывал воздействия на ФРФР 1-3 типов и FRS2 (Р>0,3 для всех сравнений), но достоверно повышал уровень фосфорилирования VEGFR1, mTOR, РБК,
МАРК (Р<0,001 для всех сравнений). Ингибитор по формуле VIII достоверно снижал
фосфорилирование FRS2 (в 14 раз, Р=0,001) и PI3K (в 10 раз, Р=0,017) и слабо - ФРФР2 (в 1,5 раза) и ФРФРЗ (в 1 ,8 раза), VEGFR1 (в 2,3 раза). На остальные киназы ингибитор не оказывал влияния. При этом уровень фосфорилирования ФРФР1 даже повысился (Фигура 8).
Следовательно, в настоящем исследовании продемонстрировано возможное влияние некоторых ингибиторов, нарушающих взаимодействие ФРФР2 и FRS2, на другие киназы и рецепторы. Это действие нельзя отрицать, и оно может быть связано с основным механизмом - нарушением связи ФРФР и FRS2, что приводит к поэтапному угнетению фосфорилирования нижележащих киназ. Действие на другие рецепторы (например, VEGFR1) не является выраженным, но также возможно как перекрестное. Повышение уровня фосфорилирования ФРФР1 может быть использовано также в дальнейшем как терапевтическое при использовании ингибиторов, описанных в настоящем изобретении.
Пример 7 (Результаты исследования): описываемые в изобретении ингибиторы могут нарушать комплекс FRS2 с Shp2 и/или Grb2
Известно, что FRS2 взаимодействует с Shp2 и Grb2, передавая через эти белки сигнал к внутриклеточным киназам. Целью настоящего исследования было доказать, что описываемые в изобретении ингибиторы могут влиять на взаимодействие FRS2 с Shp2 и Grb2, но это не является определяющим их механизмом действия: главным механизмом является нарушение взаимодействия ФРФР и FRS2.
Клетки рака желудка Snul6 были высеяны в 96-луночных пластинках, объемом 90 мкл/лунка. После 24 часов инкубации в увлажненном инкубаторе при температуре 37С, 5% С02 и 95% воздуха к клеткам были тестируемый растворенный агент, описанный формулой VI, в количестве 100 мкг/мл и затем добавлен ФРФ-2 в качестве стимулятора. После суток культивации методом автоматизированного Вестерн-блоттинга в клетках был изучен уровень фосфорилирования следующих белков: ФРФР2, FRS2a, FRS2 , Shp2, Grb2. Еще через сутки культивирования клетки были подсчитаны.
Ингибитор по формуле VI не угнетал фосфорилирования ФРФР2 (различий с контролем выявлено не было), однако, достоверно, в 12 раз, подавлял фосфорилирование FRS2a и FRS2P (1С50<10 пМ). Кроме того, в некоторых клетках было отмечено достоверное угнетение фософорилирования белков Shp2 и Grb2 (более чем в 10 раз). В связи с этим клетки были разделены на две группы по фактору фосфорилирования Shp2 и/или Grb2:
- группа с угнетением фосфорилирования FRS2 и Shp2 и/или Grb2
- группа с угнетением фосфорилирования FRS2 и без влияния на фосфорилирование Shp2 и/или Grb2.
После последующей культивации количество клеток сравнивалось между группами, а также с контролем (стимуляция ФРФ-2, без культивации с ингибитором). В таблице 1 представлены результаты. Как видно из таблицы, ингибитор достоверно угнетал пролиферацию опухолевых клеток по сравнению с контролем вне зависимости от фосфорилирования Shp2 и/или Grb2.
Следовательно, определяющих фактором, оказывающим ингибирующий эффект на пролиферацию опухолевых клеток, является нарушение взаимодействия ФРФР и FRS2, при этом уровень фосфорилирования Shp2 и/или Grb2 может быть разным, что не влияет на подавление пролиферативной активности ингибитором, описанным в изобретении.
Пример 8 (Результаты исследования): селективное нарушение взаимодействия ФРФР 2 с FRS2 при добавлении ингибитора RPT835
Целью настоящего исследования была оценка специфического действия ингибитора RPT835, обозначенного в настоящем изобретении формулой II, на взаимодействие ФРФР2 и FRS2 и оценка влияния на взаимодействие других типов ФРФР и FRS2. Для эксперимента были выбраны 2 клеточные линии:
- рака желудка КАТО III, экспрессирующая ФРФР2, и
- рака молочной железы MDA MB 134, экспрессирующая ФРФР1.
Методика исследования была такой же, как в предыдущих примерах: клетки культивировались в присутствии RPT835 с последующей стимуляцией ФРФ-2 и гепарином. Другая часть клеток культивировалась в отсутствие RPT835. Фосфорилирование ФРФР1 (в клетках рака молочной железы) и ФРФР2 (в клетках рака желудка), а также FRS2a (в обеих клеточных линиях) и некоторых внутриклеточных киназ (в клетках рака молочной железы) оценивалась с помощью ручного и автоматизированного Вестерн-блоттинга.
В клетках КАТО III ингибитор RPT835 полностью снижал фосфорилирование FRS2a (система ФРФР2) до исходного уровня в дозе 100 пМ. Угнетение фосфорилирования FRS2a более чем на 50% наблюдалось в концентрации 10 пМ (Фигура 2). При этом влияния на киназную активность самого рецептора отмечено не было (Фигура 3).
В клетках рака молочной железы MDA MB 134 ингибитор RPT835 не оказывал влияния на фосфорилирование FRS2a (система ФРФР1), равно как и на сам ФРФР1, а также на внутриклеточные киназы (Фигура 9). Как видно из рисунка, отмечено некоторое недостоверное повышение уровня фосфорилирования для некоторых внутриклеточных киназ и ФРФР 1.
Таким образом, настоящий пример подтверждает селективное действие ингибитора RPT835 и его активность в системе ФРФР2. Ингибитор RPT835 влияет только на нарушение взаимодействия ФРФР2 с FRS2 и не влияет на взаимодействие других типов ФРФР с FRS2.
Пример 9 (Результаты исследования): ингибиторы, описанные в настоящем изобретении, проявляют активность вне зависимости от изоформы ФРФР
Известно, что появление различных изоформ ФРФР может влиять на терапевтическую активность препаратов, блокирующих эти рецепторы. Задачей настоящего исследования было доказать, что ингибиторы, описываемые в настоящем изобретении, проявляют активность в отношении любых изоформ в пределах одного типа ФРФР.
В исследовании оценивалось влияние ингибитора RPT835, определенного формулой II настоящего изобретения, на нарушение взаимодействия ФРФР2, представленного двумя наиболее часто встречающимися изоформами ШЬ и Шс, с FRS2. Для этого были выбраны человеческая клеточная линия эмбриональных остеобластов (hFOB), экспрессирующая только ФРФР2 Шс, и человеческая клеточная линия рака молочной железы SUM-52, экспрессирующая ФРФР2 ШЬ (все основные варианты изоформы - CI, С2, СЗ).
Клетки были разделены на группы:
la) hFOB
lb) hFOB, стимуляция ФРФ-1 (100 нг/мл)
lc) hFOB, культивирование с RPT835 (1 мкМ) и стимуляция ФРФ-1 (100 нг/мл)
2а) SUM-52
2b) SUM-52, стимуляция ФРФ-1 (100 нг/мл)
2с) SUM-52, культивирование с RPT835 (1 мкМ) и стимуляция ФРФ-1 (100 нг/мл)
Ингибитор RPT835 достоверно подавлял пролиферацию клеток обеих линий, экспрессирующих ФРФР2 Шс или ФРФР2 ШЬ (Фигура 10). При оценке уровня фосфорилирования FRS2a значимых отличий между этими группами также не было - ингибитор полностью угнетал фосфорилирование (Фигура 11).
Следовательно, ингибитор RPT835, описываемый в настоящем изобретении, проявляет активность в отношении любых изоформ в пределах одного типа ФРФР.
Пример 10 (Результаты исследования): антиангиогенная активность ингибиторов, описываемых в настоящем изобретении
В исследовании использованы 18 мышей гибридов 1 поколения Fl (С57В1/6 х DBA/2), самцов массой 22-30 г. Мыши (номенклатурный номер м02.13.0001 1) были получены из
филиала питомника «Столбовая». В эксперименте использовали методику, описанную ранее [Passaniti А., 1992]. В контрольных и опытных группах было не менее 3 мышей. Аликвоты (1000 мкл) Матригеля (BD Biosciences), содержащие гепарин 60 У.Е./мл, VEGF (200 ng/ml, BD Biosciences) или bFGF (100 ng/ml, BD Biosciences), готовили на льду. В качестве отрицательного контроля использовали введение Матригеля, содержащего гепарин 60 У.Е./мл. Инъекцию Матригеля проводили мышам подкожно, в боковую часть спины. После инъекции Матригель быстро образует одиночный, твердый гелеобразный имплантат. Навеску ингибитора растворяли в ДМСО до концентрации 10" М. Аликвоты замораживали при -20°С и размораживали непосредственно перед введением. Повторного замораживания раствора не допускали. Испытуемый и стандартный объекты вводили внутрибрюшинно на 0, 3 и 6 день имплантации Матригеля: Бевацизумаб в дозе 10 мг/кг и RPT835 - 15 мг/кг. Описание групп представлено в Таблице 2. Через 7 дней после введения Матригеля мышей усыпляли медицинским эфиром для удаления имплантата. Имплантаты фиксировали в течение 24-36 часов в 10% нейтральном формалине и заключали в парафин для дальнейшего гистологического исследования.
Подкожная инъекция 1000 мкл Матригеля, не содержащего ростовые факторы, не индуцировала формирования микрокрососудов и миграцию стромальных и эндотелиальных клеток в имплантат.
В инъекции Матригеля, содержащего 100 нг/мл bFGF, наблюдали значительное увеличение количества микрососудов, тубулярных структур и мигрировавших клеток в целом (для всех показателей р<0,001). Аналогичный эффект показан при введении 200 нг/мл VEGF (для всех показателей р<0,001).
Наблюдали значительное снижение количества мигрировавших клеток, тубулярных структур и функциональных микрососудов при введении субстанций, блокирующих активность соответствующего стимулятора. Введение Бевацизумаба полностью блокировало ангиогенез в имплантатах, содержащих VEGF (для всех оцениваемых параметров р<0,001), но не ангиогенез, индуцированный bFGF (для всех оцениваемых параметров р>0,05). Ингибитор RPT835 эффективно ингибировал ангиогенез, стимулированный bFGF. Количество мигрировавших клеток (р<0,001); тубулярных структур (р=0,016) и функциональных микрососудов (р<0,001) снижалось по сравнению с контрольной группой более чем в 2 раза. Результаты исследования представлены на Фигурах 12 и 13.
Таким образом, в исследовании установлено, что при трехкратном введении в дозе 15 мг/кг ингибитор RPT835 статистически значимо блокирует ангиогенез, стимулированный
bFGF. Бевацизумаб в том же режиме применения был эффективен против VEGF- стимулированного ангиогенеза, но не при использовании bFGF.
Полученные данные свидетельствуют, что RPT835, как пример ингибиторов, описываемых в настоящем изобретении, является перспективным антиангиогенным веществом с самостоятельным механизмом действия.
Пример 11 (Результаты исследования): сравнение влияния ингибиторов, описываемых в настоящем изобретении, и типичного ингибитора тирозинкиназы ΦΡΦΡ/VEGFR - бриваниба на пролиферативную активность эндотелиоцитов
Эндотелиальные клетки HUVEC были высеяны в 96-луночных пластинках и культивированы в течение 24 часов. После этого к клеткам были добавлены:
1) ингибитор RPT835, или
2) ингибитор, описываемый формулой IX в настоящем изобретении, или
3) бриваниб - мультикиназный ингибитор ΦΡΦΡ/VEGFR, а также ко всем клеткам - ФРФ-2 в концентрации 25 нг/мл.
Клетки культивировались еще в целом 96 часов и затем были посчитаны.
Результаты пролиферации клеток представлены на Фигуре 14.
Ингибиторы, описываемые в настоящем изобретении (RPT835 и ингибитор по формуле IX), оказывали выраженное антипролиферативное действие на эндотелиальные клетки в отличие от типичного ингибитора тирозинкиназы ΦΡΦΡ/VEGFR бриваниба.
Пример 12 (Результаты исследования): влияние ингибитора RPT835 на пролиферацию опухолевых клеток с различной экспрессией ФРФР in vitro
Подавление пролиферативной активности опухолевых клеток при культивировании с ингибиторами, описываемыми в настоящем изобретении, уже приводилось в Примерах 4, 7, 9.
В данном исследовании были использованы дополнительные клеточные линии: SKOV3 (рак яичников, АТСС # НТВ-77), HS578T (рак молочной железы, АТСС # НТВ- 126), T47D (рак молочной железы, АТСС #НТВ-1336), модифицированная линия немелкоклеточного рака легкого А549, линия метастатической меланомы кожи Mel Ког (патент РФ N°2287578), и для дополнительного изучения антиангиогенной активности - клеточная линия SVEC-4-10 (эндотелиальные клетки, АТСС #CRL-2181).
Клетки культивировали в среде RPMI-1640 (ПАНЭКО) (SKOV3, HS578T, T47D, Mel-Kor) или DMEM (ПАНЭКО) (SVEC-4-10), содержащей 10% эмбриональной сыворотки крупного рогатого скота (FCS, HyClone), 2mM глутамина (ПАНЭКО), антибиотики (100
IU/mL пенициллина и 100 mg/ml стрептомицина (все ПАНЭКО)) при 37°С и 5% С02. Для экспериментов использовали клетки при 70-80% монослоя.
Клетки высаживали в низкой плотности (30т клеток/мл) в триплетах на 96-луночный планшет в среде, содержащей 0,1% сыворотки FCS. На следующий день в лунки добавляли тестируемый ингибитор RPT835 в необходимых концентрациях, а через 6 часов - фактор роста фибробластов (100 нг/мл bFGF (BD Bioscience)). Среду, антибиотики и препараты меняли раз в 3 дня. Рост клеток определяли модифицированным «митогенным методом по Crystal Violet» на 7 сутки после начала опыта. Клетки промывали PBS, фиксировали 1% парафармальдегидом на PBS и окрашивали 0,5% раствором Кристаллического фиолетового (Crystal Violet, Sigma Chemical Co) на этаноле. Краситель растворяли в этаноле и измеряли на спектрофотометре при 540-560 нм. Кривую ингибирования пролиферативной активности клеток строили по зависимости процента количества клеток (ось ординат) от концентрации ингибитора RPT835 в лунке планшета (ось абсцисс). Концентрацию образцов, начиная с которой процент выживших клеток становился меньше 50% (1С50) вычисляли в программе GraphPad Prizm 5. Эксперименты повторяли не менее 3 раз по три повтора в опыте.
На Фигуре 15 представлено антипролиферативное действие соединения RPT835 на культурах опухолевых и эндотелиальных клеток in vitro.
Как видно из графика, ингибитор RPT835 угнетал антипролиферативную активность опухолевых и эндотелиальных клеток в различной степени. Степень ингибирования пролиферации зависела от статуса экспрессии ФРФР2: чем сильнее был экспрессирован рецептор, тем эффективнее оказался ингибитор RPT835. Так, максимальный ингибирующий эффект отмечен при использовании линии SVEC-4-10, которая сильно экспрессирует ФРФР2. Умеренный эффект на пролиферацию выявлен в клетках с низкой ФРФР2-экспрессией SKOV3 и HS578T. Исследуемый ингибитор RPT835 не блокировал пролиферативную активность клеточной линии метастатической меланомы Mel Ког и рака молочной железы T47D, которые практически не экспрессировали ФРФР2.
Наибольший ингибирующий эффект достигнут при культивировании ингибитора RPT835 с модифицированной линия немелкоклеточного рака легкого А549, которая имела выраженную экспрессию ФРФР2 (1С 50=10 nM/L).
Таким образом, в исследовании in vitro показано, что ингибитор RPT835 оказывает антипролиферативное действие на опухолевые и эндотелиальные клетки, которое коррелирует со степенью выраженности экспрессии ФРФР2. Это подчеркивает специфическую активность ингибитора.
Пример 13 (Результаты исследования): ингибирование роста опухоли in vivo в зависимости от уровня экспрессии ФРФР2 при использовании ингибитора RPT835
Самкам иммуно дефицитных мышей (nude) возрастом 8 недель были введены 2x106 опухолевых клеток человеческих клеточных линий с различной экспрессией ФРФР2:
- клеточная линия трижды-негативного рака молочной железы SUM52PE, сильно экспрессирующая ФРФР2 (приобретена в Asterand, Inc. (Detroit, MI)
- клеточная линия трижды-негативного рака молочной железы HS578T, слабо экспрессирующая ФРФР2 (приобретена в АТСС (CRL-125)
- клеточная линия немелкоклеточного рака легкого NCI-H226, не экспрессирующая ФРФР2 (приобретена в АТСС (CRL-5826)
После того, как установился рост опухоли (1 мм ), мыши были разделены на контрольные и лечебные группы в соответствии с видом опухоли.
Контрольная группа 1 (N=10): SUM52PE, введение воды через желудочный зонд, ежедневно
Лечебная группа 1 (N=10): SUM52PE, введение ингибитора RPT835 через желудочный зонд в дозе 30 мг/кг, ежедневно
Контрольная группа 2 (N=10): HS578T, введение воды через желудочный зонд, ежедневно Лечебная группа 2 (Ν=10): HS578T, введение ингибитора RPT835 через желудочный зонд в дозе 30 мг/кг, ежедневно
Контрольная группа 3 (N=10): NCI-H226, введение воды через желудочный зонд, ежедневно
Лечебная группа 3 (N=10): NCI-H226, введение ингибитора RPT835 через желудочный зонд в дозе 30 мг/кг, ежедневно.
Размер опухоли и вес животных измеряли каждые 3 дня. Мыши подвергались эвтаназии при достижении опухоли размера 2000 мм3 или на 60 день эксперимента.
График роста опухоли в группах 1 представлен на Фигуре 16, объем опухоли - в таблице 3. У тех мышей, которым вводился ингибитор RPT835, отмечено достоверное торможение агрессивного роста опухоли SUM52PE (ΡΟ,ΟΟΟ, начиная с 16 дня эксперимента). На 31 день средний объем опухоли в группах отличался более, чем в 2 раза и составил в контрольной группе 2712.2 ± 37 ммЗ, в лечебной - 1080.7 ± 49 ммЗ. Торможение опухолевого роста было 60,2%. Согласно правилам исследования (объем опухоли >2000 ммЗ), мыши из контрольной группы были подвергнуты эвтаназии. Наблюдение за мышами из лечебной группы было продолжено. В группе выявлена стабилизация болезни (отсутствие опухолевого роста в течение последующего периода наблюдения), что расценивается как положительный эффект.
Следовательно, ингибитор RPT835 продемонстрировал противоопухолевую эффективность в эксперименте in vivo в отношении трижды-негативного рака молочной железы SUM52PE, сильно экспрессирующего ФРФР2.
График роста опухоли в группах 2 представлен на Фигуре 17.
Как видно из рисунка, ингибитор RPT835 также оказывал некоторое угнетение роста опухоли трижды-негативного рака молочной железы HS578T, слабо экспрессирующего ФРФР2. Однако, различия были менее значимыми, чем при использовании ингибитора RPT835 в модели трижды-негативного рака молочной железы с сильной экспрессией ФРФР2. Так, средний объем опухоли в лечебной и контрольной группах 2 на 31 день исследования составил 703±89.1 ммЗ и 1053±179.8 ммЗ, а на 40 день - 1104±162.2 ммЗ и 1592±335 ммЗ (Р=0.01), соответственно. Торможение роста опухоли на 31 и 40 дни было 33,2% и 30,6%.
График роста опухоли в группах 3 представлен на Фигуре 18.
На 31 день эксперимента достоверных отличий в росте немелкоклеточного рака легкого NCI-H226, не экспрессирующего ФРФР2, между контрольной и лечебной группами 3 выявлено не было: средний объем опухоли составил 1114±280.6 ммЗ и 1053±259.7 ммЗ, соответственно (Р=0.619).
По результатам исследования in vivo были сделаны основные выводы: 1) ингибитор RPT835 оказывает выраженное противоопухолевое действие; 2) Степень торможения роста опухоли при использовании ингибитора RPT835 зависит от уровня экспрессии ФРФР2.
Пример 14 (Результаты исследования in vivo): ингибитор RPT835 вызывает лечебный патоморфоз некоторых опухолей
Иммунодефицитным мышам (Nude, возраст мышей на момент начала лечения 8 недель; питомник «Пущино») были подкожно перевиты клетки ФРФР-2 экспрессирующей человеческой линии рака яичников SKOV-3. При достижении среднего объема опухоли в 200 ммЗ мыши были рандомизированы в группы контроля (стандартная химиотерапия паклитакселом и кароплатином) и лечения (ингибитор RPT835). В каждой группе было 10 мышей. Ингибитор RPT835 назначался в дозе 50 мг/кг, ежедневно. Измерение объема опухоли проводилась дважды в неделю. При достижении объема опухоли 2000 ммЗ мыши подвергались эвтаназии. Проводилось изучение патоморфоза опухолевой ткани.
В группе контроля патоморфоз выявлен не был (Фигура 19). Опухолевая ткань была без изменений.
В лечебной группе при применении ингибитора RPT835 наблюдался выраженный терапевтический патоморфоз как макроскопически, так и микроскопически (Фигура 20). В опухоли были выявлены обширные некрозы (более 75% опухоли представлены массивными некрозами, живые опухолевые клетки сохранились только по периферии опухоли). Также отмечалось раннее повреждение сосудов внутри пока еще жизнеспособной ткани. В опухолевых клетках наблюдалась дистрофия.
Таким образом, был сделан вывод, что ингибитор RPT835 вызывает лечебный патоморфоз некоторых опухолей, в частности, рака яичников.
Пример 15 (Результаты исследования in vivo): эффективность ингибиторов, описываемых в настоящем изобретении, при ревматических болезнях
В исследовании была использована модель коллаген-индуцированного артрита (Collagen- Induced Arthritis). Артрит у мышей вызывался внутридерамальными инъекциями коллагена II типа, комбинированного с адъювантом Фрейнда. Мыши были рандомизированы в 2 группы по 5 животных в каждой:
- группа 1 : контроль, вода через зонд, в течение 40 дней
- группа 2: ингибитор, описанный формулой III в настоящем изобретении, 100 мг/кг, через зонд, в течение 40 дней
На 41-й день мыши подвергались эвтаназии, ткань изучалась.
В контрольной группе был выявлен синовиит, разрушение хряща, массивная периартикулярная инфильтрация ткани Т- и В- лимфоцитами, нейтрофилами.
В лечебной группе была выявлена только незначительная инфильтрация единичными нейтрофилами.
Таким образом, ингибиторы, описываемые в изобретении, могут быть эффективны при ревматических болезнях и болезнях соединительной ткани.
Пример 16 (Результаты исследования in vivo): ингибиторы, описываемые в настоящем изобретении, не сопровождаются развитием выраженной токсичности
Токсичность ингибитора RPT835 была оценена в исследованиях in vivo, описанных в Примере 13. Согласно стандартной методике, оценивались все нежелательные явления, равно как и потеря веса животных при использовании ингибитора в сравнении с группой контроля. Ни потери веса, ни каких-либо нежелательных побочных эффектов терапии в этих исследованиях выявлено не было.
Кроме того, в отдельном исследовании проводилось изучение острой токсичности ингибитора RPT835B следующем объеме:
- исследована острая токсичность препарата. Результаты токсикометрии, данные наблюдений за экспериментальными животными на протяжении 14 дней после острого введения, а также данные некропсии позволяют отнести препарат к IV классу малотоксичных (Н. Hodge et al. Clinical Toxicology of Commercial Products. Acute Poisoning. Ed. IV, Baltimore, 1975, 427 p.; K.K. Сидоров, 1977). Состояние животных после острого введения свидетельствует о хорошей переносимости препарата в дозах, превышающих максимальные терапевтические (порядка 4,3 мкг/кг) в десятки - сотни раз. Среднесмертельная доза (ЛД50) составила более 2000,0 мг/кг;
- по результатам макро и микроскопии препарат не обладает раздражающим действием на слизистую желудочно-кишечного тракта при однократном введении;
- по результатам исследования мутагенного действия (хромосомные аберрации) препарат не обладает мутагенными свойствами;
- препарат не проявляет аллергенных свойств, выявляемых в реакции иммунных комплексов, общей анафилаксии, дегрануляции тучных клеток и конъюнктивальной пробе.
По результатам этих исследований был сделан вывод об отсутствии выраженной токсичности при применении ингибитора RPT835, как примера ингибиторов, описываемых в настоящем изобретении.