WO2015178481A1 - 吸水性樹脂粒子、これを含む吸収体および吸収性物品 - Google Patents
吸水性樹脂粒子、これを含む吸収体および吸収性物品 Download PDFInfo
- Publication number
- WO2015178481A1 WO2015178481A1 PCT/JP2015/064741 JP2015064741W WO2015178481A1 WO 2015178481 A1 WO2015178481 A1 WO 2015178481A1 JP 2015064741 W JP2015064741 W JP 2015064741W WO 2015178481 A1 WO2015178481 A1 WO 2015178481A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- group
- resin particles
- absorbent
- general formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
- C08J3/126—Polymer particles coated by polymer, e.g. core shell structures
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/58—Adhesives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/45—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the shape
- A61F13/49—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the shape specially adapted to be worn around the waist, e.g. diapers, nappies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/46—Reaction with unsaturated dicarboxylic acids or anhydrides thereof, e.g. maleinisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
- C08G81/02—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers at least one of the polymers being obtained by reactions involving only carbon-to-carbon unsaturated bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/12—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity
- C08L101/14—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity the macromolecular compounds being water soluble or water swellable, e.g. aqueous gels
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/21—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/263—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds of unsaturated carboxylic acids; Salts or esters thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/507—Polyesters
- D06M15/51—Unsaturated polymerisable polyesters
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/59—Polyamides; Polyimides
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M23/00—Treatment of fibres, threads, yarns, fabrics or fibrous goods made from such materials, characterised by the process
- D06M23/08—Processes in which the treating agent is applied in powder or granular form
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/14—Water soluble or water swellable polymers, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2453/00—Characterised by the use of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/53—Core-shell polymer
Definitions
- the present invention relates to a water absorbent resin particle, an absorbent body using the same, and an absorbent article.
- Absorbent articles paper diapers, etc.
- water-absorbing resin particles are usually produced by fixing water-absorbing resin evenly on a matrix made of cellulose fibers and including organic synthetic fibers such as nonwoven fabric.
- the water-absorbent resin particles easily fall off and move due to vibration, and cause a bias in the absorbent body.
- efficient diffusion of the absorbed material such as urine is hindered, causing problems such as urine leakage and fogging.
- a heat-fusible resin such as a polyolefin resin such as polyethylene wax, a polyolefin derivative modified with an acid anhydride, a polyester resin, a polyamide resin, or a polystyrene resin is coated on the surface of the water-absorbent resin particles in advance.
- a method of fixing water absorbent resin particles to a fibrous base material by mixing with water absorbent resin particles and heat-sealing is known.
- the above-mentioned heat-fusible resin has high affinity for either the hydrophilic water-absorbent resin particles having a polar group or the hydrophobic fibrous base material, but is not sufficiently amphiphilic.
- the water absorption which is the original characteristic of the water absorbent resin particles, is likely to be greatly impaired, which is not preferable.
- An object of the present invention is to provide water-absorbent resin particles that can obtain sufficient fixing properties without impairing the water-absorbing property that is the original property of the water-absorbent resin particles, and that can produce an absorber that does not cause the above-described problems.
- Another object of the present invention is to provide an absorbent body in which the water absorbent resin particles and a fibrous base material are fixed, and an absorbent article using the absorbent body.
- the present invention is a core-shell type water absorbent resin particle composed of a core layer (P) and a shell layer (Q), wherein (P) contains the water absorbent resin (A), Water-absorbent resin particles, wherein Q) contains a thermoplastic resin (B) having a melting point of 50 to 180 ° C., and the thermoplastic resin (B) is a polymer having a hydrophobic block containing polyolefin;
- P contains the water absorbent resin (A)
- Q) contains a thermoplastic resin (B) having a melting point of 50 to 180 ° C.
- the thermoplastic resin (B) is a polymer having a hydrophobic block containing polyolefin
- the absorbent body formed by adhering the absorbent resin particles of the present invention to the fibrous base material has excellent adhesion to the fibrous base material while maintaining excellent water absorption performance, and excellent shape retention performance even after swelling. Show.
- the core layer (P) contains the water-absorbent resin (A).
- the said core layer (P) may be substantially comprised from the water absorbing resin (A), it can also contain the other additive for resin mentioned later by a well-known compounding quantity.
- the water absorbent resin (A) is not particularly limited.
- the water absorbent resin (A) is usually a hydrophilic cross-linked polymer capable of absorbing water of 30 to 1000 times its own weight, and has a carboxylic acid as a constituent unit.
- (Salt) group refers to a carboxylic acid and / or a carboxylate group.
- Salt refers to a carboxylic acid and / or a carboxylate group.
- a water-absorbing resin having a hydrophilic group such as a sulfonic acid (salt) group, a phosphoric acid (salt) group, a tertiary amino group, a quaternary ammonium base, a hydroxyl group or a polyethylene oxide group.
- the type and production method are not particularly limited.
- Examples of the water-absorbent resin (A) that can be suitably used in the present invention include starch-acrylic acid (salt) copolymers described in JP-B-53-46199 and JP-B-53-46200, and the like.
- the absorption capacity of the water-absorbent resin (A) for physiological saline (0.9% sodium chloride aqueous solution) is preferably 30 times or more of its own weight, more preferably 35 to 100 times, still more preferably 40 to 80. Is double.
- the shell layer (Q) layer comprises a thermoplastic resin (B) having a melting point of 50 to 180 ° C. (hereinafter referred to as thermoplastic resin (B) or simply (B) Also called).
- the melting point of (B) is preferably 60 to 160 ° C. When the melting point is less than 50 ° C., a blocking problem occurs during storage or use.
- the shell layer (Q) layer may be substantially composed of the thermoplastic resin (B), but may also contain other resin additives described later in known amounts. However, when using the low molecular weight polyolefin (H) and / or the plasticizer (I) described later, (Q) also includes these components.
- thermoplastic resin (B) is a polymer having a hydrophobic block containing polyolefin, and for example, a block copolymer having a hydrophobic block containing polyolefin can be used.
- thermoplastic resin constituting the polymer has a melting point of 50 to 180 ° C., for example, polyolefin resin (polyethylene, polypropylene, low molecular weight polyethylene, low molecular weight polypropylene, etc.), polyolefin derivative (maleic acid modified polyethylene, chlorinated polyethylene).
- polyolefin resin polyethylene, polypropylene, low molecular weight polyethylene, low molecular weight polypropylene, etc.
- polyolefin derivative maleic acid modified polyethylene, chlorinated polyethylene
- polyester resins Maleic acid-modified polypropylene
- polyester resins polyamide resins, polycaprolactone resins, polystyrene resins and derivatives thereof (polystyrene, sulfonated polystyrene, styrene-maleic anhydride copolymers, etc.), thermoplastic polyurethane resins, high molecular weight Examples include polyethylene glycol, vinyl acetate resin, waxes (paraffin wax, beeswax, beef tallow, etc.), long chain fatty acid ester resins, and mixtures of two or more thereof.
- resins other than polyolefin and its derivatives can constitute portions other than the hydrophobic block containing polyolefin.
- polyolefin constituting the hydrophobic block of the thermoplastic resin (B) (hereinafter also referred to as polyolefin (a)) is preferably a polymer such as a carbonyl group (preferably a carboxyl group, the same shall apply hereinafter).
- Polyolefin (a4), polyolefin (a5) having a hydroxyl group at one end of the polymer, and polyolefin (a6) having an amino group at one end of the polymer can be used.
- polyolefins (a1) and (a4) having a carbonyl group are more preferable because of easy modification.
- both polyolefins (a0) having a polyolefin whose both ends can be modified as a main component are used. Examples thereof include those having a carbonyl group introduced at the terminal.
- (A0) is usually a mixture of a polyolefin that can be modified at both ends, a polyolefin that can be modified at one end, and a polyolefin that does not have a terminal group that can be modified. Those are preferred.
- (A0) is obtained by (co) polymerization (meaning polymerization or copolymerization; the same applies hereinafter) of one or a mixture of two or more olefins having 2 to 30 carbon atoms (hereinafter abbreviated as C).
- Polyolefin [polymerization method] and low molecular weight polyolefin [thermal degradation method] obtained by thermal degradation of high molecular weight polyolefin (polyolefin obtained by polymerization of C2-30 olefin) can be used.
- C2-30 olefins include ethylene, propylene, C4-30 (preferably 4-12, more preferably 4-10) ⁇ -olefins, and C4-30 (preferably 4-18, more preferably 4-4).
- dienes of 8 include 1-butene, 4-methyl-1-pentene, 1-pentene, 1-octene, 1-decene and 1-dodecene, and the diene includes butadiene, isoprene, 1,4- Examples include pentadiene, 1,6-hexadiene, cyclopentadiene, and 1,11-dodecadiene.
- C2-12 ethylene, propylene, C4-12 ⁇ -olefin, butadiene and / or isoprene, etc.
- C2-10 ethylene, propylene, C4-10 ⁇ -olefin is more preferable
- / or butadiene particularly preferred are ethylene, propylene and / or butadiene.
- the low molecular weight polyolefin obtained by the thermal degradation method can be easily obtained by, for example, the method described in JP-A-3-62804.
- the polyolefin obtained by the polymerization method can be produced by a known method or the like. For example, it can be easily produced by (co) polymerizing the olefin in the presence of a radical catalyst, a metal oxide catalyst, a Ziegler catalyst, a Ziegler-Natta catalyst, or the like. Obtainable.
- the thermal degradation method is preferred in terms of ease of introduction of a carbonyl group, which is a modifying group, and availability.
- the number average molecular weight (hereinafter abbreviated as Mn) of (a0) by gel permeation chromatography (GPC) is preferably 800 to 20,000, more preferably 1,000 to 10,000, and particularly preferably 1. , 200 to 6,000.
- the amount of double bonds in (a0) is preferably 1 to 40, more preferably 2 to 30, and particularly preferably 4 to 20 per 1,000 C from the viewpoint of compatibility.
- the average number of double bonds per molecule is preferably from 1.1 to 5, more preferably from 1.3 to 3, particularly preferably from 1.5 to 5 from the viewpoints of repetitive structure formation and compatibility. 2.5, most preferably 1.8 to 2.2.
- a low-molecular-weight polyolefin having an Mn in the range of 800 to 6,000 and an average number of terminal double bonds per molecule of 1.5 to 2 can be easily obtained [for example, Katsuhide Murata, See Tadahiko Makino, Journal of the Chemical Society of Japan, page 192 (1975)].
- both ends of (a0) are ⁇ , ⁇ -unsaturated carboxylic acid (anhydride) ( ⁇ , ⁇ -unsaturated carboxylic acid, its C1-4 Means an alkyl ester or an anhydride thereof.
- the Mn of the polyolefin (a1) having carbonyl groups at both ends of the polymer is preferably 800 to 25,000, more preferably 1,000 from the viewpoint of heat resistance and reactivity with the hydrophilic polymer (b) described later. 20,000 to 20,000, particularly preferably 2,500 to 10,000.
- the acid value of (a1) is preferably 4 to 280 (mg KOH / g, hereinafter, only numerical values are described) from the viewpoint of reactivity with (b), more preferably 4 to 100, particularly preferably. Is 5-50.
- (A11) can be obtained by modifying (a0) with an ⁇ , ⁇ -unsaturated carboxylic acid (anhydride).
- the ⁇ , ⁇ -unsaturated carboxylic acid (anhydride) is preferably a dicarboxylic acid, an alkyl ester thereof or an anhydride thereof, more preferably maleic acid (anhydride), from the viewpoint of reactivity with (a0).
- (A12) can be obtained by secondary modification of (a11) with lactam or aminocarboxylic acid.
- lactam and aminocarboxylic acid As the lactam and aminocarboxylic acid, caprolactam, laurolactam, glycine, leucine, ⁇ -aminocaprylic acid, 11-aminoundecanoic acid and 12-aminododecanoic acid are more preferable from the viewpoint of the reactivity of secondary modification.
- caprolactam, laurolactam, ⁇ -aminocaprylic acid, 11-aminoundecanoic acid, 12-aminododecanoic acid particularly preferably caprolactam and 12-aminododecanoic acid.
- (A13) can be obtained by oxidizing (a0) with oxygen and / or ozone or hydroformylating with an oxo method to introduce a carbonyl group. These methods are known and can be applied as appropriate.
- (A14) can be obtained by secondary modification of (a13) with lactam or aminocarboxylic acid.
- lactam and aminocarboxylic acid include those exemplified in (a12).
- the polymer in the thermoplastic resin (B) is not particularly limited as long as it has a hydrophobic block containing polyolefin.
- it may have a hydrophilic block, or may have a polymer chain other than polyolefin.
- the bonding mode between the hydrophilic block or the polymer chain other than polyolefin and the hydrophobic block containing polyolefin may be any of block, random, graft, or a mixture thereof.
- at least one selected from the group consisting of an ester bond, an amide bond, an ether bond and an imide bond for example, the hydrophobic block of the polyolefin (a) and the block of the hydrophilic polymer (b) described in detail below.
- Preferred is an alternating block copolymer (G) bonded via a.
- hydrophilic polymer (b) for example, polyether diol (b1) and polyether diamine (b2) can be used.
- the polyether diol (b1) has a structure obtained by addition reaction of an alkylene oxide (hereinafter abbreviated as AO) (C2-12) to the diol (b01) or the dihydric phenol (b02), for example, And those represented by the formula: H (OA 1 ) mO—E 1 —O (A 1 O) m′H.
- AO alkylene oxide
- E 1 represents a residue obtained by removing a hydroxyl group from (b01) or (b02)
- a 1 represents C2 to 12 (preferably 2 to 8, more preferably 2) which may contain a halogen atom.
- M) and m ′ represent an integer of 1 to 300, preferably 2 to 250, more preferably 5 to 200, particularly preferably 8 to 150, and most preferably 10 to 100, m and m ' May be the same or different. Further, m (OA 1 ) and m ′ (A 1 O) may be the same or different from each other, and the bond form in the case where these are each composed of two or more oxyalkylene groups. May be block, random or a combination thereof.
- Diol (b01) includes C2-12 (preferably 2-10, more preferably 2-8) dihydric alcohol (aliphatic, alicyclic and araliphatic dihydric alcohol) and C1-12 tertiary. Examples thereof include amino group-containing diols. Examples of the aliphatic dihydric alcohol include ethylene glycol, propylene glycol, 1,4-butanediol, 1,6-hexanediol, neopentyl glycol and 1,12-dodecanediol.
- Examples of the alicyclic dihydric alcohol include 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, 1,4-cyclooctanediol, and 1,3-cyclopentanediol.
- Examples of the araliphatic dihydric alcohol include xylylenediol, 1-phenyl-1,2-ethanediol and 1,4-bis (hydroxyethyl) benzene.
- Tertiary amino group-containing diols include aliphatic or alicyclic primary monoamines (C1-12, preferably 2-10, more preferably 2-8) bishydroxyalkyls (alkyl groups C1-12, preferably 2-10, more preferably 2-8) and bishydroxyalkyl (alkyl group C1-12) compounds of aromatic (aliphatic) primary monoamines (C6-12).
- Aliphatic primary monoamines include methylamine, ethylamine, 1- and 2-propylamine, n- and i-amylamine, hexylamine, 1,3-dimethylbutylamine, 3,3-dimethylbutylamine, 2- and 3- Examples include aminoheptane, heptylamine, nonylamine, decylamine, undecylamine, and dodecylamine.
- Examples of the alicyclic primary monoamine include cyclopropylamine, cyclopentylamine, and cyclohexylamine.
- aromatic (aliphatic) primary monoamine include aniline and benzylamine.
- dihydric phenol (b02) examples include C6-18 (preferably 8-18, more preferably 10-15), such as monocyclic dihydric phenol (hydroquinone, catechol, resorcin, urushiol, etc.), bisphenol (bisphenol A, Bisphenol F, bisphenol S, 4,4′-dihydroxydiphenyl-2,2-butane, dihydrokibiphenyl, etc.) and condensed polycyclic dihydric phenols (dihydroxynaphthalene, binaphthol, etc.).
- monocyclic dihydric phenol hydroquinone, catechol, resorcin, urushiol, etc.
- bisphenol bisphenol
- bisphenol bisphenol
- bisphenol F bisphenol F
- bisphenol S 4,4′-dihydroxydiphenyl-2,2-butane, dihydrokibiphenyl, etc.
- condensed polycyclic dihydric phenols dihydroxynaphthalene, binaphthol,
- dihydric alcohols and dihydric phenols preferred are dihydric alcohols and dihydric phenols, more preferred are aliphatic dihydric alcohols and bisphenols, and particularly preferred are ethylene glycol and bisphenol A.
- AO to be subjected to addition reaction with diol (b01) or dihydric phenol (b02) includes C2-12 AO (ethylene oxide (hereinafter referred to as EO), propylene oxide (hereinafter referred to as PO), 1,2-, 1,4-, 2). , 3- and 1,3-butylene oxide and mixtures of two or more thereof), and other AOs and substituted AOs may be used in combination as necessary.
- C2-12 AO ethylene oxide (hereinafter referred to as EO), propylene oxide (hereinafter referred to as PO), 1,2-, 1,4-, 2). , 3- and 1,3-butylene oxide and mixtures of two or more thereof), and other AOs and substituted AOs may be used in combination as necessary.
- the number of moles of AO added is preferably 1 to 300 moles, more preferably 2 to 250 moles, more preferably 2 to 250 moles per hydroxyl group of (b01) or (b02), from the viewpoint of the volume resistivity value of the hydrophilic polymer (b).
- the amount is preferably 10 to 100 mol.
- the content of the oxyalkylene unit in the polyether diol (b1) is preferably 5 to 99.8% by weight based on the weight of (b1), from the viewpoint of the volume resistivity value of the hydrophilic polymer (b). More preferably, it is 8 to 99.6% by weight, and particularly preferably 10 to 98% by weight.
- the content of the oxyethylene unit in the polyoxyalkylene chain is preferably 5 to 100% by weight, more preferably 10%, from the viewpoint of the volume resistivity value of (b), based on the weight of the polyoxyalkylene chain. -100% by weight, particularly preferably 50-100% by weight, most preferably 60-100% by weight.
- the polyether diamine (b2) has a structure in which the hydroxyl group of the polyether diol (b1) is modified to an amino group (primary or secondary amino group), for example, a general formula: RNH-A 2- (OA 1 ) mO-E 1 -O (A 1 O) m-A 2 -NHR
- E 1 in the formula represents a residue obtained by removing a hydroxyl group from (b01) or (b02)
- a 1 is C2 to 12 (preferably 2 to 8, more preferably a halogen atom).
- An alkylene group of 2 to 4); m and m ′ represent an integer of 1 to 300, preferably 2 to 250, more preferably 5 to 200, particularly preferably 8 to 150, most preferably 10 to 100; m ′ may be the same or different.
- a 2 represents a C2 to 12 (preferably 2 to 8, more preferably 2 to 4) alkylene group which may contain a halogen atom, and A 1 and A 2 may be the same or different.
- R represents H or a C1-4 (preferably 1 or 2) alkyl group.
- the volume specific resistance value of the hydrophilic polymer (b) is preferably 1 ⁇ 10 5 to 1 ⁇ 10 11 ⁇ ⁇ cm, more preferably Is 10 6 to 10 10 ⁇ ⁇ cm, more preferably 10 7 to 10 9 ⁇ ⁇ cm.
- the volume resistivity value is less than 10 5 , the resin physical properties deteriorate, and when it exceeds 10 11 , the compatibility deteriorates.
- Mn in (b) is preferably from 150 to 20,000, more preferably from 300 to 18,000, still more preferably from 1,000 to 15,000, from the viewpoint of heat resistance and reactivity with polyolefin (a). Most preferably, it is 1,200 to 8,000.
- the alternating block copolymer (G) is a group in which the block of the polyolefin (a) and the block of the hydrophilic polymer (b) are composed of an ester bond, an amide bond, an ether bond and an imide bond. It has a structure in which they are alternately and repeatedly bonded via at least one bond selected from Of these, preferred is a polymer having a repeating unit represented by the following general formula (1).
- n is an integer of 2 to 50 (preferably 3 to 40, more preferably 4 to 30); R 1 and R 2 are each independently a hydrogen atom or a methyl group. However, neither R 1 nor R 2 becomes a methyl group.
- y is an integer of 15 to 800 (preferably 20 to 500, more preferably 30 to 400).
- E 1 is an alkylene group having 1 to 11 carbon atoms or a phenylene group (residue obtained by removing a hydroxyl group from a diol (b01) or a dihydric phenol (b02)).
- a 1 is an alkylene group having 2 to 12 carbon atoms (preferably 2 to 8, more preferably 2 to 4 carbon atoms) which may contain a halogen atom.
- m and m ′ are each independently an integer of 1 to 300 (preferably 2 to 250, more preferably 5 to 200, particularly preferably 8 to 150, most preferably 10 to 100).
- X is a group represented by general formula (2) or general formula (3)
- X ′ is a group represented by general formula (2 ′) or general formula (3 ′).
- X is a group represented by the general formula (2)
- X ′ is a group represented by the general formula (2 ′)
- X is a group represented by the general formula (3)
- X ′ is a group represented by the general formula (3 ′).
- R is a hydrogen atom or an alkyl group having 1 to 4 carbon atoms (preferably 1 or 2).
- R 3 is an alkylene group having 1 to 11 carbon atoms (preferably 2 to 11, more preferably 5 to 11).
- R 4 is a hydrogen atom or an alkyl group having 1 to 10 carbon atoms (preferably 1 to 8, more preferably 1 to 6).
- a 2 is an alkylene group having 2 to 12 carbon atoms which may contain a halogen atom.
- r is an integer of 1 to 20 (preferably 1 to 15, more preferably 1 to 10), and u is 0 or 1.
- Q is a group represented by the general formula (4).
- Q ' is a group represented by the general formula (4').
- T is a group represented by the general formula (5).
- T ′ is a group represented by the general formula (5 ′).
- R 5 is a hydrogen atom or an alkyl group having 1 to 10 carbon atoms (preferably 1 to 8, more preferably 1 to 6). is there.
- R 6 is a hydrogen atom or a methyl group. t is 1 when R 6 is a methyl group, and 0 when R 6 is a hydrogen atom.
- the polyether segment ⁇ (OA 1 ) mO—E 1 —O (A 1 O) m ⁇ in ⁇ in the repeating unit represented by the general formula (1) is the polyether diol (b1) or polyether diamine. This is a structure derived from (b2), and E 1 , A 1 , m and m ′ in the formula are the same as described above.
- the block polymer in which X is a group represented by the general formula (2) and X ′ is a group represented by the general formula (2 ′) includes (a11) and / or (a12) (A-1) obtained by polymerizing (b1) and (A-2) obtained by polymerizing (a11) and / or (a12) and (b2) .
- (A-1) includes (A-11) in which (a11) and (b1) are combined, (A-12) in which (a12) and (b1) are combined, and (A-11) and (A The mixture of -12) is included.
- (A-2) is combined with (A-21), (A-12) is combined with (b2), (A-22) is combined with (b2), and (A-21) is combined with (A-21). ) And (A-22).
- (A-1) is a known method, for example, a method in which (b1) is added to (a11) and / or (a12) and a polymerization (polycondensation) reaction is usually carried out at 200 to 250 ° C. under reduced pressure, or It can be produced by a polymerization method using a single or twin screw extruder, usually at 160 to 250 ° C. and a residence time of 0.1 to 20 minutes. Reference can be made to the method described in JP 2007-146145 A.
- (A-12) may be reacted with (b1) after secondary modification of (a11) with the lactam or aminocarboxylic acid, or (a11) and lactam or An aminocarboxylic acid may be reacted in the presence of (b1) and subsequently reacted with (b1).
- (A-2) is the same as (A-1) except that the combination of (a11) and / or (a12) and (b1) is replaced with the combination of (a11) and / or (a12) and (b2) ( It can be produced by the same method as in A-1).
- (A-22) may be produced by secondarily modifying (b2) with the lactam or aminocarboxylic acid and then reacting it with (a11).
- (A-4) is combined with (A-41) in which (a13) and (b2) are combined, (A-42) in which (a14) and (b2) are combined, and (A-41) ) And (A-42).
- (A-3) and (A-4) can be produced in the same manner as (A-1) and (A-2).
- the amount of (b) constituting the alternating block copolymer (G) is preferably 20 to 90% by weight, more preferably 25% based on the total weight of (a) and (b) from the viewpoint of compatibility. -80% by weight, particularly preferably 30-70% by weight.
- the Mn of the alternating block copolymer (G) is preferably 2,000 to 60,000, more preferably 5,000 to 40,000, and particularly preferably 8,000 to 30,000. If Mn is within this range, (G) will not bleed out over time. Mn can be measured by a known method by the GPC method.
- the average number (Nn) of repeating units of the polyolefin (a) block and the hydrophilic polymer (b) block is preferably 2 to 50, more preferably 3 to 40, more preferably 4 to 30.
- Nn is obtained by a method known from Mn of (G) and 1 H-NMR analysis, for example, by observing signals attributed to several protons of interest to determine the ratio of these proton integral values. Nn can be obtained from the ratio and Mn.
- the terminal of the alternating block copolymer (G) is a carbonyl group, amino group and / or unmodified polyolefin terminal derived from (a) (polyolefin terminal without any modification, that is, alkyl group or alkenyl group), or ( b) either a derived hydroxyl group and / or an amino group.
- a carbonyl group, an amino group, and a hydroxyl group are preferable as a terminal from the viewpoint of reactivity, and a carbonyl group and a hydroxyl group are more preferable.
- the above (B) may be a thermoplastic resin having a sea-island structure composed of an island formed of a high-polarity domain and a sea formed of a low-polarity domain.
- the thermoplastic resin (B) is a thermoplastic resin having the sea-island structure
- the thermoplastic resin (B) includes an ⁇ -olefin homopolymer (C1) and / or an ethylene / ⁇ -olefin copolymer.
- the ⁇ -olefin (co) polymer (C) comprising (C2) and the low viscosity polyolefin resin (D), a styrene compound, a vinyl group-containing carboxylic acid or derivative thereof, and (meth) acrylonitrile.
- Preferable examples include resins obtained by polymerizing at least one radical polymerizable monomer (E) selected from the group consisting of in a heat-melt kneader.
- the above (C) or (D) can be a structure of a hydrophobic block containing polyolefin, which the thermoplastic resin obtained here has.
- ⁇ -olefin homopolymer (C1) examples include homopolymers such as propylene, 1-butene, 1-pentene and 4-methyl-1-pentene.
- ethylene / ⁇ -olefin copolymer (C2) examples include a copolymer of ethylene and the above ⁇ -olefin.
- the above (C1) and (C2) include graft modified products of these (co) polymers with vinyl group-containing carboxylic acids [(meth) acrylic acid, (anhydrous) maleic acid, fumaric acid, itaconic acid, etc.], and A blend of two or more of these (co) polymers or modified products is also included.
- ⁇ -olefin-based (co) polymer (C) examples include the above (C1), (C2) and vinyl group-containing carboxylic acids of these (co) polymers [(meth) acrylic acid, (anhydrous And the like), and graft blends of maleic acid, fumaric acid, itaconic acid, and the like, and blends of two or more of these (co) polymers or modifications.
- C1 acrylic acid
- C2 vinyl group-containing carboxylic acids of these (co) polymers
- vinyl group-containing carboxylic acids of these (co) polymers [(meth) acrylic acid, (anhydrous And the like), and graft blends of maleic acid, fumaric acid, itaconic acid, and the like, and blends of two or more of these (co) polymers or modifications.
- butene-1 homopolymer and ethylene / butene-1 copolymer are preferred.
- the melt index by the ASTM D1238-L method (230 ° C., 2160 g) of (C1) is preferably 1 to 100, more preferably 5 to 50. If the melt index of (C1) exceeds 100, oil resistance may be insufficient, and if it is less than 1, sufficient processability may not be obtained.
- the melt index by the ASTM D1238-L method (230 ° C., 2160 g) of (C2) above is preferably 10 to 100, more preferably 10 to 50. If the melt index of (C2) exceeds 100, the oil resistance may be insufficient, and if it is less than 10, sufficient processability may not be obtained.
- Examples of the low-viscosity polyolefin resin (D) include a low molecular weight polypropylene copolymer obtained by thermal degradation, a polypropylene (co) polymer having a crystallinity of 10% or less obtained by a known polymerization method, and these And graft modified products of vinyl group-containing carboxylic acids [(meth) acrylic acid, (anhydrous) maleic acid, fumaric acid, itaconic acid, etc.].
- the propylene unit content in (D) is preferably 30% by weight or more, more preferably 50% by weight or more. If the propylene content in (D) is less than 30% by weight, the heat resistance of the thermoplastic resin (B) may be reduced.
- the melt viscosity at 190 ° C. of the above (D) is preferably 30 to 100,000 cP, more preferably 40 to 20,000 cP. If the melt viscosity is less than 30 cP, the strength of the thermoplastic resin (B2) may be insufficient, and if it exceeds 100,000 cP, the resulting thermoplastic resin (B2) has a high viscosity, so that the workability and coatability are high. May decrease.
- radical polymerizable monomer (E) examples include at least one selected from the group consisting of styrene compounds, vinyl group-containing carboxylic acids or derivatives thereof, and (meth) acrylonitrile.
- styrenic compounds and combinations of styrenic compounds and the above-mentioned other monomers are preferable from the viewpoint of thermal stability.
- styrene compound examples include styrene, t-butylstyrene, ⁇ -methylstyrene, p-methylstyrene, chlorostyrene, bromostyrene, fluorostyrene, ethylstyrene, divinylbenzene, N, N-diethylaminostyrene, and the like. Of these, styrene is particularly preferred.
- vinyl group-containing carboxylic acids or derivatives thereof include vinyl group-containing carboxylic acids [for example, (meth) acrylic acid, maleic anhydride, fumaric acid, itaconic acid, etc.], (meth) acrylic acid esters [having 1 to 18 carbon atoms.
- the weight ratio (C) :( D) :( E) of (C), (D), (E) is 100: (30 to 300) :( 1 To 50), more preferably 100: (50 to 200): (3 to 30). If the ratio of (D) is less than 30, the resulting thermoplastic resin (B) may have a high viscosity, and if it exceeds 300, the rubber elasticity of the thermoplastic resin (B) decreases. On the other hand, when the ratio of (E) is less than 1, the cohesive force becomes weak and the strength of the thermoplastic resin (B) becomes insufficient. When it exceeds 50, the melt viscosity of the thermoplastic resin (B) becomes high and the workability is poor. It will be enough.
- thermoplastic resin (B) having the sea-island structure (liquid) hydrogenated polybutadiene (J) can be contained if necessary.
- the content of (J) is preferably 50% by weight or less, more preferably 30% by weight or less.
- the ratio of (J) exceeds 50% by weight, the cohesive force of the thermoplastic resin (B) is lowered.
- thermoplastic resin (B) having a sea-island structure has a sea-island structure composed of an island formed of a high-polarity domain and a sea formed of a low-polarity domain.
- the method to manufacture such a thermoplastic resin (B) is not specifically limited, For example, the following method is mentioned. (1) A method of previously mixing (E) in the presence of (C) and (E) in the presence of (D). (2) A method of polymerizing (E) in the presence of (D) and (E) in the presence of (C); (3) A method of polymerizing (E) in the presence of (C) and polymerizing (E) in the presence of (D); (4) A method of polymerizing (E) in the presence of (C) and (D); and the like. Among these, the method (4) is preferable.
- the thermoplastic resin (B) having a sea-island structure can be obtained, for example, by polymerizing raw materials in these methods in a heat-melt kneader.
- the hot melt kneader used for the polymerization is not particularly limited in its form and shape, for example, a mixer having a highly compressible screw having a reverse screw part or a ribbon stirrer, a kneader, Examples include an extruder and a mixer. Of these, it is preferable to use a non-open type apparatus, and it is preferable to knead in an inert gas atmosphere such as nitrogen during the polymerization.
- thermoplastic resin (B) having a sea-island structure may be obtained by demonomerization.
- a known polymerization initiator or organic solvent can be used if necessary.
- Polymerization initiators include ketone peroxides, peroxyketals, hydroperoxides, dialkyl peroxides, diacyl peroxides, peroxydicarbonates, peroxyesters and other organic peroxides, azoisobutyronitrile and other azo peroxides. System compounds and the like.
- the organic solvent include alicyclic hydrocarbon solvents, aromatic hydrocarbon solvents, alcohol solvents, halogen solvents, ketone solvents, ether solvents and the like.
- the polymerization temperature is not particularly limited, and may be a temperature at which the monomer is substantially polymerized, but is usually 80 to 260 ° C.
- thermoplastic resin (B) having a melting point of 50 to 180 ° C. contained in the shell layer (Q) layer may further optionally contain a low molecular weight polyolefin (H) and / or a plasticizer (I). Good.
- Examples of the low molecular weight polyolefin (H) include polypropylene, polyethylene, propylene and one or more other vinyl compounds [ethylene, ⁇ -olefin (C4-12, such as 1-butene, 4-methyl-1-pentene, etc.). ), Vinyl acetate, (meth) acrylic acid, etc.], graft modified products of these (co) polymers with (anhydrous) unsaturated carboxylic acids [as described above, eg (anhydrous) maleic acid], And a blend of two or more of these copolymers or modified products.
- polypropylene, polyethylene, propylene / ethylene copolymers and graft modified products of these (anhydrous) unsaturated carboxylic acids are preferable from the viewpoint of compatibility, and (anhydrous) unsaturation of polypropylene and polypropylene is more preferable. It is a graft modified product with carboxylic acid.
- the Mn of the low molecular weight polyolefin (H) is preferably 500 or more, more preferably 800 or more, particularly preferably 1,000 or more, and preferably 25,000 or less, more preferably 23,000 or less, particularly preferably. Is 20,000 or less.
- the amount of (H) used is preferably 30% or less, more preferably 0.1 to 25%, still more preferably 0.5 to 20% based on the weight of (B).
- paraffinic process oil, naphthenic process oil, and mixtures thereof are preferred from the viewpoints of thermal stability and weather resistance.
- the amount of (I) used is preferably 50% or less, more preferably 1 to 45%, still more preferably 5 to 40% based on the total weight of (B).
- the weight ratio of the thermoplastic resin (B) to the water-absorbent resin (A) is preferably 0.1 to 10% by weight.
- this range is exceeded, (A) the inherent absorption performance, absorption rate, and flexibility of the resulting absorbent body are reduced.
- it is less than this range the adherence of (A) to the fibrous base material (F) is lowered, and the shape-retaining property after water absorption of the obtained absorbent body is inferior.
- additives for resin can be used as necessary.
- examples thereof include at least one selected from the group consisting of an inhibitor, a flame retardant, an ultraviolet absorber, and an antibacterial agent.
- known additives can be used by a method such as a known blending amount.
- the method for producing the core / shell type water-absorbent resin particles of the present invention may be resin particles produced by any method and process.
- Examples of the production methods (I) to (III) are as follows.
- An aqueous dispersion (W) of resin particles comprising a thermoplastic resin (B) and a water absorbent resin (A) or a solvent solution thereof are mixed, and the water absorbent resin (A) or a solvent solution thereof is mixed in (W).
- the resin particles made of the thermoplastic resin (B) adhere to the surface of the resin particles made of the water-absorbent resin (A) at the same time as the granulation, and the core layer (P) / shell layer (Q) is formed.
- An aqueous dispersion of core-shell type resin particles is made and is made by removing the aqueous medium therefrom.
- the resin particles made of the water-absorbing resin (A) prepared in advance are coated with the coating agent (W ′) made of the thermoplastic resin (B), and if necessary, the shell layer is made into a film to form the core A method for producing shell-type resin particles.
- the coating method is not limited.
- a method of dispersing resin particles made of the water absorbent resin (A) prepared in advance in the aqueous dispersion (W ′) of the thermoplastic resin (B) examples include a method in which a solution of the thermoplastic resin (B) is sprinkled as a coating agent on the resin particles made of the produced water absorbent resin (A).
- thermoplastic resin (B) The resin particles made of the water-absorbing resin (A) prepared in advance and the thermoplastic resin (B) synthesized in advance are mixed in a mixer, and then subjected to heat treatment to obtain the thermoplastic resin (B). A method for obtaining water-absorbing resin particles coated on the surface of resin particles made of water-absorbing resin (A). Among these, the production method (III) is preferable.
- the shape of the core-shell type water-absorbent resin particles may be any shape such as granular, granular, granulated, flake shaped, lump, pearl, fine powder.
- 90% by weight or more is a granule having a particle size distribution of 1 mm or less, and particularly preferably 90% by weight or more is a granule, granule, granulated or flake having a particle size distribution of 0.1 to 0.9 mm Or bulky water-absorbing resin.
- the absorbent body of the present invention is formed by fixing the water-absorbent resin particles of the present invention to the fibrous base material (F).
- the fibrous base material (F) is preferably at least one selected from the group consisting of cellulose fibers, organic synthetic fibers, and mixtures of organic synthetic fibers and cellulose fibers.
- cellulosic fibers include natural fibers such as fluff pulp, and cellulosic chemical fibers such as viscose rayon, acetate, and cupra.
- raw materials conifers, hardwoods, etc.
- production methods chemical pulp, semi-chemical pulp, mechanical pulp, CTMP, etc.
- bleaching methods etc. of this cellulose natural fiber.
- organic synthetic fibers examples include polypropylene fibers, polyethylene fibers, polyamide fibers, polyacrylonitrile fibers, polyester fibers, polyvinyl alcohol fibers, polyurethane fibers, and heat-fusible composite fibers (for example, different melting points).
- fibrous base materials preferred are cellulose-based natural fibers, polypropylene-based fibers, polyethylene-based fibers, polyester-based fibers, heat-fusible conjugate fibers, and mixed fibers thereof, and more preferably obtained.
- the water-absorbing material is fluff pulp, heat-fusible composite fiber, and a mixture thereof in that the shape retention after water absorption is excellent.
- the length and thickness of the fibrous base material (F) are not particularly limited, and can be suitably used as long as the length is usually 1 to 200 mm and the thickness is in the range of 0.1 to 100 denier.
- the shape is not particularly limited as long as it is fibrous, and examples thereof include a thin cylindrical shape, a split yarn shape, a staple shape, a filament shape, and a web shape.
- the ratio of the water-absorbing resin particles to the fibrous base material (F) is preferably the ratio of water-absorbing resin particles: fibrous base material (F), preferably (20:80) to (95: 5). Is (30:70) to (90:10), more preferably (35:65) to (80:20). When the ratio of the water-absorbing resin particles is less than 20, the function of the obtained absorber is not sufficiently exhibited.
- the organic synthetic fiber is at least one selected from a sheath-core type, an eccentric type, and a parallel type, and includes a plurality of (two or more) components having different melting points, and a low
- a heat-fusible conjugate fiber having a melting point component of 50 to 180 ° C. is preferable because it can employ a simple method of heating in order to achieve fixation.
- the water absorbent resin particles are mixed with the fibrous base material (F) or dispersed in (F), and then the heat contained in the shell layer of the water absorbent resin particles.
- an ordinary mixing apparatus may be used as an apparatus for mixing the water-absorbent resin particles and the fibrous base material (F).
- a conical blender, a nauter mixer, a V-type mixer, a fluidized bed type mixer, an airflow type mixing apparatus examples thereof include an airflow type mixing device provided with a nozzle for spraying granular materials, and a crushing device for fibrous materials provided with a nozzle for spraying granular materials.
- the apparatus for processing at a temperature equal to or higher than the melting point of (B) include a hot air heater, a Nauter heater, a fluidized bed heater, an airflow heater, a heating calendar roll, an infrared heater, and a high frequency heater. Can be mentioned.
- the water-absorbing resin particles are fixed to the fibrous base material (F), and the water-absorbing resin particles are partially fixed, for example, 50% by weight or more to (F). It only has to be. It becomes 60 weight% or more by selection of preferable conditions.
- the fixing rate is less than 50% by weight, when applied to absorbent articles such as disposable diapers and sanitary products, the water-absorbent resin particles may move, be ubiquitous, separated, or fall off during storage or transportation of these products. is there.
- the sticking rate is a value after a vibration test.
- the vibration test can be performed using, for example, a low tap test sieve shaker and a standard sieve (JIS Z8801-1: 2006).
- the absorbent body of the present invention can be subjected to treatments usually applied to fibrous materials such as crushing, lamination, compression, cold calendering, heat calendering, needle punching, stretching and papermaking.
- an organic powder for example, pulp powder, cellulose derivative, natural polysaccharide, etc.
- an inorganic powder for example, zeolite, silica, alumina, bentonite, activated carbon, etc.
- glass fiber oxidation as an extender and additive Inhibitors, preservatives, bactericides, surfactants, colorants, fragrances and the like can be blended as necessary, and these amounts are usually 10% by weight or less, preferably 5% by weight or less based on the weight of the absorber. It is.
- the absorbent article of the present invention uses the absorbent body of the present invention.
- the absorbent article include various sanitary materials and absorbent articles such as paper diapers, sanitary products, postpartum mats, and medical underpads. In particular, it is useful for thin paper diapers and thin sanitary products having a large ratio of water-absorbing resin / fiber (pulp and / or heat-adhering fiber).
- sheet-like or tape-like water-absorbing materials such as freshness-keeping materials for fruits and vegetables, drip absorbers, moisture or humidity control sheets, anti-condensation materials, seedling sheets for paddy rice, concrete curing sheets, water-stopping materials for communication cables and optical fiber cables It is also useful when manufacturing.
- the structure and structure of these absorbent articles are well known to those skilled in the art.
- Water absorption speed 1 g of absorbent is put into a 250 mesh nylon tea bag, immersed in a large excess of 0.9% sodium chloride aqueous solution for 2 minutes, suspended for 15 minutes, drained, and then the increased weight is measured. The value obtained by dividing the measured value by 2 minutes was evaluated as the absorption rate.
- Fixing rate Using a low-tap test sieve shaker and a standard sieve (JIS Z8801-1: 2006), that is, placing the absorbent on a 850 ⁇ m sieve, and shaking with a low-tap test sieve shaker for 5 minutes.
- the fixing rate was determined from the weight (W) of the absorbent resin particles dropped off on the tray.
- the sticking rate can be obtained by the following equation.
- Adhesion rate (%) ⁇ (W0 ⁇ W) / W0 ⁇ ⁇ 100
- Shape retention After absorbing 200 ml of physiological saline in 10 g of the water-absorbing material, it was placed on a wire mesh with an opening of 4 mm. The wire mesh was vibrated, and the degree of retention of the shape of the water-absorbing material after absorption and the degree of falling off of the swollen water-absorbing resin were observed and evaluated according to the following criteria.
- ⁇ Shape retention is good and the water-absorbing resin is not easily dropped.
- ⁇ A certain shape is retained, but the water-absorbing resin.
- ⁇ The shape is not retained, and the water-absorbing resin is also frequently dropped
- this water-containing resin (Gel 1) was shredded at 25 ° C. for 5 minutes with a mincing machine (diameter hole diameter: 6 mm, made by Iizuka Kogyo Co., Ltd., 12VR-400K), and then ventilated band dryer (135 ° C. 2.0 m / sec; manufactured by Inoue Metal Industry Co., Ltd.) to obtain a dry polymer.
- a mincing machine diameter hole diameter: 6 mm, made by Iizuka Kogyo Co., Ltd., 12VR-400K
- ventilated band dryer (135 ° C. 2.0 m / sec; manufactured by Inoue Metal Industry Co., Ltd.
- a juicer mixer National MX-X53, manufactured by Matsushita Electric Industrial Co., Ltd.
- Example 1 After 20 parts by weight of the particulate water-absorbing resin (A1) produced in Production Example 1 and 0.2 part by weight of the block polymer (a3) synthesized in Production Example 2 are mixed in a V-type mixer for 20 minutes, 150 parts are mixed. A heat treatment was performed at 15 ° C. for 15 minutes to obtain water-absorbing resin particles (1) having a surface coated with a block polymer. Next, the core-sheath type polyester / polyethylene fiber (melting point of the low melting point component is 50 ° C.
- Example 2 Absorbent body (2) was obtained in the same manner except that block polymer (a3) of Example 1 was changed to block polymer (a5) synthesized in Production Example 3.
- Table 1 shows the results of water absorption rate, adhesion rate, and shape retention measured for the absorbers (1) to (6) obtained in Examples 1 and 2 and Comparative Examples 1 to 4.
- this water-containing resin (Gel 2) was shredded for 5 minutes at 25 ° C. with a mincing machine (eyepiece hole diameter: 6 mm, 12VR-400K manufactured by Iizuka Kogyo Co., Ltd.), and then ventilated band dryer (135 ° C. 2.0 m / sec; manufactured by Inoue Metal Industry Co., Ltd.) to obtain a dry polymer.
- a mincing machine eyepiece hole diameter: 6 mm, 12VR-400K manufactured by Iizuka Kogyo Co., Ltd.
- ventilated band dryer (135 ° C. 2.0 m / sec; manufactured by Inoue Metal Industry Co., Ltd.
- a juicer mixer National MX-X53, manufactured by Matsushita Electric Industrial Co., Ltd.
- thermoplastic resin (B) having a sea-island structure in the present invention.
- the melting point of the thermoplastic resin (B) was 105 ° C.
- Example 3 After 20 parts by weight of the particulate water-absorbing resin (A2) produced in Production Example 4 and 0.2 parts by weight of the thermoplastic resin (B) synthesized in Production Example 5 were mixed in a V-type mixer for 20 minutes, A heat treatment was performed at 150 ° C. for 15 minutes to obtain water-absorbing resin particles (2) having the surface coated with the thermoplastic resin (B). Next, the core-sheath type polyester / polyethylene fiber (melting point of the low melting point component is 50 ° C.
- thermoplastic resin (B) of Example 3 was acid-modified wax (molecular weight 1,500, acid value 60, melting point 104 ° C.), an absorbent body (8) was obtained.
- thermoplastic resin (B) of Example 3 was changed to ethylene vinyl acetate (viscosity 595 mPa ⁇ s, melting point 92 ° C.), an absorbent body (9) was obtained.
- Table 2 shows the results of water absorption rate, adhesion rate, and shape retention measured for the absorbers (7) to (11) obtained in Example 3 and Comparative Examples 5 to 8.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Textile Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Materials Engineering (AREA)
- Dispersion Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Absorbent Articles And Supports Therefor (AREA)
- Coating Of Shaped Articles Made Of Macromolecular Substances (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Other Resins Obtained By Reactions Not Involving Carbon-To-Carbon Unsaturated Bonds (AREA)
- Laminated Bodies (AREA)
Abstract
Description
また、上記吸水性樹脂を更に表面架橋せしめた吸水性樹脂も使用できる。
上記吸水性樹脂は2種以上併用してもよい。
(a0)中の二重結合の量は、相溶性の観点から好ましくは、C1,000当たり1~40個、さらに好ましくは2~30個、とくに好ましくは4~20個である。
1分子当たりの二重結合の平均数は、繰り返し構造の形成性の観点及び相溶性の観点から好ましくは、1.1~5、さらに好ましくは1.3~3、とくに好ましくは1.5~2.5、最も好ましくは1.8~2.2である。
熱減成法においては、Mnが800~6,000の範囲で、一分子当たりの平均末端二重結合数が1.5~2個の低分子量ポリオレフィンが容易に得られる〔例えば、村田勝英、牧野忠彦、日本化学会誌、192頁(1975)参照〕。
また、(a1)の酸価は、(b)との反応性の観点から好ましくは、4~280(mgKOH/g、以下、数値のみを記載する。)、さらに好ましくは4~100、特に好ましくは5~50である。
これらのうち、例えば、上記ポリオレフィン(a)の疎水性ブロックと下記に詳述する親水性ポリマー(b)のブロックとがエステル結合、アミド結合、エーテル結合およびイミド結合からなる群から選ばれる少なくとも一種を介して結合した交互ブロック共重合体(G)を好ましく挙げることができる。
RNH-A2-(OA1)mO-E1-O(A1O)m-A2-NHR
で示されるものが挙げられる。式中の記号E1は、(b01)又は(b02)から水酸基を除いた残基を表し、A1は、ハロゲン原子を含んでいてもよいC2~12(好ましくは2~8、さらに好ましくは2~4)のアルキレン基;m及びm’は1~300、好ましくは2~250、さらに好ましくは5~200、とくに好ましくは8~150、最も好ましくは10~100の整数を表し、mとm’とは同一でも異なっていてもよい。A2はハロゲン原子を含んでいてもよいC2~12(好ましくは2~8、さらに好ましくは2~4)のアルキレン基を表し、A1とA2とは同じでも異なってもよい。RはH又はC1~4(好ましくは1又は2)のアルキル基を表す。

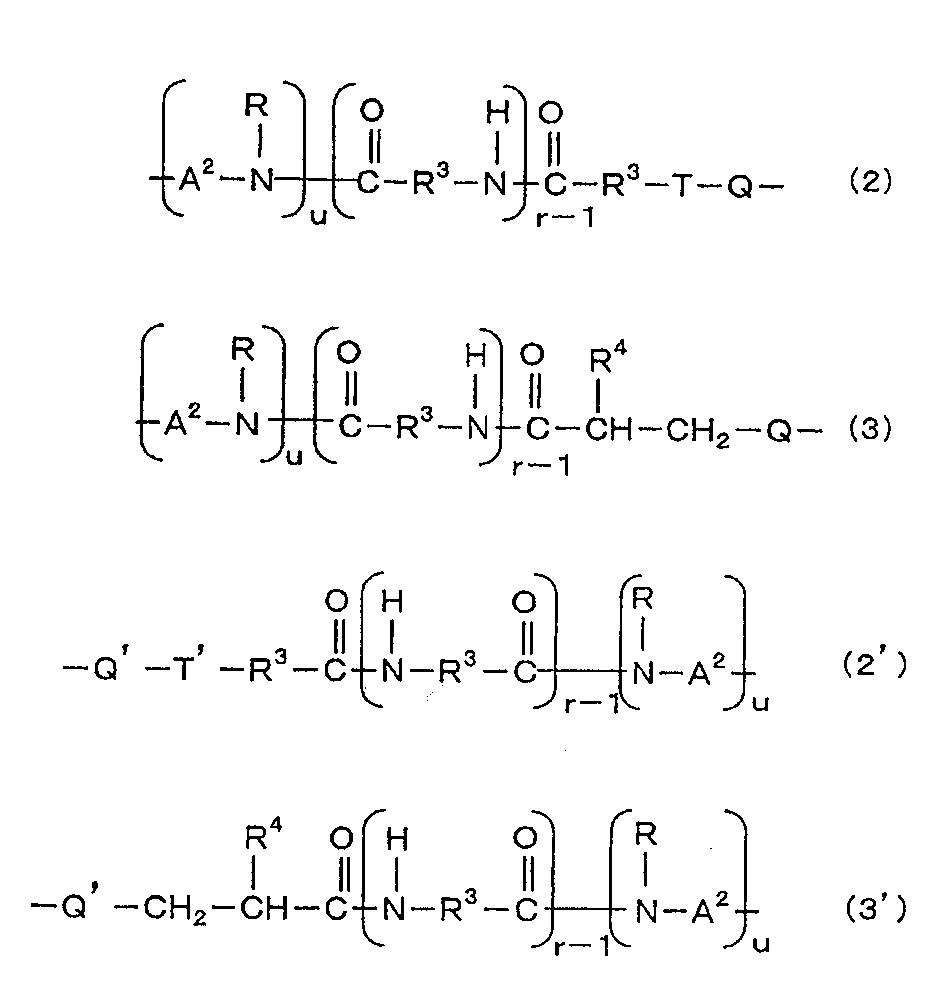

(1)あらかじめ(C)の存在下で(E)を重合したものと(D)の存在下で(E)を重合したものを混合する方法;
(2)(D)の存在下で(E)を重合したものと(C)の存在下に(E)を重合する方法;
(3)(C)の存在下で(E)を重合したものと(D)の存在下に(E)を重合する方法;
(4)(C)および(D)の存在下に(E)を重合する方法;等が挙げられる。
これらのうち好ましいものは(4)の方法である。
(I):コア粒子を作成すると同時にコア・シェル構造にする方法。
熱可塑性樹脂(B)からなる樹脂粒子の水性分散液(W)と、吸水性樹脂(A)またはその溶剤溶液とを混合し、(W)中に吸水性樹脂(A)またはその溶剤溶液を分散し、(W)中で吸水性樹脂(A)からなる樹脂粒子を形成する方法。
この場合、吸水性樹脂(A)からなる樹脂粒子の造粒と同時にその表面に熱可塑性樹脂(B)からなる樹脂粒子が付着してコア層(P)・シェル層(Q)で構成されるコア・シェル型樹脂粒子の水性分散体ができ、これからから水性媒体を除去することによって造られる。
この場合、コーティング方法には、限定はなく、例えば、熱可塑性樹脂(B)の水性分散液(W’)中にあらかじめ作製した吸水性樹脂(A)からなる樹脂粒子を分散させる方法や、あらかじめ作製した吸水性樹脂(A)からなる樹脂粒子に熱可塑性樹脂(B)の溶解液をコーティング剤としてふりかける方法などが挙げられる。
(III):あらかじめ作製した吸水性樹脂(A)からなる樹脂粒子と、予め合成した熱可塑性樹脂(B)とを混合機に入れて混合した後、加熱処理を行い、熱可塑性樹脂(B)を吸水性樹脂(A)からなる樹脂粒子の表面にコーティングした吸水性樹脂粒子を得る方法。
これらの中では(III)の製法が好ましい。
固着率、吸水速度、吸水後の形状保持性は下記の方法により測定した。以下、特に言及しない限り部は重量部、%は重量%を表す。
試験前の吸収体中に含まれる吸水性樹脂粒子の重量をW0とすると固着率は以下の式で求められる。
固着率(%)={(W0-W)/W0}×100
◎:形状保持性は良好であり、吸水性樹脂の脱落もほとんど無い
○:形状保持性は良好であり、吸水性樹脂の脱落も少ない
△:ある程度の形状は保持されているが、吸水性樹脂の脱落が多い
×:形状は保持されておらず、吸水性樹脂の脱落も多い
アクリル酸ナトリウム88部、アクリル酸22.85部、N,N’-メチレンビスアクリルアミド0.3部および脱イオン水293を攪拌・混合しながら、温度を1~2℃に保ち、この混合液中に窒素を流入して、混合液中の溶存酸素濃量を0.5ppm以下とした。引き続き、この混合液に、1重量%過酸化水素水溶液0.3部、0.2重量%アスコルビン酸水溶液0.8部及び2重量%の2,2’-アゾビスアミジノプロパンジハイドロクロライド水溶液0.8部を添加・混合して重合を開始させ、反応液が80℃に達した後、重合温度80±2℃で約5時間重合することにより、含水樹脂(ゲル1)を得た。
熱減成法[23℃における密度0.90(単位はg/cm3、以下数値のみを示す。)MFR6.0g/10分のエチレン/プロピレンランダム共重合体(エチレン含量2%)を410±0.1℃で熱減成]で得られた低分子量エチレン/プロピレンランダム共重合体(Mn3,500、密度0.89、C1,000個当たりの二重結合量7.1個、1分子当たりの二重結合の平均数1.8、両末端変性可能なポリオレフィンの含有量90%)90部、無水マレイン酸10部及びキシレン30部を混合後、窒素ガス雰囲気下(密閉下)、200℃で溶融させ、200℃で20時間反応させた。その後、過剰の無水マレイン酸とキシレンを減圧下、200℃、3時間で留去して、酸変性ポリプロピレン(a1)を得た。酸価は27.2、Mnは3,700であった。
熱減成法[23℃における密度が0.90でMFRが10(g/10分)のポリプロピレンを410±0.1℃で熱減成]で得られた低分子量ポリプロピレン(Mn10,000、密度0.89、C1,000個当たりの二重結合量1.3個、1分子当たりの二重結合の平均数1.8、両末端変性可能なポリオレフィンの含有量90重量%)94部、無水マレイン酸6部及びキシレン30部を混合後、製造例1と同様にして、酸変性ポリプロピレン(a4)を得た。(a4)の酸価は5.0、Mnは10,000であった。
製造例1で製造した粒子状の吸水性樹脂(A1)20重量部と製造例2で合成したブロックポリマー(a3)0.2重量部をV型混合機に入れて20分間混合した後、150℃で15分間過熱処理を行い、ブロックポリマーを表面にコーティングした吸水性樹脂粒子(1)を得た。
次いで芯鞘型ポリエステル/ポリエチレン繊維(低融点成分の融点は50℃以上で150℃未満)を合繊用開綿機(大和機工株式会社製)で解繊し、サンプルローラーカード機(インテック株式会社、ISC-360)でウェブ形成を行った。ウェブ状の芯鞘型ポリエステル/ポリエチレン繊維5重量部に対して吸水性樹脂粒子(1)20重量部を均一になるように播き、150℃で5分間加熱処理を行い、吸収体(1)を得た。
実施例1のブロックポリマー(a3)を製造例3で合成したブロックポリマー(a5)としたこと以外は同様に行い吸収体(2)を得た。
実施例1のブロックポリマー(a3)を酸変性ワックス(分子量1,500、酸価60、融点104℃)としたこと以外は同様に行い吸収体(3)を得た。
実施例1のブロックポリマー(a3)をエチレンビニルアセテート(粘度595mPa・s、融点92℃)としたこと以外は同様に行い吸収体(4)を得た。
比較例1の酸変性ワックスの添加量を1重量部としたこと以外は同様に行い吸収体(5)を得た。
比較例2のエチレンビニルアセテートを1重量部としたこと以外は同様に行い吸収体(6)を得た。

アクリル酸ナトリウム88部、アクリル酸22.85部、N,N’-メチレンビスアクリルアミド0.3部および脱イオン水293を攪拌・混合しながら、温度を1~2℃に保ち、この混合液中に窒素を流入して、混合液中の溶存酸素濃量を0.5ppm以下とした。引き続き、この混合液に、1重量%過酸化水素水溶液0.3部、0.2重量%アスコルビン酸水溶液0.8部及び2重量%の2,2’-アゾビスアミジノプロパンジハイドロクロライド水溶液0.8部を添加・混合して重合を開始させ、反応液が80℃に達した後、重合温度80±2℃で約5時間重合することにより、含水樹脂(ゲル2)を得た。
ジャケットの熱媒温度を160℃に設定した直径5インチ、L/D=10の連続混合装置(栗本鉄工製KRCS5)を用い、スチレン50重量部、無水マレイン酸50重量部、エチレン-ブテン1共重合体(住友化学工業製「エスプレンN0377」)350重量部、非昌質エチレン-プロピレン共重合体(宇部レキセン製「ウベタックUT2315」)350重量部、熱減成法で得られた低分子量ポリプロピレン(三洋化成工業製「ビスコール660P」)200重量部およびt-ブチルパーオキシベンゾエート0.5重量部を分散した混合物を連続混合装置の原料供給口から供給し、滞留時間が10分になるように保ちながら重合した。得られた混合物を、二軸押し出し機(池貝鉄工製PCM45、L/D=50)で脱モノマ-を行い、本発明における海島構造を有する熱可塑性樹脂(B)を得た。熱可塑性樹脂(B)の融点は105℃であった。
製造例4で製造した粒子状の吸水性樹脂(A2)20重量部と製造例5で合成した熱可塑性樹脂(B)0.2重量部をV型混合機に入れて20分間混合した後、150℃で15分間過熱処理を行い、熱可塑性樹脂(B)を表面にコーティングした吸水性樹脂粒子(2)を得た。
次いで芯鞘型ポリエステル/ポリエチレン繊維(低融点成分の融点は50℃以上で150℃未満)を合繊用開綿機(大和機工株式会社製)で解繊し、サンプルローラーカード機(インテック株式会社、ISC-360)でウェブ形成を行った。ウェブ状の芯鞘型ポリエステル/ポリエチレン繊維5重量部に対して吸水性樹脂粒子(2)20重量部を均一になるように播き、150℃で5分間加熱処理を行い、吸収体(7)を得た。
実施例3の熱可塑性樹脂(B)を酸変性ワックス(分子量1,500、酸価60、融点104℃)としたこと以外は同様に行い、吸収体(8)を得た。
実施例3の熱可塑性樹脂(B)をエチレンビニルアセテート(粘度595mPa・s、融点92℃)としたこと以外は同様に行い、吸収体(9)を得た。
比較例5の酸変性ワックスの添加量を1重量部としたこと以外は同様に行い、吸収体(10)を得た。
比較例6のエチレンビニルアセテートを1重量部としたこと以外は同様に行い、吸収体(11)を得た。

Claims (14)
- コア層(P)とシェル層(Q)とで構成されるコア・シェル型の吸水性樹脂粒子であって、(P)が吸水性樹脂(A)を含有し、(Q)が融点50~180℃の熱可塑性樹脂(B)を含有し、熱可塑性樹脂(B)が、ポリオレフィンを含む疎水性ブロックを有する重合体である吸水性樹脂粒子。
- 融点が50~180℃の熱可塑性樹脂(B)が、ポリオレフィンの疎水性ブロックと親水性ポリマーのブロックとがエステル結合、アミド結合、エーテル結合およびイミド結合からなる群から選ばれる少なくとも一種を介して結合した交互ブロック共重合体(G)である請求項1に記載の吸水性樹脂粒子。
- 親水性ポリマーの体積固有抵抗値が1×105~1×1011Ω・cmである請求項2に記載の吸水性樹脂粒子。
- 交互ブロック共重合体(G)が、下記一般式(1)で表される構造を有する共重合体である請求項2または3に記載の吸水性樹脂粒子。
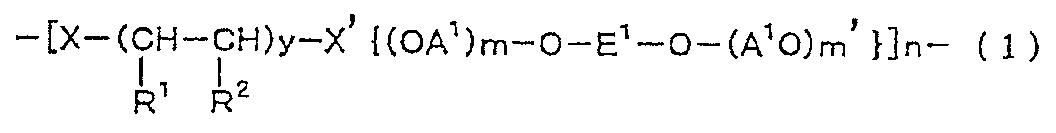
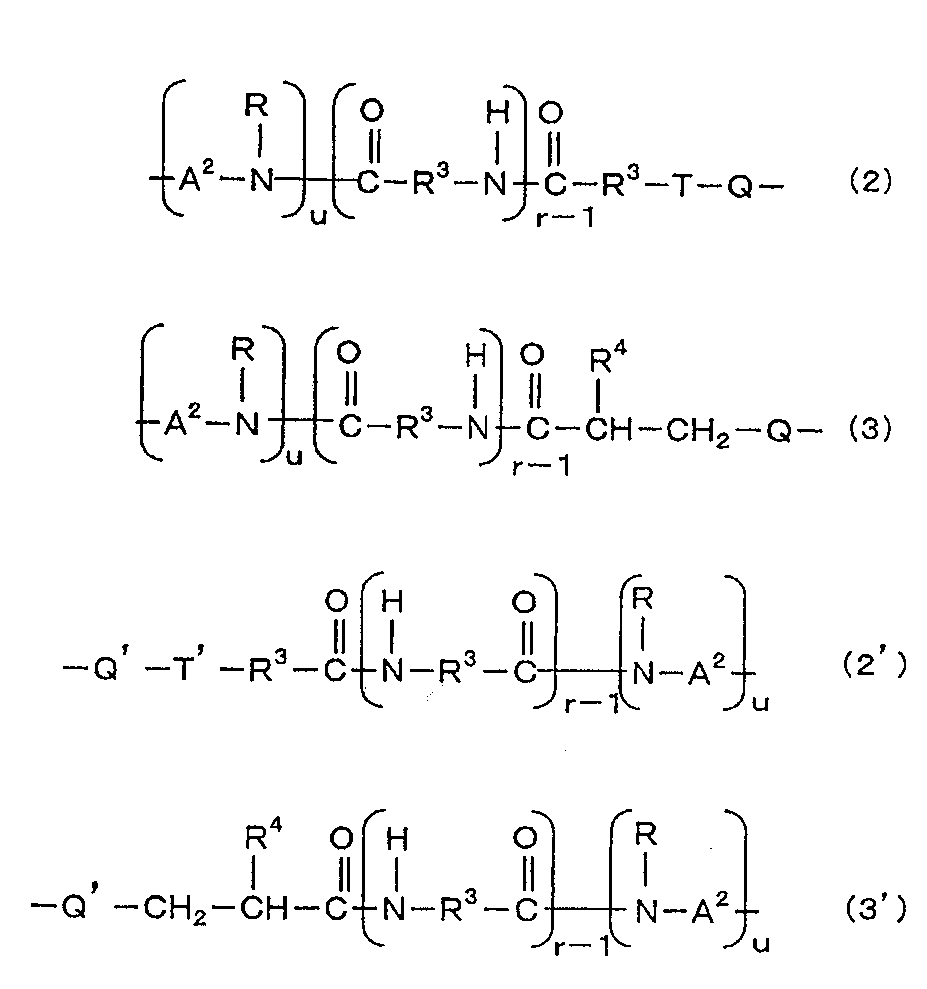
[一般式(2)、(3)、(2’)、(3’)式中、Rは水素原子又は炭素数1~4のアルキル基である;R3は炭素数1~11のアルキレン基である;R4は水素原子又は炭素数1~10のアルキル基である;A2はハロゲン原子を含んでいてもよい炭素数2~12のアルキレン基である;rは1~20の整数であり、uは0又は1である;Qは一般式(4)で表される基である;Q’は一般式(4’)で表される基である;Tは一般式(5)で表される基である;T’は一般式(5’)で表される基である。]
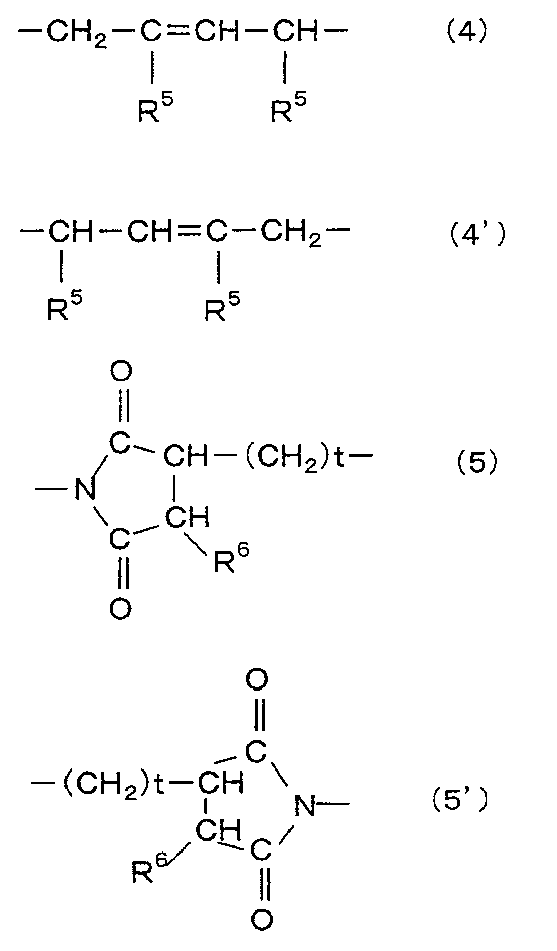
[一般式(4)、(4’)、(5)、(5’)中、R5は水素原子又は炭素数1~10のアルキル基である;R6は水素原子又はメチル基である;tは、R6がメチル基の場合は1であり、R6が水素原子の場合は0である。] - 熱可塑性樹脂(B)が高極性のドメインが成す島と低極性のドメインが成す海からなる海島構造を有する熱可塑性樹脂である請求項1~4のいずれかに記載の吸水性樹脂粒子。
- 熱可塑性樹脂(B)が、α-オレフィン単独重合体(C1)および/またはエチレン/α-オレフィン共重合体(C2)からなるα-オレフィン系(共)重合体(C)と低粘度ポリオレフィン系樹脂(D)の存在下に、スチレン系化合物、ビニル基含有カルボン酸またはその誘導体および(メタ)アクリロニトリルからなる群より選ばれる少なくとも1種のラジカル重合性単量体(E)を加熱溶融混練機中で重合して得られた樹脂である請求項5に記載の吸水性樹脂粒子。
- (C)、(D)、(E)の重量比が、(C):(D):(E)=100:(30~300):(1~50)である請求項6に記載の吸水性樹脂粒子。
- 吸水性樹脂(A)に対する熱可塑性樹脂(B)の重量比率が、0.1~10重量%である請求項1~7のいずれかに記載の吸水性樹脂粒子。
- 融点が50~180℃の熱可塑性樹脂(B)が、低分子量ポリオレフィン(H)および/または可塑剤(I)を含有する樹脂である請求項1~8のいずれかに記載の吸水性樹脂粒子。
- 請求項1~9のいずれかに記載の吸水性樹脂粒子が繊維状基材(F)に固着化されてなる吸収体。
- 振動試験後の吸水性樹脂粒子の繊維状基材(F)に対する固着率が、50重量%以上である請求項10に記載の吸収体。
- 繊維状基材(F)が、セルロース系繊維、有機系合成繊維、および有機系合成繊維とセルロース系繊維との混合物からなる群から選ばれる1種以上である請求項10または11に記載の吸収体。
- 有機系合成繊維が、鞘芯型のもの、偏芯型のものおよび並列型のものから選ばれる少なくとも1種で、融点の異なる複数成分を含み、低融点成分の融点が50~180℃の熱融着性複合繊維である請求項10~12のいずれかに記載の吸収体。
- 請求項10~13のいずれかに記載の吸収体を用いた吸収性物品。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201580027096.3A CN106459598B (zh) | 2014-05-23 | 2015-05-22 | 吸水性树脂颗粒、含有该吸水性树脂颗粒的吸收体和吸收性物品 |
| US15/313,148 US10174174B2 (en) | 2014-05-23 | 2015-05-22 | Water-absorbing resin particles, absorber comprising same, and absorbent article |
| KR1020167036024A KR101792968B1 (ko) | 2014-05-23 | 2015-05-22 | 흡수성 수지 입자, 이것을 포함하는 흡수체 및 흡수성 물품 |
| EP15795426.4A EP3147331B1 (en) | 2014-05-23 | 2015-05-22 | Water-absorbing resin particles, absorber comprising same, and absorbent article |
| JP2016521161A JP6402181B2 (ja) | 2014-05-23 | 2015-05-22 | 吸水性樹脂粒子、これを含む吸収体および吸収性物品 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-107016 | 2014-05-23 | ||
| JP2014107002 | 2014-05-23 | ||
| JP2014-107002 | 2014-05-23 | ||
| JP2014107016 | 2014-05-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015178481A1 true WO2015178481A1 (ja) | 2015-11-26 |
Family
ID=54554140
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/064741 Ceased WO2015178481A1 (ja) | 2014-05-23 | 2015-05-22 | 吸水性樹脂粒子、これを含む吸収体および吸収性物品 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10174174B2 (ja) |
| EP (1) | EP3147331B1 (ja) |
| JP (1) | JP6402181B2 (ja) |
| KR (1) | KR101792968B1 (ja) |
| CN (1) | CN106459598B (ja) |
| MY (1) | MY177389A (ja) |
| WO (1) | WO2015178481A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018132104A (ja) * | 2017-02-14 | 2018-08-23 | Jxtgエネルギー株式会社 | 高粘性流体組成物、高粘性流体組成物の製造方法及び振動エネルギー減衰装置 |
| JPWO2021117783A1 (ja) * | 2019-12-13 | 2021-06-17 | ||
| JPWO2022124137A1 (ja) * | 2020-12-09 | 2022-06-16 | ||
| WO2026005007A1 (ja) * | 2024-06-28 | 2026-01-02 | 株式会社日本触媒 | 吸水剤の製造方法および吸水剤 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6402181B2 (ja) | 2014-05-23 | 2018-10-10 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを含む吸収体および吸収性物品 |
| CN106236396A (zh) * | 2016-08-19 | 2016-12-21 | 厦门延江新材料股份有限公司 | 一种吸水薄膜及其制造方法 |
| JP2018166887A (ja) * | 2017-03-30 | 2018-11-01 | 株式会社リブドゥコーポレーション | 吸収体およびこれを備えた吸収性物品 |
| KR102002586B1 (ko) * | 2017-11-29 | 2019-10-01 | 주식회사 휴비스 | 천연섬유가 포함된 열접착 부직포 |
| JP2019171596A (ja) * | 2018-03-27 | 2019-10-10 | セイコーエプソン株式会社 | インク吸収体およびインク吸収器 |
| US20210038154A1 (en) * | 2018-03-29 | 2021-02-11 | Creative Technology Corporation | Attachment pad |
| CN109162090B (zh) * | 2018-09-13 | 2019-08-02 | 江苏三鑫纺织染整有限公司 | 一种提高纯棉纱润湿性能的处理工艺 |
| WO2021200852A1 (ja) * | 2020-03-30 | 2021-10-07 | 帝人株式会社 | 熱可塑性樹脂との相溶性に優れた重合体 |
| CN111974565B (zh) * | 2020-04-29 | 2021-12-24 | 上海电力大学 | 一种基于亲疏性组合表面协同调控的气液两相喷射器 |
| KR102407579B1 (ko) | 2020-11-25 | 2022-06-14 | 한국생산기술연구원 | 이타콘산 유래 고분자 화합물 및 이를 포함하는 흡수성 수지 |
| CN115350316B (zh) * | 2022-08-24 | 2023-04-07 | 上海睿植康医疗科技有限公司 | 一种具有缓释药物的亲水性纤维膜及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05507511A (ja) * | 1990-05-19 | 1993-10-28 | ザ・ダウ・ケミカル・カンパニー | 吸収剤構造物用水吸収性樹脂粒子 |
| JP2008538081A (ja) * | 2005-02-04 | 2008-10-09 | ザ プロクター アンド ギャンブル カンパニー | 改善された水吸収性材料を有する吸収性構造体 |
| JP2010116548A (ja) * | 2008-10-14 | 2010-05-27 | San-Dia Polymer Ltd | 吸収性樹脂粒子、この製造方法、これを含む吸収体及び吸収性物品 |
| JP2012219231A (ja) * | 2011-04-13 | 2012-11-12 | San-Dia Polymer Ltd | 吸収性樹脂粒子及びこの製造方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2909692B2 (ja) | 1993-02-24 | 1999-06-23 | 三洋化成工業株式会社 | 吸水剤 |
| JP2652316B2 (ja) | 1993-02-24 | 1997-09-10 | 三洋化成工業株式会社 | 吸水材 |
| US6090875A (en) * | 1996-02-16 | 2000-07-18 | The Dow Chemical Company | Dust control of absorbent polymers |
| JP2000212458A (ja) * | 1999-01-25 | 2000-08-02 | Sumitomo Seika Chem Co Ltd | 高吸水性樹脂粒子 |
| US7812082B2 (en) * | 2005-12-12 | 2010-10-12 | Evonik Stockhausen, Llc | Thermoplastic coated superabsorbent polymer compositions |
| US8409664B2 (en) * | 2010-06-28 | 2013-04-02 | The Procter & Gamble Company | Superabsorbent polymer particles coated with a hydrophilic elastomer and absorbent article comprising such particles |
| JP6402181B2 (ja) | 2014-05-23 | 2018-10-10 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを含む吸収体および吸収性物品 |
-
2015
- 2015-05-22 JP JP2016521161A patent/JP6402181B2/ja active Active
- 2015-05-22 MY MYPI2016703963A patent/MY177389A/en unknown
- 2015-05-22 WO PCT/JP2015/064741 patent/WO2015178481A1/ja not_active Ceased
- 2015-05-22 KR KR1020167036024A patent/KR101792968B1/ko active Active
- 2015-05-22 CN CN201580027096.3A patent/CN106459598B/zh active Active
- 2015-05-22 EP EP15795426.4A patent/EP3147331B1/en active Active
- 2015-05-22 US US15/313,148 patent/US10174174B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05507511A (ja) * | 1990-05-19 | 1993-10-28 | ザ・ダウ・ケミカル・カンパニー | 吸収剤構造物用水吸収性樹脂粒子 |
| JP2008538081A (ja) * | 2005-02-04 | 2008-10-09 | ザ プロクター アンド ギャンブル カンパニー | 改善された水吸収性材料を有する吸収性構造体 |
| JP2010116548A (ja) * | 2008-10-14 | 2010-05-27 | San-Dia Polymer Ltd | 吸収性樹脂粒子、この製造方法、これを含む吸収体及び吸収性物品 |
| JP2012219231A (ja) * | 2011-04-13 | 2012-11-12 | San-Dia Polymer Ltd | 吸収性樹脂粒子及びこの製造方法 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018132104A (ja) * | 2017-02-14 | 2018-08-23 | Jxtgエネルギー株式会社 | 高粘性流体組成物、高粘性流体組成物の製造方法及び振動エネルギー減衰装置 |
| JPWO2021117783A1 (ja) * | 2019-12-13 | 2021-06-17 | ||
| JP7337192B2 (ja) | 2019-12-13 | 2023-09-01 | 住友精化株式会社 | 被覆樹脂粒子の製造方法 |
| JPWO2022124137A1 (ja) * | 2020-12-09 | 2022-06-16 | ||
| WO2022124137A1 (ja) * | 2020-12-09 | 2022-06-16 | 住友精化株式会社 | 樹脂粒子組成物 |
| CN116568761A (zh) * | 2020-12-09 | 2023-08-08 | 住友精化株式会社 | 树脂粒子组合物 |
| WO2026005007A1 (ja) * | 2024-06-28 | 2026-01-02 | 株式会社日本触媒 | 吸水剤の製造方法および吸水剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3147331A4 (en) | 2017-12-13 |
| KR101792968B1 (ko) | 2017-11-02 |
| US10174174B2 (en) | 2019-01-08 |
| JP6402181B2 (ja) | 2018-10-10 |
| US20170190847A1 (en) | 2017-07-06 |
| CN106459598A (zh) | 2017-02-22 |
| JPWO2015178481A1 (ja) | 2017-05-25 |
| MY177389A (en) | 2020-09-14 |
| CN106459598B (zh) | 2019-04-16 |
| KR20170005491A (ko) | 2017-01-13 |
| EP3147331A1 (en) | 2017-03-29 |
| EP3147331B1 (en) | 2019-03-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6402181B2 (ja) | 吸水性樹脂粒子、これを含む吸収体および吸収性物品 | |
| US5716707A (en) | Water absorbent compositon and material | |
| US6534572B1 (en) | Compositions comprising a thermoplastic component and superabsorbent polymer | |
| CN1329445C (zh) | 超吸收性热塑性组合物和包括它的制品 | |
| JP4315680B2 (ja) | 吸収性組成物 | |
| CN100364493B9 (zh) | 吸水剂及其制备方法、使用所述吸水剂的吸收体以及吸收性物品 | |
| JP4188843B2 (ja) | 改善されたにおい制御を有するポリマー混合物 | |
| JP7278443B2 (ja) | 吸収性物品の製造方法 | |
| CZ20013129A3 (cs) | Práąkovité zesítěné absorpční polymery, způsob jejich výroby a jejich pouľití | |
| TW200640954A (ja) | ||
| MXPA01008930A (es) | Fibras superabsorbentes, de multiples componentes. | |
| TW200422330A (en) | Absorbent polymer structure with improved retention capacity and permeabilty | |
| CN107428948A (zh) | 水性液体吸收性树脂颗粒的制造方法、水性液体吸收性树脂颗粒、吸收体和吸收性物品 | |
| CN111868145B (zh) | 吸水性树脂颗粒及其制造方法 | |
| WO2020137241A1 (ja) | 吸水性樹脂粒子及びその製造方法 | |
| CN1708542B (zh) | 具有提高的保持容量和渗透性的吸收性聚合物结构 | |
| BR112019020501A2 (pt) | partículas de resina absorventes de água | |
| CN1708541A (zh) | 制备吸收性聚合物的两步混合方法 | |
| CN1939941B (zh) | 离子敏感性吸水性树脂 | |
| JPWO2019198821A1 (ja) | 吸水性シート、吸水性シートの製造方法および吸収性物品 | |
| CN111868144B (zh) | 吸水性树脂颗粒及其制造方法 | |
| WO2025231328A1 (en) | Hot melt adhesive compositions including a three polymer blend | |
| WO2024142684A1 (ja) | 創傷被覆材用粘着剤および創傷被覆材 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15795426 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016521161 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15313148 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20167036024 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015795426 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015795426 Country of ref document: EP |