WO2016029218A1 - Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation - Google Patents
Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation Download PDFInfo
- Publication number
- WO2016029218A1 WO2016029218A1 PCT/US2015/046585 US2015046585W WO2016029218A1 WO 2016029218 A1 WO2016029218 A1 WO 2016029218A1 US 2015046585 W US2015046585 W US 2015046585W WO 2016029218 A1 WO2016029218 A1 WO 2016029218A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- phenethylpiperidin
- substituted
- product
- opioid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- QYNQIJZXBWWVMO-UHFFFAOYSA-N CCC(NC1CCN(CCc2ccccc2)CC1)=O Chemical compound CCC(NC1CCN(CCc2ccccc2)CC1)=O QYNQIJZXBWWVMO-UHFFFAOYSA-N 0.000 description 1
- 0 CN(CC*(*)C1)CCC1(*)NC(*)=O Chemical compound CN(CC*(*)C1)CCC1(*)NC(*)=O 0.000 description 1
- BCEKLYJIVXGPLQ-UHFFFAOYSA-N NC1CCN(CCc2ccccc2)CC1 Chemical compound NC1CCN(CCc2ccccc2)CC1 BCEKLYJIVXGPLQ-UHFFFAOYSA-N 0.000 description 1
- BTUIFMCWPFMNRG-UHFFFAOYSA-N O=C(c1c[o]cc1)Cl Chemical compound O=C(c1c[o]cc1)Cl BTUIFMCWPFMNRG-UHFFFAOYSA-N 0.000 description 1
- WKVJYJUCSNOOAU-UHFFFAOYSA-N O=C(c1c[o]cc1)NC1CCN(CCc2ccccc2)CC1 Chemical compound O=C(c1c[o]cc1)NC1CCN(CCc2ccccc2)CC1 WKVJYJUCSNOOAU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4465—Non condensed piperidines, e.g. piperocaine only substituted in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4468—Non condensed piperidines, e.g. piperocaine having a nitrogen directly attached in position 4, e.g. clebopride, fentanyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- Substituted l-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha- adrenoreceptor modulators and method of their preparation.
- the invention relates to novel pharmacological compounds, and more specifically to the creation of a new class of small molecules which simultaneously exhibit high binding affinities to the ⁇ -, ⁇ -, and ⁇ -opioid receptors and the oc 2 - adrenoreceptor.
- the binding activity is believed to be antagonistic at least with respect to the ⁇ -opioid receptors.
- the invention also describes detailed novel methods for the preparation of representative compounds and a scheme for the synthesis of related compounds that bind to the opioid receptors and/or a 2 -adrenoreceptor.
- Opioid antagonists are drugs which bind to the opioid receptors with higher affinity than opioid agonists but do not activate the opioid receptors.
- Commonly known opioid antagonists include drugs such as, for example, naltrexone, naloxone, nelmefene, nalorphine, and nalbuphine.
- Opioid antagonists effectively block the receptor from the action of both naturally occurring agonists (e.g., morphine, codeine, thebaine) and synthetic agonists (e.g., fentanyl, pethidine, levorphanol, methadone, tramadol, dextropropoxyphene) and uses include counteracting life-threatening depression of the central nervous and respiratory systems and thus are used for emergency overdose and dependence treatment (e.g., naloxone).
- Naturally occurring agonists e.g., morphine, codeine, thebaine
- synthetic agonists e.g., fentanyl, pethidine, levorphanol, methadone, tramadol, dextropropoxyphene
- Opioid receptor antagonists are known to modulate numerous central and peripheral effects including those associated with opioid abuse, the development of opioid tolerance and dependence, opioid-induced constipation, alcohol and cocaine abuse, depression, and immune responses [1].
- the diverse therapeutic applications of ⁇ - opioid antagonists include opioid-overdose-induced respiratory depression, opioid and cocaine abuse, alcohol dependence, smoking cessation, obesity, psychosis[l ⁇ 19] and for the treatment of dyskinesia associated with Parkinson's disease [20-27].
- naloxone e.g., naltrexone, and nalorphine (a partial agonist)
- drugs e.g., naloxone, naltrexone, and nalorphine (a partial agonist)
- Alvimopan [13, 14] a peripherally acting ⁇ -opioid antagonist for the treatment of postoperative ileus— has received approval as new drug.
- some azabicyclohexane derivatives and series of bi(hetero)aryl ethers as biological tools have been proposed as new chemical entities in this class of compounds [15].
- antagonists of the adrenoreceptors bind to the adrenoreceptors and act to inhibit the action of those receptors.
- Alpha antagonists, or alpha-blockers may selectively act at the a i -adrenoreceptors or at the a 2 -adrenoreceptors, or they may non-selectively act at both receptors.
- oc-blockers include, for example, phenoxybenzamine and phentolamine (non-selective); alfuzosin and prazosin (ai -blockers); and atipamezole, idazoxan, mirtazapine and yohimbine (a 2 -blockers).
- -blockers have shown to be effective in the treatment of various medical conditions, including Raynaud's disease, hypertension, scleroderma, anxiety and panic disorders, and in the treatment of dyskinesia associated with Parkinson's disease.
- the present invention is based on the discovery of certain compounds exhibiting high binding affinity for the ⁇ -, ⁇ -, and ⁇ - opioid receptors and the 2 -adrenoreceptor.
- the compounds are believed to exhibit antagonistic activity at least with respect to ⁇ -opioid receptors.
- the compounds are structurally related to the fentanyl series of opioid receptor agonists. Processes for preparing these compounds are also included in this disclosure.
- R is H, substituted or unsubstituted Q-C10 alkyl, alkenyl, or alkynyl, or substituted or unsubstituted aryl or hetaryl;
- R 2 is H, -CH2O-C 1.
- R 3 is H, substituted or unsubstituted C1 -C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl; A is substituted or unsubstituted Q - C10 alkyl, alkylene, alkynyl; Ar or HetAr is substituted or unsubstituted monocyclic or polycyclic aromatic or heteroaromatic moiety; COOR 5 , or CON(R 6 )2, where R 5 and R 6 are H, substituted or unsubstituted C1 -C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl; the central nitrogen-containing ring is a substituted or unsubstituted 5- to 7-membered heterocyclic ring; and pharmaceutically acceptable salts of said compound.
- the prepared compound may belong to the series of N-(l -arylethyIpiperidin-4-yl)acylamides.
- the compound may be N-(l -phenethylpiperidin-4-yl)propionamide (Compound I, below).
- the compound may be the oxalate salt, or other pharmaceutically acceptable salt, of N-(l -phenethylpiperidin-4-yl)propionamide.
- a process for preparing a compound of formula III comprising the following steps: (a) reacting a cyclic ketone having a protecting group in a Grignard or Reformatsky reaction to obtain a first product; (b) reacting the product of step (a) in a Ritter reaction to obtain a second product; and (c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of formula III.
- a process for preparing a compound of formula III comprising the following steps: (a) reacting a cyclic ketone having a protecting group in a Strccker reaction to obtain a first product; (b) reacting the product of step (a) in a selective carbalkoxy group transformation to obtain a second product; and (c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of formula III.
- a process for preparing N-( 1 -phenethylpiperidin-4-yl) propionamide comprising the following steps: (a) reacting phenethylpiperidin-4-one with hydroxylamine hydrochloride in ethanol in the presence of a base, to produce l-phenethylpiperidin-4-one oxime; (b) reducing the oxime obtained in step (a) with iso-amyl alcohol and sodium metal to produce l -phenethylpipcridin-4- amine; and (c) acylating the product of step (b) with propionic acid chloride in chloroform in the presence of triethylamine to produce N-(l -phenethylpiperidin-4- y propionamide.
- N-(l -phenethylpiperidin-4-yl)propionamide may optionally be further treated with oxalic acid to obtain N-(l -phenethylpiperidin-4-yl)propionamide oxalate.
- Fig. 1 is a (LC/MS) plot of a preferred compound designated HVC-3.
- a “salt” is any acid addition salt, preferably a pharmaceutically acceptable acid addition salt, including but not limited to, halogenic acid salts such as hydrobromic, hydrochloric, hydrofluoric and hydroiodic acid salt; an inorganic acid salt such as, for example, nitric, perchloric, sulfuric and phosphoric acid salt; an organic acid salt such as, for example, sulfonic acid salts (methanesulfonic, trifluoromethan sulfonic, ethanesulfonic, benzenesulfonic or -toluenesulfonic), acetic, malic, fumaric, succinic, citric, benzoic, gluconic, lactic, mandelic, mucic, pamoic, pantothenic, oxalic and maleic acid salts; and an amino acid salt such as aspartic or glutamic acid salt.
- halogenic acid salts such as hydrobromic, hydrochloric, hydrofluor
- the acid addition salt may be a mono- or di-acid addition salt, such as a di-hydrohalogenic, di-sulfuric, di-phosphoric or di-organic acid salt.
- the acid addition salt is used as an achiral reagent which is not selected on the basis of any expected or known preference for interaction with or precipitation of a specific optical isomer of the products of this disclosure.
- “Pharmaceutically acceptable salt” is meant to indicate those salts which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of a patient without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salts are well known in the art. For example, Berge et al. (1977) J. Pharm. Sciences, vol. 6. 1 -19, which is hereby incorporated by reference in its entirety, describes pharmaceutically acceptable salts in detail.
- Modulation is meant to refer to the binding activity of a compound with respect to a particular receptor.
- the binding activity of the compound, or “modulator,” may be that of an agonist, inverse agonist, antagonist, allosteric regulator, positive allosteric modulator, negative allosteric modulator, or any other type of ligand-receptor interaction that is known in the art.
- the invention also relates to processes for preparing compounds of general formula III, which are pharmacologically active compounds:
- Representative compounds of the present invention also include the following compounds, wherein the central nitrogen containing ring is a substituted or unsubstituted 5- to 7-membered heterocyclic ring:
- R is H, substituted or unsubstituted Ci-Cio alkyl, alkenyl, alkynyl, or substituted or unsubstituted aryl or hetaryl;
- R 2 is H, -CH2O-C 1 .4 alkyl, etc.; COO-C alkyl, etc.; -CONR4 etc.;
- R is H, substituted or unsubstituted C
- A is substituted or un-substituted Ci-C 10 alkyl, alkylene, alkynyl;
- Ar or HetAr is substituted or un-substituted monocyclic or polycyclic aromatic or heteroaromatic moiety
- R 5 and R 6 are H, substituted or unsubstituted C r C 10 alkyl, alkylene, alkynyl, or substituted or un-substituted aryl or hetaryl.
- compounds of formula (III) can be prepared according to the following general schemes (wherein PG is a protecting group):
- these schemes generally include transformations of different starting cyclic ketones (e.g., a variety of piperidin-4-ones, pyrrolidin-3-ones and azepan-4-one) to desired compounds of formula (III) via, for example:
- N-(l -phenethylpiperidin-4-yl)propionamide (I) may be prepared:
- N-(l -phenethylpiperidin-4-yl)propionamide (I) may be transformed to oxalate by treating with a molar equivalent of oxalic acid in ethanol:
- Tables 1 -3) in the attached Appendix 1 incorporated herein by reference include data from binding assays performed with the N-( 1 -phenethylpiperidin- 4-yl)propionamide compound (I). This data demonstrates the high binding affinities of the compounds of the invention for ⁇ -, ⁇ -, and ⁇ - opioid receptors and for cc2-adreno- receptors.
- Table 1 illustrates the results of binding assays performed with the N-(l - phenethylpiperidin-4-yl)propionamide (compounds Rl and R2) and various receptors.
- N-( 1 - phenethylpiperidin-4-yl)propionamide demonstrated the highest percentage binding inhibition of control specific binding with respect to the a2B adrenoreceptor (74% and 55%); the ⁇ -opioid receptor (44% and 69%); the ⁇ -opioid receptor ( 107% and 104%); the ⁇ -opioid receptor (98% and 99%).
- Table 2 contains reference compound data for the various receptors used in the binding assays.
- Table 3 provides summary results of the binding assays, showing the receptors from Table 1 for which the test compound demonstrated the highest percentage binding inhibition of control specific binding.
- Appendix 2 incorporated herein by reference includes x-ray crystallography data for a representative compound of the invention, N-( l -phenethylpiperidin-4- yl)propionamide oxalate.
- Appendix 3 incorporated herein by reference includes data showing that a representative compound of the invention, N-(l -phenethylpiperidin-4-yl)propionamide, conforms to "Lipinski's rule of five," which provides a general rule of thumb for evaluating the activity of an orally administered drug.
- the compounds of the present invention may be utilized in various combinations
- the present invention also includes salts, and particularly pharmaceutically acceptable salts, of the disclosed compounds, as well as processes for preparing the salt forms of the disclosed compounds.
- l -Phenethylpiperidin-4-one oxime (6.54 g (0.03 mol)) was dissolved in 100 mL of dry i-AmOH on heating. A ten-fold excess of sodium (6.9 g (0.3 mol)) was slowly (1 hour) added to the stirred solution in small pieces, while the temperature was maintained around 1 10°. The solution was stirred on heating at 1 10° for two hours and left to cool to room temperature. 1 50 mL of ether, followed by 75 mL of water, was then added to the solution.
- HCV-3 The compound designated as HCV-3 was then subject to cellular functional assay and results reported below:
- alpha 28 (h 1814 HCV-3 1 1000233221.0E-05 -28 116.8 138.7 127.8 yohimbine 3.7E-07 4.8E-08
- Compound HCV-3 also was tested for hERG inhibition. Over the concentration range tested (up to 25 micromolar) no dose-response was obtained. Therefore the inhibition IC50 was considered as >25 micromolar. There was a hint of some inhibition at the top concentration of 25 micromolar, with 32.5% inhibition observed (insufficient to generate an IC50 value). As such, this compound is categorised as having weak or no hERG inhibition. The control compounds behaved as expected in the assay.
- Compound HCV-3 also was tested for CYP inhibition, and was found to inhibit CYP2D6, and to weakly inhibit CYP2C19. However, with CYP2C19 the inhibition was too weak to generate an IC50 value, and we observed just 36.4% inhibition at the top concentration o 25 micromolar. With CYP2D6, an IC50 of 4.2 micromolar was observed. Thus, this compound was considered to be a moderate CYP2D6 inhibitor, and a weak CYP2C 19 inhibitor. No inhibition was observed at CYP2B6, CYP2C9, CYP3A4 (with either substrate), CYP2C8 or CYP1A2. The significance of this CYP2D6 inhibition will depend on the levels of the compound in vivo.
- Compound HCV-3 also was tested in cellular and nuclear receptor functional assays, and the results reported in Appendix 4, incorporated herein by reference. Compound HCV-3 was also subjected to AMES testing, and the results reported in Appendix 5, incorporated herein by reference.
- the compound HCV-3 was negative for genotoxicity against both strains used in this assay (TA98 and TA 100) up to a maximum tested concentration of l mg/mL, in both the absence and presence of S9 metabolic activation.
- the assay controls behaved as expected.
- Compound HCV-3 also was subjected to in vitro metabolic disposition in mouse, rat, monkey and human microsomes.
- the test compound was incubated with pooled liver microsomes, since drip stability in liver microsomes can be predictive of drug stability in vivo. Aliquots were taken at 0, 5, 1 5, 30 and 45 minutes and quenched immediately. The samples were extracted and analyzed by LC-MS/MS.
- Compound HCV-3 was observed to have low clearance in human, monkey and mouse microsomes, and moderate clearance in rat.
- Compound HCV-3 also was subjected to MDCK permeability assay.
- the compound was observed to be highly permeable in the MDCK assay. There was a slight difference between the plus and minus inhibitor data in terms of the efflux ratio obtained (1.48 minus inhibitor, versus 0.929 plus inhibitor).
- a ratio of greater than 2 generally indicates that efflux, i.e., blood brain barrier permeability, is occurring.
- the control compounds behaved as expected, with prazosin (a P-gp substrate) showing efflux in the absence of Cyclosporin A, which was inhibited in its presence.
- a solution containing 6.00 g (1 equiv., 0.027 mol) of i -phenethylpiperidin-4-one oxime dissolved in 90 mL of iso-amyl alcohol was prepared and heated to approximately 1 10 °C. 6.21 g (10 equiv., 0.27 mol) of Na metal was then added slowly to the reaction mixture. After addition of Na, the reaction mixture was allowed to cool to room temperature and stirred until the reaction mixture turned into a thick slurry. The slurry was dissolved in 50 mL of ethyl acetate and 25 mL of I LO.
- the reaction mixture was then quenched with 0.5 M KHSO 4 solution followed by the addition of dichloromethane.
- the organic and aqueous layers were separated, and the aqueous layer was extracted with dichloromethane (3 x 5 mL) followed by washing with NaHCO;, solution and Brine.
- the organic extracts were then dried over anhydrous magnesium sulfate.
- the crystals submitted were small needles which diffracted poorly. Reflections were streaky, indicating that the crystal was not single but comprised of more than one not-exactly-oriented component. Diffraction was observed only to about 1 A resolution. Because of the poor resolution, A and B level cheokcif alerts are generated. The quality of the structure is such that the identity of the molecule and its conformation is confirmed, but derived parameters (bond distances, angles, thermal motion) are not reliable.
- Figure 2 shows the contents of the unit cell, which also includes an oxalate molecule.
- Figure 3 shows the unit cell viewed down the crystal lographic a axis.
- the hydrogen bonding network in the unit cell connects the organic molecule to oxalate along the crystallographic b axis (ydrogen bonds between the Nl and 02 of the oxalate and between N2 and 04 of the oxalate).
- the oxalate molecule are connected by hydrogen bonds 04 and 05 of the oxalate, along the crystallographic a axis.
- C8 has been modeled with disorder in two positions. There is unmodeled (probably rotational) disorder in the aromatic ring (C1 1 -C 1 6). Both distance and planarity restraints were applied to this ring during refinement. Not all hydrogen atoms were visible in the electron density map. Because of the low resolution and streaky diffraction pattern, the structure could not be refined without constraints. Constraints used in the refinement are included at the end of the report.
- Figure 1 The molecule with displacement ellipsoids at the 50% probability level. C8 is disordered, the minor component has been removed for clarity. An oxalate molecule also in the unit cell is not shown in this figure. 2
- Figure 2 The contents of the asymmetric unit with displacement ellipsoids at the 50% probability level. B8 is disordered, and the minor compont is not shown. Fog has been added to show depth.
- FIG. 3 The contents of the unit cell viewed down the short a axis. Hydrogen bonds are shown between the Nl and 02 of the oxalate. Hydrogen bonds also exist between N2 and 04 of the oxalate and between 04 and 05 of the oxalate, connecting the oxalate molecules along the crystallographic a axis (looking down into the page). 46585
- the APEXII DUO was purchased with funding from NSF grant CHE-0741837.
- U eq is defined as 1/3 of of the trace of the orthogonalised Uu tensor.
- Needle shaped crystals of C18H27N2O5 were submitted for structure determination. The crystals were small and were grown together. Even apparently single crystals were stacks of needles A crystal was selected and attached to a Micromount using paratone oil, then mounted on a Bruker Kappa APEX-11 Duo diffractometer. The crystal was kept at 150.0 K during data collection. Using Olex2 [1 ], the structure was solved with the ShelXS [2] structure solution program using Direct Methods and refined with the ShelXL [3] refinement package using Least Squares minimisation.
- Lipinski's rule of five also known as the Pfizer's rule of five or simply the Rule of five (R05) is a rule of thumb to evaluate druglikeness or determine if a chemical compoundwith a
- the rule describes molecular properties important for a drug's pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion ("AD E"). However, the rule does not predict if a compound is pharmacologically active.
- Lipinski's rule states that, in general, an orally active drug has no more than one violation of the following criteria:
- HCV-3 was tested at 1.0E-05 M.
- Cellular agonist effect was calculated as a % of control response to a known reference agonist for each target and cellular antagonist effect was calculated as a % inhibition of control reference agonist response for each target.
- Results showing an inhibition or stimulation higher than 50% are considered to represent significant effects of the test compounds. Such effects were not observed at any of the receptors studied.
- Results showing a stimulation or an inhibition higher than 50% are considered to represent significant effects of the test compounds. 50% is the most common cut-off value for further investigation (determination of EC 50 or IC 50 values from concentration-response curves).
- Results showing a stimulation or an inhibition between 25% and 50% are indicative of weak to moderate effects (in some assays, they may be confirmed by further testing as they are within a range where more inter-experimental variability can occur).
- Results showing a stimulation or an inhibition lower than 25% are not considered significant and mostly attributable to variability of the signal around the control level.
- control response and as a percent inhibition of control agonist response
- the EC 5 o values (concentration producing a half-maximal response) and IC 50 values (concentration causing a half-maximal inhibition of the control agonist response) were determined by non-linear regression analysis of the concentration-response curves generated with mean replicate values using Hill equation curve fitting
- the purpose of this study was to evaluate the mutagenicity potential of a test article (HCV-3) in the Ames assay using the microplate fluctuation (MPF) method.
- the top concentration in the dose response reflects the highest soluble concentration of the test article in Ames assay conditions.
- S9 fraction from the livers of Aroclor 1254-treated rats is included in the incubation at a final concentration of 4.5%.
- An NADPH-regenerating system is also included to ensure a steady supply of reducing equivalents.
- S. typhimurium TA 100 hisG45, rfa, uvrB / pKM l O l ; detects base-pair substitutions.
- Test article HCV-3 was assessed for its mutagenic potential in the Ames reverse mutation assay. This test was performed in the absence and presence of S9 metabolic activation. HCV-3 was found to be negative for genotoxicity against both strains used in this study (TA98 and TA 100) up to a maximum tested concentration of 1 mg/ml. The positive controls behaved as expected.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
The invention provides compounds that bind with high affinities to the μ-, δ- and κ- opioid receptors and α2 - adrenoreceptor. In addition to providing these compounds with novel pharmacological binding properties, the invention also describes detailed novel methods for the preparation of representative compounds and a scheme for the synthesis of related compounds that bind to the opioid receptors and/or α2 - adrenoreceptor.
Description
Substituted l-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha- adrenoreceptor modulators and method of their preparation. The invention relates to novel pharmacological compounds, and more specifically to the creation of a new class of small molecules which simultaneously exhibit high binding affinities to the μ-, δ-, and κ-opioid receptors and the oc2- adrenoreceptor. The binding activity is believed to be antagonistic at least with respect to the μ-opioid receptors. In addition to providing these compounds with novel pharmacological binding properties, the invention also describes detailed novel methods for the preparation of representative compounds and a scheme for the synthesis of related compounds that bind to the opioid receptors and/or a2-adrenoreceptor.
Opioid antagonists are drugs which bind to the opioid receptors with higher affinity than opioid agonists but do not activate the opioid receptors. Commonly known opioid antagonists include drugs such as, for example, naltrexone, naloxone, nelmefene, nalorphine, and nalbuphine. Opioid antagonists effectively block the receptor from the action of both naturally occurring agonists (e.g., morphine, codeine, thebaine) and synthetic agonists (e.g., fentanyl, pethidine, levorphanol, methadone, tramadol, dextropropoxyphene) and uses include counteracting life-threatening depression of the central nervous and respiratory systems and thus are used for emergency overdose and dependence treatment (e.g., naloxone). There are many excellent reviews dedicated to different aspects of opioid antagonists [28-46].
Opioid receptor antagonists are known to modulate numerous central and peripheral effects including those associated with opioid abuse, the development of opioid tolerance and dependence, opioid-induced constipation, alcohol and cocaine abuse, depression, and immune responses [1]. The diverse therapeutic applications of μ- opioid antagonists include opioid-overdose-induced respiratory depression, opioid and cocaine abuse, alcohol dependence, smoking cessation, obesity, psychosis[l ~19] and for the treatment of dyskinesia associated with Parkinson's disease [20-27].
The few opioid antagonists currently on the market are represented by very few drugs (e.g., naloxone, naltrexone, and nalorphine (a partial agonist)) that have been shown to have therapeutic utility in a variety of indications. During last two decades only Alvimopan [13, 14]— a peripherally acting μ-opioid antagonist for the treatment of postoperative ileus— has received approval as new drug. In addition, some
azabicyclohexane derivatives and series of bi(hetero)aryl ethers as biological tools have been proposed as new chemical entities in this class of compounds [15].
Every chemical class of compounds with opioid-agonist activity has a structurally similar opioid-antagonist pair. Agonist-antagonist transformation in any of these cases takes place as a result of a small change in the structure of the agonist. The only exceptions, where the corresponding change for agonist-antagonist transformations has not been found, are the compounds of the fentanyl series.
Since the discovery of the "army" of opioid agonists of the fentanyl series (sufentanyl, alfentanyl, carfentanyl, remifentanyl, etc.) beginning in the 1960s, a structurally corresponding antagonist has not been found for any of these compounds. Thus, for decades there has been an evident gap in the art with respect to a possible specific structural change that could make possible the transformation of powerful opioid agonist properties of compounds of fentanyl series into powerful antagonists.
Similar to the general action of the opioid antagonists, antagonists of the adrenoreceptors (adrenergic receptors) bind to the adrenoreceptors and act to inhibit the action of those receptors. Alpha antagonists, or alpha-blockers, may selectively act at the a i -adrenoreceptors or at the a2-adrenoreceptors, or they may non-selectively act at both receptors. Commonly known oc-blockers include, for example, phenoxybenzamine and phentolamine (non-selective); alfuzosin and prazosin (ai -blockers); and atipamezole, idazoxan, mirtazapine and yohimbine (a2-blockers). Generally, -blockers have shown to be effective in the treatment of various medical conditions, including Raynaud's disease, hypertension, scleroderma, anxiety and panic disorders, and in the treatment of dyskinesia associated with Parkinson's disease.
The present invention is based on the discovery of certain compounds exhibiting high binding affinity for the μ-, δ-, and κ- opioid receptors and the 2-adrenoreceptor. The compounds are believed to exhibit antagonistic activity at least with respect to μ-opioid receptors. The compounds are structurally related to the fentanyl series of opioid receptor agonists. Processes for preparing these compounds are also included in this disclosure.
In one embodiment of the disclosure, a compound having the formula
 etAr), or COOR6, or COON(R6)2 (III)
etAr), or COOR6, or COON(R6)2 (III)
is disclosed, wherein R, is H, substituted or unsubstituted Q-C10 alkyl, alkenyl, or alkynyl, or substituted or unsubstituted aryl or hetaryl; R2 is H, -CH2O-C 1. alkyl; COO- C1 -4 alkyl; -CONR4; R3 is H, substituted or unsubstituted C1 -C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl; A is substituted or unsubstituted Q - C10 alkyl, alkylene, alkynyl; Ar or HetAr is substituted or unsubstituted monocyclic or polycyclic aromatic or heteroaromatic moiety; COOR5, or CON(R6)2, where R5 and R6 are H, substituted or unsubstituted C1 -C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl; the central nitrogen-containing ring is a substituted or unsubstituted 5- to 7-membered heterocyclic ring; and pharmaceutically acceptable salts of said compound.
In a preferred embodiment, the prepared compound may belong to the series of N-(l -arylethyIpiperidin-4-yl)acylamides. In another embodiment, the compound may be N-(l -phenethylpiperidin-4-yl)propionamide (Compound I, below). In yet another embodiment the compound may be the oxalate salt, or other pharmaceutically acceptable salt, of N-(l -phenethylpiperidin-4-yl)propionamide.
In another embodiment, a process for preparing a compound of formula III is provided. The process comprising the following steps: (a) reacting a cyclic ketone having a protecting group in a Grignard or Reformatsky reaction to obtain a first product; (b) reacting the product of step (a) in a Ritter reaction to obtain a second product; and (c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of formula III.
In yet another embodiment, a process for preparing a compound of formula III is provided, comprising the following steps: (a) reacting a cyclic ketone having a protecting group in a Strccker reaction to obtain a first product; (b) reacting the product of step (a) in a selective carbalkoxy group transformation to obtain a second product; and (c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of formula III.
In another embodiment, a process for preparing N-( 1 -phenethylpiperidin-4-yl) propionamide is provided, comprising the following steps: (a) reacting
phenethylpiperidin-4-one with hydroxylamine hydrochloride in ethanol in the presence of a base, to produce l-phenethylpiperidin-4-one oxime; (b) reducing the oxime obtained in step (a) with iso-amyl alcohol and sodium metal to produce l -phenethylpipcridin-4- amine; and (c) acylating the product of step (b) with propionic acid chloride in chloroform in the presence of triethylamine to produce N-(l -phenethylpiperidin-4- y propionamide. The N-(l -phenethylpiperidin-4-yl)propionamide may optionally be further treated with oxalic acid to obtain N-(l -phenethylpiperidin-4-yl)propionamide oxalate.
These and other embodiments, features and advantages of the present invention will become more fully apparent when read in conjunction with the following detailed description taken in conjunction with the accompanying drawings, wherein
Fig. 1 is a (LC/MS) plot of a preferred compound designated HVC-3.
For the purposes of this disclosure, a "salt" is any acid addition salt, preferably a pharmaceutically acceptable acid addition salt, including but not limited to, halogenic acid salts such as hydrobromic, hydrochloric, hydrofluoric and hydroiodic acid salt; an inorganic acid salt such as, for example, nitric, perchloric, sulfuric and phosphoric acid salt; an organic acid salt such as, for example, sulfonic acid salts (methanesulfonic, trifluoromethan sulfonic, ethanesulfonic, benzenesulfonic or -toluenesulfonic), acetic, malic, fumaric, succinic, citric, benzoic, gluconic, lactic, mandelic, mucic, pamoic, pantothenic, oxalic and maleic acid salts; and an amino acid salt such as aspartic or glutamic acid salt. The acid addition salt may be a mono- or di-acid addition salt, such as a di-hydrohalogenic, di-sulfuric, di-phosphoric or di-organic acid salt. In all cases, the acid addition salt is used as an achiral reagent which is not selected on the basis of any expected or known preference for interaction with or precipitation of a specific optical isomer of the products of this disclosure.
"Pharmaceutically acceptable salt" is meant to indicate those salts which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of a patient without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well known in the art. For example, Berge et al. (1977) J. Pharm. Sciences, vol. 6. 1 -19, which is hereby incorporated by reference in its entirety, describes pharmaceutically acceptable salts in detail.
"Modulation" is meant to refer to the binding activity of a compound with respect to a particular receptor. The binding activity of the compound, or "modulator," may be
that of an agonist, inverse agonist, antagonist, allosteric regulator, positive allosteric modulator, negative allosteric modulator, or any other type of ligand-receptor interaction that is known in the art.
In this invention we disclose a new class of molecules that simultaneously bind with high affinity to opioid μ-, δ-, κ- receptors and also to -adrenoreceptors, thereby exhibiting modulation-type interactions with those receptors. The interaction of the molecules with μ-receptors is believed to have the character of antagonist action, based at least in part on the observed high affinity binding of the molecules with respect to the μ-receptors.
Although not wishing to be bound by theory, it appears that the principal structural change for agonist-antagonist transformation is the removal of a phenyl group from an N-phenylpropionamide fragment of fentanyl. This transformation is depicted below, wherein N-(l -phenethylpiperidin-4-yl)-N-phenylpropionamide (II) is transformed to N-(l -phenethylpiperidin-4-yl)-N-propionamide (I), causing a transformation of μ-agonist properties to μ-antagonist with simultaneous modulation of delta-, kappa- and alpha-receptors:

Fentanil (II) μ-Opioid Antagonist/a-agonist (I)
The invention also relates to processes for preparing compounds of general formula III, which are pharmacologically active compounds:

Ar , or (HetAr), or COOR5, or COON(R6)2 (III)
Representative compounds of the present invention also include the following compounds, wherein the central nitrogen containing ring is a substituted or unsubstituted 5- to 7-membered heterocyclic ring:

These compounds may include the following structural and functional groups:
• R] is H, substituted or unsubstituted Ci-Cio alkyl, alkenyl, alkynyl, or substituted or unsubstituted aryl or hetaryl;
• R2 is H, -CH2O-C1.4 alkyl, etc.; COO-C alkyl, etc.; -CONR4 etc.;
• R ( is H, substituted or unsubstituted C| -Cio alkyl, alkylene, alkynyl, or
substituted or unsubstituted aryl or hetaryl;
• A is substituted or un-substituted Ci-C10 alkyl, alkylene, alkynyl;
» Ar or HetAr is substituted or un-substituted monocyclic or polycyclic aromatic or heteroaromatic moiety; and
• COOR5, or CON(R6)2, where R5 and R6 are H, substituted or unsubstituted Cr C 10 alkyl, alkylene, alkynyl, or substituted or un-substituted aryl or hetaryl.
In certain embodiments of the invention, compounds of formula (III) can be prepared according to the following general schemes (wherein PG is a protecting group):

As shown above, these schemes generally include transformations of different starting cyclic ketones (e.g., a variety of piperidin-4-ones, pyrrolidin-3-ones and azepan-4-one) to desired compounds of formula (III) via, for example:
a) Grignard or Reformatsky type reactions;
b) a Strecker type reaction;
c) a Ritter type reaction;
d) selective carbalkoxy group transformations, deprotection, and further appropriate acylation or alkylation;
e) selective carbalkoxy group transformations; or
f) deprotection with further appropriate acylation or alkylation.
The process for the preparation of the first example of μ-opioid modulator/ot- modulator N-(l-phenethylpiperidin-4-yl)propionamide and its salt is described in the present invention in detail below (Scheme 1 ):

(IV) (V) (VI) (I) ( VII )
The synthetic procedure starts with the commercially available 1 - phenethylpiperidin-4-one (IV) of formula:
 which may be reacted with hydroxylamine hydrochloride in ethanol in the presence of base, to give 1 -phenethylpiperidin-4-one oxime (V) of formula:
which may be reacted with hydroxylamine hydrochloride in ethanol in the presence of base, to give 1 -phenethylpiperidin-4-one oxime (V) of formula:


By treating of compound (VI) with propionic acid chloride in chloroform in the presence of triethylamine the desired N-(l -phenethylpiperidin-4-yl)propionamide (I) may be prepared:

The obtained N-(l -phenethylpiperidin-4-yl)propionamide (I) may be transformed to oxalate by treating with a molar equivalent of oxalic acid in ethanol:

The tables (Tables 1 -3) in the attached Appendix 1 incorporated herein by reference include data from binding assays performed with the N-( 1 -phenethylpiperidin- 4-yl)propionamide compound (I). This data demonstrates the high binding affinities of the compounds of the invention for μ-, δ-, and κ- opioid receptors and for cc2-adreno- receptors. Table 1 illustrates the results of binding assays performed with the N-(l - phenethylpiperidin-4-yl)propionamide (compounds Rl and R2) and various receptors. As can be seen in the table, at a test concentration of 1 .0E-05 M, N-( 1 - phenethylpiperidin-4-yl)propionamide demonstrated the highest percentage binding inhibition of control specific binding with respect to the a2B adrenoreceptor (74% and 55%); the δ-opioid receptor (44% and 69%); the κ-opioid receptor ( 107% and 104%); the μ-opioid receptor (98% and 99%). Table 2 contains reference compound data for the various receptors used in the binding assays. Finally, Table 3 provides summary results of the binding assays, showing the receptors from Table 1 for which the test compound demonstrated the highest percentage binding inhibition of control specific binding.
Appendix 2 incorporated herein by reference includes x-ray crystallography data for a representative compound of the invention, N-( l -phenethylpiperidin-4- yl)propionamide oxalate.
Appendix 3 incorporated herein by reference includes data showing that a representative compound of the invention, N-(l -phenethylpiperidin-4-yl)propionamide, conforms to "Lipinski's rule of five," which provides a general rule of thumb for evaluating the activity of an orally administered drug.
The compounds of the present invention may be utilized in various
pharmaceutical and medical applications in which the use of a compound exhibiting high binding affinities for μ-, δ- or κ- opioid receptors and/or a2-adreno-receptors is
indicated. The compounds may be of particular use in applications in which the use of a μ-, δ- or κ- opioid receptor modulator, including particularly a μ· opioid receptor antagonist, and/or an x2 -ad rcn orece pt or modulator is indicated. Accordingly, in certain embodiments, the present invention also includes salts, and particularly pharmaceutically acceptable salts, of the disclosed compounds, as well as processes for preparing the salt forms of the disclosed compounds.
Examples
The following examples illustrate the preparation of N-(l -phenethylpiperidin-4- yl)propionamide and its oxalate salt form, N-(l -phenethylpiperidin-4-yl)propionamide oxalate, in accordance with the present disclosure.
Example 1.
l -Phenethylpiperidin-4-one oxime (Compound V)
1 -Phenethylpiperidin-4-one (10.15 g (0.05 mol) dissolved in 60 mL of ethanol) was added drop-wise at 0°C to a solution of hydroxylamine in water. The water solution of hydroxylamine was preliminarily prepared by adding at 0°C in portions 13.8 g (0.1 mol) of 2CO3 to the solution of 6.95 g (0.1 mol) hydroxylamine hydrochloride in 50 mL of water.
The mixture was set aside for a night. Ethanol was evaporated under slight vacuum. Water (-100 mL) was added, and the mixture was stirred on ice bath for an hour. The separated solid product was filtered, washed with water and allowed to air-dry. The crude oxime (10.71 g (98.25%), m.p. 132-134°C) was reserved for use in the next reaction without further purification. Analysis with electrospray ionization mass spectrometry (MS (ESI)) resulted in a peak at 219.1 (MH+).
Example 2
l -Phenethylpiperidin-4-amine (Compound VI)
l -Phenethylpiperidin-4-one oxime (6.54 g (0.03 mol)) was dissolved in 100 mL of dry i-AmOH on heating. A ten-fold excess of sodium (6.9 g (0.3 mol)) was slowly (1 hour) added to the stirred solution in small pieces, while the temperature was maintained around 1 10°. The solution was stirred on heating at 1 10° for two hours and left to cool to room temperature. 1 50 mL of ether, followed by 75 mL of water, was then added to the solution. The organic layer was separated and dried on gSCv After evaporation of solvents under slight vacuum, the product was distilled to give 4.3 g (70%) of 1-
phenethylpiperidin-4-amine (VI) with a boiling point of 138-142°/! .5mm. MS (ESI): 205.0 (MH+).
Example 3
N-(l-Phenethylpiperidin-4-yl)propionamide (Compound I)
Propionyl chloride (2.775 g (0.03 mol)) in 5.55 mL of CHC13 was added drop- wise on stirring to the cooled (0°C) solution of 4.08 g (0.02 mol) l -phenethylpiperidin-4- amine and 3.03 g (0.03 mol) of Et3N in 30 mL of CTICI3 The mixture was left to come to room temperature and stirred overnight. After working up with 5% solution of NaHCO.i (2.52 g (0.03 mol)) in 47.88 H20, the organic layer was separated, washed with water and dried on MgSCv After evaporation of solvents under slight vacuum, the residue was crystallized from hexane to give 4.9 g (94%) of
N-(l-phenethylpiperidin-4-yl)propionamide (1) with m.p.134-1350. MS (ESI): 261.2 (MH+).
• The results of proton NMR spectroscopy were as follows:
Ή NMR (600 MHz, CDC13): δ 7.27 (t, J = 7.4 Hz, 21 1), 7. 19 (m, 3H), 5.32 (d, J =
7.4 Hz, 1H), 3.82 (qt, J = 7.8, 4.2 Hz, 1H), 2.92 (dt, J = 1 1.8, 3.4 Hz, 2H), 2.79 (m*, 2H), 2.59 (m*, 2H), 2.1 9 (q, J = 7.5 Hz, 2H), 2.18 (m, 2H), 1.95 (dtd, J = 12.4, 4.4, 1.7 Hz, 2H), 1 .46 (qd, J = 1 1 .7, 3.8 Hz, 2H), 1.15 (t, J = 7.5 Hz, 3H). * these two multiplets arise from two methylene groups that have magnetically inequivalent protons AA'BB' with 2JAA- - ¾Β' = 12 Hz, 3JAB = 3JA'B' = 1 1 Hz,
3JAB' = 3JA'B = 4 Hz, consistent with a preferred anti conformation for the phenyl and piperidine rings
• The results of carbon- 1 NMR spectroscopy were as follows:
13C NMR (1 50 MHz, CDC13): δ 173.0, 140.2, 128.6, 128.4, 126.0, 60.4, 52.3, 46.3, 33.7, 32.3, 29.8, 9.9.
Example 4
N-(l-Phenethylpiperidin-4-yl)propionamide oxalate (Compound VII)
Oxalic acid (1 g (0.01 1 mol)) in 10 mL of ethanol was added drop-wise to the solution of 2.93 g (0.01 1 mol) of N-(l-phenethylpiperidin-4-yl)propionamide (I) in 29.3 mL of ethanol. The mixture was set aside overnight. The obtained crystals were then separated and dried in a desiccator over P2O5 to give 3.5 g of N-(l -phenethylpiperidin-4-
yDpropionamide oxalate (VII) with m.p. 216-218 (MS (ESI): 261 .2 ([M + 1 1]) - see Fig. 1
The compound designated as HCV-3 was then subject to cellular functional assay and results reported below:
Cellular functional assays
ExperimenlAssay Catalog Re1 Client Com Batch Compound Test Conce % of Contri % of Control Agonist Response Reference 1 EC50 Ref (M)
1st 2nd Mean
27/07/201! alpha 2B (It 1813 HCV-3 1 1000233221.0E-05 7.1 9.2 5.1 7.1 dexmedetc 1.3E-08 27/07/201! kappa (K02071 HCV-3 1 1000233221.0E-05 -4.2 -10.3 1.8 -4.2 U 50488 1.6E-09 27/07/201! mu (MOP) 1392 HCV-3 1 1000233221.0E-05 33.5 28.1 38.9 33.5 DAMGO 4.2F-09
Cellular functional assays
Experimeni Assay Catalog ReiClient Com Batch Compound Test Conce % Inhibitioi Agonist Response (% of Control) Reference ' IC50 Ref [h Kb Ref |M)
1st 2nd Mean
27/07/201! alpha 28 (h 1814 HCV-3 1 1000233221.0E-05 -28 116.8 138.7 127.8 yohimbine 3.7E-07 4.8E-08
27/07/201! kappa (KO 2072 HCV-3 1 1000233221.0E-05 6 112.4 76.5 94.4 nor-BNI 4.3E-10 7.2E-11
27/07/201! mu (MOP) 1393 HCV-3 1 1000233221.0E-05 -1 100.1 101.8 101.0 CTOP 2.1E-07 2.3E-08
Compound HCV-3 also was tested for hERG inhibition. Over the concentration range tested (up to 25 micromolar) no dose-response was obtained. Therefore the inhibition IC50 was considered as >25 micromolar. There was a hint of some inhibition at the top concentration of 25 micromolar, with 32.5% inhibition observed (insufficient to generate an IC50 value). As such, this compound is categorised as having weak or no hERG inhibition. The control compounds behaved as expected in the assay.
Compound HCV-3 also was tested for CYP inhibition, and was found to inhibit CYP2D6, and to weakly inhibit CYP2C19. However, with CYP2C19 the inhibition was too weak to generate an IC50 value, and we observed just 36.4% inhibition at the top concentration o 25 micromolar. With CYP2D6, an IC50 of 4.2 micromolar was observed. Thus, this compound was considered to be a moderate CYP2D6 inhibitor, and a weak CYP2C 19 inhibitor. No inhibition was observed at CYP2B6, CYP2C9, CYP3A4 (with either substrate), CYP2C8 or CYP1A2. The significance of this CYP2D6 inhibition will depend on the levels of the compound in vivo.
Compound HCV-3 also was tested in cellular and nuclear receptor functional assays, and the results reported in Appendix 4, incorporated herein by reference.
Compound HCV-3 was also subjected to AMES testing, and the results reported in Appendix 5, incorporated herein by reference.
In summary, the compound HCV-3 was negative for genotoxicity against both strains used in this assay (TA98 and TA 100) up to a maximum tested concentration of l mg/mL, in both the absence and presence of S9 metabolic activation. The assay controls behaved as expected.
Compound HCV-3 also was subjected to in vitro metabolic disposition in mouse, rat, monkey and human microsomes. The test compound was incubated with pooled liver microsomes, since drip stability in liver microsomes can be predictive of drug stability in vivo. Aliquots were taken at 0, 5, 1 5, 30 and 45 minutes and quenched immediately. The samples were extracted and analyzed by LC-MS/MS. Compound HCV-3 was observed to have low clearance in human, monkey and mouse microsomes, and moderate clearance in rat.
Compound HCV-3 also was subjected to MDCK permeability assay. The compound was observed to be highly permeable in the MDCK assay. There was a slight difference between the plus and minus inhibitor data in terms of the efflux ratio obtained (1.48 minus inhibitor, versus 0.929 plus inhibitor). A ratio of greater than 2 generally indicates that efflux, i.e., blood brain barrier permeability, is occurring. The control compounds behaved as expected, with prazosin (a P-gp substrate) showing efflux in the absence of Cyclosporin A, which was inhibited in its presence.
Various derivatives of the above compounds with potential opiod and alpha antagonist activity were prepared as follows, and characterized by NMR and mass-spec as reported below.
Protocols for the synthesis of substituted l-arylethyl-4-acylammopiperidine derivatives
Synthesis of l-benzylpiperidin-4-one oxime


A solution containing 6.12 g (1 equiv., 0.03 mol) of l -ben/ylpiperidin-4-one oxime dissolved in 90 mL of iso-amyl alcohol was prepared and heated to approximately 1 10 °C. 6.9 g (10 equiv., 0.3 mol) of Na metal was then added slowly to the reaction mixture. After addition of Na, the reaction mixture was allowed to cool to room temperature and stirred until the reaction mixture turned into a thick slurry. The slurry was dissolved in 50 mL of ethyl acetate and 25 mL of H20. The organic layer was separated and washed with H20 (2 x 20 mL) followed by drying over anhydrous magnesium sulfate. The solvent was removed via rotary evaporation, resulting in a yellow oil. The crude product was purified via column chromatography utilizing silica gel and a DCM:MeOH solvent system in a ratio of 4: 1 with an additional 1% of Et3N. Yield: 3.7 g (64%).
Synthesis of Ai-(l -ben

Synthesis of N-(pipcridin-4-yl)propionamide

0.7 g of N-(l -benzylpiperidin-4-yl)propionamide (1 equivalent, 0.003 moles) were added to a parr hydrogenation flask and dissolved in 30 mL of EtOH. The solution was then degassed with argon for 30 min followed by the addition of 0.07 g of 10% Pd/C (0.2 equiv., 6.58 χ 10"4 mol) and 0.07 g of 20% Pd(OH)2 (0.17 equiv., 4.98 χ 10"4 mol). The black solution was then degassed with argon for an additional 1 5 min. The reaction mixture was then charged with 50 psi of H2 gas and shaken for 24 h. The product was filtered through celite and the solvent was removed via rotary evaporation. No further purification was required. Yield: 0.467 g (99%)
Synthesis of methyl 3-(4-propionamidopiperidin-l-yl)propanoate (CRAS)

0.1 g of N-(piperidin-4-yl)propionamide (1 equiv., 5.26 χ lO"4 moles) was dissolved in 2 mL of dry acetonitrile followed by the addition of 0.071 niL of methyl acrylate (1.5 equiv., 7.89 χ 10"4 mol). The reaction mixture was re fluxed overnight. The solvent was removed via rotary evaporation. The crude product was purified by washing with hexanes followed by drying under high vacuum. Yield: 0.90 g (71 %)
Synthesis of N-(l-(2-(thiophen-2-yl)ethyl)piperidin-4-yl)propionamide (CRAS1)

0.1 g of N-(piperidin-4-yl)propionamide (1 equiv., 6.40 χ 10"4 mol), 0.145 g of 2- (thiophen-2-yl)ethyl methanesulfonate (1 .1 equiv., 7.04 10"4 moles), 0.097 g of 2C03 (1.1 equiv., 7.04 χ 10"4 mol), 0.032 g of l (1.92 χ 10"4 mol), and 0.178 mL of Et3N (2 equiv., 1.28 χ 10"3 mol) were added to a round bottom flask and dissolved in 5 mL of dry acetonitrile. The reaction mixture was stirred and refluxed overnight. The solvent was then removed via rotary evaporation followed by the addition of H20. The mixture was extracted with ethyl acetate (3 x mL), and the organic extracts were combined and dried over anhydrous magnesium sulfate. The solvent was removed via rotary evaporation. The crude product was washed with hexanes to obtain an analytically pure sample. Yield: 0.101 g (60%).
Synthesis of l-phenethylpiperidin-4-one oxime

28.35 g (1 equivalent, 0.14 mol) of 1 -benzylpiperidin-4-one were dissolved in 60 mL of EtOH and then cooled to 0° C using an ice bath. A solution containing 19.46 g (2 equivalents, 0.28 mol) of hydroxylamine hydrochloride dissolved in 75 mL of H20 was prepared and then added dropwise to the reaction mixture followed by dropwise addition of a solution containing 19.35 g (1 equivalent, 0.14 mol) of K2CO3 dissolved in 75 mL of H20. The reaction mixture was then brought to room temperature and stirred overnight. The EtOH was then removed via rotary evaporation and the reaction mixture was then cooled in an ice bath to allow the product to crystallize out of solution. The product was filtered and washed several times with H20 and recrystallized in EtOH. Yield: 25.60 g (83.77%).
Synthesis of l-phenethylpiperidin-4-amine

A solution containing 6.00 g (1 equiv., 0.027 mol) of i -phenethylpiperidin-4-one oxime dissolved in 90 mL of iso-amyl alcohol was prepared and heated to approximately 1 10 °C. 6.21 g (10 equiv., 0.27 mol) of Na metal was then added slowly to the reaction mixture. After addition of Na, the reaction mixture was allowed to cool to room temperature and stirred until the reaction mixture turned into a thick slurry. The slurry was dissolved in 50 mL of ethyl acetate and 25 mL of I LO. The organic layer was separated and washed with H20 (2 χ 20 mL) followed by drying over anhydrous magnesium sulfate. The solvent was removed via rotary evaporation, resulting in a yellow oil. The crude product was purified via column chromatography utilizing silica
gel and a DCM:MeOH solvent system in a ratio of 4: 1 containing an additional 1% of Et3N.
Synthesis of N-(l-phenethylpiperidin~4~yl)furan-2-carboxamide (CRA8)

A solution of 0.1 g of 1 -phenethylpiperidin-4-amine (1 equiv., 4.89 χ 10"4 mol) dissolved in 2 ml, of dry dichloromethane was prepared followed by the addition of 0.178 niL (2.6 equiv., 1.27 x 10"3 moles) of Et3N. The reaction mixture was then cooled to 0 °C using an ice bath, and then 0.063 mL (1.3 equiv., 6.36 χ 10"4 mol) of 2-furoyl chloride dissolved in 0.25 mL of dry dichloromethane was added dropwise to the reaction mixture. The reaction mixture was then warmed to room temperature and stirred overnight. Once the reaction was complete, 4 mL of NH4OH and 45 mL of H20 were added to the reaction mixture. The organic layer was separated, and the aqueous layer was washed with dichloromethane (3 x 5 mL) followed by NaHC03 solution and brine. The organic extracts were dried over anhydrous magnesium sulfate and the solvent was removed via rotary evaporation, resulting in a white solid. The product was washed with hexanes to obtain an analytically pure sample. Yield: 0.0845 g (58.3 %).
Synthesis of N-(l-phenethylpiperidin~4~yl)furan-3-carboxamide (CRA9)

A solution of 0.1 g of 1 -phenethylpiperidin-4-amine (1 equiv., 4.89 10"4 mol) dissolved in 2 mL of dry dichloromethane was prepared followed by the addition of 0.178 mL (2.6 equiv., 1.27 χ 10"3 mol) of Et3N. The reaction mixture was then cooled to 0 "C using an ice bath, and then 0.063 mL (1.3 equiv., 6.36 x 10"4 mol) of 3-furoyl chloride dissolved in 0.25 mL of dry dichloromethane was added dropwise to the reaction mixture. The
reaction mixture was then warmed to room temperature and stirred overnight. Once the reaction was complete, 4 niL of NH4OH and 45 mL of 1 LO were added to the reaction mixture. The organic layer was separated, and the aqueous layer was washed with dichloromethane (3 < 5 mL) followed by NaHCC solution and brine. The organic extracts were dried over anhydrous magnesium sulfate and the solvent was removed via rotary evaporation, resulting in a white solid. The product was washed with hexanes to obtain an analytically pure sample. Yield: 0.104 g (72%).
Synthesis of 8-bromo-N-(l-phenethylpiperidin-4-yl)octanamide (CRAIO)

A solution of 0.101 g of l -phenethylpiperidin-4-amine (1.1 equiv., 4.93 χ 10"4 mol), 0.1 g of 8-bromooctanoic acid (1 .0 equiv., 4.48 x 10"4 mol), 0.170 g of HATU (1.0 equiv., 4.48 x 10"4 mol), 0.061 g of HO At (1.0 equiv., 4.48 10"4 mol), and 0.314 mL of DIEPA (4.0 equiv., 0.0018 mol) in dry DMF was prepared. The reaction mixture was stirred at room temperature overnight. The reaction mixture was then quenched with 0.5 M KHSO4 solution followed by the addition of dichloromethane. The organic and aqueous layers were separated, and the aqueous layer was extracted with dichloromethane (3 x 5 mL) followed by washing with NaHCO;, solution and Brine. The organic extracts were then dried over anhydrous magnesium sulfate.
Synthesis of N-(l-phenethylpiperidin-4-yl)henzamide (CRA11)

Synthesis of 2-(thiophen-2-yl)ethyl methanes ulfonate

15 2.6 mL of 2-(thiophen-2-yl)ethanol (1 equiv., 0.023 mol) was dissolved in 45 mL of dry dichloromethane followed by the addition of 3.63 mL of Et3N (1 .13 equiv., 0.026 mol). The reaction mixture was stirred at room temperature for 1 h. It was then cooled to -5 °C using an ice bath and solid NaCl. Once cooled, 1 .92 mL of methane sulfonyl chloride was added dropwise over the course of 10 min. The reaction mixture was then warmed to
20 room temperature and stirred for 1 h. Once the reaction was complete, 30 mL of Nal ICO3 solution was added followed by separation of the organic and aqueous layers. The aqueous layer was extracted with dichloromethane (3 x 30 mL). The combined organic extracts were dried over anhydrous magnesium sulfate and the solvent was removed via rotary evaporation, resulting in a brown oil. No further purification was
25 required.
NMR (Ή & UC) & mass-spec data of novel substituted l -arylethyl-4- acylaminopiperidine derivatives (total six compounds)



(dtd, J=3.70, 11.10, 11.13, 12.61 Hz, 2H), 1.91 (m, 211), 2.17 (m, 4 H), 2.49 (m, 211). 2.68 (m, 2H), 2.81 (m, 2H), 3.67 (s, 3H), 3.78 (dddd, J=4.29, 4.36, 11.92, 15.26, IH), 5.25 (d, J= 7.96 Hz, IH).13C NMR (100 MHz, CDC ) δ 10.02, 29.99, 32.41, 46.40,
5 51.76, 52.25.55.63, 173.14, 174.05.
CRA8 NMR data: Ή NMR (400 MHz, CDC13) δ 1.63 (d, J =11.93 Hz, 211), 2.05 (d, J- 12.33 Hz, 2H), 2.26 (t, .1=11.35, 11.35 Hz, 2H), 2.64 (dd, .1=6.16.10.21 Hz, 2H), 2.83 (dd, J=6.12, 10.24 Hz, 2H), 2.99 (d, J= 12.01 Hz, 2H), 3.98 (m, IH), 6.21 (d, J=8.20 Hz, IH), 6.49 (dd, J~ 1.79.3.47 Hz, IH), 7.10 (dd, J-0.84, 3.47 Hz, IH), 7.24 (m, 5H), 7.43 0 (dd.0.84, 1.77 Hz. III). ,3C NMR (100 MHz, CDCh) δ 32.67, 34.17, 46.27.46.61, 52.76, 60.88, 112.60, 114.57, 126.56, 128.87, 129.13, 140.58, 144.16, 148.48, 158.13. CRA9 NMR data: Ή NMR (400 MHz, CDC13) δ 1.63 (m, 2H), 2.06 (d, J=l 1.35 Hz, 211), 2.26 (m, .1=11.35, 2H), 2.65 (m, 2H), 2.84 (m, 2H), 3.02 (d, J= 11.86 Hz, 2H), 3.99 (m, IH), 5.65 (d, J=7.99 Hz, IH), 6.59 (dd, .1=0.91.1.93 Hz, IH), 7.24 (m, 5H), 7.42 (dd, 5 1.58, 1.91 Hz, IH), 7.91 (dd, J=0.90, 1.59 Hz, IH).13C NMR (100 MHz, CDC13) δ 32.04, 33.49, 46.43, 52.35, 60.26, 108.25, 122.66, 126.19, 128.46, 128.68, 139.88, 143.72, 144.69, 161.95
CRAIS NMR data: Ή NMR (400 MHz, CDC13) 61.15 (t. J=7.58, 7.58 Hz, 311).1.47 (m, 2H), 1.95 (m, 2H), 2.18 (m, 4H), 2.64 (dd, J =6.87.8.62 Hz, 2H), 2.90 (m, 2H), 3.00 0 (m, 2H), 3.82 (dddd, J=4.20, 8.31, 10.86, 15.17 Hz, IH), 5.32 (d, J=7.95 Hz, IH), 6.81 (dq, J- 1.02.1.02, 1.02, 3.20 Hz, IH), 6.91 (dd, .1= 3.39.5.14 Hz, IH), 7.11 (dd, .1=1.21, 5.13 Hz, 1 H).13C NMR (100 MHz, CDC13) δ 10.04, 28.06, 30.03, 32.15.46.52, 52.42, 59.98, 123.62, 124.71, 126.69, 142.87, 173.15.
CRA10 NMR data: Ή NMR (400 MHz, CDC13) δ 1.32-2.34 (m, 14H), 2.82-3.01 (m, 5 1111), 3.16 (dd, .1=6.53, 10.91 Hz, IH), 3.49 (d, .1=11.91 Hz, IH), 4.63 (dt, J=5.61, 5.61, 8.76 Hz, IH), 7.24 (m, 4H), 7.42 (dd, .1=4.46, 8.37, 1 I I), 8.02 (m, HI).13C NMR (100
MHz, CDCI3) δ 25.17, 25.45, 27.85, 28.70, 28.75, 31.46, 36.55, 38.61, 81.47, 120.73. 128.66, 128.86, 129.19, 151.29, 162.71, 173.62.
CRA11 NMR data: \ll NMR (400 MHz, CDC13) δ 1.35 (t, J- 7.29, 7.29 Hz, 2H), 1.64 (qd, J =3.80, 11.29, 11.29, 11.35 Hz, 211).2.08 (m, 211), 2.27 (td, J=2.57, 11.61. 11.65 Hz, 2H), 2.64 (m, 2H), 2.83 (m, 2H), 3.00 (m, 3H), 4.04 (dddd, J -4.28, 8.29, 10.85, 15.24 Hz, III), 6.04 (d, J=7.94 Hz, III).7.25 (m, 511), 7.44 (m, 311), 7.75 (m, 211). C NMR (100 Mi l/.. CDCI3) δ 32.22, 33.71, 45.83.46.97.52.38, 60.42, 114.25, 126.12, 126.86, 128.42, 128.56, 128.68, 131.42, 134.75, 140.11, 166.88
Many other variations and modifications may be made to the above-described embodiments of the disclosure without departing substantially from the spirit and principles of the disclosure. All such modifications and variations are intended to be included herein within the scope of the present disclosure and protected by the following claims.
Appendix 1
Docket No. UA 15-023
Sliidy Number 3235
Table 1
Binding Assays
Snmmnry Results

8255-1 RSAlOIc I.OE-05 -21 8255-2 Rl I. E-05 74 8255-3 R2 l.OE-05 55
BZD (central) ;
8255-1 RSAlOIc I.OE-05 6 8255-2 Rl l.OE-05 17 8255-3 R2 l.OE-05 12
CORP (h)
8255-1 RSAlOIc l.OE-05 -7 8255-2 Rl •l.OE-05 -8 8255-3 R2 l.OE-05 3 "
CO, ()
8255-1 RSAlOIc l.OE-05 -3 8255-2 Rl 1, OE-05 -3 8255-3 R2 l.OE-05 12
CBi (li)
8255-1 RSAlOIc Ί.0Ε-05 5 8255-2 l l.OE-05 15 8255-3 R2 l.OE-05 25
OABAB
8255-1 RSAlOIc i .OE-05 6 8255-2 Rl l.OE-05 -1 8255-3 R2 l.OE-05 -11
Galaiiiii (lion-selective)
' 8255-1 RSAlOIc 1 ,0E-O5 -191 8255-2 R! !. OE-05 -30 8255-3 R2 J, OE-05 -30
MG,
8255-1 RSAlOIc 1.OE-05 6 8255-2 Rl l.OE-05 7 82S5-3 R2 l.OE-05 13
MO,
8255-1 RSAlOIc ! .OE-05 46 S255-2 Rl l.OE-05 I 8255-3 R2 I.OE-05 2
NK| (Ii)
8255-1 RSAlOIc l.OE-05 -7 8255-2 l 1.OE-05 11 8255-3 R2 l.OE-05 6 δ (DOP)
Study Number S255
Assny Tost % Inhibition of
Olien! Compound l.D.
Cerep Compound l.D. Concentration Control Specific Bi ding
8255-1 RSA!Olc 1.0E-05 98 8255-2 J I.OE-05 Ι 8255-3 R2 l.OE-OS 60 ic (KOP)
8255-1 RSAlOIc I.OE-05 79 S255-2 Rl I.OE-05 107 8255-3 R2 I.OE-05 104 μ 00 (MQP)

825S-3 R2 I.OE-05 18
P2Y
8255-1 RSAlOIc l.OE-05 -3
8255-2 Rl I.OE-05 -1 8255-3 R2 I.OE-05 -1 σ (iion-selectivc)
8255-1 RSAlOIc I.OE-05 33
8255-2 Rl I.OE-05 104 8255-3 R2 I.OE-05 101
K v channel
S255-I RSAlOIc l. E-05 -1
S255-2 Rl I.OE-05 -1
8255-3 R2 I .OE-05 0
NE\ transporter (V '
8255-1 RSAlOIc 1.OE-05 -7
8255-2 l l.OE-05 -16
8255-3 R2 1, OE-05 29
GABA transporter
8255-1 RSAlOIc I ,ΟΕ-05 5
8255-2 Rl 1.OE-05 -1
8255-3 R2 1. OE-05 17
2015/046585
Study Number 8255 p. 3At Table 2
Binding Assays
Reference Compound Data

dATPaS 2.4E-OS 1 .2E-08 0.8 σ (non-selective)
aiopon'dol 7.9E-OS 6.2E-OS 0.9
K*v channel
o.-dei lrotoxin 7.7E- 10 ό. Ι Ε- 10 2.8
NE transporter (It)
protrlptyline !.OE-08 7.9E-09 1.1
GABA transporter
nipecotic acid 2.SE-06 2.7E-06 0.9
Study Number 8255 p.4/4
Table'3
Binding Assnys
Summary Results
Tusl % Inhibition of
Assay Client Compound l.D, Concentration Control Specific Binding
Corcp Compound I.D.
μ (7, (MOP)
8255-2 I !.OE-05 98
8255-3 R2 !.OE-05 99
(KOP)
8255-2 l !.OE-05 107
104
8255-3 2 !.OE-05
cf (non-selective)
8255-2 R.I 1.0E-0S 104
8255-3 1.0E-05 101
82 -2 Rl 1.0E-05 7
8255-3 R2 !.OE-05 55
Appendix 2
Docket No. UA 15-023
1
X-ray Diffraction Facility
Department of Chemistry and Biochemistry
The University of Arizona
Your code: OxEtOH Our code: rvl 01
Comment
The crystals submitted were small needles which diffracted poorly. Reflections were streaky, indicating that the crystal was not single but comprised of more than one not-exactly-oriented component. Diffraction was observed only to about 1 A resolution. Because of the poor resolution, A and B level cheokcif alerts are generated. The quality of the structure is such that the identity of the molecule and its conformation is confirmed, but derived parameters (bond distances, angles, thermal motion) are not reliable.
Nevertheless, the structure could be determined and refined, and the molecule is shown in Figure 1. Figure 2 shows the contents of the unit cell, which also includes an oxalate molecule.
Figure 3 shows the unit cell viewed down the crystal lographic a axis. The hydrogen bonding network in the unit cell connects the organic molecule to oxalate along the crystallographic b axis (ydrogen bonds between the Nl and 02 of the oxalate and between N2 and 04 of the oxalate). The oxalate molecule are connected by hydrogen bonds 04 and 05 of the oxalate, along the crystallographic a axis.
C8 has been modeled with disorder in two positions. There is unmodeled (probably rotational) disorder in the aromatic ring (C1 1 -C 1 6). Both distance and planarity restraints were applied to this ring during refinement. Not all hydrogen atoms were visible in the electron density map. Because of the low resolution and streaky diffraction pattern, the structure could not be refined without constraints. Constraints used in the refinement are included at the end of the report.

Figure 1 . The molecule with displacement ellipsoids at the 50% probability level. C8 is disordered, the minor component has been removed for clarity. An oxalate molecule also in the unit cell is not shown in this figure.
2

Figure 2. The contents of the asymmetric unit with displacement ellipsoids at the 50% probability level. B8 is disordered, and the minor compont is not shown. Fog has been added to show depth.

Figure 3. The contents of the unit cell viewed down the short a axis. Hydrogen bonds are shown between the Nl and 02 of the oxalate. Hydrogen bonds also exist between N2 and 04 of the oxalate and between 04 and 05 of the oxalate, connecting the oxalate molecules along the crystallographic a axis (looking down into the page).
46585
Acknowledgement:
The APEXII DUO was purchased with funding from NSF grant CHE-0741837.
References:
APEX2 (data collection)
Bruker (2007). APEX2. Bruker AXS Inc., Madison, Wisconsin, USA.
SAINT (integration and reduction)
Bruker (2007). SAINT. Bruker AXS inc., Madison, Wisconsin, USA.
SADABS (absorption correction)
Sheldrick, G. M. (1 996). SADABS. University of Gottingen, Germany.
SHELXTL (structure solution and refinement)
Sheldrick, G. M. (2008). Acta Cryst. A64, 1 12-122.
MERCURY (molecular graphics - hydrogen bonding and packing)
Macrae, C. F., Bruno, i. J ., Chisholm, J. A., Edgington, P. R., McCabe, P. Pidcock, E„ Rodriguez - Monge, L. Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466-470.
PLATON
Spek, A. L. (2003). J, Appl. Cryst. 36, 7-13.
OLEX2
Dolomanov, O.V., Bourhis, L.J ., Gildea, R.J., Howard, J.A. ., Puschmann H.
(2008), J. Appl. Cryst. 42, 339-341.
Table 1 Crystal data and structure refinement
Identification code rvlOl
Empirical formula C18H27N205
Formula weight 351.41
Temperature/K 150.0
Crystal system triclinic
Space group P-l
a/A 5.701(3)
b/A 12.024(6)
c/A 14.105(7)
/0 100.473(14)
β/° 93.997(14)
γ/° 95.571(15)
Volume/A3 942.5(8)
Z 2
Pcalcg/Cm3 1.238
umrn"' 0.090
F(000) 378.0
Crystal size/mm3 0.3 0.08 x 0.05
Radiation ΜοΚα (λ = 0.71073)
2Θ range for data collection/0 2.948 to 41.622
Index ranges -5<h<5,-ll<k<11,-14<[< 14 Reflections collected 7408
Independent reflections 1965 [Rim = 0.0625, Rsigma = 0.0684] Data/restraints/parameters 1965/237/222
Goodness-of-fit on F2 1.592
Final R indexes [1>=2σ (I)] R, = 0.0970, w .2 = 0.2383
Final R indexes [all data] R, =0.1319, wR2 = 0.2545
Largest diff. peak/hole / e A"3 0.57/-0.46
5
Table 2 Fractional Atomic Coordinates ( ΐθ4) and Equivalent Isotropic Displacement
(A2 l03)
Ueq is defined as 1/3 of of the trace of the orthogonalised Uu tensor.
Atom x y z U(eq)
04 2499(7) 898(4) 1556(3) 26.4(12)
05 -3506(7) 78(4) 1467(3) 28.1(12)
03 -1621(7) 1811 (4) 1840(3) 34.3(13)
02 605(7) -782(4) 1662(3) 29.6(13)
Nl -2989(8) -2451(4) 1622(3) 20.6(13)
N2 -2795(9) -3369(4) -1448(4) 25.0(14)
C2 -1073(11) -3368(6) 206(5) 26.8(16)
01 -2673(8) -5260(4) -1872(3) 35.3(13)
CI -1627(11) -3398(5) 1239(5) 27.3(17)
C3 -3356(11) -3435(5) -460(5) 26.4(16)
C9 -3409(12) -2486(6) 2654(4) 28.0(17)
C4 -4744(11) -2458(6) -50(5) 25.9(16)
C18 -1623(11) 803(6) 1645(4) 16.6(14)
C5 -5248(10) -2510(6) 986(4) 22.8(16)
C17 690(11) 242(6) 1604(4) 16.3(14)
C7 -1722(13) -4117(6) -3046(5) 38.1(19)
CIO -4412(13) -1434(6) 3188(5) 36.6(19)
C6 -2419(11) -4299(6) -2080(5) 24.0(16)
Cll -4740(9) -1569(5) 4222(3) 39.1(18)
C12 -2894(8) -1194(5) 4941(4) 62(3)
C13 -3168(9) -1341(5) 5883(3) 76(3)
C14 -5288(10) -1864(5) 6107(3) 59(2)
C15 -7134(8) -2239(6) 5388(4) 120(5)
C16 -6860(8) -2091(6) 4445(4) 1.14(5)
C8 -1320(80) -5210(40) -3670(30) 66(3)
C8A 680(20) -4565(10) -3240(8) 66(3)
Table 3 Anisotropic Displacement Parameters (A2* 103).
Ui2+...]-

04 7(2) 29(3) 48(3) 18(2) 4(2) 7.9(19)
05 10(2) 29(3) 47(3) 13(2) 4(2) 2.6(19)
03 20(3) 26(3) 57(3) 6(2) 6(2) 4(2)
02 14(2) 23(2) 57(3) 15(2) 9(2) 7.7(19)
Ni 9(3) 21(3) 34(3) 11(2) -1(2) 5(2)
N2 18(3) 24(3) 36(3) 11(2) 7(2) 5(2)
C2 17(3) 24(4) 43(4) 12(3) 5(3) 11(3)
01 37(3) 23(3) 48(3) 9(2) 5(2) 6(2)
CI 12(3) 26(4) 43(4) 6(3) -1(3) 4(3)
C3 16(3) 27(4) 40(3) 13(3) 4(3) 4(3)
C9 17(4) 36(4) 33(3) 15(3) 0(3) 3(3)
C4 17(4) 29(4) 36(3) 11(3) 7(3) 7(3)
C18 9(2) 20(3) 22(3) 7(2) 3(2) 2(2)
C5 9(3) 26(4) 36(3) 13(3) -1(3) 6(3)
C17 7(2) 23(3) 22(3) 9(2) 5(2) 4(2)
C7 47(4) 39(4) 35(4) .14(3) 7(3) 15(4)
CIO 37(4) 41(4) 38(4) 14(3) 6(3) 18(4)
C6 13(4) 26(3) 36(3) 9(3) 0(3) 8(3)
Cll 32(4) 49(5) 43(4) 16(3) 9(3) 20(3)
C12 60(5) 81(7) 43(4) .17(4) -1(3) -12(5)
C13 88(6) 98(7) 39(4) 13(4) 4(4) 2(5)
C14 66(5) 72(6) 50(4) 20(4) 19(3) 42(4)
C15 55(5) 238(14) 77(5) 77(6) 11(4) -20(7)
C16 40(5) 238(14) 72(5) 78(7) -6(4) -29(6)
C8 72(7) 78(7) 59(6) 17(5) 26(5) 35(6)
C8A 72(7) 78(7) 59(6) 17(5) 26(5) 35(6)
Table 4 Bond Lengths
Atom Atom Length/A Atom Atom Length/A
04 CI 7 1.250(7) C9 CIO 1.532(9)
05 C18 1.295(7) C4 C5 1.519(8)
03 C18 1.194(7) CI 8 C17 1.538(9)
02 C17 1.245(7) C7 C6 1.493(9)
Nl CI 1.482(7) C7 C8 1.49(4) l C9 1.499(8) C7 C8A 1.542(13)
Nl C5 1.505(7) CIO CI 1 1.519(7)
N2 C3 1.465(8) C11 C12 1.3900
N2 C6 1.342(8) C11 C16 1.3900
C2 CI 1.519(9) CI 2 C13 1.3900
C2 C3 1.538(9) CI 3 CI 4 1.3900
01 C6 1.242(7) CI 4 C15 1.3900
C3 C4 1.529(8) CI 5 C16 1.3900
U 2015/046585
Table 5 Bond Angles
Atom Atom Atom Angle/0 Atom Atom Atom Angle/0
C1 Nl C9 109.4(5) 02 C17 04 126.8(6)
CI Nl C5 110.0(5) 02 C17 C18 118.2(6)
C9 Nl C5 112.8(5) C6 C7 C8A 110.5(6)
C6 N2 C3 121.5(6) C8 C7 C6 111.4(17)
CI C2 C3 111.0(5) Cll CIO C9 109.3(5)
Nl CI C2 111.3(5) N2 C6 C7 116.7(6)
N2 C3 C2 110.4(5) 01 C6 N2 121.3(6)
N2 C3 C4 110.9(5) 01 C6 C7 122.0(6)
C4 C3 C2 108.4(5) C12 Cll CIO 119.9(4)
Nl C9 CIO 114.4(5) CI 2 CI 1 C16 120.0
C5 C4 C3 110.5(5) C16 Cll CIO 120.1(4)
05 C18 CI 7 113.5(5) C13 C12 Cll 120.0
03 C18 05 124.7(6) C14 C13 C12 120.0
03 CI 8 C17 .121.7(6) C13 C14 C15 120.0
Nl C5 C4 111.1(5) C1.6 CI 5 C14 120.0
04 C17 C18 114.9(6) C15 C16 Cll 120.0
Table 6 Torsion Angles
A B c D Angle/0 A B C D Angle/'
05 C18 C17 04 164.8(5) C9 CIO Cll CI 6 -89.5(6)
05 C18 C17 02 -18.1(8) C5 Nl CI C2 57.9(7)
03 CIS C17 04 -16.5(8) C5 Nl C9 CIO -67.5(7)
03 C18 C17 02 160.6(6) CIO CM C12 C13 =178.4(5)
Nl C9 CIO CI 1 -179.6(5) CIO Cll C16 CI 5 178.4(5)
N2 C3 C4 C5 -178.1(5) C6 N2 C3 C2 85.2(7)
C2 C3 C4 C5 -56.7(7) C6 N2 C3 C4 -154.6(6)
0.0
CI Nl C9 CIO 169.7(5) Cll C12 CI 3 C14
CI Nl C5 C4 -58.3(6) C12 Cll C16 C15 0.0
CI C2 C3 N2 178.2(5) CI2 C13 C14 C15 0.0
CI C2 C3 C4 56.5(7) CI 3 C14 C15 C16 0.0
C3 N2 C6 Ol 4.4(9) CI 4 CI 5 C16 CI 1 0.0
C3 N2 C6 C7 -176.5(6) C16 Cll C12 CI 3 0.0
C3 C2 CI N -58.2(7) C8 C7 C6 N2 179(2)
C3 C4 C5 Nl 58.5(7) C8 C7 C6 01 -2(2)
C9 Nl CI C2 -177.7(5) C8A C7 C6 N2 122.0(8)
C9 Nl C5 C4 179.3(5) C8A C7 C6 01 -58.9(10)
C9 CIO CI 1 C12 88.9(6)
Table 7 Hydrogen Atom Coordinates (A*104) Isotropic Displacement Parameters (A2xl03)
Atom X y z U(eq)
H4 3698 545 1578 40
115 -3360 -442 1785 42
HI -2014 -1718 1607 25
H2 -2702 -2706 -1630 30
H2A -188 -4016 -34 32
H2B -59 -2656 191 32
HI A -2553 -4133 1259 33
MB -131 -3342 1655 33
H3 -4332 -4175 -469 32
H9A -1893 -2570 3007 34
H9B -4520 -3167 2668 34
H4A -3821 -1722 -67 31
H4B -6255 -2508 -454 31
H5A -6132 -1868 1242 27
H5B -6247 -3228 997 27
H7AA -2983 -3771 -3369 46
H7AB -255 -3582 -2958 46
H7BC -1605 -3295 -3067 46
H7BD -2948 -4520 -3558 46
HI OA -3313 -744 3188 44
H I OB -5951 -1347 2854 44
HI 2 -1446 -837 4788 75
H13 -1907 -1085 6375 91
H14 -5475 -1965 6751 71
H15 -8583 -2596 5540 144
H16 -8121 -2347 3954 137
H8A . -734 -5723 -3269 100
H8B -150 -5058 -4125 100
H8C -2812 -5564 -4039 100
H8AA 1887 -4172 -2730 100
H8AB 1128 -4424 -3869 100
H8AC 543 -5385 -3244 100
Table 8 Atomic Occupancy
Atom Occupancy Atom Occupancy Atom Occupancy H7AA 0.215(12) H7AB 0.215(12) H7BC 0.785(12) M7BD 0.785(12) C8 0.215(12) H8A 0.215(12) 118 B 0.215(12) H8C 0.215(12) C8A 0.785(12) H8AA 0.785(12) H8AB 0.785(12) H8AC 0.785(12)
IP-
Experimental Details
Needle shaped crystals of C18H27N2O5 were submitted for structure determination. The crystals were small and were grown together. Even apparently single crystals were stacks of needles A crystal was selected and attached to a Micromount using paratone oil, then mounted on a Bruker Kappa APEX-11 Duo diffractometer. The crystal was kept at 150.0 K during data collection. Using Olex2 [1 ], the structure was solved with the ShelXS [2] structure solution program using Direct Methods and refined with the ShelXL [3] refinement package using Least Squares minimisation.
1 . Dolomanov, O. V., Bourhis, L.J., Gildea, R.J, Howard, J.A.K. & Puschmann, H. (2009), J. Appl. Cryst. 42, 339-341.
2. Sheldrick, G.M. (2008). Acta Cryst. A64, 1 12-122.
3. Sheldrick, G.M. (2008). Acta Cryst. A64, 1 12-122.
Crystal Data for C18H27 2O5 (M =351 .41 g/mol): triclinic, space group P-l (no. 2), o = 5.701 (3) A, b = 12.024(6) A, c = 14.105(7) A, a = 100.473(14)°, β = 93.997(14)°, γ = 95.573 (15)°, V =
942.5(8) A3, Z= 2, T- 1 50.0 K, μ(Μο α) = 0.090 mm"1, Dcalc = 1 .238 g/cm3, 7408 reflections measured (2.948° < 2Θ < 41 .622°), 1965 unique (Rmt = 0.0625, Rsjgma = 0.0684) which were used in all calculations. The final R was 0.0970 (I > 2σ(Ι)) and wRz was 0.2545 (all data).
Refinement model description
Number of restraints - 237, number of constraints - Details:
1. Fixed Uiso
At 1 .2 times of:
All C(H) groups, All C(H,H) groups, All C(H,H,H,H) groups, All N(H) groups
At 1 .5 times of:
All C(H,H,H) groups, Ail O(H) groups
2. Restrained planarity
C1 1, C12, C 13, C14, C15, C 16, C 10
with sigma of 0, 1
3. Rigid bond restraints
02, 05, CI 8, 04, C I 7, 03
with sigma for 1 -2 distances of 0.0.1 and sigma for 1 -3 distances of 0.01.
C1 1, C16, C 15, C 14, C 13, C 12
with sigma for 1 -2 distances of 0.01 and sigma for 1 -3 distances of 0.01
4. Uiso/Uaniso restraints and constraints
Uanis(03) « Ueq, Uanis(C 1 8) « Ueq, Uanis(05) « Ueq, Uanis(02) «
Ueq, Uanis(Cl 7) « Ueq, Uanis(04) « Ueq: with sigma of 0.005 and sigma
for terminal atoms of 0.01
Uanis(C8) = Uanis(C8A)
5. Rigid body (RIGU) restrains
All non-hydrogen atoms
with sigma for 1 -2 distances of 0.004 and sigma for 1 -3 distances of 0.004
6. Others
' Sof(H7BC)=Sof(H7BD)=Sof(C8A)=Sof(H8A A)=Sof(H8AB)=Sof(H8AC)=l -FVAR(1 )
Sof(H7AA)=Sof(H7AB)=Sof(C8)=Sof(H8A)=Sof(H8B)=Sof(M8C)=FVAR( l)
7. a Ternary CIT refined with riding coordinates:
N1(H.1 ), C3(H3)
7.b Secondary CH2 refined with riding coordinates;
C2(H2A,H2B), C I (H I Α,Η Ι Β), C9(H9A,H9B), C4(H4A,H4B), C5(H5A,H5B), C7(H7AA,
13
H7AB), C7(H7BC,H7BD), C 10(H 10A,H1 OB)
7.c Aromatic/amide H refined with riding coordinates:
N2(H2), C12(HI 2), C 13(H 13), C 14(H 14), C 15(H 15), C16(H 16)
7.d Fitted hexagon refined as free rotating group:
C1 1 (C12,C13,C14,C15,C16)
7.6 Idealised Me refined as rotating group:
C8(H8A,H8B,H8C), C8A(H8AA,H8AB,H8AC)
7. f Idealised tetrahedral OH refined as rotating group:
04(H4), 05(H5)
This report has been created with 01ex2, compiled on 2014.07.22 svn.r2960 for OlexSys. Please let us know if there are any errors or if you would like to have additional features.
Appendix 3 Docket No. UA 15-023

μ-Opioid Antagonist/ -agonist (I)
Lipinski and Related Properties Value Condition Note
Freely Rotatable Bonds 5(1 )
H Acceptors 3(1 )
H Donors 1 (1 )
H Donor/Acceptor Sum 4(1 )
logP2.216 0.355Temp:25 C(1 )
Molecular Weight260.37(1 )
Lipinski's rule of five
Lipinski's rule of five also known as the Pfizer's rule of five or simply the Rule of five (R05) is a rule of thumb to evaluate druglikeness or determine if a chemical compoundwith a
certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. The rule was formulated by Christopher A. Lipinski in 1997, based on the observation that most orally administered drugs are relatively small and
moderately lipophilic molecules.11I[21
The rule describes molecular properties important for a drug's pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion ("AD E"). However, the rule does not predict if a compound is pharmacologically active.
The rule is important to keep in mind during drug discovery when a pharmacologically active lead structure is optimized step-wise to increase the activity and selectivity of the compound as well as to ensure drug-like physicochemical properties are maintained as described by Lipinski's
rule.13' Candidate drugs that conform to the R05 tend to have lower attrition rates during clinical trials and hence have an increased chance of reaching the market.'2"''1
Components of the rule
Lipinski's rule states that, in general, an orally active drug has no more than one violation of the following criteria:
• No more than 5 hydrogen bond donors (the total number of nitrogen-hydrogen and oxygen hydrogen bonds)
• Not more than 10 hydrogen bond acceptors (all nitrogen or oxygen atoms)
• A molecular mass less than 500 daltons
• An octanol-water partition coefficient'5' log P not greater than 5
Note that all numbers are multiples of five, which is the origin of the rule's name. As with many other rules of thumb, (such as Baldwin's rules for ring closure), there are many exceptions to Lipinski's Rule.
Appendix 4
Docket No. UA 15-023
. STUDY REFERENCES
Study title In Vitro Pharmacology
Study of One Compound
Study number 100023322 FINAL REPORT
Experimental period
. PERSONS INVOLVED IN THE STUDY
Investigator Eurofins Cerep Thierry JOLAS.Ph.D.
Le Bois I'Eveque Principal Scientist, Pharmacology B.P. 30001
86 600 Celle I'Evescault Tel: +33 (0)5 49 89 39 89
France Fax: +33 (0)5 49 43 21 70
Tel: +33 (0)5 49 89 30 00
Fax: +33 (0)5 49 43 21 70
Study sponsor TECH LAUNCH ARIZONA Dr. Paul EYNOTT
University of Arizona Licensing Manager, The College of Science
220 W 6th Street
4th Floor
TUCSON, AZ 85701
U.S.A.
. APPROVAL
Investigator Eurofins Cerep Thierry JOLAS.Ph.D.
Le Bois I'Eveque Principal Scientist, Pharmacology B.P. 30001
86 600 Celle I'Evescault Tel: +33 (0)5 49 89 39 89
France Fax: +33 (0)549 43 21 70
Tel: +33 (0)5 49 89 30 00
Fax: +33 (0)5 49 43 21 70
I certify that this report accurately reflects all relevant data collected in this study.
Date

Quality assurance statement Eurofins Cerep's Quality Unit certifies that results presented in this report were generated using the materials and methods mentioned and that these results accurately reflect the raw data.
Date
Signature

14. TABLE OF CONTENTS
1. STUDY REFERENCES 2
2. PERSONS INVOLVED IN THE STUDY 2
3. APPROVAL · 3
4. TABLE OF CONTENTS · - ·· - 4
5. SUMMARY ··■■ 5
5.1. Study Design ■·■■■ 5
5.2. Measurements ■■ ■ 5
5.3. Results ■ 5
6. COMPOUNDS ■ 6
6.1. Test Compounds ■ ■ 6
6.2. Reference Compounds 6
7. RESULTS 7
7.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays 7
7.1.1. Agonist Effect: Test Compound Results 7
7.1.2. Reference Compound Results 7
7.1.3. Antagonist Effect: Test Compound Results 8
7.1.4. Reference Compound Results 8
8. RESULTS INTERPRETATION GUIDE 9
9. MATERIALS AND METHODS 10
9.1. Experimental Conditions ■ 10
9.1.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays 10
9.2. Analysis and expression of results ■ 11
9.2.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays 11
10. BIBLIOGRAPHY - ■ 12
I 5. SUMMARY
The purpose of this study was to test HCV-3 in cellular and nuclear receptor functional assays.
5.1. Study Design
HCV-3 was tested at 1.0E-05 M.
5.2. Measurements
Cellular agonist effect was calculated as a % of control response to a known reference agonist for each target and cellular antagonist effect was calculated as a % inhibition of control reference agonist response for each target.
5.3. Results
Results showing an inhibition or stimulation higher than 50% are considered to represent significant effects of the test compounds. Such effects were not observed at any of the receptors studied.
6. COMPOUNDS 6.1» Test Compounds
Manufacturer: TECH LAUNCH ARIZONA
Clienf Compound ID Compound ID Reference Number Batch Number FW MW Purity Received Form Stock solution Rag
HCV-3 " 100023322-1 - 1 296.84 260.38 - Powder 1.E-02 M H2O
FW: Formula Weight - MW: Molecular Weight
6.2. Reference Compounds
In each experiment and if applicable, the respective reference compound was tested concurrently with HCV-3, and the data were compared with historical values determined at Eurofins. The experiment was accepted in accordance with Eurofins validation Standard Operating Procedure.
7. RESULTS
7.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays
7.1.1. Agonist Effect: Test Compound Results of Control Agonist esponse
-10 10 30 40 50 60 100 e;2B(h) (agonist effect)
52 (OOP) (agonist effect)
K (HOP) (agonist effect) μ (MOP) (h) (agonist effect)
I Test Concentration: 1.0E-05 M
Figure 1. Histogram for HCV-3
Compound I.D. Client Compound I.D. Test % of Control Agonist Response
Concentration 1s' 2nd Mean a2!l ^(a priist effect)
100023322-1 HCV-3 1.0E-O5 M 9.2 5.1 7.1
100023322-1 HCV-3 1.0E-05 M 4.0 4.0 4.0 (KOP) (agonist effect)
100023322-1 HCV-3 1.DE-05 -10.3 -4.2 μ (MOP) (h) (agonist effect)
100023322-1 HCV-3 1.0E-05 M 28.1 38.9 33.5
7.1.2. Reference Compound Results
Compound I.D. EC50 (M) nH t (W (agonist effect)
dexmedetomidine 1.3E-08 n/a
(DOP) (ayoiiist effect)
DPDPE 1.5E-09 M n/a
(KOP) (agonist effect)
U 50488 1.6E-09 M
μ (MOP) (h) (agonist effect)
DAM GO 4.2E-09 M n/a
7.1.3. Antagonist Effect: Test Compound Results
<M> Inhibition of Control Agonist Response
-10 0 10 20 30 40 50 60 80 90 100
«2B<h) (antagonist effect)
02 (OOP) (antagonist effect) (KOP) (antagonist effect) μ (MOP) (h) (antagonist effect)
Test Concentration: l.OE-05 M
Figure 2. Histogram for HCV-3
Compound I.D. Client Compound I.D. Test % Inhibition of Control Agonist Response
Concentration 1s1 2nd Mean ¾<an&3oniSt: ffect)
100023322-1 HCV-3 1.0E-05 -16.8 -38.7 -27.8
62 (DOP) (antagonist effect)
100023322-1 HCV-3 1.0E-05 M 0.8 5.9 3.3 (KOP) (antagonist effect)
100023322-1 HCV-3 1.0E-05 M -12.4 23.5 5.6
■i fWKJl'i ill !a :rf£,uiiist uifc-ct)
100023322-1 HCV-3 1.0E-05 M -0.1 -1.8 -1.0
7.1.4. Reference Compound Results
Compound I.D. ICso M . ^ KB ( ) ^ nH t (h) (antagonist effect)
yohimbine 3.7E-07 M 4.8E-08 M n/a o? iuur|
naltrindole 2.7E-10 M 4.6E-11 M n/a (KOP) (antagonist effect)
nor-BNI 4.3E-10 M 7.2E-11 M n/a μ (MOP) (h) (antagonist effect)
CTOP 2.1 E-07 M 2.3E-08 M n/a
8. RESULTS INTERPRETATION GUIDE
In Vitro Pharmacology
Results showing a stimulation or an inhibition higher than 50% are considered to represent significant effects of the test compounds. 50% is the most common cut-off value for further investigation (determination of EC50 or IC50 values from concentration-response curves).
Results showing a stimulation or an inhibition between 25% and 50% are indicative of weak to moderate effects (in some assays, they may be confirmed by further testing as they are within a range where more inter-experimental variability can occur).
Results showing a stimulation or an inhibition lower than 25% are not considered significant and mostly attributable to variability of the signal around the control level.
J 9. MATERIALS AND METHODS 9.1. Experimental Conditions
Minor variations to the experimental protocol described below may have occurred during the testing, they have no impact on the quality of the results obtained.
9.1.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays
Assay Source Stimulus Incubation Measured Detection Method Bibl.
Component
Receptors
a2B (l>) human recombinant none 30 min cA P HTRF 915
(agonist effect) (HEK-293 cells) (3 μ 37°C
dexrnedeiomidine
for control)
(h) human recombinant dexmedetomidine 30 min cA P HTRF 915
(antagonist effect) (HEK-293 cells) ( 100 nM ) 37°C
δ2 (OOP) NG- 10815 cells none 37°C impedance Cellular dielectric 934
(agonist effect) (endogenous) (300 nM DPDPE spectroscopy
for control)
52 (DOP) NG-10815 cells DPDPE 37°C impedance Cellular dielectric 934
(antagonist effect) (endogenous) ( 10 nM ) spectroscopy
( OP) rat recombinant none 10 min cAMP HTRF 819
(agonist effect) (CHO cells) (0.3 μΜ U 50488 37"C
for control)
(KOP) rat recombinant U 50488 10 min cAMP HTRF 819
(antagonist effect) (CHO cells) ( 3 nM ) 37°C
(MOP) (h) human recombinant none 10 min cAMP HTRF 260
(agonist effect) (CHO cells) (0.3 μΜ DAMGO 37°C
for control)
μ (MOP) (h) human recombinant DAMGO 10 min cAMP HTRF 260
(antagonist effect) (CHO cells) ( 20 nM ) 37°C
9.2. Analysis and expression of results
9.2.1. In Vitro Pharmacology: Cellular and Nuclear Receptor Functional Assays
The results are expressed as a percent of control agonist response
measured response
_. : *100
control response and as a percent inhibition of control agonist response
measured response
100-( MOO)
control response obtained in the presence of HCV-3.
The EC5o values (concentration producing a half-maximal response) and IC50 values (concentration causing a half-maximal inhibition of the control agonist response) were determined by non-linear regression analysis of the concentration-response curves generated with mean replicate values using Hill equation curve fitting
A-D
Y=D+[ -
1 +(C/C50)"H where Y = response, A = left asymptote of the curve, D = right asymptote of the curve, C = compound concentration, and C50= EC50 or IC50, and nH = slope factor.
This analysis was performed using software developed at Cerep (Hill software) and validated by comparison with data generated by the commercial software SigmaPlot ® 4.0 for Windows ® (© 1997 by SPSS Inc.).
, the apparent dissociation constants (KB ) were calculated using the modified Cheng Prusoff equation
 where A = concentration of reference agonist in the assay, and EC50A = EC50 value of the reference agonist.
where A = concentration of reference agonist in the assay, and EC50A = EC50 value of the reference agonist.
110. BIBLIOGRAPHY
260. Wang, J.B. et al, (1994), FEBS Lett., 338„: 217-222.
819. Avidor-Reiss, T. et al. (1995), FEBS Lett., 361 ; 70-74.
915. Eason, .G. et al. (1992), J. Biol. Chem., 267 : 15795-15801.
934. Law, P.Y. and Loh, H.H. (1993), Mol. Pharmacol., 43_: 684-693.
Appendix 5
Docket No. UA 15-023
Company Name: Tech Launch Arizona 2 Cyprotex Study Number: CYP 1 199-R l
Table of Contents
AUTHENTICATION STATEMENT . 3
1 PURPOSE 4
2 STUDY CONDITIONS 4
3 EXPERIMENTAL DESIGN 4
3. 1 AMES MFP: Experimental Conditions 4 RESULTS AND CONCLUS IONS 5
4.1 AMES MPF: Data Summary 5
4.2 AMES MPF: Individual Data 6 REFERENCE 8
Cyprotex Study Number: CYP1 199-R 1
AUTHENTICATION STATEMENT
I the undersigned, hereby declare that the work described in this report was performed according to the study protocol and/or standard procedures, and to the best of my knowledge, this report provides a correct record of results obtained.

Study Manager
La :

Rarry Press. Ph.D.
Principal Scientist &
Quality Manager
Company Name: Tech Launch Arizona 4 Cyprotex Study Number: CYP 1 1 99-R1
1 PURPOSE
The purpose of this study was to evaluate the mutagenicity potential of a test article (HCV-3) in the Ames assay using the microplate fluctuation (MPF) method.
STUDY CONDITIONS
This study was performed under non-GLP conditions. All work was performed with appropriate local health regulations and ethical approval.
3 EXPERIMENTAL DESIGN
3.1 AMES MFP: Experimental Conditions

*The top concentration in the dose response reflects the highest soluble concentration of the test article in Ames assay conditions.
Experimental Procedure: Approximately ten mi l lion bacteria are exposed in triplicate to test agent (six concentrations), a negative control (vehicle) and a positive control for 90 minutes i n medium containing a low concentration of histidine (sufficient for about 2 doublings.) The cultures are then diluted into indicator medium lacking histidine, and dispensed into 48 wells of a 384 wel l plate (micro-plate format, MPF). The plate is incubated for 48 hr at 37°C, and cells that have undergone a reversion will grow in a well, resulting in a color change in wells with growth. The number of wel ls showing growth are counted and compared to the vehicle control. An increase in the number of colonies of at least two-fold over baseline (mean + SD of the vehicle control) and a dose response indicates a positive response. An unpaired, one-sided Student's T-test is used to identify conditions that are significantly different from the vehicle control.
Where indicated, S9 fraction from the livers of Aroclor 1254-treated rats is included in the incubation at a final concentration of 4.5%. An NADPH-regenerating system is also included to ensure a steady supply of reducing equivalents.
Strains used in this study:
S. typhimurium TA98: hisD3052, rfa, uvrB / pK l O l ; detects frame-shift mutations.
S. typhimurium TA 100: hisG45, rfa, uvrB / pKM l O l ; detects base-pair substitutions.

Cyprotex Study Number: CYP 1 199-R1
RESULTS AND CONCLUSIONS
4.1 AMES MPF: Data Summary

*Highest soluble concentration of test article in Ames assay conditions.
Conclusion: Test article HCV-3 was assessed for its mutagenic potential in the Ames reverse mutation assay. This test was performed in the absence and presence of S9 metabolic activation. HCV-3 was found to be negative for genotoxicity against both strains used in this study (TA98 and TA 100) up to a maximum tested concentration of 1 mg/ml. The positive controls behaved as expected.
U 2015/046585
Company Name: Tech Launch Arizona
Cyprotex Study Number: CYP 1 199-R1
4.2 AMES MPF: Individual Data
Significant fold increase over baseline values (>2-fold) are indicated in bold red. Significant T-test probabilities (P < 0.05) appear red and (P<0.01 ) are indicated in bold red.
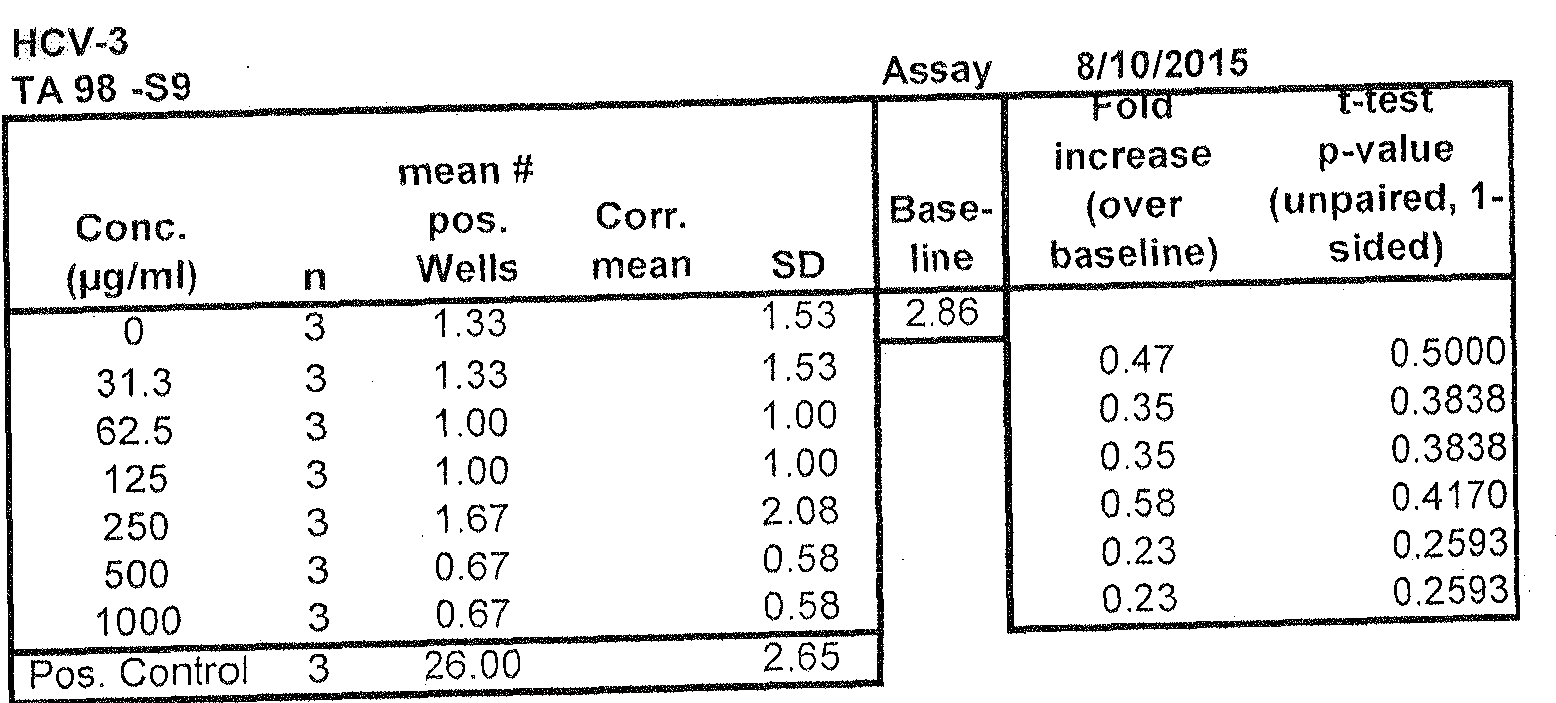
HGV-3
TA 98 +S9 Assay 8/10/2015
~— Forar— " t-test mean # increase p-value
Cone. pos. Corr. Base(over (unpaired, 1 - n Wells mean SD line baseline) sided)
0 3 4.00 1 .00 5.00
31.3 3 5.67 2.52 1 .13 0.1732
62.5 3 6.67 2.31 1.33 0.0702
125 3 6.00 3.61 1 .20 0.2035
250 3 6.00 4.36 1.20 0.2409
500 3 3.67 2.08 0.73 0.4075
1000 3 5.00 4.00 1.00 0.3480
Pos. Control 3 34.33 8.33
Cyprotex Study Number: CYPl 199-RI
HCV-3

Company Name: Tech Launch Arizona
Cyprotex Study Number: CYP1199-R]
REFERENCE
Umbuzeiro, G. et al. (2010). "Comparison of the Salmonella/microsome micro-suspension assay with the new microplate fluctuation protocol for testing the mutagenicity of environmental samples". Environ. Mol. Mutagen.51(l):31-38,
Appendix - References Cited UA 15-023 PCT
Appendix - Literature Cited
1. Thayer, A, Drugs To Fight Addictions, Chem. Eng. News, (2006), 84(39), 21 -44.
2. Heidbreder, C, Novel pharmacotherapeutic targets for the management of drug addiction, Eur. J. Pharmacol., (2005), 526 (1 -3), 101 -1 12.
3. Woods, .1.1 L, Traynor, J.R., Evaluation of new compounds for opioid activity (2000), NIDA Research Monograph, Volume Date 2000, 181 (Problems of Drug Dependence 2000), (2001 ), 140-155.
4. Husbands, S.M., Lewis, J.W., Opioid ligands having delayed long-term antagonist activity: Potential pharmacotherapies for opioid abuse, Mini-Revi. Med. Chem., (2003), 3(2), 137- 144.
5. Schmidhammer, H., Opioid receptor antagonists, Prog. Med. Chem., (1998), 35, 83- 1 32,.
6. Boothby, L.A.,Doering, P.L., Buprenorphine for the treatment of opioid dependence, Am. J. Health-System Pharm., (2007), 64(3), 266-272.
7. Lowengrub, K., Iancu, I., Aizer, A., Kotler, M., Dannon, P.N., Pharmacotherapy of pathological gambling: review of new treatment modalities, Exp. Rev. Neurotherapeut., (2006), 6(12), 1845-1 851 .
8. Krishnan-Sarin, S., O'Malley, S.S., Opioid antagonists for the treatment of nicotine dependence, Med. Treat. Nicotine Depend., (2007) 123-135.
9. White, J.M., Lopatko, O.V., Opioid maintenance: a comparative review of pharmacological strategies, Expert Opin. Pharmacotherapy, (2007), 8( 1), 1-1 1 .
10. Cunningham, C.W., Coop, A.,Therapeutic applications of opioid antagonists, Chimica Oggi, 24(3), 54-57 (2006).
1 1. Lauretti, G.R., Highlights in opioid agonists and antagonists, Expert Rev.
Neurotherapeut., 6(4), 613-622 (2006).
12. Capasso, A., D'Ursi, A., Pharmacological activity of new mu, k, delta receptor agonists and antagonists. Studies in Natur. Prod. Chem. (2005), 30, 797-823.
13. Anon, N.Z., Alvimopan: ADL 8-2698, ADL 82698, entrareg, LY 246736, Drugs in R&D, (2006), 7(4), 245-253.
14. Leslie, J. B., Alvimopan: a peripherally acting Mu-. Opioid receptor
antagonists, Drugs of Today (2007), 43(9), 61 1 -625.
15. Goodman, A. J.; Le Bourdonnec, B.; Do lie. R. E. Mu opioid receptor antagonists: recent developments, ChemMedChem (2007), 2( 1 1 ), 1 552-1570.
16. Taylor, R., Jr.; Pergolizzi, J. V., Jr.; Porreca, F.; Raffa, R. B. Opioid antagonists for pain
Exp. Opin. Invest. Drugs, (2013), 22(4), 517-525.
17. Hipkin, R. W.; Dolle, Roland E., Opioid receptor antagonists for gastrointestinal dysfunction, Ann. Rep. Med. Chem., (2010), 45, 143-155.
18. Stotts, A. L.; Dodri ll, C. L.; Kosten, T. R. Opioid dependence treatment: options in pharmacotherapy, Exp. Opin. Pharmacother., (2009), 10(1 1 ), 1727-1740.
19.Soyka, M.; Roesner, S., Opioid antagonists for pharmacological treatment of alcohol dependence - a critical review, Curr. Drug Abuse Rev., (2008), 1 (3), 280-291.
20. Hopp, M.; Trenkwalder, C, Combination of opioid agonists and opioid
antagonists for the treatment of Parkinson's disease and associated
symptoms, WO 2012089738 (2012).
21. Mouradian, M. M.; Braithwaite, S.; Voronkov, M, Method of treating dyskinesia using dual-action mu-opioid receptor antagonist /kappa- opioid receptor agonist or prodrug thereof, WO 20121491 13 (2012).
22. Bosco, D.; Plastino, M.; Colica, C; Bosco, F.; Arianna, S.; Vecchio, A.; Galati, F.; Cristiano, D.; Consoli, A.; Consoli, D., Opioid Antagonist Naltrexone for the Treatment of Pathological Gambling in Parkinson Disease, Clinical
Neuropharmacology (2012), 35(3), 1 18-120.
23. Henry, B.; Brotchie, J. M., Potential of opioid antagonists in the treatment of levodopa-induced dyskinesias in Parkinson's disease (A review and discussion), Drugs & Aging (1996), 9(3), 149-158.
24. Buck, K.; Ferger, B., The selective al adrenoceptor antagonist HEAT reduces L- DOPA-induced dyskinesia in a rat model of Parkinson's disease, Synapse
(2010), 64(2), 1 17-126.
25. Lewitt P. A; Hauser R. A; Lu M.; Nicholas A. P.; Weiner W.; Coppard N.; Leinonen M.; Savola J.-M., Randomized clinical trial of fipamezole for dyskinesia in Parkinson disease (FJORD study), Neurology (2012), 79(2), 163-9.
26. Brefel-Courbon, C; Thalamas, C; Paul, I I. P. S.: Senard, J-M.; Montastruc, J-L.; Rascol, O., a2 -Adrenoceptor antagonists. A new approach to Parkinson's disease? CNS Drugs (1998), 10(3), 189-207.
27. Millan M. J., From the cell to the clinic: a comparative review of the partial D2/D3 receptor agonist and a2-adrenoreceptor antagonists, piribedil, in the treatment of Parkinson's disease
Pharmacol. Therapeut. (2010), 128(2), 229-73.
28. 14. Kaczor, A., Matosiuk, D., Non-peptide opioid receptor ligands - recent advances. Part II. Antagonists, Curr. Med. Chem.. (2002), 9(17), 1591 - 1 603.
29. 15. Zimmerman, D. M., Leander, J. D., Opioid antagonists: structure activity relationships, NIDA Research Monograph, ( 1990), 96, 50-60 (1990).
30. Lauretti, G. R., Highlights in opioid agonists and antagonists, Exp. Rev.,
Neurotherapeut., (2006), 6(4), 613-622 (2006).
31 . van Dorp, E. L. A., Yassen, A., Dalian, A., Naloxone treatment in opioid addiction: the risks and benefits, Exp. Opin. Drug Safety, (2007), 6(2), 125- 132.
32. Comer, S. D., Sullivan, M. A,; Hulse, G. K., Sustained-release naltrexone: novel treatment for opioid dependence, Exp. Opin. Invest. Drugs, (2007), 16(8), 1285-1294.
33. Boothby, L. A., Doering, P. L., Buprenorphine for the treatment of opioid dependence, Am. J. Health-Syst. Pharm., (2007), 64(3), 266-272.
34. Raisch, D.W., Fye, C. L., Boardman, K. D., Sather, M. R. Opioid dependence treatment, including buprenorphine/naloxone. Annals Pharmacother., (2002), 36(2), 312- 321.
35. White, J. M., Lopatko, O.V., Opioid maintenance: a comparative review of pharmacological strategies, Exp. Opin. Pharmacother., (2007), 8(1), 1-1 1 .
36. Roozen, H. G., de Waart, R., van der Windt, D. A. W. M., van den Brink, W., de Jong, C. A. J., Kerkhof, A. J. F. M., A systematic review of the effectiveness of naltrexone in the maintenance treatment of opioid and alcohol dependence, Eur.
Neuropsychopharmacol., (2006), 16(5), 31 1 -323.
37. Schmidhammer, I I.. Opioid receptor antagonists, Progr. Med. Chem., (1998), 35, 83-132.
38. Yuan, C.-S., Israel, R. J., Methylnaltrexone, a novel peripheral opioid receptor antagonist for the treatment of opioid side effects, Exp. Opin. Invest. Drugs, (2006), 15(5), 541 -552.
39. Comer, S. D., Sullivan, M. A., Hulse, Gary K., Sustained-release naltrexone: novel treatment for opioid dependence, Exp. Opin. Invest. Drugs, (2007), 16(8), 1285-1294.
40. Eguchi, M., Recent advances in selective opioid receptor agonists and antagonists, Med. Res. Rev., (2004), 24(2), 182-212.
41. Portoghese, P. S., Selective nonpeptide opioid antagonists, Handbook of
Experimental Pharmacology, (1993), 104/1 (Opioids I), 279-93.
42. Takemori, A. E., Portoghese P S Selective naltrexone-derived opioid receptor antagonists, Ann. Rev. Pharm. Tox., (1992), 32, 239-69.
43. Portoghese, P. S., Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists, Trends Pharm. Sci., (1989), 10(6), 230-5.
44. Portoghese, P. S., The design of delta-selective opioid receptor antagonists, Farmaco, (1993), 48(2), 243-51.
45. Metcalf, M. D., Coop A., Kappa opioid antagonists : past successes and future prospects, The A A PS J., (2005), 7(3), E704-22.
46. Furst, S., Hosztafi, S., Friedmann, T., Structure-Activity Relationships of Synthetic and Semisynthetic Opioid Agonists and Antagonists, Curr. Med. Chem., (1995), 1, 423- 40.
Incorporation by reference
Each document, patent, patent application or patent publication cited by or referred to in this disclosure is incorporated by reference in its entirety. However, no admission is made that any such reference constitutes prior art, and the right to challenge the accuracy and pertinence of the cited documents is reserved.
Claims
1. A compound having the formula

Ar , or (HetAr), or COOR5, or COON(R6)2 (III)
wherein
Ri is I I, substituted or unsubstituted Q-Cio alkyl, alkenyl, or alkynyl, or substituted or unsubstituted aryl or hetaryl;
R2 is H, -CH20-C, _4 alkyl; COO-C, 4 alkyl; -CONR4;
R3 is H, substituted or unsubstituted Q-C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl;
A is substituted or unsubstituted Cj-C 10 alkyl, alkylene, alkynyl;
Ar or HetAr is substituted or unsubstituted monocyclic or polycyclic aromatic or heteroaromatic moiety;
COOR5, or CON(R6)2, where R5 and R6 are H, substituted or unsubstituted C r C10 alkyl, alkylene, alkynyl, or substituted or unsubstituted aryl or hetaryl;
the central nitrogen-containing ring is a substituted or unsubstituted 5- to 7- membered heterocyclic ring; and pharmaceutically acceptable salts of said compound.
2. The compound of claim 1, wherein the compound belongs to a series of N-(l- arylethylpiperidin-4-yl)acylamides.
3. The compound of claim 1, wherein the compound is N-( 1 -phenethylpiperidin-4- yl)propionamide.
4. The compound of claim 1, wherein the compound is N-( 1 -phenethylpiperidin-4- yl)propionamide oxalate.
5. A process for preparing a compound of claim 1 , comprising the following steps:
(a) reacting a cyclic ketone having a protecting group in a Grignard or
Reformatsky reaction to obtain a first product;
(b) reacting the product of step (a) in a Ritter reaction to obtain a second product; and
(c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of claim 1 .
6. A process for preparing a compound of claim 1 , comprising the following steps:
(a) reacting a cyclic ketone having a protecting group in a Strecker reaction to obtain a first product;
(b) reacting the product of step (a) in a selective carbalkoxy group
transformation to obtain a second product; and
(c) deprotecting the product of step (b) with acylation or alkylation to obtain a compound of claim 1.
A process for the preparation of N-(l -phenethylpiperidin-4-yl)propionamide, comprising the following steps:
(a) reacting phenethylpiperiding-4-one with hydroxy lam ine hydrochloride in ethanol in the presence of a base, to produce 1 -phenethylpiperidin-4-one oxime;
(b) reducing the oxime obtained in step (a) with iso-amyl alcohol and sodium metal to produce l-phenethylpiperidin-4-amine; and
(c) acylating the product of step (b) with propionic acid chloride in
chloroform in the presence of triethylamine to produce N-(l - phenethylpiperidin-4-yl)propionamide.
The process of claim 7, wherein the N-(l -phenethylpiperidin-4-yl)propionamide is further treated with oxalic acid to produce N-( 1 -phenethylpiperidin-4- yl)propionamide oxalate.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15834369.9A EP3183232B1 (en) | 2014-08-22 | 2015-08-24 | Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462040886P | 2014-08-22 | 2014-08-22 | |
| US62/040,886 | 2014-08-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2016029218A1 true WO2016029218A1 (en) | 2016-02-25 |
| WO2016029218A4 WO2016029218A4 (en) | 2016-04-14 |
Family
ID=55347719
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/046585 Ceased WO2016029218A1 (en) | 2014-08-22 | 2015-08-24 | Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation |
| PCT/US2016/030649 Ceased WO2017034631A1 (en) | 2014-08-22 | 2016-05-04 | Substituted 1-arylalkyl-4-acylaminopiperidine compounds and methods of producing and using thesame |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/030649 Ceased WO2017034631A1 (en) | 2014-08-22 | 2016-05-04 | Substituted 1-arylalkyl-4-acylaminopiperidine compounds and methods of producing and using thesame |
Country Status (3)
| Country | Link |
|---|---|
| US (5) | US9765027B2 (en) |
| EP (1) | EP3183232B1 (en) |
| WO (2) | WO2016029218A1 (en) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9765027B2 (en) * | 2014-08-22 | 2017-09-19 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation |
| PT3344248T (en) | 2015-09-02 | 2022-06-29 | Trevena Inc | 6-membered aza-heterocyclic containing delta-opioid receptor modulating compounds, methods of using and making the same |
| US10766925B2 (en) * | 2016-04-11 | 2020-09-08 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Opioid receptor modulators |
| US11225487B2 (en) | 2017-02-17 | 2022-01-18 | Trevena, Inc. | 7-membered aza-heterocyclic containing δ-opioid receptor modulating compounds, methods of using and making the same |
| BR112019016827A2 (en) | 2017-02-17 | 2020-04-07 | Trevena Inc | 5-membered azaheterocyclic delta-opioid receptor modulating compounds, methods of use and production thereof |
| WO2022119859A1 (en) * | 2020-12-01 | 2022-06-09 | Biocorrx Pharmaceuticals, Inc. | Treatment of neurological disorders |
| WO2022119862A1 (en) * | 2020-12-01 | 2022-06-09 | Biocorrx Pharmaceuticals, Inc. | Treatment of pain and neurological conditions |
| WO2023133135A2 (en) * | 2022-01-04 | 2023-07-13 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Small molecule adrenoreceptor antagonists and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4029801A (en) * | 1970-09-03 | 1977-06-14 | John Wyeth & Brother Limited | Pharmaceutical compositions and methods of treating hypertension |
| US4649144A (en) * | 1983-07-27 | 1987-03-10 | Dainippon Pharmaceutical Co., Ltd. | Antibacterial 7-(3-amino-1-pyrrolidinyl)-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid derivatives |
| US20040147503A1 (en) * | 2002-06-04 | 2004-07-29 | Sheila Zipfeil | Novel compounds and compositions as cathepsin inhibitors |
| WO2007058482A1 (en) * | 2005-11-16 | 2007-05-24 | Lg Life Sciences, Ltd. | Novel inhibitors of protein kinase |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4024147A (en) * | 1972-06-30 | 1977-05-17 | John Wyeth & Brother Limited | Heterocyclic compounds |
| US4304911A (en) * | 1980-11-19 | 1981-12-08 | Sterling Drug Inc. | Benzoylphenyl lower alkanoyl piperidines |
| US5214055A (en) * | 1990-05-18 | 1993-05-25 | Adir Et Compagnie | Aminopiperidine 4-oxo-4H-chromen-2-yl compounds |
| CA2393559C (en) * | 1999-12-06 | 2009-05-26 | Mallinckrodt Inc. | Methods for the syntheses of alfentanil, sufentanil and remifentanil |
| JP2005503351A (en) * | 2001-04-03 | 2005-02-03 | アリックス セラピューティクス | Ultra-short acting opioids for transdermal application |
| JP4740152B2 (en) * | 2003-12-23 | 2011-08-03 | セロドス アクスイェ セルスカブ | Modulator of peripheral 5-HT receptor |
| ES2289888B1 (en) | 2005-09-08 | 2008-12-16 | Consejo Superior Investig. Cientificas | DERIVATIVES OF PIRAZOLCARBOXAMIDA, ITS PROCESSING PROCEDURE AND ITS APPLICATIONS AS INVESTED ANTAGONISTS / AGONISTS OF THE CANNABINOID CB1 AND OPIOID MU. |
| AR084620A1 (en) | 2010-12-28 | 2013-05-29 | Euro Celtique Sa | A COMBINATION OF AN OPIACE AGONIST AND AN OPIACE ANTAGONIST IN THE TREATMENT OF PARKINSON'S DISEASE |
| WO2012149113A1 (en) | 2011-04-29 | 2012-11-01 | University Of Medicine And Dentistry Of New Jersey | Method of treating dyskinesia |
| US9765027B2 (en) * | 2014-08-22 | 2017-09-19 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation |
| CN105704085B (en) * | 2014-11-24 | 2018-11-02 | 国际商业机器公司 | Method and apparatus for information sharing |
-
2015
- 2015-08-24 US US14/834,185 patent/US9765027B2/en not_active Expired - Fee Related
- 2015-08-24 EP EP15834369.9A patent/EP3183232B1/en active Active
- 2015-08-24 WO PCT/US2015/046585 patent/WO2016029218A1/en not_active Ceased
-
2016
- 2016-05-04 WO PCT/US2016/030649 patent/WO2017034631A1/en not_active Ceased
- 2016-05-04 US US15/756,014 patent/US20180235949A1/en not_active Abandoned
-
2017
- 2017-09-19 US US15/709,394 patent/US20180021322A1/en not_active Abandoned
-
2018
- 2018-12-31 US US16/236,981 patent/US10617681B2/en active Active
-
2020
- 2020-03-25 US US16/829,299 patent/US11440885B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4029801A (en) * | 1970-09-03 | 1977-06-14 | John Wyeth & Brother Limited | Pharmaceutical compositions and methods of treating hypertension |
| US4649144A (en) * | 1983-07-27 | 1987-03-10 | Dainippon Pharmaceutical Co., Ltd. | Antibacterial 7-(3-amino-1-pyrrolidinyl)-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid derivatives |
| US20040147503A1 (en) * | 2002-06-04 | 2004-07-29 | Sheila Zipfeil | Novel compounds and compositions as cathepsin inhibitors |
| WO2007058482A1 (en) * | 2005-11-16 | 2007-05-24 | Lg Life Sciences, Ltd. | Novel inhibitors of protein kinase |
Non-Patent Citations (16)
| Title |
|---|
| AVIDOR-REISS, T. ET AL., FEBS LETT, vol. 361, 1995, pages 70 - 74 |
| BERGE ET AL., J. PHARM. SCIENCES, vol. 6, 1977, pages 1 - 19 |
| BRUKER: "SAINT", 2007, BRUKER AXS INC. |
| CHEMICAL ABSTRACTS, 23 October 2012, Columbus, Ohio, US; abstract no. 150462038 * |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 56006844 * |
| DOLOMANOV, O.V.BOURHIS, L.J.GILDEA, R.JHOWARD, J.A.K.PUSCHMANN, H., J. APPL. CRYST., vol. 42, 2009, pages 339 - 341 |
| EASON, M.G. ET AL., J. BIOL. CHEM., vol. 267, 1992, pages 15795 - 15801 |
| LAW, P.Y.LOH, H.H., MOL PHARMACOL., vol. 43, 1993, pages 684 - 693 |
| MACRAE, C. F.BRUNO, I. J.CHISHOLM, J. A.EDGINGTON, P. R.MCCABE, P. PIDCOCK, E.RODRIGUEZ-MONGE, L.TAYLOR, R.VAN DE STREEK, J.WOOD, , J. APPL. CRYST., vol. 42, 2008, pages 339 - 341 |
| See also references of EP3183232A4 |
| SHELDRICK, G. M., ACTA CRYST., vol. A64, 2008, pages 112 - 122 |
| SHELDRICK, G. M.: "SADABS", 1996, UNIVERSITY OF GOTTINGEN |
| SHELDRICK, G.M., ACTA CRYST. A64, 2008, pages 112 - 122 |
| SPEK, A. L., J. APPL. CRYST., vol. 36, 2003, pages 7 - 13 |
| UMBUZEIRO, G. ET AL.: "Comparison of the Salmonella/microsome micro-suspension assay with the new microplate fluctuation protocol for testing the mutagenicity of environmental samples", ENVIRON. MOL. MUTAGEN., vol. 51, no. 1, 2010, pages 31 - 38 |
| WANG, J.B. ET AL., FEBS LETT, vol. 338, 1994, pages 217 - 222 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180021322A1 (en) | 2018-01-25 |
| WO2016029218A4 (en) | 2016-04-14 |
| US20180235949A1 (en) | 2018-08-23 |
| US9765027B2 (en) | 2017-09-19 |
| WO2017034631A1 (en) | 2017-03-02 |
| US20200222380A1 (en) | 2020-07-16 |
| US20160052882A1 (en) | 2016-02-25 |
| US10617681B2 (en) | 2020-04-14 |
| EP3183232B1 (en) | 2023-05-03 |
| US20190134018A1 (en) | 2019-05-09 |
| US11440885B2 (en) | 2022-09-13 |
| EP3183232A1 (en) | 2017-06-28 |
| EP3183232A4 (en) | 2018-05-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3183232A1 (en) | Substituted 1-arylethyl-4-acylaminopiperidine derivatives as opioid/alpha-adrenoreceptor modulators and method of their preparation | |
| RS20060316A (en) | Derivatives of n-/heteroaryl(piperidine-2-yl)methyl/ benzamide, preparation method and application of same in therapeutics | |
| EP1675847B1 (en) | Spirocyclic heterocyclic derivatives and methods of their use | |
| Jia et al. | Design, synthesis and evaluation of galanthamine derivatives as acetylcholinesterase inhibitors | |
| RS20060315A (en) | Derivatives of n-/phenyl(pyrrolidine-2-yl) methyl/benzamide, and n- /(azepan-2-yl)phenylmethyl/benzamide, preparation method thereof and application of same in therapeutics | |
| EP3544972B1 (en) | Heteroarylphenoxy benzamide kappa opioid ligands | |
| CA2895129A1 (en) | Quinazoline neurotensin receptor 1 agonists and uses thereof | |
| JP2018531981A (en) | N- [2- (1-Benzylpiperidin-4-yl) ethyl] -4- (pyrazin-2-yl) -piperazine as a muscarinic receptor 4 (M4) antagonist for treating neurological diseases 1-carboxamide derivatives and related compounds | |
| RS20060317A (en) | Derivatives of n-/phenyl(alkylpiperidine-2-yl)methyl/ benzamide, preparation method thereof and application of same in therapeutics | |
| US20130158072A1 (en) | Kappa opioid receptor binding ligands | |
| Simons et al. | The synthesis and structure–activity relationship of substituted N-phenyl anthranilic acid analogs as amyloid aggregation inhibitors | |
| CN112794860B (en) | Oxazole pyrimidone amide compound or medicinal salt thereof, preparation method and application | |
| EP3470404A1 (en) | Five-membered heterocyclic derivative, method for producing same, and pharmaceutical composition comprising same | |
| Li et al. | Evaluation of N-phenyl homopiperazine analogs as potential dopamine D3 receptor selective ligands | |
| EP2539706B1 (en) | Arylpiperazine opioid receptor antagonists | |
| JP5107728B2 (en) | Kappa opioid receptor ligand | |
| HUP0400975A2 (en) | 4-(phenyl-piperidin-4-ylidene-methyl)-benzamide derivatives and their use, process for their preparation and pharmaceutical compositions containing them | |
| HUP0401285A2 (en) | 4-(phenyl-piperidin-4-ylidene-methyl)-benzamide derivatives, their use, process for their preparation and pharmaceutical compositions containing them | |
| JP2011512414A5 (en) | ||
| KR101181175B1 (en) | Novel cinnamic acid derivative and pharmaceutical composition comprising the same | |
| EP1678133A1 (en) | Sequence selective pyrrole and imidazole polyamide metallocomplexes | |
| Conroy et al. | A systematic exploration of the effects of flexibility and basicity on sigma (σ) receptor binding in a series of substituted diamines | |
| JP2013511492A (en) | Sultam derivatives | |
| Jin et al. | Synthesis and receptor binding properties of 2β-alkynyl and 2β-(1, 2, 3-triazol) substituted 3β-(substituted phenyl) tropane derivatives | |
| CA3045242C (en) | Heteroarylphenoxy benzamide kappa opioid ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15834369 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015834369 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015834369 Country of ref document: EP |