WO2016035258A1 - リビングラジカル重合開始剤、重合体の製造方法及び重合体 - Google Patents
リビングラジカル重合開始剤、重合体の製造方法及び重合体 Download PDFInfo
- Publication number
- WO2016035258A1 WO2016035258A1 PCT/JP2015/003991 JP2015003991W WO2016035258A1 WO 2016035258 A1 WO2016035258 A1 WO 2016035258A1 JP 2015003991 W JP2015003991 W JP 2015003991W WO 2016035258 A1 WO2016035258 A1 WO 2016035258A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polymer
- radical polymerization
- living radical
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- LDOXTQYWWYXYSQ-UHFFFAOYSA-N CCCCOC(Cc1ccccc1)=O Chemical compound CCCCOC(Cc1ccccc1)=O LDOXTQYWWYXYSQ-UHFFFAOYSA-N 0.000 description 1
- JVFSBOFJMYIDFN-UHFFFAOYSA-N COC(C(c(cc1)ccc1OC(CCCI)=O)I)=O Chemical compound COC(C(c(cc1)ccc1OC(CCCI)=O)I)=O JVFSBOFJMYIDFN-UHFFFAOYSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F293/00—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule
- C08F293/005—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule using free radical "living" or "controlled" polymerisation, e.g. using a complexing agent
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/63—Halogen-containing esters of saturated acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/63—Halogen-containing esters of saturated acids
- C07C69/635—Halogen-containing esters of saturated acids containing rings in the acid moiety
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/65—Halogen-containing esters of unsaturated acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/38—Polymerisation using regulators, e.g. chain terminating agents, e.g. telomerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
Definitions
- the present invention relates to a polymerization initiator used for living radical polymerization, a method for producing a polymer, and a polymer produced using them.
- a radical polymerization method has been well known as a method for obtaining a vinyl polymer by polymerizing vinyl monomers.
- the radical polymerization method has a drawback that it is difficult to control the molecular weight of the obtained vinyl polymer.
- the obtained vinyl polymer becomes a mixture of compounds having various molecular weights, and it is difficult to obtain a vinyl polymer having a narrow molecular weight distribution.
- the ratio (Mw / Mn) of the weight average molecular weight (Mw) to the number average molecular weight (Mn) could only be reduced to about 2 to 3. .
- a living radical polymerization method has been developed since around 1990 as a method for solving such drawbacks. That is, according to the living radical polymerization method, it is possible to control the molecular weight and obtain a polymer having a narrow molecular weight distribution.
- the conventional living radical polymerization method described above was a reaction system in which a polymerization reaction proceeds with respect to one initiator group of a polymerization initiator, but there are reaction systems that can perform different polymerization reactions with respect to a plurality of initiator groups. More preferably.
- the present invention has been devised in view of the above-mentioned problems, and has two halogen atoms having different reactivities, and can perform different polymerization reactions in two directions, each of which is an initiating group. It is an object to provide a living radical polymerization initiator, a method for producing a polymer, and a polymer produced using them.
- the present inventor has a living radical polymerization initiator, a heavy radical polymerization initiator, which has two halogen atoms with different reactivities and can perform different polymerization reactions in two directions with each as an initiating group.
- Invented a method for producing coalescence and a polymer produced using them.
- the living radical polymerization initiator according to the first invention is a living radical polymerization initiator consisting of the following general formula (1):
- R 1 is an organic group that can be linked to two or more other organic groups, and is an aliphatic group having 1 to 12 carbon atoms, an aromatic group, an alkylcarbonyl group having 1 to 12 carbon atoms, or 1 carbon atom.
- R 2 , R 3 , R 4 and R 5 are each a hydrogen atom, an aliphatic group having 1 to 12 carbon atoms, an aromatic group, or an aromatic group having 1 to 12 carbon atoms.
- Alkylcarbonyl group alkoxycarbonyl group having 1 to 12 carbon atoms, aminocarbonyl group, alkylaminocarbonyl group having 1 to 12 carbon atoms, dialkylaminocarbonyl having 1 to 12 carbon atoms
- the living radical polymerization initiator according to the second invention is characterized in that, in the first invention, the halogen atom is iodine, chlorine or bromine.
- a method for producing a polymer according to a third invention is a method for producing a polymer using the living radical polymerization initiator according to the first or second invention, wherein either the X or Y of the living radical polymerization initiator is used.
- the living radical polymerization initiator and a monomer having an unsaturated bond are mixed, and the living radical polymerization reaction performed under reaction conditions according to the type of the monomer is mixed.
- the first polymerization step for obtaining the first product by performing at least once while sequentially changing the above, and at least one or more types of the monomers for the X or Y halogen atoms contained in the first product A second polymerization step in which a living radical polymerization reaction is sequentially performed under reaction conditions according to the type to obtain a final product.
- the first polymerization step and the second polymerization step are performed using a catalyst, and the first polymerization step and the second polymerization step are performed at a reaction temperature. And at least one of the types of the catalyst is made different depending on the type of the monomer.
- the method for producing a polymer according to a fifth aspect of the present invention is the method according to the fourth aspect, wherein the catalyst is a transition metal complex catalyst used in atom transfer radical polymerization, phosphorus, nitrogen, carbon, oxygen, germanium used in reversible transfer catalytic polymerization, A catalyst comprising a compound comprising at least one central element selected from tin and antimony and a halogen atom bonded to the central element, an organic amine compound catalyst used in reversible complexation-mediated polymerization, or a halide ion And a non-metallic compound having a ionic bond with the non-metallic compound, wherein the non-metallic atom in the non-metallic compound is in a cation state and is a catalyst that forms an ionic bond with a halide ion.
- the catalyst is a transition metal complex catalyst used in atom transfer radical polymerization, phosphorus, nitrogen, carbon, oxygen, germanium used in reversible transfer catalytic polymer
- the method for producing a polymer according to any one of the third to fifth aspects wherein the first polymerization step and the second polymerization step are performed at 180 ° C. or lower. .
- the method for producing a polymer according to any one of the third to sixth aspects wherein the first polymerization step and the second polymerization step are performed with a reaction time of 30 minutes to 24 hours. It is characterized by being.
- the polymer according to the eighth invention is characterized by being manufactured using the method for producing a polymer according to any one of the third to seventh inventions.
- FIG. 1 is a 1H-NMR chart of a living radical polymerization initiator and each polymer obtained by the method for producing a polymer according to an example, (a) is a structural formula of an alkyl iodide initiator as a living radical polymerization initiator, (b ) Is a 1H-NMR chart of a living radical polymerization initiator, (c) is a 1H-NMR chart of polymer P, (d) is a 1H-NMR chart of polymer Q, and (e) is a 1H-NMR chart of polymer R. .
- Living radical polymerization initiator (1) Chemical formula of living radical polymerization initiator The living radical polymerization initiator according to this embodiment has a structure represented by chemical formula (1) and has two halogen atoms having different reactivities.
- R 1 is a divalent or higher organic group i.e. two or more other organic groups and connectable organic group, aliphatic group having 1 to 12 carbon atoms, an aromatic group, the number of carbon atoms
- Examples thereof include 1 to 12 alkylsulfonyl groups, arylsulfonyl groups, and organic groups obtained by combining two or more of these groups.
- R 2 , R 3 , R 4 and R 5 are each a hydrogen atom, an aliphatic group having 1 to 12 carbon atoms, an aromatic group, an alkylcarbonyl group having 1 to 12 carbon atoms, or an alkoxycarbonyl group having 1 to 12 carbon atoms.
- Group, aminocarbonyl group, alkylaminocarbonyl group having 1 to 12 carbon atoms, dialkylaminocarbonyl group having 1 to 12 carbon atoms, arylcarbonyl group, alkylsulfonyl group having 1 to 12 carbon atoms, and arylsulfonyl group X and Y are halogen atoms, and n and m each represents an integer of 1 or more.
- the structures on the left and right of the linking group R 1 in the chemical formula (1) are asymmetric, so that different reactivities are imparted to the halogen atoms X and Y.
- the connecting group R 1 is not particularly limited as long as it is an organic group that can be connected to two or more other organic groups. Specifically, an aliphatic group, an aromatic group, an alkylcarbonyl group having 1 to 12 carbon atoms, an alkoxycarbonyl group having 1 to 12 carbon atoms, an aminocarbonyl group, an alkylaminocarbonyl group having 1 to 12 carbon atoms, a carbon number Examples thereof include 1 to 12 dialkylaminocarbonyl groups, arylcarbonyl groups, alkylsulfonyl groups having 1 to 12 carbon atoms, arylsulfonyl groups, and organic groups in which two or more of these groups are combined.
- the linking group R 1 may have a substituent, and the number of substituents in this case is not particularly limited as long as substitution is possible, and is 1 or more.
- Examples of the group which may be substituted for the linking group R 1 include a halogen atom, an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms, an optionally substituted aromatic group, Examples thereof include a non-aromatic heterocyclic group which may be substituted, a carboxyl group, a linear or branched alkoxy group having 1 to 12 carbon atoms, a cyano group or a nitro group.
- Examples of the aliphatic group include an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms.
- the number of substituents is not particularly limited as long as substitution is possible, and is one or more.
- Examples of the group that may be substituted for the aliphatic group include a halogen atom, an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms, an optionally substituted aromatic group, Examples thereof include an optionally substituted non-aromatic heterocyclic group, a linear or branched alkoxy group having 1 to 12 carbon atoms, a cyano group, or a nitro group.
- aromatic group examples include an aromatic hydrocarbon ring group or an aromatic heterocyclic group. Specifically, a phenyl group, a biphenylyl group, a terphenylyl group, a naphthyl group, a binaphthyl group, an azulenyl group, an anthracenyl group, a phenanthrenyl group, a fullerenyl group.
- the aromatic group may be substituted, and the number of substituents in this case is not particularly limited as long as substitution is possible, and is one or more.
- Examples of the group that may be substituted for the aromatic group include a halogen atom, an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms, an optionally substituted aromatic group, Examples thereof include a non-aromatic heterocyclic group which may be substituted, a carboxyl group, a linear or branched alkoxy group having 1 to 12 carbon atoms, a cyano group or a nitro group.
- R 2 to R 5 examples of the aliphatic group include an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms.
- the number of substituents is not particularly limited as long as substitution is possible, and is one or more.
- Examples of the group that may be substituted for the aliphatic group include a halogen atom, an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms, an optionally substituted aromatic group, Examples thereof include an optionally substituted non-aromatic heterocyclic group, a linear or branched alkoxy group having 1 to 12 carbon atoms, a cyano group, or a nitro group.
- aromatic group examples include an aromatic hydrocarbon ring group or an aromatic heterocyclic group. Specifically, a phenyl group, a biphenylyl group, a terphenylyl group, a naphthyl group, a binaphthyl group, an azulenyl group, an anthracenyl group, a phenanthrenyl group, a fullerenyl group.
- furyl group furyl group, thienyl group, pyrrolyl group, pyrazolyl group, imidazolyl group, isoxazolyl group, thiazolyl group, thiadiazolyl group, pyridyl group, benzofuranyl group, indolyl group, benzothiazolyl group, carbazolyl group.
- the aromatic group may be substituted, and the number of substituents in this case is not particularly limited as long as substitution is possible, and is one or more.
- Examples of the group that may be substituted for the aromatic group include a halogen atom, an optionally substituted linear or branched alkyl group having 1 to 12 carbon atoms, an optionally substituted aromatic group, Examples thereof include a non-aromatic heterocyclic group which may be substituted, a carboxyl group, a linear or branched alkoxy group having 1 to 12 carbon atoms, a cyano group or a nitro group.
- arylcarbonyl group examples include benzoyl group, 1-naphthoyl group, 2-naphthoyl group, 2-pyridylcarbonyl group, 3-pyridylcarbonyl group, 4-pyridylcarbonyl group and the like.
- arylsulfonyl group examples include a benzenesulfonyl group and a toluenesulfonyl group.
- X and Y in the chemical formula (1) represent a halogen atom, preferably chlorine, bromine or iodine, and more preferably iodine.
- the living radical polymerization initiator according to the present invention described above has two halogen atoms having different reactivities as reaction initiation groups, different living radicals can be obtained for each initiation group by appropriately adjusting the reaction conditions.
- the polymerization reaction can proceed.
- FIG. 1 is a schematic diagram showing an elementary reaction in a method for producing a polymer according to an embodiment of the present invention.
- a, b, and c each represent a radical reactive monomer having an unsaturated bond, and may be different monomers or the same monomer.
- A, B, and C each represent a polymer block.
- the elementary reaction shown in FIG. 1 is carried out one or more times as the first step, with one of the halogen atoms as an initiating group, and the polymerization reaction of the radically polymerizable monomer is changed one or more times while sequentially changing the type of the monomer.
- a polymer is produced by performing a polymerization reaction using another halogen atom remaining in the skeleton of the initiator as an initiator.
- the halogen atom Y in the living radical polymerization initiator of FIG. 1 is more reactive than the halogen atom X.
- the reaction conditions 1 to 4 are different from each other, and one or more of the reaction temperature, the reaction time, the presence / absence of a catalyst, the type of catalyst, and the like are different.
- reaction condition 1 is a condition in which the living radical polymerization proceeds only to the highly reactive halogen atom Y in the living radical polymerization initiator.
- the monomer b is then subjected to a living radical polymerization reaction, but different polymers can be obtained by changing the reaction conditions at this time.
- the monomer b is reacted under the reaction condition 2 in which not only the highly reactive halogen atom Y but also the less reactive halogen atom X is reacted.
- a polymer BAB in which a plurality of monomers b are polymerized on the halogen atom Y side is generated.
- polymer A by reacting monomer b under reaction condition 3 in which only highly reactive halogen atoms Y react, polymer AB in which a plurality of monomers b are polymerized on the halogen atom Y side of polymer A is generated.
- one of the halogen atoms remains unreacted in the first stage of the reaction, and the reaction temperature is changed in the second and third stages, or a different catalyst is added.
- the remaining halogen atom reacts with the monomer as an initiating group.
- the living radical polymerization reaction may be further advanced for the polymer BAB and polymer AB thus obtained.
- the reaction can be carried out under the condition where the halogen atom Y side of the polymer AB reacts with the monomer c to obtain the polymer ABC, or a different polymerization reaction can be allowed to proceed for the polymer CABC or the polymer ABC.
- the radical polymerizable monomer used in the above-described reaction is a monomer having an unsaturated bond capable of performing radical polymerization in the presence of an organic radical.
- Such an unsaturated bond may be a double bond or a triple bond. That is, in the method for producing a polymer according to the present embodiment, any of conventionally known monomers that can perform living radical polymerization can be used.
- Such a radical polymerizable monomer is specifically a monomer called a vinyl monomer.
- the vinyl monomer is a general term for monomers represented by the general formula “CH 2 ⁇ CR 5 R 6 ”.
- a monomer in which R 5 is methyl and R 6 is carboxylate is referred to as a methacrylate monomer and can be suitably used in the present invention.
- methacrylate monomers include methyl methacrylate, ethyl methacrylate, propyl methacrylate, n-butyl methacrylate, t-butyl methacrylate, hexyl methacrylate, 2-ethylhexyl methacrylate, nonyl methacrylate, benzyl methacrylate, glycidyl methacrylate, cyclohexyl methacrylate, lauryl methacrylate.
- N-octyl methacrylate 2-methoxyethyl methacrylate, butoxyethyl methacrylate, methoxytetraethylene glycol methacrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, 3-chloro-2-hydroxypropyl methacrylate, tetrahydrofurfuryl methacrylate, 2- Hydroxy 3- E Bruno propyl methacrylate, diethylene glycol methacrylate, polyethylene glycol methacrylate, 2- (dimethylamino) ethyl methacrylate.
- methacrylic acid can be used.
- a monomer in which R 5 is hydrogen and R 6 is carboxylate in the general formula of the vinyl monomer is generally referred to as an acrylate monomer and can be suitably used in the present invention.
- acrylate monomer examples include methyl acrylate, ethyl acrylate, propyl acrylate, n-butyl acrylate, t-butyl acrylate, hexyl acrylate, 2-ethylhexyl acrylate, nonyl acrylate, benzyl acrylate, glycidyl acrylate, cyclohexyl acrylate, and lauryl acrylate.
- N-octyl acrylate 2-methoxyethyl acrylate, butoxyethyl acrylate, methoxytetraethylene glycol acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate, 3-chloro 2-hydroxypropyl acrylate, tetrahydrofurfuryl acrylate, 2- Hydroxy 3-phenoxypropyl acrylate, diethylene Recall acrylate, polyethylene glycol acrylate, 2- (dimethylamino) ethyl acrylate, and the like.
- acrylic acid can be used.
- An ionic liquid acrylate such as a salt and N-ethyl-N-methylpyrrolidinium acrylate + / fluorohydrogenation (FH) nF ⁇ ) salt can be used.
- control of living radical polymerization of acrylate is generally difficult, according to the present invention, it can be controlled.
- the polymerization of the acrylate can be suitably controlled.
- the monomer in which R 5 is hydrogen and R 6 is phenyl in the general formula of the vinyl monomer is styrene and can be suitably used in the present invention.
- a monomer in which R 6 is phenyl or phenyl derivative referred to as a styrene derivative which can be suitably used in the present invention.
- a styrene derivative which can be suitably used in the present invention.
- Specific examples include o-, m-, p-methoxystyrene, o-, m-, p-styrene sulfonic acid and the like.
- the monomer in which R 5 is hydrogen and R 6 is alkyl is alkylene and can be suitably used in the present invention.
- a monomer having two or more vinyl groups can also be used.
- diene compounds for example, butadiene, isoprene, etc.
- compounds having two allyl groups for example, diallyl phthalate
- dimethacrylates having two methacryls for example, ethylene glycol dimethacrylate
- a diacrylate having two acrylics for example, ethylene glycol diacrylate.
- vinyl monomers other than those described above can also be used.
- vinyl esters eg, vinyl acetate, vinyl propionate, vinyl benzoate, vinyl acetate
- styrene derivatives other than the above eg, ⁇ -methylstyrene
- vinyl ketones eg, vinyl methyl ketone
- Vinyl hexyl ketone methyl isopropenyl ketone
- N-vinyl compounds eg N-vinyl pyrrolidone, N-vinyl pyrrole, N-vinyl carbazole, N-vinyl indole
- (meth) acrylamide and derivatives thereof eg N -Isopropylacrylamide, N-isopropylmethacrylamide, N, N-dimethylacrylamide, N, N-dimethylmethacrylamide, N-methylolacrylamide, N-methylolmethacrylamide
- acrylonitrile methacrylonitrile
- These monomers may be used alone or in combination of two or more. Two or more monomers may be added simultaneously at the start of the first stage reaction, or may be added at each stage of the reaction.
- the catalyst of the present invention arbitrarily selected as required can be used for an arbitrarily selected monomer.
- the catalyst may be omitted, but the reaction proceeds better by adding the catalyst.
- the combination of the kind of monomer and the kind of the catalyst of the present invention is not particularly limited.
- the catalyst examples include transition metal complex-based catalysts used in atom transfer radical polymerization (Atom transfer radical polymerization, ATRP method), reversible transfer catalyst polymerization (phosphorous chain polymerization, RTCP method), phosphorus, nitrogen, and the like.
- a catalyst comprising a compound containing at least one central element selected from carbon, oxygen, germanium, tin, and antimony, and a halogen atom bonded to the central element, used in reversible complex formation mediated polymerization (RCMP)
- RCMP reversible complex formation mediated polymerization
- Non-metallic compound having an ionic bond with an organic amine compound and a halide ion, wherein the non-metallic atom in the non-metallic compound is in a cationic state and forms an ionic bond with the halide ion Mention may be made of the medium.
- transition metal complex catalysts include metal complexes formed from low-valent metals such as Group 7, Group 8, Group 9, Group 10, or Group 11 of the periodic table and organic ligands, or the periodic table.
- a metal complex composed of a combination of a low-valent metal such as Group 7, Group 8, Group 9, Group 10 or Group 11 and a high-valent metal and an organic ligand (see JP-A-2002-249505) Can be used.
- Examples of this low valent metal include cuprous chloride, cuprous bromide, cuprous iodide, cuprous cyanide, cuprous oxide, ferrous chloride, ferrous bromide, iodine
- Examples of high-valent metals such as ferrous iodide include iron dichloride, iron dibromide, iron diiodide, ruthenium dichloride, ruthenium dibromide, ruthenium diiodide, and the like.
- organic ligands examples include pyridines, bipyridines, polyamines, phosphines, and the like. Specifically, 2,2′-bipyridyl and its derivatives, 1,10-phenanthroline and its derivatives, tetra Examples include methylethylenediamine, pentamethyldiethylenetriamine, tris (dimethylaminoethyl) amine, triphenylphosphine, and tributylphosphine.
- the catalyst having a central element selected from germanium, tin, or antimony includes a compound that includes at least one central element selected from germanium, tin, or antimony and at least one halogen atom bonded to the central element.
- germanium (II) iodide, germanium (IV) iodide, tin (II) iodide, tin (IV) iodide and the like can be mentioned (see JP 2007-92014 A). .
- Examples of the catalyst having nitrogen or phosphorus as a central element include compounds containing at least one central element selected from nitrogen or phosphorus and at least one halogen atom bonded to the central element.
- Examples thereof include phosphorus halides, halogenated phosphines, nitrogen halides, halogenated phosphorous acid, halogenated amines, and halogenated imide derivatives (see International Publication WO2008 / 139980).
- organic amine compound catalyst examples include triethylamine, tributylamine, 1,1,2,2-tetrakis (dimethylamino) ethene, 1,4,8,11-tetramethyl-1,4,8,11- Examples include tetraazacyclotetradecane, ethylenediamine, tetramethylethylenediamine, tetramethyldiaminomethane, tris (2-aminoethyl) amine, tris (2- (methylamino) ethyl) amine, and hematoporphyrin (International Publication WO2011). No. 016166).
- a catalyst that is a non-metallic compound having an ionic bond with a halide ion in which the non-metallic atom in the non-metallic compound is in a cation state and forms an ionic bond with the halide ion
- Ammonium salt, imidazolium salt, pyridinium salt, phosphonium salt, sulfonium salt, iodonium salt, and the like more specifically, tetrabutylammonium iodide, tetrabutylammonium triiodide, tetrabutylammonium bromide iodide, 1-methyl-3-methyl-imidazolium iodide, 1-ethyl-3-methylimidazolium bromide, 2-chloro-1-methylpyridinium iodide, hexaphenyldiphosphazenium chloride, methyltributylphosphonium iodide, tetrafe
- reaction temperature is not particularly limited and is preferably 0 ° C to 180 ° C, more preferably 30 ° C to 120 ° C.
- reaction time can be appropriately selected from 30 minutes to 24 hours and suitable for each reaction.
- reaction solvent is not particularly limited as long as it does not inhibit the reaction, but acetonitrile, methylene chloride, tetrahydrofuran, toluene, xylene, acetone, methyl ethyl ketone, ethanol, isopropanol, ethyl acetate, butyl acetate, ethyl cellosolve, etc. may be used. preferable.
- reaction mixture was stirred under reflux for 6.5 hours under LED light irradiation.
- the obtained reaction mixture was washed with a saturated aqueous sodium sulfite solution and water, and the organic layer was extracted.
- dichloromethane was added to the obtained reaction mixture, and the organic layer was extracted by washing with a saturated aqueous sodium sulfite solution and water.
- reaction mixture was mixed with 6.5 g (30 mmol) of 2-bromophenylacetic acid and 4.2 mL (60 mmol) of thionyl chloride and stirred at 80 ° C. for 50 minutes under light shielding.
- dichloromethane was added to the reaction mixture, washed with 10 mL of an aqueous sodium sulfite solution, the aqueous phase was extracted with 20 mL of dichloromethane, and the organic layer was mixed. The extract was dried over anhydrous sodium sulfate and concentrated under reduced pressure.
- reaction mixture was washed with 5% hydrobromic acid, saturated aqueous sodium carbonate solution and water, and the organic layer was extracted.
- FIG. 2 is a schematic diagram illustrating a polymer production process in the polymer production method according to the example.
- FIG. 3 is a 1H-NMR chart of the living radical polymerization initiator and each polymer obtained by the method for producing a polymer according to the example, and (a) shows the structure of an alkyl iodide initiator as a living radical polymerization initiator.
- (B) is a 1H-NMR chart of a living radical polymerization initiator
- (c) is a 1H-NMR chart of polymer P
- (d) is a 1H-NMR chart of polymer Q
- (e) is a 1H-NMR chart of polymer R. It is a NMR chart.
- the reaction solution was added to cooled hexane, and the polymer was reprecipitated and isolated.
- the reaction solution was added to cooled hexane, and the polymer was reprecipitated and isolated.
- 90% of propionyl iodide sites did not start, and block polymers (BMA and MMA) were almost selectively generated from phenylacetyl iodide sites.
- n-butyl acrylate (BA) and tetrabutylammonium iodide (320 mM) are added to the polymer Q (160 mM) obtained by the two-step reaction, and the mixture is heated and stirred at 110 ° C. for 24 hours. It was.
- the polymerization rate of BA (monomer conversion rate) was 32%.
- the reaction solution was added to cooled hexane, and the polymer was reprecipitated and isolated.
- the polymerization started almost 100% from the propionyl iodide moiety and a BA homopolymer grew, and a triblock polymer (BMA, MMA, and BA) grew from the phenylacetyl iodide moiety.
- CABC-type asymmetric multiblock polymer in which the A chain is BMA, the B chain is MMA, and the C chain is BA.
- the concentration “M” indicates the number of moles based on 1 liter of monomer. For example, 8M means that 8 mol is contained in 1 liter of monomer. In the case of MMA, 1 liter of monomer (bulk) is 8 mol at room temperature.
- the concentration “mM” indicates the number of millimoles based on 1 liter of monomer. For example, 80 mM means that 1 milliliter of monomer contains 80 mmol.
- Mn is the number average molecular weight of the obtained polymer.
- PDI indicates a ratio of Mw / Mn.
- R 1 linking group R 2 Any one of hydrogen atom, aromatic group, aliphatic group and acyloyl group X, Y halogen atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polymerization Catalysts (AREA)
- Graft Or Block Polymers (AREA)
Abstract
Description

(1)リビングラジカル重合開始剤の化学式
本実施形態に係るリビングラジカル重合開始剤は、化学式(1)に示す構造を有するものであり、反応性が異なる2つのハロゲン原子を有している。

連結基R1は、2以上の他の有機基と連結可能な有機基であれば特に限定されない。具体的には、脂肪族基、芳香族基、炭素数1~12のアルキルカルボニル基、炭素数1~12のアルコキシカルボニル基、アミノカルボニル基、炭素数1~12のアルキルアミノカルボニル基、炭素数1~12のジアルキルアミノカルボニル基、アリールカルボニル基、炭素数1~12のアルキルスルホニル基、アリールスルホニル基及びこれらの基を2つ以上組み合わせた有機基が挙げられる。
脂肪族基としては、置換されていてもよい直鎖又は分岐鎖状の炭素数1~12のアルキル基が挙げられる。
次に、上述したリビングラジカル重合開始剤を用いてリビングラジカル重合を行うことにより得られるラジカル重合性モノマーの重合体の製造方法について説明する。
本実施形態に係る重合体の製造方法は、上述したリビングラジカル重合開始剤を用いて、図1に示す素反応を行うことにより実現される。図1は、本発明の実施形態に係る重合体の製造方法における素反応を示す模式図である。a、b、cはそれぞれ不飽和結合を有するラジカル反応性モノマーを表し、それぞれ違うモノマーであってもよく、又は同じモノマーであってもよい。また、A、B、Cはそれぞれポリマーブロックを表す。
上述した反応で用いられるラジカル重合性モノマーは、有機ラジカルの存在下でラジカル重合を行い得る不飽和結合を有するモノマーである。このような不飽和結合は二重結合の他、三重結合であってもよい。すなわち、本実施形態に係る重合体の製造方法では、従来からリビングラジカル重合を行い得る公知のモノマーのうち任意のものを用いることができる。
任意に選択されたモノマーに対し、必要に応じて任意に選択された本発明の触媒を用いることができる。触媒は無くてもよいが、触媒を加えることにより、反応はより良好に進行する。モノマーの種類と、本発明の触媒の種類との組み合わせは特に限定されない。
反応温度は特に限定されず、0℃~180℃が好ましく、30℃~120℃がより好ましい。
反応時間は30分~24時間の範囲で各反応に好適なものを適宜選択することができる。
反応は無溶媒下で行うこともできるが、溶媒を用いてもよい。反応溶媒は、反応を阻害しないものであれば特に制限は無いが、アセトニトリル、塩化メチレン、テトラヒドロフラン、トルエン、キシレン、アセトン、メチルエチルケトン、エタノール、イソプロパノール、酢酸エチル、酢酸ブチル、エチルセロソルブ等を用いることが好ましい。
(実施例1)2-ヨード-2-(4‘-(2“-ヨードプロピオニルオキシ)フェニル)酢酸メチルの製造
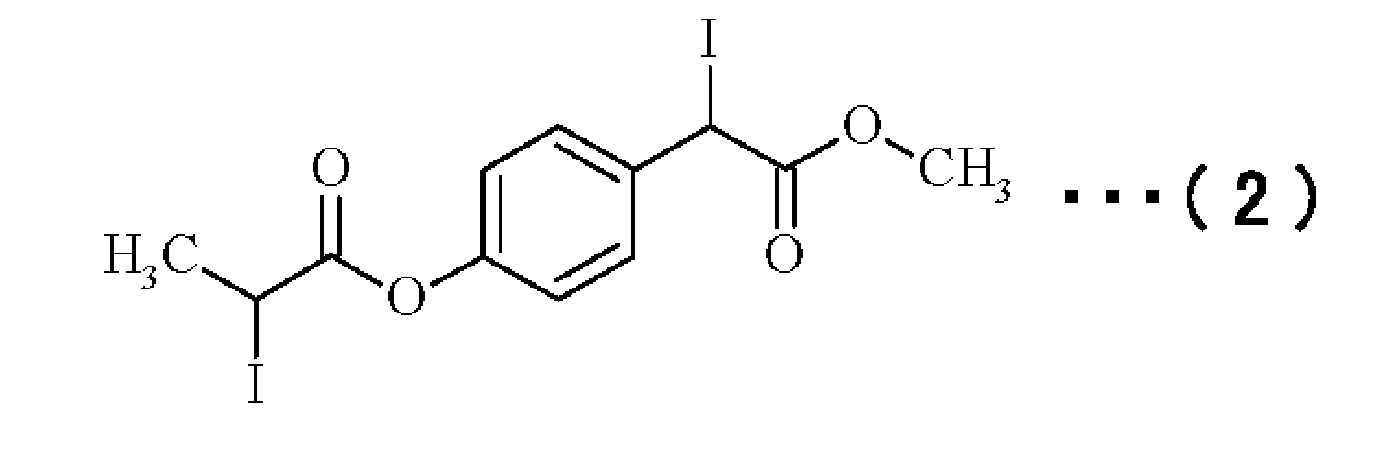




上述した素反応に基づき、以下の具体的な反応条件でリビングラジカル重合体の製造を行った。図2は、実施例に係る重合体の製造方法における重合体の生成過程を示す模式図である。図3は、実施例に係る重合体の製造方法により得られるリビングラジカル重合開始剤及び各ポリマーの1H-NMRチャートであり、(a)はリビングラジカル重合開始剤としてのヨウ化アルキル開始剤の構造式、(b)はリビングラジカル重合開始剤の1H-NMRチャート、(c)はポリマーPの1H-NMRチャート、(d)はポリマーQの1H-NMRチャート、(e)はポリマーRの1H-NMRチャートである。
R2 水素原子、芳香族基、脂肪族基及びアシロイル基のうち何れか1つ
X、Y ハロゲン原子
Claims (8)
- リビングラジカル重合開始剤であって、以下の一般式(1)からなるリビングラジカル重合開始剤:

R2、R3、R4及びR5は水素原子、炭素数1~12の脂肪族基、芳香族基、アルキルカルボニル基、アルコキシカルボニル基、アミノカルボニル基、アルキルアミノカルボニル基、炭素数1~12のジアルキルアミノカルボニル基、アリールカルボニル基、カルボキシル基、炭素数1~12のアルキルスルホニル基及びアリールスルホニル基から選ばれる有機基であり、
X、Yはハロゲン原子であり、
m、nは1以上の整数であり、
前記X及びYはモノマーに対し、互いに反応性の異なる状態である。 - 前記ハロゲン原子はヨウ素、塩素又は臭素であることを特徴とする請求項1記載のリビングラジカル重合開始剤。
- 請求項1又は2に記載のリビングラジカル重合開始剤を用いる重合体の製造方法であって、
前記リビングラジカル重合開始剤の前記X又は前記Yの何れか一方のハロゲン原子のみについて、前記リビングラジカル重合反応開始剤と不飽和結合を有するモノマーとを混合し、前記モノマーの種類に応じた反応条件で行うリビングラジカル重合反応を、混合する前記モノマーの種類を順次変えつつ1回以上行い第1生成物を得る第1重合工程と、
前記第1生成物に含まれる前記X又は前記Yの双方のハロゲン原子について、少なくとも1種類以上の前記モノマーを前記モノマーの種類に応じた反応条件で順次リビングラジカル重合反応させ最終生成物を得る第2重合工程と、
を含むことを特徴とする重合体の製造方法。 - 前記第1重合工程及び前記第2重合工程は触媒を用いて行われ、前記第1重合工程及び前記第2重合工程は反応温度及び前記触媒の種類の少なくとも一方を前記モノマーの種類に応じて異ならせて行われることを特徴とする請求項3記載の重合体の製造方法。
- 前記触媒は、原子移動ラジカル重合において用いられる遷移金属錯体系触媒、可逆移動触媒重合において用いられるリン、窒素、炭素、酸素、ゲルマニウム、スズ、及びアンチモンから選ばれる少なくとも1種の中心元素と、前記中心元素に結合したハロゲン原子と、を含む化合物からなる触媒、可逆的錯体形成媒介重合において用いられる有機アミン化合物触媒、又はハロゲン化物イオンとのイオン結合を有する非金属化合物であって、前記非金属化合物中の非金属原子がカチオンの状態であり、ハロゲン化物イオンとイオン結合を形成している触媒であることを特徴とする請求項4記載の重合体の製造方法。
- 前記第1重合工程及び前記第2重合工程は、180℃以下で行われることを特徴とする請求項3乃至5の何れか1項記載の重合体の製造方法。
- 前記第1重合工程及び前記第2重合工程は、30分以上24時間以下の反応時間で行われることを特徴とする請求項3乃至6の何れか1項記載の重合体の製造方法。
- 請求項3乃至7の何れか1項記載の方法を用いて製造されたことを特徴とする重合体。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020177005641A KR102419414B1 (ko) | 2014-09-02 | 2015-08-07 | 리빙 래디컬 중합 개시제, 중합체의 제조 방법 및 중합체 |
| EP15838864.5A EP3190131B1 (en) | 2014-09-02 | 2015-08-07 | Living radical polymerization initiator, method for producing polymer, and polymer |
| SG11201701672WA SG11201701672WA (en) | 2014-09-02 | 2015-08-07 | Living radical polymerization initiator, method for producing polymer, and polymer |
| US15/508,313 US10414848B2 (en) | 2014-09-02 | 2015-08-07 | Living radical polymerization initiator, method for producing polymer, and polymer |
| CN201580046890.2A CN106604938B (zh) | 2014-09-02 | 2015-08-07 | 活性自由基聚合引发剂、聚合物的制造方法和聚合物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-178384 | 2014-09-02 | ||
| JP2014178384A JP6528260B2 (ja) | 2014-09-02 | 2014-09-02 | 重合体の製造方法、及びリビングラジカル重合開始剤 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016035258A1 true WO2016035258A1 (ja) | 2016-03-10 |
Family
ID=55439349
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/003991 Ceased WO2016035258A1 (ja) | 2014-09-02 | 2015-08-07 | リビングラジカル重合開始剤、重合体の製造方法及び重合体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10414848B2 (ja) |
| EP (1) | EP3190131B1 (ja) |
| JP (1) | JP6528260B2 (ja) |
| KR (1) | KR102419414B1 (ja) |
| CN (1) | CN106604938B (ja) |
| SG (1) | SG11201701672WA (ja) |
| WO (1) | WO2016035258A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112175121A (zh) * | 2020-07-25 | 2021-01-05 | 青岛大学 | 基于受阻Lewis酸碱对催化极性乙烯基单体聚合的方法 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6254239B2 (ja) | 2016-02-29 | 2017-12-27 | 大日精化工業株式会社 | ポリマーの製造方法 |
| JP6833485B2 (ja) * | 2016-12-05 | 2021-02-24 | 大日精化工業株式会社 | 星形ポリマーの製造方法、星形ポリマー、及びフィラー分散体 |
| JP6245719B1 (ja) | 2017-03-24 | 2017-12-13 | 大日精化工業株式会社 | ポリマーの製造方法 |
| JP6836242B2 (ja) * | 2017-04-19 | 2021-02-24 | 大日精化工業株式会社 | ポリマーの製造方法、及びその製造方法によって得られるポリマー |
| EP3792267A4 (en) * | 2018-05-07 | 2022-03-16 | Ochanomizu University | IODINE COMPOUND |
| CN108948245A (zh) * | 2018-06-04 | 2018-12-07 | 常州大学 | N,n-二甲基丙烯酰胺的水溶液反向原子转移自由基聚合 |
| CN109251261A (zh) * | 2018-08-30 | 2019-01-22 | 常州大学 | 丙烯酰胺的水溶液反向原子转移自由基聚合 |
| CN109293810B (zh) * | 2018-09-29 | 2020-09-08 | 江苏斯德瑞克化工有限公司 | 一种非金属催化的可控自由基聚合方法 |
| CN110357992A (zh) * | 2019-07-09 | 2019-10-22 | 复旦大学 | 一种超高分子量含氟聚合物的制备方法 |
| JP7216694B2 (ja) * | 2020-10-30 | 2023-02-01 | 大日精化工業株式会社 | 顔料分散剤及びその製造方法、インクジェットインク用顔料分散液、及び水性インクジェットインク |
| CN115197379B (zh) * | 2022-07-18 | 2023-09-22 | 福州大学 | 一种调控聚合物接枝密度的方法 |
| CN116574225B (zh) * | 2023-05-17 | 2025-07-29 | 福州大学 | 一种cabc型不对称嵌段共聚物及其制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010024263A (ja) * | 2008-07-15 | 2010-02-04 | Toyota Central R&D Labs Inc | 重合開始剤、それを用いた高分子修飾材料の製造方法、および高分子修飾材料を含む成型体 |
| JP2013072069A (ja) * | 2011-09-29 | 2013-04-22 | Hitachi Chemical Co Ltd | リビングラジカル重合開始剤、星型共重合体、樹脂組成物、及びダイボンドフィルム |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009203359A (ja) | 2008-02-28 | 2009-09-10 | Nippon Chem Ind Co Ltd | リビングラジカル重合添加剤及びそれを用いたリビングラジカル重合方法 |
-
2014
- 2014-09-02 JP JP2014178384A patent/JP6528260B2/ja active Active
-
2015
- 2015-08-07 SG SG11201701672WA patent/SG11201701672WA/en unknown
- 2015-08-07 CN CN201580046890.2A patent/CN106604938B/zh active Active
- 2015-08-07 EP EP15838864.5A patent/EP3190131B1/en active Active
- 2015-08-07 KR KR1020177005641A patent/KR102419414B1/ko active Active
- 2015-08-07 WO PCT/JP2015/003991 patent/WO2016035258A1/ja not_active Ceased
- 2015-08-07 US US15/508,313 patent/US10414848B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010024263A (ja) * | 2008-07-15 | 2010-02-04 | Toyota Central R&D Labs Inc | 重合開始剤、それを用いた高分子修飾材料の製造方法、および高分子修飾材料を含む成型体 |
| JP2013072069A (ja) * | 2011-09-29 | 2013-04-22 | Hitachi Chemical Co Ltd | リビングラジカル重合開始剤、星型共重合体、樹脂組成物、及びダイボンドフィルム |
Non-Patent Citations (10)
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112175121A (zh) * | 2020-07-25 | 2021-01-05 | 青岛大学 | 基于受阻Lewis酸碱对催化极性乙烯基单体聚合的方法 |
| CN112175121B (zh) * | 2020-07-25 | 2022-07-12 | 青岛大学 | 基于受阻Lewis酸碱对催化极性乙烯基单体聚合的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10414848B2 (en) | 2019-09-17 |
| JP2016053097A (ja) | 2016-04-14 |
| CN106604938B (zh) | 2020-03-31 |
| KR102419414B1 (ko) | 2022-07-11 |
| CN106604938A (zh) | 2017-04-26 |
| KR20170048379A (ko) | 2017-05-08 |
| SG11201701672WA (en) | 2017-04-27 |
| EP3190131B1 (en) | 2020-07-29 |
| EP3190131A1 (en) | 2017-07-12 |
| JP6528260B2 (ja) | 2019-06-12 |
| US20170306073A1 (en) | 2017-10-26 |
| EP3190131A4 (en) | 2018-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016035258A1 (ja) | リビングラジカル重合開始剤、重合体の製造方法及び重合体 | |
| Ribelli et al. | Atom transfer radical polymerization: billion times more active catalysts and new initiation systems | |
| Pintauer et al. | Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes | |
| JP5995848B2 (ja) | リビングラジカル重合触媒および重合方法 | |
| US10689336B2 (en) | Radical polymerization initiator and method for producing polymers | |
| JP6552970B2 (ja) | リビングラジカル重合触媒及びそれを用いた重合体の製造方法 | |
| JP5193480B2 (ja) | リビングラジカルポリマーの製造方法およびポリマー | |
| Kamigaito | Evolutions of precision radical polymerizations from metal-catalyzed radical addition: living polymerization, step-growth polymerization, and monomer sequence control | |
| KR20050111325A (ko) | 리빙 라디칼 폴리머의 제조방법 및 폴리머 | |
| WO2018164147A1 (ja) | ポリマーの製造方法 | |
| Tsarevsky et al. | Atom transfer radical polymerization (ATRP) | |
| JP6754124B2 (ja) | 多分岐ポリマーの製造方法及び多分岐ポリマー | |
| CN110546178B (zh) | 接枝聚合物的制造方法、接枝聚合物、接枝聚合物的引发剂 | |
| WO2006001496A1 (ja) | 有機アンチモン化合物、その製造方法、リビングラジカル重合開始剤、それを用いるポリマーの製造方法及びポリマー | |
| JP2009024162A (ja) | 2官能リビングラジカル重合開始剤および重合体の製造方法 | |
| CN106046221B (zh) | 一类可逆-休眠自由基聚合的催化剂及聚合方法 | |
| JP5176120B2 (ja) | リビングラジカル重合開始剤及びそれを用いるポリマーの製造方法 | |
| JP5963516B2 (ja) | ポリマーの製造方法及び該方法により製造されたポリマー | |
| US20190092890A1 (en) | Method for producing copolymer, and method for producing latex | |
| JP2019194291A (ja) | 多分岐ポリマーの製造方法及び多分岐ポリマー、共役ジエンモノマー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15838864 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20177005641 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15508313 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015838864 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015838864 Country of ref document: EP |