WO2016035866A1 - アニオン重合体の製造方法 - Google Patents
アニオン重合体の製造方法 Download PDFInfo
- Publication number
- WO2016035866A1 WO2016035866A1 PCT/JP2015/075126 JP2015075126W WO2016035866A1 WO 2016035866 A1 WO2016035866 A1 WO 2016035866A1 JP 2015075126 W JP2015075126 W JP 2015075126W WO 2016035866 A1 WO2016035866 A1 WO 2016035866A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymerization
- solvent
- anionic polymer
- reaction
- anionic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/02—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type
- C08F297/04—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type polymerising vinyl aromatic monomers and conjugated dienes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/04—Polymerisation in solution
- C08F2/06—Organic solvent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F236/04—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F236/10—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated with vinyl-aromatic monomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F293/00—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule
- C08F293/005—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule using free radical "living" or "controlled" polymerisation, e.g. using a complexing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F36/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F36/02—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F36/04—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F36/06—Butadiene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2438/00—Living radical polymerisation
Definitions
- the present invention relates to a method for producing an anionic polymer.
- Monomers such as conjugated dienes and aromatic vinyl compounds are anionically polymerized to living polymers obtained by polymerizing conjugated dienes, aromatic vinyl compounds or mixtures thereof using organic alkali metal compounds or organic alkali metal compounds.
- an anionic polymer can be produced.
- hydrogenation catalysts such as nickel-based Ziegler catalysts and titanium-based catalysts
- the heat resistance, oxidation resistance, weather resistance, and ozone resistance of the polymer It is known that the property can be improved.
- These are industrially used as synthetic rubbers (see Patent Documents 1 to 12).
- the temperature of anionic polymerization is usually 20 to 110 ° C., and the higher the polymerization temperature, the higher the polymerization rate. However, when the temperature exceeds 110 ° C., the polymerization is stopped midway, so that a polymer with a wide molecular weight distribution is formed. As a result, the mechanical properties of the resulting polymer are reduced.
- an anionic polymer using a conjugated diene as a monomer its mechanical properties can be controlled by the bonding mode of the conjugated diene, and these bonding modes are the types and amounts of Lewis bases to coexist during the conjugated diene polymerization, It is also known that it can be controlled by the polymerization temperature. Therefore, in order to produce an anionic polymer having excellent mechanical properties, it is important to quickly remove the heat of polymerization reaction and control the temperature in the anionic polymerization reaction (see Patent Document 1).
- a cooling coil is provided inside the reactor as a complete mixing reactor that can quickly remove the heat of polymerization reaction.
- the structure of the cooling coil is complicated and the reaction liquid containing the polymer has a high viscosity, hardly soluble polymer or gel is deposited on the cooling coil.
- a molded product using such an insoluble polymer containing a sparingly soluble polymer or gel has an uneven surface or uneven transparency, and thus the value of the product is significantly impaired.
- a reactor having a cooling coil inside there is a problem that the ratio of the heat transfer area of the cooling coil to the reaction liquid volume decreases as the size of the reactor increases. For this reason, there is provided a method in which a reflux condenser is provided in a fully mixed reactor to condense the monomer or solvent and cool it (see Patent Documents 9 to 12 and Non-Patent Documents 1 and 2).
- Patent Document 9 discloses a reflux self-cooling reactor equipped with a condenser when polymerizing butadiene and styrene using an organolithium compound in a hydrocarbon solvent in the presence of an ether compound or a tertiary amine compound.
- a condenser when polymerizing butadiene and styrene using an organolithium compound in a hydrocarbon solvent in the presence of an ether compound or a tertiary amine compound.
- Patent Document 9 since the inside of the polymerization system can be cooled by evaporation and reflux of butadiene, it is easy to suppress the temperature rise due to the heat of polymerization reaction, and the maximum temperature of the polymerization system can be easily maintained at 120 ° C. or lower. Is described.
- Patent Document 10 discloses a method for producing at least one conjugated diene and at least one vinyl aromatic compound, wherein at least 60% by weight of the solvent is cyclopentane, and the heat of polymerization reaction is reduced by reflux cooling of cyclopentane. A method of recovery is disclosed.
- Patent Documents 11 to 12 when an organic metal is used as a polymerization initiator and a block copolymer composed of an aromatic vinyl compound and a conjugated diene compound is subjected to solution polymerization, solvent vapor and monomer vapor are formed above the reactor during the polymerization reaction.
- a reactor having a space constituted by inert gas the gas in the gas phase at the top of the reactor is led to a heat exchanger provided outside the reactor, and a liquid mainly composed of a solvent condensed in the heat exchanger is used as the reactor.
- the method of controlling the polymerization by forcibly returning a gas composed mainly of an inert gas, which has not been condensed, to the liquid phase part at the lower part of the reactor is disclosed. It is disclosed that heat is removed mainly by the latent heat of the evaporating solvent.
- Patent Document 9 discloses a method characterized in that a monomer having a boiling point at room temperature lower than that of a solvent is condensed and refluxed.
- the pressure in the polymerization system was kept constant by a control valve at 2.0 kg / cm 2 G (0.2 MPaG: gauge pressure, hereinafter used in the same meaning in this specification), and the temperature in the polymerization system was kept constant. Is kept constant at 70 ° C. by the latent heat of butadiene and cyclohexane, and evaporated butadiene and cyclohexane are refluxed using a condenser cooled by ammonia (boiling point ⁇ 33 ° C.) at the top of the reactor.
- Patent Document 10 describes the usefulness of using a low-boiling solvent having a boiling point lower than the polymerization reaction temperature.
- a low-boiling solvent having a boiling point lower than the polymerization reaction temperature.
- steam that is inexpensive and industrially available is used for solvent removal, and coagulation is generally employed in which the solvent is removed by contact between the reaction solution and steam.
- the condensed water reaches nearly 100 ° C., and a large condenser and a low-temperature refrigerant generator are required to recover many vaporized low boiling point solvents. Therefore, a method using a low boiling point solvent is not always economically advantageous.
- Patent Documents 11 to 12 describe that the latent heat of a solvent can be used for heat removal by forcibly returning a gas composed mainly of an inert gas that has not been condensed to the liquid phase portion at the bottom of the reactor, thereby improving the heat removal capability.
- a transfer equipment such as a blower is necessary, and the monomer is polymerized over time, thereby blocking the circulation path and fixing the blower rotating part. It is not necessarily economically advantageous.
- the present inventors have made it easy to anionic polymerize a monomer using a solvent having a boiling point at normal pressure higher than the polymerization temperature, by making the amount of inert gas present per kg of the solvent a specific amount. It was found that the heat of polymerization reaction can be quickly removed with a simple reactor. In particular, when butadiene having an atmospheric pressure boiling point of ⁇ 4.4 ° C. is subjected to anionic polymerization at 50 to 55 ° C. in a cyclohexane solvent having an atmospheric pressure boiling point of 80.7 ° C. under conditions where almost no inert gas exists.
- the present invention Anionic polymer for anionic polymerization of conjugated diene, aromatic vinyl compound, or a mixture thereof under the condition that the amount of inert gas present in 1 kg of solvent having a boiling point at normal pressure higher than the polymerization temperature is 20 mmol or less Is a manufacturing method of [2]
- an initiator for anionic polymerization any one of an organic alkali metal compound, one or more conjugated dienes, one or more aromatic vinyl compounds, or one or more conjugated dienes and one or more aromatic vinyl compounds Is a method for producing an anionic polymer according to [1], wherein a living copolymer obtained by polymerizing an organic alkali metal compound is used.
- a completely mixed reactor having a reflux condenser having an A / V ratio of 20 to 0.1 with respect to the reactor internal volume V (m 3 ) of the heat transfer area A (m 2 ) of the reflux condenser is used.
- the temperature of the anion can be controlled by quickly removing the heat of polymerization reaction using a simple apparatus.
- the polymer can be produced industrially advantageously.
- inert gas refers to a rare gas having poor reactivity, such as nitrogen gas, argon gas, or helium gas.
- the monomer which comprises the anion polymer manufactured with the manufacturing method of this invention is demonstrated.
- a conjugated diene, an aromatic vinyl compound, or a mixture thereof can be used as the monomer.
- Conjugated dienes that can be used as monomers include, for example, butadiene, isoprene, 2,3-dimethyl-1,3-butadiene, 1,3-pentadiene, 2-methyl-1,3-pentadiene, 3-methyl-1,3- Pentadiene, 1,3-hexadiene, 4,5-diethyl-1,3-butadiene, phenyl-1,3-butadiene, 4,5-diethyl-1,3-octadiene, 3-butyl-1,3-octadiene, 1,3-cyclohexadiene, 1,3,7-octatriene, myrcene (7-methyl-3-methyleneocta-1,6-diene), farnesene (3,7,11-trimethyl-1,3,6, Preferred examples include, but are not limited to, conjugated dienes having 4 to 15 carbon atoms such as 10-dodecatetraene).
- conjugated dienes may be used alone or in combination of two or more.
- butadiene or isoprene is preferably included, and butadiene, isoprene, or a mixture of butadiene and isoprene is more preferable.
- the above conjugated diene and aromatic vinyl compound may be used alone or in combination of two or more, and further diluted with a solvent that can be used for polymerization.
- Bonding mode of the conjugated diene constituting the anionic polymer for example, 1,2-bond unit and 1,4-bond unit in the case of butadiene: 1,2-bond unit, 3,4-bond in the case of isoprene
- a Lewis base can coexist during anionic polymerization.
- Lewis bases examples include acyclic monoesters such as dimethyl ether, methyl ethyl ether, diethyl ether, ethyl propyl ether, dipropyl ether, butyl methyl ether, tert-butyl methyl ether, dibutyl ether, dioctyl ether, ethyl phenyl ether, and diphenyl ether.
- acyclic monoesters such as dimethyl ether, methyl ethyl ether, diethyl ether, ethyl propyl ether, dipropyl ether, butyl methyl ether, tert-butyl methyl ether, dibutyl ether, dioctyl ether, ethyl phenyl ether, and diphenyl ether.
- Lewis bases may be used alone or in combination of two or more. There is no restriction
- the Lewis base may be diluted with a solvent that can be used for polymerization.
- an organic alkali metal compound is usually used as an initiator.
- organic alkali metal compounds that can be used include methyl lithium, ethyl lithium, propyl lithium, isopropyl lithium, butyl lithium, sec-butyl lithium, tert-butyl lithium, isobutyl lithium, pentyl lithium, hexyl lithium, butadienyl lithium, Cyclohexyl lithium, phenyl lithium, benzyl lithium, p-toluyl lithium, styryl lithium, trimethylsilyl lithium, 1,4-dilithiobutane, 1,5-dilithiopentane, 1,6-dilithiohexane, 1,10-dilithiodecane, 1,1- Dilithiodiphenylene, dilithiopolybutadiene, dilithiopolyisoprene, 1,4-dilithiobenzen
- a living polymer obtained by polymerizing one or more conjugated dienes using the organic alkali metal compound, and one or more aromatics using the organic alkali metal compound a living polymer obtained by polymerizing a vinyl compound or a living copolymer obtained by polymerizing one or more kinds of conjugated dienes and one or more kinds of aromatic vinyl compounds using the organic alkali metal compound can also be used (hereinafter, referred to as the following). These living polymers are referred to as “living polymers I”). That is, the anionic polymer may be bonded to the polymerization terminal of these living polymers I to form a copolymer.
- the bonding mode is not particularly limited and may be any of random copolymer, block copolymer, block copolymer having a tapered structure, star copolymer, and the like, but it is a block copolymer. It is preferable.
- the living polymer I the alkali metal cation derived from the organic alkali metal compound is M, the conjugated diene block composed of one or more conjugated dienes is B, and the one or more aromatic vinyl compounds are used.
- the constituted aromatic compound block is represented by S, SM, SBM, SBSM, SBSBBM, BM, B—
- a living polymer having a block structure of SM, BSSBM, or BSSBSM is preferred.
- the weight average molecular weight (Mw) in terms of polystyrene measured by gel permeation chromatography of the living polymer I is preferably less than 1,000,000, and more preferably 10,000 to 500,000.
- the molecular weight distribution (Mw / Mn) of the living polymer I is preferably 1.00 to 1.50.
- a mixture of an organic alkali metal compound and a living polymer I may be used as an initiator for producing an anionic polymer as an initiator for producing an anionic polymer.
- solvent The solvent that can be used in the production method of the present invention is not limited in type and amount as long as it contains a solvent having a boiling point at normal pressure (1 atm) higher than the polymerization temperature.
- a solvent having a boiling point at normal pressure (1 atm) higher than the polymerization temperature for example, pentane (36.1 ° C.), isopentane.
- a solvent may be used individually by 1 type, or may use 2 or more types together. Among them, it is preferable that a hydrocarbon having a boiling point at normal pressure higher than the polymerization temperature is contained as 50% by mass or more, a solvent containing 50% by mass or more of cyclohexane is more preferable, and a solvent containing 80% by mass or more of cyclohexane. Further preferred. If it is such a solvent, the removal of the solvent from the reaction liquid containing the obtained anion polymer can be implemented economically advantageously.
- the solvent examples include solvents having a boiling point lower than the polymerization temperature at normal pressure, such as butane ( ⁇ 0.5 ° C.), isobutane ( ⁇ 11.7 ° C.), and the above, within a range not impairing the effects of the present invention.
- solvents having a boiling point at normal pressure lower than the polymerization temperature may be included.
- the content of the solvent having a boiling point lower than the polymerization temperature at normal pressure is preferably less than 50% by mass, more preferably less than 30% by mass, and even more preferably less than 10% by mass.
- Polymerization terminator examples include hydrogen molecule, oxygen molecule, water; methanol, ethanol, propanol, isopropanol, butanol, heptanol, cyclohexanol, phenol, benzyl alcohol, o-cresol, m-cresol.
- ketones such as 2-heptanone, 4-methyl-2-pentanone, cyclopentanone, 2-hexanone, 2-pentanone, cyclohexanone, 3-pentanone, acetophenone, 2-butanone, acetone; methyl acetate, ethyl acetate, And esters such as butyl acetate; and epoxy compounds such as ethylene oxide and propylene oxide.
- These polymerization terminators may be used alone or in combination of two or more. Moreover, these polymerization terminators may have a function as a terminal modifier of the anionic polymer.
- the above conjugated diene, aromatic vinyl compound, or mixture thereof is obtained under the condition that the amount of the inert gas present in 1 kg of the solvent having a boiling point higher than the polymerization temperature at normal pressure is 20 mmol or less. It is characterized by anionic polymerization.
- the reactor it is preferable to use a fully mixed reactor having a reflux condenser directly or indirectly in the gas phase portion.
- a pump for exhausting the inert gas may be installed so that the inert gas can be exhausted through the reflux condenser. preferable.
- the reactor may have a jacket outside for the purpose of temperature control such as heating and cooling of the reaction solution, and the structure thereof is not particularly limited, and a known type can be used.
- a cooling baffle or a cooling coil may be provided inside the reactor for the purpose of increasing cooling heat transfer as desired.
- stirring blades of the reactor there are no particular restrictions on the stirring blades of the reactor, but examples include Max blend blades, full zone blades, paddle blades, propeller blades, turbine blades, fan turbine blades, fiddler blades, blue margin blades, and the like. It may be a combination of two or more. In particular, when the viscosity of the resulting polymer solution is high, it is preferable to use a Max blend blade or a full zone blade in terms of promoting heat removal by the jacket and controlling the molecular weight distribution of the resulting anionic polymer.
- the stirring method may be upper stirring or lower stirring, but the upper stirring blade is preferable from the viewpoint that the cleaning operation and maintenance inspection of the apparatus can be simplified.
- the structure of the reflux condenser is not particularly limited, but it is preferable to use a multitubular reflux condenser.
- the reflux condenser may have a plurality of reflux condensers connected in series or in parallel, and a different refrigerant may be passed through each reflux condenser.
- the use of a single reflux condenser is economical because the construction cost of the reactor can be reduced.
- the heat transfer area is A (m 2 )
- the sum of the heat transfer areas is A (m 2 ).
- the A / V ratio of m 2 ) to the reactor internal volume V (m 3 ) is preferably 20 to 0.1, and more preferably 10 to 0.5.
- an aqueous solution containing an antifreezing agent such as water, glycols, alcohols, glycerols, and glycerins can be preferably used.
- the temperature of the refrigerant is not particularly limited as long as the solvent to be refluxed is within a range from the temperature at which the refluxing solvent does not freeze to the temperature of the reaction solution. It is economical because it does not need.
- the refrigerant flow rate is not particularly limited as long as it is less than the pressure resistance range of the reflux condenser.
- the production method of the living polymer I is not particularly limited, and may be any of batch, semi-batch, and continuous. There are no particular restrictions on the type of reactor, and a fully mixed reactor, a tubular reactor, or two or more of these connected in series or in parallel can be used, but the same reaction as in the anionic polymerization is performed. It is preferable to use a vessel from the viewpoint that complicated operations such as liquid feeding are unnecessary.
- the production of the living polymer I is generally performed in an inert gas atmosphere.
- a living polymer I is produced by charging an organic alkali metal compound as a solvent and a polymerization initiator in a reactor substituted with an inert gas, raising the temperature to a predetermined temperature, and adding a monomer as appropriate.
- a living polymer containing a conjugated diene as a monomer unit as the living polymer I
- a Lewis base for controlling the bonding mode of the conjugated diene may be added at the same time as the monomer addition, You may charge to a reactor.
- the amount of the organic alkali metal compound used can be appropriately set according to the weight average molecular weight and polymer concentration of the desired anionic polymer, but is preferably in the range of 2 ⁇ 10 ⁇ 3 mmol to 500 mmol with respect to 1 kg of the reaction solvent.
- the polymerization temperature is not particularly limited, but can be selected from the range of ⁇ 20 to 250 ° C. above the solvent freezing point and below the thermal decomposition temperature of the polymer, preferably in the range of 20 to 110 ° C. Within this temperature range, a living polymer having a narrow molecular weight distribution can be produced in a short reaction time.
- the reaction time is not particularly limited, but can usually be selected from the range of 1 to 20 hours, preferably in the range of 2 to 10 hours. Within this time range, a high monomer conversion rate of 90% or more can be achieved, and thus the living polymer I can be produced economically advantageously.
- the concentration of the living polymer I contained in the reaction solution containing the living polymer I is not particularly limited, but it is preferably produced so as to be in the range of 1 to 50% by mass.
- the living polymer I having a narrow molecular weight distribution can be produced due to the low molecular weight distribution, and as a result, an anionic polymer exhibiting good mechanical properties having a narrow molecular weight distribution can be produced. Moreover, when using for manufacture of an anionic polymer, you may reduce the density
- Anionic polymerization method The method for producing an anionic polymer of the present invention will be described in detail. First, a fully mixed reaction vessel equipped with a reflux condenser and a pump for exhausting an inert gas is mixed with a solvent and an organic anion polymerization initiator. An alkali metal compound or the living polymer I is introduced. Note that an inert gas may be present at this stage. When anionic polymer monomer is supplied and anionic polymerization is performed, exhaust is performed until the abundance of the inert gas with respect to 1 kg of the reaction solvent is 20 mmol or less.
- the amount of the inert gas present relative to 1 kg of the reaction solvent is preferably 15 mmol or less, more preferably 9 mmol or less, and even more preferably 5 mmol or less.
- the exhaust operation for reducing the amount of the inert gas in the system may be performed before supplying the monomer of the anionic polymer, during the supply, or after the supply.
- the inert gas is exhausted out of the system by a pump through the reflux condenser, and the gas phase part is removed from the solvent. It is preferred that the inert gas is substantially absent by filling with steam, followed by monomer supply and anionic polymerization in a closed system.
- an inert gas may be supplied into the reaction system together with the monomer when supplying the monomer, but the amount of the inert gas is small and does not significantly reduce the heat removal efficiency in the reflux condenser. . Therefore, it is not always necessary to exhaust the inert gas when supplying the monomer, but it is preferable to perform an exhaust operation when the monomer supply is completed, if desired.
- the concentration of the anion derived from the organic alkali metal compound or the active terminal anion derived from the living polymer I can be appropriately set according to the weight average molecular weight and the polymer concentration of the desired anion polymer.
- the range of ⁇ 10 ⁇ 3 mmol to 500 mmol is preferred.
- the reaction temperature of the anionic polymerization is not particularly limited as long as it is a temperature equal to or lower than the boiling point at normal pressure of the solvent, and can be selected from the range of 20 to 110 ° C, and preferably in the range of 20 to 80 ° C. Within this temperature range, an anionic polymer showing good mechanical properties with a narrow molecular weight distribution can be produced in a short reaction time.
- the reaction time is not particularly limited, but can be selected from a range of 1 to 20 hours, and a range of 2 to 10 hours is preferable. Within this time range, a high monomer conversion rate of 90% or more can be achieved, so that an anionic polymer can be produced economically advantageously.
- the concentration of the resulting anionic polymer in the reaction solution is not particularly limited, but it is preferably produced so as to be in the range of 1 to 50% by mass, and produced in the range of 5 to 25% by mass. More preferred.
- living polymer I is used as the polymerization initiator, the concentration and amount of living polymer I used so that the concentration of the anionic polymer (copolymer with living polymer I) is within the above range. Is preferably adjusted as appropriate.
- the concentration of the anionic polymer is within the above range, an anionic polymer exhibiting good mechanical properties with a narrow molecular weight distribution can be produced due to the low viscosity of the reaction solution.
- the anionic polymer solution thus obtained may be allowed to act on the polymerization terminator to stop the polymerization operation, or after adding a new monomer to cause a polymerization reaction, the polymerization may be stopped.
- the production method of the present invention when producing an anionic polymer, it is necessary to carry out anionic polymerization under the condition that the amount of inert gas present in 1 kg of the solvent used for the reaction is 20 mmol or less.
- pressurization with an inert gas may be performed.
- the polymerization rate decreases as the monomer concentration in the system decreases, and the amount of heat generated per unit time decreases, so a simple jacket can be used without removing the heat of polymerization reaction with a reflux condenser. This is because the heat of the polymerization reaction can be removed using a temperature control and the like. Therefore, the polymerization termination reaction can also be performed after a pressurizing operation with an inert gas is performed.
- the reaction solution containing an anionic polymer can be subjected to a hydrogenation reaction as it is or after being diluted with a solvent used for polymerization.
- Various hydrogenation catalysts such as nickel-based and cobalt-based Ziegler catalysts and titanium-based catalysts can be used for hydrogenation without any particular limitation.
- Titanium-based catalysts use less catalyst due to their higher catalytic activity per unit metal than Ziegler catalysts, so there is no need to remove the catalyst component from the hydrogenation reaction solution or the catalyst component. Removal is simple.
- Examples of the titanium-based catalyst include, after reacting bis (cyclopentadienyl) titanium dichloride with 2 equivalents or more of an organic alkali metal compound, conjugated dienes, alkali metal alkoxides, organic aluminum compounds, organic magnesium as desired.
- alkali metal halides, alkali metal alkoxides, organoaluminum compounds, organomagnesium compounds, organozinc compounds, organotin compounds, organosilane compounds, and the like may be further used as a co-catalyst for the titanium-based catalyst.
- the amount of the titanium-based catalyst used is preferably in the range of 1.0 ⁇ 10 ⁇ 5 to 1.0 ⁇ 10 ⁇ 1 mmol as a titanium atom with respect to 1 mol of the unsaturated double bond derived from the conjugated diene.
- the hydrogenation reaction can be started at 1.0 ⁇ 10 ⁇ 5 mmol or more, and if it is 1.0 ⁇ 10 ⁇ 2 mmol or less, the yellowing of the hydrogenated anion polymer can be performed without removing the catalyst component. Can prevent changes.
- the range of 1.0 ⁇ 10 ⁇ 3 to 1.0 ⁇ 10 ⁇ 2 mmol is more preferable, and within this range, a hydrogenation reaction time sufficient for industrial implementation and a sufficient hydrogenation rate as a product can be achieved.
- the hydrogenation temperature can be selected from the range of ⁇ 20 to 250 ° C., which is higher than the solvent freezing point and lower than the thermal decomposition temperature of the anionic polymer. It is preferable in terms of production. If it is 30 ° C. or higher, the hydrogenation reaction proceeds, and if it is 150 ° C. or lower, hydrogenation can be carried out with a small amount of catalyst even if catalytic pyrolysis occurs. In particular, the range of 60 to 90 ° C. is more preferable in terms of reducing the amount of catalyst used.
- Hydrogen molecules can be used in a gaseous state, and the pressure is not particularly limited as long as the pressure is normal pressure or higher, but a range of 0 to 20 MPaG is preferable in terms of industrially advantageous production of a hydrogenated polymer.
- the range of 0.5 to 10 MPaG is more preferable in that the amount can be reduced. If it is 20 MPaG or less, hydrogenation can be carried out with a small amount of catalyst even if catalytic hydrogenolysis occurs simultaneously.
- the time required for the hydrogenation can be appropriately selected depending on the conditions, but it is preferably in the range of 10 minutes to 24 hours from the start of coexistence with the catalyst from the viewpoint of industrially advantageous production of the hydrogenated anion polymer, preferably 30 minutes to 10 minutes. A range of time is more preferred.
- reaction solution after completion of the hydrogenation reaction may be further diluted with a solvent or concentrated as necessary, and then washed with a basic aqueous solution or an acidic aqueous solution to remove the catalyst component from the reaction solution. it can.
- the anionic polymer solution or the anionic polymer solution after hydrogenation may be subjected to a concentration operation and supplied to an extruder to isolate the polymer.
- the polymer may be isolated by contacting with steam to remove the solvent.
- the polymer may be isolated, or the polymer may be isolated by contact with a heated inert gas to remove the solvent.
- MPaG as a pressure notation means a gauge pressure.
- the chemicals used are as follows. -Cyclohexane: A product dehydrated with Molecular Sieves 3A and further bubbled with nitrogen gas was used. Sec-Butyllithium: A 1.32 mmol / g cyclohexane solution was used. N, N, N ′, N′-tetramethylethylenediamine: dehydrated with neutral activated alumina, further bubbled with nitrogen gas, and diluted with cyclohexane as necessary.
- -Butadiene Water and a polymerization inhibitor were removed with molecular sieves 3A and neutral activated alumina, and nitrogen-substituted one was used.
- Styrene A product obtained by removing moisture and a polymerization inhibitor with neutral activated alumina and further substituting with nitrogen.
- the weight average molecular weight (Mw) and molecular weight distribution (Mw / Mn) of the polymers obtained in the following examples and comparative examples were measured in terms of standard polystyrene by gel permeation chromatography (hereinafter referred to as GPC) measurement. .
- GPC gel permeation chromatography
- Example 1 (1) Inside a 3 L SUS316 autoclave equipped with a 100 mL glass pressure bottle equipped with a thermometer, electric heater, electromagnetic induction stirrer, gas supply port, sampling port, raw material supply port, and water-cooled cooling pipe After replacing with nitrogen gas, 1513.0 g of cyclohexane and 1.235 g of a 1.32 mmol / g cyclohexane solution of sec-butyllithium (1.63 mmol as sec-butyllithium) were added and stirred at 500 rpm for 50 minutes at 50 ° C. The temperature was raised to.
- thermometer connected to the 100 mL glass pressure bottle showed 22 ° C. at 0.3 MPaG just before exhaust, but the temperature rose as exhausting progressed, and 48 ° C. at ⁇ 0.063 MPaG after exhaust was completed. Indicated. This confirmed that the inside of the reactor was filled with cyclohexane vapor.
- Mw of the obtained block copolymer was 267,100, and Mw / Mn was 1.04.
- the ratio of the total molar amount of butadiene polymerized in the 1,2-bonding mode bonded to the styrene block was 47.1%. .
- the analysis results are shown in Table 2.
- Example 1 (2) instead of depressurizing the pressure in the autoclave to -0.063 MPaG over 10 minutes using a vacuum pump, the process waited for 10 minutes at 0.3 MPaG. Furthermore, 176.08 g of butadiene stored in a pressure-resistant vessel whose total pressure was increased to 0.5 MPaG with nitrogen gas was supplied over 10 minutes. Otherwise, the same operation as in Example 1 was performed. Table 1 shows the change over time in the conversion of butadiene, and Table 2 shows the analysis results of the resulting block copolymer.
- Example 1 and Comparative Reference Example 1 are systems in which, after styrene polymerization, cyclohexane is used as a solvent having a boiling point higher than the polymerization temperature at normal pressure, and butadiene having a boiling point lower than the polymerization temperature is supplied as a monomer to perform anionic polymerization. . Therefore, it is a condition that butadiene as a monomer tends to stay in the gas phase.
- Example 1 in which the inert gas is removed and polymerization is performed with an inert gas content of 20 mmol or less with respect to 1 kg of the solvent in the reaction system, and polymerization is performed by dissolving the monomer in the liquid phase by pressurizing with the inert gas.
- Example 2 An anionic polymerization reaction was performed using the apparatus shown in FIG. 1 is a reactor having a volume of 2.7 m 3 , 2 is a stirrer, and the power number is 1.7 kW / m 3 in the production of the living polymer and the anion polymer, and 3 is a heat transfer area of 7.3 m
- the temperature of the refrigerant can be varied in the range of 10 to 100 ° C. as desired while keeping the refrigerant flow rate constant at 3.0 m 3 / hr. 4 is a reflux condenser having a heat transfer area of 12 m 2 , and 10 ° C.
- refrigerant is communicated at a constant 7.0 m 3 / hr
- 5 is an exhaust pump having a displacement of 8 m 3 / hr
- 6 is an inert gas supply port.
- 7 is a pressure gauge
- 8 is a pressure regulating valve
- 9 is a supply port for a solvent, a polymerization initiator, a vinylating agent, and a polymerization terminator
- 10 is a monomer supply port
- 11 is a liquid A phase thermometer
- 12 is a sampling port
- 13 is a reflux condensate shut-off valve, and is normally open except when the exhaust pump is not operated
- 14 is an exhaust shut-off valve and the exhaust pump It is normally closed except when it is in operation
- 15 is a steam thermometer
- 16 is a steam jacket, and is 10 MJ / hr only in the case of anionic polymerization in the absence of inert gas.
- Heat The A / V ratio of the heat transfer area A (m 2
- the reactor 1 purged with nitrogen was charged with 1133.3 kg of cyclohexane and stirred. While maintaining the nitrogen gas at 0.1 MPaG, the temperature was raised to 50 ° C., and 19.051 kg of sec-butyllithium 57.95 mmol / kg cyclohexane solution (1.104 mol as sec-butyllithium) was added. Then, 34.2 kg of styrene (328.4 mol as styrene) was rapidly supplied at a constant rate over 15 minutes, and polymerization was further performed for 30 minutes. Here, a part of the reaction solution was sampled, stopped with ethanol, precipitated and recovered to obtain polystyrene.
- the internal pressure was reduced to 0.03 MPaG over 10 minutes so as to maintain the liquid temperature of 50 ° C., and then a 26.88 mmol / kg cyclohexane solution of N, N, N ′, N′-tetramethylethylenediamine was obtained. 898 kg (0.508 mol as N, N, N ′, N′-tetramethylethylenediamine) was added. Subsequently, the reflux condensate shutoff valve 13 was closed, the exhaust shutoff valve 14 was opened, the exhaust pump was operated, and the internal pressure was reduced to -0.065 MPaG by exhausting nitrogen gas over 20 minutes.
- the exhaust shutoff valve 14 was quickly closed and the reflux condensate shutoff valve 13 was opened to seal the system.
- the steam thermometer 15 showed 6 ° C. at 0.03 MPaG immediately before the exhaust, but the temperature rose as the exhaust was advanced, and showed 47 ° C. at ⁇ 0.065 MPaG after the exhaust was completed. This confirmed that the inside of the reactor was filled with cyclohexane vapor. Further, according to GPC analysis, the peak of polystyrene obtained from the sampled reaction solution was single, Mw was 30,900, and Mw / Mn was 1.02.
- the heat capacity of the cyclohexane solution of the polymer was set to 2.00 kJ kg ⁇ 1 K ⁇ 1 because there was no significant difference from the heat capacity of cyclohexane itself.
- the temperature change of the polymer solution was confirmed at intervals of 1 minute, and the heat storage amount Q CM (MJ) was calculated from the temperature change.
- the refrigerant flow rate of the simple jacket 3 is 3.0 m 3 / hr, and the temperature change of the inlet temperature and the outlet temperature is confirmed at intervals of 1 minute, and the simple jacket heat removal amount Q JK (MJ) is determined from the integrated value of the temperature difference.
- the steam jacket 16 was heated at 10 MJ / hr, and the steam jacket supply heat quantity Q VAP (MJ) was calculated.
- the total heat removal amount Q CON (MJ) in the reflux condenser 4 is calculated from the polymerization reaction heat amount Q REACT (MJ) and the steam jacket supply heat amount Q VAP (MJ) to the heat storage amount Q CM (MJ) and the simple jacket heat removal amount Q JK. Calculated by subtracting (MJ).
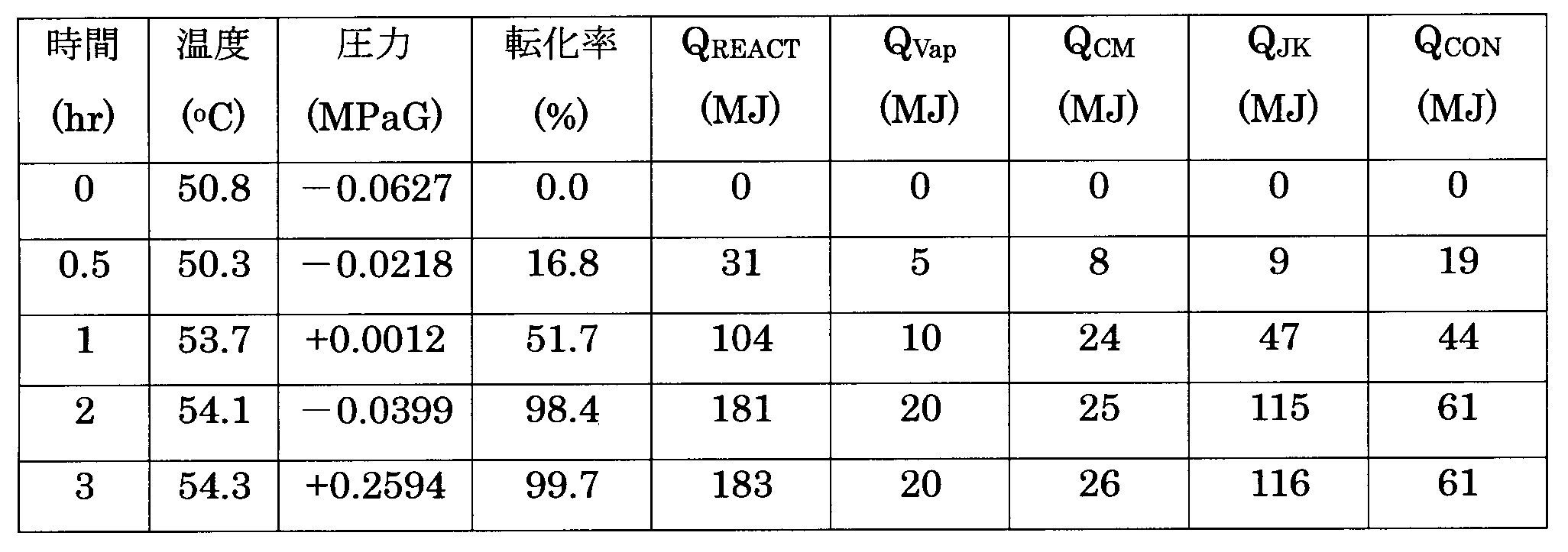
- Example 2 the inert gas in the reaction system was removed as much as possible before the start of anionic polymerization, and the amount of the inert gas with respect to 1 kg of the solvent was 20 mmol or less, so that the total heat removal amount ( Q CM + Q JK + Q CON ) Of 201 MJ, the reflux condenser removed 61 MJ of heat. Since this reaches 30% of the total heat removal amount, it is clear that the heat of polymerization reaction can be quickly removed by the method of the present invention. Further, the conversion rate was 98.4% at 2 hours after the reaction, and it was found that the reaction was almost driven. In addition, after 2 hours to 3 hours after the reaction, nitrogen gas was pressurized.
- the polymerization rate decreased with a decrease in the concentration of butadiene in the system, and the amount of polymerization reaction heat Q REACT per unit time was reduced. Because of its small size, it was possible to sufficiently control the temperature by removing heat with a simple jacket.
- butadiene having an atmospheric pressure boiling point of ⁇ 4.4 ° C. is subjected to anionic polymerization at 50 to 55 ° C. in a cyclohexane solvent having an atmospheric pressure boiling point of 80.7 ° C. in the presence of almost no inert gas. Even in this case, it is possible to achieve the same butadiene polymerization rate and conversion rate as when a large amount of an inert gas is present in the system in order to promote liquefaction of butadiene, and the polymerization reaction heat can be reduced with a simple reactor. Since it can be removed quickly, the polymerization temperature can be controlled.
- a monomer having a lower boiling point at normal pressure than the polymerization temperature can be anionically polymerized at a sufficient polymerization rate and conversion rate. It is clear that the heat of polymerization reaction can be removed quickly.
- the retention of the monomer in the gas phase reduces the polymerization rate and conversion rate and makes it difficult to remove the heat of polymerization reaction.
- the present invention can also be applied to polymerization of monomers having a boiling point at normal pressure lower than the polymerization temperature. is there. Therefore, according to the present invention, when anionic polymerization of a monomer is performed using a solvent having a boiling point higher than the polymerization temperature at normal pressure, it is possible to quickly remove the heat of polymerization reaction using a simple apparatus and control the temperature. An anionic polymer can be produced industrially advantageously.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Graft Or Block Polymers (AREA)
- Polymerisation Methods In General (AREA)
Abstract
Description
[1]重合温度よりも常圧での沸点が高い溶媒1kgに対する不活性ガスの存在量が20mmol以下となる条件で、共役ジエン、芳香族ビニル化合物、またはそれらの混合物をアニオン重合するアニオン重合体の製造方法であり、
[2]アニオン重合の開始剤として、有機アルカリ金属化合物、または1種類以上の共役ジエン、1種類以上の芳香族ビニル化合物もしくは1種類以上の共役ジエンと1種類以上の芳香族ビニル化合物のいずれかを、有機アルカリ金属化合物を用いて重合させてなるリビング共重合体を使用する、[1]のアニオン重合体の製造方法であり、
[3]還流凝縮器の伝熱面積A(m2)の反応器内容積V(m3)に対するA/V比が20~0.1である還流凝縮器を有する完全混合型反応器を用いて重合する、[1]または[2]のアニオン重合体の製造方法であり、
[4]反応温度が20~110℃の範囲でアニオン重合する、[1]~[3]のいずれかのアニオン重合体の製造方法であり、
[5]前記溶媒がシクロヘキサンを50質量%以上含むものである、[1]~[4]のいずれかのアニオン重合体の製造方法であり、
[6]前記アニオン重合体がブロック共重合体である、[1]~[5]のいずれかのアニオン重合体の製造方法である。
なお、本明細書でいう「不活性ガス」とは、窒素ガス、アルゴンガス、ヘリウムガス等の反応性に乏しい希ガスを意味する。
まず、本発明の製造方法で製造されるアニオン重合体を構成するモノマーについて説明する。モノマーとしては、共役ジエン、芳香族ビニル化合物、またはそれらの混合物を使用することができる。
かかるルイス塩基としては、例えばジメチルエーテル、メチルエチルエーテル、ジエチルエーテル、エチルプロピルエーテル、ジプロピルエーテル、ブチルメチルエーテル、tert-ブチルメチルエーテル、ジブチルエーテル、ジオクチルエーテル、エチルフェニルエーテル、ジフェニルエーテルなどの非環状モノエーテル;1,2-ジメトキシエタン、1,2-ジエトキシエタン、1,2-ジイソプロポキシエタン、1,2-ジブトキシエタン、1,2-ジフェノキシエタン、1,2-ジメトキシプロパン、1,2-ジエトキシプロパン、1,2-ジフェノキシプロパン、1,3-ジメトキシプロパン、1,3-ジエトキシプロパン、1,3-ジイソプロポキシプロパン、1,3-ジブトキシプロパン、1,3-ジフェノキシプロパンなどの非環状ジエーテル;テトラヒドロフラン、テトラヒドロピラン、1,4-ジオキサンなどの環状エーテル;ジエチレングリコールジメチルエーテル、ジプロピレングリコールジメチルエーテル、ジブチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジプロピレングリコールジエチルエーテル、ジブチレングリコールジエチルエーテル、トリエチレングリコールジメチルエーテル、トリプロピレングリコールジメチルエーテル、トリブチレングリコールジメチルエーテル、トリエチレングリコールジエチルエーテル、トリプロピレングリコールジエチルエーテル、トリブチレングリコールジエチルエーテル、テトラエチレングリコールジメチルエーテル、テトラプロピレングリコールジメチルエーテル、テトラブチレングリコールジメチルエーテル、テトラエチレングリコールジエチルエーテル、テトラプロピレングリコールジエチルエーテル、テトラブチレングリコールジエチルエーテルなどの非環状ポリエーテル;
上記モノマーをアニオン重合するに際しては、通常、有機アルカリ金属化合物を開始剤として用いる。使用できる有機アルカリ金属化合物としては、例えばメチルリチウム、エチルリチウム、プロピルリチウム、イソプロピルリチウム、ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム、イソブチルリチウム、ペンチルリチウム、ヘキシルリチウム、ブタジエニリルリチウム、シクロヘキシルリチウム、フェニルリチウム、ベンジルリチウム、p-トルイルリチウム、スチリルリチウム、トリメチルシリルリチウム、1,4-ジリチオブタン、1,5-ジリチオペンタン、1,6-ジリチオヘキサン、1,10-ジリチオデカン、1,1-ジリチオジフェニレン、ジリチオポリブタジエン、ジリチオポリイソプレン、1,4-ジリチオベンゼン、1,2-ジリチオ-1,2-ジフェニルエタン、1,4-ジリチオ-2-エチルシクロヘキサン、1,3,5-トリリチオベンゼン、1,3,5-トリリチオー2,4,6-トリエチルベンゼンなどの有機リチウム化合物;メチルナトリウム、エチルナトリウム、n-プロピルナトリウム、イソプロピルナトリウム、n-ブチルナトリウム、sec-ブチルナトリウム、tert-ブチルナトリウム、イソブチルナトリウム、フェニルナトリウム、ナトリウムナフタレン、シクロペンタジエニルナトリウムなどの有機ナトリウム化合物;などが挙げられる。中でもn-ブチルリチウム、sec-ブチルリチウムが好ましい。有機アルカリ金属化合物は1種を単独で用いても、2種以上を併用してもよい。
本発明の製造方法で使用できる溶媒は、重合温度よりも常圧(1atm)での沸点が高い溶媒を含んでいれば種類と使用量に制限はなく、例えばペンタン(36.1℃)、イソペンタン(27.9℃)、2,2,4-トリメチルペンタン(99℃)、ヘキサン(68.7℃)、ヘプタン(98.4℃)、イソヘプタン(90℃)、オクタン(125.7℃)、イソオクタン(99℃)、ノナン(150.8℃)、デカン(174.1℃)、シクロペンタン(49.3℃)、シクロヘキサン(80.7℃)、メチルシクロヘキサン(101.1℃)、エチルシクロヘキサン(132℃)、シクロヘプタン(118.1℃)、メチルシクロヘプタン(135.8℃)などの飽和脂肪族炭化水素;ベンゼン(80.1℃)、トルエン(110.6℃)、エチルベンゼン(136.2℃)、プロピルベンゼン(159.2℃)、ブチルベンゼン(183.4℃)、o-キシレン(144.4℃)、m-キシレン(139.1℃)、p-キシレン(138.4℃)などの芳香族炭化水素;などが挙げられる。これらの中でもシクロヘキサン、n-ヘキサンが特に好ましい。溶媒は1種を単独で用いても、2種以上を併用してもよい。中でも、常圧における沸点が重合温度よりも高い炭化水素を溶媒の構成成分として50質量%以上含むことが好ましく、シクロヘキサンを50質量%以上含む溶媒がより好ましく、シクロヘキサンを80質量%以上含む溶媒がさらに好ましい。このような溶媒であれば、得られるアニオン重合体を含む反応液からの溶媒の除去を経済的有利に実施できる。
なお、上記溶媒には、本発明の効果を損ねない範囲で、重合温度よりも常圧での沸点が低い溶媒、例えばブタン(-0.5℃)、イソブタン(-11.7℃)や上記で挙げられた溶媒の中で重合温度よりも常圧での沸点が低いもの、が含まれていてもよい。重合温度よりも常圧での沸点が低い溶媒の含有量は50質量%未満が好ましく、30質量%未満がより好ましく、10質量%未満がさらに好ましい。
本発明の製造方法で使用できる重合停止剤としては、例えば水素分子、酸素分子、水;メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、ヘプタノール、シクロヘキサノール、フェノール、ベンジルアルコール、o-クレゾール、m-クレゾール、p-クレゾール、エチレングリコール、プロピレングリコール、ブタンジオール、グリセリン、カテコールなどのアルコール;塩化メチル、臭化メチル、ヨウ化メチル、塩化エチル、臭化エチル、ヨウ化エチル、塩化ブチル、臭化ブチル、ヨウ化ブチル、塩化ベンジル、臭化ベンジル、ヨウ化ベンジル、弗化トリメチルシリル、塩化トリメチルシリル、臭化トリメチルシリル、ヨウ化トリメチルシリル、弗化トリエチルシリル、塩化トリエチルシリル、臭化トリエチルシリル、ヨウ化トリエチルシリル、弗化トリブチルシリル、塩化トリブチルシリル、臭化トリブチルシリル、ヨウ化トリブチルシリル、弗化トリフェニルシリル、塩化トリフェニルシリル、臭化トリフェニルシリル、ヨウ化トリフェニルシリルなどのハロゲン化合物;2-ヘプタノン、4-メチル-2-ペンタノン、シクロペンタノン、2-ヘキサノン、2-ペンタノン、シクロヘキサノン、3-ペンタノン、アセトフェノン、2-ブタノン、アセトンなどのケトン;酢酸メチル、酢酸エチル、酢酸ブチルなどのエステル;エチレンオキシド、プロピレンオキシドなどのエポキシ化合物;などが挙げられる。これらの重合停止剤は1種を単独で用いても、2種以上を併用してもよい。また、これらの重合停止剤はアニオン重合体の末端変性剤としての機能を有していてもよい。重合停止剤の使用量には特に制限はなく、所望に応じて適宜設定できるうえ、重合に使用できる溶媒で希釈して用いてもよい。
本発明の製造方法は、重合温度よりも常圧での沸点が高い溶媒1kgに対する不活性ガスの存在量が20mmol以下となる条件で、前述の共役ジエン、芳香族ビニル化合物、またはそれらの混合物をアニオン重合することを特徴とする。反応器としては、気相部分に直接式または間接式に還流凝縮器を有する完全混合型反応器を使用することが好ましい。また、不活性ガスと同伴する溶媒成分の系外流出を低減する点で、不活性ガスが還流凝縮器を介して排気できるように不活性ガスを排気するためのポンプを設置していることが好ましい。反応器は反応液の加熱冷却などの温度制御を目的に外部にジャケットを有していてもよく、その構造には特に制限なく、公知の方式のものが使用できる。また、所望に応じて冷却伝熱を増加させる目的で反応器内部に冷却バッフルまたは冷却コイルなどを付設してもよい。
還流凝縮器の構造に特に制限はないが、多管式還流凝縮器を使用することが好ましい。還流凝縮器は直列または並列で複数の還流凝縮器を連結していてもよく、各々の還流凝縮器に異なる冷媒を通じてもよい。とりわけ、1つの還流凝縮器を用いることが反応器の建設費用を低減できることから経済的である。1つの還流凝縮器を用いる場合はその伝熱面積をA(m2)、複数の還流凝縮器を連結する場合はその伝熱面積の総和をA(m2)とすると、伝熱面積A(m2)の反応器内容積V(m3)に対するA/V比が20~0.1であることが好ましく、10~0.5であることがより好ましい。還流凝縮器に通じる冷媒の種類に特に制限はないが、水、グリコール類、アルコール類、グリセロール類、グリセリン類などの凍結防止剤を含有してなる水溶液が好ましく使用できる。冷媒の温度は還流する溶媒が凍結しない温度から反応液温度の範囲であれば特に制限はないが、-20~50℃の範囲、より好ましくは5~30℃の範囲であることが大型冷凍機を必要としないことから経済的である。冷媒流通量は還流凝縮器の耐圧の範囲以下であれば特に制限はない。
リビング重合体Iの製造は、不活性ガス雰囲気下で行うことが一般的である。操作の具体例としては、不活性ガスで置換した反応器内に溶媒および重合開始剤として有機アルカリ金属化合物を仕込んで所定温度に昇温し、モノマーを適宜添加することでリビング重合体Iを製造する。なお、リビング重合体Iとして共役ジエンをモノマー単位として含むリビング重合体を製造する場合、共役ジエンの結合様式を制御するためのルイス塩基は、モノマー添加の際に同時に添加してもよいし、予め反応器に仕込んでおいてもよい。
前記有機アルカリ金属化合物の使用量は、所望のアニオン重合体の重量平均分子量および重合体濃度にあわせて適宜設定できるが、反応溶媒1kgに対して2×10-3mmol~500mmolの範囲が好ましい。重合温度に特に制限はないが、溶媒凝固点以上かつ重合体の熱分解温度以下としての-20~250℃の範囲から選択でき、20~110℃の範囲が好ましい。この温度範囲であれば短い反応時間で分子量分布の狭いリビング重合体を製造できる。圧力に特に制限はない。反応時間に特に制限はないが、通常、1~20時間の範囲から選択でき、2~10時間の範囲が好ましい。この時間範囲であれば、90%以上の高いモノマー転化率を達成できることから、経済的有利にリビング重合体Iを製造できる。
リビング重合体Iを含む反応液に含まれるリビング重合体Iの濃度に特に制限はないが、1~50質量%の範囲となるように製造することが好ましく、この範囲であれば反応液の粘度が低いことに起因して分子量分布の狭いリビング重合体Iが製造でき、ひいては分子量分布の狭い良好な力学物性を示すアニオン重合体を製造できる。また、アニオン重合体の製造に用いる場合には溶媒を追加することによってリビング重合体Iの濃度を低下させてもよい。
本発明のアニオン重合体の製造方法について詳細に説明すると、まず還流凝縮器および不活性ガスを排気するためのポンプを付設してなる完全混合型反応容器に、溶媒、およびアニオン重合開始剤として有機アルカリ金属化合物または前記リビング重合体Iを導入する。なお、この段階では不活性ガスが存在していてもよい。
アニオン重合体のモノマーを供給してアニオン重合するに際し、反応溶媒1kgに対する不活性ガスの存在量が20mmol以下となるまで排気する。反応溶媒1kgに対する不活性ガスの存在量は、好ましくは15mmol以下、より好ましく9mmol以下、さらに好ましくは5mmol以下である。反応系中の不活性ガスの量を上記範囲内とすることにより、系内を負圧にし、還流凝縮器で溶媒を還流させ、効率よく重合反応熱を除去することができる。系内の不活性ガスの存在量を低減するための排気操作は、アニオン重合体のモノマー供給前に施してもよければ、供給中に施してもよければ、供給後に施してもよい。
排気する不活性ガスに同伴するモノマー量を低減する観点から、アニオン重合体のモノマーを供給する前に還流凝縮器を介してポンプで不活性ガスを系外に排出し、気相部を溶媒の蒸気で充満させることによって実質的に不活性ガスが存在しないようにし、続いてモノマーを供給し、密閉系でアニオン重合することが好ましい。
なお、モノマー供給の際にモノマーと共に不活性ガスが反応系内に供給される場合があるが、その不活性ガスの量は僅かであり、還流凝縮器での除熱効率を著しく低下させるものではない。したがって、モノマー供給の際に必ずしも不活性ガスを排気する必要はないが、所望により、モノマー供給完了時に排気操作を施すことが好ましい。
得られるアニオン重合体の反応液中における濃度に特に制限はないが、1~50質量%の範囲となるように製造することが好ましく、5~25質量%の範囲となるように製造することがより好ましい。重合開始剤としてリビング重合体Iを使用する場合には、得られるアニオン重合体(リビング重合体Iとの共重合体)の濃度が前記範囲となるように、リビング重合体Iの濃度および使用量を適宜調整することが好ましい。アニオン重合体の濃度が上記範囲であれば反応液粘度が低いことに起因して分子量分布の狭い良好な力学物性を示すアニオン重合体を製造できる。
このようにして製造できるアニオン重合体が共役ジエンに由来する不飽和二重結合を有する場合には、アニオン重合体の耐熱性、耐酸化性、耐候性、耐オゾン性などを向上させる観点から、アニオン重合体を含む反応液をそのまま、または重合に用いる溶媒で希釈してから水素化反応に付すこともできる。水素化にはニッケル系やコバルト系のチーグラー触媒、チタン系触媒などの各種水素化触媒を特に制限なく使用することができる。なお、チタン系触媒はチーグラー触媒に比べて単位金属当たりの触媒活性が高いことに起因して触媒使用量が少ないため、水素化反応液からの触媒成分の除去が必要ないか、または触媒成分の除去が簡便である。
チタン系触媒としては、例えばビス(シクロペンタジエニル)チタニウムジクロリドと2等量以上の有機アルカリ金属化合物を反応させたのちに、所望に応じて共役ジエン、アルカリ金属アルコキシド、有機アルミニウム化合物、有機マグネシウム化合物、有機亜鉛化合物、有機錫化合物、有機シラン化合物、またはそれらの前駆体となりうる化合物などと作用させたもの;ビス(シクロペンタジエニル)チタニウムジクロリドとアルカリ金属ハライド又は2当量以上の有機アルミニウム化合物とを作用させたもの;ビス(シクロペンタジエニル)チタニウムジフルオリドを有機シラン化合物と作用させたものなどが挙げられる。所望に応じて、チタン系触媒の助触媒として、アルカリ金属ハライド、アルカリ金属アルコキシド類、有機アルミニウム化合物、有機マグネシウム化合物、有機亜鉛化合物、有機錫化合物、有機シラン化合物などをさらに併用してもよい。
チタン系触媒の使用量は、共役ジエンに由来する不飽和二重結合1モルに対してチタン原子として1.0×10-5~1.0×10-1ミリモルの範囲が好ましい。1.0×10-5ミリモル以上で水素化反応が開始でき、1.0×10-2ミリモル以下であれば触媒成分の除去作業を行わなくとも、単離してなる水素化アニオン重合体の黄変を防ぐことができる。1.0×10-3~1.0×10-2ミリモルの範囲がより好ましく、この範囲であれば工業実施に十分な水素化反応時間および製品として十分な水素化率を達成できる。
水素分子はガス状で使用でき、その圧力は常圧以上であれば特に制限はないが、0~20MPaGの範囲であることが水素化重合体を工業的有利に製造できる点で好ましく、触媒使用量を低減できる点で、0.5~10MPaGの範囲がより好ましい。20MPaG以下であれば触媒水素分解が併発しても少ない触媒使用量で水素化を実施できる。
水素化に要する時間は条件によって適宜選択できるが、触媒との共存開始から10分~24時間の範囲であることが水素化アニオン重合体を工業的有利に製造できる点で好ましく、30分~10時間の範囲がより好ましい。10分以上であれば水素化率を制御でき、24時間以下であれば共役ジエン重合体の熱分解を抑制できる。
水素化反応を終了した後の反応液は、必要に応じてさらに溶媒で希釈するかまたは濃縮してから、塩基性水溶液または酸性水溶液で洗浄することにより、反応液から触媒成分を除去することができる。
・シクロヘキサン:モレュラーシーブス3Aで脱水し、さらに窒素ガスバブリングしたものを使用した。
・sec-ブチルリチウム:1.32mmol/gのシクロヘキサン溶液を使用した。
・N,N,N’,N’-テトラメチルエチレンジアミン:中性活性アルミナで脱水し、さらに窒素ガスバブリングし、必要に応じてシクロヘキサンで希釈して使用した。
・ブタジエン:モレュラーシーブス3Aおよび中性活性アルミナによって水分および重合禁止剤を除去したうえ、窒素置換したものを使用した。
・スチレン:中性活性アルミナで水分および重合禁止剤を除去し、さらに窒素置換したものを使用した。
以下の実施例及び比較例で得られた重合体の、重量平均分子量(Mw)および分子量分布(Mw/Mn)は、ゲルパーミエーションクロマトグラフィー(以下GPCと称する)測定により標準ポリスチレン換算で測定した。測定条件は以下のとおり。
[GPC分析]
装置:東ソー株式会社製、HLC-8320GPC EcoSECシステム
試料:重合体5mgをテトラヒドロフラン10mLに溶解させた溶液
試料注入量:1μL
カラム:東ソー株式会社製TSKgel SuperHZ4000
(内径4.6mm×長さ150mm)
カラム温度:40℃
溶離液:テトラヒドロフラン
溶離液流量:1.0mL/分
検出器:UV検出器(検出波長254nm)
検量線:標準ポリスチレンにより作成
以下の実施例及び比較例で得られた重合体又は重合中間体について、1H-核磁気共鳴分光法(以下1H-NMR分析と略する)を行った。測定条件は以下のとおり。
[1H-NMR分析]
装置:ブルカー・バイオスピン株式会社製、AVANCEIII 600USPlus)
試料:重合体50mgを重クロロホルム1.0gに溶解させた溶液
基準物質:テトラメチルシラン
測定温度:32℃(305K)
積算回数:256回
(1)温度計、電気ヒーター、電磁誘導攪拌装置、ガス供給口、サンプリング口、原料供給口、および水冷式冷却管を設けた100mLガラス製耐圧ビンを備えた容量3LのSUS316製オートクレーブの内部を窒素ガスで置換した後、シクロヘキサン1513.0gおよびsec-ブチルリチウムの1.32mmol/gシクロヘキサン溶液1.235g(sec-ブチルリチウムとして1.63mmol)を加え500rpmで攪拌しながら30分かけて50℃に昇温した。次いで、スチレン45.54g(437.3mmol)をオートクレーブ内に一括添加し、窒素ガスで0.3MPaGに昇圧して液温50~52℃で1時間重合した。
重合中間体の1H-NMR分析によると、スチレンの芳香環に結合した水素原子5Hに帰属できるピークがδ6.2~7.5ppm、ブタジエンの1,2-結合単位2Hに帰属できるピークがδ4.8~5.1ppm、ブタジエンの1,4-結合単位2Hに帰属できるピークがδ5.2~5.5ppmに観測できた。
スチレンは全てスチレンブロックを形成しており、スチレンピーク積分値と各種結合様式(1,2-結合単位、1,4-結合単位)を有するブタジエン由来のピーク積分値の比からスチレンブロックに結合したブタジエンモル量を算出した。
次いで、仕込みブタジエン総モル量に対するスチレンブロックに結合したブタジエンの総モル量の割合を転化率(%)と定義し、ブタジエンの転化率を算出した。
ブタジエン供給開始の時点を反応0時間とすると、反応30分後の圧力は0.00MPaGであり転化率は68.5%であった。反応1時間後の圧力は-0.018MPaGであり転化率は87.5%であった。反応2時間後の圧力は-0.030MPaGであり転化率は99.1%であった。反応2時間のサンプリング直後に窒素ガスによって重合圧力を0.30MPaGとした。反応3時間後の圧力は0.30MPaGであり転化率は99.8%であった。結果を表1に示す。
実施例1の(2)において、真空ポンプを用いて10分かけてオートクレーブ内の圧力を-0.063MPaGに減圧操作する代わりに、0.3MPaGで10分待った。さらに、窒素ガスで全圧を0.5MPaGに加圧した耐圧容器に貯蔵していたブタジエン176.08gを10分かけて供給した。それ以外は、実施例1と同様の操作を施した。ブタジエンの転化率の経時変化を表1に、得られたブロック共重合体の分析結果を表2に示す。

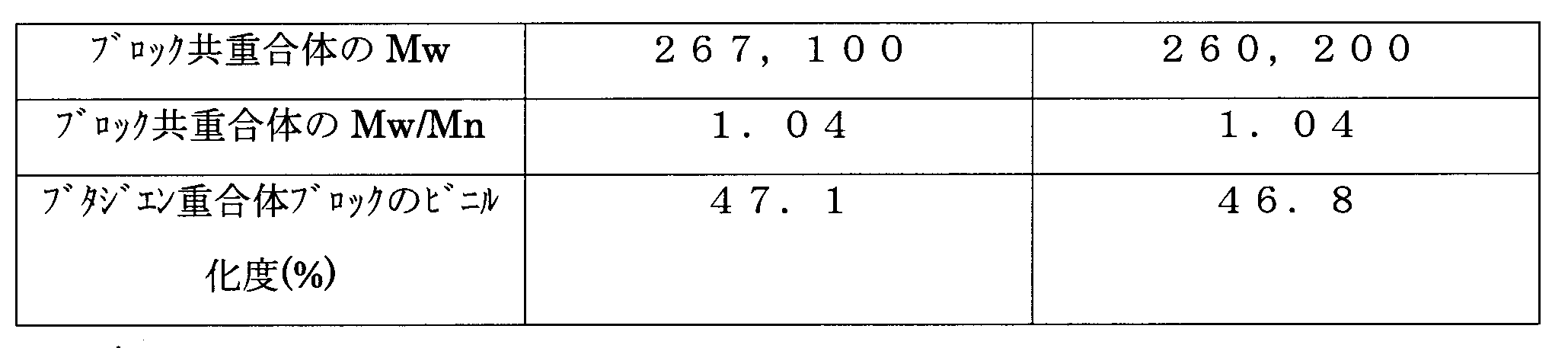
図1に示した装置を用いてアニオン重合反応を行った。1は容積2.7m3の反応器であり、2は攪拌機でありリビング重合体およびアニオン重合体の製造においてその動力数を1.7kW/m3にしており、3は伝熱面積7.3m2の簡易ジャケットであり冷媒流量は3.0m3/hrを一定としながら所望に応じて温度を10~100℃の範囲で変化させうる。4は伝熱面積12m2の還流凝縮器であり10℃の冷媒を7.0m3/hr一定で通じる、5は排気量8m3/hrの排気ポンプであり、6は不活性ガス供給口であり、7は圧力計であり、8は圧力調整弁であり、9は溶媒、重合開始剤、ビニル化剤、および重合停止剤の供給口であり、10はモノマー供給口であり、11は液相温度計であり、12はサンプリング口であり、13は還流凝縮液遮断弁であり排気ポンプを稼動しない場合を除いては通常は開の状態であり、14は排気遮断弁であり排気ポンプを稼動する場合を除いては通常は閉の状態であり、15は蒸気用温度計であり、16は蒸気用ジャケットであり不活性ガスが殆ど存在しない状態でアニオン重合する場合でのみ10MJ/hrで加熱する。なお、還流凝縮器の伝熱面積A(m2)の反応器内容積V(m3)に対するA/V比は4.4であった。
なお、蒸気用温度計15は排気直前の0.03MPaGの時に6℃を示していたが、排気を進めるに伴い温度が上昇し、排気完了後の-0.065MPaGで47℃を示した。これによって、反応器内部がシクロヘキサン蒸気で満たされていることを確認した。
また、GPC分析によればサンプリングした反応液より得たポリスチレンのピークは単一であり、Mwは30,900であり、Mw/Mnは1.02であった。
ブタジエンおよびシクロヘキサンの熱容量はルードウィッヒズ アプライド プロセス デザイン フォー ケミカル アンド ペトロケミカル プランツ(Ludwig‘s Applied Process Design for Chemical and Petrochemical Plants)、第2巻、4版、及びガルフ プロフェッショナル パブリッシング(Gulf Professional Publishing)、2010年、766~767頁などで明らかであり、ブタジエン55℃の熱容量は2.43kJ kg-1 K-1であり、シクロヘキサン55℃の熱容量は2.00kJ kg-1 K-1である。なお、重合体のシクロヘキサン溶液の熱容量はシクロヘキサンそのものの熱容量と有意な差が無かったことから、2.00kJ kg-1 K-1とした。重合体溶液の温度変化は1分間隔で確認しており、その温度変化から蓄熱量QCM(MJ)を計算した。
簡易ジャケット3の冷媒流量は3.0m3/hrであり、入口温度と出口温度の温度変化を1分間隔で確認しており、その温度差の積分値から簡易ジャケット除熱量QJK(MJ)を計算した。
蒸気用ジャケット16を用いて10MJ/hrで加熱しており、蒸気用ジャケット供給熱量QVAP(MJ)を算出した。
還流凝縮器4での除熱総量QCON(MJ)は、重合反応熱量QREACT(MJ)と蒸気用ジャケット供給熱量QVAP(MJ)から蓄熱量QCM(MJ)と簡易ジャケット除熱量QJK(MJ)を差し引くことで算出した。
反応30分後でQREACT31MJ、QVAP5MJ、QCM8MJ、QJK9MJ、QCON19MJであり、反応1時間後でQREACT104MJ、QVAP10MJ、QCM24MJ、QJK47MJ、QCON44MJであり、反応2時間後でQREACT181MJ、QVAP20MJ、QCM25MJ、QJK115MJ、QCON61MJであり、反応2時間のサンプリング直後に窒素ガスによって重合圧力を0.30MPaGするとともに蒸気用ジャケットの加熱を停止して反応3.0時間後でQREACT183MJ、QVAP20MJ、QCM26MJ、QJK116MJ、QCON61MJであった。結果を表3に示す。
GPC分析によれば、ピークは単一であり、得られた共重合体のMwは308,700であり、Mw/Mnは1.08であった。1H-NMR分析によれば、ビニル化度は41.3%であった。
従って、本発明により、重合温度よりも常圧での沸点が高い溶媒を用いてモノマーをアニオン重合するに際して、簡便な装置を用いて重合反応熱を速やかに除去して温度制御することが可能となり、アニオン重合体を工業的有利に製造できる。
2;攪拌機、常時1.7kW/m3で攪拌
3;簡易ジャケット、伝熱面積7.3m2、冷媒を3.0m3/hr一定で通じる
4;還流凝縮器、伝熱面積12m2、10℃の冷媒を7.0m3/hr一定で通じる
5;排気ポンプ8m3/hr
6;不活性ガス供給口
7;圧力計
8;圧力調整弁
9;溶媒、重合開始剤、および重合停止剤の供給口
10;モノマー供給口
11;液相温度計
12;サンプリング口
13;還流凝縮液遮断弁
14;排気遮断弁
15;蒸気用温度計
16;蒸気用ジャケット、蒸気を10MJ/hrで加熱できる
Claims (6)
- 重合温度よりも常圧での沸点が高い溶媒1kgに対する不活性ガスの存在量が20mmol以下となる条件で、共役ジエン、芳香族ビニル化合物、またはそれらの混合物をアニオン重合するアニオン重合体の製造方法。
- アニオン重合の開始剤として、有機アルカリ金属化合物、または1種類以上の共役ジエン、1種類以上の芳香族ビニル化合物もしくは1種類以上の共役ジエンと1種類以上の芳香族ビニル化合物のいずれかを、有機アルカリ金属化合物を用いて重合させてなるリビング共重合体を使用する、請求項1に記載のアニオン重合体の製造方法。
- 還流凝縮器の伝熱面積A(m2)の反応器内容積V(m3)に対するA/V比が20~0.1である還流凝縮器を有する完全混合型反応器を用いて重合する、請求項1または2に記載のアニオン重合体の製造方法。
- 反応温度が20~110℃の範囲でアニオン重合する、請求項1~3のいずれかに記載のアニオン重合体の製造方法。
- 前記溶媒がシクロヘキサンを50質量%以上含むものである、請求項1~4のいずれかに記載のアニオン重合体の製造方法。
- 前記アニオン重合体がブロック共重合体である、請求項1~5のいずれかに記載のアニオン重合体の製造方法。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2960005A CA2960005C (en) | 2014-09-04 | 2015-09-03 | Method for producing anionic polymer |
| JP2016546698A JP6733875B2 (ja) | 2014-09-04 | 2015-09-03 | アニオン重合体の製造方法 |
| SG11201701733RA SG11201701733RA (en) | 2014-09-04 | 2015-09-03 | Method for producing anionic polymer |
| KR1020177006105A KR20170051436A (ko) | 2014-09-04 | 2015-09-03 | 아니온 중합체의 제조 방법 |
| CN201580047566.2A CN106661135B (zh) | 2014-09-04 | 2015-09-03 | 阴离子聚合物的制造方法 |
| ES15838378T ES2819724T3 (es) | 2014-09-04 | 2015-09-03 | Método para producir un polímero aniónico |
| US15/508,773 US10472451B2 (en) | 2014-09-04 | 2015-09-03 | Method for producing anionic polymer |
| EP15838378.6A EP3190130B1 (en) | 2014-09-04 | 2015-09-03 | Method for producing anionic polymer |
| KR1020227036697A KR102518564B1 (ko) | 2014-09-04 | 2015-09-03 | 아니온 중합체의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014180094 | 2014-09-04 | ||
| JP2014-180094 | 2014-09-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016035866A1 true WO2016035866A1 (ja) | 2016-03-10 |
Family
ID=55439923
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/075126 Ceased WO2016035866A1 (ja) | 2014-09-04 | 2015-09-03 | アニオン重合体の製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10472451B2 (ja) |
| EP (1) | EP3190130B1 (ja) |
| JP (1) | JP6733875B2 (ja) |
| KR (2) | KR20170051436A (ja) |
| CN (1) | CN106661135B (ja) |
| CA (1) | CA2960005C (ja) |
| ES (1) | ES2819724T3 (ja) |
| SG (1) | SG11201701733RA (ja) |
| WO (1) | WO2016035866A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020007432A (ja) * | 2018-07-05 | 2020-01-16 | 株式会社クラレ | 成形体 |
| JP2020050744A (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法 |
| JP2020050743A (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法及び重合体の製造方法 |
| WO2020066174A1 (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法及び重合体の製造方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102139528B1 (ko) * | 2018-02-05 | 2020-07-30 | 주식회사 엘지화학 | 공액디엔계 중합체 제조방법 및 공액디엔계 중합체 제조장치 |
| EP4011614A4 (en) * | 2019-08-06 | 2023-08-30 | Nitto Denko Corporation | Adhesive tape |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10218953A (ja) * | 1996-12-03 | 1998-08-18 | Nippon Elastomer Kk | ゴム状重合体及びその製造方法、及び新規なアニオン重合開始剤及び樹脂組成物 |
| JP2000080115A (ja) * | 1998-09-02 | 2000-03-21 | Asahi Chem Ind Co Ltd | ブロックコポリマーの製造方法 |
| JP2003002908A (ja) * | 2001-06-18 | 2003-01-08 | Nippon Zeon Co Ltd | 共役ジエン−芳香族ビニルブロック共重合体の製造方法 |
| JP2003261740A (ja) * | 2002-03-11 | 2003-09-19 | Nippon Zeon Co Ltd | エラストマー組成物の製造方法 |
| JP2006241289A (ja) * | 2005-03-03 | 2006-09-14 | Asahi Kasei Chemicals Corp | ブロック共重合体の製造方法 |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3281383A (en) | 1962-08-09 | 1966-10-25 | Phillips Petroleum Co | Branched polymers prepared from monolithium-terminated polymers and compounds having at least three reactive sites |
| BE756177A (fr) | 1969-09-17 | 1971-03-15 | Shell Int Research | Procede de preparation de copolymeres a blocs hydrogenes |
| US4396761A (en) | 1981-08-21 | 1983-08-02 | Shell Oil Company | Method for removing hydrogenation catalyst residues from hydrogenated conjugated diene polymers |
| JPS5884809A (ja) | 1981-11-16 | 1983-05-21 | Japan Synthetic Rubber Co Ltd | スチレン−ブタジエン共重合体の製造方法 |
| JPH0617409B2 (ja) * | 1984-10-18 | 1994-03-09 | ザ ダウ ケミカル カンパニ− | アニオン重合法 |
| JPS61223181A (ja) | 1985-03-29 | 1986-10-03 | Mitsubishi Metal Corp | 表面被覆炭化タングステン基超硬合金製切削工具 |
| JPS62143194A (ja) | 1986-12-04 | 1987-06-26 | Seikosha Co Ltd | デ−タ可変型カ−ド |
| JP2639558B2 (ja) | 1988-04-28 | 1997-08-13 | 日本ゼオン株式会社 | 塩化ビニル系重合体の製造法 |
| GB8918702D0 (en) | 1989-08-16 | 1989-09-27 | Shell Int Research | Process for the preparation of random solution copolymers of conjugated dienes and vinyl aromatic compounds |
| US5354537A (en) | 1992-04-27 | 1994-10-11 | Akzo N.V. | Piercing and sampling probe |
| US5244980A (en) | 1992-12-07 | 1993-09-14 | Shell Oil Company | Selective hydrogenation of conjugated diolefin polymers with Tebbe's reagent |
| JP3400104B2 (ja) | 1993-05-31 | 2003-04-28 | 株式会社クラレ | メタクリル系樹脂組成物およびその製造方法 |
| US5436298A (en) * | 1993-09-30 | 1995-07-25 | Phillips Petroleum Company | Block copolymers of monovinylarenes and conjugated dienes and preparation thereof |
| CA2117708C (en) * | 1993-09-30 | 2002-10-22 | William J. Trepka | Block copolymers of monovinylarenes and conjugated dienes and preparation thereof |
| EP0702033B1 (en) * | 1994-09-14 | 1997-12-03 | Shin-Etsu Chemical Co., Ltd. | Process of producing vinyl chloride type polymer |
| KR0146676B1 (ko) | 1994-11-14 | 1998-08-17 | 박원배 | 염화비닐 수지의 현탁 중합용 교반기 및 이를 이용한 염화비닐 비닐 수지의 현탁 중합 방법 |
| NL1007534C2 (nl) | 1996-12-03 | 1999-05-12 | Japan Elastomer Co | Rubberachtig polymeer alsmede werkwijze voor de bereiding daarvan. |
| JP3862359B2 (ja) | 1997-05-08 | 2006-12-27 | 旭化成ケミカルズ株式会社 | 重合温度の制御方法及び該制御方法を用いたリビング重合法 |
| TW583027B (en) | 1998-10-30 | 2004-04-11 | Shell Int Research | A method for preparing a hydrogenation catalyst system |
| JP4165781B2 (ja) * | 1998-11-27 | 2008-10-15 | 旭化成ケミカルズ株式会社 | ブロック共重合体の重合方法 |
| US6313230B1 (en) | 1999-09-21 | 2001-11-06 | Industrial Technology Research Institute | Catalyst composition for hydrogenation of conjugated diene based synthetic rubbers |
| JP4912519B2 (ja) | 2000-03-24 | 2012-04-11 | 旭化成ケミカルズ株式会社 | 効率的な共役ジエン系重合体の水素添加方法 |
| JP2001316403A (ja) * | 2000-05-08 | 2001-11-13 | Asahi Kasei Corp | 加工性に優れたスチレン系樹脂の製造方法 |
| JP2003268012A (ja) | 2002-03-19 | 2003-09-25 | Nippon Shokubai Co Ltd | 水溶性重合体の製造装置 |
| DE10234746B4 (de) * | 2002-07-30 | 2004-04-29 | Sasol Germany Gmbh | Verfahren zur Herstellung von Polymerisaten unter Verwendung von konjugierten Dienen und vinylaromatischen Verbindungen, nach diesem Verfahren hergestellte Polymerisate und deren Verwendung |
| US6657028B1 (en) * | 2002-08-01 | 2003-12-02 | Albemarle Corporation | Anionic polymerization process |
| KR20080112282A (ko) | 2006-03-24 | 2008-12-24 | 크레이튼 폴리머즈 유.에스. 엘엘씨 | 고온 블록 공중합체 및 이의 제조방법 |
| US7700694B2 (en) | 2006-04-28 | 2010-04-20 | Tsrc Corporation | Catalyst composition and method for hydrogenating a polymer having a conjugated diene |
| CN100482698C (zh) * | 2007-07-13 | 2009-04-29 | 北京化工大学 | 一种阴离子聚合制备等规聚苯乙烯的方法 |
| KR20090052767A (ko) | 2007-11-21 | 2009-05-26 | 금호석유화학 주식회사 | 스티렌계 복합 블록 공중합체 혼합물의 제조방법 및 이를함유한 개질 아스팔트 조성물 |
| JP4396761B2 (ja) | 2007-11-26 | 2010-01-13 | 株式会社デンソー | 回転電機の固定子および回転電機 |
| JP5334566B2 (ja) | 2008-12-25 | 2013-11-06 | Fdk株式会社 | 蓄電モジュールの電圧補正制御方法 |
| US8598286B1 (en) * | 2012-11-05 | 2013-12-03 | The Goodyear Tire & Rubber Company | High cis diene/phenylbutadiene copolymers prepared using a Ziegler/Natta neodymium catalyst |
| GB2524245A (en) | 2014-03-17 | 2015-09-23 | Atlantic Inertial Systems Ltd | Accelerometers |
-
2015
- 2015-09-03 US US15/508,773 patent/US10472451B2/en active Active
- 2015-09-03 KR KR1020177006105A patent/KR20170051436A/ko not_active Ceased
- 2015-09-03 SG SG11201701733RA patent/SG11201701733RA/en unknown
- 2015-09-03 CN CN201580047566.2A patent/CN106661135B/zh active Active
- 2015-09-03 JP JP2016546698A patent/JP6733875B2/ja active Active
- 2015-09-03 WO PCT/JP2015/075126 patent/WO2016035866A1/ja not_active Ceased
- 2015-09-03 CA CA2960005A patent/CA2960005C/en active Active
- 2015-09-03 EP EP15838378.6A patent/EP3190130B1/en active Active
- 2015-09-03 KR KR1020227036697A patent/KR102518564B1/ko active Active
- 2015-09-03 ES ES15838378T patent/ES2819724T3/es active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10218953A (ja) * | 1996-12-03 | 1998-08-18 | Nippon Elastomer Kk | ゴム状重合体及びその製造方法、及び新規なアニオン重合開始剤及び樹脂組成物 |
| JP2000080115A (ja) * | 1998-09-02 | 2000-03-21 | Asahi Chem Ind Co Ltd | ブロックコポリマーの製造方法 |
| JP2003002908A (ja) * | 2001-06-18 | 2003-01-08 | Nippon Zeon Co Ltd | 共役ジエン−芳香族ビニルブロック共重合体の製造方法 |
| JP2003261740A (ja) * | 2002-03-11 | 2003-09-19 | Nippon Zeon Co Ltd | エラストマー組成物の製造方法 |
| JP2006241289A (ja) * | 2005-03-03 | 2006-09-14 | Asahi Kasei Chemicals Corp | ブロック共重合体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3190130A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020007432A (ja) * | 2018-07-05 | 2020-01-16 | 株式会社クラレ | 成形体 |
| JP7141027B2 (ja) | 2018-07-05 | 2022-09-22 | 株式会社クラレ | 成形体 |
| JP2020050744A (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法 |
| JP2020050743A (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法及び重合体の製造方法 |
| WO2020066174A1 (ja) * | 2018-09-26 | 2020-04-02 | 日本ゼオン株式会社 | 単量体組成物の精製方法及び重合体の製造方法 |
| JP7180241B2 (ja) | 2018-09-26 | 2022-11-30 | 日本ゼオン株式会社 | 単量体組成物の精製方法及び重合体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10472451B2 (en) | 2019-11-12 |
| SG11201701733RA (en) | 2017-04-27 |
| JPWO2016035866A1 (ja) | 2017-06-22 |
| US20170283540A1 (en) | 2017-10-05 |
| CA2960005C (en) | 2023-02-14 |
| EP3190130A4 (en) | 2018-04-04 |
| EP3190130B1 (en) | 2020-08-12 |
| ES2819724T3 (es) | 2021-04-19 |
| CN106661135A (zh) | 2017-05-10 |
| JP6733875B2 (ja) | 2020-08-05 |
| KR102518564B1 (ko) | 2023-04-05 |
| EP3190130A1 (en) | 2017-07-12 |
| KR20220148937A (ko) | 2022-11-07 |
| CN106661135B (zh) | 2020-06-19 |
| CA2960005A1 (en) | 2016-03-10 |
| KR20170051436A (ko) | 2017-05-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6733875B2 (ja) | アニオン重合体の製造方法 | |
| JP6010709B2 (ja) | 水素化重合体の製造方法 | |
| US10000585B2 (en) | Process for producing aromatic vinyl/conjugated diene copolymer and product of hydrogenation thereof | |
| JP6604961B2 (ja) | 新規陰イオン重合開始剤およびこれを用いた共役ジエン系共重合体の製造方法 | |
| JP2022075872A (ja) | 1,3,7-オクタトリエンとスチレンの共重合体およびその水素化物 | |
| JP7350927B2 (ja) | 1,3,7-オクタトリエンとブタジエンの共重合体およびその水素化物 | |
| JP2022075869A (ja) | 1,3,7-オクタトリエンとイソプレンの共重合体およびその水素化物 | |
| JP7075352B2 (ja) | 1,3,7-オクタトリエン重合体およびその水素化物、並びに該重合体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15838378 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016546698 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2960005 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20177006105 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015838378 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15508773 Country of ref document: US Ref document number: 2015838378 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |