WO2016093268A1 - 蛍光ナノ粒子用希釈液、これを用いた蛍光免疫染色用キット、蛍光免疫染色用溶液、および蛍光免疫染色法、遺伝子染色法 - Google Patents
蛍光ナノ粒子用希釈液、これを用いた蛍光免疫染色用キット、蛍光免疫染色用溶液、および蛍光免疫染色法、遺伝子染色法 Download PDFInfo
- Publication number
- WO2016093268A1 WO2016093268A1 PCT/JP2015/084500 JP2015084500W WO2016093268A1 WO 2016093268 A1 WO2016093268 A1 WO 2016093268A1 JP 2015084500 W JP2015084500 W JP 2015084500W WO 2016093268 A1 WO2016093268 A1 WO 2016093268A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fluorescent
- casein
- solution
- immunostaining
- nanoparticles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6816—Hybridisation assays characterised by the detection means
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/62—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light
- G01N21/63—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light optically excited
- G01N21/64—Fluorescence; Phosphorescence
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/62—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light
- G01N21/63—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light optically excited
- G01N21/64—Fluorescence; Phosphorescence
- G01N21/6428—Measuring fluorescence of fluorescent products of reactions or of fluorochrome labelled reactive substances, e.g. measuring quenching effects, using measuring "optrodes"
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
- G01N33/533—Production of labelled immunochemicals with fluorescent label
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
- G01N33/54313—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals the carrier being characterised by its particulate form
- G01N33/54346—Nanoparticles
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
- G01N33/54393—Improving reaction conditions or stability, e.g. by coating or irradiation of surface, by reduction of non-specific binding, by promotion of specific binding
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57575—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving oncogenic proteins
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/58—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances
- G01N33/582—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances with fluorescent label
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/62—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light
- G01N21/63—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light optically excited
- G01N21/64—Fluorescence; Phosphorescence
- G01N21/6428—Measuring fluorescence of fluorescent products of reactions or of fluorochrome labelled reactive substances, e.g. measuring quenching effects, using measuring "optrodes"
- G01N2021/6439—Measuring fluorescence of fluorescent products of reactions or of fluorochrome labelled reactive substances, e.g. measuring quenching effects, using measuring "optrodes" with indicators, stains, dyes, tags, labels, marks
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/71—Assays involving receptors, cell surface antigens or cell surface determinants for growth factors; for growth regulators
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/536—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase
Definitions
- the present invention relates to a diluted solution of fluorescent nanoparticles used in a fluorescent immunostaining method (or gene detection method), a fluorescent immunostaining kit, a fluorescent immunostaining solution using the same, a fluorescent immunostaining method, and a gene staining method .
- tissue slice the tissue sampled from the affected area is dehydrated to fix the tissue, and after processing such as blocking with paraffin, it is cut into thin slices with a thickness of 2 to 8 microns and the paraffin removed (hereinafter referred to as “tissue slice”).
- tissue slice the target biological material is stained and observed under a microscope.
- diagnosis is performed based on morphological information such as changes in the size and shape of cell nuclei and changes in pattern as tissue, and staining information.
- image digitization technology in contrast to the above-mentioned pathological diagnosis, information necessary for pathologists to perform pathological diagnosis is extracted from pathological images input as digital color images using a microscope or digital camera.
- an automated pathological diagnosis support apparatus that measures and displays.
- HE staining using a dye and DAB staining using an enzyme have been widely used as tissue staining methods, but the staining concentration depends greatly on environmental conditions such as temperature and time, and is accurate. Quantitative measurement is difficult.
- Patent Document 1 discloses an immunostaining method using fluorescent nanoparticles as a labeling reagent instead of a dye.
- fluorescent nanoparticles As a labeling reagent, it becomes possible to evaluate with high accuracy and quantification that cannot be obtained by the conventional enzyme method.
- a high-luminance phosphor When such a high-luminance phosphor is used, one particle is used. However, since it is detectable, background noise is likely to occur due to non-specific binding of fluorescent nanoparticles.
- the present invention suppresses nonspecific adsorption of fluorescent nanoparticles and suppresses background noise when performing immunostaining (or gene staining) using fluorescent nanoparticles. It is an object of the present invention to provide a means for detecting and quantifying a target biological substance with higher accuracy by suppressing it.
- the present inventor has paid attention to the composition of a diluted solution of fluorescent nanoparticles used in fluorescent immunostaining (or gene staining) using fluorescent nanoparticles, and has proceeded with research.
- fluorescent nanoparticles are dispersed in a solution containing high molecular weight (Mw ⁇ 40,000: BSA, etc.) and low molecular weight (Mw ⁇ 40,000: casein, etc.) proteins at predetermined concentrations. It has been found that non-specific adsorption of nanoparticles can be suppressed, and the present invention has been completed.
- the fluorescent nanoparticle dilution liquid reflecting one aspect of the present invention has a molecular weight of 40,000 or more used when immunostaining (or gene staining) using fluorescent nanoparticles is performed. 1 to 5% (W / W) of protein and 1 to 3% (W / W) of protein having a molecular weight of less than 40,000.
- fluorescent immunostaining or gene staining
- the fluorescent nanoparticle dilution liquid of the present invention when diluting fluorescent particles, it is possible to reduce the background noise seen at the time of detection and evaluate the stained image. In doing so, it is possible to improve accuracy and quantitativeness.
- the diluted solution for fluorescent nanoparticles of the present invention contains at least 1 to 5% (W / W) high molecular weight (Mw ⁇ 40,000) protein and 1 to 3 low molecular weight (Mw ⁇ 40,000) protein. % (W / W) content, and using this solution, fluorescent nanoparticles are dispersed to prepare a fluorescent immunostaining solution (or gene staining solution) for immunostaining (or gene staining). can do.
- a fluorescent immunostaining solution or gene staining solution
- immunostaining or gene staining solution
- the diluted solution for fluorescent nanoparticles of the present invention contains at least 1 to 5% (W / W) high molecular weight (Mw ⁇ 40,000) protein and / or 1 low molecular weight (Mw ⁇ 40,000) protein. Contains 3% (W / W).
- BSA molecular weight of about 66,000
- BSA particularly has the effect of stabilizing the protein.
- the upper limit of the molecular weight of a high molecular weight protein is not specifically limited, Usually, it is 400,000. If the high molecular weight protein concentration is below 1%, the number of observed bright spots is reduced and the signal is weakened. On the other hand, when the protein concentration exceeds 5%, the bright spots tend to aggregate and it is difficult to accurately evaluate the signal. In view of such effects, the high molecular weight protein concentration is more preferably 1.5 to 3% (W / W).
- casein molecular weight of about 23,000
- casein molecular weight of about 23,000
- the lower limit of the molecular weight of the low molecular weight protein is not particularly limited, but is usually 1,000. If the low molecular weight protein concentration is less than 1%, the background noise increases and the signal quantification decreases. On the other hand, when the protein concentration exceeds 3%, the number of bright spots observed decreases and the signal weakens. From the viewpoint of such effects, the low molecular weight protein concentration is more preferably 1.2 to 2.4% (W / W).
- casein in natural casein contained in milk and the like, four types of casein, ⁇ -casein, ⁇ -casein, ⁇ -casein and ⁇ -casein, are 50%, 35%, 13% and 2%, respectively. Contained. Each casein may be a commercially available product, or may be fractionated by various fractionation methods. By mixing them, casein having a desired composition can be prepared. .
- the ratio of ⁇ -casein: ⁇ -casein contained in the diluted solution for fluorescent nanoparticles according to the present invention is 40:60 to 60:40 (the total amount of ⁇ -casein and ⁇ -casein is 100).
- the background at the time of observation is further suppressed, and a more quantitative evaluation can be performed.
- casein is known to form various micelle structures, particularly ⁇ -casein, and the composition satisfying the above conditions is effective for the effects of the present invention through the formation of the micelle structure. It may be related to the effect.
- the fluorescent nanoparticle diluent in the present invention specifically contains 1 to 5% (W / W) of BSA and 1 to 3% (W / W) of casein.
- the content ratio of ⁇ -casein is preferably 10% (W / W) or less (particularly, the ratio of ⁇ -casein is more preferably 0.5 to 10% (W / W)). Further, it is particularly preferable that the ratio of ⁇ -casein: ⁇ -casein is 40:60 to 60:40 (the total amount of ⁇ -casein and ⁇ -casein is 100).
- buffer solution such as PBS or TBS is generally used. That is, these buffer solutions can be used as a solvent for the diluted solution for fluorescent nanoparticles of the present invention.
- the diluted solution for fluorescent nanoparticles may contain a nonionic surfactant such as Tween 20 or Digitonin, and may contain a chelating agent such as EDTA.
- the immunostaining reagent containing fluorescent nanoparticles for the target biological material is diluted to the desired concentration, so that background noise is suppressed and fluorescent immunostaining with high quantitativeness ( Or gene staining).
- kits in the present invention, a kit for performing immunostaining (or gene staining) using fluorescent nanoparticles is defined.
- This kit includes at least an immunostaining reagent (or gene staining reagent) containing fluorescent nanoparticles for a target biological material and the fluorescent nanoparticle diluent.
- This kit further includes other necessary reagents, members, etc. for immunostaining the target biological substance, and instructions for carrying out the fluorescent immunostaining (or gene staining) according to the present invention, if necessary. May be included.
- a fluorescent immunostaining solution obtained by diluting an immunostaining reagent for a target biological substance with the above-described diluent for fluorescent nanoparticles.
- the dilution factor of the immunostaining reagent can be optimized according to the affinity between the target biological substance and the immunostaining reagent.
- the target biological material in the present invention is a biological material expressed in a tissue section, particularly a protein (antigen) (or gene), and a fluorescent label is mainly used for quantification or detection from the viewpoint of pathological diagnosis. It refers to the target of immunostaining used.
- the target biological material may be selected in consideration of the use of the quantification method of the present invention such as pathological diagnosis, and is not particularly limited.
- a biological substance that is expressed in the cell membrane of various cancer tissues and can be used as a biomarker such as EGFR (HER1) (Epidermal Growth Factor Receptor: epidermal growth factor receptor)
- HER2 Human epidermal growth factor receptor
- HER3, HER4, VEGFR Vasular Endothelial Growth Factor Receptor
- IGFR Insulin-like Growth Factor Receptor) Factor receptors
- growth factor receptors such as HGFR (Hepatocyte Growth Factor Receptor), PD-1 (Programmed cell death 1), PD-L1 (Programmed cell death ligand 1) And proteins that are receptors of the immune system.
- EGFR / HER includes EGFR / HER1 (also referred to as ErbB1) that is overexpressed in cancer tissues such as colorectal cancer, EGFR2 / HER2 (also referred to as ErbB2, neu) that is overexpressed in cancer tissues such as breast cancer, and EGFR3. / HER3 and EGFR4 / HER4 are included.
- VEGFR includes VEGFR-1 (also referred to as Flt-1), VEGFR-2 (also referred to as Flt-2, KDR) and lymphatic vessels, which are upregulated in vascular endothelial cells in cancer tissues such as liver cancer and esophageal cancer.
- VEGFR-3 also called Flt-4) whose expression is upregulated in the skin cells is included.
- HER2 is suitable as a target biological material when performing the quantification method of the present invention in pathological diagnosis related to breast cancer.
- target biological substances include HER2, TOP2A, HER3, EGFR, P53, and MET as genes related to cancer growth and the response rate of molecular target drugs. Furthermore, the following are mentioned as a gene known as a cancer related gene.
- Tyrosine kinase-related genes include ALK, FLT3, AXL, FLT4 (VEGFR3, DDR1, FMS (CSF1R), DDR2, EGFR (ERBB1), HER4 (ERBB4), EML4-ALK, IGF1R, EPHA1, INSR, EPHA2, IRR (INSRR) ), EPHA3, KIT, EPHA4, LTK, EPHA5, MER (MERTK), EPHA6, MET, EPHA7, MUSK, EPHA8, NPM1-ALK, EPHB1, PDGFR ⁇ (PDGFRA), EPHB2, PDGFR ⁇ (PDGFRB), EPHEP3, T RON (MST1R), FGFR1, ROS (ROS1), FGFR2, TIE2 (TEK), FGFR3, TRKA (NTRK1), FGFR4, TRKB (NT RK2), FLT1 (VEGFR1), TRKC (NTRK3) and breast cancer-related genes such as ATM, BRCA1, BRCA2, BRCA3, CC
- genes include APC, MSH6, AXIN2, MYH, BMPR1A, p53, DCC, PMS2, KRAS2 (or Ki-ras), PTEN, MLH1, SMAD4, MS 2, STK11, MSH6 Lung cancer-related genes include ALK, PTEN, CCND1, RASSF1A, CDKN2A, RB1, EGFR, RET, EML4, ROS1, KRAS2, TP53, MYC.
- Examples of the gene include Axin1, MALAT1, b-catenin, p16 INK4A, c-ERBB-2, p53, CTNNB1, RB1, Cyclin D1, SMAD2, EGFR, SMAD4, IGFR2, TCF1, and KRAS.
- Examples include Alpha, PRCC, ASPSCR1, PSF, CLTC, TFE3, p54nrb / NONO, and TFEB
- Examples of ovarian cancer-related genes include AKT2, MDM2, BCL2, MYC, BRCA1, NCOA4, CDKN2A, p53, PIK3CA, GATA4, RB, HRAS, RET, KRAS, and RNASET2.
- Examples of prostate cancer-related genes include AR, KLK3, BRCA2, MYC, CDKN1B, NKX3.1, EZH2, p53, GSTP1, and PTEN.
- Examples of bone tumor-related genes include CDH11, COL12A1, CNBP, OMD, COL1A1, THRAP3, COL4A5, and USP6.
- an antibody that specifically recognizes and binds to a protein as a target biological substance as described above as an antigen
- an anti-EGFR antibody can be used when EGFR (expressed protein) is the target biological material
- an anti-HER2 antibody can be used when HER2 is the target biological material.
- an antibody that specifically recognizes and binds to the primary antibody as an antigen can be used.
- Both the primary antibody and the secondary antibody may be polyclonal antibodies, but a monoclonal antibody is preferable from the viewpoint of quantitative stability.
- the type of animal that produces the antibody is not particularly limited, and may be selected from mice, rats, guinea pigs, rabbits, goats, sheep, and the like as in the past.
- the primary antibody may be an antibody fragment or a derivative instead of a natural full-length antibody as long as it has the ability to specifically recognize and bind to a specific biological substance (antigen). That is, the term “antibody” in the present specification includes not only full-length antibodies but also antibody fragments such as Fab, F (ab) ′ 2, Fv, scFv and chimeric antibodies (humanized antibodies, etc.), multifunctional antibodies Derivatives such as are included.
- the fluorescent nanoparticles used in the present invention are preferably “fluorescent substance-integrated nanoparticles” that can emit fluorescence with a sufficient intensity to express the target biological substance as a bright spot one molecule at a time.
- the “fluorescent substance” in this specification is irradiated with electromagnetic waves (X-rays, ultraviolet rays, or visible rays) having a predetermined wavelength and absorbs energy to excite electrons and return from the excited state to the ground state.
- electromagnetic waves X-rays, ultraviolet rays, or visible rays
- a substance that emits surplus energy as electromagnetic waves that is, a substance that emits “fluorescence” and that can be directly or indirectly bound to a secondary antibody.
- “fluorescence” has a broad meaning and includes phosphorescence having a long emission lifetime in which emission is continued even when irradiation of electromagnetic waves for excitation is stopped, and narrow sense fluorescence having a short emission lifetime.
- the fluorescent substance-integrated nanoparticles in the present invention have a structure in which a plurality of fluorescent substances (for example, fluorescent dyes) are encapsulated in and / or adsorbed on the surface thereof, based on particles made of organic or inorganic substances. It has nano-sized particles.
- the base material for example, resin
- the fluorescent material have substituents or sites having opposite charges, and an electrostatic interaction works.
- the organic substance is a resin generally classified as a thermosetting resin such as melamine resin, urea resin, aniline resin, guanamine resin, phenol resin, xylene resin, furan resin; Resins generally classified as thermoplastic resins such as styrene resin, acrylic resin, acrylonitrile resin, AS resin (acrylonitrile-styrene copolymer), ASA resin (acrylonitrile-styrene-methyl acrylate copolymer); polylactic acid Other resins such as: polysaccharides can be exemplified, and silica, glass and the like can be exemplified as inorganic substances.
- a thermosetting resin such as melamine resin, urea resin, aniline resin, guanamine resin, phenol resin, xylene resin, furan resin
- Resins generally classified as thermoplastic resins such as styrene resin, acrylic resin, acrylonitrile resin, AS resin (acrylonitrile-styrene
- a semiconductor integrated nanoparticle has the structure where the semiconductor nanoparticle as fluorescent substance is included in the parent
- the material constituting the semiconductor nanoparticles is not particularly limited.
- a material containing a group II-VI compound, a group III-V compound, or a group IV element for example, CdSe, CdS, CdTe, ZnSe, ZnS ZnTe, InP, InN, InAs, InGaP, GaP, GaAs, Si, Ge, and the like.
- the semiconductor in the case where the semiconductor is included in the mother body, it may be dispersed within the mother body and may or may not be chemically bonded to the mother body itself.
- the fluorescent dye integrated nanoparticle has a structure in which the fluorescent dye is included in the matrix described above and / or adsorbed on the surface thereof.
- the fluorescent dye is not particularly limited, and examples thereof include rhodamine dye molecules, squarylium dye molecules, cyanine dye molecules, aromatic ring dye molecules, oxazine dye molecules, carbopyronine dye molecules, and pyromesene dye molecules. It can be illustrated.
- Alexa Fluor registered trademark, manufactured by Invitrogen
- BODIPY registered trademark, manufactured by Invitrogen
- Cy registered trademark, manufactured by GE Healthcare
- DY dye molecule registered trademark, manufactured by DYOMICS
- HiLyte registered trademark, manufactured by Anaspec
- dye molecule DyLight (registered trademark, manufactured by Thermo Scientific)
- ATTO registered trademark, manufactured by ATTO-TEC
- MFP registered trademark, manufactured by Mobitec
- the generic name of such a dye molecule is named based on the main structure (skeleton) in the compound or a registered trademark, and the range of fluorescent dyes belonging to each of them must be excessively trial and error by those skilled in the art. It can be grasped appropriately.
- the fluorescent dye when the fluorescent dye is encapsulated in the matrix, the fluorescent dye only needs to be dispersed inside the matrix and may or may not be chemically bonded to the matrix itself.
- Fluorescent substance-integrated nanoparticles can be produced according to a known method (for example, see JP2013-57937A). More specifically, for example, the fluorescent substance-encapsulating silica particles having silica as a base material and encapsulating the fluorescent substance therein are inorganic substances such as inorganic semiconductor nanoparticles, organic fluorescent dyes, and tetraethoxysilane. It can be produced by dropping a solution in which the silica precursor is dissolved into a solution in which ethanol and ammonia are dissolved and hydrolyzing the silica precursor.
- a resin solution or a dispersion of fine particles is prepared in advance.
- it can be prepared by adding a fluorescent substance such as inorganic semiconductor nanoparticles or organic fluorescent dye and stirring the mixture.
- fluorescent substance-containing resin particles can be produced by adding a fluorescent dye to the resin raw material solution and then allowing the polymerization reaction to proceed.
- thermosetting resin such as a melamine resin
- a raw material of the resin (monomer or oligomer or prepolymer, for example, methylol melamine which is a condensate of melamine and formaldehyde)
- an organic fluorescent dye
- the organic fluorescent dye-containing resin particles can be prepared by heating a reaction mixture further containing a surfactant and a polymerization reaction accelerator (such as an acid) and advancing the polymerization reaction by an emulsion polymerization method.
- thermoplastic resin such as a styrene copolymer
- an organic fluorescent dye is bonded in advance as a resin raw material monomer by a covalent bond or the like.
- the reaction mixture containing a polymerization initiator (benzoyl peroxide, azobisisobutyronitrile, etc.) is heated and the polymerization reaction proceeds by radical polymerization or ionic polymerization.
- a polymerization initiator benzoyl peroxide, azobisisobutyronitrile, etc.
- the fluorescent substance to be encapsulated in the fluorescent substance integrated nanoparticles in addition to the above-described semiconductor nanoparticles and fluorescent dyes, for example, Y 2 O 3 , Zn 2 SiO 4 or the like as a base material, Mn 2+ , Eu 3+ or the like And a “long afterglow phosphor” using the above as an activator.
- the average particle size of fluorescent substance-integrated nanoparticles is particularly limited as long as it is suitable for immunostaining (or gene staining) of a pathological specimen. However, considering easiness of detection as a bright spot, it is usually 10 to 500 nm, preferably 50 to 200 nm. The coefficient of variation indicating the variation in particle size is usually 20% or less, preferably 5 to 15%.
- the fluorescent substance integrated nanoparticles satisfying such conditions can be manufactured by adjusting the manufacturing conditions. For example, when producing fluorescent substance-integrated nanoparticles by emulsion polymerization, the particle size can be adjusted by the amount of surfactant to be added. If the amount of the active agent is relatively large, the particle size tends to be small, and if the amount is relatively small, the particle size tends to be large.
- the particle diameter of the fluorescent substance-integrated nanoparticles is obtained by taking an electron micrograph using a scanning electron microscope (SEM), measuring the cross-sectional area of the fluorescent substance-integrated nanoparticles, and assuming that the cross-sectional shape is a circle. Further, it can be calculated as the diameter of a circle corresponding to the cross-sectional area.
- the average particle size and coefficient of variation of a group consisting of a plurality of fluorescent material-integrated nanoparticles are calculated as described above for a sufficient number (for example, 1000) of fluorescent material-integrated nanoparticles, and the average particle size is The coefficient of variation is calculated by the formula: 100 ⁇ standard deviation of particle diameter / average particle diameter.
- fluorescent substance integrated nanoparticles are preferable as described above.
- immunostaining agents use primary antibodies and fluorophores indirectly, that is, antigen-antibody reaction or avidin / biotin reaction, It is preferable to use a complex linked by a bond other than a covalent bond.
- an immunostaining agent in which a probe and fluorescent nanoparticles are indirectly linked, [primary antibody against a target biological substance] ... [antibody against primary antibody (secondary antibody)] to [fluorescent nanoparticles (fluorescent substance integrated nanoparticle) Particle)].
- “...” represents binding by an antigen-antibody reaction, and the mode of binding indicated by “ ⁇ ” is not particularly limited.
- covalent bonding, ionic bonding, hydrogen bonding, coordination bonding, physical Adsorption or chemical adsorption may be mentioned, and a linker molecule may be interposed as necessary.
- a silane coupling agent that is a compound widely used for bonding an inorganic substance and an organic substance can be used.
- This silane coupling agent is a compound having an alkoxysilyl group that gives a silanol group by hydrolysis at one end of the molecule and a functional group such as a carboxyl group, an amino group, an epoxy group, an aldehyde group at the other end, Bonding with an inorganic substance through an oxygen atom of the silanol group.
- a silane coupling agent having a polyethylene glycol chain for example, PEG-silane no.SIM6492.7 manufactured by Gelest
- a known method can be used for the reaction procedure between the nanoparticles encapsulating the fluorescent substance and the silane coupling agent.
- silica nanoparticles containing the obtained fluorescent substance are dispersed in pure water, aminopropyltriethoxysilane is added, and the reaction is performed at room temperature for 12 hours.
- silica nanoparticles encapsulating a fluorescent substance whose surface is modified with an aminopropyl group can be obtained by centrifugation or filtration.
- the antibody can be bound to the silica nanoparticles encapsulating the fluorescent substance via an amide bond.
- a condensing agent such as EDC [1-Ethyl-3- [3-Dimethylaminopropy] carbodiide Hydrochloride; manufactured by Pierce] may be used.
- a linker compound having a site capable of directly binding to a silica nanoparticle encapsulating a fluorescent substance modified with an organic molecule and a site capable of binding to a molecular target substance can be used.
- sulfo-SMCC having both a site selectively reacting with an amino group and a site selectively reacting with a mercapto group [Sulfosuccinimidyl-4- [N-maleimidomethyl] cyclohexane-1-carboxylate; manufactured by Pierce ] Is used to bind the amino group of the silica nanoparticle encapsulating the fluorescent substance modified with aminopropyltriethoxysilane and the mercapto group in the antibody to obtain the silica nanoparticle encapsulating the fluorescent substance bound to the antibody. It is done.
- an immunostaining agent to which a probe and a fluorescent substance are indirectly linked, [primary antibody against target biological substance] ... [antibody against primary antibody (secondary antibody)]-[biotin] / [avidin]-[ Fluorescent substance (fluorescent nanoparticle)] (where “...” indicates binding by an antigen-antibody reaction, and “ ⁇ ” indicates binding by a covalent bond which may be mediated by a linker molecule as necessary. And a complex consisting of three molecules linked together in a manner such that “/” indicates binding by an avidin-biotin reaction).
- the secondary antibody-biotin conjugate uses, for example, a commercially available biotin labeling reagent (kit) based on a known technique that can bind biotin to a desired antibody (protein). Can be produced.
- a biotin-modified secondary antibody itself in which biotin is bound to a desired antibody is commercially available, it may be used.
- a fluorescent nanoparticle-avidin conjugate (avidin-modified phosphor) is also prepared using a commercially available avidin-labeled reagent (kit) based on a known method capable of binding avidin to the phosphor. Can do.
- the avidin in this case may be an improved type such as streptavidin or neutravidin that has a higher binding force with biotin than avidin.
- the fluorescent nanoparticle is a fluorescent substance-integrated nanoparticle based on a resin
- the functional group of the resin and the functional group of avidin (protein) have functional groups at both ends of the molecule as necessary. It can be bound via a linker molecule such as PEG.
- functional groups such as amino groups can be used for melamine resins, and monomers such as acrylic resins and styrene resins can be copolymerized with monomers having functional groups (for example, epoxy groups) in the side chains.
- the functional group itself or a functional group converted from the functional group for example, an amino group generated by reacting aqueous ammonia
- the functional group can be used, and further, the functional group can be used for another functional group.
- Groups can also be introduced.
- a desired functional group can be introduced by surface modification with a silane coupling agent, for example, aminopropyl trioxide. If methoxysilane is used, an amino group can be introduced.
- a thiol group can be introduced, for example, by reacting N-succinimidyl S-acetylthioacetate (SATA) with the amino group of avidin.
- SATA N-succinimidyl S-acetylthioacetate
- a phosphor having a group can be linked to avidin into which a thiol group is introduced.
- the secondary antibody-fluorescent nanoparticle conjugate can be obtained by using, for example, a commercially available fluorescent labeling reagent (kit) based on a known technique capable of binding a desired fluorescent dye to a desired antibody (protein). Can be produced.
- kit fluorescent labeling reagent
- a secondary antibody to fluorescent nanoparticle conjugate per se in which desired fluorescent nanoparticles are bound to a desired antibody is commercially available, it may be used.
- the section is immersed in a container containing xylene to remove paraffin.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, xylene may be exchanged during the immersion.
- the section is immersed in a container containing ethanol to remove xylene.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, ethanol may be exchanged during the immersion.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, water may be exchanged during the immersion.
- the target biological material is activated.
- the activation conditions are not particularly defined, but as the activation liquid, 0.01 M citrate buffer (pH 6.0), 1 mM EDTA solution (pH 8.0), 5% urea, 0.1 M Tris-HCl buffer A liquid etc. can be used.
- As the heating device an autoclave, a microwave, a pressure cooker, a water bath, or the like can be used.
- the temperature is not particularly limited, but can be performed at room temperature. The temperature can be 50 to 130 ° C. and the time can be 5 to 30 minutes.
- the section after the activation treatment is immersed in a container containing PBS and washed.
- the temperature is not particularly limited, but can be performed at room temperature.
- the immersion time is preferably 3 minutes or longer and 30 minutes or shorter. If necessary, PBS may be replaced during the immersion.
- fluorescent nanoparticles having a site that can be directly or indirectly bound to the target biological material are dispersed in the fluorescent nanoparticle dilution liquid of the present invention and placed on a tissue section. , React with the target biological material.
- the fluorescent immunostaining solution used in the immunostaining step or the fluorescent nanoparticle diluent and other components for preparing it are as described above, and may be prepared in advance before this step.
- the immunostaining agent is [primary antibody (probe)] ... [secondary antibody]-[biotin] / [avidin]-[fluorescent substance-containing nanoparticle (phosphor)] (" -- is bound by antigen-antibody reaction. "-" Indicates that they are bound by a covalent bond that may be mediated through a linker molecule as necessary, and "/" indicates that they are bound by an avidin-biotin reaction. ))
- primary antibody solution primary antibody solution
- secondary treatment a treatment of immersing the pathological specimen in the secondary antibody-biotin conjugate solution
- Conditions and conditions for performing the immunostaining process are the temperature and the immersion time when the tissue section as a pathological specimen is immersed in a predetermined solution (reagent). According to a conventional immunostaining method, it can be appropriately adjusted so that an appropriate signal is obtained.
- the temperature is not particularly limited, but can be performed at room temperature.
- the reaction time is preferably 30 minutes or more and 24 hours or less.
- a known blocking agent such as BSA-containing PBS or a surfactant such as Tween 20
- a surfactant such as Tween 20
- the solution for fluorescent immunostaining used in the fluorescent labeling treatment or the fluorescence used for the preparation thereof.
- the pathological specimen that has undergone the immunostaining process is preferably subjected to treatments such as immobilization / dehydration, clearing, and encapsulation so as to be suitable for observation.
- the immobilization / dehydration treatment may be performed by immersing the pathological specimen in a fixation treatment solution (crosslinking agent such as formalin, paraformaldehyde, glutaraldehyde, acetone, ethanol, methanol).
- a fixation treatment solution crosslinking agent such as formalin, paraformaldehyde, glutaraldehyde, acetone, ethanol, methanol.
- a clearing solution xylene or the like.
- the encapsulating process may be performed by immersing the pathological specimen that has undergone the penetration process in the encapsulating liquid. Conditions for performing these treatments, for example, the temperature and immersion time when the pathological specimen is immersed in a predetermined treatment solution, can be appropriately adjusted according to the conventional immunostaining method so as to obtain an appropriate signal. it can.
- a morphological observation staining step for observing the morphology of cells, tissues, organs and the like in a bright field.
- the morphological observation staining step can be performed according to a conventional method. For morphological observation of tissue specimens, staining using eosin, in which cytoplasm, stroma, various fibers, erythrocytes, and keratinocytes are stained from red to dark red, is typically used.
- hematoxylin in which cell nuclei, lime, cartilage tissue, bacteria, and mucus are stained blue-blue to light blue
- HE staining eosin staining
- each pathological specimen is irradiated with excitation light corresponding to the excitation light corresponding to the fluorescent substance labeled with the target biological substance used in the immunostaining process in the same field of view of the microscope at the desired magnification. Then, each immunostained image by fluorescence emitted from these phosphors is observed and photographed.
- the excitation light can be irradiated by using, for example, a laser light source included in the fluorescence microscope and an excitation light optical filter that selectively transmits a predetermined wavelength as required. Imaging of an immunostained image can be performed, for example, with a digital camera provided in a fluorescence microscope.
- an excitation light that contains only the desired fluorescence and causes unwanted fluorescence or noise by using a fluorescent optical filter that selectively transmits a predetermined wavelength as necessary. Further, an immunostained image in which light is excluded can be taken.
- the fluorescent label signal is preferably handled as the number of fluorescent bright spots.
- Examples of software that can be used for image processing include “ImageJ” (open source).
- image processing software By using such image processing software, bright spots with a predetermined wavelength (color) are extracted from the immunostained image and the sum of the brightness is calculated, or the number of bright spots with a predetermined brightness or higher is measured. It is possible to semi-automatically and quickly carry out the processing to be performed, particularly the processing for performing the above-described embodiment.
- the size is constant and can be recognized by microscopic observation.
- a signal whose size is larger than a certain value is determined as an aggregation bright spot. This bright spot and agglomerated bright spot can be quickly and semi-automatically distinguished using software.
- the present invention can also be applied to a method of specifically staining a gene using fluorescent nanoparticles (eg, FISH method). That is, the gene staining method according to the present invention stains a gene using a gene staining solution containing the fluorescent nanoparticle dilution solution described above, a probe, and a fluorescent nanoparticle capable of binding to or coupled to the probe. It is a method to do.
- FISH method fluorescent nanoparticles
- the probe is, for example, a probe of a nucleic acid that specifically binds to a gene that encodes a biomolecule that is specifically expressed in a disease (cancer or the like).
- the probe is a gene that encodes the target biological substance.
- a probe is a nucleic acid molecule having a sequence (probe sequence) including a part or all of a specific region on a chromosome related to a specific disease as described above.
- the nucleic acid includes naturally occurring nucleic acids such as DNA and RNA (mRNA, tRNA, miRNA, siRNA, non-coding-RNA, etc.) and artificial nucleic acids such as PNA and LNA (or BNA: Bridged Nucleic Acid). Therefore, the nucleic acid molecule is not limited as long as it can form a complementary strand with the nucleic acid sequence on the chromosome.
- the nucleic acid molecule may be a natural nucleic acid, an artificial nucleic acid, or a nucleic acid molecule in which a natural nucleic acid and an artificial nucleic acid are linked.
- Probes for the target gene can be prepared according to known methods, and can also be obtained as commercial products.
- the base length, base sequence, and GC content of the probe can be prepared so as to have appropriate stringency in consideration of hybridization conditions (binding of the probe and fluorescent nanoparticles).
- the binding between the probe and the fluorescent nanoparticle is not particularly limited as long as it does not interfere with the gene staining method (eg, FISH), and various bindings can be made.
- the binding between the probe and the fluorescent nanoparticle may be either a method in which the fluorescent nanoparticle is directly bonded to the probe, or a method in which the probe is indirectly bonded through binding between biomolecules.
- Examples of the direct binding method include, for example, introduction of a known thiol group into a hydroxyl group of phosphate bonded to the ribose C5 position at the 5 ′ end of the nucleic acid molecule or a hydroxyl group bonded to the C1 position of the ribose at the 3 ′ end of the nucleic acid molecule.
- Examples include a method in which a thiol group (SH group) is substituted with a reagent, and fluorescent nanoparticles labeled with maleimide are reacted to bond both. It can be suitably performed using a kit of “5 ′ EndTag (application trademark) Nucleic Acid Labeling System” or “3 ′ EndTag DNA Labeling System” manufactured by Vector Laboratories.
- maleimide group-modified fluorescent nanoparticles are reacted with Thiol-11-dUTP solution to obtain fluorescent nanoparticles bound with dUTP, and then incorporated into nucleic acid molecules by nick translation method.
- the phosphor-integrated nanoparticles can be directly bound to the nucleic acid molecule.
- the method of indirectly binding is a method of binding the probe and the fluorescent nanoparticle via specific binding between biomolecules (first and second binding group molecules).
- biomolecules first and second binding group molecules.
- the binding between biomolecules include, for example, an avidin-biotin binding system, a hapten-antihapten, etc., as a combination of the first and second biomolecules.
- a specific base (for example, thymine (T)) of a nucleic acid molecule is a nucleotide (for example, biotin) labeled with a first biomolecule (for example, biotin).
- Biotin-16-dUTP is substituted by nick translation, and a method in which fluorescent nanoparticles having (strept) avidin are bound to biotin of this probe can be exemplified.
- the preparation of fluorescent nanoparticles modified with the second binding group molecule can be performed, for example, as follows.
- a functional group is introduced into the fluorescent nanoparticle and the second binding group molecule by a reagent that introduces a functional group, respectively, and the second biomolecule and the fluorescent nanoparticle are bonded through the bonding between the functional groups.
- a linker may be interposed between the functional groups.
- functional group combinations include NHS ester group-amino group, thiol group-maleimide group combination, and the like.
- the linker include linkers such as EMCS (N- [Epsilon-Maleimidecaproxy] succinimide ester) (manufactured by Thermo Scientific).
- the FISH method may be performed according to standard procedures and processing conditions for each of the various methods described above.
- a specimen slide on which a tissue section is placed may be immersed in one or more types of reagents according to the FISH method under appropriate temperature and time conditions.
- Various reagents necessary for carrying out the FISH according to the present invention can be prepared according to known methods, and can also be obtained as commercial products.
- Specimen pretreatment in the FISH method includes deparaffinization, FISH pretreatment, enzyme (protease) treatment, fixation treatment, and the like.
- the staining process a process based on the FISH method (FISH staining process), that is, a DNA denaturation process, a hybridization process, a post-hybridization process, and the like, and usually a nuclear staining process (for example, by DAPI) are performed.
- FISH staining process a process based on the FISH method
- solvent replacement processing filling processing (encapsulation processing using an encapsulating agent), protection processing, and washing processing and dehydration processing performed before the solvent replacement processing as necessary are performed.
- a fluorescent nanoparticle diluent used for immunostaining using fluorescent nanoparticles contains 1 to 5% (W / W) of a protein having a molecular weight of 40,000 or more, and has a molecular weight of 40,000. If the diluted solution for fluorescent nanoparticles contains 1 to 3% (W / W) of less than 1 protein, the diluted solution for fluorescent nanoparticles of the present invention is used when the fluorescent particles are diluted in fluorescent immunostaining. As a result, non-specific adsorption of fluorescent nanoparticles can be suppressed, background noise seen during detection can be reduced, and accuracy and quantitativeness can be improved in evaluating stained images.
- the protein having a molecular weight of 40,000 or more is BSA, it is preferable from the viewpoint of stabilizing the protein other than BSA particularly when the above dilution is performed.
- the protein having a molecular weight of less than 40,000 is casein, the gap between proteins having a molecular weight of 40,000 or more attached to a non-specifically adsorbed portion (such as a section site around the antigen) is filled (closed). An effect can be acquired suitably.
- Casein has the property that oily components (hydrophobic components) that are insoluble in water are coordinated outside the casein molecules, and the casein molecules tend to gather together, so the casein molecules depend on the shape and size of the gap. Since they gather together to fill the gap, nonspecific adsorption can be suitably suppressed.
- the ratio of ⁇ -casein in the casein is in the range of 0-10% (W / W), or the ratio of ⁇ -casein: ⁇ -casein is 40: 60-60: 40 ( ⁇ -casein And the total amount of ⁇ -casein is 100.)
- the background is further suppressed and more quantitative evaluation is performed. Can do.
- the fluorescent immunostaining kit includes (i) the diluted solution for fluorescent nanoparticles according to any one of (1) to (5) above, and (ii) the immunostaining reagent containing the fluorescent nanoparticles, By simply mixing the fluorescent nanoparticle diluent and the immunostaining reagent before immunostaining and using them for fluorescent immunostaining, the effect of suppressing the non-specific adsorption described above can be obtained relatively simply.
- the container for storing the solution and the solution are sucked and discharged. Since the effect of suppressing nonspecific adsorption of the fluorescent nanoparticles to the inner surface of the pipette tip is obtained, the fluorescent nanoparticles are not unnecessarily wasted before the immunostaining step.
- the immunostaining step includes a primary reaction process for specifically binding a primary antibody to a target biological substance, and a secondary reaction process for specifically binding a secondary antibody bound to biotin to the primary antibody.
- nonspecific adsorption of avidin bound to fluorescent nanoparticles is also suppressed, resulting in an avidin-biotin binding system.
- Fluorescent labeling treatment using can be suitably performed.
- (10) 1 to 3 proteins containing 1 to 5% (W / W) of a protein having a molecular weight of 40,000 or more and having a molecular weight of less than 40,000, which are used when performing the FISH method using fluorescent nanoparticles % (W / W) -containing fluorescent nanoparticle dilution solution suppresses non-specific adsorption of fluorescent nanoparticles even when performing gene detection, and reduces noise and increases the detection signal.
- a gene staining kit comprising (i) the fluorescent nanoparticle diluent according to any one of (10) to (14) above, and (ii) a gene staining reagent containing the fluorescent nanoparticles.
- the effect described in the above (6) can be obtained in gene detection.
- the fluorescent nanoparticle dilution solution according to any one of (10) to (14) and the gene staining solution containing the fluorescent nanoparticle are used, the effect described in (7) above is effective in gene detection. Can also be obtained.
- the gene staining method includes a gene staining step using the gene staining solution, the effect described in (8) above (the same effect can be obtained with respect to an isotope signal in addition to a fluorescent signal).
- the gene staining step A primary reaction process for specifically binding a probe having a first binding group molecule to a gene to be detected;
- the diluted solution for fluorescent nanoparticles described in (10) to (14) above is used to dilute the fluorescent nanoparticles to which the second binding group molecules that specifically bind to the first binding group molecules are bound, and the diluted solution Therefore, if the gene staining method includes a step of sequentially performing a secondary reaction process in which a probe specifically bound to the gene to be detected is fluorescently labeled, the non-specificity of the second binding group molecule bound to the fluorescent nanoparticle Non-specific adsorption of fluorescent nanoparticles due to selective adsorption can be suitably suppressed.
- fluorescent nanoparticles labeled with a second binding group molecule are used, for example, when a fluorescent nanoparticle solution is stored in a tube such as an Eppendorf tube, although there arises a problem that the surface of the fluorescent nanoparticle and the portion of the second binding group molecule (avidin, etc.) are non-specifically bound, the present invention can suppress the non-specific adsorption described above, and the fluorescence Nanoparticles are hard to lose.
- the fluorescent nanoparticles when linked to the probe, the non-specific adsorption of the fluorescent nanoparticles to the tissue section is suppressed. An enhancement of the (fluorescence) signal is made.
- the linker reagent “Maleimide-PEG 2 -Biotin” (Thermo Scientific, product number 21901) was adjusted to 0.4 mM using DMSO. 8.5 ⁇ L of this linker reagent solution was added to the antibody solution, mixed, and reacted at 37 ° C. for 30 minutes to bind biotin to the anti-rabbit IgG antibody via the PEG chain.
- the reaction solution was purified through a desalting column.
- the absorbance at a wavelength of 300 nm was measured using a spectrophotometer (Hitachi “F-7000”) to calculate the concentration of the protein (biotin-modified secondary antibody) in the reaction solution.
- a solution in which the concentration of the biotin-modified secondary antibody was adjusted to 250 ⁇ g / mL using a 50 mM Tris solution was used as a biotin-modified secondary antibody (reagent II) solution.
- the particles subjected to the above surface amination treatment are adjusted to 3 nM using PBS (phosphate buffered saline) containing 2 mM of EDTA (ethylenediaminetetraacetic acid), and the final concentration of this solution is 10 mM.
- SM (PEG) 12 manufactured by Thermo Scientific, succinimidyl-[(N-maleimidopropionamido) -dodecaethyleneglycol] ester
- fluorescent substance-encapsulated melamine nanoparticles having a maleimide group at the end were obtained.
- streptavidin manufactured by Wako Pure Chemical Industries, Ltd.
- SATA N-succinimidyl S-acetylthioacetate
- the above-mentioned fluorescent substance-encapsulated melamine nanoparticles and streptavidin were mixed in PBS containing 2 mM of EDTA and reacted at room temperature for 1 hour. 10 mM mercaptoethanol was added to stop the reaction. After concentrating the obtained solution with a centrifugal filter, unreacted streptavidin and the like were removed using a gel filtration column for purification to produce streptavidin-binding fluorescent substance-encapsulated melamine nanoparticles.
- Nanoparticles having an average particle diameter of 150 nm were prepared.
- E1 Specimen preparation process
- Breast cancer tissue array slide (slide glass with tissue section) (USBiomax br243) was purchased and stained with Ventana benchmark ULTRA using Ventana I-VIEW pathway HER2 (4B5) kit, DAB method Morphologically identified the HER2 3+ region (ie, the cancer cell region) and the stromal cell region.
- the specimen was deparaffinized and washed with water.
- the washed tissue array slide was autoclaved at 121 ° C. for 15 minutes in 10 mM citrate buffer (pH 6.0) to activate the antigen.
- the tissue array slide after the activation treatment was washed with PBS, and the washed tissue array slide was subjected to blocking treatment with PBS containing 1% BSA for 1 hour.
- (E2) Immunostaining step (E2-1) Primary reaction treatment of immunostaining
- the primary reaction treatment for the first immunostaining of the target biological substance HER2 is performed using PBS containing 1 W / W% of BSA.
- a primary reaction treatment solution containing HER2 rabbit monoclonal antibody “4B5” (Ventana) at a concentration of 0.05 nM was prepared.
- the specimen prepared in the step (1) was immersed in this primary reaction treatment solution and reacted at 4 ° C. overnight.
- the specimen was irradiated with excitation light corresponding to the Texas Red dye used for the fluorescent labeling of the target biological material HER2, to emit fluorescence, and an immunostained image in that state was photographed.
- the wavelength of the excitation light was set to 575 to 600 nm using the excitation light optical filter provided in the fluorescence microscope, and the wavelength of the fluorescence to be observed was set to 612 to 692 nm using the fluorescence optical filter.
- the intensity of the excitation light at the time of observation and image photographing with a fluorescence microscope was such that the irradiation energy near the center of the visual field was 900 W / cm 2 .
- the exposure time at the time of image shooting was adjusted within a range in which the luminance of the image was not saturated, and set to, for example, 4000 ⁇ sec.
- Such immunostained images were taken in the same visual field, and then the same operation was repeated while changing the visual field, and a total of 5 visual fields were performed per specimen (first to fifth visual fields).
- the number of the bright spots representing the Texas red dye-encapsulated melamine resin particles fluorescently labeled with HER2 was counted more than a predetermined value.
- Interstitial noise number of bright spots and aggregated bright spots per cell in HER2 3+ region (namely, cancer cell region) were counted. The results are shown in Table 1. Since HER2 is not expressed in the stromal cell region, the bright spot located in the stromal cell is a nonspecific signal, that is, noise. A large number of interstitial noise bright spots means that there are many non-specific reactions, and counting this was used as an evaluation index for immune reactions.
- Example 4 Purchase a lung tissue array slide (slide glass with tissue section) (USBiomax LC241b), and morphologically identify the stromal cell region using anti-PD-L1 rabbit monoclonal antibody “SP142” Spring Bioscience (SBS) The procedure was the same as (E1) except that it was used. Then, instead of the anti-HER2 rabbit monoclonal antibody “4B5”, the anti-PD-L1 rabbit monoclonal antibody “SP142” was used by Spring Bioscience (SBS), and the preparation examples were replaced with streptavidin-modified Texas red dye-encapsulated melamine resin particles.
- Dyeing and evaluation were performed in the same manner as (E2) to (E4) except that the streptavidin-modified quantum dot-encapsulated melamine resin nanoparticles prepared in 5 were used.
- the wavelength of the excitation light to be irradiated is set to 415 to 455 nm using an excitation light optical filter (“QD655-C” manufactured by Optline Co., Ltd.) provided in the fluorescence microscope, and the fluorescence to be observed.
- the wavelength of was set to 648 to 663 nm using a fluorescent optical filter.
- Example 5 The same procedure as in Experimental Example 1 was performed except that the process of identifying the stromal cell region (E1) was omitted. That is, a breast cancer tissue array slide (slide glass carrying a tissue section) (USBiomax br243) was purchased, deparaffinized, and then washed with water. The washed tissue array slide was autoclaved at 121 ° C. for 15 minutes in 10 mM citrate buffer (pH 6.0) to activate the antigen. The tissue array slide after the activation treatment was washed with PBS, and the washed tissue array slide was subjected to blocking treatment with PBS containing 1% BSA for 1 hour.
- the reaction was performed at 15 ° C. for 4 hours, and the reaction was stopped by heating at 70 ° C. for 10 minutes. 25 ⁇ L of distilled water was added to the centrifuge tube after the reaction.
- the biotin-labeled BAC probe reaction solution was purified by a microspin column for nucleic acid purification (“MicroSpin S-200HR Column” manufactured by GE Healthcare, product number “# 27-5120-01”). To this solution, about 5.56 ⁇ L of 3M sodium acetate solution (pH 5.2) and 150 ⁇ L of 100% ethanol were added, and the mixture was allowed to stand at ⁇ 20 ° C. for 1 hour or longer. A precipitate was formed by centrifugation at 16000 rpm for 10 minutes at 4 ° C.
- FISH includes deparaffinization, specimen slide pretreatment, enzyme treatment, specimen fixing treatment, probe preparation, specimen slide DNA denaturation treatment, hybridization treatment, slide glass washing treatment, and DAPI. The staining process was performed in this order.
- Deparaffinization was performed by treating a specimen slide of a HER2-positive stained control specimen (“HER2-FISH Control Slide Code PS-09006” manufactured by Pathology Laboratories) in the order of (1) to (4) below. .
- HER2-FISH Control Slide Code PS-09006 manufactured by Pathology Laboratories
- (1) Immerse in Hemo-De for 10 minutes at room temperature.
- (2) Immerse the specimen slide in new Hemo-De for 10 minutes at room temperature. Repeat the same operation three times.
- the specimen slide is immersed in 100% ethanol at room temperature for 5 minutes, washed twice, and dehydrated.
- the specimen slide is air-dried or dried on a slide warmer at 45 to 50 ° C.
- the specimen slide was pretreated in the following order (1) to (6) to remove proteins from the cell membrane and the nuclear membrane.
- (1) Treat the specimen slide with 0.2 mol / L HCl at room temperature for 20 minutes.
- (2) Immerse the specimen slide in purified water for 3 minutes.
- (3) The specimen slide is immersed in a washing buffer solution (2 ⁇ SSC: standard saline citrate) for 3 minutes.
- (4) The specimen slide is immersed in a pretreatment solution (1N NaSCN) at 80 ° C. for 30 minutes.
- (6) The specimen slide is immersed in a washing buffer solution (2 ⁇ SSC) for 5 minutes, and this immersion operation is repeated twice.
- the sample treatment that had been pretreated was subjected to enzyme treatment by performing the following treatments (1) to (4) in this order.
- (2) The specimen slide is immersed in a protease solution heated to 37 ° C. for 10 to 60 minutes. This immersion treatment is performed with 25 mg protease (2500-3000 Units / mg) [pepsin] / 1M NaCl [pH 2.0] in 50 mL at 37 ° C. for 60 minutes) in order to decompose cell membrane and nuclear membrane proteins, particularly collagen. It is desirable to process.
- (4) The specimen slide is air-dried or dried on a slide warmer at 45 to 50 ° C. for 2 to 5 minutes.
- specimen fixation As the specimen fixing process, the following processes (137) to (3) were performed on the specimen slide that had been pretreated. (1) The specimen slide is immersed in 10% neutral buffered formalin (“4% paraformaldehyde / phosphate buffer” manufactured by Wako Pure Chemical Industries, Ltd., product number 163-1004) at room temperature for 10 minutes. (2) Immerse the specimen slide in the washing buffer for 5 minutes. Repeat the same operation twice. (3) The specimen slide is air-dried or dried on a slide warmer at 45 to 50 ° C. for 2 to 5 minutes.
- the streptavidin-binding fluorescent substance-encapsulated melamine nanoparticles of Preparation Example 3 were bound to the DNA probe bound to the HER2 gene and labeled with biotin as follows.
- the particles prepared in Preparation Example 3 were diluted with a diluent to make 0.02 nM, dropped onto a 100 ⁇ L sample slide, and a binding reaction was performed at room temperature for 60 minutes. It was immersed in PBS for 5 minutes and washed 3 times.
- DAPI staining was performed as follows. First, 10 ⁇ L of DAPI counterstain was added to the hybridization area of the specimen slide. Next, after hybridization treatment, DAPI staining (2 ⁇ g / mL PBS) is performed at 25 ° C. for 10 minutes in order to count the number of cells. The cell nucleus is stained, covered with a cover glass, and the specimen slide is covered until signal measurement. Stored protected from light. DAPI (4 ′, 6-Diamidino-2-phenylindole, Dihydrochloride) used Molecular Probes (D1306).
- ⁇ Fluorescence microscope observation> In the fluorescence microscope observation, the section subjected to FISH as described above is subjected to fluorescence microscope observation (600 times) using a fluorescence microscope Zeiss imager (Camera: MRm monochrome / with cooling function, objective lens ⁇ 60 oil immersion). The fluorescence image (fluorescence still image) and the number of bright spots were measured.
- a mixed solution of 4% and BSA 1 to 5% was used, the number of bright spots (the number of bright spots per cell in the HER2 3+ region (ie, cancer cell region)) doubled.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Pathology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Food Science & Technology (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biophysics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Nanotechnology (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Optics & Photonics (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
本発明者らは、これまでも蛍光ナノ粒子を用いた定量的な免疫染色法(さらには、遺伝子染色法)における精度をあげるため、特許文献1に記載されているように、ブロッキング工程でタンパク質を切片に添加する事などの鋭意工夫を行ってきた。しかし、蛍光ナノ粒子の希釈液の組成については、特に検討してはこなかった。例えば、特許文献2に記載されているように、これまでは、抗体希釈液として広く用いられている1%BSA含有PBSで蛍光ナノ粒子を希釈してきた。
- 蛍光ナノ粒子用希釈液 -
本発明の蛍光ナノ粒子用希釈液は、少なくとも、高分子量(Mw≧40,000)のタンパク質を1~5%(W/W)と低分子量(Mw<40,000)のタンパク質を1~3%(W/W)含有しており、この溶液を用いて蛍光ナノ粒子を分散させることで、免疫染色(または遺伝子染色)を行うための蛍光免疫染色用溶液(または遺伝子染色用溶液)を調製することができる。以下、本発明について、より詳細に説明する。
本発明の蛍光ナノ粒子用希釈液は、少なくとも、高分子量(Mw≧40,000)のタンパク質を1~5%(W/W)および/または低分子量(Mw<40,000)のタンパク質を1~3%(W/W)含有している。
さらに本発明においては、蛍光ナノ粒子を用いた免疫染色(または遺伝子染色)を行うためのキットが規定される。このキットは少なくとも、目的生体物質用の蛍光ナノ粒子を含む免疫染色用試薬(または遺伝子染色試薬)および上記蛍光ナノ粒子用希釈液を含む。このキットは更に、必要に応じて、目的生体物質を免疫染色するためのその他の必要な試薬、部材等や、本発明に係る蛍光免疫染色(または遺伝子染色)を実施するための説明書などを含んでいてもよい。
また、本発明においては、目的生体物質用の免疫染色用試薬を上記蛍光ナノ粒子用希釈液で希釈した蛍光免疫染色用溶液(または遺伝子染色用溶液)が提供される。免疫染色用試薬(または遺伝子染色用試薬)の希釈倍率については、目的生体物質と免疫染色用試薬とのアフィニティーに応じて最適化することができる。
本発明における目的生体物質は、組織切片に発現している生体物質、特にタンパク質(抗原)(や遺伝子)であって、主に病理診断の観点からの定量ないし検出のために、蛍光標識体を用いた免疫染色の対象とするものを指す。
一次抗体には、前述したような目的生体物質としてのタンパク質を抗原として特異的に認識して結合する抗体(IgG)を用いることができる。たとえば、EGFR(発現タンパク質)を目的生体物質とする場合は抗EGFR抗体を、HER2を目的生体物質とする場合は抗HER2抗体を用いることができる。
本発明において用いられる蛍光ナノ粒子としては、目的生体物質を1分子ずつ輝点として表すのに十分な強度の蛍光を発することができる「蛍光物質集積ナノ粒子」が好ましい。
本発明における蛍光物質集積ナノ粒子は、有機物または無機物でできた粒子を母体とし、複数の蛍光物質(たとえば蛍光色素)がその中に内包されているおよび/またはその表面に吸着している構造を有する、ナノサイズの粒子である。この場合、母体(たとえば樹脂)と蛍光物質は、互いに反対の電荷を有する置換基ないし部位を有しており、静電的相互作用が働くものであることが好適である。
半導体集積ナノ粒子とは、蛍光体としての半導体ナノ粒子が、上記に記載した母体の中に内包されているおよび/またはその表面に吸着している構造を有する。半導体ナノ粒子を構成する素材は特に限定されるものではなく、たとえば、II-VI族化合物、III-V族化合物、またはIV族元素を含有するもの、たとえば、CdSe、CdS、CdTe、ZnSe、ZnS、ZnTe、InP、InN、InAs、InGaP、GaP、GaAs、Si、Geなどが挙げられる。また、半導体が母体に内包されている場合、母体内部に分散されていればよく、母体自体と化学的に結合していても、していなくてもよい。
蛍光色素集積ナノ粒子とは、蛍光色素が、上記に記載した母体の中に内包されている、および/またはその表面に吸着している構造を有する。蛍光色素は特に限定されるものではなく、たとえば、ローダミン系色素分子、スクアリリウム系色素分子、シアニン系色素分子、芳香環系色素分子、オキサジン系色素分子、カルボピロニン系色素分子、ピロメセン系色素分子などを例示することができる。あるいは、Alexa Fluor(登録商標、インビトロジェン社製)系色素分子、BODIPY(登録商標、インビトロジェン社製)系色素分子、Cy(登録商標、GEヘルスケア社製)系色素分子、DY系色素分子(登録商標、DYOMICS社製)、HiLyte(登録商標、アナスペック社製)系色素分子、DyLight(登録商標、サーモサイエンティフィック社製)系色素分子、ATTO(登録商標、ATTO-TEC社製)系色素分子、MFP(登録商標、Mobitec社製)系色素分子などを用いることができる。なお、このような色素分子の総称は、化合物中の主要な構造(骨格)または登録商標に基づき命名されており、それぞれに属する蛍光色素の範囲は当業者であれば過度の試行錯誤を要することなく適切に把握できるものである。また、蛍光色素が母体に内包されている場合、蛍光色素は母体内部に分散されていればよく、母体自体と化学的に結合していても、していなくてもよい。
目的生体物質を蛍光標識するための免疫染色剤に含まれる蛍光ナノ粒子としては、前述したように「蛍光物質集積ナノ粒子」が好ましい。免疫染色剤は、蛍光標識の効率を向上させて蛍光の劣化につながる時間経過をなるべく抑えるために、一次抗体および蛍光体が間接的に、つまり抗原抗体反応やアビジン・ビオチン反応などを利用した、共有結合以外の結合によって連結される複合体を用いることが好ましい。
(免疫染色方法)
以下、本発明で用いる染色方法の一例について説明する。この染色方法が適用できる組織切片(本明細書において、単に「切片」ともいい、例えば病理切片等の切片も包含する用語として用いる。)の作製法は特に限定されず、公知の手順により作製されたものを用いることができる。
(1-1.脱パラフィン処理)
キシレンを入れた容器に、切片を浸漬させ、パラフィン除去する。温度は特に限定されるものではないが、室温で行うことができる。浸漬時間は、3分以上30分以下であることが好ましい。また必要により浸漬途中でキシレンを交換してもよい。
公知の方法に倣い、目的とする生体物質の賦活化処理を行う。賦活化条件に特に定めはないが、賦活液としては、0.01Mのクエン酸緩衝液(pH6.0)、1mMのEDTA溶液(pH8.0)、5%尿素、0.1Mのトリス塩酸緩衝液などを用いることができる。加熱機器はオートクレーブ、マイクロウェーブ、圧力鍋、ウォーターバスなどを用いることができる。温度は特に限定されるものではないが、室温で行うことができる。温度は50~130℃、時間は5~30分で行うことができる。
免疫染色工程では、生体物質を染色するために、目的生体物質に直接的または間接的に結合しうる部位を有する蛍光ナノ粒子を本発明における蛍光ナノ粒子用希釈液に分散させ、組織切片に載せ、目的とする生体物質との反応を行う。免疫染色工程に用いる蛍光免疫染色用溶液ないしそれを調製するための蛍光ナノ粒子用希釈液およびその他の成分については前述した通りであり、この工程の前にあらかじめ調製しておけばよい。
免疫染色工程を終えた病理標本は、観察に適したものとなるよう、固定化・脱水、透徹、封入などの処理を行うことが好ましい。
本発明では、もしも必要であれば、明視野において細胞、組織、臓器等の形態を観察することができるようにするための、形態観察染色工程を含めることができる。形態観察染色工程は、常法に従って行うことができる。組織標本の形態観察に関しては、細胞質・間質・各種線維・赤血球・角化細胞が赤~濃赤色に染色される、エオジンを用いた染色が標準的に用いられている。また、細胞核・石灰部・軟骨組織・細菌・粘液が青藍色~淡青色に染色される、ヘマトキシリンを用いた染色も標準的に用いられている(これら2つの染色を同時に行う方法はヘマトキシリン・エオジン染色(HE染色)として知られている)。形態観察染色工程を含める場合は、免疫染色工程の後に行うようにしてもよいし、免疫染色工程の前に行うようにしてもよい。
(5-1.観察・撮影)
観察・撮影工程では、所望の倍率における顕微鏡の同一視野において、免疫染色工程に用いられた目的生体物質を蛍光標識している蛍光体に対応した励起光に対応した励起光それぞれを病理標本に照射し、それらの蛍光体から発せられた蛍光による免疫染色像それぞれを観察・撮影する。これらの励起光の照射は、たとえば、蛍光顕微鏡が備えるレーザー光源と、必要に応じて所定の波長を選択的に透過させる励起光用光学フィルターを用いることで照射することができる。免疫染色像の撮影は、たとえば、蛍光顕微鏡が備えるデジタルカメラによって行うことができる。免疫染色像の撮影の際には、必要に応じて所定の波長を選択的に透過させる蛍光用光学フィルターを用いることで、目的とする蛍光のみを含み、目的としない蛍光やノイズとなる励起光およびその他の光を排除した免疫染色像を撮影することができる。
画像処理・計測工程では、目的生体物質に関して撮影された免疫染色像について、画像処理に基づき、目的生体物質に対応する蛍光標識シグナルに対応する蛍光標識シグナルを計測し、細胞膜の領域内にある前記目的生体物質に対応する蛍光標識シグナルを特定する。
上述した免疫染色法以外にも蛍光ナノ粒子を用いて遺伝子を特異的に染色する方法(例;FISH法)にも、本発明を適用することができる。すなわち、本発明に係る遺伝子染色法は、前述した蛍光ナノ粒子用希釈液と、プローブと、該プローブに結合可能または結合した蛍光ナノ粒子と、を含有する遺伝子染色用溶液を用いて遺伝子を染色する方法である。
プローブは、例えば、疾病(癌等)で特異的に発現する生体分子をコードした遺伝子と特異的に結合する核酸のプローブであり、例えば、前述した目的生体物質をコードする遺伝子である。
目的遺伝子に対するプローブは、公知の方法にしたがって作製することが可能であり、市販品として入手することもできる。プローブの塩基長、塩基配列、GC含量は、ハイブリダイゼーションさせる際の条件を考慮し、適切なストリンジェンシーを有するものとなるよう調製することができる
(プローブと蛍光ナノ粒子との結合)
プローブと蛍光ナノ粒子との結合は、遺伝子染色法(例;FISH)に支障がなければ特に制限なく、様々な結合とすることができる。このプローブと蛍光ナノ粒子の結合は、プローブに蛍光ナノ粒子を直接結合する方法、または生体分子同士の結合を介して間接的に結合する方法のいずれでもよい。
直接結合する方法の例としては、例えば、核酸分子の5’末端のリボースC5位に結合したリン酸の水酸基または核酸分子の3’末端のリボースのC1位に結合した水酸基を公知のチオール基導入試薬によりチオール基(SH基)に置換し、マレイミドで標識された蛍光ナノ粒子を反応させて、両者を結合する方法が挙げられる。ベクターラボラトリーズ社製「5'EndTag(出願商標)Nucleic Acid Labeling System」や「3’ EndTag DNA Labeling System」のキットを用いて好適に行うことができる。
間接的に結合する方法は、生体分子(第1,2の結合基分子)同士の特異的な結合を介してプローブと蛍光ナノ粒子とを結合させる方法である。生体分子同士の結合として、例えば第1,2の生体分子の組合せとして、アビジンービオチンの結合系、ハプテン-抗ハプテン等が挙げられる。
遺伝子染色を行う場合、(i)プローブと蛍光ナノ粒子とを染色する前に結合させてコンジュゲートを作成し、当該コンジュゲートを用いて検出対象の遺伝子を染色する方法(直接法)、または、(ii)第1結合基分子(ビオチン等)で修飾されたプローブを検出対象の遺伝子(HER2遺伝子等)にハイブリダイズ(特異的に結合)させた後に(1次反応処理後に)、第2結合基分子(アビジン等)で修飾された蛍光ナノ粒子を当該プローブに特異的に動的に結合させる(2次反応処理する)方法(間接法)がある。
FISH法は、上述したような各種の手法のそれぞれにとって標準的な手順および処理条件に従って行えばよい。一般的には、組織切片を載置した検体スライドをFISH法に応じた1種類または2種類以上の試薬に、適切な温度および時間条件の下、浸漬すればよい。本発明に係るFISHの実施に必要な各種の試薬は、公知の方法にしたがって作製することが可能であり、市販品として入手することもできる。
(1)蛍光ナノ粒子を用いた免疫染色を行う際に使用する蛍光ナノ粒子用希釈液が、分子量40,000以上のタンパク質を1~5%(W/W)含有し、かつ分子量40,000未満のタンパク質を1~3%(W/W)含有している蛍光ナノ粒子用希釈液であれば、蛍光免疫染色において、蛍光粒子を希釈する際に本発明の蛍光ナノ粒子用希釈液を用いることで、蛍光ナノ粒子の非特異的吸着が抑制され、検出時にみられるバックグラウンドノイズを低減させることが可能となり、染色画像の評価するにあたって、精度と定量性の向上を図ることができる。
(2)前記分子量40,000以上のタンパク質がBSAであれば、特に上記希釈をした際にBSA以外のタンパク質を安定化する効果を奏する観点から好ましい。
(3)前記分子量40,000未満のタンパク質がカゼインであれば、タンパク質が非特異的に吸着する部分(抗原周辺の切片部位等)に付着した分子量40000以上のタンパク質同士の隙間を埋める(塞ぐ)効果を好適に得ることができる。カゼインは、水に溶けない油性成分(疎水性成分hydrophobic)がカゼイン分子の外側に配位しており、カゼイン分子同士が集まりやすい性質があるため、上記隙間の形状・大きさに応じてカゼイン分子同士が集まって隙間を埋めるため、非特異的吸着を好適に抑制することができる。
(6) (i)上記(1)~(5)のいずれかの蛍光ナノ粒子用希釈液、および(ii)蛍光ナノ粒子を含有する免疫染色用試薬を含む蛍光免疫染色用キットであれば、免疫染色前に上記蛍光ナノ粒子用希釈液と免疫染色用試薬を混合して蛍光免疫染色に用いるだけで、上述した非特異的吸着を抑制する効果を比較的簡便に得ることができる。
(7) (1)~(5)のいずれかに記載の蛍光ナノ粒子用希釈液および蛍光ナノ粒子を含有する蛍光免疫染色用溶液であれば、該溶液を貯留する容器や該溶液を吸排するピペットチップの内表面に対する蛍光ナノ粒子の非特異的吸着を抑制する効果が得られるので、免疫染色工程の前に蛍光ナノ粒子が不必要に浪費されることがない。
(9) 上記免疫染色工程が、目的生体物質に対して一次抗体を特異的に結合させる1次反応処理、一次抗体に対し、ビオチンが結合した二次抗体を特異的に結合させる2次反応処理、請求項1~5に記載された蛍光ナノ粒子用希釈液により、アビジンが結合した蛍光ナノ粒子を希釈し、該希釈後の溶液でもって2次抗体を蛍光標識する処理、を順に行う工程を含む蛍光免疫染色法であれば、蛍光ナノ粒子の非特異的吸着が抑制されること以外にも、蛍光ナノ粒子に結合したアビジンの非特異的吸着も抑制される結果、アビジンービオチンの結合系を利用した蛍光標識処理を好適に行うことができる。
(10)蛍光ナノ粒子を用いたFISH法を行う際に使用する、分子量40,000以上のタンパク質を1~5%(W/W)含有し、かつ分子量40,000未満のタンパク質を1~3%(W/W)含有している蛍光ナノ粒子用希釈液であれば、遺伝子検出を行う場合にも蛍光ナノ粒子の非特異的吸着を抑制して、ノイズ低減や検出シグナルが強まる。
(12)前記分子量40,000未満のタンパク質がカゼインであれば、上記(3)で説明した効果が遺伝子検出においても得られる。
(13,14)前記カゼイン内のκ-カゼイン含有比率が10%(W/W)以下であれば、あるいは、α-カゼイン:β-カゼインの比率が40:60~60:40(α-カゼインおよびβ-カゼインの合計量を100とする。)であれば、上記(4,5)で説明した効果が遺伝子検出においても得られる。
(16)上記(10)~(14)のいずれかに記載の蛍光ナノ粒子用希釈液および蛍光ナノ粒子を含有する遺伝子染色用溶液であれば、上記(7)で説明した効果が遺伝子検出においても得られる。
(17)前記遺伝子染色用溶液を使用した遺伝子染色工程を含む遺伝子染色法であれば、上記(8)で説明した効果(蛍光シグナル以外にもアイソトープのシグナルについても同様の効果)が得られる。
(18)上記遺伝子染色工程が、
検出対象の遺伝子に対して、第1結合基分子を有するプローブを特異的に結合させる1次反応処理、
上記(10)~(14)に記載された蛍光ナノ粒子用希釈液により、第1結合基分子と特異的に結合する第2結合基分子が結合した蛍光ナノ粒子を希釈し、希釈された溶液でもって、検出対象の遺伝子に特異的に結合したプローブを蛍光標識する2次反応処理、を順に行う工程を含む遺伝子染色法であれば、蛍光ナノ粒子に結合した第2結合基分子の非特異的吸着に起因した蛍光ナノ粒子の非特異的吸着を好適に抑制することができる。
50mMTris溶液に、2次抗体として用いる抗ウサギIgG抗体50μgを溶解した。この溶液に、最終濃度3mMとなるようにDTT(ジチオトレイトール)溶液を添加、混合し、37℃で30分間反応させた。その後、反応溶液を脱塩カラム「Zeba Desalt Spin Columns」(サーモサイエンティフィック社、Cat.#89882)に通して、DTTで還元化した2次抗体を精製した。精製した抗体全量のうち200μLを50mMTris溶液に溶解して抗体溶液を調製した。その一方で、リンカー試薬「Maleimide-PEG2-Biotin」(サーモサイエンティフィック社、製品番号21901)を、DMSOを用いて0.4mMとなるように調整した。このリンカー試薬溶液8.5μLを前記抗体溶液に添加、混合し、37℃で30分間反応させることにより、抗ウサギIgG抗体にPEG鎖を介してビオチンを結合させた。この反応溶液を脱塩カラムに通して精製した。脱塩した反応溶液について、波長300nmにおける吸光度を分光高度計(日立製「F-7000」)を用いて測定することにより、反応溶液中のタンパク質(ビオチン修飾2次抗体)の濃度を算出した。50mMTris溶液を用いて、ビオチン修飾2次抗体の濃度を250μg/mLに調整した溶液を、ビオチン修飾2次抗体(試薬II)の溶液とした。
テキサスレッド色素分子「Sulforhodamine 101」(シグマアルドリッチ社)2.5mgを純水22.5mLに溶解した後、ホットスターラーにより溶液の温度を70℃に維持ながら20分間撹拌した。撹拌後の溶液に、メラミン樹脂「ニカラックMX-035」(日本カーバイド工業株式会社)1.5gを加え、さらに同一条件で5分間加熱撹拌した。撹拌後の溶液にギ酸100μLを加え、溶液の温度を60℃に維持しながら20分間攪拌した後、その溶液を放置して室温まで冷却した。冷却した後の溶液を複数の遠心用チューブに分注して、12,000rpmで20分間遠心分離して、溶液に混合物として含まれるテキサスレッド色素内包メラミン樹脂ナノ粒子を沈殿させた。上澄みを除去し、沈殿した粒子をエタノールおよび水で洗浄した。得られたナノ粒子の1000個についてSEM観察を行い、上述のように平均粒子径を測定したところ、平均粒子径152nmであった。
作製例2で得られた粒子0.1mgをEtOH1.5mL中に分散し、アミンプロピルトリメトキシシランLS-3150(信越化学工業社製)2μLを加えて8時間反応させて表面アミノ化処理を行なった。
アルゴン気流下、トリ-n-オクチルホスフィンオキシド7.5gに、ステアリン酸2.9g、n-テトラデシルホスホン酸620mg、および、酸化カドミウム250mgを加え、370℃に加熱混合した。これを270℃まで放冷した後、トリブチルホスフィン2.5mLにセレン200mgを溶解させた溶液を加え、減圧乾燥し、トリ-n-オクチルホスフィンオキシドで被覆されたカドミウムセレニド(CdSe)コア半導体ナノ粒子を得た。
[作製例5]ストレプトアビジン修飾量子ドット内包メラミン樹脂ナノ粒子の作製
作製例3と同様にして、作製例4で得られたナノ粒子0.1mgから、ストレプトアビジン修飾量子ドット内包メラミン樹脂ナノ粒子を得た。
(E1)標本作製工程
乳がん組織アレイスライド(組織切片を搭載したスライドグラス)を(USBiomax社 br243)を購入し、ベンタナI-VIEWパスウェーHER2(4B5)キットを用い、ベンタナベンチマークULTRAで染色、DAB法によりHER2 3+領域(すなわち癌細胞領域)と間質細胞領域を形態学的に同定した。
(E2-1)免疫染色の1次反応処理
目的生体物質HER2に係る第1免疫染色用の1次反応処理は、BSAを1W/W%含有するPBSを用いて、抗HER2ウサギモノクローナル抗体「4B5」(ベンタナ社)を0.05nMの濃度で含有する1次反応処理液を調製した。この1次反応処理液に工程(1)で作製した標本を浸漬し、4℃で1晩反応させた。
作製例1で作製したビオチン修飾抗ウサギIgG抗体の溶液を、さらにBSAを1W/W%含有するPBSを用いて6μg/mLに希釈した2次反応処理液を調製した。1次反応処理を終えた標本をPBSで洗浄した後、この2次反応処理液に浸漬し、室温で30分間反応させた。
作製例3で作製したストレプトアビジン修飾テキサスレッド色素内包メラミン樹脂粒子を、表1に示すように、カゼイン(組成=α‐カゼイン(シグマ社 c6780):50W/W%、β‐カゼイン(シグマ社 c6905):50W/W%)とBSAの含有率がそれぞれ異なった蛍光ナノ粒子用希釈液を用いて、0.02nMに希釈した蛍光標識反応処理液をそれぞれ調製した。2次反応処理を終えた標本をこの蛍光標識処理液に浸漬し、中性のpH環境下(pH6.9~7.4)、室温で3時間反応させた。
免疫染色を終えた標本に対して、純エタノールに5分間浸漬する操作を4回行う固定化・脱水処理を行った。続いて、キシレンに5分間浸漬する操作を4回行う透徹処理を行った。最後に、標本に封入剤「エンテランニュー」(メルク社)を載せて、カバーガラスを被せる封入処理を行い、観察に用いる標本とした。
(E4-1)観察・撮影工程
この工程における励起光の照射および蛍光の発光の観察には蛍光顕微鏡「BX-53」(オリンパス株式会社)を用い、免疫染色像(400倍)の撮影には、当該蛍光顕微鏡に取り付けた顕微鏡用デジタルカメラ「DP73」(オリンパス株式会社)を用いた。
この工程における画像処理には、画像処理ソフトウェア「ImageJ」(オープンソース)を用いた。
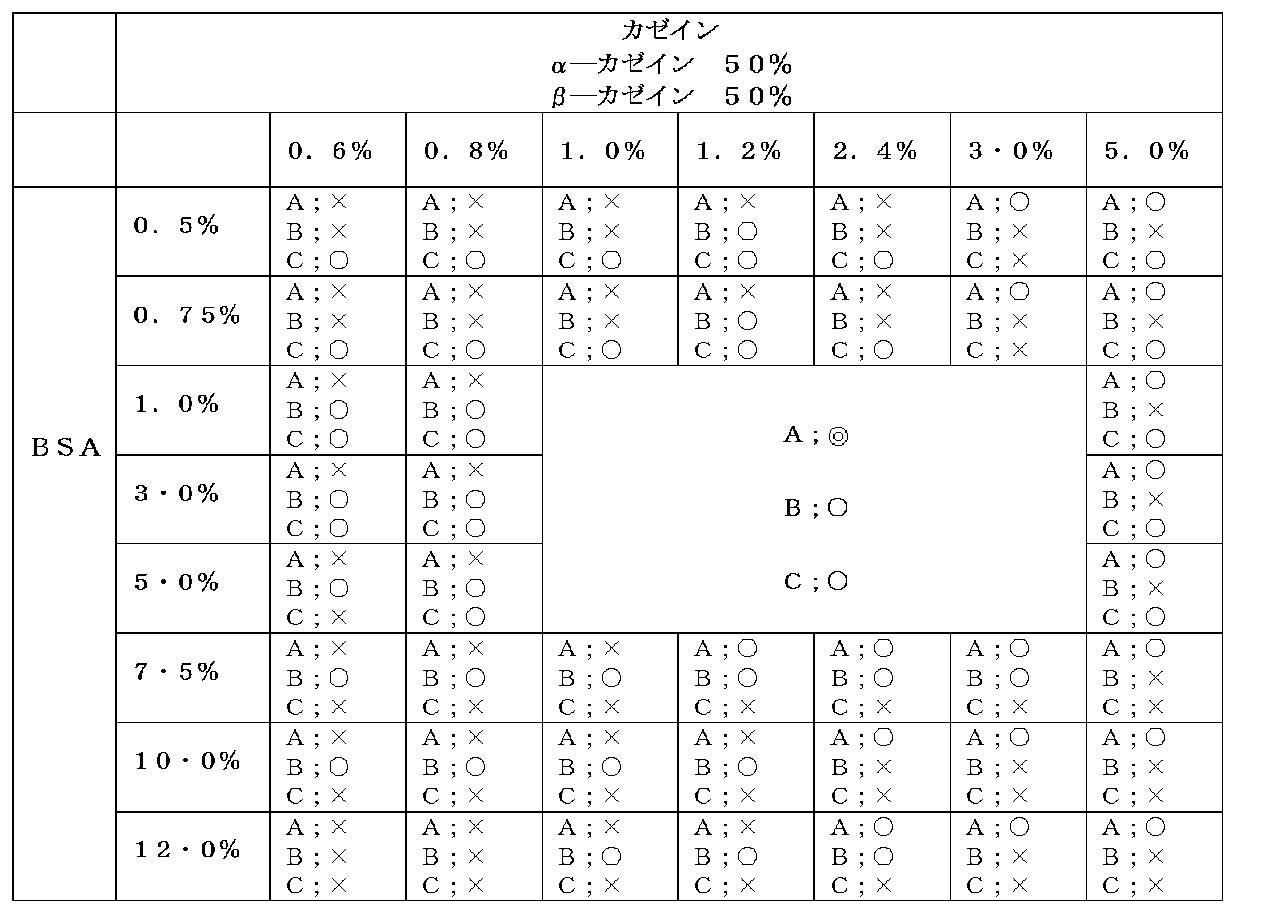
A=間質ノイズ(輝点数(以下同じ))
500以上=×
500未満300以上=○
300未満200以上=◎
200未満=☆
B=1細胞あたりの輝点数
10未満=×
10以上=○
C=凝集輝点
3個以上=×
3個以下=○
[実験例2]
(E2-3)免疫染色の蛍光標識処理で用いた蛍光ナノ粒子を分散させる溶液に含有されるカゼインの組成を変更した以外は、実施例1と同様の手順で標本作製工程、免疫染色工程および評価工程を行い、間質ノイズ、1細胞あたりの輝点数および凝集輝点数をカウントした。結果を表2~4に示す。なお、天然カゼインにおける各カゼインの含有比率は、α-カゼイン50%、β-カゼイン35%、κ-カゼイン13%、γ-カゼイン2%である。
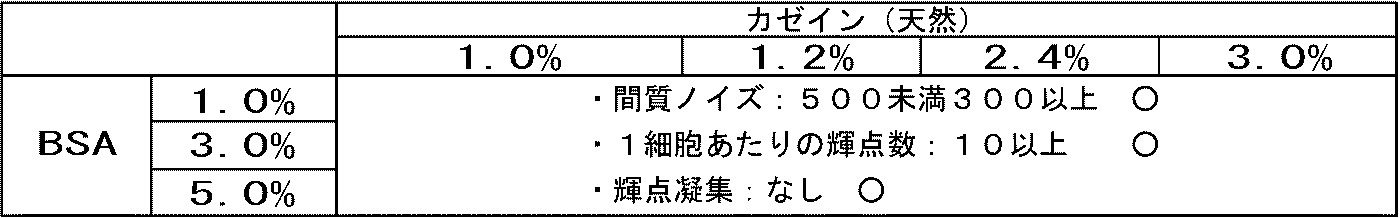



[実験例3]
間質領域の同定は(E1)と同様に、ベンタナI-VIEWパスウェーHER2(4B5)キットを用いて実施した。その後、抗HER2ウサギモノクローナル抗体「4B5」に代えて抗HER3ウサギモノクローナル抗体「SP71」Abnova社を用いたこと以外は、(E2)~(E4)同様に染色および評価を実施した。
その結果、実験例2と同様の結果を得た。
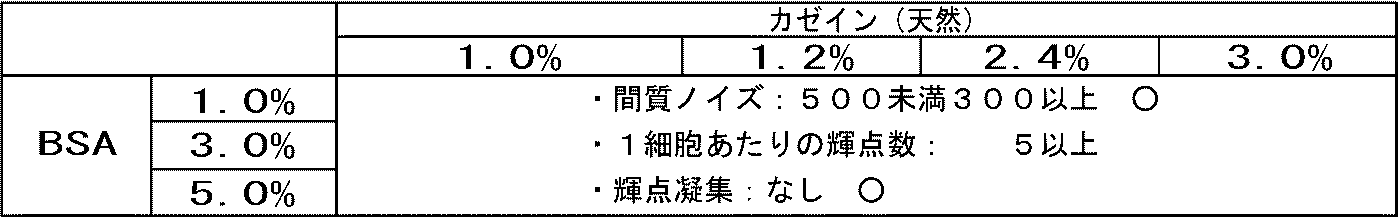



肺組織アレイスライド(組織切片を搭載したスライドグラス)を(USBiomax社 LC241b)購入し、間質細胞領域の形態学的な同定を抗PD-L1ウサギモノクローナル抗体「SP142」 Spring Bioscience(SBS)社を用いたこと以外は(E1)と同様に、実施した。その後、抗HER2ウサギモノクローナル抗体「4B5」に代えて抗PD-L1ウサギモノクローナル抗体「SP142」 Spring Bioscience(SBS)社を用いたこと、およびストレプトアビジン修飾テキサスレッド色素内包メラミン樹脂粒子に代えて作製例5で作成したストレプトアビジン修飾量子ドット内包メラミン樹脂ナノ粒子を用いた以外は、(E2)~(E4)同様に染色および評価を実施した。なお、(E)においては照射する励起光の波長は、蛍光顕微鏡が備える励起光用光学フィルター((株)オプトライン製「QD655-C」)を用いて415~455nmに設定し、観察する蛍光の波長は、蛍光用光学フィルターを用いて648~663nmに設定した。
その結果、実験例2と同様の結果を得た。
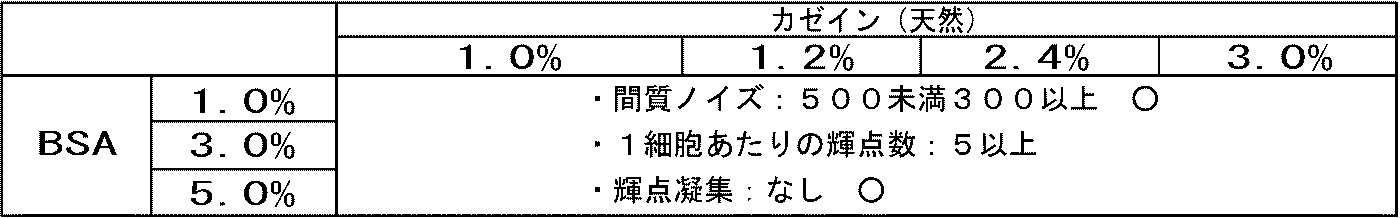



間質細胞領域を同定する(E1)のプロセスを除いたこと以外は、実験例1と同様に実施した。すなわち、乳がん組織アレイスライド(組織切片を搭載したスライドグラス)を(USBiomax社 br243)を購入し、脱パラフィン処理した後、水に置換する洗浄を行った。洗浄した組織アレイスライドを10mMクエン酸緩衝液中(pH6.0)中で121℃、15分間オートクレーブ処理することで、抗原の賦活化処理を行った。賦活化処理後の組織アレイスライドをPBSにより洗浄し、洗浄した組織アレイスライドに対してBSAを1%含有するPBSを用いて1時間ブロッキング処理を行った。
CellBiochemBiophys.2006;45(1)59の記載の方法に従って、以下のように核酸分子を調製した。GSP社から購入したHER2-DNAクローン(約150kbp)1μg(5μL)に対して、ニックトランスレーション用のキット(製品名「GSP-ニックトランスレーションキット」K-015、GSP社製)のプロトコールに従い、以下のようにニックトランスレーション法により、HER2のDNAクローン(核酸分子)のdTTPをビオチン標識dUTPで置換した。
FISHによりHER2遺伝子のコピー数を測定した。FISHは以下に示すように、脱パラフィン処理、検体スライドの前処理、酵素処理、検体の固定処理、プローブの準備、検体スライドのDNAの変性処理、ハイブリダイゼーション処理、スライドグラスの洗浄処理、およびDAPI染色処理をこの順で行うことで実施した。
HER2陽性染色対照標本の検体スライド(パソロジー研究所社製「HER2-FISHコントロールスライドCode PS-09006」)を、以下の(1)~(4)の順で処理することで脱パラフィン処理を行った。(1)ヘモディー(Hemo-De)に常温で10分間浸漬する。(2)検体スライドを新しいHemo-Deに常温10分間浸漬する。同じ操作を3回繰り返す。(3)検体スライドを100%エタノールで常温で5分間浸漬し、2回洗浄し、脱水処理を行う。(4)検体スライドを風乾または45~50℃のスライドウォーマー上で乾燥させる。
DNAプローブの到達性を向上させるために、上記検体スライドに対し以下の(1)~(6)の順で前処理を行い、細胞膜及び核膜の蛋白質の除去を行った。(1)検体スライドを0.2mоl/L HClで室温、20分間処理する。(2)検体スライドを精製水に3分間浸漬する。(3)検体スライドを洗浄緩衝液(2xSSC:standard sailine citrate)に3分間浸漬する。(4)検体スライドを80℃の前処理溶液(1N NaSCN)に30分間浸漬する。(5)検体スライドを精製水に1分間浸漬する。(6)検体スライドを洗浄緩衝液(2xSSC)に5分間浸漬し、この浸漬操作を2回繰り返す。
前処理を行った検体スライドに対して、以下の(1)~(4)の処理をこの順で行うことで酵素処理を行った。(1)前処理した検体スライドを取り出し、ペーパータオルにスライドグラスの下端をつけて余分な洗浄緩衝液を取り除く。(2)検体スライドを37℃に加温したプロテアーゼ溶液に10~60分間浸漬する。この浸漬処理は、細胞膜及び核膜のタンパク質、特にコラーゲンの分解をするために、25mg プロテアーゼ(2500-3000Units/mg)[ペプシン]/1M NaCl[pH2.0]50mLで37℃、60分間)で処理することが望ましい。(3)検体スライドを洗浄緩衝液に5分間浸漬する。この操作を2回繰り返す。(4)検体スライドを風乾または45~50℃のスライドウォーマー上で2~5分間乾燥させる。
検体の固定処理として、前処理を行った検体スライドに対して以下の(137)~(3)の処理を行った。(1)検体スライドを10%中性緩衝ホルマリン(和光純薬社製「4%パラホルムアルデヒド・リン酸緩衝液」、製品番号163-20145)に常温で10分間浸漬する。(2)検体スライドを洗浄緩衝液に5分間浸漬する。これと同じ操作を2回繰り返す。(3)検体スライドを風乾または45~50℃のスライドウォーマー上で2~5分間乾燥
させる。
冷凍保存しておいた作成例6で作成したDNAプローブの溶液を室温に戻し、正確な容量を採液可能なピペッティング操作ができる程度まで溶液の粘度を十分にさげて、ボルテックスミキサー等で溶液を混和した。
検体スライド上のDNAの変性処理として、検体の固定処理を行った検体スライドに対して以下の(1)~(8)の処理を行った。(1)検体スライドの作成前に水で湿らせたペーパータオルを底に敷いた湿潤箱(気密性の容器であり、その側面をペーパータオルでテーピングしたもの)を37℃インキュベータ内に載置して予備加熱する。(2)変性溶液(70% ホルムアミド/SSC[150mM NaCl、15mMクエン酸ナトリウム])のpHが常温でpH7.0~8.0であることを確かめる。変性溶液をコプリンジャーに入れ、溶液が72℃±1℃になるまで温浴槽で加温する(72±1℃の温浴槽に少なくとも30分間置く)。(3)ハイブリダイゼーション領域がどの部分か分かるように、検体スライドの裏側に領域を囲むようにダイアモンドペン等でマークする。(4)検体スライドを72±1℃の変性溶液の入ったコプリンジャー中に浸漬し、検体スライドのDNAを変性する。
(5)ピンセットを使って、検体スライドを変性溶液から取り出し、すぐに常温の70%エタノール中に入れる。ホルムアミドを除くためにスライドを振盪する。検体スライドを1分間浸漬する。(6)検体スライドを70%エタノールから取り出し、85%エタノール中に入れ、ホルムアミドを除くためにスライドを振盪する。検体を1分間浸漬する。100%エタノールで同じ操作を2回繰り返す。(7)ペーパータオルに検体スライドグラスの下端をつけてエタノールを取り除き、ペーパータオルでスライドグラスの裏側を拭く。(8)検体スライドをドライヤーで風乾または45~50℃のスライドウォーマーで2~5分間乾燥させる。
上記変性処理を行った検体スライドに対して以下の(1)~(3)の処理をこの順で行うことで、検体スライドに対して上述したように調製したDNAプローブ10μL(10~50ng)を用いてハイブリダイゼーション処理を行った。(1)検体スライドのハイブリダイゼーション領域に調製した上記DNAプローブを10μL添加し、すぐに、22mm×22mmのカバーグラスをプローブの上に被せ均一にプローブを広げる。ハイブリダイゼーション領域に気泡が入らないようにする。(2)ペーパーボンドでカバーグラスをシールする。(3)前もって加温した湿潤箱に検体スライドを入れて蓋をして37℃のインキュベータで14~18時間ハイブリダイゼーションを行う。
上記ハイブリダイゼーション処理を行った検体スライドに対して以下の(1)~(6)の処理をこの順で行うことで、検体スライドの洗浄処理を行った。(1)ポストハイブリダイゼーション洗浄緩衝液(2×SSC/0.3%NP-40)をコプリンジャーに入れる。ポストハイブリダイゼーション洗浄緩衝液が72℃±1℃になるまで温浴槽で予備加熱をする(72℃±1℃の温浴槽に少なくとも30分間置く)。(2)ポストハイブリダイゼーション洗浄緩衝液を入れたコプリンジャーをもうひとつ用意し、常温に維持する。(3)ピンセットでペーパーボンドのシールを取り除く。(4)検体スライドをポストハイブリダイゼーション洗浄緩衝液の中に入れる。カバーグラスは自然に溶液中で剥がれるのを待つ。(5)溶液から検体スライドを取り出し、余分な溶液を取り去り、72±1℃に加温したポストハイブリダイゼーション洗浄緩衝液に2分間浸す。ここで、73℃を超える温度や処理時間として2分を超えないようにするのが望ましい。(6)コプリンジャーから検体スライドを取り出し、遮光下(締め切った引出や締め切ったキャビネットの棚等)で風乾する。
HER2遺伝子に結合しビオチンで標識されたDNAプローブに対して、作製例3のストレプトアビジン結合蛍光物質内包メラミンナノ粒子を以下の通りに結合させた。
DAPI染色は以下のように行った。まず、10μLのDAPI対比染色液を検体スライドのハイブリダイゼーション領域に添加した。次に、ハイブリダイゼーション処理した後、細胞数をカウントするためにDAPI染色(2μg/mLPBS)を25℃、10分間行うことで細胞核を染色し、カバーガラスを被せて、シグナルの計測まで検体スライドを遮光して保存した。DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) はMolecular Probes社(D1306)を使用した。
上述のようにFISHを行った検体スライドを以下のように観察した。
蛍光顕微鏡観察は、上述のようにFISHを行った切片を、蛍光顕微鏡Zeiss imager(Camera:MRmモノクロ・冷却機能付、対物レンズ×60油浸)を用いて、蛍光顕微鏡観察(600倍)を行い、蛍光画像(蛍光静止画像)および輝点数の計測を行った。
Claims (18)
- 蛍光ナノ粒子を用いた免疫染色を行う際に使用する、分子量40,000以上のタンパク質を1~5%(W/W)含有し、かつ分子量40,000未満のタンパク質を1~3%(W/W)含有している蛍光ナノ粒子用希釈液。
- 前記分子量40,000以上のタンパク質がBSAである、請求項1に記載の蛍光ナノ粒子用希釈液。
- 前記分子量40,000未満のタンパク質がカゼインである、請求項1または2に記載の蛍光ナノ粒子用希釈液。
- 前記カゼイン内のκ-カゼイン比率が0~10%(W/W)である、請求項3に記載の蛍光ナノ粒子用希釈液。
- 前記カゼインが含有するα-カゼイン:β-カゼインの比率が40:60~60:40(α-カゼインおよびβ-カゼインの合計量を100とする。)である、請求項3または4に記載の蛍光ナノ粒子用希釈液。
- (i)請求項1~5のいずれか一項に記載の蛍光ナノ粒子用希釈液、および(ii)蛍光ナノ粒子を含有する免疫染色用試薬を含む、蛍光免疫染色用キット。
- 請求項1~5のいずれか一項に記載の蛍光ナノ粒子用希釈液および蛍光ナノ粒子を含有する蛍光免疫染色用溶液。
- 請求項7の蛍光免疫染色用溶液を使用した免疫染色工程を含む蛍光免疫染色法。
- 上記免疫染色工程が、
目的生体物質に対して一次抗体を特異的に結合させる1次反応処理、
一次抗体に対し、ビオチンが結合した二次抗体を特異的に結合させる2次反応処理、
請求項1~5に記載された蛍光ナノ粒子用希釈液により、アビジンが結合した蛍光ナノ粒子を希釈し、該希釈後の溶液でもって2次抗体を蛍光標識する処理、
を順に行う工程を含む、請求項8に記載の蛍光免疫染色法。 - 蛍光ナノ粒子を用いた遺伝子染色法を行う際に使用する、分子量40,000以上のタンパク質を1~5%(W/W)含有し、かつ分子量40,000未満のタンパク質を1~3%(W/W)含有している蛍光ナノ粒子用希釈液。
- 前記分子量40,000以上のタンパク質がBSAである、請求項10に記載の蛍光ナノ粒子用希釈液。
- 前記分子量40,000未満のタンパク質がカゼインである、請求項10または11に記載の蛍光ナノ粒子用希釈液。
- 前記カゼイン内のκ-カゼイン含有比率が10%(W/W)以下である、請求項12に記載の蛍光ナノ粒子用希釈液。
- 前記カゼインが含有するα-カゼイン:β-カゼインの比率が40:60~60:40(α-カゼインおよびβ-カゼインの合計量を100とする。)である、請求項12または13に記載の蛍光ナノ粒子用希釈液。
- (i)請求項10~14のいずれか一項に記載の蛍光ナノ粒子用希釈液、および(ii)蛍光ナノ粒子を含有する遺伝子染色用試薬を含む、遺伝子染色用キット。
- 請求項10~14のいずれか一項に記載の蛍光ナノ粒子用希釈液と、プローブと、該プローブに結合可能または結合した蛍光ナノ粒子と、を含有する遺伝子染色用溶液。
- 請求項16の遺伝子染色用溶液を使用した遺伝子染色工程を含む遺伝子染色法。
- 上記遺伝子染色工程が、
検出対象の遺伝子に対して、第1結合基分子を有するプローブを特異的に結合させる1次反応処理、
請求項10~14に記載された蛍光ナノ粒子用希釈液により、第1結合基分子と特異的に結合する第2結合基分子が結合した蛍光ナノ粒子を希釈し、希釈された溶液でもって、検出対象の遺伝子に特異的に結合したプローブを蛍光標識する2次反応処理、
を順に行う工程を含む、請求項17に記載の遺伝子染色法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20164877.1A EP3690443A1 (en) | 2014-12-12 | 2015-12-09 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
| EP15868287.2A EP3232195B1 (en) | 2014-12-12 | 2015-12-09 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
| US15/534,732 US20170342470A1 (en) | 2014-12-12 | 2015-12-09 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
| JP2016563709A JP6575533B2 (ja) | 2014-12-12 | 2015-12-09 | 蛍光ナノ粒子用希釈液、これを用いた蛍光免疫染色用キット、蛍光免疫染色用溶液、および蛍光免疫染色法、遺伝子染色法 |
| US16/778,698 US20200165666A1 (en) | 2014-12-12 | 2020-01-31 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-251979 | 2014-12-12 | ||
| JP2014251979 | 2014-12-12 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/534,732 A-371-Of-International US20170342470A1 (en) | 2014-12-12 | 2015-12-09 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
| US16/778,698 Division US20200165666A1 (en) | 2014-12-12 | 2020-01-31 | Diluent for fluorescent nano particles, kit for immunofluorescent staining which utilizes same, solution for immunofluorescent staining, immunofluorescent staining method, and gene staining method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016093268A1 true WO2016093268A1 (ja) | 2016-06-16 |
Family
ID=56107446
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/084500 Ceased WO2016093268A1 (ja) | 2014-12-12 | 2015-12-09 | 蛍光ナノ粒子用希釈液、これを用いた蛍光免疫染色用キット、蛍光免疫染色用溶液、および蛍光免疫染色法、遺伝子染色法 |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US20170342470A1 (ja) |
| EP (2) | EP3690443A1 (ja) |
| JP (2) | JP6575533B2 (ja) |
| WO (1) | WO2016093268A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6231709B1 (ja) * | 2016-05-31 | 2017-11-15 | シスメックス株式会社 | 蛍光画像分析装置および分析方法 |
| CN111830000A (zh) * | 2020-07-23 | 2020-10-27 | 南开大学 | 一种对纳米颗粒加载探针作用于斑马鱼胚胎/幼鱼的方法 |
| CN119291191A (zh) * | 2024-12-11 | 2025-01-10 | 迈杰转化医学研究(苏州)有限公司 | Pd-l1免疫组化检测质控品的制备方法及产品 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08320323A (ja) * | 1995-05-26 | 1996-12-03 | Yuka Medeiasu:Kk | サイトメガロウイルス抗原検査法 |
| JPH09196922A (ja) * | 1996-01-19 | 1997-07-31 | Shinotesuto:Kk | 複数の亜型が存在する抗原の共通抗原決定基に特異的な抗体の測定方法及び測定試薬 |
| JP2002202312A (ja) * | 2001-11-28 | 2002-07-19 | Tokuyama Corp | 診断用キット |
| JP2006053031A (ja) * | 2004-08-11 | 2006-02-23 | Kagoshima Univ | 免疫組織化学的染色による抗原の検出方法 |
| WO2012029342A1 (ja) * | 2010-08-30 | 2012-03-08 | コニカミノルタエムジー株式会社 | 組織染色方法、組織評価方法および生体物質検出方法 |
| JP2012194013A (ja) * | 2011-03-16 | 2012-10-11 | Konica Minolta Medical & Graphic Inc | 免疫組織化学染色方法及び反応試薬 |
| JP2013088296A (ja) * | 2011-10-19 | 2013-05-13 | Konica Minolta Medical & Graphic Inc | 組織評価方法 |
| JP2014066720A (ja) * | 2007-11-07 | 2014-04-17 | Toshiba Corp | 光導波路型センサチップ、光導波路型センサチップの製造方法、物質の測定方法、物質測定用キットおよび光導波路型センサ |
| JP2014142348A (ja) * | 2011-09-09 | 2014-08-07 | Konica Minolta Inc | 生体物質検出用の蛍光標識体 |
| WO2014136885A1 (ja) * | 2013-03-08 | 2014-09-12 | コニカミノルタ株式会社 | 組織染色用染色剤、組織染色用染色剤の製造方法および組織染色用染色剤を含む組織染色用キット |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4353793B2 (ja) * | 2001-06-26 | 2009-10-28 | アボット・ラボラトリーズ | Hcv抗原とhcv抗体との同時検出のための方法 |
| US7101683B2 (en) * | 2001-06-26 | 2006-09-05 | Abbott Laboratories | Methods for the simultaneous detection of HCV antigens and HCV antibodies |
| CA2477991C (en) * | 2003-08-20 | 2014-04-08 | Seikagaku Corporation | Stabilizing agent and blocking agent |
| US20050214882A1 (en) * | 2004-03-25 | 2005-09-29 | Ez Bio Inc. | Reagents, methods and kits for the universal rapid immuno-detection |
| US20090124024A1 (en) * | 2007-11-07 | 2009-05-14 | Shingo Kasai | Optical-waveguide sensor chip, method of manufacturing the same, method of measuring substance, substance-measuring kit and optical-waveguide sensor |
| KR101140029B1 (ko) * | 2010-02-23 | 2012-06-21 | 한국식품연구원 | 항원고정화 면역형광 슬라이드의 제조방법 및 그에 의해 제조되는 면역형광 슬라이드 |
| US8494401B2 (en) | 2011-09-07 | 2013-07-23 | Xerox Corporation | Active ozone scrubber |
| JP5906623B2 (ja) | 2011-09-09 | 2016-04-20 | コニカミノルタ株式会社 | 生体物質発現レベル評価システム |
| US20130224770A1 (en) * | 2012-02-29 | 2013-08-29 | General Electric Company | Antibody Diluent Buffer |
-
2015
- 2015-12-09 EP EP20164877.1A patent/EP3690443A1/en not_active Withdrawn
- 2015-12-09 EP EP15868287.2A patent/EP3232195B1/en active Active
- 2015-12-09 US US15/534,732 patent/US20170342470A1/en not_active Abandoned
- 2015-12-09 JP JP2016563709A patent/JP6575533B2/ja active Active
- 2015-12-09 WO PCT/JP2015/084500 patent/WO2016093268A1/ja not_active Ceased
-
2019
- 2019-07-04 JP JP2019125162A patent/JP6725045B2/ja active Active
-
2020
- 2020-01-31 US US16/778,698 patent/US20200165666A1/en not_active Abandoned
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08320323A (ja) * | 1995-05-26 | 1996-12-03 | Yuka Medeiasu:Kk | サイトメガロウイルス抗原検査法 |
| JPH09196922A (ja) * | 1996-01-19 | 1997-07-31 | Shinotesuto:Kk | 複数の亜型が存在する抗原の共通抗原決定基に特異的な抗体の測定方法及び測定試薬 |
| JP2002202312A (ja) * | 2001-11-28 | 2002-07-19 | Tokuyama Corp | 診断用キット |
| JP2006053031A (ja) * | 2004-08-11 | 2006-02-23 | Kagoshima Univ | 免疫組織化学的染色による抗原の検出方法 |
| JP2014066720A (ja) * | 2007-11-07 | 2014-04-17 | Toshiba Corp | 光導波路型センサチップ、光導波路型センサチップの製造方法、物質の測定方法、物質測定用キットおよび光導波路型センサ |
| WO2012029342A1 (ja) * | 2010-08-30 | 2012-03-08 | コニカミノルタエムジー株式会社 | 組織染色方法、組織評価方法および生体物質検出方法 |
| JP2012194013A (ja) * | 2011-03-16 | 2012-10-11 | Konica Minolta Medical & Graphic Inc | 免疫組織化学染色方法及び反応試薬 |
| JP2014142348A (ja) * | 2011-09-09 | 2014-08-07 | Konica Minolta Inc | 生体物質検出用の蛍光標識体 |
| JP2013088296A (ja) * | 2011-10-19 | 2013-05-13 | Konica Minolta Medical & Graphic Inc | 組織評価方法 |
| WO2014136885A1 (ja) * | 2013-03-08 | 2014-09-12 | コニカミノルタ株式会社 | 組織染色用染色剤、組織染色用染色剤の製造方法および組織染色用染色剤を含む組織染色用キット |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6725045B2 (ja) | 2020-07-15 |
| JP2019194617A (ja) | 2019-11-07 |
| US20200165666A1 (en) | 2020-05-28 |
| JP6575533B2 (ja) | 2019-09-18 |
| EP3232195A1 (en) | 2017-10-18 |
| EP3232195A4 (en) | 2018-05-30 |
| US20170342470A1 (en) | 2017-11-30 |
| EP3232195B1 (en) | 2020-04-29 |
| EP3690443A1 (en) | 2020-08-05 |
| JPWO2016093268A1 (ja) | 2017-09-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5843054B1 (ja) | 多重免疫染色法に基づく生体物質の定量方法 | |
| JP6614161B2 (ja) | 蛍光観察に使用する蛍光体集積ナノ粒子 | |
| JP6129330B2 (ja) | 蛍光体集積ナノ粒子標識剤、およびこれを用いた蛍光免疫染色法 | |
| WO2016129444A1 (ja) | 抗体結合蛍光体集積ナノ粒子、抗体結合蛍光体集積ナノ粒子の製造方法および免疫染色キット | |
| JP6504159B2 (ja) | 蛍光体集積ナノ粒子、これを用いた染色試薬、キットおよび蛍光免疫染色法 | |
| JP6725045B2 (ja) | 蛍光ナノ粒子用希釈液、これを用いた蛍光免疫染色用キット、蛍光免疫染色用溶液、および蛍光免疫染色法、遺伝子染色法 | |
| JPWO2015045961A1 (ja) | 蛍光ナノ粒子標識体、多重免疫染色剤キットおよび多重免疫染色法 | |
| JP6658330B2 (ja) | 組織切片から蛍光ナノ粒子の解離を防止する方法 | |
| WO2016072341A1 (ja) | 免疫染色法、およびこれに用いられる免疫染色試薬キット | |
| WO2017175523A1 (ja) | 蛍光免疫染色法 | |
| EP3734277A1 (en) | Method for assessing medicine | |
| JP6443581B2 (ja) | がんまたは免疫系が関係する疾患の診断または治療のための情報取得方法 | |
| EP3608669A1 (en) | Fluorescent premix particles, fluorescent stain containing same, and fluorescent staining method in which these are used | |
| JP6583011B2 (ja) | 酸性水溶液を用いた免疫染色スライドの洗浄方法 | |
| JP7001083B2 (ja) | 蛍光観察に使用する蛍光体集積ナノ粒子 | |
| WO2020148985A1 (ja) | 標的物質検出用蛍光体集積ナノ粒子 | |
| JP2020016568A (ja) | 蛍光標識体、組織染色方法、蛍光標識体の製造方法及び蛍光標識体の安定化方法 | |
| JP6398419B2 (ja) | ソラフェニブを含有する標識剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15868287 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016563709 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015868287 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15534732 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |