WO2016104144A1 - 空気入りタイヤ - Google Patents
空気入りタイヤ Download PDFInfo
- Publication number
- WO2016104144A1 WO2016104144A1 PCT/JP2015/084476 JP2015084476W WO2016104144A1 WO 2016104144 A1 WO2016104144 A1 WO 2016104144A1 JP 2015084476 W JP2015084476 W JP 2015084476W WO 2016104144 A1 WO2016104144 A1 WO 2016104144A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mass
- resin
- rubber
- parts
- less
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
- B60C1/0016—Compositions of the tread
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C19/00—Chemical modification of rubber
- C08C19/22—Incorporating nitrogen atoms into the molecule
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C19/00—Chemical modification of rubber
- C08C19/25—Incorporating silicon atoms into the molecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C19/00—Chemical modification of rubber
- C08C19/30—Addition of a reagent which reacts with a hetero atom or a group containing hetero atoms of the macromolecule
- C08C19/42—Addition of a reagent which reacts with a hetero atom or a group containing hetero atoms of the macromolecule reacting with metals or metal-containing groups
- C08C19/44—Addition of a reagent which reacts with a hetero atom or a group containing hetero atoms of the macromolecule reacting with metals or metal-containing groups of polymers containing metal atoms exclusively at one or both ends of the skeleton
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L15/00—Compositions of rubber derivatives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/04—Homopolymers or copolymers of styrene
- C08L25/08—Copolymers of styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L47/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
- C08L9/06—Copolymers with styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2227—Oxides; Hydroxides of metals of aluminium
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/006—Additives being defined by their surface area
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/32—Properties characterising the ingredient of the composition containing low molecular weight liquid component
- C08L2207/324—Liquid component is low molecular weight polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/16—Homopolymers or copolymers of alkyl-substituted styrenes
Definitions
- the present invention relates to a pneumatic tire having a tread produced using a rubber composition.
- Tire treads are required to have excellent grip performance on both wet and dry road surfaces.
- ⁇ -methylstyrene resins The method etc. which mix
- a method of blending a terpene resin such as polyterpene, terpene phenol resin, and terpene aromatic resin is also known.
- tan ⁇ at 20 ° C. it is necessary to improve tan ⁇ at 20 ° C., but in that case, tan ⁇ at 30 to 70 ° C., which is an index of rolling resistance, also increases, which is not desirable.
- tan ⁇ at 30 to 70 ° C. which is an index of rolling resistance
- a method using colesin, a coumarone indene resin, a styrene acrylic resin, or the like is known.
- the rolling resistance also increases at the same time, so that it is difficult to use in applications where low fuel consumption is important.
- resins used for improving grip performance are not only rubber's viscoelastic properties, hysteresis loss, especially the effect of increasing tan ⁇ , but also bloom on the tread surface while the resin is running, and process oil and polymer
- a resin bloom layer in combination with decomposed products, low molecular weight organic substances, etc., it also has the effect of increasing the adhesive force between the road surface and the tread, and it is thought that these together contribute to the improvement of grip performance .
- An object of the present invention is to solve the above-mentioned problems and to provide a pneumatic tire having a tread produced using a rubber composition capable of improving wet grip performance, dry grip performance, and durability in a highly balanced manner. To do.
- the present invention is a pneumatic tire having a tread produced using a rubber composition containing 90% by mass or more of a diene rubber in 100% by mass of a rubber component, wherein the rubber composition further comprises a terpene aromatic resin A hydrogenated terpene aromatic resin obtained by hydrogenating a double bond of which the hydrogenation rate of the double bond is 5 to 100%, and the hydroxyl value of the hydrogenated terpene aromatic resin is 20 mgKOH /
- the hydrogenated terpene aromatic resin content is 1 to 50 parts by mass with respect to 100 parts by mass of the diene rubber
- the rubber composition further comprises a compound represented by the following formula: consists of at least one selected from the group consisting of magnesium sulfate, and silicon carbide, a nitrogen adsorption specific surface area contains inorganic filler is 10 ⁇ 120m 2 / g, the diethyl Relative system rubber 100 parts by weight, the content of the inorganic filler is a pneumatic tire is 1 to 70 parts by mass.
- M is at least one metal selected from the group consisting of Al, Mg, Ti, Ca and Zr, an oxide or hydroxide of the metal, m is an integer of 1 to 5, and x is (An integer from 0 to 10, y is an integer from 2 to 5, and z is an integer from 0 to 10.)
- the inorganic filler is preferably aluminum hydroxide.
- the softening point of the hydrogenated terpene aromatic resin is preferably 80 to 180 ° C, and more preferably 114 to 160 ° C.
- the hydroxyl value of the hydrogenated terpene aromatic resin is preferably 0 mgKOH / g.
- the diene rubber preferably contains 60% by mass or more of styrene butadiene rubber having a styrene content of 19 to 60% by mass.
- the present invention it is obtained by hydrogenating a rubber component containing 90% by mass or more of a diene rubber and a terpene aromatic resin, and the hydrogenation rate of the double bond is 5 to 100%.
- a hydrogenated terpene aromatic resin having a hydroxyl value of 20 mgKOH / g or less and a predetermined inorganic filler having a specific nitrogen adsorption specific surface area, and the hydrogenated terpene aroma per 100 parts by mass of the diene rubber
- a pneumatic tire with improved dry grip performance and durability in a well-balanced manner can be provided.
- the pneumatic tire of the present invention is obtained by hydrogenating a double bond between a rubber component containing 90% by mass or more of a diene rubber and a terpene aromatic resin, and the hydrogenation rate of the double bond is 5 to 100%.
- the hydrogenated terpene aromatic resin having a hydrogenation rate of 5 to 100% and a hydroxyl value of 20 mgKOH / g or less has the following characteristics, the above-mentioned effects are remarkably exhibited. Inferred. Since the hydrogenated terpene aromatic resin has high flexibility of the resin structure by hydrogenation, the bloom on the tread surface is quick for the molecular weight and the softening point. Moreover, since double bonds are reduced by hydrogenation, the dispersibility of the resin in the diene rubber is greatly increased, and the crosslinking of the rubber is promoted without adsorbing sulfur of the crosslinking agent. To increase the modulus of the rubber composition after vulcanization.
- the rubber cross-linking becomes uniform and tight, durability and blowability are also improved.
- the hydroxyl value is 20 mgKOH / g or less, the self-aggregation property in the rubber is low, so the Shore hardness (Hs) at normal temperature is low and the high temperature Hs is maintained. That is, there is little dependence on Hs temperature.
- the above effect is particularly remarkable when a diene rubber is used as the rubber component.
- hydrogenation of the resin generally improves the thermal stability and can extend the storage period, so when hydrogenated terpene aromatic resin is blended with rubber, the progress of thermal decomposition and oxidation can be suppressed and odor can be suppressed. Can be suppressed.
- an inorganic filler such as aluminum hydroxide having a specific nitrogen adsorption specific surface area
- the following effects (1) to (4) are exhibited, and the above-mentioned effects (especially wet grip performance) (Improvement effect) is expected to be remarkably exhibited.
- Inorganic fillers such as blended aluminum hydroxide (Al (OH) 3 ) are partly converted into alumina (Al 2 O 3 ) having a Mohs hardness equal to or higher than that of silica during kneading, aluminum hydroxide, etc.
- Al (OH) 3 blended aluminum hydroxide
- Al 2 O 3 alumina
- Mohs hardness equal to or higher than that of silica during kneading, aluminum hydroxide, etc.
- the metal oxide lump and the inorganic filler are micro unevenness on the road surface aggregate ( It is considered that an anchor effect is exhibited at a pitch of several tens of ⁇ m, thereby improving wet grip performance.
- the silicon dioxide on the road surface and the inorganic filler such as aluminum hydroxide on the tire surface come into contact with each other (rubbing) during driving, an instantaneous covalent bond is formed and the wet grip performance is improved. It is done.
- the inorganic filler particles vibrate at high frequency during traveling, and bloom of adhesive components such as grip resins and liquid components in the adjacent rubber composition due to the high frequency vibrations As a result, the amount of the adhesive component around the inorganic filler particles is increased and wet grip performance is improved as compared with other portions not containing the inorganic filler.
- the rubber composition in the present invention is further improved in dry grip performance by adding an inorganic filler such as aluminum hydroxide having a specific nitrogen adsorption specific surface area.
- an inorganic filler such as aluminum hydroxide having a specific nitrogen adsorption specific surface area.
- the grip resin or adhesive component blooms at the interface between the inorganic filler and the rubber component, and the road surface grip is promoted.
- the inorganic filler is the surrounding silica and Bonds with carbon black physically or chemically, and no large voids are generated around the inorganic filler during running.
- Hysteresis of rubber composition is increased by fine inorganic filler with specific nitrogen adsorption specific surface area. This is considered to contribute to the improvement of dry grip performance.
- the wet grip performance can be improved even with the effects obtained by adding inorganic fillers such as conventional aluminum hydroxide, the wear resistance, the ablation appearance after wear, etc. are usually deteriorated. difficult.
- an inorganic filler such as aluminum hydroxide having a specific nitrogen adsorption specific surface area is added together with a specific hydrogenated terpene aromatic resin, so that wet grip performance, dry grip performance and durability are balanced at a high level. It is possible to improve well.
- the rubber composition in the present invention contains a diene rubber as a rubber component.
- a diene rubber as a rubber component, good grip performance can be obtained.
- wet grip performance and dry grip performance are collectively shown.
- the diene rubber is not particularly limited, and isoprene-based rubber such as natural rubber (NR), high-purity NR (UPNR), deproteinized NR (DPNR), epoxidized NR (ENR), isoprene rubber (IR), Examples thereof include styrene butadiene rubber (SBR), butadiene rubber (BR), styrene isoprene butadiene rubber (SIBR), chloroprene rubber (CR), acrylonitrile butadiene rubber (NBR), and the like.
- NR natural rubber
- UPNR high-purity NR
- DPNR deproteinized NR
- ENR epoxidized NR
- IR isoprene rubber
- SBR styrene butadiene rubber
- BR butadiene rubber
- SIBR styrene isoprene butadiene rubber
- CR acrylonitrile butadiene rubber
- SBR is essential from the viewpoint of grip performance
- NR, SBR, and BR are more preferable
- SBR and BR are more preferable
- SBR and BR are used in combination.
- the tire for trucks and buses is mainly composed of NR having a high road surface contact pressure per unit area and excellent in tensile strength and tear strength.
- the styrene butadiene rubber is not particularly limited, and examples thereof include emulsion polymerization SBR (E-SBR), solution polymerization SBR (S-SBR), and the like, whether oil-extended or not oil-extended. Good. Of these, oil-extended and high-molecular weight SBR is preferable from the viewpoint of grip performance.
- modified SBR such as terminal-modified S-SBR and main chain-modified S-SBR with increased interaction force with the filler can also be suitably used. These SBRs may be used alone or in combination of two or more.
- modified SBR those coupled with tin or silicon are preferably used.
- a coupling method of the modified SBR for example, an alkali metal (such as Li) or an alkaline earth metal (such as Mg) at the molecular chain terminal of the modified SBR is reacted with tin halide or silicon halide according to a conventional method. The method etc. are mentioned.
- a copolymer of styrene and butadiene having a primary amino group or an alkoxysilyl group is also preferable.
- the primary amino group may be bonded to any of the polymerization initiation terminal, the polymerization termination terminal, the polymer main chain, and the side chain, but it can suppress the energy loss from the polymer terminal and improve the hysteresis loss characteristics. From the viewpoint, it is preferably introduced at the polymerization initiation terminal or the polymerization termination terminal.
- modified SBRs in particular, a polymerized terminal (active terminal) of a solution-polymerized styrene butadiene rubber (S-SBR) is modified with a compound represented by the following formula (3) (modified S-SBR (JP 2010-2010-A)).
- modified SBR JP 2010-2010-A
- JP 2010-2010-A modified S-SBR
- JP 2010-2010-A modified SBR
- R 11 , R 12 and R 13 are the same or different and each represents an alkyl group or an alkoxy group (preferably having 1 to 8 carbon atoms, more preferably 1 to 6 carbon atoms, still more preferably carbon atoms).
- R 14 and R 15 are the same or different and represent a hydrogen atom or an alkyl
- n represents an integer (preferably 1 to 5, more preferably 2 to 4, more preferably 3).
- R 11 , R 12 and R 13 an alkoxy group is desirable, and as R 14 and R 15 , a hydrogen atom is desirable. As a result, excellent wet grip performance, low fuel consumption, and steering stability can be obtained.
- Specific examples of the compound represented by the above formula (3) include 3-aminopropyltrimethoxysilane, 3-aminopropyldimethylmethoxysilane, 3-aminopropylmethyldimethoxysilane, 2-dimethylaminoethyltrimethoxysilane, 3 -Diethylaminopropyltrimethoxysilane, 3- (N, N-dimethylamino) propyltrimethoxysilane and the like. These may be used alone or in combination of two or more.
- the styrene content of SBR is preferably 19% by mass or more, more preferably 21% by mass or more, still more preferably 25% by mass or more, and particularly preferably 30% by mass or more.
- the styrene content is preferably 60% by mass or less, more preferably 55% by mass or less, still more preferably 50% by mass or less, and particularly preferably 45% by mass or less. If the styrene content is less than 19% by mass, grip performance may be insufficient.
- the styrene content exceeds 60% by mass, the styrene groups are adjacent to each other, the polymer becomes too hard, the cross-linking is likely to be uneven, the durability may be deteriorated, and the temperature dependency is increased. The performance change with respect to the change becomes large, and there is a tendency that the wet grip performance and the dry grip performance cannot be obtained satisfactorily.
- the styrene content of SBR is calculated by 1 H-NMR measurement.
- the vinyl content of SBR is preferably 10% by mass or more, more preferably 15% by mass or more, still more preferably 20% by mass or more, and particularly preferably 25% by mass or more. If the vinyl content is less than 10% by mass, sufficient grip performance may not be obtained.
- the vinyl content is preferably 90% by mass or less, more preferably 80% by mass or less, still more preferably 70% by mass or less, and particularly preferably 60% by mass or less. When the vinyl content exceeds 90% by mass, the production of SBR becomes difficult, the yield is not stable, and the rubber strength is lowered, and the performance may not be stable.
- the vinyl content (1,2-bonded butadiene unit amount) of SBR can be measured by infrared absorption spectrum analysis.
- SBR also preferably has a glass transition temperature (Tg) of ⁇ 45 ° C. or higher, more preferably ⁇ 40 ° C. or higher.
- Tg is preferably 10 ° C. or less, more preferably 5 ° C. or less, and still more preferably 0 ° C. or less.
- the glass transition temperature of SBR is a value measured by performing differential scanning calorimetry (DSC) according to JIS K7121 under the condition of a heating rate of 10 ° C./min.
- SBR has a weight average molecular weight (Mw) of preferably 200,000 or more, more preferably 250,000 or more, and still more preferably 300,000 or more. For racing or high wear tires, it is particularly preferably 1.1 million or more. The Mw is preferably 2 million or less, more preferably 1.8 million or less. By using SBR having an Mw of 200,000 or more, higher grip properties, fuel efficiency and durability can be exhibited. When Mw exceeds 2 million, it is difficult to disperse the filler and the durability may be deteriorated.
- Mw weight average molecular weight
- the weight average molecular weight of SBR is gel permeation chromatography (GPC) (GPC-8000 series, manufactured by Tosoh Corporation, detector: differential refractometer, column: TSKGEL SUPERMALTPORE, manufactured by Tosoh Corporation). It can be determined by standard polystyrene conversion based on the measured value by HZ-M).
- GPC gel permeation chromatography
- the SBR content in 100% by mass of the diene rubber is preferably 60% by mass or more, more preferably 65% by mass or more, and further preferably 70% by mass or more. If it is less than 60% by mass, the grip performance tends not to be sufficient. Further, the upper limit of the SBR content is not particularly limited, and may be 100% by mass, but is preferably 90% by mass or less, and more preferably 80% by mass or less.
- the diene rubber preferably contains 60% by mass or more of SBR having a styrene content of 19 to 60% by mass, and more preferably 65% by mass or more of SBR having a styrene content of 25 to 55% by mass. Thereby, higher grip property and durability can be exhibited.
- the butadiene rubber (BR) is not particularly limited.
- Modified BR such as BR1250H, BR containing syndiotactic polybutadiene crystals such as VCR412 and VCR617 manufactured by Ube Industries, Ltd., and BR synthesized using a rare earth element-based catalyst such as BUNA-CB25 manufactured by LANXESS 1 type may be used for these BR and 2 or more types may be used together.
- BR (rare earth-based BR) synthesized using a rare earth element-based catalyst is preferable from the viewpoint of wear resistance and fuel efficiency.
- the rare earth BR is a butadiene rubber synthesized using a rare earth element-based catalyst, and has a characteristic that the cis content is high and the vinyl content is low.
- the rare earth BR those common in tire production can be used.
- the rare earth element-based catalyst used for the synthesis of the rare earth-based BR known catalysts can be used, for example, a lanthanum series rare earth element compound, an organoaluminum compound, an aluminoxane, a halogen-containing compound, and a catalyst containing a Lewis base as necessary. Etc.
- an Nd-based catalyst using a neodymium (Nd) -containing compound as the lanthanum series rare earth element compound is particularly preferable.
- the lanthanum series rare earth element compounds include halides, carboxylates, alcoholates, thioalcolates, amides, and the like of rare earth metals having an atomic number of 57 to 71.
- the Nd-based catalyst is preferable in that a BR having a high cis content and a low vinyl content can be obtained.
- the organoaluminum compound is represented by AlR a R b R c (wherein R a , R b and R c are the same or different and each represents hydrogen or a hydrocarbon group having 1 to 8 carbon atoms). Things can be used.
- the aluminoxane include a chain aluminoxane and a cyclic aluminoxane.
- halogen-containing compound examples include AlX k R d 3-k (wherein X is a halogen, R d is an alkyl group having 1 to 20 carbon atoms, an aryl group or an aralkyl group, and k is 1, 1.5, 2 or 3)
- the Lewis base is used for complexing a lanthanum series rare earth element compound, and acetylacetone, ketone, alcohol and the like are preferably used.
- Rare earth element-based catalysts may be used in the state of being dissolved in an organic solvent (n-hexane, cyclohexane, n-heptane, toluene.xylene, benzene, etc.) during the polymerization of butadiene, or may be silica, magnesia, magnesium chloride, etc. It may be used by being supported on an appropriate carrier.
- the polymerization conditions may be either solution polymerization or bulk polymerization, the preferred polymerization temperature is ⁇ 30 to 150 ° C., and the polymerization pressure may be arbitrarily selected depending on other conditions.
- the cis 1,4 bond content (cis content) of the rare earth BR is preferably 90% by mass or more, more preferably 93% by mass or more, and still more preferably 95% by mass or more. When it is less than 90% by mass, durability and wear resistance tend to deteriorate.
- the vinyl content of the rare earth BR is preferably 1.8% by mass or less, more preferably 1.5% by mass or less, still more preferably 1.0% by mass or less, and particularly preferably 0.8% by mass or less. If it exceeds 1.8% by mass, durability and wear resistance tend to deteriorate.
- the vinyl content (1,2-bond butadiene unit content) and cis content (cis 1,4-bond content) of BR can be measured by infrared absorption spectrum analysis.
- the content of BR in 100% by mass of the diene rubber is preferably 10% by mass or more, more preferably 15% by mass or more, and further preferably 20% by mass or more.
- the BR content is preferably 70% by mass or less, more preferably 60% by mass or less.
- the content of BR is less than 10% by mass or exceeds 70% by mass, sufficient wear resistance, grip performance, and low fuel consumption tend not to be obtained.
- NR those generally used in the tire industry such as SIR20, RSS # 3, TSR20 and the like can be used.
- the content of NR in 100% by mass of diene rubber is preferably 60 to 100% by mass.
- the content of NR is preferably 0 to 70% by mass. If the NR content is outside the above range, sufficient grip performance, wear resistance, and durability may not be obtained.
- the rubber component may contain a rubber component other than the diene rubber.
- examples of the other rubber component include butyl rubber (IIR).
- the content of the diene rubber is 90 mass% or more, and preferably 95 mass% or more.
- an upper limit is not specifically limited, 100 mass% may be sufficient.
- the rubber composition in the present invention is obtained by hydrogenating a double bond of a terpene aromatic resin, the hydrogenation rate of the double bond is 5 to 100%, and the hydroxyl value is 20 mgKOH / g or less Contains terpene aromatic resin.
- the “terpene aromatic resin” in the hydrogenated terpene aromatic resin is a compound obtained by copolymerizing an aromatic compound and a terpene compound by a commonly used method. Specifically, for example, each raw material can be dropped in an arbitrary order in an organic solvent such as toluene in the presence of a catalyst such as BF 3 and reacted at a predetermined temperature for a predetermined time. .
- the copolymerization ratio of the aromatic compound and the terpene compound can be appropriately set so that the hydrogenated terpene aromatic resin has physical properties described later. Further, as long as the hydrogenated terpene aromatic resin has the physical properties described later, the terpene aromatic resin may contain a copolymer unit other than the aromatic compound and the terpene compound, such as indene.
- the aromatic compound is not particularly limited as long as it is a compound having an aromatic ring.
- phenol compounds such as phenol, alkylphenol, alkoxyphenol, unsaturated hydrocarbon group-containing phenol; naphthol, alkylnaphthol, alkoxynaphthol, Examples thereof include naphthol compounds such as saturated hydrocarbon group-containing naphthol; styrene derivatives such as styrene, alkylstyrene, alkoxystyrene, and unsaturated hydrocarbon group-containing styrene. Of these, styrene derivatives are preferred.
- the number of carbon atoms of the alkyl group or alkoxy group in the above compound is preferably 1-20, and more preferably 1-12.
- the number of carbon atoms of the unsaturated hydrocarbon group in the above compound is preferably 2 to 20, and more preferably 2 to 12.
- the aromatic compound may have one substituent on the aromatic ring, may have two or more, and when there are two or more substituents on the aromatic ring, those aromatic compounds
- the substitution position may be any of o-position, m-position and p-position.
- the substitution position of the substituent may be o-position, m-position, or p-position with respect to the vinyl group derived from styrene.
- aromatic compounds may be used alone or in combination of two or more.
- alkylphenol examples include, for example, methylphenol, ethylphenol, butylphenol, t-butylphenol, octylphenol, nonylphenol, decylphenol, dinonylphenol and the like. These may be substituted at any of the o-position, m-position and p-position. Of these, t-butylphenol is preferable, and pt-butylphenol is more preferable.
- alkyl naphthol examples include compounds in which the phenol moiety of the alkyl phenol is replaced with naphthol.
- alkyl styrene examples include compounds in which the phenol part of the alkyl phenol is replaced with styrene.
- alkoxyphenol examples include compounds in which the alkyl group of the alkylphenol is replaced with a corresponding alkoxy group.
- alkoxy naphthol examples include compounds in which the alkyl group of the alkyl naphthol is replaced with a corresponding alkoxy group.
- alkoxystyrene examples include compounds in which the alkyl group of the alkylstyrene is replaced with a corresponding alkoxy group.
- Examples of the unsaturated hydrocarbon group-containing phenol include compounds in which at least one hydroxyphenyl group is contained in one molecule, and at least one of the hydrogen atoms of the phenyl group is substituted with an unsaturated hydrocarbon group. It is done.
- Examples of the unsaturated bond in the unsaturated hydrocarbon group include a double bond and a triple bond.
- Examples of the unsaturated hydrocarbon group include alkenyl groups having 2 to 20 carbon atoms.
- unsaturated hydrocarbon group-containing phenol examples include isopropenyl phenol and butenyl phenol. The same applies to the unsaturated hydrocarbon group-containing naphthol and the unsaturated hydrocarbon group-containing styrene.
- the terpene compound is a hydrocarbon represented by a composition of (C 5 H 8 ) n and an oxygen-containing derivative thereof, and includes monoterpene (C 10 H 16 ), sesquiterpene (C 15 H 24 ), diterpene (C 20 H 32 ) and other terpenes classified as basic skeletons.
- the terpene compound is not particularly limited, but is preferably a cyclic unsaturated hydrocarbon, and is preferably a compound having no hydroxyl group.
- terpene compound examples include ⁇ -pinene, ⁇ -pinene, 3-carene ( ⁇ -3-carene), dipentene, limonene, myrcene, alloocimene, ocimene, ⁇ -ferrandrene, ⁇ -terpinene, and ⁇ -terpinene.
- ⁇ -pinene, ⁇ -pinene, 3-carene ( ⁇ -3-carene), dipentene and limonene are preferred, and ⁇ -pinene and limonene are more preferred from the viewpoint of improving the grip performance and durability in a balanced manner.
- limonene may include any of d-form, l-form, and d / l-form. These terpene compounds may be used alone or in combination of two or more.
- the compound represented by following formula (I) is mentioned as a compound obtained by copolymerizing a styrene derivative and limonene, for example.
- R represents a substituent on the aromatic ring and is an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, or an unsaturated hydrocarbon group having 2 to 20 carbon atoms.
- the number of substitutions of the substituent R may be any of 1 to 5, and when the number of substitutions is 2 or more, the substituents may be the same as or different from each other.
- the substitution position is not particularly limited.
- m is 0.2-20.
- n is 2 to 10.
- terpene aromatic resin examples include, for example, YS Resin TO125, YS Resin TO115, YS Resin TO105, YS Resin TO85, YS Polystar UH115 (manufactured by Yashara Chemical Co., Ltd.).
- the hydrogenated terpene aromatic resin in the present invention can be produced by hydrogenating the double bond of the above-mentioned terpene aromatic resin by a commonly used method.
- the hydrogenation can be carried out, for example, by catalytic hydrogen reduction using a noble metal itself such as palladium, ruthenium, rhodium, nickel or the like supported on a support such as activated carbon, activated alumina, or diatomaceous earth as a catalyst. it can.
- the amount of the catalyst used is preferably 0.1 to 50% by mass and more preferably 0.2 to 40% by mass with respect to 100% by mass of the terpene aromatic resin as the raw material. If the amount of the catalyst is less than 0.1% by mass, the hydrogenation reaction tends to be slow. On the other hand, if it exceeds 50% by mass, the residual impurities cause the filler dispersion and polymer dispersion to be inhibited, and sufficient breaking strength and grip performance are obtained. There is a risk that it will not be obtained.
- the hydrogen pressure during the hydrogenation reaction is usually 5 to 200 kg / cm 2 , preferably 50 to 100 kg / cm 2 . If it is less than 5 kg / cm 2 , the reaction rate of the hydrogenation reaction tends to be slow.
- the reaction temperature during the hydrogenation reaction is usually 10 to 200 ° C., preferably 20 to 150 ° C. If the reaction temperature is less than 10 ° C, the hydrogenation reaction tends to be slow. On the other hand, if the reaction temperature exceeds 200 ° C, the reaction equipment is damaged and maintenance is difficult, resulting in poor production efficiency.
- hydrogenated terpene aromatic resin what is marketed can also be used, for example, YS polystar M80, YS polystar M105, YS polystar M115, YS polystar M125 (above, Yashara Chemical Co., Ltd. product) etc. Can be used.
- the hydrogenated terpene aromatic resin in the present invention obtained as described above is one in which double bonds are hydrogenated.
- the hydrogenation rate of the double bond is 5 to 100%, preferably 6% or more, more preferably 7% or more, and more preferably 8% or more. More preferably, it is more preferably 11% or more, and particularly preferably 15% or more.
- the upper limit of the hydrogenation rate of the double bond may be changed in the preferred range due to the pressurization and heating conditions in the hydrogenation reaction, progress in production technology such as a catalyst, improvement in productivity, etc. Although it has not been confirmed accurately at the present time, for example, it is preferably 80% or less, more preferably 60% or less, still more preferably 40% or less, and even more preferably 30% or less. 25% or less is particularly preferable.
- the hydrogenation rate is a value calculated by the following formula from each integrated value of the double bond-derived peak by 1 H-NMR (proton NMR).
- the hydrogenation rate means a hydrogenation rate of double bonds.
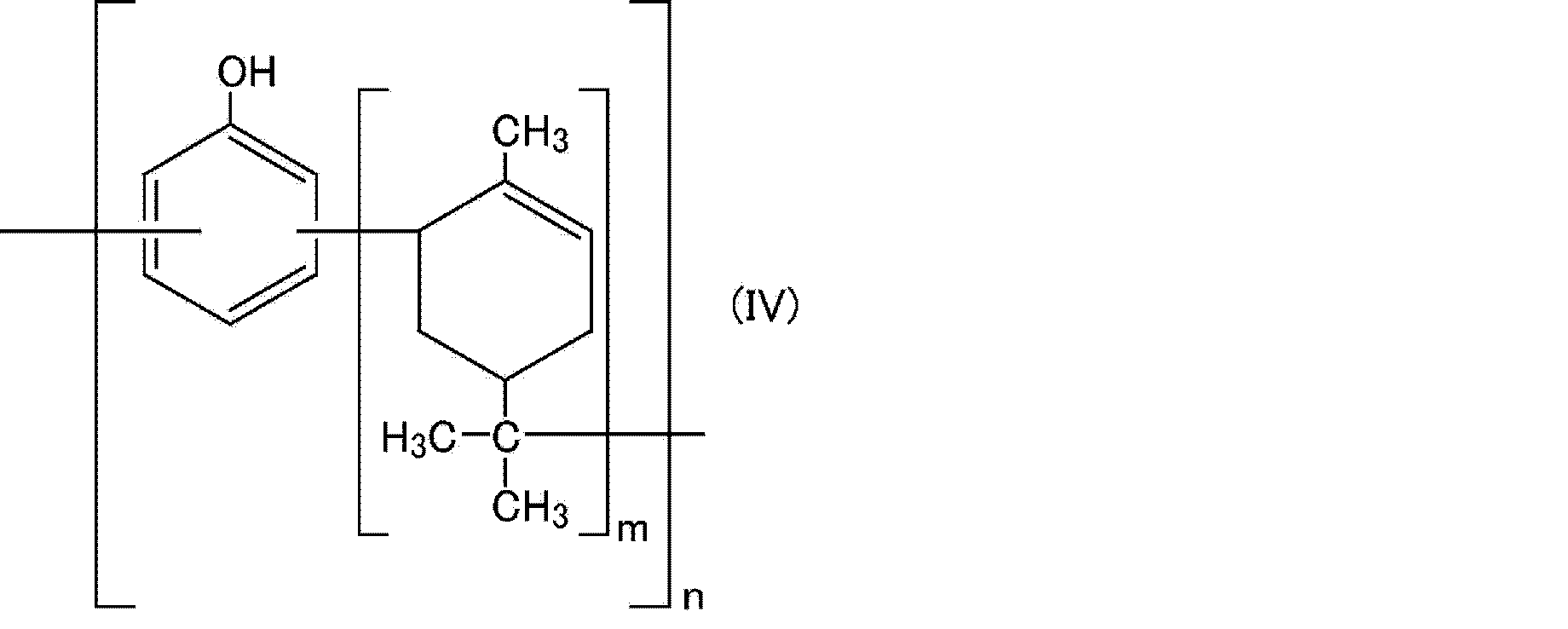
- R represents a substituent on the cyclohexane ring and is an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, or an unsaturated hydrocarbon group having 2 to 20 carbon atoms.
- the number of substitutions of the substituent R may be any of 1 to 5, and when the number of substitutions is 2 or more, the substituents may be the same as or different from each other.
- the substitution position is not particularly limited.
- m is 0.2-20.
- n is 2 to 10.
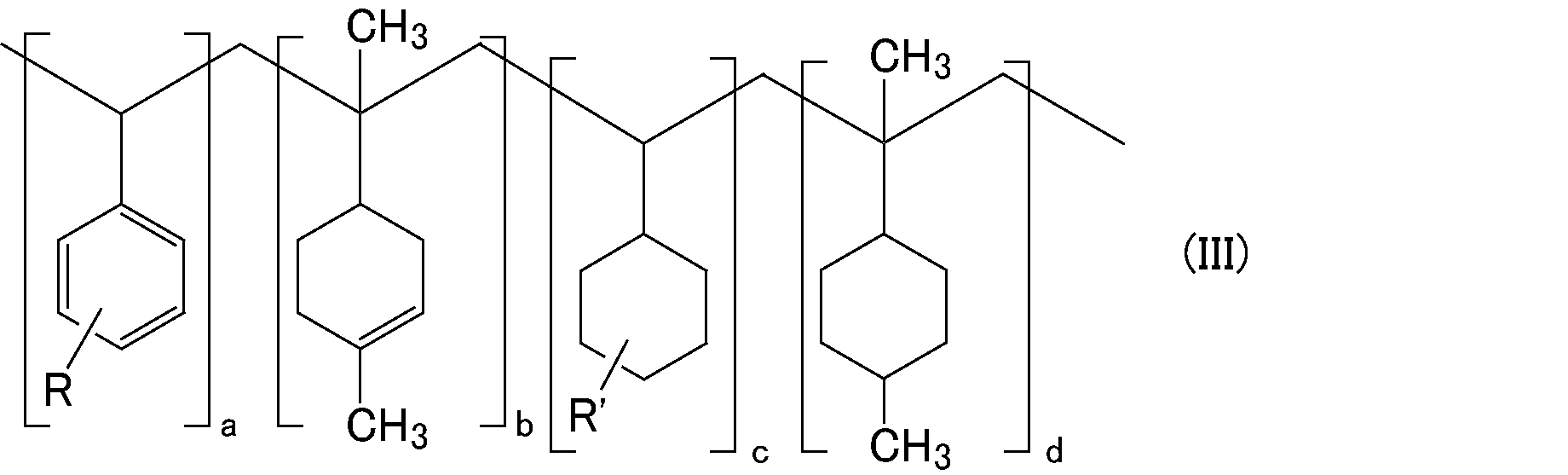
- R represents a substituent on the aromatic ring and is an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, or an unsaturated hydrocarbon group having 2 to 20 carbon atoms.
- R ′ represents a substituent on the cyclohexane ring, and is an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, or an unsaturated hydrocarbon group having 2 to 20 carbon atoms.
- the number of substituents R and R ′ may be any of 1 to 5, and when the number of substituents is 2 or more, the substituents may be the same or different. These substitution positions are not particularly limited.
- a, b, c, and d represent the number of repeating units. Note that the order in which the repeating units are combined is not particularly limited, and may be blocks, alternating, or random.
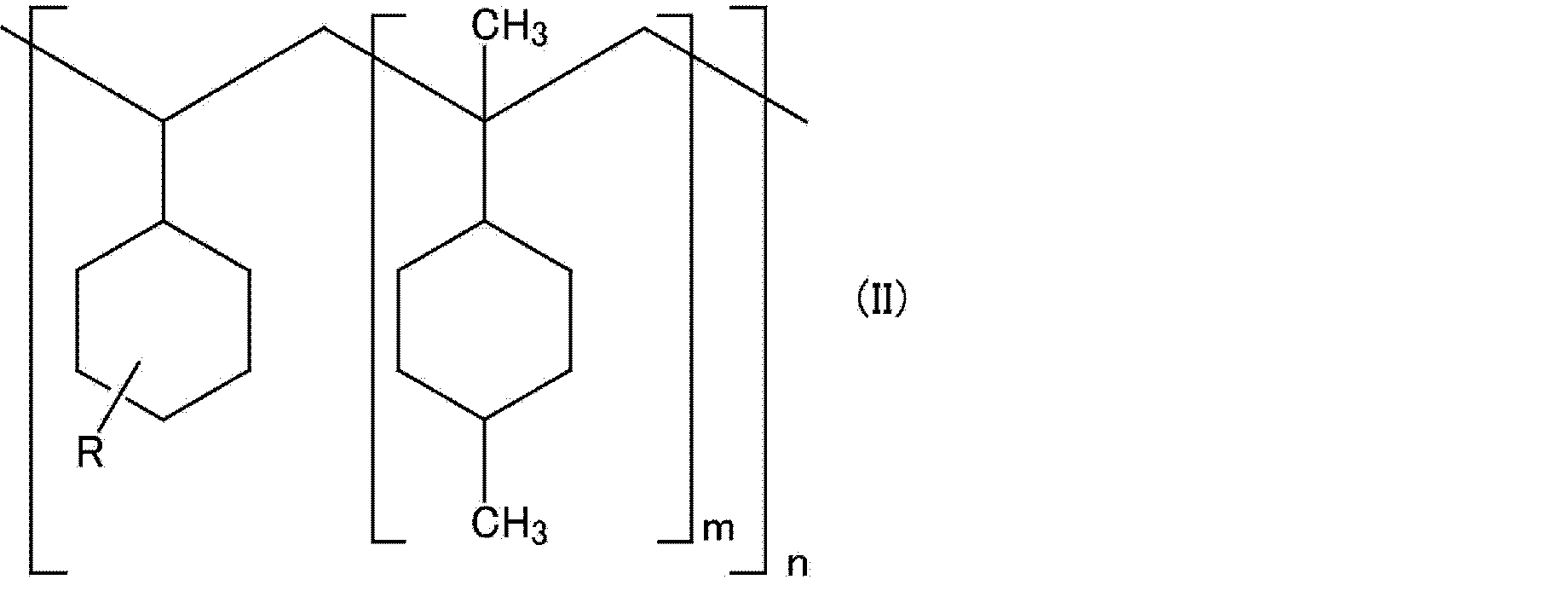
- the hydrogenated terpene aromatic resin for example, a resin containing a repeating unit represented by the above formula (II) having a cyclohexyl group (however, represented by the above formula (I) in the structure) And may contain at least one repeating unit selected from the group consisting of repeating units and repeating units represented by the following formula (IV). Note that the order in which the repeating units are combined is not particularly limited, and may be blocks, alternating, or random.
- m and n represent the number of repeating units.
- the hydroxyl value of the hydrogenated terpene aromatic resin (that is, the phenol group content) is 20 mgKOH / g or less, preferably 10 mgKOH / g or less, more preferably 5 mgKOH / g or less, and 1 mgKOH / g or less. More preferably, 0.1 mgKOH / g or less is still more preferable. In particular, 0 mg KOH / g is preferable.
- the hydroxyl value exceeds 20 mgKOH / g, the self-aggregation property of the resin is increased, the affinity with rubber and filler is lowered, and sufficient grip performance may not be obtained.
- the hydroxyl value of the hydrogenated terpene aromatic resin is expressed in milligrams of the amount of potassium hydroxide required to neutralize acetic acid bonded to the hydroxyl group when 1 g of hydrogenated terpene aromatic resin is acetylated. It is a value measured by potentiometric titration (JIS K0070: 1992).
- the softening point of the hydrogenated terpene aromatic resin is preferably 80 ° C. or higher, more preferably 90 ° C. or higher, still more preferably 100 ° C. or higher, still more preferably 114 ° C. or higher, particularly preferably 116 ° C. or higher, and 120 ° C. or higher. Is most preferred. Moreover, 180 degrees C or less is preferable, 170 degrees C or less is more preferable, 165 degrees C or less is still more preferable, 160 degrees C or less is especially preferable, and 135 degrees C or less is the most preferable.
- the softening point of the hydrogenated terpene aromatic resin is the temperature at which the sphere descends when the softening point specified in JIS K6220-1: 2001 is measured with a ring and ball softening point measuring device.
- the hydrogenated terpene aromatic resin also preferably has a glass transition temperature (Tg) of 20 ° C. or higher, more preferably 30 ° C. or higher, and still more preferably 40 ° C. or higher.
- Tg is preferably 100 ° C. or lower, more preferably 90 ° C. or lower, and still more preferably 80 ° C. or lower.
- the glass transition temperature of the hydrogenated terpene aromatic resin is a value measured by performing differential scanning calorimetry (DSC) according to JIS K7121 at a temperature rising rate of 10 ° C./min.
- the weight average molecular weight (Mw) of the hydrogenated terpene aromatic resin is not particularly limited, but is preferably 300 to 3000, more preferably 500 to 2000, and still more preferably 600 to 2000. If Mw is less than 300, G '(hardness) of the adhesive layer tends to be low and sufficient grip performance tends not to be obtained, whereas if it exceeds 3000, rubber hardness increases, sufficient grip performance and durability. There is a tendency not to get sex.
- the weight average molecular weight of the hydrogenated terpene aromatic resin is determined by gel permeation chromatography (GPC) (GPC-8000 series, manufactured by Tosoh Corporation), detector: differential refractometer, column: Tosoh Corporation ) Manufactured by TSKGEL SUPERMALTPORE HZ-M), and can be determined by standard polystyrene conversion.
- GPC gel permeation chromatography
- the rubber composition in the present invention contains 1 to 50 parts by mass of the hydrogenated terpene aromatic resin with respect to 100 parts by mass of the diene rubber.
- content of this hydrogenated terpene aromatic resin 2 mass parts or more are preferable, 3 mass parts or more are more preferable, and 5 mass parts or more are still more preferable.
- 40 mass parts or less are preferable, 35 mass parts or less are more preferable, and 30 mass parts or less are still more preferable.
- the difference in solubility parameter (SP value) between the diene rubber and the hydrogenated terpene aromatic resin is preferably 1.5 or less.
- the difference in SP value is more preferably 1.0 or less.
- the lower limit value of the difference between the SP values is not particularly limited, and it is preferably as small as possible.
- SP value of diene rubber and hydrogenated terpene aromatic resin means a solubility parameter (Solubility Parameter) calculated by the Hoy method based on the structure of the compound.
- the Hoy method is, for example, K.K. L. Hoy “Table of Solubility Parameters”, Solvent and Coatings Materials Research and Development Department, Union Carbites Corp. (1985).
- the rubber composition in the present invention contains at least one inorganic filler selected from the group consisting of a compound represented by the following formula, magnesium sulfate, and silicon carbide.
- mM ⁇ xSiO y ⁇ zH 2 O wherein M is at least one metal selected from the group consisting of Al, Mg, Ti, Ca and Zr, an oxide or hydroxide of the metal, m is an integer of 1 to 5, and x is (An integer from 0 to 10, y is an integer from 2 to 5, and z is an integer from 0 to 10.)
- Examples of the compound represented by the above formula include alumina, alumina hydrate, aluminum hydroxide, magnesium hydroxide, magnesium oxide, talc, titanium white, titanium black, calcium oxide, calcium hydroxide, magnesium aluminum oxide, clay, pyro Examples thereof include phyllite, bentonite, aluminum silicate, magnesium silicate, calcium silicate, aluminum calcium silicate, magnesium silicate, zirconium, and zirconium oxide. These inorganic compounds may be used alone or in combination of two or more.
- the above inorganic fillers has a Mohs hardness of 3 or more, water resistance, and oil resistance, and when processed into a particle size of a micron unit, a scratching effect is produced or a bloom of an adhesive component that expresses grip performance is promoted. As a result, the grip performance is improved.
- a metal of M or Al or Zr an oxide or hydroxide of the metal is preferable, and the amount of resources is abundant and inexpensive.
- Aluminum hydroxide and zirconium oxide are more preferable. Further, aluminum hydroxide is particularly preferable from the viewpoint of obtaining good kneading productivity and extrusion processability.
- the inorganic filler has a nitrogen adsorption specific surface area (N 2 SA) of 10 to 120 m 2 / g. Outside the above range, grip performance is lowered and wear resistance is deteriorated.
- the lower limit of the N 2 SA is preferably 13 m 2 / g.
- the upper limit of the N 2 SA is preferably 115 m 2 / g, more preferably 110 m 2 / g, still more preferably 80 m 2 / g, and particularly preferably 70 m 2 / g.
- the N 2 SA of the inorganic filler is a value measured by the BET method according to ASTM D3037-81.
- the amount of linseed oil absorbed by the inorganic filler is preferably 30 ml / 100 g or more, more preferably 35 ml / 100 g or more.
- the linseed oil absorption is preferably 75 ml / 100 g or less, more preferably 50 ml / 100 g or less, particularly preferably 40 ml / 100 g or less.
- the resulting pneumatic tire exhibits excellent grip performance and excellent durability.
- the lower the amount of linseed oil absorbed, the less the connection ( structure) between the particles of the inorganic filler, and it tends to exist alone in the rubber.
- the linseed oil absorption is effective for determining whether the inorganic filler single particles are reasonably fine and form agglomerates with an appropriate secondary particle size in the non-polar tire rubber composition. It is considered to be an indicator. That is, when the linseed oil absorption is less than 30 ml / 100 g, the affinity with the rubber component, softener, resin and the like is lowered, and the position of the inorganic filler in the rubber composition is considered not to be thermally stable. .
- the inorganic filler particles form an agglomerate with a large secondary particle size, and an occluded part that takes in the oil is produced inside the kneaded step. If not sufficiently mixed with the rubber component, it may cause deterioration of wear resistance and durability. Further, in this technical field, DBP oil absorption is generally used, but since linseed oil is a kind of natural oil, there is also an advantage that the burden on the environment is smaller than DBP.
- the oil absorption of linseed oil of ULTRASIL VN3 (N 2 SA: 175 m 2 / g) manufactured by Evonik, which is a typical wet silica that easily develops the particle structure, is 128 ml / 100 g.
- the linseed oil absorption is a value determined according to JIS K5101-13.
- the average particle diameter of the inorganic filler is preferably 1.5 ⁇ m or less, more preferably 0.69 ⁇ m or less, and still more preferably 0.6 ⁇ m or less.
- the average particle diameter is preferably 0.2 ⁇ m or more, more preferably 0.25 ⁇ m or more, and further preferably 0.4 ⁇ m or more. If it exceeds 1.5 ⁇ m, grip performance may be deteriorated and durability may be deteriorated. If it is less than 0.2 ⁇ m, secondary agglomerates are likely to be formed in rubber, and grip performance is reduced. Durability may deteriorate.
- the average particle diameter of an inorganic filler is a number average particle diameter, and is measured with a transmission electron microscope.
- the Mohs hardness of the inorganic filler is preferably 7 or less, comparable to silica, from 2 to 5 from the viewpoint of ensuring grip performance and durability of the tire and suppressing metal wear of a Banbury mixer or an extruder. It is more preferable.
- Mohs hardness is one of the mechanical properties of materials and is a measurement method that has been widely used in minerals for a long time. Scrub a substance you want to measure hardness (such as aluminum hydroxide) with a standard substance and check for scratches. Measure Mohs hardness.
- an inorganic filler having a Mohs hardness of less than 7 and a Mohs hardness of the inorganic filler dehydration reaction product of 8 or more.
- aluminum hydroxide has a Mohs hardness of about 3 and prevents abrasion (wear) of the banbury and rolls, and the surface layer undergoes a dehydration reaction (transition) due to vibration, heat generation and partial kneading during running and late, Since it is converted to alumina with a Mohs hardness of about 9, it has a hardness equal to or higher than that of road stone, so that excellent grip performance and durability can be obtained.
- Aluminum hydroxide and alumina are stable against water, base and acid, and do not inhibit vulcanization or promote oxidative degradation.
- the Mohs hardness after the transfer of the inorganic filler is more preferably 7 or more, and the upper limit is not particularly limited.
- Diamonds have a maximum value of 10.
- the inorganic filler preferably has a thermal decomposition starting temperature (DSC endothermic starting temperature) of 160 to 500 ° C., more preferably 170 to 400 ° C. If the temperature is lower than 160 ° C., thermal decomposition or reaggregation proceeds excessively during kneading, and metal wear such as the rotor blades of the kneader or the wall of the container may be excessive.
- the thermal decomposition start temperature of an inorganic filler is calculated
- the thermal decomposition also includes a dehydration reaction.
- the inorganic filler can be used a commercially available product having the N 2 SA, also like subjected to a treatment such as grinding an inorganic filler treated product was adjusted to a particle having the above characteristics can be used.
- a treatment such as grinding an inorganic filler treated product was adjusted to a particle having the above characteristics can be used.
- conventionally known methods such as wet pulverization and dry pulverization (jet mill, current jet mill, counter jet mill, contraplex, etc.) can be applied. Further, if necessary, it is fractionated by a membrane filter method frequently used in medicine and bio-related, and a product having a predetermined N 2 SA can be prepared and used as a rubber compounding agent.
- Content of the said inorganic filler is 1 mass part or more with respect to 100 mass parts of said diene rubbers, Preferably it is 3 mass parts or more, More preferably, it is 5 mass parts or more. If it is less than 1 part by mass, sufficient grip performance (particularly wet grip performance) cannot be obtained. Moreover, this content is 70 mass parts or less, Preferably it is 60 mass parts or less, More preferably, it is 50 mass parts or less, More preferably, it is 40 mass parts or less. If it exceeds 70 parts by mass, sufficient filler dispersibility cannot be obtained, and grip performance (particularly dry grip performance) and wear resistance are poor. In particular, for passenger car tires, 10 to 20 parts by mass is preferable with respect to 100 parts by mass of the diene rubber from the viewpoint of achieving both grip performance and wear resistance.
- the rubber composition in the present invention preferably contains carbon black from the viewpoints of reinforcement, grip performance, and prevention of ultraviolet deterioration.
- Nitrogen adsorption specific surface area (N 2 SA) of carbon black is preferably not less than 100 m 2 / g, more preferably at least 110m 2 / g, more preferably not less than 115m 2 / g, 140m 2 / g or more is particularly preferable.
- the N 2 SA is preferably 600 m 2 / g or less, more preferably 500 m 2 / g or less, and still more preferably 400 m 2 / g or less. If it is less than 100 m 2 / g, the grip performance and wear resistance tend to decrease, and if it exceeds 600 m 2 / g, good filler dispersion is difficult to obtain, and the reinforcement and durability tend to deteriorate.
- the N 2 SA of carbon black is obtained by the BET method in accordance with JIS K 6217-2: 2001.
- the carbon black content varies depending on the grip performance, wear resistance, and low fuel consumption expected of the tire. From the viewpoint of preventing ultraviolet cracks, 5 parts by mass or more is desirable with respect to 100 parts by mass of the diene rubber. When the wet grip performance is ensured by silica, the carbon black content is about 5 to 50 parts by mass with respect to 100 parts by mass of the diene rubber. Further, when the dry grip performance and wear resistance are ensured by carbon black, the carbon black content is preferably 50 to 160 parts by mass with respect to 100 parts by mass of the diene rubber.
- the rubber composition in the present invention may contain silica.
- silica By containing silica, it is possible to improve rolling resistance characteristics while improving wet grip performance and reinforcement. This effect is an effect that is synergistically prominently used in combination with the specific inorganic filler in the present invention, and this is presumed to be due to the action described in the above (1) and (B).
- Examples of the silica include silica manufactured by a wet method, silica manufactured by a dry method, and the like.
- the silica preferably has a nitrogen adsorption specific surface area (N 2 SA) of 80 m 2 / g or more, more preferably 120 m 2 / g or more, and still more preferably 150 m 2 / g or more. Further, the N 2 SA is preferably 280 m 2 / g or less, more preferably 260 m 2 / g or less, and further preferably 250 m 2 / g or less. N 2 SA of silica is a value measured by the BET method according to ASTM D3037-93.
- the silica content is preferably 30 parts by mass or more, more preferably 60 parts by mass or more, and still more preferably 75 parts by mass with respect to 100 parts by mass of the diene rubber. Above, still more preferably 85 parts by mass or more, particularly preferably 90 parts by mass or more. When the amount is less than 30 parts by mass, there is a possibility that sufficient reinforcement cannot be obtained.
- the content is preferably 150 parts by mass or less, more preferably 130 parts by mass or less, still more preferably 120 parts by mass or less, and particularly preferably 100 parts by mass or less. When it exceeds 150 parts by mass, silica is difficult to disperse, and wear resistance, durability, and fuel efficiency tend to be deteriorated.
- the rubber composition in the present invention contains the silica
- a silane coupling agent any silane coupling agent conventionally used in combination with silica in the rubber industry can be used.
- the rubber composition according to the present invention it is also one of preferred embodiments of the present invention to further blend a softener from the viewpoint of grip performance and the like.
- a softener from the viewpoint of grip performance and the like.
- Oil, liquid diene polymer, resin with a softening point of 160 degrees C or less, etc. are mentioned.
- blend resin with a softening point of 160 degrees C or less is one of the suitable forms from a viewpoint of grip performance.
- the oil examples include process oils such as paraffinic, aroma-based and naphthenic process oils. Among them, it is preferable to use a rubber having a small difference in solubility parameter (SP value) from the diene rubber. When the difference in SP value is small, the miscibility between the diene rubber and the oil becomes better.
- the difference in SP value is preferably 1.0 or less, for example. In addition, it does not specifically limit as a lower limit of the difference of this SP value, The smaller is preferable.
- the SP value of oil is calculated in the same manner as the SP value of the diene rubber and hydrogenated terpene aromatic resin.
- the oil content is preferably 2 parts by mass or more with respect to 100 parts by mass of the diene rubber, although it depends on the grip performance and fuel economy expected of the tire, that is, the amount of filler.
- it is 5 parts by mass or more.
- this content becomes like this.
- it is 85 mass parts or less, More preferably, it is 75 mass parts or less. If the amount is less than 2 parts by mass, the dispersion of the filler or polymer may be deteriorated or the dispersion of the crosslinking agent such as sulfur may be deteriorated. If the amount exceeds 85 parts by mass, the durability and wear resistance tend to deteriorate.
- the oil content includes the amount of oil contained in the oil-extended rubber. However, since tires for trucks and buses are highly expected to have wear resistance, durability and chipping properties, oil is often not blended.
- the liquid diene polymer is a diene polymer in a liquid state at normal temperature (25 ° C.).
- the liquid diene polymer preferably has a polystyrene-equivalent weight average molecular weight (Mw) measured by gel permeation chromatography (GPC) of 1.0 ⁇ 10 3 to 2.0 ⁇ 10 5 . More preferably, it is 0 ⁇ 10 3 to 1.5 ⁇ 10 4 . If it is less than 1.0 ⁇ 10 3 , the grip performance is not improved and sufficient durability may not be ensured. On the other hand, if it exceeds 2.0 ⁇ 10 5 , the viscosity of the polymerization solution becomes too high, and the productivity may be deteriorated or the fracture characteristics may be deteriorated.
- Mw of the liquid diene polymer is a polystyrene equivalent value measured by gel permeation chromatography (GPC).
- liquid diene polymer examples include a liquid styrene butadiene copolymer (liquid SBR), a liquid butadiene polymer (liquid BR), a liquid isoprene polymer (liquid IR), a liquid styrene isoprene copolymer (liquid SIR), and the like. It is done. Among these, liquid SBR is preferable because durability and grip performance can be obtained with a good balance.
- the content of the liquid diene polymer is preferably 5 parts by mass or more, more preferably 10 parts by mass or more with respect to 100 parts by mass of the diene rubber.
- the content is preferably 120 parts by mass or less, more preferably 80 parts by mass or less, still more preferably 70 parts by mass or less, and particularly preferably 30 parts by mass or less. If the amount is less than 5 parts by mass, sufficient grip performance tends not to be obtained, and if it exceeds 120 parts by mass, the durability tends to deteriorate.
- Examples of the resin having a softening point of 160 ° C. or lower used in combination with the hydrogenated terpene aromatic resin in the present invention include coumarone indene resin, pt-butylphenol acetylene resin, and styrene acrylic resin.
- the coumarone indene resin is a resin containing coumarone and indene as monomer components constituting the resin skeleton (main chain).
- monomer components contained in the skeleton include styrene and ⁇ -methylstyrene. , Methylindene, vinyltoluene and the like.
- the softening point of the coumarone indene resin is ⁇ 20 to 160 ° C.
- An upper limit becomes like this. Preferably it is 145 degrees C or less, More preferably, it is 130 degrees C or less.
- the lower limit is preferably ⁇ 10 ° C. or higher, more preferably ⁇ 5 ° C. or higher.
- the softening point exceeds 160 ° C., dispersibility during kneading deteriorates and fuel efficiency tends to deteriorate.
- the softening point is less than ⁇ 20 ° C., the production is difficult, and the migration to other members and the volatility are high, and the performance may change during use.
- a softening point when a softening point is 90 to 140 ° C., grip performance is improved.
- those having a softening point of 100 to 120 ° C. can generally increase tan ⁇ at 0 to 80 ° C. and have good durability.
- a coumarone indene resin having a softening point of 10 to 30 ° C. when a coumarone indene resin having a softening point of 10 to 30 ° C. is used, the grip property at a relatively low temperature of 10 to 40 ° C. is good, and tan ⁇ is generally lowered.
- Such a coumarone indene resin having a softening point of 10 to 30 ° C. can be used mainly for the purpose of improving durability.
- the softening point of the coumarone indene resin is a temperature at which the sphere descends when the softening point specified in JIS K6220-1: 2001 is measured with a ring and ball softening point measuring apparatus.
- the pt-butylphenol acetylene resin examples include resins obtained by condensation reaction of pt-butylphenol and acetylene.
- the resin preferably has a softening point of 120 to 160 ° C. (eg, softening point) Is 145 ° C.), and when such a pt-butylphenol acetylene resin is blended, the grip property is improved particularly at high temperatures (around 80 to 120 ° C.).
- This cholecin in combination with an ⁇ -methylstyrene resin having a softening point of around 85 ° C. excellent grip performance at low temperatures (10 to 40 ° C.)
- grip performance at a tire running temperature of 20 to 120 ° C. can be achieved.
- the softening point of the pt-butylphenol acetylene resin can be measured by the same method as that for measuring the softening point of the coumarone indene resin.
- the hydroxyl value of the pt-butylphenol acetylene resin is preferably 100 mgKOH / g or more, more preferably 150 mgKOH / g or more, and still more preferably 175 mgKOH / g or more. Moreover, 300 mgKOH / g or less is preferable, 250 mgKOH / g or less is more preferable, and 200 mgKOH / g or less is still more preferable.
- the hydroxyl value of the pt-butylphenol acetylene resin can be measured in the same manner as the hydroxyl value of the hydrogenated terpene aromatic resin described above.
- the rubber composition in the present invention may contain compounding agents generally used in the tire industry, such as waxes, zinc oxide, stearic acid, mold release agents, anti-aging agents, sulfur and the like. You may mix
- the zinc oxide used in the present invention is not particularly limited, and examples thereof include those used in the rubber field such as tires.
- fine zinc oxide can be suitably used from the viewpoint of enhancing the dispersibility and wear resistance of zinc oxide.
- zinc oxide having an average primary particle diameter of 200 nm or less, and more preferably 100 nm or less.
- the minimum of this average primary particle diameter is not specifically limited, Preferably it is 20 nm or more, More preferably, it is 30 nm or more.
- the average primary particle diameter of zinc oxide represents the average particle diameter (average primary particle diameter) converted from the specific surface area measured by the BET method by nitrogen adsorption.
- the specific surface area (N 2 SA) measured by the BET method by nitrogen adsorption is preferably 10 to 50 m 2 / g.
- the content of zinc oxide is preferably 0.5 to 10 parts by mass, more preferably 1 to 5 parts by mass with respect to 100 parts by mass of the diene rubber.
- the effect of this invention is acquired more suitably as content of zinc oxide exists in the said range.
- Examples of the vulcanization accelerator used in the present invention include sulfenamide, thiazole, thiuram, guanidine, and dithiocarbamate vulcanization accelerators. These vulcanization accelerators may be used alone or in combination of two or more. Among them, in the present invention, sulfenamide-based, thiuram-based, guanidine-based, and dithiocarbamate-based vulcanization accelerators can be suitably used, and a sulfenamide-based vulcanization accelerator and a guanidine-based vulcanization accelerator are used in combination. It is particularly preferable to do this.
- sulfenamide vulcanization accelerator examples include N-tert-butyl-2-benzothiazolylsulfenamide (TBBS), N-cyclohexyl-2-benzothiazolylsulfenamide (CBS), N, N -Dicyclohexyl-2-benzothiazolylsulfenamide (DCBS) and the like.
- thiuram vulcanization accelerator examples include tetramethylthiuram disulfide (TMTD), tetrabenzylthiuram disulfide (TBzTD), tetrakis (2-ethylhexyl) thiuram disulfide (TOT-N) and the like.
- TMTD tetramethylthiuram disulfide
- TBzTD tetrabenzylthiuram disulfide
- TOT-N tetrakis (2-ethylhexyl) thiuram disulfide
- Examples of the guanidine vulcanization accelerator include diphenyl guanidine (DPG), diortolyl guanidine, orthotolyl biguanidine and the like.
- dithiocarbamate vulcanization accelerator examples include zinc dibenzyldithiocarbamate (ZTC) and zinc ethylphenyldithiocarbamate (PX).
- the content of the vulcanization accelerator is preferably 1 part by mass or more, more preferably 2 parts by mass or more, and preferably 15 parts by mass with respect to 100 parts by mass of the diene rubber. It is 10 parts by mass or less, more preferably 10 parts by mass or less. If the amount is less than 1 part by mass, a sufficient vulcanization rate cannot be obtained and good grip performance and durability tend not to be obtained. If the amount exceeds 15 parts by mass, the crosslinking density becomes too tight or blooming occurs. The grip performance, durability, and molding adhesiveness may deteriorate.
- the rubber composition for a tread in the present invention can be produced by a conventionally known method.
- the base kneading step is not particularly limited as long as the rubber component containing the diene rubber is kneaded.
- the rubber component and the hydrogenated terpene aromatic A divided two-step base kneading step may be used in which some components such as a resin are kneaded in advance and the kneaded product and other components excluding sulfur and a vulcanization accelerator are kneaded.
- the rubber composition is used for a tread of a pneumatic tire.
- a cap tread which is a surface layer of a tread having a multilayer structure.
- a surface layer of a tread having a two-layer structure [a surface layer (cap tread) and an inner surface layer (base tread)].
- the pneumatic tire of the present invention is produced by a usual method using the rubber composition. That is, the rubber composition containing the above components is extruded in accordance with the shape of the tread at an unvulcanized stage and molded together with other tire members on a tire molding machine by a normal method. Form a vulcanized tire. The unvulcanized tire is heated and pressurized in a vulcanizer to obtain a tire.
- the pneumatic tire of the present invention is suitable for tires for passenger cars, large passenger cars, large SUV tires, heavy duty tires such as trucks and buses, and light truck tires (particularly preferably tires for passenger cars). It can be used as a tire or studless tire.
- Silica-modified SBR1 prepared by the method described below (oil- extended [containing 37.5 parts by mass of oil with respect to 100 parts by mass of rubber solid content), styrene content: 41% by mass, vinyl content: 40% by mass, Glass transition temperature: -29 ° C., weight average molecular weight: 1,190,000, SP value: 8.60)
- N9548 Nipol 9548 (E-SBR manufactured by Nippon Zeon Co., Ltd., oil- extended [containing 37.5 parts by mass of oil with respect to 100 parts by mass of rubber solid content), styrene content: 35 mass%, vinyl content: 18 mass Glass transition temperature: ⁇ 40 ° C., weight average molecular weight: 1.09 million, SP value: 8.50)
- Silica-modified SBR2 prepared by the method described below (non-oil extended, styrene content: 27 mass%, vinyl content: 58 mass%
- Aluminum hydroxide 5 dry pulverized product of ATH # B manufactured by Sumitomo Chemical Co., Ltd.
- G125 YS Polystar G125 (terpene phenol resin, hydrogenation rate: 0%, softening point: 125 ° C., Tg: 67 ° C., hydroxyl value: 140 mgKOH / g, SP value: 9.07) manufactured by Yasuhara Chemical Co., Ltd.
- TP115 Sylvares TP115 manufactured by Arizona Chemical Co., Ltd.
- SA85 SYLVARES SA85 ( ⁇ -methylstyrene resin [copolymer of ⁇ -methylstyrene and styrene], manufactured by Arizona Chemical Co., Ltd.), softening point: 85 ° C., Tg: 43 ° C., hydroxyl value: 0 mgKOH / g, SP value: 9.10)
- AH-24 Diana Process AH-24 (SP value: 8.05) manufactured by Idemitsu Kosan Co., Ltd.
- TMQ NOCRACK 224 (2,2,4-trimethyl-1,2-dihydroquinoline polymer) manufactured by Ouchi Shinsei Chemical Industry Co., Ltd.
- Stearic acid Stearic acid “ ⁇ ” manufactured by NOF Corporation
- Type 2 Zinc oxide made by Mitsui Mining & Smelting Co., Ltd.
- ⁇ vulcanizing agent> 5% oil-containing sulfur powder Hosoi Chemical Industry Co. of (Ltd.) HK-200-5 ⁇ Vulcanization accelerator>
- DPG Noxeller D (N, N-diphenylguanidine) manufactured by Ouchi Shinsei Chemical Co., Ltd.
- TBBS Noxeller NS-G (N-tert-butyl-2-benzothiazolylsulfenamide) manufactured by Ouchi Shinsei Chemical Industry Co., Ltd.
- silica-modified SBR1 In a 30-liter pressure vessel fully purged with nitrogen, add 18 liters of n-hexane, 800 g of styrene (manufactured by Kanto Chemical Co., Inc.), 1200 g of butadiene, 1.1 mmol of tetramethylethylenediamine, and warm to 40 ° C. did. Next, 1.8 mL of 1.6 M butyl lithium (manufactured by Kanto Chemical Co., Inc.) was added, and the mixture was heated to 50 ° C. and stirred for 3 hours. Next, 4.1 mL of the above terminal modifier was added and stirred for 30 minutes.
- reaction solution After adding 15 mL of methanol and 0.1 g of 2,6-tert-butyl-p-cresol to the reaction solution, the reaction solution was put into a stainless steel container containing 18 L of methanol to collect aggregates. The obtained aggregate was dried under reduced pressure for 24 hours to obtain silica-modified SBR2.
- Synthesis of hydrogenated terpene aromatic resin (Synthesis of Resin 1) Into a 3 L autoclave with stirring blades sufficiently purged with nitrogen, 200 g of 1 L of cyclohexane, 1 L of tetrahydrofuran (THF), and a raw material resin (TO125 [YS resin TO125 manufactured by Yasuhara Chemical Co., Ltd., measured softening point, 127 ° C.)] After adding 10 g of% palladium carbon and substituting with nitrogen, it was replaced with hydrogen so that the pressure became 5.0 kg / cm 2, and subjected to catalytic hydrogenation reaction at 80 ° C. for 0.5 hour to obtain Resin 1 It was. The yield was almost 100%.
- the hydrogenation rate of the double bond of Resin 1 was determined by adding resin (TO125 before hydrogenation or Resin 1 after hydrogenation) at a concentration of 15% by mass to carbon tetrachloride as a solvent, and mixing the mixture at 100 MHz.
- the proton NMR was calculated from the decrease in the spectrum corresponding to the unsaturated bond (hereinafter, the same applies to the measurement of the hydrogenation rate).
- the double bond hydrogenation rate (hydrogenation rate) of Resin 1 was approximately 2%.
- the hydroxyl value (OH value) of Resin 1 was 0 mgKOH / g.
- the softening point of Resin 1 was 123 ° C.
- the SP value of Resin 1 was 8.70.
- Resin 3 was obtained in the same manner as in (Synthesis of Resin 1) except that the reaction time of the catalytic hydrogenation reaction was 2 hours. The yield was almost 100%. Resin 3 had a double bond hydrogenation rate of approximately 8% and a hydroxyl value of 0 mgKOH / g. The softening point of the resin 3 was 124 ° C. The SP value of Resin 3 was 8.60.
- Examples and Comparative Examples> In accordance with the formulation shown in Table 2, compounding materials other than sulfur and a vulcanization accelerator were kneaded for 5 minutes at a discharge temperature of 150 ° C. using 4.0 L Banbury manufactured by Kobe Steel. Sulfur and a vulcanization accelerator were added to the obtained kneaded product, and kneaded for 4 minutes under the condition of a discharge temperature of 95 ° C. using an open roll to obtain an unvulcanized rubber composition. The obtained unvulcanized rubber composition was press vulcanized at 160 ° C. for 20 minutes to obtain a vulcanized rubber composition.
- the obtained unvulcanized rubber composition is extruded into a tread shape, bonded together with other tire members on a tire molding machine, vulcanized at 160 ° C. for 20 minutes, and a test tire (tire) Size: 215 / 45R17 summer, passenger car tire).
- the following evaluation was performed using the obtained vulcanized rubber composition and the test tire.
- the evaluation results are shown in Table 2.
- the softening point of the terpene resin was calculated according to the softening point and the composition ratio of the constituent polymer.
- the SP value of the diene rubber and the SP value of the terpene resin were calculated according to the SP value and the composition ratio of the constituting polymer.
- the test tire was mounted on a 2000 cc domestic FR vehicle, and the vehicle traveled 10 laps on a dry asphalt road test course. At that time, the test driver evaluated the stability of the control at the time of steering. The larger the index, the better the dry grip performance.
- the test tire was mounted on a 2000 cc domestic FR vehicle, and the vehicle was run for 10 laps on a wet asphalt road test course. At that time, the test driver evaluated the stability of the control at the time of steering. A larger index indicates better wet grip performance.
- the average of dry grip performance and wet grip performance targets is 103 or more.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
Abstract
Description
mM・xSiOy・zH2O
(式中、MはAl、Mg、Ti、Ca及びZrからなる群より選ばれた少なくとも1種の金属、該金属の酸化物又は水酸化物であり、mは1~5の整数、xは0~10の整数、yは2~5の整数、zは0~10の整数である。)
上記水素添加テルペン芳香族樹脂は、水素添加により樹脂の構造体のフレキシビリティーが高いため、分子量や軟化点の大きさの割にトレッド表面へのブルームが早い。また、水素添加により二重結合が少なくなるため、ジエン系ゴム中の該樹脂の分散性が大きく高まると共に、架橋剤の硫黄を吸着せずにゴムの架橋を促進し、ゴムポリマー間の架橋位置を均一化して加硫後のゴム組成物のモジュラスを大きくする。更に、ゴムの架橋が均一かつタイトになることにより耐久性、ブロー性も良好となる。また、水酸基価が20mgKOH/g以下であるためにゴム中での自己凝集性が低いことから、常温でのショア硬さ(Hs)が低く、かつ、高温Hsが維持される。すなわち、Hs温度依存が少ない。
上記効果は、ゴム成分としてジエン系ゴムを用いた場合に、特に顕著に発揮される。
ちなみに、樹脂を水素添加すると、一般的に熱安定性が向上し保管期間を長くすることができることから、水素添加テルペン芳香族樹脂をゴムに配合した場合、熱分解、酸化の進行が抑えられ臭気を抑えることができる。
(2)路面上の二酸化ケイ素とタイヤ表面上の水酸化アルミニウムなどの無機フィラーが走行中に接触する(擦れる)ことに伴って、瞬間的な共有結合が形成され、ウェットグリップ性能が向上すると考えられる。
ゴム組成物に特定の無機フィラーを配合することにより、特に小円旋回時、横滑り走行時にトレッドゴム表面に大きな張力が発生し、トレッドゴムの高周波振動が起こる。この高周波振動が1000Hz以上となると、(A)無機フィラーとゴム成分との界面にグリップレジンや粘着成分がブルームし、路面グリップを促進される、(B)好ましくは、無機フィラーが周辺のシリカ及びカーボンブラックと物理的又は化学的に結合し、走行中も無機フィラー周辺に大きなボイドが生じないこと、(C)特定の窒素吸着比表面積を有する微粒子状の無機フィラーによりゴム組成物のヒステリシスが上昇することが、ドライグリップ性能の向上に寄与するものと考えられる。
なお、本明細書において、単に「グリップ性能」という場合には、ウェットグリップ性能とドライグリップ性能とをまとめて表している。

なお、本明細書において、SBRのスチレン含量は、1H-NMR測定により算出される。
なお、本明細書において、SBRのビニル含量(1,2-結合ブタジエン単位量)は、赤外吸収スペクトル分析法によって測定できる。
なお、本明細書において、SBRのガラス転移温度は、JIS K7121に従い、昇温速度10℃/分の条件で示差走査熱量測定(DSC)を行って測定される値である。
なお、本明細書において、SBRの重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)(東ソー(株)製GPC-8000シリーズ、検出器:示差屈折計、カラム:東ソー(株)製のTSKGEL SUPERMALTPORE HZ-M)による測定値を基に標準ポリスチレン換算により求めることができる。
なお、本明細書において、BRのビニル含量(1,2-結合ブタジエン単位量)及びシス含量(シス1,4結合含有率)は、赤外吸収スペクトル分析法によって測定できる。
なお、芳香族化合物とテルペン化合物との共重合割合は、水素添加テルペン芳香族樹脂が後述する物性を有するものとなるように適宜設定することができる。また、水素添加テルペン芳香族樹脂が後述する物性を有するものとなる限り、上記テルペン芳香族樹脂は、芳香族化合物及びテルペン化合物以外の共重合単位、例えばインデン等、を含んでいてもよい。
なお、上記芳香族化合物は、芳香環上に置換基を1つ有していてもよいし、2つ以上有していてもよく、芳香環上の置換基が2つ以上の場合、それらの置換位置は、o位、m位、p位のいずれであってもよい。更に芳香環上に置換基を有するスチレン誘導体においては、該置換基の置換位置はスチレン由来のビニル基に対してo位であってもよいし、m位、又はp位であってもよい。
これら芳香族化合物は、単独で用いてもよいし、2種以上を併用してもよい。
上記不飽和炭化水素基としては、炭素数2~20のアルケニル基が挙げられる。
これらテルペン化合物は、単独で用いてもよいし、2種以上を併用してもよい。

なお、該水素添加率(水添率)は、1H-NMR(プロトンNMR)による二重結合由来ピークの各積分値から、下記式により、算出される値である。本明細書において、水素添加率(水添率)とは、二重結合の水素添加率を意味する。
(水添率〔%〕)={(A-B)/A}×100
A:水素添加前の二重結合のピークの積分値
B:水素添加後の二重結合のピークの積分値
なお、上記水素添加テルペン芳香族樹脂の水酸基価は、水素添加テルペン芳香族樹脂1gをアセチル化するとき、水酸基と結合した酢酸を中和するのに要する水酸化カリウムの量をミリグラム数で表したものであり、電位差滴定法(JIS K0070:1992)により測定した値である。
なお、水素添加テルペン芳香族樹脂の軟化点は、JIS K6220-1:2001に規定される軟化点を環球式軟化点測定装置で測定し、球が降下した温度である。
なお、本明細書において、水素添加テルペン芳香族樹脂のガラス転移温度は、JIS K7121に従い、昇温速度10℃/分の条件で示差走査熱量測定(DSC)を行って測定される値である。
なお、本明細書において、水素添加テルペン芳香族樹脂の重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)(東ソー(株)製GPC-8000シリーズ、検出器:示差屈折計、カラム:東ソー(株)製のTSKGEL SUPERMALTPORE HZ-M)による測定値を基に標準ポリスチレン換算により求めることができる。
なお、ジエン系ゴム、水素添加テルペン芳香族樹脂のSP値は、化合物の構造に基づいてHoy法によって算出された溶解度パラメーター(Solubility Parameter)を意味する。Hoy法とは、例えば、K.L.Hoy “Table of Solubility Parameters”,Solvent and Coatings Materials Reserch and Development Department,Union Carbites Corp.(1985)に記載された計算方法である。
mM・xSiOy・zH2O
(式中、MはAl、Mg、Ti、Ca及びZrからなる群より選ばれた少なくとも1種の金属、該金属の酸化物又は水酸化物であり、mは1~5の整数、xは0~10の整数、yは2~5の整数、zは0~10の整数である。)
なお、無機フィラーのN2SAは、ASTM D3037-81に準じてBET法で測定される値である。
参考に、粒子ストラクチャーが発達しやすい代表的湿式シリカであるEvonik社製のULTRASIL VN3(N2SA:175m2/g)の亜麻仁油吸油量は128ml/100gである。
なお、上記亜麻仁油吸油量は、JIS K5101-13に従って求められる値である。
なお、無機フィラーの平均粒子径は、数平均粒子径であり、透過型電子顕微鏡により測定される。
モース硬度は、材料の機械的性質の一つで古くから鉱物関係で汎用されている測定法であり、硬さを計りたい物質(水酸化アルミニウム等)を標準物質でこすり、ひっかき傷の有無でモース硬度を測定する。
また、必要に応じて、医薬、バイオ関係で頻用されるメンブランフィルター法にて分取し、所定のN2SAを有するものを作製し、ゴム配合剤として使用することもできる。
中でも、乗用車用タイヤでは、グリップ性能と耐摩耗性の両立の観点から、上記ジエン系ゴム100質量部に対して、10~20質量部が好ましい。
シリカとしては、例えば、湿式法で製造されたシリカ、乾式法で製造されたシリカなどが挙げられる。
なお、シリカのN2SAは、ASTM D3037-93に準じてBET法で測定される値である。
なお、オイルのSP値は、上記ジエン系ゴム、水素添加テルペン芳香族樹脂のSP値と同様にして算出される。
なお、本明細書において、オイルの含有量には、油展ゴムに含まれるオイル量も含まれる。
ただし、トラック・バス用タイヤでは、耐摩耗性、耐久性、欠け性が高度に期待される為、オイルを配合しない場合が多い。
液状ジエン系重合体は、ゲルパーミエーションクロマトグラフィー(GPC)で測定したポリスチレン換算の重量平均分子量(Mw)が、1.0×103~2.0×105であることが好ましく、3.0×103~1.5×104であることがより好ましい。1.0×103未満では、グリップ性能の向上効果がなく、充分な耐久性が確保できないおそれがある。一方、2.0×105を超えると、重合溶液の粘度が高くなり過ぎ生産性が悪化したり、破壊特性が低下したりするおそれがある。
なお、本明細書において、液状ジエン系重合体のMwは、ゲルパーミエーションクロマトグラフィー(GPC)で測定したポリスチレン換算値である。
上記クマロンインデン樹脂のなかでも、軟化点が90~140℃のものを用いた場合には、グリップ性能が向上する。なかでも軟化点が100~120℃のものは、0~80℃におけるtanδを全般に高めることができ、耐久性も良い。
軟化点が10~30℃のクマロンインデン樹脂を用いた場合には、10~40℃の比較的低温におけるグリップ性は良く、tanδを全般に下げる。このような軟化点が10~30℃のクマロンインデン樹脂は、主に耐久性の向上を目的に用いることができる。
なお、クマロンインデン樹脂を用いることにより耐久性が改善する理由は、架橋ポリマー鎖に適度な滑りを付与し、均一な伸びを生じさせるためと考えられる。
なお、本明細書において、上記クマロンインデン樹脂の軟化点は、JIS K6220-1:2001に規定される軟化点を環球式軟化点測定装置で測定し、球が降下した温度である。
なお、p-t-ブチルフェノールアセチレン樹脂の軟化点は、上記クマロンインデン樹脂の軟化点の測定方法と同様の方法で測定することができる。
なお、上記p-t-ブチルフェノールアセチレン樹脂の水酸基価は、上述した水素添加テルペン芳香族樹脂の水酸基価と同様にして測定することができる。
なお、酸化亜鉛の平均一次粒子径は、窒素吸着によるBET法により測定した比表面積から換算された平均粒子径(平均一次粒子径)を表す。また、窒素吸着によるBET法により測定した比表面積(N2SA)は10~50m2/gであることが好ましい。
先ず、バンバリーミキサー、オープンロールなどのゴム混練装置に硫黄及び加硫促進剤以外の成分を配合(添加)して混練りした後(ベース練り工程)、得られた混練物に、更に硫黄及び加硫促進剤を配合(添加)して混練りしその後加硫する方法などにより製造できる。
すなわち、前記成分を配合したゴム組成物を、未加硫の段階でトレッドの形状にあわせて押出し加工し、他のタイヤ部材とともに、タイヤ成型機上にて通常の方法で成形することにより、未加硫タイヤを形成する。この未加硫タイヤを加硫機中で加熱加圧することによりタイヤを得る。
<SBR>
シリカ変性SBR1:以下で説明する方法により調製したもの(油展〔ゴム固形分100質量部に対してオイル分37.5質量部含有〕、スチレン含量:41質量%、ビニル含量:40質量%、ガラス転移温度:-29℃、重量平均分子量:119万、SP値:8.60)
N9548:日本ゼオン(株)製のNipol 9548(E-SBR、油展〔ゴム固形分100質量部に対してオイル分37.5質量部含有〕、スチレン含量:35質量%、ビニル含量:18質量%、ガラス転移温度:-40℃、重量平均分子量:109万、SP値:8.50)
シリカ変性SBR2:以下で説明する方法により調製したもの(非油展、スチレン含量:27質量%、ビニル含量:58質量%、ガラス転移温度:-27℃、重量平均分子量:72万、SP値:8.55)
NS612:日本ゼオン(株)製のNipol NS612(S-SBR、非油展、スチレン含量:15質量%、ビニル含量:30質量%、ガラス転移温度:-65℃、重量平均分子量:78万、SP値:8.40)
<BR>
CB25:ランクセス社製のBUNA-CB25(Nd系触媒を用いて合成した希土類系BR、ビニル含量:0.7質量%、シス含量:97質量%、ガラス転移温度:-110℃、SP値:8.20)
<NR>
TSR20(SP値:8.10)
<カーボンブラック>
HP180:オリオンエンジニアドカーボンズ社製のHP180(N2SA:175m2/g)
<シリカ>
VN3:Evonik社製のULTRASIL VN3(N2SA:175m2/g、亜麻仁油吸油量:128ml/100g)
<水酸化アルミニウム>
水酸化アルミニウム1:戸田工業(株)製の湿式合成品(平均粒子径:2.7μm、N2SA:274m2/g、亜麻仁油吸油量:104ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム2:戸田工業(株)製の湿式合成品(平均粒子径:2.7μm、N2SA:122m2/g、亜麻仁油吸油量:78ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム3:住友化学(株)製のATH#Bの乾式粉砕品(平均粒子径:0.5μm、N2SA:95m2/g、亜麻仁油吸油量:42ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム4:住友化学(株)製のATH#Bの乾式粉砕品(平均粒子径:0.5μm、N2SA:75m2/g、亜麻仁油吸油量:42ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム5:住友化学(株)製のATH#Bの乾式粉砕品(平均粒子径:0.3μm、N2SA:35m2/g、亜麻仁油吸油量:37ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム6:住友化学(株)製のATH#B(平均粒子径:0.6μm、N2SA:15m2/g、亜麻仁油吸油量:40ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
水酸化アルミニウム7:昭和電工(株)製のハイジライトH-43(平均粒子径:0.75μm、N2SA:7m2/g、亜麻仁油吸油量:33ml/100g、モース硬度:3、熱分解物(アルミナ)のモース硬度:9、熱分解開始温度:200℃)
<テルペン系樹脂>
M125:ヤスハラケミカル(株)製のYSポリスターM125(水添率:11%、軟化点:123℃、Tg:69℃、水酸基価:0mgKOH/g、SP値:8.52)
M115:ヤスハラケミカル(株)製のYSポリスターM115(水添率:12%、軟化点:115℃、Tg:59℃、水酸基価:0mgKOH/g、SP値:8.52)
M105:ヤスハラケミカル(株)製のYSポリスターM105(水添率:12%、軟化点:105℃、Tg:48℃、水酸基価:0mgKOH/g、SP値:8.52)
M80:ヤスハラケミカル(株)製のYSポリスターM80(水添率:12%、軟化点:80℃、Tg:23℃、水酸基価:0mgKOH/g、SP値:8.52)
樹脂1~4:後述する方法により製造されたもの
TO125:ヤスハラケミカル(株)製のYSレジンTO125(芳香族変性テルペン樹脂、水添率:0%、軟化点:125℃、Tg:64℃、水酸基価:0mgKOH/g、SP値:8.73)
TO115:ヤスハラケミカル(株)製のYSレジンTO115(芳香族変性テルペン樹脂、水添率:0%、軟化点:115℃、Tg:54℃、水酸基価:0mgKOH/g、SP値:8.73)
TO105:ヤスハラケミカル(株)製のYSレジンTO105(芳香族変性テルペン樹脂、水添率:0%、軟化点:105℃、Tg:45℃、水酸基価:0mgKOH/g、SP値:8.73)
TO85:ヤスハラケミカル(株)製のYSレジンTO85(芳香族変性テルペン樹脂、水添率:0%、軟化点:85℃、Tg:25℃、水酸基価:0mgKOH/g、SP値:8.73)
T160:ヤスハラケミカル(株)製のYSポリスターT160(テルペンフェノール樹脂、水添率:0%、軟化点:160℃、Tg:100℃、水酸基価:60mgKOH/g、SP値:8.81)
G125:ヤスハラケミカル(株)製のYSポリスターG125(テルペンフェノール樹脂、水添率:0%、軟化点:125℃、Tg:67℃、水酸基価:140mgKOH/g、SP値:9.07)
TP115:アリゾナケミカル社製のSylvaresTP115(テルペンフェノール樹脂、水添率:0%、軟化点:115℃、Tg:55℃、OH価:50mgKOH/g、SP値:8.77)
<石油系樹脂>
コレシン:BASF社製のKoresinの乾式粉砕品(p-t-ブチルフェノールアセチレン樹脂〔p-t-ブチルフェノールとアセチレンの縮合樹脂〕、軟化点:145℃、Tg:98℃、水酸基価:193mgKOH/g、N2SA:4.1m2/g、SP値:9.10)
V-120:日塗化学(株)製のニットレジン クマロン V-120(クマロンインデン樹脂、軟化点:120℃、水酸基価:30mgKOH/g、SP値:9.00)
SA85:アリゾナケミカル社製のSYLVARES SA85(αメチルスチレン系樹脂〔α-メチルスチレンとスチレンとの共重合体〕、軟化点:85℃、Tg:43℃、水酸基価:0mgKOH/g、SP値:9.10)
<オイル>
AH-24:出光興産(株)製のダイアナプロセスAH-24(SP値:8.05)
<液状ジエン系重合体>
L-SBR-820:(株)クラレ製のL-SBR-820(液状SBR、Mw:10,000)
<シランカップリング剤>
Si75:Evonik社製のシランカップリング剤Si75(ビス(3-トリエトキシシリルプロピル)ジスルフィド)
<ワックス>
Ozoace355:日本精鑞(株)製のOzoace355
<老化防止剤>
6PPD:住友化学(株)製のアンチゲン6C(N-フェニル-N’-(1,3-ジメチル)-p-フェニレンジアミン)
TMQ:大内新興化学工業(株)製のノクラック224(2,2,4-トリメチル-1,2-ジヒドロキノリン重合体)
<ステアリン酸>
ステアリン酸:日油(株)製のステアリン酸「椿」
<酸化亜鉛>
第2種:三井金属鉱業(株)製の第2種酸化亜鉛
<加硫剤>
5%オイル含有粉末硫黄:細井化学工業(株)製のHK-200-5
<加硫促進剤>
DPG:大内新興化学工業(株)製のノクセラーD(N,N-ジフェニルグアニジン)
TBBS:大内新興化学工業(株)製のノクセラーNS-G(N-tert-ブチル-2-ベンゾチアゾリルスルフェンアミド)
窒素雰囲気下、250mlメスフラスコに3-(N,N-ジメチルアミノ)プロピルトリメトキシシラン(アヅマックス(株)製)を20.8g入れ、さらに無水ヘキサン(関東化学(株)製)を加え、全量を250mlにして作製した。
充分に窒素置換した30L耐圧容器にn-ヘキサンを18L、スチレン(関東化学(株)製)を800g、ブタジエンを1200g、テトラメチルエチレンジアミンを1.1mmol加え、40℃に昇温した。次に、1.6Mブチルリチウム(関東化学(株)製)を1.8mL加えた後、50℃に昇温させ3時間撹拌した。次に上記末端変性剤を4.1mL追加し30分間撹拌を行った。反応溶液にメタノール15mL及び2,6-tert-ブチル-p-クレゾール(大内新興化学工業(株)製)0.1gを添加後、TDAE1200g添加し10分間撹拌を行った。その後、スチームストリッピング処理によって重合体溶液から凝集体を回収した。得られた凝集体を24時間減圧乾燥させ、シリカ変性SBR1を得た。
充分に窒素置換した30L耐圧容器にn-ヘキサンを18L、スチレン(関東化学(株)製)を740g、ブタジエンを1260g、テトラメチルエチレンジアミンを10mmol加え、40℃に昇温した。次に、ブチルリチウムを10mL加えた後、50℃に昇温させ3時間撹拌した。次に、上記末端変性剤を11mL追加し30分間撹拌を行った。反応溶液にメタノール15mL及び2,6-tert-ブチル-p-クレゾール0.1gを添加後、反応溶液を18Lのメタノールが入ったステンレス容器に入れて凝集体を回収した。得られた凝集体を24時間減圧乾燥させ、シリカ変性SBR2を得た。
(樹脂1の合成)
充分に窒素置換した撹拌翼つきの3Lオートクレーブに、シクロヘキサン1L、テトラヒドロフラン(THF)1L、原料樹脂(TO125〔ヤスハラケミカル(株)製のYSレジンTO125〕の実測軟化点127℃のロット品)を200g、10%パラジウムカーボン10gを加え、窒素置換した後、圧力が5.0kg/cm2となるように水素置換して、80℃で、0.5時間、接触水素添加反応に付し、樹脂1を得た。収率は、ほぼ100%であった。
接触水素添加反応の反応時間を1時間とした以外は、(樹脂1の合成)と同様にして、樹脂2を得た。収率は、ほぼ100%であった。樹脂2の二重結合の水素添加率はほぼ5%、水酸基価は0mgKOH/gであった。樹脂2の軟化点は、123℃であった。樹脂2のSP値は、8.68であった。
接触水素添加反応の反応時間を2時間とした以外は、(樹脂1の合成)と同様にして、樹脂3を得た。収率は、ほぼ100%であった。樹脂3の二重結合の水素添加率はほぼ8%、水酸基価は0mgKOH/gであった。樹脂3の軟化点は、124℃であった。樹脂3のSP値は、8.60であった。
接触水素添加反応の反応時間を4時間とした以外は、(樹脂1の合成)と同様にして、樹脂4を得た。収率は、ほぼ100%であった。樹脂4の二重結合の水素添加率はほぼ20%、水酸基価は0mgKOH/gであった。樹脂4の軟化点は、127℃であった。樹脂4のSP値は、8.48であった。

表2に示す配合処方に従い、神戸製鋼(株)製4.0Lバンバリーを用いて硫黄及び加硫促進剤以外の配合材料を排出温度150℃の条件下で5分間混練りした。得られた混練り物に硫黄及び加硫促進剤を添加し、オープンロールを用いて排出温度95℃の条件下で4分間練り込み、未加硫ゴム組成物を得た。
得られた未加硫ゴム組成物を160℃の条件下で20分間プレス加硫し、加硫ゴム組成物を得た。
また、得られた未加硫ゴム組成物をトレッドの形状に押出し成形し、タイヤ成形機上で他のタイヤ部材とともに貼り合わせ、160℃の条件下で20分間加硫し、試験用タイヤ(タイヤサイズ:215/45R17サマー、乗用車用タイヤ)を得た。
なお、テルペン系樹脂の軟化点は、構成するポリマーの軟化点と構成比率に応じて計算した。また、ジエン系ゴムのSP値及びテルペン系樹脂のSP値は、構成するポリマーのSP値と構成比率に応じて計算した。
上記試験用タイヤを排気量2000ccの国産FR車に装着し、ドライアスファルト路面のテストコースにて10周の実車走行を行った。その際における、操舵時のコントロールの安定性をテストドライバーが評価し、比較例1を100として指数表示をした。指数が大きいほどドライグリップ性能に優れることを示す。
上記試験用タイヤを排気量2000ccの国産FR車に装着し、ウェットアスファルト路面のテストコースにて10周の実車走行を行った。その際における、操舵時のコントロールの安定性をテストドライバーが評価し、比較例1を100として指数表示をした。指数が大きいほどウェットグリップ性能に優れることを示す。
なお、ドライグリップ性能及びウェットグリップ性能の目標はそれらの平均が103以上である。
上記加硫ゴム組成物からなる3号ダンベル型試験片を用いて、JIS K6251「加硫ゴム及び熱可塑性ゴム-引張特性の求め方」に準じて、室温にて引張試験を実施して、破断時伸びEB(%)を測定し、比較例1を100として指数表示した。指数が大きいほど耐久性に優れることを示す。

そして、ポリマー系はSBR、BR、NR量、フィラー系はカーボンブラック量、シリカ量によらず、性能向上できると判明した。
Claims (6)
- ゴム成分100質量%中ジエン系ゴムを90質量%以上含有するゴム組成物を用いて作製したトレッドを有する空気入りタイヤであって、
前記ゴム組成物は、更に、テルペン芳香族樹脂の二重結合を水素添加して得られる水素添加テルペン芳香族樹脂を含有し、
前記二重結合の水素添加率が5~100%であり、
前記水素添加テルペン芳香族樹脂の水酸基価が20mgKOH/g以下であり、
前記ジエン系ゴム100質量部に対して、前記水素添加テルペン芳香族樹脂の含有量が1~50質量部であり、
前記ゴム組成物は、更に、下記式で表される化合物、硫酸マグネシウム、及び炭化ケイ素からなる群より選択される少なくとも1種からなり、窒素吸着比表面積が10~120m2/gである無機フィラーを含有し、
前記ジエン系ゴム100質量部に対して、前記無機フィラーの含有量が1~70質量部である空気入りタイヤ。
mM・xSiOy・zH2O
(式中、MはAl、Mg、Ti、Ca及びZrからなる群より選ばれた少なくとも1種の金属、該金属の酸化物又は水酸化物であり、mは1~5の整数、xは0~10の整数、yは2~5の整数、zは0~10の整数である。) - 前記無機フィラーが、水酸化アルミニウムである請求項1記載の空気入りタイヤ。
- 前記水素添加テルペン芳香族樹脂の軟化点が、80~180℃である請求項1又は2記載の空気入りタイヤ。
- 前記水素添加テルペン芳香族樹脂の軟化点が、114~160℃である請求項3記載の空気入りタイヤ。
- 前記水素添加テルペン芳香族樹脂の水酸基価が、0mgKOH/gである請求項1~4のいずれかに記載の空気入りタイヤ。
- 前記ジエン系ゴムが、スチレン含量19~60質量%のスチレンブタジエンゴムを60質量%以上含む請求項1~5のいずれかに記載の空気入りタイヤ。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016512126A JP6072979B2 (ja) | 2014-12-24 | 2015-12-09 | 空気入りタイヤ |
| EP15872713.1A EP3228658B1 (en) | 2014-12-24 | 2015-12-09 | Pneumatic tire |
| US15/534,831 US10266008B2 (en) | 2014-12-24 | 2015-12-09 | Pneumatic tire |
| CN201580066580.7A CN107001712B (zh) | 2014-12-24 | 2015-12-09 | 充气轮胎 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-260947 | 2014-12-24 | ||
| JP2014260947 | 2014-12-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016104144A1 true WO2016104144A1 (ja) | 2016-06-30 |
Family
ID=56150174
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/084476 Ceased WO2016104144A1 (ja) | 2014-12-24 | 2015-12-09 | 空気入りタイヤ |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10266008B2 (ja) |
| EP (1) | EP3228658B1 (ja) |
| JP (1) | JP6072979B2 (ja) |
| CN (1) | CN107001712B (ja) |
| WO (1) | WO2016104144A1 (ja) |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018024844A (ja) * | 2016-07-29 | 2018-02-15 | 株式会社ブリヂストン | ゴム組成物及びそれを用いた更生タイヤ及びその更生タイヤの製造方法 |
| JP2018048294A (ja) * | 2016-09-22 | 2018-03-29 | クムホ タイヤ カンパニー インコーポレーテッド | タイヤトレッド用ゴム組成物 |
| EP3323630A1 (en) * | 2016-11-16 | 2018-05-23 | Sumitomo Rubber Industries, Ltd. | Rubber composition for treads and pneumatic tire |
| JP2018095877A (ja) * | 2016-12-15 | 2018-06-21 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| EP3345775A1 (en) * | 2017-01-04 | 2018-07-11 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tread and pneumatic tire |
| JP2018135410A (ja) * | 2017-02-20 | 2018-08-30 | 住友ゴム工業株式会社 | ゴム組成物、タイヤトレッドおよび空気入りタイヤ |
| EP3385319A1 (en) * | 2017-04-07 | 2018-10-10 | Sumitomo Rubber Industries, Ltd. | Tire |
| US20180291190A1 (en) * | 2017-04-07 | 2018-10-11 | Sumitomo Rubber Industries, Ltd. | Tire |
| JP2018168217A (ja) * | 2017-03-29 | 2018-11-01 | 住友ベークライト株式会社 | 封止用樹脂組成物及びこれを用いた半導体装置 |
| US10266008B2 (en) | 2014-12-24 | 2019-04-23 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JP2019073580A (ja) * | 2017-10-12 | 2019-05-16 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| JP2019073579A (ja) * | 2017-10-12 | 2019-05-16 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| CN109843942A (zh) * | 2016-10-14 | 2019-06-04 | 科腾化学品有限责任公司 | 含有改进的胎面补强添加剂的橡胶组合物及其用途 |
| JP2019112598A (ja) * | 2017-12-26 | 2019-07-11 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2019137835A (ja) * | 2018-02-05 | 2019-08-22 | ハンコック タイヤ カンパニー リミテッド | タイヤトレッド用ゴム組成物、及びそれを用いて製造したタイヤ |
| JP2019524950A (ja) * | 2016-08-17 | 2019-09-05 | コンティネンタル・ライフェン・ドイチュラント・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | 硫黄架橋性ゴム混合物および車両用タイヤ |
| JP2019218118A (ja) * | 2018-06-21 | 2019-12-26 | 三井化学株式会社 | 医療用容器および医療用容器用環状オレフィン系樹脂組成物 |
| JP2020023600A (ja) * | 2018-08-06 | 2020-02-13 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| JP2020055980A (ja) * | 2018-10-04 | 2020-04-09 | 住友ゴム工業株式会社 | トレッドゴム組成物及び空気入りタイヤ |
| JP2020070330A (ja) * | 2018-10-30 | 2020-05-07 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2020070329A (ja) * | 2018-10-30 | 2020-05-07 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2020076110A (ja) * | 2018-10-04 | 2020-05-21 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| WO2020100492A1 (ja) * | 2018-11-12 | 2020-05-22 | 住友ゴム工業株式会社 | ゴム組成物及び空気入りタイヤ |
| JP2020105380A (ja) * | 2018-12-27 | 2020-07-09 | Toyo Tire株式会社 | タイヤ用ゴム組成物、及びそれを用いた空気入りタイヤ |
| WO2021033713A1 (ja) * | 2019-08-21 | 2021-02-25 | 横浜ゴム株式会社 | 空気入りタイヤ |
| JP2021523959A (ja) * | 2018-05-04 | 2021-09-09 | ブリヂストン アメリカズ タイヤ オペレーションズ、 エルエルシー | タイヤトレッドゴム組成物 |
| US11499038B2 (en) | 2017-12-26 | 2022-11-15 | Bridgestone Corporation | Tread composition and tire produced by using the same |
| JPWO2023112364A1 (ja) * | 2021-12-14 | 2023-06-22 | ||
| WO2024038812A1 (ja) * | 2022-08-15 | 2024-02-22 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| WO2024080242A1 (ja) * | 2022-10-14 | 2024-04-18 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| US12054614B2 (en) | 2018-11-12 | 2024-08-06 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| US12173140B2 (en) | 2018-11-12 | 2024-12-24 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| US12410308B2 (en) | 2018-11-12 | 2025-09-09 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3228657B1 (en) * | 2014-12-24 | 2019-10-23 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JP6733223B2 (ja) * | 2016-03-07 | 2020-07-29 | 住友ゴム工業株式会社 | 高性能タイヤ |
| US12227651B2 (en) | 2016-08-17 | 2025-02-18 | Continental Reifen Deutschland Gmbh | Rubber blend, sulfur-crosslinkable rubber mixture, and vehicle tire |
| JP7009768B2 (ja) * | 2017-04-07 | 2022-01-26 | 住友ゴム工業株式会社 | ゴム組成物およびタイヤ |
| JP2021523260A (ja) | 2018-05-04 | 2021-09-02 | ブリヂストン アメリカズ タイヤ オペレーションズ、 エルエルシー | タイヤトレッドゴム組成物 |
| US12365202B2 (en) | 2018-05-04 | 2025-07-22 | Bridgestone Americas Tire Operations, Llc | Tire tread rubber composition |
| WO2019213226A1 (en) | 2018-05-04 | 2019-11-07 | Bridgestone Americas Tire Operations, Llc | Tire tread rubber composition |
| CN109822776A (zh) * | 2019-02-18 | 2019-05-31 | 哈尔滨博实自动化股份有限公司 | 低分子液体橡胶制备装置 |
| DE102019203924A1 (de) * | 2019-03-22 | 2020-09-24 | Continental Reifen Deutschland Gmbh | Fahrzeugluftreifen |
| JP2022535725A (ja) | 2019-05-29 | 2022-08-10 | ブリヂストン アメリカズ タイヤ オペレーションズ、 エルエルシー | タイヤトレッドゴム組成物及び関連する方法 |
| JP2022534568A (ja) | 2019-05-29 | 2022-08-02 | ブリヂストン アメリカズ タイヤ オペレーションズ、 エルエルシー | タイヤトレッドゴム組成物及びその関連方法 |
| EP3976393A1 (en) | 2019-05-29 | 2022-04-06 | Bridgestone Americas Tire Operations, LLC | Tire tread rubber composition and related methods |
| WO2020246218A1 (ja) * | 2019-06-04 | 2020-12-10 | 住友ゴム工業株式会社 | ゴム組成物及びタイヤ |
| JP7392356B2 (ja) * | 2019-09-27 | 2023-12-06 | 住友ゴム工業株式会社 | タイヤ |
| JP6874809B2 (ja) * | 2019-10-23 | 2021-05-19 | 住友ゴム工業株式会社 | ゴム組成物及びタイヤ |
| US20230043282A1 (en) * | 2019-12-19 | 2023-02-09 | Bridgestone Corporation | Rubber composition and tire |
| JP7478754B2 (ja) * | 2019-12-19 | 2024-05-07 | 株式会社ブリヂストン | タイヤ |
| US12006436B2 (en) | 2020-11-13 | 2024-06-11 | The Goodyear Tire & Rubber Company | Rubber composition and a tire |
| JP7168029B1 (ja) * | 2021-05-12 | 2022-11-09 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| EP4349613A4 (en) * | 2021-05-28 | 2024-09-11 | Bridgestone Corporation | Rubber composition for tire, tread rubber, and tire |
| US20240270945A1 (en) * | 2021-05-28 | 2024-08-15 | Bridgestone Corporation | TIRE RUBBER COMPOSITION, TREAD RUBBER, and TIRE |
| JP7815685B2 (ja) * | 2021-10-28 | 2026-02-18 | 住友ゴム工業株式会社 | 重荷重用タイヤ |
| US11987705B2 (en) | 2022-05-26 | 2024-05-21 | The Goodyear Tire & Rubber Company | Rubber composition and a tire comprising a rubber composition |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002020655A1 (en) * | 2000-09-06 | 2002-03-14 | Jsr Corporation | Diene rubber/inorganic compound composite and method for producing the same and rubber composition |
| JP2007177209A (ja) * | 2005-11-29 | 2007-07-12 | Sumitomo Rubber Ind Ltd | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2008184505A (ja) * | 2007-01-29 | 2008-08-14 | Bridgestone Corp | タイヤ用ゴム組成物及びそれを用いた空気入りタイヤ。 |
| JP2009138025A (ja) * | 2007-12-03 | 2009-06-25 | Bridgestone Corp | タイヤ用ゴム組成物及びそれを用いたタイヤ |
| JP2013166826A (ja) * | 2012-02-14 | 2013-08-29 | Sumitomo Rubber Ind Ltd | タイヤ用ゴム組成物及びウインタータイヤ |
| WO2015104955A1 (ja) * | 2014-01-07 | 2015-07-16 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| WO2016002506A1 (ja) * | 2014-07-02 | 2016-01-07 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2594809B2 (ja) | 1988-04-02 | 1997-03-26 | 日本ゼオン株式会社 | タイヤトレッド用ゴム組成物 |
| EP0807603B2 (en) * | 1996-05-16 | 2007-08-29 | Sumitomo Chemical Company, Limited | Aluminum hydroxide, method for producing the same, and use of the same |
| JP3628105B2 (ja) | 1996-05-22 | 2005-03-09 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| ES2283494T3 (es) * | 1997-07-11 | 2007-11-01 | Bridgestone Corporation | Neumatico. |
| EP1514901B1 (en) * | 2002-06-19 | 2011-06-01 | Bridgestone Corporation | Rubber composition for tire and tire made therefrom |
| JP4955191B2 (ja) | 2002-08-21 | 2012-06-20 | 住友ゴム工業株式会社 | タイヤトレッド用ゴム組成物 |
| JP4615874B2 (ja) | 2004-02-02 | 2011-01-19 | 株式会社ブリヂストン | ゴム組成物、これを用いたタイヤ及びゴム組成物の製造方法 |
| JP2005350535A (ja) | 2004-06-09 | 2005-12-22 | Sumitomo Rubber Ind Ltd | タイヤ用ゴム組成物 |
| DE602006004826D1 (de) * | 2005-11-29 | 2009-03-05 | Sumitomo Rubber Ind | Kautschukzusammensetzung und Luftreifen, der diese verwendet |
| JP5334234B2 (ja) | 2007-02-27 | 2013-11-06 | 株式会社ブリヂストン | ゴム組成物及びそれを用いた空気入りタイヤ |
| EP2103650B1 (en) | 2007-01-11 | 2012-08-08 | Bridgestone Corporation | Rubber composition and tire using the same |
| JP2011088998A (ja) | 2009-10-21 | 2011-05-06 | Bridgestone Corp | ゴム組成物及びタイヤ |
| JP4849176B2 (ja) | 2010-02-26 | 2012-01-11 | 横浜ゴム株式会社 | タイヤトレッド用ゴム組成物およびそれを用いた空気入りタイヤ |
| JP4835769B2 (ja) * | 2010-05-26 | 2011-12-14 | 横浜ゴム株式会社 | タイヤトレッド用ゴム組成物 |
| JP5658098B2 (ja) | 2011-06-16 | 2015-01-21 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| JP5234203B2 (ja) * | 2011-07-14 | 2013-07-10 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP5913188B2 (ja) | 2013-04-30 | 2016-04-27 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及び空気入りタイヤ |
| JP5829734B2 (ja) | 2014-08-15 | 2015-12-09 | 株式会社ブリヂストン | タイヤ用ゴム組成物及びそれを用いたタイヤ |
| EP3228657B1 (en) | 2014-12-24 | 2019-10-23 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| WO2016104144A1 (ja) | 2014-12-24 | 2016-06-30 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| JP6355581B2 (ja) * | 2015-03-20 | 2018-07-11 | 住友ゴム工業株式会社 | 高分子複合体及びその製造方法、並びに、タイヤ用ゴム組成物及び空気入りタイヤ |
-
2015
- 2015-12-09 WO PCT/JP2015/084476 patent/WO2016104144A1/ja not_active Ceased
- 2015-12-09 EP EP15872713.1A patent/EP3228658B1/en not_active Not-in-force
- 2015-12-09 CN CN201580066580.7A patent/CN107001712B/zh not_active Expired - Fee Related
- 2015-12-09 JP JP2016512126A patent/JP6072979B2/ja active Active
- 2015-12-09 US US15/534,831 patent/US10266008B2/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002020655A1 (en) * | 2000-09-06 | 2002-03-14 | Jsr Corporation | Diene rubber/inorganic compound composite and method for producing the same and rubber composition |
| JP2007177209A (ja) * | 2005-11-29 | 2007-07-12 | Sumitomo Rubber Ind Ltd | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2008184505A (ja) * | 2007-01-29 | 2008-08-14 | Bridgestone Corp | タイヤ用ゴム組成物及びそれを用いた空気入りタイヤ。 |
| JP2009138025A (ja) * | 2007-12-03 | 2009-06-25 | Bridgestone Corp | タイヤ用ゴム組成物及びそれを用いたタイヤ |
| JP2013166826A (ja) * | 2012-02-14 | 2013-08-29 | Sumitomo Rubber Ind Ltd | タイヤ用ゴム組成物及びウインタータイヤ |
| WO2015104955A1 (ja) * | 2014-01-07 | 2015-07-16 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| WO2016002506A1 (ja) * | 2014-07-02 | 2016-01-07 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3228658A4 * |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10266008B2 (en) | 2014-12-24 | 2019-04-23 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JP2018024844A (ja) * | 2016-07-29 | 2018-02-15 | 株式会社ブリヂストン | ゴム組成物及びそれを用いた更生タイヤ及びその更生タイヤの製造方法 |
| JP7129764B2 (ja) | 2016-07-29 | 2022-09-02 | 株式会社ブリヂストン | 更生タイヤ及び更生タイヤの製造方法 |
| JP2019524950A (ja) * | 2016-08-17 | 2019-09-05 | コンティネンタル・ライフェン・ドイチュラント・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | 硫黄架橋性ゴム混合物および車両用タイヤ |
| JP2018048294A (ja) * | 2016-09-22 | 2018-03-29 | クムホ タイヤ カンパニー インコーポレーテッド | タイヤトレッド用ゴム組成物 |
| CN109843942A (zh) * | 2016-10-14 | 2019-06-04 | 科腾化学品有限责任公司 | 含有改进的胎面补强添加剂的橡胶组合物及其用途 |
| EP3323630A1 (en) * | 2016-11-16 | 2018-05-23 | Sumitomo Rubber Industries, Ltd. | Rubber composition for treads and pneumatic tire |
| CN108070123A (zh) * | 2016-11-16 | 2018-05-25 | 住友橡胶工业株式会社 | 用于胎面的橡胶组合物和充气轮胎 |
| CN108070123B (zh) * | 2016-11-16 | 2021-10-08 | 住友橡胶工业株式会社 | 用于胎面的橡胶组合物和充气轮胎 |
| JP2018095877A (ja) * | 2016-12-15 | 2018-06-21 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| EP3345775A1 (en) * | 2017-01-04 | 2018-07-11 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tread and pneumatic tire |
| JP2018135410A (ja) * | 2017-02-20 | 2018-08-30 | 住友ゴム工業株式会社 | ゴム組成物、タイヤトレッドおよび空気入りタイヤ |
| JP2018168217A (ja) * | 2017-03-29 | 2018-11-01 | 住友ベークライト株式会社 | 封止用樹脂組成物及びこれを用いた半導体装置 |
| JP2018177908A (ja) * | 2017-04-07 | 2018-11-15 | 住友ゴム工業株式会社 | タイヤ |
| US20180291190A1 (en) * | 2017-04-07 | 2018-10-11 | Sumitomo Rubber Industries, Ltd. | Tire |
| JP7013669B2 (ja) | 2017-04-07 | 2022-02-01 | 住友ゴム工業株式会社 | タイヤ |
| EP3385319A1 (en) * | 2017-04-07 | 2018-10-10 | Sumitomo Rubber Industries, Ltd. | Tire |
| US10752757B2 (en) * | 2017-04-07 | 2020-08-25 | Sumitomo Rubber Industries, Ltd. | Tire |
| JP7119329B2 (ja) | 2017-10-12 | 2022-08-17 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| JP2019073579A (ja) * | 2017-10-12 | 2019-05-16 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| JP2019073580A (ja) * | 2017-10-12 | 2019-05-16 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| JP7119330B2 (ja) | 2017-10-12 | 2022-08-17 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物 |
| US11499038B2 (en) | 2017-12-26 | 2022-11-15 | Bridgestone Corporation | Tread composition and tire produced by using the same |
| JP2019112598A (ja) * | 2017-12-26 | 2019-07-11 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2019137835A (ja) * | 2018-02-05 | 2019-08-22 | ハンコック タイヤ カンパニー リミテッド | タイヤトレッド用ゴム組成物、及びそれを用いて製造したタイヤ |
| JP2021523959A (ja) * | 2018-05-04 | 2021-09-09 | ブリヂストン アメリカズ タイヤ オペレーションズ、 エルエルシー | タイヤトレッドゴム組成物 |
| JP2019218118A (ja) * | 2018-06-21 | 2019-12-26 | 三井化学株式会社 | 医療用容器および医療用容器用環状オレフィン系樹脂組成物 |
| US12031040B2 (en) | 2018-08-06 | 2024-07-09 | Sumitomo Rubber Industries, Ltd. | Tread rubber composition and pneumatic tire |
| JP7434703B2 (ja) | 2018-08-06 | 2024-02-21 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| JP2020023600A (ja) * | 2018-08-06 | 2020-02-13 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| JP2020076110A (ja) * | 2018-10-04 | 2020-05-21 | 住友ゴム工業株式会社 | トレッド用ゴム組成物及び空気入りタイヤ |
| JP2020055980A (ja) * | 2018-10-04 | 2020-04-09 | 住友ゴム工業株式会社 | トレッドゴム組成物及び空気入りタイヤ |
| JP7255137B2 (ja) | 2018-10-30 | 2023-04-11 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2020070329A (ja) * | 2018-10-30 | 2020-05-07 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2020070330A (ja) * | 2018-10-30 | 2020-05-07 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| JP7255136B2 (ja) | 2018-10-30 | 2023-04-11 | 日本ゼオン株式会社 | ゴム組成物およびそれを用いた空気入りタイヤ |
| WO2020100492A1 (ja) * | 2018-11-12 | 2020-05-22 | 住友ゴム工業株式会社 | ゴム組成物及び空気入りタイヤ |
| US12187894B2 (en) | 2018-11-12 | 2025-01-07 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| JP2020079336A (ja) * | 2018-11-12 | 2020-05-28 | 住友ゴム工業株式会社 | ゴム組成物及び空気入りタイヤ |
| US12173140B2 (en) | 2018-11-12 | 2024-12-24 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| US12054614B2 (en) | 2018-11-12 | 2024-08-06 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| US12410308B2 (en) | 2018-11-12 | 2025-09-09 | Sumitomo Rubber Industries, Ltd. | Rubber composition and pneumatic tire |
| JP7174621B2 (ja) | 2018-12-27 | 2022-11-17 | Toyo Tire株式会社 | タイヤ用ゴム組成物、及びそれを用いた空気入りタイヤ |
| JP2020105380A (ja) * | 2018-12-27 | 2020-07-09 | Toyo Tire株式会社 | タイヤ用ゴム組成物、及びそれを用いた空気入りタイヤ |
| WO2021033713A1 (ja) * | 2019-08-21 | 2021-02-25 | 横浜ゴム株式会社 | 空気入りタイヤ |
| JP7159999B2 (ja) | 2019-08-21 | 2022-10-25 | 横浜ゴム株式会社 | 空気入りタイヤ |
| JP2021031557A (ja) * | 2019-08-21 | 2021-03-01 | 横浜ゴム株式会社 | 空気入りタイヤ |
| WO2023112364A1 (ja) * | 2021-12-14 | 2023-06-22 | 株式会社ブリヂストン | タイヤ用ゴム組成物、トレッドゴム及びタイヤ |
| JPWO2023112364A1 (ja) * | 2021-12-14 | 2023-06-22 | ||
| JP7847155B2 (ja) | 2021-12-14 | 2026-04-16 | 株式会社ブリヂストン | タイヤ用ゴム組成物、トレッドゴム及びタイヤ |
| JP2024025862A (ja) * | 2022-08-15 | 2024-02-28 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP7473829B2 (ja) | 2022-08-15 | 2024-04-24 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| WO2024038812A1 (ja) * | 2022-08-15 | 2024-02-22 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| WO2024080242A1 (ja) * | 2022-10-14 | 2024-04-18 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP2024058155A (ja) * | 2022-10-14 | 2024-04-25 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP7485976B2 (ja) | 2022-10-14 | 2024-05-17 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3228658A4 (en) | 2018-07-04 |
| JP6072979B2 (ja) | 2017-02-01 |
| CN107001712A (zh) | 2017-08-01 |
| EP3228658A1 (en) | 2017-10-11 |
| EP3228658B1 (en) | 2019-11-06 |
| CN107001712B (zh) | 2020-03-17 |
| JPWO2016104144A1 (ja) | 2017-04-27 |
| US10266008B2 (en) | 2019-04-23 |
| US20170341468A1 (en) | 2017-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6072979B2 (ja) | 空気入りタイヤ | |
| JP6074113B2 (ja) | 空気入りタイヤ | |
| JP5559234B2 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP5913188B2 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP6030697B1 (ja) | タイヤ外層用ゴム組成物及び空気入りタイヤ | |
| JP6189976B2 (ja) | トレッド用ゴム組成物及び空気入りタイヤ | |
| JP5097862B1 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP5650690B2 (ja) | トレッド用ゴム組成物及び空気入りタイヤ | |
| JP6215959B2 (ja) | 冬用タイヤ | |
| JP5767753B2 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| WO2015076049A1 (ja) | 空気入りタイヤ | |
| JP2015232114A (ja) | ゴム組成物及び空気入りタイヤ | |
| JP2015013974A (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| WO2015104955A1 (ja) | 空気入りタイヤ | |
| JP2016020427A (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP2016037532A (ja) | 空気入りタイヤ | |
| JP2023168930A (ja) | タイヤ | |
| JP2020169265A (ja) | タイヤトレッド用ゴム組成物及びタイヤ | |
| WO2021024666A1 (ja) | タイヤ用ゴム組成物及びタイヤ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016512126 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15872713 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15534831 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015872713 Country of ref document: EP |