WO2016152786A1 - 細胞傷害性t細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬 - Google Patents
細胞傷害性t細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬 Download PDFInfo
- Publication number
- WO2016152786A1 WO2016152786A1 PCT/JP2016/058721 JP2016058721W WO2016152786A1 WO 2016152786 A1 WO2016152786 A1 WO 2016152786A1 JP 2016058721 W JP2016058721 W JP 2016058721W WO 2016152786 A1 WO2016152786 A1 WO 2016152786A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- tumor
- ecv
- cell
- therapeutic agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0663—Bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2503/00—Use of cells in diagnostics
- C12N2503/02—Drug screening
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/178—Oligonucleotides characterized by their use miRNA, siRNA or ncRNA
Definitions
- the present invention relates to a therapeutic agent that can be used for the treatment of cell proliferative diseases such as cancer, comprising a cytotoxic T cell-released exosome or miRNA contained in the exosome as an active ingredient.
- the tumor stroma is formed by extracellular matrices such as fibronectin, laminin and collagen, and many other cells. These cells include cancer-associate fibroblasts (CAFs, fibroblast markers and platelet-derived growth factor receptor ⁇ (CD140a) + , ⁇ -smooth muscle actin ( ⁇ -SMA) + ), stroma Stem cells (mesenchymal stem cells (MSCs) (Non-patent Document 1. The literature is summarized at the end): CD140a + stem cell antigen Sca-1 + ) are observed.
- CAFs cancer-associate fibroblasts
- CD140a fibroblast markers and platelet-derived growth factor receptor ⁇
- ⁇ -SMA ⁇ -smooth muscle actin
- MSCs meenchymal stem cells
- Non-patent Documents 3 and 4 an interstitium is stretched to fill the gap between the cancer cells to be tightly bound by E-cadherin and the like (Non-patent Documents 3 and 4) and the cancer cells, and angiogenesis (Sca-1 + CD31 + (Non-Patent Document 2).
- EMT epithelial to mesenchymal transition
- Non-patent Document 5 There are some reports as molecules involved in epithelial-mesenchymal transition (Non-Patent Documents 3, 6, and 7).
- Endosomal membrane-derived microvesicles (100-200 nm in diameter) are released from various cells including tumor cells and tumor stromal cells, and are known to transmit information between cells using proteins and RNA contained therein.
- Tumor cells release a variety of extracellular vesicles (ECVs, sometimes referred to as exosomes or exosomes, referred to herein as “exosomes” or “ECVs”), which are self-proliferating, immune-tolerant, There are reports that it is involved in the adjustment of the tumor environment (Non-Patent Documents 8, 10 to 13).
- Non-Patent Documents 9, 14, and 15 activated CD8 + T cells that infiltrate tumors sometimes infiltrate tumors and tumor stroma.
- CD8 + T cells containing cytotoxic T lymphocytes (CTL) accumulate in tumor tissues or proliferate in tumors in immunotherapy using monoclonal antibodies against tumor-associated antigens (non-patented) References 17, 18).
- CTLs are not tumor specific but infiltrate from blood vessels to tumors through remodeling of the basement membrane (Non-Patent Document 19), so CD8 + T cells are various in response to tumor progression and progression Presumed to be involved.
- the first aspect of the present invention relates to a therapeutic agent for a cell proliferative disease containing extracellular vesicles (exosomes) released from cytotoxic T cells.
- the second embodiment of the present invention is an extracellular vesicle released from at least one or more of human CD4 + , CD8 + , CD9 + , CD63 + and TCR + T cells among the cytotoxic T cells.
- the present invention relates to a therapeutic agent for cell proliferative diseases according to the first aspect, which comprises (exosome).
- the third aspect of the present invention is the cell according to the second aspect, which includes extracellular vesicles (exosomes) that are extracellular vesicles (exosomes) released from CD8 + T cells among the cytotoxic T cells. It relates to therapeutic agents for proliferative diseases.
- the fourth aspect of the present invention is the therapeutic agent for a cell proliferative disease according to the second aspect or the third aspect, characterized in that the extracellular vesicle (exosome) contains an miRNA effective for suppressing cell growth. About.
- a fifth aspect of the present invention relates to a therapeutic agent for a cell proliferative disease according to the fourth aspect, which contains the miRNA effective for suppressing cell proliferation.
- the therapeutic agent for cell proliferative diseases is a bactericidal agent, mucosal removing agent, isotonic agent, pH adjuster, stabilizer, thickener, preservative, adhesive, or
- the present invention relates to a therapeutic agent for cell proliferative diseases according to the second aspect, further comprising one or more selected from among immunopotentiators.
- the therapeutic agent in the treatment for a cell proliferative disease according to the second aspect, is administered into tumor tissue, mesenchymal cells in the tumor tissue, intravenously or subcutaneously.
- the eighth aspect of the present invention relates to a method for extracting miRNA for cell proliferative disease treatment, which collects exosomes released from cytotoxic T cells and identifies miRNAs effective for suppressing cell proliferation from the exosomes.
- a ninth aspect of the present invention relates to a therapeutic agent for a cell proliferative disease, comprising miRNA effective for suppressing cell growth.
- a miRNA having the same base sequence as an miRNA contained in an exosome released from a cytotoxic T cell is added to a cultured human mesenchymal stem cell (MSC) and cultured to damage the MSC.
- MSC human mesenchymal stem cell
- the present invention relates to a method for identifying MSC-damaging miRNA, wherein the MSC toxicity of miRNA is evaluated by examining the activity. After MSC-damaging miRNA is identified, it can be used for cell proliferative diseases by synthesizing an exosome containing the miRNA or miRNA having the same sequence as the miRNA and reconstructing the synthesized miRNA as it is or in an exosome-like manner. It is used as a therapeutic drug.
- the eleventh aspect of the present invention relates to a therapeutic agent for cell proliferative diseases comprising MSC-damaging miRNA.
- Another embodiment relates to a method for treating a cell proliferative disease, wherein the therapeutic agent of each of the above embodiments is administered to a patient.
- the administration method is preferably within the tumor tissue, mesenchymal cells in the tumor tissue, vein or subcutaneous.
- An exosome means a vesicle formed of a lipid bilayer secreted to the outside from various cells, and has a diameter of about 40 nm to 200 nm. In a living body, it is observed in body fluids such as saliva, blood, urine, amniotic fluid, and malignant ascites.
- the exosome contains various proteins and RNAs, and it has been pointed out that it may play a role in transmitting information between cells.
- the exosome kills mesenchymal cells surrounding the cancer cell (cancer stroma collapse), and as a result, Proliferation / metastasis can be suppressed.
- CD8 + T cell-derived exosomes are particularly effective for suppressing proliferation / metastasis.
- the exosome is taken up by both cancer cells and mesenchymal cells, and only the mesenchymal cells are killed (apoptotic). As a result, the cancer cells are interstitial necessary for proliferation and metastasis. Loses cells, isolates and suppresses proliferation / metastasis. Similarly, the specific miRNA contained in the exosome can also have the effect of suppressing the growth / metastasis of cancer cells.
- a therapeutic agent for cell proliferative diseases containing an extracellular vesicle (exosome) derived from cytotoxic T cells as an active ingredient can be provided.
- a therapeutic agent for cell proliferative diseases comprising miRNA effective for suppressing cell growth.
- These therapeutic agents according to the present invention can suppress cell growth and metastasis of tumors such as cancer.
- DUC18 ECV immobilized latex beads control monoclonal antibodies CD4 -, CD8 -, TCRVb -, CD9 - is a graph showing and CD63- results of flow cytometric analysis after staining with specific monoclonal antibodies.
- C Graph showing the results of intratumoral administration of DUC18, CMS5aTB or BLAB / c ECV to CMS5a-treated wild-type mice and nude BALB / c mice on the 12th day after CMS5a administration, and the results of observing tumor growth Yes (* ⁇ 0.05, ** ⁇ 0.001, ns: no significant difference, respectively) in the graph.
- CMS5a tumor 3 days after ECV treatment is a photographic diagram showing the appearance of spheroid formation observed with a microscope when cultured for 24 hours.
- DUC18 CD8 + T cells are graphs showing specific lytic effects on corresponding tumors. The results of cytotoxicity tests using mutant ERK2 peptide-stimulated DUC18 CD8 + T cells and CMS5a, H-2Kd-neutralized CMS5a, CT26, CMS7 and mERK2 peptide pulse CMS7 as target cells are shown.
- a and B It is a graph which shows the result of having compared the expression of CD140a with CMS5a ECV or a BALB / c ECV processing group when two lots of DUC18 ECV was administered in a CMS5a tumor by a flow cytometry analysis.
- A shows a dot plot
- B shows a histogram.
- C Fluorescence micrograph showing the result of staining a section of CMS5a tumor on day 3 after ECV treatment with a CD140a-specific monoclonal antibody and DAPI.
- D Fluorescence micrograph showing the result of staining a section of CMS5a tumor on day 3 after ECV treatment with a CD140a-specific monoclonal antibody and DAPI.
- ECV prepared from culture medium of B6 spleen cells stimulated with TRP-2 and gp100 peptide (5th, 7th, 10th and 15th day) CMS5a administered BALB / c mice or B16 administered 12 days after tumor administration B6 mice were administered intratumorally. It is a graph which shows the result of having applied the flow cytometry analysis about the tumor cell suspension which dye
- CD8 + T cell-release ECV causes BM-MSC to undergo apoptosis and suppress MB-MSC-mediated tumor growth.
- EMT epithelial-mesenchymal transition
- BM-MSCs were prepared by culturing cells obtained from crushed femur for one month. DUC18, B6 and hPBMC ECV were added to the BM-MSC culture solution at the concentrations shown in the figure, and the remaining BM-MSC on the third day was stained with annexin V monoclonal antibody and then subjected to flow cytometry analysis to determine the total number It is a graph of time.
- DUC18 ECV was added at the concentration shown in the figure and cultured for 4 days. The obtained tumor cells were stained with a CD140a-specific monoclonal antibody and subjected to flow cytometry analysis.
- DUC18 ECV or CMS5a TB ECV is added and CMS5a, 4T1, CT26 and B16 spheroid numbers cultured with MSC are observed with a microscope (marks in the graph are * ⁇ 0.05, **, respectively) ⁇ 0.001, ns: means no significant difference).
- D. CD90.1 + BM-MSC chimeric mice were prepared by introducing normal BM cells of BLAB / c mice and cultured CD90.1 + BM-MSCs into irradiated BALB / c mice. Two months after the introduction of CD90.1 + BM-MSC, DUC18 ECV, hPBMC ECV or BLAB / c ECV was administered into the CMS5a tumor on the 12th day after administration.
- CD90.1 BM-MSC cultured for 2 months with PE-conjugated CD140a monoclonal antibody, APC-conjugated Sca-1 monoclonal antibody, FITC-conjugated CD90.1, CD29 or CD105 monoclonal antibody, or a mixture of CD14, CD34 and CD45 monoclonal antibodies was stained. The obtained cells are graphs when subjected to flow cytometry analysis.
- FIG. 3 is a photographic diagram when BM-MSC differentiated into adipocytes or bone cells is stained with Oil Red O or Alizarin Red S.
- 1 is a schematic diagram of a strategy for creating CD90.1 BM-MSC chimeric BALB / c mice used to clarify the role of CD8 + T cell-releasing ECV on tumor-infiltrating MSC apoptosis.
- CD90.1 BM-MSC and BALB / c bone marrow cells were intravenously administered to BALB / c mice irradiated with 6 gray radiation.
- CMS5a tumor cells were subcutaneously administered to CD90.1 BM-MSC chimeric mice.
- DUC18 ECV, BALB / c ECV or hPBMC ECV was administered into the CMS5a tumor (approximately 1 cm diameter), and CD90.1 + cell defects in the tumor were obtained on day 3 after ECV administration.
- Splenocytes were examined by flow cytometry. Shows functional uptake of ECV-derived RNA by BM-MSC but not tumor cells.
- B16 cells or CMS5a cells and BM-MSC were mixed or cultured separately for 3 days, and SYTO RNASelect stained DUC18, CMS5a TB or hPBMC ECV was added. It is a graph when the green fluorescence intensity
- B16 or CMS5a tumor suspension obtained from tumors 30 minutes after administration of SYTO RNASelect stained DUC18 ECV was stained with CD140a-, Sca-1-specific monoclonal antibody and subjected to flow cytometry analysis It is a graph of time.
- FIG. The result of the total mRNA microarray analysis of the tumor cell which contacted cultured BM-MSC is shown.
- B16 or CMS5a cells were cultured for 3 days alone or with cultured BM-MSC. Tumor cells were separated from BM-MSCs with a fluorescence activated cell sorter. It is a graph when total RNA is extracted using a RNeasy mini kit from tumor cells cultured in a mixed culture with BM-MSC or only from tumor cells, and subjected to total mRNA microarray analysis.
- B Table showing the top 9 genes with increased expression levels in both B16 and CMS5a tumors cultured in a mixture with BM-MSC. The involvement of miRNA in the deficiency of cultured BM-MSC is shown.
- miRNAs dominant in DUC18 ECV were selected.
- the selected 15 miRNAs genetically formed two clusters.
- miR-298, miR-1943, and miR-5099 (shown in red) were highly expressed and were obtained from unknowns in PubMed.
- FIG. 7 is a pie chart showing that 70% of the reported reported DUC18 ECV dominant miRNAs were down-regulating tumor promotion and growth. This shows that our search method is accurate.
- C Selected miR-298, -1943 and -5099 were synthesized and transfected into cultured BM-MSCs alone or mixed. Negative control miR and synthetic CMS5a TB ECV dominant miR (miR-150, -223 or -3470b) were used as controls. It is a graph when the total number of BM-MSC remaining on the third day after transfection is counted by flow cytometry.
- FIG. 11 is a table showing 14 miRNAs dominant in DUC18 ECV selected by comparison with BLAB / c, CMS5a TB or CD4 BALB / c miRNA. Over 100 indicators of miRNA extracted by the global normalization method are shown in orange. Three miRNAs shown in gray (miR-298-5p, -1943-5p, -5099) were used to study BM-MSC deficiency. It shows that administration of DUC18 B ECV and BALB / c ECV to primary tumor suppresses infiltration and metastasis of B16F10. A.
- DUC18 or BALB / c ECV was administered to the primary B16F10 tumor at 50 ⁇ g / tumor / site.
- B A micrograph showing the degree of invasion of the removed B16F10 tumor on the 18th day after tumor administration by HE staining. The figure is representative of 6 tumor sections in the untreated group.
- C Fluorescence micrograph when a B16F10 tumor section was prepared and stained with CD140a, Sca-1, and DAPI on the 18th day after tumor administration. The photo is representative of 3 samples in each group.
- CD90.1 DUC18 CD8 + T cells treated with DMSO or GW4869 were intravenously administered to BALB / c mice on day 12 after administration of CMS5a. 24 hours later, a section of the obtained tumor was prepared, and a fluorescence micrograph when stained with CD90.1 monoclonal antibody, Sca-1 monoclonal antibody and DAPI, or CD31 monoclonal antibody, Sca-1 monoclonal antibody and DAPI is there.
- BALB / c wild-type or nude mice were administered CMS5a tumor, and on day 12, DMSO-treated or GW4869-treated CD90.1 DUC18 CD8 + T cells were administered.
- ECV from the supernatant of cultured CD90.1 DUC18 CD8 + T cells is bound to latex beads and stained with control monoclonal antibody, CD8 monoclonal antibody, CD9 monoclonal antibody or CD90.1 monoclonal antibody, and flow site It is a graph which shows the result which used for the measurement analysis.
- FIG. 4 is a fluorescence microscopic photograph when stained with an antibody and DAPI, or a CD8 monoclonal antibody, a CD9 monoclonal antibody and DAPI, and observed with a fluorescence microscope.
- the cells were stained with Sca-1 monoclonal antibody and DAPI, and observed with a two-photon confocal microscope.
- the MSC region stained purple was surrounded by a dot circle.
- Yellow arrows are CD90.1 DUC18 CD8 + T cells infiltrating the tumor, and white arrows are fluorescence micrographs showing CD90.1 ECV uptake CD140a + Sca-1 + MSC.
- the figure is representative of some of the six photos. It is a graph which shows the result of having investigated the T cell population after culturing the mononuclear cell isolate
- each graph indicates the fluorescence intensity, and the vertical axis indicates the ratio (%) of beads.
- numerator was shown on the upper side of the graph. It is a photograph figure which shows the result of having investigated the influence which various miRNA which the exosome which the human T cell released has has on MSC.
- the vertical direction of the photo shows the amount of miRNA added, and the horizontal direction shows the type of miRNA added. As a control, miRNA was not added. Of 40 types of miRNA, 2 types, miR-6089 and miR-6090, showed MSC damage activity. Moreover, miR-204-3p was shown as what does not show MSC disorder activity. It is a graph which shows the result of having investigated the influence which each miRNA has on the survival of MSC. miRNA was added at a final concentration of 40 nM.
- the therapeutic agent according to the present invention comprises extracellular vesicles (exosomes) released from cytotoxic T cells as active ingredients.
- the cytotoxic T cells may be derived from humans, monkeys, mice, rats, cows, horses, camels, sheep, birds (including chickens and ostriches).
- the cytotoxic T cells used for treatment do not necessarily have to be derived from the same species, but if possible, it is preferable to use cytotoxic T cells derived from the same species.
- cytotoxic T cells derived from the same species are not necessarily limited to the cytotoxic T cells of the party to be treated (including humans and animals other than humans). It may be extracted from cytotoxic T cells.
- the therapeutic agent includes extracellular vesicles (exosomes) released from at least one or more of CD4 + , CD8 + , CD9 + , CD63 + and TCR + T cells among the cytotoxic T cells.
- a cell proliferative disease means a disease having the property of abnormally proliferating cells beyond a normal region. For example, a malignant tumor (cancer) or a precancerous condition (occurs a malignant tumor). A state with a significantly increased risk, or a precancerous lesion with a morphological change that is likely to cause a malignant tumor compared to normal tissue).
- the therapeutic agent is effective not only for cell proliferation but also for suppressing metastasis.
- the therapeutic agent is preferably effective as a treatment target from a precancerous to cancerous tumor, but is effective in suppressing tumor growth / metastasis regardless of benign or malignant.
- the administration method of the therapeutic agent according to the present invention include, but are not limited to, direct administration to a tumor, administration method to mesenchymal cells around the tumor, intravenous administration, and subcutaneous administration.
- the therapeutic agent according to the present invention is characterized in that it contains miRNA derived from extracellular vesicles (exosomes) released from cytotoxic T cells as an active ingredient. It is preferable that this miRNA has an activity effective for suppressing cell growth.
- miRNA is a kind of ncRNA (non-coding RNA) that is present in cells and has a short length of about 20-25 bases and is thought to have a function to regulate gene expression. means. Moreover, such miRNA can be encapsulated in an endoplasmic reticulum (including exosomes and artificial liposomes).
- additives such as bactericides, isotonic agents, pH adjusters, stabilizers, thickeners, preservatives, fragrances, adhesives, immune enhancers and the like mentioned in the sixth aspect of the present invention An example of this is described below.
- the disinfectant iodine agent, alcohols, and pronase are specific examples used as a mucous membrane removing agent.
- tonicity agents include sodium chloride and glycerin
- pH regulators include citric acid, gluconic acid, succinic acid, potassium carbonate, and lactic acid
- stabilizers and thickeners include citric acid, gluconic acid, succinic acid, potassium carbonate, and lactic acid
- preservatives such as carrageenan, sodium carboxymethylcellulose, xanthan gum, guar gum, pectin, etc.
- preservatives such as carrageenan, sodium carboxymethylcellulose, xanthan gum, guar gum, pectin, etc.
- adhesives include gelatin, starch, and casein.
- immunopotentiators include chemotherapeutic agents such as taxane compounds, signal transduction inhibitors, etc., in addition to Toll-like receptor agonists such as CpG oligo DNA and poly IC RNA. The substance is not limited to this as long as it can be safely used for living cells.
- CMS5a, CMS7, CT26, 4T1, B16 and B16F10 tumor cell lines were passaged using D-MEM medium containing 10% FCS.
- the experimental protocol was evaluated by the Animal Ethics Committee at Mie University. 2.
- ECV extracellular microvesicles
- FCS containing ECV ECV-free FCS
- ECV-free FCS ECV-free FCS
- Spleen cells (2 ⁇ 10 7 cells / ml) prepared from DUC18 mice or CD90.1 DUC18 mice were cultured in RPMI-1640 medium containing 10% ECV-free FCS and 1 ⁇ g / ml mERK2 peptide.
- Spleen cells (2 x 10 7 cells / ml) prepared from B6 mice were treated with 10% ECV-free FCS and 1 ⁇ g / ml TRP-2 (SEQ ID NO: SVYDFFVWL) and 1 ⁇ g / ml gp100 (SEQ ID NO: 3 EGSRNQDWL) peptide
- the cells were cultured in RPMI-1640 medium containing (Non-patent Document 21).
- Spleen cells (2 x 10 7 cells / ml) from BALB / c mice, CMS5a tumor-bearing BALB / c mice, or CD8 + T cell-removed BALB / c mice (2C11: 2 ⁇ g / ml: Biolegend) was cultured in RPMI-1640 medium containing 10% ECV-free FCS and 1 ⁇ g / ml anti-CD28 monoclonal antibody (37.51: eBioscience).
- hPBMC human peripheral blood mononuclear cell
- GE healthcare Ficoll-Paque PLUS (GE healthcare) gradient in a 12-well plate with an OKT3 monoclonal antibody (2 ⁇ g / ml: Biolegend) immobilized on a 10-well plate.
- the cells were cultured in RPMI-1640 medium containing% ECV-free FCS and 1 ⁇ g / ml CD28 monoclonal antibody (Biolegend).
- CD8 + T cell-depleted BALB / c mice were prepared by intravenous administration of Lyt-2.2-specific monoclonal antibody (400 ⁇ g / mouse) to increase CD4 + T cells in vitro.
- each medium was changed to an RPMI-1640 medium containing 10% ECV-free FCS and recombinant IL-2 (rIL-2) (100 IU / ml) and cultured for 3 days.
- the resulting supernatant was used as an ECV source.
- the obtained cells were mouse CD4 (GK1.5) specific monoclonal antibody, CD8 (53-6.7) specific monoclonal antibody, TCRV ⁇ (H57-597) specific monoclonal antibody and V ⁇ 8.3 (8C1) specific monoclonal antibody.
- the concentrated culture supernatant was treated with a 0.22 ⁇ m filter and then ultracentrifuged at 120,000 ⁇ g for 90 minutes (SW28 rotor: Beckman Courter).
- the obtained ECV precipitate was suspended in 30 ml of PBS and washed by ultracentrifugation at 120,000 ⁇ g. Finally, the ECV precipitate was suspended in 1-2 ml of PBS and stored at 4 ° C.
- the protein concentration of purified ECV was measured with a bicinchoninic acid (BCA) protein assay kit (Pirece). The average number and average diameter of purified ECV were measured using a nano-tracking assay (LM10-HS: Nanosight).
- the ECV surface protein is composed of anti-CD4 monoclonal antibody, anti-CD8 monoclonal antibody, anti-CD9 monoclonal antibody (MZ3), anti-CD63 monoclonal combined with fluorescein isothiocyanate (FITC) or phycoerythrin (PE) after binding ECV to latex beads.
- the antibody (NVG-2) and anti-V ⁇ 8.3 monoclonal antibody (all Biokegend) were stained and detected by flow cytometry analysis.
- BM-MSCs bone marrow mesenchymal stem cells
- BD of MSC stained with PE-conjugated anti-CD140a monoclonal antibody and FITC-conjugated anti-Sca-1 monoclonal antibody.
- Cultured MSCs were further evaluated for the presence of CD29, CD90.1 and CD105 and the absence of CD14, CD34 and CD45 by flow cytometric analysis using monoclonal antibodies against each molecule.
- mice were maintained for 2 weeks using 1 mg / ml neomycin (Calbiochem) -containing autoclave water and X-ray irradiated food.
- CD90.1 + BM-MSC chimeric BALB / c mice were used to confirm tumor MSCs.
- CD8 + T cells and CD8 + T cell released ECV were administered intravenously.
- CMS5a tumor cells CD90.1 DUC18 CD8 + T cells (1 x 10 7 cells / mouse) alone or GW4869 (ECV release) 7 days after culture in CLB5c tumor-bearing BALB / c mice or BALB / c nude mice with a tumor diameter of about 10 mm Inhibitor
- Treated CD90.1 DUC18 CD8 + T cells (1 ⁇ 10 7 cells / mouse) were administered intravenously.
- an anti-mouse glucocorticoid-induced TNF receptor-related protein (GITR) monoclonal antibody (DTA-1) (2 ⁇ g / tumor) was intratumorally administered (inter tumor: it).
- GITR glucocorticoid-induced TNF receptor-related protein
- DTA-1 was used to enhance the accumulation of CD8 + T cells at the tumor site.
- GW4869 was added at 20 ⁇ g / ml 24 hours before the end of the culture.
- CMS5a tumor tissues were collected 1, 2, 3, 5 and 7 days after administration of cultured CD90.1 DUC18 CD8 + T cells, and immunohistochemically stained.
- CMS5a cells and B16 cells (1 ⁇ 10 6 cells / mouse) were subcutaneously administered to the back skin of BALB / c and B6 mice, respectively. About 2 weeks later, mice having a tumor diameter of 1.2 to 1.5 cm were selected from mice administered with the tumor and used for ECV treatment. The ECV obtained from each culture supernatant was intratumorally administered with a protein amount of 1, 5 or 10 ⁇ g to CMS5a or B16, and then the tumor diameter was measured. Further, 3 and 5 days after ECV administration, the tumor was cut with scissors and then incubated at 37 ° C. for 60 minutes in PBS containing 0.5% trypsin and 1 mM EDTA.
- the obtained tumor cell suspension was passed through a wool column and washed 3 times with 1% FCS-containing PBS, and subjected to flow cytometry analysis and spheroid formation confirmation test.
- flow cytometric analysis a FITC-binding monoclonal antibody specific for Sca-1, I-Ad, CD11b CD11c, CD73 or CD206 and a PE-binding monoclonal antibody specific for F4 / 80, Gr-1 or CD140a were used.
- As a confirmation test for spheroid formation 1 ⁇ 10 5 cells / ml were cultured in RPMI-1640 containing 10% FCS.
- CD90.1 + BM-MSC chimeric BALB / c mice transplanted subcutaneously with CMS5a were intratumorally administered with 5 ⁇ g of DUC18 CD8 + T cell-release ECV ((ECV protein amount) / tumor) 2 weeks after tumor cell administration .
- Tumor cell suspension obtained 3 days after ECV administration, stained with FITC-conjugated CD90.1 specific monoclonal antibody, PE-conjugated CD140a specific monoclonal antibody, and allophycocyanin (APC) -conjugated Sca-1 specific monoclonal antibody Later, 7-aminoactinomycin D (7-ADD) stained cells were subjected to flow cytometry analysis.
- B16F10 was administered subcutaneously, and 50 ⁇ g of DUC18 CD8 + T cell ECV, CMS5a-bearing BALB / c spleen cell ECV or BALB / c spleen cell ECV was intratumorally administered after 7, 10 and 13 days.
- B16F10-derived tumors (about 2 cm in diameter) were carefully excised with scissors, and the skin was stitched with surgical sutures after observing tumor invasion. 45 days after administration of the tumor cells, the presence or absence of B16F10 lung metastasis was observed.
- ECV Treatment of CD8 + T cell release ECV in vitro ECV obtained from cultured spleen cells of DUC18, CMS5a-bearing BALB / c, B16-bearing B6, BALB / c and B6 was 5 ⁇ 10 4 cells / ml CMS5a, B16, CT26 Or it added in the cell culture solution of BM-MSC. Each cell was cultured using 10% FCS-containing RPMI-1640 medium or BM-MSC medium (20% MSC stimulating substance-containing MesenCult MSC basal medium).
- Fluorescence immunoassay Frozen specimens of CMS5a and B16F10 tumors embedded in OCT compound were cut at a thickness of 3 ⁇ m, air-dried for 2 hours, then fixed with ice-cold acetone for 15 minutes, and the immune tissue Used for chemistry. After washing 3 times with PBS, the tissue section was combined with blocking solution (PBS containing 1% BSA, 5% Blocking One Histo (Nacalai Tesque), 0.2 ⁇ g / ml anti-mouse CD16 / CD32 monoclonal antibody (Biolegend)) at 4 ° C. Incubated for 30 minutes.
- blocking solution PBS containing 1% BSA, 5% Blocking One Histo (Nacalai Tesque), 0.2 ⁇ g / ml anti-mouse CD16 / CD32 monoclonal antibody (Biolegend)
- the tumor section on the slide was double-labeled for 1 hour at room temperature using PE-conjugated monoclonal antibody and FITC-conjugated monoclonal antibody dissolved in PBS containing 1% BSA and 5% Blocking One Histo. .
- the slides were treated with DAPI-containing ProLong Gold antifade reagent (Invitrogen Life Technologies) and observed with a fluorescence microscope (Olympus BX53F). Images of PE, FITC and DAPI stained tissues were overlaid using Photoshop elements software (Adobe Systems).
- Fluorescence immunoassays include PE8 monoclonal antibodies against CD8, CD140a, Ki-67, CD31, CD11b, ER-TR7 and TGF- ⁇ 1, and ER-TR7, Sca-1, F4 / 80, Gr-1, CD90.
- FITC-conjugated monoclonal antibodies against .1 and ⁇ -smooth muscle actin ( ⁇ -SMA) were used.
- Cytotoxicity test CMS5a, CMS7 and CT26 cells were labeled with 2.5 mM carboxyfluorescein diacetate succinimidyl ester (CFSE) at 37 ° C. for 6 minutes. After washing 3 times with RPMI-1640 containing 10% FCS, CFSE-labeled CMS5a was used as target cells.
- CFSE-labeled CMS5a was used as target cells.
- mERK2 peptide-stimulated DUC18 spleen cells were mixed with CFSE-labeled CMS5a, CMS7 or B16 cells (1 ⁇ 10 5 cells) at a ratio of 1, 5 and 10 using a 24-well plate. After incubation for 12 hours, the remaining cells were analyzed by flow cytometry.
- Non-patent Document 18 For each sample, 20,000 non-CFSE labeled cells were collected and the number of viable cells labeled with CFSE was counted. The survival rate was determined as an average value of two wells, and the% cytotoxic activity was calculated according to the literature (Non-patent Document 18).
- mouse miR298-5p (SEQ ID NO: 4: GGC AGA GGA GGG CUG UUC UUC CC), miR-298-3p (SEQ ID NO: 5: GAG GAA CUA GCC UUC UCU CAG C), miR1943- 5p (SEQ ID NO: 6: AAG GGA GGA UCU GGG CAC CUG GA), miR-1943-3p (SEQ ID NO: 7: CAG GUG CCA GCU CCU CCC UUC), miR-5099-5p (SEQ ID NO: 8: GUU AGA AAU UAC AUU GAU UUA A), miR5099-3p (SEQ ID NO: 9: UUA GAU CGA UGU GGU GCU CC), miR-150-5p (SEQ ID NO: 10: UCU CCC AAC CCU UGU ACC AGU G), miR-150-3p (SEQ ID NO: 11: CUG GUA CAG GCC UGG GGG AUA G
- ECV obtained from human PBMC was used as a temporary vesicle for functional analysis of synthetic miRNA.
- anti-CD3 and anti-CD28 monoclonal antibodies were added to RPMI-1640 medium containing 10% FCS and 100 IU / ml rIL-2, and human PBMC were cultured and stimulated for 3 days.
- stimulated human PBMC (1 x 10 8 ) is suspended in 1.5 ml of 1% DMSO-containing RPMI-1640 medium, miR-298 (50 ⁇ g), miR Mix with RNA pools -1943 (50 ⁇ g) and miR-5099 (50 ⁇ g) or miR-150 (50 ⁇ g), miR-223 (50 ⁇ g) and miR-3470b (50 ⁇ g) went.
- the resulting cells are cultured in 10% FCS-containing RPMI-1640 medium without ECV for 20 hours, and the supernatant is passed through 0.45 and 0.22 filters, followed by ultracentrifugation (120,000 g), thereby producing synthetic miRNA. Contained ECV was obtained.
- PBMC miRNA Mononuclear cells
- RNAs Forty identified miRNAs were synthesized in descending order of abundance.
- the synthesized miRNA was added to cultured human adipose tissue-derived mesenchymal stem cells (MSC), and the cytotoxic effect was examined. Cytotoxic activity is determined by (1) adding the synthesized miRNA to the cultured MSC and then Giemsa staining (Wako) of the cultured MSC, and (2) using an xCELLigence (ACEA BioSciences) instrument that measures cell survival using electrical resistance values.
- the MSC viability after adding each miRNA to MSC cultured on a dedicated plate was measured. 11.
- Two groups of data were analyzed by Mann-Whitney U test.
- the obtained culture supernatant was subjected to ultracentrifugation, and ECV was purified from each cell (DUC18, CMS5aTB, BALB / c, CD4 BALB / c and hPBMC). All ECVs were present in the culture supernatant at a 0.6-1.0 ⁇ g / ml protein concentration, about 4-8 ⁇ 10 9 cells / ml, and an average diameter of 110-140 nm (FIGS. 3A and 3B).
- the surface marker of DUC18 ECV was examined. Consistent with the parental cell phenotype, DUC18 ECV slightly expressed CD8, TCRV ⁇ 8.3, and CD63, and also highly expressed CD9, an already known ECV marker (FIG. 1B).
- DUC18 ECV, CMS5aTB ECV and BALB / c ECV were administered intratumorally (1.2 to 1.5 cm tumor diameter) in BABL / c mice subcutaneously administered with CMS5a.
- the growth of CMS5a treated with DUC18 ECV and BALB / c ECV was arrested and significantly attenuated compared to the CMS5aTB ECV treated or untreated group, respectively (FIG. 1C).
- spheroid formation after culturing the CMS5a suspension for 1 day was not observed in the DUC18 ECV-treated group, but was observed in the untreated group or the CMS5a TB ECV-treated group (FIGS. 1C and 1D).
- CD8 + T cell release ECV reduces tumor CD140a expression level Activated CD8 + T cell release ECV directly suppresses tumor cell growth or suppresses tumor growth by acting on tumor-related cells, One of them was guessed. To solve this problem, we examined changes in tumor-infiltrating cell populations that have been reported to regulate tumor growth. Three days after administration of DUC18 ECV and BABL / c ECV to CMS5a tumor, F4 / 80 + CD206 + macrophages or F4 / 80 + IA d + macrophages and CD11c + dendritic cells CD11b + Gr-1 + MDSC or CD4 + And CD8 + lymphocyte ratio did not change (Fig.
- TRP-2 specific CD8 + lymphocytes and gp100 specific CD8 + lymphocytes gradually increased during the culture, reaching 95% and 3%, respectively, on day 15 (FIG. 7).
- the peak decrease in CD140a expression by intratumoral administration occurs in ECV obtained on day 7 of culture, and ECV released by TRP-2 and gd100-specific CD8 + T cells is related to irrelevant CMS5a and related B16 Acted similarly (FIG. 6D).
- the decrease in functional ECV production by peptide-specific CD8 + T cells 15 days after administration was considered to be associated with lymphocyte decline. From these results, it was shown that the growth and progression of the tumor were suppressed without specificity by the influence of the CD8 + T cell release ECV.
- BM-MSC mesenchymal tumor stromal cell
- CD8 + T cell release ECV The number of cultured BM-MCS was dramatically reduced by apoptosis after 3 days when DUC18 ECB and B6 ECV were added (FIG. 10), but no such response was observed with human PBMC released ECV. (FIG. 9B). Furthermore, when DUC18 ECV was added to the mixture of cultured BM-MSC and tumor cells, the expression of CD140a in CMS5a and B16 cells and the spheroid formation of CMS5a, 4T1, CT26 and B16 were compared with those in the CMS5aTB ECV treatment group.
- BM-MSC differentiated cells eg, CAF, cancer associated fibroblasts, pericytes, etc.
- ECV is taken up by both BM-MSC and tumor cells, but B16 cells and CMS5a cells mixed with BM-MSC take SYTO RNASelect-labeled DUC18, CMS5a TB or hPBMC ECV and immediately intensify green fluorescence. Decrease was confirmed. Tumor cells in contact with BM-MSC appear to degrade ECV-derived miRNA immediately after ECV uptake (FIG. 12A). According to the total RNA microarray analysis (Toray 3D gene), a strong increase in the amount of lysozyme mRNA was observed in the tumor cells in contact with BM-MSC (FIG. 13).
- DUC18 ECV-derived miR-298, miR-141, miR-1249, miR-23b, and miR-370 have been reported to have tumor self-suppressing effects and that the selected miRNA is correct. It was confirmed (FIG. 14B, FIG. 15).
- 5p and 3p oligonucleotides of three miRNAs (miR-298, miR-1943 and miR-5099) predicted to be the most effective of DUC18 ECV were synthesized, annealed, and cultured BM-MSC Introduced. When miR-298 was introduced, the number of BM-MSCs was markedly reduced compared to when miR-1943 and miR-5099 were introduced (FIG. 14C).
- miR-298 was first discovered as a miRNA that kills BM-MSC. Considering the similarity of the immune system and basic research on cancer, there are specific miRNAs having such effects in miRNAs contained in exosomes obtained from human cytotoxic T cells.
- Tumor invasion and metastasis is an indicator of tumor epithelial-mesenchymal transition and exacerbation. Therefore, we investigated the effects of intramuscular administration of DUC18 ECV, CMS5a TB ECV or BALB / c ECV on the 10th, 13th, and 16th days of mice administered subcutaneously with B16F10 cells. . On day 18, the state of tumor invasion was observed, the B16F10 tumor was surgically excised, and the skin was sutured. Although tumor removal was possible in 50% in the untreated group and 33% in the CMS5a ECV-treated group, infiltration into fascia was observed in all the tumors that could not be removed (Table 1).
- CD90.1 + CD8 + T cells become 24 hours after administration in the Sca-1 + CD31 + angiogenic region of the CMS5a tumor stroma composed of endothelial progenitor cells and BM-MSC It was observed (FIG. 17B). Surprisingly, the Sca-1 + or CD140a + region of the CMS5a tumor disappeared on day 3 of CD90.1 + CD8 + T cell transfer and persisted until day 7 (FIG. 17C).
- Interstitial destruction effect is greater in wild-type BALB / c mice than in BALB / c nude mice, which shows that BALB / c-derived CD8 + T cells are tumors along with administered CD90.1 + CD8 + T cells This is thought to be the result of internal infiltration.
- the disappearance of Sca-1 + or CD140a + stroma was not observed in the GW4869-treated CD90.1 + CD8 + T cell administration group (FIG. 17C), similar to the result using purified ECV, It was suggested that intratumoral infiltrating CD8 + T cells produce ECV to destroy the tumor stromal structure.
- CD90.1 + CD8 + T cell-released ECV showed strong CD9 and CD90.1 expression, with very little CD8 (FIG. 18D).
- CDV-derived CD9 and CD90.1 (FIG. 18E) and their fusion signals (FIG. 18F) are observed in the CD140a + Sca-1 + stromal region.
- MSC-damaging miRNAs can be identified from exosomes released by cultured human T cells.
- CD8 was a dominant T cell population (FIG. 19).
- Nanotracking analysis of the diameter of the exosome released by the T cell into the culture supernatant was about 150 nm (FIG. 20).
- tetraspanin molecules CD9, CD63, CD81
- CD8 and HLA class I molecules as exosome markers were expressed (FIG. 21).
- Boelens MC Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman- Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J Mi Ex to breast cancer cells regulates therapy resistance pathways. Cell. (2014) 159: 499-513. Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. (2010) 70: 9621-9630.
- Bone marrow multipotent mesenchymal stroma cells act as pericyte like migratorysment 2009 -190.
- Apoptosis of antigen-specific CTLs contributes to low immune response in gut-associated lymphoid tissue post vaccination.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- Developmental Biology & Embryology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Dermatology (AREA)
- Biophysics (AREA)
- Rheumatology (AREA)
- Physics & Mathematics (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
マウスモデルやヒトの研究によれば、腫瘍に浸潤する活性化CD8+ T細胞が、腫瘍や腫瘍間質に浸潤することがある(非特許文献16)。また、腫瘍関連抗原に対するモノクローナル抗体を用いた免疫療法において、細胞傷害性Tリンパ球(CTL)を含むCD8+ T細胞が、腫瘍組織に集積もしくは腫瘍内増殖することが知られている(非特許文献17,18)。CTLは、腫瘍特異的ではなく、基底膜のリモデリングを介して、血管から腫瘍へと浸潤する(非特許文献19)ので、CD8+ T細胞は、腫瘍の進展や増悪化に対して様々に関与すると推定されている。
そこで、本発明者らは、細胞傷害性T細胞が放出するエキソソームが、腫瘍の悪性化に対して与える影響を詳細に検討した。その結果、各種の細胞傷害性T細胞のなかでも、とくにCD8+ T細胞由来エキソソームは、腫瘍組織の癌部ではなく、周囲の間葉系細胞を死滅させ、癌の増殖・転移を含めた癌の進行を抑制するという事実を見出し、基本的には本発明を完成するに至った。
本発明の第1の態様は、細胞傷害性T細胞から放出された細胞外小胞(エキソソーム)を含む細胞増殖性疾病用の治療薬に関する。
本発明の第2の態様は、前記細胞傷害性T細胞のなかでもヒトCD4+、CD8+、CD9+、CD63+、 TCR+T細胞のうち少なくとも1または2以上から放出された細胞外小胞(エキソソーム)を含む第1の態様の細胞増殖性疾病用の治療薬に関する。
本発明の第3の態様は、前記細胞傷害性T細胞のなかでもCD8+T細胞から放出された細胞外小胞(エキソソーム)である細胞外小胞(エキソソーム)を含む第2の態様の細胞増殖性疾病用の治療薬に関する。
本発明の第4の態様は、前記細胞外小胞(エキソソーム)が細胞増殖抑制に有効なmiRNAを含むことを特徴とする第2の態様または第3の態様の細胞増殖性疾病用の治療薬に関する。
本発明の第6の態様は、前記細胞増殖性疾病用治療薬が、殺菌剤、粘膜除去剤、等張化剤、pH調節剤、安定化剤、増粘剤、防腐剤、粘着剤、又は免疫強化剤の中から選択される1または複数をさらに含有する第2の態様の細胞増殖性疾病用の治療薬に関する。
本発明の第7の態様は、前記治療薬は腫瘍組織内、腫瘍組織内の間葉系細胞、静脈または皮下に投与されることを特徴とする第2の態様の細胞増殖性疾病用の治療薬に関する。
本発明の第8の態様は、細胞傷害性T細胞から放出されたエキソソームを回収し、そのエキソソームから細胞増殖抑制に有効なmiRNAを特定する細胞増殖性疾病治療用miRNAの抽出方法に関する。
本発明の第9の態様は、細胞増殖抑制に有効なmiRNAを含むことを特徴とする細胞増殖性疾病用の治療薬に関する。
MSC傷害性miRNAが同定された後には、そのmiRNAを含むエキソソーム又は、miRNAと同じ配列を持つmiRNAを合成し、合成されたmiRNAをそのまま又はエキソソーム様に再構成することで、細胞増殖性疾患用の治療薬として用いられる。こうして、本発明の第11の態様は、MSC傷害性miRNAを含む細胞増殖性疾患用の治療薬に関する。
また、別の態様は、細胞増殖性疾病の治療方法であって、患者に対して、上記各態様の治療薬を投与する方法に関する。このとき、投与方法は、腫瘍組織内、腫瘍組織内の間葉系細胞、静脈または皮下のいずれかであることが好ましい。
エキソソームとは、各種の細胞から外部に分泌された脂質二重膜で形成される小胞を意味しており、直径が約40nm~200nm程度のものである。生体では、唾液、血液、尿、羊水、悪性腹水等の体液中で観察される。また、培養細胞から培養液中に分泌される。エキソソームには、様々のタンパク質、RNAが含まれており、細胞間の情報伝達を行う役割を担っている可能性が指摘されている。
また、本発明によれば、細胞傷害性T細胞から取得したエキソソームを投与することにより、前記エキソソームが癌細胞周囲の間葉系細胞を死滅(癌間質崩壊)させ、その結果、癌細胞の増殖/転移を抑制することができる。前記細胞傷害性T細胞のなかでもCD8+ T細胞由来のエキソソームがとくに増殖/転移抑制効果がある。作用機序としては、前記エキソソームが、癌細胞と間葉系細胞の両方に取り込まれ、前記間葉系細胞のみを死滅(アポトーシス)させ、その結果、癌細胞が増殖や転移に必要な間質細胞を失い、孤立し、増殖/転移を抑制する。また、前記エキソソームに含まれる特定のmiRNAでも同様に癌細胞の増殖/転移抑制効果が得られる。
本発明による治療薬は、細胞傷害性T細胞から放出された細胞外小胞(エキソソーム)を有効成分として含有してなることを特徴とする。前記細胞傷害性T細胞の由来は、ヒト、サル、マウス、ラット、ウシ、ウマ、ラクダ、ヒツジ、トリ(ニワトリ、ダチョウを含む)でも良い。なお、治療に用いられる前記細胞傷害性T細胞としては、必ずしも同一種由来のものでなくても良いが、可能であれば、同一種由来の細胞傷害性T細胞を用いることが好ましい。また、同一種由来の細胞傷害性T細胞を用いる場合であっても、必ずしも治療される当事者(ヒトの他、ヒト以外の動物を含む)の細胞傷害性T細胞には限られず、他の者の細胞傷害性T細胞から抽出されても良い。
前記治療薬は、前記細胞傷害性T細胞のなかでもCD4+、CD8+、CD9+、CD63+、 TCR+T細胞のうち少なくとも1または2以上から放出された細胞外小胞(エキソソーム)を含むことを特徴とするが、これらの細胞傷害性T細胞に限定されない。
細胞増殖性疾病とは、正常の領域を超えて、細胞が異常に増殖する性質を備えた疾病のことを意味しており、例えば、悪性腫瘍(癌)、前癌状態(悪性腫瘍を発生する危険性が有意に増加した状態、または正常組織に比べて、悪性腫瘍を発生しやすい形態学的な変化を伴う前癌病変)などが含まれる。
本発明による治療薬の投与方法は、腫瘍への直接投与、腫瘍周辺の間葉系細胞への投与方法、静脈投与、皮下投与が挙げられるが、これらに限定はされない。
さらに本発明による治療薬は、細胞傷害性T細胞から放出された細胞外小胞(エキソソーム)由来のmiRNAを有効成分として含有してなることを特徴とする。このmiRNAが、細胞増殖抑制に有効な活性を持つことが好ましい。
miRNA(マイクロRNA)とは、細胞内に存在し、長さが20~25塩基程度の短いRNAであり、遺伝子発現を調節する機能を有すると考えられているncRNA(ノンコーディングRNA)の一種を意味する。
また、そのようなmiRNAは、小胞体(エキソソーム、人工的なリポソームを含む)に封入して用いることができる。
また、前記本発明の第6の態様に挙げた、殺菌剤、等張化剤、pH調節剤、安定化剤、増粘剤、防腐剤、香料、粘着剤、又は免疫強化剤等の添加物の例を下記に記載する。殺菌剤としては、ヨウ素剤、アルコール類、プロナーゼは粘膜除去剤として用いられる具体例である。等張化剤の具体例としては塩化ナトリウムやグリセリン等、pH調節剤の具体例としてはクエン酸、グルコン酸、コハク酸、炭酸カリウム、乳酸等、安定化剤や増粘剤の具体例としてはカラギナン、カルボキシメチルセルロースナトリウム、キサンタンガム、グァーガム、ペクチン等、防腐剤(保存料)の具体例としては安息香酸など、粘着剤の具体例としてはゼラチン、デンプン、カゼインなどがある。免疫強化剤の具体例としては、CpGオリゴDNAやポリIC RNAなどのToll様受容体のアゴニスト以外にも、タキサン系化合物などの化学療法剤、シグナル伝達阻害剤などがある。生体細胞に対し安全に用いることができる物質であればこれに限定されない。
1.マウス及び腫瘍細胞株
BALB/c (CD90.2) 及び C57BL/6 (B6) 雌性マウスは、6-8 週齢のものを日本SLCより購入した。CD90.1コンジェニックBALB/cマウス、変異ERK2(mERK2)136-144(配列番号1:QYIHSANVL)特異的H-2Kd拘束性TCR(Vβ10.1/Jβ48 及び Vβ8.3/Dβ2.1/Jβ2.6)遺伝子導入DUC18マウス(非特許文献20)及びCD90.1コンジェニックDUC18マウスは、三重大学の動物研究施設において維持した。CMS5a, CMS7, CT26, 4T1, B16及びB16F10 腫瘍細胞株は、10%FCS含有D-MEM培地を用いて継代した。DUC18マウス脾細胞から培養したCD8+ T細胞は、mREK2+ CMS5aを特異的に溶解したが、mREK2- CMS7 、CT26 (BALB/c バックグラウンドに対し)、B16 または B16F10 メラノーマ(B6 バックグラウンドに対し)を溶解しなかった。実験プロトコールは、三重大学の動物倫理委員会において評価した。
2.培養液からの細胞外微小胞(ECV)の調製
FCSを100,000×gにて4時間超遠心し、フィルター処理(0.45及び0.22μm)することで、ECVを含まないFCS(ECV-free FCS)を調製した。DUC18マウスまたはCD90.1 DUC18マウスから調製した脾臓細胞(2 x 107 個/ml) を10%ECV-free FCS及び1μg/ml mERK2ペプチド含有RPMI-1640培地中にて培養した。B6マウスから調製した脾臓細胞 (2 x 107 個/ml) を10%ECV-free FCSと1μg/ml TRP-2 (配列番号2:SVYDFFVWL) 及び 1μg/ml gp100 (配列番号3:EGSRNQDWL)ペプチド(非特許文献21)を含有するRPMI-1640培地中にて培養した。BALB/cマウス、 CMS5a担癌BALB/cマウス、又はCD8+ T細胞除去BALB/cマウスの脾臓細胞 (2 x 107 cells/ml) を抗CD3モノクローナル抗体(2C11: 2 μg/ml: Biolegend)を固相化した12穴プレート中にて、10%ECV-free FCS及び1μg/ml 抗CD28モノクローナル抗体(37.51: eBioscience )含有RPMI-1640培地中にて培養した。
ECVは、超遠心を用いたプロトコールに従って精製した。培養上清(約500ml)を10,000×gにて40分間遠心し、0.45μm及び0.22μmフィルターを用いて処理した後、100mlになるまで限外ろ過にて濃縮した(Kvick Lab Packet 50 KD: GE Healthcare)。濃縮した培養上清は、0.22μmフィルターにて処理した後、120,000×gにて90分間、超遠心処理した(SW28 rotor: Beckman Courter)。得られたECV沈殿を30mlのPBSに懸濁し、120,000×gの超遠心にて洗浄した。最後にECV沈殿を1~2mlのPBSに懸濁し、4℃にて保存した。
インビボ及びインビトロでのECVの動態を評価するために、100~300μgのECVを10μM SYTO RNASelect グリーン蛍光細胞染色(Molecular Probes)にて37℃で20分間染色した後、セファデックスG25スピンカラムを用いて結合していない染色物質を除いた。
BM-MSCは、添付書類の指示に従って、大腿骨から調製した(StemCell Technologies Inc.)。BALB/cまたはCD90.1+ BALB/cから得た10本の大腿骨の両端部分を切断し、乳鉢中に5mlの1%BSA含有PBSと共に移した。大腿骨を乳鉢で弱い力で5分間擦って粉砕し、このとき認められた赤い脊髄細胞は捨て去った。粉砕した大腿骨から赤い脊髄細胞を1%BSA含有PBSを新しいものと交換しながら5回取り去った後、粉々になった白い大腿骨を集め、0.2%コラゲナーゼ・タイプI(Sigma)含有PBSと共にインキュベートした。水浴中で37℃にて40分間激しく振盪した後、MSCを含む上清を70μmフィルターに通過させた。3回洗浄後に、培養プレートの壁面に接着したMSCをマウス20%MSC刺激物質含有MesenCult MSC基礎培地(StemCell Technologie Inc.)中で30日間培養した。培養中は、3日毎に半量の培地を交換した。得られたMSCが脂肪細胞及び骨細胞に分化する能力を確認するために、20%脂肪細胞形成及び骨細胞形成刺激物質を含有するMesenCult MSC基礎培地を用いて、70%コンフルエントMSCを2週間培養した後、脂肪細胞についてはOil Red O(Sigma-Aldrich)を、骨細胞についてはAlizarin Red S(和光ピュアケミカル)とヘマトキシリン(武藤ピュアケミカル)をそれぞれ用いて染色した。MSC刺激物質含有培地で得られた初期MSCコロニーをギムザ(和光ピュアケミカル)染色した。培養されたBM-MSCの純度は、PEを結合した抗CD140aモノクローナル抗体及びFITC結合抗Sca-1モノクローナル抗体で染色したMSCをフローサイトメトリー分析(FACScant II. BD)にて確認した。培養されたMSCは、更にCD29, CD90.1及びCD105の存在と、CD14, CD34及びCD45の非存在とに関し、各分子に対するモノクローナル抗体を用いたフローサイトメトリー分析によって評価した。
BALB/cマウス(CD90.2)の大腿骨をPBSで洗浄し、骨髄細胞を調製した。培養後BM-MSC移植する前に、BALB/cマウスに6-Gyの放射線を照射した。CD90.1+ BALB/cマウスから得た培養MSC(1 x 106/マウス)をBALB/cの大腿骨から得た骨髄細胞(5 x 106 個/マウス)と混合し、放射線を照射したBALB/cマウスに静脈内投与した。得られたキメラマウスは、1mg/mlネオマイシン(Calbiochem社)含有オートクレーブ水と、X線照射餌とを用いて2週間維持した。MSC移植から60日後に、CD90.1+ BM-MSCキメラBALB/ cマウスを腫瘍MSCの確認に用いた。
腫瘍浸潤CD8+T細胞放出ECVと腫瘍間質構造の変化との関係を調べるために、皮下にCMS5a腫瘍細胞を投与して10日後(腫瘍直径が約10mm)のCMS5a担癌BALB/cマウスまたはBALB/cヌードマウスに培養後7日目のCD90.1 DUC18 CD8+ T 細胞(1 x 107 個/マウス)単独またはGW4869(ECV放出阻害剤)処理したCD90.1 DUC18 CD8+ T 細胞(1 x 107 個/マウス)を静脈内投与した。このとき同時に、抗マウス・グルココルチコイド誘導TNFレセプター関連タンパク質(GITR)モノクローナル抗体(DTA-1)(2μg/腫瘍)を腫瘍内投与(inter tumor: i.t.)した。DTA-1は、非特許文献18に示すように、腫瘍部位へのCD8+T細胞の集積を高めるために使用した。GW4869は、培養終了の24時間前から20μg/ml添加した。培養したCD90.1 DUC18 CD8+ T細胞の投与後1,2,3,5及び7日後にCMS5a腫瘍組織を回収し、免疫組織染色した。
CMS5aを皮下移植したCD90.1+ BM-MSCキメラBALB/cマウスに対し、腫瘍細胞投与から2週間後にDUC18 CD8+ T細胞放出ECVを5μg((ECVタンパク質量)/腫瘍)で腫瘍内投与した。ECV投与から3日後に得られた腫瘍細胞懸濁液に関し、FITC結合CD90.1特異的モノクローナル抗体、PE結合CD140a特異的モノクローナル抗体、及びアロフィコシアニン(APC)結合Sca-1特異的モノクローナル抗体で染色後に、7-アミノアクチノマイシンD(7-ADD)染色細胞を除いたものをフローサイトメトリー分析に供した。
B16F10を皮下投与し、7, 10及び13日後に50μgのDUC18 CD8+ T細胞ECV, CMS5a担癌BALB/c脾臓細胞ECVまたはBALB/c脾臓細胞ECVを腫瘍内投与した。腫瘍細胞の投与から16日後に、B16F10由来の腫瘍(直径が約2cm)をハサミで注意深く切除し、腫瘍の浸潤を観察した後に外科用縫合糸で皮膚を縫い合わせた。腫瘍細胞の投与から45日後に、B16F10の肺転移の有無を観察した。
DUC18, CMS5a担癌BALB/c , B16担癌B6, BALB/c及びB6の培養脾臓細胞から得られたECVを5x104個/ml のCMS5a, B16, CT26またはBM-MSCの細胞培養液中に添加した。各細胞の培養には、それぞれ10%FCS含有RPMI-1640培地またはBM-MSC培地(20%MSC刺激物質含有MesenCult MSC基礎培地)を用いた。また、5x104 個/mlのCMS5a, CT26又はB16細胞と5x104 個/mlのBM-MSC(10%FCS含有RPMI-1640培地にて培養)の混合培養後中に1又は5μg(ECVタンパク質)/mlで添加した。
培養開始から4日後に、得られた細胞について、スフェロイド形成の確認及び全細胞数とCD140a及び/またはSca-1の発現確認のフローサイトメトリー分析を行った。
OCTコンパウンド(サクラ・ファインテクニカル)に埋め込まれたCMS5a及びB16F10腫瘍の凍結標本を3μmの厚さで切断し、2時間風乾した後、氷冷アセトンで15分間固定しものを免疫組織化学に供した。PBSで3回洗浄後に組織切片をブロッキング溶液(1%BSA, 5% Blocking One Histo(ナカライテスク社)含有PBS, 0.2μg/mlの抗マウスCD16/CD32モノクローナル抗体(Biolegend))と共に4℃にて30分間インキューベートした。更に、加湿チャンバー内において、スライド上の腫瘍切片を1%BSA・5% Blocking One Histo含有PBSに溶解したPE結合モノクローナル抗体及びFITC結合モノクローナル抗体を用いて、室温にて1時間、2重標識した。0.02% Tween-20含有PBSにて3回洗浄後、スライドをDAPI含有ProLong Gold退色防止試薬(インビトロージェン・ライフテクノロジーズ)で処理し、蛍光顕微鏡(オリンパス製BX53F)にて観察した。PE、FITC及びDAPI染色組織の画像は、Photoshop elementsソフトウエア(アドビ・システムズ)を用いて重ね合わせた。蛍光免疫測定法には、CD8, CD140a, Ki-67, CD31, CD11b, ER-TR7及びTGF-β1に対するPE結合モノクローナル抗体、並びにER-TR7, Sca-1, F4/80, Gr-1, CD90.1 及びα-smooth muscle actin (α-SMA)に対するFITC結合モノクローナル抗体を用いた。
CMS5a, CMS7及びCT26細胞を2.5mMカルボキシフルオレセイン・ジアセテート・スクシンイミジル・エステル(CFSE)を用いて、37℃にて6分間標識した。10%FCS含有RPMI-1640を用いて3回洗浄後、CFSE標識CMS5aを標的細胞として用いた。mERK2ペプチド刺激DUC18脾臓細胞を24穴プレートを用いて、CFSE標識CMS5a, CMS7またはB16細胞(1x105個)と1, 5 及び10の比率で混合した。12時間インキュベーション後、残りの細胞をフローサイトメトリーにて分析した。各サンプルについて、20,000個の非CFSE標識細胞を回収し、CFSE標識された生存している細胞数をカウントした。生存率は、2個のウエルの平均値として求め、%細胞傷害活性は文献に従い計算した(非特許文献18)。
培養したDUC18(2ロット分),CMS5a担癌BALB/c及びBALB/c脾細胞から得られた100μgのCD8+T細胞放出ECV、並びにCD8+ T細胞除去BALB/cマウス脾臓細胞から得られた100μgのCD4+T細胞放出ECVを3Dジーン・マイクロアッセイ・システム(東レ・インダストリーズ)によって解析しmiRNAを同定した。マイクロアレイの正規化された生データについては、各サンプルを比較した。CMS5a担癌BALB/cとCD4+BALB/cのECVと比較して、DUC18のECVにおいて優位に存在するmiRNAを検出し、特定された3個のmiRNAを機能解析にかけた。
フィコールを用いて、ヒト末梢血から単核球(PBMC)を分離した。PBMC(2 x 105 cells/ml)を0.6%自己血漿、0.2%ヒト血清アルブミン(CSL Behring)、600 IU/ml rIL-2入りのGT-T503培地(タカラバイオ)で2週間培養した。培養用プレートとして、5μg/ml OKT3抗体(バイオレジェンド)と25μg/ml RetroNectin (タカラバイオ)をコートしたものを用いた。培養後の細胞集団は、フローサイトメトリーにより、CD4及びCD8の有無を分析した。また、培養後の培養上清を10,000 gにて20 minの遠心処理した後、0.45μm及び0.22μmのフィルターで処理することにより、細胞破片や凝集タンパク質を除去した。更に、120,000 gにて70 minの超遠心処理することにより、ヒト培養T細胞放出エキソソームを分離した。
得られたエキソソームの直径をナノトラッキング解析(Nano-Tracking Analysis (NTA))により測定した。また、各種抗体をラテックスビーズ(4μm径: Life Technologies)と静電気的に結合させた物を用いて、フローサイトメトリー分析(BD: FACSCant)することにより、T細胞及びエキソソームの表面分子を調べた。
培養ヒトT細胞が放出するエキソソームが有するmiRNAをマイクロアレイ(東レ: 3D-Gene)により解析した。特定された40種類のmiRNAを存在量の多いものから順に合成した。合成したmiRNAを培養ヒト脂肪組織由来間葉系幹細胞(MSC)に添加し、細胞傷害作用を調べた。細胞傷害活性は、(1)培養MSCに合成したmiRNAを添加した後に培養MSCをギムザ染色(和光)する方法、及び(2)電気抵抗値で細胞生存を測定するxCELLigence (ACEA BioSciences)機器を用い、専用のプレートで培養したMSCに各miRNAを添加した後のMSC生存率を計測する方法により行った。
11.統計解析
2群のデータをマン・ホイットニーU検定にて解析した。分散の同等性をレビンの試験によって確認し、2群間のデータ比較はスチューデントのt検定により解析した。 p < 0.05を統計的に有意とした。統計計算は、SPSS統計ソフトウエアv21.0(IBM)にて行った。
1. CD8+T細胞放出ECVによる腫瘍増殖抑制
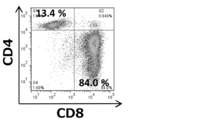
まず、我々はTCR刺激リンパ球由来ECVが腫瘍増殖に与える影響を調べた。TCR遺伝子トランスジェニックDUC18マウスの変異ERK2ペプチド刺激脾臓細胞、CD3特異的モノクローナル抗体及びCD28特異的モノクローナル抗体の両者で刺激したCMS5a担癌BALB/cマウス、BALB/cマウス、CD8+T細胞欠損BALB/cマウス脾細胞またはhPBMCを4日間培養した後、rIL-2(100IU/ml)を含有させて更に3日間培養した。培養されたDUC18, CMS5a担癌 BALB/c及びBALB/cの脾臓細胞は、4日後には全てCD4-CD8+の表現型を示した。CD8+T細胞欠損BALB/cマウスの脾臓細胞は、65%がCD4+及びCD8-であった。培養したhPBMCでは、それぞれCD8が70%、CD4が30%を示した(図1A)。
更に、培養したCD8+ DUC18脾臓細胞は、mERK2+ CMS5aに対して細胞毒性を示し、mERK2- CMS7 又は CT26には毒性を示さず、mERK2ペプチド刺激CMS7は溶解した(図2)。得られた培養上清を超遠心処理し、各細胞(DUC18, CMS5aTB, BALB/c, CD4 BALB/c及び hPBMC)からECVを精製した。すべてのECVは、培養上清中に0.6~1.0μg/mlタンパク質濃度、約4~8x109個/ml、及び110~140nmの平均直径で存在した(図3A及び3B)。DUC18 ECVの表面マーカを調べた。親細胞の表現型と一致して、DUC18 ECVは、僅かにCD8、TCRVβ8.3及びCD63を発現し、更に既に知られているECVマーカであるCD9を高く発現した(図1B)。
活性化CD8+ T細胞放出ECVが腫瘍細胞の増加を直接に抑制するか、腫瘍関連細胞に作用することによって腫瘍の増殖を抑制するか、のいずれかが推測された。この問題を解決するために、我々は、腫瘍増殖を調節すると報告されている腫瘍浸潤細胞集団の変動について調べた。CMS5a腫瘍に対しDUC18 ECV 及びBABL/c ECVを投与して3日後に、F4/80+ CD206+マクロファージまたはF4/80+ I-Ad+マクロファージと、CD11c+樹状細胞CD11b+Gr-1+MDSC又はCD4+及びCD8+リンパ球との比には変化が認められなかったが(図5)、増殖中の腫瘍細胞、BM-MSC及び/またはCAF細胞を含むCD140+間葉系マーカー陽性細胞数は、DUC18 ECV投与によって大きく減少した(図6A及び6B)。これらのデータは、DUC18 ECV及びBALB/c ECV を投与したCMS5aではCD140aの発現量が減少すること(図6C)によっても確認された。別に、ECV発現動態を、TRP-2及びGP100ペプチド刺激B6脾臓細胞の5, 7, 10, 15日目の培養上清から得られたECVによって確認した。TRP-2特異的CD8+リンパ球及びgp100特異的CD8+リンパ球は、培養中に徐々に増加し、15日目には、それぞれ95%及び3%に至った(図7)。興味深いことに、腫瘍内投与によるCD140a発現の減少のピークは培養7日目に得られるECVで起こり、TRP-2及びgd100特異的CD8+T細胞が放出するECVは、無関係なCMS5a及び関連するB16に対して同様に作用した(図6D)。投与後15日目のペプチド特異的CD8+T細胞による機能的ECV産生の減少は、リンパ球の衰退と関連すると考えられた。
これらの結果より、腫瘍の増殖と進行は、CD8+T細胞放出ECVの影響によって、特異性無く抑制されることが示された。
DUC18 ECVは、CMS5a細胞だけでなくCD26, 4T1及びB16細胞においても、CD140a発現量に対して直接には影響を与えず、インビトロでのアネクシンV染色によって、CMS5a、CT26, 4T1又はCMS7のアポトーシスを誘導できなかったことから、CD8+T細胞放出ECVによる腫瘍細胞の直接的な抑制効果は否定された(図8)。更に、免疫組織学においても、DUC18 ECV処理後のCMS5a腫瘍について、BM-MSC(CD140a+ Sca-1+)及びCAF(ER-TR7+ α-平滑筋アクチン[SMA]+)の消失、TGF-β1発現減少が認められた(図9A)。そこで我々は、間葉系腫瘍間質細胞の代表としてのBM-MSCとCD8+T細胞放出ECVとの関係について調べた。培養したBM-MCSの数は、DUC18 ECV及びB6 ECVを添加すると、3日後にはアポトーシスによって、劇的に減少した(図10)が、ヒトPBMC放出ECVでは、そのような反応は認められなかった(図9B)。更に、DUC18 ECVを培養BM-MSCと腫瘍細胞の混合物に加えたところ、CMS5aTB ECV処理群に比べると、CMS5a及びB16細胞のCD140aの発現並びにCMS5a, 4T1, CT26及びB16のスフェロイド形成は、DUC18 ECVの存在によってダウンレギュレートされた(図9C)ことから見て、BM-MSCとの相互作用による腫瘍の間葉系への遷移は、BM-MSCがCD8+ T細胞放出ECVによって損失を受けたことによって妨害されたものと考えられた。CD8+T細胞由来のECVによって、腫瘍浸潤BM-MSCが消失することをインビボで確認するために、DUC18 ECV, BALB/c ECV及びhPBMC ECVをCMS5a担癌CD90.1+ BM-MCSキメラBALB/cマウスの腫瘍内に投与した(図11)。
ECVは、BM-MSC及び腫瘍細胞のいずれに対しても取り込まれるが、BM-MSCと混合培養したB16細胞及びCMS5a細胞は、SYTO RNASelect標識DUC18, CMS5a TBまたはhPBMC ECVを取り込んで直ぐに緑色蛍光強度の減少が確認された。BM-MSCと接触した腫瘍細胞はECVを取り込んだ後すぐにECV由来のmiRNAを分解するようだ(図12A)。全RNAマイクロアレイ解析(東レ社製3Dジーン)によれば、BM-MSCに接触している腫瘍細胞では、リゾチームmRNA量の強い増大が認められた(図13)。SYTO RNASelect標識DUC18 ECV処理したCMS5aのフローサイトメトリー分析及び免疫組織化学解析でも、ECV処理から30分後には、CD140a+ またはSca-1+ 間質領域に緑色蛍光標識が認められたが、癌部には認められなかった(図12B、12C)。
BM-MSCの死滅に対し、hPMBC ECVが不応答で、自己及び同種CD8+ T細胞由来ECVが応答することから、ECV内のmiRNAの関与が示唆された。そこで、DUC18(2ロット), CMS5a TB及びCD4 Balb/c ECVから得られた全RNAをmiRNAマイクロアレイ(東レ社製3Dジーン)によって解析した。グローバル正規化数値及びmiRNA間のヒートマッピングデータを比較することによって、DUC18 ECVでは、miR-298, miR-351, miR-700, miR-141, miR-1943, miR-1249, miR-344g, miR-23b, miR-370, miR-1199, miR-5113, miR-5114, miR-6347, miR-6392及び miR-5099が、CMS5a ECVでは、 miR-150, miR-223 及び miR-3470bが、それぞれ優位に存在すること分かった(図14A)。予想された通り、DUC18 ECV由来のmiR-298, miR-141,miR -1249, miR-23b, 及び miR-370については腫瘍の自己抑制効果が報告されており、選択されたmiRNAが正しいことが確認された(図14B、図15)。併せて、DUC18 ECVのうちで最も効果的であると予測された3種類のmiRNA(miR-298, miR-1943及びmiR-5099)の5p及び3pオリゴヌクレオチドを合成、アニールし、培養BM-MSCに導入した。miR-1943及びmiR-5099を導入した場合に比べ、miR-298を導入した場合には、BM-MSCの数が際立って減少した(図14C)。CMD5a TB ECVに優位なmiR-150, miR-223及びmiR-3470bを導入した場合には、BM-MSC死滅作用は見られなかった。これらの結果から、BM-MSCを死滅させるmiRNAとして、miR-298が初めて見出された。免疫系の類似性及び癌の基礎研究から考えて、ヒトの細胞傷害性T細胞から取得したエキソソームに含まれるmiRNAにおいても、このような効果のある特定のmiRNAが存在している。
腫瘍の浸潤や転移は、腫瘍の上皮間葉転換及び増悪の指標となる。そこで我々は、B16F10細胞を皮下投与したマウスに対し、DUC18 ECV, CMS5a TB ECVまたはBALB/c ECVを10, 13 及び16日目に腫瘍内投与することで、浸潤と転移に与える影響を調べた。18日目に腫瘍浸潤の状態を観察し、B16F10腫瘍を外科的に切除し、皮膚を縫合した。無処理群では50%、CMS5a ECV処理群では33%で腫瘍の除去が可能であったものの、除去できなかった全ての腫瘍において筋膜への浸潤が認められた(表1)。

最後に、CD8+ T細胞が腫瘍に浸入し、ECVを放出しながら腫瘍間質構造を破壊するか否かを調べた。BALB/cまたはBALB/cヌードマウスにCMS5aを皮下投与し、約1cm径となったところで、GW4869(エキソソーム産生阻害剤)で処理または未処理の培養CD90.1+ DUC18 CD8+ T細胞(図17A)を腫瘍内に投与し、腫瘍に浸潤したCD90.1+ DUC18 CD8+ T細胞と間質の状態を経時的に腫瘍切片の免疫染色によって調べた。GW4869処理の有無に依らず、CD90.1+ CD8+ T細胞は、投与24時間後には、内皮前駆細胞およびBM-MSCによって構成されたCMS5a腫瘍間質のSca-1+ CD31+血管新生領域に認められた(図17B)。驚いたことに、CMS5a腫瘍のSca-1+または CD140a+ 領域は、CD90.1+ CD8+ T細胞の移入3日目には消失し、その状態が7日目まで持続した(図17C)。間質破壊効果は、BALB/cヌードマウスに比べ、野生型BALB/cマウスの方が大きく、これはBALB/c由来CD8+T細胞が、投与されたCD90.1+ CD8+ T細胞と共に腫瘍内浸潤した結果と考えられる。また、Sca-1+または CD140a+ 間質の消失は、GW4869処理CD90.1+ CD8+ T細胞の投与群では認められなかった(図17C)ことから、精製ECVを用いた結果と同様に、腫瘍内浸潤CD8+ T細胞は腫瘍間質構造を破壊するためのECVを産生していることが示唆された。CD90.1+ CD8+ T細胞放出ECVは強いCD9及びCD90.1発現を示し、CD8はほとんど出ていない(図18D)。CD90.1+ CD8+T細胞の投与24時間後のCMS5a腫瘍では、CD140a+ Sca-1+ 間質領域におけるECV由来CD9及びCD90.1(図18E)並びにその融合シグナル(図18F)が認められるものの、GW4869処理したCD90.1+ CD8+ T細胞処理群腫瘍では認められなかった。
ヒト末梢血から分離した単核球を2週間培養した後のT細胞集団をフローサイトメトリーで分析した結果、CD8が優位なT細胞集団となった(図19)。このT細胞が培養上清中に放出したエキソソームの直径をナノトラッキング解析したところ、約150nmであった(図20)。
エキソソーム及びヒト培養T細胞の表面抗原をフローサイトメトリーで分析した結果、エキソソームマーカーとしてのテトラスパニン分子(CD9, CD63, CD81)とCD8及びHLA class I分子を表現していた(図21)。
エキソソームが有する40種類のmiRNAを培養中のMSCに添加し、細胞傷害活性を調べた結果、2種類のmiRNA(miR-6089及びmiR-6090)を同定できた(図22,図23)。
こうして得られたMSC傷害性miRNAは、細胞増殖性疾患用の治療薬として応用できる。
ヒトとマウスとは、免疫系において、ほぼ同様のシステムを有していることから、マウスのインビボ及びインビトロで得られた知見は、そのままヒトにおいて転用できる。
このように、本実施形態によれば、細胞傷害性T細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬を提供できた。
次に、本発明に関する先行技術文献を示す。なお、明細書中に番号を付して説明しなかったが、本願発明の先行技術と成りうるものを示してある。
Claims (11)
- 細胞傷害性T細胞から放出された細胞外小胞(エキソソーム)を含む細胞増殖性疾病用の治療薬。
- 前記細胞傷害性T細胞は、ヒトCD4+、CD8+、CD9+、CD63+、 TCR+T細胞のうち少なくとも1または2以上から放出された細胞外小胞(エキソソーム)を含む請求項1記載の細胞増殖性疾病用の治療薬。
- 前記細胞傷害性T細胞は、CD8+T細胞から放出された細胞外小胞(エキソソーム)である細胞外小胞(エキソソーム)を含む請求項2記載の細胞増殖性疾病用の治療薬。
- 前記細胞外小胞(エキソソーム)は、細胞増殖抑制に有効なmiRNAを含むことを特徴とする請求項2または3記載の細胞増殖性疾病用の治療薬。
- 前記細胞増殖抑制に有効なmiRNAを含むことを特徴とする請求項4記載の細胞増殖性疾病用の治療薬。
- 前記細胞増殖性疾病用治療薬が、殺菌剤、粘膜除去剤、等張化剤、pH調節剤、安定化剤、増粘剤、防腐剤、粘着剤、又は免疫強化剤の中から選択される1または複数をさらに含有する請求項2記載の細胞増殖性疾病用の治療薬。
- 前記治療薬は腫瘍組織内、腫瘍組織内の間葉系細胞、静脈または皮下に投与されることを特徴とする請求項2に記載の細胞増殖性疾病用の治療薬。
- 細胞傷害性T細胞から放出されたエキソソームを回収し、そのエキソソームから細胞増殖抑制に有効なmiRNAを特定する細胞増殖性疾病治療用miRNAの抽出方法。
- 請求項8に記載の方法により得られた細胞増殖抑制に有効なmiRNAを含むことを特徴とする細胞増殖性疾病用の治療薬。
- 請求項8に記載の方法により特定されたmiRNAと同じ塩基配列を持つmiRNAを培養ヒト間葉系幹細胞(MSC)に添加して培養し、MSCに対する障害活性を調べることにより、miRNAのMSC傷害性を評価するMSC傷害性miRNAの同定方法。
- 請求項10に記載の方法により同定されたMSC傷害性miRNAを含む細胞増殖性疾患用の治療薬。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017508320A JP7292638B2 (ja) | 2015-03-20 | 2016-03-18 | 細胞傷害性t細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬 |
| EP16768689.8A EP3287135A4 (en) | 2015-03-20 | 2016-03-18 | THERAPEUTICS RELATED TO THE SUPPRESSION OF PROLIFERATION AND METASTASIC FORMATION OF CANCER WITH CYTOTOXIC T CELLS RELEASED EXOSOMES AND FACED AGAINST ELECTRICITY / MESENCHYMAL CANCER CELLS |
| US15/559,771 US11083744B2 (en) | 2015-03-20 | 2016-03-18 | Therapeutic agent associated with suppression of proliferation and metastasis of tumor, which comprises exosomes released from cytotoxic T cells and targets cancer stromal/mesenchymal cells |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-058467 | 2015-03-20 | ||
| JP2015058467 | 2015-03-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016152786A1 true WO2016152786A1 (ja) | 2016-09-29 |
Family
ID=56978400
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/058721 Ceased WO2016152786A1 (ja) | 2015-03-20 | 2016-03-18 | 細胞傷害性t細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11083744B2 (ja) |
| EP (1) | EP3287135A4 (ja) |
| JP (1) | JP7292638B2 (ja) |
| WO (1) | WO2016152786A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018106648A1 (en) * | 2016-12-05 | 2018-06-14 | The Penn State Research Foundation | Lipid-based probes for extracellular isolation |
| JP2020529477A (ja) * | 2017-08-04 | 2020-10-08 | シーダーズ—シナイ メディカル センター | がんの治療及び防止のためのカルディオスフィア由来細胞及びその細胞外小胞 |
| US11660355B2 (en) | 2017-12-20 | 2023-05-30 | Cedars-Sinai Medical Center | Engineered extracellular vesicles for enhanced tissue delivery |
| US11759482B2 (en) | 2017-04-19 | 2023-09-19 | Cedars-Sinai Medical Center | Methods and compositions for treating skeletal muscular dystrophy |
| US12146137B2 (en) | 2018-02-05 | 2024-11-19 | Cedars-Sinai Medical Center | Methods for therapeutic use of exosomes and Y-RNAS |
| US12544409B2 (en) | 2014-10-03 | 2026-02-10 | Cedars-Sinai Medical Center | Cardiosphere-derived cells and exosomes secreted by such cells in the treatment of muscular dystrophy |
| US12584127B2 (en) | 2012-08-13 | 2026-03-24 | Cedars-Sinai Medical Center | Exosomes and micro-ribonucleic acids for tissue regeneration |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102770425B1 (ko) | 2016-02-08 | 2025-02-18 | 고쿠리츠다이가쿠호진 미에다이가쿠 | 면역체크포인트 저해제 저항성 종양에 대한 t세포 수주요법의 전처치약 |
| ES2971550T3 (es) * | 2016-09-02 | 2024-06-05 | Univ Cornell | Receptor de antígeno quimérico de dominio I específico para ICAM-1 |
| WO2018183930A1 (en) * | 2017-03-30 | 2018-10-04 | Carson Dennis A | Methods for isolating, expanding and administering cancer specific cd8+ t cells |
| US11497768B2 (en) | 2017-06-05 | 2022-11-15 | Mie University | Antigen-binding protein that recognizes MAGE-A4-derived peptide |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014507140A (ja) * | 2011-02-11 | 2014-03-27 | エイジェンシー・フォー・サイエンス,テクノロジー・アンド・リサーチ | 治療用エキソソームを検出する方法 |
-
2016
- 2016-03-18 US US15/559,771 patent/US11083744B2/en active Active
- 2016-03-18 EP EP16768689.8A patent/EP3287135A4/en not_active Withdrawn
- 2016-03-18 WO PCT/JP2016/058721 patent/WO2016152786A1/ja not_active Ceased
- 2016-03-18 JP JP2017508320A patent/JP7292638B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014507140A (ja) * | 2011-02-11 | 2014-03-27 | エイジェンシー・フォー・サイエンス,テクノロジー・アンド・リサーチ | 治療用エキソソームを検出する方法 |
Non-Patent Citations (4)
| Title |
|---|
| ANGELA MONTECALVO ET AL.: "Methods of Purification of CTL-Derived Exosomes", METHODS IN MOLECULAR BIOLOGY, vol. 1186, 2014, pages 87 - 102, XP009506705 * |
| NAOHIRO SEO ET AL.: "Future possibility of tumor immunotherapy by exosomes", THE CELL, vol. 48, no. 1, January 2016 (2016-01-01), pages 21 - 24, XP009506711 * |
| NAOHIRO SEO ET AL.: "Tumor- and Immune Cell - Derived Exosomes", DRUG DELIVERY SYSTEM, vol. 29, no. 2, 2014, pages 152 - 159, XP055317754 * |
| See also references of EP3287135A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12584127B2 (en) | 2012-08-13 | 2026-03-24 | Cedars-Sinai Medical Center | Exosomes and micro-ribonucleic acids for tissue regeneration |
| US12544409B2 (en) | 2014-10-03 | 2026-02-10 | Cedars-Sinai Medical Center | Cardiosphere-derived cells and exosomes secreted by such cells in the treatment of muscular dystrophy |
| WO2018106648A1 (en) * | 2016-12-05 | 2018-06-14 | The Penn State Research Foundation | Lipid-based probes for extracellular isolation |
| US11759482B2 (en) | 2017-04-19 | 2023-09-19 | Cedars-Sinai Medical Center | Methods and compositions for treating skeletal muscular dystrophy |
| JP2020529477A (ja) * | 2017-08-04 | 2020-10-08 | シーダーズ—シナイ メディカル センター | がんの治療及び防止のためのカルディオスフィア由来細胞及びその細胞外小胞 |
| US11660355B2 (en) | 2017-12-20 | 2023-05-30 | Cedars-Sinai Medical Center | Engineered extracellular vesicles for enhanced tissue delivery |
| US12146137B2 (en) | 2018-02-05 | 2024-11-19 | Cedars-Sinai Medical Center | Methods for therapeutic use of exosomes and Y-RNAS |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3287135A4 (en) | 2019-03-27 |
| US20180177816A1 (en) | 2018-06-28 |
| US11083744B2 (en) | 2021-08-10 |
| EP3287135A1 (en) | 2018-02-28 |
| JP7292638B2 (ja) | 2023-06-19 |
| JPWO2016152786A1 (ja) | 2018-03-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7292638B2 (ja) | 細胞傷害性t細胞放出エキソソームによる癌間質間葉系細胞を標的とした腫瘍増殖及び転移抑制に係る治療薬 | |
| JP7724504B2 (ja) | 骨格筋ジストロフィーを治療する方法及び組成物 | |
| Seo et al. | Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells | |
| JP6716668B2 (ja) | リンパ組織に幹細胞および前駆細胞が結合することを阻害する組成および方法、ならびにリンパ組織の胚中心を再生させるための組成および方法 | |
| AU2017219415B2 (en) | Combination immune therapy and cytokine control therapy for cancer treatment | |
| AU2016221305B2 (en) | Combination immune therapy and cytokine control therapy for cancer treatment | |
| WO2021053667A2 (en) | Combination cancer therapy and cytokine control therapy for cancer treatment | |
| BR112019020214A2 (pt) | substituição do pré-condicionamento citotóxico antes da imunoterapia celular | |
| Wan et al. | Peptide hydrogels loaded with irradiated tumor cell secretions enhance cancer immunotherapy | |
| CN110072534A (zh) | 外泌体的医学应用 | |
| KR20220026575A (ko) | Pdl1 양성 nk 세포 암 치료 | |
| CN114729314A (zh) | 用于癌症治疗的组合癌症疗法和细胞因子控制疗法 | |
| EP4031655A2 (en) | Combination cancer therapy and cytokine control therapy for cancer treatment | |
| EP2649995A2 (en) | Inhibition of post-radiation tumor growth | |
| WO2023044039A1 (en) | Compositions and methods for treating cancer | |
| US20250213687A1 (en) | Therapeutic t cell product | |
| JP2021523359A (ja) | 併用療法のための患者の選択 | |
| WO2018075856A2 (en) | Methods related to breaking t cell exhaustion | |
| HK40008076A (en) | Replacement of cytotoxic preconditioning before cellular immunotherapy | |
| HK1186117B (zh) | 抑制幹細胞和祖細胞與淋巴樣組織結合和用於使淋巴組織中的生髮中心再生的組合物和方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16768689 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017508320 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016768689 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15559771 Country of ref document: US |