WO2016195059A1 - マイトマイシンcの精製方法 - Google Patents
マイトマイシンcの精製方法 Download PDFInfo
- Publication number
- WO2016195059A1 WO2016195059A1 PCT/JP2016/066540 JP2016066540W WO2016195059A1 WO 2016195059 A1 WO2016195059 A1 WO 2016195059A1 JP 2016066540 W JP2016066540 W JP 2016066540W WO 2016195059 A1 WO2016195059 A1 WO 2016195059A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mitomycin
- methanol
- purity
- purification method
- ppb
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/005—Selection of auxiliary, e.g. for control of crystallisation nuclei, of crystal growth, of adherence to walls; Arrangements for introduction thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D2009/0086—Processes or apparatus therefor
Definitions
- the present invention relates to a method for purifying mitomycin C and the like.
- Mitomycin C is an antitumor antibiotic obtained by culturing a strain of Streptomyces kespitose, and is widely used clinically.
- a method of purifying mitomycin C from a culture solution obtained by culturing the above strain activated carbon adsorption method in which activated carbon is added to a culture filtrate obtained by separating cells and adsorbed, and eluted with an organic solvent.
- a concentrated solution of mitomycin C obtained by transferring the culture filtrate into an organic solvent is purified by subjecting it to an alumina chromatography method or a countercurrent distribution method, and then concentrated until the eluted mitomycin C solution is solidified.
- Patent Document 1 A method of obtaining crystals by adding a small amount of acetone to this is known (Patent Document 1).
- the culture solution is adsorbed on a reverse-phase adsorption resin, it is eluted with a solvent such as acetone, methanol or ethanol, the eluate is concentrated to remove the solvent, saturated with sodium chloride, and dissolved in chloroform.
- the extract was subjected to alumina column chromatography, separated and eluted, and the eluate was concentrated to a concentrated methanol solution, and then ether, petroleum ether, benzine or ligroin was added to obtain pure mitomycin C crystals, A method of obtaining mitomycin C pure crystals by washing the coarse crystals obtained by further concentrating the mother liquor and performing the same operation with a 10% methanol / ether mixture is known (Patent Document 2).
- Patent Document 4 a method for producing mitomycin C crystals from a solvent system comprising a combination of a solvent capable of dissolving mitomycin C and a poor solvent is known (Patent Document 4).
- mitomycin C for injection and mitomycin C for injection the number of insoluble microparticles derived from mitomycin C increased rapidly, saying, ⁇ No more than 6000 particles of 10 ⁇ m or more and 600 particles of 25 ⁇ m or more per container.
- mitomycin C was produced that deviated from the standard for injections (16th Japanese Pharmacopoeia-Insoluble fine particle test method for injections). Therefore, in order to produce mitomycin C for injection and mitomycin C for injection, it was necessary to improve the purification method of mitomycin C crude crystals.
- An object of the present invention is to provide a method for purifying mitomycin C and the like in which the increase in insoluble fine particles over time is suppressed.
- the present inventors have found that mitomycin C in which the increase in insoluble fine particles over time is suppressed can be stably obtained by using high-purity methanol when purifying mitomycin C crude crystals.
- the present invention relates to the following (1) to (23), for example.
- (1) A method for purifying mitomycin C comprising a step of crystallizing crude crystals of mitomycin C using high-purity methanol.
- the purification method according to (1), wherein the purity of the high-purity methanol is 99.50 to 99.99%.
- a method for reducing insoluble fine particle production comprising a step of crystallizing crude crystals of mitomycin C using high-purity methanol.
- a method for purifying mitomycin C crude crystals comprising the following steps (A) to (E): (A) A step of adding high-purity methanol to mitomycin C crude crystals and dissolving them. (B) A step of cooling the filtrate obtained in (A) above. (C) A step of adding a poor solvent to the solution obtained in (B) above. (D) A step of filtering the mixture obtained in (C) to obtain crystals.
- the method for purifying crude crystals of mitomycin C of the present invention will be specifically described.
- the crude crystal of mitomycin C used in the present invention can be obtained, for example, by the method described in JP-B-35-17897, JP-B-36-9094, JP-A-4-87092, and the like, specifically, obtained by culturing microorganisms.
- a resin for example, reverse-phase adsorption resin
- alumina chromatography method for example, alumina chromatography method, a countercurrent distribution method, or the like.
- the above confirmation test, content and loss on drying shall be measured, for example, according to the methods described in each item of “Confirmation Test”, “Quantitative Method” and “Loss on Drying” of “Mitomycin C” of the 16th revised Japanese Pharmacopoeia Can do.
- the unit v / w in the present invention represents the volume (mL) of liquid with respect to 1 g of the object.
- Examples of the poor solvent in the present invention include ethyl ether and petroleum ether.
- Examples of the metal in the present invention include organometallic compounds, metals, metal salts, and the like.
- the metal include nickel, lead, cadmium, iron, and zinc.
- the metal salt include nickel salts such as nickel chloride and nickel oxide, lead salts such as lead chloride and lead oxide, cadmium salts such as cadmium chloride and cadmium oxide, iron salts such as iron chloride and iron oxide, zinc chloride And zinc salts such as zinc oxide.
- each metal is detected as each metal ion by, for example, the metal analysis method described in Inductively Coupled Plasma Emission Spectroscopy and Inductively Coupled Plasma Mass Spectrometry ⁇ 2.63> of the 16th revision Japanese Pharmacopoeia
- the total sum of each metal content is shown.
- Examples of zinc in the present invention include organic zinc compounds, metallic zinc, zinc salts and the like.
- Examples of the zinc salt include zinc chloride and zinc oxide.
- Production method Add 50-100 v / w, preferably 60-70 v / w methanol to the crude crystal of mitomycin C, and stir at a temperature between 50-70 ° C, preferably between 55-67 ° C. And dissolve.
- the obtained solution can be filtered as necessary to remove insolubles and the like.
- the methanol include methanol having a low sum of each metal concentration, and examples of methanol having a low sum of each metal concentration include, for example, methanol obtained by distilling commercially available methanol, and the sum of each metal concentration. Examples include low commercial high-purity methanol. Examples of the commercially available high-purity methanol having a low total concentration of each metal include commercially available products such as purified methanol (manufactured by Toyo Gosei Co., Ltd.).
- the purity of the methanol is, for example, 99.00 to 99.99%, preferably 99.50 to 99.99%, more preferably 99.60 to 99.99%, especially 99.80 to 99.99%.
- the total concentration of each metal in the methanol is preferably as low as possible, for example, 0.01 to 300 ppb, preferably 0.01 to 100 ppb, more preferably 0.01 to 30 ppb, especially 0.01 to 10 ppb.
- the zinc concentration in the methanol is preferably as low as possible, for example, 0.01 to 100 ppb, preferably 0.01 to 10 ppb, more preferably 0.01 to 7 ppb, and particularly preferably 0.01 to 6 ppb.
- the solution obtained above or the filtrate after completion of filtration is cooled, 70 to 110 v / w, preferably 85 to 95 v / w of ethyl ether is added to the crude crystals, and 200 to 200 to the crude crystals.
- the resulting mixture is left at -10 to 15 ° C, preferably -5 to 10 ° C for 0.5 to 2 hours, preferably 0.8 to 1.2 hours without stirring or stirring, and the precipitated crystals are collected by filtration. Dry and obtain purified mitomycin C.
- Mitomycin C (1.51 g) was obtained in the same manner as in Example 1 using crude crystals of mitomycin C (1.8 g) and commercially available methanol (Wako Pure Chemicals, first grade).
- Mitomycin C (1.56 g) was prepared in the same manner as in Example 1, using mitomycin C crude crystals (1.8 g) and commercially available methanol (Wako Pure Chemicals, first grade) concentrated 5-fold using a rotary evaporator.
- Table 2 shows the measured values of zinc concentration in mitomycin C obtained by purification in Example 1 and Comparative Examples 1 and 2.
- Test Example 2 Change in the number of microparticles in mitomycin C under severe stability test 300 mg of mitomycin C obtained in Example 1 and Comparative Examples 1 and 2 was put in vials, respectively, under conditions of 50 ° C and humidity. Samples were stored and sampled at 0, 191 and 300 hours after the start of storage, and the increase in fine particles was observed according to the following “quantification of fine particles in mitomycin C”.
- the mitomycin C obtained in Example 1 had a particle size of 10 ⁇ m or more and a particle size of 25 ⁇ m or more even after 300 hours of severe conditions, and the number of fine particles was almost all. It did not increase.
- the mitomycin C obtained in Comparative Examples 1 and 2 and containing more zinc than Example 1 had a particle size of 10 ⁇ m or more and a particle size of 25 ⁇ m after a total severe condition time of 300 hours. In all of the above, the number of fine particles increased significantly.
- the present invention provides a method for purifying mitomycin C and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Crystallography & Structural Chemistry (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description
従来、上記菌株を培養して得られる培養液からマイトマイシンCを精製する方法としては、菌体を分離して得られる培養ろ液に活性炭を加えて吸着させ、有機溶媒で溶出させる活性炭吸着法、あるいは培養ろ液から有機溶媒に転溶して得られるマイトマイシンCの濃縮液を、アルミナクロマトグラフィー法もしくは向流分配法に付して精製した後に、溶出したマイトマイシンCの溶液を乾固するまで濃縮し、これに少量のアセトンを添加することにより結晶を得る方法が知られている(特許文献1)。
本発明は、例えば以下の(1)~(23) に関する。
(1) 高純度メタノールを用いてマイトマイシンCの粗結晶の結晶化を行う工程を含むことを特徴とするマイトマイシンCの精製方法。
(2) 高純度メタノールの純度が、99.00~99.99%である (1) 記載の精製方法。
(3) 高純度メタノールの純度が、99.50~99.99%である (1) 記載の精製方法。
(4) 高純度メタノール中の各金属濃度の総和が0.01~300 ppbである(1)~(3) のいずれかに記載の精製方法。
(5) 高純度メタノール中の各金属濃度の総和が0.01~100 ppbである(1)~(3) のいずれかに記載の精製方法。
(6) 高純度メタノール中の亜鉛濃度が0.01~100 ppbである(1)~(3) のいずれかに記載の精製方法。
(7) 高純度メタノール中の亜鉛濃度が0.01~10 ppbである(1)~(3) のいずれかに記載の精製方法。
(8) (1)~(7)のいずれかに記載の精製方法により得られるマイトマイシンC。
(9) 高純度メタノールを用いてマイトマイシンCの粗結晶の結晶化を行う工程を含むことを特徴とする不溶性微粒子生成の低減方法。
(10) 高純度メタノールの純度が、99.00~99.99%である (9) 記載の低減方法。
(11) 高純度メタノールの純度が、99.50~99.99%である (9) 記載の低減方法。
(12) 高純度メタノール中の各金属濃度の総和が0.01~300 ppbである(9)~(11) のいずれかに記載の低減方法。
(13) 高純度メタノール中の各金属濃度の総和が0.01~100 ppbである(9)~(11) のいずれかに記載の低減方法。
(14) 高純度メタノール中の亜鉛濃度が0.01~100 ppbである(9)~(11) のいずれかに記載の低減方法。
(15) 高純度メタノール中の亜鉛濃度が0.01~10 ppbである(9)~(11) のいずれかに記載の低減方法。
(16) 下記 (A)~(E) の工程を含むマイトマイシンCの粗結晶の精製方法。
(A) マイトマイシンCの粗結晶に高純度メタノールを加えて溶解させる工程。
(B) 上記 (A) で得られたろ液を冷却する工程。
(C) 上記 (B) で得られた溶液に貧溶媒を添加する工程。
(D) 上記 (C) で得られた混合液のろ過を行い、結晶を得る工程。
(E) 上記 (D) で得られた結晶を乾燥させる工程。
(17) 高純度メタノールの純度が、99.00~99.99%である(16) 記載の精製方法。
(18) 高純度メタノールの純度が、99.50~99.99%である(16) 記載の精製方法。
(19) 高純度メタノール中の各金属濃度の総和が0.01~300 ppbである(16) 記載の精製方法。
(20) 高純度メタノール中の各金属濃度の総和が0.01~100 ppbである(16) 記載の精製方法。
(21) 高純度メタノール中の亜鉛濃度が0.01~100 ppbである(16) 記載の精製方法。
(22) 高純度メタノール中の亜鉛濃度が0.01~10 ppbである(16) 記載の精製方法。
(23) (16)~(22) のいずれかに記載の精製方法により得られるマイトマイシンC。
本発明で用いるマイトマイシンCの粗結晶は、例えば、特公昭35-17897、特公昭36-9094、特開平4-187092等に記載の方法により得られ、具体的には、微生物の培養で得られた培養液に対して、例えば、活性炭または樹脂(例えば、逆相吸着樹脂等)への吸着を利用した精製法、アルミナクロマトグラフィー法、向流分配法等の処理を行って得られる粗結晶であり、
i) 確認試験に適合する。 ii) マイトマイシンCの粗結晶 1 mg中のマイトマイシンCの標準品 1 mgに対する含量が950 μg以上である。およびiii) 乾燥減量が1.0%以下である。
という条件を満たすマイトマイシンCを示す。
本願発明における単位v/wは、対象1 gに対する液体の体積 (mL) を表す。
本願発明における貧溶媒としては、例えば、エチルエーテル、石油エーテル等が挙げられる。
本願発明における亜鉛としては、例えば、有機亜鉛化合物、金属亜鉛、亜鉛塩等が挙げられる。該亜鉛塩としては、例えば、塩化亜鉛、酸化亜鉛等が挙げられる。
製造法
マイトマイシンCの粗結晶に対して50~100 v/w、好ましくは60~70 v/wのメタノールを加えて、50~70 ℃の間、好ましくは55~67 ℃の間の温度で撹拌し、溶解させる。
該メタノールとしては、例えば、各金属濃度の総和が低いメタノール等が挙げられ、各金属濃度の総和が低いメタノールとしては、例えば、市販のメタノールを蒸留して得られるメタノール、各金属濃度の総和が低い市販の高純度メタノール等が挙げられる。
該各金属濃度の総和が低い市販の高純度メタノールとしては、例えば、精製メタノール(東洋合成工業社製) 等の市販品が挙げられる。
・比較例1
マイトマイシンCの粗結晶(1.8 g)および市販品のメタノール(和光純薬、一級品)を用いて実施例1と同様にしてマイトマイシンC(1.51 g)を得た。
・比較例2
マイトマイシンCの粗結晶(1.8 g)および市販品のメタノール(和光純薬、一級品)をロータリーエバポレーターを用いて5倍に濃縮したメタノールを用いて、実施例1と同様にしてマイトマイシンC(1.56 g)を得た。
・試験例1
メタノール中およびマイトマイシンC中の亜鉛の定量
1) メタノール
ナスフラスコにメタノール500 mLを入れ、エバポレーターで濃縮乾固した。ナスフラスコを特級硝酸(和光純薬)を薄めて得られた4%硝酸水溶液で洗いこみ50 mLとし、試料溶液とした。
2) マイトマイシンC
容器に、上記実施例1ならびに比較例1および2で得られたマイトマイシンCを0.2 g量りとり、特級硝酸(和光純薬)3 mLおよび特級過酸化水素水(純正化学)3 mLを加えた後、水を加えて正確に50 mLとし、試料溶液とした。
・試験条件
波長:亜鉛213.856 nm
高周波出力:1.2 kW
キャリヤーガス:アルゴン
キャリヤーガス流量:0.7 L/min
補助ガス流量:0.6 L/min
冷却ガス流量:10.0 L/min
上記実施例1ならびに比較例1および2のマイトマイシンCの粗結晶の精製に用いたメタノール中の亜鉛濃度の測定値を表1に示す。
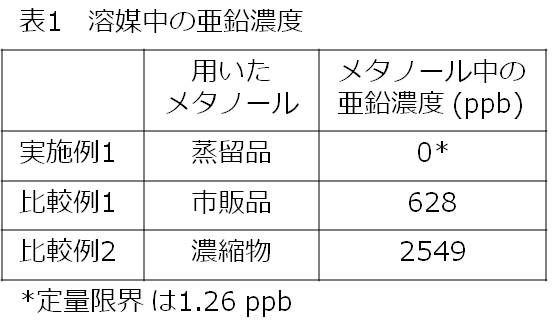
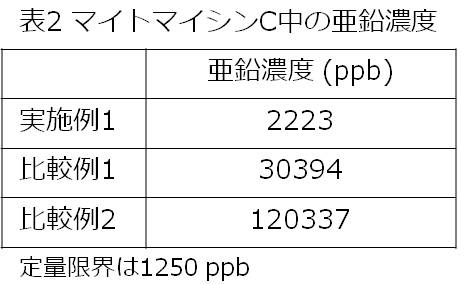
試験例2 苛酷安定性試験下でのマイトマイシンC中の微粒子数の変化
実施例1ならびに比較例1および2で得られたマイトマイシンCの300 mgをそれぞれバイアルに取り、50 ℃、湿度成り行きの条件でサンプルを保管し、保管開始からの延べ時間で0、191および300時間後にサンプリングを行い、下記「マイトマイシンC中の微粒子の定量」に従い、微粒子の増加を観察した。
マイトマイシンC 10 mgを精密に量り、注射用水を用いて正確に25 mLとし、試料溶液とした。
マイトマイシンC中の微粒子はMFI DPA5200(Protein Simple)を用いて分析した。試料溶液分析前に、機器流路を注射用水で洗浄し、微粒子フリーのベースラインを得た。試料溶液1 mLをピペットチップで採取し、サンプルホルダーに導入した。流速0.1 mL/minで流路を試料溶液で置換後、試料溶液(分析容量0.61 mL)を分析した。
以上の操作を3回繰り返し、その平均を定量値とした。
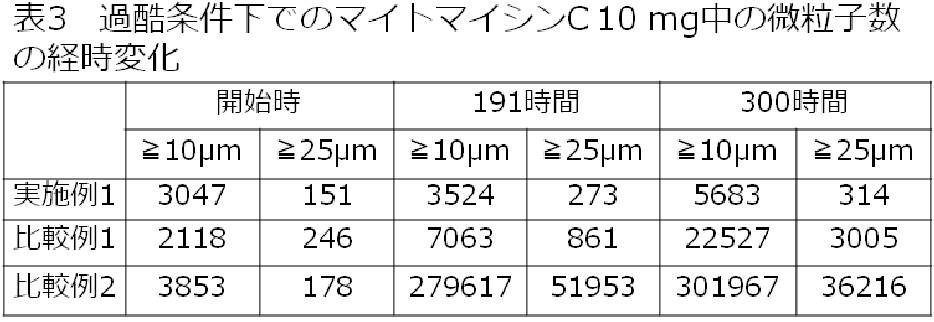
Claims (8)
- 高純度メタノールを用いてマイトマイシンCの粗結晶の結晶化を行う工程を含むことを特徴とするマイトマイシンCの精製方法。
- 高純度メタノールの純度が、99.00~99.99%である請求項1記載の精製方法。
- 高純度メタノールの純度が、99.50~99.99%である請求項1記載の精製方法。
- 高純度メタノール中の各金属濃度の総和が0.01~300 ppbである請求項1~3のいずれかに記載の精製方法。
- 高純度メタノール中の各金属濃度の総和が0.01~100 ppbである請求項1~3のいずれかに記載の精製方法。
- 高純度メタノール中の亜鉛濃度が0.01~100 ppbである請求項1~3のいずれかに記載の精製方法。
- 高純度メタノール中の亜鉛濃度が0.01~10 ppbである請求項1~3のいずれかに記載の精製方法。
- 請求項1~7のいずれかに記載の精製方法により得られるマイトマイシンC。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680031732.4A CN107709329A (zh) | 2015-06-05 | 2016-06-03 | 丝裂霉素c的纯化方法 |
| JP2017522270A JPWO2016195059A1 (ja) | 2015-06-05 | 2016-06-03 | マイトマイシンcの精製方法 |
| US15/579,230 US10125142B2 (en) | 2015-06-05 | 2016-06-03 | Method for purifying mitomycin C |
| EP16803490.8A EP3305791A4 (en) | 2015-06-05 | 2016-06-03 | Purification method for mitomycin c |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015114495 | 2015-06-05 | ||
| JP2015-114495 | 2015-06-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016195059A1 true WO2016195059A1 (ja) | 2016-12-08 |
Family
ID=57440529
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/066540 Ceased WO2016195059A1 (ja) | 2015-06-05 | 2016-06-03 | マイトマイシンcの精製方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10125142B2 (ja) |
| EP (1) | EP3305791A4 (ja) |
| JP (1) | JPWO2016195059A1 (ja) |
| CN (1) | CN107709329A (ja) |
| WO (1) | WO2016195059A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001031680A (ja) * | 1999-07-19 | 2001-02-06 | Mercian Corp | 結晶質マイトマイシンcおよびその製造方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2971128B2 (ja) | 1990-11-20 | 1999-11-02 | 協和醗酵工業株式会社 | マイトマイシンcの精製法 |
| US5180670A (en) * | 1990-12-28 | 1993-01-19 | Kyowa Hakko Kogyo, Ltd. | Method for purification of mitomycin C |
-
2016
- 2016-06-03 EP EP16803490.8A patent/EP3305791A4/en not_active Withdrawn
- 2016-06-03 US US15/579,230 patent/US10125142B2/en not_active Expired - Fee Related
- 2016-06-03 WO PCT/JP2016/066540 patent/WO2016195059A1/ja not_active Ceased
- 2016-06-03 CN CN201680031732.4A patent/CN107709329A/zh active Pending
- 2016-06-03 JP JP2017522270A patent/JPWO2016195059A1/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001031680A (ja) * | 1999-07-19 | 2001-02-06 | Mercian Corp | 結晶質マイトマイシンcおよびその製造方法 |
Non-Patent Citations (5)
| Title |
|---|
| "Seihin Kikakusho Methanol Wako Ikkyu", 2006, Retrieved from the Internet <URL:http://www.siyaku.com/uh/Sks.do ? pcode=13-0183&now=1470633046137&JE=J&pop_ zenkai_gst=1> [retrieved on 20160805] * |
| HIDEYUKI AZUMA: "ICH Guideline Q3D (Kinzoku Fujunbutsu) ni Taio suru Iyakuhin To no Kinzoku Fujunbutsu Bunseki", SCAS NEWS, vol. 40, 2014, pages 11 - 14, XP009507742 * |
| IYENGAR, BHASHYAM S. ET AL.: "Metal complexes of mitomycins", JOURNAL OF MEDICINAL CHEMISTRY, vol. 29, no. 1, 1986, pages 144 - 147, XP002272032, ISSN: 0022-2623 * |
| See also references of EP3305791A4 * |
| SEIHIN KIKAKUSHO METHANOL SC, 2011, Retrieved from the Internet <URL:http://www.siyaku.com/uh/Sks. do?pcode=13-1639&now=1470632958914&JE=J&pop_ zenkai gst=1> [retrieved on 20160805] * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107709329A (zh) | 2018-02-16 |
| JPWO2016195059A1 (ja) | 2018-03-22 |
| EP3305791A4 (en) | 2018-11-07 |
| US20180179219A1 (en) | 2018-06-28 |
| EP3305791A1 (en) | 2018-04-11 |
| US10125142B2 (en) | 2018-11-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10214552B2 (en) | Method for purifying beta-nicotinamide mononucleotide | |
| CN107353201B (zh) | 一种高含量天然莽草酸提取物及其制备方法 | |
| US20160340378A1 (en) | Method for purifying oxidized form of beta-nicotinamide adenine dinucleotide phosphate | |
| CN119335103B (zh) | 一种奥拉氟及其相关组分的检测方法 | |
| CN103149312A (zh) | 一种利用超高效液相色谱串联四级杆质谱分析婴儿乳粉中唾液酸的方法 | |
| EP3378868A1 (en) | 6'-sialyl lactose sodium salt crystal and method for manufacturing same | |
| CN106093254A (zh) | 一种玉米赤霉烯酮类毒素的富集净化方法 | |
| CN115340650A (zh) | 一种磁性方酸基功能化COFs材料、制备方法及其应用 | |
| CN114249711A (zh) | 一种拆分制备尼古丁的方法 | |
| CN107417749B (zh) | 一种辅酶i的树脂填料分离方法 | |
| RU2456233C2 (ru) | Способ получения фуллерена с60 | |
| WO2016195059A1 (ja) | マイトマイシンcの精製方法 | |
| CN107589187B (zh) | 一种烟草中残留抑芽丹的提取方法及测定方法 | |
| Cullen et al. | The effect of arsenicals on cell suspension cultures of the Madagascar periwinkle (Catharanthus roseus) | |
| EP3118207B1 (en) | Inclusion compound of 3',5'-cyclicdiadenylic acid, and method for producing same | |
| CN109206486B (zh) | 一种硫酸多黏菌素b的杂质及其制备方法 | |
| CN103399104B (zh) | 一种茶叶中邻苯二甲酸酯检测前处理试剂盒及其处理方法 | |
| CN107033114B (zh) | 一种二氢杨梅素的分离纯化方法 | |
| CN113087774A (zh) | 棘白菌素b母核降解杂质的去除方法 | |
| CN117085653A (zh) | 一种用于特异性富集抗氧化肽的Cu-MOF材料及其制备和应用方法 | |
| CN101367728B (zh) | 从紫锥菊提取物纯化菊苣酸和单咖啡酰酒石酸的方法 | |
| CN113759048A (zh) | 一种十八烷二酸单叔丁酯检验方法 | |
| CN102219793B (zh) | D(-)-磺苄西林钠的提纯方法 | |
| KR20200123711A (ko) | 클로린 e6염 광민감제의 제조방법 | |
| CN101671383A (zh) | 单体7-乙氧基灵芝酸o及单体灵芝酸t分离纯化方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16803490 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017522270 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15579230 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016803490 Country of ref document: EP |