WO2017018476A1 - キナゾリン誘導体の塩またはその結晶およびそれらの製造方法 - Google Patents
キナゾリン誘導体の塩またはその結晶およびそれらの製造方法 Download PDFInfo
- Publication number
- WO2017018476A1 WO2017018476A1 PCT/JP2016/072129 JP2016072129W WO2017018476A1 WO 2017018476 A1 WO2017018476 A1 WO 2017018476A1 JP 2016072129 W JP2016072129 W JP 2016072129W WO 2017018476 A1 WO2017018476 A1 WO 2017018476A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- compound
- formula
- powder
- ray diffraction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- WHIBYUKSMBAUAY-RXMQYKEDSA-N NOC[C@@H]1NCCOC1 Chemical compound NOC[C@@H]1NCCOC1 WHIBYUKSMBAUAY-RXMQYKEDSA-N 0.000 description 2
- IBCIAMOTBDGBJN-NRLRZRKLSA-N CC#C/C(/c1cc2c(Nc(cc3)cc(Cl)c3OCc3cc(F)ccc3)ncnc2cc1)=N\OC[C@@H]1NCCOC1 Chemical compound CC#C/C(/c1cc2c(Nc(cc3)cc(Cl)c3OCc3cc(F)ccc3)ncnc2cc1)=N\OC[C@@H]1NCCOC1 IBCIAMOTBDGBJN-NRLRZRKLSA-N 0.000 description 1
- JCMZTBHMGPQRRV-UHFFFAOYSA-N CC#CC(c1cc2c(Nc(cc3)cc(Cl)c3OCc3cc(F)ccc3)ncnc2cc1)=O Chemical compound CC#CC(c1cc2c(Nc(cc3)cc(Cl)c3OCc3cc(F)ccc3)ncnc2cc1)=O JCMZTBHMGPQRRV-UHFFFAOYSA-N 0.000 description 1
- NNNUVMKUCXQAPC-ZCFIWIBFSA-N C[C@@]1(CON)NCCOC1 Chemical compound C[C@@]1(CON)NCCOC1 NNNUVMKUCXQAPC-ZCFIWIBFSA-N 0.000 description 1
Images





















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/30—1,4-Oxazines; Hydrogenated 1,4-oxazines not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the present invention relates to an acid addition salt or acid addition salt crystal of a quinazoline derivative and a pharmaceutical composition containing them. Furthermore, it is related with the manufacturing method of the acid addition salt, the crystal
- Tyrosine kinase is an enzyme that phosphorylates tyrosine residues in proteins, and is known to play an important role in cell differentiation / proliferation and intracellular signal transduction system.
- HER2 also referred to as ErbB2 or Neu
- receptor tyrosine kinase growth factor receptor tyrosine kinase
- EGF receptor growth factor receptor tyrosine kinase
- Non-patent Document 4 it is known that co-expression of EGF receptor and HER2 further accelerates canceration by EGF receptor alone (Non-patent Document 4), and dual that inhibits tyrosine kinases of both EGF receptor and HER2. Inhibitors are broader in indications than compounds that act only on a single kinase and are superior in that a stronger therapeutic effect can be obtained by the dual inhibition synergy.
- Patent Document 1 the following formula: It is described that the quinazoline derivative represented by the formula has a dual inhibitory action on the EGF receptor and HER2, and is useful as a therapeutic and / or prophylactic agent for cancer.
- the examples also include the following compound (VIII-102): Are disclosed in the form of the free base, but their acid addition salts and / or solvates are not specifically disclosed. Further, the crystal is not specifically disclosed.
- the examples include the following compound (VI-15): Are disclosed in the form of the free base, but their acid addition salts and / or solvates are not specifically disclosed. Further, the crystal is not specifically disclosed.
- Patent Documents 3 and 4 have the following formula having a dual inhibitory action of EGF receptor and HER2: Lapatinib ditosylate, ditosylate salt of N- ⁇ 3-Chloro-4-[(3-fluorobenzyl) oxy] phenyl ⁇ -6- [5- Hydrate crystals and anhydride crystals of ( ⁇ [2- (methanesulphonyl) ethyl] amino ⁇ methyl) -2-furyl] -4-quinazolinamine) are disclosed.
- Patent Document 5 discloses N- ⁇ 3-Chloro-4-[(3-fluorobenzyl) oxy] phenyl ⁇ -6- [5-( ⁇ [2- (methanesulphonyl) ethyl] amino ⁇ methyl) -2- furyl] -4-quinazolinamine free base anhydride crystals are disclosed. In drug delivery, crystalline forms with useful chemical and / or physical properties that are useful are desired.
- the pharmaceutically active ingredient may have substantially different physical properties depending on the respective solid form. Such a difference in physical properties can affect, for example, a method for producing or administering a pharmaceutically active ingredient, or a pharmaceutical composition containing the pharmaceutically active ingredient.
- the present invention relates to an acid addition salt of a compound represented by formula (I), which is very useful as compared with other solid forms in a method for producing or administering a pharmaceutically active ingredient, or in a pharmaceutical composition containing a pharmaceutically active ingredient. Or a solvate thereof, or a crystal thereof. Furthermore, intermediates useful for producing the acid addition salts or solvates thereof, or crystals thereof are provided.
- the compound represented by the formula (I) has already been disclosed, it has been desired to establish a suitable solid form as well as a more preferable production method for use as a pharmaceutical or industrial production as a pharmaceutical.
- monohydrochloride forms I, II, III, V, VI, VII and solvates of the hydrochloride of the compound represented by formula (I) It was found that a crystalline form exists. In addition, there are crystal forms of monop-toluenesulfonate of the compound of formula (I); monosulfate and monosulfate hydrate; monophosphate and monophosphate hydrate; monofumarate I found out. Furthermore, it has been found that monohydrochloride Form I, Form V and Form VI and monop-toluenesulfonate crystals are more thermodynamically stable than the other crystal forms.
- the present inventors also have a hydrochloride or hydrochloride salt of a compound represented by the formula (I) that is useful as a drug substance, or a monop-toluenesulfonate or a monop-toluenesulfonate.
- the present inventors have found a compound represented by the formula (II) or a pharmaceutically acceptable salt thereof, a solvate thereof, or a crystal thereof that is useful in producing a crystal.
- the present invention relates to the following items 1) to 36).
- the crystal according to item 4) having a peak at 25.8 ⁇ 0.2 °.
- a pharmaceutical composition for parenteral administration comprising the crystal according to any one of items 3) to 10) above.
- Formula (II) A crystal of dip-toluenesulfonate of the compound represented by 30) In powder X-ray diffraction spectrum, diffraction angle (2 ⁇ ): 6.4 ⁇ 0.2 °, 7.3 ⁇ 0.2 °, 21.1 ⁇ 0.2 °, 24.7 ⁇ 0.2 ° The crystal according to item 29) having a peak at 25.3 ⁇ 0.2 °.
- Formula (II) A crystal of dip-toluenesulfonate dihydrate of the compound represented by the formula: 33) In powder X-ray diffraction spectrum, diffraction angle (2 ⁇ ): 7.3 ⁇ 0.2 °, 17.0 ⁇ 0.2 °, 18.5 ⁇ 0.2 °, 22.6 ⁇ 0.2 ° The crystal according to item 32) having a peak at 24.0 ⁇ 0.2 °.
- the present invention also relates to the following items 1A) to 105A).
- 13A ′ A pharmaceutical composition comprising the crystal according to any one of the above items 4A) to 11A), wherein the content of impurities is 2% by weight or less.
- 15A The salt or solvate thereof according to item 1A) or 14A), which is a mono-p-toluenesulfonate.
- 21A A pharmaceutical composition comprising the crystal according to any one of the above items 16A) to 19A).
- 21A ′ A pharmaceutical composition comprising the crystal according to any one of the above items 16A) to 19A), wherein the content of impurities is 2% by weight or less.
- 22A The pharmaceutical composition according to item 12A) or 20A), which is used for treatment and / or prevention of cancer.
- 23A The pharmaceutical composition according to the above item 13A) or 21A), which is used for treatment and / or prevention of cancer.
- 24A The pharmaceutical composition according to item 22A) above, wherein the cancer is breast cancer.
- 25A The pharmaceutical composition according to item 23A), wherein the cancer is breast cancer.
- 26A The pharmaceutical composition according to item 12A) or 20A) above, which has an EGF receptor inhibitory action and a HER2 inhibitory action.
- 27A The pharmaceutical composition according to the above item 13A) or 21A), which has an EGF receptor inhibitory action and a HER2 inhibitory action.
- 28A A therapeutic and / or prophylactic agent for cancer, comprising the crystal according to any one of the above items 4A) to 11A).
- 29A A therapeutic and / or prophylactic agent for cancer, comprising the crystal according to any one of the above items 16A) to 19A).
- 30A A method for treating and / or preventing cancer, comprising administering a pharmaceutical composition comprising the crystal according to any of the above items 4A) to 11A).
- 30A ′ The method for treating and / or preventing cancer according to item 30A) above, wherein the cancer is breast cancer.
- 31A) A method for treating and / or preventing cancer, comprising administering a pharmaceutical composition comprising the crystal according to any one of the above items 16A) to 19A).
- 31A ′ The method for treating and / or preventing cancer according to item 31A) above, wherein the cancer is breast cancer.
- 32A) Use of the crystal according to any of the above items 4A) to 11A) for the manufacture of a medicament for the treatment and / or prevention of cancer.
- 32A ′ Use of a crystal according to item 32A) above, wherein the cancer is breast cancer.
- 33A Use of the crystal according to any of the above items 16A) to 19A) for the manufacture of a medicament for the treatment and / or prevention of cancer.
- 33A ′ Use of a crystal according to item 33A) above, wherein the cancer is breast cancer.
- 34A The crystal according to any one of the above items 4A) to 11A) for treatment and / or prevention of cancer.
- 34A ′ The crystal according to item 34A) above, wherein the cancer is breast cancer.
- 35A The crystal according to any one of the above items 16A) to 19A), for the treatment and / or prevention of cancer.
- 35A ′ The crystal according to the above item 35A), wherein the cancer is breast cancer.
- 36A) A pharmaceutical composition for oral administration comprising the crystal according to any one of the above items 4A) to 11A).
- 37A A pharmaceutical composition for oral administration comprising the crystal according to any of the above items 16A) to 19A).
- 39A Tablets, powders, granules, capsules, pills, films, suspensions, emulsions, elixirs, syrups, limonades, spirits, fragrances, extracts, decoctions or tinctures, The pharmaceutical composition according to the above item 37A).
- 40A Sugar-coated tablets, film-coated tablets, enteric-coated tablets, sustained-release tablets, troche tablets, sublingual tablets, buccal tablets, chewable tablets, orally disintegrating tablets, dry syrups, soft capsules, microcapsules or sustained-release capsules A pharmaceutical composition according to item 38A) above.
- 44A The pharmaceutical composition according to the above item 42A) for transdermal, subcutaneous, intravenous, intraarterial, intramuscular, intraperitoneal, transmucosal, inhalation, nasal, eye drop, ear drop or intravaginal administration.
- 45A The pharmaceutical composition according to the above item 43A), for transdermal, subcutaneous, intravenous, intraarterial, intramuscular, intraperitoneal, transmucosal, inhalation, nasal, eye drop, ear drop or intravaginal administration.
- Formula (II) A crystal of dip-toluenesulfonate dihydrate of the compound represented by the formula: 56A) In powder X-ray diffraction spectrum, diffraction angle (2 ⁇ ): 7.3 ⁇ 0.2 °, 17.0 ⁇ 0.2 °, 18.5 ⁇ 0.2 °, 22.6 ⁇ 0.2 ° The crystal of the above item 55A) having a peak at 24.0 ⁇ 0.2 °.
- the present invention provides an acid addition salt of the compound represented by formula (I) and a crystal thereof.
- the crystal body has good stability and solubility, and can be used as an active ingredient for pharmaceutical production.
- crystallization of the acid addition salt of the compound shown by the formula (I) of this invention can be used as an anticancer agent.
- the present invention provides a compound represented by the formula (II) or a pharmaceutically acceptable salt thereof, a solvate thereof, or a crystal thereof.
- the compound is useful in producing an acid addition salt of the compound represented by the formula (I) and a crystal thereof.
- the free base of the acid addition salt or solvate of the compound represented by the formula (I) of the present invention is a compound having utility as a medicine.
- the usefulness as a medicine includes good solubility, good metabolic stability, little induction of drug metabolizing enzymes, small inhibition of drug metabolizing enzymes that metabolize other drugs, oral absorption
- the compound includes a highly active compound, a small hERG inhibition, a small clearance, and / or a sufficiently long half-life for exhibiting a medicinal effect.
- the acid addition salt of the compound represented by the formula (I) of the present invention or a solvate thereof, or a crystal thereof has utility as a medicine.
- the compound in solid form includes (1) packaging properties such as molar volume, density and hygroscopicity, (2) thermodynamic properties such as melting temperature, vapor pressure and solubility, (3) dissolution rate and stability, etc.
- 1 shows a powder X-ray diffraction pattern of a monohydrochloride Form I crystal (Form I) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of monohydrochloride V-form crystals (Form V) of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a monohydrochloride VI type crystal (Form VI) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- the TG / DTA analysis result of the monohydrochloride type I crystal (Form I) of the compound represented by the formula (I) is shown.
- crystallization (Form V) of the compound shown by a formula (I) is shown.
- crystallization (Form VI) of the compound shown by a formula (I) is shown.
- the DSC analysis result of the monohydrochloride type I crystal (Form I) of the compound represented by the formula (I) is shown.
- crystallization (Form V) of the compound shown by a formula (I) is shown.
- the DSC analysis result of the monohydrochloride VI form crystal (Form VI) of the compound shown by a formula (I) is shown.
- 2 shows a powder X-ray diffraction pattern of a 2-p-toluenesulfonate crystal of the compound represented by formula (II).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of dip-toluenesulfonate dihydrate crystals of the compound represented by formula (II).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of monohydrochloride type II crystal (Form II) of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of monohydrochloride type III crystal (Form III) of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a monohydrochloride form VII crystal (Form VII) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a monohydrochloride ethanolate crystal of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a free base monohydrate crystal of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a free base trihydrate crystal of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- FIG. 1 shows a powder X-ray diffraction pattern of a 1-p-toluenesulfonic acid salt form I crystal (Form I) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of monosulfate crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of monosulfate monohydrate crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of monophosphate crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of monophosphate dihydrate form I crystal (Form I) of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a crystal of a monofumarate form I crystal (Form I) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- FIG. 1 shows a powder X-ray diffraction pattern of a crystal of a monofumarate type II crystal (Form II) of a compound represented by the formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- the TG / DTA analysis result of the monohydrochloride type II crystal (Form II) of the compound represented by the formula (I) is shown.
- the TG / DTA analysis result of the monohydrochloride type III crystal (Form III) of the compound represented by the formula (I) is shown.
- the TG / DTA analysis result of the monohydrochloride type VII crystal (Form VII) of the compound represented by the formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- the TG / DTA analysis result of the 1-p-toluenesulfonic acid salt form I crystal (Form I) of the compound represented by the formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- crystallization of the compound shown by a formula (I) is shown.
- the TG / DTA analysis result of the monophosphate dihydrate form I crystal (Form I) of the compound represented by the formula (I) is shown.
- the TG / DTA analysis result of the mono-fumarate Form I crystal (Form I) of the compound represented by the formula (I) is shown.
- the TG / DTA analysis result of the mono-fumarate type II crystal (Form II) of the compound represented by the formula (I) is shown.
- 2 shows a powder X-ray diffraction pattern of dihydrochloride crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a crystal of one p-toluenesulfonate salt form II crystal (Form II) of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a single benzenesulfonate crystal of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of monophosphate dihydrate form II crystal (Form II) of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of monocitrate crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 2 shows a powder X-ray diffraction pattern of monotartrate crystals of the compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 shows a powder X-ray diffraction pattern of a free base crystal of a compound represented by formula (I).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- 1 is a moisture adsorption / desorption isotherm of a monohydrochloride Form I crystal (Form I) of a compound represented by the formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm
- the curve plotted with ⁇ is the moisture desorption isotherm.
- 2 is a moisture adsorption / desorption isotherm of a monohydrochloride V-form crystal (Form V) of the compound represented by formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm, and the curve plotted with ⁇ is the moisture desorption isotherm.
- 2 is a moisture adsorption / desorption isotherm of a monohydrochloride VI type crystal (Form VI) of a compound represented by the formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm
- the curve plotted with ⁇ is the moisture desorption isotherm.
- 2 is a moisture adsorption / desorption isotherm of a monohydrochloride type II crystal (Form II) of a compound represented by the formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm, and the curve plotted with ⁇ is the moisture desorption isotherm.
- 1 is a moisture adsorption / desorption isotherm of a p-toluenesulfonate Form I crystal (Form I) of a compound represented by formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm
- the curve plotted with ⁇ is the moisture desorption isotherm.
- crystallization of the compound shown by a formula (I) is shown.
- 2 is a moisture adsorption / desorption isotherm of a free base crystal of a compound represented by formula (I).
- the vertical axis represents the ratio of increased mass to the mass at 0% Target% P / P 0 [Change In Mass, unit:%], and the horizontal axis represents the relative humidity [Target% P / P 0 , unit:%].
- the curve plotted with ⁇ is the moisture adsorption isotherm, and the curve plotted with ⁇ is the moisture desorption isotherm.
- 2 shows a powder X-ray diffraction pattern of a dibenzenesulfonate crystal of the compound represented by the formula (II).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- anticancer agent and “cancer therapeutic agent” means brain tumor (eg glioblastoma), urinary cancer (eg bladder cancer, kidney cancer), genital cancer (eg prostate cancer, ovarian cancer, uterus). Cancer), lymphoid tumor, digestive organ cancer (eg gastric cancer, esophageal cancer, colon cancer, colon cancer), throat cancer, lung cancer (eg lung adenocarcinoma, small cell lung cancer, non-small cell lung cancer), pancreatic cancer, breast cancer, head Includes therapeutic agents for cervical cancer and thyroid cancer.
- brain tumor eg glioblastoma
- urinary cancer eg bladder cancer, kidney cancer
- genital cancer eg prostate cancer, ovarian cancer, uterus
- cancer lymphoid tumor
- digestive organ cancer eg gastric cancer, esophageal cancer, colon cancer, colon cancer
- throat cancer eg lung adenocarcinoma, small cell lung cancer, non-small cell lung cancer
- pancreatic cancer breast cancer
- the present invention encompasses a method of treating or preventing cancer in a mammal in need of treating or preventing cancer, the method comprising adding a therapeutically effective amount of a compound of formula (I) to said mammal. It consists of administering a salt crystal or a pharmaceutical composition containing it.
- Preferred cancers to treat are brain tumors (eg glioblastoma types), urological cancers (eg bladder cancer, kidney cancer), genital cancers (eg prostate cancer, ovarian cancer, uterine cancer), lymphoid tumors, digestive organ cancers (Eg, stomach cancer, esophageal cancer, colon cancer, colon cancer), throat cancer, lung cancer (eg lung adenocarcinoma, small cell lung cancer, non-small cell lung cancer), pancreatic cancer, breast cancer, head and neck cancer, thyroid cancer. More preferred are breast cancer, brain tumor, bladder cancer, kidney cancer, prostate cancer, ovarian cancer, uterine cancer, lung cancer, pancreatic cancer, and head and neck cancer. More preferably, it is breast cancer.
- urological cancers eg bladder cancer, kidney cancer
- genital cancers eg prostate cancer, ovarian cancer, uterine cancer
- lymphoid tumors eg, digestive organ cancers (Eg, stomach cancer, esophageal cancer,
- One or more hydrogen, carbon and / or other atoms of the compound of formula (I) or formula (II) may be replaced with hydrogen, carbon and / or isotopes of other atoms, respectively.
- Examples of such isotopes are 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O, 31 P, 32 P, 35 S, 18 F, 123 I and
- hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, iodine and chlorine are included.
- the compound represented by the formula (I) or the formula (II) includes a compound substituted with such an isotope.
- the compound substituted with the isotope is also useful as a pharmaceutical, and includes all radiolabeled compounds of the compound represented by formula (I) or formula (II).
- a “radiolabeling method” for producing the “radiolabeled substance” is also included in the present invention, and the “radiolabeled substance” is useful as a metabolic pharmacokinetic study, a research in a binding assay, and / or a diagnostic tool. Is
- the radiolabeled compound of the compound represented by formula (I) or formula (II) can be prepared by a method well known in the art.
- a tritium-labeled compound represented by the formula (I) or the formula (II) is introduced into a specific compound represented by the formula (I) or the formula (II) by a catalytic dehalogenation reaction using tritium.
- This method comprises a precursor and tritium in which a compound of formula (I) or (II) is appropriately halogen-substituted in the presence of a suitable catalyst such as Pd / C, in the presence or absence of a base. It includes reacting with a gas.
- the 14 C-labeled compound can be prepared by using a raw material having 14 C carbon.
- Examples of the pharmaceutically acceptable salt of the compound represented by the formula (I) or the formula (II) include a compound represented by the formula (I) or the formula (II) and an inorganic acid (for example, hydrochloric acid, sulfuric acid, nitric acid).
- an inorganic acid for example, hydrochloric acid, sulfuric acid, nitric acid.
- Organic acids eg formic acid, acetic acid, propionic acid, trifluoroacetic acid, citric acid, lactic acid, tartaric acid, oxalic acid, maleic acid, fumaric acid And salts with mandelic acid, glutaric acid, malic acid, benzoic acid, phthalic acid, ascorbic acid, benzenesulfonic acid, p-toluenesulfonic acid, methanesulfonic acid, ethanesulfonic acid and the like.
- organic acids eg formic acid, acetic acid, propionic acid, trifluoroacetic acid, citric acid, lactic acid, tartaric acid, oxalic acid, maleic acid, fumaric acid And salts with mandelic acid, glutaric acid, malic acid, benzoic acid, phthalic acid, ascorbic acid, benzenesulfonic acid, p-toluenesulfonic acid, methanesulf
- salts with hydrochloric acid, p-toluenesulfonic acid, sulfuric acid, phosphoric acid, fumaric acid, tartaric acid, methanesulfonic acid and the like can be mentioned. These salts can be formed by a commonly performed method.
- the hydrochloride, p-toluenesulfonate or other pharmaceutically acceptable salt of the compound represented by the formula (I) of the present invention, or the pharmaceutically acceptable salt of the compound represented by the formula (II) is a solvent. It may form solvates (eg, hydrates, ethanol solvates, etc.), co-crystals and / or crystal polymorphs, and the present invention also includes such various solvates, co-crystals and crystal polymorphs. To do.
- a “solvate” is a hydrochloride, p-toluenesulfonate or other pharmaceutically acceptable salt of a compound of formula (I), or a pharmaceutically acceptable salt of a compound of formula (II)
- the salt may be coordinated with any number of solvent molecules (for example, water molecules).
- the hydrochloride, p-toluenesulfonate or other pharmaceutically acceptable salt of the compound represented by formula (I), or the pharmaceutically acceptable salt of the compound represented by formula (II) is left in the atmosphere. By doing so, moisture may be absorbed and adsorbed water may adhere or a hydrate may be formed.
- Crystal polymorph refers to a hydrochloride, p-toluenesulfonate or other pharmaceutically acceptable salt of a compound of formula (I), or a pharmaceutically acceptable salt of a compound of formula (II)
- Counter molecules are present in the same crystal lattice, and may be formed with any number of counter molecules.
- a prodrug is a derivative of a compound of the present invention having a group that can be chemically or metabolically degraded, and is a compound that becomes a pharmaceutically active compound of the present invention by solvolysis or under physiological conditions in vivo.
- a prodrug is a compound that is enzymatically oxidized, reduced, hydrolyzed, etc. under physiological conditions in vivo to be converted into a compound represented by formula (I), hydrolyzed by gastric acid, etc. The compound etc. which are converted into the compound shown are included.
- a method for selecting and producing an appropriate prodrug derivative is described in, for example, “Design of Prodrugs, Elsevier, Amsterdam, 1985”. Prodrugs may themselves have activity.
- Formula (I) The compound represented by is an EGF receptor / HER2 dual inhibitor described in Patent Document 1, and a pharmaceutical composition containing the compound is useful for the prevention or treatment of cancer.
- the compound represented by the formula (I) can be prepared according to the method described in Patent Document 1 or 2.
- a compound represented by the formula (I) can be produced by reacting a compound represented by the formula (I) under acidic conditions and neutralizing the resulting crude product.
- the following formula: And a compound represented by the following formula: The compound represented by the formula (I) can be produced by reacting the compound represented by the formula (I) or a salt thereof or a solvate thereof under acidic conditions and neutralizing the obtained crude product.
- free base A of compound (I) and free base B of compound (I) can be produced as a compound represented by formula (I) by the method described in the examples of the present specification.
- An acid addition salt of the compound represented by the formula (I) or a crystal thereof can be produced by dissolving the compound represented by the formula (I) in various organic solvents and crystallization under acidic conditions.
- Acids include inorganic acids (eg, hydrochloric acid, sulfuric acid, nitric acid, carbonic acid, hydrobromic acid, phosphoric acid, hydroiodic acid, etc.) and organic acids (eg, formic acid, acetic acid, propionic acid, trifluoroacetic acid, citric acid, etc.
- Acid lactic acid, tartaric acid, oxalic acid, maleic acid, fumaric acid, mandelic acid, glutaric acid, malic acid, benzoic acid, phthalic acid, ascorbic acid, benzenesulfonic acid, p-toluenesulfonic acid, methanesulfonic acid, ethanesulfonic acid Etc.).
- hydrochloric acid and p-toluenesulfonic acid are mentioned.
- crystal forms of Form I, Form II, Form III, Form V, Form VI, Form VII and ethanolate there are crystal forms of monop-toluenesulfonate of the compound of formula (I); monosulfate and monosulfate hydrate; monophosphate and monophosphate hydrate; monofumarate To do.
- crystal polymorphs include the type of organic solvent used for crystallization and the free base A of the compound represented by formula (I) or the release of the compound represented by formula (I) as the compound represented by formula (I). It is possible to make differently depending on which of bases B is used.
- Form I crystals of the monohydrochloride of the compound of formula (I) can be prepared by dissolving the free base A of the compound of formula (I) in methanol and crystallizing it in the presence of hydrochloric acid.
- the monohydrochloride V-form crystal of the compound of formula (I) is prepared by dissolving the free base B of the compound of formula (I) in 2-propanol and crystallizing it in the presence of an acid. Can do.
- the monohydrochloride form VI of the compound of formula (I) is prepared by dissolving the free base A of the compound of formula (I) in 2-propanol and crystallizing it in the presence of hydrochloric acid. Can do.
- the monohydrochloride form VII of the compound of formula (I) is obtained by dissolving the monohydrochloride form VI of the compound of formula (I) in 1,2-dimethoxyethane and allowing it to crystallize. Can be manufactured.
- the monohydrochloride ethanolate crystals of the compound of formula (I) can be prepared by adding monohydrochloride Form I crystals as seed crystals in a mixed solution of ethyl acetate and ethanol.
- the free base crystal of the compound represented by the formula (I) can be produced by dissolving the free base B of the compound represented by the formula (I) in a mixed solution of hexane and ethyl acetate and allowing it to crystallize.
- One p-toluenesulfonic acid salt form I crystal of the compound represented by the formula (I) is prepared by purifying the free base A of the compound represented by the formula (I) in ethyl acetate and dissolving it in ethyl acetate. It can be produced by adding p-toluenesulfonic acid methanol solution and crystallization.
- the monosulfate crystals of the compound represented by the formula (I) can be produced by dissolving the free base A of the compound represented by the formula (I) in acetonitrile, adding 1 mol / L methanol sulfate and crystallizing. it can.
- the monosulfate monohydrate crystal of the compound represented by the formula (I) is obtained by dissolving the trihydrate crystal of the compound represented by the formula (I) in a mixed solution of acetonitrile and 2-propanol, and adding 0.1 mol / Add L sulfuric acid and concentrate. Furthermore, it can manufacture by adding the mixed solution of methanol and water, and concentrating after shaking.
- Monophosphate crystals of the compound represented by the formula (I) are obtained by dissolving trihydrate crystals of the compound represented by the formula (I) in a mixed solution of acetonitrile and 2-propanol, and adding 0.1 mol / L phosphoric acid. Is added and concentrated. Furthermore, it can manufacture by adding the mixed solution of ethanol and water, and concentrating after shaking.
- Monophosphate dihydrate Form I of the compound represented by the formula (I) is obtained by dissolving the trihydrate crystal of the compound represented by the formula (I) in a mixed solution of acetonitrile and 2-propanol. Add 1 mol / L phosphoric acid and concentrate. Furthermore, it can manufacture by adding the mixed solution of methanol and water, and concentrating after shaking.
- Monofumarate Form I crystals of the compound of formula (I) are obtained by dissolving trihydrate crystals of the compound of formula (I) in a mixed solution of acetonitrile and 2-propanol to give 0.1 mol / L Add a solution of fumaric acid in methanol and water and concentrate. Furthermore, it can manufacture by adding methanol and water and concentrating after shaking.
- Monofumarate Form II crystals of the compound of formula (I) are obtained by dissolving trihydrate crystals of the compound of formula (I) in a mixed solution of acetonitrile and 2-propanol to give 0.1 mol / L Add a solution of fumaric acid in methanol and water and concentrate. Further, it can be produced by adding acetonitrile and water and concentrating after shaking.
- crystal means a structure in which atoms, ions, molecules, etc. constituting a solid are regularly arranged, and as a result, has periodicity and anisotropy.
- the crystallinity of a crystalline form should be measured by a number of techniques including, for example, powder X-ray diffraction measurement, moisture adsorption / desorption measurement, differential scanning calorimetry, differential thermothermal gravimetric measurement, solution colorimetry, and dissolution characteristics. Can do.
- X-ray powder diffraction In general, a crystalline organic compound is composed of a large number of atoms periodically arranged in a three-dimensional space. Structural periodicity typically develops physical properties that are clearly distinguishable by most spectroscopic probes (eg, X-ray diffraction, infrared spectra, Raman spectra, and solid state NMR). Among these, powder X-ray diffraction (XRPD) is one of the most sensitive analytical methods for measuring the crystallinity of solids.
- Amorphous solids When X-rays are irradiated onto the crystal, it reflects off the crystal lattice plane, interferes with each other, and the intensity of only the diffraction lines in the direction that satisfies the conditions predicted by the Bragg law increases, and the ordered diffraction corresponding to the period of the structure Show the line.
- a wide range of ordered diffraction lines is not observed for amorphous solids.
- Amorphous solids usually do not have an orderly repeating period in their structure, and therefore exhibit a broad XRPD pattern with no features and no diffraction phenomenon.
- the crystalline form of the acid addition salt of the compound of formula (I) disclosed in this application preferably has a distinguishable powder X-ray diffraction profile.
- crystals containing the monohydrochloride or monop-toluenesulfonate salt of the compound of formula (I) can be distinguished from other crystal forms, preferably by the presence of a characteristic diffraction peak.
- a characteristic diffraction peak as used herein is a peak selected from the observed diffraction pattern.
- the characteristic peaks are selected from about 20 in the diffraction pattern, more preferably about 10 and most preferably about 5.
- an error may occur in the diffraction angle (2 ⁇ ) in the powder X-ray diffraction within a range of ⁇ 0.2 °. It needs to be understood as including. Therefore, the present invention includes not only a crystal in which the diffraction angle of the peak in powder X-ray diffraction completely coincides but also a crystal in which the diffraction angle of the peak coincides with an error of about ⁇ 0.2 °.
- the absolute and relative intensities of the peaks shown in the tables and figures below generally depend on a number of factors, such as the effect of selective orientation of the crystal on the x-ray beam, the influence of coarse particles, the purity of the substance being analyzed or the sample It is known that it can vary depending on the degree of crystallinity.
- the peak position can also be shifted based on the variation in sample height.
- TG / DTA (simultaneous measurement of differential thermothermal weight)
- TG / DTA is one of the main measurement methods of thermal analysis, and is a method for measuring the weight and thermal properties of a substance as an aggregate of atoms and molecules.
- TG / DTA is a method for measuring changes in weight and calorie with temperature or time of a pharmaceutically active ingredient. By plotting the obtained data against temperature or time, TG (thermogravimetric) and DTA (differential) A heat) curve is obtained. From the TG / DTA curve, it is possible to obtain information on the change in weight and calorie regarding decomposition, dehydration, oxidation, reduction, sublimation, and evaporation of the pharmaceutically active ingredient.
- the “melting point” in TG / DTA refers to an onset temperature that is not easily influenced by the sample preparation technique. In the identification of crystal identity, not only the melting point but also the overall pattern is important, and may vary somewhat depending on the measurement conditions and measurement equipment.
- DSC differential scanning calorimetry
- DSC is one of the main measurement methods of thermal analysis, and is a method of measuring the thermal properties of a substance as an aggregate of atoms and molecules.
- DSC measures the change in calorie with temperature or time of the pharmaceutically active ingredient and plots the obtained data against temperature or time to obtain a differential scanning calorie curve. From the differential scanning calorimetry curve, it is possible to obtain information on the onset temperature when the pharmaceutically active ingredient melts, the maximum value of the endothermic peak curve accompanying melting, and the enthalpy.
- the observed temperature can depend on the rate of temperature change as well as the sample preparation technique and the particular equipment used.
- the “melting point” in DSC refers to an onset temperature that is not easily affected by sample preparation techniques.
- the error range in onset temperature obtained from the differential scanning calorimetry curve is approximately ⁇ 2 ° C.
- the overall pattern is important, and may vary somewhat depending on the measurement conditions and the measurement equipment.
- Moisture adsorption / desorption isotherm measurement is a measurement method that measures the adsorption and desorption behavior of moisture by measuring the weight change of each solid to be measured under each relative humidity condition.
- the relative humidity is increased every 5% or 10%, and the weight is stable at each relative humidity. After conversion, the amount of adsorbed water can be determined from the weight increase from the reference value. Similarly, the amount of desorption of water can be measured by decreasing the relative humidity every 5% or 10% from 100% Target% P / P 0 .
- the photostability test is one of methods for measuring chemical or physicochemical changes in drug substances and preparations by exposure to evaluate the characteristics of a sample with respect to light.
- samples such as drug substance and drug product are irradiated with light for a certain period of time with a specified output.
- the sample for light is analyzed by analyzing the impurities, related substances, crystal form, color difference, etc. of the sample using scientific methods (high-performance liquid chromatography, X-ray crystal diffraction, color difference meter, etc.) Can be evaluated.
- exposure dose management using a radiometer or illuminometer and a test using a chemical light measurement system are performed.
- the pharmaceutical composition containing the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention is very useful as a therapeutic or prophylactic agent for cancer.
- the acid addition salt or acid addition salt crystals of the compound of formula (I) of the present invention can be administered to human patients per se, or the crystals can be mixed with a suitable carrier or excipient. Can be administered as a pharmaceutical composition. Techniques for drug formulation and administration can be appropriately selected and used by combining pharmaceutical formulations and techniques known to those skilled in the art.
- the route of administration of the acid addition salt of the compound represented by formula (I) or the crystal of the acid addition salt of the present invention or the pharmaceutical composition containing them is not limited, but oral, rectal, transmucosal or intestinal administration Alternatively, intramuscular, subcutaneous, intrathecal, intrathecal, direct intraventricular, intravenous, intravitreal, intraperitoneal, intranasal, intraocular, injection may be included.
- Preferred routes of administration include oral or injection (intramuscular, subcutaneous, intrathecal, intrathecal, intravenous). A particularly preferred route of administration is oral.
- the pharmaceutical composition of the present invention is manufactured by a method well known in the art, for example, conventional mixing, dissolving, granulating, sugar-coating, powdering, emulsifying, encapsulating, inclusion, lyophilization process. Can do.
- the acid addition salt or acid addition salt crystal of the compound of formula (I) of the present invention or a pharmaceutical composition containing them is an aqueous solution, preferably a physiologically compatible buffer such as Ringer's solution or physiological saline.
- the solution can be administered by injection.
- the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention or a pharmaceutical composition containing them can be transmucosally administered using a penetrant suitable for the barrier to be permeated. .
- the penetrant those generally known in the art can be used.
- the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention or a pharmaceutical composition in which they are combined with a pharmaceutically acceptable carrier well known in the art should be administered orally.
- a pharmaceutically acceptable carrier well known in the art
- the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention is converted into tablets, pills, lozenges, dragees, capsules, solutions, gels, syrups, suspensions. It can be administered as an agent.
- Pharmaceutical compositions for oral administration use solid excipients and, if desired, add other suitable adjuvants, then grind the resulting mixture and process the granule mixture to produce a tablet or dragee core Can be created by
- Useful excipients include fillers such as sugars including lactose, sucrose, mannitol or sorbitol, for example, cellulose preparations such as corn starch, wheat starch, rice starch and potato starch, gelatin, tragacanth gum, methylcellulose, Such as hydroxypropylmethylcellulose and / or sodium carboxymethylcellulose. If necessary, disintegrating agents such as agar and alginic acid can be added. A salt such as sodium alginate can also be used.
- sugars including lactose, sucrose, mannitol or sorbitol
- cellulose preparations such as corn starch, wheat starch, rice starch and potato starch, gelatin, tragacanth gum, methylcellulose, Such as hydroxypropylmethylcellulose and / or sodium carboxymethylcellulose.
- disintegrating agents such as agar and alginic acid can be added.
- a salt such as sodium alginate can also be used.
- compositions that can be used for oral administration include push-fit capsules made of gelatin, sealed capsules made of gelatin and a plasticizer such as glycerol or sorbitol.
- Push-fit capsules can contain a filler such as lactose, a binder such as starch and / or a lubricant such as talc or magnesium stearate and optionally an active ingredient mixed with a stabilizer.
- the acid addition salt crystals of the compound of formula (I) of the present invention can be dissolved or suspended in a suitable liquid such as fatty oil, liquid paraffin or liquid polyethylene glycol.
- Stabilizers can also be added to these formulations.
- the pharmaceutical composition may also contain a suitable solid or gel phase carrier or excipient.
- suitable solid or gel phase carrier or excipients include calcium carbonate, calcium phosphate, various sugars, starch, cellulose derivatives, polymers such as gelatin and polyethylene glycol, and the like.
- the therapeutically effective amount of the acid addition salt or acid addition salt crystal of the compound of formula (I) of the present invention can be estimated initially from cell culture assays. Then, the acid addition salt or crystal of the acid addition salt of the compound of formula (I) of the present invention that achieves half-maximal inhibition of PK activity determined in cell culture (ie, pharmaceutical composition comprising them) High dosages can be formulated for use in animal models to achieve a circulating concentration range that includes product concentrations. Such information can then be used to more accurately determine useful amounts in humans.
- the therapeutic effect of the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention or a pharmaceutical composition containing them can be measured by standard pharmaceutical techniques in cell culture or experimental animals. it can. For example, the evaluation may be performed according to the biological test method described in International Publication No. 2006/090717. The data obtained from these cell culture assays and animal studies can be used to formulate a range of dosage for use in humans.
- the dosage can vary depending on the dosage form used and the route of administration utilized. The exact route of administration and dosage can be chosen by the individual physician in view of the patient's condition.
- the acid addition salt or acid addition salt crystal of the compound of formula (I) of the present invention or a pharmaceutical composition containing them can be combined with other anticancer agents and the like.
- the acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention or a pharmaceutical composition containing them can be used in combination with or in combination with other anticancer agents.
- trastuzumab for example, trastuzumab, microtubule inhibitors [vinorelbine, taxane drugs (eg, paclitaxel, docetaxel, etc.), irinotecan, eribulin mesylate], platinum drugs (eg, cisplatin, carboplatin, oxaliplatin, nedaplatin, etc.), 5- FU drugs (eg, capecitabine, 5-fluorouracil, etc.), breast cancer hormone therapy, HER2 inhibitor (trastuzumab, pertuzumab, lapatinib tosylate hydrate, neratinib, margetuimab), HER2 antibody conjugate drug (trastuzumab emtansine (T- DM1), MM-302), HDAC inhibitors (entinostat), PARP inhibitors (tarazoparib, niraparib, olaparib, veriparib), immunotherapy vaccines (eg
- the dose of the acid addition salt of the compound represented by the formula (I) or the crystal of the acid addition salt of the present invention is orally administered to an adult, although it varies depending on the disease state, administration route, patient age, or body weight. In this case, it is usually 10 to 1600 mg / person / day, preferably 100 to 1200 mg / person / day, and most preferably 200 to 800 mg / person / day.
- Measurement method Reflection method Light source type: Cu tube Use wavelength: CuK ⁇ ray tube current: 40 mA Tube voltage: 40kV Sample plate: Glass, aluminum X-ray incident angle: 3-40 ° (Method B) (apparatus) Rigaku MiniFlex600 (Method of operation) The sample was measured under the following conditions. Measurement method: Reflection method Light source type: Cu tube Use wavelength: CuK ⁇ ray tube current: 15 mA Tube voltage: 40 kV Sample plate: Circular non-reflective sample plate X-ray incident angle: 4-40 °
- TG / DTA analysis results are shown in FIG. An onset temperature of about 234 ° C. was observed. The weight loss with TG was about 0.91%.
- the moisture adsorption / desorption isotherm measurement results are shown in FIG. In the measurement of the moisture adsorption isotherm, assuming that 95% Target% P / P 0 , the ratio of the increased mass to the mass at 0% Target% P / P 0 was about 0.8. In the measurement of the moisture desorption isotherm, when 0% Target% P / P 0 , the ratio was about 0.08.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 238 ° C. was observed. The weight loss with TG was about 1.34%.
- the DSC analysis results are shown in FIG. An onset temperature of about 234 ° C. was observed.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 225 ° C. was observed. The weight loss with TG was about 0.98%.
- the DSC analysis results are shown in FIG. An onset temperature of about 225 ° C. was observed.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 210.9 ° C. was observed. No weight loss with TG was observed.
- the moisture adsorption / desorption isotherm measurement results are shown in FIG.
- the ratio of the increased mass to the mass at 0% Target% P / P 0 was about 0.3.
- the ratio was about 0.05.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 223.9 ° C. was observed. No weight loss with TG was observed.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 171.7 ° C. was observed. The weight loss with TG was about 0.31%.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 231 ° C. was observed. The weight loss with TG was about 1.01%.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 208.5 ° C. was observed. No weight loss with TG was observed.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 190.5 ° C. was observed. No weight loss with TG was observed.
- the moisture adsorption / desorption isotherm measurement results are shown in FIG.
- the ratio of the increased mass to the mass at 0% Target% P / P 0 was about 3.3.
- the ratio was about 0.13 when 0% Target% P / P 0 .
- TG / DTA analysis results are shown in FIG. An onset temperature of about 188.1 ° C. was observed. The weight loss with TG was about 1.76%.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 191.3 ° C. was observed. No weight loss with TG was observed.
- FIG. 37 shows the TG / DTA analysis results. An onset temperature of about 195.6 ° C. was observed. The weight loss with TG was about 1.42%.
- TG / DTA analysis results are shown in FIG. An onset temperature of about 134.1 ° C. was observed. No weight loss with TG was observed.
- the moisture adsorption / desorption isotherm measurement results are shown in FIG.
- the ratio of the increased mass to the mass at 0% Target% P / P 0 was about 1.0.
- the ratio was about 0.16.
- a salt can be formed if the difference in pKa value from a counter (eg, hydrochloric acid) is 3 or more, and hydrochloric acid has a pKa value of ⁇ 6.
- Dihydrochloride can also be formed with the free base of (I) (Reference: Evaluation of physical properties of poorly water-soluble drugs and new development of formulation design, 2010, 111-121, Structure, solubility, screening, and synthesis of molecular salts, JOURNAL OF PHARMACEUTICAL SCIENCES, VOL.
- the monohydrochloride Form I crystal, monohydrochloride Form II crystal, monohydrochloride Form V crystal, monohydrochloride Form VI crystal and monop-toluenesulfonate Form I crystal have high solubility. It has been found to be a crystalline form suitable for use as an active pharmaceutical ingredient.
- the free base crystals of compound (I) have high concentrations (wt%) in various organic solvents (about 0.5 wt% to about 8 wt%) and high solubility.
- the monohydrochloride I-form crystals and monohydrochloride V-form crystals of compound (I) are hardly dissolved in various organic solvents (both 0.1% by weight or less). That is, when the compound (I) is produced according to the description in Reference Example 1 and Example 18 or 25 below, if the solubility of the produced compound (I) in the organic solvent is high, the ratio of the product precipitated from the organic solvent is high. It will be lower and the yield will decrease. Therefore, it was found that the monohydrochloride I crystal or monohydrochloride V crystal of compound (I) is a suitable crystal form for use as a drug substance for pharmaceuticals.
- Step 1 Preparation of ethyl acetate solution of compound (I)
- Compound 4 (30.04 g, 67.4 mmol) was dissolved in N-methylpyrrolidone (70.86 g) and tetrahydrofuran (18.68 g) to obtain compound (IIA) (36.85 g, 77.3 mmol), p-toluenesulfonic acid monohydrate (15.37 g, 80.8 mmol), tetrahydrofuran (53.41 g), water (5.40 g), and a slurry solution at 57 ° C. For 5 hours. After cooling to room temperature, compound 4 (0.25 g) was added.
- Step 2-1 Preparation of free base crystals of compound (I) A solution of compound (I) in ethyl acetate (89.92 g) was concentrated to 22.97 g, heptane (17.67 g), ethyl acetate (13.27 g). ) And then heated to 60 ° C., a solid precipitated.
- the monohydrochloride VI crystal and monohydrochloride I crystal of compound (I) can be said to be crystal forms suitable for mass synthesis because high-purity crystals can be obtained by one crystallization. That is, it has been found that the monohydrochloride VI crystal and monohydrochloride I crystal of compound (I) are crystal forms suitable for use as an active pharmaceutical ingredient.
- the monosulfate crystals of Compound (I) showed an increase in water of about 3.3%, whereas the monohydrochloride Form I crystals, monohydrochloride II of Compound (I) Form crystal, monohydrochloride V-form crystal, monohydrochloride VI-form crystal and mono-p-toluenesulfonate I-form crystal were found to have a low moisture increase ratio.
- the crystal is more susceptible to moisture absorption by making it into a salt, and it is said that the absorbability varies depending on the type of salt (Reference: Evaluation of physical properties and preparations of poorly water-soluble drugs) New design development, 2010, 117-118 pages).
- Table 23 shows the exposure test results of the monohydrochloride Form I crystal, monohydrochloride Form II crystal, monohydrochloride Form V crystal, monohydrochloride Form VI crystal and monop-toluenesulfonate Form I crystal of Compound (I). Shown in Quality evaluation of each crystal was performed using HPLC (Method B).
- the monohydrochloride I crystal, monohydrochloride II crystal, monohydrochloride V crystal and monohydrochloride VI crystal of compound (I) have good photostability under exposure conditions and are active pharmaceutical ingredients. It was found to be a suitable crystalline form for use as.
- the preparation solution of compound 3 was concentrated to 114 g and stirred at 0 ° C. for 2 hours. After filtering the precipitated insoluble matter, toluene (56 mL) was added to the filtrate. This toluene solution was added to a solution of p-toluenesulfonic acid monohydrate (28.2 g, 148.1 mmol) in tetrahydrofuran (42 mL), and the mixture was stirred at 50 ° C. for 2 hours. The mixture was further cooled to 0 ° C., stirred for 2 hours, and filtered to obtain Compound (IIA) (27.2 g, yield 88.4%).
- the crystal of the compound (II ′) described in Reference Example 12 has a deliquescent property and low crystallinity. Further, the crystal of the compound (II ′′) described in Reference Example 13 shows a weak peak and low crystallinity as shown in FIG. As described in Example 15 above, crystals with low crystallinity are difficult to handle. In contrast, the crystals of compound (IIA) and the dihydrate crystals of compound (IIA) described in Examples 21 and 22 are crystals with good crystallinity as shown in FIGS. It was found to be a crystalline form that can be selected as a drug intermediate.
- Formulation Example 1 Tablet Acid addition salt or acid addition salt crystal of compound of formula (I) of the present invention, lactose and calcium stearate are mixed, crushed and granulated, dried, and granule of appropriate size And Next, calcium stearate is added and compressed to form tablets.
- Formulation Example 2 Capsule The acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention, lactose and calcium stearate are uniformly mixed to form a powder as a powder or fine granules. It is filled into a capsule container to form a capsule.
- Formulation Example 3 Granules Acid addition salt or acid addition salt crystals of the compound represented by the formula (I) of the present invention, lactose and calcium stearate are uniformly mixed, compression molded, pulverized, granulated, sieved Separately, the granule is appropriately sized.
- Formulation Example 4 Orally disintegrating tablet Acid addition salt or acid addition salt crystals of the compound represented by the formula (I) of the present invention and crystalline cellulose are mixed and tableted after granulation to obtain an orally disintegrating tablet.
- Formulation Example 5 Dry syrup The acid addition salt or acid addition salt crystal of the compound represented by the formula (I) of the present invention and lactose are mixed, pulverized, sized and sieved to obtain a dry syrup of an appropriate size.
- Formulation Example 6 Inhalant An acid addition salt of the compound represented by the formula (I) of the present invention or a crystal of acid addition salt and lactose are mixed and finely pulverized to obtain an inhalant.
- the hydrochloride or hydrochloride crystal of the compound represented by the formula (I) and the crystal of 1-toluenesulfonate or 1-toluenesulfonate of the present invention are useful as a drug substance.
- a pharmaceutical composition containing a hydrochloride or hydrochloride crystal of a compound represented by the formula (I), or a crystal of 1-p-toluenesulfonate or 1-toluenesulfonate is a therapeutic agent for cancer. Or it is very useful as a preventive agent.
- the compound represented by the formula (II) or a pharmaceutically acceptable salt thereof or a solvate thereof is an intermediate useful for producing a hydrochloride or hydrochloride crystal of the compound represented by the formula (I). It is.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
また、EGF受容体とHER2の共発現によりEGF受容体単独による癌化がさらに加速されることが知られており(非特許文献4)、EGF受容体とHER2の両方のチロシンキナーゼを阻害するデュアル阻害剤は単独のキナーゼにのみ作用する化合物と比較して、適応疾患が広く、デュアル阻害の相乗作用によってより強い治療効果が得られる点で優れている。
特許文献1には、下式:
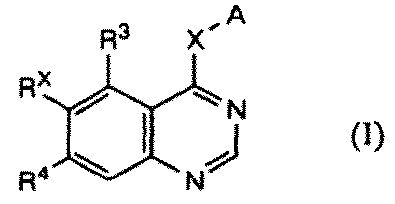
で表されるキナゾリン誘導体が、EGF受容体およびHER2のデュアル阻害作用を有し、癌の治療および/または予防剤として有用であることが記載されている。またその実施例には、以下の化合物(VIII-102):
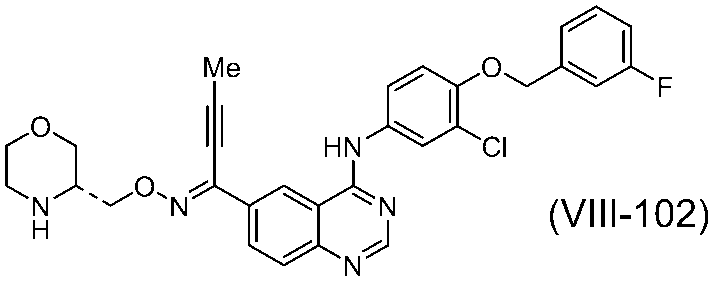
が遊離塩基の形態で開示されているが、その酸付加塩および/または溶媒和物は、具体的に開示されていない。また、その結晶についても具体的に開示されていない。
特許文献2には、式(VI’):
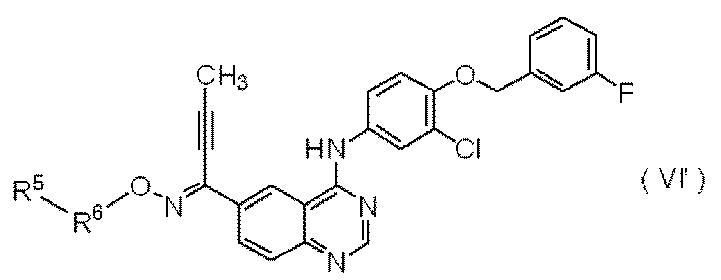
で表されるキナゾリン誘導体の製造方法が記載されている。また、その実施例には、以下の化合物(VI-15):
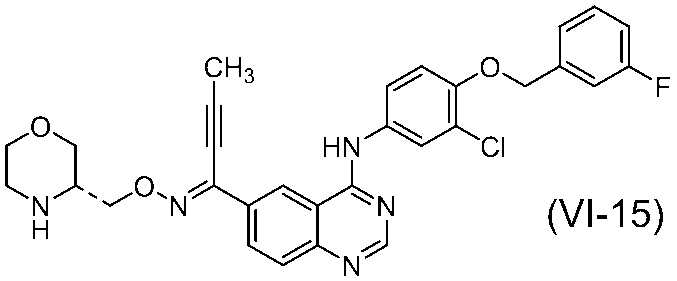
が遊離塩基の形態で開示されているが、その酸付加塩および/または溶媒和物は、具体的に開示されていない。また、その結晶についても具体的に開示されていない。
特許文献3および4には、EGF受容体およびHER2のデュアル阻害作用を有する、下式:
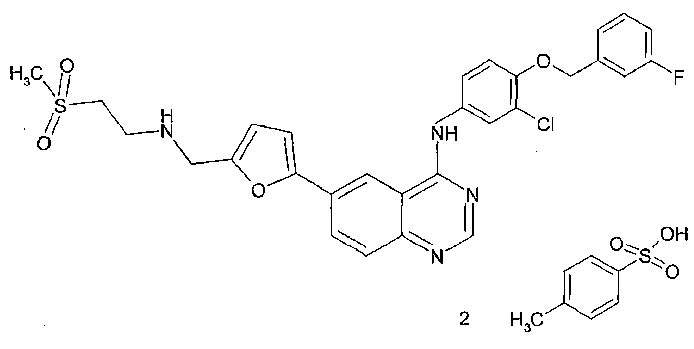
で表される、ラパチニブ二トシル酸塩(Lapatinib ditosylate、ditosylate salt of N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-
({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine)の水和物結晶および無水物結晶が開示されている。また、特許文献5には、N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamineの遊離塩基の無水物結晶が開示されている。
薬物デリバリーにおいては、有用な優れた化学的および/または物理的特性を有する結晶形態が望まれている。
式(I)で示される化合物は既に開示されているものの、医薬品として使用または医薬品として工業的に製造するために、好適な固体形態ならびにより好ましい製造方法の確立が望まれていた。
また、本発明者らは、医薬原体として有用である式(I)で示される化合物の塩酸塩もしくは塩酸塩の結晶、または、一p-トルエンスルホン酸塩もしくは一p-トルエンスルホン酸塩の結晶を製造するにあって有用な、式(II)で示される化合物もしくはその製薬上許容される塩、それらの溶媒和物、またはそれらの結晶を見出した。
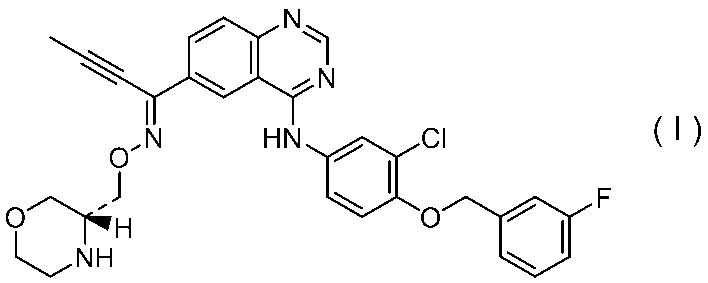
1)式(I):

で示される化合物の塩酸塩またはその溶媒和物。
2)一塩酸塩である、上記項目1)記載の塩酸塩またはその溶媒和物。
3)式(I):

で示される化合物の塩酸塩の結晶。
4)一塩酸塩である、上記項目3)記載の結晶。
5)粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、14.1±0.2°、20.6±0.2°、21.0±0.2°、25.8±0.2°にピークを有する上記項目4)記載の結晶。
6)粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、8.0±0.2°、14.1°±0.2°、17.9°±0.2°、18.5°±0.2°、20.6°±0.2°、21.0°±0.2°、22.5°±0.2°、25.8°±0.2°、28.4°±0.2°にピークを有する上記項目4)記載の結晶。
7)粉末X線回折スペクトルにおいて、回折角度(2θ):23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する上記項目4)記載の結晶。
8)粉末X線回折スペクトルにおいて、回折角度(2θ):7.9°±0.2°、9.7±0.2°、11.9±0.2°、15.8±0.2°、18.5±0.2°、23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する上記項目4)記載の結晶。
9)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、16.3±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°にピークを有する上記項目4)記載の結晶。
10)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、8.9±0.2°、11.7±0.2°、13.8±0.2°、16.3±0.2°、20.9±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°、26.6±0.2°にピークを有する上記項目4)記載の結晶。
11)上記項目1)または2)に記載の塩酸塩またはその溶媒和物を含む医薬組成物。
11’)上記項目3)~10)のいずれかに記載の結晶を含む医薬組成物。
12)癌の治療および/または予防に用いられる、上記項目11)記載の医薬組成物。
12’)癌が乳癌である、上記項目12)記載の医薬組成物。
12’’)EGF受容体阻害作用およびHER2阻害作用を有する、上記項目11)記載の医薬組成物。
13)上記項目3)~10)のいずれかに記載の結晶を含有することを特徴とする、癌の治療および/または予防剤。
14)上記項目3)~10)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、癌の治療および/または予防方法。
15)癌の治療および/または予防のための医薬を製造するための、上記項目3)~10)のいずれかに記載の結晶の使用。
16)癌の治療および/または予防のための、上記項目3)~10)のいずれかに記載の結晶。
17)上記項目3)~10)のいずれかに記載の結晶を含有する、経口投与のための医薬組成物。
18)錠剤、散剤、顆粒剤、カプセル剤、丸剤、フィルム剤、懸濁剤、乳剤、エリキシル剤、シロップ剤、リモナーデ剤、酒精剤、芳香水剤、エキス剤、煎剤またはチンキ剤である、上記項目17)記載の医薬組成物。
19)糖衣錠、フィルムコーティング錠、腸溶性コーティング錠、徐放錠、トローチ錠、舌下錠、バッカル錠、チュアブル錠、口腔内崩壊錠、ドライシロップ、ソフトカプセル剤、マイクロカプセル剤または徐放性カプセル剤である、上記項目18)記載の医薬組成物。
20)上記項目3)~10)のいずれかに記載の結晶を含有する、非経口投与のための医薬組成物。
21)経皮、皮下、静脈内、動脈内、筋肉内、腹腔内、経粘膜、吸入、経鼻、点眼、点耳または膣内投与のための、上記項目20)記載の医薬組成物。
22)注射剤、点滴剤、点眼剤、点鼻剤、点耳剤、エアゾール剤、吸入剤、ローション剤、注入剤、塗布剤、含嗽剤、浣腸剤、軟膏剤、硬膏剤、ゼリー剤、クリーム剤、貼付剤、パップ剤、外用散剤または坐剤である、上記項目20)または21)記載の医薬組成物。
23)上記項目3)~10)のいずれかに記載の結晶を含有する、小児用または高齢者用の医薬組成物。
24)図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目4)記載の結晶。
25)図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目4)記載の結晶。
26)図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目4)記載の結晶。
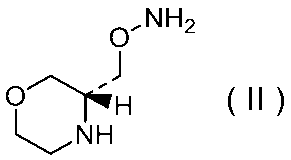
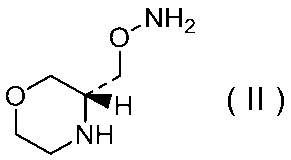
27)式(II):
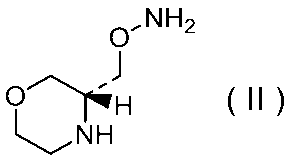
で示される化合物もしくはその製薬上許容される塩またはそれらの溶媒和物。
28)二p-トルエンスルホン酸塩である、上記項目27)記載の塩またはそれらの溶媒和物。
29)式(II):
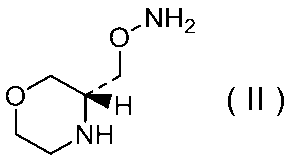
で示される化合物の二p-トルエンスルホン酸塩の結晶。
30)粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、21.1±0.2°、24.7±0.2°、25.3±0.2°にピークを有する上記項目29)記載の結晶。
31)粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、11.4±0.2°、15.2±0.2°、17.6±0.2°、20.1±0.2°、21.1±0.2°、21.7±0.2°、24.7±0.2°、25.3±0.2°にピークを有する上記項目29)記載の結晶。
32)式(II):
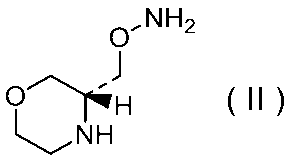
で示される化合物の二p-トルエンスルホン酸塩二水和物の結晶。
33)粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、22.6±0.2°、24.0±0.2°にピークを有する上記項目32)記載の結晶。
34)粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、19.7±0.2°、21.7±0.2°、22.6±0.2°、22.8±0.2°、24.0±0.2°、24.8±0.2°、29.7±0.2°にピークを有する上記項目32)記載の結晶。
35)図10に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目29)記載の結晶。
36)図11に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目32)記載の結晶。
また、本発明は、以下の項目1A)~105A)に関する。
1A)式(I):

で示される化合物の塩またはその溶媒和物。
2A)塩酸塩である、上記項目1A)記載の塩またはその溶媒和物。
3A)一塩酸塩である、上記項目1A)または2A)記載の塩またはその溶媒和物。
4A)式(I):

で示される化合物の塩酸塩の結晶。
5A)一塩酸塩である、上記項目4A)記載の結晶。
6A)粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、14.1±0.2°、20.6±0.2°、21.0±0.2°、25.8±0.2°にピークを有する上記項目5A)記載の結晶。
7A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、8.0±0.2°、14.1°±0.2°、17.9°±0.2°、18.5°±0.2°、20.6°±0.2°、21.0°±0.2°、22.5°±0.2°、25.8°±0.2°、28.4°±0.2°にピークを有する上記項目5A)記載の結晶。
8A)粉末X線回折スペクトルにおいて、回折角度(2θ):23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する上記項目5A)記載の結晶。
9A)粉末X線回折スペクトルにおいて、回折角度(2θ):7.9°±0.2°、9.7±0.2°、11.9±0.2°、15.8±0.2°、18.5±0.2°、23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する上記項目5A)記載の結晶。
10A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、16.3±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°にピークを有する上記項目5A)記載の結晶。
11A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、8.9±0.2°、11.7±0.2°、13.8±0.2°、16.3±0.2°、20.9±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°、26.6±0.2°にピークを有する上記項目5A)記載の結晶。
12A)上記項目2A)または3A)に記載の塩酸塩またはその溶媒和物を含む医薬組成物。
13A)上記項目4A)~11A)のいずれかに記載の結晶を含む医薬組成物。
13A’)不純物の含有量が、2重量%以下である、上記項目4A)~11A)のいずれかに記載の結晶を含む医薬組成物。
13A’’)不純物が、式(I)で示される化合物のE異性体である、上記13A’)記載の医薬組成物。
14A)p-トルエンスルホン酸塩である、上記項目1A)記載の塩またはその溶媒和物。
15A)一p-トルエンスルホン酸塩である、上記項目1A)または14A)記載の塩またはその溶媒和物。
16A)式(I):
で示される化合物のp-トルエンスルホン酸塩の結晶。
17A)一p-トルエンスルホン酸塩である、上記項目16A)記載の結晶。
18A)粉末X線回折スペクトルにおいて、回折角度(2θ):13.7±0.2°、15.7±0.2°、20.0±0.2°、22.7±0.2°、25.3±0.2°にピークを有する上記項目17A)記載の結晶。
19A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.1±0.2°、6.4±0.2°、10.8±0.2°、13.7±0.2°、15.7±0.2°、16.3±0.2°、20.0±0.2°、22.7±0.2°、24.6±0.2°、25.3±0.2°にピークを有する上記項目17A)記載の結晶。
20A)上記項目14A)または15A)に記載のp-トルエンスルホン酸塩またはその溶媒和物を含む医薬組成物。
21A)上記項目16A)~19A)のいずれかに記載の結晶を含む医薬組成物。
21A’)不純物の含有量が、2重量%以下である、上記項目16A)~19A)のいずれかに記載の結晶を含む医薬組成物。
21A’’)不純物が、式(I)で示される化合物のE異性体である、上記21A’)記載の医薬組成物。
22A)癌の治療および/または予防に用いられる、上記項目12A)または20A)記載の医薬組成物。
23A)癌の治療および/または予防に用いられる、上記項目13A)または21A)記載の医薬組成物。
24A)癌が乳癌である、上記項目22A)記載の医薬組成物。
25A)癌が乳癌である、上記項目23A)記載の医薬組成物。
26A)EGF受容体阻害作用およびHER2阻害作用を有する、上記項目12A)または20A)記載の医薬組成物。
27A)EGF受容体阻害作用およびHER2阻害作用を有する、上記項目13A)または21A)記載の医薬組成物。
28A)上記項目4A)~11A)のいずれかに記載の結晶を含有することを特徴とする、癌の治療および/または予防剤。
29A)上記項目16A)~19A)のいずれかに記載の結晶を含有することを特徴とする、癌の治療および/または予防剤。
30A)上記項目4A)~11A)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、癌の治療および/または予防方法。
30A’)癌が乳癌である、上記項目30A)記載の、癌の治療および/または予防方法。
31A)上記項目16A)~19A)のいずれかに記載の結晶を含む医薬組成物を投与することを特徴とする、癌の治療および/または予防方法。
31A’)癌が乳癌である、上記項目31A)記載の、癌の治療および/または予防方法。
32A)癌の治療および/または予防のための医薬を製造するための、上記項目4A)~11A)のいずれかに記載の結晶の使用。
32A’)癌が乳癌である、上記項目32A)記載の、結晶の使用。
33A)癌の治療および/または予防のための医薬を製造するための、上記項目16A)~19A)のいずれかに記載の結晶の使用。
33A’)癌が乳癌である、上記項目33A)記載の、結晶の使用。
34A)癌の治療および/または予防のための、上記項目4A)~11A)のいずれかに記載の結晶。
34A’)癌が乳癌である、上記項目34A)記載の、結晶。
35A)癌の治療および/または予防のための、上記項目16A)~19A)のいずれかに記載の結晶。
35A’)癌が乳癌である、上記項目35A)記載の、結晶。
36A)上記項目4A)~11A)のいずれかに記載の結晶を含有する、経口投与のための医薬組成物。
37A)上記項目16A)~19A)のいずれかに記載の結晶を含有する、経口投与のための医薬組成物。
38A)錠剤、散剤、顆粒剤、カプセル剤、丸剤、フィルム剤、懸濁剤、乳剤、エリキシル剤、シロップ剤、リモナーデ剤、酒精剤、芳香水剤、エキス剤、煎剤またはチンキ剤である、上記項目36A)記載の医薬組成物。
39A)錠剤、散剤、顆粒剤、カプセル剤、丸剤、フィルム剤、懸濁剤、乳剤、エリキシル剤、シロップ剤、リモナーデ剤、酒精剤、芳香水剤、エキス剤、煎剤またはチンキ剤である、上記項目37A)記載の医薬組成物。
40A)糖衣錠、フィルムコーティング錠、腸溶性コーティング錠、徐放錠、トローチ錠、舌下錠、バッカル錠、チュアブル錠、口腔内崩壊錠、ドライシロップ、ソフトカプセル剤、マイクロカプセル剤または徐放性カプセル剤である、上記項目38A)記載の医薬組成物。
41A)糖衣錠、フィルムコーティング錠、腸溶性コーティング錠、徐放錠、トローチ錠、舌下錠、バッカル錠、チュアブル錠、口腔内崩壊錠、ドライシロップ、ソフトカプセル剤、マイクロカプセル剤または徐放性カプセル剤である、上記項目39A)記載の医薬組成物。
42A)上記項目4A)~11A)のいずれかに記載の結晶を含有する、非経口投与のための医薬組成物。
43A)上記項目16A)~19A)のいずれかに記載の結晶を含有する、非経口投与のための医薬組成物。
44A)経皮、皮下、静脈内、動脈内、筋肉内、腹腔内、経粘膜、吸入、経鼻、点眼、点耳または膣内投与のための、上記項目42A)記載の医薬組成物。
45A)経皮、皮下、静脈内、動脈内、筋肉内、腹腔内、経粘膜、吸入、経鼻、点眼、点耳または膣内投与のための、上記項目43A)記載の医薬組成物。
46A)注射剤、点滴剤、点眼剤、点鼻剤、点耳剤、エアゾール剤、吸入剤、ローション剤、注入剤、塗布剤、含嗽剤、浣腸剤、軟膏剤、硬膏剤、ゼリー剤、クリーム剤、貼付剤、パップ剤、外用散剤または坐剤である、上記項目42A)または44A)記載の医薬組成物。
47A)注射剤、点滴剤、点眼剤、点鼻剤、点耳剤、エアゾール剤、吸入剤、ローション剤、注入剤、塗布剤、含嗽剤、浣腸剤、軟膏剤、硬膏剤、ゼリー剤、クリーム剤、貼付剤、パップ剤、外用散剤または坐剤である、上記項目43A)または45A)記載の医薬組成物。
48A)上記項目4A)~11A)のいずれかに記載の結晶を含有する、小児用または高齢者用の医薬組成物。
49A)上記項目16A)~19A)のいずれかに記載の結晶を含有する、小児用または高齢者用の医薬組成物。
50A)式(II):
で示される化合物もしくはその製薬上許容される塩またはそれらの溶媒和物。
51A)二p-トルエンスルホン酸塩である、上記項目50A)記載の塩またはそれらの溶媒和物。
52A)式(II):

で示される化合物の二p-トルエンスルホン酸塩の結晶。
53A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、21.1±0.2°、24.7±0.2°、25.3±0.2°にピークを有する上記項目52A)記載の結晶。
54A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、11.4±0.2°、15.2±0.2°、17.6±0.2°、20.1±0.2°、21.1±0.2°、21.7±0.2°、24.7±0.2°、25.3±0.2°にピークを有する上記項目52A)記載の結晶。
55A)式(II):
で示される化合物の二p-トルエンスルホン酸塩二水和物の結晶。
56A)粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、22.6±0.2°、24.0±0.2°にピークを有する上記項目55A)記載の結晶。
57A)粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、19.7±0.2°、21.7±0.2°、22.6±0.2°、22.8±0.2°、24.0±0.2°、24.8±0.2°、29.7±0.2°にピークを有する上記項目55A)記載の結晶。
58A)図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
59A)図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
60A)図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
61A)粉末X線回折スペクトルにおいて、回折角度(2θ):11.3±0.2°、17.1±0.2°、25.5±0.2°、25.8±0.2°、26.4±0.2°にピークを有する上記項目5A)記載の結晶。
62A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.3±0.2°、11.3±0.2°、17.1±0.2°、18.8±0.2°、21.7±0.2°、23.2±0.2°、25.5±0.2°、25.8±0.2°、26.4±0.2°、29.4±0.2°にピークを有する上記項目5A)記載の結晶。
63A)図12に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
64A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、9.9±0.2°、15.3±0.2°、21.4±0.2°、23.3±0.2°にピークを有する上記項目5A)記載の結晶。
65A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、5.6±0.2°、9.2±0.2°、9.9±0.2°、14.4±0.2°、15.3±0.2°、21.4±0.2°、22.6±0.2°、23.3±0.2°にピークを有する上記項目5A)記載の結晶。
66A)図13に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
67A)粉末X線回折スペクトルにおいて、回折角度(2θ):7.0±0.2°、12.3±0.2°、16.0±0.2°、19.1±0.2°、21.2±0.2°にピークを有する上記項目5A)記載の結晶。
68A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.7±0.2°、7.0±0.2°、11.4±0.2°、12.3±0.2°、16.0±0.2°、17.3±0.2°、19.1±0.2°、21.2±0.2°、23.0±0.2°にピークを有する上記項目5A)記載の結晶。
69A)図14に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目5A)記載の結晶。
70A)式(I)で示される化合物の一塩酸塩のエタノール和物の結晶。
71A)粉末X線回折スペクトルにおいて、回折角度(2θ):8.3±0.2°、8.9±0.2°、12.9±0.2°、13.7±0.2°、14.7±0.2°にピークを有する上記項目70A)記載の結晶。
72A)粉末X線回折スペクトルにおいて、回折角度(2θ):7.6±0.2°、8.3±0.2°、8.9±0.2°、12.9±0.2°、13.7±0.2°、14.7±0.2°、21.1±0.2°、21.5±0.2°、23.0±0.2°、23.7±0.2°にピークを有する上記項目70A)記載の結晶。
73A)図15に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目70A)記載の結晶。
82A)図18に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目17A)記載の結晶。
83A)式(I)で示される化合物の一硫酸塩の結晶。
84A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.2±0.2°、14.0±0.2°、14.5±0.2°、16.8±0.2°、22.9±0.2°にピークを有する上記項目83A)記載の結晶。
85A)粉末X線回折スペクトルにおいて、回折角度(2θ):6.2±0.2°、12.1±0.2°、14.0±0.2°、14.5±0.2°、15.9±0.2°、16.2±0.2°、16.8±0.2°、21.0±0.2°、22.9±0.2°、26.9±0.2°にピークを有する上記項目83A)記載の結晶。
86A)図19に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目83A)記載の結晶。
87A)式(I)で示される化合物の一硫酸塩一水和物の結晶。
88A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.0±0.2°、9.9±0.2°、13.8±0.2°、14.7±0.2°、17.0±0.2°にピークを有する上記項目87A)記載の結晶。
89A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.0±0.2°、7.4±0.2°、9.9±0.2°、10.1±0.2°、13.8±0.2°、14.7±0.2°、17.0±0.2°、21.4±0.2°にピークを有する上記項目87A)記載の結晶。
90A)図20に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目87A)記載の結晶。
91A)式(I)で示される化合物の一リン酸塩の結晶。
92A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.2±0.2°、6.7±0.2°、9.8±0.2°、12.3±0.2°にピークを有する上記項目91A)記載の結晶。
93A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.2±0.2°、6.7±0.2°、9.8±0.2°、12.3±0.2°、13.3±0.2°、16.6±0.2°、21.1±0.2°にピークを有する上記項目91A)記載の結晶。
94A)図21に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目91A)記載の結晶。
95A)式(I)で示される化合物の一リン酸塩の二水和物の結晶。
96A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.5±0.2°、9.6±0.2°、12.9±0.2°、18.6±0.2°にピークを有する上記項目95A)記載の結晶。
97A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.5±0.2°、9.6±0.2°、10.0±0.2°、11.9±0.2°、12.2±0.2°、12.9±0.2°、16.9±0.2°、18.6±0.2°にピークを有する上記項目95A)記載の結晶。
98A)図22に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目95A)記載の結晶。
99A)式(I)で示される化合物の一フマル酸の結晶。
100A)粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、9.1±0.2°、16.1±0.2°、19.5±0.2°、19.9±0.2°にピークを有する上記項目99A)記載の結晶。
101A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4±0.2°、6.0±0.2°、8.0±0.2°、9.1±0.2°、10.0±0.2°、12.3±0.2°、14.8±0.2°、16.1±0.2°、19.5±0.2°、19.9±0.2°にピークを有する上記項目99A)記載の結晶。
102A)図23に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目99A)記載の結晶。
103A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4±0.2°、9.1±0.2°、13.3±0.2°、13.7±0.2°、18.1±0.2°にピークを有する上記項目99A)記載の結晶。
104A)粉末X線回折スペクトルにおいて、回折角度(2θ):5.4±0.2°、8.0±0.2°、9.1±0.2°、13.3±0.2°、13.7±0.2°、16.4±0.2°、17.1±0.2°、18.1±0.2°、19.9±0.2°、21.8±0.2°にピークを有する上記項目99A)記載の結晶。
105A)図24に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、上記項目99A)記載の結晶。
また、本発明の式(I)で示される化合物の酸付加塩の結晶を含む医薬組成物は、抗癌剤として使用しうる。
さらに、本発明によって、式(II)で示される化合物もしくはその製薬上許容される塩、それらの溶媒和物、またはそれらの結晶が提供される。当該化合物等は、式(I)で示される化合物の酸付加塩およびその結晶体を製造するにあって有用である。
本発明の式(I)で示される化合物の酸付加塩もしくはその溶媒和物、またはそれらの結晶は、医薬としての有用性を備えている。ここで、医薬としての有用性としては、水に対する溶解度が高い点、熱安定性が高い点、吸湿性が低い点、光安定性が高い点、溶液安定性が高い点、保存安定性が高い点、着色安定性が高い点、曝光安定性が高い点、遮光時安定性が高い点、比容積が低い点、帯電性が低い点、凝縮性が高い点、流動性が良い点、結晶化度が高い点、ろ過/遠心分離の操作性が高い点、溶媒除去効率が高い点、工業的に有利な溶媒が使用できる点、錠剤の圧縮形成性が高い点などが含まれる。
また、固体形態の化合物は、(1)モル体積、密度及び吸湿性等の包装特性、(2)融解温度、蒸気圧及び溶解性等の熱力学的特性、(3)溶解速度及び安定性等の動態学的特性(例えば、湿度に対する安定性、保存条件下での周囲条件における安定性等)、(4)表面積、湿潤性、界面張力及び形状等の表面特性、(5)例えば硬度、引っ張り強さ、成形性、取り扱い、流動性(flow)及び混合性(blend)等の機械的特性、又は(6)濾過特性を包含する化合物の物理的特性に実質的に影響を与え得る。固体形態の選択及び制御は、特に薬物となる化合物にとって重要である。固体形態の慎重な選択及び制御は、化合物に関する製造、処方又は投与の問題を減らすことができる。
「からなる」という用語は、構成要件のみを有することを意味する。
「含む」という用語は、構成要件に限定されず、記載されていない要素を排除しないことを意味する。
本発明は、癌を治療又は予防する必要がある哺乳動物において癌を治療又は予防する方法を包含し、当該方法は、前記哺乳動物に治療有効量の式(I)で示される化合物の酸付加塩の結晶またはそれを含有する医薬組成物を投与することからなる。治療するのに好ましい癌は、脳腫瘍(例えば神経膠芽細胞種)、泌尿器癌(例えば膀胱癌、腎臓癌)、生殖器癌(例えば前立腺癌、卵巣癌、子宮癌)、リンパ系腫瘍、消化器癌(例えば胃癌、食道癌、大腸癌、結腸癌)、咽喉癌、肺癌(例えば肺腺癌、小細胞肺癌、非小細胞肺癌)、膵臓癌、乳癌、頭頚部癌、甲状腺癌から選択される。より好ましくは、乳癌、脳腫瘍、膀胱癌、腎臓癌、前立腺癌、卵巣癌、子宮癌、肺癌、膵臓癌、および頭頚部癌である。さらに好ましくは、乳癌である。

で示される化合物は、特許文献1に記載されている、EGF受容体/HER2デュアル阻害剤であり、当該化合物を含有する医薬組成物は、癌の予防または治療に有用である。式(I)で示される化合物は、特許文献1または2記載の方法に従い調整することができる。
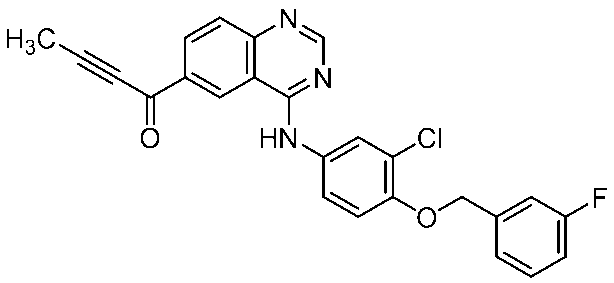
で示される化合物と、下式:

で示される化合物を、酸性条件下で反応させ、得られた粗生成物を中和することにより、式(I)で示される化合物を製造することができる。
別法としては、下式:
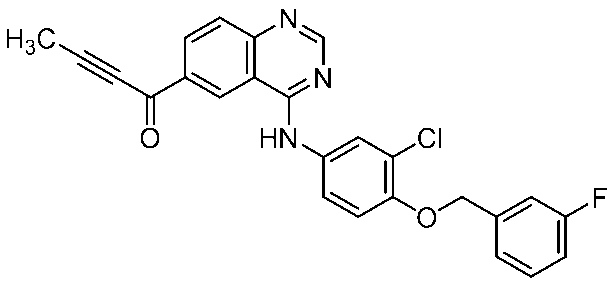
で示される化合物と、下式:

で示される化合物もしくはその塩またはそれらの溶媒和物を酸性条件下で反応させ、得られた粗生成物を中和することにより、式(I)で示される化合物を製造することができる。

で示される化合物を各種有機溶媒に溶解させ、酸性条件下において結晶化させることにより、式(I)で示される化合物の酸付加塩またはその結晶を製造することができる。
酸としては、無機酸(例えば、塩酸、硫酸、硝酸、炭酸、臭化水素酸、リン酸、ヨウ化水素酸等)、および有機酸(例えば、ギ酸、酢酸、プロピオン酸、トリフルオロ酢酸、クエン酸、乳酸、酒石酸、シュウ酸、マレイン酸、フマル酸、マンデル酸、グルタル酸、リンゴ酸、安息香酸、フタル酸、アスコルビン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸等)が挙げられる。特に塩酸およびp-トルエンスルホン酸が挙げられる。
式(I)で示される化合物の一塩酸塩のI形結晶は、式(I)で示される化合物の遊離塩基Aをメタノールに溶解し、塩酸存在下で結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のII形結晶は、式(I)で示される化合物の遊離塩基Aをメタノールと酢酸エチルの混合溶媒(メタノール:酢酸エチル=1:1)に溶解し、塩酸存在下で結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のIII形結晶は、式(I)で示される化合物の遊離塩基Aをメタノールと酢酸エチルの混合溶媒(メタノール:酢酸エチル=1:4)に溶解し、塩酸存在下で結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のV形結晶は、式(I)で示される化合物の遊離塩基Bを2-プロパノールに溶解し、酸存在下で結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のVI形結晶は、式(I)で示される化合物の遊離塩基Aを2-プロパノールに溶解し、塩酸存在下で結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のVII形結晶は、式(I)で示される化合物の一塩酸塩のVI形結晶を1,2-ジメトキシエタンに溶解し、結晶化させることにより製造することができる。
式(I)で示される化合物の一塩酸塩のエタノール和物結晶は、酢酸エチルおよびエタノール混合溶液中に、一塩酸塩I形結晶を種晶として加えることにより製造することができる。
式(I)で示される化合物の遊離塩基結晶は、式(I)で示される化合物の遊離塩基Bをヘキサンおよび酢酸エチルの混合溶液に溶解し、結晶化させることにより製造することができる。
式(I)で示される化合物の一p-トルエンスルホン酸塩I形結晶は、式(I)で示される化合物の遊離塩基Aを、常法により精製後、酢酸エチルに溶解し、1mol/L p-トルエンスルホン酸メタノール溶液を加え、結晶化することにより製造することができる。
式(I)で示される化合物の一硫酸塩結晶は、式(I)で示される化合物の遊離塩基Aをアセトニトリルに溶解し、1mol/L硫酸メタノールを加え、結晶化することにより製造することができる。
式(I)で示される化合物の一硫酸塩一水和物結晶は、式(I)で示される化合物の三水和物結晶をアセトニトリルおよび2-プロパノールの混合溶液に溶解し、0.1mol/L硫酸を加えた後、濃縮する。さらにメタノールおよび水の混合溶液を加え、振とう後に濃縮することにより製造することができる。
式(I)で示される化合物の一リン酸塩結晶は、式(I)で示される化合物の三水和物結晶をアセトニトリルおよび2-プロパノールの混合溶液に溶解し、0.1mol/Lリン酸を加えた後、濃縮する。さらにエタノールおよび水の混合溶液を加え、振とう後に濃縮することにより製造することができる。
式(I)で示される化合物の一リン酸塩二水和物I形結晶は、式(I)で示される化合物の三水和物結晶をアセトニトリルおよび2-プロパノールの混合溶液に溶解し、0.1mol/Lリン酸を加えた後、濃縮する。さらにメタノールおよび水の混合溶液を加え、振とう後に濃縮することにより製造することができる。
式(I)で示される化合物の一フマル酸塩I形結晶は、式(I)で示される化合物の三水和物結晶をアセトニトリルおよび2-プロパノールの混合溶液に溶解し、0.1mol/Lフマル酸のメタノールおよび水の混合溶液を加え、濃縮する。さらにメタノールおよび水を加え、振とう後に濃縮することにより製造することができる。
式(I)で示される化合物の一フマル酸塩II形結晶は、式(I)で示される化合物の三水和物結晶をアセトニトリルおよび2-プロパノールの混合溶液に溶解し、0.1mol/Lフマル酸のメタノールおよび水の混合溶液を加え、濃縮する。さらにアセトニトリルおよび水を加え、振とう後に濃縮することにより製造することができる。
特に言及がなければ、本明細書中及び特許請求の範囲記載の数値は、おおよその値である。数値の変動は、装置キャリブレーション、装置エラー、物質の純度、結晶サイズ、サンプルサイズ、その他の因子に起因する。
一般に結晶性有機化合物は、3次元空間に周期的に配列された多数の原子よりなる。構造周期性は、通例、ほとんどの分光学的プローブ(例えば、X線回折、赤外スペクトル、ラマンスペクトル及び固体NMR)によって明確に区別可能な物理的特性を発現する。中でも粉末X線回折(XRPD)は、固体の結晶性を測定するための最も感度の良い分析法のうちの1つである。X線が結晶に照射されると、結晶格子面で反射し、互いに干渉しあい、ブラッグ則よって予測される条件を満たす方向の回折線のみ強度が増大し、構造の周期に対応した秩序だった回折線を示す。一方、非晶質固体については広範囲の秩序だった回折線は認められない。非晶質固体は、通常、その構造の中に秩序だった繰返し周期をもたないため、回折現象は起こらず特徴のないブロードなXRPDパターンを示す。
TG/DTAは、熱分析の主要な測定方法のひとつで、原子・分子の集合体としての物質の重量及び熱的性質を測定する方法である。
TG/DTAは医薬活性成分の温度又は時間に係る重量及び熱量の変化を測定する方法であり、得られたデータを温度又は時間に対してプロットすることにより、TG(熱重量)及びDTA(示差熱)曲線が得られる。TG/DTA曲線より、医薬活性成分の分解、脱水、酸化、還元、昇華、蒸発に関する重量及び熱量変化の情報を得ることができる。
TG/DTAについて、観察される温度、重量変化は、温度変化速度ならびに用いる試料調製技法及び特定の装置に依存し得ることが知られている。したがって、TG/DTAにおける「融点」とは試料の調製技法の影響を受けにくいオンセット温度のことを指す。結晶の同一性の認定においては、融点のみならず全体的なパターンが重要であり、測定条件や測定機器によって多少変化し得る。
DSCは、熱分析の主要な測定方法のひとつで、原子・分子の集合体としての物質の熱的性質を測定する方法である。DSCにより、医薬活性成分の温度又は時間に係る熱量の変化を測定し、得られたデータを温度又は時間に対してプロットすることにより示差走査熱量曲線が得られる。示差走査熱量曲線より、医薬活性成分が融解する際のオンセット温度、融解に伴う吸熱ピーク曲線の最大値ならびにエンタルピーに関する情報を得ることができる。
DSCについて、観察される温度は、温度変化速度ならびに用いる試料調製技法及び特定の装置に依存し得ることが知られている。したがって、DSCにおける「融点」とは試料の調製技法の影響を受けにくいオンセット温度のことを指す。示差走査熱量曲線から得られるオンセット温度における誤差範囲はおよそ±2℃である。結晶の同一性の認定においては、融点のみならず全体的なパターンが重要であり、測定条件や測定機器によって多少は変化し得る。
水分吸脱着等温線測定は、測定対象の固体について各相対湿度条件下での重量変化を測定する事で、水分の吸着、脱着挙動を計測する測定法である。
基本的な測定法として、0%Target % P/P0(相対湿度0%)での乾燥重量を基準とし、5%、もしくは10%ごとに相対湿度を上げ、それぞれの相対湿度での重量安定化後、基準値からの重量増加から、吸着水の量を求める事が出来る。同様に、100%Target % P/P0から5%、もしくは10%ごとに相対湿度を下げる事で、水の脱着量を測定する事が可能である。
各相対湿度での重量変化値をプロットする事で、吸脱着等温線を得る事ができる。この結果から、各湿度における付着水分の吸着、脱着現象の考察が可能である。また、無水物結晶が湿度により水和物結晶と相互に結晶転移する場合には、結晶転移が起こる湿度、並びに結晶水の量を計算する事が可能である。
付着水,及び結晶水の吸脱着は粒子径、結晶化度、晶癖等の影響を受けるため、測定結果は多少変化し得る。
光安定性試験は、曝光による原薬や製剤の化学的または物理化学的な変化を測定し、光に対する試料の特性を評価する方法の一つである。光安定性試験では、原薬や製剤などの試料に規定の出力により一定期間光を照射する。規定の総照度に到達した時点で、試料の不純物や類縁物質、結晶形、色差などを科学的手法(高速液体クロマトグラフィ―、X線結晶回折、色差計など)により解析することで、光に対する試料の特性を評価することができる。規定された曝光量が得られていることを確認するため、放射計または照度計を用いた曝光量の管理や化学光量測定システムを用いた試験が行われる。
本発明の式(I)で示される化合物の酸付加塩もしくは酸付加塩の結晶またはそれらを含む医薬組成物は、浸透させるべきバリアーに適した浸透剤を用いて、経粘膜投与することができる。該浸透剤は一般に当該分野で知られているものを用いることができる。
特筆しない限り、%は成分の重量%及び組成物の全重量の重量%であり、圧力は大気圧か又はそれに近い圧力である。
本明細書で使用する他の略語は以下のように定義される:
g:グラム
L:リットル
mg:ミリグラム
mL:ミリリットル
Boc:tert-ブトキシカルボニル
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(メソッドA)
(装置)
Bruker社製D-8Discover
(操作方法)
試料について、以下の条件で測定を行った。
測定法:反射法
光源の種類:Cu管球
使用波長:CuKα線
管電流:40mA
管電圧:40kV
試料プレート:ガラス、アルミ
X線の入射角:3-40°
(メソッドB)
(装置)
リガク社製MiniFlex600
(操作方法)
試料について、以下の条件で測定を行った。
測定法:反射法
光源の種類:Cu管球
使用波長:CuKα線
管電流:15mA
管電圧:40kV
試料プレート:円形無反射試料板
X線の入射角:4-40°
各実施例で得られた各結晶約1mgを量り、金メッキスチール高圧パンにつめ、密閉系にて測定した。測定条件は以下のとおりである。
(測定条件)
装置:ティー・エイ・インスツルメント社製DSCDiscovery
測定温度範囲:25℃-250℃
昇温速度:10℃/分
各実施例で得られた各結晶約10mgを量り、アルミニウムパンにつめ、開放系にて測定した。測定条件は以下のとおりである。
(測定条件)
装置:日立ハイテクサイエンス社製TG/DTA6300
測定温度範囲:30℃-300℃
昇温速度:10℃/分
NMRデータを示す場合は、測定した全てのピークを記載していない場合が存在する。
(メソッドA)
装置:Agilent 1290 Infinity
検出波長:232nm
カラム:ZORBAX SB-C18、 1.8μm(2.1mm×30mm)
カラム温度:60℃付近
移動相:0.1%トリフルオロ酢酸水溶液/アセトニトリル混液(90:10から10:90にグラジエント)
流速:1.2mL/min
注入量:1μL
装置:島津2010シリーズまたは島津10A VPシリーズ
カラム:CAPCELL PAK C18 MGII 3μm 内径4.6mm 長さ150mm
移動相:[A]10mM酢酸アンモニウム水溶液/[B]アセトニトリル-メタノール混合液(1:1)
グラジエントプログラムを表1に示す。
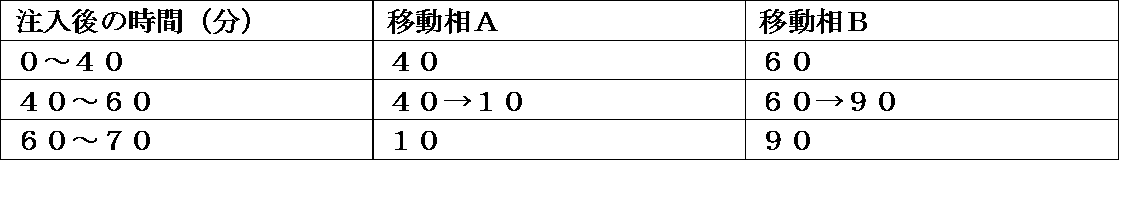
流速:1.0mL/min
検出波長:225nm
注入量:10μL
カラム温度:35℃
各実施例で得られた結晶の水分吸脱着等温線測定を行った。サンプルパンに試料約10mgを測り取り測定を行った。測定条件を以下に示す。
装置:Surface Measurement Systems Ltd.社製 DVS Advantage
測定ポイント:0%Target% P/P0から5%ごとに95%Target% P/P0まで。その後95%Target% P/P0から5%ごとに0%Target% P/P0まで
温度:25℃
ナガノサイエンス社製 光安定性試験装置(LTL200A5-15WCD型)を用いて、実施した。光の照射線源にはD65ランプを使用し、4000lx・hrの条件で総照度として約120万lx・hrまで光を照射した。
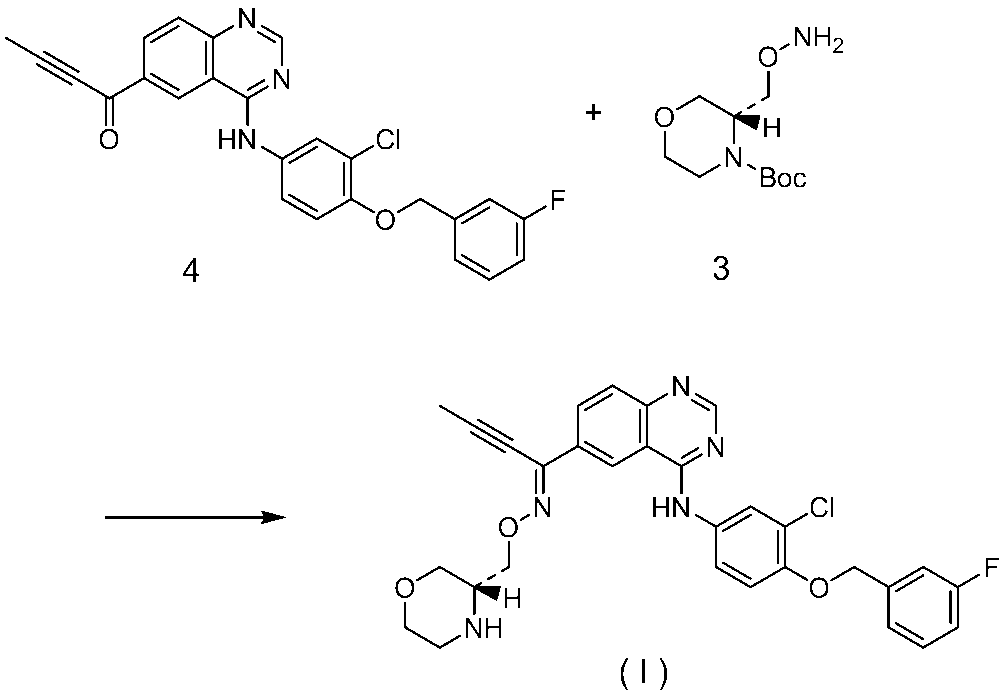
化合物4(8.23g、18.5mmol)、化合物3(6.43g、27.7mmol)をジオキサン(326mL)に懸濁させ、2mol/Lメタンスルホン酸メタノール溶液(23.3mL)を加えた。60℃で4時間攪拌し、その後2mol/Lメタンスルホン酸(14.1mL)を追加して、60℃で17時間攪拌した。反応液を酢酸エチル(815mL)と水(200mL)で希釈し、炭酸カリウム水溶液(炭酸カリウム20.65g、水150mL)を加え抽出した。有機層を食塩水(飽和食塩水50mL、水250mL)で洗浄した。次いで、有機層を硫酸マグネシウムで乾燥し、濾過した後に、濾液を濃縮して、化合物(I)の遊離塩基A(11.83g)を褐色油状物として得た。
化合物(I)の遊離塩基A(1.18g)を酢酸エチル(8mL)に溶解し濾過後、濾液を半分まで減圧濃縮して、4mol/L塩酸酢酸エチル溶液(0.42mL)を加えた。ジエチルエーテル(2mL)を加え生じた沈殿を濾過し、ジエチルエーテル:酢酸エチル混液(2:3)、次いでジエチルエーテルで洗浄した。濾取した固体(817mg)をメタノール(20mL)に加温溶解して、全量が3.6gとなるまで減圧濃縮し、室温で放置した。析出物を濾過し、冷メタノール、次いでジエチルエーテルで洗浄し、乾燥して、化合物(I)の一塩酸塩I形結晶(701mg)を黄色結晶として得た。
1H-NMR(300 MHz、DMSO-d6)δ 2.28 (6H, s), 3.08-3.28 (2H, m), 3.58-3.75 (3H, m), 3.90-3.94 (1H, m), 4.03 (1H, dd, J = 12, 2.7 Hz), 4.44 (2H, d, J = 5.4 Hz), 5.27 (2H, s), 7.15-7.22 (1H, m), 7.26-7.35 (3H, m), 7.44-7.51 (1H, m), 7.72 (1H, dd, J = 9.0, 2.4 Hz), 7.82 (1H, d, J = 8.7 Hz), 7.99 (1H, d, J = 2.4 Hz), 8.26 (1H, dd, J = 8.7, 1.8 Hz), 8.60 (1H, s), 8.88 (1H, d, J = 1.5 Hz), 9.29 (1H, s), 10.20 (1H, s).
元素分析:
計算値: C, 60.41; H, 4.73; Cl, 11.89; F, 3.19; N, 11.74
実測値: C, 60.17; H, 4.79; Cl, 11.62; F, 3.06; N, 11.81
粉末X線回折の結果を、図1および表2に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、14.1±0.2°、20.6±0.2°、21.0±0.2°、25.8±0.2°にピークが認められた。
化合物(I)の遊離塩基B(500mg)に2-プロパノール(5.0mL)を加え、65℃に加熱して溶解した。冷却後、4mol/L塩酸酢酸エチル溶液(212μL)を加えた。室温で5分攪拌後、全体量が3.09gとなるまで減圧濃縮した。生じた沈殿を濾過し、冷2-プロパノール(3mL)で洗浄した。得られた固体をメタノール(13mL)で加温溶解し、全量が3.03gとなるまで減圧濃縮した。酢酸エチル(6.0mL)で希釈して、全量が2.84gとなるまで再び減圧濃縮した。析出物を濾取し、冷酢酸エチル(5mL)で洗浄し、乾燥して、化合物(I)の一塩酸塩V形結晶(382mg)を黄色結晶として得た。
粉末X線回折の結果を、図2および表3に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークが認められた。
化合物(I)の遊離塩基A(1.00g)を2-プロパノール(8.0mL)に溶解し、4mol/L塩酸酢酸エチル溶液(318μL)を加えて、室温で1時間、0℃で1時間攪拌した。室温で30分攪拌後、析出物を濾取した。2-プロパノールで洗浄し、乾燥して、化合物(I)の一塩酸塩VI形結晶(592mg)を淡黄色結晶として得た。
粉末X線回折の結果を、図3および表4に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、16.3±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°にピークが認められた。
化合物(I)の遊離塩基A(50.0mg)をメタノール(100μL)、酢酸エチル(100μL)に溶解し、4mol/L塩酸酢酸エチル溶液(21.2μL)を加えた。室温で1時間攪拌後、酢酸エチル(500μL)で希釈して析出物を濾取した。酢酸エチル(500μL)で洗浄し、乾燥して、化合物(I)の一塩酸塩II形結晶(14.4mg)を無色結晶として得た。
粉末X線回折の結果を、図12および表5に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):11.3±0.2°、17.1±0.2°、25.5±0.2°、25.8±0.2°、26.4±0.2°にピークが認められた。
化合物(I)の遊離塩基A(50.0mg)をメタノール(50μL)、酢酸エチル(200μL)に溶解し、4mol/L塩酸酢酸エチル溶液(21.2μL)を加えた。直後に固体が析出した。酢酸エチル(300μL)で希釈して析出物を濾取した。酢酸エチル(500μL)で洗浄し、乾燥して、化合物(I)の一塩酸塩III形結晶(25.0mg)を淡黄色結晶として得た。
粉末X線回折の結果を、図13および表6に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、9.9±0.2°、15.3±0.2°、21.4±0.2°、23.3±0.2°にピークが認められた。
化合物(I)の一塩酸塩VI形結晶(10mg)に、1,2-ジメトキシエタン(2mL)を加えて加温溶解させた後、液体窒素で冷却して生じた沈殿を濾過し、化合物(I)の一塩酸塩VII形結晶を黄色結晶として得た。
粉末X線回折の結果を、図14および表7に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):7.0±0.2°、12.3±0.2°、16.0±0.2°、19.1±0.2°、21.2±0.2°にピークが認められた。
化合物(IIA)(35.27g、74.0mmol)、p-トルエンスルホン酸一水和物(13.41g、80.7mmol)、テトラヒドロフラン(150mL)、水(13mL)を混合し、50℃で化合物4(30.00g、67.3mmol)を加えて5時間撹拌した。室温に冷却後、126.8gまで濃縮し、水酸化ナトリウム水溶液でpH9.5に調整し、酢酸エチル(240mL×2)で抽出した。抽出液を81.5gまで濃縮し、酢酸エチル(240mL)を加えて再度79.7gまで濃縮した。さらに酢酸エチル(240mL)を加えて61.6gまで濃縮し、エタノール(240mL)を加えた。60℃に加温して実施例1で得た一塩酸塩I形結晶(9mg)を種晶として加えたのち、濃塩酸5.83mLを加えると固体が析出した。25℃で2時間撹拌し濾過すると、種晶とは異なる化合物(I)の一塩酸塩エタノール和物結晶(36.31g、87.8%)を得た。
粉末X線回折の結果を、図15および表8に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):8.3±0.2°、8.9±0.2°、12.9±0.2°、13.7±0.2°、14.7±0.2°にピークが認められた。
化合物4(1.01g)を原料として、参考例1に従い合成した遊離塩基A(1.42g)をシリカゲルクロマトグラフィーで精製(クロロホルム:メタノール=100:1~95:5)して、目的物のフラクションを減圧濃縮した。残渣をアセトンで溶解して減圧濃縮し、ジエチルエーテル(4mL)、ヘキサン(1mL)を加えた。生じた固体を濾過してヘキサン:ジエチルエーテル(1:1)混液、ヘキサンで洗浄して、遊離塩基体(981mg)を得た。このうち112mgを酢酸エチル(1mL)に溶解し、1mol/Lパラトルエンスルホン酸メタノール溶液(190μL)を加えた。酢酸エチル(2mL)を追加して、1時間室温で攪拌した。生じた固体を濾取し、酢酸エチル、ジエチルエーテルで洗浄し、乾燥して、化合物(I)の一p-トルエンスルホン酸塩I形結晶(136mg)を無色結晶として得た。
1H-NMR(300 MHz、DMSO-d6)δ 2.29 (6H, s), 3.14-3.30 (2H, m), 3.52-3.81 (3H, m), 3.88-3.89 (1H, m), 4.00-4.08 (1H, m), 4.41 (2H, d, J = 5.5 Hz), 5.28 (2H, s), 7.11 (2H, d, J = 7.9 Hz), 7.15-7.24 (1H, m), 7.26-7.36 (3H, m), 7.45-7.52 (3H, m), 7.68 (1H, dd, J = 8.8, 2.6 Hz), 7.83 (1H, d, J = 8.8 Hz), 7.95 (1H, d, J = 2.5 Hz), 8.27 (1H, dd, J = 8.8, 1.7 Hz), 8.61 (1H, s), 8.77 (1H, d, J = 1.7 Hz), 8.90 (1H, s), 10.11 (1H, s).
.
元素分析:
計算値: C, 60.69; H, 4.82; Cl, 4.84; F, 2.59; N, 9.56; S, 4.38
実測値: C, 60.45; H, 4.79; Cl, 4.47; F, 2.42; N, 9.46; S, 4.11
粉末X線回折の結果を、図18および表9に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):13.7±0.2°、15.7±0.2°、20.0±0.2°、22.7±0.2°、25.3±0.2°にピークが認められた。
化合物(I)の遊離塩基A(50.0mg)をアセトニトリル(400μL)に溶解し、1mol/L硫酸メタノール溶液(84.8μL)を加えた。アセトニトリル(1mL)を追加して0℃で攪拌し、生じた固体を濾取し、冷アセトニトリルで洗浄した。乾燥し、黄色晶(15.1mg)を得た。これを種結晶として使用した。
化合物(I)の遊離塩基A(100mg)をアセトニトリル(400μL)に溶解し、1mol/L硫酸メタノール溶液(179μL)、上記で調製した種結晶を少量加えた。アセトニトリル(1mL)を追加して0℃で攪拌した。生じた固体を濾取し、冷アセトニトリルで洗浄、乾燥して、化合物(I)の一硫酸塩結晶(47.9mg)を得た。
1H-NMR(400 MHz、DMSO-d6)δ 2.30 (3H, s), 3.12-3.24 (1H, m), 3.24-3.32 (1H, m), 3.62-3.90 (3H, m), 3.93 (1H, d, , J = 11.4 Hz), 4.05 (1H, d, J = 11.4 Hz), 4.49-4.52 (2H, m), 5.32 (2H, s), 7.16-7.22 (1H, m), 7.30-7.39 (3H, m), 7.45-7.52 (1H, m), 7.65-7.70 (1H, m), 7.91 (1H, d, J = 2.0 Hz), 8.00 (1H, d, J = 8.8 Hz), 8.47 (1H, d, J = 8.8 Hz), 8.95 (1H, s), 9.28 (1H, s), 9.53 (1H, s), 9.64 (1H, s), 12.11 (1H, s).
元素分析:
計算値: C, 54.08; H, 4.66; Cl, 5.32; F, 2.85; N, 10.51; S, 4.33 (0.9H2SO4 1.0H2O)
実測値: C, 54.11; H, 4.78; Cl, 5.68; F, 2.65; N, 10.06; S, 4.32
粉末X線回折の結果を、図19および表10に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):6.2±0.2°、14.0±0.2°、14.5±0.2°、16.8±0.2°、22.9±0.2°にピークが認められた。
参考例5に記載の方法で製造した化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/L硫酸(0.056mL)を加えた後,減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一硫酸塩一水和物結晶を得た。
粉末X線回折の結果を、図20および表11に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.0±0.2°、9.9±0.2°、13.8±0.2°、14.7±0.2°、17.0±0.2°にピークが認められた。
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lリン酸(0.056mL)を加えた後、減圧濃縮した。エタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一リン酸塩結晶を得た。
粉末X線回折の結果を、図21および表12に示す。(測定条件:メソッドA)
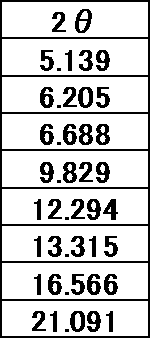
粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.2±0.2°、6.7±0.2°、9.8±0.2°、12.3±0.2°にピークが認められた。
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lリン酸(0.056mL)を加えた後,減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一リン酸塩二水和物I形結晶を得た。
粉末X線回折の結果を、図22および表13に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.1±0.2°、6.5±0.2°、9.6±0.2°、12.9±0.2°、18.6±0.2°にピークが認められた。
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lフマル酸のメタノール:水混液(1:1)(0.056mL)を加えた後、減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一フマル酸塩I形結晶を得た。
粉末X線回折の結果を、図23および表14に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、9.1±0.2°、16.1±0.2°、19.5±0.2°、19.9±0.2°にピークが認められた。
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lフマル酸のメタノール:水混液(1:1)(0.056mL)を加えた後、減圧濃縮した。アセトニトリル:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一フマル酸塩II形結晶を得た。
粉末X線回折の結果を、図24および表15に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.4±0.2°、9.1±0.2°、13.3±0.2°、13.7±0.2°、18.1±0.2°にピークが認められた。
化合物(I)の遊離塩基B(50.0mg)をヘキサン(1mL)、酢酸エチル(0.7mL)に懸濁させ、加温溶解した。室温で放置し、生じた固体を濾取し、乾燥して、式(I)の遊離塩基結晶(39.9mg)を無色結晶として得た。
1H-NMR(400 MHz、DMSO-d6)δ 2.28 (3H, s), 2.74-2.83 (2H, m), 3.09-3.30 (3H, m), 3.68 (1H, d, J = 10.6 Hz), 3.82 (1H, d, J = 10.6 Hz), 4.10-4.22 (2H, m), 5.28 (2H, s), 7.19 (1H, t, J = 8.2 Hz), 7.25-7.38 (3H, m), 7.45-7.52 (1H, m), 7.71 (1H, d, J = 8.8 Hz), 7.81 (1H, d, J = 8.8 Hz), 7.99 (1H, s), 8.22 (1H, d, J = 8.8 Hz), 8.61 (1H, s), 8.76 (1H, s).
粉末X線回折の結果を、図44および表16に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.4±0.2°、13.9±0.2°、17.2±0.2°、20.4±0.2°、24.9±0.2°にピークが認められた。
実施例25における酢酸エチル抽出液を15℃下で撹拌しながら化合物(I)の遊離塩基種晶を溶けなくなるまで加えた。多量に析出した結晶を濾取して化合物(I)の遊離塩基の一水和物結晶を得た。
水分値:3.23%(一水和物の理論値:3.12%)
粉末X線回折の結果を、図16および表17に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):6.0±0.2°、8.3±0.2°、20.1±0.2°、25.5±0.2°、26.1±0.2°にピークが認められた。
実施例25における抽出液を濃縮した際に析出した結晶を濾取して通気乾燥し、化合物(I)の遊離塩基の三水和物結晶を得た。
水分値:9.87%(三水和物の理論値:8.8%)
粉末X線回折の結果を、図17および表18に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):5.9±0.2°、8.7±0.2°、10.9±0.2°、24.7±0.2°、25.4±0.2°にピークが認められた。
化合物(I)の遊離塩基(約50g)を含むエタノール溶液(約400g)のうち、
16gの溶液を分取し、実施例1で得た一塩酸塩I形結晶(2.5mg)を加えたのちに濃塩酸0.838g(2.5当量)を加えた。2時間撹拌後、析出した固体を濾取し、化合物(I)の二塩酸塩結晶を得た。
元素分析:
計算値: C, 57.59; H, 4.64; Cl, 15.87; F, 3.04; N, 11.19(1.8HCl salt)
実測値: C, 57.99; H, 5.51; Cl, 16.74; F, 2.86; N, 11.47
粉末X線回折の結果を、図38に示す。(測定条件:メソッドA)
化合物(I)の遊離塩基A(50.0mg)をアセトニトリル(500μL)に溶解し、1mol/Lパラトルエンスルホン酸メタノール溶液(84.8μL)を加えた。室温で2時間攪拌後、アセトニトリル(500μL)を追加してさらに1時間攪拌し、酢酸エチルで希釈して、生じた固体を濾取した。乾燥して、化合物(I)の一p-トルエンスルホン酸塩II形結晶(29.7mg)を無色結晶として得た。
粉末X線回折の結果を、図39に示す。(測定条件:メソッドA)
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lベンゼンスルホン酸の水溶液(0.056mL)を加えた後、減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一ベンゼンスルホン酸塩結晶を得た。
粉末X線回折の結果を、図40に示す。(測定条件:メソッドA)
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lリン酸(0.056mL)を加えた後、減圧濃縮した。酢酸メチル::水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一リン酸塩二水和物II型結晶を得た。
粉末X線回折の結果を、図41に示す。(測定条件:メソッドA)
化合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/Lクエン酸の水溶液(0.056mL)を加えた後、減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一クエン酸塩結晶を得た。
粉末X線回折の結果を、図42に示す。(測定条件:メソッドA)
合物(I)の遊離塩基の三水和物結晶(3mg)をアセトニトリル:2-プロパノール混液(1:1)(0.15mL)に溶解し、0.1mol/L酒石酸の水溶液(0.056mL)を加えた後、減圧濃縮した。メタノール:水混液(95:5)(0.3mL)を加え、15℃で1時間振とうした後、減圧濃縮し、化合物(I)の一酒石酸塩結晶を得た。
粉末X線回折の結果を、図43に示す。(測定条件:メソッドA)
本願の化合物(I)と構造が類似した化合物が、特許文献2の実施例2に化合物(VI-1)として開示されている。当該化合物(VI-1)は、二塩酸塩結晶として得られたことが記載されている。

次に、特許文献2記載の化合物(VI-1)および本願記載の化合物(I)の遊離塩基の塩基性を有する窒素原子のpKa値を示す。pKa値の計算には、ACD/Labs(Technical Computing Solution ACD/Labs、富士通社製)を使用した。

上記から明らかな通り、モルホリン部分の塩基性窒素原子およびキナゾリン部分の塩基性窒素原子の各々のpKa値はほぼ同じ値を示している。
一般的に、カウンター(例:塩酸)とのpKa値の差が3以上あれば、塩を形成可能であると言われており、塩酸はpKa値=-6であることから、本願記載の化合物(I)の遊離塩基についても、二塩酸塩を形成することが可能である(参考文献:難水溶性薬物の物性評価と製剤設計の新展開、2010年、111~121ページ、Structure, solubility, screening, and synthesis of molecular salts、JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 96, NO. 5, MAY 2007)。
ここで、参考例6に従い、化合物(I)の二塩酸塩結晶を調製したところ、図38に示す通り、ブロードなピークを示し、化合物(I)の二塩酸塩結晶は結晶性が低いことが判明した。
一般的に、結晶性が低い結晶は、物理的安定性が低い、化学的安定性が低い等の特徴が知られており、原薬のハンドリングが難しいと言われている(参考文献:難水溶性薬物の物性評価と製剤設計の新展開、2010年、215~216ページ)。例えば、結晶性の低い結晶を原薬として使用する場合、製造のスケールアップを行った際に、結晶性の良好な結晶に転移することがあり、一定の品質の原薬を提供することが難しい。また、安定性が低いため、長期保存には向かない。
実施例1~7に記載の化合物(I)の一塩酸塩の各種結晶は、図1~図3および図12~図15に記載の通り、結晶性の良好な結晶であり、化合物(I)を一塩酸塩とすることで、原薬に適した結晶多形が得られることは、予期せぬ効果である。
また、参考例7~11に記載の、化合物(I)の一p-トルエンスルホン酸塩II形結晶、一ベンゼンスルホン酸塩結晶、一リン酸塩二水和物II形結晶、一クエン酸結晶および一酒石酸塩結晶は、図39~43に示す通り、ブロードなピークを示し、結晶性が低いことが判明した。
以上より、化合物(I)の二塩酸塩のみならず、酸が一分子付加した塩の一部は、結晶性が低い結晶しか得られなかったにもかかわらず、化合物(I)の一塩酸塩の結晶は、いずれの結晶形においても結晶性が良好であり、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
1.検量線の作成
化合物(I)の一塩酸塩I形結晶5 mgを精密に量り取り、アセトニトリル:水混液(1:1)に溶解させ、500μg/mLの溶液を得た。得られた溶液をそれぞれ、化合物の濃度が5 、50μg/mLになるようにアセトニトリル:水混液(1:1)で希釈し、調製した。これによりHPLC測定条件(メソッドA)にしたがって標準の検量線を作成した。一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶、一p-トルエンスルホン酸塩I形結晶および遊離塩基結晶についても、同様の操作を行った。
2.試料溶液の作成
化合物(I)の一塩酸塩I形結晶1mgを精密に量り取り、4mL容のバイアル瓶に移した。水(注射用水)1mLを添加し、37℃で1時間攪拌した。攪拌後、この懸濁液を濾過し、濾液をアセトニトリル:水混液(1:1)で2倍に希釈した溶液を試料として、HPLC測定条件(メソッドA)でピーク面積を測定した。濃度は、ピーク面積及び先に作成した検量線を用いて算出した。一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶、一p-トルエンスルホン酸塩I形結晶および遊離塩基結晶についても、同様の操作を行った。
(結果)
注射用水に対する化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶、一p-トルエンスルホン酸塩I形結晶および遊離塩基結晶の各溶解度を表19に示す。

(N.D.:Not Detected、単位:μg/mL)
上記表から明らかな通り、化合物(I)の遊離塩基結晶は、注射用水に対して全く溶解しないのに対し、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶および一p-トルエンスルホン酸塩I形結晶は、注射用水に対する高い溶解度を示した。
一般的に、薬剤の溶解度は体内動態に深く関与しており、原薬は高い溶解度を有することが望まれる。従って、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶および一p-トルエンスルホン酸塩I形結晶は、高い溶解度を有し、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
化合物(I)の一塩酸塩I形結晶、一塩酸塩V形結晶、および遊離塩基結晶を、それぞれ2-プロパノール、アセトン、酢酸エチルに懸濁させて22℃下で4時間撹拌し、上澄み液の濃度を測定した(HPLC:メソッドB)。
(結果)2-プロパノール、アセトンおよび酢酸エチルに対するに溶解度を表20に示す。

表20から明らかな通り、化合物(I)の遊離塩基結晶は、各種有機溶媒に対する濃度(重量%)が高く(約0.5重量%~約8重量%)、高い溶解度を示しているのに対し、化合物(I)の一塩酸塩I形結晶および一塩酸塩V形結晶は、各種有機溶媒にほとんど溶解していないことがわかる(いずれも0.1重量%以下)。つまり、化合物(I)を上記参考例1、下記実施例18または25の記載に従い製造する際、生成した化合物(I)の有機溶媒に対する溶解度が高いと、生成物が有機溶媒から析出する割合が低くなり、収量が減少することになる。従って、化合物(I)の一塩酸塩I形結晶または一塩酸塩V形結晶は、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
化合物(I)の酢酸エチル溶液を調製し、当該溶液から、化合物(I)の遊離塩基結晶、一塩酸塩I形結晶および一塩酸塩VI形結晶をそれぞれ晶析した場合の、不純物除去効果を比較した。

(工程1)化合物(I)の酢酸エチル溶液の製造
化合物4(30.04g、67.4mmol)をN-メチルピロリドン(70.86g)、テトラヒドロフラン(18.68g)に溶解し、化合物(IIA)(36.85g、77.3mmol)、p-トルエンスルホン酸一水和物(15.37g、80.8mmol)、テトラヒドロフラン(53.41g)、水(5.40g)のスラリー液に加え、57℃で5時間撹拌した。室温に冷却後、化合物4(0.25g)を加えた。その後、水酸化ナトリウム水溶液でpH9.0に調整し、酢酸エチル(651.52g)で抽出した。抽出液を107.39gまで濃縮し、酢酸エチル(162.40g)を加えることで、化合物(I)の酢酸エチル溶液(269.76g)を得た。
(工程2-1)化合物(I)の遊離塩基結晶の製造
化合物(I)の酢酸エチル溶液(89.92g)を22.97gまで濃縮し、ヘプタン(17.67g)、酢酸エチル(13.27g)を加えたのち、60℃に加温すると固体が析出した。酢酸エチル(12.31g)を加えて室温に冷却し、ヘプタン(41.26g)、酢酸エチル(6.0g)を加えて49.70gまで濃縮した。ヘプタン(49.83g)、酢酸エチル(27.0g)を加えて一晩静置したのちに濾過することで、化合物(I)の遊離塩基結晶(10.91g、86.7%)を得た。
(工程2-2)化合物(I)の一塩酸塩I形結晶の製造
化合物(I)の酢酸エチル溶液(40.72g)に水(0.13g)、2-プロパノール(16.26g)を加え45℃に加温した。I形結晶の種晶(225.7mg)を加えたのちに35%塩酸でpH4.07に調整した。25℃で約30分撹拌した後に濾過することで、化合物(I)の一塩酸塩I形結晶(5.50g、90.6%)を得た。
(工程2-3)化合物(I)の一塩酸塩VI形結晶の製造
化合物(I)の酢酸エチル溶液(45.20g)に2-プロパノール(7.85g)を加え60℃に加温した。35%塩酸でpH3.5に調整後、25℃で約30分撹拌し濾過することで、化合物(I)の一塩酸塩VI形結晶(5.84g、86.7%)を得た。
上記製造法により得られた化合物(I)の遊離塩基結晶、一塩酸塩I形結晶および一塩酸塩VI形結晶について、HPLCを用いて品質を評価した(HPLC:メソッドB)。
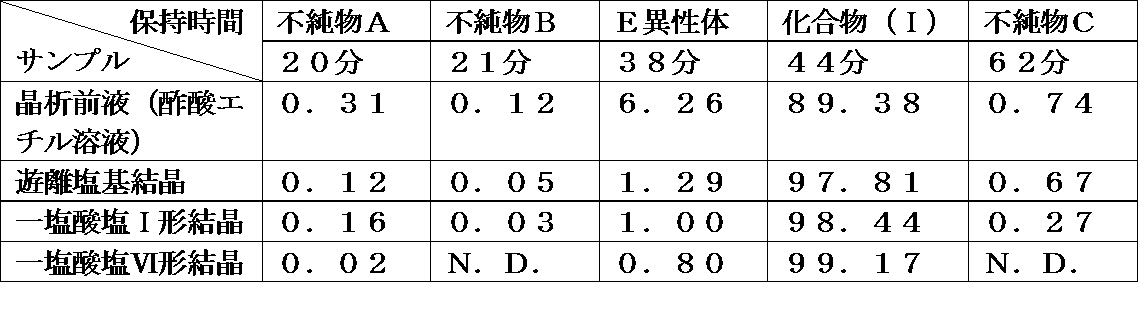
(N.D.:Not Detected、単位:面積%)
上記で得られた各結晶の純度を比較すると、表21より明らかな通り、一塩酸塩VI形結晶、一塩酸塩I形結晶、遊離塩基結晶の順で、化合物(I)のピーク面積%の割合が大きく(順に、約%99.2、約98.4%、約97.8%)、含有される各種不純物の量が少量であることがわかる。従って、不純物が多く含まれる晶析前の溶液(酢酸エチル溶液)から晶析を行う際は、遊離塩基結晶として結晶を得るより、一塩酸塩VI形結晶または一塩酸塩I形結晶として結晶を得る方が、各種不純物を除去することができ、より高純度の結晶が得られることがわかった。
一般的に、不純物が多く含まれる結晶は、再結晶の工程を繰り返し、結晶の純度を上げる必要がある。また、再結晶工程を繰り返すと、母液に溶出する結晶の量が増え、収量が減少することになる。不純物によっては再結晶工程により除去される割合が小さいため、実用的な繰り返し回数では純度を上げることができない場合も多い。
従って、化合物(I)の一塩酸塩VI形結晶および一塩酸塩I形結晶は、一度の晶析で高純度の結晶が得られることから、大量合成に好適な結晶形であると言える。つまり、化合物(I)の一塩酸塩VI形結晶および一塩酸塩I形結晶は、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
化合物(I)の一塩酸塩I形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶、遊離塩基結晶、一p-トルエンスルホン酸塩結晶および一硫酸塩結晶の水分増加質量比率を表22に示す。

表22より明らかな通り、化合物(I)の一硫酸塩結晶は、約3.3%の水分増加が見られたのに対し、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶および一p-トルエンスルホン酸塩I形結晶は、水分増加比率が少ないことが分かった。
一般的に、結晶を塩にすることでより吸湿の影響を受けやすくなることが考えられ、塩の種類により、吸収性は異なるといわれている(参考文献:難水溶性薬物の物性評価と製剤設計の新展開、2010年、117~118ページ)。また、水分が吸着しやすい結晶は潮解等の現象がみられ、結晶の取り扱いが困難である。さらに、そのような結晶は長期保存には適しておらず、原薬として選択されることはほとんどない。従って、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶および一p-トルエンスルホン酸塩I形結晶は、水分増加比率が少ないことから、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶、一塩酸塩VI形結晶および一p-トルエンスルホン酸塩I形結晶の曝光試験結果を表23に示す。HPLC(メソッドB)を用いて、各結晶の品質評価を実施した。

(単位は面積%。*その他のピークについて曝光による変化はほとんど認められない。)
表23に示す通り、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶および一塩酸塩VI形結晶を曝光試験に付すと、Z異性体からE異性体への変換がほとんど見られないか、または多くても約3.8%ほどしか増加しない。それに対し、化合物(I)の一p-トルエンスルホン酸塩I形結晶は、曝光後にE異性体が約33%増加していることから、光安定性が低いことがわかった。
一般的に、光安定性が低い結晶は、光によって分解等の現象が見られ、曝光によって許容できない変化が起きる場合がある。また、そのような結晶は、保存方法に細心の注意を払う必要があり、結晶の取り扱いが困難である。
従って、化合物(I)の一塩酸塩I形結晶、一塩酸塩II形結晶、一塩酸塩V形結晶および一塩酸塩VI形結晶は、曝光条件下において光安定性が良く、医薬品の原薬として使用するのに好適な結晶形であることが判明した。
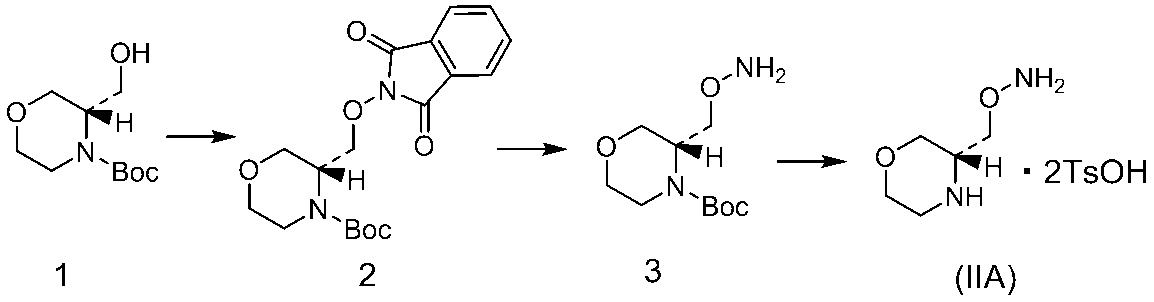
トリフェニルホスフィン(20.3g、77.3mmol)、アゾジカルボン酸ジイソプロピル(16.6g、81.8mmol)、化合物1(14.0g、64.4mmol)、N-ヒドロキシフタルイミド(4.2g、70.9mmol)をテトラヒドロフラン(98mL)、トルエン(77mL)中、5乃至15℃で2時間撹拌し化合物2の調製液を得た。そこに35%モノメチルヒドラジン水溶液(9.3g、70.9mmol)を加え、15℃で2時間撹拌することで化合物3の調製液を得た。化合物3の調製液を114gまで濃縮し、0℃で2時間撹拌した。析出した不溶物を濾過後、濾液にトルエン(56mL)を加えた。このトルエン溶液をp-トルエンスルホン酸一水和物(28.2g、148.1mmol)のテトラヒドロフラン(42mL)溶解液に加え、50℃で2時間撹拌した。さらに、0℃に冷却し2時間撹拌後、濾過することで化合物(IIA)(27.2g、収率88.4%)を得た。
1H-NMR (CD3OD) δ: 7.67-7.73 (4H, m), 7.20-7.26 (4H, m), 4.92 (5H, bs), 4.30-4.34 (2H, m), 3.91-4.03 (2H, m), 3.63-3.79 (3H, m), 3.18-3.38 (2H, m), 2.36 (6H, s).
粉末X線回折の結果を、図10および表24に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、21.1±0.2°、24.7±0.2°、25.3±0.2°にピークが認められた。
粉末X線回折の結果を、図11および表25に示す。(測定条件:メソッドA)

粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、22.6±0.2°、24.0±0.2°にピークが認められた。

化合物3(4.12g、17.7mmol)に酢酸エチル(8mL)を加えて撹拌し、4mol/L 塩化水素酢酸エチル溶液(16mL)を加えると、白色固体が析出した。酢酸エチル(8mL)を加えたのちに約4時間室温で撹拌した。固体を濾取し、吸湿性が高い化合物(II’)を得た。
1H-NMR (DMSO-d6) δ: 9.60-11.50 (4H, bs), 4.20-4.31 (2H, m), 3.86-3.93 (2H, m), 3.21-3.77 (4H, m), 3.01-3.10 (1H, m).
得られた化合物は吸湿して潮解する為、粉末X線などの測定はできなかった。
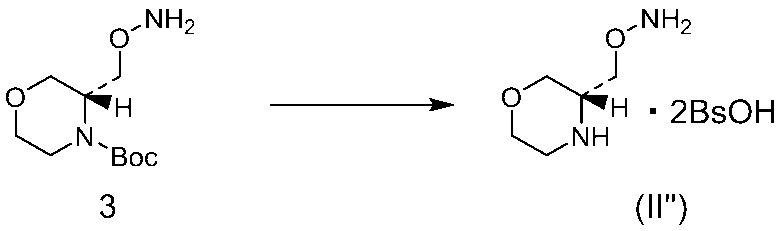
ベンゼンスルホン酸(78.57mg)とテトラヒドロフラン(0.2mL)を50℃で撹拌し、化合物3(51.54mg、0.222mmol)のテトラヒドロフラン(0.2mL)溶液を滴下した。約20分後に室温まで冷却すると固体が析出した。テトラヒドロフラン0.15mLを加えて50℃で10分撹拌し、10℃まで冷却した。固体を窒素雰囲気下で濾取し、吸湿性が高い化合物(II’’)を57.3mg得た。
1H-NMR (CD3OD) δ: 7.84-7.87 (4H, m), 7.45-7.47 (6H, m), 4.34-4.36 (2H, m), 3.98-4.08 (2H, m) , 3.71-3.80 (3H, m), 3.28-3.41 (2H, m).
粉末X線回折の結果を、図53および表26に示す。(測定条件:メソッドB)

参考例12に記載の化合物(II’)の結晶は潮解する性質を有し、結晶性が低い。また、参考例13に記載の化合物(II’’)の結晶は、図53に示す通り、強度の弱いピークを示し、結晶性が低い。上記実施例15に記載の通り、結晶性が低い結晶はハンドリングが困難である。それに対して、実施例21および22に記載の化合物(IIA)の結晶および化合物(IIA)の二水和物結晶は、図10および図11に示す通り、結晶性の良好な結晶であり、原薬の中間体として選択可能な結晶形であることが判明した。
実施例21で得られた化合物(IIA)の結晶を、50℃で8時間加熱し、NMRデータを測定した。その結果、加熱前と比較してピークに変化は観測されず、化合物(IIA)の結晶は安定性が高い結晶であることが判明した。

化合物4(97.01g、217.6mmol)をN-メチルピロリドン(223mL)、テトラヒドロフラン(116mL)に溶解させたものと、化合物(IIA)(119.25g、250.2mmol)、p-トルエンスルホン酸一水和物(49.67g、261.1mmol)、テトラヒドロフラン(194mL)、水(17mL)を混合し、57℃で5時間撹拌した。室温に冷却後、水酸化ナトリウム水溶液でpH9.0に調整し、酢酸エチル(776mL)で抽出した。抽出液を357mLまで濃縮し、酢酸エチル(582mL)を加えて希釈液882.12gを得た。
希釈液44.11gに2-プロパノール(10mL)を加え60℃に加温した。35%塩酸でpH4.3に調整後、25℃で4時間撹拌し濾過することで、化合物(I)の一塩酸塩VI形結晶(5.6g、86.8%)を得た。
製剤例1: 錠剤
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶、乳糖及びステアリン酸カルシウムを混合し、破砕造粒して乾燥し、適当な大きさの顆粒剤とする。次にステアリン酸カルシウムを添加して圧縮成形して錠剤とする。
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶、乳糖及びステアリン酸カルシウムを均一に混合して粉末又は細粒状として散剤をつくる。それをカプセル容器に充填してカプセル剤とする。
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶、乳糖及びステアリン酸カルシウムを均一に混合し、圧縮成型した後、粉砕、整粒し、篩別して適当な大きさの顆粒剤とする。
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶及び結晶セルロースを混合し、造粒後打錠して口腔内崩壊錠とする。
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶及び乳糖を混合し、粉砕、整粒、篩別して適当な大きさのドライシロップとする。
本発明の式(I)で示される化合物の酸付加塩または酸付加塩の結晶及び乳糖を混合し細かく粉砕することにより、吸入剤とする。
また、式(II)で示される化合物もしくはその製薬上許容される塩またはそれらの溶媒和物は、式(I)で示される化合物の塩酸塩または塩酸塩の結晶を製造するにあたって有用な中間体である。
Claims (29)
- 式(I):
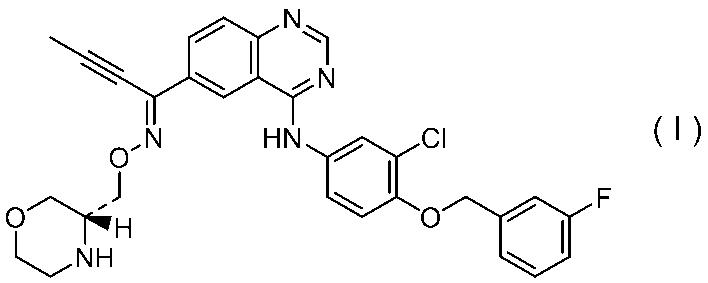
で示される化合物の塩またはその溶媒和物。 - 塩酸塩である、請求項1記載の塩またはその溶媒和物。
- 一塩酸塩である、請求項1または2記載の塩またはその溶媒和物。
- 式(I):

で示される化合物の塩酸塩の結晶。 - 一塩酸塩である、請求項4記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):8.0±0.2°、14.1±0.2°、20.6±0.2°、21.0±0.2°、25.8±0.2°にピークを有する請求項5記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):6.8°±0.2°、8.0±0.2°、14.1°±0.2°、17.9°±0.2°、18.5°±0.2°、20.6°±0.2°、21.0°±0.2°、22.5°±0.2°、25.8°±0.2°、28.4°±0.2°にピークを有する請求項5記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する請求項5記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):7.9°±0.2°、9.7±0.2°、11.9±0.2°、15.8±0.2°、18.5±0.2°、23.9±0.2°、25.9±0.2°、26.2±0.2°、26.7±0.2°、28.4±0.2°にピークを有する請求項5記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、16.3±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°にピークを有する請求項5記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):5.4°±0.2°、8.9±0.2°、11.7±0.2°、13.8±0.2°、16.3±0.2°、20.9±0.2°、21.6±0.2°、23.2±0.2°、23.7±0.2°、26.6±0.2°にピークを有する請求項5記載の結晶。
- 請求項2または3に記載の塩酸塩またはその溶媒和物を含む医薬組成物。
- 請求項4~11のいずれかに記載の結晶を含む医薬組成物。
- p-トルエンスルホン酸塩である、請求項1記載の塩またはその溶媒和物。
- 一p-トルエンスルホン酸塩である、請求項1または14記載の塩またはその溶媒和物。
- 式(I):
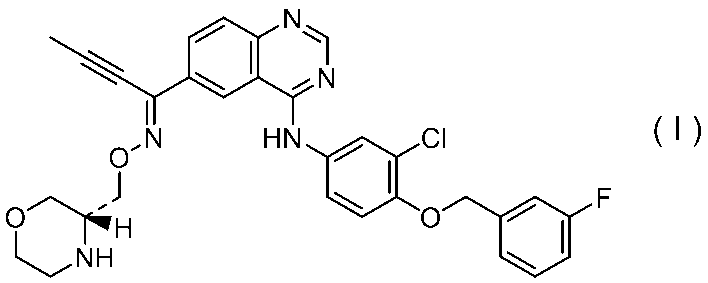
で示される化合物のp-トルエンスルホン酸塩の結晶。 - 一p-トルエンスルホン酸塩である、請求項16記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):13.7±0.2°、15.7±0.2°、20.0±0.2°、22.7±0.2°、25.3±0.2°にピークを有する請求項17記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):6.1±0.2°、6.4±0.2°、10.8±0.2°、13.7±0.2°、15.7±0.2°、16.3±0.2°、20.0±0.2°、22.7±0.2°、24.6±0.2°、25.3±0.2°にピークを有する請求項17記載の結晶。
- 請求項14または15に記載のp-トルエンスルホン酸塩またはその溶媒和物を含む医薬組成物。
- 請求項16~19のいずれかに記載の結晶を含む医薬組成物。
- 式(II):
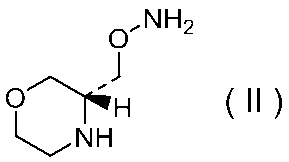
で示される化合物もしくはその製薬上許容される塩またはそれらの溶媒和物。 - 二p-トルエンスルホン酸塩である、請求項22記載の塩またはそれらの溶媒和物。
- 式(II):

で示される化合物の二p-トルエンスルホン酸塩の結晶。 - 粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、21.1±0.2°、24.7±0.2°、25.3±0.2°にピークを有する請求項24記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):6.4±0.2°、7.3±0.2°、11.4±0.2°、15.2±0.2°、17.6±0.2°、20.1±0.2°、21.1±0.2°、21.7±0.2°、24.7±0.2°、25.3±0.2°にピークを有する請求項24記載の結晶。
- 式(II):

で示される化合物の二p-トルエンスルホン酸塩二水和物の結晶。 - 粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、22.6±0.2°、24.0±0.2°にピークを有する請求項27記載の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):7.3±0.2°、17.0±0.2°、18.5±0.2°、19.7±0.2°、21.7±0.2°、22.6±0.2°、22.8±0.2°、24.0±0.2°、24.8±0.2°、29.7±0.2°にピークを有する請求項27記載の結晶。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES16830581T ES2908349T3 (es) | 2015-07-29 | 2016-07-28 | Sal de derivado de quinazolina o cristal de la misma, y método para producir sal de derivado de quinazolina o cristal de la misma |
| JP2017530920A JP6334065B2 (ja) | 2015-07-29 | 2016-07-28 | キナゾリン誘導体の塩またはその結晶およびそれらの製造方法 |
| PL16830581T PL3330267T3 (pl) | 2015-07-29 | 2016-07-28 | Sól pochodnej chinazoliny lub jej kryształ oraz sposób wytwarzania soli pochodnej chinazoliny lub jej kryształu |
| US15/748,283 US10329285B2 (en) | 2015-07-29 | 2016-07-28 | Salts of quinazoline derivative or crystals thereof, and the process for producing thereof |
| EP16830581.1A EP3330267B1 (en) | 2015-07-29 | 2016-07-28 | Salt of quinazoline derivative or crystal thereof, and method for producing salt of quinazoline derivative or crystal thereof |
| DK16830581.1T DK3330267T3 (da) | 2015-07-29 | 2016-07-28 | Salt af quinazolinderivat eller crystal deraf samt fremgangsmåde til fremstilling af salt af quinazolinderivat eller crystal deraf |
| US16/401,342 US10513513B2 (en) | 2015-07-29 | 2019-05-02 | Salts of quinazoline derivative or crystals thereof, and the process for producing thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015149259 | 2015-07-29 | ||
| JP2015-149259 | 2015-07-29 | ||
| JP2015219635 | 2015-11-09 | ||
| JP2015-219635 | 2015-11-09 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/748,283 A-371-Of-International US10329285B2 (en) | 2015-07-29 | 2016-07-28 | Salts of quinazoline derivative or crystals thereof, and the process for producing thereof |
| US16/401,342 Continuation US10513513B2 (en) | 2015-07-29 | 2019-05-02 | Salts of quinazoline derivative or crystals thereof, and the process for producing thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017018476A1 true WO2017018476A1 (ja) | 2017-02-02 |
Family
ID=57885722
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/072129 Ceased WO2017018476A1 (ja) | 2015-07-29 | 2016-07-28 | キナゾリン誘導体の塩またはその結晶およびそれらの製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US10329285B2 (ja) |
| EP (1) | EP3330267B1 (ja) |
| JP (1) | JP6334065B2 (ja) |
| DK (1) | DK3330267T3 (ja) |
| ES (1) | ES2908349T3 (ja) |
| PL (1) | PL3330267T3 (ja) |
| TW (1) | TWI705962B (ja) |
| WO (1) | WO2017018476A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018139626A1 (ja) * | 2017-01-30 | 2018-08-02 | 塩野義製薬株式会社 | キナゾリン誘導体を含有する固形製剤 |
| US10287353B2 (en) | 2016-05-11 | 2019-05-14 | Huya Bioscience International, Llc | Combination therapies of HDAC inhibitors and PD-1 inhibitors |
| US10385131B2 (en) | 2016-05-11 | 2019-08-20 | Huya Bioscience International, Llc | Combination therapies of HDAC inhibitors and PD-L1 inhibitors |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019208422A (ja) * | 2018-06-04 | 2019-12-12 | コリア・インスティテュート・オブ・サイエンス・アンド・テクノロジー | ヒアルロニダーゼを含む新規な組換えエキソソーム及びその用途 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006090717A1 (ja) * | 2005-02-23 | 2006-08-31 | Shionogi & Co., Ltd. | チロシンキナーゼ阻害作用を有するキナゾリン誘導体 |
| WO2010074150A1 (ja) * | 2008-12-25 | 2010-07-01 | 塩野義製薬株式会社 | キナゾリン誘導体の製造方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2001273071B2 (en) | 2000-06-30 | 2005-09-08 | Glaxo Group Limited | Quinazoline ditosylate salt compounds |
| EP1854798A3 (en) * | 2000-09-19 | 2007-11-28 | Bristol-Myers Squibb Company | Fused heterocyclic succinimide compounds and analogs thereof, modulators of nuclear hormone receptor function |
| WO2009079541A1 (en) | 2007-12-18 | 2009-06-25 | Smithkline Beecham (Cork) Limited | Quinazoline ditosylate anhydrate forms |
| WO2009079547A1 (en) | 2007-12-18 | 2009-06-25 | Smithkline Beecham (Cork) Limited | Quinazoline anhydrate forms |
-
2016
- 2016-07-28 ES ES16830581T patent/ES2908349T3/es active Active
- 2016-07-28 EP EP16830581.1A patent/EP3330267B1/en active Active
- 2016-07-28 US US15/748,283 patent/US10329285B2/en active Active
- 2016-07-28 JP JP2017530920A patent/JP6334065B2/ja active Active
- 2016-07-28 WO PCT/JP2016/072129 patent/WO2017018476A1/ja not_active Ceased
- 2016-07-28 DK DK16830581.1T patent/DK3330267T3/da active
- 2016-07-28 PL PL16830581T patent/PL3330267T3/pl unknown
- 2016-07-29 TW TW105124192A patent/TWI705962B/zh active
-
2019
- 2019-05-02 US US16/401,342 patent/US10513513B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006090717A1 (ja) * | 2005-02-23 | 2006-08-31 | Shionogi & Co., Ltd. | チロシンキナーゼ阻害作用を有するキナゾリン誘導体 |
| WO2010074150A1 (ja) * | 2008-12-25 | 2010-07-01 | 塩野義製薬株式会社 | キナゾリン誘導体の製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| BRADLEY D. ANDERSON; KARL P. FLORA: "[Preparation of water-soluble organic compound by salt formation]", SAISHIN SOYAKU KAGAKU LAST VOLUME [THE PRACTICE OF MEDICINAL CHEMISTRY], 1999, JAPAN, pages 347 - 365, XP009508529 * |
| See also references of EP3330267A4 * |
| YUKI KAGOBUTSU KESSHO SAKUSEI HANDBOOK -GENRI TO KNOW-HOW, 25 July 2008 (2008-07-25), pages 57 - 84, XP 008183996 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10287353B2 (en) | 2016-05-11 | 2019-05-14 | Huya Bioscience International, Llc | Combination therapies of HDAC inhibitors and PD-1 inhibitors |
| US10385131B2 (en) | 2016-05-11 | 2019-08-20 | Huya Bioscience International, Llc | Combination therapies of HDAC inhibitors and PD-L1 inhibitors |
| US10385130B2 (en) | 2016-05-11 | 2019-08-20 | Huya Bioscience International, Llc | Combination therapies of HDAC inhibitors and PD-1 inhibitors |
| US11535670B2 (en) | 2016-05-11 | 2022-12-27 | Huyabio International, Llc | Combination therapies of HDAC inhibitors and PD-L1 inhibitors |
| US12122833B2 (en) | 2016-05-11 | 2024-10-22 | Huyabio International, Llc | Combination therapies of HDAC inhibitors and PD-1 inhibitors |
| WO2018139626A1 (ja) * | 2017-01-30 | 2018-08-02 | 塩野義製薬株式会社 | キナゾリン誘導体を含有する固形製剤 |
| US10953016B2 (en) | 2017-01-30 | 2021-03-23 | Shionogi & Co., Ltd. | Solid dosage form containing quinazoline derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| US10329285B2 (en) | 2019-06-25 |
| PL3330267T3 (pl) | 2022-04-04 |
| TWI705962B (zh) | 2020-10-01 |
| EP3330267B1 (en) | 2022-01-05 |
| US20180215744A1 (en) | 2018-08-02 |
| ES2908349T3 (es) | 2022-04-28 |
| US10513513B2 (en) | 2019-12-24 |
| US20190256506A1 (en) | 2019-08-22 |
| DK3330267T3 (da) | 2022-03-14 |
| JP6334065B2 (ja) | 2018-05-30 |
| EP3330267A1 (en) | 2018-06-06 |
| JPWO2017018476A1 (ja) | 2018-05-17 |
| EP3330267A4 (en) | 2018-12-26 |
| TW201718530A (zh) | 2017-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7383652B2 (ja) | B-rafキナーゼのマレイン酸塩、結晶形、調整方法、及びその使用 | |
| CN114728899B (zh) | 新型三苯基化合物盐 | |
| EP2643314B1 (en) | Novel salts and polymorphic forms of afatinib | |
| JP6971390B2 (ja) | 重水素化azd9291の結晶形、製造方法および使用 | |
| JP2011515370A (ja) | 4−アミノ−5−フルオロ−3−[5−(4−メチルピペラジン−1−イル)−1h−ベンズイミダゾール−2−イル]キノリン−2(1h)−オン乳酸塩の結晶形態及び2つの溶媒和物形態 | |
| KR102142797B1 (ko) | 피리디닐아미노피리미딘 유도체의 메실레이트 염의 결정질 형태, 그의 제조 방법, 및 그의 용도 | |
| US10513513B2 (en) | Salts of quinazoline derivative or crystals thereof, and the process for producing thereof | |
| CN113968843B (zh) | 一种环己烷甲酰胺的新晶型及其制备方法 | |
| JP2019526605A (ja) | 置換2−h−ピラゾール誘導体の結晶形、塩型及びその製造方法 | |
| JP2023512471A (ja) | Pde3/pde4二重阻害剤の結晶及びその使用 | |
| TW202124373A (zh) | 1,3,5-三衍生物或其溶劑合物之結晶及其製造方法 | |
| JP2019500370A (ja) | キナゾリン誘導体の結晶及びその調製方法 | |
| US20250026773A1 (en) | Mono-p-toluenesulfonate of axl kinase inhibitor and crystal form thereof | |
| JP2018538348A (ja) | キナゾリン誘導体の結晶及びその調製方法 | |
| CN111315748B (zh) | 3-(5-氟苯并呋喃-3-基)-4-(5-甲基-5H-[1,3]间二氧杂环戊烯并[4,5-f]吲哚-7-基)吡咯-2,5-二酮晶体 | |
| JP7750535B2 (ja) | 化合物の結晶形態 | |
| WO2019154273A1 (zh) | 喹啉衍生物的结晶 | |
| TWI535724B (zh) | 埃克替尼磷酸鹽的新晶型及其用途 | |
| CN120344512A (zh) | 氧异吲哚-5-甲酰胺类化合物或其盐、溶剂合物的结晶形式或无定形形式 | |
| TWI596098B (zh) | 埃克替尼馬來酸鹽的晶型及其用途 | |
| HK40072576B (zh) | 用作激酶抑制剂的化合物及其应用 | |
| KR20250174948A (ko) | 질소 함유 테트라시클릭 화합물의 결정 형태 및 이의 제조 방법 | |
| CN119841853A (zh) | 一种嘧啶甲酰胺类化合物的晶型及其制备方法 | |
| CN121079430A (zh) | 唾液酸衍生物的结晶形式及其使用方法 | |
| HU231012B1 (hu) | Lapatinib sók |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16830581 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017530920 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15748283 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016830581 Country of ref document: EP |