WO2017057642A1 - 光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸の製造法 - Google Patents
光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸の製造法 Download PDFInfo
- Publication number
- WO2017057642A1 WO2017057642A1 PCT/JP2016/078939 JP2016078939W WO2017057642A1 WO 2017057642 A1 WO2017057642 A1 WO 2017057642A1 JP 2016078939 W JP2016078939 W JP 2016078939W WO 2017057642 A1 WO2017057642 A1 WO 2017057642A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- compound represented
- mol
- nickel
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *[C@]1N=C(C(*)(*)C2=N[C@@](*)CO2)OC1 Chemical compound *[C@]1N=C(C(*)(*)C2=N[C@@](*)CO2)OC1 0.000 description 4
- CVAWKJKISIPBOD-UHFFFAOYSA-N CC(C(OC(C)(C)C)=O)Br Chemical compound CC(C(OC(C)(C)C)=O)Br CVAWKJKISIPBOD-UHFFFAOYSA-N 0.000 description 1
- IRBZORXEHCJFTO-UHFFFAOYSA-N Fc(cc(cc1)[Zn])c1-c1ccccc1 Chemical compound Fc(cc(cc1)[Zn])c1-c1ccccc1 IRBZORXEHCJFTO-UHFFFAOYSA-N 0.000 description 1
- HTRNHWBOBYFTQF-UHFFFAOYSA-N Fc1cc(Br)ccc1-c1ccccc1 Chemical compound Fc1cc(Br)ccc1-c1ccccc1 HTRNHWBOBYFTQF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/08—Halides
- B01J27/10—Chlorides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0234—Nitrogen-, phosphorus-, arsenic- or antimony-containing compounds
- B01J31/0235—Nitrogen containing compounds
- B01J31/0244—Nitrogen containing compounds with nitrogen contained as ring member in aromatic compounds or moieties, e.g. pyridine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/52—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing halogen
- C07C57/58—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms containing halogen containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a novel process for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid.
- 2- (2-Fluorobiphenyl-4-yl) propanoic acid or a pharmaceutically acceptable salt thereof is known to have widespread use as a drug having anti-inflammatory action, analgesic action and the like.
- a method for producing this 2- (2-fluorobiphenyl-4-yl) propanoic acid for example, it can be obtained by using a metal salt of 2- (2-fluorobiphenyl-4-yl) magnesium bromide and 2-bromopropionic acid.
- Method (Patent Document 1), 2- (2-fluorobiphenyl-4-yl) magnesium bromide, 2-bromopropionic acid ester and nickel catalyst are used to obtain an ester intermediate (Patent Document 2) Has been.
- optical resolution method for example, a method of preferentially crystallizing a salt of one optical isomer by forming a salt with an optically active amine has been reported.
- fluorobiphenyl-4-yl) propanoic acid a plurality of recrystallization steps are essential, and the recovery rate is as low as about 60% (Patent Document 3).
- Non-patent Document 1 asymmetric hydrogenation of 2- (2-fluorobiphenyl-4-yl) acrylic acid has been reported (Non-patent Document 1), but the number of steps is long and the final Since a rhodium catalyst having toxicity is used in the process, it is not practical.
- Non-Patent Document 2 uses a nickel catalyst and an optically active bisoxazoline ligand, and an asymmetric kumada using an ⁇ -haloketone as a substrate. Reactions have been reported, and Non-Patent Document 3 reports an asymmetric Negishi reaction using ⁇ -haloamide and ⁇ -haloketone as substrates, respectively.
- the asymmetric Kumada reaction requires a low reaction temperature of ⁇ 60 to ⁇ 40 ° C., and the optically active compound obtained by multi-step synthesis in both the asymmetric Kumada reaction and the asymmetric Negishi reaction.
- Uses 6.5 to 13% bisoxazoline ligand is not suitable for industrial production, and reports on the synthesis of profenes and reports on the synthesis of ⁇ -haloesters that can be easily derived into profens There is no.
- Patent Document 4, Patent Document 5, and Non-Patent Document 5 show an asymmetric Kumada reaction using a cobalt catalyst and an optically active bisoxazoline ligand.
- An object of the present invention is to provide a novel process for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid.
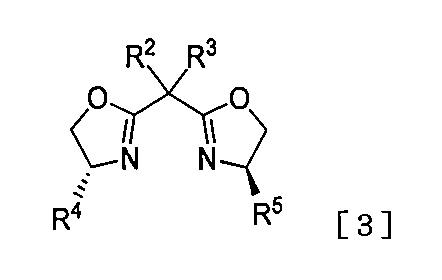
- a compound represented by the formula [5] (hereinafter also referred to as the compound [5]) or a pharmaceutically acceptable product thereof.
- optically active 2- (2-fluoro It found to be able to produce a phenyl-4-yl) propanoic acid.
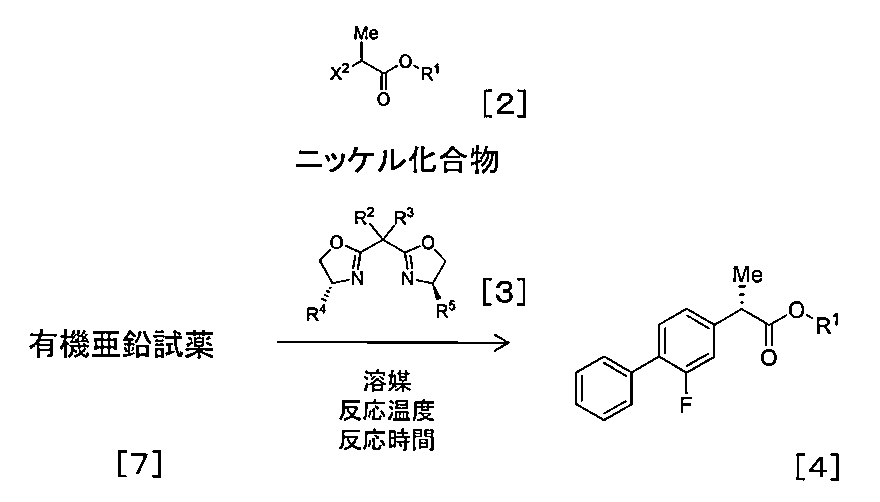
- the present inventors have found that the above production method is less in the presence of a smaller amount of the nickel compound and a smaller amount of the optically active compound represented by the formula [3] than the conventional method. It has been found that a compound represented by the formula [4] can be produced by reacting the compound represented by the formula [2] with the prepared organometallic reagent.
- the inventors of the present invention have a higher reaction temperature than the conventional method in the presence of a catalytic amount of the nickel compound and a catalytic amount of the optically active compound represented by the formula [3]. It has been found that a compound represented by the formula [4] can be produced by reacting the compound represented by the formula [2] with the prepared organometallic reagent.
- the method for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid or a pharmaceutically acceptable salt thereof includes the following steps (a) to (c): Manufacturing method to: (a) preparing an organometallic reagent comprising reacting the compound represented by the formula [1] with magnesium;
- step (b) an organometallic reagent prepared in step (a) in the presence of a catalytic amount of a nickel compound and a catalytic amount of an optically active compound represented by formula [3]; Reacting to obtain a compound represented by the formula [4], and
- X 1 represents a halogen atom
- X 2 represents a halogen atom
- R 1 may be substituted with 1 to 2 groups selected from tert-butyldiphenylsilyl, C 1-6 alkyl, C 2-6 alkenyl, C 3-8 cycloalkyl, phenyl, or substituent group A1.
- R 2 and R 3 independently represent C 1-6 alkyl, Or R 2 , R 3 and the carbon atom adjacent to the substituent may be taken together to form a C 3-6 cycloalkane;
- R 4 and R 5 independently represent C 1-6 alkyl, benzyl, phenethyl, or phenyl optionally substituted by 1 to 2 groups selected from the substituent group A2,
- the substituent group A1 represents a group consisting of C 1-6 alkyl and phenyl
- the substituent group A2 represents a group consisting of a halogen atom, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, and phenyl.
- step (a) after reacting the compound represented by the formula [1] with magnesium, further reacting with zinc chloride or zinc bromide to prepare an organometallic reagent, described in (1) Manufacturing method.
- step (b) The production method according to (1) or (2), wherein R 1 is C 1-6 alkyl for the compound represented by formula [2] in step (b).
- step (b) The production method according to (3), wherein R 1 is tert-butyl for the compound represented by formula [2] in step (b).
- the catalyst amount of the nickel compound used is 0.03 to 1.00 mol% with respect to the compound represented by the formula [2], and is represented by the formula [3] used.
- the catalyst amount of the nickel compound used is 0.50 to 1.00 mol% with respect to the compound represented by the formula [2], and is represented by the formula [3] used.
- the production method according to (1), (3) or (4), wherein the catalyst amount of the optically active compound is 0.60 to 1.20 mol%.
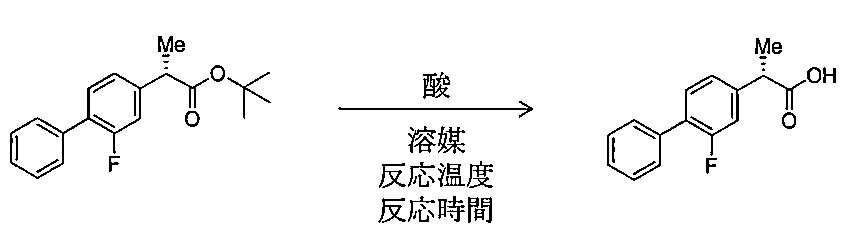
- step (c) In any one of (1) to (8), in the step (c), the step of converting the compound represented by the formula [4] into the compound represented by the formula [5] is conversion under acidic conditions.
- the manufacturing method of crab. (10)
- R 2 and R 3 are both methyl,
- step (a) the molar ratio of the organomagnesium reagent prepared by reacting the compound represented by the formula [1] with magnesium and the zinc chloride or zinc bromide to be further reacted is 2: 1 to 3
- the amount of the nickel compound used is 0.10 mol% or less with respect to the compound represented by the formula [2], and the optical activity represented by the formula [3] used
- step (b) the amount of catalyst of the nickel compound used is 1.00 mol% or less with respect to the compound represented by formula [2], and the optical activity represented by formula [3] used The production method according to (1), wherein the catalyst amount of the compound is 1.20 mol% or less.
- step (c) the acid used for conversion to the compound represented by formula [5] under acidic conditions is hydrochloric acid, sulfuric acid, formic acid, trifluoroacetic acid, methanesulfonic acid, and p-toluene.
- step (b) an organometallic reagent prepared in step (a) in the presence of a catalytic amount of a nickel compound and a catalytic amount of an optically active compound represented by formula [3]; Reacting to obtain a compound represented by the formula [4], and
- a process for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid comprising: here,
- X 1 represents a halogen atom
- X 2 represents a halogen atom
- R 1 may be substituted with 1 to 2 groups selected from tert-butyldiphenylsilyl, C 1-6 alkyl, C 2-6 alkenyl, C 3-8 cycloalkyl, phenyl, or substituent group A1.
- R 2 and R 3 independently represent C 1-6 alkyl, Or R 2 , R 3 and the carbon atom adjacent to the substituent may be taken together to form a C 3-6 cycloalkane;
- R 4 and R 5 independently represent C 1-6 alkyl, benzyl, phenethyl, or phenyl optionally substituted by 1 to 2 groups selected from the substituent group A2,
- the substituent group A1 represents a group consisting of C 1-6 alkyl and phenyl
- Substituent group A2 represents a group consisting of a halogen atom, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, and phenyl.
- step (a) after reacting the compound represented by the formula [1] with magnesium, further reacting with zinc chloride or zinc bromide, the production method according to (17), (19) In the step (a), after reacting the compound represented by the formula [1] with magnesium, zinc chloride or zinc bromide is not reacted, The production method according to (17), (20) The production method according to (17), wherein R 1 is C 1-6 alkyl for the compound represented by formula [2] in step (b), (21) The optical activity represented by the formula [3] used, wherein the catalyst amount of the nickel compound used is 1.00 mol% or less with respect to the compound represented by the formula [2] in the step (b).
- the production method according to (17), wherein the catalyst amount of the compound is 1.20 mol% or less, (22) The production method according to (21), wherein the reaction temperature in step (b) is 0 ° C. or higher, (23) The production method according to (17), wherein in step (c), the step of converting the compound represented by formula [4] into the compound represented by formula [5] is conversion under acidic conditions, (24) About the optically active compound represented by the formula [3] in the step (b), R 2 and R 3 are both methyl, The production method according to (17), wherein R 4 and R 5 are both phenyl; (25) For the compound represented by formula [2] in step (b), The production method according to (20), wherein R 1 is tert-butyl.
- step (a) the molar ratio of the organomagnesium reagent prepared by reacting the compound represented by the formula [1] with magnesium and the zinc chloride or zinc bromide to be further reacted is 2: 1 to 3 1:
- step (b) the amount of catalyst of the nickel compound used is 0.10 mol% or less relative to the compound represented by formula [2], and the optical activity represented by formula [3] used
- the catalyst amount of the nickel compound to be used is 0.03 to 0.10 mol% with respect to the compound represented by the formula [2], and is represented by the formula [3] to be used.
- step (27) wherein the catalytic amount of the optically active compound is 0.036 to 0.12 mol%
- step (b) the catalytic amount of the nickel compound used is 1.00 mol% or less relative to the compound represented by formula [2], and the optical activity represented by formula [3] used
- step (b) the catalyst amount of the nickel compound used is 0.50 to 1.00 mol% with respect to the compound represented by the formula [2], and is represented by the formula [3] used.
- the acid used for conversion to the compound represented by formula [5] under acidic conditions is hydrochloric acid, sulfuric acid, formic acid, trifluoroacetic acid, methanesulfonic acid, and p-toluene.
- 2- (2-fluorobiphenyl-4-yl) propanoic acid can be produced with high optical purity.
- the use of a smaller amount of a catalytic amount of a nickel compound and a smaller amount of a catalytic amount of the optically active compound represented by the formula [3], the formula [4] The compound represented by this can be manufactured.
- the compound represented by the formula [4] can be produced at a higher reaction temperature than in the conventional method.
- the present invention can provide a useful method as an industrial process for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid, which is a drug having anti-inflammatory and analgesic effects.
- optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid that can be produced according to the present invention has the following structure.
- the “optically active compound” represented by the formulas [4] and [5] indicates either the (S) isomer or the (R) isomer.
- n is normal, “i” is iso, “s” and “sec” are secondary, “t” and “tert” are tertiary, “c” is cyclo, “o” "” Means ortho, “m” means meta, and “p” means para.
- halogen atom is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- C 1-6 alkyl refers to a straight-chain or branched alkyl having 1 to 6 carbon atoms. For example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, 2-methylbutyl, tert-amyl, n-hexyl, isohexyl, etc. It is done.
- HaloC 1-6 alkyl refers to a linear or branched alkyl having 1 to 6 carbon atoms, substituted with a halogen atom.
- the preferred number of substitution of the halogen atom is 1 to 5, and the preferred halogen atom is a fluorine atom.
- C 2-6 alkenyl refers to a straight-chain or branched alkenyl having 2 to 6 carbon atoms.
- C 1-6 alkoxy refers to straight or branched alkoxy having 1 to 6 carbon atoms. For example, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, tert-butoxy, n-pentyloxy, isopentyloxy, neopentyloxy, 2-methylbutoxy, n-hexyloxy, And isohexyloxy.
- Halo C 1-6 alkoxy refers to a straight or branched alkoxy having 1 to 6 carbon atoms, substituted with a halogen atom.
- the preferred number of substitution of the halogen atom is 1 to 5, and the preferred halogen atom is a fluorine atom.
- C 3-6 cycloalkane means a cyclic alkane having 3 to 6 carbon atoms, and is cyclopropane, cyclobutane, cyclopentane or cyclohexane.
- C 3-8 cycloalkyl means a cyclic alkyl having 3 to 8 carbon atoms, and is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl.
- organometallic reagent is a reagent prepared by reacting an organic compound with a metal reagent.
- the organometallic reagent may be a reagent prepared by reacting the obtained organometallic reagent with another metal reagent.
- an organomagnesium reagent prepared by reacting an organic compound with magnesium hereinafter sometimes referred to as a Grignard reagent
- an organozinc-containing reagent prepared by further reacting a “zinc compound” (hereinafter referred to as “ "Organic zinc reagent”).
- the “zinc compound” may be anything that can be prepared as an organic zinc reagent when reacted with an organic magnesium reagent, and examples thereof include zinc halide. Specific examples include zinc chloride, zinc bromide, or zinc iodide.
- nickel compound means a halogen-containing nickel compound or a nickel complex.
- ether solvent examples include tetrahydrofuran, 1,4-dioxane, methyltetrahydrofuran, diethyl ether, dimethoxyethane, and cyclopentylmethyl ether.
- the “benzene solvent” includes, for example, benzene, toluene and xylene.
- hydrocarbon solvent examples include toluene, xylene, benzene, heptane and hexane.
- alcohol solvent examples include methanol, ethanol, 1-propanol, 2-propanol, and tert-butyl alcohol.
- ester solvent examples include ethyl acetate.
- “Pharmaceutically acceptable salt” means, for example, glycine salt, lysine salt, arginine salt, histidine salt, ornithine salt, glutamate, aspartate, amino acid salt, lithium salt, sodium salt, potassium salt , Inorganic salts such as calcium salts and magnesium salts, or salts with organic bases such as ammonium salts, triethylamine salts, diisopropylamine salts and cyclohexylamine salts.
- the salt includes hydrated salt.
- optically active compound represented by the formula [3] will be described below.
- a compound represented by the formula [2] is produced in the presence of a catalytic amount of a nickel compound using a catalytic amount of an optically active compound represented by the formula [3].
- the compound represented by the formula [4] that is optically active can be produced by reacting with the organometallic reagent prepared in (1).
- optically active compound represented by the formula [3] can be obtained commercially or by a known method described in the literature (for example, J. Am. Chem. Soc., 2014, 136, 17662.).
- (R) and (S) in the present specification indicate the configuration of a chiral molecule having a central chirality.
- (Z) and (E) are those in which the higher rank rule appears on the opposite side among the groups bonded to the atoms that form a double bond in the planar portion of the molecule connected by the double bond.
- the stereochemistry is displayed as (E) when it is present and (Z) when it is on the same side.
- Catalyst refers to a substance that does not change itself but accelerates or slows down the reaction of other substances.
- the amount of catalyst used may be equal to or less than the amount of other substances (substrates) to be reacted.
- the “catalytic amount” means, for example, when the compound represented by the formula [2] is reacted with the organomagnesium reagent, the amount of nickel compound used relative to the compound represented by the formula [2].
- the amount of the optically active compound represented by the formula [3] used is 12 mol% or less, preferably It means 1.20 mol% or less, more preferably in the range of 0.60 to 1.20 mol%.
- the usage-amount of a nickel compound is 10 mol% or less with respect to the compound represented by Formula [2], Preferably it is 1.00 mol%.
- the optical system represented by the formula [3] is more preferably in the range of 0.03 to 1.00 mol%, more preferably 0.10 mol% or less, and particularly preferably in the range of 0.03 to 0.10 mol%.
- the amount of active compound used is 12 mol% or less, preferably 0.036 to 1.20 mol%, more preferably 0.12 mol% or less, particularly preferably 0.036 to 0.12 mol%. Means.
- the upper limit of the amount of compound [3] used is not particularly limited, but the reaction can proceed well if it is used in excess relative to the nickel compound.
- the amount thereof is 12 mol% or less with respect to the compound [2], and it is more preferable when the compound represented by the formula [2] is reacted with the organomagnesium reagent. Is 1.20 mol% or less, more preferably 0.60 to 1.20 mol%, and more preferably 0.12 mol% or less when the compound represented by the formula [2] is reacted with the organozinc reagent. More preferably, it is 0.036 to 0.12 mol%.
- the upper limit of the amount of nickel compound is not particularly limited.
- the amount thereof is 10 mol% or less relative to the compound [2]. More preferably, the compound represented by the formula [2] is reacted with an organomagnesium reagent. 0.000 mol% or less, more preferably 0.50 to 1.00 mol%, and more preferably 0.10 mol% or less when the compound represented by the formula [2] is reacted with an organozinc reagent. More preferably, it is 0.03 to 0.10 mol%.
- Preferred embodiments of the present invention are as follows.
- Preferred X 1 is a chlorine atom, a bromine atom, or an iodine atom, and more preferred X 1 is a bromine atom.
- Preferred X 2 is a chlorine atom, bromine atom or iodine atom, and more preferred X 2 is a chlorine atom or bromine atom.
- R 1 may be substituted with 1 to 2 groups selected from tert-butyldiphenylsilyl, C 1-6 alkyl, C 2-6 alkenyl, C 3-8 cycloalkyl, phenyl, or substituent group A1. More preferred R 1 is benzyl optionally substituted by 1 to 2 groups selected from tert-butyldiphenylsilyl, C 1-6 alkyl, C 3-8 cycloalkyl, or substituent group A1.
- R 1 is tert-butyldiphenylsilyl, tert-butyl, neopentyl, tert-amyl, cyclohexyl, 1-methyl-1-phenylethyl, or benzhydryl, and particularly preferred R 1 is tert-butyl. It is.
- Preferred R 2 is C 1-6 alkyl, more preferred R 2 is methyl or ethyl, and further preferred R 2 is methyl.
- Preferred R 3 is C 1-6 alkyl, more preferred R 3 is methyl or ethyl, and further preferred R 3 is methyl.
- a preferred C 3-6 cycloalkane formed by combining R 2 , R 3 and the carbon atom adjacent to the substituent is cyclopropane.
- Preferred R 4 is C 1-6 alkyl or phenyl optionally substituted by 1 to 2 groups selected from the substituent group A2, and more preferred R 4 is phenyl.
- Preferred R 5 is C 1-6 alkyl or phenyl optionally substituted with 1 to 2 groups selected from the substituent group A2, and more preferred R 5 is phenyl.
- a preferred zinc compound that is further reacted with an organomagnesium reagent (Grignard reagent) obtained by reacting the compound represented by the formula [1] with magnesium is zinc chloride or zinc bromide.
- Preferred nickel compounds are nickel chloride (II), nickel iodide (II), nickel chloride (II) / DME complex, acetylacetone nickel (II), or bis (triphenylphosphine) nickel (II).
- one preferred embodiment for producing the compound represented by the formula [4] is a production method shown in the following scheme 2.
- X 1 is a bromine atom
- X 2 is a chlorine atom or a bromine atom
- R 1 is tert-butyl
- R 2 and R 3 are both methyl, This is the case when R 4 and R 5 are both phenyl.
- the zinc compound represents zinc chloride or zinc bromide.
- X 1 is a bromine atom
- X 2 is a bromine atom
- R 1 is tert-butyldiphenylsilyl, tert-butyl, neopentyl, tert-amyl, cyclohexyl, 1-methyl-1-phenylethyl, or benzhydryl
- R 2 and R 3 are both methyl or ethyl, Or C 2-6 cycloalkane formed by the combination of R 2 , R 3 and the carbon atom adjacent to the substituent is cyclopropane
- R 4 and R 5 are both phenyl, This is the case where the zinc compound is zinc bromide.
- X 1 is a bromine atom
- X 2 is a bromine atom
- R 1 is tert-butyl, tert-amyl, 1-methyl-1-phenylethyl, or benzhydryl
- R 2 and R 3 are both methyl or ethyl, Or C 2-6 cycloalkane formed by the combination of R 2 , R 3 and the carbon atom adjacent to the substituent is cyclopropane
- R 4 and R 5 are both phenyl, This is the case where the zinc compound is zinc bromide.
- one preferred embodiment is conversion under acidic conditions.
- a preferable acid used for the conversion to the compound represented by the formula [5] is an acid selected from the group consisting of hydrochloric acid, sulfuric acid, formic acid, trifluoroacetic acid, methanesulfonic acid, and p-toluenesulfonic acid.
- a more preferred acid is formic acid.
- Step (a) Preparation Method of Organomagnesium Reagent [6]
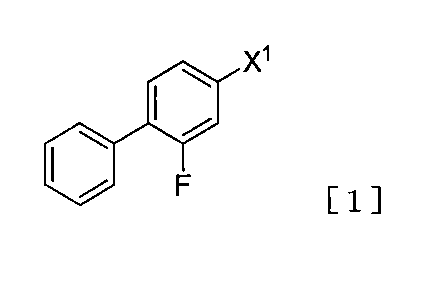
- compound [1] wherein X 1 is a halogen atom can be used as compound [1].
- the use of compound [1] wherein X 1 is a chlorine atom, bromine atom or iodine atom is preferred, and the use of compound [1] wherein X 1 is a bromine atom is more preferred.
- the amount of magnesium used may theoretically be equimolar with respect to compound [1], but can be about 1.0 to 1.3 equivalents.
- solvents are ether solvents, more preferred solvents are tetrahydrofuran, 1,4-dioxane, methyltetrahydrofuran and diethyl ether, and a more preferred solvent is tetrahydrofuran.
- the reaction temperature is preferably adjusted to be 0 to 60 ° C., and may be cooled or heated if necessary.
- additives such as iodine and 1,2-dibromoethane may be used for the preparation of the Grignard reagent.
- the reaction time may be determined while confirming the remaining amount of the compound [1] as a raw material, but can usually be 0.5 hours or more and 10 hours or less after the addition of the compound [1]. Preferably it is 2 to 3 hours.
- Step (b) a method for producing a compound represented by the formula [4] from an organomagnesium reagent [6]
- a compound in which X 2 is a halogen atom can be used as the compound [2].
- the use of the compound [2] wherein X 2 is a chlorine atom or a bromine atom is preferred, and the use of the compound [2] wherein X 2 is a bromine atom is more preferred.
- R 1 is to use a compound C 1-6 alkyl [2].
- the use of compound [2] wherein R 1 is tert-butyl is preferred.
- the Grignard reagent to be reacted with the compound [2] is used so that the amount of the starting compound [1] used is preferably 1.0 to 1.5 equivalents with respect to the compound [2].
- compound [3] wherein R 2 and R 3 are independently C 1-6 alkyl can be used as compound [3].
- R 2 and R 3 are both methyl.
- R 4 and R 5 are each independently phenyl optionally substituted by 1 to 2 groups selected from C 1-6 alkyl, benzyl, phenethyl, or substituent group A2.
- Compound [3] can be used.
- the upper limit of the amount of compound [3] used is not particularly limited, but the reaction can proceed well if it is used in excess relative to the nickel compound.
- the amount thereof is 12 mol% or less, more preferably 1.20 mol% or less, further preferably 0.60 to 1.20 mol, based on the compound [2]. %.
- the upper limit of the amount of the nickel compound is not particularly limited, but the reaction can proceed favorably if the amount is 0.5 mol% or more based on the compound [2].
- the amount thereof is 10 mol% or less, more preferably 1.00 mol% or less, further preferably 0.50 to 1.00 mol%, relative to compound [2]. is there.
- Nickel compounds to be used are generally known as nickel chloride (II) / DME complex, nickel chloride (II), bis (triphenylphosphine) nickel (II) chloride, nickel (II) iodide, nickel acetylacetone (II), etc.
- Nickel chloride (II) / DME complex and acetylacetone nickel (II) are more preferable, and acetylacetone nickel (II) is more preferable.
- solvent that can be used in this step, it is sufficient that the solvent itself does not inhibit the progress of the reaction.
- Preferred solvents are ether and benzene solvents, and a more preferred solvent is tetrahydrofuran.
- the solvent may be used alone or as a mixture of a plurality of solvents.
- the amount used is usually affected by the properties of the compound used in the reaction and the reagent, and can be arbitrarily set according to the type of the compound.
- the concentration of compound [2] is preferably 0.05 to 1M, more preferably 0.1 to 0.5M.
- the reaction can be carried out at any temperature from ⁇ 78 ° C. to the boiling point of the reaction solvent, preferably from ⁇ 20 ° C. to room temperature, more preferably from ⁇ 20 ° C. to 0 ° C., still more preferably 0 ° C.
- the reaction time may be determined while confirming the remaining amount of the compound [2], but is usually 0.5 hours or more and 24 hours or less after the addition of the organomagnesium reagent [6].
- the compound [4] is heated and dissolved in a predetermined organic solvent, and then allowed to cool to precipitate crystals.
- the precipitated crystals are separated from the solvent by filtration, centrifugation, and the like, and then dried. Can be obtained.
- the recrystallization may be repeated twice or more, but is usually performed only once.
- the cooling time is not particularly limited, but is usually 10 minutes to 24 hours, preferably 1 to 5 hours.
- the solvent that can be used for recrystallization of the compound [4] is an alcohol solvent and water.
- Preferred solvents are methanol, ethanol, 2-propanol, and water, and more preferred solvents are ethanol and water.
- a seed crystal of the compound [4] can be used as necessary.
- the seed crystal can be obtained by a method well known to those skilled in the art, such as rubbing the wall of a container containing a solution for crystallization with a spatula.
- the crystallization temperature is in the range of ⁇ 20 ° C. to 60 ° C. unless otherwise specified.
- compound [1] wherein X 1 is a halogen atom can be used as compound [1].
- the use of compound [1] wherein X 1 is a chlorine atom, bromine atom or iodine atom is preferred, and the use of compound [1] wherein X 1 is a bromine atom is more preferred.
- the amount of magnesium used may theoretically be equimolar, but can be about 1.0 to 1.3 equivalents relative to compound [1].
- zinc compound zinc bromide and zinc chloride can be used, and zinc bromide is preferred.
- the amount of the zinc compound used can be 0.3 equivalent to 1.0 equivalent relative to compound [1], preferably 0.3 equivalent to 0.5 equivalent, more preferably Is the use of 0.5 equivalents.
- solvents are ether solvents, more preferred solvents are tetrahydrofuran, 1,4-dioxane, 2-methyltetrahydrofuran and diethyl ether, and a more preferred solvent is tetrahydrofuran.
- the solvent may be used alone or as a mixture of a plurality of solvents.
- the amount used is usually affected by the properties of the compound used in the reaction and the reagent, and can be arbitrarily set according to the type of the compound.
- the concentration of compound [2] is preferably 0.05 to 1M, more preferably 0.1 to 0.5M.
- the reaction temperature is preferably adjusted to be 0 to 60 ° C., and may be cooled or heated if necessary.
- additives such as iodine and 1,2-dibromoethane may be used for the preparation of the Grignard reagent.
- the reaction time may be determined while confirming the remaining amount of the compound [1] as a raw material for the preparation of the Grignard reagent, but is usually 0.5 hours or more and 10 hours or less after the addition of the compound [1]. be able to. Preferably it is 2 to 3 hours.
- the preparation of the organozinc reagent 10 minutes to 1 hour is preferable after adding zinc bromide or zinc chloride to the Grignard reagent. 4).
- Step (b) a process for producing a compound represented by the formula [4] from an organozinc reagent [7]
- a compound in which X 2 is a halogen atom can be used as the compound [2].
- X 2 is a bromine atom.
- R 1 is substituted with 1 to 2 groups selected from tert-butyldiphenylsilyl, C 1-6 alkyl, C 2-6 alkenyl, C 3-8 cycloalkyl, phenyl, or substituent group A1.
- Compound [2] which is also a good benzyl, can be used.
- R 1 is tert-butyldiphenylsilyl, C 1-6 alkyl, C 3-8 cycloalkyl, or benzyl optionally substituted by 1 to 2 groups selected from the substituent group A1
- R 1 is tert-butyldiphenylsilyl, tert-butyl, neopentyl, tert-amyl, cyclohexyl, 1-methyl-1-phenylethyl, or benzhydryl is preferred. More preferred is the use of compound [2] wherein R 1 is tert-butyl.
- the organozinc reagent to be reacted with the compound [2] is used so that the amount of the raw material compound [1] used is preferably 1.0 to 1.5 equivalents with respect to the compound [2].
- compound [3] wherein R 2 and R 3 are independently C 1-6 alkyl can be used as compound [3].
- R 2 and R 3 are both methyl or ethyl
- a compound [3] in which R 2 , R 3 and a carbon atom adjacent to the substituent together form a C 3-6 cycloalkane can also be used.
- R 2 , R 3 and the carbon atom adjacent to the substituent are combined to form cyclopropane.
- R 4 and R 5 are each independently phenyl optionally substituted by 1 to 2 groups selected from C 1-6 alkyl, benzyl, phenethyl, or substituent group A2.
- Compound [3] can be used.
- the upper limit of the amount of compound [3] used is not particularly limited, but the reaction can proceed well if it is used in excess relative to the nickel compound.
- the amount thereof is 12 mol% or less, preferably in the range of 0.036 to 1.20 mol%, more preferably 0.12 mol%, relative to compound [2]. Or less, more preferably 0.036 to 0.12 mol%.
- the upper limit of the amount of nickel compound is not particularly limited.
- the amount thereof is 10 mol% or less, preferably in the range of 0.03 to 1.00 mol%, more preferably 0.10 mol% or less, relative to compound [2]. More preferably 0.03 to 0.10 mol%.
- Nickel compounds used include nickel (II) chloride / DME complexes, nickel (II) chloride, bis (triphenylphosphine) nickel (II), nickel iodide (II), nickel acetylacetone (II), and other known nickels.
- a catalyst is mentioned.
- solvents are ether, benzene, and ester solvents, more preferred solvents are tetrahydrofuran, 1,4-dioxane, 2-methyltetrahydrofuran, toluene, and ethyl acetate, and a more preferred solvent is tetrahydrofuran. .
- the solvent may be used alone or as a mixture of a plurality of solvents.
- the amount used is usually affected by the properties of the compound used in the reaction and the reagent, and can be arbitrarily set according to the type of the compound.
- the concentration of compound [2] is preferably 0.05 to 1M, more preferably 0.1 to 0.5M.
- an additive When an additive is used, it may be used alone or a plurality of additives may be mixed and used.
- the amount of the additive used when used can be arbitrarily set according to the type of compound used in the reaction, and is 0.1 to 50 equivalents, preferably 0.5 to 20 equivalents, relative to compound [1]. More preferably, it is 1 to 5 equivalents.
- the reaction temperature can be any of ⁇ 78 ° C. to the boiling point of the reaction solvent, preferably ⁇ 20 to 25 ° C., more preferably 0 to 25 ° C.
- the reaction time may be determined while confirming the remaining amount of the raw material compound, but can usually be 0.5 hours or more and 24 hours or less after the addition of the organozinc reagent [7].
- Preferred R 1 is as described above, and more preferred R 1 is tert-butyl.
- the conversion to the compound [5] can be performed under acidic conditions.
- hydrochloric acid sulfuric acid, formic acid, acetic acid, trifluoroacetic acid, phosphoric acid, methanesulfonic acid, or p-toluenesulfonic acid
- hydrochloric acid, sulfuric acid, formic acid, trifluoroacetic acid, methanesulfonic acid, or p-toluenesulfonic acid is preferred, and use of formic acid is more preferred.
- solvent used in this step it is sufficient if the reaction is not inhibited.
- Preferred solvents are heptane, toluene, acetic acid, 1,4-dioxane.
- the reaction temperature can be any from room temperature to the boiling point of the reaction solvent, but it is preferably performed at room temperature to 80 ° C.
- the reaction time may be determined while confirming the remaining amount of the compound [4] as a raw material, but can usually be 0.5 hours or more and 40 hours or less.
- the compound [5] is dissolved by heating in a predetermined organic solvent, and then allowed to cool to precipitate crystals.
- the precipitated crystals are separated from the solvent by filtration, centrifugation, and the like, and then dried. Can be obtained.
- the recrystallization may be repeated twice or more, but is usually performed only once.
- the cooling time is not particularly limited, but is usually 10 minutes to 24 hours, preferably 1 to 5 hours.
- Solvents that can be used for recrystallization of compound [5] are benzene-based solvents, hydrocarbon-based solvents, alcohol-based solvents, and water.
- Preferred solvents are toluene, heptane, methanol, ethanol, 2-propanol, Water and more preferred solvents are toluene and heptane.
- the solvent used for the recrystallization may be used alone or as a mixture of a plurality of solvents.
- a seed crystal of the compound [5] can be used as necessary.
- the seed crystal can be obtained by a method well known to those skilled in the art, such as rubbing the wall of a container containing a solution for crystallization with a spatula.
- the seed crystal of compound [5] can also be obtained, for example, by the method described in Example 9-1 described later.
- the crystallization temperature is in the range of ⁇ 20 ° C. to 80 ° C. unless otherwise specified.
- the compound obtained by the above production method can be obtained by a known means, for example, purification operations such as recrystallization and various chromatography.
- phase separator in the following examples, ISOLUTE (registered trademark) Phase Separator manufactured by Biotage Corporation was used.
- Nuclear magnetic resonance (NMR) spectra were measured at room temperature at 600 MHz (JNM-ECA600, JEOL) and 400 MHz (AVANCEIII-HD400, BRUKER).
- the chemical shift value in the present specification is shown as a parts per million ( ⁇ ) value with respect to the internal standard substance (tetramethylsilane).
- Optical purity was measured by high-performance liquid chromatography (HPLC) using Agilent 1100 manufactured by Agilent Technologies and supercritical fluid chromatography (SFC) using Acquity UPC2 manufactured by Waters.
- the optical rotation was measured with an AUTOPOLV polarimeter manufactured by Rudolf Research Analytical.
- room temperature refers to 20 to 30 ° C.
- Ice-cooled refers to 0 to 5 ° C.
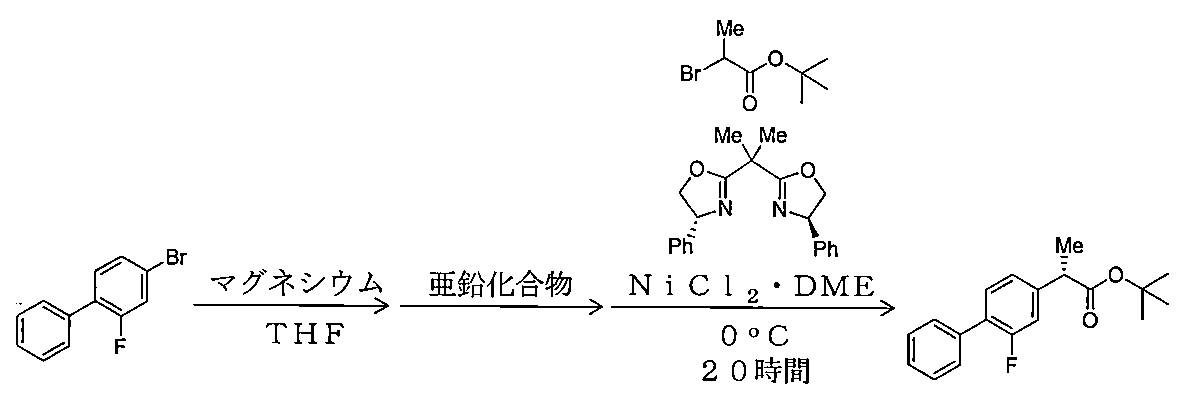
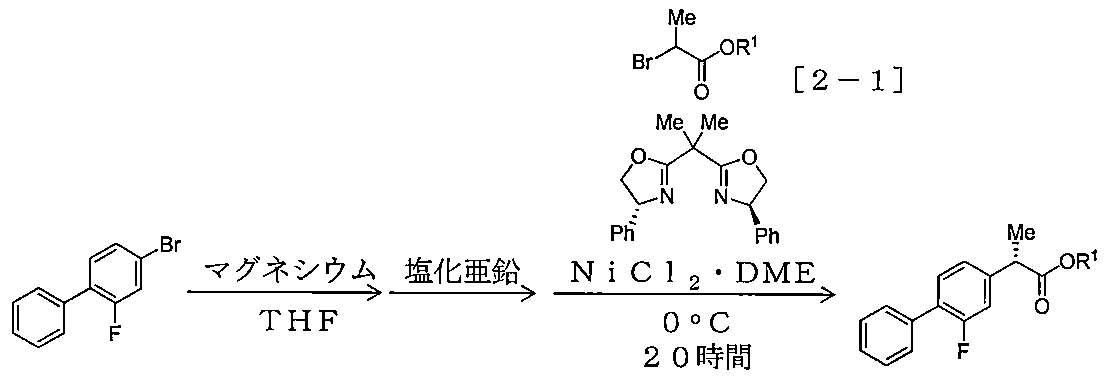
- Example 1-1 (S) Process for producing -2- (2-fluorobiphenyl-4-yl) propanoic acid tert-butyl ester
- nickel chloride (II) / DME complex 11 mg, 0.0501 mmol
- (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) 20 mg, 0.0598 mmol
- THF 5 mL
- nickel chloride (II) chloride / DME complex 0.0031 mmol
- nickel (II) chloride / DME complex 0.0037 mmol of (R, R) -2,2′-isopropylidenebis) (4-phenyl-2-oxazoline
- 0.1 mol% nickel (II) chloride / DME complex 0.12 mol% (R, R) R-2,2 with respect to 2-bromopropionic acid tert-butyl ester '-Isopropylidenebis (4-phenyl-2-oxazoline)) and 2-bromopropionic acid tert-butyl ester (640 mg, 3 0.06 m
- Example 2-2 2-bromopropionic acid tert-butyl ester (641 mg, 3.07 mmol), nickel (II) chloride.DME complex (0.031 mmol, 1.0 mol% with respect to 2-bromopropionic acid tert-butyl ester), (R , R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (0.037 mmol, 1.2 mol% relative to 2-bromopropionic acid tert-butyl ester)
- the reaction was carried out under the same conditions as in Example 1-1, except that metal magnesium pieces (101 mg, 4.14 mmol) were used instead of 116 mg, 4.78 mmol) under the same conditions as in Example 1-1.
- Table 1-1 shows the compounds used and their amounts used, yield, and optical purity.
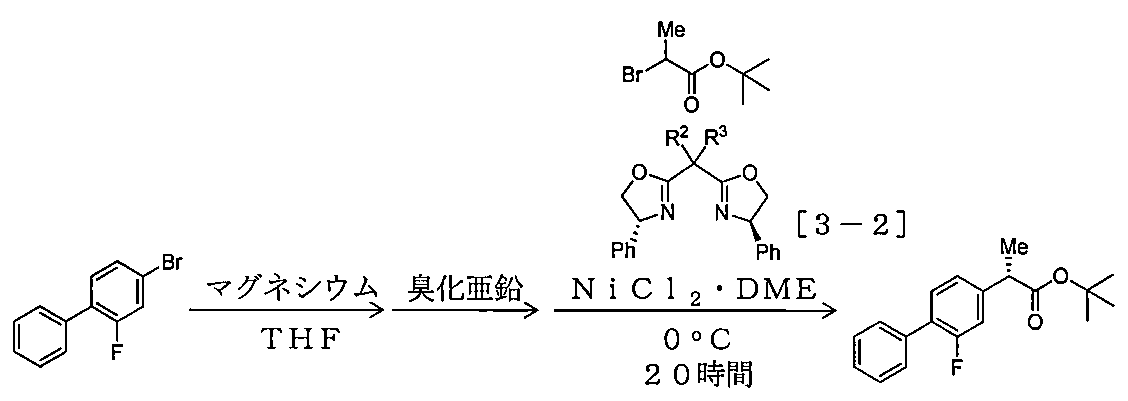
- Example 1- Using 4-bromo-2-fluorobiphenyl (1.007 g, 3.99 mmol), 2-bromopropionic acid tert-butyl ester (641 mg, 3.07 mmol), zinc bromide (1.99 mmol), Example 1- Under the same conditions as in 1, the reaction temperature was changed and the reaction was carried out. Table 2-1 shows the compounds used and their amounts used, yield, and optical purity.
- Example 3-3 and Example 3-4 Using 4-bromo-2-fluorobiphenyl (1.007 g, 3.99 mmol), 2-bromopropionic acid tert-butyl ester (641 mg, 3.07 mmol), zinc bromide (1.99 mmol), Example 1- Under the same conditions as in Example 1, the nickel (II) chloride / DME complex and (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (in Table 2-1, the compound [3-1 ] was used (respectively expressed in mol% with respect to 2-bromopropionic acid t-butyl ester), and the reaction was carried out. Table 2-1 shows the compounds used and their amounts used, yield, and optical purity.
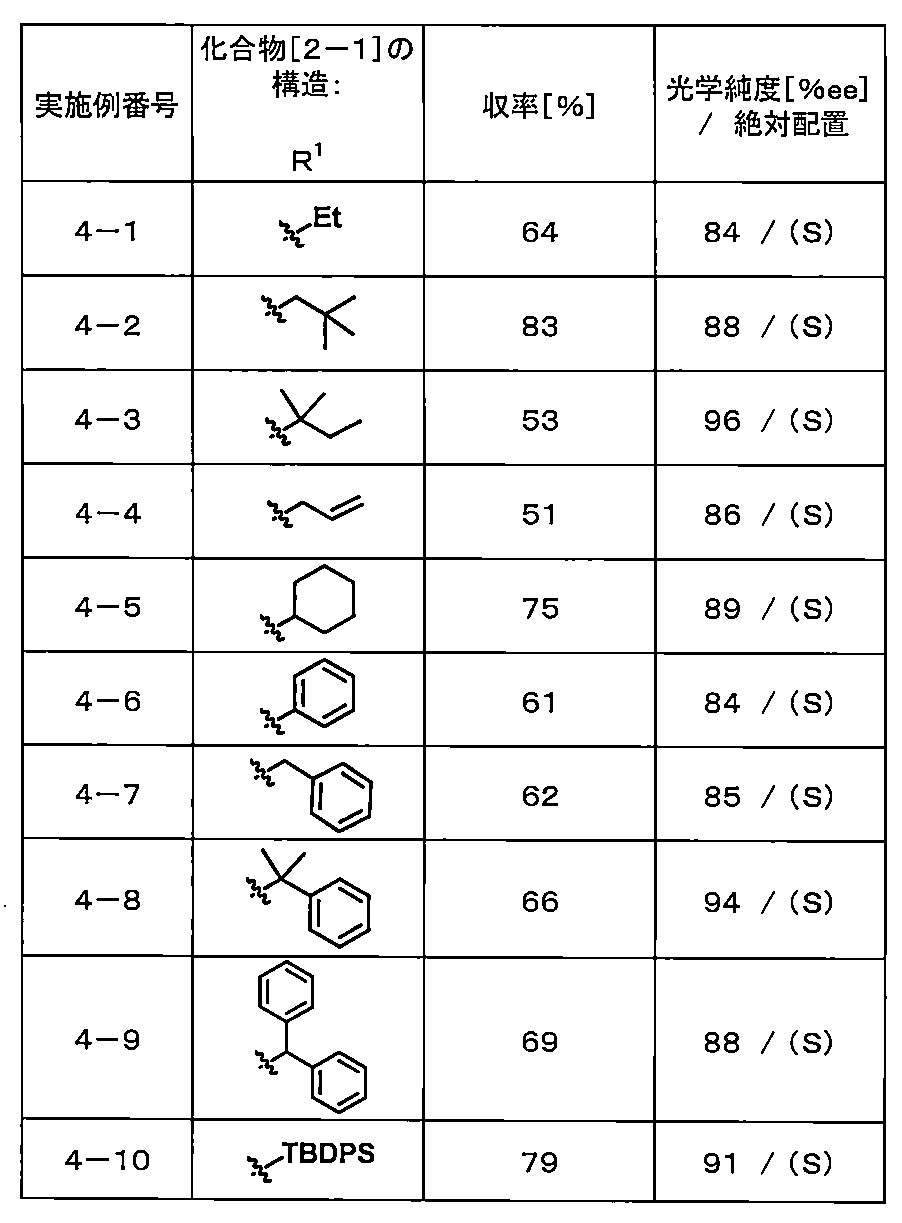
- the obtained compound was obtained by the method of Example 9-6 described later for Examples 4-1 to 4-6, and for Examples 4-7 to 4-10. Each was induced to 2- (2-fluorobiphenyl-4-yl) propanoic acid by the method described below, and then determined to be S-form by the result of HPLC analysis using the chiral column described in Example 1-1. .
- 2- (2-Fluorobiphenyl-4-yl) propanoic acid 66 mg
- Example 4-9 The compound (57 mg) obtained in Example 4-9 was dissolved in chloroform (0.3 mL), TFA (0.106 mL) was added, and the reaction solution was stirred at room temperature for 3 hours and at 60 ° C. for 3 hours. Water was added to the reaction mixture, and the mixture was extracted twice with chloroform. The organic layer was separated with a phase separator, and the solvent was distilled off under reduced pressure.
- Example 4-10 The compound (30 mg) obtained in Example 4-10 was dissolved in tetrahydrofuran, tetra-n-butylammonium fluoride / 1M tetrahydrofuran solution (0.124 mL) was added, and the mixture was stirred at room temperature for 8 hours. Water and 1M hydrochloric acid were added to the reaction solution to adjust the pH to 3 to 4, followed by extraction with chloroform twice. After separating the organic layer with a phase separator, the solvent was distilled off under reduced pressure to obtain 2- (2-fluorobiphenyl-4-yl) propanoic acid (30 mg) as a colorless solid.
- Example 1-1 Under the same conditions as in Example 1-1, 6.0 mL of an organic zinc reagent in THF (1.36 mmol, 0.90 equivalent to 2-bromopropionic acid t-butyl ester), and (R, R)- 2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (shown as compound [3-1] in Table 4-1, the amount used is mol% relative to 2-bromopropionic acid t-butyl ester, The reaction was carried out by changing the nickel chloride (II) .DME complex and the amount used (in mol% with respect to 2-bromopropionic acid t-butyl ester) as the compound to be used. . Table 4-1 shows the compounds used and their amounts used, yield, and optical purity.
- II nickel chloride
- Table 4-1 shows the compounds used and their amounts used, yield, and optical purity.
- the nickel chloride (II) / DME complex and compound [3-2] were prepared in advance in a THF solution of these compounds in the same manner as in Example 1-1, and the amount required for the reaction. Weighed and used.
- the nickel chloride (II) / DME complex and compound [3-2] were prepared in advance in a THF solution of these compounds in the same manner as in Example 1-1, and the amount required for the reaction. Weighed and used.
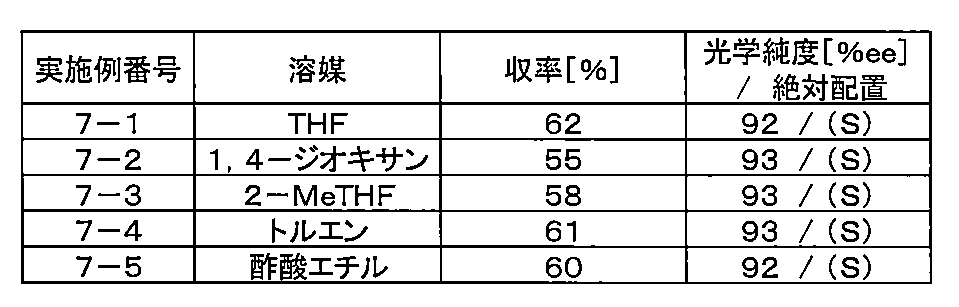
- the amount of 2-bromopropionic acid tert-butyl ester used was 209 mg (1.0 mmol), and 1.5 ml of a THF solution of an organozinc reagent (compound [7-1]) ( 0.341 mmol, 0.68 equivalent based on 2-bromopropionic acid tert-butyl ester), nickel (II) chloride / DME complex (0.0010 mmol, 0.1 mol based on 2-bromopropionic acid tert-butyl ester) %), (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (0.0012 mmol, 0.12 mol% relative to 2-bromopropionic acid tert-butyl ester)
- the solvent used was changed and the reaction was carried out. Table 6-1 shows the solvent used, yield, and optical purity.
- Compound [7-1] was prepared using 0.5 equivalent of zinc chloride with respect to the prepared organomagnesium reagent as described above.
- Example 8-1 In the above scheme, “additive” is ethyl acetate or lithium bromide.
- Example 8-2 After preparing an organozinc reagent under the same conditions as in Example 1-1, lithium bromide (1 equivalent to 2-bromopropionic acid tert-butyl ester) was added to give 2-bromopropionic acid tert-butyl ester. The coupling reaction with was carried out for 2 hours.
- Example 9-1 (S) Process for producing -2- (2-fluorobiphenyl-4-yl) propanoic acid
- Heptane (111 mL) and formic acid (111 mL) were added to (S) ⁇ ⁇ ⁇ ⁇ -2- (2-fluorobiphenyl-4-yl) propanoic acid tert-butyl ester (37.0 g, 123 mmol), and the mixture was stirred at room temperature for 20 hours.
- Toluene (74 mL) and water (111 mL) were added, and the mixture was stirred at an internal temperature of 50 ° C. for 1 hour.
- the organic layer was separated and washed with water (111 mL, 50 ° C.).
- Heptane (333 mL) was added to the organic layer, and the internal temperature was raised to 70 ° C. and dissolved.
- Example 9-1 the seed crystal used in Example 9-1 was obtained by the following method.
- Example 9-2 to Example 9-9 The reaction was carried out under the same conditions as in Example 9-1 except that the reaction temperature, reaction time, acid and solvent to be used were changed. The compounds used, yield, and optical purity are shown in Table 7-1.
- Example 10-1 A Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide under the same conditions as in Example 1-1 (1).
- 2-bromopropionic acid tert-butyl ester 10 g, 47.8 mmol
- nickel chloride (II) / DME complex 0.478 mmol, 2-bromopropionic acid tert-butyl ester 1 mol
- (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) 0.574 mmol, 1.2 mol% relative to 2-bromopropionic acid tert-butyl ester
- the coupling reaction was carried out under the same conditions as in Example 1-1 (2) at a reaction temperature of ⁇ 20 ° C. and a reaction time of 24 hours.
- Example 10-2 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide. To the prepared Grignard reagent (12 mmol), 2-bromopropionic acid tert-butyl ester (2.09 g, 10 mmol), nickel chloride (II) .DME complex (0.1 mmol, 2-bromopropionic acid tert-butyl ester 1 mol%), (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (0.12 mmol, 1.2 mol% relative to 2-bromopropionic acid tert-butyl ester). The coupling reaction was carried out under the same conditions as in Example 1-1 (2), at a reaction temperature of ⁇ 20 ° C. and for a reaction time of 24 hours.
- Example 10-3 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide.
- a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide.
- the coupling reaction was carried out under the same conditions as in Example 1-1 (2) at a reaction temperature of ⁇ 10 ° C. and a reaction time of
- Example 10-4 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide. To the prepared Grignard reagent (10 mmol), 2-bromopropionic acid tert-butyl ester (2.09 g, 10 mmol), nickel (II) chloride / DME complex (0.1 mmol, 2-bromopropionic acid tert-butyl ester) 1 mol%), (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (0.12 mmol, 1.2 mol% relative to 2-bromopropionic acid tert-butyl ester). The coupling reaction was carried out under the same conditions as in Example 1-1 (2), at a reaction temperature of ⁇ 10 ° C. and for a reaction time of 3 hours.
- Example 10-5 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide. To the prepared Grignard reagent (10 mmol), 2-bromopropionic acid tert-butyl ester (2.09 g, 10 mmol), nickel (II) chloride / DME complex (0.1 mmol, 2-bromopropionic acid tert-butyl ester) 1 mol%), (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) (0.12 mmol, 1.2 mol% relative to 2-bromopropionic acid tert-butyl ester). The coupling reaction was carried out under the same conditions as in Example 1-1 (2), with a reaction time of 3 hours.
- Example 10-6 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide.
- Example 10-7 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide.
- -Coupling reaction was performed using 2-chloropropionic acid ethyl ester (1.37 g, 10 mmol) in place of bromopropionic acid tert-butyl ester and a reaction time of 24 hours.
- Example 10-8 Under the same conditions as in Example 1-1 (1), a Grignard reagent was prepared in place of the organic zinc reagent without adding zinc bromide.
- Prepared Grignard reagent (2.0 mmol), 2-bromopropionic acid tert-butyl ester (0.42 g, 2.0 mmol), nickel (II) chloride / DME complex (0.1 mmol, tert-butyl 2-bromopropionic acid) (R, R) -2,2′-isopropylidenebis (4-phenyl-2-oxazoline) under the same conditions as in Example 1-1 (2)
- (R, R) -2,2 ′-(pentane-3,3-diyl) bis (4-phenyl-2-oxazoline) (0.12 mmol, 6 mol% relative to 2-bromopropionic acid tert-butyl ester) Was used to carry out a coupling reaction with 2-bromopropionic acid tert-
- optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid useful as a pharmaceutical can be produced with high optical purity.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description

(1)光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸又はその製薬学的に許容される塩の製造法において、下記(a)~(c)工程を含むことを特徴とする製造法:
(a)式[1]で表される化合物をマグネシウムと反応させることを含む、有機金属試薬を調製する工程、


式[5]で表される化合物又はその製薬学的に許容される塩に変換する工程

X1はハロゲン原子を表し、
X2はハロゲン原子を表し、
R1はtert-ブチルジフェニルシリル、C1-6アルキル、C2-6アルケニル、C3-8シクロアルキル、フェニル、又は置換基群A1から選ばれる1から2個の基で置換されてもよいベンジルを表し、
R2及びR3は独立してC1-6アルキルを表し、
またはR2、R3、及び該置換基に隣接する炭素原子は一緒になってC3-6シクロアルカンを形成してもよく、
R4及びR5は独立してC1-6アルキル、ベンジル、フェネチル、又は置換基群A2から選ばれる1から2個の基で置換されてもよいフェニルを表し、
ここで、置換基群A1は、C1-6アルキル及びフェニルからなる群を表し、
置換基群A2は、ハロゲン原子、C1-6アルキル、ハロC1-6アルキル、C1-6アルコキシ、ハロC1-6アルコキシ、及びフェニルからなる群
を表す。
(2)工程(a)において、式[1]で表される化合物をマグネシウムと反応させた後、さらに塩化亜鉛又は臭化亜鉛を反応させて、有機金属試薬を調製する、(1)に記載の製造法。
(3)工程(b)における式[2]で表される化合物について、R1がC1-6アルキルである、(1)又は(2)に記載の製造法。
(4)工程(b)における式[2]で表される化合物について、R1がtert-ブチルである、(3)に記載の製造法。
(5)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.03から1.00mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.036から1.20mol%である、(2)~(4)のいずれかに記載の製造法。
(6)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.50から1.00mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.60から1.20mol%である(1)、(3)又は(4)に記載の製造法。
(7)工程(b)において、反応温度が0から25℃である、(2)~(5)のいずれかに記載の製造法。
(8)工程(b)において、反応温度が-20から0℃である(1)、(3)、(4)又は(6)に記載の製造法。
(9)工程(c)において、式[4]で表される化合物を式[5]で表される化合物に変換する工程が酸性条件下における変換である、(1)~(8)のいずれかに記載の製造法。
(10)工程(b)における式[3]で表される光学活性な化合物について、
R2及びR3がともにメチルであり、
R4及びR5がともにフェニルである、(1)~(9)のいずれかに記載の製造法、
(11)工程(a)において、式[1]で表される化合物とマグネシウムと反応させて調製した有機マグネシウム試薬と、さらに反応させる塩化亜鉛又は臭化亜鉛のモル比が、2:1から3:1である、(2)~(5)、〈7〉~(9)のいずれかに記載の製造法。
(12)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.10mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が0.12mol%以下である、(2)に記載の製造法。
(13)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が1.00mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が1.20mol%以下である、(1)に記載の製造法。
(14)工程(c)について、酸性条件下において、式[5]で表される化合物への変換に使用する酸が、塩酸、硫酸、ギ酸、トリフルオロ酢酸、メタンスルホン酸、及びp-トルエンスルホン酸からなる群から選ばれる酸である、(9)に記載の製造法。
(15)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が1.00mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が1.20mol%以下である、(2)に記載の製造法。
(16)工程(b)において、反応温度が0℃以上である、(15)に記載の製造法。
(17)光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸の製造法であって、
(a)式[1]で表される化合物をマグネシウムと反応させ、場合によってはさらに塩化亜鉛又は臭化亜鉛を反応させて、有機金属試薬を調製する工程、

式[5]で表される化合物又はその製薬学的に許容される塩に変換する工程

ここで、
上記式[1]から[5]において、
X1はハロゲン原子を表し、
X2はハロゲン原子を表し、
R1はtert-ブチルジフェニルシリル、C1-6アルキル、C2-6アルケニル、C3-8シクロアルキル、フェニル、又は置換基群A1から選ばれる1から2個の基で置換されてもよいベンジルを表し、
R2及びR3は独立してC1-6アルキルを表し、
またはR2、R3、及び該置換基に隣接する炭素原子は一緒になってC3-6シクロアルカンを形成してもよく、
R4及びR5は独立してC1-6アルキル、ベンジル、フェネチル、又は置換基群A2から選ばれる1から2個の基で置換されてもよいフェニルを表し、
ここで、置換基群A1は、C1-6アルキル及びフェニルからなる群を表し、
置換基群A2は、ハロゲン原子、C1-6アルキル、ハロC1-6アルキル、C1-6アルコキシ、ハロC1-6アルコキシ、及びフェニルからなる群
を表す、
(18)工程(a)において、式[1]で表される化合物をマグネシウムと反応させた後、さらに塩化亜鉛又は臭化亜鉛を反応させる、(17)に記載の製造法、
(19)工程(a)において、式[1]で表される化合物をマグネシウムと反応させた後、塩化亜鉛又は臭化亜鉛を反応させない、(17)に記載の製造法、
(20)工程(b)における式[2]で表される化合物について、R1がC1-6アルキルである、(17)に記載の製造法、
(21)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が1.00mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が1.20mol%以下である、(17)に記載の製造法、
(22)工程(b)において、反応温度が0℃以上である、(21)に記載の製造法、
(23)工程(c)において、式[4]で表される化合物を式[5]で表される化合物に変換する工程が酸性条件下における変換である、(17)に記載の製造法、
(24)工程(b)における式[3]で表される光学活性な化合物について、
R2及びR3がともにメチルであり、
R4及びR5がともにフェニルである、(17)に記載の製造法、
(25)工程(b)における式[2]で表される化合物について、
R1がtert-ブチルである、(20)に記載の製造法、
(26)工程(a)において、式[1]で表される化合物とマグネシウムと反応させて調製した有機マグネシウム試薬と、さらに反応させる塩化亜鉛又は臭化亜鉛のモル比が、2:1から3:1である、(18)に記載の製造法、
(27)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.10mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が0.12mol%以下である、(18)に記載の製造法、
(28)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.03から0.10mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.036から0.12mol%である、(27)に記載の製造法、
(29)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が1.00mol%以下であり、使用する式[3]で表される光学活性な化合物の触媒量が1.20mol%以下である、(19)に記載の製造法、
(30)工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.50から1.00mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.60から1.20mol%以下である、(29)に記載の製造法、
(31)工程(b)において、反応温度が0から25℃である、(22)に記載の製造法、
(32)工程(c)について、酸性条件下において、式[5]で表される化合物への変換に使用する酸が、塩酸、硫酸、ギ酸、トリフルオロ酢酸、メタンスルホン酸、及びp-トルエンスルホン酸からなる群から選ばれる酸である、(23)に記載の製造法。


X1が臭素原子であり、
X2が塩素原子又は臭素原子であり、
R1がtert-ブチルであり、
R2及びR3がともにメチルであり、
R4及びR5がともにフェニルである場合である。

X1が臭素原子であり、
X2が臭素原子であり、
R1がtert-ブチルジフェニルシリル、tert-ブチル、ネオペンチル、tert-アミル、シクロへキシル、1-メチル-1-フェニルエチル、又はベンズヒドリルであり、
R2及びR3がともにメチル又はエチルであり、
または、R2、R3、及び該置換基に隣接する炭素原子が一緒になって形成するC3-6シクロアルカンがシクロプロパンであり、
R4及びR5がともにフェニルであり、
亜鉛化合物が臭化亜鉛である場合である。
R2及びR3がともにメチルである場合である。
R1がtert-ブチルである場合である。
X1が臭素原子であり、
X2が臭素原子であり、
R1がtert-ブチル、tert-アミル、1-メチル-1-フェニルエチル、又はベンズヒドリルであり、
R2及びR3がともにメチル又はエチルであり、
または、R2、R3、及び該置換基に隣接する炭素原子が一緒になって形成するC3-6シクロアルカンがシクロプロパンであり、
R4及びR5がともにフェニルであり、
亜鉛化合物が臭化亜鉛である場合である。
R2及びR3がともにメチルである場合である。
R1がtert-ブチルである場合である。

R1がtert-ブチルである場合である。
1.工程(a)、有機マグネシウム試薬[6]の調製法

2.工程(b)、有機マグネシウム試薬[6]から式[4]で表される化合物の製造法

3.工程(a)、有機亜鉛試薬[7]の調製法

4.工程(b)、有機亜鉛試薬[7]から式[4]で表される化合物の製造法
5.工程(c)、式[5]で表される化合物の製造法

s : シングレット(singlet)
d : ダブレット(doublet)
t : トリプレット(triplet)
q : クァルテット(quartet)
m : マルチプレット(multiplet)
J : カップリング定数(coupling constant)
Hz : ヘルツ(Hertz)
CHLOROFORM-d、CDCl3 : 重クロロホルム
NMR:核磁気共鳴
MS:マススペクトル
ee:鏡像体過剰率
HPLC:高速液体クロマトグラフィー
Et:エチル
Me:メチル
Ph:フェニル
MsOH:メシル酸、メタンスルホン酸
TsOH:トシル酸、p-トルエンスルホン酸
TFA:トリフルオロ酢酸
acac:アセチルアセトナト
PPh3:トリフェニルホスフィン
TBDPS:tert-ブチルジフェニルシリル
THF:テトラヒドロフラン
DME:1,2-ジメトキシエタン
MgSO4:無水硫酸マグネシウム
rt:室温
(S) -2-(2-フルオロビフェニル-4-イル)プロパン酸tert-ブチルエステルの製造法

化学収率:84%
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.43 (s, 9 H), 1.48 (d, J=7.0 Hz, 3 H), 3.64 (q, J=7.0 Hz, 1 H), 7.08 - 7.16 (m, 2 H), 7.33 - 7.40 (m, 2 H), 7.43 (t, J=7.6 Hz, 2 H), 7.54 (d, J=8.3 Hz, 2 H).
MS(ESI/APCI Dual pos.)m/z: 323[M+Na]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=4.19分、S体=4.60分
光学純度:93%ee(S)
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.52 (d, J=7.0 Hz, 3 H), 3.75 (q, J=7.0 Hz, 1 H), 7.08 - 7.16 (m, 2 H), 7.29 - 7.43 (m, 4 H), 7.45 - 7.51 (m, 2 H).
MS(ESI/APCI Dual neg.)m/z: 243[M-H]-
[α]20 D = +44.9±0.05(c=1.01、EtOH)
カラム名:DAICEL CHIRALPAK AY-H/SFC(4.6mmΦ x 250mmL)
溶離液:メタノール:二酸化炭素=10:90
流速:3.0mL/分
カラム温度:40℃
保持時間:R体=2.37分、S体=3.13分
光学純度:97%ee(S)
2-ブロモプロピオン酸tert-ブチルエステル(641mg、3.07mmol)、塩化ニッケル(II)・DME錯体(0.031mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.0mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.037mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、金属マグネシウム片(116mg、4.78mmol)の代わりに金属マグネシウム片(101mg、4.14mmol)を用いた以外は、実施例1-1と同様の条件下、臭化亜鉛の使用量を変更し、反応を実施した。使用した化合物及びその使用量、収率、並びに光学純度について表1-1に示す。


4-ブロモ-2-フルオロビフェニル(1.007g、3.99mmol)、2-ブロモプロピオン酸tert-ブチルエステル(641mg、3.07mmol)、臭化亜鉛(1.99mmol)を用い、実施例1-1と同様の条件下、塩化ニッケル(II)・DME錯体及び(R,R)-2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(表2-1では化合物[3-1]と表記)の使用量(各々2-ブロモプロピオン酸t-ブチルエステルに対するmol%で表記)を変更し、反応を実施した。使用した化合物及びその使用量、収率、並びに光学純度について表2-1に示す。

1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.25 (t, J=7.2 Hz, 3 H), 1.53 (d, J=7.2 Hz, 3 H), 3.74 (q, J=7.2 Hz, 1 H), 4.11 - 4.24 (m, 2 H), 7.09 - 7.18 (m, 2 H), 7.35 - 7.47 (m, 4 H), 7.50 - 7.57 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 273[M+H]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=5.50分、S体=6.18分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 0.88 (s, 9 H), 1.56 (d, J=7.4 Hz, 3 H), 3.73 - 3.85 (m, 3 H), 7.10 - 7.19 (m, 2 H), 7.34 - 7.47 (m, 4 H), 7.49 - 7.58 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 337[M+Na]+
カラム名:DAICEL CHIRALPAK AD-3 x 2(4.6mmΦ x 150mmL x 2)
溶離液:ヘキサン:エタノール=96:4
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=4.44分、S体=5.54分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 0.80 (t, J=7.6 Hz, 3 H), 1.36 - 1.43 (m, 6 H), 1.49 (d, J=7.2 Hz, 3 H), 1.70 - 1.83 (m, 2 H), 3.66 (q, J=7.2 Hz, 1 H), 7.09 - 7.17 (m, 2 H), 7.34 - 7.47 (m, 4 H), 7.49 - 7.59 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 315[M+H]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=4.02分、S体=4.31分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.55 (d, J=7.2 Hz, 3 H), 3.79 (q, J=7.2 Hz, 1 H), 4.55 - 4.68 (m, 2 H), 5.19 - 5.30 (m, 2 H), 5.82 - 5.95 (m, 1 H), 7.11 - 7.19 (m, 2 H), 7.34 - 7.47 (m, 4 H), 7.50 - 7.57 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 285[M+H]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=6.18分、S体=6.91分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.14 - 1.90 (m, 10 H), 1.50 - 1.55 (m, 3 H), 3.72 (q, J=7.3 Hz, 1 H), 4.74 - 4.87 (m, 1 H), 7.10 - 7.18 (m, 2 H), 7.27 - 7.47 (m, 4 H), 7.49 - 7.59 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 327[M+H]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=4.61分、S体=5.15分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.66 (d, J=7.0 Hz, 3 H), 4.00 (q, J=7.0 Hz, 1 H), 7.01 - 7.06 (m, 2 H), 7.19 - 7.28 (m, 3 H), 7.33 - 7.48 (m, 6 H), 7.54 - 7.58 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 343[M+Na]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:R体=22.7分、S体=26.9分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.54 - 1.57 (m, 3 H), 3.81 (q, J=7.3 Hz, 1 H), 5.05 - 5.21 (m, 2 H), 7.09 - 7.16 (m, 2 H), 7.26 - 7.39 (m, 7 H), 7.42 - 7.47 (m, 2 H), 7.51 - 7.56 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 357[M+Na]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:S体=10.5分、R体=12.4分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.49 (d, J=7.2 Hz, 3 H), 1.71 (s, 3H), 1.77 (s, 3H), 3.73 (q, J=7.2 Hz, 1 H), 7.06 - 7.15 (m, 2 H), 7.16 - 7.29 (m, 5 H), 7.32 - 7.49 (m, 4 H), 7.53 - 7.60 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 385[M+Na]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:S体=8.23分、R体=9.19分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.56 (d, J=7.2 Hz, 3 H), 3.87 (q, J=7.2 Hz, 1 H), 6.85 (s, 1 H), 7.06 - 7.17 (m, 4 H), 7.20 - 7.40 (m, 10 H), 7.42 - 7.48 (m, 2 H), 7.51 - 7.57 (m, 2 H).
MS(ESI/APCI Dual pos.)m/z: 433[M+Na]+
カラム名:DAICEL CHIRALPAK OJ-3(4.6mmΦ x 250mmL)
溶離液:ヘキサン:エタノール=84:16
流速:1.0mL/分
カラム温度:40℃
保持時間:S体=16.2分、R体=17.7分
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.01 (s, 9 H), 1.58 (d, J=7.2 Hz, 3 H), 3.88 (q, J=7.2 Hz, 1 H), 7.12 - 7.21 (m, 2 H), 7.29 - 7.73 (m, 16 H).
MS(ESI/APCI Dual pos.)m/z: 505[M+Na]+
光学純度:84%ee(S)
光学純度:95%ee(S)
光学純度:89%ee(S)
光学純度:91%ee(S)





化学収率:75%
光学純度:99%ee(S)
実施例1-1と同様の条件下、有機亜鉛試薬を調製した後に臭化リチウム(2-ブロモプロピオン酸tert-ブチルエステルに対して1当量)を加えて、2-ブロモプロピオン酸tert-ブチルエステルとのカップリング反応を2時間実施した。
化学収率:55%
得られた化合物の光学純度を以下に記載する。
光学純度:89%ee(S)
(S) -2-(2-フルオロビフェニル-4-イル)プロパン酸の製造法
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.52 (d, J=7.0 Hz, 3 H), 3.75 (q, J=7.0 Hz, 1 H), 7.08 - 7.16 (m, 2 H), 7.29 - 7.43 (m, 4 H), 7.45 - 7.51 (m, 2 H).
MS(ESI/APCI Dual neg.)m/z: 243[M-H]-
カラム名:DAICEL CHIRALPAK AY-H/SFC(4.6mmΦ x 250mmL)
溶離液:メタノール:二酸化炭素=10:90
流速:3.0mL/分
カラム温度:40℃
実施例9-1と同様の条件下、反応温度、反応時間、使用する酸と溶媒を変更し、反応を実施した。使用した化合物、収率、並びに光学純度について表7-1に示す。

実施例例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(47.8mmol)、2-ブロモプロピオン酸tert-ブチルエステル(10g、47.8mmol)、塩化ニッケル(II)・DME錯体(0.478mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.574mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、反応温度-20℃、反応時間24時間にて、カップリング反応を実施した。
化学収率:72%
光学純度:86%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(12mmol)、2-ブロモプロピオン酸tert-ブチルエステル(2.09g、10mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、反応温度-20℃、反応時間24時間にて、カップリング反応を実施した。
化学収率:89%
光学純度:87%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(10mmol)、2-ブロモプロピオン酸tert-ブチルエステル(2.09g、10mmol)、塩化ニッケル(II)・DME錯体(0.05mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して0.5mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.06mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して0.6mol%)を用い、実施例1-1(2)と同様の条件下、反応温度-10℃、反応時間3時間にて、カップリング反応を実施した。
化学収率:72%
光学純度:87%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(10mmol)、2-ブロモプロピオン酸tert-ブチルエステル(2.09g、10mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、反応温度-10℃、反応時間3時間にて、カップリング反応を実施した。
化学収率:73%
光学純度:89%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(10mmol)、2-ブロモプロピオン酸tert-ブチルエステル(2.09g、10mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、反応時間3時間にて、カップリング反応を実施した。
化学収率:72%
光学純度:86%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(10mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-クロロプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-クロロプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、2-ブロモプロピオン酸tert-ブチルエステルの代わりに2-クロロプロピオン酸tert-ブチルエステル(1.65g、10mmol)を用い、反応時間24時間にてカップリング反応を実施した。
化学収率:60%
光学純度:94%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(10mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-クロロプロピオン酸tert-ブチルエステルに対して1mol%)、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-クロロプロピオン酸tert-ブチルエステルに対して1.2mol%)を用い、実施例1-1(2)と同様の条件下、2-ブロモプロピオン酸tert-ブチルエステルの代わりに2-クロロプロピオン酸エチルエステル(1.37g、10mmol)を用い、反応時間24時間にてカップリング反応を実施した。
化学収率:79%
光学純度:85%ee(S)
実施例1-1(1)と同様の条件下、臭化亜鉛を加えずに、有機亜鉛試薬の代わりにグリニャール試薬を調製した。調製したグリニャール試薬(2.0mmol)、2-ブロモプロピオン酸tert-ブチルエステル(0.42g、2.0mmol)、塩化ニッケル(II)・DME錯体(0.1mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して5mol%)を用い、実施例1-1(2)と同様の条件下、(R,R) -2,2'-イソプロピリデンビス(4-フェニル-2-オキサゾリン)の代わりに(R,R) -2,2'-(ペンタン-3,3-ジイル)ビス(4-フェニル-2-オキサゾリン)(0.12mmol、2-ブロモプロピオン酸tert-ブチルエステルに対して6mol%)を用い、反応温度-20℃、反応時間3時間にて、2-ブロモプロピオン酸tert-ブチルエステルとのカップリング反応を実施した。
化学収率:53%
光学純度:90%ee(S)
Claims (9)
- 光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸又はその製薬学的に許容される塩の製造法において、下記(a)~(c)工程を含むことを特徴とする製造法:
(a)式[1]で表される化合物をマグネシウムと反応させることを含む有機金属試薬を調製する工程、

(b)触媒量のニッケル化合物、及び触媒量の式[3]で表される光学活性な化合物の存在下、式[2]で表される化合物を工程(a)において調製した有機金属試薬と反応させ、式[4]で表される化合物を得る工程、及び


X1はハロゲン原子を表し、
X2はハロゲン原子を表し、
R1はtert-ブチルジフェニルシリル、C1-6アルキル、C2-6アルケニル、C3-8シクロアルキル、フェニル、又は置換基群A1から選ばれる1から2個の基で置換されてもよいベンジルを表し、
R2及びR3は独立してC1-6アルキルを表し、
またはR2、R3、及び該置換基に隣接する炭素原子は一緒になってC3-6シクロアルカンを形成してもよく、
R4及びR5は独立してC1-6アルキル、ベンジル、フェネチル、又は置換基群A2から選ばれる1から2個の基で置換されてもよいフェニルを表し、
ここで、置換基群A1は、C1-6アルキル及びフェニルからなる群を表し、
置換基群A2は、ハロゲン原子、C1-6アルキル、ハロC1-6アルキル、C1-6アルコキシ、ハロC1-6アルコキシ、及びフェニルからなる群
を表す。 - 工程(a)において、式[1]で表される化合物をマグネシウムと反応させた後、さらに塩化亜鉛又は臭化亜鉛を反応させて、有機金属試薬を調製する、請求項1に記載の製造法。
- 工程(b)における式[2]で表される化合物について、R1がC1-6アルキルである請求項1又は2に記載の製造法。
- 工程(b)における式[2]で表される化合物について、R1がtert-ブチルである、請求項3に記載の製造法。
- 工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.03から1.00mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.036から1.20mol%である請求項2~4のいずれか1項に記載の製造法。
- 工程(b)において、式[2]で表される化合物に対して、使用するニッケル化合物の触媒量が0.50から1.00mol%であり、使用する式[3]で表される光学活性な化合物の触媒量が0.60から1.20mol%である請求項1、3又は4に記載の製造法。
- 工程(b)において、反応温度が0から25℃である請求項2~5のいずれか1項に記載の製造法。
- 工程(b)において、反応温度が-20から0℃である請求項1、3、4又は6に記載の製造法。
- 工程(c)において、式[4]で表される化合物を式[5]で表される化合物に変換する工程が酸性条件下における変換である請求項1~8のいずれか1項に記載の製造法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680057083.5A CN108026014B (zh) | 2015-09-30 | 2016-09-29 | 光学活性的2-(2-氟联苯-4-基)丙酸的制备方法 |
| PH1/2018/500677A PH12018500677B1 (en) | 2015-09-30 | 2016-09-29 | Method for producing optically active 2- (2-fluorobiphenyl-4-yl) propanoic acid |
| US15/764,598 US10207976B2 (en) | 2015-09-30 | 2016-09-29 | Method for producing optically active 2-(2-fluorobiphenyl-4-yl) propanoic acid |
| EP16851813.2A EP3357901A4 (en) | 2015-09-30 | 2016-09-29 | PROCESS FOR PREPARING OPTICALLY ACTIVE 2- (2-FLUOROBIPHENYL-4-YL) PROPIONIC ACID |
| MYPI2018701271A MY194792A (en) | 2015-09-30 | 2016-09-29 | Method for producing optically active 2-(2-fluorobiphenyl-4-yl) propanoic acid |
| JP2017543596A JP6504530B2 (ja) | 2015-09-30 | 2016-09-29 | 光学活性な2−(2−フルオロビフェニル−4−イル)プロパン酸の製造法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-194284 | 2015-09-30 | ||
| JP2015194284 | 2015-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017057642A1 true WO2017057642A1 (ja) | 2017-04-06 |
Family
ID=58423941
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/078939 Ceased WO2017057642A1 (ja) | 2015-09-30 | 2016-09-29 | 光学活性な2-(2-フルオロビフェニル-4-イル)プロパン酸の製造法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10207976B2 (ja) |
| EP (1) | EP3357901A4 (ja) |
| JP (1) | JP6504530B2 (ja) |
| CN (1) | CN108026014B (ja) |
| MY (1) | MY194792A (ja) |
| PH (1) | PH12018500677B1 (ja) |
| TW (1) | TWI730989B (ja) |
| WO (1) | WO2017057642A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108558651A (zh) * | 2018-06-13 | 2018-09-21 | 上海峰林生物科技有限公司 | 一种氟比洛芬的制备方法及氟比洛芬酯的制备方法 |
| IT201800003382A1 (it) * | 2018-03-08 | 2019-09-08 | Taisho Pharmaceutical Co Ltd | Crystal of tert-butyl (2S)-2-(2-fluorobiphenyl-4-yl) propanoate and process for producing the same |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112778127A (zh) * | 2020-06-25 | 2021-05-11 | 北京澳合药物研究院有限公司 | 一种氟比洛芬的制备方法 |
| CN112457182A (zh) * | 2020-12-16 | 2021-03-09 | 江苏慧聚药业有限公司 | 一种氟比洛芬杂质的制备方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS51127042A (en) * | 1975-04-04 | 1976-11-05 | Boots Co Ltd | Production of phenylpropionic acid derivatives |
| JPS6011439A (ja) * | 1983-06-29 | 1985-01-21 | Daikin Ind Ltd | 2−アリ−ルアルキルカルボン酸化合物の製法 |
| JPS60112735A (ja) * | 1983-11-22 | 1985-06-19 | シンテツクス・フア−マシユ−テイカルズ・インタ−ナシヨナル・リミテツド | 光学活性なα−アリ−ルアルカン酸類の製造法およびその前駆体 |
| JPS61210049A (ja) * | 1984-11-21 | 1986-09-18 | Daicel Chem Ind Ltd | 光学活性なα↓−芳香族基置換アルカンカルボン酸類の製造方法 |
| JPH07506808A (ja) * | 1991-12-27 | 1995-07-27 | ヘーミッシュ・ビオテヒノロギッシェ・ラボラトリエン・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | キラル・オキサゾリンによる高光学純度の2−アリール−プロピオン酸の立体選択的合成 |
| JP2004339085A (ja) * | 2003-05-13 | 2004-12-02 | Asahi Kasei Pharma Kk | α−置換フェニル−アルカンカルボン酸エステル誘導体の製法 |
| JP2005255577A (ja) * | 2004-03-10 | 2005-09-22 | Asahi Kasei Pharma Kk | 管型反応装置を用いた連続的な製造方法 |
| WO2013073596A1 (ja) * | 2011-11-14 | 2013-05-23 | 住友化学株式会社 | 光学活性ビスオキサゾリン化合物、不斉触媒およびそれを用いた光学活性シクロプロパン化合物の製造方法 |
| JP2014065693A (ja) * | 2012-09-27 | 2014-04-17 | Daikin Ind Ltd | チオノカルボン酸エステルの製造方法 |
| JP2014193833A (ja) * | 2013-03-29 | 2014-10-09 | Yamaguchi Univ | 光学活性α−ブロモベンゾイル酢酸エステル類の製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1459084A (en) | 1973-05-24 | 1976-12-22 | Boots Co Ltd | Preparation of arylalkanoic acid |
| EP0110671B1 (en) | 1982-11-24 | 1986-05-21 | Syntex Pharmaceuticals International Limited | Preparation of optically active alpha-arylalkanoic acids and precursors thereof |
| JP4191829B2 (ja) | 1998-11-05 | 2008-12-03 | 大日本住友製薬株式会社 | 光学活性カルボン酸の製造方法 |
| CN103755554B (zh) | 2014-01-17 | 2015-02-18 | 中国农业大学 | 不对称催化合成(s)-非诺洛芬的方法 |
| CN103755566B (zh) | 2014-01-17 | 2015-04-29 | 中国农业大学 | 一种不对称催化合成(s)-2-芳基丙酸酯的方法 |
-
2016
- 2016-09-29 US US15/764,598 patent/US10207976B2/en active Active
- 2016-09-29 PH PH1/2018/500677A patent/PH12018500677B1/en unknown
- 2016-09-29 JP JP2017543596A patent/JP6504530B2/ja active Active
- 2016-09-29 MY MYPI2018701271A patent/MY194792A/en unknown
- 2016-09-29 EP EP16851813.2A patent/EP3357901A4/en not_active Withdrawn
- 2016-09-29 CN CN201680057083.5A patent/CN108026014B/zh active Active
- 2016-09-29 WO PCT/JP2016/078939 patent/WO2017057642A1/ja not_active Ceased
- 2016-09-29 TW TW105131363A patent/TWI730989B/zh active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS51127042A (en) * | 1975-04-04 | 1976-11-05 | Boots Co Ltd | Production of phenylpropionic acid derivatives |
| JPS6011439A (ja) * | 1983-06-29 | 1985-01-21 | Daikin Ind Ltd | 2−アリ−ルアルキルカルボン酸化合物の製法 |
| JPS60112735A (ja) * | 1983-11-22 | 1985-06-19 | シンテツクス・フア−マシユ−テイカルズ・インタ−ナシヨナル・リミテツド | 光学活性なα−アリ−ルアルカン酸類の製造法およびその前駆体 |
| JPS61210049A (ja) * | 1984-11-21 | 1986-09-18 | Daicel Chem Ind Ltd | 光学活性なα↓−芳香族基置換アルカンカルボン酸類の製造方法 |
| JPH07506808A (ja) * | 1991-12-27 | 1995-07-27 | ヘーミッシュ・ビオテヒノロギッシェ・ラボラトリエン・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | キラル・オキサゾリンによる高光学純度の2−アリール−プロピオン酸の立体選択的合成 |
| JP2004339085A (ja) * | 2003-05-13 | 2004-12-02 | Asahi Kasei Pharma Kk | α−置換フェニル−アルカンカルボン酸エステル誘導体の製法 |
| JP2005255577A (ja) * | 2004-03-10 | 2005-09-22 | Asahi Kasei Pharma Kk | 管型反応装置を用いた連続的な製造方法 |
| WO2013073596A1 (ja) * | 2011-11-14 | 2013-05-23 | 住友化学株式会社 | 光学活性ビスオキサゾリン化合物、不斉触媒およびそれを用いた光学活性シクロプロパン化合物の製造方法 |
| JP2014065693A (ja) * | 2012-09-27 | 2014-04-17 | Daikin Ind Ltd | チオノカルボン酸エステルの製造方法 |
| JP2014193833A (ja) * | 2013-03-29 | 2014-10-09 | Yamaguchi Univ | 光学活性α−ブロモベンゾイル酢酸エステル類の製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| LOU, SHA ET AL.: "Nickel/Bis(oxazoline)- Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic a- Bromoketones", J. AM. CHEM. SOC., vol. 132, no. 4, pages 1264 - 1266, XP055385368 * |
| MAO, JIANYOU ET AL.: "Cobalt-Bisoxazoline-Catalyzed Asymmetric Kumada Cross-Coupling of Racemic a-Bromo Esters with Aryl Grignard Reagents", J. AM. CHEM. SOC., vol. 136, no. 50, 5 December 2014 (2014-12-05), pages 17662 - 17668, XP055385369 * |
| See also references of EP3357901A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT201800003382A1 (it) * | 2018-03-08 | 2019-09-08 | Taisho Pharmaceutical Co Ltd | Crystal of tert-butyl (2S)-2-(2-fluorobiphenyl-4-yl) propanoate and process for producing the same |
| CN108558651A (zh) * | 2018-06-13 | 2018-09-21 | 上海峰林生物科技有限公司 | 一种氟比洛芬的制备方法及氟比洛芬酯的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6504530B2 (ja) | 2019-04-24 |
| TW201726593A (zh) | 2017-08-01 |
| CN108026014A (zh) | 2018-05-11 |
| US20180282252A1 (en) | 2018-10-04 |
| PH12018500677A1 (en) | 2018-10-15 |
| MY194792A (en) | 2022-12-15 |
| JPWO2017057642A1 (ja) | 2018-05-24 |
| EP3357901A1 (en) | 2018-08-08 |
| EP3357901A4 (en) | 2019-06-19 |
| CN108026014B (zh) | 2021-03-16 |
| PH12018500677B1 (en) | 2023-11-10 |
| US10207976B2 (en) | 2019-02-19 |
| TWI730989B (zh) | 2021-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100978970B1 (ko) | Hmg-coa 환원효소 저해 메발론산 유도체의 제조방법 | |
| NO327958B1 (no) | Pregabalin og metode for fremstilling av slike forbindelser | |
| JP6504530B2 (ja) | 光学活性な2−(2−フルオロビフェニル−4−イル)プロパン酸の製造法 | |
| EP2123661A1 (en) | Chiral iridium aqua complex and method for producing optically active hydroxy compound by using the same | |
| JP5108777B2 (ja) | 軸不斉を有する光学活性な4級アンモニウム塩およびそれを用いたα−アミノ酸およびその誘導体の製造方法 | |
| WO2003064420A1 (en) | Novel optically active compounds, method for kinetic optical resolution of carboxylic acid derivatives and catalysts therefor | |
| EP2792664A1 (en) | Optical resolution method for bicyclic compound using asymmetric catalyst | |
| JP5396578B2 (ja) | 光学活性テトラアミノホスホニウム塩、不斉合成反応用触媒、及び光学活性β−ニトロアルコールの製造方法 | |
| US20100286442A1 (en) | Novel method for preparing pregabalin | |
| CA3169869A1 (en) | Process for preparing s-beflubutamid by resolving 2-bromobutanoic acid | |
| KR20130090360A (ko) | 물 또는 다양한 산을 첨가제로 이용한 새로운 마이클-첨가 반응을 통하여 화합물을 제조하는 방법 | |
| US8957233B2 (en) | Method for producing optically active 1,2-bis(dialkylphosphino)benzene derivative | |
| JP5569938B2 (ja) | ピロリジン誘導体及びその製造方法 | |
| JP2017014109A (ja) | 光学活性2,6−ジメチルチロシン誘導体の製造法 | |
| KR20130107698A (ko) | 고순도 ⒮―메토프롤롤의 제조방법 | |
| JP5829529B2 (ja) | 光学活性4級アンモニウム塩、および光学活性化合物の製造方法 | |
| US20030236429A1 (en) | Process for the production of chiral compounds | |
| JP2008115178A (ja) | ジフェニルアラニン−Ni(II)錯体の製造方法 | |
| Błocka et al. | Structural and electronic effects of oxazolidine ligands derived from (1R, 2S)-ephedrine in the asymmetric addition of diethylzinc to aldehydes | |
| US11623913B2 (en) | Method of synthesizing (1R,2R)-nitroalcohol compound | |
| HK1249498B (zh) | 光学活性的2-(2-氟联苯-4-基)丙酸的制备方法 | |
| HK1249498A1 (en) | Method for producing optically active 2- (2-fluorobiphenyl-4-yl)propanoic acid | |
| JP3432880B2 (ja) | 光学活性アザスピロ化合物の製法 | |
| JP5757022B2 (ja) | 光学活性アルキルアミノスルホンアミド誘導体の製造方法及び光学活性β−アミノアルコール誘導体 | |
| WO2012176830A1 (ja) | 1,2-ビス(ジアルキルホスフィノ)-4,5-(メチレンジオキシ)ベンゼン誘導体、その製造方法及び該1,2-ビス(ジアルキルホスフィノ)-4,5-(メチレンジオキシ)ベンゼン誘導体を配位子とする金属錯体並びに不斉水素化方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16851813 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017543596 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12018500677 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15764598 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016851813 Country of ref document: EP |