WO2017082263A1 - 酸化黒鉛誘導体及びその製造方法 - Google Patents
酸化黒鉛誘導体及びその製造方法 Download PDFInfo
- Publication number
- WO2017082263A1 WO2017082263A1 PCT/JP2016/083143 JP2016083143W WO2017082263A1 WO 2017082263 A1 WO2017082263 A1 WO 2017082263A1 JP 2016083143 W JP2016083143 W JP 2016083143W WO 2017082263 A1 WO2017082263 A1 WO 2017082263A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- graphite oxide
- oxide derivative
- graphite
- mass
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
































Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/20—Graphite
- C01B32/21—After-treatment
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/20—Graphite
- C01B32/21—After-treatment
- C01B32/23—Oxidation
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/15—Nano-sized carbon materials
- C01B32/182—Graphene
- C01B32/198—Graphene oxide
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/82—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by IR- or Raman-data
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10M—LUBRICATING COMPOSITIONS; USE OF CHEMICAL SUBSTANCES EITHER ALONE OR AS LUBRICATING INGREDIENTS IN A LUBRICATING COMPOSITION
- C10M103/00—Lubricating compositions characterised by the base-material being an inorganic material
- C10M103/02—Carbon; Graphite
Definitions
- the present invention relates to a method for producing a graphite oxide derivative. More specifically, catalysts, battery and capacitor electrode active materials, thermoelectric conversion materials, conductive materials, luminescent materials, lubricant additives (additives for lubricating oil for machinery), polymer additives (addition to resins) Agent), a permeable membrane material, an antibacterial material, a water repellent material, an adsorbing material and the like.
- Graphite oxide is obtained by oxidizing graphite with a layered structure in which carbon atoms bonded by sp 2 bonds are arranged in a plane and adding oxygen-containing functional groups.
- Graphite oxide is a catalyst, battery and capacitor electrode active materials, thermoelectric conversion materials, conductive materials, luminescent materials, lubricant additives, polymer additives, permeable membrane materials, antibacterial materials, water repellent materials, adsorbent materials, etc.
- it is desired to be used by being dispersed in oil as an additive for lubricating oil for machinery.
- a method of derivatizing graphite oxide can be considered.
- graphite oxide is used as a lubricating additive by being dispersed in oil, or combined with a polymer or other resin. Therefore, as it is, hydrophilic graphite oxide cannot be sufficiently dispersed in nonpolar, low polarity dispersion media (hydrophobic dispersion media) such as oil and resin. It is conceivable to reduce the contained functional group or to modify graphite oxide with a hydrophobic substituent.
- graphite oxide reduction reactions and substituent modification (introduction) reactions see Patent Documents 1 to 3 and Non-Patent Documents 1 to 8).
- Non-Patent Document 3 has been studied for the purpose of improving the dispersibility of graphite oxide in a non-polar dispersion medium, but actively removes reactive oxygen by purification and drying as a pretreatment and reduction treatment. In addition, treatment with a strongly basic reagent as pre-reaction treatment is performed.
- graphite oxide in order for graphite oxide to exhibit a sufficient effect as a lubricant additive for machinery, etc., it is necessary to have sufficient dispersibility in oil, so graphite oxide excellent in dispersibility in nonpolar dispersion media Is required. Furthermore, in order for graphite oxide to exhibit a sufficient effect as an additive to various resins, graphite oxide having excellent dispersibility in an amphiphilic dispersion medium is required.
- the present invention has been made in view of the above situation, and an object thereof is to provide graphite oxide having excellent dispersibility in a nonpolar dispersion medium or an amphiphilic dispersion medium.
- the present invention has been made in view of the above situation, and provides a method for producing a graphite oxide derivative capable of easily producing a high-quality graphite oxide derivative, and a high-quality graphite oxide derivative. With the goal.
- the present inventors have studied various methods for easily producing high-quality graphite oxide derivatives, and various methods for improving the dispersibility of graphite oxide in nonpolar dispersion media or amphiphilic dispersion media.
- the present invention has been achieved by studying and conceiving that the above-mentioned problems can be solved brilliantly.
- the present invention is a method for producing a graphite oxide derivative, which comprises a step of oxidizing graphite, graphite oxide in a graphite oxide-containing composition obtained in the oxidation step, and oxygen in the graphite oxide.
- It is a manufacturing method of the graphite oxide derivative characterized by not containing.
- the present invention is also a graphite oxide derivative having an alkyl group and having a grade of 8 grade as measured by a drop sensitivity test defined in JIS K 4810.
- the present invention is a graphite oxide derivative having a functional group having a hydrocarbon group having 13 or more carbon atoms.
- the present invention is also a graphite oxide derivative characterized by having a functional group having a hydrocarbon group having 6 to 10 carbon atoms.
- the method for producing a graphite oxide derivative of the present invention By the method for producing a graphite oxide derivative of the present invention, a high quality graphite oxide derivative can be easily obtained. Further, when the graphite oxide derivative of the present invention has a functional group having a hydrocarbon group having 13 or more carbon atoms, it is sufficiently dispersed in a nonpolar dispersion medium and has a hydrocarbon group having 6 to 10 carbon atoms. When it has a functional group, it is sufficiently dispersed in an amphiphilic dispersion medium.
- 3 is an FT-IR chart of graphite oxide produced in Preparation Example 1-2.
- 2 is an FT-IR chart of graphite oxide derivative A produced in Example 1-1.
- 3 is an XRD chart of graphite oxide derivative A produced in Example 1-1.
- 3 is an FT-IR chart of graphite oxide derivative B produced in Example 1-2.
- 4 is an FT-IR chart of graphite oxide derivative C produced in Example 1-3.
- 4 is an FT-IR chart of graphite oxide derivative D produced in Example 1-4.
- 6 is an FT-IR chart of graphite oxide derivative E produced in Example 1-5.
- 6 is an XRD chart of graphite oxide derivative E produced in Example 1-5.
- 6 is an FT-IR chart of graphite oxide derivative F produced in Example 1-6.
- FIG. 7 is an FT-IR chart of graphite oxide derivative G produced in Example 1-7.
- 6 is an FT-IR chart of graphite oxide derivative H produced in Example 1-8.
- 2 is an FT-IR chart of graphite oxide derivative I produced in Example 1-9.
- 2 is an XRD chart of graphite oxide derivative I produced in Example 1-9.
- 2 is an FT-IR chart of graphite oxide derivative J produced in Example 1-10.
- 3 is an XRD chart of graphite oxide derivative J produced in Example 1-10.
- 3 is an FT-IR chart of graphite oxide derivative K produced in Example 1-23.
- 2 is an FT-IR chart of graphite oxide derivative L produced in Example 1-24.
- FIG. 6 is a view showing an XRD measurement result of the graphite oxide powder obtained in Reference Example 2-1.
- FIG. 6 is a view showing an XRD measurement result of the graphite oxide powder obtained in Reference Example 2-2.
- FIG. 6 is a view showing an XRD measurement result of reduced graphite oxide powder obtained in Reference Example 2-3.
- FIG. 6 is a view showing an XRD measurement result of reduced graphite oxide powder obtained in Reference Example 2-4.
- 3 is an FT-IR chart of graphite oxide as a raw material.
- 3 is an FT-IR chart of the graphite oxide derivative produced in Example 3-1.
- 4 is an FT-IR chart of the graphite oxide derivative produced in Example 3-2.
- 3 is an FT-IR chart of the graphite oxide derivative produced in Example 3-3.
- 4 is an FT-IR chart of the graphite oxide derivative produced in Example 3-4.
- 4 is an FT-IR chart of a graphite oxide derivative produced in Comparative Example 3-1.
- 4 is an FT-IR chart of a graphite oxide derivative produced in Comparative Example 3-2. It is an actual image of hexane-methanol separation of Example 3-1, Example 3-3, and Comparative Example 3-1 (Example 3-1, Example 3-3, Comparative Example 3-1 from the left).
- . 6 is an FT-IR chart of the graphite oxide derivative produced in Example 3-5.
- 4 is an FT-IR chart of the graphite oxide derivative produced in Example 3-6.
- 7 is an FT-IR chart of the graphite oxide derivative produced in Example 3-7.
- Non-Patent Document 3 when oxygen oxide is purified and dried, reduction treatment, treatment with a strongly basic reagent as pre-reaction treatment, etc., reactive oxygen is removed from the graphite oxide. Therefore, it is considered that sufficient modification reaction does not proceed and the dispersibility is poor.
- using the method of the present invention it was found that a graphite oxide derivative with good dispersibility can be obtained even in ethanol or decane, which was not sufficiently dispersible with the graphite oxide derivative obtained by the method described in Non-Patent Document 3. It was. Further, when a cation of a cationic organic compound is used as a pretreatment as described in Patent Document 1, the cation-derived component is then strongly bonded to the graphite oxide derivative as an impurity, and it becomes difficult to remove.
- Graphite oxide is oxygen bonded by oxidizing a graphitic carbon material such as graphene, graphite (graphite) (oxygen bonded to the carbon material), and the oxygen is added to the graphitic carbon material.
- a graphitic carbon material such as graphene, graphite (graphite) (oxygen bonded to the carbon material), and the oxygen is added to the graphitic carbon material.
- graphite exists as a substituent such as a carboxyl group, a carbonyl group, a hydroxyl group (hydroxyl group), or an epoxy group.
- the graphite oxide is preferably graphene oxide in which oxygen is bonded to carbon of graphene.
- graphene refers to a sheet composed of one layer in which carbon atoms bonded by sp 2 bonds are arranged in a plane, and a stack of graphene sheets is referred to as graphite.
- graphene oxide In addition to a sheet composed of only one carbon atom, those having a structure in which about 2 to 100 layers are laminated are included.
- the graphene oxide is preferably a sheet composed of only one carbon atom layer or has a structure in which about 2 to 20 layers are laminated.
- the graphite oxide may further have a functional group such as a sulfur-containing group and a nitrogen-containing group, but the content of carbon, hydrogen, and oxygen as constituent elements with respect to all constituent elements is 97 mol% or more. It is preferable that it is 99 mol% or more, and it is still more preferable that the graphite oxide contains only carbon, hydrogen, and oxygen as constituent elements.
- the graphite oxide derivative of the present invention further has a structure in which a functional group having a group derived from a compound that reacts with an oxygen-containing functional group of graphite oxide is bonded to a carbon atom of graphite oxide.
- a functional group having a hydrocarbon group having 13 or more carbon atoms or a functional group having a hydrocarbon group having 6 to 10 carbon atoms is bonded to the carbon atom of graphite oxide. It has a structure.
- the graphite oxide derivative of the present invention further includes a group derived from a compound that reacts with the oxygen-containing functional group of graphite oxide on the carbon atom of graphene oxide. It has a structure in which functional groups possessed
- the graphite oxide derivative may further have a functional group such as a sulfur-containing group or a nitrogen-containing group, but is composed of only carbon, hydrogen, and oxygen as constituent elements, or carbon, It is more preferable that only hydrogen, oxygen, and nitrogen are constituent elements. Preferred examples of the graphite oxide derivative will be described later.
- Examples of analysis showing the characteristics of the graphite oxide derivative of the present invention include mass spectrometry and FT-IR. Fragments ionized by mass spectrometry can be easily observed. For example, since the graphite oxide derivative of the present invention has a hydrocarbon group at the end or the like, fragments ionized by mass spectrometry can be easily observed. Since graphite oxide itself has a large mass, ions are not detected by mass spectrometry, that is, only a site derived from a compound introduced into graphite oxide is observed.
- the graphite oxide derivative when the graphite oxide derivative has a hydrocarbon group at the end, the graphite oxide derivative appears as a CH peak (around 2900 cm ⁇ 1 ) derived from the hydrocarbon group. Can be easily analyzed.
- One preferred form (also referred to as a first preferred form) of the method for producing a graphite oxide derivative of the present invention includes a step of oxidizing graphite and a step of obtaining a graphite oxide derivative, and a step of oxidizing the graphite And a step of purifying and drying the graphite oxide-containing composition between the step of obtaining the graphite oxide derivative and the step of obtaining the graphite oxide derivative.
- the first preferred form of the production method of the present invention may be one in which the graphite oxide-containing composition is not purified and dried between the oxidation step and the step of obtaining the graphite oxide derivative. Only one of purification and drying of the graphite-containing composition may be performed.
- the above-mentioned “excludes the steps of purifying and drying the graphite oxide-containing composition” means that the graphite oxide-containing composition is used when the graphite oxide-containing composition is subjected to a reaction in the step of obtaining a graphite oxide derivative. It means that the composition is not purified and dried.
- the present inventors have found that the reactive oxygen-containing functional group possessed by graphite oxide is reduced and / or inactivated during the purification step and drying step of graphite oxide from the graphite oxide-containing composition, By omitting at least one of refining and drying, graphite oxide in a composition containing graphite oxide with a sufficient number of reactive oxygen-containing functional groups is applied to the reaction (modification reaction) in the process of obtaining a graphite oxide derivative. Found that you can. As a result, it is possible to efficiently obtain a high-quality graphite oxide derivative in which a group having a desired functional group is sufficiently introduced by a modification reaction. Moreover, since at least one of refining and drying is omitted, the production process becomes simpler, and there is a merit on the process.
- the step of purifying the graphite oxide-containing composition refers to solid impurities (for example, an oxidizing agent such as permanganate) from the graphite oxide-containing composition and / or a solvent (such as sulfuric acid) during the oxidation reaction.
- the graphite oxide-containing composition is made into a graphite oxide aqueous dispersion.
- the role and significance of purification include, for example, obtaining graphite oxide or a graphite oxide aqueous dispersion by setting the sulfuric acid concentration to 1% by mass or less with respect to graphite oxide in the graphite oxide-containing composition.
- Patent Document 6 When an aqueous dispersion is diluted in water as described in Patent Document 6 (for example, when it is made into a 1 to 10% graphite oxide aqueous dispersion), the total ion concentration (mostly sulfuric acid concentration) is described in Patent Document 6. It has been found that the ionization of graphite oxide itself is promoted below the threshold concentration, and the interaction with water molecules becomes stronger and the dispersibility of graphite oxide is dramatically improved. Moreover, if refinement
- the purification step is essential, and many studies have been made.
- the graphite oxide itself is not applied to the material as in the present invention.
- a chemical reaction such as modification is performed and a derivative is applied as the material
- the reactivity of the graphite oxide itself is more important than the dispersibility of the graphite oxide. It becomes.
- the total ion concentration (sulfuric acid concentration) as described in Patent Document 6 is ionized, the present inventors ionize or decompose highly active oxygen substituents, which is disadvantageous in the subsequent modification reaction.
- the advanced purification step that can be omitted in the present invention can be said to reduce acidity, promote ionization and dissociation of graphite oxide, and impart strong dispersibility to water, as described in Patent Document 6. . That is, the purification step that can be omitted in the present invention is a step of making sulfuric acid less than 1% by mass with respect to the mass of graphite oxide.
- the separation step is as described later from the reaction composition to the extent that it does not correspond to the step of purifying and drying the reaction composition containing graphite oxide.
- one or two methods such as washing with water, centrifugation after dissolving solid impurities in water, and filtration are used.
- a flocculant such as a surfactant or an organic solvent may be added during filtration as described later.
- the separation step may be performed in air or in an inert gas atmosphere such as nitrogen, helium, or argon.
- the production method of the present invention preferably does not include a step of purifying the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- a step of purifying the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- the step of oxidizing graphite, the graphite oxide in the graphite oxide-containing composition obtained in the oxidation step, and the oxygen-containing function of the graphite oxide And a step of obtaining a graphite oxide derivative by reacting with a compound that reacts with a group, and the step of purifying the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- a method for producing a graphite oxide derivative, wherein the step of purifying the graphite oxide-containing composition is a step of reducing sulfuric acid to less than 1% by mass with respect to the mass of graphite oxide contained in the graphite oxide-containing composition. Is the method.
- the process of drying a graphite oxide containing composition means the process of removing volatile solvents, such as water used in the oxidation process at least from the graphite oxide containing composition, and making it a dried material.
- the present inventors can remove even the water coordinated to the highly active oxygen functional group of graphite oxide when the water concentration relative to the graphite oxide in the graphite oxide-containing composition is at least 3% by mass. As a result, it was found that the oxygen functional group, which was stable in coordination, was eliminated, which was disadvantageous in the subsequent modification reaction. That is, by using a graphite oxide-containing composition containing 3% by mass or more of water with respect to graphite oxide, an active oxygen functional group superior in modification reaction can be stably maintained.
- the drying step that can be omitted in the present invention is a step of making water less than 3% by mass with respect to the mass of graphite oxide.
- the drying step can be performed using one or two or more methods such as centrifugation, filtration, evaporation, and the like during the filtration, An aggregating agent such as an organic solvent may be added.
- the evaporation can be performed, for example, under reduced pressure or under heating, but is preferably performed under reduced pressure and under heating. Centrifugation and filtration may be performed in air or in an inert gas atmosphere such as nitrogen, helium, or argon.
- the first preferred embodiment of the production method of the present invention includes, for example, a step of oxidizing graphite, a graphite oxide in a graphite oxide-containing composition obtained in the oxidation step, and a reaction with an oxygen-containing functional group of the graphite oxide. And a step of obtaining a graphite oxide derivative by reacting with a compound to be produced, and a step of drying the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- the step of drying the graphite oxide-containing composition is a method for producing a graphite oxide derivative, which is a step of reducing the water to less than 3% by mass with respect to the mass of the graphite oxide contained in the graphite oxide-containing composition. May be.
- the production method of the present invention is preferably one in which neither the purification step of the graphite oxide-containing composition nor the drying step is performed.
- the first preferable form of the production method of the present invention is more preferably a step of oxidizing graphite (oxidation step), graphite oxide in the graphite oxide-containing composition obtained in the oxidation step, and the oxidation Including a step of reacting a compound that reacts with an oxygen-containing functional group of graphite to obtain a graphite oxide derivative, and purifying the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- the method includes a step and a step of drying the reaction liquid containing graphite oxide, and the step of purifying the graphite oxide-containing composition is a mass of graphite oxide contained in the graphite oxide-containing composition. And the step of drying the graphite oxide-containing composition with respect to the mass of graphite oxide contained in the graphite oxide-containing composition is less than 3% by weight of water.
- Graphite oxide A method for producing a conductor.
- the manufacturing method of this invention includes the process (concentration process) of removing a part of sulfuric acid and a solvent from a reaction composition. More preferably, the production method of the present invention includes a step of concentrating the graphite oxide-containing composition between the oxidation step and the step of obtaining the graphite oxide derivative.
- the concentration step in the present invention is a step of obtaining a graphite oxide-containing composition by removing a part of sulfuric acid and water from the reaction composition, and the composition contains 1% by mass of sulfuric acid with respect to the mass of graphite oxide. It means a step of remaining above and / or leaving 3% by mass or more of water with respect to the mass of graphite oxide.
- the acid relative to the amount of the raw material compound is appropriately removed by the concentration step.
- the amount of can be adjusted within a suitable range described later, and the modification reaction can proceed with higher efficiency.
- the graphite oxide-containing composition subjected to the reaction in the step of obtaining the graphite oxide derivative separated through the concentration step or the like has a water content of 3 to 10,000 masses with respect to 100 mass% of the graphite oxide mass in the composition. % Is preferable.
- the content is more preferably 5% by mass or more, further preferably 10% by mass or more, and particularly preferably 50% by mass or more.
- the content is more preferably 5000% by mass or less, still more preferably 3000% by mass or less, still more preferably 2000% by mass or less, and particularly preferably 1000% by mass or less. Most preferably, it is 500 mass% or less.
- an acid is present in excess of graphite oxide immediately after the oxidation step (for example, about 2000 to 5000% by mass of sulfuric acid with respect to 100% by mass of graphite oxide). Excessive with respect to graphite (for example, about 500 to 10000% by mass with respect to 100% by mass of graphite oxide). It is preferable to concentrate the acid and water in the concentration step so that the amount of water falls within the above range. Thereby, reactive oxygen can exist stably until the process of obtaining a graphite oxide derivative, and can be efficiently used for the modification reaction.
- the graphite oxide containing composition after a concentration process may contain 0.01 mass% or more of manganese in the composition.
- the graphite oxide-containing composition after the concentration step may contain 0.01% by mass or more of potassium in the composition.
- the concentration step in the present invention is to adjust the amount of water or the like in the graphite oxide-containing composition.
- centrifugal separation, water, etc. are added to redisperse, filter, and agglomerate. It can carry out using methods, such as filtration using an agent, and vacuum concentration.
- these processes may be repeated, it is preferable to complete by one process, without repeating.
- the flocculant there is no particular limitation on the flocculant in the case of concentrating with the flocculant, but those that do not interfere with the subsequent modification reaction are preferred.
- the flocculant include polymeric flocculants that do not react with oxygen functional groups and sulfuric acid of graphite oxide, and volatile flocculants that can be easily removed at the reaction temperature during the reaction.
- a graphite oxide-containing composition In the production method of the present invention, a graphite oxide-containing composition, typically a graphite oxide-containing composition, typically an oxidation containing a higher concentration of graphite oxide, is obtained from a reaction liquid containing graphite oxide obtained in the oxidation step. It is preferable to include a step of separating the graphite-containing composition. That is, in still another preferred embodiment of any one of the methods for producing a graphite oxide derivative of the present invention, the solubility in water in the reaction composition is 0.01 between the oxidation step and the step of obtaining the graphite oxide derivative. %, And after adding a solvent which is not optionally miscible with water, a step of separating the graphite oxide-containing composition is included.
- the present invention is also a method for producing a graphite oxide derivative, which comprises a step of oxidizing graphite, graphite oxide in a graphite oxide-containing composition obtained in the oxidation step, and oxygen of the graphite oxide.
- It is also a method for producing a graphite oxide derivative comprising a step of separating a graphite oxide-containing composition containing a high concentration of graphite oxide.
- This production method may include a step of purifying and drying the graphite oxide-containing composition, but
- the added solvent When such a solvent that does not completely separate from water and is not completely miscible with water when added to water is added to a reaction solution containing graphite oxide, the added solvent is oxidized in the reaction solution.
- the graphite oxide particles exhibiting affinity with graphite adhere to the graphite oxide particles, and the graphite oxide particles to which the solvent adheres easily aggregate.
- the added solvent acts as a flocculant for aggregating the graphite oxide particles. As a result, the separation of the graphite oxide particles and the reaction liquid in the reaction liquid is promoted, and the graphite oxide is more efficiently separated. Is possible.
- the production method of the present invention includes the separation step, the present inventors can more efficiently produce a graphite oxide-containing composition and a graphite oxide derivative, and have a desired functional group. It has been found that high quality graphite oxide or graphite oxide derivatives in which groups are sufficiently introduced can be obtained.
- the method for producing a graphite oxide derivative is also one of the preferred embodiments of the present invention (also referred to as a second preferred embodiment).
- a solvent having a solubility in water of 0.01% or more and optionally immiscible with water is added to a reaction solution containing graphite oxide. More preferably, the method further includes a step of separating the graphite oxide-containing composition.
- the solvent added to the reaction solution containing graphite oxide in the separation step may be any solvent that has a solubility in water of 0.01% or more and is not arbitrarily miscible with water.
- the solubility is preferably 0.5% or more. With such a solubility, the effect of agglomerating graphite oxide by being appropriately mixed with water is more sufficiently exhibited.
- the solubility of the solvent in water is more preferably 0.7% or more, still more preferably 1% or more, and particularly preferably 1.5% or more. Further, the solubility of the solvent is preferably 30% or less, more preferably 20% or less, still more preferably 15% or less, and particularly preferably 10% or less.
- the solubility of the solvent in water can be measured by a solution precipitation method.
- the solubility in water is 0.01% or more
- the solvent dissolves 0.01% by mass or more with respect to 100% by mass of water, in other words, 0.01 g or more of the solvent dissolves with respect to 100 g of water. Represents what to do.
- the amount of the solvent added to the reaction solution containing graphite oxide in the separation step may be set as appropriate, but is 1 to 1000% by mass with respect to 100% by mass of graphite oxide in the reaction solution containing graphite oxide. Is preferred. By adding such an amount of the solvent, the graphite oxide can be more sufficiently aggregated and the separation can proceed more efficiently.
- the amount of the solvent added is more preferably 1 to 950% by mass, still more preferably 10 to 900% by mass, particularly preferably 100% by mass with respect to 100% by mass of graphite oxide in the reaction liquid containing graphite oxide. 100 to 800% by mass.
- Solvents having a solubility in water of 0.01% or more and not arbitrarily miscible with water include cyclic alkanones such as cyclopentanone and cyclohexanone; carbon numbers such as butanol, pentanol, hexanol and heptanol. Alcohols of 7 to 7; ketones such as acetylacetone, methyl ethyl ketone, methyl propyl ketone, methyl butyl ketone, and the like, and one or more of these can be used.
- the method of separating the graphite oxide-containing composition after adding a solvent having a predetermined solubility to the reaction liquid containing graphite oxide in the separation step is particularly effective as long as the graphite oxide-containing composition is separated from the reaction liquid.
- it is preferably performed by any one of filtration, decantation, centrifugation, and liquid separation extraction. By using these methods, the graphite oxide-containing composition can be more efficiently separated from the reaction solution.
- these operations may be performed once or a plurality of times. Moreover, only any one of these may be performed and it may carry out in combination of 2 or more.
- Filtration is the simplest method among the above separation methods, but the conventional method for producing graphite oxide and its derivatives is prone to clogging of the filter due to graphite oxide, and it takes time to filter. .
- the separation step aggregation of graphite oxide particles is promoted in the reaction liquid by the action of a solvent having a solubility in water within a predetermined range, and thus the filter is clogged even if the reaction liquid is filtered.
- the time required for filtration is much shorter than the filtration step in the conventional method for producing graphite oxide and its derivatives. For this reason, by using filtration as a method for separating the graphite oxide-containing composition, separation of the reaction solution and graphite oxide can be easily and efficiently proceeded.
- a graphite oxide-containing composition is separated after adding a solvent that has a solubility in water of 0.01% or more and is not miscible with water to a reaction solution containing graphite oxide.
- the graphite oxide-containing composition separated from the reaction liquid may further include another separation process that is suitable for the subsequent reaction process.
- Other separation steps include washing with water and the like.
- the above-described preferred embodiments of the concentration step can be applied as long as the effects of the present invention are exhibited.
- the preferred embodiment of the graphite oxide-containing composition after the separation step is the same as the preferred embodiment of the graphite oxide-containing composition after the concentration step described above.
- the step of oxidizing the graphite is not particularly limited as long as the graphite is oxidized, and any method such as the above-described Hummers method, Brodie method, or Staudenmaier method may be used. It may be a step of adding permanganate to a mixed solution containing graphite and sulfuric acid, which employs the oxidation method in the Hummers method, as in the method described in the examples described later. Thus, it is one of the preferred embodiments of the present invention that the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid.
- the oxidation step oxidizes graphite using an acid.
- the production method of the present invention is used for the step of obtaining a graphite oxide derivative in order to omit at least one of purification and drying of the graphite oxide-containing composition obtained in the oxidation step.
- the acid remains in the graphite oxide-containing composition to be formed, and the acid stabilizes the oxygen functional group contained in the graphite oxide.
- the modification reaction can proceed with high efficiency.
- the step of obtaining the graphite oxide derivative it is possible to use it for the modification reaction while stably presenting the oxygen functional group contained in the graphite oxide, and the production method of the present invention is simpler and more efficient. can do.
- the said oxidation process is a process of adding permanganate to the liquid mixture containing graphite and a sulfuric acid.
- the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid
- the amount of sulfuric acid used is such that the mass ratio of sulfuric acid to graphite (sulfuric acid / graphite) is 25-60.
- the mass ratio is 25 or more
- graphite oxide can be efficiently produced by sufficiently preventing the reaction solution (mixed solution) from becoming highly viscous during the oxidation reaction.
- the mass ratio is 60 or less, the amount of waste liquid can be sufficiently reduced.
- the mass ratio is more preferably 26 or more, further preferably 27 or more, and particularly preferably 28 or more.
- the mass ratio is more preferably 54 or less, still more preferably 48 or less, and particularly preferably 42 or less.
- the graphite used in the oxidation step preferably has an average particle size of 3 ⁇ m or more and 80 ⁇ m or less. By using a material having such an average particle size, the oxidation reaction can be advanced more efficiently.
- the average particle diameter of graphite is more preferably 3.2 ⁇ m or more and 70 ⁇ m or less. The average particle diameter can be measured by a particle size distribution measuring device.
- the shape of the graphite used for the oxidation step is not particularly limited, and examples thereof include fine powder, powder, granule, granule, scale, polyhedron, rod, and curved surface-containing shape.
- the particles having the average particle diameter in the above range are produced by, for example, a method of pulverizing the particles with a pulverizer, a method of selecting the particle diameter by sieving the particles, a combination of these methods, or the like. It is possible to manufacture by optimizing the preparation conditions at the stage of obtaining and obtaining particles having a desired particle diameter.
- the graphite content in the mixed solution containing graphite and sulfuric acid is preferably 0.5% by mass or more, more preferably 1% by mass or more, with respect to 100% by mass of the mixed solution.
- the content is more preferably 5% by mass or more, and particularly preferably 2% by mass or more.
- the graphite content is preferably 10% by mass or less, more preferably 8% by mass or less, still more preferably 7% by mass or less, and particularly preferably 6% by mass or less. Only one type of graphite may be used in the oxidation step in the production method of the present invention, or two or more types different in any of the average particle diameter, shape, and the like may be used.
- the permanganate added in the oxidation step includes sodium permanganate, potassium permanganate, permanganate.
- Ammonium acid, silver permanganate, zinc permanganate, magnesium permanganate, calcium permanganate, barium permanganate, and the like can be used, and one or more of these can be used, among which sodium permanganate , Potassium permanganate is preferred, and potassium permanganate is more preferred.
- the total amount of the permanganate added in the oxidation step is 100% by mass of graphite in the mixed solution.
- the content is preferably 50 to 500% by mass.
- graphite oxide can be manufactured safely and efficiently.
- the amount of oxygen atoms introduced into the graphite oxide can be adjusted by changing the total amount of the oxidizing agent added.
- the total addition amount is more preferably 100% by mass or more, further preferably 150% by mass or more, further preferably 200% by mass or more, and particularly preferably 240% by mass or more. Further, the total addition amount is more preferably 450% by mass or less, further preferably 400% by mass or less, further preferably 350% by mass or less, and particularly preferably 300% by mass or less. preferable.
- the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid
- the permanganate may be added all at once or added in multiple times. Alternatively, it may be added continuously, but it is preferable to add in multiple portions or add continuously. Thereby, it is possible to control the reaction more easily by suppressing the rapid progress of the oxidation reaction.
- the number of times of addition is preferably 3 times or more, more preferably 5 times or more, further preferably 7 times or more, 9 times The above is particularly preferable.
- the amount added per time may be the same or different.
- the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid
- permanganic acid is maintained while maintaining the temperature of the mixed solution within a range of 10 to 50 ° C. It is preferred to add a salt. By maintaining in such a temperature range, it is possible to sufficiently proceed while controlling the oxidation reaction.
- the temperature is preferably maintained at 12 ° C or higher, more preferably maintained at 15 ° C or higher, further preferably maintained at 18 ° C or higher, and particularly preferably maintained at 20 ° C or higher.
- the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid
- the oxidation step adds permanganate while maintaining the temperature change of the mixed solution at 25 ° C. or lower. It is preferable that it is a process to perform. Thereby, an oxidation process can be performed more stably.
- the temperature change is more preferably maintained at 20 ° C. or less, further preferably maintained at 15 ° C. or less, and particularly preferably maintained at 10 ° C. or less.
- the oxidation step is a step of adding permanganate to a mixed solution containing graphite and sulfuric acid
- the permanganate is added for 10 minutes to 10 hours from the viewpoint of performing the oxidation step stably. It is preferable to add over the period. More preferably, the permanganate is added over 30 minutes or more, more preferably over 1 hour or more, particularly preferably over 2 hours or more. It is. Further, from the viewpoint of efficiently producing graphite oxide, the addition time of permanganate is preferably 8 hours or less, preferably 7 hours or less, and more preferably 6 hours or less.
- the oxidation step is preferably performed while stirring using a known stirrer or the like.
- the oxidation step can be performed, for example, in air or in an inert gas atmosphere such as nitrogen, helium, or argon.
- the said pressure process does not specifically limit the said oxidation process, For example, it is preferable to carry out on normal-pressure conditions.
- the time for the oxidation step is preferably 0.5 hours to 120 hours, more preferably 1 hour to 15 hours, and further preferably 2 hours to 10 hours.
- the oxidation step may be performed continuously or intermittently.
- the liquid mixture can be obtained by mixing graphite, sulfuric acid, and other components as required.
- the mixing can be appropriately performed by a known method. For example, it is preferable to uniformly disperse graphite by performing ultrasonic treatment or using a known disperser.
- the method for producing a graphite oxide derivative of the present invention includes a step of oxidizing graphite and a step of obtaining a graphite oxide derivative described later, and does not include a step of purifying and drying the graphite oxide-containing composition between both steps. As long as it includes other steps such as an aging step and an oxidation reaction stop (quenching) step after the oxidation step.
- the temperature and time for aging the reaction solution obtained in the oxidation step may be appropriately selected, but the reaction solution is preferably maintained at a temperature of 0 to 90 ° C., more preferably 20 to 80 ° C. To maintain a temperature of °C.
- the aging time is preferably 0.1 to 24 hours. More preferably, it is 0.5 to 5 hours.
- the oxidation reaction stopping step may be performed in air or in an inert gas atmosphere such as nitrogen, helium, or argon. Moreover, you may carry out in a vacuum.
- the oxidation reaction stopping step can be performed, for example, by setting the temperature of the reaction solution to 5 to 15 ° C., adding water to the reaction solution, and then adding hydrogen peroxide as a reducing agent. Alternatively, the reaction solution may be added to water or hydrogen peroxide set at 5 to 25 ° C.
- the time for the oxidation reaction stopping step can be, for example, 0.01 to 5 hours.
- a graphite oxide derivative is obtained by reacting graphite oxide in the graphite oxide-containing composition obtained in the oxidation step with a compound that reacts with the oxygen-containing functional group of the graphite oxide.
- the compound that reacts with the oxygen-containing functional group of graphite oxide is preferably at least one selected from the group consisting of alcohol, silane compound, fatty acid (salt), fatty acid ester, isocyanate compound, and amine, for example. Of these, alcohol and / or amine are more preferable.
- the silane compound preferably has a siloxy group and / or an alkoxy group directly bonded to a silicon atom from the viewpoint of improving the reactivity with the oxygen-containing functional group of graphite oxide.
- generation of a graphite oxide derivative is confirmed by measuring an infrared absorption spectrum along the method of an Example.
- the compound that reacts with the oxygen-containing functional group of graphite oxide preferably has a hydrocarbon group from the viewpoint of further improving the dispersibility of the graphite oxide derivative in the nonpolar dispersion medium.
- the hydrocarbon group preferably has 5 or more carbon atoms, more preferably 6 or more, still more preferably 7 or more, and particularly preferably 8 or more.
- the hydrocarbon group has a sufficiently high dispersion rate in the non-polar dispersion medium of the graphite oxide derivative, and has a carbon number of 50 or less from the viewpoint of suitably producing the graphite oxide derivative. Is preferably 36 or less, more preferably 24 or less.
- the hydrocarbon group is preferably linear.
- the straight chain hydrocarbon group is easily introduced into the graphite oxide, so that the production method of the present invention becomes more efficient.
- the hydrocarbon group is a branched chain.
- the dispersibility of the graphite oxide derivative in the nonpolar dispersion medium is further improved.
- the compound as a raw material tends to become liquid at room temperature (25 ° C.), and in that case, handling at the time of producing the derivative is easy.
- the compound that reacts with the oxygen-containing functional group of the graphite oxide is preferably an alcohol and / or an amine. More preferably, the alcohol is an aliphatic alcohol. The amine is more preferably an aliphatic amine.
- Examples of the aliphatic alcohol include n-octyl alcohol, sec-octyl alcohol, n-nonyl alcohol, sec-nonyl alcohol, n-decyl alcohol, sec-decyl alcohol, n-undecyl alcohol, sec-undecyl alcohol.
- Examples of the aliphatic amine include n-octylamine, sec-octylamine, n-nonylamine, sec-nonylamine, n-decylamine, sec-decylamine, n-undecylamine, sec-undecylamine, and n-dodecyl.
- a compound that reacts with the oxygen-containing functional group of graphite oxide in the graphite oxide-containing composition obtained in the oxidation step;
- other components for example, a solvent
- the mixing can be appropriately performed by a known method, but it is preferable to uniformly disperse graphite oxide by, for example, performing ultrasonic treatment or using a known disperser.
- the amount of the compound that reacts with the oxygen-containing functional group of the graphite oxide is preferably 300 to 10,000% by mass with respect to 100% by mass of the graphite oxide in the mixed solution.
- the amount used is more preferably 350% by mass or more, still more preferably 400% by mass or more, still more preferably 450% by mass or more, and particularly preferably 500% by mass or more.
- the amount used is more preferably 8000% by mass or less, further preferably 6000% by mass or less, further preferably 3000% by mass or less, and particularly preferably 1000% by mass or less. .
- a known catalyst or the like can be applied.
- an acid catalyst such as sulfuric acid, an alkali metal, etc.
- a base catalyst such as hydroxide, amine, pyridine or the like can be used.
- the compound that reacts with the oxygen-containing functional group of graphite oxide is an amine
- the amine itself can be used as a catalyst.
- the compound that reacts with the oxygen-containing functional group of graphite oxide is a compound other than an amine and an acid catalyst is used as a catalyst in the process of obtaining a graphite oxide derivative, such as a compound that reacts with the oxygen-containing functional group of graphite oxide or graphite oxide
- a preferable content of the acid catalyst with respect to 100% by mass of graphite oxide in the composition is, for example, 0.01 to 1000% by mass. .
- the modification reaction can proceed efficiently. Further, when the content is 1000% by mass or less, the amount of waste (amount of waste liquid) can be sufficiently reduced and the modification reaction can be efficiently advanced.
- the acid catalyst an acid residue such as sulfuric acid used in the oxidation step can be effectively used.
- the content of the acid catalyst is more preferably 0.1% by mass or more, further preferably 1% by mass or more, further preferably 10% by mass or more, and 20% by mass or more. Is more preferably 30% by mass or more, still more preferably 40% by mass or more, and particularly preferably 50% by mass or more. Further, the content is more preferably 700% by mass or less, further preferably 500% by mass or less, and further preferably 200% by mass or less.
- the acid of the acid catalyst is sulfuric acid
- the graphite oxide-containing composition used for the reaction in the step of obtaining the graphite oxide derivative has 1 sulfuric acid with respect to 100% by mass of the graphite oxide in the composition. It is preferable to contain at least 1000 mass%.
- the compound that reacts with the oxygen-containing functional group of graphite oxide is a compound other than an amine, and an acid catalyst is used as a catalyst in the step of obtaining a graphite oxide derivative
- the graphite oxide contained in the reaction of the step of obtaining the graphite oxide derivative It is preferable that the composition contains 0.1% by mass or more and 50% by mass or less of the acid catalyst with respect to 100% by mass of the compound that reacts with the oxygen-containing functional group of graphite oxide in the composition.
- the modification reaction can proceed more efficiently.
- the side reaction resulting from an acid catalyst can fully be suppressed by containing an acid catalyst 50 mass% or less.
- the content of the acid catalyst is more preferably 0.5% by mass or more. Further, the content of the acid catalyst is more preferably 20% by mass or less.
- graphite oxide is essentially an acidic substance, and the reaction proceeds autocatalytically.
- a catalyst it is preferable to use a catalyst as described above, it is possible to allow the reaction to proceed autocatalytically without using a catalyst.
- the amount of the base catalyst with respect to the amount of the reaction raw material such as graphite oxide or a compound that reacts with the oxygen-containing functional group of graphite oxide is preferably in the range. By adjusting inward, the modification reaction can proceed more efficiently.
- the preferable content of the base catalyst with respect to 100 mass% of the graphite oxide is 100 mass% of the graphite oxide in the above-described graphite oxide-containing composition. This is the same as the preferred acid content.
- the graphite oxide-containing composition subjected to the reaction in the step of obtaining a graphite oxide derivative is an alkali with respect to 100% by mass of the graphite oxide in the composition. It is preferable to contain 1% by mass or more and 1000% by mass or less of metal hydroxide and amine.
- a base catalyst neutralizes with the acid component in a graphite oxide containing composition, it is preferable that the quantity of the part except the neutralized part corresponds to the said range among the used base catalysts.
- the reaction temperature in the step of obtaining the graphite oxide derivative is the reaction temperature.
- the reaction temperature includes a step of obtaining graphite oxide derivatives having a functional group having a hydrocarbon group by reacting graphite oxide with alcohol and / or amine at a reaction temperature of 120 ° C. or higher.
- the reaction temperature is more preferably 130 ° C. or higher, and still more preferably 140 ° C. or higher. 150 ° C. or higher is particularly preferable. From the viewpoint of suppressing side reactions, the reaction temperature is preferably 200 ° C. or lower. That is, a graphite oxide derivative having a functional group having a hydrocarbon group is obtained by reacting graphite oxide with alcohol and / or an amine at a reaction temperature of 120 ° C.
- the first preferred embodiment of the production method of the present invention or the second preferred embodiment of the production method of the present invention is a hydrocarbon group obtained by reacting graphite oxide with alcohol and / or amine at a reaction temperature of 120 ° C. or higher. It is more preferable to include a step of obtaining a graphite oxide derivative having a functional group having.
- a method for producing a graphite oxide derivative comprising a step of obtaining a graphite oxide derivative having a functional group having a hydrocarbon group by reacting graphite oxide and alcohol at a reaction temperature of 120 ° C. or higher, the graphite oxide is obtained in the oxidation step.
- it may be obtained by purifying and drying a graphite oxide-containing composition containing graphite oxide, it is preferable that at least one of purification and drying is omitted, and purification is omitted. More preferred.
- the reduction reaction can be sufficiently suppressed to increase the amount of alcohol and / or amine introduced into graphite oxide, and a high-quality graphite oxide derivative can be obtained with high efficiency.
- the graphite oxide derivative obtained by this production method has high crystallinity due to the interaction between the introduced alcohol and / or amine-derived hydrocarbon groups.
- the crystallinity of the graphite oxide derivative obtained in the said manufacturing method, there exists a tendency for the crystallinity of the graphite oxide derivative obtained to become higher, so that there are many carbon numbers of alcohol and / or amine, but even if there are few carbon numbers of alcohol and / or amine.
- the number of hydrocarbon groups introduced into graphite oxide can be made sufficiently large, and a high-quality graphite oxide derivative can be obtained.
- the present invention is also a graphite oxide derivative having an alkyl group and having a grade of 8 grade measured by a drop sensitivity test defined in JIS K 4810.
- a graphite oxide derivative having an alkyl group and having a grade of 8 grade measured by a drop sensitivity test defined in JIS K 4810.
- the alkyl group has an alkyl group and the sensitivity of the sensitivity by the sensitivity test is sufficiently reduced. It is possible to obtain (disappeared) high quality graphite oxide derivatives.
- the present invention further shows at least one peak at 9-13 ° and 21-24 ° in the X-ray diffraction spectrum, and the crystallite diameter calculated by the Scherrer equation of the peak at 9-13 ° is 100 mm or more. It is also a graphite oxide derivative characterized in that it is 500 ⁇ or less. For example, in the production method of the present invention, it is possible to obtain a high quality graphite oxide derivative exhibiting such a peak by reacting graphite oxide with alcohol and / or amine.
- the crystallite diameter calculated by Scherrer's formula of the peak at 9-13 ° is 100 ⁇ or more and 500 ⁇ or less” means that when there are a plurality of peaks at 9-13 °, any one of the peaks
- the crystallite diameter calculated by Scherrer's formula may be 100 to 500 mm.
- the graphite oxide derivative of the present invention preferably exhibits one peak at 9-13 ° and 21-24 ° in the X-ray diffraction spectrum.
- the crystallite diameter is more preferably 200 mm or more. Furthermore, it is more preferable that the crystallite diameter is 400 mm or less.
- the X-ray diffraction spectrum can be measured by the “XRD measurement method” in the examples.
- the reaction time in the step of obtaining the graphite oxide derivative is, for example, preferably 1 hour or more, more preferably 3 hours or more, and further preferably 5 hours or more.
- the reaction time is preferably 120 hours or less, more preferably 100 hours or less, and more preferably 80 hours or less from the viewpoint of proceeding with the modification reaction while suppressing the reduction reaction sufficiently. Further preferred.
- the said reaction process can be performed, stirring using a well-known stirrer etc.
- the said reaction process can be performed in air or inert gas atmosphere, such as nitrogen, helium, and argon, for example.
- the pressure condition of the reaction step is not particularly limited, and can be performed under a pressurized condition, a normal pressure condition, and a reduced pressure condition. For example, it is preferably performed under a normal pressure condition.
- a volatile flocculant is used in the concentration step, it is preferable to perform distillation of impurities such as the flocculant by heating under reduced pressure before the reaction step. Thereby, bumping of a flocculant etc. and side reaction can fully be prevented in a reaction process, and it is possible to advance a reaction process stably.
- the graphite oxide derivative obtained by the production method of the present invention has a functional group derived from a compound that reacts with the oxygen-containing functional group of graphite oxide (preferably a functional group having a hydrocarbon group).
- the functional group is not particularly limited, and includes an oxygen-containing group such as an alkoxycarbonyl group (—COOR) or an alkoxyl group (—OR); a sulfur-containing group; an alkylamino group (—NHR, —NRR ′), an alkylamide group ( Nitrogen-containing groups such as —CONHR, —CONRR ′); phosphorus-containing groups, etc., including oxygen-containing groups such as alkoxycarbonyl groups (—COOR) and alkoxyl groups (—OR), alkylamino groups (—NHR, — NRR ′) and a nitrogen-containing group such as an alkylamide group (—CONHR, —CONRR ′) are preferable.
- R and R ' are the same or different and each represents an organic
- the graphite oxide derivative obtained by reacting graphite oxide with alcohol contains carbon atoms, hydrogen atoms, and other atoms other than oxygen atoms in 100% by mass of the graphite oxide derivative. Is preferably 10% by mass or less, more preferably 7% by mass or less, and still more preferably 5% by mass or less.
- the graphite oxide derivative is particularly preferably free from other atoms. In other words, it is preferable that the graphite oxide derivative has only carbon atoms, hydrogen atoms, and oxygen atoms as constituent elements. Examples of other atoms include a nitrogen atom, a phosphorus atom, and a halogen atom.
- the graphite oxide derivative preferably has a nitrogen atom content of 0.1% by mass or less in 100% by mass of the graphite oxide derivative.
- the graphite oxide derivative obtained by reacting graphite oxide with an amine is a carbon atom, a hydrogen atom, an oxygen atom, and other than nitrogen atoms in 100% by mass of the graphite oxide derivative.
- the content of atoms is preferably 10% by mass or less, more preferably 7% by mass or less, and still more preferably 5% by mass or less.
- the graphite oxide derivative is particularly preferably free from other atoms. In other words, it is preferable that the graphite oxide derivative has only carbon atoms, hydrogen atoms, oxygen atoms, and nitrogen atoms as constituent elements. Examples of other atoms include phosphorus atoms and halogen atoms.
- the graphite oxide derivative preferably has an average particle size of 0.01 ⁇ m or more and 100 ⁇ m or less.
- the average particle diameter is more preferably 0.1 ⁇ m or more, further preferably 1 ⁇ m or more, and further preferably 3 ⁇ m or more.
- the average particle diameter is more preferably 60 ⁇ m or less.
- the average particle diameter can be measured by a particle size distribution measuring device.
- Examples of the shape of the graphite oxide derivative include fine powder, powder, granule, granule, scale, polyhedron, rod, and curved surface.
- the particles having the average particle size in the above range are, for example, pulverized by a ball mill or the like, and the coarse particles obtained by the pulverization are dispersed in a dispersant to obtain a desired particle size, and then dried.
- a method of screening the particle size by sieving the particle a combination of these methods, a method for obtaining (nano) particles having a desired particle size by optimizing the preparation conditions at the stage of particle production, etc. It is possible.
- the graphite oxide derivative has a functional group having a hydrocarbon group having 13 or more carbon atoms.
- This preferred embodiment can be applied to each of the first to third preferred embodiments.
- the present invention is also a graphite oxide derivative having a functional group having a hydrocarbon group having 13 or more carbon atoms.
- the hydrophilic dispersoid is most difficult to disperse, and the dispersion of graphite oxide and its derivatives has been reported so far.
- the hydrocarbon group having 13 or more carbon atoms is not particularly limited, and is a saturated aliphatic hydrocarbon group such as an alkyl group or a cycloalkyl group; an acyclic unsaturated aliphatic hydrocarbon group such as an alkynyl group or an alkenyl group; Any of aromatic hydrocarbon groups such as an aryl group may be used, but among them, a saturated aliphatic hydrocarbon group is preferable, and an alkyl group is more preferable.
- alkyl group examples include n-tetradecyl group, sec-tetradecyl group, n-hexadecyl group, sec-hexadecyl group, n-octadecyl group, sec-octadecyl group, n-eicosyl group, sec-eicosyl group, 2- Octyldodecyl, n-docosyl, sec-docosyl, 2-octyltetradecyl, n-tetracosyl, sec-tetracosyl, 2-octylhexadecyl, n-hexacosyl, sec-hexacosyl, n- Octacosyl group, sec-octacosyl group, n-triacontyl group, sec-triacontyl group, n-dotriacontyl group, sec-dotriacontyl group, n-tetratriacon
- the hydrocarbon group in the graphite oxide derivative of the present invention preferably has 14 or more carbon atoms, and 16 or more. More preferably, it is more preferably 18 or more.
- the hydrocarbon group in the graphite oxide derivative of the present invention has a sufficiently high dispersion rate in the nonpolar dispersion medium of the graphite oxide derivative of the present invention, and the graphite oxide derivative of the present invention is preferably produced.
- the number of carbon atoms is preferably 50 or less, and more preferably 36 or less.
- the hydrocarbon group in the graphite oxide derivative of the present invention preferably has 20 to 28 carbon atoms. Thereby, the above-described effects can be achieved in a balanced manner.
- the carbon number is more preferably 21 or more, and still more preferably 22 or more. Further, the carbon number is more preferably 24 or less, and most preferably 24.
- the present invention is also a graphite oxide derivative having a functional group having a hydrocarbon group having 6 to 10 carbon atoms. It has been found that when the number of carbon atoms is 6 or more and 10 or less, affinity for an amphiphilic dispersion medium such as a ketone-based, ester-based, or amide-based solvent can be imparted, and good dispersibility can be achieved. Therefore, the graphite oxide derivative can be applied as an additive to various resins.
- the hydrocarbon group having 6 to 10 carbon atoms is not particularly limited, and is a saturated aliphatic hydrocarbon group such as an alkyl group or a cycloalkyl group; an acyclic unsaturated aliphatic carbon group such as an alkynyl group or an alkenyl group. Any of a hydrogen group; an aromatic hydrocarbon group such as an aryl group may be used, but among them, a saturated aliphatic hydrocarbon group is preferable, and an alkyl group is more preferable.
- alkyl group examples include n-hexyl group, sec-hexyl group, n-heptyl group, sec-heptyl group, n-octyl group, sec-octyl group, n-nonyl group, sec-nonyl group, 2- An octyldodecyl group, an n-decyl group, a sec-decyl group and the like can be mentioned, and one or more of these can be used.
- the hydrocarbon group in the graphite oxide derivative of the present invention preferably has 7 to 9 carbon atoms.
- the hydrocarbon group in the graphite oxide derivative of the present invention is most preferably 8 carbon atoms.
- the graphite oxide derivative having a functional group having a hydrocarbon group having 13 or more carbon atoms and the graphite oxide derivative having a functional group having a hydrocarbon group having 6 to 10 carbon atoms have been described above.
- the graphite oxide derivative of the present invention preferably has a functional group having the above-mentioned hydrocarbon group at the terminal.
- the hydrocarbon group in the graphite oxide derivative of the present invention is preferably linear.
- the hydrocarbon group is easily introduced into the graphite oxide, so that the graphite oxide derivative of the present invention can be suitably obtained.
- the hydrocarbon group in the graphite oxide derivative of the present invention is a branched chain.
- the hydrocarbon group is a branched chain, the dispersibility of the graphite oxide derivative of the present invention in a nonpolar dispersion medium is further improved.
- the hydrocarbon group-containing compound as a raw material tends to become liquid at room temperature (25 ° C.), and in that case, handling during production of the derivative is easy.
- the functional group having a hydrocarbon group having 13 or more carbon atoms or 6 to 10 carbon atoms is not particularly limited, and oxygen-containing groups such as alkoxycarbonyl groups (—COOR) and alkoxyl groups (—OR); Groups; nitrogen-containing groups such as alkylamino groups (—NHR, —NRR ′), alkylamide groups (—CONHR, —CONRR ′); phosphorus-containing groups, etc., including alkoxycarbonyl groups (—COOR), alkoxyl groups Oxygen-containing groups such as (—OR), nitrogen-containing groups such as alkylamino groups (—NHR, —NRR ′) and alkylamide groups (—CONHR, —CONRR ′) are preferred.
- oxygen-containing groups such as alkoxycarbonyl groups (—COOR) and alkoxyl groups (—OR); Groups; nitrogen-containing groups such as alkylamino groups (—NHR, —NRR ′), alkylamide groups (—CONHR
- R and R ′ are the same or different and each represents a hydrocarbon group having 13 or more carbon atoms or 6 or more and 10 or less carbon atoms. That is, the portion other than the hydrocarbon group in the functional group is preferably, for example, —COO—, —O—, —NH—, —N—, —CONH—, or —CON—.
- the preferred average particle size and shape of the graphite oxide derivative and the method for obtaining particles having a desired particle size are the preferred average particle size, shape and desired particle size of the graphite oxide derivative obtained by the production method of the present invention described above. It is the same as the method of obtaining.
- a method particularly suitable for producing the graphite oxide derivative of the present invention will be briefly described. However, the following method can also be suitably applied to the above-described method for producing the graphite oxide derivative of the present invention. .
- the present invention is also a method for producing a graphite oxide derivative that obtains a graphite oxide derivative by reacting graphite oxide with a hydrocarbon group-containing compound having 13 or more carbon atoms.
- the present invention is also a method for producing a graphite oxide derivative, which is obtained by reacting graphite oxide with a hydrocarbon group-containing compound having 6 to 10 carbon atoms.
- the hydrocarbon group-containing compound include amines, isocyanate group-containing compounds, carbonyl group-containing compounds (for example, carboxylic acid halides), alcohols, and the like.
- the hydrocarbon group-containing compound is preferably an alcohol and / or an amine.
- alcohol what corresponds to the hydrocarbon group mentioned above can be used suitably.
- amine one corresponding to the above-described hydrocarbon group (one having a hydrocarbon group bonded to a nitrogen atom) can be used as appropriate.
- any known catalyst can be applied.
- sodium hydroxide, potassium hydroxide, calcium hydroxide can be used as a catalyst.
- a base catalyst such as amine or pyridine or an acid catalyst such as sulfuric acid can be used.
- generation of the graphite oxide derivative of this invention is confirmed by measuring an infrared absorption spectrum along the method of an Example.
- the amount of the base catalyst used is 100 mass of graphite oxide in the mixed solution used in the reaction step. % To 0.01 to 1000% by mass. Thereby, a graphite oxide derivative can be manufactured efficiently.
- the amount of the base catalyst used is more preferably 0.1% by mass or more, and further preferably 1% by mass or more.
- the amount used is more preferably 500% by mass or less.
- the usage-amount of a base catalyst means the preparation amount of the base catalyst used in order to produce the said liquid mixture.
- the amount of the acid catalyst used is the same as the preferable content of the acid catalyst with respect to 100% by mass of graphite oxide in the above-described graphite oxide-containing composition.
- the usage-amount of an acid catalyst means the preparation amount of the acid catalyst used in order to produce the said liquid mixture.
- Graphite oxide is essentially an acidic substance, and the reaction proceeds in an autocatalytic manner. Although it is more preferable to add a catalyst as mentioned above, it is also possible to advance the reaction in an autocatalytic manner without adding a catalyst.
- the preferred use amount of the hydrocarbon group-containing compound having 13 or more carbon atoms or 6 or more and 10 or less carbon atoms in the reaction step is as described above. It is the same as the preferable usage-amount of the compound which reacts with the oxygen containing functional group of the graphite oxide in this invention.
- the amount of the hydrocarbon group-containing compound having 13 or more carbon atoms or 6 or more and 10 or less carbon atoms in the mixed solution is the 13 or more carbon atoms or the carbon number used for preparing the mixed solution.
- the charge amount of the hydrocarbon group-containing compound of 6 or more and 10 or less is said.
- the mixed liquid can be obtained by mixing the graphite oxide, a hydrocarbon group-containing compound such as the alcohol, and the catalyst.
- the graphite oxide, the hydrocarbon group-containing compound, and the catalyst can be obtained using known methods, and commercially available products can also be used.
- the mixing can be appropriately performed by a known method, but it is preferable to uniformly disperse graphite oxide by, for example, ultrasonic treatment or using a known disperser.
- the said reaction process can be suitably performed by the method similar to the manufacturing method of this invention mentioned above.
- the reaction temperature may be, for example, 60 ° C. or higher. By setting the reaction temperature to 60 ° C. or higher, the reaction proceeds efficiently, but the reaction temperature may be the same as that of the production method of the present invention described above. preferable. The same applies to the reaction time.
- the graphite oxide derivative of the present invention has a functional group having a hydrocarbon group having 13 or more carbon atoms, it has excellent dispersibility in a nonpolar dispersion medium, and therefore, an additive for mechanical lubricating oil and an additive for resin Etc. can be used particularly preferably.
- an additive for mechanical lubricating oil and an additive for resin Etc. can be used particularly preferably.
- the graphite oxide derivative of the present invention has a functional group having a hydrocarbon group having 6 to 10 carbon atoms, it has excellent dispersibility in an amphiphilic dispersion medium, and therefore, as an additive to various resins. It can be particularly preferably used.
- the present invention is also a dispersion obtained by dispersing a graphite oxide derivative in a dispersion medium.
- the dispersion of the present invention can be obtained by dispersing the graphite oxide derivative of the present invention in a dispersion medium such as a nonpolar dispersion medium.
- Nonpolar dispersion media include, for example, aromatic hydrocarbon dispersion media having 6 to 14 carbon atoms such as benzene, xylene, toluene, cyclohexylbenzene, dihydrobenzofuran, trimethylbenzene, tetramethylbenzene, naphthalene, anthracene, pyridine, pyrazine C4-C6 aromatic heterocyclic compound dispersion media such as furan, pyrrole, thiophene, etc., fats having 5 or more carbon atoms such as pentane, hexane, heptane, octane, decane, dodecane, tetradecane, hexadecane, octadecane, cyclohexane Group hydrocarbon dispersion media and the like, and dispersion media such as mineral oil and synthetic oil. What mixed these 1 type (s) or 2 or more types can be used.
- the dispersion of the present invention can also be obtained by dispersing the graphite oxide derivative of the present invention in a dispersion medium such as an amphiphilic dispersion medium.
- Amphiphilic dispersion media include alcoholic dispersion media such as methanol, ethanol and propanol, amides such as N, N-dimethylformamide (DMF) and N-methylpyrrolidone (NMP), lactam dispersion media, acetone, butanone and pentanone.
- Examples thereof include ketone dispersion media such as ethylene glycol, ethylene glycol methyl ether, propylene glycol, glycols such as propylene glycol methyl ether, and glycol ether dispersion media.
- the mass ratio of the graphite oxide derivative to 100% by mass of the dispersion medium is preferably 0.0001% by mass or more, and more preferably 0.001% by mass or more. Moreover, it is preferable that this mass ratio is 10 mass% or less, It is more preferable that it is 1 mass% or less, It is still more preferable that it is 0.1 mass% or less.
- the present invention is a method for producing reduced graphite oxide, which comprises a step of oxidizing graphite and reducing graphite oxide in the graphite oxide-containing composition obtained in the oxidation step to reduce the graphite oxide.
- Reducing graphite oxide characterized by not including a step of purifying a graphite oxide-containing composition and drying between the oxidation step and the step of obtaining reduced graphite oxide It is also a manufacturing method. Thereby, reduced graphite oxide can be easily manufactured.
- a method for producing such reduced graphite oxide that is, a method for producing reduced graphite oxide obtained by reducing graphite oxide, the method comprising oxidizing graphite and oxidation obtained in the oxidation step A step of separating the graphite or graphite oxide-containing composition and a step of reducing the graphite oxide obtained in the separation step, wherein the separation step has a solubility in water of 0.01 in a reaction liquid containing graphite oxide. %, And after adding a solvent which is not optionally miscible with water, a method for producing reduced graphite oxide, comprising the step of separating graphite oxide or a graphite oxide-containing composition, It is one of the present inventions.
- the step of reducing graphite oxide is not particularly limited as long as the hydrophilic functional group is eliminated from graphite oxide and reduced, and NaBH 4 ,
- a method using a known reducing agent such as LiAlH 4 or L-ascorbic acid, electrolytic reduction, or the like can be used, but a method of reducing graphite oxide by heating is preferable.
- the temperature for heating the graphite oxide is preferably 100 ° C. or higher. More preferably, it is 120 ° C. or higher. Although there is no upper limit in particular in the heating temperature of graphite oxide, it is normally performed at 2000 degrees C or less.
- the time for heating the graphite oxide is preferably 0.1 to 100 hours. More preferably, it is 0.2 to 50 hours.
- the graphite oxide may be heated in the air or in an inert gas atmosphere such as nitrogen, helium, or argon. Moreover, you may carry out in a vacuum.
- a preferred embodiment of the step of oxidizing graphite in the method for producing reduced graphite oxide of the present invention and the step of separating the graphite oxide or graphite oxide-containing composition obtained in the oxidation step is the production of the above-described graphite oxide derivative of the present invention.
- the preferred forms of these steps in the method are the same.
- the method for producing reduced graphite oxide of the present invention includes a step of oxidizing graphite, a step of separating graphite oxide or a graphite oxide-containing composition obtained in the oxidation step, and reducing the graphite oxide obtained in the separation step. As long as a process is included, the other process may be included. Examples of the other steps include the oxidation reaction stopping step described above.
- the graphite oxide derivative obtained by the first to third preferred embodiments of the production method of the present invention and the graphite oxide derivative of the present invention are used as a catalyst, an electrode active material of a battery or a capacitor, a thermoelectric conversion material, a conductive material, and a light emitting material. It can be suitably used as a lubricant additive, a polymer additive, a permeable membrane material, an antibacterial material, a water repellent material, an adsorbing material, and the like.
- the graphite oxide derivative of the present invention has a hydrocarbon group having 13 or more carbon atoms introduced therein, it is excellent in dispersibility in a nonpolar dispersion medium, so that it is an additive for mechanical lubricating oil.
- the graphite oxide derivative of the present invention has a hydrocarbon group having 6 to 10 carbon atoms introduced therein, it is excellent in dispersibility in an amphiphilic dispersion medium, and thus can be applied to various resins. It can be particularly preferably used as an additive for the above.
- the battery include a lithium ion secondary battery, a polymer electrolyte fuel cell, and a metal-air battery.
- thermoelectric conversion device examples include, for example, a geothermal / hot spring thermal power generator, a solar heat power generator, a waste heat power generator such as a factory or an automobile, a power generator such as a body temperature power generator, Examples include various electric products, electric motors, artificial satellites, and the like used as one.
- graphite oxide obtained by the method for producing graphite oxide of the present invention and reduced graphite oxide obtained by the method for producing reduced graphite oxide of the present invention can also be suitably used for the aforementioned applications.
- ⁇ Method for measuring XPS> XPS measurement was performed using a photoelectron spectrometer (JPS-9000MX, manufactured by JEOL Ltd.). The peak separation in the narrow scan of C1s was performed by background fitting using the Shirley method and peak fitting using a Gauss-Lorentz function as a fitting function.
- ⁇ Measurement method of FT-IR> It measured by mixing a graphite oxide derivative with KBr and pelletizing it using Nicolet NEXTUS670 FTIR by Thermo Fisher Scientific Co., Ltd. Measurement range resolution at 900 ⁇ 4000 cm -1 was 1 cm -1.
- ⁇ Analysis of sulfuric acid concentration The concentration of sulfuric acid in the graphite oxide-containing composition was confirmed by neutralizing and titrating an aqueous graphite oxide dispersion with an aqueous sodium hydroxide solution.
- ⁇ Analysis of oxygen content> The amount of oxygen in graphite oxide or a derivative thereof was analyzed using JPS-9000MX manufactured by JEOL. The result was given as a ratio to carbon (C / O ratio).
- ⁇ Measurement method of FT-IR> It measured by mixing a graphite oxide derivative with KBr and pelletizing it using Nicolet NEXTUS670 FTIR by Thermo Fisher Scientific Co., Ltd. Measurement range resolution at 900 ⁇ 4000 cm -1 was 1 cm -1.
- ⁇ Dispersibility evaluation method> Using an APEL colorimeter AP-1000M, the time transition of the light transmittance at a wavelength of 660 nm in a dispersion of a 0.1 mg / ml graphite oxide derivative was measured. It was determined that the dispersibility was good when the time during which light transmission could be confirmed (the time when the transmittance was 0% ⁇ 1%) was 1 hour or longer.
- reaction liquid containing graphite oxide After the reaction, 890 ml of water and 30% hydrogen peroxide water (88 ml, manufactured by Wako Pure Chemical Industries, Ltd.) were added to stop the reaction while cooling the reactor in an ice bath to obtain a graphite oxide-containing composition (reaction liquid containing graphite oxide). It was.
- FIG. 1 is an FT-IR chart of graphite oxide produced in Preparation Example 1-2.
- a graphite oxide derivative was produced by the following steps.
- Example 1-1 The graphite oxide-containing composition obtained in Preparation Example 1-5 was transferred to a reactor, and to this was added 500% of 2-decyl-1-tetradecanol (Shin Nippon Rika Co., Ltd., Engecol 240A) with respect to graphite oxide. , And reacted at 150 ° C. for 5 hours. After the reaction, hexane was poured, filtered, washed with water and acetone, and the obtained solid was vacuum dried at 100 ° C. to obtain graphite oxide derivative A. The oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative A was 8.5.
- FIG. 2 is an FT-IR chart of graphite oxide derivative A produced in Example 1-1.
- FIG. 3 is an XRD chart of the graphite oxide derivative A produced in Example 1-1. As shown in FIG. 3, the graphite oxide derivative A has one peak at 9-13 ° and 21-24 ° in the X-ray diffraction spectrum, and is calculated by the Scherrer equation of the peak at 9-13 °. The crystallite diameter was 313 mm.
- Example 1-2 The graphite oxide derivative B was prepared by the method described in Example 1-1 except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-2, and 10% sulfuric acid was added to the graphite oxide as a catalyst. Obtained.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative B was 7.5.
- the modification amount was small and the C / O ratio was low. It was thought that the oxygen functional groups were reduced by the purification, and the amount of modification was small compared to Example 1-1.
- FIG. 4 is an FT-IR chart of graphite oxide derivative B produced in Example 1-2.
- Example 1-3 The graphite oxide derivative C was prepared by the method described in Example 1-1 except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-4, and 10% sulfuric acid was added to the graphite oxide as a catalyst. Obtained.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative C was 7.1.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional groups were reduced by purification and drying, and the amount of modification was small compared to Example 1-1.
- FIG. 5 is an FT-IR chart of graphite oxide derivative C produced in Example 1-3.
- Example 1-4 The graphite oxide derivative D was prepared by the method described in Example 1-1 except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-6, and 10% sulfuric acid was added to the graphite oxide as a catalyst. Obtained.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative D was 7.3.
- the modification amount was small and the C / O ratio was low. This was probably because oxygen functional groups were reduced by drying, and the amount of modification was small compared to Example 1-1.
- FIG. 6 is an FT-IR chart of graphite oxide derivative D produced in Example 1-4.
- Example 1-5 A graphite oxide derivative E was obtained by the method described in Example 1-1 except that 2-ethyl-1-hexanol (manufactured by Wako Pure Chemical Industries, Ltd.) was used instead of 2-decyl-1-tetradecanol. .
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative E was 6.8. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- FIG. 7 is an FT-IR chart of graphite oxide derivative E produced in Example 1-5.
- FIG. 7 is an FT-IR chart of graphite oxide derivative E produced in Example 1-5.
- FIG. 8 is an XRD chart of the graphite oxide derivative E produced in Example 1-5. As shown in FIG. 8, the graphite oxide E shows one peak at 9-13 ° and 21-24 ° in the X-ray diffraction spectrum, and is calculated by the Scherrer equation of the peak at 9-13 °. The crystallite diameter was 111.6 mm.
- Example 1-6 A graphite oxide derivative F was obtained by the method described in Example 1-2 except that 2-ethyl-1-hexanol (manufactured by Wako Pure Chemical Industries, Ltd.) was used instead of 2-decyl-1-tetradecanol. .
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative F was 6.2.
- the modification amount was small and the C / O ratio was low. It was thought that the oxygen functional groups were reduced by the purification, and the amount of modification was small compared to Example 1-5.
- FIG. 9 is an FT-IR chart of graphite oxide derivative F produced in Example 1-6.
- Example 1--7 A graphite oxide derivative G was obtained by the method described in Example 1-3 except that 2-ethyl-1-hexanol (manufactured by Wako Pure Chemical Industries, Ltd.) was used instead of 2-decyl-1-tetradecanol. .
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative G was 6.0.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional group was reduced by purification and drying, and the amount of modification was small compared to Example 1-5.
- FIG. 10 is an FT-IR chart of graphite oxide derivative G produced in Example 1-7.
- Example 1-8 A graphite oxide derivative H was obtained by the method described in Example 1-4 except that 2-ethyl-1-hexanol (manufactured by Wako Pure Chemical Industries, Ltd.) was used instead of 2-decyl-1-tetradecanol. .
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative H was 6.1.
- the modification amount was small and the C / O ratio was low. This was probably because oxygen functional groups were reduced by drying and the amount of modification was small compared to Example 1-5.
- FIG. 11 is an FT-IR chart of graphite oxide derivative H produced in Example 1-8.
- Example 1-9 Except that the reaction temperature was 100 ° C. and the reaction time was 24 hours, graphite oxide derivative I was obtained by the method described in Example 1-1. The oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative I was 7.1. Comparing the XRD patterns of Example 1-1 and Example 1-9, the graphite oxide derivative A of Example 1-1 is sharper and has higher crystallinity than the graphite oxide derivative I of Example 1-9. It was a thing. Moreover, the sensitivity by the calm sensitivity test was 7th grade.
- FIG. 12 is an FT-IR chart of graphite oxide derivative I produced in Example 1-9.
- FIG. 13 is an XRD chart of the graphite oxide derivative I produced in Example 1-9. As shown in FIG. 13, the graphite oxide derivative I shows one peak at 9-13 ° and 21-24 ° in the X-ray diffraction spectrum, and is calculated by the Scherrer equation of the peak at 9-13 °. The crystallite diameter was 61.8 mm.
- Example 1-10 Except that the reaction temperature was 100 ° C. and the reaction was performed for 24 hours, graphite oxide derivative J was obtained by the method described in Example 1-5.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative J was 6.1. Comparing the XRD patterns of Examples 1-5 and 1-10, the graphite oxide derivative E of Example 1-5 is sharper and has higher crystallinity than the graphite oxide derivative J of Example 1-10. there were.
- the sensitivity by the calm sensitivity test was 7th grade. As described above, when Example 1-1 and Example 1-9, and Example 1-5 and Example 1-10 were compared, it was found that the sensitivity could be lowered by setting the reaction temperature to 150 ° C. .
- FIG. 14 is an FT-IR chart of graphite oxide derivative J produced in Example 1-10.
- FIG. 15 is an XRD chart of the graphite oxide derivative J produced in Example 1-10.
- Graphite oxide derivative J showed one peak at 21-24 ° in the X-ray diffraction spectrum as shown in FIG. 15, but it did not show a peak at 9-13 °.
- Example 1-11 The graphite oxide-containing composition obtained in Preparation Example 1-5 was transferred to a reactor, and to this was added 500% of 2-decyl-1-tetradecanol (Nippon Rika Co., Ltd., Engecol 240A) with respect to graphite oxide. Water and 1-butanol were distilled off at 100 ° C. under reduced pressure. Thereafter, the temperature was raised and the reaction was carried out at 150 ° C. for 5 hours. After the reaction, hexane was poured, filtered, washed with water and acetone, and the obtained solid was vacuum dried at 100 ° C. to obtain graphite oxide derivative X.
- 2-decyl-1-tetradecanol Nippon Rika Co., Ltd., Engecol 240A
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative X was 8.7. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-12 A graphite oxide derivative Y was obtained by the method described in Example 1-11 except that 2-ethyl-1-hexanol (manufactured by Wako Pure Chemical Industries, Ltd.) was used instead of 2-decyl-1-tetradecanol. .
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative Y was 6.9. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. From the results of Examples 1-11 and 1-12, when a volatile compound such as 1-butanol is used as a flocculant, it is also a preferred form to undergo a vacuum distillation step in advance. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-13 A graphite oxide derivative AA was obtained by the method described in Example 1-1 except that stearylamine (manufactured by Tokyo Chemical Industry Co., Ltd.) was used instead of 2-decyl-1-tetradecanol.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AA was 8.0. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-14 A graphite oxide derivative AB was obtained by the method described in Example 1-13, except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-2.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AB was 7.2.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional group was reduced by the purification, and the amount of modification was small compared to Example 1-13.
- Example 1-15 A graphite oxide derivative AC was obtained by the method described in Example 1-13, except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-4.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AC was 6.6.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional group decreased due to purification and drying, and the amount of modification was small compared to Example 1-13.
- Example 1-16 A graphite oxide derivative AD was obtained by the method described in Example 1-13 except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-6.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AD was 6.9.
- the modification amount was small and the C / O ratio was low. This was probably because oxygen functional groups were reduced by drying, and the amount of modification was small compared to Example 1-13.
- Example 1-17 A graphite oxide derivative AE was obtained by the method described in Example 1-1 except that 2-ethylhexylamine (manufactured by Tokyo Chemical Industry Co., Ltd.) was used instead of 2-decyl-1-tetradecanol.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AE was 6.7. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-18 A graphite oxide derivative AF was obtained by the method described in Example 1-17, except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-2.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AF was 6.2.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional group was reduced by the purification, and the amount of modification was small compared to Example 1-17.
- Example 1-19 A graphite oxide derivative AG was obtained by the method described in Example 1-17, except that the graphite oxide raw material used was the graphite oxide obtained in Preparation Example 1-4.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AG was 6.0.
- the modification amount was small and the C / O ratio was low. It was considered that the oxygen functional group was reduced by purification and drying, and the amount of modification was small compared to Example 1-17.
- Example 1-20 A graphite oxide derivative AH was obtained by the method described in Example 1-17, except that the graphite oxide material used was the graphite oxide obtained in Preparation Example 1-6.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AH was 6.1.
- the modification amount was small and the C / O ratio was low. This was probably because oxygen functional groups were reduced by drying, and the amount of modification was small compared to Example 1-17.
- Example 1-21 Example except that NMP was added as a reaction solvent to 10000% of graphite oxide and dispersed by ultrasonic treatment, and stearylamine (manufactured by Tokyo Chemical Industry Co., Ltd.) was used instead of 2-decyl-1-tetradecanol.
- a graphite oxide derivative AI was obtained by the method described in 1-1.
- the oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative AI was 8.3. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-22 Except that NMP was added as a reaction solvent to 10000% of graphite oxide and dispersed by ultrasonic treatment, and 2-ethylhexylamine (manufactured by Tokyo Chemical Industry Co., Ltd.) was used instead of 2-decyl-1-tetradecanol.
- Graphite oxide derivative AJ was obtained by the method described in Example 1-1. In the obtained graphite oxide derivative AJ, the oxygen-carbon ratio (C / O ratio) was 7.0. Further, in terms of dispersibility, it was also confirmed that the dispersibility was good for chloroform, acetone, DMF, ethanol, and decane. Further, the sensitivity according to the sensitivity test was grade 8.
- Example 1-23 Except that the reaction temperature was 100 ° C. and the reaction time was 24 hours, graphite oxide derivative K was obtained by the method described in Example 1-3. The oxygen-carbon ratio (C / O ratio) in the obtained graphite oxide derivative K was 6.0. It was confirmed that the modification amount was small as compared with Examples 1-1 to 1-4.
- FIG. 16 is an FT-IR chart of graphite oxide derivative K produced in Example 1-23. Note that the graphite oxide derivative of Example 1-23 was added with 24-decyl-1-tetradecanol having 24 carbon atoms, and thus can be dispersed in a nonpolar dispersion medium. .
- FIG. 17 is an FT-IR chart of graphite oxide derivative L produced in Example 1-24.
- the graphite oxide derivative of Example 1-24 was added with 2-ethyl-1-hexanol having 8 carbon atoms, and thus can be dispersed in an amphiphilic dispersion medium.
- Preparation Example 2-1 Concentrated sulfuric acid (reagent grade, manufactured by Wako Pure Chemical Industries, Ltd.) 28.75 parts and natural graphite (Z-5F, flake graphite, manufactured by Ito Graphite Industries Co., Ltd.) 1.00 parts were added to the corrosion resistant reactor to obtain a mixed solution. . While stirring the mixed solution, potassium permanganate (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd.) was introduced 20 times into the mixed solution at 15 minute intervals. A single charge of potassium permanganate was 0.125 parts, and the total charge was 2.50 parts. After the addition of potassium permanganate, the mixture was heated to 35 ° C. and aged for 2 hours while maintaining the liquid temperature.
- potassium permanganate special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd.
- post-reaction slurry The graphite oxide-containing slurry obtained by this method is hereinafter referred to as “post-reaction slurry”.
- Comparative Example 2-1 30 g of the slurry after the reaction (containing 0.45 g of graphite oxide) was filtered using a suction filter having a diameter of 55 mm to separate graphite oxide. Teflon (registered trademark) filter paper (manufactured by Advantech, PF050) was used as the filter paper. It took 104 seconds for the cake-like graphite oxide layer to be exposed in the filter.
- Example 2-1 A comparison was made except that 0.13 g of cyclohexanone (special grade reagent, Wako Pure Chemical Industries, solubility in water; 5% by weight) was added to 30 g of the slurry after the reaction (containing 0.45 g of graphite oxide) and shaken well.
- the graphite oxide was separated under the same conditions as in Example 2-1.
- the time required for the filtration was 65 seconds, and it was confirmed that the filtration rate was extremely increased by adding cyclohexanone.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours. After the reaction, the reaction solution was filtered, washed with water and acetone, and dried to obtain 1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid
- Example 2-2 Under the same conditions as in Comparative Example 2-1, 0.39 g of cyclohexanone (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., solubility in water; 5% by weight) was added to 30 g of the post-reaction slurry and shaken well. Separation of graphite oxide was performed. The time required for the filtration was 73 seconds, and it was confirmed that the filtration rate was extremely increased by adding cyclohexanone.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours. After the reaction, the reaction solution was filtered, washed with water and acetone, and dried to obtain 1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- Example 2-3 Under the same conditions as in Comparative Example 2-1, 1.30 g of cyclohexanone (special grade reagent, manufactured by Wako Pure Chemical Industries, solubility in water; 5% by weight) was added to 30 g of the post-reaction slurry and shaken well. Separation of graphite oxide was performed. The time required for the filtration was 47 seconds, and it was confirmed that the filtration rate was extremely increased by adding cyclohexanone.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours. After the reaction, the reaction solution was filtered, washed with water and acetone, and dried to obtain 1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- Example 2-4 Comparative Example 2-1 and Comparative Example 2-1 except that 0.39 g of 1-butanol (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., solubility in water; 6.4 wt%) was added to 30 g of the post-reaction slurry and well shaken.
- the graphite oxide was separated under the same conditions. The time required for the filtration was 80 seconds, and it was confirmed that the filtration rate was extremely increased by adding 1-butanol.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours.
- the reaction solution was filtered, washed with water, washed with acetone, and dried to obtain 1.1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- Example 2-5 Comparative Example 2-1 and Comparative Example 2-1 except that 1.3 g of 1-butanol (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., solubility in water; 6.4% by weight) was added to 30 g of the slurry after the reaction and shaken well.
- the graphite oxide was separated under the same conditions. The time required for the filtration was 70 seconds, and it was confirmed that the filtration rate was extremely increased by adding 1-butanol.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours.
- the reaction solution was filtered, washed with water, washed with acetone, and dried to obtain 1.1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- the graphite oxide was separated under the same conditions. The time required for the filtration was 18 seconds, and it was confirmed that the filtration rate was extremely increased by adding 1-butanol.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours.
- the reaction solution was filtered, washed with water, washed with acetone, and dried to obtain 1.1 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- Example 2-7 Comparative Example 2-1 and Comparative Example 2-1 except that 2.6 g of 1-butanol (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., solubility in water; 6.4 wt%) was added to 30 g of the slurry after the above reaction and shaken well.
- the graphite oxide was separated under the same conditions. The time required for the filtration was 1 second, and it was confirmed that the filtration rate was extremely increased by adding 1-butanol.
- the graphite oxide-containing composition on the filter paper (both purified and dried) was dispersed in 30 g of NMP, 1.5 g of stearylamine was added, and the mixture was reacted at 120 ° C. for 5 hours. After the reaction, the reaction solution was filtered, washed with water and acetone, and dried to obtain 1.2 g of graphite oxide derivative.
- the obtained graphite oxide derivative showed good dispersibility in nonpolar solvents such as decane and liquid paraffin.
- Comparative Example 2-2 The same as Comparative Example 2-1 except that 1.3 g of 2-propanol (special grade reagent, manufactured by Wako Pure Chemical Industries, arbitrarily mixed with water) was added to 30 g of the slurry after the reaction, and the mixture was shaken well. The graphite oxide was separated under the conditions. The time required for the filtration was 146 seconds.
- 2-propanol special grade reagent, manufactured by Wako Pure Chemical Industries, arbitrarily mixed with water
- Comparative Example 2-3 Graphite oxide under the same conditions as in Comparative Example 2-1, except that 1.3 g of hexane (special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., insoluble in water) was added to 30 g of the slurry after the reaction and shaken well. Separation. The time required for the filtration was 586 seconds.
- hexane special grade reagent, manufactured by Wako Pure Chemical Industries, Ltd., insoluble in water
- FIG. 18 is a diagram showing the XRD measurement results of the graphite oxide powder obtained in Reference Example 2-1. Since a characteristic signal derived from the layered structure of graphite oxide was observed, it was confirmed that the graphite oxide could be separated without any problem by the separation method.
- FIG. 19 shows the XRD measurement results of the graphite oxide powder obtained in Reference Example 2-2. Since a characteristic signal derived from the layered structure of graphite oxide was observed, it was confirmed that the graphite oxide could be separated without any problem by the purification method.
- FIG. 20 is a diagram showing the XRD measurement results of the reduced graphite oxide powder obtained in Reference Example 2-3.
- Table 2 shows the C1s XPS measurement results before and after firing. As shown in FIG. 20, the peak derived from the layer structure of graphite oxide has disappeared, and the bond derived from the oxygen-containing functional group has disappeared remarkably by firing, indicating that reduced graphite oxide has been produced. It was done.
- FIG. 21 is a diagram showing the XRD measurement results of the reduced graphite oxide powder obtained in Reference Example 2-4.
- Table 3 shows the C1s XPS measurement results before and after firing. As shown in FIG. 21, the peak derived from the layer structure of graphite oxide has disappeared, and the bond derived from the oxygen-containing functional group has disappeared remarkably by firing, indicating that reduced graphite oxide has been produced. It was done.
- Example 3-1 Synthesis of OGO20-B
- Graphite oxide as a raw material was synthesized with reference to a method described in non-patent literature (Karthikeyan K, et.al, Carbon, 53, (2013), 38-49). This graphite oxide (200 mg), 2-octyl-1-dodecanol (manufactured by Tokyo Chemical Industry Co., Ltd., 10 ml) and sulfuric acid (manufactured by Wako Pure Chemical Industries, 200 mg) were mixed and reacted at 100 ° C. for 24 hours. After the reaction, acetone was poured into the reaction solution and filtered. The obtained solid was dispersed in hexane and washed with water.
- FIG. 23 shows an FT-IR chart. Compared to the starting material of the FT-IR chart (Fig.
- FIG. 22 is an FT-IR chart of graphite oxide as a raw material
- FIG. 23 is an FT-IR chart of the graphite oxide derivative produced in Example 3-1.
- the total mass concentration of CHO was 95.66%
- the mass concentration of N was 0.01%.
- Example 3-2 [Synthesis of OGO20-S] Graphite oxide (200 mg), 1-eicosanol (manufactured by Tokyo Chemical Industry Co., Ltd., 8 g), and sulfuric acid (200 mg) were mixed. The obtained solid was dispersed in hexane and washed with water. The organic layer was filtered to obtain OG020-S. When the obtained solid was added to hexadecane and ultrasonic waves were applied for 1 hour to evaluate dispersibility, the time during which light transmission could be confirmed was 6 hours or more, and good dispersibility was confirmed.
- FIG. 24 shows an FT-IR chart.
- FIG. 24 is an FT-IR chart of the graphite oxide derivative produced in Example 3-2.
- the total mass concentration of CHO was 96.12%, and the mass concentration of N was 0.00%.
- Example 3-3 [Synthesis of OGO14-S]
- OGO14-S was synthesized by the same method as described in Example 1 except that the alcohol used was changed from 2-octyl-1-dodecanol to 1-tetradecanol (manufactured by Tokyo Chemical Industry Co., Ltd., 10 ml).
- the obtained solid was added to hexadecane and ultrasonic waves were applied for 1 hour to evaluate dispersibility, the time during which light transmission could be confirmed was 6 hours or more, and good dispersibility was confirmed.
- the mixture was added to a mixed solvent of hexane-methanol and sonicated for 1 hour to confirm whether it was dispersed in the hexane layer or the methanol layer.
- FIG. 25 shows an FT-IR chart.
- FIG. 25 is an FT-IR chart of the graphite oxide derivative produced in Example 3-3.
- the total mass concentration of CHO was 96.95%, and the mass concentration of N was 0.00%.
- Example 3-4 [Synthesis of OGO20-B-BA] Graphite oxide (200 mg), 2-octyl-1-dodecanol (manufactured by Tokyo Chemical Industry Co., Ltd., 10 ml) and potassium hydroxide (manufactured by Wako Pure Chemical Industries, Ltd., 200 mg) were mixed and reacted at 100 ° C. for 24 hours. After the reaction, acetone was poured into the reaction solution and filtered. The obtained solid was dispersed in hexane and washed with 1% aqueous sulfuric acid.
- FIG. 26 shows an FT-IR chart.
- FIG. 26 is an FT-IR chart of the graphite oxide derivative produced in Example 3-4.
- the total mass concentration of CHO was 95.51%
- the mass concentration of N was 0.01%.
- FIG. 27 shows an FT-IR chart.
- FIG. 27 is an FT-IR chart of the graphite oxide derivative produced in Comparative Example 3-1.
- FIG. 29 shows an actual image of hexane-methanol separation. That is, FIG. 29 is an actual image of the hexane-methanol separation of Example 3-1, Example 3-3, and Comparative Example 3-1. It can be seen that only Comparative Example 3-1 has no dispersibility in the hexane layer.
- FIG. 30 shows an FT-IR chart.
- FIG. 30 is an FT-IR chart of the graphite oxide derivative produced in Example 3-5. As a result of elemental analysis, the total mass concentration of CHO was 96.73%, and the mass concentration of N was 0.00%.
- FIG. 31 shows an FT-IR chart.
- FIG. 31 is an FT-IR chart of the graphite oxide derivative produced in Example 3-6. As a result of elemental analysis, the total mass concentration of CHO was 96.30%, and the mass concentration of N was 0.00%.
- FIG. 32 shows an FT-IR chart.
- FIG. 32 is an FT-IR chart of the graphite oxide derivative produced in Example 3-7. As a result of elemental analysis, the total mass concentration of CHO was 96.66%, and the mass concentration of N was 0.00%.
- Example 3-1 to 3-7 and Comparative Example 3-1 the alcohol was introduced into the carboxyl group, epoxy group, and hydroxyl group of graphite oxide based on the results of the FT-IR chart, and the alkoxycarbonyl group and the alkoxy group were It is thought that it is formed.
- the comparison between Example 3-3 and Comparative Example 3-1 proves that the hydrophobic characteristics are dramatically different when the hydrocarbon group has 14 carbon atoms and when the hydrocarbon group has 12 carbon atoms.
- the dispersion characteristics in the amphiphilic dispersion medium are dramatically different depending on whether the hydrocarbon group has 8 carbon atoms or 12 carbon atoms. Proved.
- the graphite oxide derivatives obtained in Examples 3-1 to 3-6 are sufficiently dispersed in a nonpolar dispersion medium, they are added to lubricating oil additives for machines and resins that can be combined with various resins. It is thought that it can be suitably used as an agent or the like. Since the graphite oxide derivative obtained in Example 3-7 is sufficiently dispersed in an amphiphilic dispersion medium, it is considered that it can be suitably used as an additive or the like to a resin that can be combined with various resins.
- the graphite oxide derivatives obtained in Examples 3-1 to 3-7 are obtained by using graphite oxide obtained through a purification process and a drying process that require a long time as raw materials. Compared to the first preferred embodiment, there is room for improvement in terms of efficiently obtaining a high-quality raw material. For example, as a raw material for producing a graphite oxide derivative on an industrial scale, it is practically impossible to prepare a large amount of graphite oxide obtained through the purification process and the drying process.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Nanotechnology (AREA)
- Carbon And Carbon Compounds (AREA)
Abstract
本発明は、高品質な酸化黒鉛誘導体を簡便に製造することを可能とする酸化黒鉛誘導体の製造方法、及び、高品質な酸化黒鉛誘導体を提供する。本発明は、酸化黒鉛誘導体を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛を含む反応液中の酸化黒鉛又は該反応液から分離した酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛を含む反応液を精製し、かつ乾燥する工程を含まないことを特徴とする酸化黒鉛誘導体の製造方法である。
Description
本発明は、酸化黒鉛誘導体の製造方法に関する。より詳しくは、触媒、電池やキャパシタの電極活物質、熱電変換材料、導電性材料、発光材料、潤滑用添加剤(機械用の潤滑油の添加剤)、高分子用添加剤(樹脂への添加剤)、透過膜材料、抗菌材料、撥水材料、吸着材料等として好適に用いることができる酸化黒鉛誘導体の製造方法に関する。
酸化黒鉛は、sp2結合で結合した炭素原子が平面的に並んだ層状構造をもつ黒鉛を酸化し、酸素含有官能基を付与したものであり、その特異な構造や物性のために数多くの研究がなされている。酸化黒鉛は、触媒、電池やキャパシタの電極活物質、熱電変換材料、導電性材料、発光材料、潤滑用添加剤、高分子用添加剤、透過膜材料、抗菌材料、撥水材料、吸着材料等の種々の用途に用いられることが期待されており、例えば、機械用の潤滑油の添加剤として油中に分散させて用いることが望まれている。
このような酸化黒鉛に対し、更に所望の機能を付与するために、酸化黒鉛を誘導体化する手法が考えられる。例えば、酸化黒鉛の使用態様として、潤滑用添加剤として油中に分散させて用いることや、高分子等の樹脂と複合化させて用いることが望まれている。そこで、そのままでは親水性の酸化黒鉛を油、樹脂等の非極性、低極性分散媒(疎水性分散媒)中で充分には分散できないため、分散させる処理として熱や化学反応で親水性の酸素含有官能基を還元したり、疎水性の置換基で酸化黒鉛を修飾したりする手法が考えられる。なお、これまでにも、酸化黒鉛の還元反応や置換基の修飾(導入)反応については報告例がある(特許文献1~3、非特許文献1~8参照。)。その中でも非特許文献3において酸化黒鉛の非極性分散媒中での分散性の向上を目的とした検討はされているが、前処理としての精製乾燥、還元処理による積極的な反応性酸素の除去、反応前処理としての強塩基性の試薬による処理等を行っている。
ところで、酸化黒鉛の製造方法としては、黒鉛を酸溶媒中で強力な酸化剤と作用させることで酸化黒鉛を合成した後、生成した酸化黒鉛を溶液中から精製する方法が一般的であり、酸化剤として硫酸と過マンガン酸カリウムを用いるHummers法が知られている(非特許文献9、特許文献4~6参照)。またその他の方法として、硝酸と塩素酸カリウムを用いるBrodie法、酸化剤として硫酸、硝酸と塩素酸カリウムを用いるStaudenmaier法等が知られている。黒鉛を酸化した後、酸化黒鉛を含む反応液からの酸化黒鉛の分離、精製は、反応液の遠心分離又は濾過により行われることが一般的であるが、気体で加圧しながら濾過を行う方法が効率的な酸化黒鉛の分離、精製方法として報告されている(非特許文献10、11参照)。
仁科勇太、「酸化グラフェンの表面修飾技術の開発」、新学術領域研究「原子層科学」中間成果報告(2014)公募研究合成班、71-74
Daniel R. Dreyer, et.al, "Reduction of graphite oxide using alcohols"J. Mater. Chem., 2011, 21, 3443-3447
Jean-Philippe, et.al, Langmuir, 2012, 28. 6691-6697
岡田祐樹ら、他1名、「酸化グラフェンの触媒的エステル化反応の開発」、2014年、日本化学会年会、2E4-29
Chun Kiang Chua, et.al, Chem. Soc. Rev., 2014, 43, 291-312
Xiaobin Fan, et.al, Adv. Mater., 2008, 20, 4490-4493 and Supporting Information
Daniel R. Dreyer, et.al, "Reduction of graphite oxide using alcohols"J. Mater. Chem., 2011, 21, 3443-3447
Daniel R. Dreyer, et.al, Chem. Soc. Rev., 2010, 39, 228-240
William S. Hummers, et.al, Journal of American Chemical Society, 1958, 80, 1339
Gabriel Ceriotti, et.al, RSC Advances, 2015, 5, 50365
Gabriel Ceriotti, et.al, Nanoscale, 2015, 00, SI, pp.1-8
上記のとおり、酸化黒鉛の製造方法として種々の方法が知られている。しかしながら、これらのいずれの方法も、黒鉛を酸化した後、酸化黒鉛を含む反応液からの酸化黒鉛の分離の際に、遠心分離を繰り返して行うとある程度手間がかかるとともに廃液の量が多くなってしまう。また、後述するがある一定の精製度まで精製が進むと加圧下であっても反応液を濾過するとフィルターが目詰まりを起こすおそれがあるため、酸化黒鉛やその誘導体の効率的な製造を妨げる要因となっている。現状では酸化黒鉛やその誘導体を工業的規模で製造することは殆ど不可能である。
また酸化黒鉛が機械用の潤滑油の添加剤等として充分な効果を発揮するためには、油中への充分な分散性が必要であるため、非極性分散媒への分散性に優れる酸化黒鉛が求められている。更に、酸化黒鉛が様々な樹脂への添加剤等として充分な効果を発揮するために、両親媒性分散媒への分散性に優れる酸化黒鉛が求められている。
本発明は、上記現状に鑑みてなされたものでもあり、非極性分散媒や両親媒性分散媒への分散性に優れる酸化黒鉛を提供することを目的とする。
本発明は、上記現状に鑑みてなされたものであり、高品質な酸化黒鉛誘導体を簡便に製造することを可能とする酸化黒鉛誘導体の製造方法、及び、高品質な酸化黒鉛誘導体を提供することを目的とする。
本発明者らは、高品質な酸化黒鉛誘導体を簡便に製造する方法について種々検討し、更に、酸化黒鉛の非極性分散媒や両親媒性分散媒中での分散性を向上させる方法についても種々検討し、上記課題をみごとに解決することができることに想到し、本発明に到達したものである。
すなわち本発明は、酸化黒鉛誘導体を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことを特徴とする酸化黒鉛誘導体の製造方法である。
本発明はまた、アルキル基を有し、JIS K 4810に規定される落つい感度試験で測定される等級が8級である酸化黒鉛誘導体でもある。
本発明はそして、炭素数13以上の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体である。
本発明はまた、炭素数6以上、10以下の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体でもある。
本発明はまた、アルキル基を有し、JIS K 4810に規定される落つい感度試験で測定される等級が8級である酸化黒鉛誘導体でもある。
本発明はそして、炭素数13以上の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体である。
本発明はまた、炭素数6以上、10以下の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体でもある。
本発明の酸化黒鉛誘導体の製造方法により、高品質な酸化黒鉛誘導体を簡便に得ることができる。また、本発明の酸化黒鉛誘導体は、炭素数13以上の炭化水素基をもつ官能基を有する場合、非極性分散媒中で充分に分散し、炭素数6以上、10以下の炭化水素基をもつ官能基を有する場合、両親媒性分散媒中で充分に分散する。
以下に本発明を詳述する。
なお、以下において段落に分けて記載される個々の本発明の好ましい特徴を2つ以上組み合わせた形態も、本発明の好ましい形態である。
なお、以下において段落に分けて記載される個々の本発明の好ましい特徴を2つ以上組み合わせた形態も、本発明の好ましい形態である。
なお、非特許文献3に記載のように、酸化黒鉛の精製乾燥、還元処理、反応前処理としての強塩基性の試薬による処理等を行った場合は、酸化黒鉛から反応性酸素が除去されてしまい、充分な修飾反応が進まず、分散性に乏しいと考えられる。一方、本発明の手法を用いれば、非特許文献3に記載の手法で得られる酸化黒鉛誘導体では分散性が充分でなかったエタノールやデカン中でも分散性が良好な酸化黒鉛誘導体が得られることが分かった。
また、特許文献1に記載のように前処理としてカチオン性有機化合物のカチオンを用いるとその後、該カチオン由来の成分が不純物として酸化黒鉛誘導体と強く結合し、除去することが困難となる。
また、特許文献1に記載のように前処理としてカチオン性有機化合物のカチオンを用いるとその後、該カチオン由来の成分が不純物として酸化黒鉛誘導体と強く結合し、除去することが困難となる。
(酸化黒鉛誘導体)
酸化黒鉛は、グラフェン、黒鉛(グラファイト)等の黒鉛質の炭素材料を酸化することにより酸素が結合したもの(該炭素材料に酸素が結合したもの)であり、該酸素は黒鉛質の炭素材料に対しカルボキシル基、カルボニル基、ヒドロキシル基(水酸基)、エポキシ基等の置換基として存在している。
上記酸化黒鉛は、グラフェンの炭素に酸素が結合した酸化グラフェンであることが好ましい。
なお、一般的にグラフェンとは、sp2結合で結合した炭素原子が平面的に並んだ1層からなるシートをいい、グラフェンシートが多数積層されたものはグラファイトといわれるが、本発明における酸化グラフェンには、炭素原子1層のみからなるシートだけではなく、2層~100層程度積層した構造を有するものも含まれる。該酸化グラフェンは、炭素原子1層のみからなるシートであるか、又は、2層~20層程度積層した構造を有するものであることが好ましい。
上記酸化黒鉛は、更に、硫黄含有基、窒素含有基等の官能基を有していてもよいが、全構成元素に対する炭素、水素、及び、酸素の構成元素としての含有率が97モル%以上であることが好ましく、99モル%以上であることがより好ましく、酸化黒鉛が炭素、水素、及び、酸素のみを構成元素とするものであることが更に好ましい。
酸化黒鉛は、グラフェン、黒鉛(グラファイト)等の黒鉛質の炭素材料を酸化することにより酸素が結合したもの(該炭素材料に酸素が結合したもの)であり、該酸素は黒鉛質の炭素材料に対しカルボキシル基、カルボニル基、ヒドロキシル基(水酸基)、エポキシ基等の置換基として存在している。
上記酸化黒鉛は、グラフェンの炭素に酸素が結合した酸化グラフェンであることが好ましい。
なお、一般的にグラフェンとは、sp2結合で結合した炭素原子が平面的に並んだ1層からなるシートをいい、グラフェンシートが多数積層されたものはグラファイトといわれるが、本発明における酸化グラフェンには、炭素原子1層のみからなるシートだけではなく、2層~100層程度積層した構造を有するものも含まれる。該酸化グラフェンは、炭素原子1層のみからなるシートであるか、又は、2層~20層程度積層した構造を有するものであることが好ましい。
上記酸化黒鉛は、更に、硫黄含有基、窒素含有基等の官能基を有していてもよいが、全構成元素に対する炭素、水素、及び、酸素の構成元素としての含有率が97モル%以上であることが好ましく、99モル%以上であることがより好ましく、酸化黒鉛が炭素、水素、及び、酸素のみを構成元素とするものであることが更に好ましい。
本発明の酸化黒鉛誘導体は、更に、酸化黒鉛の炭素原子に、酸化黒鉛の酸素含有官能基と反応する化合物由来の基をもつ官能基が結合した構造を有する。例えば本発明の酸化黒鉛誘導体の一形態では、酸化黒鉛の炭素原子に、炭素数13以上の炭化水素基をもつ官能基や炭素数6以上、10以下の炭化水素基をもつ官能基が結合した構造を有する。また、本発明の酸化黒鉛誘導体の製造方法の好ましい一実施形態では、本発明の酸化黒鉛誘導体は、更に、酸化グラフェンの炭素原子に、酸化黒鉛の酸素含有官能基と反応する化合物由来の基をもつ官能基が結合した構造を有する。
上記酸化黒鉛誘導体は、更に、硫黄含有基、窒素含有基等の官能基を有していてもよいが、炭素、水素、及び、酸素のみを構成元素とするものであるか、又は、炭素、水素、酸素、及び、窒素のみを構成元素とするものであることが更に好ましい。酸化黒鉛誘導体の好ましいものについては、後述する。
上記酸化黒鉛誘導体は、更に、硫黄含有基、窒素含有基等の官能基を有していてもよいが、炭素、水素、及び、酸素のみを構成元素とするものであるか、又は、炭素、水素、酸素、及び、窒素のみを構成元素とするものであることが更に好ましい。酸化黒鉛誘導体の好ましいものについては、後述する。
本発明の酸化黒鉛誘導体の特徴を示す分析として質量分析法とFT-IR法が挙げられる。質量分析法によってイオン化されたフラグメントが容易に観測できる。例えば本発明の酸化黒鉛誘導体は末端等に炭化水素基を有することから、質量分析法によってイオン化されたフラグメントが容易に観測できる。酸化黒鉛自身は大質量であることから質量分析法ではイオン検出されず、つまり、酸化黒鉛に導入された化合物由来の部位のみが観測される。
FT-IR法では後述する実施例の通り、例えば酸化黒鉛誘導体が末端に炭化水素基を有する場合は、酸化黒鉛誘導体は炭化水素基に由来するC-Hのピーク(2900cm-1付近)の出現により容易に分析できる。
FT-IR法では後述する実施例の通り、例えば酸化黒鉛誘導体が末端に炭化水素基を有する場合は、酸化黒鉛誘導体は炭化水素基に由来するC-Hのピーク(2900cm-1付近)の出現により容易に分析できる。
本発明の酸化黒鉛誘導体の製造方法の好ましい形態の一つ(第1の好ましい形態とも言う)は、黒鉛を酸化する工程と酸化黒鉛誘導体を得る工程とを含み、かつ、該黒鉛を酸化する工程と該酸化黒鉛誘導体を得る工程との間に酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことである。
すなわち、本発明の製造方法の第1の好ましい形態は、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製せず、かつ乾燥しないものであってもよく、酸化黒鉛含有組成物の精製及び乾燥のいずれか一方のみを行うものであってもよい。なお、上述した「酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まない」とは、該酸化黒鉛含有組成物を、酸化黒鉛誘導体を得る工程の反応に供する場合に、該酸化黒鉛含有組成物が、精製され、かつ乾燥されたものではないことを意味する。本発明者らは、酸化黒鉛含有組成物からの酸化黒鉛の精製工程、乾燥工程の際に、酸化黒鉛が有する反応性の酸素含有官能基が還元及び/又は不活化されてしまうことを見出し、精製及び乾燥の少なくとも一方を省くことにより、反応性の酸素含有官能基の数を充分に維持した酸化黒鉛含有組成物中の酸化黒鉛を、酸化黒鉛誘導体を得る工程の反応(修飾反応)に適用することができることを見出した。その結果、修飾反応により所望の官能基を有する基が充分に導入された高品質な酸化黒鉛誘導体を効率的に得ることができる。また、精製及び乾燥の少なくとも一方を省くので、製法工程がより簡便なものとなり、工程上のメリットがある。
すなわち、本発明の製造方法の第1の好ましい形態は、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製せず、かつ乾燥しないものであってもよく、酸化黒鉛含有組成物の精製及び乾燥のいずれか一方のみを行うものであってもよい。なお、上述した「酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まない」とは、該酸化黒鉛含有組成物を、酸化黒鉛誘導体を得る工程の反応に供する場合に、該酸化黒鉛含有組成物が、精製され、かつ乾燥されたものではないことを意味する。本発明者らは、酸化黒鉛含有組成物からの酸化黒鉛の精製工程、乾燥工程の際に、酸化黒鉛が有する反応性の酸素含有官能基が還元及び/又は不活化されてしまうことを見出し、精製及び乾燥の少なくとも一方を省くことにより、反応性の酸素含有官能基の数を充分に維持した酸化黒鉛含有組成物中の酸化黒鉛を、酸化黒鉛誘導体を得る工程の反応(修飾反応)に適用することができることを見出した。その結果、修飾反応により所望の官能基を有する基が充分に導入された高品質な酸化黒鉛誘導体を効率的に得ることができる。また、精製及び乾燥の少なくとも一方を省くので、製法工程がより簡便なものとなり、工程上のメリットがある。
本明細書中、酸化黒鉛含有組成物を精製する工程とは、該酸化黒鉛含有組成物から固形不純物(例えば、過マンガン酸塩等の酸化剤等)及び/又は酸化反応時の溶媒(硫酸等の強酸等)を除き、該酸化黒鉛含有組成物を酸化黒鉛水分散体とする工程を言う。
精製の役割及び意義として、例えば該酸化黒鉛含有組成物中の酸化黒鉛に対する硫酸濃度を1質量%以下とすることで、酸化黒鉛又は酸化黒鉛水分散体を得ることであり、酸化黒鉛又は酸化黒鉛水分散体を特許文献6に記載の通り、水に希釈した場合(たとえば1~10%酸化黒鉛水分散液とした場合)、総イオン濃度(大部分は硫酸濃度)が特許文献6に記載の閾値濃度を下回り酸化黒鉛自身の電離を促し、水分子との相互作用が強くなり酸化黒鉛の分散性が飛躍的に向上することが分かっている。また、この水準まで精製が進むと、その分散性により濾過性が非常に悪くなってしまう。酸化黒鉛を材料として使用する場合、この分散性が大きく物性に作用することも分かっている。すなわち酸化黒鉛単独の物性を充分に発揮させるためには該精製工程が必須であり、多くの検討がなされている。しかし、本発明のように酸化黒鉛自身を材料に適用するわけではなく、修飾等の化学反応を行い、誘導体を材料として適用する場合は酸化黒鉛の分散性よりも酸化黒鉛自身の反応性が重要となってくる。本発明者らは、特許文献6に記載のような総イオン濃度(硫酸濃度)では酸化黒鉛が電離することで、高活性な酸素置換基も電離もしくは分解してしまい、続く修飾反応で不利となってしまうことを見出した。本発明においては、酸化工程後の反応液から、適切な硫酸濃度の酸化黒鉛含有組成物を分離する工程のみを行うことができるが、より高度な精製工程を行わないことが好ましい。すなわち本発明において省略することができる高度な精製工程とは、特許文献6に記載のような、酸性度を下げ、酸化黒鉛の電離、解離を促し、水に対する強い分散性を付与することと言える。つまり、本発明において省略することができる精製工程とは、酸化黒鉛の質量に対して硫酸を1質量%未満にする工程のことである。
本発明の酸化黒鉛含有組成物を分離する工程を行う場合、該分離工程は、酸化黒鉛を含む反応組成物を精製し、かつ乾燥する工程には該当しない程度に、反応組成物から後述する通り、後段の反応工程に適するように酸化黒鉛含有組成物を分離するものであればよく、水洗や、固形不純物を水に溶解させたうえでの遠心分離、濾過等の手法の1種又は2種以上を用いて行うことができ、また後述するような濾過等の際に界面活性剤、有機溶媒等の凝集剤を添加しても構わない。また、上記分離工程は、空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。また、分離工程を行う際に、硫酸や、水等の溶媒の一部を除去してもよい。
精製の役割及び意義として、例えば該酸化黒鉛含有組成物中の酸化黒鉛に対する硫酸濃度を1質量%以下とすることで、酸化黒鉛又は酸化黒鉛水分散体を得ることであり、酸化黒鉛又は酸化黒鉛水分散体を特許文献6に記載の通り、水に希釈した場合(たとえば1~10%酸化黒鉛水分散液とした場合)、総イオン濃度(大部分は硫酸濃度)が特許文献6に記載の閾値濃度を下回り酸化黒鉛自身の電離を促し、水分子との相互作用が強くなり酸化黒鉛の分散性が飛躍的に向上することが分かっている。また、この水準まで精製が進むと、その分散性により濾過性が非常に悪くなってしまう。酸化黒鉛を材料として使用する場合、この分散性が大きく物性に作用することも分かっている。すなわち酸化黒鉛単独の物性を充分に発揮させるためには該精製工程が必須であり、多くの検討がなされている。しかし、本発明のように酸化黒鉛自身を材料に適用するわけではなく、修飾等の化学反応を行い、誘導体を材料として適用する場合は酸化黒鉛の分散性よりも酸化黒鉛自身の反応性が重要となってくる。本発明者らは、特許文献6に記載のような総イオン濃度(硫酸濃度)では酸化黒鉛が電離することで、高活性な酸素置換基も電離もしくは分解してしまい、続く修飾反応で不利となってしまうことを見出した。本発明においては、酸化工程後の反応液から、適切な硫酸濃度の酸化黒鉛含有組成物を分離する工程のみを行うことができるが、より高度な精製工程を行わないことが好ましい。すなわち本発明において省略することができる高度な精製工程とは、特許文献6に記載のような、酸性度を下げ、酸化黒鉛の電離、解離を促し、水に対する強い分散性を付与することと言える。つまり、本発明において省略することができる精製工程とは、酸化黒鉛の質量に対して硫酸を1質量%未満にする工程のことである。
本発明の酸化黒鉛含有組成物を分離する工程を行う場合、該分離工程は、酸化黒鉛を含む反応組成物を精製し、かつ乾燥する工程には該当しない程度に、反応組成物から後述する通り、後段の反応工程に適するように酸化黒鉛含有組成物を分離するものであればよく、水洗や、固形不純物を水に溶解させたうえでの遠心分離、濾過等の手法の1種又は2種以上を用いて行うことができ、また後述するような濾過等の際に界面活性剤、有機溶媒等の凝集剤を添加しても構わない。また、上記分離工程は、空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。また、分離工程を行う際に、硫酸や、水等の溶媒の一部を除去してもよい。
本発明の製造方法は、中でも、上記酸化工程と上記酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製する工程を含まないものであることが好ましい。これにより、反応性の酸素含有官能基の数をより充分に維持した酸化黒鉛を修飾反応に用いることができ、得られる酸化黒鉛誘導体が、所望の官能基を有する基がより充分に導入された高品質なものとなる。更に、本発明の製造方法が非常に簡便なものとなる。
すなわち、本発明の製造方法の第1の好ましい形態は、より好ましくは、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に酸化黒鉛含有組成物を精製する工程を含まない製造方法であって、該酸化黒鉛含有組成物を精製する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して硫酸を1質量%未満にする工程である、酸化黒鉛誘導体の製造方法である。
すなわち、本発明の製造方法の第1の好ましい形態は、より好ましくは、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に酸化黒鉛含有組成物を精製する工程を含まない製造方法であって、該酸化黒鉛含有組成物を精製する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して硫酸を1質量%未満にする工程である、酸化黒鉛誘導体の製造方法である。
また酸化黒鉛含有組成物を乾燥する工程とは、酸化黒鉛含有組成物から少なくとも酸化工程において用いた水等の揮発性溶媒を除き、乾燥物とする工程を言う。
また、本発明者らは、該酸化黒鉛含有組成物中の酸化黒鉛に対する水濃度を少なくとも3質量%未満とすると、酸化黒鉛の高活性な酸素官能基に対し配位していた水まで除かれることで、配位安定していた酸素官能基が脱離し、続く修飾反応で不利となってしまうことを見出した。すなわち、酸化黒鉛に対し、水が3質量%以上含まれる酸化黒鉛含有組成物とすることで、修飾反応に優位な活性な酸素官能基を安定に維持させることができる。つまり、本発明において省略することができる乾燥工程とは、酸化黒鉛の質量に対して水を3質量%未満にする工程のことである。
酸化黒鉛含有組成物を乾燥する工程を行う場合、乾燥工程は、遠心分離、濾過、蒸発等の手法の1種又は2種以上を用いて行うことができ、濾過等の際に界面活性剤、有機溶媒等の凝集剤を添加しても構わない。
また、本発明者らは、該酸化黒鉛含有組成物中の酸化黒鉛に対する水濃度を少なくとも3質量%未満とすると、酸化黒鉛の高活性な酸素官能基に対し配位していた水まで除かれることで、配位安定していた酸素官能基が脱離し、続く修飾反応で不利となってしまうことを見出した。すなわち、酸化黒鉛に対し、水が3質量%以上含まれる酸化黒鉛含有組成物とすることで、修飾反応に優位な活性な酸素官能基を安定に維持させることができる。つまり、本発明において省略することができる乾燥工程とは、酸化黒鉛の質量に対して水を3質量%未満にする工程のことである。
酸化黒鉛含有組成物を乾燥する工程を行う場合、乾燥工程は、遠心分離、濾過、蒸発等の手法の1種又は2種以上を用いて行うことができ、濾過等の際に界面活性剤、有機溶媒等の凝集剤を添加しても構わない。
上記蒸発は、例えば減圧条件下で行ったり、加熱条件下で行ったりすることができるが、減圧条件下かつ加熱条件下で行うことが好ましい。また、遠心分離や濾過は、空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。
また本発明の製造方法の第1の好ましい形態は、例えば、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に酸化黒鉛含有組成物を乾燥する工程を含まない製造方法であって、該酸化黒鉛含有組成物を乾燥する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して水を3質量%未満にする工程である、酸化黒鉛誘導体の製造方法であってもよい。
また本発明の製造方法の第1の好ましい形態は、例えば、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に酸化黒鉛含有組成物を乾燥する工程を含まない製造方法であって、該酸化黒鉛含有組成物を乾燥する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して水を3質量%未満にする工程である、酸化黒鉛誘導体の製造方法であってもよい。
本発明の製造方法は、中でも、酸化黒鉛含有組成物の精製工程、乾燥工程のいずれも行わないものであることが好ましい。これにより、高品質な酸化黒鉛誘導体を簡便に得ることができる本発明の効果が顕著なものとなる。
すなわち、本発明の製造方法の第1の好ましい形態は、更に好ましくは、黒鉛を酸化する工程(酸化工程)、及び、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程を含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製する工程と、酸化黒鉛を含む反応液を乾燥する工程とをいずれも含まない製造方法であって、該酸化黒鉛含有組成物を精製する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して硫酸を1質量%未満にする工程であり、該酸化黒鉛含有組成物を乾燥する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して水を3質量%未満にする工程である、酸化黒鉛誘導体の製造方法である。
すなわち、本発明の製造方法の第1の好ましい形態は、更に好ましくは、黒鉛を酸化する工程(酸化工程)、及び、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程を含み、かつ、該酸化工程と該酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製する工程と、酸化黒鉛を含む反応液を乾燥する工程とをいずれも含まない製造方法であって、該酸化黒鉛含有組成物を精製する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して硫酸を1質量%未満にする工程であり、該酸化黒鉛含有組成物を乾燥する工程は、該酸化黒鉛含有組成物に含まれる酸化黒鉛の質量に対して水を3質量%未満にする工程である、酸化黒鉛誘導体の製造方法である。
(酸化黒鉛含有組成物を濃縮する工程)
本発明の製造方法は、反応組成物から硫酸や溶媒の一部を除去する工程(濃縮工程)を含むことが好ましい。より好ましくは、本発明の製造方法は、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を濃縮する工程を含む。本発明における濃縮工程とは、反応組成物から硫酸や水の一部を除去して酸化黒鉛含有組成物を得る工程であって、組成物中、酸化黒鉛の質量に対して硫酸を1質量%以上残存させるか、及び/又は、酸化黒鉛の質量に対して水を3質量%以上残存させる工程を意味する。例えば酸化工程直後の反応組成物中に、酸が、酸化黒鉛誘導体を得る工程における原料化合物量に対して過剰に存在する場合、濃縮工程により酸を適度に除去することで、原料化合物量に対する酸の量を後述する好適な範囲内に調節することができ、修飾反応をより高効率で進行させることができる。
本発明の製造方法は、反応組成物から硫酸や溶媒の一部を除去する工程(濃縮工程)を含むことが好ましい。より好ましくは、本発明の製造方法は、酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を濃縮する工程を含む。本発明における濃縮工程とは、反応組成物から硫酸や水の一部を除去して酸化黒鉛含有組成物を得る工程であって、組成物中、酸化黒鉛の質量に対して硫酸を1質量%以上残存させるか、及び/又は、酸化黒鉛の質量に対して水を3質量%以上残存させる工程を意味する。例えば酸化工程直後の反応組成物中に、酸が、酸化黒鉛誘導体を得る工程における原料化合物量に対して過剰に存在する場合、濃縮工程により酸を適度に除去することで、原料化合物量に対する酸の量を後述する好適な範囲内に調節することができ、修飾反応をより高効率で進行させることができる。
例えば上記濃縮工程等を経て分離した、上記酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物は、組成物中の酸化黒鉛の質量100質量%に対し、水を3~10000質量%含むことが好ましい。水分量が3質量%以上であることで、活性な酸素官能基を配位し、安定化させ、続く修飾反応に効率よく反応させることができる。また、10000質量%以下とすることで水が修飾反応の妨げとならないようにすることができる。
上記含有量は、5質量%以上であることがより好ましく、10質量%以上であることが更に好ましく、50質量%以上であることが特に好ましい。また、該含有量は、5000質量%以下であることがより好ましく、3000質量%以下であることが更に好ましく、2000質量%以下であることが一層好ましく、1000質量%以下であることが特に好ましく、500質量%以下であることが最も好ましい。
上記含有量は、5質量%以上であることがより好ましく、10質量%以上であることが更に好ましく、50質量%以上であることが特に好ましい。また、該含有量は、5000質量%以下であることがより好ましく、3000質量%以下であることが更に好ましく、2000質量%以下であることが一層好ましく、1000質量%以下であることが特に好ましく、500質量%以下であることが最も好ましい。
なお、通常は、酸化工程直後では酸が酸化黒鉛に対し過剰に存在しており(例えば、酸化黒鉛100質量%に対し硫酸2000~5000質量%程度)、反応停止工程直後であれば水が酸化黒鉛に対し過剰に存在する(例えば、酸化黒鉛100質量%に対し、500~10000質量%程度)。
この水の量が上記範囲内となるように上記濃縮工程で酸及び水を濃縮することが好ましい。これにより、反応性の酸素を酸化黒鉛誘導体を得る工程まで安定的に存在させ効率的に修飾反応に供することができる。
この水の量が上記範囲内となるように上記濃縮工程で酸及び水を濃縮することが好ましい。これにより、反応性の酸素を酸化黒鉛誘導体を得る工程まで安定的に存在させ効率的に修飾反応に供することができる。
また後述するように酸化工程において酸化剤として過マンガン酸塩を用いる場合、濃縮工程後の酸化黒鉛含有組成物は、組成物中、マンガンを0.01質量%以上含んでいてもよい。
更に、後述するように酸化工程において酸化剤として過マンガン酸カリウムを用いる場合、濃縮工程後の酸化黒鉛含有組成物は、組成物中、カリウムを0.01質量%以上含んでいてもよい。
更に、後述するように酸化工程において酸化剤として過マンガン酸カリウムを用いる場合、濃縮工程後の酸化黒鉛含有組成物は、組成物中、カリウムを0.01質量%以上含んでいてもよい。
本発明における濃縮工程とは酸化黒鉛含有組成物中の水量等を調節することであり、この工程は水量が上述した下限値未満とならない限り、遠心分離、水等を加え再分散、濾過、凝集剤を用いた濾過、減圧濃縮等の方法を使用して行うことができる。また、これらの工程は繰り返してもよいが、繰り返すことなく、1工程で完了することが好ましい。更に、凝集剤を用いて濃縮する場合の凝集剤については特に限定はされないが、続く修飾反応の妨げにならないものが好ましい。凝集剤としては、例えば、ポリマー性の凝集剤で酸化黒鉛の酸素官能基や硫酸と反応しないもの、揮発性の凝集剤で、反応時の反応温度で容易に取り除けるものが挙げられる。
(分離工程)
本発明の製造方法においては、酸化黒鉛含有組成物、典型的には酸化工程で得られた酸化黒鉛を含む反応液から酸化黒鉛含有組成物、典型的にはより高濃度の酸化黒鉛を含む酸化黒鉛含有組成物を分離する工程を含むことが好ましい。すなわち、本発明の酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい一実施形態では、酸化工程と酸化黒鉛誘導体を得る工程との間に、反応組成物に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含む。
本発明はまた、酸化黒鉛誘導体を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程を含み、該酸化工程と該酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物、典型的には該酸化工程で得られる酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物、典型的にはより高濃度の酸化黒鉛を含む酸化黒鉛含有組成物を分離する工程を含むことを特徴とする酸化黒鉛誘導体の製造方法でもある。この製造方法は、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含んでいてもよいが、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことが好ましく、酸化黒鉛含有組成物を精製する工程を含まないことがより好ましい。
本発明の製造方法においては、酸化黒鉛含有組成物、典型的には酸化工程で得られた酸化黒鉛を含む反応液から酸化黒鉛含有組成物、典型的にはより高濃度の酸化黒鉛を含む酸化黒鉛含有組成物を分離する工程を含むことが好ましい。すなわち、本発明の酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい一実施形態では、酸化工程と酸化黒鉛誘導体を得る工程との間に、反応組成物に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含む。
本発明はまた、酸化黒鉛誘導体を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程を含み、該酸化工程と該酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物、典型的には該酸化工程で得られる酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物、典型的にはより高濃度の酸化黒鉛を含む酸化黒鉛含有組成物を分離する工程を含むことを特徴とする酸化黒鉛誘導体の製造方法でもある。この製造方法は、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含んでいてもよいが、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことが好ましく、酸化黒鉛含有組成物を精製する工程を含まないことがより好ましい。
水に添加したときに水と完全に層分離せず、かつ、水と完全に混和もしないこのような溶媒を酸化黒鉛を含む反応液に添加した場合、添加された溶媒が反応液中で酸化黒鉛と親和性を発揮して酸化黒鉛粒子に付着し、溶媒が付着した酸化黒鉛粒子同士は凝集し易くなる。このように、添加された溶媒が酸化黒鉛粒子を凝集させる凝集剤としてはたらく結果、反応液中での酸化黒鉛粒子と反応液との分離が促進され、酸化黒鉛の分離をより効率的に行うことが可能となる。
なお、酸化黒鉛を含む反応液等からの酸化黒鉛含有組成物の分離を行うには、通常は遠心分離を繰り返して行ったり、加圧下で反応液を濾過したりする。前者の場合、工程が煩雑になるとともに廃液の量が多くなる場合があるといった課題があり、より効率的な生産の面から改善の余地があった。後者の場合、フィルターが目詰まりして時間がかかる場合があり、やはり、より効率的な生産の面から改善の余地があった。
本発明者らは、本発明の製造方法が上記分離工程を含むことにより、酸化黒鉛含有組成物や、酸化黒鉛誘導体を、より効率的に製造することが可能となり、また所望の官能基を有する基が充分に導入された高品質な酸化黒鉛又は酸化黒鉛誘導体を得ることができることを見出した。
よって、酸化黒鉛を含む反応液等に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛を分離する工程を含む酸化黒鉛又は酸化黒鉛誘導体の製造方法も、本発明の好ましい実施形態の一つ(第2の好ましい形態とも言う)である。
なお、上記本発明の製造方法の第1の好ましい形態は、酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含むことがより好ましい。
本発明者らは、本発明の製造方法が上記分離工程を含むことにより、酸化黒鉛含有組成物や、酸化黒鉛誘導体を、より効率的に製造することが可能となり、また所望の官能基を有する基が充分に導入された高品質な酸化黒鉛又は酸化黒鉛誘導体を得ることができることを見出した。
よって、酸化黒鉛を含む反応液等に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛を分離する工程を含む酸化黒鉛又は酸化黒鉛誘導体の製造方法も、本発明の好ましい実施形態の一つ(第2の好ましい形態とも言う)である。
なお、上記本発明の製造方法の第1の好ましい形態は、酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含むことがより好ましい。
上記分離工程において酸化黒鉛を含む反応液等に添加する溶媒は、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒であればよいが、溶媒の水に対する溶解度は0.5%以上であることが好ましい。このような溶解度であると、水と適度に混和して酸化黒鉛を凝集させる作用がより充分に発揮されることになる。溶媒の水に対する溶解度は、より好ましくは、0.7%以上であり、更に好ましくは、1%以上であり、特に好ましくは、1.5%以上である。また、溶媒の溶解度は、30%以下であることが好ましく、より好ましくは、20%以下であり、更に好ましくは、15%以下であり、特に好ましくは、10%以下である。
なお、溶媒の水に対する溶解度は、溶液沈降法により測定することができる。
なお、「水に対する溶解度が0.01%以上」とは、水100質量%に対して溶媒が0.01質量%以上溶解すること、言い換えれば、水100gに対して溶媒が0.01g以上溶解することを表す。
なお、溶媒の水に対する溶解度は、溶液沈降法により測定することができる。
なお、「水に対する溶解度が0.01%以上」とは、水100質量%に対して溶媒が0.01質量%以上溶解すること、言い換えれば、水100gに対して溶媒が0.01g以上溶解することを表す。
上記分離工程において酸化黒鉛を含む反応液等に添加する溶媒の量は適宜設定すればよいが、酸化黒鉛を含む反応液等中の酸化黒鉛100質量%に対して1~1000質量%であることが好ましい。このような量の溶媒を添加することで、酸化黒鉛をより充分に凝集させ、分離をより効率的に進めることができる。溶媒の添加量は、より好ましくは、酸化黒鉛を含む反応液中の酸化黒鉛100質量%に対して1~950質量%であり、更に好ましくは、 10~900質量%であり、特に好ましくは、100~800質量%である。
上記水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒としては、シクロペンタノン、シクロヘキサノン等の環状アルカノン;ブタノール、ペンタノール、ヘキサノール、ヘプタノール等の炭素数4~7のアルコール;アセチルアセトン、メチルエチルケトン、メチルプロピルケトン、メチルブチルケトン等のケトン類等が挙げられ、これらの1種又は2種以上を用いることができる。
上記分離工程において酸化黒鉛を含む反応液等に所定の溶解度の溶媒を添加した後、酸化黒鉛含有組成物を分離する方法は、酸化黒鉛含有組成物が反応液から分離されることになる限り特に制限されないが、濾過、デカンテーション、遠心分離、及び、分液抽出のいずれかにより行われることが好ましい。これらの方法を用いることで、反応液から酸化黒鉛含有組成物をより効率的に分離することができる。分離工程をこれらのいずれかにより行う場合、これらの操作を1回行ってもよく、複数回行ってもよい。また、これらのいずれか1つのみを行ってもよく、2つ以上を組み合わせて行ってもよい。なお、酸化黒鉛を含む反応液等に対して、これらの方法を用いたものであっても、酸化黒鉛の質量に対して硫酸が1質量%以上残存している限り、精製したものには該当せず、酸化黒鉛の質量に対して水が3質量%以上残存している限り、乾燥したものには該当しない。また、酸化黒鉛を含む反応液等に対してこれらの方法のみを用いたものは、通常、酸化黒鉛の質量に対して水が3質量%以上、硫酸が1質量%以上残存しており、精製したものや乾燥したものには該当しない。
これらの分離方法の中でも、より好ましくは、濾過、分液抽出のいずれかであり、最も好ましくは、濾過である。
これらの分離方法の中でも、より好ましくは、濾過、分液抽出のいずれかであり、最も好ましくは、濾過である。
濾過は上記分離方法の中でも最も簡便な方法であるが、従来の酸化黒鉛やその誘導体の製造方法では、酸化黒鉛によるフィルターの目詰まりが起こりやすく、濾過に時間がかかることが問題となっていた。これに対し、上記分離工程によれば、水に対する溶解度が所定の範囲の溶媒の作用により反応液中で酸化黒鉛粒子の凝集が促進され、これにより、反応液を濾過してもフィルターの目詰まりが起こりにくく、濾過に要する時間が従来の酸化黒鉛やその誘導体の製造方法における濾過工程に比べて大幅に短い。このため、酸化黒鉛含有組成物を分離する方法として濾過を用いることで、反応液と酸化黒鉛との分離を簡便かつ効率的に進めることができる。
上記分離工程は、酸化黒鉛を含む反応液等に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含むうえで、反応液から分離された酸化黒鉛含有組成物を更に後段の反応工程に適するものとするその他の分離工程を更に含んでいてもよい。その他の分離工程としては、水洗等が挙げられる。
上記分離工程のその他の好ましい実施形態としては、本発明の効果を発揮する限り、上述した濃縮工程の好ましい実施形態を適用することができる。例えば、分離工程後の酸化黒鉛含有組成物の好ましい実施形態は、上述した濃縮工程後の酸化黒鉛含有組成物の好ましい実施形態と同様である。
上記分離工程のその他の好ましい実施形態としては、本発明の効果を発揮する限り、上述した濃縮工程の好ましい実施形態を適用することができる。例えば、分離工程後の酸化黒鉛含有組成物の好ましい実施形態は、上述した濃縮工程後の酸化黒鉛含有組成物の好ましい実施形態と同様である。
(黒鉛を酸化する工程)
上記黒鉛を酸化する工程は、黒鉛が酸化されることになる限り、その方法は特に制限されず、上述したHummers法、Brodie法、Staudenmaier法等のいずれの方法における黒鉛の酸化方法を用いてもよく、後述する実施例に記載の方法のように、Hummers法における酸化方法を採用した、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であってもよい。このように、酸化工程が、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であることは、本発明の好適な実施形態の1つである。
上記黒鉛を酸化する工程は、黒鉛が酸化されることになる限り、その方法は特に制限されず、上述したHummers法、Brodie法、Staudenmaier法等のいずれの方法における黒鉛の酸化方法を用いてもよく、後述する実施例に記載の方法のように、Hummers法における酸化方法を採用した、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であってもよい。このように、酸化工程が、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であることは、本発明の好適な実施形態の1つである。
上述した酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい実施形態では、上記酸化工程は、酸を用いて黒鉛を酸化する。
上記酸化工程が酸を用いて黒鉛を酸化する場合、本発明の製造方法は、酸化工程で得られる酸化黒鉛含有組成物の精製及び乾燥の少なくとも一方を省くため、酸化黒鉛誘導体を得る工程に供される酸化黒鉛含有組成物中に酸が残留し、該酸は、酸化黒鉛に含まれる酸素官能基を安定化させる。なお、この場合、酸化黒鉛誘導体を得る工程に供される酸化黒鉛含有組成物中に、酸化工程で用いた酸化剤由来の成分や、酸化工程や反応停止工程で用いた水等が残留していてもよく、修飾反応を高効率で進行させることができる。その結果、酸化黒鉛誘導体を得る工程において、該酸化黒鉛に含まれる酸素官能基を安定に存在させたまま修飾反応に供することが可能となり、本発明の製造方法をより簡便で高効率なものとすることができる。
上記酸化工程が酸を用いて黒鉛を酸化する場合、本発明の製造方法は、酸化工程で得られる酸化黒鉛含有組成物の精製及び乾燥の少なくとも一方を省くため、酸化黒鉛誘導体を得る工程に供される酸化黒鉛含有組成物中に酸が残留し、該酸は、酸化黒鉛に含まれる酸素官能基を安定化させる。なお、この場合、酸化黒鉛誘導体を得る工程に供される酸化黒鉛含有組成物中に、酸化工程で用いた酸化剤由来の成分や、酸化工程や反応停止工程で用いた水等が残留していてもよく、修飾反応を高効率で進行させることができる。その結果、酸化黒鉛誘導体を得る工程において、該酸化黒鉛に含まれる酸素官能基を安定に存在させたまま修飾反応に供することが可能となり、本発明の製造方法をより簡便で高効率なものとすることができる。
中でも、上記酸化工程が、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であることは、本発明の好適な実施形態の1つである。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、硫酸の使用量は、黒鉛に対する硫酸の質量比(硫酸/黒鉛)が25~60となる量であることが好ましい。該質量比が25以上であることにより、酸化反応中に反応液(混合液)の高粘度化を充分に防止して酸化黒鉛を効率的に製造することができる。また、該質量比が60以下であることにより、廃液量を充分に少なくすることができる。
上記質量比は、26以上であることがより好ましく、27以上であることが更に好ましく、28以上であることが特に好ましい。また、該質量比は、54以下であることがより好ましく、48以下であることが更に好ましく、42以下であることが特に好ましい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、硫酸の使用量は、黒鉛に対する硫酸の質量比(硫酸/黒鉛)が25~60となる量であることが好ましい。該質量比が25以上であることにより、酸化反応中に反応液(混合液)の高粘度化を充分に防止して酸化黒鉛を効率的に製造することができる。また、該質量比が60以下であることにより、廃液量を充分に少なくすることができる。
上記質量比は、26以上であることがより好ましく、27以上であることが更に好ましく、28以上であることが特に好ましい。また、該質量比は、54以下であることがより好ましく、48以下であることが更に好ましく、42以下であることが特に好ましい。
上記酸化工程に用いる黒鉛は、平均粒子径が3μm以上、80μm以下であることが好ましい。このような平均粒子径のものを用いることで、酸化反応をより効率的に進めることができる。黒鉛の平均粒子径は、より好ましくは3.2μm以上、70μm以下である。
上記平均粒子径は、粒度分布測定装置により測定することができる。
上記平均粒子径は、粒度分布測定装置により測定することができる。
上記酸化工程に用いる黒鉛の形状は特に制限されず、微粉状、粉状、粒状、顆粒状、鱗片状、多面体状、ロッド状、曲面含有状等が挙げられる。なお、平均粒子径が上述のような範囲の粒子は、例えば、粒子を粉砕機等により粉砕する方法や、粒子をふるい等にかけて粒子径を選別する方法、これら方法の組み合わせのほか、粒子を製造する段階で調製条件を最適化し、所望の粒子径の粒子を得る方法等により製造することが可能である。
上記黒鉛と硫酸とを含む混合液中における黒鉛の含有量は、混合液100質量%に対して0.5質量%以上であることが好ましく、1質量%以上であることがより好ましく、1.5質量%以上であることが更に好ましく、2質量%以上であることが特に好ましい。該黒鉛の含有量は、10質量%以下であることが好ましく、8質量%以下であることがより好ましく、7質量%以下であることが更に好ましく、6質量%以下であることが特に好ましい。
本発明の製造方法における酸化工程に用いる黒鉛は、1種のみであってもよく、上記平均粒子径、形状等のいずれかにおいて異なる2種以上のものを用いてもよい。
本発明の製造方法における酸化工程に用いる黒鉛は、1種のみであってもよく、上記平均粒子径、形状等のいずれかにおいて異なる2種以上のものを用いてもよい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程で添加する過マンガン酸塩としては、過マンガン酸ナトリウム、過マンガン酸カリウム、過マンガン酸アンモニウム、過マンガン酸銀、過マンガン酸亜鉛、過マンガン酸マグネシウム、過マンガン酸カルシウム、過マンガン酸バリウム等が挙げられ、これらの1種又は2種以上を使用できるが、中でも過マンガン酸ナトリウム、過マンガン酸カリウムが好ましく、過マンガン酸カリウムがより好ましい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程における上記過マンガン酸塩の全添加量は、上記混合液中の黒鉛量100質量%に対し、50~500質量%であることが好ましい。これにより、酸化黒鉛を安全かつ効率的に製造することができる。なお、酸化剤の全添加量を変化させることで、酸化黒鉛に導入される酸素原子の量を調節することができる。
該全添加量は、100質量%以上であることがより好ましく、150質量%以上であることが更に好ましく、200質量%以上であることが一層好ましく、240質量%以上であることが特に好ましい。また、該全添加量は、450質量%以下であることがより好ましく、400質量%以下であることが更に好ましく、350質量%以下であることが一層好ましく、300質量%以下であることが特に好ましい。
該全添加量は、100質量%以上であることがより好ましく、150質量%以上であることが更に好ましく、200質量%以上であることが一層好ましく、240質量%以上であることが特に好ましい。また、該全添加量は、450質量%以下であることがより好ましく、400質量%以下であることが更に好ましく、350質量%以下であることが一層好ましく、300質量%以下であることが特に好ましい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程では、過マンガン酸塩を一括で添加してもよく、複数回に分けて添加してもよく、また連続的に添加しても良いが、複数回に分けて添加するか連続的に添加することが好ましい。これにより、酸化反応が急激に進行することを抑えて反応の制御をよりしやすくすることができる。過マンガン酸塩を複数回に分けて添加する場合、添加する回数は、3回以上であることが好ましく、5回以上であることがより好ましく、7回以上であることが更に好ましく、9回以上であることが特に好ましい。
上記過マンガン酸塩を複数回に分けて添加する場合、1回当たりの添加量は、それぞれ同じであってもよく、異なっていてもよい。
上記過マンガン酸塩を複数回に分けて添加する場合、1回当たりの添加量は、それぞれ同じであってもよく、異なっていてもよい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程では、上記混合液の温度を10~50℃の範囲内に維持しながら過マンガン酸塩を添加することが好ましい。このような温度範囲に維持することで、酸化反応を制御しながら充分に進行させることができる。
上記温度は、12℃以上に維持することが好ましく、15℃以上に維持することがより好ましく、18℃以上に維持することが更に好ましく、20℃以上に維持することが特に好ましい。
上記温度は、12℃以上に維持することが好ましく、15℃以上に維持することがより好ましく、18℃以上に維持することが更に好ましく、20℃以上に維持することが特に好ましい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程は、上記混合液の温度変化を25℃以下に維持しながら過マンガン酸塩を添加する工程であることが好ましい。これにより、より安定的に酸化工程を行うことができる。
上記酸化工程は、該温度変化を20℃以下に維持することがより好ましく、15℃以下に維持することが更に好ましく、10℃以下に維持することが特に好ましい。
上記酸化工程は、該温度変化を20℃以下に維持することがより好ましく、15℃以下に維持することが更に好ましく、10℃以下に維持することが特に好ましい。
上記酸化工程が黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程である場合、上記酸化工程では、安定的に酸化工程を行う観点から、過マンガン酸塩を10分~10時間の間にわたって添加することが好ましい。より好ましくは、過マンガン酸塩を30分以上の間にわたって添加することであり、更に好ましくは、1時間以上の間にわたって添加することであり、特に好ましくは、2時間以上の間にわたって添加することである。
また、酸化黒鉛を効率的に製造する点から、過マンガン酸塩の添加時間は、8時間以下であることが好ましく、7時間以下であることが好ましく、6時間以下であることが更に好ましい。
また、酸化黒鉛を効率的に製造する点から、過マンガン酸塩の添加時間は、8時間以下であることが好ましく、7時間以下であることが好ましく、6時間以下であることが更に好ましい。
上記酸化工程は、公知の撹拌機等を用いて撹拌しながら行うことが好ましい。
上記酸化工程は、例えば空気中、又は、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行うことができる。また、上記酸化工程は、その圧力条件は特に限定されないが、例えば常圧条件下で行うことが好ましい。
また上記酸化工程の時間は、0.5時間~120時間とすることが好ましく、1時間~15時間とすることがより好ましく、2時間~10時間とすることが更に好ましい。
上記酸化工程は、連続的に行ってもよいし、断続的に行ってもよい。
上記酸化工程は、例えば空気中、又は、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行うことができる。また、上記酸化工程は、その圧力条件は特に限定されないが、例えば常圧条件下で行うことが好ましい。
また上記酸化工程の時間は、0.5時間~120時間とすることが好ましく、1時間~15時間とすることがより好ましく、2時間~10時間とすることが更に好ましい。
上記酸化工程は、連続的に行ってもよいし、断続的に行ってもよい。
上記混合液は、黒鉛、硫酸、及び、必要に応じてその他の成分を混合して得ることができる。上記混合は、公知の方法で適宜行うことが可能であるが、例えば、超音波処理を行ったり、公知の分散機を用いたりして黒鉛を均一に分散させることが好ましい。
本発明の酸化黒鉛誘導体の製造方法は、黒鉛を酸化する工程と、後述する酸化黒鉛誘導体を得る工程とを含み、両工程の間に酸化黒鉛含有組成物を精製しかつ乾燥する工程を含まない限り、熟成工程、酸化工程の後の酸化反応停止(クエンチ)工程等のその他の工程を含んでいてもよい。
上記熟成工程において、酸化工程で得られた反応液を熟成させる温度及び時間は適宜選択すればよいが、反応液を0~90℃の温度に維持することが好ましく、より好ましくは、20~80℃の温度に維持することである。
また熟成させる時間は、0.1~24時間であることが好ましい。より好ましくは、0.5~5時間である。
また熟成させる時間は、0.1~24時間であることが好ましい。より好ましくは、0.5~5時間である。
上記酸化反応停止工程は、空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。また真空中で行っても良い。
上記酸化反応停止工程は、例えば、反応液の温度を5~15℃に設定し、反応液に水を添加し、次いで還元剤として過酸化水素水を添加して行うことができる。また、反応液を、5~25℃に設定した、水又は過酸化水素水に添加して行ってもよい。
上記酸化反応停止工程の時間は、例えば0.01~5時間とすることができる。
上記酸化反応停止工程は、例えば、反応液の温度を5~15℃に設定し、反応液に水を添加し、次いで還元剤として過酸化水素水を添加して行うことができる。また、反応液を、5~25℃に設定した、水又は過酸化水素水に添加して行ってもよい。
上記酸化反応停止工程の時間は、例えば0.01~5時間とすることができる。
(酸化黒鉛誘導体を得る工程)
本発明の製造方法では、酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させることにより酸化黒鉛誘導体を得る。上記酸化黒鉛の酸素含有官能基と反応する化合物は、例えば、アルコール、シラン化合物、脂肪酸(塩)、脂肪酸エステル、イソシアネート化合物、及び、アミンからなる群より選択される少なくとも1種であることが好ましく、中でもアルコール及び/又はアミンであることがより好ましい。なお、シラン化合物は、酸化黒鉛の酸素含有官能基との反応性を良好なものとする観点から、珪素原子と直接結合したシロキシ基及び/又はアルコキシ基を有することが好ましい。なお、酸化黒鉛誘導体の生成は、実施例の方法に沿って赤外線吸収スペクトルを測定することにより確認される。
本発明の製造方法では、酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させることにより酸化黒鉛誘導体を得る。上記酸化黒鉛の酸素含有官能基と反応する化合物は、例えば、アルコール、シラン化合物、脂肪酸(塩)、脂肪酸エステル、イソシアネート化合物、及び、アミンからなる群より選択される少なくとも1種であることが好ましく、中でもアルコール及び/又はアミンであることがより好ましい。なお、シラン化合物は、酸化黒鉛の酸素含有官能基との反応性を良好なものとする観点から、珪素原子と直接結合したシロキシ基及び/又はアルコキシ基を有することが好ましい。なお、酸化黒鉛誘導体の生成は、実施例の方法に沿って赤外線吸収スペクトルを測定することにより確認される。
上記酸化黒鉛の酸素含有官能基と反応する化合物は、酸化黒鉛誘導体の非極性分散媒中での分散性をより向上する観点からは、炭化水素基を有することが好ましい。炭化水素基は、炭素数が5以上であることが好ましく、6以上であることがより好ましく、7以上であることが更に好ましく、8以上であることが特に好ましい。また、該炭化水素基は、酸化黒鉛誘導体の非極性分散媒中での分散速度を充分に速いものとし、また、酸化黒鉛誘導体を好適に製造する観点からは、炭素数が50以下であることが好ましく、36以下であることがより好ましく、24以下であることが更に好ましい。
上記炭化水素基は、直鎖であることが好ましい。炭化水素基が直鎖であると、直鎖の炭化水素基が酸化黒鉛に導入されやすいため、本発明の製造方法がより効率的なものとなる。
上記炭化水素基は、分岐鎖であることもまた好ましい。上記炭化水素基が分岐鎖であると、酸化黒鉛誘導体の非極性分散媒中での分散性がより向上する。また、原料である化合物が常温(25℃)で液体となる傾向にあり、その場合は、誘導体製造時のハンドリングも容易である。
上記酸化黒鉛誘導体を得る工程において、酸化黒鉛が酸化黒鉛の酸素含有官能基と反応する化合物と反応して修飾される反応と、酸化黒鉛自身の還元反応とが同時進行して競合するところ、還元反応を充分に抑制して酸化黒鉛を修飾する反応を優位的に進める観点からは、上記酸化黒鉛の酸素含有官能基と反応する化合物がアルコール及び/又はアミンであることが好ましい。アルコールは、脂肪族アルコールであることがより好ましい。また、アミンは、脂肪族アミンであることがより好ましい。
上記脂肪族アルコールとしては、例えば、n-オクチルアルコール、sec-オクチルアルコール、n-ノニルアルコール、sec-ノニルアルコール、n-デシルアルコール、sec-デシルアルコール、n-ウンデシルアルコール、sec-ウンデシルアルコール、n-ドデシルアルコール、sec-ドデシルアルコール、n-トリデシルアルコール、sec-トリデシルアルコール、n-テトラデシルアルコール、sec-テトラデシルアルコール、n-ヘキサデシルアルコール、sec-ヘキサデシルアルコール、n-オクタデシルアルコール、sec-オクタデシルアルコール、n-エイコシルアルコール、sec-エイコシルアルコール、2-オクチルドデシルアルコール、n-ドコシルアルコール、sec-ドコシルアルコール、2-オクチルテトラデシルアルコール、n-テトラコシルアルコール、sec-テトラコシルアルコール、2-オクチルヘキサデシルアルコール、n-ヘキサコシルアルコール、sec-ヘキサコシルアルコール、n-オクタコシルアルコール、sec-オクタコシルアルコール、n-トリアコンチルアルコール、sec-トリアコンチルアルコール、n-ドトリアコンチルアルコール、sec-ドトリアコンチルアルコール、n-テトラトリアコンチルアルコール、sec-テトラトリアコンチルアルコール、n-ヘキサトリアコンチルアルコール、sec-ヘキサトリアコンチルアルコール等が挙げられ、これらの1種又は2種以上を使用できる。また、2種以上の脂肪族アルコールを使用する場合、これら脂肪族アルコールの混合物を使用してもよい。
上記脂肪族アミンとしては、例えば、n-オクチルアミン、sec-オクチルアミン、n-ノニルアミン、sec-ノニルアミン、n-デシルアミン、sec-デシルアミン、n-ウンデシルアミン、sec-ウンデシルアミン、n-ドデシルアミン、sec-ドデシルアミン、n-トリデシルアミン、sec-トリデシルアミン、n-テトラデシルアミン、sec-テトラデシルアミン、n-ヘキサデシルアミン、sec-ヘキサデシルアミン、n-オクタデシルアミン(ステアリルアミン)、sec-オクタデシルアミン、n-エイコシルアミン、sec-エイコシルアミン、2-オクチルドデシルアミン、n-ドコシルアミン、sec-ドコシルアミン、2-オクチルテトラデシルアミン、n-テトラコシルアミン、sec-テトラコシルアミン、2-オクチルヘキサデシルアミン、n-ヘキサコシルアミン、sec-ヘキサコシルアミン、n-オクタコシルアミン、sec-オクタコシルアミン、n-トリアコンチルアミン、sec-トリアコンチルアミン、n-ドトリアコンチルアミン、sec-ドトリアコンチルアミン、n-テトラトリアコンチルアミン、sec-テトラトリアコンチルアミン、n-ヘキサトリアコンチルアミン、sec-ヘキサトリアコンチルアミン等が挙げられ、これらの1種又は2種以上を使用できる。また、2種以上の脂肪族アミンを使用する場合、これら脂肪族アミンの混合物を使用してもよい。
上記酸化黒鉛誘導体を得る工程において、酸化工程で得られる酸化黒鉛含有組成物(中でも、好ましくは、濃縮工程後の酸化黒鉛含有組成物)に、酸化黒鉛の酸素含有官能基と反応する化合物、及び、必要に応じてその他の成分(例えば溶媒)を混合して混合液を得ることができる。混合は、公知の方法で適宜行うことが可能であるが、例えば、超音波処理を行ったり、公知の分散機を用いたりして酸化黒鉛を均一に分散させることが好ましい。
上記酸化黒鉛の酸素含有官能基と反応する化合物の使用量は、混合液中の酸化黒鉛量100質量%に対し、300~10000質量%であることが好ましい。該酸化黒鉛の酸素含有官能基と反応する化合物を大過剰に用いることにより、酸化黒鉛誘導体を効率的に製造することができる。また、これにより、還元反応を抑制して酸化黒鉛の修飾反応を優位に進めることができる。
該使用量は、350質量%以上であることがより好ましく、400質量%以上であることが更に好ましく、450質量%以上であることが一層好ましく、500質量%以上であることが特に好ましい。また、該使用量は、8000質量%以下であることがより好ましく、6000質量%以下であることが更に好ましく、3000質量%以下であることが一層好ましく、1000質量%以下であることが特に好ましい。
該使用量は、350質量%以上であることがより好ましく、400質量%以上であることが更に好ましく、450質量%以上であることが一層好ましく、500質量%以上であることが特に好ましい。また、該使用量は、8000質量%以下であることがより好ましく、6000質量%以下であることが更に好ましく、3000質量%以下であることが一層好ましく、1000質量%以下であることが特に好ましい。
また酸化黒鉛と酸化黒鉛の酸素含有官能基と反応する化合物との反応では、公知の触媒等を適用することができるが、本発明の好適な形態として、触媒として硫酸等の酸触媒、アルカリ金属水酸化物、アミン、ピリジン等の塩基触媒を用いることができ、また酸化黒鉛の酸素含有官能基と反応する化合物がアミンの場合はアミン自身を触媒として用いることができる。
酸化黒鉛の酸素含有官能基と反応する化合物がアミン以外の化合物であり、酸化黒鉛誘導体を得る工程の触媒として酸触媒を用いる場合、酸化黒鉛や酸化黒鉛の酸素含有官能基と反応する化合物等の反応原料の量に対する酸触媒の量を好適な範囲内に調節することにより、修飾反応をより効率的に進行させることができる。
酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物における、組成物中の酸化黒鉛の質量100質量%に対する酸触媒の好ましい含有量は、例えば、0.01~1000質量%である。該酸触媒の含有量が0.01質量%以上であることにより、効率よく修飾反応を進行させることができる。また、該含有量が1000質量%以下であることにより、廃棄物量(廃液量)を充分に少なくするとともに、効率よく修飾反応を進行させることができる。ここで、酸触媒としては、酸化工程で用いた硫酸等の酸の残留物を有効に活用できる。
酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物における、組成物中の酸化黒鉛の質量100質量%に対する酸触媒の好ましい含有量は、例えば、0.01~1000質量%である。該酸触媒の含有量が0.01質量%以上であることにより、効率よく修飾反応を進行させることができる。また、該含有量が1000質量%以下であることにより、廃棄物量(廃液量)を充分に少なくするとともに、効率よく修飾反応を進行させることができる。ここで、酸触媒としては、酸化工程で用いた硫酸等の酸の残留物を有効に活用できる。
上記酸触媒の含有量は、0.1質量%以上であることがより好ましく、1質量%以上であることが更に好ましく、10質量%以上であることが一層好ましく、20質量%以上であることがより一層好ましく、30質量%以上であることが更に一層好ましく、40質量%以上であることが特に好ましく、50質量%以上であることが特に一層好ましい。また、該含有量は、700質量%以下であることがより好ましく、500質量%以下であることが更に好ましく、200質量%以下であることが一層好ましい。
例えば、酸触媒の酸が硫酸である場合、上記酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物は、組成物中の酸化黒鉛の質量100質量%に対して、硫酸を1質量%以上、1000質量%以下含むことが好ましい。
例えば、酸触媒の酸が硫酸である場合、上記酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物は、組成物中の酸化黒鉛の質量100質量%に対して、硫酸を1質量%以上、1000質量%以下含むことが好ましい。
また酸化黒鉛の酸素含有官能基と反応する化合物がアミン以外の化合物であり、酸化黒鉛誘導体を得る工程の触媒として酸触媒を用いる場合、酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物が、組成物中の酸化黒鉛の酸素含有官能基と反応する化合物の質量100質量%に対して、酸触媒を0.1質量%以上、50質量%以下含むことが好ましい。酸触媒を0.1質量%以上含むものとすることにより、修飾反応をより効率的に進めることができる。また、酸触媒を50質量%以下含むものとすることにより、酸触媒に起因する副反応を充分に抑制することができる。該酸触媒の含有量は、0.5質量%以上であることがより好ましい。また、該酸触媒の含有量は、20質量%以下であることがより好ましい。
なお、酸化黒鉛の酸素含有官能基と反応する化合物がアミン以外の化合物である場合、酸化黒鉛は本質的に自身が酸性の物質であり、自触媒的に反応が進行する。前述のとおり触媒を用いることが好ましいが、触媒を用いなくても自触媒的に反応を進行させることも可能である。
酸化黒鉛誘導体を得る工程の触媒として塩基触媒(アミン等を含む)を用いる場合、酸化黒鉛や酸化黒鉛の酸素含有官能基と反応する化合物等の反応原料の量に対する塩基触媒の量を好適な範囲内に調節することにより、修飾反応をより効率的に進行させることができる。酸化黒鉛誘導体を得る工程に供される酸化黒鉛含有組成物における、酸化黒鉛の質量100質量%に対する塩基触媒の好ましい含有量は、上述した酸化黒鉛含有組成物における、酸化黒鉛の質量100質量%に対する酸の好ましい含有量と同様である。
例えば、塩基がアルカリ金属水酸化物及びアミンである場合、酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物が、組成物中の酸化黒鉛の質量100質量%に対して、アルカリ金属水酸化物及びアミンを1質量%以上、1000質量%以下含むことが好ましい。なお塩基触媒は酸化黒鉛含有組成物中の酸成分と中和するため、使用した塩基触媒のうち、中和した部分を除いた部分の量が上記範囲に該当することが好ましい。
例えば、塩基がアルカリ金属水酸化物及びアミンである場合、酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物が、組成物中の酸化黒鉛の質量100質量%に対して、アルカリ金属水酸化物及びアミンを1質量%以上、1000質量%以下含むことが好ましい。なお塩基触媒は酸化黒鉛含有組成物中の酸成分と中和するため、使用した塩基触媒のうち、中和した部分を除いた部分の量が上記範囲に該当することが好ましい。
上述した酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい実施形態(本発明の製造方法の第3の好ましい形態とも言う。)では、上記酸化黒鉛誘導体を得る工程における反応温度は、反応が進行する限り特に限定されないが、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含む。反応温度を120℃以上とすることにより、還元反応を充分に抑制して修飾反応を優位的に進めることができ、更に酸素が脱離しやすいため落つい感度試験による落つい感度を低減(消失)させるためにも優位である。このように修飾反応をより効率的に進行させる観点及び落つい感度を低減(消失)させる観点から、該反応温度は、130℃以上であることがより好ましく、140℃以上であることが更に好ましく、150℃以上であることが特に好ましい。
また、副反応を抑制する観点からは、反応温度を200℃以下とすることが好ましい。
すなわち、本発明の製造方法の第3の好ましい形態である、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含む酸化黒鉛誘導体の製造方法により、高品質な酸化黒鉛誘導体を簡便に製造することが可能となることが見出された。
なお、本発明の製造方法の第1の好ましい形態又は本発明の製造方法の第2の好ましい形態は、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含むことがより好ましい。
また、副反応を抑制する観点からは、反応温度を200℃以下とすることが好ましい。
すなわち、本発明の製造方法の第3の好ましい形態である、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含む酸化黒鉛誘導体の製造方法により、高品質な酸化黒鉛誘導体を簡便に製造することが可能となることが見出された。
なお、本発明の製造方法の第1の好ましい形態又は本発明の製造方法の第2の好ましい形態は、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含むことがより好ましい。
酸化黒鉛及びアルコールを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含む酸化黒鉛誘導体の製造方法において、酸化黒鉛は、上記酸化工程で得られる酸化黒鉛を含む酸化黒鉛含有組成物を精製及び乾燥して得たものであってもよいが、精製及び乾燥の少なくとも一方を省いたものであることが好ましく、精製を省いたものであることがより好ましい。上記製造方法により、還元反応を充分に抑制して酸化黒鉛へのアルコール及び/又はアミンの導入量を増やすことができ、高品質の酸化黒鉛誘導体を高効率で得ることが可能である。また、この製造方法により得られる酸化黒鉛誘導体は、導入されたアルコール及び/又はアミン由来の炭化水素基同士の相互作用により、結晶性が高いものとなる。なお、上記製造方法では、アルコール及び/又はアミンの炭素数が多いほど、得られる酸化黒鉛誘導体の結晶性がより高いものとなる傾向があるが、アルコール及び/又はアミンの炭素数が少なくても、酸化黒鉛に導入される炭化水素基の数を充分に多いものとすることができ、高品質の酸化黒鉛誘導体を得ることができる。
なお、上述した特許文献及び非特許文献には、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得ることについて何ら開示されていない。
なお、上述した特許文献及び非特許文献には、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得ることについて何ら開示されていない。
本発明はまた、アルキル基を有し、JIS K 4810に規定される落つい感度試験で測定される等級が8級である酸化黒鉛誘導体でもある。例えば、本発明の製造方法において、酸化黒鉛とアルコール及び/又はアミンとを上述したような高反応温度で反応させることにより、アルキル基を有し、落つい感度試験による落つい感度が充分に低減(消失)された、高品質の酸化黒鉛誘導体を得ることが可能である。
本発明は更に、X線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを少なくとも1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が100Å以上、500Å以下であることを特徴とする酸化黒鉛誘導体でもある。例えば、本発明の製造方法において、酸化黒鉛とアルコール及び/又はアミンとを反応させることにより、このようなピークを示す高品質の酸化黒鉛誘導体を得ることが可能である。上記「9-13°にあるピークのScherrerの式により算出される結晶子径が100Å以上、500Å以下である」とは、9-13°にピークが複数ある場合は、そのいずれか1つのピークのScherrerの式により算出される結晶子径が100Å以上、500Å以下であればよい。中でも、本発明の酸化黒鉛誘導体は、X線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示すことが好ましい。また、上記結晶子径が200Å以上であることがより好ましい。更に、上記結晶子径が400Å以下であることがより好ましい。
上記X線回折スペクトルは、実施例の「XRDの測定方法」により測定することができる。
上記X線回折スペクトルは、実施例の「XRDの測定方法」により測定することができる。
上記酸化黒鉛誘導体を得る工程における反応時間は、例えば1時間以上であることが好ましく、3時間以上であることがより好ましく、5時間以上であることが更に好ましい。該反応時間は、還元反応を充分に抑制しつつ修飾反応を優位的に進める観点からは、120時間以下であることが好ましく、100時間以下であることがより好ましく、80時間以下であることが更に好ましい。
上記反応工程は、公知の撹拌機等を用いて撹拌しながら行うことができる。
上記反応工程は、例えば空気中、又は、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行うことができる。また、上記反応工程は、その圧力条件は特に限定されず、加圧条件下、常圧条件下、減圧条件下で行うことができるが、例えば常圧条件下で行うことが好ましい。また、濃縮工程において揮発性の凝集剤を用いた場合は特に、減圧加熱による凝集剤等の不純物留去を反応工程前に行うことが好ましい。これにより、反応工程において凝集剤等の突沸や、副反応を充分に防ぐことができ、反応工程を安定的に進めることが可能である。
上記反応工程は、例えば空気中、又は、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行うことができる。また、上記反応工程は、その圧力条件は特に限定されず、加圧条件下、常圧条件下、減圧条件下で行うことができるが、例えば常圧条件下で行うことが好ましい。また、濃縮工程において揮発性の凝集剤を用いた場合は特に、減圧加熱による凝集剤等の不純物留去を反応工程前に行うことが好ましい。これにより、反応工程において凝集剤等の突沸や、副反応を充分に防ぐことができ、反応工程を安定的に進めることが可能である。
本発明の製造方法により得られる酸化黒鉛誘導体は、酸化黒鉛の酸素含有官能基と反応する化合物由来の官能基(好ましくは、炭化水素基をもつ官能基)を有する。該官能基は、特に限定されず、アルコキシカルボニル基(-COOR)、アルコキシル基(-OR)等の酸素含有基;硫黄含有基;アルキルアミノ基(-NHR、-NRR’)、アルキルアミド基(-CONHR、-CONRR’)等の窒素含有基;リン含有基等が挙げられるが、アルコキシカルボニル基(-COOR)、アルコキシル基(-OR)等の酸素含有基、アルキルアミノ基(-NHR、-NRR’)、アルキルアミド基(-CONHR、-CONRR’)等の窒素含有基であることが好ましい。なお、上記R及びR’は、同一又は異なって、有機基を表し、中でも、炭化水素基を表すことが好ましい。
本発明の製造方法において、酸化黒鉛とアルコールとを反応させることにより得られる酸化黒鉛誘導体は、酸化黒鉛誘導体100質量%中、炭素原子、水素原子、及び、酸素原子以外のその他の原子の含有量が、10質量%以下であることが好ましく、7質量%以下であることがより好ましく、5質量%以下であることが更に好ましい。上記酸化黒鉛誘導体は、その他の原子を有しないことが特に好ましい。言い換えれば、上記酸化黒鉛誘導体は、炭素原子、水素原子、及び、酸素原子のみを構成元素とするものであることが好ましい。その他の原子としては、窒素原子、リン原子、ハロゲン原子等が挙げられる。特に、上記酸化黒鉛誘導体は、酸化黒鉛誘導体100質量%中、窒素原子の含有量が0.1質量%以下であることが好ましい。
また本発明の製造方法において、酸化黒鉛とアミンとを反応させることにより得られる酸化黒鉛誘導体は、酸化黒鉛誘導体100質量%中、炭素原子、水素原子、酸素原子、及び、窒素原子以外のその他の原子の含有量が、10質量%以下であることが好ましく、7質量%以下であることがより好ましく、5質量%以下であることが更に好ましい。上記酸化黒鉛誘導体は、その他の原子を有しないことが特に好ましい。言い換えれば、上記酸化黒鉛誘導体は、炭素原子、水素原子、酸素原子、及び、窒素原子のみを構成元素とするものであることが好ましい。その他の原子としては、リン原子、ハロゲン原子等が挙げられる。
また本発明の製造方法において、酸化黒鉛とアミンとを反応させることにより得られる酸化黒鉛誘導体は、酸化黒鉛誘導体100質量%中、炭素原子、水素原子、酸素原子、及び、窒素原子以外のその他の原子の含有量が、10質量%以下であることが好ましく、7質量%以下であることがより好ましく、5質量%以下であることが更に好ましい。上記酸化黒鉛誘導体は、その他の原子を有しないことが特に好ましい。言い換えれば、上記酸化黒鉛誘導体は、炭素原子、水素原子、酸素原子、及び、窒素原子のみを構成元素とするものであることが好ましい。その他の原子としては、リン原子、ハロゲン原子等が挙げられる。
上記酸化黒鉛誘導体は、平均粒子径が0.01μm以上、100μm以下であることが好ましい。
上記平均粒子径は、0.1μm以上であることがより好ましく、1μm以上であることが更に好ましく、3μm以上であることが更に好ましい。該平均粒子径は、60μm以下であることがより好ましい。
上記平均粒子径は、粒度分布測定装置により測定することができる。
上記平均粒子径は、0.1μm以上であることがより好ましく、1μm以上であることが更に好ましく、3μm以上であることが更に好ましい。該平均粒子径は、60μm以下であることがより好ましい。
上記平均粒子径は、粒度分布測定装置により測定することができる。
上記酸化黒鉛誘導体の形状としては、微粉状、粉状、粒状、顆粒状、鱗片状、多面体状、ロッド状、曲面含有状等が挙げられる。なお、平均粒子径が上述のような範囲の粒子は、例えば、粒子をボールミル等により粉砕し、該粉砕により得られた粗粒子を分散剤に分散させて所望の粒子径にした後に乾固する方法や、粒子をふるい等にかけて粒子径を選別する方法、これら方法の組み合わせのほか、粒子を製造する段階で調製条件を最適化し、所望の粒子径の(ナノ)粒子を得る方法等により製造することが可能である。
本発明の酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい一実施形態では、酸化黒鉛誘導体は、炭素数13以上の炭化水素基をもつ官能基を有する。この好ましい一実施形態は、上記第1~第3の好ましい形態にそれぞれ適用することができる。
また本発明は、炭素数13以上の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体でもある。
本発明者らは、上記酸化黒鉛誘導体を炭素数13以上の炭化水素基をもつ官能基を有する誘導体とすると、親水性分散質が最も分散しにくく、これまで酸化黒鉛やその誘導体の分散が報告されていない単純アルカン中でも分散可能な新規な酸化黒鉛誘導体が得られることを見出した。炭素数13以上の長い炭化水素基を用いることにより、単純に疎水性を向上するだけでなく、酸化黒鉛誘導体に残存する親水性の酸素官能基を外部から覆うため、非極性分散媒への分散性が飛躍的に向上し、酸化黒鉛の誘導体の非極性分散媒に対する期待以上の分散性向上効果が発揮されたものと考えられる。
また本発明は、炭素数13以上の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体でもある。
本発明者らは、上記酸化黒鉛誘導体を炭素数13以上の炭化水素基をもつ官能基を有する誘導体とすると、親水性分散質が最も分散しにくく、これまで酸化黒鉛やその誘導体の分散が報告されていない単純アルカン中でも分散可能な新規な酸化黒鉛誘導体が得られることを見出した。炭素数13以上の長い炭化水素基を用いることにより、単純に疎水性を向上するだけでなく、酸化黒鉛誘導体に残存する親水性の酸素官能基を外部から覆うため、非極性分散媒への分散性が飛躍的に向上し、酸化黒鉛の誘導体の非極性分散媒に対する期待以上の分散性向上効果が発揮されたものと考えられる。
上記炭素数13以上の炭化水素基としては、特に限定されず、アルキル基やシクロアルキル基等の飽和脂肪族炭化水素基;アルキニル基やアルケニル基等の非環式不飽和脂肪族炭化水素基;アリール基等の芳香族炭化水素基等のいずれであってもよいが、中でも飽和脂肪族炭化水素基であることが好ましく、アルキル基であることがより好ましい。
上記アルキル基としては、例えば、n-テトラデシル基、sec-テトラデシル基、n-ヘキサデシル基、sec-ヘキサデシル基、n-オクタデシル基、sec-オクタデシル基、n-エイコシル基、sec-エイコシル基、2-オクチルドデシル基、n-ドコシル基、sec-ドコシル基、2-オクチルテトラデシル基、n-テトラコシル基、sec-テトラコシル基、2-オクチルヘキサデシル基、n-ヘキサコシル基、sec-ヘキサコシル基、n-オクタコシル基、sec-オクタコシル基、n-トリアコンチル基、sec-トリアコンチル基、n-ドトリアコンチル基、sec-ドトリアコンチル基、n-テトラトリアコンチル、sec-テトラトリアコンチル、n-ヘキサトリアコンチル、sec-ヘキサトリアコンチル等が挙げられ、これらの1種又は2種以上を使用できる。
上記アルキル基としては、例えば、n-テトラデシル基、sec-テトラデシル基、n-ヘキサデシル基、sec-ヘキサデシル基、n-オクタデシル基、sec-オクタデシル基、n-エイコシル基、sec-エイコシル基、2-オクチルドデシル基、n-ドコシル基、sec-ドコシル基、2-オクチルテトラデシル基、n-テトラコシル基、sec-テトラコシル基、2-オクチルヘキサデシル基、n-ヘキサコシル基、sec-ヘキサコシル基、n-オクタコシル基、sec-オクタコシル基、n-トリアコンチル基、sec-トリアコンチル基、n-ドトリアコンチル基、sec-ドトリアコンチル基、n-テトラトリアコンチル、sec-テトラトリアコンチル、n-ヘキサトリアコンチル、sec-ヘキサトリアコンチル等が挙げられ、これらの1種又は2種以上を使用できる。
本発明の酸化黒鉛誘導体における上記炭化水素基は、本発明の酸化黒鉛誘導体の非極性分散媒中での分散性をより向上する観点からは、炭素数が14以上であることが好ましく、16以上であることがより好ましく、18以上であることが更に好ましい。また、本発明の酸化黒鉛誘導体における上記炭化水素基は、本発明の酸化黒鉛誘導体の非極性分散媒中での分散速度を充分に速いものとし、また、本発明の酸化黒鉛誘導体を好適に製造する観点からは、炭素数が50以下であることが好ましく、36以下であることがより好ましい。
本発明の酸化黒鉛誘導体における上記炭化水素基は、炭素数が20以上、28以下であることが好ましい。これにより、上述した効果をバランスよく両立することができる。該炭素数は、21以上であることがより好ましく、22以上であることが更に好ましい。また、該炭素数は24以下であることがより好ましく、24であることが最も好ましい。
本発明はまた、炭素数6以上、10以下の炭化水素基をもつ官能基を有する酸化黒鉛誘導体でもある。
炭素数6以上、10以下とすることでケトン系、エステル系、アミド系溶媒等の両親媒性分散媒に対する親和性を付与することができ、良好な分散性を達成できることを見出した。よって、上記酸化黒鉛誘導体は、様々な樹脂への添加剤として適用が可能である。
上記炭素数6以上、10以下の炭化水素基としては、特に限定されず、アルキル基やシクロアルキル基等の飽和脂肪族炭化水素基;アルキニル基やアルケニル基等の非環式不飽和脂肪族炭化水素基;アリール基等の芳香族炭化水素基等のいずれであってもよいが、中でも飽和脂肪族炭化水素基であることが好ましく、アルキル基であることがより好ましい。
上記アルキル基としては、例えば、n-ヘキシル基、sec-ヘキシル基、n-ヘプチル基、sec-ヘプチル基、n-オクチル基、sec-オクチル基、n-ノニル基、sec-ノニル基、2-オクチルドデシル基、n-デシル基、sec-デシル基等が挙げられ、これらの1種又は2種以上を使用できる。
炭素数6以上、10以下とすることでケトン系、エステル系、アミド系溶媒等の両親媒性分散媒に対する親和性を付与することができ、良好な分散性を達成できることを見出した。よって、上記酸化黒鉛誘導体は、様々な樹脂への添加剤として適用が可能である。
上記炭素数6以上、10以下の炭化水素基としては、特に限定されず、アルキル基やシクロアルキル基等の飽和脂肪族炭化水素基;アルキニル基やアルケニル基等の非環式不飽和脂肪族炭化水素基;アリール基等の芳香族炭化水素基等のいずれであってもよいが、中でも飽和脂肪族炭化水素基であることが好ましく、アルキル基であることがより好ましい。
上記アルキル基としては、例えば、n-ヘキシル基、sec-ヘキシル基、n-ヘプチル基、sec-ヘプチル基、n-オクチル基、sec-オクチル基、n-ノニル基、sec-ノニル基、2-オクチルドデシル基、n-デシル基、sec-デシル基等が挙げられ、これらの1種又は2種以上を使用できる。
本発明の酸化黒鉛誘導体における上記炭化水素基は、本発明の酸化黒鉛誘導体の両親媒性分散媒への分散性をより向上する観点からは、炭素数が7以上、9以下であることがより好ましい。本発明の酸化黒鉛誘導体における上記炭化水素基は、この観点からは、炭素数が8であることが最も好ましい。
炭素数13以上の炭化水素基をもつ官能基を有する酸化黒鉛誘導体と、炭素数6以上、10以下の炭化水素基をもつ官能基を有する酸化黒鉛誘導体とについて上述した。いずれの場合であっても、本発明の酸化黒鉛誘導体は、末端に上記炭化水素基をもつ官能基を有することが好ましい。
本発明の酸化黒鉛誘導体における上記炭化水素基は、直鎖であることが好ましい。上記炭化水素基が直鎖であると、直鎖の炭化水素基が酸化黒鉛に導入されやすいため、本発明の酸化黒鉛誘導体を好適に得ることができる。
本発明の酸化黒鉛誘導体における上記炭化水素基は、分岐鎖であることもまた好ましい。上記炭化水素基が分岐鎖であると、本発明の酸化黒鉛誘導体の非極性分散媒中での分散性がより向上する。また、原料である炭化水素基含有化合物が常温(25℃)で液体となる傾向にあり、その場合は、誘導体製造時のハンドリングも容易である。
上記炭素数13以上又は炭素数6以上、10以下の炭化水素基をもつ官能基は、特に限定されず、アルコキシカルボニル基(-COOR)、アルコキシル基(-OR)等の酸素含有基;硫黄含有基;アルキルアミノ基(-NHR、-NRR’)、アルキルアミド基(-CONHR、-CONRR’)等の窒素含有基;リン含有基等が挙げられるが、アルコキシカルボニル基(-COOR)、アルコキシル基(-OR)等の酸素含有基、アルキルアミノ基(-NHR、-NRR’)、アルキルアミド基(-CONHR、-CONRR’)等の窒素含有基であることが好ましい。なお、上記R及びR’は、同一又は異なって、炭素数13以上又は炭素数6以上、10以下の炭化水素基を表す。すなわち、上記官能基における炭化水素基以外の部分は、例えば、-COO-、-O-、-NH-、-N-、-CONH-、又は、-CON-であることが好ましい。
上記酸化黒鉛誘導体中の、炭素原子、水素原子、及び、酸素原子以外のその他の原子の好ましい含有量や炭素原子、水素原子、酸素原子、及び、窒素原子以外のその他の原子の好ましい含有量は、それぞれ、上述した本発明の製造方法により得られる酸化黒鉛誘導体中の、その他の原子の好ましい含有量と同様である。
上記酸化黒鉛誘導体の好ましい平均粒子径、形状、所望の粒子径の粒子を得る方法は、上述した本発明の製造方法により得られる酸化黒鉛誘導体の好ましい平均粒子径、形状、所望の粒子径の粒子を得る方法と同様である。
以下では、本発明の酸化黒鉛誘導体を製造する際に特に好適な方法について簡単に説明するが、以下の方法は、上述した本発明の酸化黒鉛誘導体の製造方法にも好適に適用することができる。
以下では、本発明の酸化黒鉛誘導体を製造する際に特に好適な方法について簡単に説明するが、以下の方法は、上述した本発明の酸化黒鉛誘導体の製造方法にも好適に適用することができる。
本発明は、酸化黒鉛と炭素数13以上の炭化水素基含有化合物とを反応させることにより酸化黒鉛誘導体を得る酸化黒鉛誘導体の製造方法でもある。本発明はまた、酸化黒鉛と炭素数6以上、10以下の炭化水素基含有化合物とを反応させることにより酸化黒鉛誘導体を得る酸化黒鉛誘導体の製造方法でもある。該炭化水素基含有化合物としては、アミン、イソシアネート基含有化合物、カルボニル基含有化合物(例えば、カルボン酸ハライド)、アルコール等が挙げられる。ここで、上記反応において、酸化黒鉛が特定炭素数の炭化水素基含有化合物と反応して修飾される反応と、酸化黒鉛自身の還元反応とが同時進行して競合するところ、還元反応を充分に抑制して酸化黒鉛を修飾する反応を優位的に進める観点からは、上記炭化水素基含有化合物がアルコール及び/又はアミンであることが好ましい。アルコールとしては、上述した炭化水素基に対応するものを適宜使用できる。アミンとしては、上述した炭化水素基に対応するもの(窒素原子に炭化水素基が結合されたもの)を適宜使用できる。また、酸化黒鉛と炭化水素基含有化合物との反応では、公知の触媒等、何れも適応することができるが、本発明の好適な形態として、触媒として水酸化ナトリウム、水酸化カリウム、水酸化カルシウム、アミン、ピリジン等の塩基触媒や、硫酸等の酸触媒を用いることができる。
なお、本発明の酸化黒鉛誘導体の生成は、実施例の方法に沿って赤外線吸収スペクトルを測定することにより確認される。
なお、本発明の酸化黒鉛誘導体の生成は、実施例の方法に沿って赤外線吸収スペクトルを測定することにより確認される。
上述した酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい実施形態では、上記反応工程において塩基触媒を用いる場合、塩基触媒の使用量は、反応工程に用いる混合液中の酸化黒鉛量100質量%に対し、0.01~1000質量%である。これにより、酸化黒鉛誘導体を効率的に製造することができる。
該塩基触媒の使用量は、0.1質量%以上であることがより好ましく、1質量%以上であることが更に好ましい。また、該使用量は、500質量%以下であることがより好ましい。
本明細書中、塩基触媒の使用量とは、上記混合液を作製するために用いられた塩基触媒の仕込み量を言う。
該塩基触媒の使用量は、0.1質量%以上であることがより好ましく、1質量%以上であることが更に好ましい。また、該使用量は、500質量%以下であることがより好ましい。
本明細書中、塩基触媒の使用量とは、上記混合液を作製するために用いられた塩基触媒の仕込み量を言う。
上記反応工程において酸触媒を用いる場合、酸触媒の使用量は、上述した酸化黒鉛含有組成物における、酸化黒鉛の質量100質量%に対する酸触媒の好ましい含有量と同様である。
本明細書中、酸触媒の使用量とは、上記混合液を作製するために用いられた酸触媒の仕込み量を言う。
本明細書中、酸触媒の使用量とは、上記混合液を作製するために用いられた酸触媒の仕込み量を言う。
また酸化黒鉛は本質的に自身が酸性の物質であり、自触媒的に反応が進行する。前述のとおり触媒を添加することがより好ましいが、触媒を添加せずとも自触媒的に反応を進行させることも可能である。
上述した酸化黒鉛誘導体の製造方法のいずれかの更に別の好ましい実施形態では、上記反応工程における炭素数13以上又は炭素数6以上、10以下の炭化水素基含有化合物の好ましい使用量は、上述した本発明における酸化黒鉛の酸素含有官能基と反応する化合物の好ましい使用量と同様である。
本明細書中、混合液中の炭素数13以上又は炭素数6以上、10以下の炭化水素基含有化合物の使用量とは、混合液を作製するために用いられた炭素数13以上又は炭素数6以上、10以下の炭化水素基含有化合物の仕込み量を言う。
本明細書中、混合液中の炭素数13以上又は炭素数6以上、10以下の炭化水素基含有化合物の使用量とは、混合液を作製するために用いられた炭素数13以上又は炭素数6以上、10以下の炭化水素基含有化合物の仕込み量を言う。
上記混合液は、上記酸化黒鉛、上記アルコール等の炭化水素基含有化合物、及び、上記触媒等を混合して得ることができる。上記酸化黒鉛、上記炭化水素基含有化合物、及び、上記触媒は、公知の方法を用いて得ることができ、市販品を使用することも可能である。上記混合は、公知の方法で適宜行うことが可能であるが、例えば、超音波処理を行ったり、公知の分散機を用いたりして酸化黒鉛を均一に分散させることが好ましい。
上記反応工程は、上述した本発明の製造方法と同様の方法により好適に行うことができる。
上記反応温度は、例えば60℃以上とすればよく、反応温度を60℃以上とすることにより、反応が効率的に進行するが、上述した本発明の製造方法と同様の反応温度とすることが好ましい。反応時間についても同様である。
上記反応温度は、例えば60℃以上とすればよく、反応温度を60℃以上とすることにより、反応が効率的に進行するが、上述した本発明の製造方法と同様の反応温度とすることが好ましい。反応時間についても同様である。
本発明の酸化黒鉛誘導体を製造するために、上記反応工程の後、その他の工程を適宜行うことができる。
本発明の酸化黒鉛誘導体は、炭素数13以上の炭化水素基をもつ官能基を有する場合、非極性分散媒中での分散性に優れるため機械用の潤滑油の添加剤、樹脂への添加剤等として特に好適に使用できる。
本発明の酸化黒鉛誘導体はまた、炭素数6以上、10以下の炭化水素基をもつ官能基を有する場合、両親媒性分散媒中での分散性に優れるため、様々な樹脂への添加剤として特に好適に使用できる。
本発明の酸化黒鉛誘導体はまた、炭素数6以上、10以下の炭化水素基をもつ官能基を有する場合、両親媒性分散媒中での分散性に優れるため、様々な樹脂への添加剤として特に好適に使用できる。
<分散体>
本発明はまた、酸化黒鉛誘導体が分散媒中に分散してなる分散体でもある。
本発明の分散体は、本発明の酸化黒鉛誘導体を、非極性分散媒等の分散媒中に分散させて得ることができる。
非極性分散媒としては、例えば、ベンゼン、キシレン、トルエン、シクロヘキシルベンゼン、ジヒドロベンゾフラン、トリメチルベンゼン、テトラメチルベンゼン、ナフタレン、アントラセン等の炭素数6~14の芳香族炭化水素系分散媒、ピリジン、ピラジン、フラン、ピロール、チオフェン等の炭素数4~6の芳香族複素環化合物系分散媒、ペンタン、ヘキサン、ヘプタン、オクタン、デカン、ドデカン、テトラデカン、ヘキサデカン、オクタデカン、シクロヘキサン等の炭素数5以上の脂肪族炭化水素系分散媒等が挙げられ、また、鉱物油、合成油等の分散媒も挙げられる。これらの1種又は2種以上を混合したものを使用できる。
本発明はまた、酸化黒鉛誘導体が分散媒中に分散してなる分散体でもある。
本発明の分散体は、本発明の酸化黒鉛誘導体を、非極性分散媒等の分散媒中に分散させて得ることができる。
非極性分散媒としては、例えば、ベンゼン、キシレン、トルエン、シクロヘキシルベンゼン、ジヒドロベンゾフラン、トリメチルベンゼン、テトラメチルベンゼン、ナフタレン、アントラセン等の炭素数6~14の芳香族炭化水素系分散媒、ピリジン、ピラジン、フラン、ピロール、チオフェン等の炭素数4~6の芳香族複素環化合物系分散媒、ペンタン、ヘキサン、ヘプタン、オクタン、デカン、ドデカン、テトラデカン、ヘキサデカン、オクタデカン、シクロヘキサン等の炭素数5以上の脂肪族炭化水素系分散媒等が挙げられ、また、鉱物油、合成油等の分散媒も挙げられる。これらの1種又は2種以上を混合したものを使用できる。
本発明の分散体はまた、本発明の酸化黒鉛誘導体を、両親媒性分散媒等の分散媒中に分散させて得ることができる。
両親媒性分散媒としてはメタノール、エタノール、プロパノール等のアルコール系分散媒、N,N-ジメチルホルムアミド(DMF)、N-メチルピロリドン(NMP)等のアミド、ラクタム系分散媒、アセトン、ブタノン、ペンタノン等のケトン系分散媒、エチレングリコール、エチレングリコールメチルエーテル、プロピレングリコール、プロピレングリコールメチルエーテル等のグリコ―ル、グリコールエーテル系分散媒等が挙げられる。これらの1種又は2種以上を混合したものを使用できる。
分散には、公知の撹拌機、公知の超音波発生装置等を使用できる。例えば、酸化黒鉛誘導体と分散媒との混合物に30分~2時間超音波をかけて分散体を調製することが好ましい。
両親媒性分散媒としてはメタノール、エタノール、プロパノール等のアルコール系分散媒、N,N-ジメチルホルムアミド(DMF)、N-メチルピロリドン(NMP)等のアミド、ラクタム系分散媒、アセトン、ブタノン、ペンタノン等のケトン系分散媒、エチレングリコール、エチレングリコールメチルエーテル、プロピレングリコール、プロピレングリコールメチルエーテル等のグリコ―ル、グリコールエーテル系分散媒等が挙げられる。これらの1種又は2種以上を混合したものを使用できる。
分散には、公知の撹拌機、公知の超音波発生装置等を使用できる。例えば、酸化黒鉛誘導体と分散媒との混合物に30分~2時間超音波をかけて分散体を調製することが好ましい。
本発明の分散体は、分散媒100質量%に対する酸化黒鉛誘導体の質量割合が、0.0001質量%以上であることが好ましく、0.001質量%以上であることがより好ましい。また、該質量割合が、10質量%以下であることが好ましく、1質量%以下であることがより好ましく、0.1質量%以下であることが更に好ましい。
(還元型酸化黒鉛)
上述した酸化黒鉛は、誘導体化反応に供する代わりに、更に還元して親水性の官能基を脱離させることで、より疎水性の強い還元型酸化黒鉛とすることができる。すなわち、本発明は、還元型酸化黒鉛を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛を還元して還元型酸化黒鉛を得る工程とを含み、酸化工程と還元型酸化黒鉛を得る工程との間に、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことを特徴とする還元型酸化黒鉛の製造方法でもある。これにより、還元型酸化黒鉛を簡便に製造することができる。
このような還元型酸化黒鉛の製造にも、上述した本発明の酸化黒鉛誘導体の製造方法の酸化工程、分離工程と同じ工程を用いることができ、そのような工程を用いることで、還元型酸化黒鉛をより効率的に製造することができる。
このような還元型酸化黒鉛の製造方法、すなわち、酸化黒鉛が還元された還元型酸化黒鉛を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛又は酸化黒鉛含有組成物を分離する工程と、該分離工程で得られた酸化黒鉛を還元する工程とを含み、該分離工程は、酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛又は酸化黒鉛含有組成物を分離する工程を含むことを特徴とする還元型酸化黒鉛の製造方法もまた、本発明の1つである。
上述した酸化黒鉛は、誘導体化反応に供する代わりに、更に還元して親水性の官能基を脱離させることで、より疎水性の強い還元型酸化黒鉛とすることができる。すなわち、本発明は、還元型酸化黒鉛を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛を還元して還元型酸化黒鉛を得る工程とを含み、酸化工程と還元型酸化黒鉛を得る工程との間に、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことを特徴とする還元型酸化黒鉛の製造方法でもある。これにより、還元型酸化黒鉛を簡便に製造することができる。
このような還元型酸化黒鉛の製造にも、上述した本発明の酸化黒鉛誘導体の製造方法の酸化工程、分離工程と同じ工程を用いることができ、そのような工程を用いることで、還元型酸化黒鉛をより効率的に製造することができる。
このような還元型酸化黒鉛の製造方法、すなわち、酸化黒鉛が還元された還元型酸化黒鉛を製造する方法であって、該製造方法は、黒鉛を酸化する工程と、該酸化工程で得られる酸化黒鉛又は酸化黒鉛含有組成物を分離する工程と、該分離工程で得られた酸化黒鉛を還元する工程とを含み、該分離工程は、酸化黒鉛を含む反応液に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛又は酸化黒鉛含有組成物を分離する工程を含むことを特徴とする還元型酸化黒鉛の製造方法もまた、本発明の1つである。
本発明の還元型酸化黒鉛の製造方法において、酸化黒鉛を還元する工程は、酸化黒鉛から親水性の官能基が脱離して還元されることになる限りその方法は特に制限されず、NaBH4、LiAlH4、L-アスコルビン酸等の公知の還元剤を使用する方法や電解還元等も用いることができるが、酸化黒鉛を加熱することで還元する方法が好ましい。
酸化黒鉛を加熱する温度は、100℃以上が好ましい。より好ましくは、120℃以上である。酸化黒鉛の加熱温度に特に上限はないが、通常、2000℃以下で行われる。酸化黒鉛を加熱する時間は、0.1~100時間が好ましい。より好ましくは、0.2~50時間である。
酸化黒鉛の加熱は空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。また真空中で行っても良い。
酸化黒鉛を加熱する温度は、100℃以上が好ましい。より好ましくは、120℃以上である。酸化黒鉛の加熱温度に特に上限はないが、通常、2000℃以下で行われる。酸化黒鉛を加熱する時間は、0.1~100時間が好ましい。より好ましくは、0.2~50時間である。
酸化黒鉛の加熱は空気中で行ってもよく、窒素、ヘリウム、アルゴン等の不活性ガス雰囲気中で行ってもよい。また真空中で行っても良い。
本発明の還元型酸化黒鉛の製造方法における黒鉛を酸化する工程、該酸化工程で得られる酸化黒鉛又は酸化黒鉛含有組成物を分離する工程の好ましい形態は、上述した本発明の酸化黒鉛誘導体の製造方法におけるこれらの工程の好ましい形態と同様である。
本発明の還元型酸化黒鉛の製造方法は、黒鉛を酸化する工程、該酸化工程で得られる酸化黒鉛又は酸化黒鉛含有組成物を分離する工程、及び、分離工程で得られた酸化黒鉛を還元する工程を含む限り、その他の工程を含んでいてもよい。その他の工程としては、上述した酸化反応停止工程等が挙げられる。
本発明の還元型酸化黒鉛の製造方法は、黒鉛を酸化する工程、該酸化工程で得られる酸化黒鉛又は酸化黒鉛含有組成物を分離する工程、及び、分離工程で得られた酸化黒鉛を還元する工程を含む限り、その他の工程を含んでいてもよい。その他の工程としては、上述した酸化反応停止工程等が挙げられる。
本発明の製造方法の第1~第3の好ましい形態により得られる酸化黒鉛誘導体や、本発明の酸化黒鉛誘導体は、触媒、電池やキャパシタの電極活物質、熱電変換材料、導電性材料、発光材料、潤滑用添加剤、高分子用添加剤、透過膜材料、抗菌材料、撥水材料、吸着材料等として好適に用いることができる。例えば、本発明の酸化黒鉛誘導体が、炭素数13以上の炭化水素基が導入されたものである場合、非極性分散媒中での分散性に優れるものであるため機械用の潤滑油の添加剤、樹脂への添加剤等として特に好適に使用できる。また、本発明の酸化黒鉛誘導体が、炭素数6以上、10以下の炭化水素基が導入されたものである場合、両親媒性分散媒中での分散性に優れるものであるため様々な樹脂への添加剤等として特に好適に使用できる。
なお、上記電池としては、例えば、リチウムイオン二次電池、固体高分子型燃料電池、金属-空気電池等が挙げられる。
上記熱電変換材料が用いられる熱電変換装置としては、例えば、地熱・温泉熱発電機、太陽熱発電機、工場や自動車等の廃熱発電機、体温発電機等の発電機や、該発電機を電源の少なくとも一つとして用いた各種電気製品、電動機、人工衛星等が挙げられる。
また本発明の酸化黒鉛の製造方法により得られる酸化黒鉛や、本発明の還元型酸化黒鉛の製造方法により得られる還元型酸化黒鉛もまた、上述した用途に好適に使用できる。
なお、上記電池としては、例えば、リチウムイオン二次電池、固体高分子型燃料電池、金属-空気電池等が挙げられる。
上記熱電変換材料が用いられる熱電変換装置としては、例えば、地熱・温泉熱発電機、太陽熱発電機、工場や自動車等の廃熱発電機、体温発電機等の発電機や、該発電機を電源の少なくとも一つとして用いた各種電気製品、電動機、人工衛星等が挙げられる。
また本発明の酸化黒鉛の製造方法により得られる酸化黒鉛や、本発明の還元型酸化黒鉛の製造方法により得られる還元型酸化黒鉛もまた、上述した用途に好適に使用できる。
以下に実施例を掲げて本発明を更に詳細に説明するが、本発明はこれらの実施例のみに限定されるものではない。なお、特に断りのない限り、「部」は「質量部」を、「%」は「質量%」を意味するものとする。
下記実施例及び比較例においては、次のようにして分析し、評価を行った。
<XRDの測定方法>
XRD測定は、全自動水平型X線回折装置(リガク社製、SMART LAB)を用いて、以下の条件により行った。
CuKα1線:0.15406nm
走査範囲:5°-45°
X線出力設定:45kV-200mA
ステップサイズ:0.010°
スキャン速度:0.5°min-1-4°min-1
なお、XRD測定は、試料をグローブボックス中にて気密試料台に装填することにより、不活性雰囲気を保った状態で行った。
<XRDの測定方法>
XRD測定は、全自動水平型X線回折装置(リガク社製、SMART LAB)を用いて、以下の条件により行った。
CuKα1線:0.15406nm
走査範囲:5°-45°
X線出力設定:45kV-200mA
ステップサイズ:0.010°
スキャン速度:0.5°min-1-4°min-1
なお、XRD測定は、試料をグローブボックス中にて気密試料台に装填することにより、不活性雰囲気を保った状態で行った。
<XPSの測定方法>
XPS測定は、光電子分光装置(JPS-9000MX,日本電子株式会社製)を用いて行った。C1sのナロースキャンに於けるピーク分離は、バックグラウンド補正をShirley法で行い、フィッティング関数としてGauss-Lorentz関数を用いたピークフィットにより行った。
<FT-IRの測定方法>
サーモフィッシャーサイエンティフィック株式会社製Nicolet NEXUS670 FTIRを用いて、酸化黒鉛誘導体をKBrと混合しペレット化することで測定した。測定範囲は900~4000cm-1で分解能は1cm-1とした。
<硫酸濃度の分析>
酸化黒鉛含有組成物中の硫酸の濃度は、酸化黒鉛水分散液を水酸化ナトリウム水溶液により中和滴定することで確認した。
<水分の分析>
酸化黒鉛含有組成物中の水濃度は平沼産業株式会社製微量水分測定装置AQV-2200Aを用いて測定した。
<酸素含有率の分析>
JEOL社製JPS-9000MXを用いて、酸化黒鉛又はその誘導体中の酸素量を分析した。結果は炭素に対する比(C/O比)で出した。
なお、酸化黒鉛の酸素炭素比(C/O比)が小さいほど、酸化黒鉛中の酸素官能基がより多く維持されていると考えられる。また、酸化黒鉛誘導体の酸素炭素比(C/O比)が大きいほど、酸化黒鉛誘導体に炭化水素基が充分に導入されていると考えられる。
XPS測定は、光電子分光装置(JPS-9000MX,日本電子株式会社製)を用いて行った。C1sのナロースキャンに於けるピーク分離は、バックグラウンド補正をShirley法で行い、フィッティング関数としてGauss-Lorentz関数を用いたピークフィットにより行った。
<FT-IRの測定方法>
サーモフィッシャーサイエンティフィック株式会社製Nicolet NEXUS670 FTIRを用いて、酸化黒鉛誘導体をKBrと混合しペレット化することで測定した。測定範囲は900~4000cm-1で分解能は1cm-1とした。
<硫酸濃度の分析>
酸化黒鉛含有組成物中の硫酸の濃度は、酸化黒鉛水分散液を水酸化ナトリウム水溶液により中和滴定することで確認した。
<水分の分析>
酸化黒鉛含有組成物中の水濃度は平沼産業株式会社製微量水分測定装置AQV-2200Aを用いて測定した。
<酸素含有率の分析>
JEOL社製JPS-9000MXを用いて、酸化黒鉛又はその誘導体中の酸素量を分析した。結果は炭素に対する比(C/O比)で出した。
なお、酸化黒鉛の酸素炭素比(C/O比)が小さいほど、酸化黒鉛中の酸素官能基がより多く維持されていると考えられる。また、酸化黒鉛誘導体の酸素炭素比(C/O比)が大きいほど、酸化黒鉛誘導体に炭化水素基が充分に導入されていると考えられる。
<FT-IRの測定方法>
サーモフィッシャーサイエンティフィック株式会社製Nicolet NEXUS670 FTIRを用いて、酸化黒鉛誘導体をKBrと混合しペレット化することで測定した。測定範囲は900~4000cm-1で分解能は1cm-1とした。
<分散性の評価方法>
APEL社製比色計AP-1000Mを用いて、0.1mg/mlの酸化黒鉛誘導体の分散液における、波長660nmの光の透過率の時間推移を測定した。光の透過が確認できる時間(透過率が0%→1%となる時間)が1時間以上のものに対して分散性が良好であるとした。最大測定時間を6時間とした。
<元素分析の分析方法>
elementer社製vario EL cube CHNSを用いてCHNOの質量濃度を測定した。
<落つい感度試験>
JIS K 4810に規定される落つい感度試験方法による。
サーモフィッシャーサイエンティフィック株式会社製Nicolet NEXUS670 FTIRを用いて、酸化黒鉛誘導体をKBrと混合しペレット化することで測定した。測定範囲は900~4000cm-1で分解能は1cm-1とした。
<分散性の評価方法>
APEL社製比色計AP-1000Mを用いて、0.1mg/mlの酸化黒鉛誘導体の分散液における、波長660nmの光の透過率の時間推移を測定した。光の透過が確認できる時間(透過率が0%→1%となる時間)が1時間以上のものに対して分散性が良好であるとした。最大測定時間を6時間とした。
<元素分析の分析方法>
elementer社製vario EL cube CHNSを用いてCHNOの質量濃度を測定した。
<落つい感度試験>
JIS K 4810に規定される落つい感度試験方法による。
(酸化黒鉛の合成)
酸化黒鉛を以下の工程で合成した。反応容器にあらかじめ黒鉛(伊藤黒鉛株式会社製Z-5F、5.76g)、硫酸(和光純薬工業株式会社製、167ml)を入れ、25℃に調整しながら過マンガン酸カリウム(和光純薬工業株式会社製、14.4g)を入れた。投入後、30分、35℃に昇温し2時間反応させた。反応後反応器を氷浴しながら水890ml、30%過酸化水素水(和光純薬工業株式会社製、88ml)を加え反応停止させ、酸化黒鉛含有組成物(酸化黒鉛を含む反応液)を得た。
酸化黒鉛を以下の工程で合成した。反応容器にあらかじめ黒鉛(伊藤黒鉛株式会社製Z-5F、5.76g)、硫酸(和光純薬工業株式会社製、167ml)を入れ、25℃に調整しながら過マンガン酸カリウム(和光純薬工業株式会社製、14.4g)を入れた。投入後、30分、35℃に昇温し2時間反応させた。反応後反応器を氷浴しながら水890ml、30%過酸化水素水(和光純薬工業株式会社製、88ml)を加え反応停止させ、酸化黒鉛含有組成物(酸化黒鉛を含む反応液)を得た。
(調製例1-1)
得られた酸化黒鉛を含む反応液を遠心分離用遠沈管にとり、10000G、10分間遠心分離にかけた。遠心分離後、上澄みを除去し酸化黒鉛含有組成物を得た。この組成物中の硫酸は酸化黒鉛に対し300%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.37であった。硫酸の濃度は遠心分離前に適宜水を追加することで容易に調整ができる。
得られた酸化黒鉛を含む反応液を遠心分離用遠沈管にとり、10000G、10分間遠心分離にかけた。遠心分離後、上澄みを除去し酸化黒鉛含有組成物を得た。この組成物中の硫酸は酸化黒鉛に対し300%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.37であった。硫酸の濃度は遠心分離前に適宜水を追加することで容易に調整ができる。
(調製例1-2)
調製例1-1で得られた酸化黒鉛含有組成物を遠心分離、上澄み除去、水への再分散を計8回繰り返すことで精製された酸化黒鉛を得た。この組成物中の硫酸は酸化黒鉛に対し0.01%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.59であった。調製例1と比較して、精製によって酸素量が減少することが確認された。図1は、調製例1-2で作製した酸化黒鉛のFT-IRチャートである。
調製例1-1で得られた酸化黒鉛含有組成物を遠心分離、上澄み除去、水への再分散を計8回繰り返すことで精製された酸化黒鉛を得た。この組成物中の硫酸は酸化黒鉛に対し0.01%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.59であった。調製例1と比較して、精製によって酸素量が減少することが確認された。図1は、調製例1-2で作製した酸化黒鉛のFT-IRチャートである。
(調製例1-3)
調製例1-2で得られた酸化黒鉛含有組成物を減圧乾燥により1時間乾燥させた。この組成物中の水分は酸化黒鉛に対し10%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.62であった。調製例1-2と比較しても乾燥によって顕著に酸素量が減少することはなく、水分の存在により酸素官能基が安定化されていることがわかった。
調製例1-2で得られた酸化黒鉛含有組成物を減圧乾燥により1時間乾燥させた。この組成物中の水分は酸化黒鉛に対し10%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.62であった。調製例1-2と比較しても乾燥によって顕著に酸素量が減少することはなく、水分の存在により酸素官能基が安定化されていることがわかった。
(調製例1-4)
調製例1-2で得られた酸化黒鉛含有組成物を50℃の減圧乾燥により2日間乾燥させた。この組成物中の水分は酸化黒鉛に対し3%未満であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は2.60であった。調製例1-2と比較して、加熱乾燥によって顕著に酸素量が減少することが確認された。
調製例1-2で得られた酸化黒鉛含有組成物を50℃の減圧乾燥により2日間乾燥させた。この組成物中の水分は酸化黒鉛に対し3%未満であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は2.60であった。調製例1-2と比較して、加熱乾燥によって顕著に酸素量が減少することが確認された。
(調製例1-5)
得られた酸化黒鉛を含む反応液に対し、水を95%、1-ブタノール(和光純薬工業株式会社製)を凝集剤として16%添加した。凝集した酸化黒鉛含有組成物を濾過により濃縮し、ペースト状の酸化黒鉛含有組成物を得た。この組成物中の硫酸は酸化黒鉛に対し3%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.42であった。凝集による濃縮も酸素官能基を維持する手法として好適であることがわかった。また硫酸の濃度は凝集前に適宜水を追加することで容易に調整ができる。
得られた酸化黒鉛を含む反応液に対し、水を95%、1-ブタノール(和光純薬工業株式会社製)を凝集剤として16%添加した。凝集した酸化黒鉛含有組成物を濾過により濃縮し、ペースト状の酸化黒鉛含有組成物を得た。この組成物中の硫酸は酸化黒鉛に対し3%であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は1.42であった。凝集による濃縮も酸素官能基を維持する手法として好適であることがわかった。また硫酸の濃度は凝集前に適宜水を追加することで容易に調整ができる。
(調製例1-6)
調製例1-5で得られた酸化黒鉛含有組成物を50℃の減圧乾燥により2日間乾燥させた。この組成物中の水分は酸化黒鉛に対し3%未満であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は2.24であった。調製例1-5と比較して、加熱乾燥によって酸素量が減少することが確認された。
調製例1-5で得られた酸化黒鉛含有組成物を50℃の減圧乾燥により2日間乾燥させた。この組成物中の水分は酸化黒鉛に対し3%未満であった。得られた酸化黒鉛中の酸素炭素比(C/O比)は2.24であった。調製例1-5と比較して、加熱乾燥によって酸素量が減少することが確認された。
(酸化黒鉛誘導体の製造)
酸化黒鉛誘導体を以下の工程で製造した。
酸化黒鉛誘導体を以下の工程で製造した。
(実施例1-1)
調製例1-5で得られた酸化黒鉛含有組成物を反応器に移し、ここへ2-デシル-1-テトラデカノール(新日本理化株式会社製、エヌジェコール240A)を酸化黒鉛に対し500%加え、150℃で5時間反応させた。反応後ヘキサンを注ぎ、濾過し、水洗、アセトン洗浄を行い、得られた固体を100℃で真空乾燥することで酸化黒鉛誘導体Aを得た。得られた酸化黒鉛誘導体A中の酸素炭素比(C/O比)は8.5であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。図2は、実施例1-1で作製した酸化黒鉛誘導体AのFT-IRチャートである。図3は、実施例1-1で作製した酸化黒鉛誘導体AのXRDチャートである。酸化黒鉛誘導体Aは、図3に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が313Åであった。
調製例1-5で得られた酸化黒鉛含有組成物を反応器に移し、ここへ2-デシル-1-テトラデカノール(新日本理化株式会社製、エヌジェコール240A)を酸化黒鉛に対し500%加え、150℃で5時間反応させた。反応後ヘキサンを注ぎ、濾過し、水洗、アセトン洗浄を行い、得られた固体を100℃で真空乾燥することで酸化黒鉛誘導体Aを得た。得られた酸化黒鉛誘導体A中の酸素炭素比(C/O比)は8.5であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。図2は、実施例1-1で作製した酸化黒鉛誘導体AのFT-IRチャートである。図3は、実施例1-1で作製した酸化黒鉛誘導体AのXRDチャートである。酸化黒鉛誘導体Aは、図3に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が313Åであった。
(実施例1-2)
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Bを得た。得られた酸化黒鉛誘導体B中の酸素炭素比(C/O比)は7.5であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図4は、実施例1-2で作製した酸化黒鉛誘導体BのFT-IRチャートである。
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Bを得た。得られた酸化黒鉛誘導体B中の酸素炭素比(C/O比)は7.5であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図4は、実施例1-2で作製した酸化黒鉛誘導体BのFT-IRチャートである。
(実施例1-3)
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Cを得た。得られた酸化黒鉛誘導体C中の酸素炭素比(C/O比)は7.1であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図5は、実施例1-3で作製した酸化黒鉛誘導体CのFT-IRチャートである。
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Cを得た。得られた酸化黒鉛誘導体C中の酸素炭素比(C/O比)は7.1であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図5は、実施例1-3で作製した酸化黒鉛誘導体CのFT-IRチャートである。
(実施例1-4)
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Dを得た。得られた酸化黒鉛誘導体D中の酸素炭素比(C/O比)は7.3であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図6は、実施例1-4で作製した酸化黒鉛誘導体DのFT-IRチャートである。
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと、触媒として硫酸を酸化黒鉛に対し10%加えたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Dを得た。得られた酸化黒鉛誘導体D中の酸素炭素比(C/O比)は7.3であった。実施例1-1と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-1と比較して少ないためと考えられた。図6は、実施例1-4で作製した酸化黒鉛誘導体DのFT-IRチャートである。
(実施例1-5)
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体Eを得た。得られた酸化黒鉛誘導体E中の酸素炭素比(C/O比)は6.8であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。図7は、実施例1-5で作製した酸化黒鉛誘導体EのFT-IRチャートである。図8は、実施例1-5で作製した酸化黒鉛誘導体EのXRDチャートである。酸化黒鉛誘導体Eは、図8に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が111.6Åであった。
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体Eを得た。得られた酸化黒鉛誘導体E中の酸素炭素比(C/O比)は6.8であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。図7は、実施例1-5で作製した酸化黒鉛誘導体EのFT-IRチャートである。図8は、実施例1-5で作製した酸化黒鉛誘導体EのXRDチャートである。酸化黒鉛誘導体Eは、図8に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が111.6Åであった。
(実施例1-6)
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-2に記載の方法で酸化黒鉛誘導体Fを得た。得られた酸化黒鉛誘導体F中の酸素炭素比(C/O比)は6.2であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図9は、実施例1-6で作製した酸化黒鉛誘導体FのFT-IRチャートである。
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-2に記載の方法で酸化黒鉛誘導体Fを得た。得られた酸化黒鉛誘導体F中の酸素炭素比(C/O比)は6.2であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図9は、実施例1-6で作製した酸化黒鉛誘導体FのFT-IRチャートである。
(実施例1-7)
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-3に記載の方法で酸化黒鉛誘導体Gを得た。得られた酸化黒鉛誘導体G中の酸素炭素比(C/O比)は6.0であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図10は、実施例1-7で作製した酸化黒鉛誘導体GのFT-IRチャートである。
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-3に記載の方法で酸化黒鉛誘導体Gを得た。得られた酸化黒鉛誘導体G中の酸素炭素比(C/O比)は6.0であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図10は、実施例1-7で作製した酸化黒鉛誘導体GのFT-IRチャートである。
(実施例1-8)
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-4に記載の方法で酸化黒鉛誘導体Hを得た。得られた酸化黒鉛誘導体H中の酸素炭素比(C/O比)は6.1であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図11は、実施例1-8で作製した酸化黒鉛誘導体HのFT-IRチャートである。
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-4に記載の方法で酸化黒鉛誘導体Hを得た。得られた酸化黒鉛誘導体H中の酸素炭素比(C/O比)は6.1であった。実施例1-5と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-5と比較して少ないためと考えられた。図11は、実施例1-8で作製した酸化黒鉛誘導体HのFT-IRチャートである。
(実施例1-9)
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Iを得た。得られた酸化黒鉛誘導体I中の酸素炭素比(C/O比)は7.1であった。実施例1-1、実施例1-9のXRDパターンを比較すると、実施例1-1の酸化黒鉛誘導体Aが実施例1-9の酸化黒鉛誘導体Iに比べよりシャープであり、結晶性の高いものであった。また落つい感度試験による感度は7級であった。図12は、実施例1-9で作製した酸化黒鉛誘導体IのFT-IRチャートである。図13は、実施例1-9で作製した酸化黒鉛誘導体IのXRDチャートである。酸化黒鉛誘導体Iは、図13に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が61.8Åであった。
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-1に記載の方法で酸化黒鉛誘導体Iを得た。得られた酸化黒鉛誘導体I中の酸素炭素比(C/O比)は7.1であった。実施例1-1、実施例1-9のXRDパターンを比較すると、実施例1-1の酸化黒鉛誘導体Aが実施例1-9の酸化黒鉛誘導体Iに比べよりシャープであり、結晶性の高いものであった。また落つい感度試験による感度は7級であった。図12は、実施例1-9で作製した酸化黒鉛誘導体IのFT-IRチャートである。図13は、実施例1-9で作製した酸化黒鉛誘導体IのXRDチャートである。酸化黒鉛誘導体Iは、図13に示すようにX線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が61.8Åであった。
(実施例1-10)
反応温度を100℃、反応を24時間にしたこと以外は実施例1-5に記載の方法で酸化黒鉛誘導体Jを得た。得られた酸化黒鉛誘導体J中の酸素炭素比(C/O比)は6.1であった。実施例1-5、1-10のXRDパターンを比較すると、実施例1-5の酸化黒鉛誘導体Eが実施例1-10の酸化黒鉛誘導体Jに比べよりシャープであり、結晶性の高いものであった。また落つい感度試験による感度は7級であった。以上、実施例1-1と実施例1-9、実施例1-5と実施例1-10を比較すると、反応温度を150℃とすることで落つい感度を消失させることができるとわかった。図14は、実施例1-10で作製した酸化黒鉛誘導体JのFT-IRチャートである。図15は、実施例1-10で作製した酸化黒鉛誘導体JのXRDチャートである。酸化黒鉛誘導体Jは、図15に示すようにX線回折スペクトルにおいて21-24°にピークを1つ示すが、9-13°にピークを示すものではなかった。
反応温度を100℃、反応を24時間にしたこと以外は実施例1-5に記載の方法で酸化黒鉛誘導体Jを得た。得られた酸化黒鉛誘導体J中の酸素炭素比(C/O比)は6.1であった。実施例1-5、1-10のXRDパターンを比較すると、実施例1-5の酸化黒鉛誘導体Eが実施例1-10の酸化黒鉛誘導体Jに比べよりシャープであり、結晶性の高いものであった。また落つい感度試験による感度は7級であった。以上、実施例1-1と実施例1-9、実施例1-5と実施例1-10を比較すると、反応温度を150℃とすることで落つい感度を消失させることができるとわかった。図14は、実施例1-10で作製した酸化黒鉛誘導体JのFT-IRチャートである。図15は、実施例1-10で作製した酸化黒鉛誘導体JのXRDチャートである。酸化黒鉛誘導体Jは、図15に示すようにX線回折スペクトルにおいて21-24°にピークを1つ示すが、9-13°にピークを示すものではなかった。
(実施例1-11)
調製例1-5で得られた酸化黒鉛含有組成物を反応器に移し、ここへ2-デシル-1-テトラデカノール(新日本理化株式会社製、エヌジェコール240A)を酸化黒鉛に対し500%加え、100℃で減圧下、水及び1-ブタノールを留去した。その後昇温し、150℃で5時間反応させた。反応後ヘキサンを注ぎ、濾過し、水洗、アセトン洗浄を行い、得られた固体を100℃で真空乾燥することで酸化黒鉛誘導体Xを得た。得られた酸化黒鉛誘導体X中の酸素炭素比(C/O比)は8.7であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
調製例1-5で得られた酸化黒鉛含有組成物を反応器に移し、ここへ2-デシル-1-テトラデカノール(新日本理化株式会社製、エヌジェコール240A)を酸化黒鉛に対し500%加え、100℃で減圧下、水及び1-ブタノールを留去した。その後昇温し、150℃で5時間反応させた。反応後ヘキサンを注ぎ、濾過し、水洗、アセトン洗浄を行い、得られた固体を100℃で真空乾燥することで酸化黒鉛誘導体Xを得た。得られた酸化黒鉛誘導体X中の酸素炭素比(C/O比)は8.7であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
(実施例1-12)
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-11に記載の方法で酸化黒鉛誘導体Yを得た。得られた酸化黒鉛誘導体Y中の酸素炭素比(C/O比)は6.9であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。実施例1-11及び1-12の結果から、1-ブタノール等の揮発性化合物を凝集剤として用いた場合にあらかじめ減圧留去工程を経ることもまた好ましい形態である。また落つい感度試験による感度は8級であった。
2-デシル-1-テトラデカノールの代わりに2-エチル-1-ヘキサノール(和光純薬工業株式会社製)を用いた以外は実施例1-11に記載の方法で酸化黒鉛誘導体Yを得た。得られた酸化黒鉛誘導体Y中の酸素炭素比(C/O比)は6.9であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。実施例1-11及び1-12の結果から、1-ブタノール等の揮発性化合物を凝集剤として用いた場合にあらかじめ減圧留去工程を経ることもまた好ましい形態である。また落つい感度試験による感度は8級であった。
(実施例1-13)
2-デシル-1-テトラデカノールの代わりにステアリルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Aを得た。得られた酸化黒鉛誘導体A-A中の酸素炭素比(C/O比)は8.0であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
2-デシル-1-テトラデカノールの代わりにステアリルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Aを得た。得られた酸化黒鉛誘導体A-A中の酸素炭素比(C/O比)は8.0であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
(実施例1-14)
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Bを得た。得られた酸化黒鉛誘導体A-B中の酸素炭素比(C/O比)は7.2であった。実施例1-13と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Bを得た。得られた酸化黒鉛誘導体A-B中の酸素炭素比(C/O比)は7.2であった。実施例1-13と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
(実施例1-15)
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Cを得た。得られた酸化黒鉛誘導体A-C中の酸素炭素比(C/O比)は6.6であった。実施例1-13と比較して、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Cを得た。得られた酸化黒鉛誘導体A-C中の酸素炭素比(C/O比)は6.6であった。実施例1-13と比較して、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
(実施例1-16)
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Dを得た。得られた酸化黒鉛誘導体A-D中の酸素炭素比(C/O比)は6.9であった。実施例1-13と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと以外は実施例1-13に記載の方法で酸化黒鉛誘導体A-Dを得た。得られた酸化黒鉛誘導体A-D中の酸素炭素比(C/O比)は6.9であった。実施例1-13と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-13と比較して少ないためと考えられた。
(実施例1-17)
2-デシル-1-テトラデカノールの代わりに2-エチルヘキシルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Eを得た。得られた酸化黒鉛誘導体A-E中の酸素炭素比(C/O比)は6.7であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
2-デシル-1-テトラデカノールの代わりに2-エチルヘキシルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Eを得た。得られた酸化黒鉛誘導体A-E中の酸素炭素比(C/O比)は6.7であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
(実施例1-18)
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Fを得た。得られた酸化黒鉛誘導体A-F中の酸素炭素比(C/O比)は6.2であった。実施例1-17と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-2で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Fを得た。得られた酸化黒鉛誘導体A-F中の酸素炭素比(C/O比)は6.2であった。実施例1-17と比較してやはり、修飾量が少なく、C/O比が低いものであった。精製により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
(実施例1-19)
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Gを得た。得られた酸化黒鉛誘導体A-G中の酸素炭素比(C/O比)は6.0であった。実施例1-17と比較して、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-4で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Gを得た。得られた酸化黒鉛誘導体A-G中の酸素炭素比(C/O比)は6.0であった。実施例1-17と比較して、修飾量が少なく、C/O比が低いものであった。精製及び乾燥により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
(実施例1-20)
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Hを得た。得られた酸化黒鉛誘導体A-H中の酸素炭素比(C/O比)は6.1であった。実施例1-17と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
用いる酸化黒鉛原料を調製例1-6で得られた酸化黒鉛としたこと以外は実施例1-17に記載の方法で酸化黒鉛誘導体A-Hを得た。得られた酸化黒鉛誘導体A-H中の酸素炭素比(C/O比)は6.1であった。実施例1-17と比較してやはり、修飾量が少なく、C/O比が低いものであった。乾燥により酸素官能基が減り、修飾量が実施例1-17と比較して少ないためと考えられた。
(実施例1-21)
反応溶媒としてNMPを酸化黒鉛に対し10000%加え超音波処理により分散させたこと、2-デシル-1-テトラデカノールの代わりにステアリルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Iを得た。得られた酸化黒鉛誘導体A-I中の酸素炭素比(C/O比)は8.3であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
反応溶媒としてNMPを酸化黒鉛に対し10000%加え超音波処理により分散させたこと、2-デシル-1-テトラデカノールの代わりにステアリルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Iを得た。得られた酸化黒鉛誘導体A-I中の酸素炭素比(C/O比)は8.3であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
(実施例1-22)
反応溶媒としてNMPを酸化黒鉛に対し10000%加え超音波処理により分散させたこと、2-デシル-1-テトラデカノールの代わりに2-エチルヘキシルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Jを得た。得られた酸化黒鉛誘導体A-J中の酸素炭素比(C/O比)は7.0であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
反応溶媒としてNMPを酸化黒鉛に対し10000%加え超音波処理により分散させたこと、2-デシル-1-テトラデカノールの代わりに2-エチルヘキシルアミン(東京化成工業株式会社製)を用いた以外は実施例1-1に記載の方法で酸化黒鉛誘導体A-Jを得た。得られた酸化黒鉛誘導体A-J中の酸素炭素比(C/O比)は7.0であった。また分散性においても、クロロホルム、アセトン、DMF、エタノール、デカンに対し良好な分散性を持つことも確認された。また落つい感度試験による感度は8級であった。
(実施例1-23)
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-3に記載の方法で酸化黒鉛誘導体Kを得た。得られた酸化黒鉛誘導体K中の酸素炭素比(C/O比)は6.0であった。修飾量が実施例1-1~1-4と比較して少ないことが確認された。図16は、実施例1-23で作製した酸化黒鉛誘導体KのFT-IRチャートである。なお、実施例1-23の酸化黒鉛誘導体は、炭素数24の2-デシル-1-テトラデカノールが付加されたものであるため、非極性分散媒中で分散することが可能なものである。
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-3に記載の方法で酸化黒鉛誘導体Kを得た。得られた酸化黒鉛誘導体K中の酸素炭素比(C/O比)は6.0であった。修飾量が実施例1-1~1-4と比較して少ないことが確認された。図16は、実施例1-23で作製した酸化黒鉛誘導体KのFT-IRチャートである。なお、実施例1-23の酸化黒鉛誘導体は、炭素数24の2-デシル-1-テトラデカノールが付加されたものであるため、非極性分散媒中で分散することが可能なものである。
(実施例1-24)
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-7に記載の方法で酸化黒鉛誘導体Lを得た。得られた酸化黒鉛誘導体L中の酸素炭素比(C/O比)は5.6であった。修飾量が実施例1-5~1-8と比較して少ないことが確認された。図17は、実施例1-24で作製した酸化黒鉛誘導体LのFT-IRチャートである。なお、実施例1-24の酸化黒鉛誘導体は、炭素数8の2-エチル-1-ヘキサノールが付加されたものであるため、両親媒性分散媒中で分散することが可能なものである。
反応温度を100℃、反応時間を24時間にしたこと以外は実施例1-7に記載の方法で酸化黒鉛誘導体Lを得た。得られた酸化黒鉛誘導体L中の酸素炭素比(C/O比)は5.6であった。修飾量が実施例1-5~1-8と比較して少ないことが確認された。図17は、実施例1-24で作製した酸化黒鉛誘導体LのFT-IRチャートである。なお、実施例1-24の酸化黒鉛誘導体は、炭素数8の2-エチル-1-ヘキサノールが付加されたものであるため、両親媒性分散媒中で分散することが可能なものである。
実施例1-1~1-24の結果を下記表1に示す。
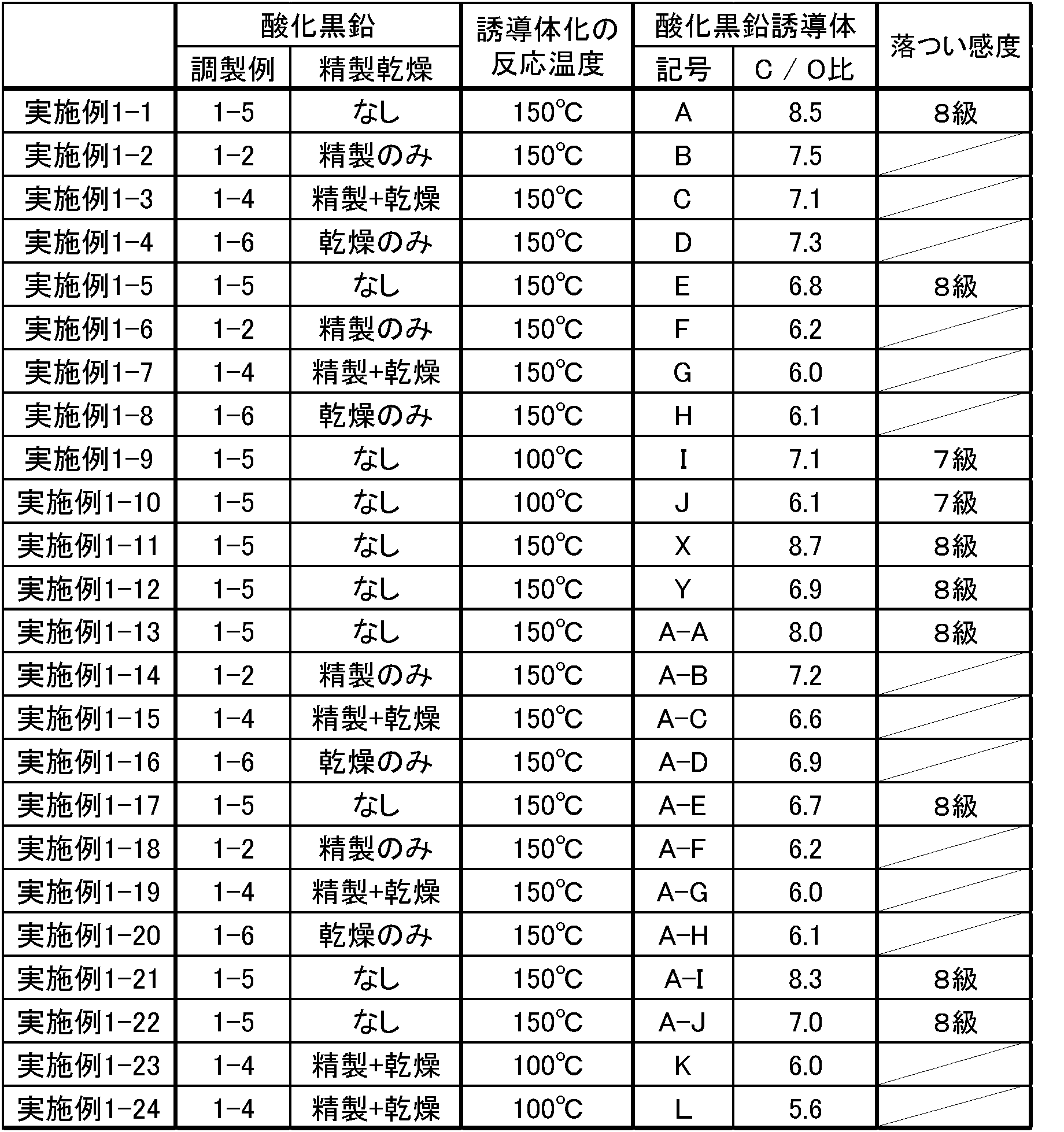
調製例2-1
耐食性反応器に濃硫酸(試薬特級、和光純薬工業社製)28.75部と天然黒鉛(Z-5F、鱗片状黒鉛、伊藤黒鉛工業社製)1.00部を加えて混合液とした。混合液を撹拌しながら過マンガン酸カリウム(試薬特級、和光純薬工業社製)を15分間隔で混合液中へ20回投入した。過マンガン酸カリウムの一回の投入量は0.125部であり、投入量の合計は2.50部であった。過マンガン酸カリウムの投入終了後、混合液を35℃まで昇温し液温を維持したまま2時間熟成を行った。その後60℃以下の液温を維持したままイオン交換水63.45部と30%過酸化水素水(試薬特級、和光純薬工業社製)1.77部を投入し反応を停止させた。当該手法により得られた酸化黒鉛含有スラリーを以下から“反応後スラリー”と呼称する。
耐食性反応器に濃硫酸(試薬特級、和光純薬工業社製)28.75部と天然黒鉛(Z-5F、鱗片状黒鉛、伊藤黒鉛工業社製)1.00部を加えて混合液とした。混合液を撹拌しながら過マンガン酸カリウム(試薬特級、和光純薬工業社製)を15分間隔で混合液中へ20回投入した。過マンガン酸カリウムの一回の投入量は0.125部であり、投入量の合計は2.50部であった。過マンガン酸カリウムの投入終了後、混合液を35℃まで昇温し液温を維持したまま2時間熟成を行った。その後60℃以下の液温を維持したままイオン交換水63.45部と30%過酸化水素水(試薬特級、和光純薬工業社製)1.77部を投入し反応を停止させた。当該手法により得られた酸化黒鉛含有スラリーを以下から“反応後スラリー”と呼称する。
比較例2-1
上記反応後スラリー30g(酸化黒鉛を0.45g含む)を直径55mmの吸引型濾過器を用いて濾過することで酸化黒鉛の分離を行った。濾紙としてテフロン(登録商標)濾紙(アドバンテック社製、PF050)を使用した。濾過器中でケーキ状の酸化黒鉛層が露出するまでに104秒の時間を要した。
上記反応後スラリー30g(酸化黒鉛を0.45g含む)を直径55mmの吸引型濾過器を用いて濾過することで酸化黒鉛の分離を行った。濾紙としてテフロン(登録商標)濾紙(アドバンテック社製、PF050)を使用した。濾過器中でケーキ状の酸化黒鉛層が露出するまでに104秒の時間を要した。
実施例2-1
上記反応後スラリー30g(酸化黒鉛を0.45g含む)に対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を0.13g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は65秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30g(酸化黒鉛を0.45g含む)に対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を0.13g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は65秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-2
上記反応後スラリー30gに対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を0.39g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は73秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を0.39g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は73秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-3
上記反応後スラリー30gに対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を1.30g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は47秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対してシクロヘキサノン(試薬特級、和光純薬工業社製、水に対する溶解度;5重量%)を1.30g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は47秒であり、シクロヘキサノンを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-4
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を0.39g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は80秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を0.39g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は80秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-5
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は70秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は70秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-6
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を1.9g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は18秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を1.9g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は18秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.1g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
実施例2-7
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を2.6g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は1秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.2g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
上記反応後スラリー30gに対して1-ブタノール(試薬特級、和光純薬工業社製、水に対する溶解度;6.4重量%)を2.6g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は1秒であり、1-ブタノールを添加することで濾過速度が非常に早くなることを確認出来た。濾紙上の酸化黒鉛含有組成物(精製、乾燥工程ともに行っていない)を30gのNMPに分散させ、ステアリルアミン1.5gを加えた後、120℃で5時間反応させた。反応後、反応液を濾過し、水洗、アセトン洗浄を行い、乾燥させることで酸化黒鉛誘導体が1.2g得られた。得られた酸化黒鉛誘導体はデカン、流動パラフィン等の非極性溶媒中で良好な分散性を示した。
比較例2-2
上記反応後スラリー30gに対して2-プロパノール(試薬特級、和光純薬工業社製、水に対して任意に混和)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は146秒であった。
上記反応後スラリー30gに対して2-プロパノール(試薬特級、和光純薬工業社製、水に対して任意に混和)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は146秒であった。
比較例2-3
上記反応後スラリー30gに対してヘキサン(試薬特級、和光純薬工業社製、水に対して不溶)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は586秒であった。
上記反応後スラリー30gに対してヘキサン(試薬特級、和光純薬工業社製、水に対して不溶)を1.3g添加しよく振盪させた以外は比較例2-1と同様の条件で酸化黒鉛の分離を行った。濾過に要した時間は586秒であった。
参考例2-1
実施例2-3と同様の手法にて酸化黒鉛の分離を行った後に少量のイオン交換水で洗浄を行い、50℃真空下にて一晩乾燥を行うことで酸化黒鉛乾燥粉末を得た。XRD測定結果を図18に示す。図18は、参考例2-1で得られた酸化黒鉛粉末のXRD測定結果を示した図である。酸化黒鉛の層状構造に由来する特徴的なシグナルが認められたことから当該分離手法にて問題無く酸化黒鉛の分離が可能であることが確認出来た。
実施例2-3と同様の手法にて酸化黒鉛の分離を行った後に少量のイオン交換水で洗浄を行い、50℃真空下にて一晩乾燥を行うことで酸化黒鉛乾燥粉末を得た。XRD測定結果を図18に示す。図18は、参考例2-1で得られた酸化黒鉛粉末のXRD測定結果を示した図である。酸化黒鉛の層状構造に由来する特徴的なシグナルが認められたことから当該分離手法にて問題無く酸化黒鉛の分離が可能であることが確認出来た。
参考例2-2
実施例2-7と同様の手法にて酸化黒鉛の分離を行った後に少量のイオン交換水で洗浄を行い、50℃真空下にて一晩乾燥を行うことで酸化黒鉛乾燥粉末を得た。XRD測定結果を図19に示す。図19は、参考例2-2で得られた酸化黒鉛粉末のXRD測定結果を示した図である。酸化黒鉛の層状構造に由来する特徴的なシグナルが認められたことから当該精製手法にて問題無く酸化黒鉛の分離が可能であることが確認出来た。
実施例2-7と同様の手法にて酸化黒鉛の分離を行った後に少量のイオン交換水で洗浄を行い、50℃真空下にて一晩乾燥を行うことで酸化黒鉛乾燥粉末を得た。XRD測定結果を図19に示す。図19は、参考例2-2で得られた酸化黒鉛粉末のXRD測定結果を示した図である。酸化黒鉛の層状構造に由来する特徴的なシグナルが認められたことから当該精製手法にて問題無く酸化黒鉛の分離が可能であることが確認出来た。
参考例2-3
参考例2-1にて得られた酸化黒鉛乾燥粉末を昇温速度10℃/分にて800℃まで昇温させ、窒素流通下にて5時間焼成を実施した。得られた粉末のXRD測定結果を図20に示す。図20は、参考例2-3で得られた還元型酸化黒鉛粉末のXRD測定結果を示した図である。また焼成前後のC1s XPS測定結果を表2に示す。図20に示すように酸化黒鉛の層構造に由来するピークが消失しており、また酸素含有官能基に由来する結合が焼成により著しく消失していることから還元型酸化黒鉛が生成したことが示された。
参考例2-1にて得られた酸化黒鉛乾燥粉末を昇温速度10℃/分にて800℃まで昇温させ、窒素流通下にて5時間焼成を実施した。得られた粉末のXRD測定結果を図20に示す。図20は、参考例2-3で得られた還元型酸化黒鉛粉末のXRD測定結果を示した図である。また焼成前後のC1s XPS測定結果を表2に示す。図20に示すように酸化黒鉛の層構造に由来するピークが消失しており、また酸素含有官能基に由来する結合が焼成により著しく消失していることから還元型酸化黒鉛が生成したことが示された。
参考例2-4
参考例2-2にて得られた酸化黒鉛乾燥粉末を昇温速度10℃/分にて800℃まで昇温させ、窒素流通下にて5時間焼成を実施した。得られた粉末のXRD測定結果を図21に示す。図21は、参考例2-4で得られた還元型酸化黒鉛粉末のXRD測定結果を示した図である。また焼成前後のC1s XPS測定結果を表3に示す。図21に示すように酸化黒鉛の層構造に由来するピークが消失しており、また酸素含有官能基に由来する結合が焼成により著しく消失していることから還元型酸化黒鉛が生成したことが示された。
参考例2-2にて得られた酸化黒鉛乾燥粉末を昇温速度10℃/分にて800℃まで昇温させ、窒素流通下にて5時間焼成を実施した。得られた粉末のXRD測定結果を図21に示す。図21は、参考例2-4で得られた還元型酸化黒鉛粉末のXRD測定結果を示した図である。また焼成前後のC1s XPS測定結果を表3に示す。図21に示すように酸化黒鉛の層構造に由来するピークが消失しており、また酸素含有官能基に由来する結合が焼成により著しく消失していることから還元型酸化黒鉛が生成したことが示された。
(実施例3-1)
〔OGO20-Bの合成〕
非特許文献(Karthikeyan K, et.al, Carbon, 53, (2013), 38-49)に記載の方法を参考に原料となる酸化黒鉛を合成した。この酸化黒鉛(200mg)と2-オクチル-1-ドデカノール(東京化成工業社製、10ml)、硫酸(和光純薬社製、200mg)を混合し、100℃で24時間反応させた。反応後、反応液にアセトンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO20-Bを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図23にFT-IRチャートを示した。原料のFT-IRチャート(図22)と比較し、C-Hに由来するピーク(2900cm-1前後)の出現、及びC-O-Cに由来するピーク(1200cm-1前後)のシフトが確認できることから、アルコールが導入されたことが確認できた。なお、図22は、原料である酸化黒鉛のFT-IRチャートであり、図23は、実施例3-1で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は95.66%であり、Nの質量濃度は0.01%であった。
〔OGO20-Bの合成〕
非特許文献(Karthikeyan K, et.al, Carbon, 53, (2013), 38-49)に記載の方法を参考に原料となる酸化黒鉛を合成した。この酸化黒鉛(200mg)と2-オクチル-1-ドデカノール(東京化成工業社製、10ml)、硫酸(和光純薬社製、200mg)を混合し、100℃で24時間反応させた。反応後、反応液にアセトンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO20-Bを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図23にFT-IRチャートを示した。原料のFT-IRチャート(図22)と比較し、C-Hに由来するピーク(2900cm-1前後)の出現、及びC-O-Cに由来するピーク(1200cm-1前後)のシフトが確認できることから、アルコールが導入されたことが確認できた。なお、図22は、原料である酸化黒鉛のFT-IRチャートであり、図23は、実施例3-1で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は95.66%であり、Nの質量濃度は0.01%であった。
(実施例3-2)
〔OGO20-Sの合成〕
酸化黒鉛(200mg)、1-エイコサノール(東京化成工業社製、8g)、硫酸(200mg)を混合し、反応後、反応液に50℃に加温したヘキサンを投入し、熱時ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO20-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図24にFT-IRチャートを示した。なお、図24は、実施例3-2で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.12%であり、Nの質量濃度は0.00%であった。
〔OGO20-Sの合成〕
酸化黒鉛(200mg)、1-エイコサノール(東京化成工業社製、8g)、硫酸(200mg)を混合し、反応後、反応液に50℃に加温したヘキサンを投入し、熱時ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO20-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図24にFT-IRチャートを示した。なお、図24は、実施例3-2で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.12%であり、Nの質量濃度は0.00%であった。
(実施例3-3)
〔OGO14-Sの合成〕
用いたアルコールを2-オクチル-1-ドデカノールから1-テトラデカノール(東京化成工業社製、10ml)に変えた以外は実施例1に記載の方法と同様に合成しOGO14-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図25にFT-IRチャートを示した。なお、図25は、実施例3-3で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.95%であり、Nの質量濃度は0.00%であった。
〔OGO14-Sの合成〕
用いたアルコールを2-オクチル-1-ドデカノールから1-テトラデカノール(東京化成工業社製、10ml)に変えた以外は実施例1に記載の方法と同様に合成しOGO14-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、ヘキサン層に分散したことから非常に良好な疎水特性があることがわかった。
図25にFT-IRチャートを示した。なお、図25は、実施例3-3で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.95%であり、Nの質量濃度は0.00%であった。
(実施例3-4)
〔OGO20-B-BAの合成〕
酸化黒鉛(200mg)と2-オクチル-1-ドデカノール(東京化成工業社製、10ml)、水酸化カリウム(和光純薬社製、200mg)を混合し、100℃で24時間反応させた。反応後、反応液にアセトンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち1%硫酸水で洗浄した。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。有機層をろ過し、OGO20-B―BAを得た。
図26にFT-IRチャートを示した。なお、図26は、実施例3-4で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は95.51%であり、Nの質量濃度は0.01%であった。
〔OGO20-B-BAの合成〕
酸化黒鉛(200mg)と2-オクチル-1-ドデカノール(東京化成工業社製、10ml)、水酸化カリウム(和光純薬社製、200mg)を混合し、100℃で24時間反応させた。反応後、反応液にアセトンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち1%硫酸水で洗浄した。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。有機層をろ過し、OGO20-B―BAを得た。
図26にFT-IRチャートを示した。なお、図26は、実施例3-4で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は95.51%であり、Nの質量濃度は0.01%であった。
(比較例3-1)
〔OGO12-Sの合成〕
用いたアルコールを2-オクチル-1-ドデカノールから1-ドデカノール(東京化成工業社製、10ml)に変えた以外は実施例3-1に記載の方法と同様に合成しOGO12-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、エマルジョンになり、すぐに2層には分離しなかったこと、疎水性が充分でないため、ヘキサン層に良好に分散せず界面に被膜が生じたことから、実施例3-1、3-3に比較して疎水特性に劣ることが確認できた。この点、特に実施例3-3と比較例3-1との比較から、疎水特性は炭化水素基が炭素数14の場合と炭素数12の場合とで劇的に異なることが証明された。また得られた固体をNMPへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が10分以下であり、両親媒性分散媒への良好な分散性は確認できなかった。
図27にFT-IRチャートを示した。なお、図27は、比較例3-1で作製した酸化黒鉛誘導体のFT-IRチャートである。
図29にヘキサン-メタノール分液の実際の画像を示した。すなわち、図29は、実施例3-1、実施例3-3、比較例3-1のヘキサン-メタノール分液の実際の画像である。比較例3-1のみヘキサン層への分散性がないことがわかる。また、界面に泡状の被膜が生じ、疎水性が低いことがうかがえる(上部がヘキサン層、下部がメタノール層、左から実施例3-1、実施例3-3、比較例3-1)。
また元素分析の結果、CHOの質量濃度の合計は97.47%であり、Nの質量濃度は0.00%であった。
〔OGO12-Sの合成〕
用いたアルコールを2-オクチル-1-ドデカノールから1-ドデカノール(東京化成工業社製、10ml)に変えた以外は実施例3-1に記載の方法と同様に合成しOGO12-Sを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。また、ヘキサン-メタノールの混合溶媒へ加え1時間超音波をかけ、ヘキサン層に分散するかメタノール層へ分散するかを確認したところ、エマルジョンになり、すぐに2層には分離しなかったこと、疎水性が充分でないため、ヘキサン層に良好に分散せず界面に被膜が生じたことから、実施例3-1、3-3に比較して疎水特性に劣ることが確認できた。この点、特に実施例3-3と比較例3-1との比較から、疎水特性は炭化水素基が炭素数14の場合と炭素数12の場合とで劇的に異なることが証明された。また得られた固体をNMPへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が10分以下であり、両親媒性分散媒への良好な分散性は確認できなかった。
図27にFT-IRチャートを示した。なお、図27は、比較例3-1で作製した酸化黒鉛誘導体のFT-IRチャートである。
図29にヘキサン-メタノール分液の実際の画像を示した。すなわち、図29は、実施例3-1、実施例3-3、比較例3-1のヘキサン-メタノール分液の実際の画像である。比較例3-1のみヘキサン層への分散性がないことがわかる。また、界面に泡状の被膜が生じ、疎水性が低いことがうかがえる(上部がヘキサン層、下部がメタノール層、左から実施例3-1、実施例3-3、比較例3-1)。
また元素分析の結果、CHOの質量濃度の合計は97.47%であり、Nの質量濃度は0.00%であった。
(比較例3-2)
〔rGOの合成〕
アルコールの代わりにヘキサデカン(東京化成工業社製、10ml)に変えた以外は実施例1に記載の方法と同様に合成しrGOを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が20分であり、良好な分散性が確認できなかった。
図28にFT-IRチャートを示した。なお、図28は、比較例3-2で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.94%であり、Nの質量濃度は0.01%であった。
〔rGOの合成〕
アルコールの代わりにヘキサデカン(東京化成工業社製、10ml)に変えた以外は実施例1に記載の方法と同様に合成しrGOを得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が20分であり、良好な分散性が確認できなかった。
図28にFT-IRチャートを示した。なお、図28は、比較例3-2で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.94%であり、Nの質量濃度は0.01%であった。
(実施例3-5)
〔OGO24-B-100の合成〕
酸化黒鉛(2g)と2-デシル-1-テトラデカノール(新日本理化社製、10g)、硫酸(和光純薬社製、2g)を混合し、100℃で24時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO24-B-100を得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。
図30にFT-IRチャートを示した。なお、図30は、実施例3-5で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.73%であり、Nの質量濃度は0.00%であった。
〔OGO24-B-100の合成〕
酸化黒鉛(2g)と2-デシル-1-テトラデカノール(新日本理化社製、10g)、硫酸(和光純薬社製、2g)を混合し、100℃で24時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO24-B-100を得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。
図30にFT-IRチャートを示した。なお、図30は、実施例3-5で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.73%であり、Nの質量濃度は0.00%であった。
(実施例3-6)
〔OGO24-B-150の合成〕
酸化黒鉛(2g)と2-デシル-1-テトラデカノール(新日本理化社製、10g)、硫酸(和光純薬社製、2g)を混合し、150℃で5時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO24-B-150を得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。
図31にFT-IRチャートを示した。なお、図31は、実施例3-6で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.30%であり、Nの質量濃度は0.00%であった。
〔OGO24-B-150の合成〕
酸化黒鉛(2g)と2-デシル-1-テトラデカノール(新日本理化社製、10g)、硫酸(和光純薬社製、2g)を混合し、150℃で5時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO24-B-150を得た。得られた固体をヘキサデカンへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。
図31にFT-IRチャートを示した。なお、図31は、実施例3-6で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.30%であり、Nの質量濃度は0.00%であった。
(実施例3-7)
〔OGO8-B-150の合成〕
酸化黒鉛(2g)と2-エチル-1-ヘキサノール(和光純薬社製、10g)、硫酸(和光純薬社製、2g)を混合し、150℃で5時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO8-B-150を得た。得られた固体をNMPへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。比較例3-1と比較しアルキル鎖が短いことで両親媒性分散媒への分散性が向上することがわかった。この点、特に実施例3-7と比較例3-1との比較から、両親媒性分散媒への分散特性は炭化水素基が炭素数12の場合と炭素数8の場合とで劇的に異なることが証明された。
図32にFT-IRチャートを示した。なお、図32は、実施例3-7で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.66%であり、Nの質量濃度は0.00%であった。
〔OGO8-B-150の合成〕
酸化黒鉛(2g)と2-エチル-1-ヘキサノール(和光純薬社製、10g)、硫酸(和光純薬社製、2g)を混合し、150℃で5時間反応させた。反応後、反応液にヘキサンを注ぎ、ろ過した。得られた固体をヘキサンに分散させたのち水洗した。有機層をろ過し、OGO8-B-150を得た。得られた固体をNMPへ加え、1時間超音波をかけ、分散性を評価したところ、光の透過が確認できる時間が6時間以上であり、良好な分散性が確認できた。比較例3-1と比較しアルキル鎖が短いことで両親媒性分散媒への分散性が向上することがわかった。この点、特に実施例3-7と比較例3-1との比較から、両親媒性分散媒への分散特性は炭化水素基が炭素数12の場合と炭素数8の場合とで劇的に異なることが証明された。
図32にFT-IRチャートを示した。なお、図32は、実施例3-7で作製した酸化黒鉛誘導体のFT-IRチャートである。
また元素分析の結果、CHOの質量濃度の合計は96.66%であり、Nの質量濃度は0.00%であった。
上述した実施例3-1~3-7及び比較例3-1では、FT-IRチャートの結果から、酸化黒鉛のカルボキシル基、エポキシ基や水酸基にアルコールが導入され、アルコキシカルボニル基やアルコキシ基が形成されていると考えられる。特に実施例3-3と比較例3-1との比較から、疎水特性は炭化水素基が炭素数14の場合と炭素数12の場合とで劇的に異なることが証明された。また、実施例3-7と比較例3-1との比較から、両親媒性分散媒への分散特性は炭化水素基が炭素数8の場合と炭素数12の場合とで劇的に異なることが証明された。
実施例3-1~3-6で得られた酸化黒鉛誘導体は、非極性分散媒中で充分に分散するため、機械用の潤滑油の添加剤、さまざまな樹脂と複合化できる樹脂への添加剤等として好適に使用できると考えられる。実施例3-7で得られた酸化黒鉛誘導体は、両親媒性分散媒中で充分に分散するため、さまざまな樹脂と複合化できる樹脂への添加剤等として好適に使用できると考えられる。
実施例3-1~3-6で得られた酸化黒鉛誘導体は、非極性分散媒中で充分に分散するため、機械用の潤滑油の添加剤、さまざまな樹脂と複合化できる樹脂への添加剤等として好適に使用できると考えられる。実施例3-7で得られた酸化黒鉛誘導体は、両親媒性分散媒中で充分に分散するため、さまざまな樹脂と複合化できる樹脂への添加剤等として好適に使用できると考えられる。
なお、実施例3-1~3-7で得られた酸化黒鉛誘導体は、長時間を要する精製工程及び乾燥工程を経て得られる酸化黒鉛を原料として用いたものであるが、本発明の製造方法の第1の好ましい形態と比較すると高品質の原料を効率よく得る点では工夫の余地があった。例えば、工業的規模で酸化黒鉛誘導体を製造するための原料として、このように精製工程及び乾燥工程を経て得られる酸化黒鉛を大量に用意することは、現実にはほとんど不可能である。しかし、本発明の酸化黒鉛誘導体のような特定炭素数の炭化水素基をもつ官能基を有する酸化黒鉛誘導体の製造において、本発明の製造方法の第1の好ましい形態で上述した酸化黒鉛の精製工程及び乾燥工程の少なくとも一方を省略する方法を適用することにより、上述した本発明の製造方法の第1の好ましい形態の実施例で示した通り高品質な酸化黒鉛を簡便に得ることが可能となり、工業的規模で酸化黒鉛誘導体を製造するうえで特に好適なものとなる。
Claims (26)
- 酸化黒鉛誘導体を製造する方法であって、
該製造方法は、黒鉛を酸化する工程と、
該酸化工程で得られる酸化黒鉛含有組成物中の酸化黒鉛と、該酸化黒鉛の酸素含有官能基と反応する化合物とを反応させて酸化黒鉛誘導体を得る工程とを含み、
酸化工程と酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製し、かつ乾燥する工程を含まないことを特徴とする酸化黒鉛誘導体の製造方法。 - 前記酸化工程と前記酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物を精製する工程を含まないことを特徴とする請求項1に記載の酸化黒鉛誘導体の製造方法。
- 前記酸化工程と前記酸化黒鉛誘導体を得る工程との間に、酸化黒鉛含有組成物に、水に対する溶解度が0.01%以上であって、かつ、水と任意には混和しない溶媒を添加した後、酸化黒鉛含有組成物を分離する工程を含むことを特徴とする請求項1又は2に記載の酸化黒鉛誘導体の製造方法。
- 前記酸化工程は、酸を用いて黒鉛を酸化することを特徴とする請求項1~3のいずれかに記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛の酸素含有官能基と反応する化合物は、アルコール及び/又はアミンであることを特徴とする請求項1~4のいずれかに記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛誘導体を得る工程は、酸化黒鉛と、酸化黒鉛の酸素含有官能基と反応する化合物とを120℃以上の反応温度で反応させる工程を含むことを特徴とする請求項1~5のいずれかに記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物は、組成物中の酸化黒鉛の質量100質量%に対して、硫酸を1質量%以上、1000質量%以下含むことを特徴とする請求項1~6のいずれかに記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛誘導体を得る工程の反応に供される酸化黒鉛含有組成物は、組成物中の酸化黒鉛の質量100質量%に対して、水を3質量%以上、10000質量%以下含むことを特徴とする請求項1~7のいずれかに記載の酸化黒鉛誘導体の製造方法。
- 酸化黒鉛誘導体を製造する方法であって、
該製造方法は、酸化黒鉛とアルコール及び/又はアミンとを120℃以上の反応温度で反応させて炭化水素基をもつ官能基を有する酸化黒鉛誘導体を得る工程を含むことを特徴とする酸化黒鉛誘導体の製造方法。 - 前記アルコールは、脂肪族アルコールであり、
前記アミンは、脂肪族アミンであることを特徴とする請求項5又は9に記載の酸化黒鉛誘導体の製造方法。 - 前記溶媒の水に対する溶解度は0.5%以上であることを特徴とする請求項3に記載の酸化黒鉛誘導体の製造方法。
- 前記溶媒の添加量は、酸化黒鉛含有組成物中の酸化黒鉛100質量%に対して1~1000質量%であることを特徴とする請求項3又は11に記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛含有組成物の分離は、濾過、デカンテーション、遠心分離、及び、分液抽出のいずれかにより行われることを特徴とする請求項3、11、又は、12に記載の酸化黒鉛誘導体の製造方法。
- 前記酸化黒鉛含有組成物の分離は、濾過により行われることを特徴とする請求項13に記載の酸化黒鉛誘導体の製造方法。
- 前記酸化工程は、黒鉛と硫酸とを含む混合液に過マンガン酸塩を添加する工程であることを特徴とする請求項1~14のいずれかに記載の酸化黒鉛誘導体の製造方法。
- アルキル基を有し、
JIS K 4810に規定される落つい感度試験で測定される等級が8級であることを特徴とする酸化黒鉛誘導体。 - X線回折スペクトルにおいて9-13°及び21-24°にそれぞれピークを少なくとも1つずつ示し、9-13°にあるピークのScherrerの式により算出される結晶子径が100Å以上、500Å以下であることを特徴とする酸化黒鉛誘導体。
- 炭素数13以上の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体。
- 前記炭化水素基は、炭素数が36以下であることを特徴とする請求項18に記載の酸化黒鉛誘導体。
- 炭素数20以上、28以下の炭化水素基をもつ官能基を有することを特徴とする請求項19に記載の酸化黒鉛誘導体。
- 炭素数6以上、10以下の炭化水素基をもつ官能基を有することを特徴とする酸化黒鉛誘導体。
- 前記炭化水素基は、アルキル基であることを特徴とする請求項18~21のいずれかに記載の酸化黒鉛誘導体。
- 前記炭化水素基は、直鎖であることを特徴とする請求項18~22のいずれかに記載の酸化黒鉛誘導体。
- 前記炭化水素基は、分岐鎖であることを特徴とする請求項18~22のいずれかに記載の酸化黒鉛誘導体。
- 窒素原子の含有量が0.1質量%以下であることを特徴とする請求項18~24のいずれかに記載の酸化黒鉛誘導体。
- 請求項16~25のいずれかに記載の酸化黒鉛誘導体が分散媒中に分散してなることを特徴とする分散体。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/775,526 US10766774B2 (en) | 2015-11-12 | 2016-11-08 | Oxidized graphite derivative and method for producing same |
| EP16864224.7A EP3375756A4 (en) | 2015-11-12 | 2016-11-08 | OXIDIZED GRAPHITE DERIVATIVE AND METHOD FOR THE PRODUCTION THEREOF |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015222306 | 2015-11-12 | ||
| JP2015-222306 | 2015-11-12 | ||
| JP2016005309A JP6749099B2 (ja) | 2016-01-14 | 2016-01-14 | 酸化黒鉛の製造方法 |
| JP2016-005309 | 2016-01-14 | ||
| JP2016-033326 | 2016-02-24 | ||
| JP2016033326 | 2016-02-24 | ||
| JP2016-060680 | 2016-03-24 | ||
| JP2016060680 | 2016-03-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017082263A1 true WO2017082263A1 (ja) | 2017-05-18 |
Family
ID=58695398
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/083143 Ceased WO2017082263A1 (ja) | 2015-11-12 | 2016-11-08 | 酸化黒鉛誘導体及びその製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US10766774B2 (ja) |
| EP (1) | EP3375756A4 (ja) |
| WO (1) | WO2017082263A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107879338A (zh) * | 2017-12-29 | 2018-04-06 | 天津市天波科达科技有限公司 | 一种工业级氧化石墨烯生产装置 |
| JP2019064867A (ja) * | 2017-09-29 | 2019-04-25 | 株式会社日本触媒 | 酸化黒鉛の製造方法 |
| WO2019208636A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社日本触媒 | 炭素材料複合体の製造方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7752819B2 (ja) | 2019-07-08 | 2025-10-14 | アルター ビオタ インコーポレイテッド | コンクリート混和剤として使用するための水性酸化グラフェンの製造 |
| KR102420365B1 (ko) * | 2020-04-07 | 2022-07-14 | 주식회사 한솔케미칼 | 나노입자의 대량 정제 시스템 및 이를 이용한 나노입자의 대량 정제방법 |
| EP4296226A1 (en) | 2022-06-24 | 2023-12-27 | Espio, s.r.o. | The method of preparing functionalized reduced graphene oxide layer and the temperature sensor comprising such a layer |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5234325B2 (ja) * | 1972-02-28 | 1977-09-02 | ||
| JP2006233017A (ja) * | 2005-02-24 | 2006-09-07 | Toray Ind Inc | 熱可塑性樹脂組成物およびその製造方法 |
| JP2007009205A (ja) * | 2005-06-28 | 2007-01-18 | Hilti Ag | ポリウレタン−グラファイト酸化物複合材料、その製造方法、耐火材料としての使用方法及び防火障壁への使用方法 |
| JP4798411B2 (ja) * | 2000-08-09 | 2011-10-19 | 三菱瓦斯化学株式会社 | 炭素からなる骨格を持つ薄膜状粒子の合成方法 |
| JP2013006909A (ja) * | 2011-06-22 | 2013-01-10 | Denso Corp | 熱輸送流体 |
| JP2013079176A (ja) * | 2011-10-05 | 2013-05-02 | Dic Corp | 修飾グラフェン、膜、及び成形体 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6596396B2 (en) | 2000-08-09 | 2003-07-22 | Mitsubishi Gas Chemical Company, Inc. | Thin-film-like particles having skeleton constructed by carbons and isolated films |
| JP2003176117A (ja) | 2001-12-07 | 2003-06-24 | Mitsubishi Gas Chem Co Inc | 炭素からなる骨格を持つ薄膜状粒子の積層集合体 |
| US20030186059A1 (en) | 2002-02-08 | 2003-10-02 | Masukazu Hirata | Structure matter of thin film particles having carbon skeleton, processes for the production of the structure matter and the thin-film particles and uses thereof |
| JP5234325B2 (ja) | 2008-03-31 | 2013-07-10 | 株式会社豊田中央研究所 | 有機化グラファイト材料の製造方法 |
| CN101935030B (zh) | 2010-08-31 | 2012-12-12 | 南京理工大学 | 利用有机链段调控溶剂分散性能的功能化氧化石墨烯及其制备方法 |
| JP2012153590A (ja) | 2011-01-28 | 2012-08-16 | Mitsubishi Gas Chemical Co Inc | 凝集物及び当該凝集物を溶媒に分散してなる分散液 |
| CN103402914B (zh) | 2011-03-23 | 2015-08-12 | 积水化学工业株式会社 | 氧化薄片化石墨衍生物、其树脂复合材料以及该树脂复合材料的制造方法 |
| JP5339099B2 (ja) | 2011-05-09 | 2013-11-13 | 三菱瓦斯化学株式会社 | 炭素からなる骨格を持つ薄膜状粒子の合成方法 |
| CN102431997B (zh) * | 2011-09-07 | 2013-05-08 | 南京师范大学 | 具有抗菌、抗凝血功能的氧化石墨烯及其制备方法 |
| WO2014148763A1 (ko) * | 2013-03-19 | 2014-09-25 | 서울대학교산학협력단 | 이차전지 양극용 다공성 그래핀 및 이의 제조 방법 |
| KR101510806B1 (ko) * | 2013-10-22 | 2015-04-08 | 현대자동차주식회사 | 산화 그래핀-세라믹 하이브리드 코팅막 및 이의 제조 방법 |
-
2016
- 2016-11-08 US US15/775,526 patent/US10766774B2/en active Active
- 2016-11-08 EP EP16864224.7A patent/EP3375756A4/en active Pending
- 2016-11-08 WO PCT/JP2016/083143 patent/WO2017082263A1/ja not_active Ceased
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5234325B2 (ja) * | 1972-02-28 | 1977-09-02 | ||
| JP4798411B2 (ja) * | 2000-08-09 | 2011-10-19 | 三菱瓦斯化学株式会社 | 炭素からなる骨格を持つ薄膜状粒子の合成方法 |
| JP2006233017A (ja) * | 2005-02-24 | 2006-09-07 | Toray Ind Inc | 熱可塑性樹脂組成物およびその製造方法 |
| JP2007009205A (ja) * | 2005-06-28 | 2007-01-18 | Hilti Ag | ポリウレタン−グラファイト酸化物複合材料、その製造方法、耐火材料としての使用方法及び防火障壁への使用方法 |
| JP2013006909A (ja) * | 2011-06-22 | 2013-01-10 | Denso Corp | 熱輸送流体 |
| JP2013079176A (ja) * | 2011-10-05 | 2013-05-02 | Dic Corp | 修飾グラフェン、膜、及び成形体 |
Non-Patent Citations (4)
| Title |
|---|
| DANIEL R. DREYER ET AL.: "The chemistry of graphene oxide", CHEMICAL SOCIETY REVIEWS, vol. 39, no. 1, 1 January 2010 (2010-01-01), pages 228 - 240, XP055541986, DOI: 10.1039/B917103G * |
| SASHA STANKOVICH ET AL.: "Synthesis and exfoliation of isocyanate-treated graphene oxide nanoplatelets", CARBON, vol. 44, 2006, pages 3342 - 3347, XP 002648478 * |
| See also references of EP3375756A4 * |
| YANWU ZHU ET AL.: "Graphene and Graphene Oxide: Synthesis, Properties, and Applications", ADVANCED MATERIALS, vol. 22, no. 35, 2010, pages 3906 - 3924, XP 055100171 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019064867A (ja) * | 2017-09-29 | 2019-04-25 | 株式会社日本触媒 | 酸化黒鉛の製造方法 |
| CN107879338A (zh) * | 2017-12-29 | 2018-04-06 | 天津市天波科达科技有限公司 | 一种工业级氧化石墨烯生产装置 |
| WO2019208636A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社日本触媒 | 炭素材料複合体の製造方法 |
| EP3786111A4 (en) * | 2018-04-27 | 2022-02-16 | Nippon Shokubai Co., Ltd. | PROCESS FOR MAKING A CARBON MATERIAL COMPLEX |
| US11945723B2 (en) | 2018-04-27 | 2024-04-02 | Nippon Shokubai Co., Ltd. | Method for producing carbon material complex |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180327269A1 (en) | 2018-11-15 |
| EP3375756A4 (en) | 2019-08-14 |
| US10766774B2 (en) | 2020-09-08 |
| EP3375756A1 (en) | 2018-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017082263A1 (ja) | 酸化黒鉛誘導体及びその製造方法 | |
| JP6762850B2 (ja) | 酸化黒鉛誘導体の製造方法 | |
| JP5574761B2 (ja) | 被覆銀超微粒子とその製造方法 | |
| CN113677458B (zh) | 混合银粉和包含其的导电糊 | |
| Tigwere et al. | Transition metal (Ni, Cu and Fe) doped MnS nanostructures: effect of doping on supercapacitance and water splitting | |
| US7785392B2 (en) | Method for manufacturing metal nanoparticles | |
| JP4978242B2 (ja) | 銀超微粒子の製造方法 | |
| US10737942B2 (en) | Zeta positive hydrogenated nanodiamond powder, zetapositive single digit hydrogenated nanodiamond dispersion, and methods for producing the same | |
| JP5770675B2 (ja) | 硫化リチウムの製造方法 | |
| US9051179B2 (en) | Continuous method and apparatus for functionalizing carbon nanotube | |
| JP2008095195A (ja) | 銅ナノ粒子の製造方法及びこれにより製造された銅ナノ粒子 | |
| Zou et al. | A new method for synthesis of ZnO flower-like nanostructures and their photocatalytic performance | |
| JP2009242209A (ja) | 有機化グラファイト材料の製造方法 | |
| JP2009270146A (ja) | 銀超微粒子の製造方法 | |
| JP2022136081A (ja) | 銅酸化物粒子組成物、導電性ペースト及び導電性インク | |
| Pham et al. | Synthesis of highly concentrated suspension of chemically converted graphene in organic solvents: effect of temperature on the extent of reduction and dispersibility | |
| KR20110108088A (ko) | 금속 나노입자의 제조방법, 이를 이용한 잉크 조성물 및 그의 제조 방법 | |
| JP2006118010A (ja) | Agナノ粒子及びその製造方法、Agナノ粒子の分散溶液 | |
| JP6765831B2 (ja) | 還元型酸化黒鉛の製造方法 | |
| JP6832134B2 (ja) | 酸化黒鉛誘導体 | |
| JP2011195951A (ja) | 金属ナノ粒子の製造方法、これを用いたインク組成物及びその製造方法 | |
| JP6943715B2 (ja) | 酸化黒鉛の製造方法 | |
| JP5776993B2 (ja) | 化合物半導体超微粒子の製造方法 | |
| JP6695129B2 (ja) | 酸化黒鉛誘導体 | |
| JP5822983B2 (ja) | 被覆銀超微粒子とその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16864224 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15775526 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016864224 Country of ref document: EP |