WO2017110851A1 - 1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 - Google Patents
1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 Download PDFInfo
- Publication number
- WO2017110851A1 WO2017110851A1 PCT/JP2016/088052 JP2016088052W WO2017110851A1 WO 2017110851 A1 WO2017110851 A1 WO 2017110851A1 JP 2016088052 W JP2016088052 W JP 2016088052W WO 2017110851 A1 WO2017110851 A1 WO 2017110851A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- base
- reactor
- production method
- ion
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/25—Preparation of halogenated hydrocarbons by splitting-off hydrogen halides from halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/013—Preparation of halogenated hydrocarbons by addition of halogens
- C07C17/04—Preparation of halogenated hydrocarbons by addition of halogens to unsaturated halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C19/00—Acyclic saturated compounds containing halogen atoms
- C07C19/08—Acyclic saturated compounds containing halogen atoms containing fluorine
- C07C19/10—Acyclic saturated compounds containing halogen atoms containing fluorine and chlorine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C21/00—Acyclic unsaturated compounds containing halogen atoms
- C07C21/02—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds
- C07C21/18—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds containing fluorine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a method for producing 1-chloro-2,3,3,3-tetrafluoropropene.
- HFO hydrofluoroolefin
- HFC hydrofluorocarbon
- Hydrochlorofluoroolefins such as hydrochlorofluoropropene and chlorofluoroolefins (CFO) having a bond.
- Patent Document 1 discloses 1,2-dichloro-2,3,3,3-tetrafluoropropane (CClH 2 -CFCl—CF 3 , HCFC-234bb), such as acid washing.
- a method of dehydrochlorinating at a temperature of 200 to 500 ° C. using carbon (activated carbon) that has been pretreated and supported with an alkali metal salt as a catalyst is disclosed.
- the present invention has been made to solve the above-mentioned problems, and efficiently uses 1-chloro-2,3,3-tetrafluoropropane (HCFC-234bb) as a raw material.
- An object is to provide an economically advantageous production method capable of obtaining 3,3,3-tetrafluoropropene (HCFO-1224yd).
- the present invention provides a method for producing 1224yd having the following configurations [1] to [12].
- [1] A process for producing 1224yd, wherein 234bb is dehydrochlorinated in the liquid phase in the presence of a base.
- [2] The process according to [1], wherein 234bb is dehydrochlorinated in a liquid phase in the presence of the solvent for dissolving the base and the base.
- [3] The method according to [1] or [2], wherein the base is at least one base selected from the group consisting of metal hydroxides, metal oxides, and metal carbonates.
- phase transfer catalyst is a quaternary ammonium salt.
- the quaternary ammonium salt is at least one selected from the group consisting of tetra-n-butylammonium chloride, tetra-n-butylammonium bromide, and methyltri-n-octylammonium chloride.
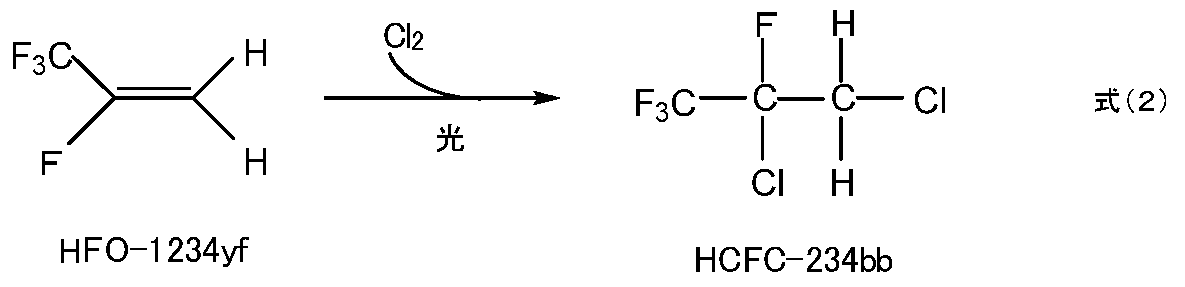
- Manufacturing method [11] The production method of [1] to [10], comprising a step of reacting 2,3,3,3-tetrafluoropropene (HCFO-1234yf) with chlorine to obtain 234bb.
- HCFO-1234yf 2,3,3,3-tetrafluoropropene
- 224bb can be used as a raw material to produce 1224yd with high conversion and high selectivity.
- a dehydrochlorination reaction can be stably maintained with few processes.
- the dehydrochlorination reaction can be performed at a relatively low temperature, the energy cost is low, and there is no need to use an inert gas for supplying the raw material, so the inert gas is separated and recovered from the product. This is economically advantageous because it does not require any process or equipment.
- the production method of 1224yd of the present invention is a method in which 234bb is dehydrochlorinated in the liquid phase in the presence of a base.
- the dehydrochlorination reaction is preferably carried out by bringing 234bb into contact with a base dissolved in a solvent in the presence of the base and a solvent dissolving the base. Moreover, it is preferable to make it react in the state in which the raw material and the product are uniformly distributed in the reaction system.
- the 1224yd obtained by the production method of the present invention may be a Z body, an E body, or a mixture of a Z body and an E body.
- Z-form 1224yd (Z) has higher chemical stability than E-form 1224yd (E), and is more preferable as a working medium for a heat cycle system.
- 1224yd which makes 1224yd (Z) essential can be manufactured efficiently.
- 1224yd having a higher content ratio of 1224yd (Z) than 1224yd (E) can be obtained.
- 234bb used in the production method of the present invention is a known compound known as a raw material or intermediate for producing a fluorine-containing compound, and can be produced by a known method.
- 234bb can be produced by reacting 2,3,3,3-tetrafluoropropene (HFO-1234yf) with chlorine.
- reaction (2) Since 1234yf, which is a starting material for the reaction represented by the formula (2) (hereinafter referred to as reaction (2)), has a very low global warming potential, demand for a working medium for a heat cycle system is increasing in recent years. . 1234yf can be produced by a known production method. Examples of the method for producing 1234yf include the method described in Japanese Patent No. 5713016.
- Reaction (2) is preferably performed under light irradiation from the viewpoint of increasing the reaction rate.
- the light used for irradiation is 1,1,2-trichloro-2,3,3,3-tetrafluoropropane (HCFC-224ba), which is a perchlorinated product of 1234yf, and 1,1,1,2-tetrachloro.
- HCFC-224ba 1,1,2-trichloro-2,3,3,3-tetrafluoropropane
- CFC-214bb 1,1,1,2-tetrachloro.
- CFC-214bb -2,3,3,3-tetrafluoropropane
- Visible light is light having a short wavelength limit of 360 to 400 nm and a long wavelength limit of 760 to 830 nm.
- the wavelength of light used for irradiation is preferably 400 to 750 nm, and more preferably 420 to 730 nm. Note that the light used for irradiation may include part of light having a wavelength of less than 400 nm or light having a wavelength of more than 750 nm.
- the reaction when a high energy ray having a wavelength of less than 400 nm is used for the reaction (2) using a high pressure mercury lamp, a low pressure mercury lamp, a metal halide lamp, etc., the reaction is easily activated excessively, and the reaction is controlled. Tends to be difficult. For this reason, it is preferable to use light having a wavelength of 400 nm or more.
- the light used for the reaction (2) may be light excluding light having a wavelength of less than 400 nm.
- the conversion rate refers to the ratio (mol%) of the amount of raw material consumed in the reaction to the total amount of raw material used in the reaction
- the selectivity refers to the amount of the target product generated relative to the total amount of product. It refers to the ratio (mol%).
- the reaction is activated and the reaction tends to proceed efficiently. Since light having a wavelength exceeding 750 nm hardly affects the selectivity of the target product 234bb, the light used for the reaction may include light having a wavelength exceeding 750 nm.
- examples of the light source that can efficiently irradiate the raw material with light having a wavelength of 400 to 750 nm include a fluorescent lamp, an incandescent lamp, and an LED light.
- Light having a wavelength of less than 400 nm included in light obtained from a fluorescent lamp or an incandescent lamp may be removed using a filter or the like.
- the method of irradiating the raw material of reaction (2) with light is not particularly limited as long as it can irradiate the reaction liquid including the raw material, the solvent and the product uniformly throughout the reaction time.
- a method of inserting a light source equipped with a jacket into the reaction solution and irradiating the raw material in the reaction solution with light from the inside of the reaction solution can be mentioned.
- the jacket is preferably made of a material that transmits at least light having a wavelength useful for the above reaction, is inert to the components contained in the reaction solution, and is not easily corroded by these components.
- the jacket preferably has a cooling means depending on the reaction temperature.
- 1234yf and chlorine may be separately supplied to the reactor, or may be supplied in a premixed state. Moreover, 1234yf and chlorine may be supplied in a gas state or in a liquid state, respectively.
- the ratio of the supplied 1234yf and chlorine is the ratio of the molar amount of chlorine to the molar amount of 1234yf (hereinafter referred to as “from the viewpoint of activating the reaction and increasing the selectivity of 234bb by suppressing by-products”).
- Chlorine / 1234yf is preferably 0.5 to 2.0, more preferably 0.8 to 1.2.
- Reaction (2) is usually carried out in a reactor containing a mixed solution in which raw materials are dissolved in a solvent.
- the reaction temperature is preferably from 0 to 100 ° C., more preferably from 5 to 60 ° C., from the viewpoint of increasing the reaction rate.
- the pressure in the reactor is preferably from 0 to 1 MPa, more preferably from 0.05 to 0.5 MPa, because it can be produced efficiently. In order to improve productivity, it is preferable to perform the reaction under pressurized conditions. In the present specification, the pressure is a gauge pressure unless otherwise specified.
- the material of the reactor is not particularly limited as long as the material is inert to the components contained in the reaction solution and is not easily corroded by these components.
- Examples of the material for the reactor include iron, nickel, alloys containing these as main components, glass, and resins. From the viewpoint of pressure resistance and corrosion resistance, a reaction vessel made of the above alloy in which the inner surface of the reactor is lined with a resin is preferable.
- Reaction (2) may be performed by any of semi-continuous, batch, and continuous methods.
- the reaction time can be appropriately adjusted by a general method according to each method.
- the feed of the raw material to the reactor may be a method of supplying each predetermined amount for each component, or a method of supplying each component as a mixture containing each predetermined amount.
- the supply of the raw material may be performed by diluting with an inert gas such as nitrogen as necessary.
- the raw material is added and supplied at a constant rate as each component of the raw material during the reaction or as a mixture obtained by mixing each component of the raw material.
- the addition of the raw material may be intermittent or continuous.
- the raw material is charged into the reactor together with a solvent before the reaction and used for the reaction.
- the raw material is continuously supplied from, for example, the lower part of the reactor charged with the solvent during the reaction.
- the product after completion of the reaction is continuously taken out from the upper part of the reactor, for example, by overflow.
- reaction (2) it is preferable to stir using a normal method, apparatus or the like in any of the semi-continuous method, batch method, and continuous method.
- the product obtained as described above contains 234bb as the target product, unreacted raw materials, solvent, by-products and the like.
- By-products include 1,1,2-trichloro-2,3,3,3-tetrafluoropropane (224ba), 1,1,1,2-tetrachloro-2,3,3,3-tetrafluoro. Examples include propane (214bb).
- a normal separation method such as a method in which chlorine is removed by washing with alkali and then a solvent and a by-product are removed by distillation.
- 234bb can be purified by distillation, and 234bb having a desired purity can be obtained by repeating distillation.
- the method for producing 1224yd according to the present invention is characterized in that 234bb is subjected to dehydrochlorination reaction in the liquid phase in the presence of a base according to the above reaction formula (1).
- 234bb obtained by the above-described method can be used as the starting material 234bb of the reaction represented by the formula (1) (hereinafter referred to as reaction (1)).
- reaction (1) the starting material 234bb of the reaction represented by the formula (1)
- 234bb obtained by the above-described method can be used.
- the acquisition method of 234bb is not limited to this.
- the starting material for the reaction (1) is conceptually preferable not containing impurities in addition to 234bb, but may contain impurities from the viewpoint of economy.
- the impurity is preferably a compound that does not inhibit the 234bb dehydrochlorination reaction.
- Examples of the impurity include 1234yf chlorinated substances other than 234bb, and examples thereof include 224ba and 214bb.
- the ratio of 234bb to the total amount of impurities and 234bb is preferably 85% by mass or more and less than 100% by mass, and more preferably 90% by mass or more and 99% by mass or less.
- the starting material for the reaction (1) is preferably one containing 234bb as a main component and containing at least one compound selected from 224ba and 214bb.
- the ratio of the total amount of impurities 224ba and 214bb is preferably more than 0 mol% and not more than 15 mol%, preferably 0.1 mol% or more and 7 mol% with respect to the total amount of impurities and 234bb. The following is more preferable.
- the base in the reaction (1) is not particularly limited as long as it is a base capable of executing the dehydrochlorination reaction in the reaction (1).
- the base is preferably at least one selected from the group consisting of metal hydroxides, metal oxides and metal carbonates.
- metal hydroxides examples include alkaline earth metal hydroxides and alkali metal hydroxides.
- alkaline earth metal hydroxide magnesium hydroxide, calcium hydroxide, strontium hydroxide, and barium hydroxide are preferable, and as the alkali metal hydroxide, lithium hydroxide, sodium hydroxide, and potassium hydroxide are preferable.
- the base may be one kind or a combination of two or more kinds.
- the metal oxide examples include alkali metal oxides and alkaline earth metal oxides.
- the alkali metal oxide sodium oxide is preferable, and as the alkaline earth metal oxide, calcium oxide is preferable.
- the metal oxide may be an oxide of one kind of metal or a composite oxide of two or more kinds of metals.
- metal carbonates examples include alkaline earth metal carbonates and alkali metal carbonates.
- Alkaline earth metal carbonates include beryllium, magnesium, calcium, strontium, barium or radium carbonate.
- Alkali metal carbonates include lithium, sodium, potassium, rubidium, cesium or francium carbonate.
- the base is preferably at least one selected from metal hydroxides, and more preferably potassium hydroxide, sodium hydroxide or a combination of potassium hydroxide and sodium hydroxide.
- the ratio of the base to 234bb is preferably 0.2 to 2.5 mol, more preferably 0.5 to 2.0 mol, relative to 1 mol of 234bb. preferable.
- the above base is present in the liquid phase in which the reaction (1) is performed.
- Reaction (1) is preferably carried out in a liquid phase in the presence of a base and a solvent.
- the solvent is not particularly limited as long as it can dissolve a predetermined amount of the base and does not contribute to the dehydrochlorination reaction.
- Water is preferable as the solvent for dissolving the base because it has high solubility in the base and is inactive to the dehydrochlorination reaction. That is, in the reaction (1), the base is preferably used as an aqueous solution of a base.
- the aqueous base solution is preferably an aqueous alkali metal hydroxide solution, more preferably an aqueous sodium hydroxide solution or an aqueous potassium hydroxide solution.
- the ratio of the mass of the base to the total mass of the solvent and the base is preferably 10 to 50% by mass, and more preferably 20 to 40% by mass.
- the amount of the base is not less than the above lower limit, a sufficient reaction rate can be easily obtained, and the target product can be easily separated by two-layer separation. If it is below the above upper limit value, the base is likely to be sufficiently dissolved, and the metal salt is difficult to precipitate, which is advantageous in an industrial process.
- a solution in which a base is dissolved in a solvent, 234bb, and a compound (shown by reference numeral 2) involved in other reaction used as necessary are used. 1 to carry out the reaction.
- generated composition containing 1224yd is collect
- the reactor 1 a known reactor used for a dehydrochlorination reaction in a liquid phase reaction is preferable.
- the material of the reactor 1 include iron, nickel, alloys containing these as main components, and glass. If necessary, the reactor 1 may be subjected to lining treatment such as resin lining and glass lining.
- the reaction temperature is the temperature in the reactor 1, and is preferably 40 to 100 ° C, more preferably 50 to 80 ° C. By setting the reaction temperature within the above range, the reaction rate and the reaction rate are improved, and by-products are easily suppressed.
- the pressure in the reactor during the reaction is preferably 0 to 10 MPa, more preferably 0.05 to 5 MPa, and further preferably 0.15 to 1 MPa.
- the pressure in the reactor is preferably not less than the vapor pressure of 234bb at the reaction temperature.
- Reaction (1) can be performed by any of semi-continuous, batch, and continuous methods.
- reaction time can be suitably adjusted with a general method by each system.
- the reaction time is preferably 1 to 50 hours for the batch type and 1 to 3000 seconds for the continuous type because the conversion rate of the raw material 234bb and the selectivity of 1224yd are easily controlled.
- Reaction (1) may be performed in the presence of a phase transfer catalyst as long as the reaction is not affected.
- a water-soluble organic solvent such as tetraglyme may be used as long as it does not affect the reaction.
- phase transfer catalyst examples include quaternary ammonium salts, quaternary phosphonium salts, quaternary arsonium salts, sulfonium salts, crown ethers, and the like. Quaternary ammonium salts, quaternary phosphonium salts, quaternary arsonium Salts and sulfonium salts are preferred, and quaternary ammonium salts are more preferred.
- Examples of the quaternary ammonium salt include compounds represented by the following formula (i).
- R 11 to R 14 each independently represents a monovalent hydrocarbon group or a monovalent hydrocarbon group to which a functional group inert to the reaction is bonded, and Y ⁇ represents Represents an anion.
- R 11 to R 14 are hydrocarbon groups, examples thereof include an alkyl group, a cycloalkyl group, an alkenyl group, a cycloalkenyl group, and an aryl group, and an alkyl group and an aryl group are preferable.
- R 11 carbon atoms ⁇ R 14 is preferably 4 to 100, 6 to 30 is more preferred.
- R 11 to R 14 may be the same group or different groups.
- R 11 to R 14 are a monovalent hydrocarbon group bonded with a functional group inert to the reaction is appropriately selected depending on the reaction conditions, but a halogen atom, an alkoxycarbonyl group, an acyloxy group Group, nitrile group, acyl group, carboxyl group, alkoxyl group and the like.
- Fluorine ion, chlorine ion, bromine ion, iodine ion, hydrogen sulfate ion, hydroxide ion are preferable, fluorine ion, chlorine ion, bromine ion Ions, iodine ions, and hydroxide ions are more preferable, and chlorine ions or bromine ions are more preferable.
- the compound represented by the above formula (i) is preferably a combination of the following quaternary ammonium (R 11 R 12 R 13 R 14 N + ) and the following Y ⁇ from the viewpoint of versatility and reactivity.
- Y ⁇ Fluorine ion, chlorine ion, bromine ion, iodine ion, hydroxide ion.
- the quaternary ammonium salt is at least one selected from the group consisting of tetra-n-butylammonium chloride (TBAC), tetra-n-butylammonium bromide (TBAB), and methyltri-n-octylammonium chloride (TOMAC). Preferably there is.
- TBAC tetra-n-butylammonium chloride
- TBAB tetra-n-butylammonium bromide
- TOMAC methyltri-n-octylammonium chloride
- Examples of the quaternary phosphonium salt include compounds represented by the following formula (ii).
- R 21 to R 24 each independently represents a monovalent hydrocarbon group, and Y ⁇ represents an anion.
- R 21 ⁇ R 24 may each be the same group or may be different groups.
- Examples of the hydrocarbon group for R 21 to R 24 include an alkyl group, a cycloalkyl group, an alkenyl group, a cycloalkenyl group, an aryl group, and the like, and an alkyl group and an aryl group are preferable.
- tetraethylphosphonium tetra-n-butylphosphonium, ethyltri-n-octylphosphonium, cetyltriethylphosphonium, cetyltri-n- Examples thereof include butyl phosphonium, n-butyl triphenyl phosphonium, n-amyl triphenyl phosphonium, methyl triphenyl phosphonium, benzyl triphenyl phosphonium, and tetraphenyl phosphonium.
- Y ⁇ includes chlorine ion, fluorine ion, bromine ion, iodine ion, sulfate ion, nitrate ion, phosphate ion, perchlorate ion, hydrogen sulfate ion, hydroxide ion, acetate ion, benzoate ion, and benzenesulfone. Acid ion, p-toluenesulfonic acid ion and the like, and fluorine ion, chlorine ion and bromine ion are preferable.
- Examples of the quaternary arsonium salt include compounds represented by the following formula (iii).
- R 31 to R 34 are the same as R 21 to R 24 in formula (ii), and the preferred embodiments are also the same.
- Y ⁇ represents an anion.
- Y ⁇ is preferably a halogen ion, more preferably a fluorine ion, a chlorine ion or a bromine ion.
- Examples of the quaternary arsonium salt represented by the above formula (iii) include triphenylmethylarsonium fluoride, tetraphenylarsonium fluoride, triphenylmethylarsonium chloride, tetraphenylarsonium chloride, tetraphenylarsonium bromide and the like. Is mentioned.
- triphenylmethylarsonium chloride is preferable.
- Examples of the sulfonium salt include compounds represented by the following formula (iv).
- R 41 to R 43 and Y ⁇ are the same as R 31 to R 34 and Y ⁇ in formula (iii), and preferred embodiments are also the same.
- Examples of the sulfonium salt represented by the above formula (iv) include di-n-butylmethylsulfonium iodide, tri-n-butylsulfonium tetrafluoroborate, dihexylmethylsulfonium iodide, dicyclohexylmethylsulfonium iodide, dodecylmethylethylsulfonium. Examples include chloride and tris (diethylamino) sulfonium difluorotrimethyl silicate. As the sulfonium salt, dodecylmethylethylsulfonium chloride is preferable.
- crown ether examples include 18-crown-6, dibenzo-18-crown-6, and dicyclohexyl-18-crown-6.
- the amount of the phase transfer catalyst used is preferably 0.01 to 10 parts by weight, more preferably 0.05 to 5.0 parts by weight, and more preferably 0.1 to 1.0 parts by weight with respect to 100 parts by weight of 234bb. Further preferred. When the amount of the phase transfer catalyst is within the above range, a sufficient reaction rate is easily obtained. If it is out of the above range, the reaction promoting effect is difficult to obtain, and it tends to be disadvantageous in terms of cost. When using a phase transfer catalyst, it is preferable to mix a phase transfer catalyst with 234bb beforehand and to supply to a reactor in the state of a liquid mixture with 234bb.
- the materials of the reaction process, the reaction apparatus, and the reactor may be the same as those in the case where the phase transfer catalyst is not used. Further, the reaction conditions such as the concentration of the base, the amount used, and the reaction temperature may be the same as in the case where no phase transfer catalyst is used.
- reaction (1) for example, 234bb, a base, a solvent as necessary, and a compound involved in the reaction such as a phase transfer catalyst as needed are supplied to the reactor, and stirred so that they are uniform. It is possible to proceed by setting the temperature and pressure conditions as desired.
- the reaction system is separated into an aqueous phase and an organic phase.
- the reaction (1) is performed by compatibilizing the aqueous phase containing the base and the organic phase using, for example, a water-soluble organic solvent such as tetraglyme instead of the phase transfer catalyst. Can do.
- a water-soluble organic solvent it is preferable to sufficiently stir in order to make the compounds involved in the reaction in the reaction system uniform.
- reaction liquid after completion of the reaction is allowed to stand and is separated into an organic phase and an aqueous phase
- by-products other than unreacted 234bb and target product 1224yd may be contained in the organic phase.
- 1-chloro-3,3,3-trifluoropropyne obtained by further dehydrochlorination of 1224yd can be mentioned.
- 1,1-dichloro-2,3,3,3-tetrafluoropropene (CFO-1214ya) or the like may be contained as a by-product. .
- the product obtained in the reaction (1) includes unreacted 234bb, the by-product and the like in addition to the target product 1224yd. Except for the target product, 1224yd, it can be easily removed by a method such as distillation and separation.
- 1224yd useful as a working medium for a thermal cycle system having a low global warming potential is obtained from 234bb in an economically advantageous manner industrially feasible. Can be manufactured.
- Production Examples 1 to 6 are production examples of 234bb.
- Examples 1 to 4 are examples in the production of 1224yd.
- composition analysis of the obtained products was performed using gas chromatography (GC).
- GC gas chromatography
- DB-1301 trade name, manufactured by Agilent Technologies, length 60 m ⁇ inner diameter 250 ⁇ m ⁇ thickness 1 ⁇ m
- the analysis results of the obtained product are shown in Table 1 or Table 2.
- 1234yf was supplied into the reactor at a flow rate of 245 g / hr, and chlorine gas was supplied into the reactor at a flow rate of 152 g / hr.
- reaction heat was generated, the temperature in the reactor rose to 7.6 ° C., and the pressure in the reactor rose to 0.08 MPa.
- the reaction was continued for 1 hour while supplying 1234yf and chlorine gas at the above flow rates.
- the supply of 1234yf and chlorine was stopped, and the pressure in the reactor The light irradiation was continued until became normal pressure.
- reaction solution was neutralized with a 20% by mass aqueous potassium hydrogen carbonate solution, and then a liquid separation operation was performed. After standing, 1734 g of product (1) was recovered from the separated lower layer. The product (1) was distilled by a normal operation to obtain 234bb with a purity of 99.8%.
- Production Example 2 A solenoid valve that can be automatically opened and closed by a timer was connected to the reactor used in Production Example 1. 1800 g of 234bb (purity 99.8 g) obtained in Production Example 1 was used as a solvent.
- 1234yf and chlorine gas were supplied in the same manner as in Production Example 1 while irradiating visible light with the fluorescent lamp of Production Example 1.
- 1234yf was adjusted so that the flow rate was gradually increased to 1070 g / hour over several minutes to 10 and several minutes after the start of supply, and supply was continued at this flow rate after reaching 1070 g / hour.
- the chlorine gas was adjusted stepwise so that the flow rate was 666 g / hour, and the supply was continued at this flow rate after reaching 666 g / hour.
- the reaction was carried out for 8 hours while continuously extracting the product so that the amount of liquid in the reactor was constant.
- the total supply amount of 1234yf was 8327 g, and the total supply amount of chlorine gas was 5028 g.
- the temperature in the reactor was 36 to 39 ° C., and the pressure in the reactor was 0.18 MPa.
- the total supply amount of 1234yf was 6420 g, and the total supply amount of chlorine gas was 3990 g.
- the temperature in the reactor was 14 to 15 ° C., and the pressure in the reactor was 0.06 MPa.
- the same operation as in Production Example 2 was performed to obtain 10110 g of product (5).
- the reaction was continued for 1 hour while supplying 1234yf and chlorine gas at the above flow rates. After confirming that the total supply amount of 1234yf was 245g and the total supply amount of chlorine gas was 152g, the supply was stopped, and the reactor Irradiation with ultraviolet light was continued until the internal pressure reached normal pressure.
- Table 1 shows the GC analysis results of the compositions (1) to (6) obtained in Production Examples 1 to 6.
- the conversion rate of 1234yf is the ratio (unit: mol%) of the amount of 1234yf consumed in the reaction to the total amount of 1234yf supplied to the reactor.
- the selectivity of each compound is the ratio of each compound to the total amount of all products (unit: mol%).
- the conversion rate of 1234yf and the selectivity of each compound in Production Examples 2 to 5 in which the reaction mode is a continuous type were calculated from the GC analysis results of the extracted liquid after 5 hours from the start of the reaction.
- the selectivity of 234bb was determined as the amount (mol) of 234bb produced by removing the amount of 234bb initially supplied as a solvent from 234bb in the product.
- Example 1 A tube reactor made of a fluororesin having an outer diameter of 1/2 inch and a length of 30 m (inner volume: 1 L) equipped with a Noritake resin static mixer was used as a reactor. The reactor was installed in a thermostat and the reaction temperature was 60 ° C. A 20% by mass aqueous KOH solution was supplied to the reactor at a flow rate of 4550 g / h, and a 234bb mixed liquid in which tetra-n-butylammonium bromide (TBAB) was mixed so as to be 1% by mass was supplied to the reactor at a flow rate of 1500 g / h. Supplied.
- TBAB tetra-n-butylammonium bromide
- 234bb used in Example 1 is 234bb (purity 99.8%) obtained by distillation in Production Example 1.
- the product obtained from the reactor was extracted into a tank with a normal pressure jacket kept at 60 ° C.
- the extracted product was gasified, taken out from the gas phase portion, and collected in a jacketed tank cooled to ⁇ 20 ° C.
- the collected product was gasified at 60 ° C., sampled, and subjected to GC analysis.
- Example 2 The reaction was carried out in the same manner as in Example 1 except that the temperature in the reactor was changed from 60 ° C to 70 ° C. The product obtained from the reactor was recovered by the same operation as in Example 1, and then GC analysis was performed.
- Example 3 The temperature in the reactor is 70 ° C., the supply rate of 20% by mass KOH aqueous solution to the reactor is 6304 g / h, and the supply rate to the 234bb reactor mixed with TBAB at 1% by mass is 2160 g / h.
- the reaction was conducted in the same manner as in Example 1 except that.
- the obtained product was recovered by the same operation as in Example 1, and then GC analysis was performed.
- Table 2 shows the reaction conditions of Examples 1 to 3 and the GC analysis results of the obtained compositions.
- the conversion rate of 234bb is the ratio (mol%) of the amount of 234bb consumed in the reaction to the total amount of 234bb supplied to the reactor.
- the selectivity of each compound is the ratio (mol%) of each component generated to the converted 234bb, and is calculated from the GC analysis result of the gas vaporized at 40 ° C.
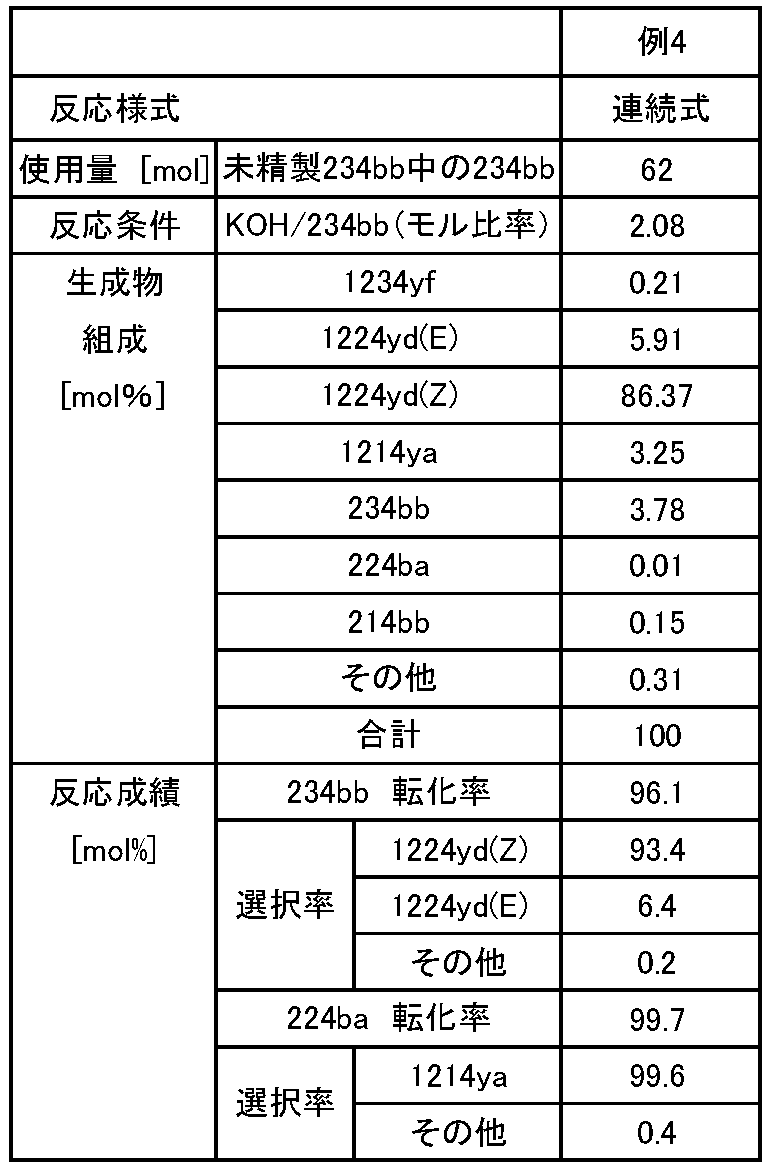
- Example 4 The product (3) obtained in Production Example 3 was used in place of 234bb of Example 1 and reacted in the same manner as in Example 1. The obtained product was recovered in the same manner as in Example 1 and subjected to GC analysis.
- Table 3 shows the reaction conditions of Example 4 and the GC analysis results of the resulting composition.
- the selectivity of 1224yd was calculated as a product derived from 234bb
- the selectivity of 1214ya was calculated as a product derived from 224ba.
- the desired 1224yd can be produced with high selectivity and high yield by suppressing the formation of by-products.
- the selectivity of 1224yd (Z) which is high in chemical stability and useful as a working medium for a heat cycle system, is higher than that of 1224yd (E)
- the method for producing 1224yd of the present invention is suitable for the operation for a heat cycle system. Industrially advantageous as a method for producing the medium.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description
また、1224ydは、二重結合を有する炭素に結合された置換基の位置により、幾何異性体であるZ体とE体が存在する。本明細書中で、Z体とE体が存在する化合物について、特に断らずに化合物名や化合物の略称を用いた場合には、Z体もしくはE体、またはZ体とE体の任意の割合の混合物を示す。化合物名や化合物の略称の後ろに(Z)または(E)を付した場合には、それぞれの化合物のZ体またはE体であることを示す。
このように、特許文献1に記載の方法は、経済的に有利な製造方法とはいえなかった。
また、1224ydは、二重結合を有する炭素に結合された置換基の位置により、幾何異性体であるZ体とE体が存在する。本明細書中で、Z体とE体が存在する化合物について、特に断らずに化合物名や化合物の略称を用いた場合には、Z体もしくはE体、またはZ体とE体の任意の割合の混合物を示す。化合物名や化合物の略称の後ろに(Z)または(E)を付した場合には、それぞれの化合物のZ体またはE体であることを示す。
[1]234bbを、塩基の存在下に液相中で脱塩化水素反応させる1224ydの製造方法。
[2]234bbを、前記塩基を溶解する溶媒、および、前記塩基の存在下に液相中で脱塩化水素反応させる、[1]の製造方法。
[3]前記塩基は、金属水酸化物、金属酸化物、および金属炭酸塩からなる群より選ばれる少なくとも1種の塩基である、[1]または[2]の製造方法。
[4]前記塩基を、前記234bbの1モルに対して0.2~2.5モルの割合で用いる、[1]~[3]の製造方法。
[5]前記脱塩化水素反応の反応温度は40~100℃である、[1]~[4]の製造方法。
[6]前記溶媒が、水である、[2]~[5]の製造方法。
[7]前記塩基の量が、前記溶媒と前記塩基の総質量に対して、10~50質量%である[2]~[6]の製造方法。
[8]前記脱塩化水素反応を相間移動触媒の存在下に行う、[1]~[7]の製造方法。
[9]前記相間移動触媒は第4級アンモニウム塩である、[8]の製造方法。
[10]前記第4級アンモニウム塩は、テトラ-n-ブチルアンモニウムクロリド、テトラ-n-ブチルアンモニウムブロミド、およびメチルトリ-n-オクチルアンモニウムクロリドからなる群より選ばれる少なくとも1種である、[9]の製造方法。
[11]2,3,3,3-テトラフルオロプロペン(HCFO-1234yf)と塩素を反応させて、234bbを得る工程を有する、[1]~[10]の製造方法。
[12]前記1234yfと塩素との反応を、400~750nmの波長域の光の照射下で行わせる、[11]の製造方法。
本発明の製造方法に係る234bbの脱塩化水素反応は、下式(1)で示される。

本発明の製造方法に用いる234bbは、含フッ素化合物の製造原料または中間体として知られる公知の化合物であり、公知の方法により製造できる。例えば、下式(2)に示すように、2,3,3,3-テトラフルオロプロペン(HFO-1234yf)と塩素を反応させることにより、234bbを製造できる。
バッチ式の場合は、原料は反応前に反応器に溶媒などとともに仕込まれ、反応に供される。
本発明の1224ydの製造方法は、塩基の存在下に液相中で234bbを、前記反応式(1)にしたがって脱塩化水素反応させることを特徴とする。
本発明の製造方法において、式(1)で示される反応(以下、反応(1)という。)の出発物質である234bbとしては、前述の方法で得られた234bbを用いることができる。なお、234bbの入手方法はこれに限定されない。
また金属酸化物は、1種の金属の酸化物であってもよく、2種以上の金属の複合酸化物であってもよい。

第4級アンモニウム(R11R12R13R14N+):テトラメチルアンモニウム、テトラエチルアンモニウム、テトラ-n-プロピルアンモニウム、テトラ-n-ブチルアンモニウム、メチルトリ-n-オクチルアンモニウム。
Y-:フッ素イオン、塩素イオン、臭素イオン、ヨウ素イオン、水酸化物イオン。


第4級アルソニウム塩としては、トリフェニルメチルアルソニウムクロライドが好ましい。

スルホニウム塩としては、ドデシルメチルエチルスルホニウムクロライドが好ましい。
以下の製造例および例において、得られた生成物の組成分析は、ガスクロマトグラフィー(GC)を用いて行った。カラムは、DB-1301(商品名、アジレント・テクノロジー株式会社製、長さ60m×内径250μm×厚み1μm)を用いた。得られた生成物の分析結果を表1または表2に示す。
公知の方法で得られた1234yfを、塩素化して234bbを製造した。
まず、光源からの光を透過する石英管およびジャケットを取り付けたステンンレス製反応器(内容積2.3L)を、0℃に冷却した。この反応器内に、溶媒として1395gの四塩化炭素(CCl4)を入れた後、蛍光灯(東芝社製、商品名:ネオコンパクト、電球型:EFP12EL、出力12W)からの可視光を照射しながら、1234yfを毎時245gの流量で反応器内に供給し、塩素ガスを毎時152gの流量で、反応器内に供給した。反応の進行に伴って反応熱が生じるとともに、反応器内の温度は7.6℃に上昇し、反応器内の圧力は0.08MPaに上昇した。上記流量で1234yfおよび塩素ガスをそれぞれ供給しながら1時間反応を続け、1234yfの245gおよび塩素ガスの152gが供給されたことを確認した後、1234yfおよび塩素の供給を停止し、反応器内の圧力が常圧となるまで、光照射を継続した。
製造例1で使用した反応器に、タイマーによって自動的に開閉可能な電磁弁を接続した。製造例1で得られた234bb(純度99.8g)の1800gを溶媒として用いた。
溶媒として、製造例2で得られた生成物(2)の1917gを用いた。製造例2と同様の方法で流量を調整した1234yfと塩素ガスを反応器内に8.5時間供給した。この時の1234yfの供給速度は、毎時1221gであり、塩素ガスの供給速度は毎時760gであった。1234yfの全供給量は9991gであり、塩素ガスの全供給量は6154gであった。反応器内の温度は49~51℃、反応器内の圧力は0.24MPaであった。製造例2と同様にして17884gの生成物(3)を得た。
溶媒として、製造例3で得られた生成物(3)の2468gを用いた。製造例2と同様の方法で流量を調整した1234yfと塩素ガスを反応器内に6時間供給した。この時の1234yfの供給速度は、毎時1068gであり、塩素ガスの供給速度は毎時660gであった。1234yfと塩素ガスの供給量を前記の通りとする以外は、製造例2と同様の操作を行った。1234yfの全供給量は6669gであり、塩素ガスの全供給量は4209gであった。反応器内の温度は53~57℃、反応器内の圧力は0.24MPaであった。製造例2と同様の操作を行い、13098gの生成物(4)を得た。
反応器の内容積を4.5Lとし、使用する光源をLEDランプ(三菱電機社製、電球型:LHT15D-G-E39、出力15W)とし、溶媒として、製造例4で得られた生成物(4)の3000gを用いた。製造例2と同様の方法で流量を調整した1234yfと塩素ガスを反応器内に6時間供給した。この時の1234yfの供給速度は、毎時1069gであり、塩素ガスの供給速度は毎時666gであった。1234yfと塩素ガスの供給速度を前記の通りとする以外は、製造例2と同様の操作を行った。1234yfの全供給量は6420gであり、塩素ガスの全供給量は3990gであった。反応器内の温度は14~15℃、反応器内の圧力は0.06MPaであった。製造例2と同様の操作を行い、10110gの生成物(5)を得た。
製造例1で用いたものと同じ反応器に、CCl4の1323gを入れた後、高圧水銀灯(英光社製、出力400W)からの、波長250~320nmおよび波長360nmに特徴的な線スペクトルを持つ紫外光を照射しながら、1234yfを毎時245gの流量で反応器内に供給し、塩素ガスを毎時152gの流量で反応器内に供給した。反応の進行に伴い、反応熱が生じるとともに、反応器内の温度は9.8℃に上昇し、反応器内の圧力は0.14MPaに上昇した。上記流量で1234yfおよび塩素ガスをそれぞれ供給しながら1時間反応を続け、1234yfの全供給量が245gおよび塩素ガスの全供給量が152gとなったことを確認した後、供給を停止し、反応器内の圧力が常圧となるまで、紫外光の照射を継続した。
表1中、1234yfの転化率は、反応器に供給した1234yfの全量に対する、反応で消費された1234yfの量の割合(単位:モル%)である。また、各化合物の選択率は、全ての生成物の合計量に対する各化合物の割合(単位:モル%)である。

ノリタケ社製樹脂スタティックミキサーを設置した、外径1/2インチ、長さ30m(内容積1L)のフッ素系樹脂製のチューブリアクターを反応器として用いた。恒温槽内に反応器を設置し、反応温度を60℃とした。20質量%のKOH水溶液を毎時4550gの流量で反応器に供給し、1質量%となるようにテトラ-n-ブチルアンモニウムブロミド(TBAB)を混合した234bb混合液を毎時1500gの流量で反応器に供給した。次いで、反応器の出口に設置した圧力調節弁で、反応器内の圧力を0.2MPaとなるように調整し、8時間連続的に反応させた。例1で用いた234bbは、製造例1で蒸留して得られた234bb(純度99.8%)である。
反応器内の温度を、60℃から70℃へ変更したこと以外は、例1と同様にして、反応させた。反応器から得られた生成物を、例1と同様の操作により回収した後、GC分析を行った。
反応器内の温度を70℃として、20質量%のKOH水溶液の反応器への供給速度を毎時6304gとし、TBABを1質量%となるように混合した234bbの反応器への供給速度を毎時2160gとした以外は実施例1と同様にして反応させた。得られた生成物を、例1と同様の操作により回収した後、GC分析を行った。

製造例3で得られた生成物(3)を例1の234bbの代わりに使用し、例1と同様に反応させた。得られた生成物を、例1と同様にして回収し、GC分析を行った。
表3においては、234bbに由来する生成物として1224ydの選択率を算出し、224baに由来する生成物として1214yaの選択率を算出した。
また、1224yd(E)に比べて、化学的安定性が高く熱サイクルシステム用作動媒体として有用な1224yd(Z)の選択率が高いので、本発明の1224ydの製造方法は、熱サイクルシステム用作動媒体の製造方法として、工業的に有利である。
Claims (12)
- 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンを、塩基の存在下に液相中で脱塩化水素反応させる1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法。
- 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンを、前記塩基を溶解する溶媒、および、前記塩基の存在下に液相中で脱塩化水素反応させる、請求項1に記載の製造方法。
- 前記塩基は、金属水酸化物、金属酸化物、および金属炭酸塩からなる群より選ばれる少なくとも1種の塩基である、請求項1または2に記載の製造方法。
- 前記塩基を、前記1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンの1モルに対して0.2~2.5モルの割合で用いる、請求項1~3のいずれか1項に記載の製造方法。
- 前記脱塩化水素反応の反応温度は40~100℃である、請求項1~4のいずれか1項に記載の製造方法。
- 前記溶媒が、水である請求項2~5のいずれか1項に記載の製造方法。
- 前記塩基の量が、前記溶媒と前記塩基の総質量に対して、10~50質量%である請求項2~6のいずれか1項に記載の製造方法。
- 前記脱塩化水素反応を相間移動触媒の存在下に行う、請求項1~7のいずれか1項に記載の製造方法。
- 前記相間移動触媒は第4級アンモニウム塩である、請求項8に記載の製造方法。
- 前記第4級アンモニウム塩は、テトラ-n-ブチルアンモニウムクロリド、テトラ-n-ブチルアンモニウムブロミド、およびメチルトリ-n-オクチルアンモニウムクロリドからなる群より選ばれる少なくとも1種である、請求項9に記載の製造方法。
- 2,3,3,3-テトラフルオロプロペンと塩素を反応させて、1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンを得る工程を有する、請求項1~10のいずれか1項に記載の製造方法。
- 前記2,3,3,3-テトラフルオロプロペンと塩素との反応を、400~750nmの波長域の光の照射下で行わせる、請求項11に記載の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16878738.0A EP3395789B1 (en) | 2015-12-25 | 2016-12-21 | Manufacturing method of 1-chloro-2,3,3,3-tetrafluoropropene |
| JP2017558178A JP6812988B2 (ja) | 2015-12-25 | 2016-12-21 | 1−クロロ−2,3,3,3−テトラフルオロプロペンの製造方法 |
| CN201680075717.XA CN108473397A (zh) | 2015-12-25 | 2016-12-21 | 1-氯-2,3,3,3-四氟丙烯的制造方法 |
| US15/929,024 US10399915B2 (en) | 2015-12-25 | 2018-06-20 | Manufacturing method of 1-chloro-2,3,3,3-tetrafluoropropene |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015254136 | 2015-12-25 | ||
| JP2015-254136 | 2015-12-25 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/929,024 Continuation US10399915B2 (en) | 2015-12-25 | 2018-06-20 | Manufacturing method of 1-chloro-2,3,3,3-tetrafluoropropene |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017110851A1 true WO2017110851A1 (ja) | 2017-06-29 |
Family
ID=59089444
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/088052 Ceased WO2017110851A1 (ja) | 2015-12-25 | 2016-12-21 | 1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10399915B2 (ja) |
| EP (1) | EP3395789B1 (ja) |
| JP (1) | JP6812988B2 (ja) |
| CN (1) | CN108473397A (ja) |
| WO (1) | WO2017110851A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018079726A1 (ja) * | 2016-10-28 | 2018-05-03 | 旭硝子株式会社 | テトラフルオロプロペンの製造方法 |
| WO2019098337A1 (ja) | 2017-11-20 | 2019-05-23 | Agc株式会社 | (z)-1-クロロ-2,3,3,3-テトラフルオロプロペンの精製方法 |
| WO2019168115A1 (ja) * | 2018-03-02 | 2019-09-06 | Agc株式会社 | 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンの製造方法及び1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 |
| WO2019189024A1 (ja) * | 2018-03-30 | 2019-10-03 | Agc株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| WO2019203318A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2019203319A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2020026990A1 (ja) * | 2018-07-30 | 2020-02-06 | Agc株式会社 | 精製含フッ素不飽和炭化水素の製造方法 |
| WO2020036836A1 (en) | 2018-08-13 | 2020-02-20 | The Chemours Company Fc, Llc | Compositions and processes for producing chlorofluoroalkenes |
| WO2020066754A1 (ja) * | 2018-09-25 | 2020-04-02 | セントラル硝子株式会社 | 化合物の製造方法 |
| CN112125776A (zh) * | 2020-10-20 | 2020-12-25 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018021275A1 (ja) | 2016-07-29 | 2018-02-01 | 旭硝子株式会社 | 熱サイクル用作動媒体 |
| JP6958596B2 (ja) * | 2019-07-01 | 2021-11-02 | ダイキン工業株式会社 | アルカンの製造方法 |
| JP7722167B2 (ja) * | 2020-12-22 | 2025-08-13 | Agc株式会社 | ハイドロハロオレフィン組成物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56150027A (en) * | 1980-03-28 | 1981-11-20 | Hoechst Ag | 2,3-dichloro-2-trifluoromethyl-1,1,1,3,4,4,5,5,5- nonafluoropentane and its manufacture |
| JP2005504097A (ja) * | 2001-09-25 | 2005-02-10 | ハネウェル・インターナショナル・インコーポレーテッド | フルオロオレフィンの製造方法 |
| JP2014513673A (ja) * | 2011-02-21 | 2014-06-05 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | ヒドロクロロフルオロカーボンの触媒接触脱塩化水素化 |
| JP2014237627A (ja) * | 2013-05-09 | 2014-12-18 | セントラル硝子株式会社 | 2−クロロ−1,3,3,3−テトラフルオロプロペンの製造方法 |
| JP2015120670A (ja) * | 2013-12-24 | 2015-07-02 | 旭硝子株式会社 | 1−クロロ−1,2−ジフルオロエチレンの製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6037015B2 (ja) | 1980-06-27 | 1985-08-23 | 横河電機株式会社 | 移送量制御装置 |
| US5316690A (en) * | 1991-04-18 | 1994-05-31 | Allied Signal Inc. | Hydrochlorofluorocarbons having OH rate constants which do not contribute substantially to ozone depletion and global warming |
| US20080076950A1 (en) * | 2004-12-22 | 2008-03-27 | Velliyur Nott Mallikarjuna Rao | Photochlorination And Dehydrohalogenation Process For Preparation Of Olefinic Compounds |
| US8002968B2 (en) * | 2005-11-14 | 2011-08-23 | Statoil Canada Ltd. | Process for treating a heavy hydrocarbon feedstock and a product obtained therefrom |
| KR100771636B1 (ko) * | 2006-07-04 | 2007-10-31 | 엘지전자 주식회사 | 프로젝션 시스템 |
| GB0625214D0 (en) * | 2006-12-19 | 2007-01-24 | Ineos Fluor Holdings Ltd | Process |
| US7700815B2 (en) * | 2007-09-28 | 2010-04-20 | Honeywell International Inc | Method for producing fluorinated organic compounds |
| EP2586762B1 (en) | 2010-06-23 | 2019-08-07 | AGC Inc. | Production method for 1,1-dichloro-2,3,3,3-tetra-fluoropropene and 2,3,3,3-tetrafluoropropene |
-
2016
- 2016-12-21 WO PCT/JP2016/088052 patent/WO2017110851A1/ja not_active Ceased
- 2016-12-21 CN CN201680075717.XA patent/CN108473397A/zh active Pending
- 2016-12-21 EP EP16878738.0A patent/EP3395789B1/en active Active
- 2016-12-21 JP JP2017558178A patent/JP6812988B2/ja active Active
-
2018
- 2018-06-20 US US15/929,024 patent/US10399915B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56150027A (en) * | 1980-03-28 | 1981-11-20 | Hoechst Ag | 2,3-dichloro-2-trifluoromethyl-1,1,1,3,4,4,5,5,5- nonafluoropentane and its manufacture |
| JP2005504097A (ja) * | 2001-09-25 | 2005-02-10 | ハネウェル・インターナショナル・インコーポレーテッド | フルオロオレフィンの製造方法 |
| JP2014513673A (ja) * | 2011-02-21 | 2014-06-05 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | ヒドロクロロフルオロカーボンの触媒接触脱塩化水素化 |
| JP2014237627A (ja) * | 2013-05-09 | 2014-12-18 | セントラル硝子株式会社 | 2−クロロ−1,3,3,3−テトラフルオロプロペンの製造方法 |
| JP2015120670A (ja) * | 2013-12-24 | 2015-07-02 | 旭硝子株式会社 | 1−クロロ−1,2−ジフルオロエチレンの製造方法 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018079726A1 (ja) * | 2016-10-28 | 2018-05-03 | 旭硝子株式会社 | テトラフルオロプロペンの製造方法 |
| WO2019098337A1 (ja) | 2017-11-20 | 2019-05-23 | Agc株式会社 | (z)-1-クロロ-2,3,3,3-テトラフルオロプロペンの精製方法 |
| US10934235B2 (en) | 2017-11-20 | 2021-03-02 | AGC Inc. | Method of purifying (Z)-1-chloro-2,3,3,3-tetrafluoropropene |
| WO2019168115A1 (ja) * | 2018-03-02 | 2019-09-06 | Agc株式会社 | 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンの製造方法及び1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 |
| WO2019189024A1 (ja) * | 2018-03-30 | 2019-10-03 | Agc株式会社 | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 |
| JPWO2019189024A1 (ja) * | 2018-03-30 | 2021-04-08 | Agc株式会社 | 1−クロロ−2,3,3−トリフルオロプロペンの製造方法 |
| WO2019203318A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2019203319A1 (ja) * | 2018-04-19 | 2019-10-24 | Agc株式会社 | フルオロオレフィンの製造方法 |
| WO2020026990A1 (ja) * | 2018-07-30 | 2020-02-06 | Agc株式会社 | 精製含フッ素不飽和炭化水素の製造方法 |
| US11565987B2 (en) | 2018-08-13 | 2023-01-31 | The Chemours Company Fc, Llc | Compositions and processes for producing chlorofluoroalkenes |
| WO2020036836A1 (en) | 2018-08-13 | 2020-02-20 | The Chemours Company Fc, Llc | Compositions and processes for producing chlorofluoroalkenes |
| EP4714933A2 (en) | 2018-08-13 | 2026-03-25 | The Chemours Company FC, LLC | Compositions and processes for producing chlorofluoroalkenes |
| WO2020066754A1 (ja) * | 2018-09-25 | 2020-04-02 | セントラル硝子株式会社 | 化合物の製造方法 |
| CN112125776B (zh) * | 2020-10-20 | 2021-06-29 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
| US12304879B2 (en) | 2020-10-20 | 2025-05-20 | Zibo Rhema International Inc. | Preparation method of 1-chloro-2,3,3-trifluoropropene |
| CN112125776A (zh) * | 2020-10-20 | 2020-12-25 | 淄博雷玛国际贸易有限公司 | 一种1-氯-2,3,3-三氟丙烯的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2017110851A1 (ja) | 2018-10-11 |
| JP6812988B2 (ja) | 2021-01-13 |
| US10399915B2 (en) | 2019-09-03 |
| EP3395789B1 (en) | 2020-11-18 |
| CN108473397A (zh) | 2018-08-31 |
| US20180297918A1 (en) | 2018-10-18 |
| EP3395789A1 (en) | 2018-10-31 |
| EP3395789A4 (en) | 2019-08-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6812988B2 (ja) | 1−クロロ−2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP7081596B2 (ja) | 2-クロロ-1,1,1,2-テトラフルオロプロパンおよび/または3-クロロ-1,1,1,2-テトラフルオロプロパンの製造方法、ならびに2,3,3,3-テトラフルオロプロペンの製造方法 | |
| JP6620815B2 (ja) | 1−クロロ−2,3,3−トリフルオロプロペンの製造方法 | |
| JP7631408B2 (ja) | ヒドロクロロフルオロカーボンの脱ハロゲン化水素 | |
| JP2015120670A (ja) | 1−クロロ−1,2−ジフルオロエチレンの製造方法 | |
| JP2017001990A (ja) | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| WO2019189024A1 (ja) | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP7331700B2 (ja) | 1-クロロ-2,3,3,4,4,5,5-ヘプタフルオロペンテンの製造方法 | |
| JP2021120351A (ja) | フルオロオレフィンの製造方法 | |
| JP7287391B2 (ja) | 含フッ素プロペンの製造方法 | |
| WO2019168115A1 (ja) | 1,2-ジクロロ-2,3,3,3-テトラフルオロプロパンの製造方法及び1-クロロ-2,3,3,3-テトラフルオロプロペンの製造方法 | |
| JP7259764B2 (ja) | 5-クロロ-1,1,2,2,3,3,4,4-オクタフルオロペンタンの製造方法及び1-クロロ-2,3,3,4,4,5,5-ヘプタフルオロペンテンの製造方法 | |
| JP7647581B2 (ja) | 1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP7070220B2 (ja) | 2-クロロ-3,3-ジフルオロプロペンの製造方法、2-クロロ-1,1,2-トリフルオロプロパンの製造方法、2,3,3-トリフルオロプロペンの製造方法、1,2-ジクロロ-2,3,3-トリフルオロプロパンの製造方法、1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP2016079101A (ja) | 1,1−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| WO2022163745A1 (ja) | 3-クロロ-1,1,2,2-テトラフルオロプロパンの製造方法および1-クロロ-2,3,3-トリフルオロプロペンの製造方法 | |
| JP2013018722A (ja) | 1−クロロ−3,3,3−トリフルオロプロピンの製造方法 | |
| JP2015224236A (ja) | (e)−1−クロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| WO2014208452A1 (ja) | トリフルオロエチレンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16878738 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017558178 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016878738 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016878738 Country of ref document: EP Effective date: 20180725 |